

Abstract
Nonalcoholic steatohepatitis (NASH) is a severe liver disorder characterized by triglyceride accumulation, severe inflammation, and fibrosis. With the recent increase in prevalence, NASH is now the leading cause of liver transplant, with no approved therapeutics available. Although the exact molecular mechanism of NASH progression is not well understood, a widely held hypothesis is that fat accumulation is the primary driver of the disease. Therefore, diacylglycerol O-acyltransferase 2 (DGAT2), a key enzyme in triglyceride synthesis, has been explored as a NASH target. RNAi-based therapeutics is revolutionizing the treatment of liver diseases, with recent chemical advances supporting long-term gene silencing with single subcutaneous administration. Here, we identified a hyper-functional, fully chemically stabilized GalNAc-conjugated small interfering RNA (siRNA) targeting DGAT2 (Dgat2-1473) that, upon injection, elicits up to 3 months of DGAT2 silencing (>80%–90%, p < 0.0001) in wild-type and NSG-PiZ “humanized” mice. Using an obesity-driven mouse model of NASH (ob/ob-GAN), Dgat2-1473 administration prevents and reverses triglyceride accumulation (>85%, p < 0.0001) without increased accumulation of diglycerides, resulting in significant improvement of the fatty liver phenotype. However, surprisingly, the reduction in liver fat did not translate into a similar impact on inflammation and fibrosis. Thus, while Dgat2-1473 is a practical, long-lasting silencing agent for potential therapeutic attenuation of liver steatosis, combinatorial targeting of a second pathway may be necessary for therapeutic efficacy against NASH.
Keywords: Non-alcoholic steatohepatitis, NASH, RNAi therapeutics, Oligonucleotide therapy, non-alcoholic fatty liver disease, NAFLD
Graphical abstract
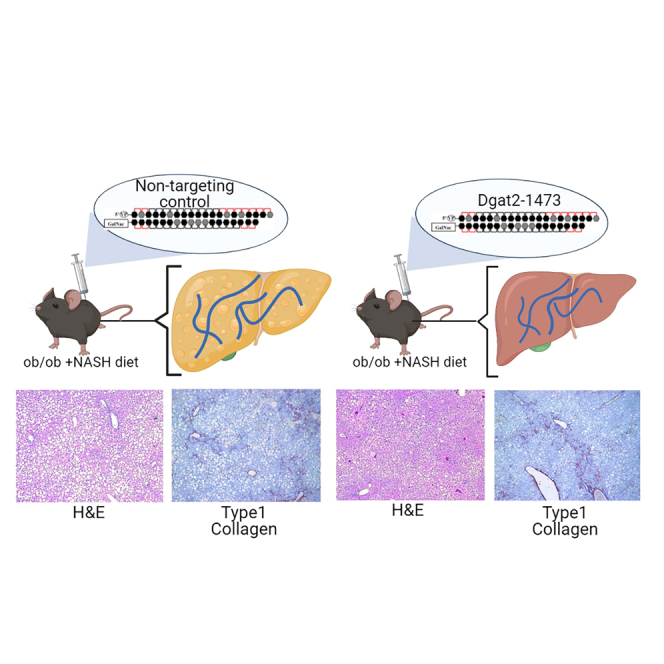
Liver fat overload in obesity and type 2 diabetes can proceed to severe inflammation, fibrosis, and liver failure. A chemically stabilized, GalNAc-conjugated siRNA targeting lipid synthesis enzyme DGAT2 was identified that decreases liver fat by 85% following single subcutaneous injection in mice; however, inflammation and scarring are surprisingly unaffected.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is characterized by triglyceride accumulation within hepatocytes (hepatic steatosis) and affects up to 1 billion humans worldwide.1 NAFLD by itself can be relatively benign but is often part of a sequel of liver conditions in obesity and type 2 diabetes (T2D) varying in severity of injury. Most notably, NAFLD can precede the occurrence of nonalcoholic steatohepatitis (NASH), which is associated with increased liver inflammation through resident and infiltrating immune cell activation as well as scarring and fibrosis through activation of resident stellate cells to produce collagen.2 Currently, it is estimated that ∼20%–25% of NAFLD patients will progress to develop NASH and, if left untreated, the risk of developing cirrhosis, severe liver failure, and hepatocellular carcinoma greatly increases.3, 4, 5, 6 Collectively, the various pathologies associated with dysfunctional liver lipid metabolism, inflammation, and fibrosis represent a huge and burgeoning burden on the health care system, with increasing rates among young adults and even children projected to continue.3,4 While lifestyle improvements, including weight loss, can alleviate NASH, behavioral modifications are difficult to execute and have not provided a solid solution to the problem.4, 5, 6 Despite the high prevalence of NASH, there is no US Food and Drug Administration (FDA)-approved therapeutic agent that can specifically alleviate it,7, 8, 9 indicating an urgent unmet medical need.9
One of the major unresolved questions in this field is whether the hepatic steatosis in NAFLD alone drives the inflammation and fibrosis that occur in NASH, independent of other liver perturbations that occur in obesity and T2D. If this is the case, might therapeutic targeting of the hepatic steatosis in NAFLD alleviate its progression to NASH? Strong support for the idea that hepatic steatosis is the major independent initiator of NASH has been derived from human genome-wide association studies showing its strong link with single-nucleotide polymorphisms in genes related to lipid metabolism. These include PNLP3, TM6SF2, LYPLAL1, GCKR, and PPP1R3B, which can harbor polymorphisms that not only track with steatosis but also with steatohepatitis and hepatic fibrosis.10, 11, 12 These findings strengthen the idea that hepatic steatosis drives the inflammation/fibrosis aspects of the pathology and have resulted in major investment toward therapeutic targets within lipid metabolism pathways. On the other hand, clinical trials13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 with agents that target such proteins, for example thiazolidinediones (TZDs), FXR agonists,18, 19, 20, 21 and acetyl coenzyme A (CoA) carboxylase (ACC) inhibitors,26, 27, 28, 29 have not succeeded, due to either adverse effects or lack of improvement in inflammation and fibrosis.13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 These failures suggest the possibility that additional perturbations of gene products in the inflammation and fibrosis pathways, independent of those initiated by hepatic steatosis, are critical to the progression of NASH. Thus, hepatic steatosis may be required but not sufficient to initiate NASH.
On a similar note, candidate therapeutics designed to attack the end-stage inflammation and fibrosis pathways of NASH, such as C-C chemokine receptor (CCR)2/5 antagonists,23,24 ASK1 inhibitors,25 or caspase inhibitors,31,32 have also failed to achieve FDA approval.7,33,34 Together, these unfavorable results raise the likelihood that the multiple pathways contributing to NASH must be simultaneously targeted by therapeutics to be successful.
Based on the above considerations, the aims of the present study were 2-fold: first, we addressed the unsolved question of whether specific inhibition of liver triglyceride synthesis would also diminish inflammation and fibrosis in the livers of a novel mouse model whereby NASH is rapidly developed by high fat-high cholesterol-high fructose (GAN) diet feeding. For this aim, we targeted diacylglycerol (DAG) acyltransferase 2 (DGAT2), which catalyzes the last step in the synthesis of triacylglycerol through the esterification of fatty acyl-CoA to DAG.35 DGAT2 is expressed prominently in liver and adipose tissue, while the DGAT1 isoform is expressed mostly in intestine and much less so in other tissues.36 It has been shown previously that depletion of liver DGAT2 by antisense oligonucleotide (ASO) does alleviate hepatic steatosis in mouse37,38 and rat39 models, but the effects on inflammation and fibrosis were not evaluated in these studies. In a study using a choline deficient diet-induced mouse NASH model, liver damage appeared to actually be increased by liver DGAT2 loss.38 In two short-term human trials, pharmalogical40 or ASO41 targeting of DGAT2 diminished liver triglycerides and suggested lower liver damage and fibrosis,40,41 but the degree to which targeting DGAT2 alone will yield effective therapy for the severe inflammation and fibrosis of NASH is still not clear.
To approach the above question, a second aim of this study was to identify and advance the therapeutic potential of a chemically modified short interfering, double-stranded RNA compound to silence Dgat2.42,43 RNA interference (RNAi) is an extremely efficient tool for gene silencing that leverages the ability of a designed small interfering RNA (siRNA) to bind to a target mRNA and cause its cleavage by the cellular RNA-induced silencing complex (RISC).42,44 Advances in siRNA design and chemistry combined with suitable delivery platforms have overcome many of the previous limitations associated with non-modified siRNAs.45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 This has led to a remarkable potential for RNAi therapeutics.46,47,51,52,63, 64, 65, 66, 67 In particular, the use of a synthetic triantennary GalNAc conjugation to the double-stranded siRNA has allowed delivery of the GalNAc-siRNAs safely and efficiently to the liver via the GalNAc conjugate's high affinity for the asialoglycoprotein receptor (ASGPR).48,50, 51, 52,57,65, 66, 67, 68 A single subcutaneous injection of GalNAc-siRNA can achieve efficacy for >12 months in humans.46,47,65,67 We demonstrate here the identification of a therapeutically promising, highly stable, and active GalNAc-siRNA targeting Dgat2 mRNA that directs strong alleviation of hepatic steatosis in obese mice. The data suggest that advancement of this compound may lead to an effective therapy against NAFLD. However, our findings also suggest that the advent of inflammation and fibrosis in response to high-fat, high-cholesterol, high-fructose (GAN) diet-induced NASH in mice is not alleviated simply by substantial inhibition of triglyceride accumulation.
Results
Screening of chemically modified siRNAs targeting both human and mouse Dgat2 transcripts
The chemical modifications and their locations among the nucleotides in the RNA compounds used in this study are shown in Table S1. The chemically modified siRNAs are asymmetric compounds composed of 18-nucleotide, modified RNA duplexed with a single-stranded 3′, two-base extension on the guide strand. Modifications at the 2′ OH position of ribose in the siRNA are modified with 2′-O-methyl or 2′-fluoro to provide stability, and the 3′ end of the passenger strand is conjugated to a hydrophobic cholesterol through a tetraethylene glycol linker to promote membrane binding. The single-stranded tail also contains phosphorothioate linkages to promote cellular uptake by a mechanism similar to that of ASOs.37,39,42 The above chemical modifications (phosphorothioates, ribose modifications) contribute to overall hydrophobicity and are essential for compound stabilization and efficient cellular internalization. The construct used for the in vitro screening in Figure 1 is conjugated to cholesterol for added hydrophobicity. Such hydrophobic siRNAs have been shown to bind to a wide range of cells and are readily internalized without the requirement of a transfection reagent.42,53,54,56,58,59,69,70
Figure 1.
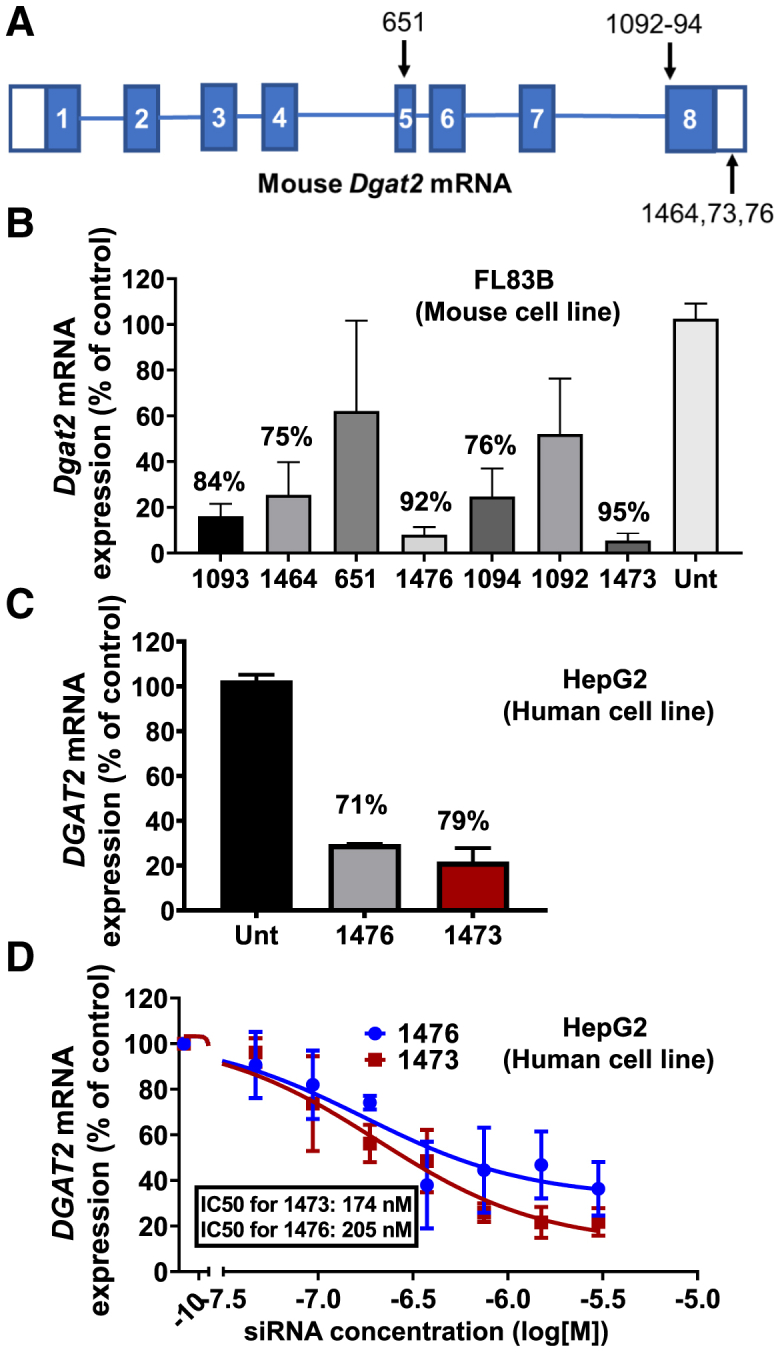
Identification of two potent chemically modified siRNAs that silence both human and mouse Dgat2 transcripts in cultured hepatocytes
(A) Targeted locations on the mouse Dgat2 transcript by the candidate compounds. (B) FL83B hepatocytes were treated with siRNA compounds (1.5 μM) for 72 h prior to analysis of Dgat2 mRNA levels to identify the most active siRNA sequences. (C) HepG2 human hepatocytes were treated with two best siRNA compounds (1.5 μM), from the previous mouse cell line screening, for 72 h prior to analysis of human DGAT2 mRNA levels to identify the most active siRNA sequences. (D) Dose response relationships for the two siRNAs (compounds 1476 and 1473) that showed the strongest silencing using human HepG2 cell line. The IC50 values were determined by using eight point serially diluted concentrations of the compounds starting from 1.5 μM.
To identify the siRNA sequences that have strong and potent silencing capability in vitro, several test sequences targeting different regions of mouse Dgat2 mRNA were generated by a custom algorithm designed to optimize predicted silencing efficiency of chemically modified RNAs (Figure 1A). Of the sequences identified, only those predicted to silence both mouse and human Dgat2 mRNA were then selected. This panel of siRNAs was synthesized and initially screened for silencing efficacy in FL83B mouse hepatocytes by their direct addition to the culture medium to a final concentration of 1.5 μM. Silencing effects on levels of Dgat2 mRNA and housekeeping (Hprt) gene mRNA after 72 h of treatment was performed with the QuantiGene assay. Five siRNAs were identified (Table S1) that effectively reduce mouse Dgat2 transcript in the mouse cell line compared with untreated control (Figure 1B). Additionally, to confirm the activity of the best two hits from previous mouse cell line screening against human DGAT2 transcript, human HepG2 cell lines were treated with these two siRNAs (denoted 1473 and 1476) with final concentration of 1.5 μM (Figure 1C). Further, to investigate the potency of 1473 and 1476 that showed the highest activity in this initial screening, a dose response relationship assay was carried out with HepG2 cells treated with eight concentrations of either compound 1473 or 1476 (1.5 μM–0.023 μM) for 72 h. These potency tests revealed half maximal inhibitory concentration (IC50) values of 174 nM and 205 nM for siRNAs 1473 and 1476 for silencing human DGAT2 mRNA, respectively (Figure 1D).
Strength and potency of silencing with GalNAc-conjugated 1473 in vivo
Next, the in vivo silencing capability and longevity of siRNA1473 were investigated. For this, non-targeting control (NTC) siRNA and siRNA 1473 were synthesized and conjugated to a GalNAc construct (Dgat2-1473)48,50, 51, 52,57,68 for liver-specific targeting (Figure S1) with a final chemical moiety as shown in Figure 2A. Groups of 8-week-old wild-type male C57BL6/J mice fed with chow diet were subcutaneously injected once with either NTC (10 mg/kg) or various concentrations of Dgat2-1473 (1 mg/kg, 3 mg/kg, 10 mg/kg) and were sacrificed 4, 8, and 12 weeks after the single injection (Figure 2B). Strikingly, at the 10 mg/kg dose of Dgat2-1473, silencing of Dgat2 mRNA in liver was nearly complete (87%, p = 0.0001) at 4 weeks and remained strong at 8 weeks and 12 weeks (79%, p < 0.0001) post single injection. Even at 3mg/kg, Dgat2-1473 provided 73% (p; 0.0002) silencing at 4 weeks and 60% (p:0.0013) at 8 weeks post injection. The lowest dose of 1 mg/kg Dgat2-1473 caused only about 50% (p = 0.02) or less silencing in the 4- to 8-week time period post injection and this was lost by 12 weeks (Figure 2B).
Figure 2.

Dgat2-1473 provides strong silencing for at least 12 weeks after a single subcutaneous injection in the livers of male C57BL/6 mice
(A) Representative cartoon of chemically modified, GalNAc-conjugated siRNAs that were used for in vivo studies. (B) Dose response relationships and longevity of silencing elicited by Dgat2-1473 that was subcutaneously injected into wild-type C57BL/6 mice (n = 3) on chow diet. The mice were treated with the indicated doses (10, 3, 1 mg/kg) of Dgat2-1473 or NTC compound (10 mg/kg) and sacrificed at the indicated time points (4, 8, 12 weeks) after injections. The mRNA levels of Dgat2 were normalized against the NTC group and 18S was used as the housekeeping gene for the calculations. (C) DNL-related gene expression changes upon Dgat2 silencing in wild-type C57BL6 mice. ∗p < 0.05, ∗∗p < 0.005.
While loss of DGAT2 protein by itself is expected to greatly decrease triglyceride synthesis and accumulation in liver, it has been previously reported that DGAT2 depletion also causes a surprising decrease in expression of enzymes in the de novo fatty acid synthesis pathway.37,39,40,71 Moreover, this extended transcriptional effect of DGAT2 loss was attributed to decreased Srebf1, a transcription factor that controls expression of enzymes involved in fatty acid metabolism. It is not known how Dgat2 silencing leads to loss of Srebf1. We therefore investigated this extended effect of DGAT2 in the above experiment and could confirm a strong reduction in expression of de novo lipogenic genes such as fatty acid synthase (Fasn) and stearoyl-CoA desaturase-1 (Scd1) (Figure 2C). In addition, expression of Srebf1 mRNA was blunted, consistent with the previous work, suggesting important signaling mechanisms at play in response to DGAT2 loss.
Dgat2-1473 silencing in human hepatocytes engrafted in a mouse model
Since Dgat2-1473 was designed to silence Dgat2 mRNA in both human and mouse cells, it was important to determine if this predicted dual targeting actually occurs in vivo. We employed a “humanized” immunocompromised mouse model (NSG-PiZ) that takes advantage of a genetic modification that blunts the mouse hepatocytes’ ability to regenerate following injury, and therefore provides an advantage for engrafted normal human hepatocytes to predominate.72 Four-week-old NSG-PiZ male mice that had been intravenously treated with a monoclonal mouse-specific anti-Fas antigen (CD95) antibody (2 μg) were injected with 1 million human hepatocytes into the spleen. After 5 weeks on chow diet (Figure 3A), a human serum albumin ELISA and human albumin immunoblotting were performed to validate the human hepatocyte engraftment (Figures 3B and 3C). Animals were then subcutaneously injected once with 10 mg/kg NTC or Dgat2-1473 and sacrificed after 1 week on chow diet (Figure 3A). Human and mouse transcript-specific qPCR primers were used to assess the Dgat2 mRNA levels in total RNA samples isolated from livers of these mice. The results shown in Figure 3 demonstrate that Dgat2-1473 provided substantial silencing of both mouse (Figure 3D) and a strong silencing trend in human (Figure 3E) DGAT2 mRNA levels by 85% in the livers of the human hepatocyte-engrafted NSG-PiZ mice.
Figure 3.
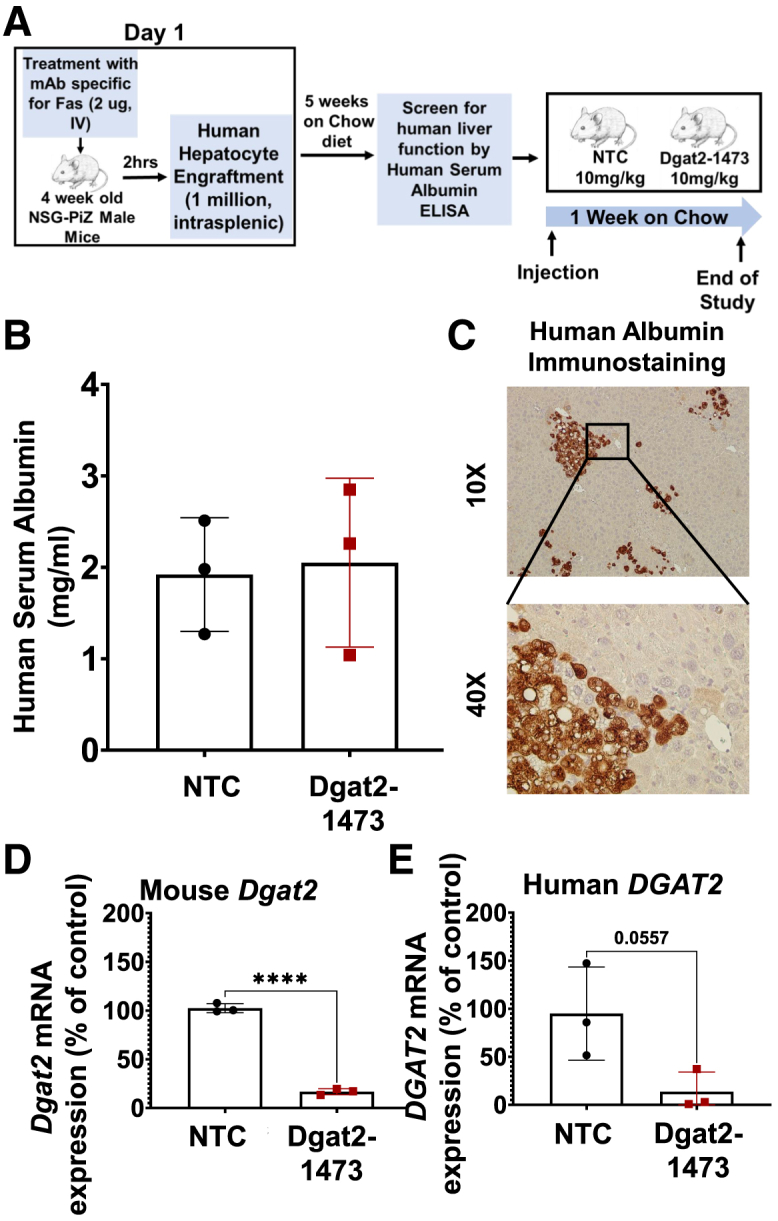
Dgat2-1473 silences the human Dgat2 transcript in human hepatocyte-engrafted NSG-PiZ mice
(A) Experimental procedure for generating human hepatocyte-engrafted NSG-PiZ mouse model. (B) Human serum albumin levels in the plasma of these engrafted mice were determined after being randomized from the study groups. (C) Immunostaining of liver sections for human albumin for confirmation of engraftment. (D) Mouse Dgat2 mRNA levels 1 week after injection (E) HumanDGAT2 mRNA levels 1 week after the injection of mice(n = 3) with Dgat2-1473. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005.
Testing of Dgat2-1473 in a genetically obese NASH mouse model
We next investigated whether Dgat2 silencing has a beneficial effect in the livers of a severe obese, NASH mouse model. Genetically obese C57BL6/J (ob/ob) male mice were subcutaneously injected with the either NTC (10 mg/kg) or Dgat2-1473 (10 mg/kg) and fed with Research Diet D09100310 (GAN diet) to induce NASH. After 3 weeks, the mice were sacrificed, and the tissues were harvested (Figure 4A). The virtually complete DGAT2 protein knockdown in the livers of the Dgat2-1473-treated mice was documented by immunoblotting (Figure 4B) and Dgat2 mRNA loss was confirmed by qPCR (Figure 4C). The body weights of the mice at the start and the end of the study revealed decreased weight gain in the Dgat2-1473-injected group (Figure 4D). Hematoxylin-eosin (H&E) staining of liver sections from these mice showed that the Dgat2-1473-injected group had fewer and smaller lipid droplets compared with the control NTC group (Figure 4E). This was also supported by a significantly lower liver weight to body weight ratio (Figure 4F) and lower triglyceride content per milligram of protein (Figure 4G) in the livers of the Dgat2-1473-injected group without changing plasma lipids (Figure S2). Interestingly, this triglyceride lowering did not cause an accumulation of diglycerides in the pathway, but rather it shunted fatty acids toward phospholipid synthesis (Figure S3). Thus, the decreased liver triglyceride and liver weights in the Dgat2-1473-treated, obese mice can account for much or all of the loss of body weight in these mice.
Figure 4.
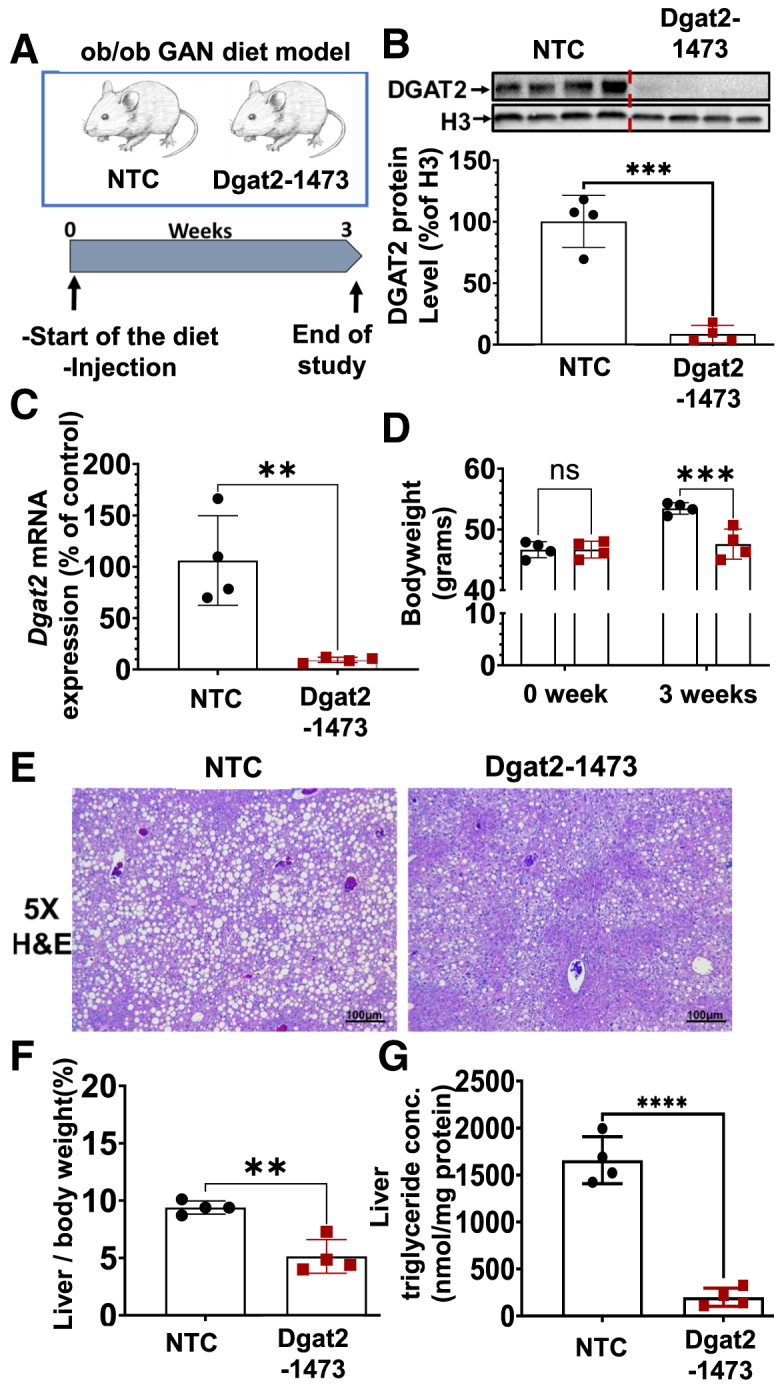
Single subcutaneous injection of Dgat2-1473 alleviates liver fat accumulation and decreased liver-to-body weight ratio in a genetically obese NASH mouse model
(A) Ten-week-old male genetically obese C57BL6/J (ob/ob) mice (n = 4) were injected subcutaneously with either NTC (10 mg/kg) or Dgat2-1473 (10 mg/kg) and provided a NASH-inducing diet (GAN diet) for 3 weeks. After 3 weeks, mice were sacrificed. (B) DGAT2 protein levels in liver 3 weeks after NTC or Dgat2-1473 injection. (C) Dgat2 mRNA level changes in liver in response to either Dgat2-1473 or NTC. (D) Body-weight difference comparison (start versus 3 weeks on GAN diet). (E) Histological comparison of liver H&E sections of the groups. (F) Liver weight to body weight ratio between groups. (G) Liver total triglyceride measurements via lipidomics. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005.
Gene expression in livers of NASH mice injected with Dgat2-1473
To investigate the transcriptional changes in the ob/ob NASH mice subcutaneously injected either with NTC or Dgat2-1473, total RNA was isolated from the livers of these mice and assessed for changes in gene expression profiles by RNA sequencing (RNA-seq). A poly A selection of mRNA species was used for rRNA removal to enrich our dataset. Principle component analysis (PCA) for RNA-seq showed a clear segregation and clustering of NTC and Dgat2-1473-injected groups (Figure S4A). Next, it was important to assess the possible off-target silencing of Dgat2-1473. For that, the seed enrichment p value was calculated using a Fisher's exact test comparing the prevalence of the seed (guide 2–8) target in the 3′ UTR of genes that were downregulated with the prevalence of the seed target in the 3′ UTR of genes that were not downregulated. The analysis revealed that Dgat2-1473 did not elicit detectable off-target silencing among all the reads that were collected by RNA-seq (Figure S4B).
To better visualize the upregulated (Figure 5A) and downregulated (Figure 5B) differentially expressed (DE) genes and pathways, heatmaps were generated. The data generated by RNA-seq was first filtered to eliminate genes whose expression level was not above 10 reads in any sample and then DESeq2 was employed to determine DE genes, using an adjusted p value of 0.05 as the cutoff and requiring at least a 1.5-fold change, up or down. The list of DE genes was then analyzed using the enrichGO function in the clusterProfiler package. The pathways were analyzed using the simplify function with options of a p value cutoff of 0.05. This was followed by manually specified merging of similar pathways to produce heatmaps. DE genes were clustered according to the pathways they were involved in and the pathways displayed on the left column alongside the heatmap. Color scheme for the DE gene presentation was produced by Z-scoring method (−2 to +2 standard deviation from the mean). In these heat maps, DE genes were clustered depending on the pathways that are shown in a color-coded manner. This analysis revealed 34 upregulated (Figure 5A) and 43 downregulated (Figure 5B) pathways that are significantly altered in the livers of ob/ob NASH mice treated with Dgat2-1473.
Figure 5.

Dgat2 silencing by Dgat2-1473 elicits significant changes in many pathways and genes in the livers of genetically obese mice with NASH
DEBrowser data that came from the DolphinNext RNA-seq pipeline (Figure 5) was first filtered to eliminate genes whose expression level was not above 10 in any sample, and then DESeq2 was employed to determine DE genes, using an adjusted p value of 0.05 as the cutoff and requiring at least a 1.5-fold change, up or down. The list of DE genes was then analyzed using the enrichGO function in the clusterProfiler package. The pathways were simplified using its simplify function with options of p value cutoff of 0.05. This was followed by manually specified merging of similar pathways to produce heatmaps. DE genes were clustered according to the pathways they were involved in, and the pathways (color coded) were displayed on the left column alongside the heatmap. Color scheme for the DE gene presentation was produced by Z-scoring method (−2 to +2 standard deviation from the mean) on expression levels normalized for sample depth by DESeq2. Heatmap representation of (A) upregulated and (B) downregulated DE genes clustered by the pathways in which they are involved.
Next, further analysis was performed on the fatty acid metabolism pathway-related genes in livers of ob/ob NASH mice injected with either NTC or Dgat2-1473. Major genes that play a role in fatty acid biosynthesis were downregulated significantly, indicated by black arrows on the pathway heat map such as Chrebp, Fasn, Acly, and Elovl6 (Figure 6A). Changes in the expression of genes either controlling or within the de novo lipogenic pathway, such as Srebf1, Srebf2, Fasn, Scd1, Chrebp, were confirmed with qPCR analysis. Importantly, although the de novo lipogenesis (DNL)-related genes such as Fasn and Scd1 were downregulated, the gene expression of transcription factor Srebf1c did not change in the livers of ob/ob mice that were fed with GAN diet and treated with Dgat2-1473 compared with the control NTC-injected group (Figure 6B). This differs from what was observed in lean mice, as noted above (Figure 2C), and in previous reports.37,39,40,71 When this phenomenon was further investigated by immunoblotting of the processed, transcriptionally active, nuclear SREBP1c fragment versus the unprocessed, cytosolic SREBP1c protein levels, a remarkable correlation between downregulation of DNL gene expression and the decrease in processed, nuclear SREBP1c protein levels (Figure 6C) was detected. In addition, protein levels of the transcription factor Chrebp, known also to regulate genes in the lipogenic pathway, were reduced in correlation with SREBP1c processing (Figure 6D). Noteworthy is the fact that it has been reported that Chrebp expression is strongly correlated with SREBP1c processing in liver.73
Figure 6.
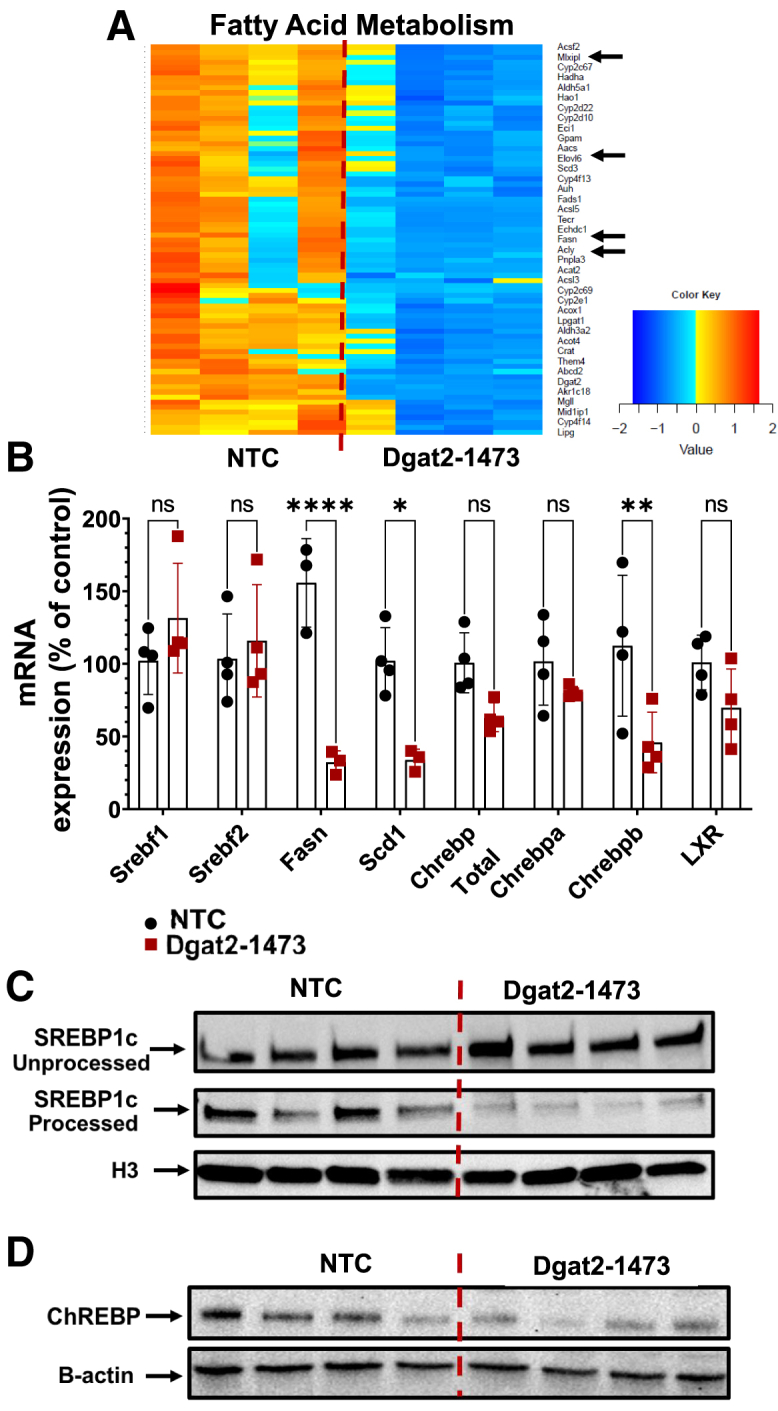
Targeting liver DGAT2 by Dgat2-1473 in genetically obese NASH mice causes downregulation of major genes in the fatty acid metabolism pathway, correlating with a decrease in SREBP1c processing and ChREBP protein expression
(A) Heatmap representation of DE liver genes in fatty acid metabolism pathway (major DNL genes in the pathway highlighted by the black arrows). (B) qPCR confirmation of the RNA-seq data for important genes in fatty acid metabolism pathway. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005. (C) Measurements of both unprocessed (cytosolic) and processed (nuclear) SREBP1c protein levels in liver. (D) ChREBP protein level measurements in liver. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.00005.
Further investigation of other major metabolic pathways such as acyl-CoA metabolism (Figure 7A), cholesterol metabolism (Figure 7B), and carbohydrate metabolism (Figure 7C) showed that Dgat2 silencing in the liver of ob/ob NASH mice resulted in downregulation of major genes that play a role in the biosynthesis of the metabolites in these pathways (black arrows).
Figure 7.
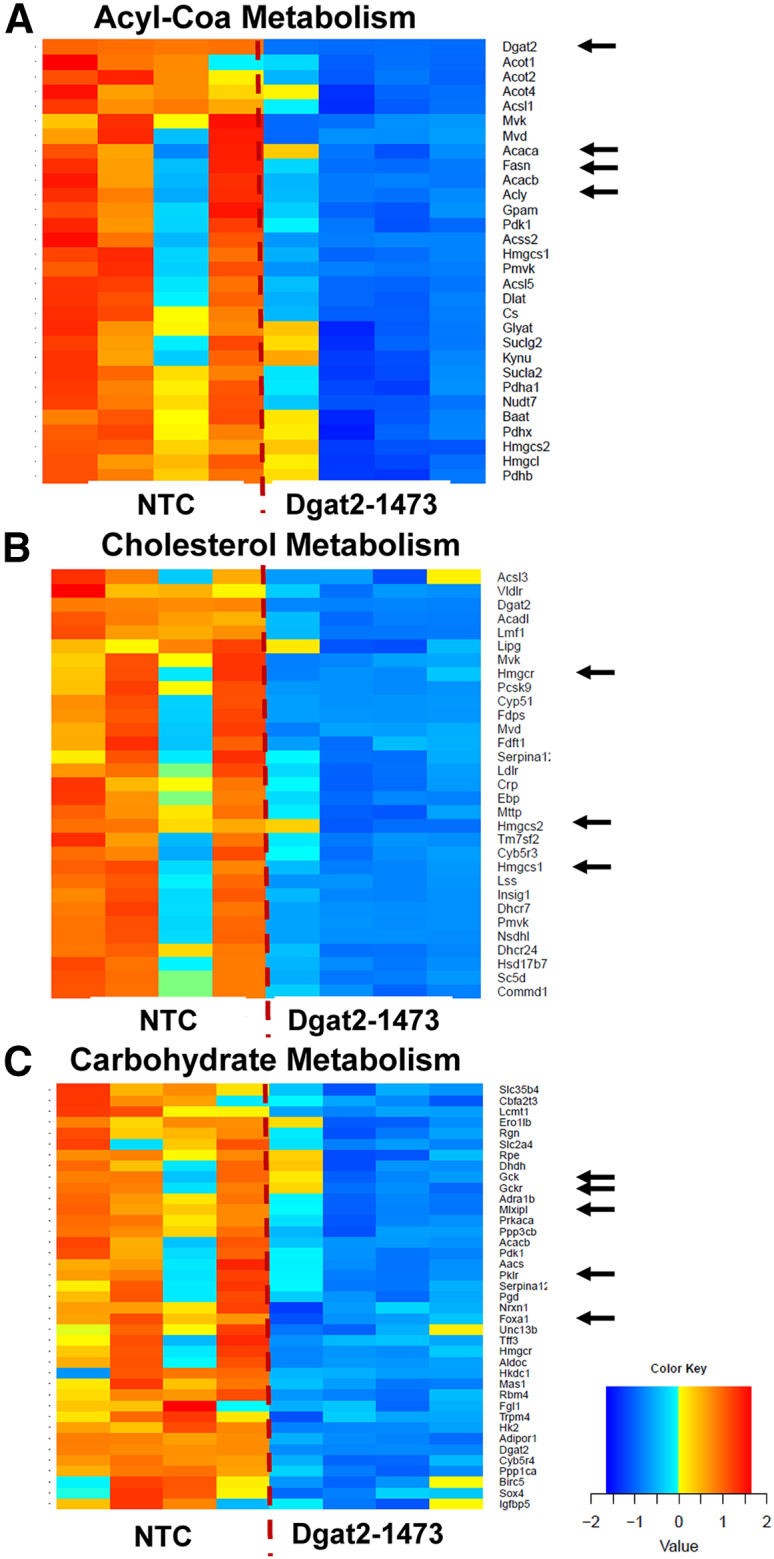
Dgat2 silencing in liver of genetically obese NASH mice causes downregulation of major metabolic pathways
Heatmap representation of DE genes in (A) acyl-CoA metabolism, (B) cholesterol metabolism, and (C) carbohydrate metabolism (major genes in the pathways that are involved with the synthesis are highlighted by the black arrows).
Liver inflammation and fibrosis in Dgat2-1473-treated NASH mice
To investigate the inflammation and fibrosis states in our NASH mouse model, the histological presence of collagen fibers in liver sections from ob/ob NASH mice injected with NTC versus Dgat2-1473 were assessed by both trichrome staining and immunohistochemistry (IHC) analysis for type 1 collagen. The histological analysis revealed no significant alleviation of fibrosis in the livers of Dgat2-1473-injected ob/ob NASH mice (Figures 8A and S5). The collagen protein levels in the livers were measured by immunoblotting and this analysis also showed no significant difference in Dgat2-1473-injected mice (Figure 8B). The extent of hepatocyte cell death and damage as assessed by plasma alanine aminotransferase (ALT) activity also indicated no difference due to the Dgat2-1473 treatment (Figure 8C). The gene expression levels of genes in the inflammation and fibrosis pathways, such as CCL2, IL1b, Opn, Timp1, Acta2, and Col1a1, showed no improvements in Dgat2-1473-injected ob/ob NASH mice compared with the NTC-injected ob/ob NASH mice (Figure 8D). Further analysis of the expression levels of genes in the inflammation pathway, such as M1 macrophage markers (Figure S6A), M2 macrophage markers (Figure S6B), as well as chemokines and their receptors (Figure S6C), showed no improvements in Dgat2-1473-injected ob/ob mice compared with the NTC-injected ob/ob mice. Finally, no improvements in plasma cytokine levels upon Dgat2 silencing were observed (Figure S7).
Figure 8.
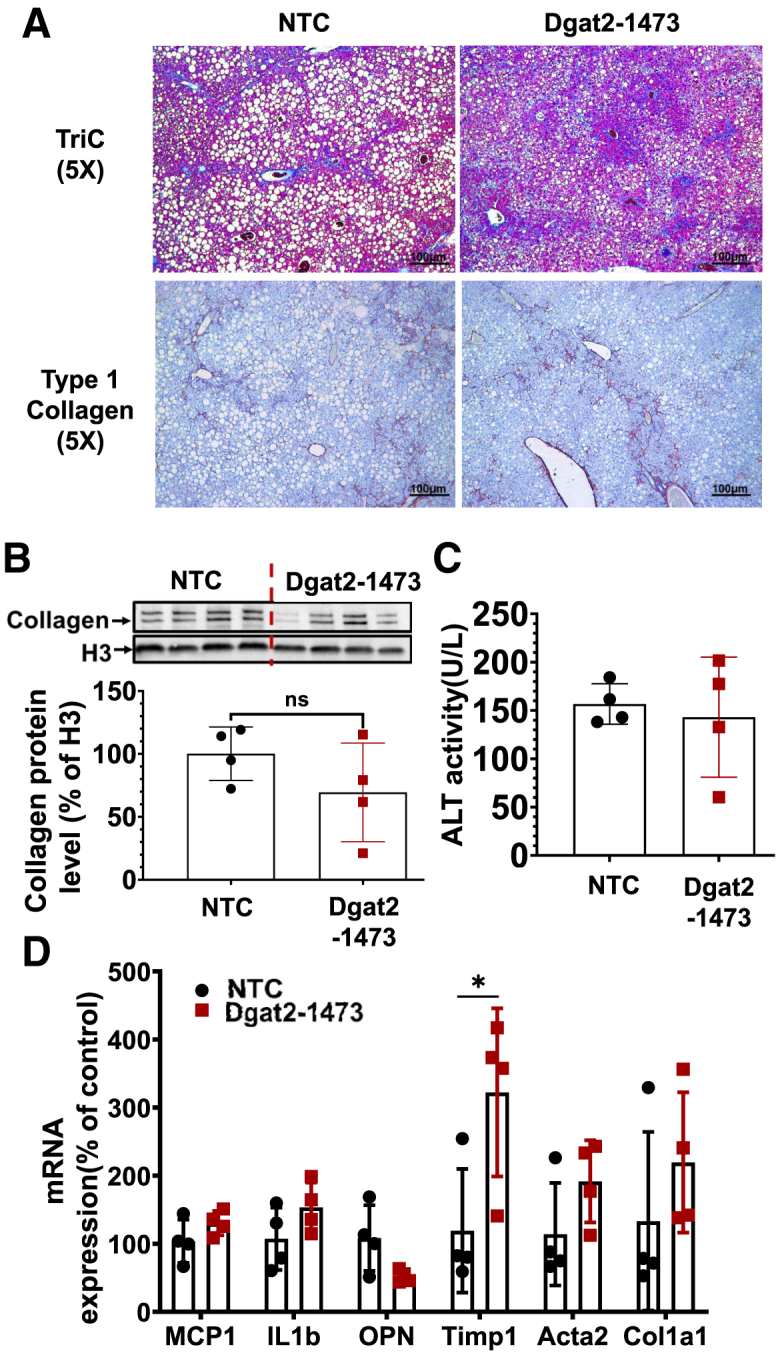
Dgat2 silencing does not significantly alleviate the inflammation and fibrosis in the liver of genetically obese NASH mice
Ten-week-old genetically obese ob/ob mice (n = 4) were injected subcutaneously with either NTC (10 mg/kg) or Dgat2-1473 (10 mg/kg) and provided a NASH-inducing diet (GAN diet) for 3 weeks. After 3 weeks, mice were sacrificed. (A) Histological examination of fibrosis via trichrome staining and type 1 collagen IHC. (B) Type I collagen protein levels in whole-liver tissue. (C) Plasma ALT measurements. (D) Inflammation and fibrosis related gene expression levels. ∗p < 0.05, ∗∗p < 0.005.
Discussion
A major finding of this study is the identification and characterization of Dgat2-1473, a therapeutically promising siRNA targeting Dgat2 mRNA in liver. This compound was derived from screening siRNA sequences complementary to both mouse and human Dgat2 in a mouse hepatocyte cell line, revealing that at 1.5 μM the 1473 sequence caused 95% silencing of Dgat2 mRNA and in human hepatocyte cell line it exhibited an IC50 of less than 200 nM (Figure 1). Remarkably, a single subcutaneous injection of Dgat2-1473 at 10 mg/kg elicited a 79% loss of Dgat2 mRNA for at least 12 weeks in the livers of male C57BL/6J mice (Figure 2B), which is the longest duration of silencing by single dosing of a siRNA or ASO reported in mice.37,52,74, 75, 76 Dgat2-1473 was also effective in silencing human DGAT2 mRNA in a humanized mouse model (Figure 3). Dgat2-1473 embodies many advancements in applying chemical modifications into native siRNA, including 2′ ribose modifications, a 5′-(E)-vinylphosphonate moiety, and phosphorothioate linkages, which result in greater stability and potency of silencing compounds.42,45,53,54,56,59,61,62,77 A particularly important feature of Dgat2-1473 is the pattern of 2-O-methyl and fluoro modifications at the 2' ribose positions, which are designed to optimize stability without compromising silencing potency by using limited 2'fluoro content.45,53,54,58,59,61,62,77 It has been shown that the higher 2′-OMe/2′-F content ratio results in higher potency of chemically modified RNAs.53,62 These improvements in chemical modification patterns for siRNA have enabled FDA approval of the first RNAi therapeutics and potentially open the way for many others in the future. Considering the comparatively long-lasting efficacy of current GalNAc-conjugated oligonucleotide therapeutics in human trials versus their relatively short-lived effects in mouse studies,41,46,47,66 our Dgat2-1473 compound may also be expected to elicit long-term silencing with single administration in humans.
An important question we addressed is whether the strong siRNA-mediated silencing that is observed in livers of lean mice is attenuated in fatty livers with damaged, inflamed hepatocytes and increased fibrotic extracellular matrix, as occurs in human NASH. For this purpose, we used an extremely obese, steatotic mouse model in which mice reach 50 g or more in body weight (Figure 4D) with heavy liver fibrosis (Figure 8A) during the course of a short study. A single injection of Dgat2-1473 indeed did achieve a similar strong and potent silencing of liver Dgat2 in this ob/ob NASH model (Figures 4B and 4C). Importantly, such silencing of liver Dgat2 by Dgat2-1473 in mice caused the expected marked inhibition of hepatic triglyceride levels and alleviation of hepatic steatosis as measured by histology (Figure 4G). This level of liver triglyceride reduction is similar to what has been reported in rodent models with liver-specific DGAT2 KO,71 AAV-shRNA silencing of Dgat2,78 silencing of Dgat2 with ASOs,37, 38, 39 or inhibition of DGAT2 with a small molecule inhibitor.40,78 These data show that not just prevention but actual reversal of hepatic steatosis occurs upon DGAT2 loss, as the obese mice that were treated with Dgat2-1473 in this study were already afflicted with severe hepatic steatosis and metabolic syndrome (Figure 4). The large attenuation of liver triglyceride levels and liver weight (Figure 4F) in these Dgat2-1473-injected mice were indeed sufficient to account for the reduced body-weight gain in this group compared with mice treated with NTC (Figure 4D). Taken together, Dgat2-1473 is effective at both strong Dgat2 silencing and alleviation of hepatic steatosis (NAFLD) in an extreme model of mouse obesity and NASH.
A previous unexpected finding associated with hepatic DGAT2 loss was an apparent decrease in DNL based on greatly decreased expression of genes encoding enzymes such as Fasn and Scd1 in this pathway.39,40,71 This phenomenon was linked to a reduction in expression of Srebf1, encoding a key transcription factor that regulates genes related to lipid synthesis and metabolism.79, 80, 81, 82 While we did not observe this effect in the obese mouse NASH model, conversion of unprocessed cytosolic SREBP1c protein to its transcriptionally active fragment that locates to the nucleus was greatly inhibited. This conversion is catalyzed by proteolytic cleavage (proteases S1P and S2P) in the Golgi membrane, preceding translocation of the cleaved protein into the nucleus to regulate lipid metabolism-related gene expression.79,81,82 Our data (Figure 6) thus reveal a powerful mechanism to explain the connection between DGAT2 loss and the downregulation of many genes that control lipid synthesis.
Adding to a role for SREBP1c processing in how Dgat2 silencing causes marked decreases in expression of genes that control many metabolic pathways is our finding that Chrebp expression is also downregulated in the Dgat2-1473-treated obese mice. The essential role of ChREBP in lipid metabolism has been investigated in rodents by gene deletion83 and silencing with adenovirus84 as well as ASO.85 In summary, these published studies showed that ChREBP is required for the increased lipogenic gene expression exemplified by Fasn, Scd1, and Acc1 in response to high carbohydrate intake. It also has been shown that Chrebp plays an important role in enhancing both Srebf1 gene expression and processing by an unknown mechanism.73 In contrast to a previous study in a different mouse model, we did not observe a decrease in Srebf1 mRNA in the ob/ob-GAN diet model even though we did detect a similar downregulation of DNL genes in the Dgat2-1473-injected group(Figures 6A and 6B). However, we unexpectedly observed a marked inhibition by Dgat2-1473 of the processing of SREBP1c leading to a decreased level of transcriptionally active fragment (Figure 6C). This downregulation of processed SREBP1c strongly correlated with a decreased Chrebp protein expression pattern, suggesting that the downregulation of DNL gene expression upon Dgat2 silencing,37, 38, 39 deficiency,71 or inhibition40,78 is closely linked to these changes in the major transcription factors controlling lipid metabolism (Figures 6C and 6D). Additionally, in a previous report, a genetically driven reduction of phosphatidylcholine (PC) levels induced the processing of cytosolic SREBP1c and increased lipid synthesis.86 Here a remarkable increase in PC levels (Figure S3B) in the Dgat2-1473-injected group was observed and this may explain the lower levels of processing of SREBP1c protein.
A critical question in liver metabolism is whether the hepatic steatosis in NAFLD is sufficient to drive the inflammation and fibrosis that develops in NASH independent of any other factors in obesity that might directly activate Kupffer or stellate cells. This unresolved issue raises another crucial question related to strategies for drug development against NASH: might hepatic steatosis as sole therapeutic target be effective in alleviating NASH? In the studies presented here, the marked reduction in hepatic steatosis did not prevent the development of inflammation and fibrosis in the livers of Dgat2-1473-injected ob/ob mice on the GAN diet. Our assessments of inflammation and fibrosis showed no significant change in collagen accumulation (Figures 8A, 8B, and S5), plasma ALT levels (Figure 8C), or in expression of genes in the inflammation pathway (Figures 8D and S6). There are conflicting reports in the literature about the effect of Dgat2 silencing or inhibition on inflammation and fibrosis pathology in liver.37,38,40,41,78 One study showed that DGAT2 loss actually elevated inflammation and fibrosis in the livers of MCD diet-fed mice in which NASH is induced rapidly.38 Another study showed no change in plasma ALT levels in placebo versus Dgat2 targeting ASO-treated human subjects.41 On the other hand, a small molecule inhibitor of DGAT2 elicited alleviation of high plasma ALTs and aspartate transaminase (ASTs) in early human clinical trials. However, based on the modest size and time frame of this latter study, further work is needed to confirm these findings.40 Interestingly, while our study was in the review process, a phase 2 clinical trial of a DGAT2 small molecule inhibitor was published supporting our conclusions on inflammation and fibrosis not changing upon DGAT2 inhibition.87 It is possible that mouse models do not exactly reflect the mechanisms at play in the human liver physiology of NASH.
Another possibility is that although our mouse model seems to mimic human NASH more than other models due to the presence of obesity as well as metabolic syndromes such as strong insulin resistance, hyperglycemia, and hyperinsulinemia, it is a metabolic model where mice are extremely hyperphagic. Our use of a genetic model of leptin deficiency is also unlike the typical human obesity condition in which leptin levels are high.86 To this point, leptin itself has metabolic effects on whole-body metabolism beyond its ability to suppress appetite.87 Finally, our NASH model is a very rapidly developing NASH model that also does not fully align with human NASH progression. This issue of rapid development could also explain the Dgat2-1473’s inability to prevent the development of fibrosis. Although the general outcomes and phenotype in terms of NASH in this mouse model are similar to the human disease state, clearly it is not an exact model of human NASH. This issue may explain the fact that although Dgat2-1473 treatment alleviated fatty liver, it failed to prevent the development of fibrosis because there is a constant overwhelming intake of toxic metabolites such as cholesterol from the GAN diet and these mice do not have enough time to recover from already fat-accumulated liver.
In summary, the present studies have yielded a promising siRNA compound, Dgat2-1473, which is highly effective in alleviating NAFLD in a model of extreme obesity in mice. Silencing Dgat2 expression by Dgat2-1473 is long lasting after a single subcutaneous injection and leads to strong downregulation of fatty acid synthesis as well as triglyceride synthesis in liver. Our data also indicate that attenuating NAFLD alone may not be fully effective in alleviating the inflammation and fibrosis in NASH, and that targeting multiple pathways in parallel may be necessary for an optimal therapeutic effect.
Materials and methods
Animals and siRNA administration
Animal experiments were performed in accordance with animal care ethics approval and guidelines of University of Massachusetts Medical School Institutional Animal Care and Use Committee (IACUC, protocol number A-1600-19). For in vivo dose response and KD longevity of Dgat2-1473, wild-type C57BL6/J male mice (n = 3) were injected doses of 1, 3, or 10 mg/kg or 10 mg/kg NTC subcutaneously once and sacrificed at time points of 4, 8, and 12 weeks after injections. For NASH studies, 10-week-old genetically obese (ob/ob) male mice (n = 4) were injected subcutaneously either with NTC or Dgat2-1473 and put on a NASH-inducing GAN diet (Research Diets D09100310) for 3 weeks. After 3 weeks, mice were sacrificed with CO2 and double-killed with a cervical dislocation.
Oligonucleotide synthesis
Oligonucleotides were synthesized using modified (2ʹ-F, 2ʹ-O-Me) phosphoramidites with standard protecting groups. 5′-vinyl tetra-phosphonate (pivaloyloxymethyl) 2′-O-methyl uridine 3′-CE phosphoramidite (VP) was used for the 5′-vinyl-phosphonate coupling when needed. All amidites were purchased from (Chemgenes, Wilmington, MA). Phosphoramidite solid-phase synthesis was done on a MerMade12 (Biosearch Technologies, Novato, CA) using modified protocols. Unconjugated oligonucleotides were synthesized on 500-Å long-chain alkyl amine (LCAA) controlled pore glass (CPG) functionalized with Unylinker terminus (Chemgenes, Wilmington, MA). Cholesterol-conjugated oligonucleotides were made on a 500-Å LCAA-CPG support, where the cholesterol moiety is bound to tetraethylene glycol through a succinate linker (Chemgenes, Wilmington, MA). GalNAc-conjugated oligonucleotides were grown on a 500-Å LCAA custom aminopropanediol-based trivalent GalNAc-CPG (Centernauchsnab, Minsk, Belarus). Phosphoramidites were prepared at 0.1 M in anhydrous acetonitrile (ACN), with added dry 15% dimethylformamide in the 2′-OMe-uridine amidite. 5-(benzylthio)-1H-tetrazole (BTT) was used as the activator at 0.25 M. Detritylations were performed using 3% trichloroacetic acid in dichloromethane. Capping reagents used were CAP A, 20% n-methylimidazole in ACN and CAP B, 20% acetic anhydride, 30% 2,6-lutidine in ACN (synthesis reagents were purchased at AIC, Westborough, MA). Sulfurization was performed with 0.1 M solution of 3-[(dimethylaminomethylene)amino]-3H-1,2,4-dithiazole-5-thione (DDTT) in pyridine (Chemgenes, Wilmington, MA) for 3 min. Phosphoramidite coupling times were 4 min.
Deprotection and purification of oligonucleotides
Conjugated oligonucleotides were cleaved and deprotected 28%–30% ammonium hydroxide and 40% aq. methylamine (AMA) in a 1:1 ratio, for 2 h at room temperature. The VP-containing oligonucleotides were cleaved and deprotected as described previously.88 Briefly, CPG with VP-oligonucleotides was treated with a solution of 3% diethylamine in 28%–30% ammonium hydroxide at 35⁰C for 20 h.
The solutions containing cleaved oligonucleotides were filtered to remove the CPG and dried under vacuum. The resulting pellets were resuspended in 5% ACN in water. Purifications were performed on an Agilent 1290 Infinity II HPLC system. VP- and GalNAc-conjugated oligonucleotides were purified using a custom 20 × 150-mm column packed with Source 15Q anion exchange resin (Cytiva, Marlborough, MA); run conditions were eluent A, 10 mM sodium acetate in 20% ACN in water; eluent B, 1 M sodium perchlorate in 20% ACN in water; linear gradient, 10%–35% B 20 min at 40°C. Cholesterol-conjugated oligonucleotides were purified using a 21.2 × 150-mm PRP-C18 column (Hamilton Co, Reno, NV); run conditions were eluent A, 50 mM sodium acetate in 5% ACN in water; eluent B, 100% ACN; linear gradient, 40%–60% B 20 min at 60°C. Flow was 40mL/min in both methods and peaks were monitored at 260 nm. Fractions were analyzed by liquid chromatography mass spectrometry (LC-MS), and pure fractions were dried under vacuum. Oligonucleotides were resuspended in 5% ACN and desalted by size exclusion on a 50 × 250-mm custom column packed with Sephadex G-25 media (Cytiva, Marlborough, MA), and lyophilized.
LC-MS analysis of oligonucleotides
The identity of oligonucleotides was verified by LC-MS analysis on an Agilent 6530 accurate mass Q-TOF using the following conditions: buffer A, 100 mM 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and 9 mM triethylamine (TEA) in LC-MS grade water; buffer B,100 mM HFIP and 9 mM TEA in LC-MS grade methanol; column, Agilent AdvanceBio oligonucleotides C18; linear gradient 0%–30% B 8 min (VP and GalNAc); 50%–100% B 8 min (cholesterol); temperature, 60°C; flow rate, 0.5 mL/min. LC peaks were monitored at 260 nm. MS parameters: source, electrospray ionization; ion polarity, negative mode; range, 100–3,200 m/z; scan rate, two spectra/second; capillary voltage, 4,000; fragmentor, 180 V.
Deprotection, purification, and LC-MS reagents were purchased from Fisher Scientific, Sigma-Aldrich, and Oakwood Chemicals.
Humanized liver
A human hepatocyte-engrafted NSG-PiZ mouse model was generated in collaboration with Michael Brehm as previously explained.72 Cryopreserved plateable human hepatocytes were purchased from BioIVT (https://bioivt.com/liverpool-cryoplateable-hepatocytes). The hepatocytes were collected from a female cadaveric donor and deidentified. The hepatocytes were stored in liquid nitrogen and thawed for transplant. The 4-week-old NSG-PiZ male mice were intravenously treated with a monoclonal mouse-specific anti-Fas antigen (CD95) antibody (2 μg). After 2 h, 1 million human hepatocytes were injected into the spleen by the intrasplenic injection. After 5 weeks on chow diet, a human serum albumin ELISA was performed to screen for human hepatocyte engraftment. Animals were then subcutaneously injected with 10 mg/kg NTC (n = 3) or Dgat2-1473 (n = 3) and sacrificed after 1 week on chow diet. Human- and mouse-specific qpcr primers were used to assess the transcript levels.
Histological analysis
For the IHC, one lobe of the liver was fixed in 4% paraformaldehyde and embedded in paraffin. Sectioned slides were then stained with trichrome and type I collagen (Southern Biotech) at the UMass Medical School Morphology Core. Photos from the liver sections were taken with an Axiovert 35 Zeiss microscope (Zeiss) equipped with an Axiocam CCI camera at the indicated magnification.
RNA isolation and qRT-PCR
Frozen liver tissue punches (25–50 mg) were homogenized in Trizol using the Qiagen TissueLyser II. Chloroform was added to the homogenate and centrifuged for 15 min at maximum speed. The clear upper layer was added to an equal volume of 100% isopropanol and incubated for 1 h at 4°C. After 10 min of centrifugation at maximum speed, the supernatant was discarded and 70%–75% ethanol was added to wash the pellet. After 15 min of centrifugation at maximum speed, the supernatant was discarded and the pellet was briefly dried in the hood before being resuspended in double-distilled H2O (ddH2O). RNA concentration was then measured by the Thermo Scientific NanoDrop2000 spectrophotometer. cDNA was synthesized from 1 μg of total RNA using iScript cDNA Synthesis Kit (Bio-Rad) and Bio-Rad T100 thermocycler. Quantitative RT-PCR was performed using iQ SybrGreen Supermix on the Bio-Rad CFX96 C1000 Touch Thermal Cycler and analyzed as previously described.89 18S served as a housekeeping gene for analysis. Primer sequences for qRT-PCR analysis can be found in Table S2.
In vitro screening of chemically modified siRNAs
The initial in vitro screening was carried out by plating 10,000 cells/well in a 96-well plate. The cells were treated with the candidate compounds in final concentration of 1.5 μM. Cells were treated for 72 h at 37°C, 5% CO2 in 3% FBS F-12K medium. After 72 h, mRNA levels were quantified using the QuantiGene 2.0 assay kit (Affymetrix, QS0011) as previously described.58,69,70 Briefly, cells were lysed in 300 μL of diluted lysis mixture composed of one part lysis mixture (Affymetrix, QG0503), two parts H2O, and 0.167 μg/μL proteinase K (Thermo Scientific, EO0491) for 1 h at 55°C. Cell lysates were mixed thoroughly, and 20 μL/well of each lysate was added to the capture plate with 60 μL of diluted lysis mixture without proteinase K. Probe sets for Dgat2 and HPRT (Affymetrix; 80246, 80003) were used according to the manufacturer's recommended protocol. Datasets were normalized to HPRT. The dose response investigation was performed using serially diluted doses of the two candidate compounds (1.500, 0.750, 0.375, 0.188, 0.094, 0.047, 0.023, 0.012 μM), each condition in triplicate, and then the QuantiGene 2.0 assay was carried out.
Immunoblotting
For protein expression analyses, frozen liver tissue (∼25 mg) was homogenized by the Qiagen TissueLyser II in a sucrose buffer (250 mM sucrose, 50 mM Tris-Cl pH 7.4) with 1:100 protease inhibitor (Sigma-Aldrich). The tissue lysates were denatured by boiling, separated on a 4%–15% sodium dodecyl sulfate/polyacrylamide gel electrophoresis gel (Bio-Rad), and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked with Tris-buffered saline with Tween (TBST) containing 5% milk for 1 h at room temperature and incubated with primary antibodies; DGAT2 (generously provided by Farese & Walther Lab, Harvard), histone H3, β-actin purchased from Cell Signaling, SREBP1c antibody purchased from Millipore, type I collagen antibody purchased from Southern Biotech, and ChREBP antibody purchased from Novus Bio. The blot was washed in TBST for 1 h, incubated at room temperature with corresponding second antibody at room temperature for 30 min, washed again, incubated with ECL (Perkin Elmer), and visualized with the ChemiDox XRS+ image-forming system.
Nuclear extraction protocol
A 50-mg sample of frozen liver tissue was homogenized in cold DPBS with 1:100 protease inhibitor in the Qiagen TissueLyser II for 3 min. The sample was then centrifuged at 1850 × g for 15 min. The fat cake was discarded, and the supernatant was saved as the cytoplasmic fraction. The pellet was resuspended in 500 μL of CER I buffer with 1:100 protease inhibitor and incubated for 10 min. After incubation, 55 μL of CER II buffer was added and then centrifuged at 14,000 × g for 5 min. The supernatant was discarded, and the pellet was resuspended in 250 μL of NER buffer with 1:100 protease inhibitor and vortexed every 10 min for a total of 40 min. The supernatant was then collected as the nuclear fraction.
Lipidomics analysis
Lipid species in liver samples were analyzed using multidimensional MS-based shotgun lipidomic analysis.90 In brief, each liver tissue sample homogenate containing 0.5 mg of protein (determined with a Pierce BCA assay) was accurately transferred to a disposable glass culture test tube. A pre-mixture of lipid internal standards (IS) was added prior to conducting lipid extraction for quantification of the targeted lipid species. Lipid extraction was performed using a modified Bligh and Dyer procedure,91 and each lipid extract was reconstituted in chloroform:methanol (1:1, v/v) at a volume of 400 μL/mg protein.
For shotgun lipidomics, the lipid extract was further diluted to a final concentration of ∼500 fM total lipids per microliter. Mass spectrometric analysis was performed on a triple-quadrupole mass spectrometer (TSQ Altis, Thermo Fisher Scientific, San Jose, CA) and a Q Exactive mass spectrometer (Thermo Scientific, San Jose, CA), both of which were equipped with an automated nanospray device (TriVersa NanoMate, Advion Bioscience, Ithaca, NY) as described.92 Identification and quantification of lipid species were performed using an automated software program.93 Data processing (e.g., ion peak selection, baseline correction, data transfer, peak intensity comparison, and quantitation) was performed as described.93 The results were normalized to the protein content (nanomoles of lipid per milligram of protein).
Plasma measurements
Alanine transaminase quantification was determined using the Alanine Transaminase Colorimetric Activity Assay Kit from Cayman Chemical following the user manual using plasma collected via eye bleed prior to sacrifice. Absorbance was read by the Tecan safire2 microplate reader. Plasma cytokine and lipid level measurements were carried out by UMass Metabolic Core.
RNA-seq analysis
Ten-week-old genetically obese ob/ob mice (n = 4/group) were injected subcutaneously with either NTC (10 mg/kg) or Dgat2-1473 (10 mg/kg) and provided with a NASH-inducing diet (GAN diet) for 3 weeks. After 3 weeks, mice were sacrificed. Total RNA samples were isolated from whole-liver tissues from the study explained in Figure 4 and sent out for next-generation RNA-seq. PolyA selection of mRNA species was used for the method of rRNA removal. The depth of the sequencing was 20–30 million reads/sample. The RNA-seq pipeline in DolphinNext94,95 was used to convert the fastq files into gene counts. The three parts of that pipeline that were used were the FastQC, trimmomatic96, and RSEM modules.
-
1
FastQC (v0.11.8) was used to verify the quality of the data.
-
2
trimmomatic (v0.39) was used to improve the analysis by removing Illumina adapter sequences via the ILLUMINACLIP:<fastaWithAdaptersEtc>:<seed mismatches>:<palindrome clip threshold>:<simple clip threshold>:<minAdapterLength > option. Specifically, we specifiedILLUMINACLIP:GATCGGAAGAGCACACGTCTGAACTCCAGTCA, GATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT,AATGATACGGCGACCACCGAGATCTACACTCTT:2:30:5:10.
-
3
RSEM (v1.2.28) was used to align the fastq reads (using –star for the STAR aligner) and quantify the gene expression levels.
The resulting estimates for gene expression were passed to DEBrowser.umassmed.edu94. Within DEBrowser, data were first filtered to eliminate genes whose expression level did not get above 10 in any sample and then DESeq2 was called to normalize expression levels and determine DE genes97, using an adjusted p value of 0.05 as the cutoff and requiring at least a 1.5-fold change, up or down.
The list of DE genes was then analyzed with using the enrichGO function in the clusterProfiler package. It was used with options ont = "BP”, pAdjustMethod = "fdr", pvalueCutoff = 0.05, universe = default, and minGSSize = 20. The pathways were simplified using its simplify function with options of cutoff = 0.75, by = "p value", select_fun = min. This was followed by manually specified merging of similar pathways to produce heatmaps.
The presence of siRNA seed complementarity (6-mer complementary to siRNA AS strand positions 2–8) in all genes with annotated 3′ UTRs in Ensembl GRMCm38.p6 annotations was determined with a custom Python script. siRNA seed enrichment in downregulated versus unchanged transcripts was calculated using a Fisher exact test. Data were plotted using Matplotlib.
Software and statistics
All statistical analyses were performed using the GraphPad Prism 8 (GraphPad Software). The data are presented as mean ± SEM. For analysis of the statistical significance between four or more groups, two-way ANOVA and multiple comparison t tests were used. NS is nonsignificant (p > 0.05), ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.0005.
Data availability
The RNA-seq data generated in this study have been deposited in GEO with accession number: GSE178987 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178987)
Acknowledgments
We wish to thank members of the Czech and Khvorova laboratories for helpful discussions, and Kerri Miller and Mary Beth Dziewietin for excellent assistance throughout the project. We thank the University of Massachusetts Morphology Core Facility for assistance in the histological preparations, stains, and analysis. We also wish to thank Robert Farese and Tobias Walther for generously providing the anti-DGAT2 antibody. This work was supported by National Institutes of Health grants DK103047 (to M.P.C.), R35GM131839 (to A.K.), and S10OD020012 (to A.K.). We also gratefully acknowledge generous funding through the Isadore and Fannie Foxman Chair in Medical Science (to M.P.C.), and pre-doctoral fellowship support to B.Y. by the American Heart Association (AHA Award #19PRE34460013).
Author contributions
B.Y., M.P.C., and A.K. designed the study, analyzed the data and wrote the manuscript. B.Y., N.W., M.K., K.M., and C.D. performed most of the experiments and analyzed the data. D.E., N.M., L.V., and J.S. contributed to the synthesis of the oligonucleotides. L.L. performed bioinformatics analysis of RNA-seq database. S.H. performed the off-target analysis. M.A.B. provided the NSG-PiZ humanized liver mouse model. A.K., J.F.A., M.R.H., and D.E. provided guidance in oligonucleotide technology. M.P. and X.H. performed the lipidomics analysis.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.11.007.
Contributor Information
Anastasia Khvorova, Email: anastasia.khvorova@umassmed.edu.
Michael P. Czech, Email: michael.czech@umassmed.edu.
Supplemental information
7 and Tables S1 and S2
References
- 1.Younossi Z.M., Blissett D., Blissett R., Henry L., Stepanova M., Younossi Y., Racila A., Hunt S., Beckerman R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016;64:1577–1586. doi: 10.1002/hep.28785. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z.M., Koenig A.B., Abdelatif D., Fazel Y., Henry L., Wymer M. Global epidemiology of nonalcoholic fatty liver disease—meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Alexander M., Loomis A.K., van der Lei J., Duarte-Salles T., Prieto-Alhambra D., Ansell D., Pasqua A., Lapi F., Rijnbeek P., Mosseveld M., et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18 million patients in four European cohorts. BMC Med. 2019;17:95. doi: 10.1186/s12916-019-1321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lomonaco R., Ortiz-Lopez C., Orsak B., Webb A., Hardies J., Darland C., Finch J., Gastaldelli A., Harrison S., Tio F., Cusi K. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:1389–1397. doi: 10.1002/hep.25539. [DOI] [PubMed] [Google Scholar]
- 5.Estes C., Razavi H., Loomba R., Younossi Z., Sanyal A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67:123–133. doi: 10.1002/hep.29466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diehl A.M., Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. New Engl. J. Med. 2017;377:2063–2072. doi: 10.1056/NEJMra1503519. [DOI] [PubMed] [Google Scholar]
- 7.Friedman S.L., Neuschwander-Tetri B.A., Rinella M., Sanyal A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018:24 908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romeo S., Sanyal A., Valenti L. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speliotes E.K., Yerges-Armstrong L.M., Wu J., Hernaez R., Kim L.J., Palmer C.D., Gudnason V., Eiriksdottir G., Garcia M.E., Launer L.J., et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valenti L., Al-Serri A., Daly A.K., Galmozzi E., Rametta R., Dongiovanni P., Nobili V., Mozzi E., Roviaro G., Vanni E., et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin i148m polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y.L., Reeves H.L., Burt A.D., Tiniakos D., McPherson S., Leathart J.B., Allison M.E., Alexander G.J., Piguet A.C., Anty R., et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014;5:1. doi: 10.1038/ncomms5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong V.W.S., Singal A.K. Emerging medical therapies for non-alcoholic fatty liver disease and for alcoholic hepatitis. Translational Gastroenterol. Hepatol. 2019;4:53. doi: 10.21037/tgh.2019.06.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh S., Khera R., Allen A.M., Murad M.H., Loomba R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: a systematic review and network meta-analysis. Hepatology. 2015;62:1417–1432. doi: 10.1002/hep.27999. [DOI] [PubMed] [Google Scholar]
- 14.Musso G., Cassader M., Rosina F., Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55:885–904. doi: 10.1007/s00125-011-2446-4. [DOI] [PubMed] [Google Scholar]
- 15.Mahady S.E., Webster A.C., Walker S., Sanyal A., George J. The role of thiazolidinediones in non-alcoholic steatohepatitis - a systematic review and meta analysis. J. Hepatol. 2011;55:1383–1390. doi: 10.1016/j.jhep.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Macauley M., Hollingsworth K.G., Smith F.E., Thelwall P.E., Al-Mrabeh A., Schweizer A., Foley J.E., Taylor R. Effect of vildagliptin on hepatic steatosis. J. Clin. Endocrinol. Metab. 2015;100:1578–1585. doi: 10.1210/jc.2014-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui J., Philo L., Nguyen P., Hofflich H., Hernandez C., Bettencourt R., Richards L., Salotti J., Bhatt A., Hooker J., et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J. Hepatol. 2016;65:369–376. doi: 10.1016/j.jhep.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mudaliar S., Henry R.R., Sanyal A.J., Morrow L., Marschall H.U., Kipnes M., Adorini L., Sciacca C.I., Clopton P., Castelloe E., et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 19.Neuschwander-Tetri B.A., Loomba R., Sanyal A.J., Lavine J.E., Van Natta M.L., Abdelmalek M.F., Chalasani N., Dasarathy S., Diehl A.M., Hameed B., et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pockros P.J., Fuchs M., Freilich B., Schiff E., Kohli A., Lawitz E.J., Hellstern P.A., Owens-Grillo J., Van Biene C., Shringarpure R., et al. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019;39:2082–2093. doi: 10.1111/liv.14209. [DOI] [PubMed] [Google Scholar]
- 21.Younossi Z.M., Ratziu V., Loomba R., Rinella M., Anstee Q.M., Goodman Z., Bedossa P., Geier A., Beckebaum S., Newsome P.N., et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–2196. doi: 10.1016/S0140-6736(19)33041-7. [DOI] [PubMed] [Google Scholar]
- 22.Harrison S.A., Alkhouri N., Davison B.A., Sanyal A., Edwards C., Colca J.R., Lee B.H., Loomba R., Cusi K., Kolterman O., et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol. 2020;72:613–626. doi: 10.1016/j.jhep.2019.10.023. [DOI] [PubMed] [Google Scholar]
- 23.Friedman S.L., Ratziu V., Harrison S.A., Abdelmalek M.F., Aithal G.P., Caballeria J., Francque S., Farrell G., Kowdley K.V., Craxi A., et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754–1767. doi: 10.1002/hep.29477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ratziu V., Sanyal A., Harrison S.A., Wong V.W., Francque S., Goodman Z., Aithal G.P., Kowdley K.V., Seyedkazemi S., Fischer L., et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. 2020;72:892–905. doi: 10.1002/hep.31108. [DOI] [PubMed] [Google Scholar]
- 25.Harrison S.A., Wong V.W., Okanoue T., Bzowej N., Vuppalanchi R., Younes Z., Kohli A., Sarin S., Caldwell S.H., Alkhouri N., Shiffman M.L., et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J. Hepatol. 2020;73:26–39. doi: 10.1016/j.jhep.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 26.Lawitz E.J., Coste A., Poordad F., Alkhouri N., Loo N., McColgan B.J., Tarrant J.M., Nguyen T., Han L., Chung C., et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2018;16:1983–1991.e3. doi: 10.1016/j.cgh.2018.04.042. [DOI] [PubMed] [Google Scholar]
- 27.Loomba R., Kayali Z., Noureddin M., Ruane P., Lawitz E.J., Bennett M., Wang L., Harting E., Tarrant J.M., McColgan B.J., et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155:1463–1473.e6. doi: 10.1053/j.gastro.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goedeke L., Bates J., Vatner D.F., Perry R.J., Wang T., Ramirez R., Li L., Ellis M.W., Zhang D., Wong K.E., et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68:2197–2211. doi: 10.1002/hep.30097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim C.W., Addy C., Kusunoki J., Anderson N.N., Deja S., Fu X., Burgess S.C., Li C., Ruddy M., Chakravarthy M., et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26:394–406.e6. doi: 10.1016/j.cmet.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison S.A., Abdelmalek M.F., Caldwell S., Shiffman M.L., Diehl A.M., Ghalib R., Lawitz E.J., Rockey D.C., Schall R.A., Jia C., et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155:1140–1153. doi: 10.1053/j.gastro.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Harrison S.A., Goodman Z., Jabbar A., Vemulapalli R., Younes Z.H., Freilich B., Sheikh M.Y., Schattenberg J.M., Kayali Z., Zivony A., et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J. Hepatol. 2020;72:816–827. doi: 10.1016/j.jhep.2019.11.024. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Tsao G., Bosch J., Kayali Z., Harrison S.A., Abdelmalek M.F., Lawitz E., Satapathy S.K., Ghabril M., Shiffman M.L., Younes Z.H., et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020;72:885–895. doi: 10.1016/j.jhep.2019.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Muthiah M.D., Sanyal A.J. Current management of non-alcoholic steatohepatitis. Liver Int. 2020;40:89–95. doi: 10.1111/liv.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodcock J., Griffin J.P., Behrman R.E. Development of novel combination therapies. New Engl. J. Med. 2011;364:985–987. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
- 35.Yen C.L.E., Stone S.J., Koliwad S., Harris C., Farese R.V. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008;49:2283–2301. doi: 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zammit V.A. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. Biochem. J. 2013;451:1–12. doi: 10.1042/BJ20121689. [DOI] [PubMed] [Google Scholar]
- 37.Yu X.X., Murray S.F., Pandey S.K., Booten S.L., Bao D., Song X.Z., Kelly S., Chen S., McKay R., Monia B.P., Bhanot S. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–371. doi: 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi K., Yang L., McCall S., Huang J., Yu X.X., Pandey S.K., Bhanot S., Monia B.P., Li Y.X., Diehl A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 39.Choi C.S., Savage D.B., Kulkarni A., Yu X.X., Liu Z.X., Morino K., Kim S., Distefano A., Samuel V.T., Neschen S., et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- 40.Amin N.B., Carvajal-Gonzalez S., Purkal J., Zhu T., Crowley C., Perez S., Chidsey K., Kim A.M., Goodwin B. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci. Transl. Med. 2019;11:9701. doi: 10.1126/scitranslmed.aav9701. [DOI] [PubMed] [Google Scholar]
- 41.Loomba R., Morgan E., Watts L., Xia S., Hannan L.A., Geary R.S., Baker B.F., Bhanot S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020;5:829–838. doi: 10.1016/S2468-1253(20)30186-2. [DOI] [PubMed] [Google Scholar]
- 42.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017:35 238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 44.Zamore P.D. RNA interference: listening to the sound of silence. Nat. Struct. Biol. 2001;8:746–750. doi: 10.1038/nsb0901-746. [DOI] [PubMed] [Google Scholar]
- 45.Rajeev K.G., Zimmerman T., Manoharan M., Maier M., Kuchimanchi K., Charisse K. 2013. Modified RNAI Agents. [Google Scholar]
- 46.Fitzgerald K., White S., Borodovsky A., Bettencourt B.R., Strahs A., Clausen V., Wijngaard P., Horton J.D., Taubel J., Brooks A., et al. A highly durable RNAi therapeutic inhibitor of PCSK9. New Engl. J. Med. 2017;376:41–51. doi: 10.1056/NEJMoa1609243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coelho T., Adams D., Silva A., Lozeron P., Hawkins P.N., Mant T., Perez J., Chiesa J., Warrington S., Tranter E., et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. New Engl. J. Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 48.Springer A.D., Dowdy S.F. GalNAc-siRNA conjugates: leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018:28 109–118. doi: 10.1089/nat.2018.0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel'in A.V., Milstein S., et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014;136:16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 50.Parmar R., Willoughby J.L., Liu J., Foster D.J., Brigham B., Theile C.S., Charisse K., Akinc A., Guidry E., Pei Y., et al. 5′-(E)-vinylphosphonate: a stable phosphate mimic can improve the RNAi activity of siRNA-GalNAc conjugates. ChemBioChem. 2016;17:985–989. doi: 10.1002/cbic.201600130. [DOI] [PubMed] [Google Scholar]
- 51.Zimmermann T.S., Karsten V., Chan A., Chiesa J., Boyce M., Bettencourt B.R., Hutabarat R., Nochur S., Vaishnaw A., Gollob J. Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol. Ther. 2017;25:71–78. doi: 10.1016/j.ymthe.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nair J.K., Attarwala H., Sehgal A., Wang Q., Aluri K., Zhang X., Gao M., Liu J., Indrakanti R., Schofield S., et al. Impact of enhanced metabolic stability on pharmacokinetics and pharmacodynamics of GalNAc-siRNA conjugates. Nucleic Acids Res. 2017;45:10969–10977. doi: 10.1093/nar/gkx818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schlegel M.K., Foster D.J., Kel'in A.V., Zlatev I., Bisbe A., Jayaraman M., Lackey J.G., Rajeev K.G., Charissé K., Harp J., et al. Chirality dependent potency enhancement and structural impact of glycol nucleic acid modification on siRNA. J. Am. Chem. Soc. 2017;139:8537–8546. doi: 10.1021/jacs.7b02694. [DOI] [PubMed] [Google Scholar]
- 54.Hassler M.R., Turanov A.A., Alterman J.F., Haraszti R.A., Coles A.H., Osborn M.F., Echeverria D., Nikan M., Salomon W.E., Roux L., et al. Comparison of partially and fully chemically-modified siRNA in conjugate-mediated delivery in vivo. Nucleic Acids Res. 2018;46:2185–2196. doi: 10.1093/nar/gky037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nikan M., Osborn M.F., Coles A.H., Godinho B.M., Hall L.M., Haraszti R.A., Hassler M.R., Echeverria D., Aronin N., Khvorova A. Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol. Ther. Nucleic Acids. 2016;5:e344. doi: 10.1038/mtna.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haraszti R.A., Roux L., Coles A.H., Turanov A.A., Alterman J.F., Echeverria D., Godinho B.M.D.C., Aronin N., Khvorova A. 5’-Vinylphosphonate improves tissue accumulation and efficacy of conjugated siRNAs in vivo. Nucleic Acids Res. 2017;45:7581–7592. doi: 10.1093/nar/gkx507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janas M.M., Schlegel M.K., Harbison C.E., Yilmaz V.O., Jiang Y., Parmar R., Zlatev I., Castoreno A., Xu H., Shulga-Morskaya S., et al. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 2018;9:1. doi: 10.1038/s41467-018-02989-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alterman J.F., Hall L.M., Coles A.H., Hassler M.R., Didiot M.C., Chase K., Abraham J., Sottosanti E., Johnson E., Sapp E., et al. Hydrophobically modified siRNAs silence huntingtin mRNA in primary neurons and mouse brain. Mol. Ther. Nucleic Acids. 2015;4:e266. doi: 10.1038/mtna.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choung S., Kim Y.J., Kim S., Park H.O., Choi Y.C. Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine. 2019;43:460–472. doi: 10.1016/j.ebiom.2019.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wittrup A., Lieberman J. Early blood-brain barrier dysfunction predicts neurological outcome following aneurysmal subarachnoid hemorrhage. EBioMedicine. 2019;43:460–472. doi: 10.1016/j.ebiom.2019.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Czauderna F., Fechtner M., Dames S., Aygün H., Klippel A., Pronk G.J., Giese K., Kaufmann J. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003;31:2705–2716. doi: 10.1093/nar/gkg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maier M., Foster D., Milstein S., Kuchimanchi S., Jadhav V., Rajeev K.G., Manoharan M., Parmar R. 2016. Modified Double-Stranded RNA Agents. [Google Scholar]
- 63.Yasuda M., Gan L., Chen B., Kadirvel S., Yu C., Phillips J.D., New M.I., Liebow A., Fitzgerald K., Querbes W., Desnick R.J. RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc. Natl. Acad. Sci. U S A. 2014;111:7777–7782. doi: 10.1073/pnas.1406228111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fitzgerald K., Frank-Kamenetsky M., Shulga-Morskaya S., Liebow A., Bettencourt B.R., Sutherland J.E., Hutabarat R.M., Clausen V.A., Karsten V., Cehelsky J., et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. The Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ray K.K., Landmesser U., Leiter L.A., Kallend D., Dufour R., Karakas M., Hall T., Troquay R.P., Turner T., Visseren F.L., et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. New Engl. J. Med. 2017;376:1430–1440. doi: 10.1056/NEJMoa1615758. [DOI] [PubMed] [Google Scholar]
- 66.Adams D., Gonzalez-Duarte A., O'Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. New Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 67.Solomon S.D., Adams D., Kristen A., Grogan M., González-Duarte A., Maurer M.S., Merlini G., Damy T., Slama M.S., Brannagan T.H., 3rd, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis: analysis of the APOLLO study. Circulation. 2019;139:431–443. doi: 10.1161/CIRCULATIONAHA.118.035831. [DOI] [PubMed] [Google Scholar]
- 68.Huang Y. Preclinical and clinical advances of GalNAc-decorated nucleic acid therapeutics. Mol. Ther. Nucleic Acids. 2017;6:116–132. doi: 10.1016/j.omtn.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coles A.H., Osborn M.F., Alterman J.F., Turanov A.A., Godinho B.M., Kennington L., Chase K., Aronin N., Khvorova A. A high-throughput method for direct detection of therapeutic oligonucleotide-induced gene silencing in vivo. Nucleic Acid Ther. 2016;26:86–92. doi: 10.1089/nat.2015.0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nikan M., Osborn M.F., Coles A.H., Godinho B.M., Hall L.M., Haraszti R.A., Hassler M.R., Echeverria D., Aronin N., Khvorova A. Docosahexaenoic acid conjugation enhances distribution and safety of siRNA upon local administration in mouse brain. Mol. Ther. Nucleic Acids. 2016;5:e344. doi: 10.1038/mtna.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gluchowski N.L., Gabriel K.R., Chitraju C., Bronson R.T., Mejhert N., Boland S., Wang K., Lai Z.W., Farese R.V., Jr., Walther T.C. Hepatocyte deletion of triglyceride-synthesis enzyme acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology. 2019;70:1972–1985. doi: 10.1002/hep.30765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borel F., Tang Q., Gernoux G., Greer C., Wang Z., Barzel A., Kay M.A., Shultz L.D., Greiner D.L., Flotte T.R., et al. Survival advantage of both human hepatocyte xenografts and genome-edited hepatocytes for treatment of α-1 antitrypsin deficiency. Mol. Ther. 2017;25:2477–2489. doi: 10.1016/j.ymthe.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linden A.G., Li S., Choi H.Y., Fang F., Fukasawa M., Uyeda K., Hammer R.E., Horton J.D., Engelking L.J., Liang G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 2018;59:475–487. doi: 10.1194/jlr.M081836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X., Sommerfeld M.R., Jahn-Hofmann K., Cai B., Filliol A., Remotti H.E., Schwabe R.F., Kannt A., Tabas I. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol. Commun. 2019;3:1221–1234. doi: 10.1002/hep4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U S A. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan A., Liebow A., Yasuda M., Gan L., Racie T., Maier M., Kuchimanchi S., Foster D., Milstein S., Charisse K., et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther. Nucleic Acids. 2015;4:e263. doi: 10.1038/mtna.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Allerson C.R., Sioufi N., Jarres R., Prakash T.P., Naik N., Berdeja A., Wanders L., Griffey R.H., Swayze E.E., Bhat B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- 78.McLaren D.G., Han S., Murphy B.A., Wilsie L., Stout S.J., Zhou H., Roddy T.P., Gorski J.N., Metzger D.E., Shin M.K., et al. DGAT2 inhibition alters aspects of triglyceride metabolism in rodents but not in non-human primates. Cell Metab. 2018;27:1236–1248.e6. doi: 10.1016/j.cmet.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 79.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li S., Brown M.S., Goldstein J.L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc. Natl. Acad. Sci. U S A. 2010;107:3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tian J., Goldstein J.L., Brown M.S. Insulin induction of SREBP-1c in rodent liver requires LXRα-c/EBPβ complex. Proc. Natl. Acad. Sci. U S A. 2016;113:8182–8187. doi: 10.1073/pnas.1608987113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brown M.S., Goldstein J.L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. U S A. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iizuka K., Miller B., Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am. J. Physiol. - Endocrinol. Metab. 2006;291:358–364. doi: 10.1152/ajpendo.00027.2006. [DOI] [PubMed] [Google Scholar]
- 84.Dentin R., Benhamed F., Hainault I., Fauveau V., Foufelle F., Dyck J.R., Girard J., Postic C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 85.Erion D.M., Popov V., Hsiao J.J., Vatner D., Mitchell K., Yonemitsu S., Nagai Y., Kahn M., Gillum M.P., Dong J., et al. The role of the carbohydrate response element-binding protein in male fructose-fed rats. Endocrinology. 2013;154:36–44. doi: 10.1210/en.2012-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walker A.K., Jacobs R.L., Watts J.L., Rottiers V., Jiang K., Finnegan D.M., Shioda T., Hansen M., Yang F., Niebergall L.J., et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell. 2011;147:840–852. doi: 10.1016/j.cell.2011.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Calle R.A., Amin N.B., Carvajal-Gonzalez S., Ross T.T., Bergman A., Aggarwal S., Crowley C., Rinaldi A., Mancuso J., Aggarwal N., et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021;27:1836–1848. doi: 10.1038/s41591-021-01489-1. [DOI] [PubMed] [Google Scholar]
- 88.O’Shea J., Theile C.S., Das R., Babu R., Charisse K., Manoharan M., Maier M.A., Zlatev I. An efficient deprotection method for 5′-[O,O-bis(pivaloyloxymethyl)]-(E)-vinylphosphonate containing oligonucleotides. Tetrahedron. 2018;74:6182–6186. [Google Scholar]
- 89.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 90.Han X. Wiley-Blackwell; 2016. Lipidomics: Comprehensive Mass Spectrometry of Lipids. Lipidomics: Comprehensive Mass Spectrometry of Lipids. [DOI] [Google Scholar]
- 91.Wang M., Han X. Multidimensional mass spectrometry-based shotgun lipidomics. Methods Mol. Biol. 2014;1198:203–220. doi: 10.1007/978-1-4939-1258-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han X., Yang K., Gross R.W. Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high-throughput platform for shotgun lipidomics. Rapid Commun. Mass Spectrom. 2008;22:2115–2124. doi: 10.1002/rcm.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang K., Cheng H., Gross R.W., Han X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kucukural A., Yukselen O., Ozata D.M., Moore M.J., Garber M. DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics. 2019;20:6. doi: 10.1186/s12864-018-5362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yukselen O., Turkyilmaz O., Ozturk A.R., Garber M., Kucukural A. DolphinNext: a distributed data processing platform for high throughput genomics. BMC Genom. 2020;21:310. doi: 10.1186/s12864-020-6714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
7 and Tables S1 and S2
Data Availability Statement
The RNA-seq data generated in this study have been deposited in GEO with accession number: GSE178987 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178987)