

Abstract
DNA double strand breaks (DSBs) pose a serious threat to genome stability. In vertebrates, these breaks are predominantly repaired by non-homologous end joining (NHEJ), which pairs DNA ends in a multi-protein synaptic complex to promote their direct ligation. NHEJ is a highly versatile pathway that utilizes an array of processing enzymes to modify damaged DNA ends and enable their ligation. The mechanisms of end synapsis and end processing have important implications for genome stability. Rapid and stable synapsis is necessary to limit chromosome translocations that result from the mispairing of DNA ends. Furthermore, end processing must be tightly regulated to minimize mutations at the break site. Here we review our current mechanistic understanding of vertebrate NHEJ, with a particular focus on end synapsis and processing.
Keywords: DNA repair, DNA double-strand break, non-homologous end joining, DNA end synapsis, DNA end processing
1. INTRODUCTION
DNA double strand breaks (DSBs) are extremely toxic, and elaborate pathways exist to repair them. Failure to repair even a single DSB can lead to cell death, as persistent DSBs can trigger apoptosis. Moreover, misrepair of breaks can lead to pathological genomic alterations. For example, chromosome translocations, a major driver of oncogenesis, result from improper joining of DSBs from different chromosomes (1). In addition, error-prone polymerases and nucleases can generate localized mutations at the repair junction (2). Thus, DSB repair pathways must be tightly regulated to maximize fidelity.
Two major pathways, non-homologous end joining (NHEJ) and homologous recombination (HR), are responsible for repairing the majority of DSBs. NHEJ is the predominant pathway in human cells and repairs up to ~80% of all DSBs (3). During NHEJ, DNA ends are brought together by a multi-protein synaptic complex and directly ligated (2). In comparison, during HR, 5´→3´ resection at the DSB generates a 3´ single-stranded DNA overhang that invades the sister chromatid, which acts as a template for repair (4, 5). Disruption of either NHEJ or HR dramatically sensitizes cells to DSB-inducing agents and leads to spontaneous chromosomal aberrations, suggesting that the two pathways are complementary (6). The requirement of a sister chromatid in HR means that in most organisms, this repair pathway is restricted to the S and G2 phases of the cell cycle, whereas NHEJ is active throughout the cell cycle.
In addition to NHEJ and HR, the minor pathways of alternative end joining (alt-EJ) and single-strand annealing (SSA) also contribute to DSB repair. Both of these pathways act on resected DNA substrates and therefore compete with HR. Alt-EJ is a mutagenic repair pathway involving the multi-functional polymerase Pol θ that directly ligates partially resected DNA ends together. Alt-EJ relies on microhomologies near the DSB and is characterized by insertions and deletions (7). SSA anneals homologous repetitive sequences flanking a DSB. It requires extensive resection and results in a deletion of the sequence between the annealed repeats (4, 8).
How cells choose among these pathways to repair a given DSB choice is a central question in genome maintenance. Whether or not a DSB undergoes 5´→3´ resection is a critical determinant of pathway choice, as the resulting 3′ ssDNA overhang is a poor substrate for the NHEJ machinery but is required for HR, alt-EJ, and SSA (9, 10). The decision to resect appears to be a dynamic process governed by opposing factors both at the DSB itself (e.g., the anti-resection NHEJ factor Ku vs. the resection initiator MRN) and the surrounding chromatin (e.g., the anti-resection factors 53BP1 and Shieldin vs. pro-resection factor BRCA1) (9, 10). Here, we focus on our current understanding of the NHEJ pathway and direct readers to excellent reviews of other pathways and pathway choice, cited above.
2. AN OVERVIEW OF THE NHEJ PATHWAY
NHEJ proceeds through a set of mechanistically distinct steps to join DNA ends (Figure 1). Upon formation of a DSB, the DNA ends are rapidly bound by the Ku70/80 heterodimer, an extremely abundant ring-shaped molecule that tightly encircles DNA (11). Due to its role as a recruitment hub for many downstream NHEJ factors, Ku binding is a critical DNA end detection step that initiates the assembly of the NHEJ machinery (Table 1). One downstream NHEJ factor is DNA-PKcs, a large protein kinase belonging to the phosphoinositide 3-kinase (PI3K)-related kinase family. DNA-PKcs recognizes DNA-bound Ku, forming the DNA-PK holoenzyme (12). DNA binding stimulates DNA-PKcs kinase activity, leading to the phosphorylation of numerous NHEJ and DNA repair factors (13). Although the functional consequences of many of these phosphorylation events remain unclear, autophosphorylation of DNA-PKcs appears to be critical for DSB repair (Sections 4.3 and 5.4).
Figure 1: An overview of the NHEJ pathway.
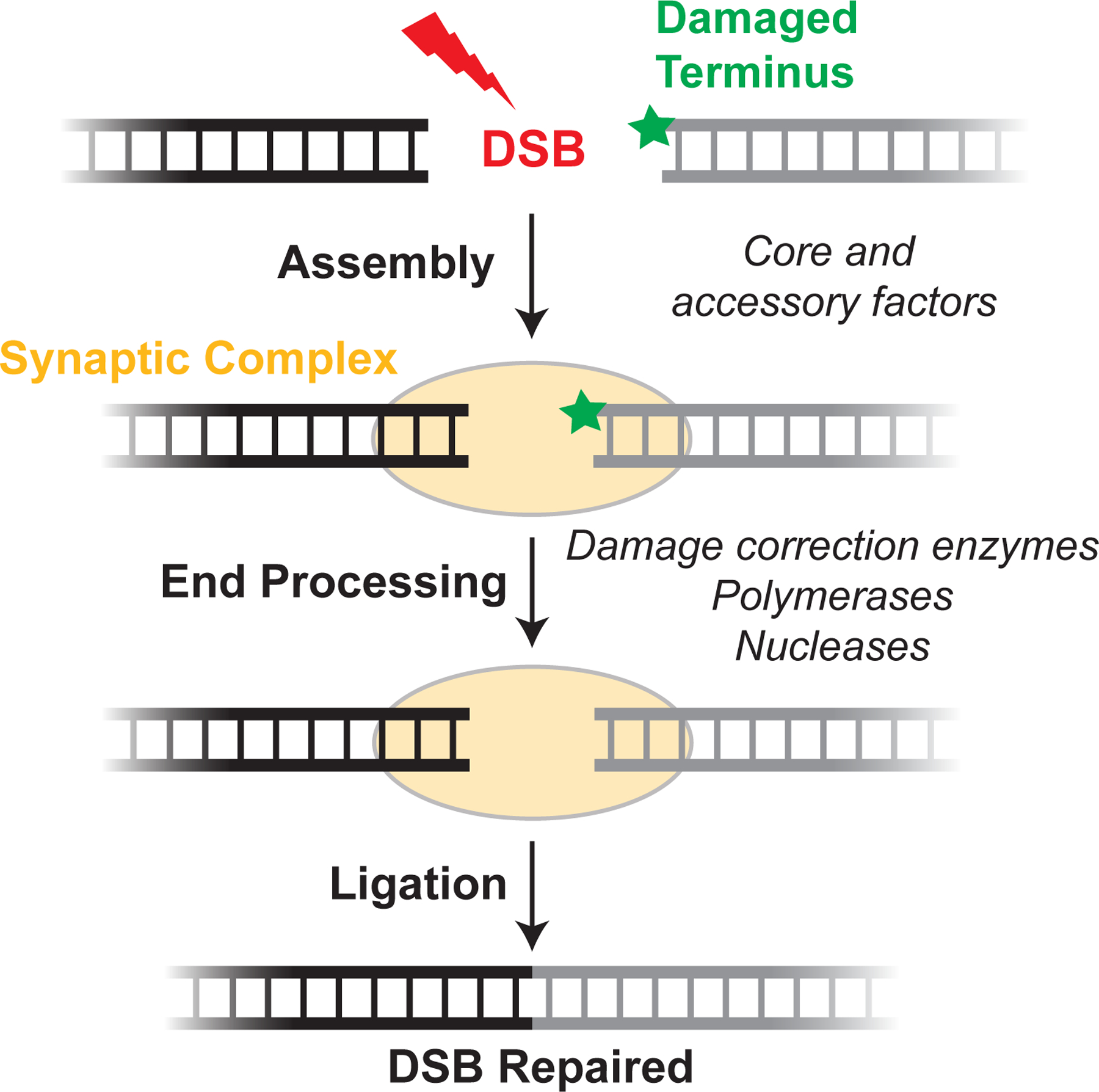
Upon DSB formation, core and accessory NHEJ factors (Table 1) recognize DNA ends and tether them together in a synaptic complex (see Section 4 for details). If the DNA ends are not compatible for immediate ligation, they are modified by processing enzymes (Table 1). until ligation can occur (see Section 5 for details).
Table 1. NHEJ proteins and their proposed roles.
Three classes of proteins comprise the NHEJ machinery: core factors, whose loss results in severe NHEJ defects; accessory factors, which interact with core factors but whose loss results in mild defects; and end processing enzymes, which chemically modify damaged DNA ends to prepare them for ligation.
| NHEJ PROTEIN | PROPOSED ROLE(S) IN NHEJ |
|---|---|
| Core factors | |
| Ku70/80 | Initial DSB sensor and interaction hub (11), 5′dRP/AP lyase (175) |
| DNA-PKcs | Kinase activity (13), end synapsis (53, 56, 69), and molecular “gate” (13) |
| LIG4 | Catalytic role in ligation (19), structural role in synapsis (53, 56, 58, 72) |
| XRCC4 | Constitutive LIG4 interactor (19), XLF interactor for synapsis (53, 55, 56, 58, 77) |
| XLF | XRCC4 interactor for synapsis (53, 55, 56, 58, 77, 80) |
| Accessory factors | |
| PAXX | Synapsis (56), redundant roles with XLF (95, 96) |
| APLF | Scaffolding factor (98) |
| WRN | Helicase and exonuclease (99), role in repair pathway choice (101) |
| MRI/CYREN | Cell cycle regulator of NHEJ (102, 103) |
| End processing enzymes | |
| Damage correction | |
| PNKP | 5′ kinase, 3′ phosphatase (130) |
| TDP1 | Removal of TOP1 (136) and other 3′ adducts (138) |
| TDP2 | Removal of TOP2 5′ adducts (137) |
| Aprataxin | Removal of 5′ adenylate (132) |
| Polymerases | |
| Pol λ | Templated synthesis, paired primer terminus (127) |
| Pol μ | Templated synthesis, unpaired primer terminus (127) |
| TdT | Untemplated synthesis during V(D)J recombination (35) |
| Nucleases | |
| Artemis | V(D)J hairpin opening (32), processing of some IR DSBs (114) |
| SETMAR/Metnase | ssDNA endonuclease (117) |
Ligation requires close alignment of DNA ends in a synaptic complex (Section 4). Many NHEJ factors have been implicated in synapsis, including the paralogs XRCC4 (14), XRCC4-like factor (XLF) (15, 16) and PAralog of XRCC4 and XLF (PAXX) (17, 18). Ultimately, DNA ligase IV (LIG4), which forms a constitutive complex with XRCC4, catalyzes ligation (19). Notably, LIG4 can tolerate certain terminal mismatches and damaged bases (20, 21), a unique feature among vertebrate ligases. Nonetheless, many DNA end structures are incompatible for direct ligation. Accordingly, a large number of end processing factors including polymerases and nucleases are recruited to DSBs and act on the ends to prepare them for ligation (2) (Table 1).
Decades of investigation have established the importance of many NHEJ factors and outlined the major steps of the pathway (Figure 1). Current questions in the field include how these factors cooperate with each other at the molecular level, and how the distinct steps of NHEJ are coordinated and regulated. These topics and their implications for genome stability are the focus of this review.
3. PHYSIOLOGICAL ROLES OF NHEJ
NHEJ is responsible for repairing spontaneous DSBs arising from myriad sources, as well as some developmentally programmed DSBs. Given its importance in genome maintenance, misregulation of NHEJ is associated with cancer and other human diseases. Moreover, inhibition of NHEJ has emerged as a means to bolster DSB-inducing cancer therapies or alter genome engineering outcomes. Here, we provide an overview of the various physiological roles of NHEJ.
3.1. Spontaneous DNA double strand breaks
Spontaneous DSBs occur roughly 50 times per day per somatic cell in mammals (22). Major causes of spontaneous DSBs are reactive oxygen species (ROS), a byproduct of aerobic respiration, and environmental ionizing radiation (IR). Both ROS and IR generate single-strand DNA breaks (SSBs) (23), which may become DSBs if there is another SSB nearby on the opposite strand. Such two-ended DSBs are readily repaired by NHEJ. One-ended DSBs are generated when a replication fork encounters an unrepaired SSB (24). In contrast to two-ended DSBs, single-ended DSBs require HR for accurate repair, and repair by NHEJ is associated with toxicity (25). Other sources of DSBs include nuclease activity and abortive topoisomerase activity. Finally, deprotected telomeres are erroneously recognized as DSBs, potentially resulting in NHEJ-mediated chromosome fusions (26).
3.2. The role of NHEJ in V(D)J recombination and class switch recombination
The vast diversity of the vertebrate adaptive immune system is achieved in part through programmed rearrangements of antigen receptor genes. These rearrangements—V(D)J recombination and class switch recombination (CSR)—proceed through DSB intermediates that are ultimately repaired by NHEJ. Here, we provide a brief overview of the involvement of NHEJ in V(D)J recombination and CSR and refer readers to excellent in-depth reviews of these processes (27–31).
V(D)J recombination selects one V, one D, and one J “coding” segment and joins them together to assemble the antigen-binding variable region of the immunoglobulin (Ig) heavy chain and T cell receptor (TCR) β chain (Figure 2A). Ig light and TCR α chains lack D segments and undergo V-to-J rearrangement. Because the germline contains multiple copies of each segment type (e.g., 44 V segments, 23 D segments, and 6 J segments in the human immunoglobulin heavy chain locus), the many possible gene segment permutations augment immune receptor diversity. V(D)J recombination is initiated by the RAG recombinase, which binds to a pair of short recombination signal sequences (RSSs) that flank V, D, and J coding segments (Figure 2B) (30). RAG introduces DSBs between the RSSs and coding segments, generating blunt “signal ends” and hairpin-capped “coding ends.” DNA cleavage by RAG follows the RSS “12/23 rule.” RSSs consist of conserved heptamer and nonamer sequences separated by spacers of either 12 or 23 basepairs (12RSS and 23RSS). RAG promotes appropriate V(D)J exon assembly by initiating cleavage only when bound to both a 12RSS and 23RSS (30). For example, at the Ig heavy chain locus (IgH), V, D, and J segments are flanked by 23RSSs, 12RSSs, and 23RSSs, respectively. Thus, the 12/23 rule allows V-to-D joining and D-to-J joining while precluding direct V-to-J joining.
Figure 2: NHEJ and programmed DSBs.
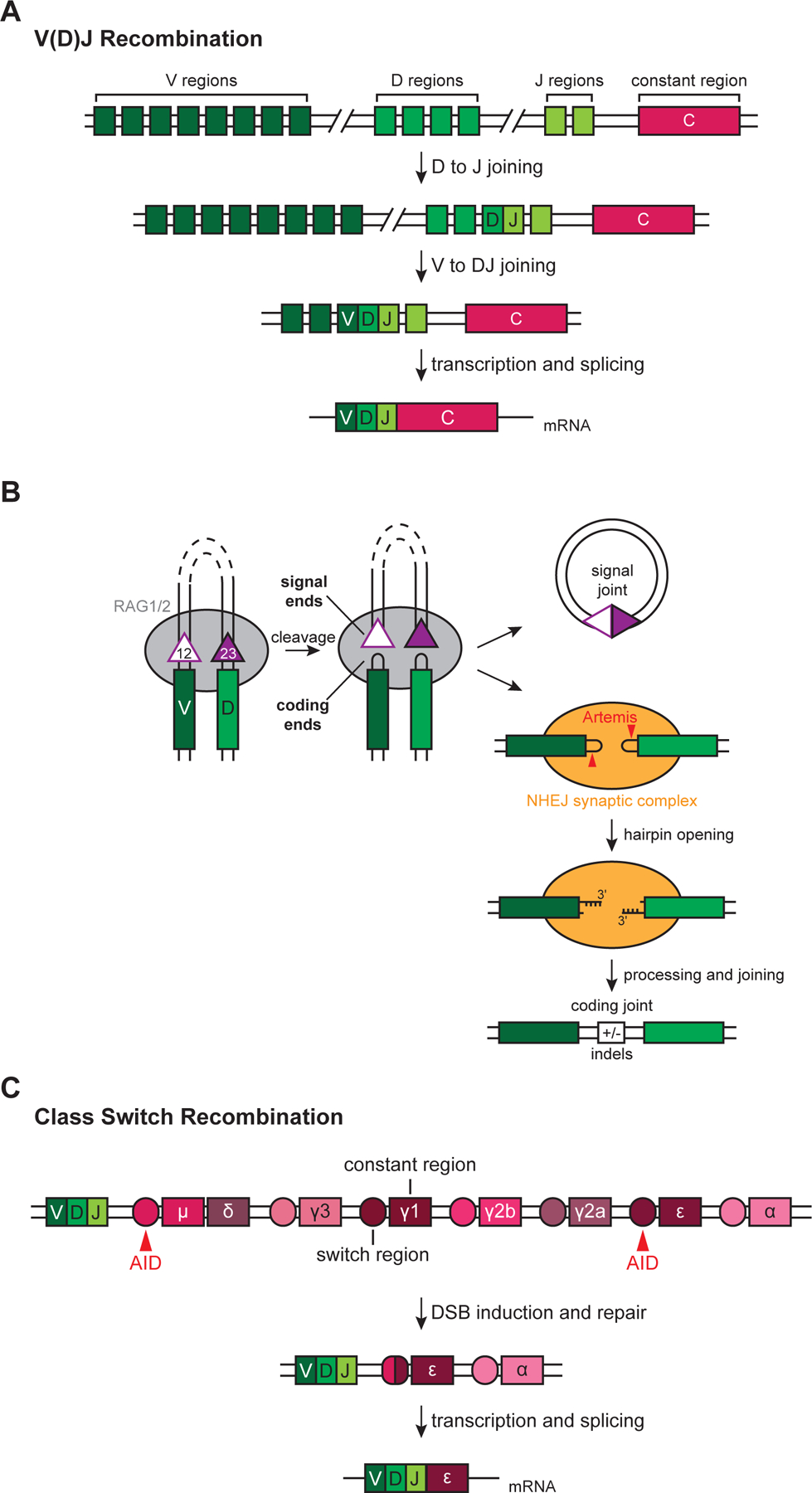
(A) Simplified example of V(D)J recombination at the IgH locus. RAG introduces DSBs adjacent to a D segment and J segment according to the 12/23 rule (see text), and NHEJ facilitates D to J joining. Subsequent V to DJ joining occurs through a similar process and assembles the V(D)J exon.
(B) Generation and repair of DSBs during V(D)J recombination. V-to-D joining is shown as an example. See text for details. 12- and 23-RSSs, open and filled purple triangles, respectively.
(C) Simplified example of CSR at the IgH locus. AID activity at the μ and ε switch regions leads to DSBs that are repaired by NHEJ in a productive class switching event to the IgE isotype.
Following DSB induction, RAG channels the DNA ends to the NHEJ pathway, which is strictly required for repair (Figure 2B) (30). NHEJ generates both “signal joints” (ligation of two signal ends) and “coding joints” (ligation of two coding ends). NHEJ directly ligates blunt signal ends, forming “excision circles” that contain the intervening sequence between two coding segments. Hairpin-capped coding ends must be opened by the Artemis nuclease to allow subsequent joining (32). Artemis cleaves one to four nucleotides 3′ of the hairpin tip, generating a palindromic 3′ overhang of variable length. These overhangs then undergo templated extension by the NHEJ-associated polymerases pol λ and pol μ (see Section 5) at Ig heavy and light chain loci, respectively (33, 34), and template-independent extension by TdT (35). In contrast to the relatively uniform and conservative repair of spontaneous DSBs (see Section 5), heterogenous processing by Artemis and the NHEJ polymerases results in coding joint variability that greatly expands the repertoire of antigen binding receptors (29). We will discuss the mechanistic underpinnings of NHEJ fidelity in Section 5.
NHEJ also repairs DSB intermediates programmed during CSR, which rearranges IgH constant (CH) regions that define immunoglobulin isotype (Figure 2C) (28). Upon B cell activation, DSBs are induced at “switch” regions, and subsequent repair alters which CH region is juxtaposed with the expressed V(D)J segment. This process replaces the default μ CH region with γ, ε, or α CH regions, resulting in an isotype switch from IgM to IgG, IgE, or IgA, respectively. DSB induction at switch regions relies on activation-induced cytidine deaminase (AID) (36), which deaminates cytosine bases to generate uracil, leaving a mismatched U:G basepair. Repair of this lesion by either DNA mismatch repair (MMR) or base excision repair (BER) proceeds through nicked intermediates, and nicks on opposite strands within a few basepairs result in a DSB (31). Although NHEJ is the primary pathway responsible for joining DSBs during CSR, alt-EJ can serve as a back-up pathway (37), unlike in V(D)J recombination.
Productive class switching requires that a DNA end arising from a DSB at the μ switch region (Sμ) be joined to a DNA end arising from a DSB at a downstream acceptor switch region. Unlike V(D)J recombination, CSR does not make use of a sequence-specific recombinase to direct joining of the appropriate gene segments. Instead, CSR appears to rely on transcription and three-dimensional genome architecture (27, 31). Transcription at switch regions exposes single-stranded DNA tracts required for AID activity, thereby ensuring DSBs are induced at the appropriate locations. Moreover, chromatin looping juxtaposes Sμ and acceptor S regions to promote productive CSR (31).
3.3. NHEJ and human health
Given the critical role in repairing both spontaneous and programmed DSBs, deficiencies in NHEJ have various adverse effects on human health. Mutations in the genes encoding XLF, LIG4, DNA-PKcs, and Artemis confer severe combined immunodeficiency (SCID) as a result of defective V(D)J recombination (reviewed in (38)). A subset of SCID patients also exhibit radiosensitivity (RS-SCID), and human cells with deficiencies in core NHEJ factors are generally hypersensitive to IR. Some SCID patients also present with developmental abnormalities, including microcephaly and/or growth delays. The molecular origins of these developmental abnormalities are less clear but may be caused by neuronal apoptosis, which is observed in NHEJ-deficient LIG4-null mice (39–41).
Whereas inactivation of NHEJ is associated with SCID and radiosensitivity, the misregulation or hyperactivation of NHEJ machinery has been linked to cancer and resistance to cancer therapy. Chromosome translocations, a frequent driver of tumorigenesis, are generated by NHEJ in human cells (42). NHEJ has also been implicated in chromothripsis, a mutational phenomenon in cancer that involves the shattering and rearrangement of a chromosome (43). Moreover, several studies have reported that core NHEJ factors are overexpressed in certain tumor tissues (44), and overly active NHEJ is associated with resistance to DSB-inducing chemotherapy and radiotherapy (45). As such, NHEJ components have emerged as drug targets for cancer therapy (45), and DNA-PKcs inhibitors have entered clinical trials (46).
Many laboratories have also disrupted NHEJ to improve genome editing efficiency (47–50). CRISPR-Cas9 and related technologies introduce sequence-specific DSBs to disrupt or edit a target gene. Such DSBs are primarily repaired by NHEJ, and error-prone NHEJ may result in a frameshift mutation that disrupts the targeted gene. However, precise gene editing typically requires addition of a homologous DNA template and repair of the DSB by HR. Although efforts to inhibit NHEJ have yielded modest improvements, the low efficiency of homology-directed repair (HDR) remains the key barrier to precise genome editing.
4. END SYNAPSIS DURING NHEJ
The NHEJ machinery has no means to determine whether a pair of DNA ends arose from the same DSB. Instead, NHEJ relies on end proximity to direct repair. Upon formation of a chromosomal DSB, the motion of DNA ends is largely constrained (51). However, loss of the NHEJ machinery through depletion of Ku results in a significant increase in the local diffusion of DNA ends (52). Therefore, the NHEJ machinery appears to rapidly and stably synapse DNA ends until they are joined, thereby suppressing chromosome translocations. The following sections describe our emerging understanding of the NHEJ synaptic complex.
4.1. Single-molecule methods to probe end synapsis during repair
Investigators have employed a variety of approaches to determine how DNA ends are synapsed during NHEJ. DNA pulldown experiments have been widely used to identify factors involved in end synapsis, and structural approaches have elucidated how NHEJ sub-complexes could bridge DNA ends. These studies, discussed in subsequent sections, have implicated all core NHEJ factors in end synapsis. Single molecule methods, described in this section, are beginning to reveal how these factors interact with each other and DNA in a highly dynamic synaptic complex, with transient intermediates and changing composition.
Single-molecule Förster resonance energy transfer (smFRET) is an especially powerful approach to study synapsis because it monitors the distance between paired DNA ends in real time during a physiological repair reaction (53, 54). In these experiments, DNAs are differentially labeled near their ends with donor and acceptor fluorophores. When the DNA ends are close together (i.e. less than 80 angstroms apart), excitation of the donor dye leads to energy transfer to the acceptor dye and acceptor fluorescence. FRET efficiency is highly sensitive to the distance between the two dyes and can report on distance changes as small as a few angstroms. smFRET has been used to observe intermolecular synapsis by tethering donor-labeled DNAs to the surface of a cover slip via a biotin-streptavidin linkage and providing acceptor-labeled DNAs in solution (Figure 3A). Alternatively, a long (~2 kilobase pair) DNA substrate labeled at each end with donor or acceptor dyes can be tethered to the surface at an internal site to observe intramolecular synapsis (Figure 3B). Furthermore, NHEJ factors labeled with distinct fluorophores can be added to the NHEJ reaction in order to correlate the stoichiometry and dynamics of these factors with DNA end joining (Figure 3B) (55).
Figure 3: Single-molecule assays for synapsis.
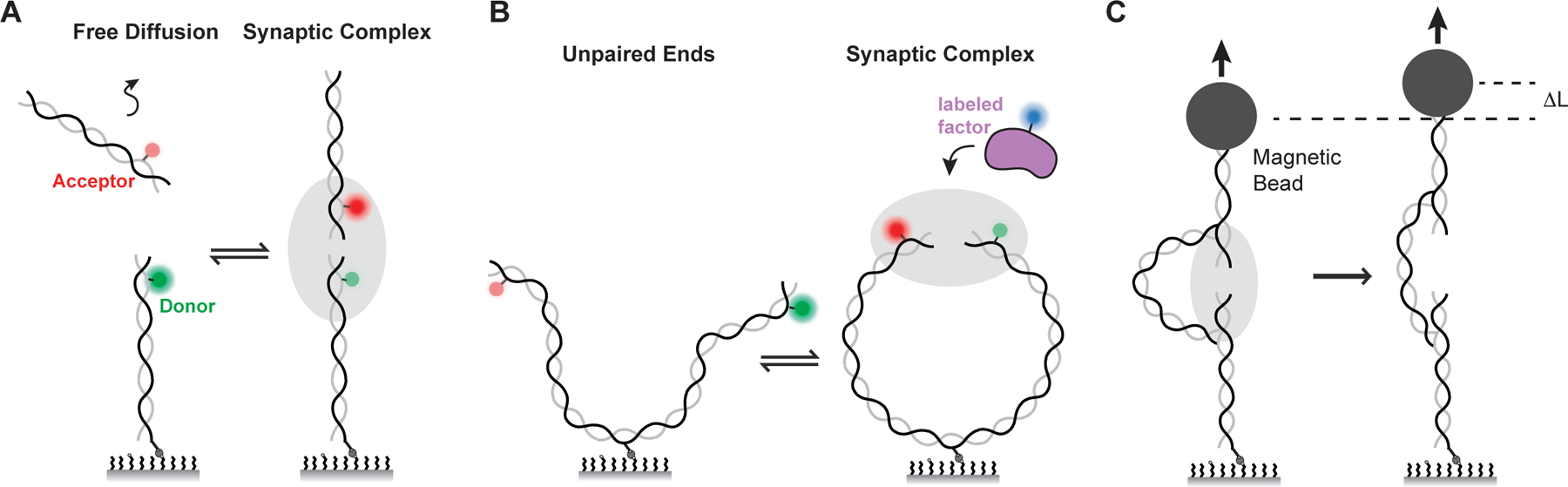
(A) Inter-molecular synapsis single-molecule FRET (smFRET) assay. Green, donor fluorophore; red, acceptor fluorophore.
(B) Intra-molecular synapsis smFRET assay. Proteins labeled with unique fluorophores (blue) can be imaged to quantify their stoichiometry and dynamics with the synaptic complex.
(C) Magnetic forceps allow application of force to a DNA scaffold during synapsis.
Nanomanipulation-based single-molecule methods have been used to characterize the stability of the synaptic complex under force (56). These experiments utilize a DNA scaffold of two linear dsDNA molecules, each anchored at one end to either a magnetic bead or a glass surface, and the other end loosely tethered by a dsDNA leash (Figure 3C). The free ends undergo synapsis, and force is applied to the DNA scaffold through the magnetic bead by a standard magnetic tweezers instrument. Rupture of the synaptic complex under force leads to an increase in the measured DNA length. Subsequent relaxation of the force allows for reformation of the synaptic complex and permits multiple rupture cycles to be studied on a single DNA substrate. Additionally, optical tweezers-based approaches have been used to study how sub-complexes of the NHEJ reaction interact with DNA (57).
4.2. End synapsis is maintained by a dynamic multi-protein complex that evolves during repair
Single-molecule imaging from our laboratory and others has shown that the NHEJ synaptic complex is not a static structure but instead evolves during repair (Figure 4). smFRET experiments in Xenopus egg extracts showed that DNA ends are initially tethered together in a long-range synaptic complex, in which DNA ends are separated by >80 angstroms (53). This long-range complex requires the core NHEJ factors Ku and DNA-PKcs and only persists for ~ 5 seconds on average. DNA-PKcs kinase activity, along with the factors XRCC4-LIG4 and XLF, are required for the transition to a short-range synaptic complex in which the ends are closely aligned for ligation. Importantly, formation of the short-range complex does not require the catalytic activity of LIG4, indicating that LIG4 plays a structural role in synapsis in addition to its enzymatic role in ligation. In contrast to the long-range complex, the short-range complex is quite stable and can persist for ~100 seconds or more. Formation of the long-range complex is required to proceed to the short-range complex, demonstrating that a structural remodeling of the NHEJ machinery occurs during repair.
Figure 4: End synapsis during NHEJ.
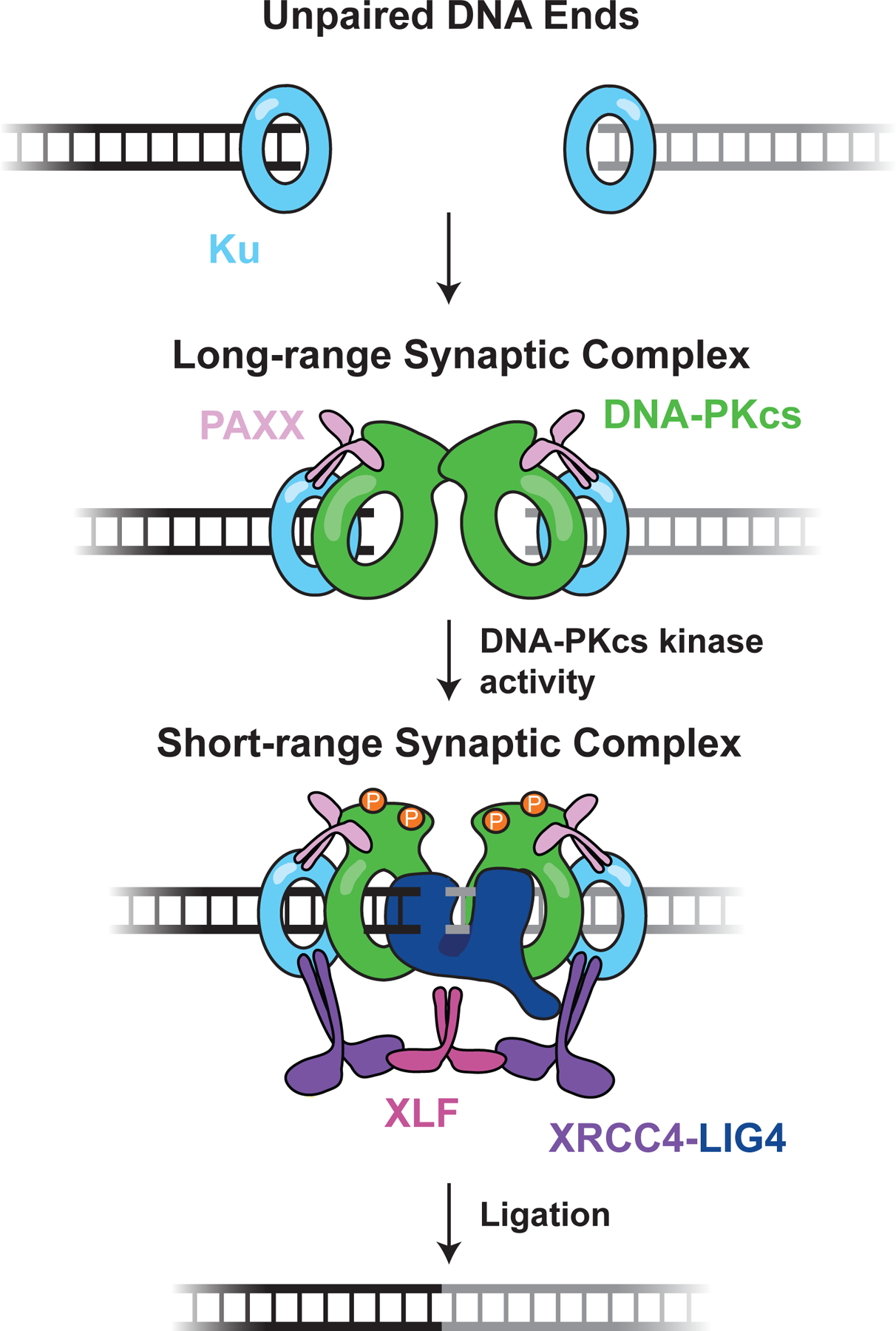
DNA ends are rapidly bound by Ku, which recruits further NHEJ factors. Ku, DNA-PKcs, and PAXX enable formation of the long-range synaptic complex, in which the DNA ends are tethered together but separated by >80 Å. Transition to the short-range synaptic complex, in which DNA ends are directly juxtaposed, requires XLF, XRCC4-LIG4, and DNA-PKcs kinase activity. Putative DNA-PKcs autophosphorylation in the short-range complex is shown as orange circles. Here, LIG4 is shown directly engaging both DNA ends to promote short-range synapsis, but other structural contributions are possible (see text). The figure depicts the protein and enzymatic requirements for formation of each synaptic state; the precise composition and factor stoichiometry of each state is largely unknown, beyond the observation that a single XLF dimer promotes short range synapsis (55).
Single-molecule magnetic tweezers experiments using a reconstitution of human NHEJ proteins provided further evidence that distinct synaptic complexes form during DSB repair (56). While these studies lacked the spatial resolution to directly measure small distance changes between DNA ends, two synaptic complexes were identified that showed similar stabilities and compositions to the long-range and short-range complexes described above. Ku and DNA-PKcs alone supported fleeting synapsis of DNA ends, with a half-life of ~100 ms. Addition of PAXX dramatically increased the lifetime of this synaptic complex to ~2 seconds, similar to that of the long-range complex observed in Xenopus egg extracts. Thus, PAXX (which is also present in egg extracts) and the DNA-PK holoenzyme appear to mediate long-range synapsis. Inclusion of XLF and XRCC4-LIG4 further increased the lifetime of the synaptic complex to ~60 seconds, consistent with the short-range complex observed in Xenopus egg extracts. Omission of any one of these factors resulted in a substantial drop in the stability of synapsis and the frequency of end joining. Other single-molecule studies involving reconstitutions of human NHEJ proteins similarly show that end synapsis is a dynamic process and that persistent and close alignment of DNA ends requires XLF and XRCC4-LIG4, although they challenge the requirement of DNA-PKcs in end synapsis (54, 58), a topic that will be described in detail below (Section 4.3).
4.3. Catalytic and non-catalytic roles of DNA-PKcs in end synapsis and joining
DNA-PKcs is a core NHEJ factor whose deficiency severely disrupts NHEJ. Loss of DNA-PKcs or the inhibition of its kinase activity with selective, small-molecule inhibitors sensitizes human cells to DSB inducing agents (59, 60) and blocks end joining in cell extracts (53, 61). DNA-PKcs inhibition also disrupts repair of Cas9-generated breaks by NHEJ (50), and mice expressing catalytically inactive DNA-PKcs display an embryonic lethal phenotype similar to that of LIG4−/− and Xrcc4−/− mice (62). Beyond its role in NHEJ, DNA-PKcs contributes to telomere maintenance, ribosomal RNA processing, and mitosis (13, 63, 64).
DNA-PKcs is rapidly recruited to DSBs in cells (65). This recruitment occurs through an interaction with Ku that depends, in part, on a flexible C-terminal extension of Ku80 (12, 66). Formation of the DNA-PK holoenzyme activates its kinase activity (12). DNA-PK typically phosphorylates a serine or threonine followed by a glutamine, although this sequence preference is not strict (13). DNA-PKcs phosphorylates many NHEJ and DSB repair factors, but DNA-PKcs itself may be the most critical substrate for NHEJ. DNA-PKcs is autophosphorylated at numerous sites including two distinct clusters known as ABCDE (residues 2609–2647) and PQR (residues 2023–2056). Individual phosphoablating mutations within these clusters have at most a modest effect on sensitivity to DSBs. However, combining mutations, particularly within the ABCDE cluster, leads to profound sensitization to DSB inducing agents in cells (67, 68). We further discuss the roles of DNA-PKcs autophosphorylation in Section 5.4.
Multiple lines of evidence indicate that DNA-PKcs also plays a critical role in end synapsis. DNA-PKcs greatly stimulates the ability of Ku to bridge DNA ends in biochemical assays (69). Consistent with this biochemical activity, cryo-EM and x-ray structures suggest that the N-terminal region of the DNA-PKcs self-associates to bridge DNA ends (70, 71). In addition, depletion of DNA-PKcs from human cell and Xenopus egg extracts abolished DNA end synapsis (53, 72). As DNA-PKcs autophosphorylation is believed to occur largely in trans (73), this suggests that DNA-PKcs is likely a component of the synaptic complex. However, other observations suggest DNA-PKcs is not universally essential for NHEJ. Reconstitutions of human NHEJ proteins have provided conflicting results, with some studies concluding that DNA-PKcs is critical for synapsis and end joining (56) and others suggesting that it is dispensable (58). These discrepancies may highlight the difficulty of faithfully recapitulating physiological DNA repair using purified components. Still, IR sensitivity of cells lacking DNA-PKcs is less pronounced than in cells lacking XRCC4 (62). It is therefore possible that, compared to DSB substrates in biochemical experiments, chromosomal DSBs have less stringent requirements for synapsis in certain cellular contexts. For example, chromosomal breaks may be more easily synapsed because they are limited in mobility by the large size of chromosomes and higher order chromatin structure. Nonetheless, the bulk of the evidence across a number of model systems suggests that DNA-PKcs and its kinase activity are broadly important for end synapsis and joining. Future studies will be necessary to dissect the specific contributions of DNA-PKcs at each stage of the repair reaction.
4.4. XLF contributes to the close alignment of DNA ends
XLF was first linked to NHEJ when human patients exhibiting immunodeficiency, radiosensitivity and developmental abnormalities were found to carry mutations in XLF (15, 16). In cultured human cells, loss of XLF substantially attenuates NHEJ and V(D)J recombination (16). XLF is a paralog of XRCC4, and although it has little sequence conservation with XRCC4, it is structurally quite similar (74). Both XLF and XRCC4 exist as homodimers with a globular N-terminal head domain, a coiled-coil domain that mediates dimerization, and an intrinsically disordered C-terminal tail (74–76). XLF and XRCC4 interact through their head domains, and disrupting this interaction severely attenuates NHEJ (55, 77). The C-terminal tail of XLF plays a dual role in end synapsis. First, the extreme C-terminus contains a Ku binding motif (KBM), a short peptide that recruits XLF to DSBs (78). Removing the KBM disrupts end joining in both biochemical and cell-based experiments (79, 80). Second, while the KBM anchors XLF to Ku, the flexible tail allows XLF to explore the NHEJ complex and form interactions with XRCC4 that are required for synapsis. XLF mutants with a shortened tail but intact KBM are deficient in end joining in both Xenopus egg extracts and in mouse embryonic stem cells (80). This deficiency is not attributable to known and putative phosphorylation sites in the tail, which are dispensable for joining (81, 82). Furthermore, XLF variants with shuffled tail sequences are competent for NHEJ (80). Thus, the length and likely the flexibility of the XLF tail, rather than its specific sequence, are critical for efficient joining. In support of this model, the C-terminal tail of XLF has poor sequence conservation outside of the KBM but is highly conserved in its length. Because asymmetric XLF mutants containing only a single KBM are proficient in end joining (80), it is unlikely that long XLF tails are required to interact with Ku molecules on both DNA ends. Rather, XLF variants with internally truncated tails are efficiently recruited to Ku but fail to stabilize LIG4-XRCC4 at DSBs (80), supporting the idea that sufficient XLF tail length appears to allow interaction with XRCC4.
Numerous studies have implicated XLF in DNA end synapsis. Alternating filaments of XLF and XRCC4 have been proposed to be a key structure that mediates synapsis. These filaments have been observed in structural studies (83, 84) and can bridge DNA ends in vitro (75). In superresolution imaging of fixed cells, XLF and XRCC4 puncta have been interpreted as continuous XLF-XRCC4 filaments (54). However, it is unclear how these filaments could allow LIG4 or other end processing factors access to DNA ends and how the interior channel could accommodate DNA-PKcs. By directly imaging fluorescently labeled XLF in Xenopus egg extracts, we showed that, while Xenopus XLF and XRCC4 can form filaments in vitro, only a single XLF dimer is present within the short-range complex during end joining (55). Short-range complex formation requires the interaction between XLF and XRCC4, and asymmetric XLF mutants that are deficient in XRCC4-interaction in only one monomer were also severely deficient in end synapsis and joining. These results support a model in which a complex of XLF interacting with XRCC4-LIG4 through each of its head domains is necessary to closely align DNA ends. This ternary complex is consistent with structural work suggesting that LIG4 may prevent extensive XLF-XRCC4 filaments (85). Subsequent work in a reconstitution of human NHEJ factors found that end synapsis is not particularly sensitive to XLF concentration, further suggesting that filament formation is not necessary for end synapsis (58). Collectively, these biochemical studies may explain genetic observations that mutations (e.g. XLF L115A) that ablate filament formation in vitro (75, 86), but do not significantly attenuate the XLF–XRCC4 interaction (55), also do not affect end joining in cells (86). Therefore, we propose that a stoichiometric complex of a single XLF dimer interacting with two copies of XRCC4-LIG4 is likely the physiological complex essential for mediating synapsis.
4.5. Ligase 4 is an essential structural factor in DNA end synapsis
LIG4 is a multi-domain enzyme comprised of an N-terminal DNA binding domain, catalytic and OB domains and C-terminal tandem BRCT domains. The region between the two BRCT domains interacts with XRCC4 (87) while the first BRCT domain interacts with Ku (88). In cells, LIG4 interaction with Ku targets XRCC4-LIG4 to DSBs and dramatically stimulates end joining activity (89). As a result, loss of Ku results in a total loss of NHEJ (79, 90, 91).
In addition to its catalytic role in ligation, accumulating evidence implicates LIG4 in DNA end synapsis. LIG4 is required for stable synapsis in both human cell and Xenopus egg extracts and in reconstitutions of human NHEJ factors (53, 56, 72). Importantly, the catalytic activity of LIG4 is not required for synapsis. Thus, LIG4 structurally participates in a network of intermolecular interactions that hold DNA ends together. How LIG4 facilitates DNA end synapsis during NHEJ remains unclear. LIG4 engagement of both DNA ends may be necessary to closely align DNA ends (53, 58). Consistent with this possibility, a LIG4-specific structural motif, insert1, facilitates LIG4 engagement of ends with diverse structures (92), and DNA ends are poised to be ligated within the short-range complex (53). Alternatively, before it engages DNA ends, LIG4 may act as an essential “connector” within the synaptic complex through recruitment of XRCC4. Once present at DNA ends, XLF and XRCC4-LIG4 could form a protein “bridge” that spans DNA ends. These models need not be mutually exclusive. Indeed, a number of distinct connections across the DNA break likely stabilize the synaptic complex and provide it with the flexibility necessary to carry out the diverse set of enzymatic activities required to join complex DNA end structures.
4.6. NHEJ accessory factors and their potential role in end synapsis
Chromatin isolation and mass spectrometry experiments have shown that along with the core NHEJ factors, many “accessory” proteins are recruited to DSBs (93, 94)(Table 1). In contrast to core NHEJ factors, deficiency in accessory factors is characterized by mild impairment of cellular NHEJ. The accessory factor PAXX was identified as an interactor of Ku and a structural homolog of XRCC4 and XLF (17, 18). Accumulating evidence suggests that PAXX is functionally redundant with XLF in certain cellular contexts. The loss of both XLF and PAXX is embryonically lethal in the mouse (95) and leads to profound defects in V(D)J recombination and the response to ionizing radiation in murine G1-arrested pro-B-cells (96). Aprataxin and PNKP-Like Factor (APLF) is an accessory factor recruited to DSBs through its interaction with Ku, XRCC4, and poly (ADP-ribose). Although it possesses nuclease activity in vitro (97), its predominant role in NHEJ appears to be stimulation of repair via stabilization of XRCC4-LIG4 and XLF at DSBs (98). The Werner syndrome helicase (WRN), another accessory factor, is a RecQ-like helicase with 3´ → 5´ exonuclease activity. WRN interacts tightly with Ku, which stimulates WRN exonuclease activity in vitro (99) and is required for optimal repair kinetics in cells (100). WRN has also been implicated in DSB repair pathway choice due to its role in suppressing DNA end resection (101). Finally, the modulator of retrovirus infection (MRI, also known as CYREN) interacts with Ku and plays a complex role in regulating NHEJ during the cell cycle. MRI suppresses NHEJ on deprotected telomeres in the S and G2 phases of the cell cycle (102) yet is required for efficient NHEJ in other contexts, including resistance to ionizing radiation and for cells lacking XLF(103).
Each of these accessory factors possesses at least one KBM for recruitment to DSBs. It remains unclear how all of these factors compete for a limited number of Ku binding sites and if this competition impacts the assembly of the NHEJ synaptic complex (104, 105). Three distinct KBMs have been identified that interact with unique sites on Ku. The X-KBM from XLF and the A-KBM motif from APLF interact with Ku80 (98, 100), whereas the P-KBM of PAXX interacts with Ku70 (106). The A-KBM binds to a hydrophobic cleft near the periphery of the von Willebrand factor A-like (vWA) domain of Ku80 (107). Surprisingly, the X-KBM binds the same vWA domain at a site that is buried in the absence of the X-KBM peptide. Thus, a large outward rotation of the vWA domain is required to expose the XLF binding site. It is unknown whether this conformational transition in Ku is regulated by other NHEJ factors. The Ku binding site of the P-KBM remains poorly defined. Biochemical evidence of a ternary complex of Ku-XLF and PAXX on DNA demonstrates that the binding of PAXX and XLF to Ku is not mutually exclusive (106). Furthermore, PAXX binds to the Ku heterodimer and the Ku70 homodimer with similar affinities, suggesting that binding is mediated through Ku70.
In most cases the mechanistic role of accessory factors in NHEJ remains unclear. One possibility is that they influence assembly of the synaptic complex by excluding or stabilizing other NHEJ factors. In the former case, accessory factors may compete with each other and core NHEJ factors for Ku binding. For example, structural studies have shown that the A-KBMs from APLF, WRN, and MRI all bind the same site (108). Therefore, competitive binding by mass action could result in substantial heterogeneity among individual NHEJ synaptic complexes. Alternatively, regulatory mechanisms such as phosphorylation by DNA-PKcs could modulate Ku-KBM affinity and direct an ordered progression of synaptic complex composition. Accessory proteins may also act to stabilize other factors at DNA ends. APLF has been proposed to act as a scaffold protein that recruits a number of DSB repair factors to breaks including LIG4 (98). Similarly, PAXX has been implicated in recruiting and stabilizing the NHEJ polymerase Pol λ at DNA ends (109).
NHEJ accessory factors may also directly contribute to DNA end synapsis. Single-molecule nanomanipulation experiments demonstrated that PAXX increases the stability of the DNA-PK synaptic complex by ~20-fold, although the mechanistic origin of this effect remains unknown (56). Notably, some accessory proteins have multiple KBMs that could span the DNA break and therefore directly synapse DNA ends. Such proteins include MRI and WRN, which contain different types of KBMs at their N- and C-termini. MRI has an A-KBM and an X-KBM at its N- and C-termini, respectively. However, the C-terminal X-KBM fails to bind Ku on its own (100) and may instead play a role in recruiting other DSB repair proteins (103). WRN possesses an N-terminal A-KBM and C-terminal A- and X-KBMs (100). Whether WRN simultaneously utilizes all of these interaction sites within the synaptic complex is currently unknown. However, the vast number of potential interactions between core and accessory factors undoubtedly contributes to the stability of the NHEJ synaptic complex.
In addition to protein factors, noncoding RNAs (ncRNAs) may also contribute to DNA end synapsis during NHEJ. The long ncRNA LINP1 interacts with Ku80 and acts as a scaffold to stabilize Ku and DNA-PKcs at DSBs (110). Moreover, ncRNAs contribute to DSB repair by modulating the activity of p53, sequestering regulators of DSB repair and altering the expression of DNA repair proteins (111). Uncovering additional ncRNAs that influence NHEJ and articulating their mechanism in synaptic complex formation and stability will be important areas of future investigation.
5. THE REGULATION OF ERROR PRONE END PROCESSING
DSB induction frequently yields damaged DNA ends that are not suitable for immediate ligation by LIG4. As such, NHEJ employs a variety of end-processing enzymes that modify DNA ends until they are compatible for ligation (Table 1). NHEJ end-processing enzymes include polymerases and nucleases, which can result in insertions and/or deletions (indels) near the DSB site, and “damage correction” enzymes, which prepare termini for ligation but do not alter sequence information. Although end processing endows NHEJ with important flexibility, it is also potentially mutagenic. In this section, we provide a brief overview of NHEJ end-processing enzymes and focus on how they are regulated to maintain versatility while minimizing mutagenicity.
5.1. Overview of end-processing enzymes and their cognate DNA substrates
The nuclease most clearly implicated in NHEJ is Artemis, which has defined interactions with both LIG4 and DNA-PKcs (112, 113). During V(D)J recombination, Artemis cleaves hairpin intermediates generated by the RAG recombinase at coding ends (Figure 2B, Figure 5), thereby allowing their subsequent joining (32). Artemis-deficient cells are unable to repair a subset of DSBs induced by ionizing radiation, indicating that Artemis also contributes to the repair of some spontaneous DSBs (114). It is unclear which non-hairpin DNA end structures Artemis resolves in vivo, but it may remove 5′-aldehyde structures formed by certain radiomimetics (115). Furthermore, the purified protein cleaves various overhang and flap structures in vitro (116). APLF, WRN, and primate-specific SETMAR (Metnase) have also been proposed as NHEJ-associated nucleases (117–119). In addition, each of these proteins has non-nuclease activities (Section 4.6) (120–122), and their contributions as nucleases during physiological NHEJ are unclear.
Figure 5: Processing enzymes and their DNA substrates.
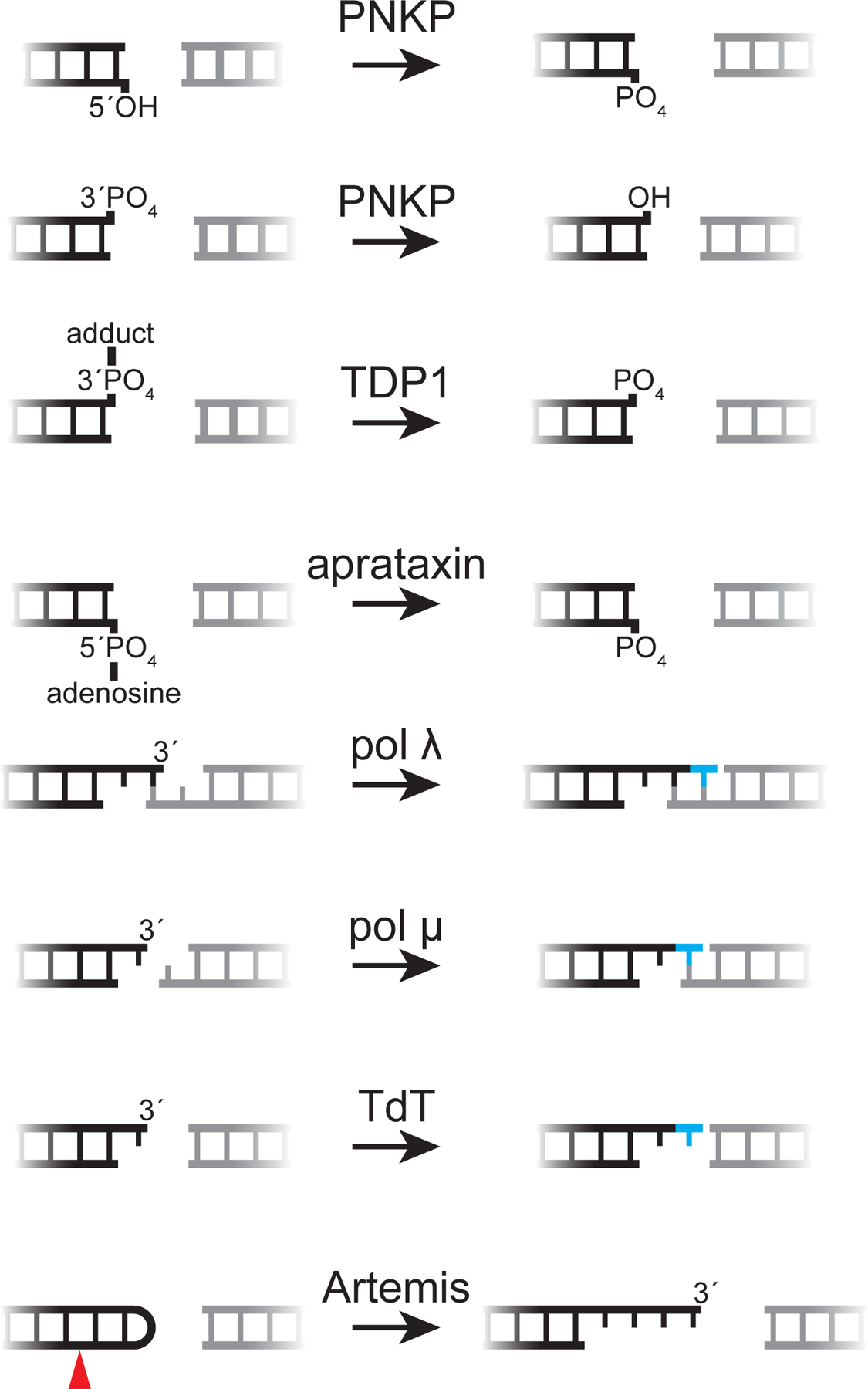
An array of end processing enzymes enables NHEJ to repair DSBs with diverse end structures, as shown here. Artemis is shown opening a V(D)J recombination hairpin intermediate (red arrow, cleavage site); its cognate substrates during repair of spontaneous breaks are unclear.
The NHEJ polymerases pol λ, pol μ, and terminal deoxynucleotidyl-transferase (TdT) belong to the X family of DNA polymerases. Each is recruited to DSBs via an N-terminal BRCT domain that allows interaction with Ku and XRCC4-LIG4 (123, 124). Pol λ and pol μ are expressed ubiquitously, whereas TdT is expressed only in developing lymphocytes, where it is utilized during V(D)J recombination (125). The other X family polymerase, pol β, functions primarily in base excision repair, with limited proposed roles in NHEJ (126). The NHEJ polymerases are biochemically specialized for particular DNA end structures: pol λ acts preferentially on 3′ termini that are base-paired to the template strand, whereas pol μ does not require a paired 3′ terminus for templated synthesis (Figure 5) (127). Loss of either pol λ or pol μ alone has distinct effects on repair fidelity, suggesting that polymerase specialization occurs during cellular NHEJ (128). TdT incorporates deoxynucleotides at 3′ termini in a template-independent fashion and contributes to junctional diversification during V(D)J recombination (Figure 5) (29). Surprisingly, pol μ and TdT primarily incorporate ribonucleotides when they are engaged during NHEJ (129). This activity affords NHEJ additional flexibility and likely highlights the evolutionary imperative for rapid DSB repair.
NHEJ utilizes several “damage correction” enzymes that resolve specific chemical blocks to enable ligation without altering sequence information (Figure 5). The bifunctional polynucleotide kinase 3′-phosphatase (PNKP), recruited to DSBs via interaction with XRCC4, phosphorylates 5′-hydroxyl termini and removes 3′-phosphates generated by IR (130). Aprataxin, which also interacts with XRCC4 (131), reverses damage caused by abortive ligation (132). Ligation reactions involve transfer of an AMP moiety to the DNA 5′-phosphate terminus, and this intermediate persists when damaged DNA ends cannot be aligned to complete ligation (133). Aprataxin removes AMP to regenerate the 5′-phosphate terminus for subsequent ligation attempts. Similarly, tyrosyl-DNA phosphodiesterases 1 and 2 (TDP1 and TDP2) regenerate ligatable termini after abortive topoisomerase activity. Topoisomerases cleave and re-ligate DNA to relieve topological stress, proceeding through a phosphotyrosine intermediate that covalently crosslinks the protein to DNA (134). Topoisomerases can stall in the presence of topoisomerase inhibitors or when they cleave within distorted DNA, thereby leaving a covalent block to re-ligation (135). Thus, TDP1 hydrolyzes 3′-phosphotyrosine topoisomerase I intermediates (136) and TDP2 reverses 5′-phosphotyrosine topoisomerase II intermediates (137) to allow re-ligation by NHEJ or other pathways. In addition, TDP1 has been implicated in removal of diverse 3′-phosphodiester adducts (138), including 3′-phosphoglycolates, which commonly arise at DSBs induced by ionizing radiation (139). Finally, beyond its role as a DSB sensor and recruitment hub, Ku acts as a 5′-deoxyribose-5-phosphate/apurinic-apyrimidinic (5′dRP/AP) lyase near DNA ends (140). This activity removes abasic sites—generated by IR or as intermediates during CSR, among other sources—that would otherwise block repair by NHEJ.
5.2. Coordination of end processing and end synapsis minimizes NHEJ errors
Accumulating evidence indicates that NHEJ employs end-processing enzymes only when necessary and repairs DSBs with minimal indels. Compatible (i.e., blunt or sticky) ends are typically joined by NHEJ without errors in mammalian cells (141–143), human cell extracts (61, 144), and Xenopus egg extracts (145, 146). Furthermore, incompatible ends are typically processed as conservatively as possible in the minimum number of steps (128, 146, 147). For example, 5′-hydroxyl ends in principle could be converted to 5′-phosphate ends through either direct phosphorylation by PNKP or nuclease activity, but the former is strongly preferred (146). Thus, end processing during NHEJ appears to be a carefully regulated process that has evolved to minimize errors during DSB repair.
How end processing is regulated directly impacts the fidelity of genome maintenance and the outcomes of genome engineering techniques that introduce DSBs (e.g., CRISPR-Cas9). Indeed, a deeper understanding of end processing regulation could inform efforts to modulate the fidelity of CRISPR-Cas9-generated DSBs. One model of end processing regulation, based on biochemical reconstitutions of purified proteins, posits that each DNA end is processed independently, with modification occurring stochastically and iteratively by a variety of processing enzymes until the ends become compatible for ligation (124, 148). It is unclear, however, how such a model can account for the conservative properties of NHEJ outlined above. Recent work from our laboratory in Xenopus egg extracts has elucidated how end processing is coordinated with end synapsis (Section 4.2) and provides a model for how NHEJ minimizes errors (146) (Figure 6). This model comprises three features that promote conservative end joining: first, end processing is blocked by Ku and DNA-PKcs (see section 5.4) until DNA ends enter the short-range synaptic complex. Second, because DNA ends are poised for ligation in the short-range complex, compatible ends are ligated immediately without processing, as observed in multiple experimental systems (61, 141–146). Third, end processing occurs directly within the short-range synaptic complex, which ensures that incompatible DNA ends are rapidly ligated after undergoing the minimal number of processing steps required for compatibility. In support of this idea, perturbations of short-range synapsis (LIG4-XRCC4 deficiency, XLF deficiency, DNA-PKcs inhibition) block a range of end processing activities in human cells, human cell extracts, and Xenopus egg extracts (146, 147, 149–152). Moreover, single-molecule FRET experiments that simultaneously monitor end synapsis and end processing in real time directly demonstrate that pol λ and Tdp1 act preferentially in the short-range synaptic complex (146). It is likely that other end processing enzymes exhibit a similar preference. Thus, end processing in the short-range synaptic complex could promote repair with minimal errors without sacrificing versatility.
Figure 6: Model of error-prone processing regulation.
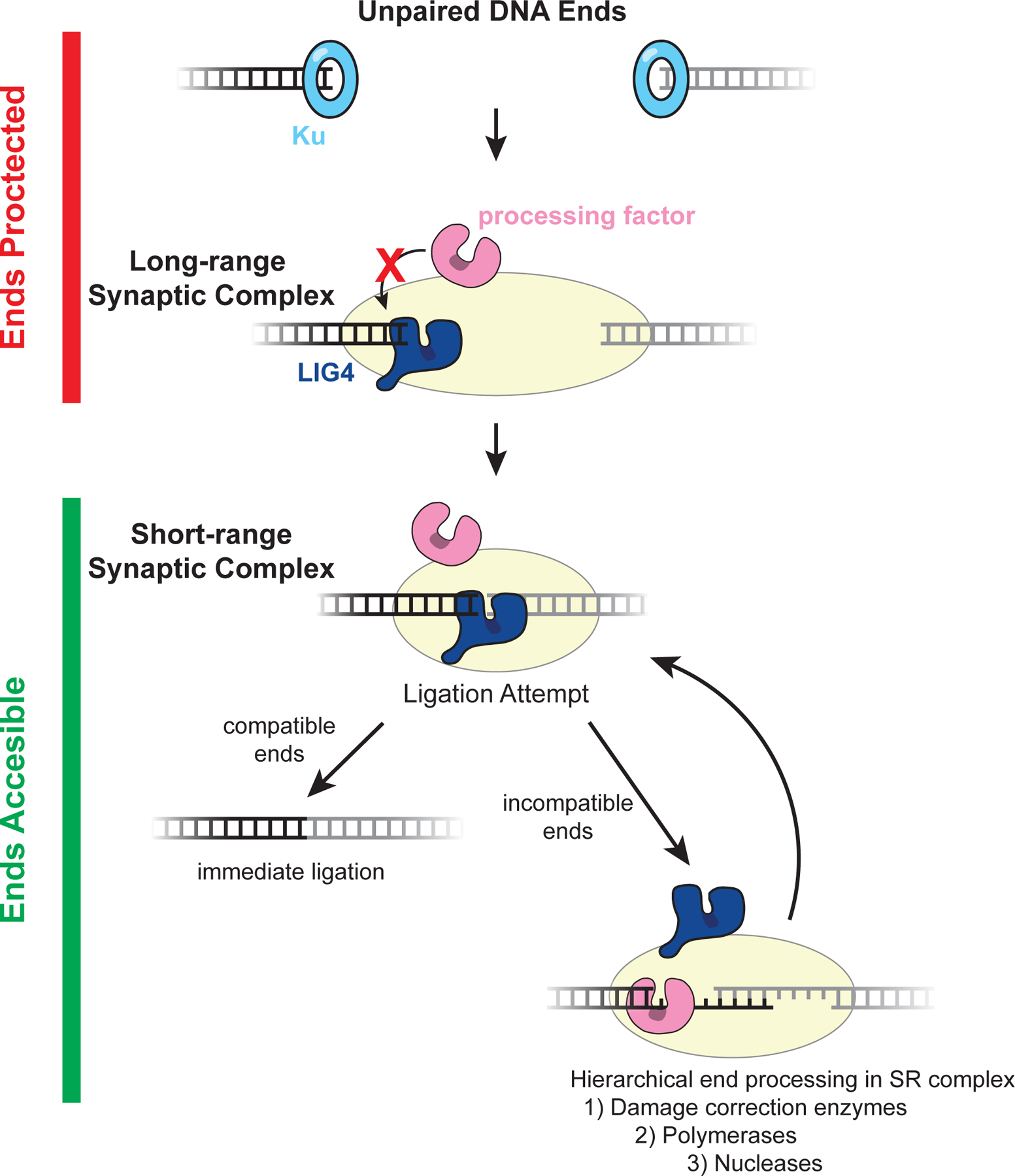
Unpaired ends and those in the long-range synaptic complex are protected from aberrant or premature processing by Ku and DNA-PKcs. Although NHEJ processing factors are recruited, they are unable to act on DNA ends in these states (146). It is possible that other core NHEJ factors also protect DNA ends from processing. For example, as depicted here, LIG4 may directly engage a single DNA end prior to short-range synapsis, thereby preventing processing through steric occlusion. A second LIG4 molecule (not depicted) could protect the second end. Subsequently, direct juxtaposition of ends in the LIG4-dependent short-range synaptic enables a ligation attempt. If DNA ends are compatible, they can be ligated immediately without processing. If DNA ends are incompatible for ligation, they undergo processing in the short-range synaptic complex. Processing enzymes act hierarchically with the depicted priority to promote conservative modifications, and LIG4 frequently re-engages DNA ends to ligate them after the minimum number of processing events required for compatibility. Together, these features allow NHEJ to repair diverse DNA ends and minimize unnecessary genomic alterations.
End processing in the short-range synaptic complex likely requires that LIG4 and processing enzymes alternate in engaging incompatible DNA ends. Although the precise molecular choreography remains unclear, LIG4 may disengage DNA ends to allow processing yet remain associated with the NHEJ complex through its interaction with XRCC4 (153), thereby allowing rapid ligation of newly compatible ends. Moreover, LIG4 engagement of incompatible ends may result in an unproductive “open” conformation or other remodeling of the synaptic complex that facilitates end processing factor recruitment (92, 143, 154). Further biochemical and structural studies are needed to address these possibilities. Finally, an interesting but unresolved question is how end processing is regulated during V(D)J recombination, which is characterized by higher junctional diversity than that observed for non-programmed breaks (29). Unique features of V(D)J recombination, including the involvement of TdT and contributions by RAG, may account for these differences.
5.3. A hierarchical model of end processing
The processing enzymes, if any, that act on a given pair of DNA ends determine the fidelity of NHEJ. Accumulating evidence suggests a model in which end-modifying enzymes are employed hierarchically as an additional layer of regulation to maximize fidelity (Figure 6) (146, 155, 156). Under this model, direct ligation by LIG4 is given highest priority, followed by conservative damage correction activity, and finally sequence-altering polymerase and nuclease activity. Consistent with this idea and as noted above, compatible ends are typically directly ligated without processing. Moreover, LIG4 has the unique ability to join ends with mild nucleotide mismatches or oxidative damage (e.g., 8-oxoguanine), thereby acting as a “translesion ligase” (20, 21, 143). If LIG4 is unable to repair the DSB, conservative damage correction enzymes are prioritized over sequence altering events. For example, ends requiring only 5′-phosphorylation by PNKP for ligation are usually joined without insertions or deletions, and Tdp1 activity is preferred over nuclease activity for the removal of 3′-phosphodiester adducts (146). Among sequence-altering enzymes, gap-filling polymerase activity appears to be prioritized over nuclease activity. In mammalian cells and Xenopus egg extracts, non- or partially-complementary ends that could in principle be resolved by either polymerase or nuclease activity are typically subjected only to polymerase activity (143, 146, 157, 158). Gap-filling limited by the availability of single-stranded template may allow for greater preservation of genetic information than heterogeneous nuclease activity. Overall, hierarchical organization of processing enzymes appears to play an important role in restraining the potential mutagenicity of NHEJ.
The molecular mechanisms enforcing such a hierarchy remain unclear but may include the relative abundance of processing enzymes and their affinity for the NHEJ complex. The extent of DNA end accessibility may also regulate processing hierarchy. For example, damage correction enzymes PNKP and TDP1 make minimal contacts with DNA substrates outside the extreme terminus, whereas pol λ and pol μ make more extensive DNA contacts. Thus, a gradual loosening of end protection, perhaps mediated by multiple DNA-PKcs phosphorylation events (see next section), could allow some conservative enzymes to access DNA ends before polymerases. Moreover, the availability of appropriate DNA end chemistry may also contribute to hierarchical end processing. For example, pol λ and pol μ must extend from an undamaged 3′-hydroxyl terminus and are stimulated by the presence of a 5′-phosphate on the downstream DNA end (159). Thus, efficient polymerase activity may depend on prior preparation of termini by conservative damage correction enzymes.
5.4. Ku and DNA-PKcs are regulators of DNA end accessibility
Protection of DNA ends from unrestricted modification provides another layer of end processing regulation. Initial Ku binding to DNA ends blocks both long-range resection, which would disfavor NHEJ and promote homologous recombination or alternative end joining (160), as well as premature processing by NHEJ enzymes (146). It is unclear, however, how this initial Ku block is alleviated to allow regulated processing in later stages of the NHEJ reaction. An interesting possibility suggested by footprinting studies is that the Ku ring slides inward upon DNA-PKcs association, thereby exposing the DNA end (161). Linking Ku deprotection to association of DNA-PKcs could limit end processing prior to the formation of a synaptic complex. Posttranslational modification of Ku by phosphorylation (162) or ubiquitination (163–168) may also regulate end accessibility.
DNA-PKcs has also long been implicated in regulating DNA end accessibility (reviewed in (169)). Initial association of DNA-PKcs contributes to the protection of ends from aberrant processing (146, 170). Subsequent phosphorylation of DNA-PKcs at two distinct clusters of residues (Section 4.3) has been reported to have opposing effects on end accessibility. Sequence analysis of V(D)J coding joints and repaired I-SceI-induced DSBs in cells expressing mutant DNA-PKcs reveals that phosphorylation at the ABCDE cluster promotes end processing, whereas phosphorylation at the PQR cluster restricts end processing (67, 68, 171). Phosphorylation of DNA-PKcs induces its dissociation from DNA ends (172). Notably, a DNA-PKcs mutant with alanine substitutions at both the ABCDE and PQR clusters shows no defect in dissociation from DNA ends (173). Thus, it appears that phosphorylation of the ABCDE and PQR clusters modulate the interaction of DNA-PKcs with DNA ends independently of complete dissociation, which is mediated by phosphorylation of other unidentified sites. DNA-PKcs autophosphorylation in trans (73) links regulation of end accessibility to synapsis. Further biochemical and structural studies are required to elucidate the molecular details of how phosphorylation of the ABCDE and PQR clusters, as well as of dissociation-inducing sites, regulates access to DNA ends.
The extent to which other NHEJ factors contribute to DNA end protection is unclear. Extensive XLF-XRCC4 filaments have been proposed to protect DNA ends (174), but such filaments are not likely to form during physiological NHEJ (55). An intriguing possibility is that LIG4 directly engages DNA ends, thereby prohibiting access by processing enzymes before ligation has been attempted (Figure 6). To address this possibility, it will be important to determine whether engagement of a given DNA end by LIG4 and other end-modifying enzymes is mutually exclusive.
6. Conclusion and outlook
Here, we have reviewed how DNA end synapsis and regulated end processing during NHEJ promote efficient and faithful DSB repair. Synapsis is carried out by a dynamic multi-protein complex that evolves during repair. DNA ends are initially weakly tethered together in a long-range complex that depends on the core NHEJ factors Ku and DNA-PKcs and is stabilized by the accessory factor PAXX. Close and stable alignment of DNA ends in a short-range complex requires XLF and XRCC4-LIG4 along with the kinase activity of DNA-PKcs. Future work is necessary to determine the full complement of molecular interactions required for end synapsis. Outstanding issues include the precise structural role of LIG4 in short-range synapsis; whether transition from the long-range to short-range synaptic complexes is due to changes in protein composition or instead a structural remodeling of pre-bound factors; and the roles of NHEJ accessory factors in the assembly and stability of the synaptic complex.
End processing during NHEJ is finely tuned to minimize mutagenesis. DNA ends are initially protected from processing, which is limited to the ligation-poised short-range synaptic complex. This mechanism prioritizes ligation over end processing and promotes rapid ligation after the minimum number of processing events. In addition, processing appears to occur hierarchically, with conservative end processing activities prioritized over mutagenic ones. Unanswered questions in the regulation of end processing include how end protection is relieved to allow processing and ligation, and how hierarchical processing is enforced. Answers to these and other mechanistic questions will be important to fully understand how NHEJ contributes to genome maintenance.
ACKNOWLEDGEMENTS
We apologize to our many colleagues whose work we were unable to cite due to space limitations. We thank current and former members of the Loparo laboratory and Marie Bao for their critical reading of the manuscript. This work was supported by a grant from the National Institutes of Health grant (R01GM115487 to J.J.L.) and a Damon Runyon Postdoctoral Research Fellowship (to B.M.S).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Bunting SF, Nussenzweig A. 2013. End-joining, translocations and cancer. Nature reviews Cancer 13(7):443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79:181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karanam K, Kafri R, Loewer A, Lahav G. 2012. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Molecular cell 47(2):320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haber JE. 2018. DNA Repair: The Search for Homology. Bioessays 40(5):e1700229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5(11):a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, et al. 1998. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17(18):5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sfeir A, Symington LS. 2015. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends in biochemical sciences 40(11):701–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhargava R, Onyango DO, Stark JM. 2016. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends in Genetics 32(9):566–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ceccaldi R, Rondinelli B, D’Andrea AD. 2016. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends in Cell Biology 26(1):52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scully R, Panday A, Elango R, Willis NA. 2019. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol 20(11):698–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker JR, Corpina RA, Goldberg J. 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412(6847):607–14 [DOI] [PubMed] [Google Scholar]
- 12.Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434(7033):605–11 [DOI] [PubMed] [Google Scholar]
- 13.Jette N, Lees-Miller SP. 2015. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol 117(2–3):194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Otevrel T, Gao Y, Cheng H-L, Seed B, et al. 1995. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 83(7):1079–89 [DOI] [PubMed] [Google Scholar]
- 15.Ahnesorg P, Smith P, Jackson SP. 2006. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 124(2):301–313 [DOI] [PubMed] [Google Scholar]
- 16.Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche M-C, et al. 2006. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 124(2):287–299 [DOI] [PubMed] [Google Scholar]
- 17.Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, et al. 2015. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science (New York, NY) 347(6218):185–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing M, Yang M, Huo W, Feng F, Wei L, et al. 2015. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nature communications 6:6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, et al. 1997. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 388(6641):492–95 [DOI] [PubMed] [Google Scholar]
- 20.Gu J, Lu H, Tsai AG, Schwarz K, Lieber MR. 2007. Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Res 35(17):5755–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai CJ, Kim SA, Chu G. 2007. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl. Acad. Sci. U.S.A 104(19):7851–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilenchik MM, Knudson AG. 2003. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences 100(22):12871–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caldecott KW. 2008. Single-strand break repair and genetic disease. Nat. Rev. Genet 9(8):619–31 [DOI] [PubMed] [Google Scholar]
- 24.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. 2000. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol 20(11):3977–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balmus G, Pilger D, Coates J, Demir M, Sczaniecka-Clift M, et al. 2019. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat Commun 10(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Lange T. 2018. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet 52:223–47 [DOI] [PubMed] [Google Scholar]
- 27.Alt FW, Zhang Y, Meng F-L, Guo C, Schwer B. 2013. Mechanisms of Programmed DNA Lesions and Genomic Instability in the Immune System. Cell 152(3):417–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaudhuri J, Alt FW. 2004. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 4(7):541–52 [DOI] [PubMed] [Google Scholar]
- 29.Gellert M. 1992. Molecular analysis of V(D)J recombination. Annu. Rev. Genet 26:425–46 [DOI] [PubMed] [Google Scholar]
- 30.Schatz DG, Swanson PC. 2011. V(D)J Recombination: Mechanisms of Initiation. Annu. Rev. Genet 45(1):167–202 [DOI] [PubMed] [Google Scholar]
- 31.Stavnezer J, Schrader CE. 2014. IgH Chain Class Switch Recombination: Mechanism and Regulation. J.I 193(11):5370–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma Y, Pannicke U, Schwarz K, Lieber MR. 2002. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 108(6):781–94 [DOI] [PubMed] [Google Scholar]
- 33.Bertocci B, De Smet A, Berek C, Weill J-C, Reynaud C-A. 2003. Immunoglobulin kappa light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu. Immunity 19(2):203–11 [DOI] [PubMed] [Google Scholar]
- 34.Bertocci B, De Smet A, Weill J-C, Reynaud C-A. 2006. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 25(1):31–41 [DOI] [PubMed] [Google Scholar]
- 35.Komori T, Okada A, Stewart V, Alt FW. 1993. Lack of N regions in antigen receptor variable region genes of TdT-deficient lymphocytes. Science 261(5125):1171–75 [DOI] [PubMed] [Google Scholar]
- 36.Okazaki I, Kinoshita K, Muramatsu M, Yoshikawa K, Honjo T. 2002. The AID enzyme induces class switch recombination in fibroblasts. 416:6. [DOI] [PubMed] [Google Scholar]
- 37.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, et al. 2007. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 449(7161):478–82 [DOI] [PubMed] [Google Scholar]
- 38.Woodbine L, Gennery AR, Jeggo PA. 2014. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst.) 16:84–96 [DOI] [PubMed] [Google Scholar]
- 39.Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. 1998. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol 8(25):1395–98 [DOI] [PubMed] [Google Scholar]
- 40.Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, et al. 1998. Late embryonic lethality and impaired V (D)J recombination in mice lacking DNA ligase IV. Nature 396(6707):173–77 [DOI] [PubMed] [Google Scholar]
- 41.Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, et al. 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95(7):891–902 [DOI] [PubMed] [Google Scholar]
- 42.Ghezraoui H, Piganeau M, Renouf B, Renaud J-B, Sallmyr A, et al. 2014. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 55(6):829–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, et al. 2011. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 144(1):27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sishc BJ, Davis AJ. 2017. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers (Basel) 9(7): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kefala Stavridi A, Appleby R, Liang S, Blundell TL, Chaplin AK. 2020. Druggable binding sites in the multicomponent assemblies that characterise DNA double-strand-break repair through non-homologous end joining. Essays Biochem [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohiuddin IS, Kang MH. 2019. DNA-PK as an Emerging Therapeutic Target in Cancer. Front Oncol 9:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu VT, Weber T, Wefers B, Wurst W, Sander S, et al. 2015. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33(5):543–48 [DOI] [PubMed] [Google Scholar]
- 48.Li G, Liu D, Zhang X, Quan R, Zhong C, et al. 2018. Suppressing Ku70/Ku80 expression elevates homology-directed repair efficiency in primary fibroblasts. The International Journal of Biochemistry & Cell Biology 99:154–60 [DOI] [PubMed] [Google Scholar]
- 49.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. 2015. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33(5):538–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robert F, Barbeau M, Éthier S, Dostie J, Pelletier J. 2015. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med 7(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, et al. 2006. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. The Journal of cell biology 172(6):823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, et al. 2007. Positional stability of single double-strand breaks in mammalian cells. Nature cell biology 9(6):675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graham TGW, Walter JC, Loparo JJ. 2016. Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Molecular cell 61(6):850–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, et al. 2015. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America 112(20):E2575–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Graham TGW, Carney SM, Walter JC, Loparo JJ. 2018. A single XLF dimer bridges DNA ends during nonhomologous end joining. Nat. Struct. Mol. Biol 25(9):877–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang JL, Duboc C, Wu Q, Ochi T, Liang S, et al. 2018. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nature structural & molecular biology 25(6):482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, et al. 2016. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature [DOI] [PubMed] [Google Scholar]
- 58.Zhao B, Watanabe G, Morten MJ, Reid DA, Rothenberg E, Lieber MR. 2019. The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nature communications 10(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fok JHL, Ramos-Montoya A, Vazquez-Chantada M, Wijnhoven PWG, Follia V, et al. 2019. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nature communications 10(1):1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, et al. 2006. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer research 66(10):5354–5362 [DOI] [PubMed] [Google Scholar]
- 61.Baumann P, West SC. 1998. DNA end-joining catalyzed by human cell-free extracts. Proc. Natl. Acad. Sci. U.S.A 95(24):14066–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, et al. 2015. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Molecular cell 58(1):172–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruis BL, Fattah KR, Hendrickson EA. 2008. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Molecular and Cellular Biology 28(20):6182–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shao Z, Flynn RA, Crowe JL, Zhu Y, Liang J, et al. 2020. DNA-PKcs has KU-dependent function in rRNA processing and haematopoiesis. Nature 79:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, et al. 2007. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. The Journal of cell biology 177(2):219–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gell D, Jackson SP. 1999. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Research 27(17):3494–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui X, Yu Y, Gupta S, Cho Y-M, Lees-Miller SP, Meek K. 2005. Autophosphorylation of DNA-Dependent Protein Kinase Regulates DNA End Processing and May Also Alter Double-Strand Break Repair Pathway Choice. MCB 25(24):10842–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ding Q, Reddy YVR, Wang W, Woods T, Douglas P, et al. 2003. Autophosphorylation of the Catalytic Subunit of the DNA-Dependent Protein Kinase Is Required for Efficient End Processing during DNA Double-Strand Break Repair. MCB 23(16):5836–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeFazio LG, Stansel RM, Griffith JD, Chu G. 2002. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J 21(12):3192–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. 2017. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 355(6324):520–24 [DOI] [PubMed] [Google Scholar]
- 71.Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. 2006. Three-Dimensional Structure of the Human DNA-PKcs/Ku70/Ku80 Complex Assembled on DNA and Its Implications for DNA DSB Repair. Molecular cell 22(4):511–519 [DOI] [PubMed] [Google Scholar]
- 72.Cottarel J, Frit P, Bombarde O, Salles B, Négrel A, et al. 2013. A noncatalytic function of the ligation complex during nonhomologous end joining. The Journal of cell biology 200(2):173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP. 2007. trans Autophosphorylation at DNA-Dependent Protein Kinase’s Two Major Autophosphorylation Site Clusters Facilitates End Processing but Not End Joining. MCB 27(10):3881–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andres SN, Modesti M, Tsai CJ, Chu G, Junop MS. 2007. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Molecular cell 28(6):1093–1101 [DOI] [PubMed] [Google Scholar]
- 75.Andres SN, Vergnes A, Ristic D, Wyman C, Modesti M, Junop M. 2012. A human XRCC4-XLF complex bridges DNA. Nucleic acids research 40(4):1868–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, Chirgadze DY, Bolanos-Garcia VM, Sibanda BL, Davies OR, et al. 2008. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. The EMBO journal 27(1):290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roy S, Andres SN, Vergnes A, Neal JA, Xu Y, et al. 2012. XRCC4’s interaction with XLF is required for coding (but not signal) end joining. Nucleic acids research 40(4):1684–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yano K, Morotomi-Yano K, Lee K-J, Chen DJ. 2011. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS letters 585(6):841–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhargava R, Sandhu M, Muk S, Lee G, Vaidehi N, Stark JM. 2018. C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nature communications 9(1):2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carney SM, Moreno AT, Piatt SC, Cisneros-Aguirre M, Lopezcolorado FW, et al. 2020. XLF acts as a flexible connector during non-homologous end joining. Biochemistry [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Normanno D, Négrel A, de Melo AJ, Betzi S, Meek K, Modesti M. 2017. Mutational phospho-mimicry reveals a regulatory role for the XRCC4 and XLF C-terminal tails in modulating DNA bridging during classical non-homologous end joining. eLife 6:e22900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yu Y, Mahaney BL, Yano K, Ye R, Fang S, et al. 2008. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA repair 7(10):1680–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hammel M, Rey M, Yu Y, Mani RS, Classen S, et al. 2011. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. Journal of Biological Chemistry 286(37):32638–32650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, et al. 2011. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proceedings of the National Academy of Sciences of the United States of America 108(31):12663–12668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ochi T, Wu Q, Chirgadze DY, Grossmann JG, Bolanos-Garcia VM, Blundell TL. 2012. Structural insights into the role of domain flexibility in human DNA ligase IV. Structure (London, England : 1993) 20(7):1212–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roy S, de Melo AJ, Xu Y, Tadi SK, Négrel A, et al. 2015. XRCC4/XLF Interaction Is Variably Required for DNA Repair and Is Not Required for Ligase IV Stimulation. Mol. Cell. Biol 35(17):3017–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu P-Y, Frit P, Meesala S, Dauvillier S, Modesti M, et al. 2009. Structural and functional interaction between the human DNA repair proteins DNA ligase IV and XRCC4. Molecular and Cellular Biology 29(11):3163–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. 2007. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA repair 6(6):712–722 [DOI] [PubMed] [Google Scholar]
- 89.Nick McElhinny SA, Snowden CM, McCarville J, Ramsden DA. 2000. Ku recruits the XRCC4-ligase IV complex to DNA ends. Molecular and Cellular Biology 20(9):2996–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. 2010. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet 6(2):e1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xing M, Bjørås M, Daniel JA, Alt FW, Oksenych V. 2017. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair (Amst.) 57:133–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conlin MP, Reid DA, Small GW, Chang HH, Watanabe G, et al. 2017. DNA Ligase IV Guides End-Processing Choice during Nonhomologous End Joining. Cell Rep 20(12):2810–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berthelot V, Mouta-Cardoso G, Hégarat N, Guillonneau F, François J-C, et al. 2016. The human DNA ends proteome uncovers an unexpected entanglement of functional pathways. Nucleic acids research 44(10):4721–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haahr P, Hoffmann S, Tollenaere MAX, Ho T, Toledo LI, et al. 2016. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nature cell biology, pp. 1–27 [DOI] [PubMed] [Google Scholar]
- 95.Balmus G, Barros AC, Wijnhoven PWG, Lescale C, Hasse HL, et al. 2016. Synthetic lethality between PAXX and XLF in mammalian development. Genes & development 30(19):2152–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar V, Alt FW, Frock RL. 2016. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proceedings of the National Academy of Sciences of the United States of America 113(38):10619–10624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kanno S, Kuzuoka H, Sasao S, Hong Z, Lan L, et al. 2007. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. The EMBO journal 26(8):2094–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, et al. 2013. APLF promotes the assembly and activity of non-homologous end joining protein complexes. The EMBO journal 32(1):112–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. 2000. Ku complex interacts with and stimulates the Werner protein. Genes & development 14(8):907–912 [PMC free article] [PubMed] [Google Scholar]
- 100.Grundy GJ, Rulten SL, Arribas-Bosacoma R, Davidson K, Kozik Z, et al. 2016. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nature communications 7:11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shamanna RA, Lu H, de Freitas JK, Tian J, Croteau DL, Bohr VA. 2016. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat Commun 7(1):13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arnoult N, Correia A, Ma J, Merlo A, Garcia-Gomez S, et al. 2017. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549(7673):548–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hung PJ, Johnson B, Chen B-R, Byrum AK, Bredemeyer AL, et al. 2018. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Molecular cell 71(2):332–342.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frit P, Ropars V, Modesti M, Charbonnier J-B, Calsou P. 2019. Plugged into the Ku-DNA hub: The NHEJ network. Progress in biophysics and molecular biology [DOI] [PubMed] [Google Scholar]
- 105.Rulten SL, Grundy GJ. 2017. Non-homologous end joining: Common interaction sites and exchange of multiple factors in the DNA repair process. BioEssays : news and reviews in molecular, cellular and developmental biology, p. 1600209. [DOI] [PubMed] [Google Scholar]
- 106.Tadi SK, Tellier-Lebègue C, Nemoz C, Drevet P, Audebert S, et al. 2016. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell reports 17(2):541–555 [DOI] [PubMed] [Google Scholar]
- 107.Nemoz C, Ropars V, Frit P, Gontier A, Drevet P, et al. 2018. XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining. Nature structural & molecular biology 25(10):971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim K, Min J, Kirby TW, Gabel SA, Pedersen LC, London RE. 2020. Ligand binding characteristics of the Ku80 von Willebrand domain. DNA repair 85:102739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Craxton A, Munnur D, Jukes-Jones R, Skalka G, Langlais C, et al. 2018. PAXX and its paralogs synergistically direct DNA polymerase λ activity in DNA repair. Nature communications 9(1):3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang Y, He Q, Hu Z, Feng Y, Fan L, et al. 2016. Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer. Nature structural & molecular biology 23(6):522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chowdhury D, Choi YE, Brault ME. 2013. Charity begins at home: non-coding RNA functions in DNA repair. Nat. Rev. Mol. Cell Biol 14(3):181–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.De Ioannes P, Malu S, Cortes P, Aggarwal AK. 2012. Structural basis of DNA ligase IV-Artemis interaction in nonhomologous end-joining. Cell Rep 2(6):1505–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Niewolik D, Pannicke U, Lu H, Ma Y, Wang L-CV, et al. 2006. DNA-PKcs Dependence of Artemis Endonucleolytic Activity, Differences between Hairpins and 5′ or 3′ Overhangs. J. Biol. Chem 281(45):33900–909 [DOI] [PubMed] [Google Scholar]
- 114.Riballo E, Kühne M, Rief N, Doherty A, Smith GCM, et al. 2004. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 16(5):715–24 [DOI] [PubMed] [Google Scholar]
- 115.Kawale AS, Akopiants K, Valerie K, Ruis B, Hendrickson EA, et al. 2018. TDP1 suppresses mis-joining of radiomimetic DNA double-strand breaks and cooperates with Artemis to promote optimal nonhomologous end joining. Nucleic Acids Research 46(17):8926–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma Y, Schwarz K, Lieber MR. 2005. The Artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair 4(7):845–51 [DOI] [PubMed] [Google Scholar]
- 117.Beck BD, Lee S-S, Williamson E, Hromas RA, Lee S-H. 2011. Biochemical Characterization of Metnase’s Endonuclease Activity and Its Role in NHEJ Repair. Biochemistry 50(20):4360–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kusumoto R, Dawut L, Marchetti C, Wan Lee J, Vindigni A, et al. 2008. Werner Protein Cooperates with the XRCC4-DNA Ligase IV Complex in End-Processing †. Biochemistry 47(28):7548–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li S, Kanno S, Watanabe R, Ogiwara H, Kohno T, et al. 2011. Polynucleotide Kinase and Aprataxin-like Forkhead-associated Protein (PALF) Acts as Both a Single-stranded DNA Endonuclease and a Single-Stranded DNA 3′ Exonuclease and Can Participate in DNA End Joining in a Biochemical System. J. Biol. Chem 286(42):36368–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grundy GJ, Rulten SL, Zeng Z, Arribas-Bosacoma R, Iles N, et al. 2012. APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO J 32(1):112–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee JW, Harrigan J, Opresko PL, Bohr VA. 2005. Pathways and functions of the Werner syndrome protein. Mechanisms of Ageing and Development 126(1):79–86 [DOI] [PubMed] [Google Scholar]
- 122.Tellier M, Chalmers R. 2019. The roles of the human SETMAR (Metnase) protein in illegitimate DNA recombination and non-homologous end joining repair. DNA Repair 80:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fan W, Wu X. 2004. DNA polymerase lambda can elongate on DNA substrates mimicking non-homologous end joining and interact with XRCC4-ligase IV complex. Biochem. Biophys. Res. Commun 323(4):1328–33 [DOI] [PubMed] [Google Scholar]
- 124.Ma Y, Lu H, Tippin B, Goodman MF, Shimazaki N, et al. 2004. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 16(5):701–13 [DOI] [PubMed] [Google Scholar]
- 125.Uchiyama Y, Takeuchi R, Kodera H, Sakaguchi K. 2009. Distribution and roles of X-family DNA polymerases in eukaryotes. Biochimie 91(2):165–70 [DOI] [PubMed] [Google Scholar]
- 126.Xia W, Ci S, Li M, Wang M, Dianov GL, et al. 2019. Two‐way crosstalk between BER and c‐NHEJ repair pathway is mediated by Pol‐β and Ku70. FASEB j 33(11):11668–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juárez R, Bebenek K, et al. 2005. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell 19(3):357–66 [DOI] [PubMed] [Google Scholar]
- 128.Pryor JM, Waters CA, Aza A, Asagoshi K, Strom C, et al. 2015. Essential role for polymerase specialization in cellular nonhomologous end joining. Proc. Natl. Acad. Sci. U.S.A 112(33):E4537–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pryor JM, Conlin MP, Carvajal-Garcia J, Luedeman ME, Luthman AJ, et al. 2018. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 361(6407):1126–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, et al. 2004. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J 23(19):3874–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Clements PM, Breslin C, Deeks ED, Byrd PJ, Ju L, et al. 2004. The ataxia-oculomotor apraxia 1 gene product has a role distinct from ATM and interacts with the DNA strand break repair proteins XRCC1 and XRCC4. DNA Repair (Amst.) 3(11):1493–1502 [DOI] [PubMed] [Google Scholar]
- 132.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, et al. 2006. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443(7112):713–16 [DOI] [PubMed] [Google Scholar]
- 133.Ellenberger T, Tomkinson AE. 2008. Eukaryotic DNA Ligases: Structural and Functional Insights. Annu. Rev. Biochem 77(1):313–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Champoux JJ. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 70:369–413 [DOI] [PubMed] [Google Scholar]
- 135.Cho J-E, Jinks-Robertson S. 2018. Topoisomerase I and Genome Stability: The Good and the Bad. Methods Mol. Biol 1703:21–45 [DOI] [PubMed] [Google Scholar]
- 136.Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA. 1996. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. U.S.A 93(21):11534–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461(7264):674–78 [DOI] [PubMed] [Google Scholar]
- 138.Interthal H, Chen HJ, Champoux JJ. 2005. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J. Biol. Chem 280(43):36518–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. 2002. Conversion of phosphoglycolate to phosphate termini on 3’ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J. Biol. Chem 277(30):27162–68 [DOI] [PubMed] [Google Scholar]
- 140.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, et al. 2010. Ku is a 5’-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 464(7292):1214–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bétermier M, Bertrand P, Lopez BS. 2014. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10(1):e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin WY, Wilson JH, Lin Y. 2013. Repair of chromosomal double-strand breaks by precise ligation in human cells. DNA Repair (Amst.) 12(7):480–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Waters CA, Strande NT, Pryor JM, Strom CN, Mieczkowski P, et al. 2014. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat Commun 5:4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Feldmann E, Schmiemann V, Goedecke W, Reichenberger S, Pfeiffer P. 2000. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res 28(13):2585–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Labhart P. 1999. Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol. Cell. Biol 19(4):2585–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stinson BM, Moreno AT, Walter JC, Loparo JJ. 2020. A Mechanism to Minimize Errors during Non-homologous End Joining. Molecular Cell 77(5):1080–1091.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Budman J, Chu G. 2005. Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J 24(4):849–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ma Y, Lu H, Schwarz K, Lieber MR. 2005. Repair of Double-Strand DNA Breaks by the Human Nonhomologous DNA End Joining Pathway: The Iterative Processing Model. Cell Cycle 4(9):1193–1200 [DOI] [PubMed] [Google Scholar]
- 149.Akopiants K, Zhou R-Z, Mohapatra S, Valerie K, Lees-Miller SP, et al. 2009. Requirement for XLF/Cernunnos in alignment-based gap filling by DNA polymerases lambda and mu for nonhomologous end joining in human whole-cell extracts. Nucleic Acids Res 37(12):4055–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Celli GB, de Lange T. 2005. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol 7(7):712–18 [DOI] [PubMed] [Google Scholar]
- 151.Lee JW, Yannone SM, Chen DJ, Povirk LF. 2003. Requirement for XRCC4 and DNA ligase IV in alignment-based gap filling for nonhomologous DNA end joining in vitro. Cancer Res 63(1):22–24 [PubMed] [Google Scholar]
- 152.Lee JW, Blanco L, Zhou T, Garcia-Diaz M, Bebenek K, et al. 2004. Implication of DNA Polymerase λ in Alignment-based Gap Filling for Nonhomologous DNA End Joining in Human Nuclear Extracts. J. Biol. Chem 279(1):805–11 [DOI] [PubMed] [Google Scholar]
- 153.Perry JJP, Cotner-Gohara E, Ellenberger T, Tainer JA. 2010. Structural dynamics in DNA damage signaling and repair. Curr. Opin. Struct. Biol 20(3):283–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Reid DA, Conlin MP, Yin Y, Chang HH, Watanabe G, et al. 2017. Bridging of double-stranded breaks by the nonhomologous end-joining ligation complex is modulated by DNA end chemistry. Nucleic Acids Res 45(4):1872–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Strande NT, Waters CA, Ramsden DA. 2012. Resolution of complex ends by Nonhomologous end joining - better to be lucky than good? Genome Integr 3(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA. 2014. Nonhomologous end joining: A good solution for bad ends. DNA Repair 17:39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pfeiffer P, Thode S, Hancke J, Vielmetter W. 1994. Mechanisms of overlap formation in nonhomologous DNA end joining. Mol. Cell. Biol 14(2):888–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Roth DB, Wilson JH. 1986. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol. Cell. Biol 6(12):4295–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moon AF, Garcia-Diaz M, Batra VK, Beard WA, Bebenek K, et al. 2007. The X family portrait: structural insights into biological functions of X family polymerases. DNA Repair (Amst.) 6(12):1709–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet 45:247–71 [DOI] [PubMed] [Google Scholar]
- 161.Yoo S, Dynan WS. 1999. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res 27(24):4679–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lee K-J, Saha J, Sun J, Fattah KR, Wang S-C, et al. 2016. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res 44(4):1732–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brown JS, Lukashchuk N, Sczaniecka-Clift M, Britton S, le Sage C, et al. 2015. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep 11(5):704–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Feng L, Chen J. 2012. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol 19(2):201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ishida N, Nakagawa T, Iemura S-I, Yasui A, Shima H, et al. 2017. Ubiquitylation of Ku80 by RNF126 Promotes Completion of Nonhomologous End Joining-Mediated DNA Repair. Mol. Cell. Biol 37(4): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ismail IH, Gagné J-P, Genois M-M, Strickfaden H, McDonald D, et al. 2015. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol 17(11):1446–57 [DOI] [PubMed] [Google Scholar]
- 167.Postow L, Funabiki H. 2013. An SCF complex containing Fbxl12 mediates DNA damage-induced Ku80 ubiquitylation. Cell Cycle 12(4):587–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, Chait BT, Funabiki H. 2008. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol 182(3):467–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Meek K, Dang V, Lees-Miller SP. 2008. DNA-PK: the means to justify the ends? Adv. Immunol 99:33–58 [DOI] [PubMed] [Google Scholar]
- 170.Weterings E, Verkaik NS, Brüggenwirth HT, Hoeijmakers JHJ, van Gent DC. 2003. The role of DNA dependent protein kinase in synapsis of DNA ends. Nucleic Acids Res 31(24):7238–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Reddy YVR, Ding Q, Lees-Miller SP, Meek K, Ramsden DA. 2004. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J. Biol. Chem 279(38):39408–13 [DOI] [PubMed] [Google Scholar]
- 172.Chan DW, Lees-Miller SP. 1996. The DNA-dependent Protein Kinase Is Inactivated by Autophosphorylation of the Catalytic Subunit. J. Biol. Chem 271(15):8936–41 [DOI] [PubMed] [Google Scholar]
- 173.Douglas P, Cui X, Block WD, Yu Y, Gupta S, et al. 2007. The DNA-Dependent Protein Kinase Catalytic Subunit Is Phosphorylated In Vivo on Threonine 3950, a Highly Conserved Amino Acid in the Protein Kinase Domain. MCB 27(5):1581–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. 2013. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem. Cell Biol 91(1):31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, et al. 2010. Ku is a 5’-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 464(7292):1214–17 [DOI] [PMC free article] [PubMed] [Google Scholar]