

Abstract
Bladder cancer is the ninth most frequently diagnosed cancer worldwide, and there is a need to develop new biomarkers for staging and prognosis of this disease. Here we report that cell lines derived from low-grade and high-grade bladder cancers exhibit major differences in expression of glycans in surface glycoproteins. We analyzed protein glycosylation in three low-grade bladder cancer cell lines RT4 (grade-1-2), 5637 (grade-2), and SW780 (grade-1), and three high-grade bladder cancer cell lines J82COT (grade-3), T24 (grade-3) and TCCSUP (grade-4), with primary bladder epithelial cells, A/T/N, serving as a normal bladder cell control. Using a variety of approaches including flow cytometry, immunofluorescence, glycomics and gene expression analysis, we observed that the low-grade bladder cancer cell lines RT4, 5637 and SW780 express high levels of the fucosylated Lewis-X antigen (Lex, CD15) (Galβ1–4(Fucα1–3)GlcNAcβ1-R), while normal bladder epithelial A/T/N cells lack Lex expression. T24 and TCCSUP cells also lack Lex, whereas J82COT cells express low levels of Lex. Glycomics analyses revealed other major differences in fucosylation and sialylation of N-glycans between these cell types. O-glycans are highly differentiated, as RT4 cells synthesize core 2-based O-glycans that are lacking in the T24 cells. These differences in glycan expression correlated with differences in RNA expression levels of their cognate glycosyltransferases, including α1–3/4-fucosyltransferase genes. These major differences in glycan structures and gene expression profiles between low- and high-grade bladder cancer cells suggest that glycans and glycosyltransferases are candidate biomarkers for grading bladder cancers.
Keywords: bladder cancer, fucosyltransferase, glycan marker, glycomics, Lewis-X
Introduction
The majority of membrane-bound and secreted proteins in mammalian cells are glycosylated, and the attached glycans facilitate critical cellular functions. As aberrant glycosylation is a hallmark of cancer (Munkley and Elliott 2016), cancer-specific glycan determinants may serve as diagnostic biomarkers as well as potential therapeutic targets (Varki 1993; Christiansen et al. 2014; Stowell et al. 2015; Cervoni et al. 2020). In addition, changes in glycosylation have been linked to functional aspects of carcinogenesis and metastasis (Stowell et al. 2015; Oliveira-Ferrer et al. 2017). Therefore, it is critical to investigate glycosylation changes associated with specific types of cancer. Unfortunately, among the many types of cancers in people, few of them have been analyzed in detail in terms of their glycomes, glycan determinants, and transcriptional expression of genes that facilitate glycan biosynthesis.
One such cancer that is not well studied in regard to glycan expression is bladder cancer, a disease causing ~165,000 deaths globally every year and which ranks ninth among all causes of deaths due to cancer (Colombel et al. 2008; Burger et al. 2013; Ferlay et al. 2015; Antoni et al. 2017). Notably, disease recurrence with metastasis is the major cause of bladder cancer related mortalities; prognosis is determined by grade and stage of disease at diagnosis (Grignon 2009). Presently, bladder cancer is broadly classified as low-grade, i.e., minimally invasive disease with moderate recurrence rate and lower metastatic potential; and high grade, i.e., bladder tumors with higher progression rates and greater metastatic potential (Grignon 2009). However, heterogeneity of grade within a tumor is common and leads to lower prognostic accuracy (Cheng et al. 2000). Therefore, more precise information about grade-specific malignant cells within a tumor is critical for accurate diagnosis and treatment of bladder cancer (Batista et al. 2020).
Prior studies using immunohistochemical approaches suggested that the glycan epitope Lewis-X (Lex; also designated as CD15 and stage-specific embryonic antigen-1 [SSEA-1]; Heimburg-Molinaro et al. 2011) is expressed by bladder cancer cells (Golijanin et al. 1995), and that Lex expression may correlate with tumor grade and stage (Shirahama et al. 1992; Konety et al. 1997). However, there have been only a few studies assessing the structures of glycans and glycan epitopes in bladder cancer (Ishimura et al. 2006; Guo et al. 2014; Yang et al. 2015; Zhou et al. 2020). Such studies have indicated that dysregulation of expression of the glycan-related genes and accompanying changes in cell surface glycan expression may affect bladder cancer progression. One such example is the study by Zhang et al. (2018), which provides evidence that microRNA-induced alterations in expression of fucosyltransferase FUT4 can suppress bladder cancer progression.
Thus, to gain more insights into the glycome of bladder cancer, we extensively analyzed glycan structures and expression of genes shaping glycan biosynthesis in bladder cancer cell lines that represent a variety of stages and grades of disease. We employed flow cytometry with specific anti-carbohydrate monoclonal antibodies (mAbs) and lectins, and glycomic analysis in order to elucidate the glycan structures expressed by these cell lines. We utilized quantitative RT-PCR to assess transcript levels of glycosyltransferase enzymes in these cell lines. Our results demonstrate that different grades of bladder cancer cells express different types of glycan structures and differentially express a number of glycosyltransferase genes including those encoding fucosyltransferases and sialyltransferases. These differences suggest the potential to develop novel glycan-based biomarkers for the identification of low-grade and high-grade bladder cancers and also as targets for the development of therapeutics.
Results
Anti-Lex mAbs HI98 and F8A1.1 bind uniformly to low-grade bladder cancer cell lines
To explore potential differences in glycan expression by bladder cancer cells of different disease stages and clinical grades, we utilized the following six bladder cancer cell lines, each of which were derived from a specific stage and grade (Table I); namely, low-grade bladder papilloma cell lines RT4 (stage-2, grade-1-2), 5637 (grade-2) and SW780 (grade-1), and high-grade cell lines J82COT (stage-3, grade-3), T24 (metastatic, undefined stage, grade-3) and TCCSUP (stage-4, grade-4). Primary human normal bladder epithelial cells A/T/N were analyzed as a control. To explore Lex expression on the cell surface, we performed a combination of flow cytometry and immunofluorescence studies using anti-Lex antibodies HI98 and F8A1.1 (Figure 1, Supplementary Figure S1).
Table I.
Cell lines used in this study
| Cell lines | ATCC Cat# | P. Stage/Grade | Use |
|---|---|---|---|
| A/T/N | PCS-420-010 | Normal bladder epithelia | Normal cell control |
| RT4 | HTB-2 | T2/G1 papilloma | Investigational-low grade |
| 5637 | HTB-9 | T?/G1 | Investigational-low grade |
| SW780 | CRL-2169 | T?/G2 | Investigational-low grade |
| T24 | HTB-4 | T?/G3 TCCBC | Investigational-high grade |
| J82COT | HTB-1 | T3/G3 TCCBC | Investigational-high grade |
| TCCSUP | HTB-5 | T4/G4 TCCBC | Investigational-high grade |
| HL60 | CCL-240 | Acute promyelocytic leukemia | Positive control |
TCCBC, transitional cell carcinoma bladder cancer.
Fig. 1.
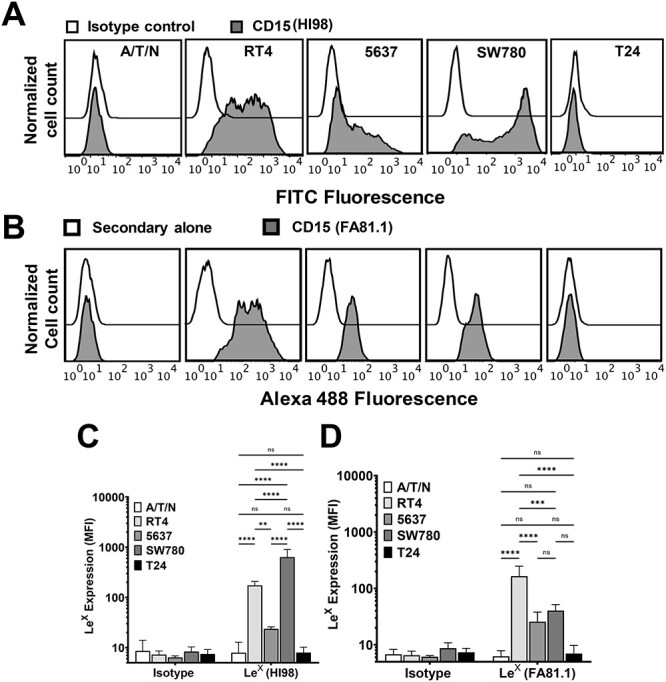
Analysis of bladder cancer cell lines for LeX expression. (A–D) Flow cytometry analysis of normal bladder epithelial cell line A/T/N, low-grade bladder cancer cell lines RT4, 5637 and SW780, and high-grade bladder cancer cell line T24 to assess expression of Lex. (A) Flow cytometry histograms showing isotype control staining (Open histograms) and staining with anti-CD15 mAb (HI98) (Gray histograms) for (left to right) A/T/N, RT4, 5637, SW780 and T24 cells. (B) Flow cytometry histograms showing secondary alone control staining (Open histograms) and staining with anti-CD15 mAb (FA81.1) (Gray histograms) for (left to right) A/T/N, RT4, 5637, SW780 and T24 cells. (C) Bar plot presenting aggregate data of mean fluorescence intensities (MFI) of mAb HI98 binding. N = 3–5, error bar indicates standard deviation. Statistics, two-way ANOVA followed by Tukey’s multiple comparison test comparing mean values for A/T/N, RT4, 5637, SW780 and T24. ns, not statistically significant, ****P < 0.0001, **P < 0.01. (D) Bar plot presenting aggregate data of MFI of mAb FA81.1 binding. N = 3, error bar indicates standard deviation. Statistics, two-way ANOVA followed by Tukey’s multiple comparison test comparing mean values for A/T/N, RT4, 5637, SW780 and T24. ns, not statistically significant, ****P < 0.0001, ***P < 0.001.
Using flow cytometry, we observed robust expression of Lex by the low grade cell lines RT4, 5637 and SW780, as assessed by both mAbs HI98 (Figure 1A and C) and F8A1.1 (Figure 1B and D), whereas, neither healthy bladder epithelial cell line A/T/N nor high-grade bladder cancer cell line T24 (Figure 1A–D) express Lex on the cell surface. We also performed immunofluorescence studies using mAb F8A1.1 to examine Lex expression pattern by bladder cancer cells (Supplementary Figure S1A). Similar to flow cytometry, immunofluorescence studies using mAb F8A1.1 also reveal that A/T/N cells and T24 cells lack Lex expression, whereas RT4 cells exhibit strong Lex expression. We tested Lex expression in two additional high-grade bladder cancer cell lines, J82COT and TCCSUP. We observed no binding of F8A1.1 to TCCSUP cells, but this mAb bound at low levels to J82COT cells. HL-60 cells, which are known to express Lex (Mondal et al. 2015), served as a positive control for measurements of mAb F8A1.1 binding specificity.
To confirm the pattern of Lex expression observed in the flow cytometry and the immunofluorescence studies, and to concomitantly visualize the Lex-bearing glycoproteins in the outer membranes of bladder cancer cells, we performed immunoprecipitation studies using mAb F8A1.1 (Supplementary Figure S1C). To examine only surface glycoproteins, we biotinylated the intact cell surface glycoproteins of J82COT, RT4 and T24 cells, prepared detergent soluble extracts from the cells, immunoprecipitated the material with mAb F8A1.1, separated the immunoprecipitated material by SDS-PAGE, transferred the bands onto PVDF membrane, and probed the immunoprecipitated, biotinylated glycoproteins from each cell line with streptavidin-peroxidase. We observed that RT4 cells expressed many Lex-bearing glycoproteins on the cell surface (sizes ranging from ~15 to ~250 kDa), whereas J82COT cells exhibit intermediate levels of Lex-expressing glycoproteins, and T24 lack such glycoproteins. Together, these data demonstrate that normal bladder epithelial cells lack expression of surface glycoproteins containing the Lex antigen, while low grade bladder cancer cells express high levels of Lex in a variety of surface glycoproteins, and high-grade bladder tumor cells lack expression of this epitope detectable with these mAbs. Together, these data demonstrate that normal bladder epithelial cells lack expression of Lex, low-grade bladder cancer cells express high levels of Lex on a variety of glycoproteins, and high-grade bladder tumor cells lack expression of this glycan determinant detectable by the aforementioned mAbs.
Low- and high-grade bladder cancer cells lack expression of sialyl Lex (sLex)
To evaluate whether the observed absence of Lex on the surface of high-grade bladder cancer cells might be due to capping of the galactose residue by sialic acid, which would prevent anti-Lex mAb binding, we evaluated expression of sialyl Lex (sLex; CD15s) (Neu5Acα2–3Galβ1–4(Fucα1–3)GlcNAcβ1-R). To this end, we employed flow cytometry (Figure 2A, Supplementary Figure S2) and immunofluorescence (Supplementary Figure S1B) to assess binding of rat-derived mAb HECA452, which recognizes sLex (Koszik et al. 1994) to the aforementioned cell lines. The results demonstrate that normal bladder epithelial cells (A/T/N), low-grade cancer lines (RT4, 5637 and SW780), and high-grade tumor cell line (T24) do not express detectable levels of sLex. Our immunofluorescence studies confirmed these observations and demonstrated that HECA452 does not bind to either RT4 or T24 cells, while the positive control cell line HL-60 exhibits high binding, as expected, to HECA452. Together, our results indicate that bladder cancer cells do not express sLex and therefore the lack of Lex epitope expression on the T24 high-grade bladder cancer cell line is not due to masking by sialylation.
Fig. 2.
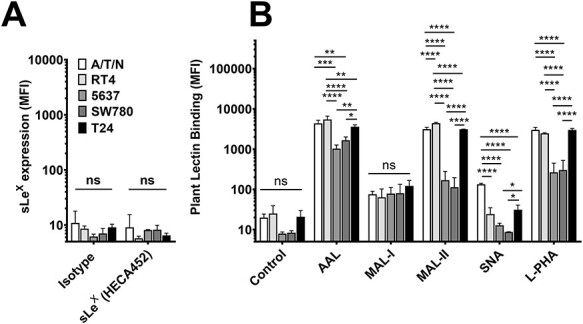
Analysis of bladder cancer cells to assess expression of sialo-fucosylated lactosaminyl glycans. (A, B) Flow cytometry analysis of normal bladder epithelial cell line A/T/N, and bladder cancer cell lines RT4, 5637, SW780 and T24 stained with antibody against (A) sialyl Lewis-X (sLex, clone HECA452), and (B) plant lectins AAL (binds fucose residues), MAL-I (binds α(2,3)-sialylated or unsialylated type 2 lactosamine residue), MAL-II (binds α(2,3)-linked sialic acid), SNA (binds α(2,6)-linked sialic acid), and L-PHA (binds β1–6-branched N-glycans). (A) Mean fluorescence intensities (MFI) for binding of either isotype control (rat IgM) or sLex reactive antibody HECA452. n = 3–5, error bars represent standard deviation. Statistics, two-way ANOVA for each measurement comparing means of individual cell lines. ns, not significant. (B) MFIs for binding of plant lectins AAL, MAL-I, MAL-II, SNA and L-PHA. Control measurements represent MFIs for binding of secondary detection reagent alone (streptavidin). n = 3–5, error bar indicates standard deviation, statistics for each lectin, ordinary one-way ANOVA followed by Tukey’s multiple comparison test comparing mean values for A/T/N, RT4, 5637, SW780 and T24. ns, not statistically significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Low-grade bladder cancer cell line exhibits characteristic hyper-fucosylation
We assessed the overall levels of fucosylated glycans on the bladder cancer cell lines by measuring binding of Aleuria aurantia lectin (AAL), which binds to α1–2, α1–3, α1–4 and α1–6-linked fucose residues (Matsumura et al. 2007). Binding of AAL to A/T/N, RT4, 5637, SW780 and T24 cell lines was assessed by flow cytometry (Figure 2B, Supplementary Figure S3). The results indicate that AAL binds extensively to all five cell lines. However, the RT4 cells displayed significantly higher levels of AAL binding compared to all other cell types, suggesting higher overall levels of fucosylation on RT4 cells. We also performed immunofluorescence experiments to assess binding of AAL to RT4, J82COT, T24 and TCCSUP cell lines, while A/T/N cells were used as normal bladder epithelial cell control (Supplementary Figure S4A). Similar to flow cytometry results, we observed that AAL binds to all tested cell lines, i.e., RT4, J82COT, T24, TCCSUP and A/T/N cells. AAL binding was inhibited by addition of free fucose, thus confirming fucose binding-specificity of AAL to these cells.
To assess the diversity of fucosylated glycoproteins, we used gel electrophoresis with identical amounts of cell lysate protein from each tested cell line and performed lectin blot analysis of A/T/N, RT4, J82COT, T24 and TCCSUP cells using AAL as a probe (Supplementary Figure S4C). Many glycoproteins in the lysates of each of the tested cell lines were bound by AAL; importantly, binding of AAL was inhibited by free fucose. Notably, we found that band patterns of the glycoproteins from low-grade and high-grade cancer cells were markedly different, indicating differential expression of glycoproteins and/or their glycosylation. Lysates of A/T/N and RT4 cells exhibited intense staining with AAL, whereas lower intensity staining was observed with J82COT, T24 and TCCSUP cells (Supplementary Figure S4C, left). While high molecular weight glycoprotein bands (>250 kDa) were visible in A/T/N and RT4 lines, these glycoprotein bands were absent in the high-grade J82COT, T24 and TCCSUP cell lines. A few glycoprotein bands between 37 and 150 kDa were visible in all cell lines. These findings indicate low-grade and high-grade bladder cancer cell lines have distinct but partly overlapping expression of fucosylated glycoproteins.
Treatment of cell lysates with PNGase F, an enzyme that removes most N-glycans from glycoproteins, prior to AAL blotting, led to partial loss of AAL reactivity within RT4 cells, and significant loss of AAL reactivity in the extracts of J82COT, T24, and TCCSUP cells (Supplementary Figure S4C, middle). These results demonstrate that N-glycans express only a minor portion of fucosylated glycans on low-grade bladder cancer line RT4, while the majority of the fucosylated glycans of the high-grade lines J82COT, T24 and TCCSUP are N-glycans. In a complementary experiment, wherein O-glycans from glycoproteins were released by β-elimination using NaOH treatment, we observed partial loss of AAL binding in all tested cell lines (Supplementary Figure S4C, right). Together, these results indicate that fucose residues are present on both N- and O-glycans in bladder cancer cells. Collectively, our results indicate that the bladder cancer cell lines express highly fucosylated glycans on the cell surface, while the Lex antigen is only expressed by the low-grade tumor cell line RT4.
SNA lectin differentially recognizes low-grade bladder cancer cells from normal bladder epithelial cells
We next assessed whether low-grade and high-grade bladder cancer lines show differential expression of α2,3-sialylated glycans, α2,6-sialylated glycans and β1–6-branched N-glycans. To this end, we evaluated binding of Maackia amurensis lectin-I (MAL-I), a lectin that binds to sialylated N-glycans (NeuAcα2–3Galβ1–4GlcNAc-R) and to certain branched non-sialylated N-glycans (Galβ1–4GlcNAc-R; Wang and Cummings 1988; Gao et al. 2019); M. amurensis lectin-II (MAL-II), a lectin that binds to α2–3-linked sialic acids on core 1 O-glycans (Geisler and Jarvis 2011); Sambucus nigra agglutinin (SNA), a lectin that binds to α2,6-linked sialic acid primarily on N-glycans (NeuAcα2–6Galβ1–4GlcNAc-R) (Goldstein and Poretz 1986; Mach et al. 1991); and Phaseolus vulgaris, l-phytohemagglutinin (L-PHA) that binds β1–6-branched N-glycan structures (Cummings and Kornfeld 1982) (Figure 2B, Supplementary Figure S3). We observed that MAL-I bound to A/T/N, RT4, 5637, SW780 and T24 cell lines equally and at moderate levels. MAL-II bound at high levels to A/T/N, RT4 and T24 cells but at lower levels to 5637 and SW780 cell lines. RT4 cell line exhibited the highest MAL-II binding. We observed a major variation among the cell lines in terms of SNA binding, which bound at moderate levels to the normal bladder epithelial cell line A/T/N. Interestingly, we did not detect SNA binding to the low-grade RT4, 5637, or SW780 cells, while T24 cells showed low levels of binding to SNA. L-PHA bound equally to A/T/N, RT4, and T24 cell lines, while 5637 and SW780 cells showed lower L-PHA binding. Our immunofluorescence studies (Supplementary Figure S4B) also indicate that T24 and TCCSUP bound L-PHA in a hapten-sensitive fashion, whereas low-grade RT4 cells exhibit lower binding to L-PHA. Interestingly, the high-grade bladder cancer cell line J82COT bound weakly to L-PHA.
Fig. 3.
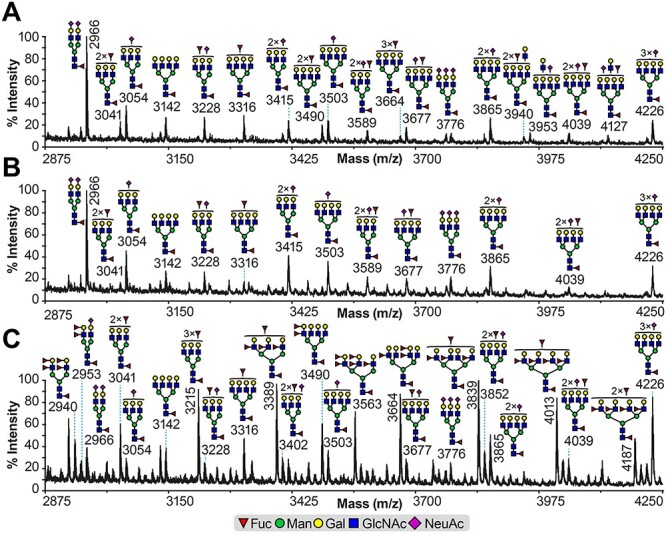
Partial MALDI-TOF MS spectra of permethylated N-glycans. N-glycans derived from (A) A/T/N, (B) T24 and (C) RT4 cell lines. Permethylated N-glycans were eluted at the 50% acetonitrile fraction (Materials and methods). Main structures are depicted. Structures above a bracket have not had their location unequivocally defined. Putative structures are based on composition, tandem MS and knowledge of biosynthetic pathways. All molecular ions are [M + Na]+. Full spectra can be found in Supplementary Figure S5A–C.
Collectively, these results indicate that all high-grade and low-grade bladder cancer lines as well as normal bladder epithelial cells display comparable levels of α2–3-sialylation based on the binding of MAL-I and MAL-II. However, low-grade (RT4, 5637 and SW780) bladder cancer cell lines are conspicuously deficient in expression of α2–6-linked sialic acid based on SNA binding, compared to both normal bladder epithelial cells and high-grade bladder cancer cells. Additionally, low binding of L-PHA to the low-grade bladder cancer cell lines RT4, 5637 and SW780 compared to the high-grade cell line T24, indicates that low-grade bladder cancer cells may be distinguishable from high-grade cell lines via expression of the β1–6-branched N-glycan structures that are recognized by L-PHA.
Glycomics analysis of N-glycans of bladder cancer cell lines
The above studies using anti-carbohydrate reagents are indirect, thus we complemented these analyses through a detailed structural analysis by mass spectrometry of N- and O-glycans of three of these bladder cancer cell lines, representing low-grade (RT4), high-grade (T24 cells) and control (A/T/N cells). Overall, glycoproteins in all cell lines express high mannose- and complex-type N-glycans, while no hybrid-type N-glycans were detected (Supplementary Figure S5). Comparison of the complex N-glycans from the A/T/N (Supplementary Figure S5A) and T24 (Supplementary Figure S5B) cell lines indicate that they consist of relatively similar structures. Middle-mass range (m/z 2875–4250) N-glycans have structures and compositions that correspond mainly to bi- (m/z 2966), tri- (m/z 3041, 3054, 3228, 3415, 3589, 3776) and tetra-antennary (m/z 3142, 3316, 3677, 3865, 4039 and 4226) structures, all core-fucosylated and non-bisected (Figure 3A and B). Their non-reducing capping groups include occasional NeuAc and/or fucose residues, which do not appear to be present on Lex- or Ley-type structures, as there was no detectable presence of the indicative fragment ion corresponding to the elimination of a fucose residue (when present as Fucα1–3) (Supplementary Figure S6A and B).
By contrast, structural analysis of RT4 cell N-glycans demonstrate the presence of a very different profile when compared to the A/T/N and T24 cells (Figure 3C; Supplementary Figure S5C). In the middle-mass range, the complex N-glycans with the highest relative abundance correspond mainly to structures with their antennas capped with multiple fucose residues and lacking NeuAc residues (Figure 3C; m/z 3215, 3389, 3490, 3563, 3664, 3839 and 4013). These structural features were mostly absent from the A/T/N and T24 cell lines (Figure 3A and B). By contrast, structures similar to the ones found on the A/T/N and T24 cell lines were also detected in the RT4 cell line, but the majority of these structures are of minor relative abundance (Figure 3C).
The RT4 cells express fucosylated N-glycans of high relative abundance that extend to the high-mass range spectrum (Figure 4A). Comprehensive analysis showed that they are mixtures of isomers, with the most abundant corresponding to mainly tri-antennary N-glycans extended with LacNAc repeats (-Galβ1–4GlcNAc-) modified with multiple fucose residues per antenna (m/z 5708, 5883, 6332, 6781 and 6955). Two examples are the structures detected for the molecular ions at m/z 5708 and 6332 (Figure 4B). When the molecular ion at m/z 5708 (Figure 4A) was subjected to Matrix-assisted laser desorption/ionization-Time of flight/Time of flight (MALDI-TOF/TOF) mass spectrometer (MS)/MS analysis (Figure 4B, left panel), fragment ions were detected corresponding to 3 LacNAc repeats attached with fucoses ranging from 2 to 4 residues (Figure 4B, left panel; m/z 3999, 3824 and 3650, respectively). Similarly, for the molecular ion at m/z 6332, fragment ions were detected corresponding to 3 LacNAc repeats attached with fucoses ranging from 2 to 4 residues (Figure 4B, right panel; m/z 4622, 4448 and 4274, respectively). The latter molecular ion also exhibited fragments ions of minor relative abundance that corresponded to 4 LacNAc repeats attached with either 3 or 4 fucose residues (Figure 4B, right panel; m/z 3999 and 3824, respectively).
Fig. 4.
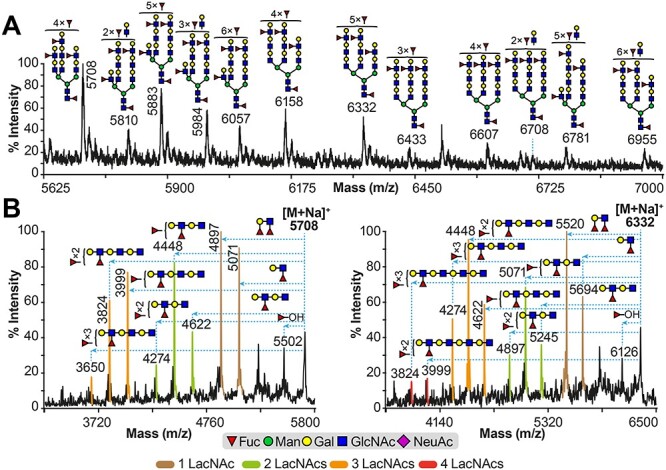
Glycomics analysis of high-mass RT4 cell line. (A) Partial MALDI-TOF MS spectrum of permethylated N-glycans derived from RT4 cell line; (B) partial MALDI-TOF/TOF MS/MS of molecular ions at m/z 5708 (left panel) and 6332 (right panel) found in (A). Permethylated N-glycans were eluted at the 50% acetonitrile fraction (Materials and methods). Main structures are depicted. Structures above/outside bracket have not had their location unequivocally defined. Putative structures are based on composition, tandem MS and knowledge of biosynthetic pathways. All molecular ions are [M + Na]+. In (B), horizontal blue dashed lines with arrowheads indicate losses of the corresponding structures from the molecular ions. Fragment ion peaks in brown, green, yellow and red colors correspond to losses of a single, two, three and four LacNAc repeats attached to various fucose residues from the molecular ion, respectively. Full spectra for MALDI-TOF MS can be found in Supplementary Figure S5C, while full spectra for MALDI-TOF/TOF MS/MS, and additional spectra, can be found in Supplementary Figure S7.
The above results indicate that the N-glycans of RT4 cells contain multiple fucose residues on various positions within each antenna, although the antennae vary in terms of LacNAc extensions. Also, it is important to note that in the aforementioned MALDI-TOF/TOF MS/MS spectra, we detected fragment ions with compositions corresponding to Lea and Lex epitopes (m/z 5071 and 5694 for left and right panels, respectively) and Leb and Ley epitopes (m/z 4897 and 5520). Moreover, fragment ions were also detected corresponding to an elimination of a fucose residue, indicating an α1–3 linkage (Figure 4B; m/z 5502 and 6126 for left and right panels respectively). From the above data, it is evident that at least a portion of the fucosylated epitopes corresponded to Lex and/or Ley epitopes. Therefore, the high-mass RT4 N-glycans contained antennae extended with mixtures of, at least partially, Lex and/or Ley epitopes. Similar results were obtained from other MALDI-TOF/TOF MS/MS spectra (Supplementary Figure S7A–F). Taken together, the results indicate that the A/T/N and T24 cell lines express N-glycans with NeuAc and fucose residues restricted to a single fucose per antenna, whereas RT4 express N-glycans lacking sialic acid and containing highly fucosylated antennae.
Glycomics analysis of O-glycans of bladder cancer cell lines
O-glycans were prepared from low-grade RT4 and high-grade T24 cell lines and their structures analyzed by MALDI-TOF mass spectrometry. Analysis of NaOH/NaBH4-released glycans revealed that the RT4 and T24 cells synthesize an assortment of common and distinct O-glycan structures (Figure 5). The common O-glycan structures include the core 1 O-glycan Galβ1–3GalNAcα1-Ser/Thr (T antigen), and mono- and di-sialylated core 1 O-glycan (Figure 5A and B). However, the RT4 cells synthesize many distinctive core 2-type O-glycans R-Galβ1–3(R-GlcNAcβ1–6)GalNAcα1-Ser/Thr, including mono-fucosylated and mono- or di-sialylated structures (Figure 5B). Interestingly, the di-sialylated core 2 structure at m/z 1705.9 appeared in both RT4 and T24 cell lines, but at much lower levels in T24.
Fig. 5.
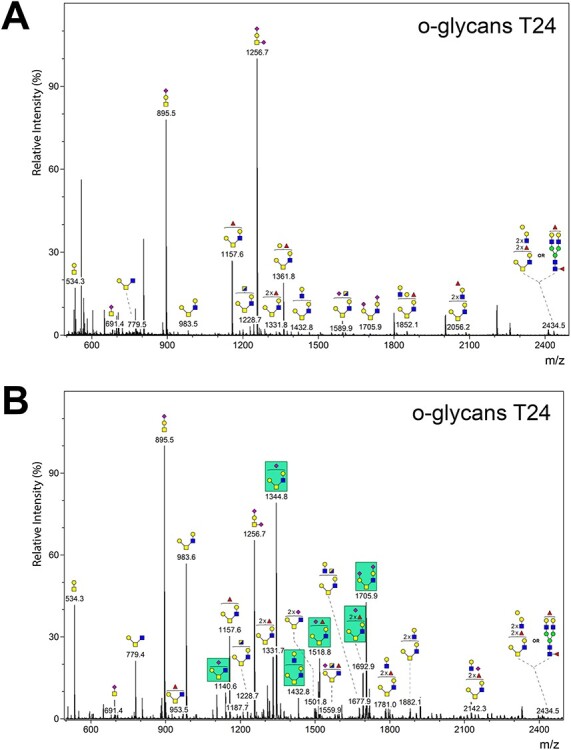
Analysis of O-glycans from T24 and RT4 cells. Bound glycopeptides recovered from Sep-Pak columns after recovery of released N-glycans were treated with base/borohydride to release O-glycans. Released O-glycans were permethylated and structures were determined by mass spectrometry. (A) Structures of O-glycans from T24 cells. (B) Structures of O-glycans from RT4 cells. Green boxes in panel B indicate O-glycans that show higher relative abundance in RT4 cells compared to T24 cells or are uniquely present in RT4 cells and not in T24 cells.
Specialized glycosyltransferase genes are differentially expressed between low-grade versus high-grade bladder cancer cell lines
In order to assess the molecular bases of cell surface glycan display, we used real time PCR (RT-qPCR) to evaluate expression of a number of glycosyltransferase genes (Figure 6) implicated by the structural analyses described above in the glycomics analysis section and encode enzymes that synthesize the terminal lactosaminyl glycan structures on N-linked or O-linked glycans (listed in Table II). We examined expression of these genes in the five cell lines A/T/N, RT4, 5637, SW780 and T24. In addition, we measured expression of fucosyltransferase enzymes in J82COT and TCCSUP cell lines (Supplementary Figure S8).
Fig. 6.
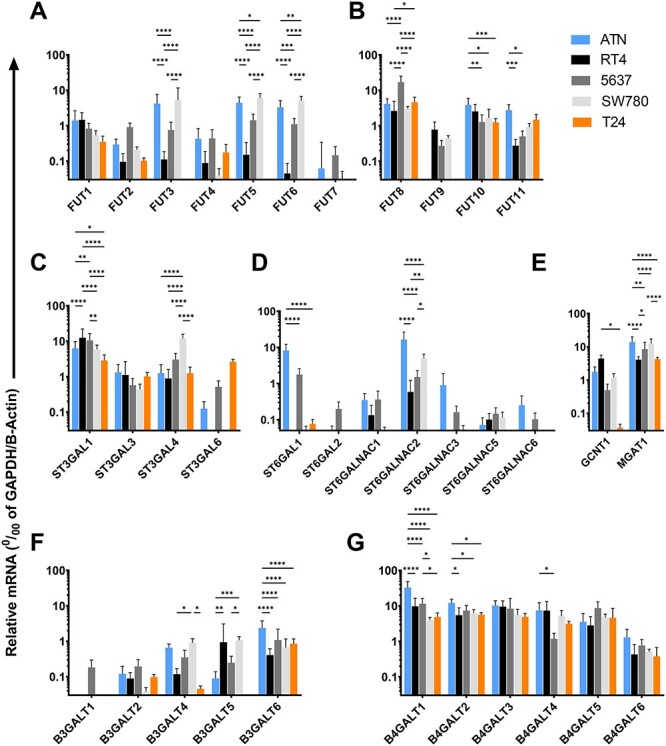
Quantitative RT-PCR analysis to assess expression levels of genes encoding glycosyltransferase enzymes. Relative mRNA levels of glycosyltransferase enzymes were measured compared to housekeeping control, i.e., geometric mean of the transcript levels of Glyceraldehyde 6phosphate dehydrogenase (GAPDH) and β-Actin. (A–G), Bar plots present relative mRNA levels of genes encoding human (A) fucosyltransferase enzymes FUTs 1–7, (B) FUTs 8–11, (C) human α(2,3)-sialyltransferases ST3GAL1, 3, 4, and 6, (D) α(2,6)-sialyltransferases ST6GAL1–2, and ST6GALNAC1–3, 5–6. (E) GlcNActransferases GCNT1 and MGAT1, (F) β(1,3)-galactosyltransferases β3GALT1, 2, 4, 5, 6 and (G) β(1,4)-galactosyltransferases β4GALT1–6, in A/T/N (blue bars), RT4 (black bars), 5637 (dark gray bars), SW780 (light gray bars) and T24 (orange bars) cell lines. N = 3 independent qRT-PCR experiments done on cells harvested at three different time points. Data are presented as mean ± SD. Statistics, Two-way ANOVA followed by Tukey’s multiple comparison tests comparing the five cell lines with each other. ****P < 0.0001, ***P < 0.001, **P < 0.01 and *P < 0.05, indicate statistical significance.
Table II.
Glycosyltransferase enzymes and their product specificities
| Enzyme | Monosaccharide-linkage | Product | References |
|---|---|---|---|
| FUT1–2 | Fucα1–2 | H antigen | Kelly et al. (1994) |
| FUT3 | Fucα1–3/4 | LeX, sLeX, Lea, sLea | Kukowska-Latallo et al. (1990) |
| FUT4 | Fucα1–3 | LeX | Goelz et al. (1990), Lowe et al. (1991) |
| FUT5 | Fucα1–3/4 | LeX, sLeX, Lea, sLea | Weston et al. (1992) |
| FUT6 | Fucα1–3 | LeX, sLeX | Mondal et al. (2018) |
| FUT7 | Fucα1–3 | sLeX | Mondal et al. (2018) |
| FUT8 | Fucα1–6 | α1–6 fucosylated chitobiose core | Yanagidani et al. (1997) |
| FUT9 | Fucα1–3 | LeX | Mondal et al. (2018) |
| FUT10 | Fucα1–3 | LeX | Mollicone et al. (2009), Kumar et al. (2013) |
| FUT11 | Fucα1–3 | LeX | Mollicone et al. (2009) |
| ST3GAL1 | NeuAcα2–3 | Sialyl T antigen | Kitagawa and Paulson (1994) |
| ST3GAL3 | NeuAcα2–3 | α2–3 Sialyl type 1/2 LacNAc | Mondal et al. (2015) |
| ST3GAL4 | NeuAcα2–3 | α2–3 Sialyl type 2 LacNAc | Mondal et al. (2015) |
| ST3GAL6 | NeuAcα2–3 | α2–3 Sialyl type 2 LacNAc | Mondal et al. (2015) |
| ST6GAL1 | NeuAcα2–6 | α2–6 Sialyl type 2 LacNAc | Grundmann et al. (1990) |
| ST6GALNAC1–6 | NeuAcα2–6 | Sialyl Tn antigen | Kurosawa et al. (1994), Sjoberg et al. (1996), Ikehara et al. (1999), Lee et al. (1999), Kono et al. (2000) |
| GCNT1 | GlcNAcβ1–6 | Core 2 O-glycan | Bierhuizen and Fukuda (1992) |
| MGAT1 | GlcNAcβ1–2 | GlcNAc addition to Manα1–3 branch of N-glycans | Kornfeld and Kornfeld (1985) |
| B3GALT1, 2, 4, 5,6 | Galβ1–4 | Type 1 LacNAc | Isshiki et al. (1999) |
| B4GALT1–6 | Galβ1–4 | Type 2 LacNAc | Amado et al. (1999) |
Glycan sequences:
H antigen, Fucα1–2Galβ1–3/4GlcNAc
LeX, Galβ1–4(Fucα1–3)GlcNAc
sLeX, NeuAcα2–3Galβ1–4(Fucα1–3)GlcNAc
Lea, Galβ1–3(Fucα1–4)GlcNAc
sLea, NeuAcα2–3Galβ1–3(Fucα1–4)GlcNAc
Type 1 LacNAc, Galβ1–3GlcNAc
Type 1 sialyl LacNAc, NeuAcα2–3Galβ1–3GlcNAc
Type 2 LacNAc, Galβ1–4GlcNAc
Type 2 sialyl LacNAc, NeuAcα2–3Galβ1–4GlcNAc
Sialyl T antigen, NeuAcα2–3Galβ1–3GalNAc-O-Ser/Thr
Sialyl Tn antigen, NeuAcα2–6GalNAc-O-Ser/Thr
Core 2 O-glycan, GlcNAcβ1–6(Galβ1–3)GalNAc-O-Ser/Thr
We first measured expression of human fucosyltransferase enzymes (FUTs) (Figure 6A and B, Supplementary Figure S8). We observed that the α1,2-FUTs FUT1 and FUT2 were expressed with only minor variations across cell lines. FUT2 expression also showed negligible variation across the five tested cell lines. Notably, FUTs 3, 5 and 6 were expressed at high levels in the A/T/N cells, while RT4 and T24 cells do not express any appreciable amounts of FUTs 3, 5 and 6 transcripts. However, the low-grade bladder cancer cell line 5637 displayed moderate level and SW780 cell line displayed high level expression of FUTs 3, 5 and 6. Low levels of FUT4 transcript were detected in all five cell types with negligible variation across cell lines. FUT7 transcript was not expressed by any of the tested cell lines. FUT8 was ubiquitously and highly expressed by all five tested cell lines.
Interestingly, FUT9 was only expressed by low-grade bladder cancer cell lines RT4, 5637 and SW780, the only cell lines that display high levels of Lex on cell surface. FUT10 was highly expressed by A/T/N, RT4, 5637 and SW780 cells, while T24 cells displayed lower levels of FUT10 transcript. FUT11 was highly expressed by A/T/N and T24 cells, while RT4, 5637 and SW780 cells displayed slightly lower levels of FUT11 gene product. J82COT cells expressed a low level of FUT1 and a high level of FUT2. This cell line expressed a moderate level of FUT4, while lacking expression of FUTs 3, 5, 6 and 7. J82COT cells also expressed moderate levels of FUT8, FUT10 and FUT11. Interestingly, this cell line expressed a low level of FUT9. The TCCSUP cell line expressed a high level of FUT1 and a modest level of FUT2. Among α1,3 FUTs, this cell line expressed a modest level of FUT4 and a high level of FUT11, while FUTs 5, 6, 7, 9 and 10 were not expressed at any appreciable amounts. FUT8 was expressed highly by this cell line.
We also examined expression of sialyltransferase genes in the five cells lines, A/T/N, RT4, 5637, SW780 and T24. Expression of genes for α2–3-sialylation (Figure 6C) is relatively conserved across the cell types. ST3GAL1 was very highly expressed by all five cell lines with minor differences across cell lines. RT4 cells expressed the highest levels of this gene. ST3GAL3 and ST3GAL4 were expressed by all five tested cell lines at similar levels. SW780 cells expressed significantly higher levels of ST3GAL4 compared to the other four cell lines. By contrast, ST3GAL6 expression was detected at negligible levels within A/T/N, RT4, 5637 and SW780 cells, while T24 cells displayed high degree of ST3GAL6 expression.
Strikingly, significant cell line-specific variations were observed in terms of expression of the α2,6-sialyltransferases (Figure 6D). ST6GAL1 was highly expressed by A/T/N cells, while ST6GAL1 expression was not detectable in RT4, SW780, or T24 cells. 5637 cells displayed moderate expression of ST6GAL1. ST6GAL2 was not detected in A/T/N, RT4, SW780, and T24 cell lines, while 5637 cells express very low level of ST6GAL2. ST6GALNAC1, ST6GALNAC2, and ST6GALNAC3 are the only ST6GALNAC genes expressed by cell lines tested. The normal bladder epithelial cells A/T/N expressed all three ST6GALNAC genes at moderate to high levels. While RT4 cells expressed low transcript levels of ST6GALNAC1 and ST6GALNAC2. ST6GALNAC3 was not expressed by RT4 cells. 5637 and SW780 cell lines showed low expression of ST6GALNAC1 and ST6GALNAC2, and ST6GALNAC3. Remarkably, T24 cells lacked expression of all three ST6GALNAC (1, 2 or 3) genes, indicating that these cells in general lack expression of the enzymes to synthesize α2,6-sialic acid linkages.
We also examined expression of GlcNAc-transferase genes GCNT1 and MGAT1 (Figure 6E). Interestingly, we did not detect expression of the GCNT1 transcript in the T24 cell line, while AT/N, RT4, 5637 and SW780 cell lines expressed significant amounts of this transcript. MGAT1 was highly expressed by all five tested cell lines. Highest levels of MGAT1 were expressed by A/T/N cells. 5637 and SW780 cells expressed MGAT1 at equal levels as that of A/T/N cells. RT4 and T24 cells displayed equal expression of this enzyme transcript at significantly lower levels compared to A/T/N, 5637 and SW780 cell lines.
The β1–4-galactosyltransferase (β4GALTs) transcripts were found in equivalent levels across all cell lines, indicating that the synthesis of β1–4-Gal linkages is conserved across the five tested cell types (Figure 6F). In general, these transcripts were expressed at high levels in these cells (~10% of housekeeping control). By contrast, the β1–3-galactosyltransferase (β3GALT) genes were expressed at low levels by the five tested cell lines (Figure 6G). β3GALT1 transcripts could not be detected in either A/T/N, RT4, 5637 or T24 cell lines. 5637 cells displayed low level expression of β3GALT1. β3GALT2 was expressed at very low levels by all five cell lines. β3GALT4 was expressed at modest levels by A/T/N and SW780 cell lines, while RT4, 5637, and T24 cell lines expressed very low levels of β3GALT4 transcript. β3GALT5 was expressed at very low levels by A/T/N and T24 cell lines, while RT4, 5637 and SW780 cell lines showed moderate expression of this enzyme transcript. β3GALT6 is the only β3GALT that was found to be expressed at moderate levels in all five cell lines, with A/T/N cells displaying significantly higher level of expression compared to RT4, 5637, SW780 or T24 cells.
Discussion
Our results demonstrate that enzymatic pathways that shape expression of the cell surface sialylated and/or fucosylated lactosaminyl glycan determinants are distinctly different between low-grade (RT4, 5637 and SW780) and high-grade (J82COT, T24 and TCCSUP) bladder cancer cell lines. Importantly, both low- and high-grade cancer lines diverge considerably from the normal bladder epithelial cell line A/T/N, highlighting the fact that malignant transformation is associated with marked alterations in the glycosylation machinery. Surface expression of Lex, the trisaccharide glycan determinant, was evident in the low-grade (RT4, 5637 and SW780 cells) and highly diminished or absent in the high-grade bladder cancer cells (J82COT, T24 and TCCSUP cells). These results are consistent across a variety of analytical approaches we employed, including flow cytometry, western blotting, immunoprecipitation, glycan structural analysis and gene expression analyses.
Several prior studies employing a variety of mAbs against the Lex antigen (Table III) in bladder cancer tissues, cells from bladder washings, and cells recovered from voided urine, report expression of Lex by bladder cancer cells, but not by normal bladder epithelial cells (Parham et al. 1990; Sheinfeld et al. 1990; Golijanin et al. 1995; Planz et al. 2001; Kajiwara et al. 2005). It should be noted, however, that mAbs for Lex used in various studies may have differential binding specificities, thus complicating the interpretation of results (Konety et al. 1997). Notable in this regard is that the Lex trisaccharide motif may reside at the termini of any of the three major classes of glycoconjugate backbones, i.e., either N-glycans and/or O-glycans of glycoproteins or glycosphingolipids (Mondal et al. 2018). In the context of a cell, glycoconjugate backbones that express Lex may differentially influence antibody binding. This observation is supported by our recent findings that the murine IgG mAb F8A1.1 binds to Lex epitopes on N-glycans, but not O-glycans, as assessed by analysis on glycan microarrays (Mandalasi et al. 2013). The limitations of such monoclonal antibody measurements illustrate the need for additional structural studies on the glycans under study, as we have performed here.
Table III.
Antibody clones used for staining of CD15 in bladder cancer
| Antibody clone | Grade/stage-specific expression | References |
|---|---|---|
| P-12 | Moderate correlation with tumor grade | Konety et al. (1997) |
| Anti-SSEA-1 | Moderate correlation with tumor grade | Konety et al. (1997) |
| Anti-SSEA-1 | Detection in higher grade tumors | Shirahama et al. (1992) |
| P-12 | Detection of low-grade tumors | Sheinfeld et al. (1990) |
| MC2 | No significant correlation | Kajiwara et al. (2005) |
SSEA-1, stage-specific embryonic antigen-1.
In general, the normal bladder epithelial, and the low- and high-grade bladder cancer cell lines, each lack expression of β1–3-galactosyltransferase enzymes, which catalyze synthesis of type 1-based lactosaminyl glycans Galβ1–3GlcNAcβ-R. By contrast, the β1–4-galactosyltransferase family isoenzymes (B4GALT1–6), that catalyze the formation of type 2-based lactosaminyl glycans Galβ1–4GlcNAcβ-R are expressed at similar levels by both high- and low-grade cell lines. Thus, none of the cell lines appear to be capable of synthesizing type 1 glycans and can only synthesize type 2-based glycans. Therefore, although cells may express FUT3 and FUT5, which can utilize both type 1 and type 2 chains (Kukowska-Latallo et al. 1990; Weston et al. 1992), the low expression of the β1–3 galactosyltransferases in the bladder cancer cells suggest that they primarily synthesize the Lex antigen, and not the Lea antigen Galβ1–3(Fucα1–4)GlcNAcβ-R.
We observed major differences among the bladder cancer cell lines in expression of the human fucosyltransferase family. While the normal bladder epithelial cells A/T/N express high levels of FUTs 3, 4, 5, 6, encoding enzymes that catalyze formation of Lewis structures (Mondal et al. 2018), transcript levels of these enzymes are very low in the cancer cells. Interestingly, FUT9, an enzyme that synthesizes Lex (Mondal et al. 2018), is highly expressed by the low-grade tumor cells RT4, 5637 and SW780; however, it is conspicuously absent in the normal bladder cells as well as the high-grade cancer (T24, TCCSUP) cell lines. Interestingly, the high-grade cell line J82COT, which expresses very low levels of LeX glycan also expresses modest levels of FUT9 transcript, indicating that LeX may have an impact on the aggressiveness of bladder cancer cells, but that remains to be studied. This finding correlates with our observation that Lex is detectable only on the surface of RT4, 5637 and SW780 cells. In addition, the RT4 cells are exceptional in expressing extremely highly fucosylated N-glycans of relatively intermediate to large sizes and lacking terminal α2–6-linked sialic acid. Importantly, FUT10, an enzyme that is associated with expression of Lex in the brain (Nishihara et al. 1999; Kumar et al. 2013), is highly expressed by normal bladder cells as well as the low- and the high-grade cancer cell lines. On the other hand, FUT11, an enzyme that catalyzes addition of α1–3-Fuc to the chitobiosyl core of N-glycans (Mollicone et al. 2009), is expressed by all tested cell lines, although the low-grade cell lines RT4 and 5637 express very low levels of FUT11 mRNA. Importantly, upregulation of cellular fucosylation and fucosyltransferase gene expression have been previously associated with increased metastatic potential of colorectal (Osuga et al. 2016), hepatocellular (Liu et al. 2001), prostate (Li et al. 2013) and breast cancers (Yan et al. 2015). Here, we report for the first time to our knowledge that hyperfucosylation and high expression of FUT9 may be associated with low-grade bladder cancer, but further studies with clinical specimens are needed to address this possibility.
The α2–3-sialylTs were found to be expressed at relatively similar levels between the low- and high-grade cancer cell lines, with the exception of ST3GAL6, which exhibits very low transcript levels within the RT4 and SW780 cell lines, modest transcript levels line A/T/N and 5637 cell lines, and very high transcript levels by the high-grade T24 line. Interestingly, both cancer cell lines were deficient in expression of α2–6-sialylTs. Importantly, ST6GAL1 transcript levels are very low in all of the cancer lines, compared with the normal bladder cells. These findings are consistent with our results demonstrating very little staining of the cancer cells with SNA lectin. This lack of α2–6-sialylation is unusual and perhaps significant for bladder cancer, as most normal cells and human cell lines express this common modification.
We observed that the O-glycans in all cell types were primarily core 1-type with or without core 2 branching by GCNT1, the enzyme that synthesizes the core 2 branching of O-glycans (Bierhuizen and Fukuda 1992). GCNT1 is expressed at very low levels in the high-grade bladder cancer cell line T24, while normal bladder epithelial cell A/T/N and low-grade cancer lines RT4, 5637 and SW780 express GCNT1 transcripts at higher and similar levels. Therefore, the high-grade line T24 is predicted to be deficient in core 2-derived O-glycans. This finding is consistent with our glycomics analyses that demonstrate reduced abundance of core 2 branching of O-glycans in T24 cell line compared to RT4 cells.
We found that T24 cells express extended core 1 O-glycans (R-Galβ1–3GalNAcα1-Ser/Thr), but do not express core 2 branched O-glycans. In addition, prior studies suggested that altered expression of O-glycan initiation enzyme GALNT1 in T24 cells leads to aberrantly glycosylated integrin α3β1, which is associated with bladder tumorigenesis (Li et al. 2014). While we did not explore the expression of truncated O-glycans such as Tn (GalNAcα1-Ser/Thr) and sialyl Tn (NeuAcα2–6GalNAcα1-Ser/Thr) antigens, it has been reported that those antigens can be induced in some bladder cancer cell lines, including T24 cells, by hypoxic conditions (Peixoto et al. 2016). It would be interesting in future studies to explore the consequences of hypoxia on overall glycan expression and glycogene expression.
Using western blot approaches and surface biotinylation and pull-down immunoprecipitation, we observed a wide variety of glycoproteins expressing the Lex antigen in low- (RT4) and high-grade (J82COT) bladder cancer cells, suggesting that the modifications by fucosyltransferases are not limited to a few glycoproteins. Prior studies on the N-glycans of integrin α3β1 in T24 bladder cancer cell line demonstrated the presence of many highly sialylated N-glycans (Pochec et al. 2006), although the sites of N-glycosylation were not identified. Our results indicate that this cell line does express sialylated N-glycans, but the linkages must be α2–3, as this cell line displays very low SNA binding, and the enzyme ST6GAL1, which is responsible for expression of α2–6 linkages, is not expressed by T24 cells. Notably, SNA binding and ST6GAL1 expression significantly distinguishes low-grade bladder cancer cell lines (RT4, 5637 and SW780 cells) from normal bladder epithelial cells (A/T/N). In the future it would be interesting to map out both the glycoproteins and the sites of glycosylation carrying the Lex antigen and other modifications, including branching and presence of sialic acid.
Collectively, our results provide new insights into the molecular bases of expression of sialo/fucosylated lactosaminyl glycan determinants and highlight key differences that may be utilized to distinguish between low-grade and high-grade bladder cancer cells. Our results demonstrate unique N- and O-glycan expression profiles at both the RNA and glycomic levels between bladder cancer cell lines. The findings may be useful for identifying and developing novel biomarkers in bladder cancer using urinary glycoproteins and immunohistochemical-based cell markers.
In this study, using bladder cancer cell lines derived from different disease stages and grades, we provide initial evidence that bladder cancer progression may be accompanied by distinct changes in the cellular glyco-genome and the cell surface glycome. These findings highlight the necessity for further investigation of the glycome in primary patient derived bladder cancer tissues. Conceptually, urinary glycoproteins should be easily accessible for analysis and can be easily utilized for diagnostics. Furthermore, our results underscore the need for orthogonal methods of analysis to better define and correlate protein glycosylation and gene expression in human tumor cells. It should also be noted that often changes in protein glycosylation may affect protein functions, and such changes as observed here may be related to the differential clinical outcomes for these tumor cell phenotypes.
Methods
Unless otherwise stated, all the chemical compounds used to prepare buffer solutions were obtained from either Thermo-Fisher Scientific (Rockford, IL), Acros Organics (Geel, Belgium) or Sigma-Aldrich (St. Louis, MO). Peroxidase-conjugated streptavidin, polyvinylidenefluoride (PVDF) membrane, neuraminidase and pre-stained molecular weight markers were obtained from Bio-Rad Laboratories (Richmond, CA). Precast polyacrylamide gels, Alexa-488-conjugated goat anti-mouse IgG, Alexa-488-conjugated streptavidin and Protein G-conjugated Dynabeads kit were purchased from Invitrogen (Carlsbad, CA). SuperSignal West Pico chemiluminescent substrate for horse radish peroxidase, bicinchoninic acid (BCA) protein assay kit, bovine serum albumin (BSA) and sulfo-N-hydroxysuccinimide-conjugated biotin (sulfo-NHS-biotin) were obtained from Thermo-Fisher Scientific (Rockford, IL). Fetal Bovine Serum (FBS), Hanks balanced salt solution (HBSS), Iscoves media, Eagle’s minimum essential media (EMEM) and l-glutamine were bought from Media Tech (Manassas, VA). Tissue culture plates and flasks were obtained from Corning Life Sciences (Lowell, MA). Protease inhibitor cocktail was purchased from Roche Applied Sciences (Indianapolis, IN). HECA452 monoclonal antibody was purchased from BD Bioscience (Franklin Lakes, NJ).
Preparation of IgG anti-Lex mAb F8A1.1 from hybridoma cells
Anti-Lex monoclonal antibody F8A1.1 (mouse IgG) was prepared from a hybridoma clone secreting the antibody using protocols described previously (Mandalasi et al. 2013). Briefly, frozen hybridoma stocks were thawed and grown in Iscoves media supplemented with 20% FBS, 4 mM l-glutamine in 24-well tissue culture plates at 37°C in 5% CO2 atmosphere to 80% confluence density and sequentially expanded into T-25, T-75 and T-150 flasks respectively and grown in Iscoves media as described above. Passage of the cells from one flask to another was conducted when the cell densities reached 80% confluency. The cells from T-150 flasks were weaned from dependency on FBS at 80% confluent density by splitting and diluting the contents of each T-150 cell culture flask into two T-150 cell culture flasks containing equal volume of hybridoma serum-free medium (SFM) to derive two cell cultures containing media with 10% FBS. The cells were grown to 80% confluent density and split once again into SFM as described above to yield cell cultures containing 5% FBS and grown to about 80% confluence. The splitting and dilution of the contents of each T-150 flask into SFM was carried out one last time to derive cell cultures containing 2.5% FBS and grown once again to 80% confluence density. Weaned hybridoma cells from 16 T-150 flasks were harvested by centrifugation at 1200 rpm for 5 min, suspended in 500 mL of SFM supplemented with 4 mM l-glutamine, transferred into roller bottles and grown in Wheaton roller incubator Model IO57606 (Millville, NJ) at 37°C in 5% CO2 atmosphere. The cell culture was periodically sampled and monitored for cell viability. The hybridoma cell culturing was stopped when cell viability declined to about 5%. The contents of the roller bottles were transferred to 50 mL Falcon tubes and centrifuged at 1200 rpm for 5 min at 4°C to remove cells. The supernatants were collected, pooled and centrifuged further at 14,000 rpm at 4°C to remove cell debris. The supernatant was collected and filtered through Whatman filter paper. Thimerisol and benzamidine were added to final concentrations of 0.02% and 1 mM respectively and the media was stored at 4°C or used for purification of mAb F8A1.1 as described below.
Purification of mAb F8A1.1 by affinity chromatography over MEP HyperCel column
The media recovered from the roller bottles was applied to a 5 mL column of MEP HyperCel (Pall Life Sciences, Ann Arbor, MI) and 5 mL fractions of the run through were collected. Unbound material was washed from the column with 10 mM HEPES/150 mM NaCl buffer, pH 7.2, and 5 mL fractions were collected. The column run-through and wash fractions were monitored for protein by absorbance measurements at 280 nm. The column washing was continued until no protein was detected in the wash fractions. Bound F8A1.1 mAb was batch-eluted from the column by applying 5 mL of 50 mM sodium acetate buffer, pH 4.0. A pipette bulb was used to apply pressure at the top of the column after application of each batch of acetate buffer to facilitate quick and efficient elution of bound mAb. The eluted fractions were neutralized immediately by mixing the fractions with 1 mL of 1.0 M HEPES/150 mM NaCl pH 7.4 buffer. The neutralized eluted fractions were monitored for protein by absorbance at 280 nm and fractions containing protein were pooled and dialyzed against 0.1 M HEPES/150 mM NaCl buffer pH 7.4. The protein content of the purified mAb F8A1.1 was determined by BCA protein assay as described previously (Mandalasi et al. 2013) using BSA as standard protein. The purity of the mAb preparation was determined by SDS-PAGE and Coomassie blue staining. The binding specificity of purified IgG F8A1.1 was determined by ELISA and Western blot respectively using Schistosoma mansoni soluble egg antigen (SEA) and LNFPIII-BSA as antigenic targets. LNnT-BSA and LDNF-BSA were used as negative controls. Bound mAb F8A1.1 were determined by incubation with horseradish peroxidase-conjugated goat anti-mouse IgG followed by incubation with ABTS/H2O2 substrate and absorbance measurements at 405 nm. The F8A1.1 preparation was filter-sterilized using a 0.22 μm filter and stored at 4°C until used.
Cell culture
Bladder cancer cell lines RT4, J82COT, T24 and TCCSUP, normal bladder epithelial cells, A/T/N, HL-60 cells, and the cell culture media were obtained from American Type Culture Collection (ATCC; Manassas, VA). J82COT, T24 and TCCSUP cell lines were grown in Eagles minimum essential medium (EMEM) while the RT4 cell line was maintained in McCoy 5A Medium (ATCC, Manassas, VA). The HL-60 cells were grown in Iscoves media. All the media were supplemented with 10% fetal bovine serum (FBS), 100 μg/mL penicillin–streptomycin and 4 mM l-glutamine respectively. The primary bladder epithelial cells, A/T/N, were grown in prostate epithelial cell basal medium supplemented with prostate epithelial cell growth factors using a growth factor kit and instructions provided by ATCC. 5637 (Cat#HTB-9™) and SW780 (Cat#CRL-2169™) were purchased from ATCC. 5637 was cultured in RPMI1640 medium (Cat#10–041-CV, Corning®) supplemented with 10% (v/v) fetal bovine serum and 200 units/mL penicillin–streptomycin. All the above cells were cultured at 37°C in 5% CO2. SW780 was cultured in Leibovitz’s L-15 (Cat#30-2008™, ATCC) supplemented with 10% (v/v) fetal bovine serum and 200 units/mL penicillin–streptomycin at 37°C without CO2. The cells were passed or harvested for experimental analysis or cryopreservation at about 85% confluent density. Adherent cells were detached for experimental analyses by incubating the cells at 37°C in 5% CO2 atmosphere for 5 min with Dulbecco’s phosphate buffered saline (PBS) containing 5 mM EDTA. Detached cells were transferred into Falcon tubes and washed 4× with cold PBS to remove tissue culture material. The cells were either used immediately for cytological staining, preparation of detergent extracts, preparation of biotinylated detergent extracts or stored at −20°C for use in preparation of released glycans for structural analyses.
Flow cytometry
Cells were harvested from tissue culture flasks by incubation with 5 mM EDTA for 15 min at 37°C. Harvested cells were then washed three times with staining buffer (Hank’s Buffered Salt Solution (HBSS) with 2% FBS) and resuspended at 1 × 106/mL cell concentration in staining buffer. Cell surface human Fc-receptors were blocked by incubating the cells with staining buffer containing 1 μg/mL TrueStain human Fc-Receptor blocking reagent (Biolegend, San Diego, CA) for 5 min at 4°C. Cells were then incubated with FITC-conjugated antibodies against sLex (HECA452) or Lex (CD15; HI98) (Biolegend) at 5 μg/mL concentration for 30 min at 4°C. For lectin staining, cells were incubated with 1 μg/mL biotinylated plant lectins for 30 min at 4°C. Binding of the following lectins were assessed. 1. Aleuria aurantia lectin (AAL; binds to α1,3 and α1,6-linked fucose residues), 2. M. amurensis lectin-I (MAL-I; binds to sialylated and unsialylated Galβ1,4-GlcNAc), 3. M. amurensis lectin-II (MAL-II; binds to α2,3-linked sialic acid) and 4. S. nigra agglutinin (SNA; binds to α2,6-linked sialic acid) (Vector Labs, Burlingame, CA). Lectin binding to cells was detected by flow cytometry after incubation with 1 μg/mL Alexa-488-conjugated streptavidin in staining buffer for 30 min at 4°C. After 30 min cells were washed twice and analyzed using BD FACSCalibur (BD Biosciences, San Jose, CA) flow cytometry analyzer.
Quantitative RT-qPCR
Total cellular RNA was purified from cell lines cultured as described in the cell culture section using RNeasy microkit (QIAGEN, Hilden, Germany) as per the manufacturer’s instructions. The RNA thus isolated, was reverse transcribed using SuperScript IV VILO cDNA conversion kit (Invitrogen, Carlsbad, CA), Quantitative RT-qPCR was performed with specific primers to amplify target genes (listed in Supplementary Table SI) using SYBR Select master mix (Applied Biosystems, Foster City, CA) and ViiA7 PCR detection system (Applied Biosystems, Waltham, MA) according to the following conditions: 95°C for 2 min and 40 cycles of 95°C for 1 min and 60°C for 30 s. Post-amplification melt curve analysis was performed to ensure primer-binding specificity according to the following conditions: 95°C for 15 s, 60°C for 1 min, 3°C increment every 15 s to reach 95°C. Expression of glycosyltransferase genes in each cell type were calculated relative to geometric mean of GAPDH and β-Actin levels (Vandesompele et al. 2002) according to the following formula: Relative mRNA level (0/00) = 2-ΔCt X 1000, where Ct is the threshold cycle of amplification for each gene, and ΔCt = Cttarget gene – (Geometric mean of CtGAPDH and Ctβ-Actin).
Cell extracts and western blot
Cell extracts from bladder cell lines were prepared in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.4) containing protease inhibitors (Cat#11836170001, Sigma) followed by sonication. Proteins (~30 μg) were separated on SDS-PAGE gel (Cat#M42012, GenScript), and stained with Colloidal Coomassie Brilliant Blue G-250 (Cat#161-0803, Bio-Rad), or transferred to a PVDF membrane (Cat#IPVH00010, EMD Millipore). After blocking with 5% (w/v) BSA (Cat#BP1600-1, Fraction V, Fisher BioReagents™) in Tris-buffer saline with 0.05% Tween 20 (TBST) for 1 h at room temperature, western blots were analyzed with anti-Lex antibody (FA81.1 in house (Mandalasi et al. 2013), diluted to 10 μg/mL), or anti-β-actin (C4, Cat#sc-47778, Santa Cruz, diluted at 1:1000) as a primary staining, and horseradish peroxidase (HRP)-labeled goat anti-mouse IgG (Cat#115-035-062, Jackson ImmunoResearch Laboratories, Inc.) at 1:5000 dilution in TBST, using SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) on an Amersham™ Imager 600 (GE Healthcare Life Sciences), or onto the autoradiography films (Cat#1141 J52, HyBlot CL®, Thomas Scientific).
Biotinylation of cells
RT4, J82COT, T24 bladder cancer cells were harvested at approximately 85% confluent density and washed 4× with cold PBS. The cells were then incubated with sulfo-NHS-biotin reagent to biotinylate cell surface glycoproteins using instructions supplied by the manufacturer. Briefly, 20 mM solution of sulfo-NHS-biotin reagent was prepared by dissolving 9 mg of sulfo-NHS-biotin reagent in 1 mL dimethylformamide (DMF). Approximately 2 × 108 cells from each cell line were suspended in 1 mL PBS in 15 mL polypropylene tubes and 200 μL of the 20 mM sulfo-NHS-biotin solution was added to each cell suspension. The volume of each tube was adjusted to 2 mL with PBS to yield a final concentration of 2 mM sulfo-NHS-biotin in each tube. The sulfo-NHS-biotin/cell suspensions were incubated at room temperature for 30 min and washed 3× with PBS/100 mM glycine solution to quench the reaction and remove excess biotin. The cells were further washed 2× with PBS to remove residual glycine. As controls, a batch of the cells were mock biotinylated by adding 200 μL DMF solution without sulfo-NHS-biotin and the incubations and washings were carrying out essentially as described for the biotinylation process. The biotinylated and mock biotinylated cells were used to prepare detergent extracts as described below.
Preparation of cell extracts
Detergent extracts of non-biotinylated, biotinylated and mock biotinylated cells were prepared by suspending approximately 1 × 108 cells in 1 mL of lysis buffer made up of PBS containing 1 tablet of protease inhibitor cocktail (10× concentrated protease inhibitor cocktail) and 0.1% Triton X-100. The cell suspensions were sonicated on ice using Branson sonifier (Branson Ultrasonic Corporation, Danbury, CT) and the homogenates were left at room temperature for 30 min to solubilize membrane proteins. The homogenates were centrifuged at 16,000 × g in an Eppendorf centrifuge (Model 5415 R) at 4°C for 30 min and the supernatant fractions were recovered. The protein concentrations of the supernatant were determined by BCA protein assay method using instructions provided by the manufacturer and bovine serum albumin (BSA) as standard protein. Protein concentration of the extracts was reported as mg protein per mL of solution (mg/mL). The biotinylated and mock biotinylated cells were used for immunoprecipitation experiments while non-biotinylated cell extracts were used for lectin blot analyses.
SDS-PAGE and transblotting
Separation of proteins by SDS-PAGE was carried out under reducing conditions. The samples were boiled in reducing SDS sample buffer for 10 min and applied to 4–20% precast polyacrylamide gradient gels and ran in 20 mM Tris/192 mM Glycine/0.1% SDS (w/v) running buffer at 125 V for 2 h to effect separation of proteins. The separated proteins were transblotted from the gel onto PVDF membrane (Bio-Rad, Richmond, CA) at room temperature at 100 V for 1 h or 15 V for 12 h using 25 mM Tris/192 mM glycine/20% methanol transblot buffer. The PVDF membranes were pre-soaked in methanol and transblot buffer respectively before being used for the blotting. The PVDF membranes were removed after blotting and washed with water to remove traces of transblot buffer. The membranes were then stained with Ponceau S stain to visualize the efficiency and integrity of the transblot process. The membranes were washed with 20 mM Tris, 150 mM NaCl pH 7.4 (TBS) solution to remove the Ponceau S stain and the membranes were used for the lectin blot analyses described below.
Immunoprecipitation of glycoproteins from cell extracts using mAb F8A1.1
Biotinylated and mock biotinylated cell extracts were immunoprecipitated with mAb F8A1.1 using protein G-conjugated Dynabeads magnetic system and a modification of the instructions provided by the manufacturer. Briefly, protein G-Dynabeads were gently mixed by repeated pipetting over a period of 5 min to thoroughly suspend the magnetic beads and 50 μL aliquots representing 1.5 mg protein G were transferred to microfuge tubes. The tubes were placed on a DynaMag™-2 magnetic particle concentrator (Invitrogen Dynal AS, Oslo, Norway) to separate the beads from the storage solution. The supernatant was discarded and the protein G Dynabeads were mixed with 20 μg of mAb F8A1.1 in PBS and rotated gently on a rotating platform for 10 min at room temperature to capture the mAb onto the protein G Dynabeads. The mAb F8A1.1-protein G-Dynabead complex was pulled down with the magnetic particle concentrator and unbound mAb was removed and discarded. The mAb-protein G-Dynabead complex was washed 3× with PBS/0.02% Tween-20 to remove residual unbound antibody. The washings were carried out by suspending the beads in PBS/Tween-20 solution, pulling down the beads on the magnetic particle concentrator and removing the supernatant. Approximately 150 μg of either biotinylated or mock biotinylated RT4, J82COT or T24 cell extracts prepared as described above were added to the F8A1.1-Dynabead complex and incubated at room temperature for 10 min with gentle rotation on a rotating platform to capture any Lex-bearing glycoproteins onto the beads. The resulting protein G-Dynabeads-F8A1.1-Lex-bearing glycoprotein complex was pulled down using the magnetic particle concentrator and the supernatant was removed. The magnetic beads with captured immune complexes were washed 3× with PBS/0.02% Tween-20 wash buffer to remove unbound extracts using the washing protocol described above. Reducing SDS-PAGE sample buffer was added to the complex and boiled for 10 min to dissociate the immune complex. The boiled samples were cooled at room temperature for 10 min and centrifuged at 1200 rpm for 5 min. The supernatant fractions containing released immunoprecipitated glycoproteins and mAb were recovered for analysis. Mock immunoprecipitations in which mAb F8A1.1 was omitted were also carried out for each cell extract and the Dynabeads were boiled in reducing SDS sample buffer as described above and the supernatant fractions were also recovered for the analysis described below.
Detection of immunoprecipitated proteins
The supernatant fractions recovered from the immunoprecipitation experiments above were analyzed essentially as described previously (Mandalasi et al. 2013). Briefly, the supernatant fractions recovered from the immunoprecipitations and mock precipitations after boiling the Dynabead-immune complexes in reducing sample buffer were separated by SDS-PAGE as described above and blotted onto PVDF membrane. After staining with Ponceau S to assess the integrity of the transblot, the membrane was incubated with 5% solution of BSA in TBS at room temperature for 30 min to block areas of the membrane lacking bound proteins. The membrane was washed 3× with TBS/0.3% Tween-20 (TTBS; 20 mM Tris, 150 mM NaCl pH 7.4, 0.3% Tween-20) solution and incubated with a 1:10,000 dilution of horseradish peroxidase (HRP) conjugated streptavidin in a dilution solution of TBS/0.3% Tween-20/1% BSA, for 30 min at room temperature. The membrane was washed 3× with TTBS and the immunoprecipitated Lex-bearing glycoprotein bands were revealed by incubating the PVDF membrane with West Pico enhance chemiluminescent substrate for HRP (Pierce, Rockford, IL) for 10 min at room temperature followed by imaging on a UVP EC-3 imager (UVP Bioimaging Systems, Upland, CA).
Lectin blotting with biotinylated Aleuria aurantia lectin
Approximately 150 μg of detergent extracts of A/T/N, RT4, J82COT, T24 and TCCSUP cell lines were separated on 4–20% SDS-PAGE gradient gels in duplicate under reducing conditions, and transblotted onto PVDF membranes. The membranes were stained with Ponceau S to determine the integrity of blotting and then blocked by incubation in 5% BSA solution in TBS as described above. The membranes were washed 3× with TTBS and 2 μg/mL solution of biotinylated-AAL (Vector Labs, Burlingame, CA) in TTBS/1% BSA dilution solution was added to one membrane while 2 μg/mL biotinylated-AAL in TTBS/1% BSA dilution solution containing 10 mM free fucose was added to the second membrane. Both membranes were incubated at room temperature for 30 min and washed 3× with TTBS. The membranes were incubated with 1:5000 dilution of HRP-streptavidin conjugate in TTBS/1% BSA dilution solution for 30 min at room temperature and washed 3× with TTBS. Reactive bands were revealed by incubation with West Pico chemiluminescent substrate for 10 min followed by imaging on a UVP EC-3 imager.
PNGase F treatment and lectin blotting with biotinylated-AAL
Approximately 150 μg aliquots of A/T/N, RT4, J82COT, T24 and TCCSUP cell extracts were either mock treated by omitting PNGase F from the reaction mixture or were treated with 10 U of PNGase F using buffer solutions and instructions provided by the manufacturer (New England Biolabs, Ipswich, MA). The mock and PNGase F treated extracts were boiled in reducing sample buffer for 10 min and separated by SDS-PAGE on 4–20% polyacrylamide gradient gels. The separated protein bands were blotted onto PVDF membranes as described previously and the blotting efficiency was assessed by Ponceau S staining. The membranes were washed with TBS and blocked by incubation in 5% BSA/TBS solution for 30 min. The membranes were washed 3× with TTBS and incubated with 2 μg/mL solution of biotinylated AAL in TTBS/1% BSA solution for 30 mins at room temperature. The membranes were washed 3× with TTBS and incubated with 1:5000 dilution of HRP-streptavidin at room temperature for 30 min. The membranes were washed 3× with TTBS and reactive bands were revealed by incubation with Pico West chemiluminescent substrate for 10 min followed by imaging on a UVP EC-3 imager.
β-Elimination and lectin blotting with biotinylated-AAL
Approximately, 150 μg of detergent extracts of A/T/N, RT4, J82COT, T24, TCCSUP cell lines were separated in duplicate by SDS-PAGE on 4–20% polyacrylamide gradient gels under reducing conditions and transblotted onto PVDF membranes as described above. One of the membranes was incubated overnight at 40°C in 55 mM NaOH in water with constant shaking to effect release of O-glycans by β-elimination while the other membrane was mock treated by carrying out the incubation with water without NaOH. The membranes were washed 3× with TBS for 10 min/wash and blocked in 5% BSA solution for 30 min. The membranes were washed 3× with TTBS wash solution and incubated for 30 min at room temperature with 2 μg/mL solution of biotinylated-AAL in TBS/0.3% Tween-20/1% BSA. The membranes were washed 4× with TTBS to remove unbound biotinylated-AAL and incubated with 1:5000 dilution HRP-streptavidin conjugate in dilution solution at room temperature for 30 min. The membranes were washed 4× with TTBS wash solution and reactive bands were revealed by incubating the membranes in West Pico enhanced chemiluminescent substrate system for 10 min followed by imaging on a UVP imager as previously described.
Immunocytological staining
Two sets of approximately 1 × 106 intact, live bladder cancer cell lines, RT4, J82COT, T24, TCCSUP and normal bladder epithelial cell A/T/N were incubated at 4°C for 30 min with either 20 μg/mL mAb F8A1.1 or 10 μg/mL HECA452 antibody solutions in Hanks buffer/1% BSA respectively. Two sets of intact live HL-60 were similarly incubated with F8A1.1 or HECA452 as positive controls respectively. Additionally, two sets of all the bladder cancer cells used above, together with the control HL-60 cells were mock treated by incubations in Hanks/1% BSA solution without mAb F8A1.1 or HECA452 antibody to assess the specificity of the immunostaining procedure. The cells were washed 4× with cold Hanks buffer and one set of the mAb F8A1.1-treated cells and one set of mock treated cells were incubated with 1:1000 dilution of Alexa Fluor 488-labeled goat-anti-mouse IgG diluted in Hanks buffer/1% BSA while one set of the HECA452-treated cells and a second set of mock treated cells were incubated with 10 μg/mL solution of Alexa-488 conjugated goat anti-rat IgM in Hanks buffer/1% BSA. The other set of F8A1.1, HECA452 and mock-treated cells were incubated in Hanks/1% BSA solution without any fluorescently labeled secondary antibody conjugates. The incubations were carried out at 4°C for 30 min and the cells were washed 4× with cold Hanks buffer to remove unbound antibodies. The cells were then imaged for bound antibodies on a Nikon Eclipse Ti-E inverted fluorescence microscope (Nikon Instruments Inc., Melville, NY).
Cytological staining with biotinylated-AAL
One set of intact, live RT4, J82COT, T24, TCCSUP bladder cancer cell lines and normal bladder epithelial A/T/N cells were incubated at 4°C for 30 min with 2 μg/mL solution of biotinylated-AAL (Vector Labs, Burlingame, CA) in Hanks buffer/1% BSA. A second set of the bladder cancer cells and the normal bladder epithelial A/T/N cells were incubated under similar conditions with solution of 2 μg/mL biotinylated-AAL in Hanks/1% BSA containing 10 mM free fucose to assess specificity of the lectin binding. The cells were washed with cold Hanks buffer to remove unbound biotinylated-lectin and incubated with 10 μg/mL solution of Alexa-488 conjugated streptavidin (Invitrogen, Carlsbad, CA) in Hanks buffer/1% BSA at 4°C for 30 min to detect bound lectins. The cells were washed 4× with cold Hanks buffer and imaged by fluorescence microscopy as described previously.
Conjugation of l-phytohemagglutinin, M. amurensis lectin and S. nigra agglutinin with Alexa-488
L-PHA, MAL and SNA were purchased from EY Laboratories (San Mateo, CA) and conjugated to NHS-activated Alexa-488 fluorescent probe (Invitrogen, Carlsbad, CA) using instructions provided by the manufacturer. Briefly, 2 mg of L-PHA, MAL or SNA in 1 mL PBS solution was mixed with 100 μL of 1 M sodium bicarbonate solution to adjust the pH of the lectin solutions to 7.5 to 8.3. The lectin solutions were added to vials of supplied Alexa-488 dye and stirred at room temperature for 1 h. The Alexa-488 conjugated lectins were purified away from unconjugated dye and reaction by-products by chromatography over the manufacturer’s supplied gel filtration columns and the protein content of the Alexa-488-lectin conjugates recovered from the gel filtration column void fractions were determined by BCA protein assay.
Cytological staining of cells with Alexa-488 conjugated L-PHA
Approximately 1 × 106 RT4, J82COT, T24 and TCCSUP cells were suspended in 20 μg/mL solution Alexa-488 conjugated L-PHA, MAL or SNA in a dilution solution of Hanks/1% BSA and incubated at 4°C for 30 min. Another set of the bladder cancer cells were also incubated with the three lectins in the presence of their respective haptens to determine the specificity of the lectin binding. The L-PHA/hapten incubations were carried out in the presence of 100 mM GalNAc. The cells were washed 4× with cold PBS after the incubations and imaged by fluorescence microscopy as described previously.
Isolation of total RNA and determination of α1–3/4-fucosyltransferase gene expression by RT-qPCR
RT4, J82COT, T24, TCCSUP and A/T/N cells were grown in quadruplicates in 6-well tissue culture plates to 80% confluent density using media and cell culture conditions described previously. The media were removed, and the wells were washed 3× with cold PBS and used for extraction of total RNA by the TRIzol method using instructions provided by the manufacturer (Invitrogen, Grand Island, NY). Briefly, 500 μL of TRIzol was added to each of the 4 wells to lyse and solubilize the cells. The cell lysates were transferred into microcentrifuge tubes and 100 μL of chloroform was added to each tube. The contents of the tubes were mixed by gentle tapping for 15 s and then incubated at room temperature for 10 min. The tubes were centrifuged at 12,000 × g for 15 min at 4°C to effect phase separation. The upper phase containing total RNA was recovered into 1.5 mL microcentrifuge tubes, and 250 μL of absolute isopropanol was added to each tube. The contents of the tubes were mixed by gentle tapping and allowed to stand at room temperature for 10 min to precipitate total RNA, the tubes were centrifuged at 20,000 × g for 10 min at 4°C and the supernatant fraction was discarded. The RNA pellet was suspended in 500 μL 75% ethanol and centrifuged at 7500 × g for 5 min at 4°C. The supernatant was discarded, and the pellet was left to air-dry at room temperature for 2 h. The total RNA pellet was dissolved in 15 μL of RNAse-free water and the RNA concentration was determined using Nano Vue Plus spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Approximately 2 μg of the RNA preparations were used as a template to synthesize single stranded cDNA employing iScript cDNA synthesis kit (BioRad, Richmond CA). The single stranded cDNA prepared from the different bladder cancer cell lines were used to amplify and quantify the expression levels of 11 human fucosyltransferase genes (FUTs) by RT-qPCR using iTag Universal SYBR green supermix (BioRad, Richmond, CA) and sets of primers for the eight human FUTs (Supplementary Figure S8C) The primer sequences were kindly provided by Dr. Kelley Moremen (the Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia). The RT-qPCR reactions were carried out on an iCycler instrument that uses the MyiQ2 two color RT-qPCR detection system (Bio-Rad, Richmond, CA). FUT gene expression in the samples from the different bladder cancer cell lines were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene expression to cancel out the probable differences in the concentration of RNA used. Each RT-qPCR reaction included a non-template control. All the analyses were performed in quadruplicate and the results represent averages of the four determinations. The analysis of the target amplicon/message from each sample was estimated from a threshold number (CT), which has inverse relationship with the abundance of its initial mRNA.
Preparation of cell extracts for release of N- and O-glycans from glycoproteins of RT4 and T24 cells
Approximately 2.5 mL of packed cell volume was prepared by centrifuging thawed pooled aliquots of frozen RT4 and T24 at 1200 rpm for 5 min in 15 mL Falcon tubes. Residual supernatant buffer was removed, and the cell paste was suspended in 5 mL of lysis buffer composed of 25 mM Tris/acetic acid pH 7.4, 1% CHAPS, 5 mM EDTA. The suspension was sonicated on ice with a Branson sonifier using 6, 10 s pulses at 50% amplitude with 30 s cooling internals. The homogenate was incubated at room temperature for 30 min to solubilize membrane proteins and then centrifuged at 16,000 rpm at 4°C for 30 min to remove insoluble material. The supernatant fraction containing released membrane glycoproteins were recovered and dialyzed against 50 mM ammonium bicarbonate buffer, pH 8.4 at 4°C using regenerated cellulose dialysis tubing. The dialysate was recovered into 15 mL Falcon tubes and dried in Centrivap speedvac evaporator (Labconco, Kansas City, MO).
Preparation of glycopeptides
The dried glycoprotein extracts were dissolved in 1.5 mL of 2 mg/mL dithiothreitol (DTT) solution prepared in degassed 0.6 M Tris/acetic acid buffer pH 8.5 and incubated at 37°C for 45 min to effect reduction of disulfide bonds. Then, 1.5 mL of 12 mg/mL of solution of iodoacetamide in degassed 0.6 M Tris/acetic acid, pH 8.5 was added and incubated further in the dark at room temperature for 90 min to alkylate thiol groups. The sample was dialyzed against 50 mM ammonium bicarbonate pH 8.4 buffer at 4°C to remove salts and dried in Centrivap speedvac evaporator as described previously. The dried sample was mixed with 2 mg TPCK treated bovine trypsin in 1.5 mL 50 mM ammonium bicarbonate buffer, pH 8.4, and incubated at 37°C for 8 h to effect digestion of the glycoproteins to glycopeptides. The sample was dried in Centrivap speedvac, dissolved in 500 μL of 5% acetic acid in deionized water and applied to Sep-Pak C18 cartridges to purify glycopeptides using propanol/5% acetic acid solvent system.
Purification of glycopeptides by chromatography on Sep-Pak C-18 cartridges
Sep-Pak C18 cartridges were conditioned by successive washes with 5 mL each of methanol, 5% acetic acid in water, and n-propanol. The conditioning was concluded by washing the cartridges 3× with 5% acetic acid. The tryptic digest in 5% acetic acid was applied directly to the conditioned Sep-Pak cartridge and washed with 30 mL of 5% acetic acid solution and the column flow through discarded. The cartridge was washed stepwise with 4 mL each of 20% n-propanol in 5% acetic acid, 40% n-propanol in 5% acetic acid, 60% n-propanol in 5% acetic acid and 100% n-propanol and collected into culture tubes. The volumes of the fractions were reduced by evaporation under a gentle stream of nitrogen gas, and pooled. The pooled fractions were subsequently evaporated to dryness in a CentriVap speedvac concentrator and treated with PNGase F to release N-glycans as described below.
Release and purification of N-glycans
The dried glycopeptides were dissolved in 200 μL of 50 mM ammonium bicarbonate buffer, pH 8.4 and incubated with 500 U of PNGase F at 37°C for 20 h. The digest was dried in a CentriVap speedvac and the dried PNGase F digest was dissolved in 200 μL 5% acetic acid solution and applied to a conditioned Sep-Pak C18 cartridge. The unbound material was washed from the cartridge with 5 mL of 5% acetic acid and recovered as the N-glycan fraction. The cartridge was subsequently eluted sequentially with 5 mL each of 20%, 40%, 60% n-propanol in 5% acetic acid respectively and the released material were collected separately into tubes. The cartridge was finally eluted with 5 mL of 100% n-propanol and the eluted material was recovered. The volumes of the eluted fractions were reduced by evaporation under a gentle stream of nitrogen gas and pooled as the O-linked glycopeptide fraction. The acetic acid fraction, representing released N-glycans, and the pooled propanol fractions representing O-linked glycopeptides were dried by evaporation in CentriVap speedvac concentrator.
Release and purification of O-glycans
The dried O-linked glycopeptides recovered from the Sep-Pak C-18 cartridge were mixed with 400 μL of 55 mg/mL solution of NaBH4 in 0.1 M NaOH and incubated overnight at 45°C to release O-glycans by β-elimination reaction. The reaction was terminated by addition of 5–7 drops of glacial acetic acid until no bubbling was observed. Dowex 50 W X8 ion exchange column (mesh size 200–400) prepared in 4 M HCl (Sigma, St. Louis, MO) was rinsed with Milli Q water and 5% acetic acid successively and then washed with 20 mL of 5% acetic acid. The NaOH/NaBH4 treated sample was applied to the column and the column was washed with 3 mL of 5% acetic acid. The flow through and wash fractions were collected, pooled and lyophilized. The dried sample was mixed with 1 mL of acetic acid:methanol solution (1:9, v/v), vortexed and evaporated under a stream of nitrogen gas. The mixing and evaporation in acetic acid:methanol step was repeated three more times. The sample was suspended into 200 μL of 50% methanol and loaded onto a conditioned 50 mg Sep-Pak C18 column. The unbound material was washed with 5% acetic acid and the flow through fraction was recovered and lyophilized.
Permethylation and analysis of N- and O-glycan structures by mass spectrometry
The dimethyl sulfoxide (DMSO), NaOH, methyl iodide, chloroform, acetonitrile, 2,5-dihydroxybenzoic acid (DHB) and trifluoroacetic acid (TFA) used in the permethylation and MS analysis were obtained from Sigma (St Louis, MO). A slurry of NaOH in DMSO was prepared by quickly grinding 7 pellets of NaOH in 4 mL of anhydrous DMSO with a pestle and mortar to avoid excessive water absorption from the atmosphere. Lyophilized N- and O-glycan samples were mixed with 1 mL of the NaOH/DMSO slurry solution, 500 μL of methyl iodide was added and the mixture was incubated at room temperature for 30 min under vigorous shaking to effect deprotonation and methylation of free hydroxyl groups. The reaction was stopped by slow, dropwise addition of 1 mL of Milli Q water with shaking and 1 mL of chloroform was added to the mixture and the permethylated glycans were extracted out in the chloroform fraction. The chloroform fraction was mixed with 3 mL of Milli Q water, vortexed briefly to wash the chloroform fraction and the water was separated from chloroform by centrifugation and discarded. This wash step was repeated three times and the chloroform fraction was finally dried under a gentle stream of nitrogen gas. The sample was redissolved in 200 μL of 50% methanol. A 200 mg Sep-Pak C18 column was conditioned by successive washes with one column volume each of methanol, Milli Q water, acetonitrile and Milli Q water. The permethylated sample was applied to the conditioned Sep-Pak C18 column and the column was washed with 6 mL of 15% acetonitrile. Bound permethylated glycans were eluted with 6 mL of 50% acetonitrile. The eluted fraction was lyophilized and redissolved in 10 μL of 75% methanol from which 1 μL was mixed with 1 μL of 5 mg/mL solution of DHB in 50% acetonitrile/0.1% TFA and spotted on a MALDI polished steel target plate (Bruker). MS data were acquired on a Bruker UltraFlex II MALDI-TOF MS instrument. Reflective positive mode was used, and data were recorded between 500 and 8000 m/z. N-glycan MS profiles represent the aggregation of at least 20,000 laser shots. Mass peaks were then annotated and assigned to N-glycan composition when a match was found. Subsequent MS data and MS profile analyses were performed using mMass (Strohalm et al. 2010).
Glycomics analysis
All cell lines were treated as described previously (Jang-Lee et al. 2006). Each sample was sonicated in the presence of detergent CHAPS (Roche; 10810118001), reduced with DTT (Roche; 10197777001) in 4 M guanidine-HCl (Thermo; 24115), carboxymethylated with IAA (Sigma; I4386), and trypsin digested (Sigma; T0303). The digested glycoproteins were then purified by HLB plus Sep-Pak (Waters; 186000132). N-glycans were released by PNGase F (E.C. 3.5.1.52; Roche) digestion. Released N-glycans were permethylated using the sodium hydroxide procedure and purified by classic C18−Sep-Pak (Waters, WAT051910). Permethylated N-glycans were eluted at the 50% acetonitrile fraction. MS and MS/MS data were acquired using a 4800 MALDI-TOF/TOF (Applied Biosystems) mass spectrometer. Permethylated samples were dissolved in 10 μL of methanol and 1 μL of dissolved sample was premixed with 1 μL of matrix (10 mg/mL 3,4-diaminobenzophenone in 75% (v/v) aqueous acetonitrile), spotted onto a target plate and dried under vacuum. For the MS/MS studies the collision energy was set to 1 kV, and Argon was used as the collision gas. The 4700 Calibration standard kit, calmix (Applied Biosystems), was used as the external calibrant for the MS mode and [Glu1] fibrinopeptide B human (Sigma) was used as an external calibrant for the MS/MS mode. MS and MS/MS data were processed using Data Explorer 4.9 Software (Applied Biosystems). The processed spectra were subjected to manual assignment and annotation with the aid of a glycobioinformatics tool, GlycoWorkBench (Ceroni et al. 2008). The proposed assignments for the selected peaks were based on 12C isotopic composition together with knowledge of the biosynthetic pathways. The proposed structures were then confirmed by data obtained from MS/MS experiments.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Dr Kelley Moremen of Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia for providing the qRT-PCR primers used in Supplementary Figure S8.
Contributor Information
Bernadette Ezeabikwa, Department of Natural Sciences, University of Maryland Eastern Shore, Princess Anne, MD, USA.
Nandini Mondal, Department of Surgery, Beth Israel Deaconess Medical Center—Harvard Medical School, Boston, MA, USA.
Aristotelis Antonopoulos, Department of Life Sciences, Imperial College London, London SW7 2AZ, UK.
Stuart M Haslam, Department of Life Sciences, Imperial College London, London SW7 2AZ, UK.
Yasuyuki Matsumoto, Department of Surgery, Beth Israel Deaconess Medical Center—Harvard Medical School, Boston, MA, USA.
Miguel Martin-Caraballo, Department of Pharmaceutical Sciences, School of Pharmacy, University of Maryland Eastern Shore, Princess Anne, MD, USA.
Sylvain Lehoux, Department of Surgery, Beth Israel Deaconess Medical Center—Harvard Medical School, Boston, MA, USA; Novab Inc., Atlanta, GA, USA.
Msano Mandalasi, Department of Natural Sciences, University of Maryland Eastern Shore, Princess Anne, MD, USA; Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA, USA.
Ali Ishaque, Department of Natural Sciences, University of Maryland Eastern Shore, Princess Anne, MD, USA.
Jamie Heimburg-Molinaro, Department of Surgery, Beth Israel Deaconess Medical Center—Harvard Medical School, Boston, MA, USA.
Richard D Cummings, Department of Surgery, Beth Israel Deaconess Medical Center—Harvard Medical School, Boston, MA, USA.
Anthony K Nyame, Department of Natural Sciences, University of Maryland Eastern Shore, Princess Anne, MD, USA.
Conflict of interest statement
The authors declare no conflict of interest.
Funding
National Institute of Health grants to R.D.C. (P41GM103694, R24GM137763, R01AI101982, P01HL085607, U01CA168930).
References
- Amado M, Almeida R, Schwientek T, Clausen H. 1999. Identification and characterization of large galactosyltransferase gene families: Galactosyltransferases for all functions. Biochim Biophys Acta. 1473:35–53. [DOI] [PubMed] [Google Scholar]
- Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. 2017. Bladder cancer incidence and mortality: A global overview and recent trends. Eur Urol. 71(1):96–108. [DOI] [PubMed] [Google Scholar]
- Batista R, Vinagre N, Meireles S, Vinagre J, Prazeres H, Leao R, Maximo V, Soares P. 2020. Biomarkers for bladder cancer diagnosis and surveillance: A comprehensive review. Diagnostics (Basel). 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhuizen MF, Fukuda M. 1992. Expression cloning of a cDNA encoding UDP-GlcNAc:Gal beta 1-3-GalNAc-R (GlcNAc to GalNAc) beta 1-6GlcNAc transferase by gene transfer into CHO cells expressing polyoma large tumor antigen. Proc Natl Acad Sci USA. 89(19):9326–9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger M, Catto JW, Dalbagni G, Grossman HB, Herr H, Karakiewicz P, Kassouf W, Kiemeney LA, La Vecchia C, Shariat S, et al. 2013. Epidemiology and risk factors of urothelial bladder cancer. Eur Urol. 63(2):234–241. [DOI] [PubMed] [Google Scholar]
- Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. 2008. Glyco Workbench: A tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res. 7(4):1650–1659. [DOI] [PubMed] [Google Scholar]
- Cervoni GE, Cheng JJ, Stackhouse KA, Heimburg-Molinaro J, Cummings RD. 2020. O-glycan recognition and function in mice and human cancers. Biochem J. 477:1541–1564. [DOI] [PubMed] [Google Scholar]
- Cheng L, Neumann RM, Nehra A, Spotts BE, Weaver AL, Bostwick DG. 2000. Cancer heterogeneity and its biologic implications in the grading of urothelial carcinoma. Cancer. 88:1663–1670. [DOI] [PubMed] [Google Scholar]
- Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. 2014. Cell surface protein glycosylation in cancer. Proteomics. 14:525–546. [DOI] [PubMed] [Google Scholar]
- Colombel M, Soloway M, Akaza H, Böhle A, Palou J, Buckley R, Lamm D, Brausi M, Witjes JA, Persad R. 2008. Epidemiology, staging, grading, and risk stratification of bladder cancer. Eur Urol Suppl. 7:618–626. [Google Scholar]
- Cummings RD, Kornfeld S. 1982. Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J Biol Chem. 257:11230–11234. [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. 2015. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. [DOI] [PubMed] [Google Scholar]
- Gao C, Hanes MS, Byrd-Leotis LA, Wei M, Jia N, Kardish RJ, McKitrick TR, Steinhauer DA, Cummings RD. 2019. Unique Binding Specificities of Proteins toward Isomeric Asparagine-Linked Glycans. Cell Chem Biol. 26:535, e534–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler C, Jarvis DL. 2011. Effective glycoanalysis with Maackia amurensis lectins requires a clear understanding of their binding specificities. Glycobiology. 21:988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goelz SE, Hession C, Goff D, Griffiths B, Tizard R, Newman B, Chi-Rosso G, Lobb R. 1990. ELFT: A gene that directs the expression of an ELAM-1 ligand. Cell. 63:1349–1356. [DOI] [PubMed] [Google Scholar]
- Goldstein IJ, Poretz RD. 1986. 2—Isolation, physicochemical characterization, and carbohydrate-binding specificity of lectins. In: Liener IE, Sharon N, Goldstein IJ, editors. The Lectins. Cambridge, MA: Academic Press. p. 33–247. [Google Scholar]
- Golijanin D, Sherman Y, Shapiro A, Pode D. 1995. Detection of bladder tumors by immunostaining of the Lewis X antigen in cells from voided urine. Urology. 46:173–177. [DOI] [PubMed] [Google Scholar]
- Grignon DJ. 2009. The current classification of urothelial neoplasms. Mod Pathol. 22(Suppl 2):S60–S69. [DOI] [PubMed] [Google Scholar]
- Grundmann U, Nerlich C, Rein T, Zettlmeissl G. 1990. Complete cDNA sequence encoding human beta-galactoside alpha-2,6-sialyltransferase. Nucleic Acids Res. 18:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Li X, Tan Z, Lu W, Yang G, Guan F. 2014. Alteration of N-glycans and expression of their related glycogenes in the epithelial-mesenchymal transition of HCV29 bladder epithelial cells. Molecules. 19:20073–20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg-Molinaro J, Lum M, Vijay G, Jain M, Almogren A, Rittenhouse-Olson K. 2011. Cancer vaccines and carbohydrate epitopes. Vaccine. 29:8802–8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikehara Y, Kojima N, Kurosawa N, Kudo T, Kono M, Nishihara S, Issiki S, Morozumi K, Itzkowitz S, Tsuda T, et al. 1999. Cloning and expression of a human gene encoding an N-acetylgalactosamine-alpha2,6-sialyltransferase (ST6GalNAc I): A candidate for synthesis of cancer-associated sialyl-Tn antigens. Glycobiology. 9:1213–1224. [DOI] [PubMed] [Google Scholar]
- Ishimura H, Takahashi T, Nakagawa H, Nishimura S, Arai Y, Horikawa Y, Habuchi T, Miyoshi E, Kyan A, Hagisawa S, et al. 2006. N-acetylglucosaminyltransferase V and beta1-6 branching N-linked oligosaccharides are associated with good prognosis of patients with bladder cancer. Clin Cancer Res. 12:2506–2511. [DOI] [PubMed] [Google Scholar]
- Isshiki S, Togayachi A, Kudo T, Nishihara S, Watanabe M, Kubota T, Kitajima M, Shiraishi N, Sasaki K, Andoh T, et al. 1999. Cloning, expression, and characterization of a novel UDP-galactose:beta-N-acetylglucosamine beta1,3-galactosyltransferase (beta3Gal-T5) responsible for synthesis of type 1 chain in colorectal and pancreatic epithelia and tumor cells derived therefrom. J Biol Chem. 274:12499–12507. [DOI] [PubMed] [Google Scholar]
- Jang-Lee J, North SJ, Sutton-Smith M, Goldberg D, Panico M, Morris H, Haslam S, Dell A. 2006. Glycomic profiling of cells and tissues by mass spectrometry: Fingerprinting and sequencing methodologies. Methods Enzymol. 415:59–86. [DOI] [PubMed] [Google Scholar]
- Kajiwara H, Yasuda M, Kumaki N, Shibayama T, Osamura Y. 2005. Expression of carbohydrate antigens (SSEA-1, sialyl-Lewis X, DU-PAN-2 and CA19-9) and E-selectin in urothelial carcinoma of the renal pelvis, ureter, and urinary bladder. Tokai J Exp Clin Med. 30:177–182. [PubMed] [Google Scholar]
- Kelly RJ, Ernst LK, Larsen RD, Bryant JG, Robinson JS, Lowe JB. 1994. Molecular basis for H blood group deficiency in Bombay (Oh) and para-Bombay individuals. Proc Natl Acad Sci USA. 91:5843–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa H, Paulson JC. 1994. Cloning of a novel alpha 2,3-sialyltransferase that sialylates glycoprotein and glycolipid carbohydrate groups. J Biol Chem. 269:1394–1401. [PubMed] [Google Scholar]
- Konety BR, Ballou B, Jaffe R, Singh J, Reiland J, Hakala TR. 1997. Expression of SSEA-1 (Lewis(x)) on transitional cell carcinoma of the bladder. Urol Int. 58:69–74. [DOI] [PubMed] [Google Scholar]
- Kono M, Tsuda T, Ogata S, Takashima S, Liu H, Hamamoto T, Itzkowitz SH, Nishimura S, Tsuji S. 2000. Redefined substrate specificity of ST6GalNAc II: A second candidate sialyl-Tn synthase. Biochem Biophys Res Commun. 272:94–97. [DOI] [PubMed] [Google Scholar]
- Kornfeld R, Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 54:631–664. [DOI] [PubMed] [Google Scholar]
- Koszik F, Strunk D, Simonitsch I, Picker LJ, Stingl G, Payer E. 1994. Expression of monoclonal antibody HECA-452-defined E-selectin ligands on Langerhans cells in normal and diseased skin. J Invest Dermatol. 102:773–780. [DOI] [PubMed] [Google Scholar]
- Kukowska-Latallo JF, Larsen RD, Nair RP, Lowe JB. 1990. A cloned human cDNA determines expression of a mouse stage-specific embryonic antigen and the Lewis blood group alpha(1,3/1,4)fucosyltransferase. Genes Dev. 4:1288–1303. [DOI] [PubMed] [Google Scholar]
- Kumar A, Torii T, Ishino Y, Muraoka D, Yoshimura T, Togayachi A, Narimatsu H, Ikenaka K, Hitoshi S. 2013. The Lewis X-related alpha1,3-fucosyltransferase, Fut10, is required for the maintenance of stem cell populations. J Biol Chem. 288:28859–28868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosawa N, Hamamoto T, Lee YC, Nakaoka T, Kojima N, Tsuji S. 1994. Molecular cloning and expression of GalNAc alpha 2,6-sialyltransferase. J Biol Chem. 269:1402–1409. [PubMed] [Google Scholar]
- Lee YC, Kaufmann M, Kitazume-Kawaguchi S, Kono M, Takashima S, Kurosawa N, Liu H, Pircher H, Tsuji S. 1999. Molecular cloning and functional expression of two members of mouse NeuAcalpha2,3Galbeta1,3GalNAc GalNAcalpha2,6-sialyltransferase family, ST6GalNAc III and IV. J Biol Chem. 274:11958–11967. [DOI] [PubMed] [Google Scholar]
- Li C, Yang Z, Du Y, Tang H, Chen J, Hu D, Fan Z. 2014. BCMab1, a monoclonal antibody against aberrantly glycosylated integrin alpha3beta1, has potent antitumor activity of bladder cancer in vivo. Clin Cancer Res. 20:4001–4013. [DOI] [PubMed] [Google Scholar]
- Li J, Guillebon AD, Hsu JW, Barthel SR, Dimitroff CJ, Lee YF, King MR. 2013. Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br J Cancer. 109:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Qi HL, Chen HL. 2001. Regulation of differentiation- and proliferation-inducers on Lewis antigens, alpha-fucosyltransferase and metastatic potential in hepatocarcinoma cells. Br J Cancer. 84:1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JB, Kukowska-Latallo JF, Nair RP, Larsen RD, Marks RM, Macher BA, Kelly RJ, Ernst LK. 1991. Molecular cloning of a human fucosyltransferase gene that determines expression of the Lewis x and VIM-2 epitopes but not ELAM-1-dependent cell adhesion. J Biol Chem. 266:17467–17477. [PubMed] [Google Scholar]
- Mach L, Scherf W, Ammann M, Poetsch J, Bertsch W, Marz L, Glossl J. 1991. Purification and partial characterization of a novel lectin from elder (Sambucus nigra L.) fruit. Biochem J. 278(Pt 3):667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandalasi M, Dorabawila N, Smith DF, Heimburg-Molinaro J, Cummings RD, Nyame AK. 2013. Development and characterization of a specific IgG monoclonal antibody toward the Lewis x antigen using splenocytes of Schistosoma mansoni-infected mice. Glycobiology. 23:877–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura K, Higashida K, Ishida H, Hata Y, Yamamoto K, Shigeta M, Mizuno-Horikawa Y, Wang X, Miyoshi E, Gu J, et al. 2007. Carbohydrate binding specificity of a fucose-specific lectin from Aspergillus oryzae: A novel probe for core fucose. J Biol Chem. 282:15700–15708. [DOI] [PubMed] [Google Scholar]
- Mollicone R, Moore SE, Bovin N, Garcia-Rosasco M, Candelier JJ, Martinez-Duncker I, Oriol R. 2009. Activity, splice variants, conserved peptide motifs, and phylogeny of two new alpha1,3-fucosyltransferase families (FUT10 and FUT11). J Biol Chem. 284:4723–4738. [DOI] [PubMed] [Google Scholar]
- Mondal N, Buffone A Jr, Stolfa G, Antonopoulos A, Lau JT, Haslam SM, Dell A, Neelamegham S. 2015. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 125:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal N, Dykstra B, Lee J, Ashline DJ, Reinhold VN, Rossi DJ, Sackstein R. 2018. Distinct human α(1,3)-fucosyltransferases drive Lewis-X/sialyl Lewis-X assembly in human cells. J Biol Chem. 293:7300–7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkley J, Elliott DJ. 2016. Hallmarks of glycosylation in cancer. Oncotarget. 7:35478–35489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara S, Iwasaki H, Kaneko M, Tawada A, Ito M, Narimatsu H. 1999. Alpha1,3-fucosyltransferase 9 (FUT9; Fuc-TIX) preferentially fucosylates the distal GlcNAc residue of polylactosamine chain while the other four alpha1,3FUT members preferentially fucosylate the inner GlcNAc residue. FEBS Lett. 462:289–294. [DOI] [PubMed] [Google Scholar]
- Oliveira-Ferrer L, Legler K, Milde-Langosch K. 2017. Role of protein glycosylation in cancer metastasis. Semin Cancer Biol. 44:141–152. [DOI] [PubMed] [Google Scholar]
- Osuga T, Takimoto R, Ono M, Hirakawa M, Yoshida M, Okagawa Y, Uemura N, Arihara Y, Sato Y, Tamura F, et al. 2016. Relationship between increased fucosylation and metastatic potential in colorectal cancer. J Natl Cancer Inst. 108:djw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham DM, Morton K, Coghill G, Robertson AJ, Kerr MA. 1990. Expression of CD15 antigen in urinary bladder transitional cell carcinoma. J Clin Pathol. 43:541–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto A, Fernandes E, Gaiteiro C, Lima L, Azevedo R, Soares J, Cotton S, Parreira B, Neves M, Amaro T, et al. 2016. Hypoxia enhances the malignant nature of bladder cancer cells and concomitantly antagonizes protein O-glycosylation extension. Oncotarget. 7:63138–63157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planz B, Synek C, Deix T, Bocking A, Marberger M. 2001. Diagnosis of bladder cancer with urinary cytology, immunocytology and DNA-image-cytometry. Anal Cell Pathol. 22:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pochec E, Litynska A, Bubka M, Amoresano A, Casbarra A. 2006. Characterization of the oligosaccharide component of alpha3beta1 integrin from human bladder carcinoma cell line T24 and its role in adhesion and migration. Eur J Cell Biol. 85:47–57. [DOI] [PubMed] [Google Scholar]
- Sheinfeld J, Reuter VE, Melamed MR, Fair WR, Morse M, Sogani PC, Herr HW, Whitmore WF, Cordon-Cardo C. 1990. Enhanced bladder cancer detection with the Lewis X antigen as a marker of neoplastic transformation. J Urol. 143:285–288. [DOI] [PubMed] [Google Scholar]
- Shirahama T, Ikoma M, Muramatsu T, Ohi Y. 1992. Expression of SSEA-1 carbohydrate antigen correlates with stage, grade and metastatic potential of transitional cell carcinoma of the bladder. J Urol. 148:1319–1322. [DOI] [PubMed] [Google Scholar]
- Sjoberg ER, Kitagawa H, Glushka J, Halbeek H, Paulson JC. 1996. Molecular cloning of a developmentally regulated N-acetylgalactosamine alpha2,6-sialyltransferase specific for sialylated glycoconjugates. J Biol Chem. 271:7450–7459. [DOI] [PubMed] [Google Scholar]
- Stowell SR, Ju T, Cummings RD. 2015. Protein glycosylation in cancer. Annu Rev Pathol. 10:473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohalm M, Kavan D, Novak P, Volny M, Havlicek V. 2010. mMass 3: A cross-platform software environment for precise analysis of mass spectrometric data. Anal Chem. 82:4648–4651. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. 1993. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology. 3:97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WC, Cummings RD. 1988. The immobilized leukoagglutinin from the seeds of Maackia amurensis binds with high affinity to complex-type Asn-linked oligosaccharides containing terminal sialic acid-linked alpha-2,3 to penultimate galactose residues. J Biol Chem. 263:4576–4585. [PubMed] [Google Scholar]
- Weston BW, Nair RP, Larsen RD, Lowe JB. 1992. Isolation of a novel human alpha (1,3)fucosyltransferase gene and molecular comparison to the human Lewis blood group alpha (1,3/1,4)fucosyltransferase gene. Syntenic, homologous, nonallelic genes encoding enzymes with distinct acceptor substrate specificities. J Biol Chem. 267:4152–4160. [PubMed] [Google Scholar]
- Yan X, Lin Y, Liu S, Aziz F, Yan Q. 2015. Fucosyltransferase IV (FUT4) as an effective biomarker for the diagnosis of breast cancer. Biomed Pharmacother. 70:299–304. [DOI] [PubMed] [Google Scholar]
- Yanagidani S, Uozumi N, Ihara Y, Miyoshi E, Yamaguchi N, Taniguchi N. 1997. Purification and cDNA cloning of GDP-L-Fuc:N-acetyl-beta-D-glucosaminide:alpha1-6 fucosyltransferase (alpha1-6 FucT) from human gastric cancer MKN45 cells. J Biochem. 121:626–632. [DOI] [PubMed] [Google Scholar]
- Yang G, Tan Z, Lu W, Guo J, Yu H, Yu J, Sun C, Qi X, Li Z, Guan F. 2015. Quantitative glycome analysis of N-glycan patterns in bladder cancer vs normal bladder cells using an integrated strategy. J Proteome Res. 14:639–653. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang D, Lv J, Wang S, Zhang Q. 2018. MiR-125a-5p suppresses bladder cancer progression through targeting FUT4. Biomed Pharmacother. 108:1039–1047. [DOI] [PubMed] [Google Scholar]
- Zhou X, Zhai Y, Liu C, Yang G, Guo J, Li G, Sun C, Qi X, Li X, Guan F. 2020. Sialidase NEU1 suppresses progression of human bladder cancer cells by inhibiting fibronectin-integrin alpha5beta1 interaction and Akt signaling pathway. Cell Commun Signal. 18:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.