

Abstract
Influenza viral infection causes acute upper respiratory diseases in humans, posing severe risks to global public health. However, current vaccines provide limited protection against mismatched circulating influenza A viruses. Here, the immune responses induced in mice by novel double-layered protein nanoparticles were investigated. The nanoparticles were composed of influenza nucleoprotein (NP) cores and hemagglutinin (HA) or matrix 2 protein ectodomain (M2e) shells. Vaccination with the nanoparticles significantly enhanced M2e-specific serum antibody titers and concomitant ADCC responses. Robust NP-specific T cell responses and robust HA neutralization were also detected. Moreover, vaccination with a trivalent nanoparticle combination containing two routinely circulated HA, conserved M2e, and NP reduced lung virus titers, pulmonary pathologies, and weight loss after homologous virus challenge. This combination also improved survival rates against heterologous and heterosubtypic influenza virus challenges. Our results demonstrate that the trivalent combination elicited potent and long-lasting immune responses conferring influenza viral cross-protection.
Keywords: Protein nanoparticles, Cross-protection, Influenza A viruses, Antibody-dependent cellular cytotoxicity, Long-lasting immune responses
Graphical Abstract

The double-layered protein nanoparticles combination composed of conserved NP as core and two routinely circulated HAs and conserved M2e as shell induces broad immune protection against divergent influenza A viruses.
Background
The influenza virus is a significant respiratory pathogen that can cause acute upper respiratory diseases in humans. Infection with the influenza virus is a major public health burden worldwide, with symptoms potentially leading to hospitalization or death. Over 300,000 people die annually due to seasonal influenza viruses worldwide [1, 2]. Influenza A viruses are a leading cause of seasonal epidemics and occasional global pandemics [3–6]. Current subtypes of influenza A viruses that routinely circulate in humans are H1N1 and H3N2 [7, 8]. In addition, human cases of fatal zoonotic infections with avian influenza viruses H5N1 and H7N9, and influenza B viruses are also severe public health threats and represent potential pandemics or seasonal epidemics [3, 9–13]. Our current best defense against influenza is the seasonal vaccine.
The seasonal influenza vaccines need to be reformulated annually based on surveillance and prediction. Vaccine effectiveness depends on the accuracy of the forecasts for circulating strains [14–19]. The seasonal influenza vaccines have no protection against a zoonotic strain of influenza crossing over into humans. Therefore, a universal vaccine is needed to induce broad cross-protection against divergent viruses to prevent influenza epidemics and potential pandemics.
The multi-antigen vaccine approach is a strategy to elicit broad cross-protection. As the main viral membrane protein, hemagglutinin (HA) is the primary protective antigen for current influenza vaccines because of its immunodominant induction of high neutralizing antibody titers [20, 21]. However, HA-induced immunity provides limited efficacies against heterologous or heterosubtypic influenza strains.
The M2 protein is a type III integral membrane protein that forms a pH-activated tetrameric proton-selective ion channel [22–25]. Compared to HA, the ectodomain of M2 (M2e) is relatively conserved across human influenza A viruses [26, 27]. M2e antibodies cannot prevent influenza virus infection but enable viral clearance by interacting with virus-infected cells and triggering immune effector cell activation through the Fc region [28]. Nevertheless, different M2e vaccine constructs have been demonstrated to elicit protective immunity to various extents, indicating that a well-optimized M2e can be a synergistic component of a future broadly protective or universal influenza vaccine [29–33]. Due to M2e’s small size and scarcity on the virion surface, viral infections or seasonal influenza vaccinations rarely result in M2e-specific antibody titers [34–38].
As a major internal protein, influenza nucleoprotein (NP) plays essential roles in RNA packing, nuclear trafficking, and RNA replication [39, 40]. Due to the high level of conservation across influenza A viruses (>90%) and cross-protective NP-specific CD8+ T cell responses, NP has been studied as a universal influenza vaccine immunogen [41–46].
HA, M2e and NP have different advantages as vaccine immunogens and induce complementary immune responses. Therefore, by taking advantage of the various protective effects, combining multiple viral antigens in vaccine formulations have been more effective in eliciting multifaceted immune responses and broad protection.
The use of nanoplatforms to display relevant antigens is a promising approach for developing new influenza vaccines. The high density and structurally ordered antigens in nanoparticles promote antigen recognition by B-cell receptors and elicit potent cellular and humoral immune responses [47, 48]. Nanoparticles have also been proven to protect antigens from proteolytic degradation, improve antigen delivery, and prolong antigen presentation by antigen-presenting cells (APCs) [49, 50].
In the current study, double-layered protein nanoparticles composed of influenza NP cores with HA or M2e shells (designated NP/HA or NP/M2e, core/shell) were generated to test as influenza vaccine candidates against divergent influenza A viruses. Our results indicated that vaccination with the trivalent nanoparticle combination of NP/HA1, NP/HA3, and NP/M2e resulted in robust NP-specific T cell responses, potent M2e-specific serum antibody responses, elevated HA neutralization titers, reduced lung virus titers, decreased pulmonary pathology, and increased survival rates in BALB/c mice during viral challenges. The immunity conferred broad cross-protection against a spectrum of influenza A strains and long-lasting immune responses without apparent side effects.
Materials and methods
Ethics statement
We faithfully adhered to the “Guide for the Care and Use of Laboratory Animals” by the National Institutes of Health. All mouse studies were approved by Georgia State University Institutional Animal Care and Use Committee (IACUC) under protocol number A19025.
Generation and characterization of recombinant proteins HA1, HA3, M2e, and NP
Trimeric HA (HA1 from A/Puerto Rico/8/1934 (PR8, H1N1) and HA3 from A/Aichi/2/1968 (Aichi, H3N2)) were stabilized by fusing the GCN4 trimerization sequence [51]. A gene encoding tetrameric M2e [32, 33] was constructed by fusing honeybee melittin, M2e repeats, the tetrabrachion tetramerization region (tetra-), and His-tag coding sequences in frame. The M2e repeats contain four tandem M2e sequences from human (hu), swine (swn), avian (avi), and domestic fowl (fwl) influenza viruses. Recombinant NP [32] was constructed by fusing NP and His-tag encoding sequences in frame. The recombinant protein constructs were shown in Supplementary Figure S1A. EMBL-EBI Clustal Omega [52] was used to analyze the phylogram of HA, M2, and NP —these sequence details are shown in Supplementary Table S1.
Sequence-confirmed recombinant protein-encoding genes were used to generate recombinant baculoviruses for protein purification from Spodoptera frugiperda (Sf9) cells (ATCC, CRL-1711) [32]. Purified proteins were characterized by SDS-PAGE and Western blots (probed with anti-His antibody, Cat. No. ab18184, Abcam). The approximate yield by one-step purification was 5 mg of purified protein per liter of culture.
Bis [sulfosuccinimidyl] (BS3) crosslinking
The polymeric states of trimeric HA1 and HA3 were confirmed by Bis [sulfosuccinimidyl] (BS3) (Thermo Scientific, Waltham, MA) crosslinking and subsequent SDS-PAGE/Western blotting analysis, as described previously (Supplementary Figure S2 A, B) [33].
Nanoparticles fabrication
Nanoparticles were made as previously described with modification [33]. For the generation of NP protein nanoparticles (cores), NP protein solution in DPBS (Thermo Scientific, Waltham, MA) was desolvated with ethanol. Absolute ethanol (400 μl) was dripped into 100 μl NP solution (0.5 μg/μl) at a constant rate of 1 ml/min under continuous stirring (magnetic stir bar at 600 rpm) at room temperature for 0.5 hrs. NP particle pellets were collected by centrifugation at 15,000 × g for 20 min at room temperature. The particle pellets were resuspended in shell protein (HA1, HA3, or M2e) solutions (100 μl, 0.5 μg/μl in DPBS) by sonication. Crosslinker 3,3’-dithiobis [sulfosuccinimidylpropionate] (DTSSP, Thermo Scientific, Waltham, MA) was used to stabilize the particles. Crosslinking reactions were performed in 5 mM DTSSP for 1 h while stirring at 4 °C and were quenched with 30 mM Tris-HCl solution at pH 7.4 for 15 min. The resulting double-layered protein nanoparticles were collected by centrifugation at 15,000 × g at 4 °C for 30 min. Nanoparticles were resuspended by sonication in 50 μl DPBS.
Nanoparticle Characterization
The double-layered nanoparticles’ size and zeta potential were measured by dynamic light scattering analysis with a Malvern Zetasizer Nano ZS (Malvern Instruments, Westborough, MA. The nanoparticles were imaged with a Jeol JEM-100CX II at 100 kV, and images were acquired with an Apogee Imaging Systems CCD camera system.
Protein nanoparticles were analyzed to confirm the antigen retention of the comprising proteins by ELISA. Briefly, ELISA plates (Corning Inc., NY, USA) were coated with the nanoparticles overnight at 4 °C in carbonate coating buffer (pH 9.5). After blocking with 1% BSA for 1 hour at 37 °C, different antibodies were added to the plates for 1 h at 37 °C. C179 (Cat No. M145, TaKaRa), F49 (Cat No. M146, TaKaRa), 14C2 (Cat No. MA1-082, Invitrogen), and C43 (Cat No. ab128193, Abcam) were used to recognize HA1, HA3, M2e, and NP, respectively. HRP-conjugated goat anti-mouse IgG (Cat No. G-21040, Invitrogen) was used for color development. The absorbance was read by Epoch Microplate Spectrophotometer (BioTek).
Immunization and Influenza A Virus Challenges
Female BALB/c mice of 6–8 weeks of age were grouped (n=5), intramuscularly (i.m.) immunized at day 0, and boosted on day 28 with 1 μg of double-layered protein nanoparticles (NP/HA1, NP/HA3, or NP/M2e), soluble protein mixtures (NP+HA1, NP+HA3, or NP+M2e), double-layered protein nanoparticle combinations (NP/HA1 with NP/M2e designated as N 1+M; NP/HA3 with NP/M2e designated as N 3+M) or trivalent nano (N 1+3+M) in 50 μl of PBS. PBS was given as a negative control, while 1 μg of formalin-inactivated PR8 or Aichi virus (FI-PR8 and FI-Aichi) in 50 μl of PBS were positive controls.
The mice were divided into four experimental cohorts (Figure 2A). For cohort 1, mice were challenged with 3 × LD50 of the influenza viruses PR8 or Aichi at day 42. The lung virus titer and histopathology were analyzed 5 days after the challenge (day 47).
Figure 2. Animal experiment schedule and humoral immune responses.
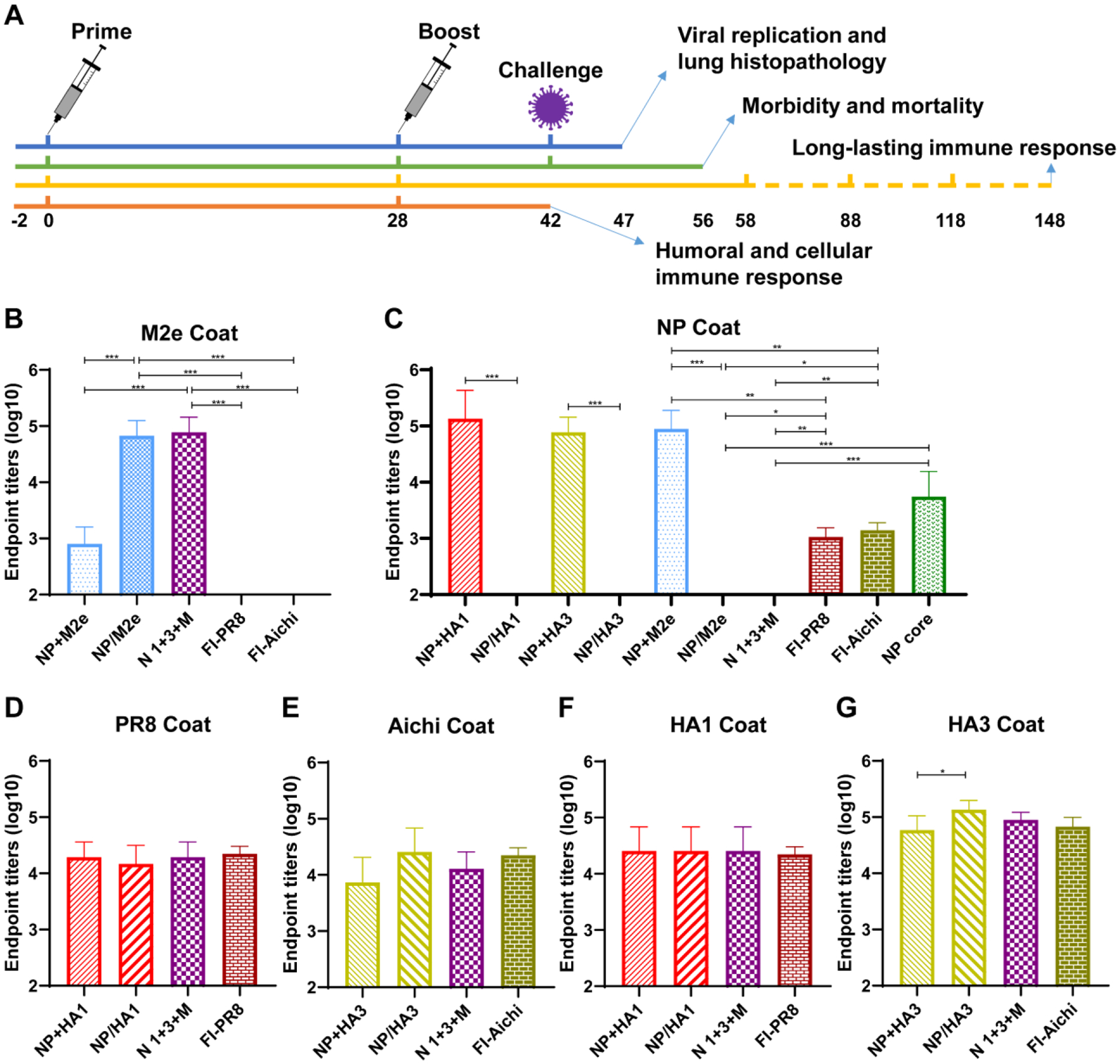
A Animal experiment schedule. Groups of mice were immunized via intramuscular (i.m.) route in thigh quadriceps with 1 μg of nanoparticles (NP/HA1, NP/HA3, NP/M2e, and NP core), soluble protein (NP+HA1, NP+HA3, and NP+M2e), and formalin-inactivated influenza viruses (FI-PR8 and FI-Aichi) in PBS and boosted at 4-week intervals. B-G Specific serum IgG antibody titers against M2e (B), NP (C), PR8 (D), Aichi (E), HA1 (F), and HA3 (G), two weeks post the boost-immunization. Data represent mean ± SD. The statistical significance was analyzed with one-way ANOVA followed by Tukey’s test (n = 5, * P < 0.05, ** P < 0.01, *** P < 0.001).
Cohort 2 was used to analyze morbidity and mortality. Two weeks after the boost immunization, immunized mice were challenged with 3 × LD50 influenza viruses: PR8, Aichi, A/California/07/2009 (California, H1N1), A/Philippines/2/1982 (Philippines, H3N2), A/Vietnam/1203/2004 (Vietnam, H5N1), and A/Shanghai/02/2013 (Shanghai, H7N9), respectively. The body weight changes and murine survival were recorded for 14 days.
Cohort 3 was used to evaluate long-lasting immune responses. After the immunization, blood samples from the mice were collected four times at 30 days intervals.
Cohort 4 was used for assessing humoral and cellular immune responses. On day 42, samples were collected, and the T-cell responses, serum antibody titers, HAI titers, and neutralization titers were analyzed.
Humoral and Cellular Immune Response Assays
Immune serum IgG specific to PR8 and Aichi viruses, HA1, HA3, M2e, and NP were assessed by ELISA [32]. Briefly, ELISA plates (Corning Inc., NY, USA) were coated with the purified viruses or proteins overnight at 4 °C in carbonate coating buffer (pH 9.5). Four-fold serial dilutions of murine sera were added to the antigen-coated plates for 1 h at 37 °C. After washing, HRP-conjugated anti-mouse Ab was added. Finally, HRP substrate TMB solution (Prod# 34029, Thermo Scientific, US) was added, and the absorbance at 450 nm was measured.
Hemagglutinin Inhibition (HAI) titers of murine sera were determined as previously described [51]. Samples were treated with receptor destroying enzyme (RDE II, Denka Seiken Co., Ltd) overnight at 37 °C and then heated at 56°C for 30 min before the test. The viruses of PR8 and Aichi were mixed with serial dilutions of mouse sera at room temperature for 1 hour. Turkey red blood cells (LAMPIRE Biological Laboratories, USA) were used to detect the HAI titer. The HAI was the highest dilution of serum capable of inhibiting virus hemagglutination.
The Antibody-dependent cellular cytotoxicity (ADCC) surrogate assay was performed according to the kit protocol (Promega) with modification. Briefly, monolayer cultures of HEK293T cells were transfected with plasmids that encode for M2e, HA1, or HA3 (Sino Biological. Inc.). After 48 hours of transfection, cells were harvested and seeded in sterile white 96-well culture plates (Costar) and incubated for 24 hours. Heat-inactivated serum samples were serially diluted in the assay buffer and mixed with transfected cells. The mFcγRIV effector cells were added and incubated for 6 h at 37 °C. Bio-Glo Luciferase assay substrate (Promega) was added. Luminescence was read out after 5 min on a GloMax (Promega). Data are expressed as fold induction using the following formula: fold induction = RLU (induced – background) / RLU (no antibody control – background).
Immune serum antibody neutralizing titers were assessed as described previously [32]. Serum samples were heat-inactivated at 56 °C for 30 min. Serial dilutions of the mouse sera mixed with 100 × TCID50 viruses and incubated at 37 °C for 2 hours. Then the mixtures were added to the MDCK cells and incubated at 37 °C, 5% CO2 for 72 h. Turkey red blood cells were used to measure neutralization, defined as the highest dilution capable of inhibiting virus hemagglutination.
IFN-γ ELISPOT assays were performed to detect the cellular immune responses as previously described [33]. Briefly, ELISPOT plates (BD Biosciences) were coated overnight with murine IFN-γ specific monoclonal antibodies. Splenocytes were added into plates and stimulated with NP protein. The spots were visualized by a biotinylated IFN-γ antibody. IFN-γ spot-forming cells were counted using a Bioreader-6000-E (Biosys, Germany).
Lung Viral Titration and Histological Analysis
Mice were sacrificed on day five post-challenge. Murine lung tissues were homogenized and centrifuged at 2000 rpm for 10 min at 4 °C. The supernatant was ten-fold serially diluted and added to MDCK cells (1.5 × 104 cells/well) cultured in 96-well plates for five days. A standard hemagglutination assay was carried out to determine viral titers in the supernatants.
For histological analysis, lung tissues from immunized mice were harvested on day five post-challenge and fixed in 10% formalin, embedded in dorsoventral position in paraffin. Subsequently, 10 μm thick sections were obtained and stained with hematoxylin and eosin (H&E). Using a semiquantitative scale (0 to 5) (0 = absent and 5 = maximum), sections stained for inflammatory infiltration around peribronchiolar, perivascular, and interstitial regions were evaluated.
Safety of nanoparticles in mice
The BALB/c mice were grouped (n=3) and intramuscularly (i.m.) immunized with 1 μg of double-layered protein nanoparticles (Nano), or soluble protein mixtures (Solu) in 50 μl of PBS. PBS was given as a negative control. The physical injury or edema of injection site were recorded daily for 3 days after injection, as well as body weight change.
The sera were collected before or 24, 48 hours after injection. Alanine Transaminase (ALT) Activity assay kit (ab105134, Abcam), Creatinine Assay kit (ab65340, Abcam), Mouse IL-6 Uncoated ELISA kit (88-7064-88, Invitrogen), and Mouse TNF-α Uncoated ELISA kit (88-7324-88, Invitrogen) were used to detect the change of ALT, Creatinine, IL-6, and TNF-α concentration respectively after immunization. The inner thigh muscle tissues of injected site of mice were harvested and fixed in 10% formalin. After trimming and paraffin embedding, 10 μm thick sections were stained with H&E and observed for inflammation.
Statistical analysis
All data plotted with error bars are expressed as means with standard derivation. The statistical significance was analyzed with one-way ANOVA followed by Tukey’s test. The body weight change and the survival curves were analyzed by the Student’s test and log-rank test, respectively. P values < 0.05 were regarded as statistically significant.
Results
Characterization of nanoparticles
Codon-optimized trimeric HA1, trimeric HA3, tetrameric M2e, and NP sequence compositions are shown in Figure S1A. Based on Gene bank information (Supplementary Table S1), NP and M2 are highly conserved across influenza A viruses compared with HA in the phylogram (Supplementary Fig. S1B, S1D). Purified proteins were confirmed by both their molecular weights and binding to specific polyclonal antibodies (Fig. 1B). After BS3 crosslinking, the trimeric states of HA1 and HA3 were shown by Western blots (Supplementary Fig. S2A, S2B).
Figure 1. Fabrication and characterization of double-layered protein nanoparticles.
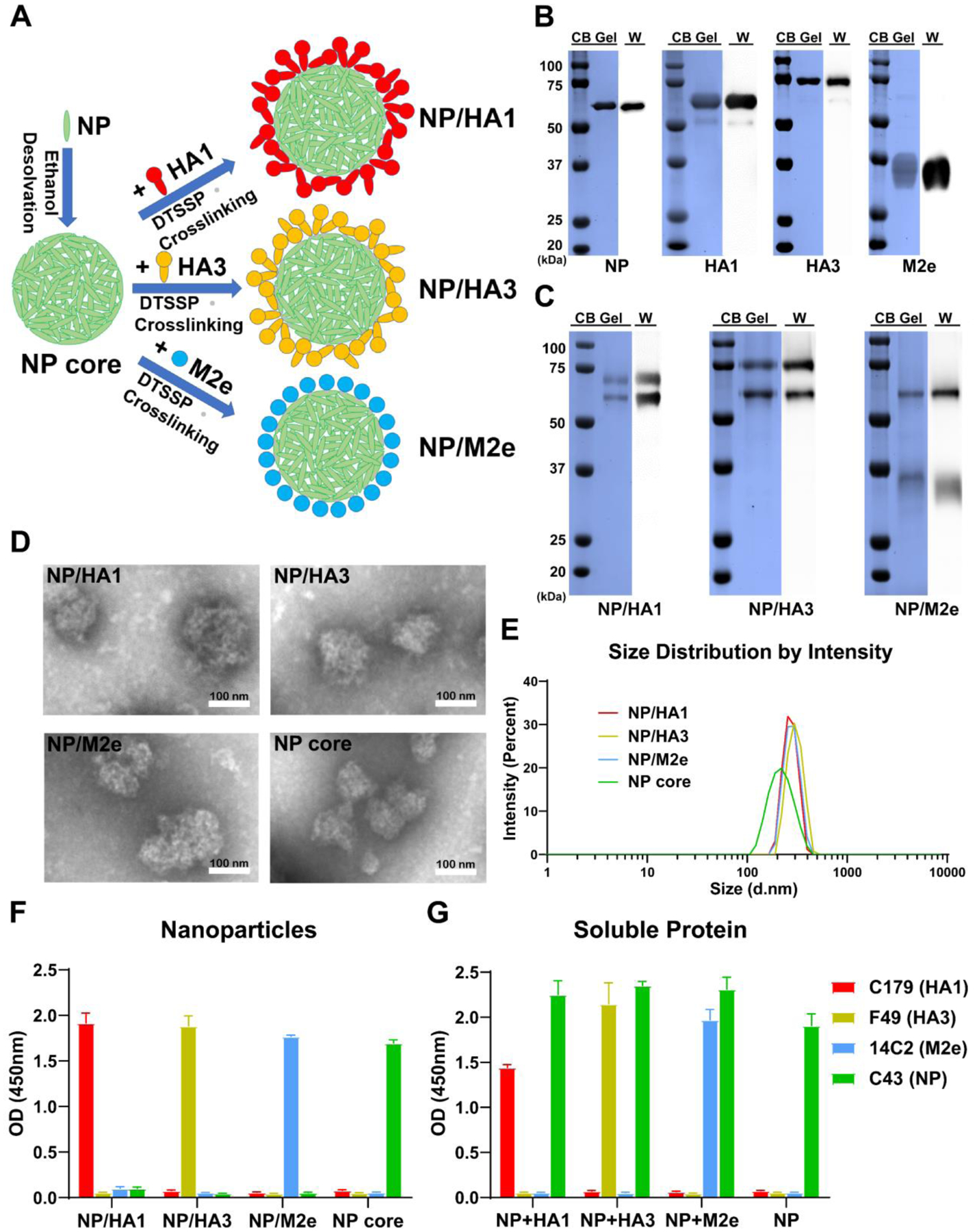
A Fabrication process of double-layered protein nanoparticles by respectively crosslinking shell proteins HA1, HA3, or M2e on desolvated NP cores. B, C Identification of purified recombinant proteins (B) and double-layered nanoparticles (C) with Coomassie blue staining (CB Gel) and Western blot (W). D TEM image of NP core, double-layered NP/HA1, NP/HA3, and NP/M2e nanoparticles. E Size distribution of NP/HA1, NP/HA3, NP/M2e, and NP core. F, G Identification of nanoparticles (F) and soluble proteins (G) by ELISA with monoclonal antibodies C179, F49, 14C2, and C43, which could specifically bind with HA1, HA3, M2e, and NP, respectively. Data are presented as mean ± SD.
The purified proteins were used to fabricate double-layered nanoparticles through ethanol desolvation and DTSSP crosslinking (Fig. 1A). Nanoparticle compositions were confirmed by SDS-PAGE, followed by Coomassie blue staining and Western blotting analysis (Fig. 1C). Nanoparticles were relatively spherical with irregular surface morphology (Fig. 1D). The diameters of nanoparticles were approximately 200–300 nm (Fig. 1E). NP/HA1 (272.2 nm), NP/HA3 (303.7 nm), and NP/M2e (275.7 nm) are larger than the NP particles (208.6 nm) due to the shell protein coatings (Fig. 1E). Meanwhile, the double-layered protein nanoparticles showed lower apparent zeta potential than NP nanoparticles (Fig. S2C).
To demonstrate the nanoparticles’ double-layered structures, ELISA plates were coated with NP/HA1, NP/HA3, NP/M2e, and NP core nanoparticles. Soluble proteins were used as positive controls (NP+HA1, NP+HA3, NP+M2e, and NP). The monoclonal antibodies C179, F49, 14C2, and C43, were used to detect HA1, HA3, M2e, and NP, respectively. As shown in Fig. 2F and 2G, C43 bound NP core nanoparticles but not double-layered NP/HA1, NP/HA3, or NP/M2e nanoparticles, demonstrating the NP nanoparticles’ full coverage by the shell proteins. Our results showed that C179, F49, and 14C2 could bind with the respective shell proteins on corresponding double-layered nanoparticles.
Humoral and cellular immune responses
The immunogenicity of the double-layered protein nanoparticles was determined by measuring antigen-specific humoral immune responses and comparing them with equivalent soluble protein mixtures and inactivated influenza vaccines (Fig. 2). After the boost immunization, nanoparticles significantly enhanced M2e-specific antibody titers of immune sera compared with the soluble protein or the inactivated influenza vaccine groups (Fig. 2B). M2e-specific antibodies were not detected from the inactivated vaccine groups. Furthermore, double-layered nanoparticle groups elicited lower NP-specific antibody levels than NP nanoparticles (without a shell layer) (Fig. 2C). Interestingly, the soluble protein mixture groups induced more robust NP-specific antibodies than the inactivated influenza vaccine groups and double-layered nanoparticle groups. Additionally, similar serum antibody titers against PR8 (Fig. 2D), Aichi (Fig. 2E), or HA1 (Fig. 2F) were observed in all groups. The NP/HA3 elicited significantly higher HA3-specific serum antibodies than the soluble mixture NP+HA3 (Fig. 2G).
Neutralizing antibodies are critical to controlling influenza virus replication. Our results demonstrated that inactivated PR8 induced higher HAI and neutralization titers against PR8 than other groups (Fig. 3A and 3C). The trivalent nano group produced more neutralizing antibodies than the soluble mixture NP+HA1 group. Meanwhile, the trivalent nano and inactivated Aichi elicited better Aichi-specific HAI and neutralization titers rather than the soluble mixture NP+HA3 group (Fig. 3B and 3D).
Figure 3. Humoral and cellular immune responses of vaccinated mice.
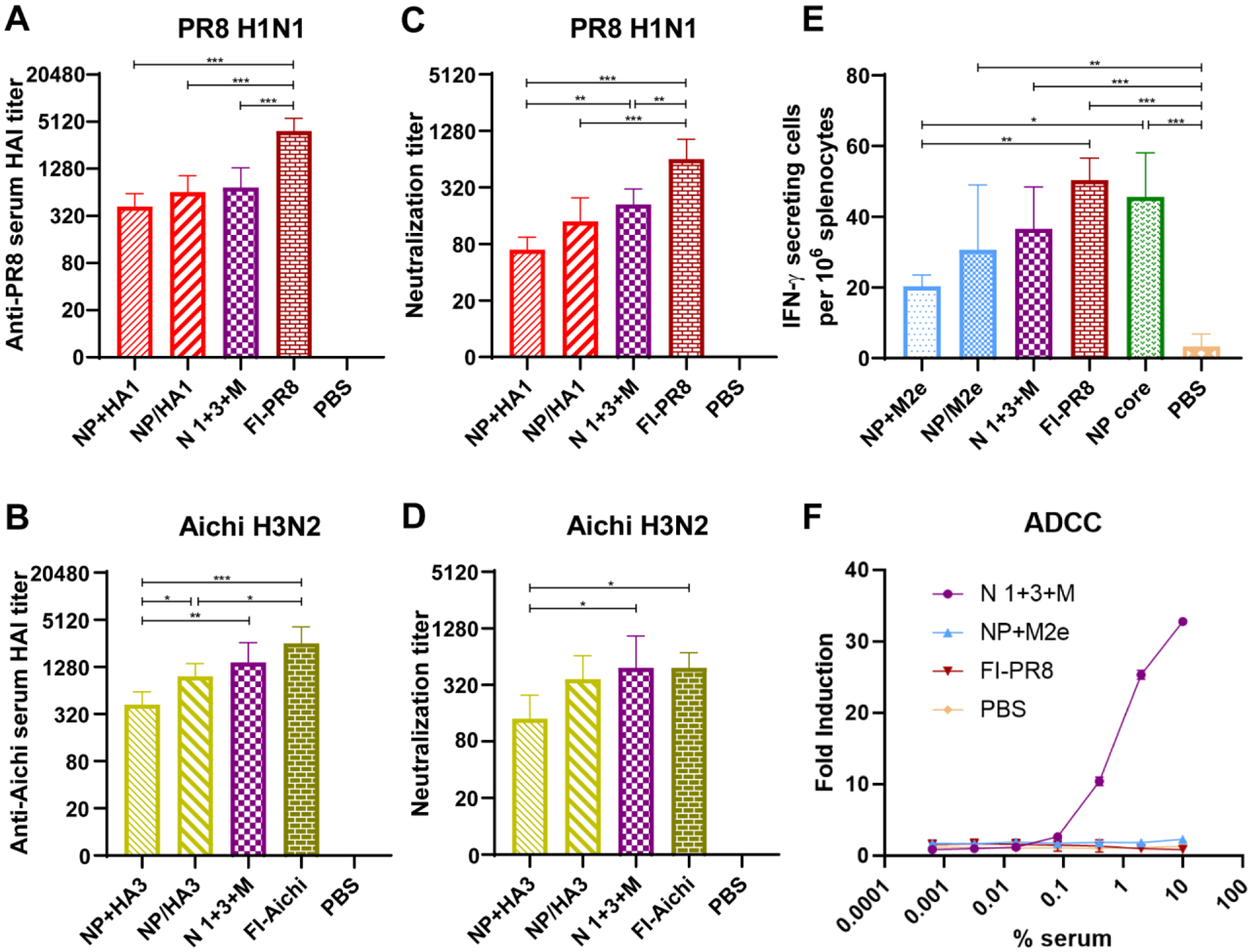
A-D Virus-specific HAI and neutralization titers against PR8 (A, C) and Aichi (B, D) 2 weeks post-boosting immunization (n=5). E NP-specific T-cell immune responses. NP stimulated interferon-gamma (IFN-γ) secreting cell clones 14 days after the last immunization (n=5). F ADCC surrogate assay results. M2 transiently transfected HEK293T cells were used as target cells. (n = 3). Data information: In (A–E), data are presented as mean ± SD. The statistical significance was analyzed with one-way ANOVA followed by Tukey’s test (* P < 0.05, ** P < 0.01, *** P < 0.001).
T-cell immune responses are important effectors in virus clearance. The population of IFN-γ-secreting splenocytes stimulated with NP protein were investigated by ELISPOT (Fig. 3E). The results showed that the double-layered nanoparticle groups, the NP nanoparticle group (without a shell layer), and the inactivated influenza vaccine (FI-PR8) group showed comparable IFN-γ-secreting cell frequency. The soluble protein mixture NP+M2e group demonstrated fewer IFN-γ-secreting cells than the inactivated PR8 and NP nanoparticle groups.
Antibody-dependent cellular cytotoxicity (ADCC) is an immune effect that contributes substantially to limiting and containing influenza infections. Immune sera from the trivalent nanoparticle group demonstrated the greatest ADCC activity against M2-expressing target cells (Fig. 3F). Meanwhile, HA-coated double-layered protein nanoparticles induced HA head domain-dominant neutralizing antibodies but not ADCC antibodies (Supplementary Fig. S3A, S3B).
Immune protection against virus challenge
Compared with the PBS group, the immunized mouse groups demonstrated various virus replication-inhibition capacities after PR8 and Aichi challenge infections (Fig. 4A and 4B). The double-layered nanoparticle groups and inactivated influenza vaccine groups showed significantly lower virus levels than the soluble protein mixture groups.
Figure 4. Lung viral titers and physiology after virus challenge.
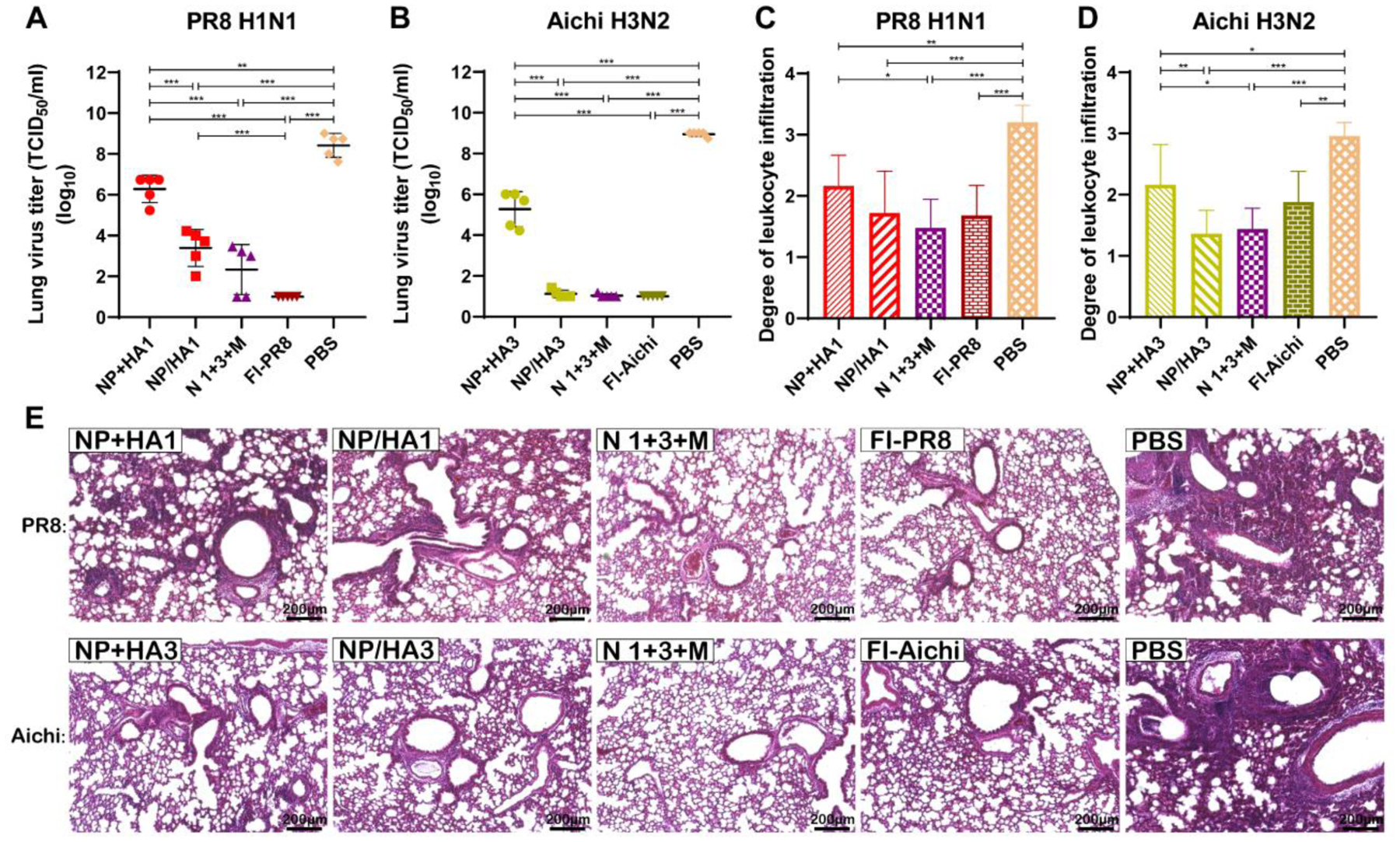
A, B Viral titers of lungs five days after the infection of PR8 H1N1 (A) or Aichi H3N2 (B). C-E Pulmonary histopathology examination by using H&E staining. H&E-stained lungs from mice infected with PR8 H1N1 (C) or Aichi H3N2 (D) were scored for inflammation with a semi-quantitative scale from 0 to 5 (0 = absent and 5 = maximum/severe) base on inflammation of the peribronchiolar region, the perivascular region, and the interstitial region. Data information: In (A–D), data represent mean ± SD. The statistical significance was analyzed with one-way ANOVA followed by Tukey’s test for comparison of groups (n = 5, *P < 0.05, **P < 0.01, ***P < 0.001).
As shown in Fig 4C–4E, compared with the PBS group, markedly lower leukocyte infiltration and tissue damage levels were found after viral challenges in nanoparticle-immunized mice. Notably, mice in the trivalent nanoparticle group showed lower leukocyte infiltration and tissue damage levels upon PR8 and Aichi challenges than mice vaccinated with soluble proteins.
Mouse morbidity and mortality were evaluated after homologous (PR8 and Aichi), heterologous (California and Philippines), or heterosubtypic (Vietnam and Shanghai) viral challenges in immunized mice. Upon PR8 and Aichi challenges, double-layered protein nanoparticle and inactivated vaccine groups showed completed protection (100% survival, Fig. 5B and 5D). Mice in the soluble protein mixture groups suffered ~10% body weight loss and had a 60% survival rate (Fig. 5A–5D).
Figure 5. Immune protection against homologous, heterologous, and heterosubtypic virus challenges.
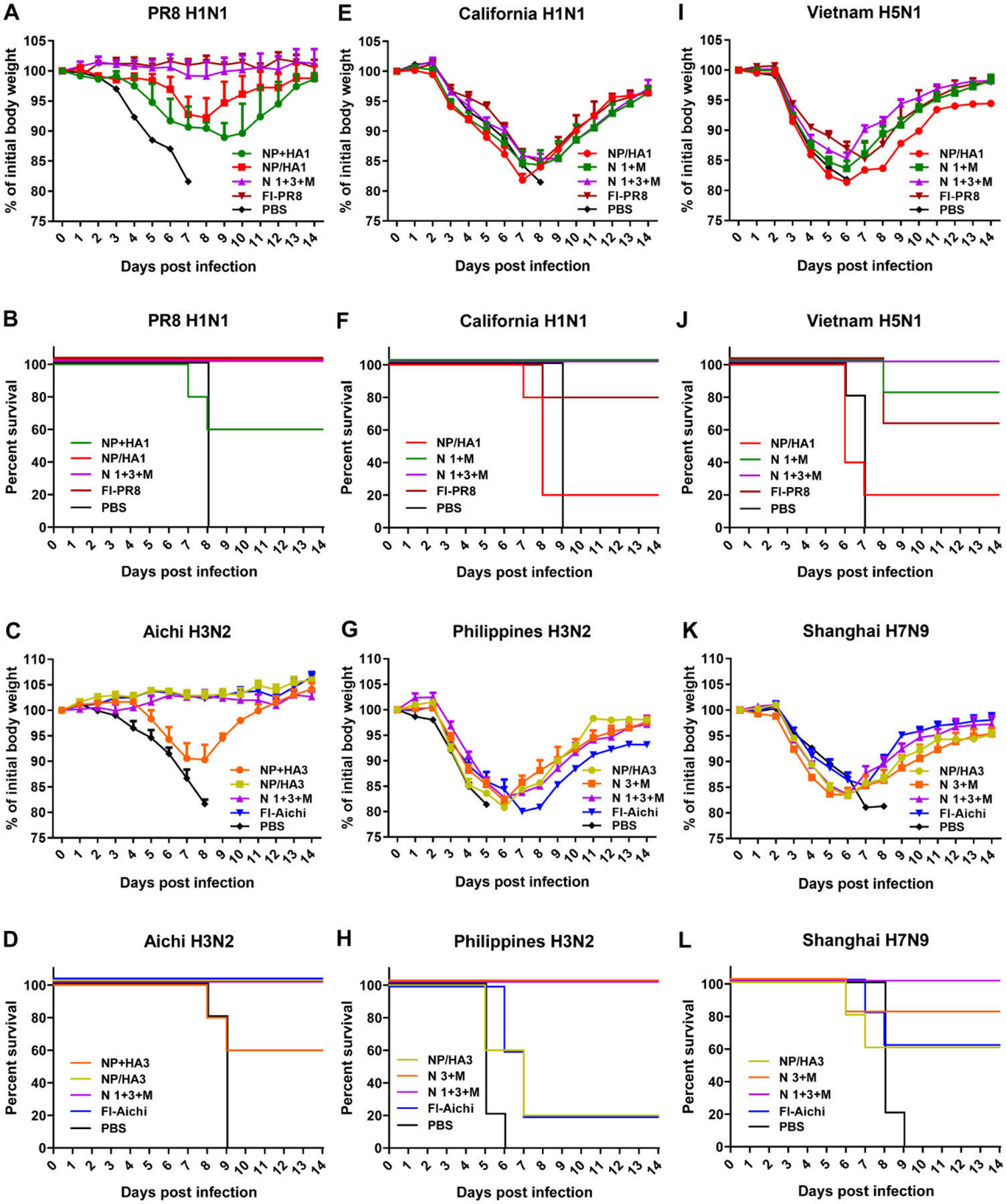
A-F Mean body weight changes and survivals upon lethal dose (3 × LD50) challenges (n = 5). Challenge viruses: PR8 (A and B), Aichi (C and D), California (E and F), Philippines (G and H), Vietnam (I and J), and Shanghai (K and L).
In comparison, severe body weight loss was observed after heterologous and heterosubtypic viral challenges (Fig. 5E, 5G, 5I, and 5K). Notably, nanoparticle combination groups NP/HA1+NP/M2e (N 1+M), NP/HA3+NP/M2e (N 3+M) or trivalent nanoparticle combination NP/HA1+NP/HA3+ NP/M2e (N 1+3+M) showed 100% survival rates in heterologous viral challenges (California and Philippines) in comparison with the partial survival rates in the single nanoparticle groups (NP/HA1 or NP/HA3; Fig. 5F and 5H). Inactivated vaccine groups showed 80% and 20% survival rates after California and Philippines virus challenges. Upon heterosubtypic viral challenges (reassortant Vietnam and Shanghai), mice in the trivalent group showed 100% survival rates in both challenges (Fig. 5J and 5L). It was observed that the trivalent nanoparticles induced efficient protection against homologous, heterologous, and heterosubtypic viral challenges.
Long-lasting immunity
A long-lasting immune response is an important index to evaluate the effectiveness of vaccines. An antibody titer follow-up study demonstrated that nanoparticle-induced antibodies specific to PR8, Aichi, and M2e were durable up to 4 months after the last immunization (Fig. 6A–6C). Interestingly, although the trivalent nanoparticle group showed decreases of PR8-specific serum antibody titers four months after vaccination (Fig. 6A), there was no significant HAI titer reduction (Fig. 6D and 6E), demonstrating the extended durability of the neutralizing antibody responses. The trivalent nanoparticle group maintained a high M2e antibody titer, although the titer and concomitant ADCC activity were reduced after four months.
Figure 6. Long-lasting immunity.
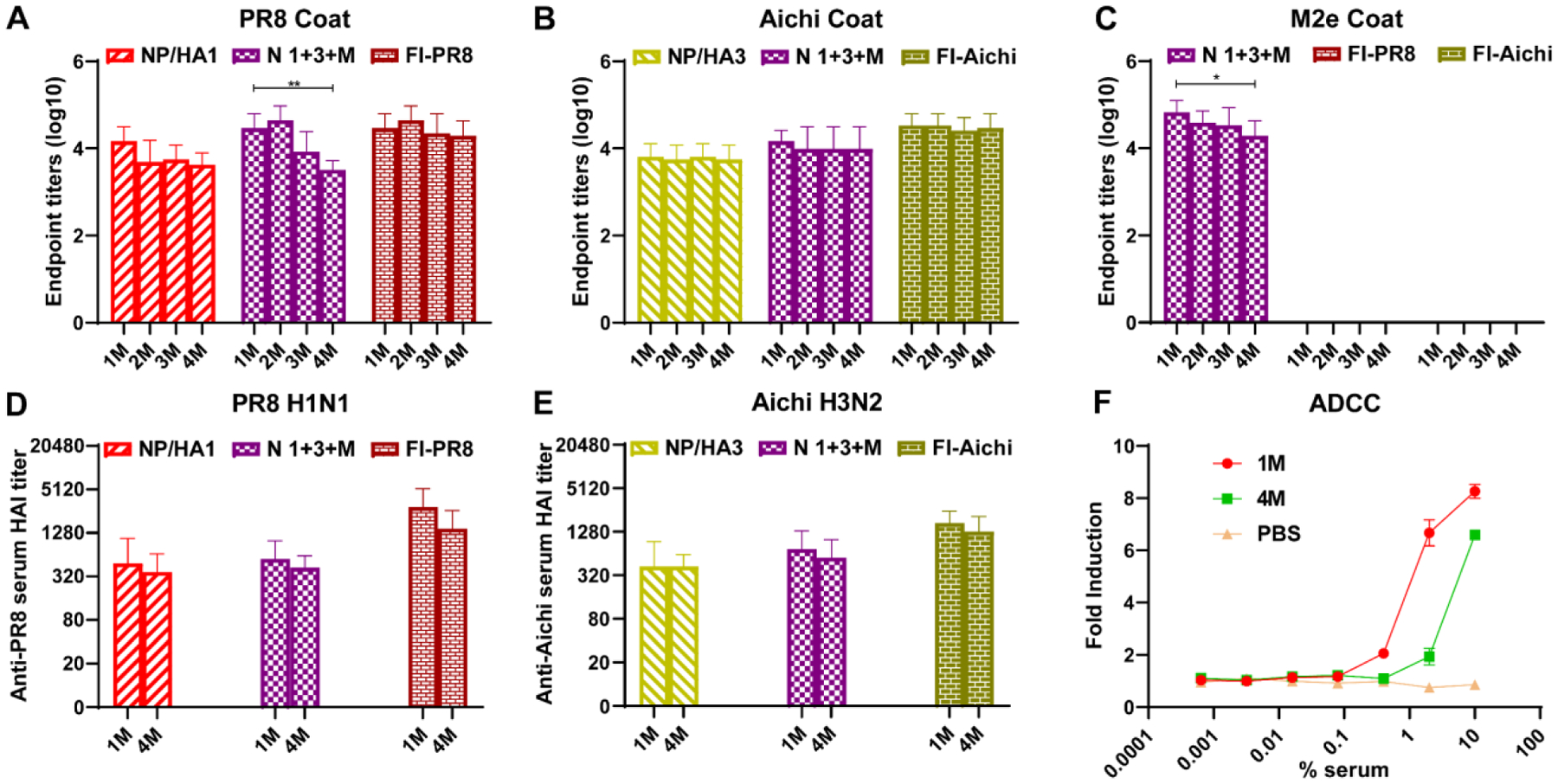
A-E Specific serum IgG antibody titers against PR8 (A), Aichi (B), and M2e (C) at 1, 2, 3, and 4 months (1, 2, 3, and 4M) after the boost (n=5). The 1M and 4M sera were used to measured virus-specific HAI titers against PR8 (D) and Aichi (E) (n=5). F ADCC surrogate assay results with five mice pooled pre-challenge sera from N 1+3+M 1M and 4M sera. M2 transiently transfected HEK293T cells were used as target cells, and PBS immunized mice sera were used as a negative control. (n = 3). Data information: In (A-F), Data are presented as mean ± SD. The statistical significance was analyzed with one-way ANOVA followed by Tukey’s test (A-E) and Student’s test (F) (* P < 0.05, ** P < 0.01, *** P < 0.001).
Nanoparticle safety in mice
The injection site and body weight were recorded to test the side effects of nanoparticle injection. No detrimental physical consequence or edema of administration was observed after immunization with nanoparticles (Fig. S4A). Meanwhile, there was no significant change in bodyweights of mice immunized with either Nano, Solu, or PBS (Supplementary Fig. S4B). ALT and creatinine were tested to evaluate the liver and kidney function after immunization. Similar levels of ALT and creatinine have been detected between pre-immunization and 24 or 48 hours after vaccination (Supplementary Fig. S4C, S4D). The major inflammatory cytokines IL-6 and TNF-α also maintained at a constant level in sera (Supplementary Fig. S4E, S4F). Meanwhile, no sign of inflammation was observed in the muscle tissue section from the site of injection (Supplementary Fig. S4G). Therefore, no apparent side effects were found after immunization with nanoparticles.
Discussion
Influenza is one of the most common seasonal respiratory infectious diseases, causing a significant health burden worldwide with symptoms potentially leading to hospitalization or death. Vaccination is the most effective way to reduce the incidence of influenza infections. However, the influenza virus is always rapidly evolving by antigenic drift and antigenic shift [53–56]. Current influenza vaccines must be reformulated each year based on surveillance and prediction. The effectiveness of these vaccines is highly variable and depends on the accuracy of the circulating strain forecasts [14–19]. Therefore, a universal vaccine is required against pandemics of genetically novel influenza viruses.
Several antigens in our double-layered protein nanoparticles have elicited broad protection against divergent influenza A viruses. As demonstrated previously, the NP in the nanoparticle formulations induced protective T cell responses, which are the critical effectors in viral clearance, rather than specific antibody responses [41–46]. Humoral and cellular immune responses suggest that the NP core preferentially produced cellular immune responses as a previous study [32]. Therefore, NP was used as the core of double-layered protein nanoparticles, which induced favorable T-cell immune responses and limited antibody responses. The HAs (HA1 and HA3) and M2e were used as shell proteins to elicit robust humoral immune responses. The trivalent nanoparticle combinations efficiently activated multiple facets of the immune system to protect vaccinees against matched and mismatched virus strains.
HAs from the two HA phylogenic groups in the double-layered protein nanoparticle formulations elicited higher levels of neutralizing antibodies than soluble HAs. As a potent alternative to current influenza vaccines, more HAs from circulating strains could be used as shell proteins to fabricate double-layered protein nanoparticles capable of targeting any strain of interest.
As M2 is a small protein with a few copies on the virion surface, only 33–44% of individuals have detectable M2e-specific serum IgG antibodies in humans. Viral infection or seasonal flu vaccination result in very weak M2e-specific antibody titers [34–38]. This phenomenon is consistent with our experimental results. The soluble protein mixtures and inactivated influenza vaccines produced low M2e-specific antibody titers. The immunogenicity of M2e was significantly enhanced by crosslinking it to the shell of NP nanoparticle cores and creating a high-density presentation of the antigen on the particles’ surfaces. Due to the nanoparticle immunogen depot effect, nanoparticle vaccination extended antigen release and presentation [47–50]. Double-layered NP/M2e nanoparticles also induced long-lasting, robust M2e-specific immune responses. So, the results of this study provide insights into the practical format of nanoparticle influenza vaccines.
Nanotechnology provides a promising approach for the development of new generations of influenza vaccines. The nanoparticles were composed almost entirely of antigens of interest in this study, avoiding possible off-target immune responses or pre-existing immunity to carrier materials. Our method of nanoparticle fabrication enabled high antigen loads, preservation of antigenic structures, and control of surface protein crosslinking density without the encapsulating agent. Additionally, this study provided insight into a novel format of nanoparticulate influenza vaccine, which modulates antigen release, stimulates antigen-presenting cells (APCs) maturation, exhibits adjuvant effects, and avoids immune tolerance upon binding and internalization of the antigens [49, 50].
In conclusion, double-layered protein nanoparticles were fabricated with desolvated NP as the core antigen and HAs and M2e as the shell antigens. The immune responses and protective efficacy were systematically investigated in mice. The nanoparticles significantly enhanced M2e-specific serum antibody levels that contributed to protection via ADCC. Meanwhile, the nanoparticles also elicited robust HA-specific neutralizing antibody responses and NP-specific T cell responses. The nanoparticle combination examined here displayed broad immune protection against divergent influenza A viruses and exemplify a better alternative to current seasonal influenza vaccines.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) under grants R01AI101047, R01AI116835, and R01AI143844 to B.-Z.W. The electron microscopy study was performed at the Georgia Institute of Technology for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI) supported by the National Science Foundation (Grant ECCS-1542174). The content in this study is solely the authors’ responsibility and does not necessarily represent the official views of the funders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All authors have declared no financial or other potential conflicts of interest.
References
- 1.Paules C, Subbarao K: Influenza. Lancet 2017, 390:697–708. [DOI] [PubMed] [Google Scholar]
- 2.Iuliano AD, Roguski KM, Chang HH, Muscatello DJ, Palekar R, Tempia S, Cohen C, Gran JM, Schanzer D, Cowling BJ, et al. : Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 2018, 391:1285–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palese P: Influenza: old and new threats. Nat Med 2004, 10:S82–87. [DOI] [PubMed] [Google Scholar]
- 4.McCullers JA: The Role of Punctuated Evolution in the Pathogenicity of Influenza Viruses. Microbiol Spectr 2016, 4. [DOI] [PubMed] [Google Scholar]
- 5.Broecker F, Liu STH, Sun W, Krammer F, Simon V, Palese P: Immunodominance of Antigenic Site B in the Hemagglutinin of the Current H3N2 Influenza Virus in Humans and Mice. J Virol 2018, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson AJ, Cao L, Ma Y, Wang X, Diedrich JK, Kikuchi C, Willis S, Worth C, McBride R, Yates JR 3rd, Paulson JC: Human Influenza Virus Hemagglutinins Contain Conserved Oligomannose N-Linked Glycans Allowing Potent Neutralization by Lectins. Cell Host Microbe 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, Gallagher JR, Wang L, Zhang Y, Joyce MG, et al. : Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med 2015, 21:1065–1070. [DOI] [PubMed] [Google Scholar]
- 8.Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, Garcia-Sastre A: Influenza. Nat Rev Dis Primers 2018, 4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan Y, Vijaykrishna D, Bahl J, Zhu H, Wang J, Smith GJ: The emergence of pandemic influenza viruses. Protein Cell 2010, 1:9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ran Z, Shen H, Lang Y, Kolb EA, Turan N, Zhu L, Ma J, Bawa B, Liu Q, Liu H, et al. : Domestic pigs are susceptible to infection with influenza B viruses. J Virol 2015, 89:4818–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subbarao K: Avian influenza H7N9 viruses: a rare second warning. Cell Res 2018, 28:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis ME, King ML, Kelvin AA: Back to the Future for Influenza Preimmunity-Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cardenas-Garcia S, Caceres CJ, Rajao D, Perez DR: Reverse genetics for influenza B viruses and recent advances in vaccine development. Curr Opin Virol 2020, 44:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Jong JC, Beyer WE, Palache AM, Rimmelzwaan GF, Osterhaus AD: Mismatch between the 1997/1998 influenza vaccine and the major epidemic A(H3N2) virus strain as the cause of an inadequate vaccine-induced antibody response to this strain in the elderly. J Med Virol 2000, 61:94–99. [PubMed] [Google Scholar]
- 15.Gerdil C: The annual production cycle for influenza vaccine. Vaccine 2003, 21:1776–1779. [DOI] [PubMed] [Google Scholar]
- 16.Taubenberger JK, Morens DM: 1918 Influenza: the mother of all pandemics. Emerg Infect Dis 2006, 12:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC: The genomic and epidemiological dynamics of human influenza A virus. Nature 2008, 453:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Memoli MJ, Jagger BW, Dugan VG, Qi L, Jackson JP, Taubenberger JK: Recent human influenza A/H3N2 virus evolution driven by novel selection factors in addition to antigenic drift. J Infect Dis 2009, 200:1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterholm MT, Kelley NS, Sommer A, Belongia EA: Efficacy and effectiveness of influenza vaccines: a systematic review and meta-analysis. Lancet Infect Dis 2012, 12:36–44. [DOI] [PubMed] [Google Scholar]
- 20.Krammer F, Palese P: Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 2015, 14:167–182. [DOI] [PubMed] [Google Scholar]
- 21.Krammer F, Fouchier RAM, Eichelberger MC, Webby RJ, Shaw-Saliba K, Wan H, Wilson PC, Compans RW, Skountzou I, Monto AS: NAction! How Can Neuraminidase-Based Immunity Contribute to Better Influenza Virus Vaccines? mBio 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heijne G: The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J 1986, 5:3021–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinto LH, Holsinger LJ, Lamb RA: Influenza virus M2 protein has ion channel activity. Cell 1992, 69:517–528. [DOI] [PubMed] [Google Scholar]
- 24.Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ: Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol 1996, 494 (Pt 2):329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mould JA, Drury JE, Frings SM, Kaupp UB, Pekosz A, Lamb RA, Pinto LH: Permeation and activation of the M2 ion channel of influenza A virus. J Biol Chem 2000, 275:31038–31050. [DOI] [PubMed] [Google Scholar]
- 26.Liu W, Zou P, Ding J, Lu Y, Chen YH: Sequence comparison between the extracellular domain of M2 protein human and avian influenza A virus provides new information for bivalent influenza vaccine design. Microbes Infect 2005, 7:171–177. [DOI] [PubMed] [Google Scholar]
- 27.Mezhenskaya D, Isakova-Sivak I, Rudenko L: M2e-based universal influenza vaccines: a historical overview and new approaches to development. J Biomed Sci 2019, 26:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X: Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J Immunol 2011, 186:1022–1031. [DOI] [PubMed] [Google Scholar]
- 29.Kim MC, Song JM, O E, Kwon YM, Lee YJ, Compans RW, Kang SM: Virus-like particles containing multiple M2 extracellular domains confer improved cross-protection against various subtypes of influenza virus. Mol Ther 2013, 21:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Hess A, Chang TZ, Wang YC, Champion JA, Compans RW, Wang BZ: Nanoclusters self-assembled from conformation-stabilized influenza M2e as broadly cross-protective influenza vaccines. Nanomedicine 2014, 10:473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L, Mohan T, Chang TZ, Gonzalez GX, Wang Y, Kwon YM, Kang SM, Compans RW, Champion JA, Wang BZ: Double-layered protein nanoparticles induce broad protection against divergent influenza A viruses. Nat Commun 2018, 9:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deng L, Chang TZ, Wang Y, Li S, Wang S, Matsuyama S, Yu G, Compans RW, Li JD, Prausnitz MR, et al. : Heterosubtypic influenza protection elicited by double-layered polypeptide nanoparticles in mice. Proc Natl Acad Sci U S A 2018, 115:E7758–E7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Deng L, Gonzalez GX, Luthra L, Dong C, Ma Y, Zou J, Kang SM, Wang BZ: Double-Layered M2e-NA Protein Nanoparticle Immunization Induces Broad Cross-Protection against Different Influenza Viruses in Mice. Adv Healthc Mater 2020, 9:e1901176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Black RA, Rota PA, Gorodkova N, Klenk HD, Kendal AP: Antibody response to the M2 protein of influenza A virus expressed in insect cells. J Gen Virol 1993, 74 (Pt 1):143–146. [DOI] [PubMed] [Google Scholar]
- 35.Feng J, Zhang M, Mozdzanowska K, Zharikova D, Hoff H, Wunner W, Couch RB, Gerhard W: Influenza A virus infection engenders a poor antibody response against the ectodomain of matrix protein 2. Virol J 2006, 3:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutchinson EC, Charles PD, Hester SS, Thomas B, Trudgian D, Martinez-Alonso M, Fodor E: Conserved and host-specific features of influenza virion architecture. Nat Commun 2014, 5:4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhong W, Reed C, Blair PJ, Katz JM, Hancock K, Influenza Serology Working G: Serum antibody response to matrix protein 2 following natural infection with 2009 pandemic influenza A(H1N1) virus in humans. J Infect Dis 2014, 209:986–994. [DOI] [PubMed] [Google Scholar]
- 38.Kolpe A, Schepens B, Fiers W, Saelens X: M2-based influenza vaccines: recent advances and clinical potential. Expert Rev Vaccines 2017, 16:123–136. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, Ozawa M, Kakugawa S, Shimojima M, Yamada S, Neumann G, Kawaoka Y: Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J Virol 2009, 83:4153–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisfeld AJ, Neumann G, Kawaoka Y: At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 2015, 13:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Townsend AR, Skehel JJ, Taylor PM, Palese P: Recognition of influenza A virus nucleoprotein by an H-2-restricted cytotoxic T-cell clone. Virology 1984, 133:456–459. [DOI] [PubMed] [Google Scholar]
- 42.Yewdell JW, Bennink JR, Smith GL, Moss B: Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc Natl Acad Sci U S A 1985, 82:1785–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shu LL, Bean WJ, Webster RG: Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J Virol 1993, 67:2723–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Portela A, Digard P: The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 2002, 83:723–734. [DOI] [PubMed] [Google Scholar]
- 45.LaMere MW, Lam HT, Moquin A, Haynes L, Lund FE, Randall TD, Kaminski DA: Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J Immunol 2011, 186:4331–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vemula SV, Sayedahmed EE, Sambhara S, Mittal SK: Vaccine approaches conferring cross-protection against influenza viruses. Expert Rev Vaccines 2017, 16:1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sahdev P, Ochyl LJ, Moon JJ: Biomaterials for nanoparticle vaccine delivery systems. Pharm Res 2014, 31:2563–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lopez-Sagaseta J, Malito E, Rappuoli R, Bottomley MJ: Self-assembling protein nanoparticles in the design of vaccines. Comput Struct Biotechnol J 2016, 14:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fredriksen BN, Grip J: PLGA/PLA micro- and nanoparticle formulations serve as antigen depots and induce elevated humoral responses after immunization of Atlantic salmon (Salmo salar L.). Vaccine 2012, 30:656–667. [DOI] [PubMed] [Google Scholar]
- 50.Pachioni-Vasconcelos Jde A, Lopes AM, Apolinario AC, Valenzuela-Oses JK, Costa JS, Nascimento Lde O, Pessoa A, Barbosa LR, Rangel-Yagui Cde O: Nanostructures for protein drug delivery. Biomater Sci 2016, 4:205–218. [DOI] [PubMed] [Google Scholar]
- 51.Weldon WC, Wang BZ, Martin MP, Koutsonanos DG, Skountzou I, Compans RW: Enhanced immunogenicity of stabilized trimeric soluble influenza hemagglutinin. PLoS One 2010, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, Lopez R: The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 2019, 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Both GW, Sleigh MJ, Cox NJ, Kendal AP: Antigenic drift in influenza virus H3 hemagglutinin from 1968 to 1980: multiple evolutionary pathways and sequential amino acid changes at key antigenic sites. J Virol 1983, 48:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferguson NM, Galvani AP, Bush RM: Ecological and immunological determinants of influenza evolution. Nature 2003, 422:428–433. [DOI] [PubMed] [Google Scholar]
- 55.Bouvier NM: The Future of Influenza Vaccines: A Historical and Clinical Perspective. Vaccines (Basel) 2018, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madsen A, Cox RJ: Prospects and Challenges in the Development of Universal Influenza Vaccines. Vaccines (Basel) 2020, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.