

Abstract
Diet and nutrition are intricately related to cancer prevention, growth, and treatment response. Preclinical rodent models are a cornerstone to biomedical research and remain instrumental in our understanding of the relationship between cancer and diet and in the development of effective therapeutics. However, the success rate of translating promising findings from the bench to the bedside is suboptimal. Well-designed rodent models will be crucial to improving the impact basic science has on clinical treatment options. This review discusses essential experimental factors to consider when designing a preclinical cancer model with an emphasis on incorporating these models into studies interrogating diet, nutrition, and metabolism. The aims of this review are to (a) provide insight into relevant considerations when designing cancer models for obesity, nutrition, and metabolism research; (b) identify common pitfalls when selecting a rodent model; and (c) discuss strengths and limitations of available preclinical models.
Keywords: rodent models, cancer, diet, nutrition, obesity, metabolism
INTRODUCTION
Diet and nutrition modulate cancer incidence, tumor progression, and treatment response (210). Consequently, understanding the biological processes and mechanisms by which different diets, metabolic disturbances, or targeted nutrients either accelerate or retard tumor growth is of significant importance. Preclinical cancer models are the realistic starting point to manipulate cellular pathways, investigate tumor and whole-body responses to dietary perturbations, and test drug efficacy. Largely due to their relatively low cost, ease of use, and short life span, mice constitute 60%—and rats 12%—of all preclinical models and are therefore the focus of this review (33). Rodent models have been at the core of discovering many cancer therapies and will remain foundational in biomedical research. However, reports have exposed a sobering reality that 95% of preclinical drugs fail during clinical trials, emphasizing that there is ample room to improve how we use rodent models in research (171). Carefully designing the rodent model that maximizes the biological or pharmacological relevance to the question being interrogated while minimizing limitations should ultimately improve translational potential. Thus, this review discusses key considerations when designing a rodent model. We then also discuss the advantages and limitations of current preclinical cancer models in the context of obesity, nutrition, and metabolism research.
EXPERIMENTAL FACTORS TO CONSIDER WHEN CHOOSING A RODENT MODEL
Despite the availability of thousands of rodent models of cancer, there is no single model that fully replicates human cancer due to inherent genetic, anatomical, and physiological differences between the species. Instead, the strength of rodent models comes from being able to genetically, nutritionally, or pharmacologically manipulate specific biological pathways. Consequently, the rodent model should be carefully selected to recapitulate key, measurable features of human cancer that are relevant to the research question (187). Here we consider some critical variables in model design. These include variables related to the rodent—diet, age, sex, genetics, and immune system status—as well as variables related to modeling cancer such as tumor progression and mode of tumor induction (Figure 1).
Figure 1.
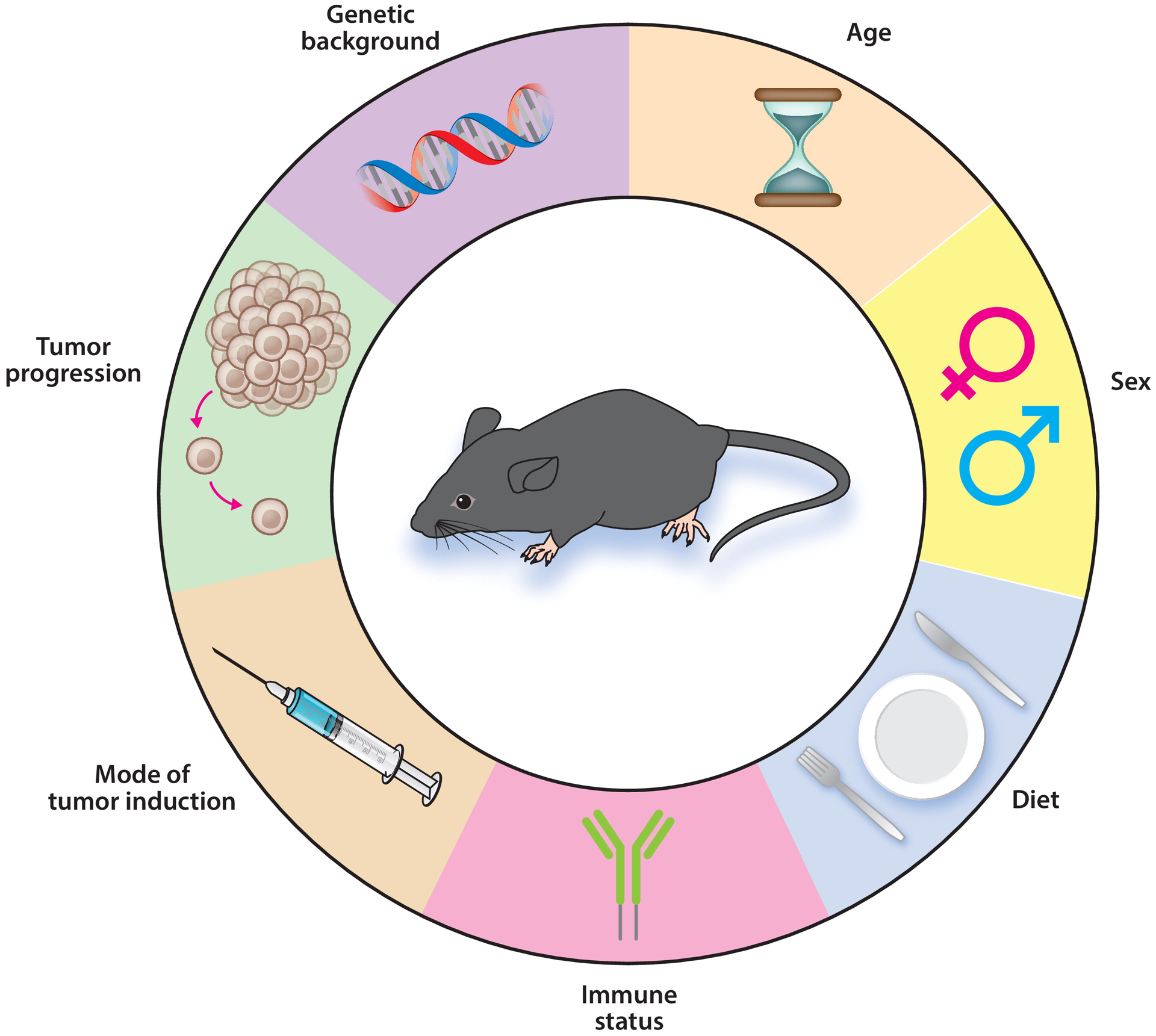
Experimental factors to consider when designing rodent cancer models. Numerous variables are considered in designing appropriate rodent cancer models for diet, nutrient, and metabolism studies. Adapted from figure originally created with BioRender.com.
Diet Provided to the Rodent
There are two major diet types available for rodent studies: grain based (chow) and purified. Grain based diets are formulated using agricultural and animal by-products such as ground corn, ground oats, and soybean meal with ingredient proportions and nutrient content that will vary between lots (154). Nontrivial concentrations of pesticides and heavy metals such as lead and arsenic are present in these diets (127). Conversely, purified diets use highly refined ingredients, where each ingredient contributes to a single macronutrient (e.g., lard for fat), with minimal variation between batches but at a significantly greater financial cost to the investigator. Fiber content is another important distinction, as grain based diets are composed of approximately 20–25% soluble and insoluble fibers (approximately 100–125 g/2,000 kcal), while purified diets generally only include 5% (approximately 25 g/2,000 kcal) cellulose (an insoluble fiber) (154). Gut atrophy is a significant consequence of the low fiber content in purified diets but can be remedied with the addition of inulin (a soluble fiber) to the diet (27). It is estimated that the typical US adult only consumes an average of 17 g soluble/insoluble fiber each day (190) despite the US Department of Agriculture (USDA) recommendation for adults to consume a daily diet that is composed of 28 g/2,000 kcal fiber (191). Mice on a grain based diet therefore consume approximately 6–7 times more fiber than the average adult. Conversely, the amount of fiber in a purified diet is more comparable to the ideal adult diet, but insoluble cellulose is the sole fiber source.
Sex of the Rodent
Male rodents have been historically overrepresented in biomedical research. One metanalysis reported that 80% of articles published across four surgical journals used only male animals and only 3% of articles used both male and female animals (215). Female rodents are commonly avoided because of a notion that the estrous cycle introduces a variable that is difficult to control for and will therefore increase variability (157). However, an analysis collating 293 studies found that male and female mice display remarkably similar intragroup variation of quantitative behavioral, morphological, and physiological traits, indicating that the estrous cycle does not increase variability (157). Moreover, the National Institutes of Health now require grant proposals to account for sex as a biological variable in animal research and, in the absence of strong scientific justification (e.g., prostate cancer research), both sexes must be included (139). Clinical oncology research has demonstrated that men clear doxorubicin more quickly than do women (44) and that women are at higher risk for 5-fluorouracil toxicity than are men (25), further highlighting the importance of detecting sexual dimorphism in preclinical studies. In obesity-related research, numerous groups have observed that female mice, more so than female rats, are less susceptible to diet-induced obesity (DIO) regimens relative to males unless they are ovariectomized (68, 87, 101, 143, 168).
Age of the Rodent
Cancer is an age-related disease where the median age for a cancer diagnosis is 66 years (89). It is well established that aging leads to many physiological changes, many of which could be oncogenic, including gradual accumulation of mutations, chronic low-grade inflammation [known as inflammaging (63)], cellular senescence, and epigenetic alterations (55). Despite acknowledgment that aging contributes to cancer development, young mice (8–12 weeks old) are predominantly used in preclinical cancer research (93). Importantly, studies have highlighted marked differences in immunotherapy response between young and aged mice. Bouchlaka et al. (17) demonstrated that aged mice (greater than 16 months old) were less able to tolerate an αCD40/interleukin-2 drug cocktail relative to young mice (2–3 months old), while another study has shown that middle-aged mice (greater than 10 months old) have improved anti-programmed cell death-1 antibody (αPD1) therapy response compared with young mice (6–10 weeks old) (103). As a reference point, 18–24-month-old mice (62) and 22–27-month-old rats (169) are in an equivalent stage of the aging process as 56–69-year-old humans.
Genetic Background of the Rodent
Selecting an appropriate genetic background is arguably one of the most important factors to consider when designing a preclinical cancer study. The central study question will dictate using either an inbred or outbred rodent model as well as whether the rodent should be immunodeficient or immunologically intact.
Common genetic backgrounds.
Common rodent strains used in laboratory research are differentially predisposed to experimental exposures and challenges. For example, C57BL/6 inbred mice are widely used for obesity research given their proclivity to gain weight and develop glucose intolerance on a high-fat diet (HFD), while BALB/c inbred mice are much more resistant to the same diet (131). Conversely, FVB/N and A/J inbred mice are more susceptible to azoxymethane—a carcinogen administered to induce colorectal tumors—than are C57BL/6 mice and therefore predominate in such studies (138). It can become a balancing act to choose the appropriate rodent model when studying obesity and cancer interactions, as C57BL/6 mice are often preferred for obesity studies but are poorly responsive to certain modes of tumor induction. Though less common in cancer studies, various rat cancer models exist using both Wistar and Sprague-Dawley outbred strains, as we have previously reviewed (69).
Collaborative cross and diversity outbred mice.
In contrast to outbred strains, inbred strains control for genetic diversity as an experimental variable. However, these genetically homogenous rodents ignore the considerable role that genetic diversity plays in complex human diseases, including cancer. In response to this limitation, an international effort has emerged to generate fully sequenced recombinant inbred collaborative cross (CC) mouse lines by systematically crossing 8 inbred founder lines (188). The culmination of this project has resulted in approximately 70 new isogenic lines that have significantly increased the available genetic diversity without surrendering control over the genetic information (179). CC mice are particularly useful when multiple strains are screened to detect differential responses to challenges such as dietary interventions, carcinogens, or chemotherapeutic drugs. These observations can be paired with quantitative trait locus analyses to map the phenotypic variation back to specific genomic loci. Wang et al. (202) identified Nfκb1 as a candidate gene target for spontaneous gastric tumor development by implementing this strategy.
Diversity outbred (DO) mice originated from randomly mating 180 partially inbred CC lines to produce the most genetically diverse set of heterogenous mice available for research (184). This immense genetic heterogeneity is especially relevant when studying diseases with complex genetic interactions. DO mice have already been successfully used in cancer and nutrition research to identify susceptibility genes for outcomes such as prostate metastasis (208) or atherosclerosis development (177).
Immunocompetent and immunodeficient mice.
When engrafting nonsyngeneic material—such as cancer cells or an intact tumor—into mice, immunodeficient mice are required to minimize transplant rejection. A variety of immunodeficient mice exist for this purpose, with the most commonly used strains being nude [no functional T cells (59)], nonobese diabetic/severe combined immunodeficient (NOD/SCID) [no functional B or T cells (15)], and NOD/SCID gamma mice [no functional B, T, or natural killer cells (92)]. The optimal immunodeficient mouse depends on the experiment, and essential considerations have been discussed by Shultz et al. (174). In the context of obesity research, immunodeficient mice are resistant to weight gain on a HFD unless housed at thermoneutrality (69). Additionally, a functional immune system is a critical component of the tumor microenvironment and is intricately involved in all aspects of tumor development as well as treatment response. Humanized mice—mice engrafted with human hematopoietic stem cells to produce a functional human immune system—may provide a solution to this limitation, but generation of these mice is expensive and technically difficult (200, 204).
General Recommendations for Factors Related to the Rodent
The experimental question must be critically evaluated to determine the most appropriate and feasible rodent model. Herein we discuss considerations related to diet, sex, age, and genetic background of the rodent when designing preclinical studies.
Studies using a purified HFD (40–60% kcal fat), a commonly used DIO regimen, should use a matched purified diet that is lower in fat content for the lean control group, where the reduced fat content is replaced with cornstarch but all other macro- and micronutrients are constant. Improper control diets are still commonly used. An analysis performed by Pellizzon & Ricci (154) found that of 69 studies using HFDs and published in 2016 in prominent journals, 41% used a grain based diet as the control group and another 41% did not report sufficient diet information. While it is tempting to use a less expensive grain based chow diet for the control group, any differences observed between the HFD and control rodent groups cannot be attributed to the fat content because other diet variables such as fiber or phytochemicals could also be contributors. Accounting for fiber amount and fiber type may be especially pertinent to preclinical breast and colon cancer studies where increased dietary fiber intake has been associated with reduced cancer risk in humans (8, 28). Given that diet manipulation studies intentionally alter specific micro- and/or macronutrients, purified diets, as opposed to grain based diets, should be used for these experiments (154). Moreover, pharmaceutical studies that use diet to administer a drug also require the use of purified diets, and the placebo group should be provided an identical diet that incorporates a vehicle control.
Given the extent to which sexual dimorphisms exist in both humans and rodents as well as the National Institutes of Health’s (139) commitment to addressing sex as a biological variable, experiments will be strengthened by incorporating both sexes unless there is strong scientific justification otherwise (e.g., an ovarian cancer study).
For experimental questions in which age-associated physiology may be an important variable, investigators should consider using aged rodents [18–24 months old for mice (62) and 22–27 months old for rats (169)]. Critically, not all cancers are age-related diseases, including cancers that are inheritable or develop early in life such as early-onset leukemia. If needed, a young control group should be at the age when rapid growth tapers off and the rodents are considered mature adults: 3 months in mice (62) and 4–5 months in rats (68).
Finally, careful research to select the rodent strain that is susceptible to the unique experimental conditions is essential. If an ideal strain is unavailable, other options such as CC or DO mice may be useful. Two considerations for DO studies include recognizing that all DO mice are unique, which means exact cohorts are impossible to reproduce, and that the sample size will need to be carefully considered and likely significantly increased, compared with using inbred strains, to achieve appropriate statistical power.
Features of the Tumor
Cancer is a collection of related diseases, and each person’s cancer has a unique combination of genetic alterations. Tumorigenesis is a multistage process which effects numerous tissues and can be modeled in several ways. Consideration of the cancer stage to be investigated and the means of tumor induction is important to incorporate into the study design.
Models to study different stages of cancer.
Cancer is a progressive disease that passes through discrete stages—initiation, growth and progression, and metastasis—and rodent models are rarely able to capture all stages of the disease. For example, spontaneous rodent models, including genetically engineered rodents, are appropriate for studying stages that include tumor prevention, initiation, progression, and response of an established tumor to an intervention. In contrast, tumor transplant models, often involving subcutaneously or orthotopically injecting human cell lines or patient-derived xenografts into immunodeficient mice, are best suited for investigating tumor growth or therapeutic response (37).
To study metastasis, primary tumors must grow slowly enough to promote the spread of cancer cells from the primary site into a secondary site without growing large enough to necessitate euthanasia. This is often not feasible, and experimental metastasis is therefore more often studied by directly injecting cancer cells either intravenously to promote lung colonization, intraportally to promote liver colonization, or intracardially to promote brain and/or bone colonization (165). While effective and relatively simple, this approach does not address therapeutically important steps in the metastatic cascade (e.g., local invasion and intravasation) (180). To model metastatic spread via lymph, cancer cells can be injected into a lymphatic vessel or lymph node (10, 189). Surgical resection of the primary tumor after micrometastasis formation retains the natural metastatic process for mammary tumors (66). While this is not feasible for all cancers, and there are inherent procedural/biological complications with surgical resection (150), primary tumor resection is an attractive option for studying the full metastatic cascade and may also closely reflect metastatic progression as commonly occurs in humans following surgical resection of primary tumors. All models of metastasis involving a surgical procedure may be confounded by the alteration of metastasis by the wound healing process induced following surgery unless experimental design accounts for such effects (150). Obesity is associated with increased tumor progression in several cancer types (210). Lung inflammation and expansion of metastasis-initiating cell populations in the tumor are both implicated in preclinical models of breast cancer metastasis and obesity (18, 158). However, there remains limited work in this area, highlighting an important knowledge gap.
Mode of tumor induction.
Murine tumor models have contributed importantly to our understanding of the biological mechanisms involved in tumor formation, and for identifying strategies for preventing cancer. The three most common means to induce a tumor in a rodent are either chemical carcinogen exposure, genetic manipulation, or transplantation of tumor cells. Each mode of tumor induction has its strengths and limitations, depending on the question being addressed.
Carcinogen-induced rodent models.
Carcinogen-induced models permit studies investigating the tumor progression process from preneoplastic lesion to various stages of cancer by administering a carcinogen known to induce tumor formation. The advantages of carcinogen-induced tumors include the presence of a tumor microenvironment, the gradual development of the tumor within its native tissue, and the frequent acquisition of multiple mutations to drive tumor growth (71). Limitations can include unpredictability with respect to tumor latency period, the number of tumors that are formed, and the location(s) of tumor formation. As discussed above (see the section titled Common Genetic Backgrounds), it is critical to select a rodent strain that is susceptible to the carcinogen.
Genetically engineered models.
Genetically engineered models (GEMs) have been used in cancer research since the 1980s to either activate oncogenes or delete tumor suppressor genes (76, 182). Similar to carcinogen-induced models, GEM rodents also have the potential to model all stages of cancer within the native tissue. The advent of the Cre recombinase/loxP recombination system in the late 1980s has greatly expanded the pool of available transgenic models by permitting either the conditional deletion of a tumor suppressor gene or the activation of an oncogene in a specific tissue and/or at a specific time (45, 186). For example, the adenomatous polyposis coli (Apc)Min/+ mouse is a spontaneous intestinal tumor model where a mutation in the Apc gene drives biallelic Apc inactivation and adenoma formation predominantly in the small intestine (133). However, most human intestinal cancers occur in the colon or rectum. ApcMin/+ mice expressing Cre recombinase using colon-specific promoters (84, 161) or inducible Cre recombinase driven by epithelial-specific promoters combined with tamoxifen administration to the colonic mucosa (162) can overcome this limitation.
Transplantation Models.
Tumor transplant models can vary dramatically in the species, amount and type of neoplastic material being transplanted as well as the site of transplantation. Transplant models often involve subcutaneously or orthotopically engrafting cancer cells into rodents (37, 75). As transplanted tumors are fully transformed upon introduction to the rodent, it is not possible to study carcinogenesis with a transplantation model (37). Instead, this model is frequently used to investigate tumor response to potential drugs, therapies, or dietary manipulations. Subcutaneous injection is technically much easier than orthotopic transplantation and creates a tumor that is visible to the eye and measurable with a caliper to track growth over time (37, 75). However, subcutaneously transplanted cells or tissues do not grow in their native environment. Consequently, it is not possible for this model to adequately recapitulate the relevant immune, stromal, and vascular components of the tumor microenvironment that are often intricately related to tumor growth and behavior (75). These limitations are thought to largely explain why promising outcomes observed in subcutaneously transplanted experiments poorly predict drug success in clinical trials (37). Orthotopic transplants provide the native tissue for tumor engraftment and can offer more consistent tumor multiplicity and growth rates. However, depending on the type of cancer, orthotopic transplantation may be technically challenging, and relatively quick tumor growth in these experiments may prohibit sufficient stromal remodeling (75). Therefore, while orthotopic transplantation models are generally accepted to be superior to subcutaneous injection models, the experimental question should be carefully evaluated to determine the most appropriate and feasible model.
The most commonly transplanted materials are either cancerous cells or intact tumor tissue. Human or rodent cancer cell lines are easy to genetically manipulate and are the most cost-effective option. However, because cell lines are inherently homogenous cell populations, transplantation of cell lines fails to recapitulate the cellular heterogeneity observed in tumors (75). Furthermore, cells grown in artificial cell culture conditions (e.g., plastic surface, 21% O2, and growth factor–rich media) acquire mutations over time, which could affect in vivo behavior and growth (104). Patient-derived xenografts (PDXs) are intact tumor tissues directly implanted into immunodeficient mice. The PDX model may hold promise for personalized medicine where transplantation of a patient’s tumor could facilitate a drug screen in recipient rodents to identify candidate therapies (176). Indeed, this strategy successfully predicted responders and nonresponders to cetuximab, an epidermal growth factor receptor monoclonal antibody, in patients with metastatic colorectal cancer (14). Despite the excitement surrounding PDXs, the human-derived tumor microenvironment, which initially accompanies the tumor into the mouse, appears to be largely replaced with murine-derived stroma and vasculature network (91). Selection pressures can also cause acquisition of different mutations as the tumor grows and is transplanted into new mice (12).
EXPERIMENTAL INTERVENTIONS TO STUDY NUTRITION AND METABOLISM IN CANCER RODENT MODELS
Diet and nutrition are intricately intertwined with cancer, and investigations are ongoing to understand how diet, individual nutrients, pharmacological agents, and surgical interventions influence cancer risk and cancer growth. Moreover, clear dietary recommendations are sorely needed but do not currently exist for patients undergoing cancer treatment (96). Therefore, exploiting pathways critical for cancer initiation or progression via nutrition modulation has great potential to either promote the prevention of cancer or support existing therapy paradigms.
Diet and Obesity
Obesity is defined as the accumulation of excess body fat and is frequently accompanied by whole-body metabolic impairments (42, 101). This metabolic dysfunction generally presents as low-grade chronic inflammation, elevated circulating growth hormones, abnormal blood lipid profile, and/or elevated blood glucose levels (101). Overweight and obesity [body mass index (BMI) of more than 25 kg/m2 and more than 30 kg/m2, respectively] are positively associated with risk for at least 12 different cancers (107, 210) and account for 14–20% of all cancer deaths (23). Understanding the mechanisms and biological processes by which excess fat promotes tumor growth is an active area of investigation. In preclinical rodent models, obesity is generally studied using a calorically dense diet or a monogenic hyperphagic rodent model.
Defining and measuring obesity in rodents.
There is currently no consensus among researchers on a quantitative measurement of obesity in rodents like there is for humans. To address this, we developed obesity criteria in mice that are reflective of the BMI cut points used in humans. In brief, we compared the body fat percentage of female C57BL/6 mice following 10 weeks on diets of varying fat content (up to 60% kcal fat) and energy densities to the body fat percentage of women within each BMI category, using data from the National Health and Nutrition Examination Survey III study (143). We defined lean, overweight, and obese nonovariectomized female mice as having percent body fat ranges of less than 26%, 26%–35%, and more than 35%, respectively (143). We observed greater and more rapid body fat accumulation in ovariectomized female mice fed the same diets and thus recommended less than 30%, 30–44%, and more than 45% body fat ranges for lean, overweight, and obese, respectively (143). Alternatively, Enriori et al. (53) defined mouse obesity as body weight more than 3 standard deviations above the mean body weight of the lean control group. In studies where HFD-fed rodents have a propensity to either gain or not gain weight, we have used percent body fat measured at an intermediate time point to determine a priori placement into the control or DIO group (69). Other specific criteria that can be used to ascertain whether a rodent is recapitulating key features of obesity include (a) hyperphagia, (b) hyperglycemia in a fasted state, (c) hyperinsulinemia in a fasted state, (d) dyslipidemia, and (e) elevated markers of inflammation, especially in the liver and adipose tissue (101, 120).
Monogenic models of obesity.
Signaling via leptin, a peptide hormone that regulates appetite by inducing satiety, is disrupted in the common monogenic rodent models of obesity—ob/ob (indicating obese) and db/db (indicating diabetic) (30, 31, 83). Defective leptin production (ob/ob) or leptin sensing (db/db) leads to severe hyperphagia and obesity in these mice. Both mouse models develop hyperinsulinemia, insulin resistance, hyperphagia, and dyslipidemia, while the C57BLKS/J background of db/db mice also promotes marked hyperglycemia and pancreatic islet cell dysfunction (101). Zucker fatty rats also carry a functionally ineffective leptin receptor due to a homozygous fa/fa mutation (155). This rat model develops hyperglycemia in addition to the other obesity-associated characteristics present in ob/ob mice (155).
Monogenic models of obesity reproducibly develop extreme obesity when fed a normal rodent diet. In order to disentangle whether interesting phenotypes are driven by the obesity or the impaired leptin signaling, food availability can be carefully controlled to prevent weight gain (111). However, a major drawback of these models is their limited translatability to the human population given that only 3–6% of humans are estimated to carry a homozygous genetic mutation in the leptin signaling pathway (56). Additionally, rather than using diet to induce obesity in a time-controlled manner, hyperphagia and metabolic disturbances begin at birth. Due to these model limitations, genetic hyperphagia models are not widely used in preclinical cancer research, although there is limited evidence that ob/ob mice have greater tumor mass than do wild-type controls in a subcutaneous pancreatic cancer model (159).
High-fat diets.
Relative to monogenic rodent models, dietary manipulation to induce obesity in polygenic rodents is considered to more accurately recapitulate the human condition (101, 120). HFDs are purified diets containing 45–60% kcal fat (Table 1). C57BL/6 mice provided ad libitum access to a 60% HFD for ≥15 weeks progressively develop obesity, insulin resistance, and dyslipidemia (101). A 60% HFD is more commonly used because the higher fat content accelerates excess lipid deposition and promotes a more pronounced obese phenotype. However, the USDA reports that the average American diet is ~37% fat (190), and therefore a 45% HFD may be more relevant if matching dietary composition between humans and mice is critical to the study question (120, 178). Interestingly, approximately 50% of outbred Sprague-Dawley rats are susceptible to DIO on a HFD while the other 50% are resistant (111); Wistar rats show a similar response (68). This differential response to diet permits DIO investigations where diet is not a variable between control and obese groups. Preclinical studies indicate that DIO results in accelerated tumor growth and progression across multiple obesity-associated cancer types, further reinforcing the validity of DIO as a useful preclinical model (19, 52, 67, 121, 151).
Table 1.
Comparison of human and rodent obesity-promoting and dietary energy–restriction diets
| Diet category | Diet name | Description of human diet | Description of rodent diet | References (cancer related if available) |
|---|---|---|---|---|
| Obesity promoting | High-fat diet | Western-style diet: calorically dense refined foods and beverages that are high in fat and salt but low in fiber | 45–60% caloric content from lard | 19, 52, 67, 121 |
| Dietary energy restriction | Calorie restriction | 10–25% daily calorie restriction without malnutrition; routine meal schedule is maintained | 30–40% daily calorie restriction without malnutrition; single meal presented daily | 13, 95 |
| 5:2 intermittent energy restriction | 5 days per week: Mediterranean-style diet; 2 nonconsecutive days per week: 75% reduction in caloric intake with low-carbohydrate foods | 5 days per week: 14% daily calorie restriction; 2 nonconsecutive days per week: 70% daily calorie restriction | ND | |
| Time-restricted feeding | Consistent ≤10 h daily timeframe when food can be consumed ad libitum | Consistent 8–12 h daily timeframe when food can be consumed ad libitum | 24, 81, 170 | |
| Fasting-mimicking diet | Minimal caloric intake (300–1,000 calories per day) while consuming foods low in carbohydrates and proteins and high in unsaturated fats; 5 consecutive days, twice per month coinciding with cancer treatment | 50–90% calorie restriction for 4 consecutive days 2 times per month in combination with cancer therapy administration; food consists of broth powders, glycerol, and essential fatty acids | 22, 43 | |
| Ketogenic diet | High-fat, low-carbohydrate diet (≥85% kcal fat); no calorie restriction required | Ad libitum access to a diet that is generally >85% calories from fat and <5% calories from carbohydrate with the remaining calories from protein, with slight variation between studies | 88, 90 |
Abbreviation: ND, no data.
Overcoming resistance to diet-induced obesity.
Many mouse strains display varying degrees of resistance to DIO, even when placed on a very high-fat, high-calorie diet. One recent approach to overcome this challenge has been to alter the housing temperature of these obesity-resistant mice so that they are maintained closer to their thermoneutral zone (69). While the exact temperature to maintain thermoneutrality has been debated, typical vivarium temperatures that are comfortable for humans (20–23°C) are known to cause chronic cold stress to rodents. Because of this, mice and rats housed at these temperatures can experience adaptive thermogenesis (181), expending energy to maintain their body temperature. The thermoneutral zone has been reported to be approximately 28–31°C based on metabolic phenotyping data (57). Warming the rodents can be accomplished by increasing the temperature in the vivarium or more directly by placing cages on warming mats, as we previously described (69). We have shown that within a few days of warming, mice on a HFD begin to gain weight and increase fat deposition, while those on a low-fat diet do not (69).
Dietary Energy–Restriction Approaches
Dietary energy-restriction (DER) encompasses diets where (a) caloric intake is limited, (b) food is restricted for specific periods of time, and/or (c) specific nutrients are restricted. DER approaches are associated with a prolonged health span and have been implemented in individuals who are normoweight as well as in individuals who are overweight or obese and are seeking to lose weight (38). Biological mechanisms by which DER approaches delay the onset of age-related diseases are expertly reviewed elsewhere (38, 128, 140, 149) and include improved insulin sensitivity, reduced growth factor signaling, cellular metabolic reprogramming, reduced adiposity, and decreased inflammatory signaling. While many DER approaches exist, this review focuses on calorie restriction (CR), 5:2 intermittent energy restriction (IER), time-restricted feeding (TRF), a fasting-mimicking diet (FMD), and a ketogenic diet (Table 1).
Calorie restriction.
CR chronically reduces macronutrient consumption but maintains adequate micronutrient intake and confers health benefits in species ranging from Caenorhabditis elegans to humans (61). Collectively, CR appears to confer health benefits that are conserved across species, making rodents a reasonable preclinical model for these experiments. In rodents, a metanalysis revealed that a 30% CR diet (i.e., rodents received 30% fewer calories than ad libitum controls) results in a 14–45% median life span increase in rats and a 4–27% increase in mice (185). Other rodent studies have highlighted the benefits of CR in preventing obesity and cancer (13, 95, 128), and rhesus monkeys on a 30% CR regimen have better overall health, experience delayed onset of age-related conditions, and generally live longer compared with controls (126). In addition to delaying tumor initiation, rodent studies have reproducibly demonstrated that 20–30% CR suppresses tumor growth across multiple cancer types and models of tumor induction (128, 130). Rodents on a CR dietary regimen generally receive a single meal each day, which is then rapidly consumed within 2–4 h (1). Therefore, this study design makes it inherently difficult to decipher whether the observed health benefits are a consequence of the 20–30% calorie deficit or the 20–22-h prolonged daily fast (see the section titled Time-Restricted Feeding).
In humans, CR improves many metabolic biomarkers, including blood pressure, lipid profiles, and insulin sensitivity in men and women who are not obese (60). CR is generally more moderate in humans (20–30% versus 30–40% in rodents) and can be difficult to achieve due to limited compliance (47, 194). Moreover, the risk of unwanted weight loss, specifically from adipose tissue and muscle, in patients with cancer undergoing treatment or at risk of cachexia is concerning. In response to these limitations, alternative dietary regimens (discussed below) have emerged with the intention of providing the same metabolic health benefits while increasing feasibility of diet adherence and limiting risks for patients with cancer.
5:2 Intermittent energy restriction.
A person adhering to the 5:2 IER diet consumes a Mediterranean-style diet 5 days a week and then decreases caloric intake by approximately 75% (500–800 kcal/day total intake) with a low-carbohydrate intake on 2 nonconsecutive days (79). Over the course of a week, this diet results in an average daily restriction of approximately 20–25%. Harvie et al. (79) directly compared 25% CR with 5:2 IER in overweight women and reported that women consuming the 5:2 IER diet for 4 months demonstrated greater reductions in insulin resistance and waist circumference. We have translated the human 5:2 IER diet to mice by restricting calories at 14% for 5 days and at 70% on 2 nonconsecutive days each week, resulting in an average 30% daily caloric reduction. In a syngeneic mammary tumor transplant model, weight loss in DIO mice imparted by either 5:2 IER or 30% CR results in smaller mammary tumor mass compared with mice always fed a low-fat diet (L.W. Bowers; unpublished data). Further studies should (a) seek to understand whether the mechanisms by which tumor growth is attenuated are conserved across 5:2 IER and 30% CR and (b) test the effectiveness of the 5:2 IER diet in different cancer types and stages.
Time-restricted feeding.
The rationale behind TRF in rodents emerged from two primary observations: (a) Many metabolic-related genes operate on a circadian rhythm, and (b) CR rodents are routinely subject to a prolonged fast after finishing their daily meal within 2–4 h (1, 149). A recent clinical trial enrolling participants diagnosed with metabolic syndrome demonstrated that consuming food within a daily 10-h timeframe for 12 weeks is sufficient to promote many health benefits, including weight loss, body fat reduction, and metabolic biomarker improvements (207). Epidemiological data support an association between nightly fasts (more than 13 h) and decreased risk of breast cancer recurrence in women (124). Similar to clinical trials, rodents on a TRF regimen generally have daily ad libitum access to food for 8–12 h (24, 81). Hatori et al. (81) observed that TRF mice provided a HFD for 9 h each day ate the same amount of food as unrestricted HFD controls but gained less body weight, had lower percent body fat, and displayed better glucose control. There is some evidence that TRF may improve several metabolic features known to support tumor growth (170). These promising findings warrant investigations in preclinical cancer models and suggest that these beneficial metabolic health outcomes may be independent of CR.
Fasting-mimicking diet.
Fasting diets have been broadly defined as consuming no or minimal food and caloric beverages for a period of time between 12 h and 3 weeks (116). People adhering to a FMD reduce caloric intake into a range of 300–1,000 calories per day by consuming foods and beverages that are low in carbohydrates and proteins but high in unsaturated fats (205). Suggested foods include soups, juices, energy bars, chips, and herbal teas. The equivalent rodent FMD restricts 50–90% of caloric intake for 4 consecutive days twice per month (20). The food presented is a combination of broth powders, glycerol, and essential fatty acids bound together with hydrogel (20).
A FMD was shown to be safe and feasible in a pilot study of patients receiving platinum-based chemotherapies (46) and was associated with an improved treatment response in patients undergoing chemo- or radiotherapy cancer treatment when adhered to for 2–5 consecutive days per month (39, 140). The biology underpinning FMD synergy with cytotoxic therapies is that a fast causes noncancerous proliferative cells to exit the cell cycle (140). Consequently, these cells transiently in cell cycle arrest should confer protection from chemotherapy-related damage, while continuously proliferating cancer cells will remain susceptible (140). Rodent studies have suggested that a FMD improves immunosurveillance by increasing intratumoral CD8+ T cells in mammary tumors (43) and promotes mammary tumor regression in combination with antineoplastic agents by suppressing growth hormone pathways (22).
Ketogenic diet.
The ketogenic diet is a high-fat, low-carbohydrate diet that has been successfully used to both reduce seizure occurrences in children with epilepsy and promote weight loss (4). Rodent ketogenic diets vary slightly across studies but are generally more than 85% fat and less than 5% carbohydrate (88, 90). Importantly, the ketogenic diet does not necessitate caloric restriction and therefore may be safer than other DER approaches for patients undergoing cancer treatment (54). A ketogenic diet imposes severe glucose restriction, forcing the liver to produce ketone bodies (β-hydroxybutyrate, acetoacetate, and acetate) as an alternative energy source for use by extrahepatic tissues. In the context of cancer, this fuel switch may impede tumor growth due to both decreased insulin-phosphoinositide 3-kinase (PI3K)-AKT growth factor signaling and increased reactive oxygen species production (4).
Many clinical trials are currently underway to determine the safety, efficacy, and tolerability of the ketogenic diet in cancer therapy (54). Meanwhile, rodent studies have offered conflicting evidence and suggest that both the implementation of the diet and cancer subtype may be critical. Hsieh et al. (90) convincingly demonstrated that a ketogenic diet slows tumor progression in a xenograft mouse model of squamous cell carcinoma but not in nonsquamous cell carcinoma and that these contradictory results are likely due to differential basal activity of the PI3K-AKT pathway. In a separate study, a ketogenic diet in combination with PI3K inhibitors slowed tumor growth across multiple tumor types, while a ketogenic diet alone accelerated acute myeloid leukemia progression (88). Collectively, preclinical studies aimed at better understanding the circumstances under which implementing the ketogenic diet are beneficial could be instrumental in informing future clinical trials.
General recommendations for incorporating diet energy restriction approaches in rodent studies.
While seemingly straightforward, implementing DER diets to obtain meaningful data requires significant forethought. For diet regimens that are not ad libitum, the first consideration is the time at which food should be presented. Given that rodents are nocturnal, it is generally preferable to present food at the beginning of the dark cycle (1). However, this may not be feasible depending on facility light cycle restrictions and the time of day that rodents will be euthanized for tissue collection. Whether rodents will be euthanized in a fed or fasted state is a critical consideration, especially for study outcomes focused on nutrient-dependent and growth factor signaling pathways. For studies that involve significant daily fasting periods, euthanizing rodents (and their respective controls) following both a feed and fast period should be considered to properly discern outcomes that are a consequence of the postprandial period (80). Finally, deciding at what stage of tumor progression to implement a DER approach heavily depends on the rodent cancer model being used and the primary study question.
To improve translatability, the fasting and feeding periods should be critically evaluated as well as the obese status of the rodents being subjected to DER approaches. For example, a 24-h fast is more drastic for rodents than for humans, and so outcomes may be more pronounced in rodents that are less able to withstand equivalent periods of food deprivation. It has also been observed that rodents on TRF regimens remain isocaloric to controls by compensating for the calorie deficit during feed cycles (7, 81) while humans do not compensate (207). Therefore, body weight loss in humans, but not rodents, is at least partially attributed to the reduced calorie consumption. CR and 5:2 IER rodent diets where food consumption is tightly controlled each day in both human and rodent studies may be easier to translate. Finally, most DER clinical trials are conducted in participants who are overweight or obese (79, 194, 207). Therefore, investigators should consider first placing rodents on a 40–60% HFD for over 15 weeks to induce obesity prior to beginning a DER diet.
Targeted Nutrient Interventions
In addition to interventions modulating large macronutrient groups such as DER, clinical and preclinical investigations have also targeted individual or limited sets of metabolites as potentially tractable cancer interventions. These approaches are particularly advantageous in dissecting the contribution of individual nutrients to a cancer outcome or when targeting metabolic liabilities of cancer. This section discusses important amino acids (AAs), folate, and choline metabolism and experimental approaches to pharmacologically, genetically, and nutritionally manipulate these pathways.
Amino acids in cancer.
Cancer cells, like all cells, balance dietary protein consumption with de novo biosynthesis of AAs to maintain sufficient AA pools. Nonessential amino acids (NEAAs) can easily be overlooked due to their categorization as nonessential. However, the ready interchange of these AAs with other metabolites positions them as interconnected nodes between otherwise disparate metabolic pathways (29) (Figure 2). The rapid growth, dysregulated tissue vasculature, and dysregulated metabolism of tumors (77) create metabolic vulnerabilities that restrict supply of both essential amino acids (EAAs) and NEAAs while sustaining high metabolic demand (29, 196). Strategies to deplete AAs are comprehensively reviewed elsewhere (97). This section describes basic biochemistry and key considerations for studying some AAs commonly targeted in preclinical studies.
Figure 2.
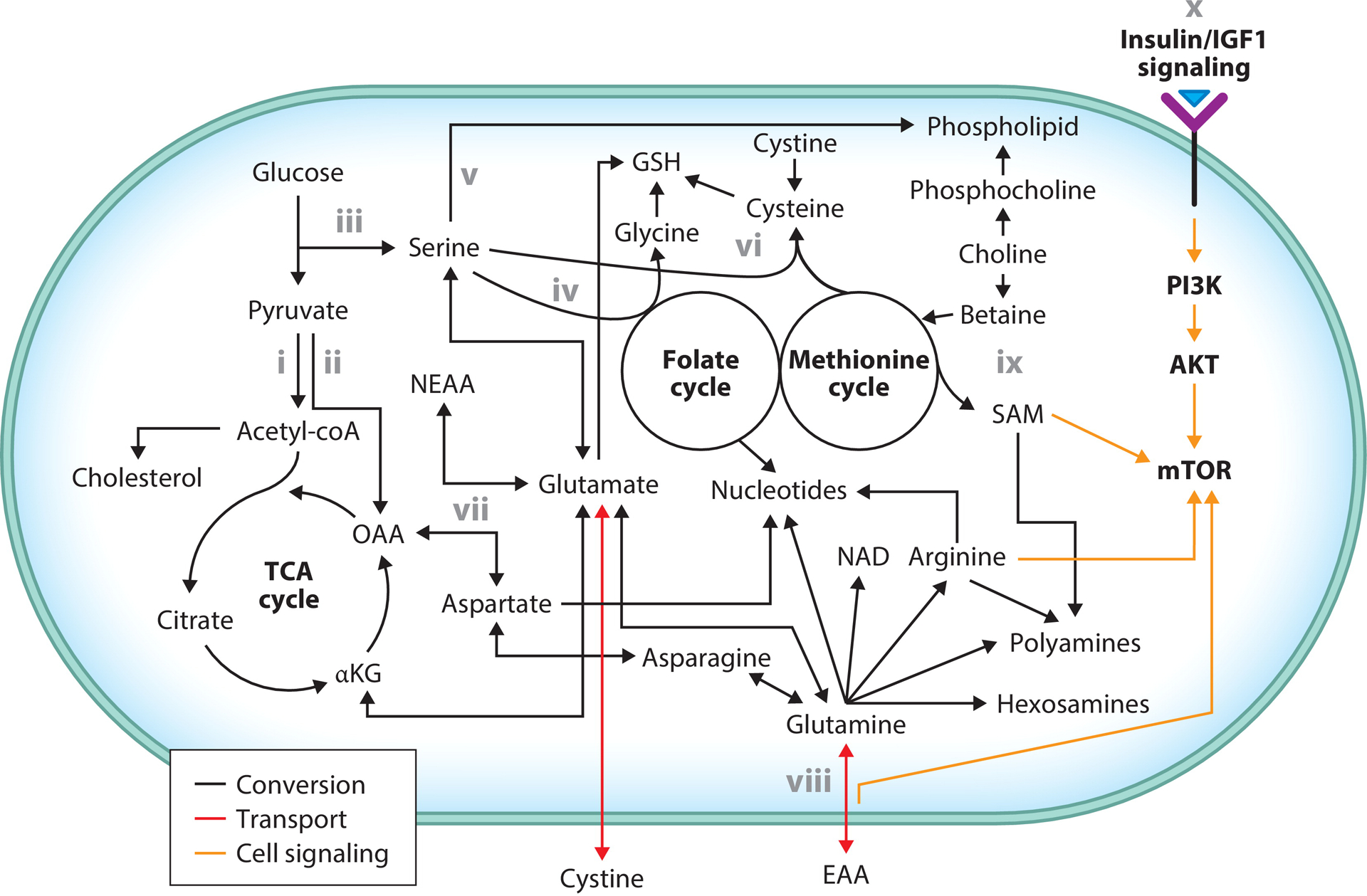
Interconnectivity in nutrient metabolism and mTOR signaling. Glucose-derived pyruvate either is converted to acetyl-coA to enter the TCA cycle or cholesterol biosynthesis or can be converted to OAA. Glucose-derived carbon combined with an amino group from glutamate can produce serine. Serine catabolism to glycine provides one-carbon units to the folate cycle, which ultimately contributes to nucleotide biosynthesis. Alternatively, serine is incorporated into phospholipids. Glycine, cysteine, and glutamate are required for the production of GSH. Cysteine is produced from intermediates of the methionine cycle and serine in the transsulfuration pathway. Alternatively, cysteine is produced from cystine, which is taken up via antiport with glutamate. Glutamate interconversion with αKG via transamination produces various NEAAs. OAA transamination produces aspartate, which is critical for nucleotide synthesis. Glutamine is also essential for nucleotide synthesis as well as NAD, arginine, polyamine, hexosamine, and asparagine synthesis. Glutamine antiports with EAAs to promote their uptake. SAM, produced in the methionine cycle, is critical for polyamine synthesis. Choline-derived betaine contributes methyl groups to the methionine cycle. Alternatively, choline is metabolized into phospholipids. Extracellular metabolic state is communicated to the cell via insulin/IGF1 signaling to activate mTOR. mTOR is also regulated by SAM, EAAs, and arginine. Major metabolic contributions of key metabolites and their interconversion with other relevant metabolites are depicted. Bolded text reflects cell signaling and metabolic cycles; regular text reflects metabolic intermediates. Abbreviations: αKG, α-ketoglutarate; EAA, essential amino acid; GSH, glutathione; IGF1, insulin-like growth factor 1; mTOR, mechanistic target of rapamycin; NAD, nicotinamide adenine dinucleotide; NEAA, nonessential amino acid; OAA, oxaloacetate; SAM, S-adenosyl methionine; TCA, tricarboxylic acid. Adapted from figure originally created with BioRender.com.
Glutamine/glutamate.
Glutamine is largely produced in muscle and liver and is the most abundant AA in plasma. It is considered nonessential under physiological states but can become conditionally essential during a critical illness (36). Glutamine contributes its terminal amino group to de novo purine, pyrimidine, nicotinamide adenine dinucleotide, hexosamine, and arginine biosynthesis (16). Glutamine regulates cellular metabolism via mechanistic target of rapamycin (mTOR) activation (152), in part by importing EAAs (141). Tumors can adapt to insufficient extracellular glutamine by converting asparagine to glutamine as well as de novo synthesis of glutamine (153).
Glutamine deamination produces another NEAA, glutamate. Glutamate directly supports glutathione metabolism by reacting with cysteine to form glutamyl-cysteine and via antiport with cystine. Such metabolism supports the high cysteine demand of KRAS (115) and Kelch-like ECH-associated protein (KEAP 1) (108, 166) mutant cells but may be deleterious in some cell types where oxidative stress is less severe (173). Indeed, extracellular NEAA supply is essential for survival of cells with high levels of oxidative stress, as competition for glutamate limits transamination and thus de novo biogenesis (108). To help replenish depleted tricarboxylic acid (TCA) cycle intermediates, glutamate can be deaminated to produce α-ketoglutarate (αKG). This deamination reaction also provides a nitrogen group for the production of other NEAAs (5). As these reactions are reversible, glucose-derived αKG is converted to glutamate/glutamine in conditions of low glutamate/glutamine (5).
While dietary restriction of glutamine reduces tumor growth in some models of cancer (142), there remains significant interest in pharmacological inhibition of glutamine metabolism. CB-839 is currently the most promising intervention that is part of several ongoing phase I and II trials (5). Thus, tumor genetics, redox homeostasis, and the availability of other nutrients are important factors that investigators interested in modulating glutamine metabolism should consider.
Cysteine.
Cysteine contains a reactive thiol group, which is critical for redox homeostasis, protein synthesis, and protein folding (32). Thus, cancer requires sufficient cysteine for survival. Most extracellular cystine is produced from hepatic cysteine. Cystine is readily reduced to cysteine in a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent fashion (32). System xC− transporter (xCT) imports cystine by antiporting glutamate (40) and is regulated by a range of stressors, including oxidative stress, AA starvation, and inflammation (112). xCT is also regulated by several common cancer mutations and can be pharmacologically inhibited (112). Pharmacological inhibition of xCT induces oxidative stress and ferroptosis and promotes hypoxia-inducible factor 1-alpha (HIF1α) activation via glutamate accumulation (217). Depletion of extracellular cystine levels via recombinant cystine-degrading enzyme and inhibition of glutaminase activity to limit glutamate availability are alternative direct and indirect methods, respectively, to target cystine import (9, 34, 64). Cysteine can be synthesized de novo via the transsulfuration pathway (TSP). However, the TSP is generally incapable of fully satisfying cysteine requirements for cancer cells due to a limited methionine pool (32). Hepatic regulation of circulating cysteine levels may limit the utility of cysteine dietary restriction if other sulfur-containing AAs (e.g., methionine) remain in excess. Indeed, cysteine supplementation reverses several effects of methionine restriction (201). Hence, investigations of cysteine metabolism in cancer should include appropriate consideration of other AAs, both as competitors for glutamate and as biosynthetically intertwined metabolites.
Arginine.
While dietary arginine is directly absorbed into the blood from the intestine, significant amounts of arginine are derived from dietary citrulline or enterocyte production of citrulline from glutamate, glutamine, or proline (3). These substrates are then converted to arginine in the kidney (3). Indeed, dietary citrulline increases circulating arginine levels to a greater extent than arginine supplementation (3).
Arginine is rapidly consumed by tumors for polyamine biosynthesis and nitric oxide production rather than urea (114). Arginine regulates cancer metabolism through direct incorporation into biomolecules and by activating mTOR via enhanced lysosomal EAA release (211) and antagonizing cellular arginine sensor for mTORC1 (CASTOR) (26). Nitrogen-containing urea cycle intermediates, required for de novo arginine biosynthesis, are often diverted toward other anabolic processes (99, 105). For example, carbamoyl phosphate is directed to pyrimidine metabolism by overexpression of carbamoyl phosphate synthetase I (CPS1) in some lung cancers (99, 100). Conversely, ornithine transcarbamylase (OTC) expression is frequently suppressed in cancer to reduce citrulline production and ultimately increase the availability of ornithine and carbamoyl phosphate (109). Similarly, conversion of aspartate and citrulline to argininosuccinate is often reduced in cancers via suppression of argininosuccinate synthase 1 (ASS1), which increases aspartate supply for pyrimidine synthesis (105). However, ASS1 overexpression can benefit cancers by supporting arginine supply for nitric oxide synthesis and limiting autophagy induction (99). OTC or ASS1 suppression results in many cancer cells being auxotrophic for arginine (99).
Arginine restriction has been studied via dietary restriction and enzymatic degradation, the latter of which has proven clinically tractable in recent years (119, 160). Depletion of circulating arginine levels via enzymatic degradation with arginase 1 (ARG1), arginine deiminase (ADI), or arginine decarboxylase (ADC) effectively reduces tumor growth (160). PEGylation is critical to extend serum half-life of ADI (ADI-PEG20) and ARG1 (PEG-rhARG1) and to reduce immunogenicity of ADI given its bacterial origin (160). However, PEGylation ablates ADC activity, which limits its utility (160). Suppression of de novo arginine biosynthesis is an important determinant of cell sensitivity to arginine depletion. As such, ADI-PEG20 is effective in ASS1-deficient tumors, while PEG-rhARG1 is effective in both ASS1- and OTC-deficient tumors (99).
Additionally, T cells rely on arginine to mediate effective antitumor immunity (74), which further complicates arginine-depletion models because arginine deprivation blunts the T cell response in part via myeloid-derived suppressor cell (MDSC) recruitment (58). Indeed, tumors harboring CPS1 overexpression, or OTC or ASS1 suppression, may be more responsive to immune checkpoint inhibitors. Neoantigen load driven by increased genomic transversions, which arises from an increased pyrimidine:purine ratio, in part underlies this phenomenon (109). Thus, consideration of immune function and tumoral urea cycle activation are critical to design effective arginine modulatory studies.
Serine.
Serine is a critical regulator of nucleotide biosynthesis, redox homeostasis, and one-carbon metabolism. The metabolic fate of serine is manifold with anabolic incorporation into proteins, phospholipids, and cystathionine biosynthesis (114). Through its contribution of one-carbon groups to the folate cycle, serine also supports nucleotide biosynthesis (51), mitochondrial translation (129, 132), cytosolic and mitochondrial redox homeostasis (212, 214), and the methionine cycle (51). Serine-derived redox equivalents are particularly important for survival when intratumoral conditions such as hypoxia impair mitochondrial function (212).
The serine synthesis pathway (SSP) is frequently upregulated in cancer, and suppression of key SSP enzymes has been shown to inhibit tumor growth and metastasis in multiple cancers (113). Despite increased flux through the SSP, several cancer models are highly sensitive to dietary serine restriction (73, 122). Thus, dietary serine and the SSP likely contribute to cancer metabolism in a nonredundant manner. Glycine can also interconvert with serine, but glycine consumption only plays an important role in very rapidly proliferating cancer cells (94). While most studies restrict both serine and glycine, in vitro studies have indicated that serine, rather than glycine, is the key mediator (106).
Genetic targeting of various SSP enzymes such as phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase 1 (PSAT1) blunts production of serine, αKG, and NADH, which consequently delays the growth and progression of various cancer types (113). Mitochondrial serine hydroxymethyltransferase (SHMT)2 mediates the conversion of serine to glycine and predominates over cytosolic SHMT1 serine catabolism in cancer (51). Serine catabolism contributes one-carbon units to the folate cycle (51), maintains effective mitochondrial translation (129, 132), and enables redox buffering following disruption of mitochondrial respiration (212, 214). However, disruption of SHMT2-mediated serine catabolism may be compensated for by the cytosolic pathway (49, 50). Therefore, while targeting of SHMT2 has proven effective in some rodent models (50, 206, 213, 214), cytosolic compensation via SHMT1 may be critical. Pharmacological inhibitors of PHGDH (113, 135, 148) and SHMT1/2 (50) have also demonstrated efficacy in rodent models. Thus, while serine is a metabolic lynch pin in many cancers, considering the potential compensatory mechanisms via diet, alternative metabolic routes, or intratumoral metabolic stressors is important to effectively target serine metabolism.
Methionine.
Similar to serine, methionine plays a crucial role in the maintenance of effective one-carbon metabolism, epigenetic methylation reactions, redox control, nucleotide biosynthesis, protein synthesis, and polyamine biosynthesis in both cancerous and normal cells (164). While methionine is classified as an EAA and therefore dietary methionine is required, methionine can also be regenerated from homocysteine plus a methyl group derived from either betaine or the folate cycle or via the methionine salvage pathway (51). Dietary restriction of methionine reduces tumor growth and promotes chemotherapy and radiotherapy response (65). Importantly, hepatic and blood biochemical profiles obtained from cancer-bearing mice undergoing methionine restriction mirror those observed in methionine-restricted humans (65). Methionine restriction also promotes antitumor immunosurveillance by reducing MDSCs, increasing macrophage M1 polarization, and potentiating immune checkpoint blockade (146). T cells require methionine to maintain both epigenetic and protein synthesis requirements for activation (175), yet they are still able to promote antitumor immunity under methionine-restrictive conditions (146). Methionine degradation via recombinant methioninase administration is an alternative to dietary methionine restriction with demonstrated efficacy in reducing prostate cancer (117), melanoma (98), and sarcoma (134) growth. In addition to targeting methionine abundance, pharmacological targeting of methionine metabolism is a promising area of research (164).
Folate.
Folate represents a diverse pool of metabolites with a common central structure that exist in various oxidation states and bind various one-carbon units (51). Cancer relies on effective maintenance of reduced folate pools for numerous metabolic pathways (51) (Figure 2). Folic acid (FA) is a synthetic folate commonly added to processed grain products in the United States, but numerous other folates are also found in the diet. Dietary FA supplementation protects against cancer initiation for several cancers, with prostate cancer risk being a notable exception (156). However, dietary FA may exacerbate growth or progression of established tumors. For example, FA depletion blunts epithelial to mesenchymal transition and cancer cell lung colonization (145), and mouse mammary tumor virus-polyomavirus middle T (MMTV-PyMT) mammary tumor growth is accelerated by FA supplementation (78). Rodent studies limiting or withdrawing FA from the diet can effectively reduce total folate pools in vivo. However, complete folate elimination can only be achieved by also ablating folate-producing intestinal microbiota (102). Pharmacological inhibitors of folate metabolism (i.e., antifolates) have existed for over 60 years (198) and effectively disrupt folate metabolism in rodents. However, antifolates exhibit significant toxicity, particularly to the hematopoietic system, which may become a limiting factor in preclinical studies (198).
Choline.
Choline either forms essential phospholipids following phosphorylation or contributes to the maintenance of S-adenosyl methionine (SAM) through betaine (216) (Figure 2). Inhibition of choline kinase to block phosphatidylcholine production reduces breast tumor growth and progression in vivo and synergizes with cytotoxic chemotherapy in vitro (125). Similarly, inhibition of choline uptake reduces pancreatic cancer (85) and glioma growth (203). Chronic dietary choline deficiency coupled with a HFD promotes nonalcoholic fatty liver disease and hepatocellular carcinoma (209). Conversely, choline deficiency alone may reduce experimental hepatic metastasis (137). Hence, particularly when metastasis is of interest, hepatic and extrahepatic effects of choline restriction should be considered. Given the contribution of betaine to SAM, studies restricting dietary choline may benefit from also restricting methionine to limit the contribution of methionine to the SAM pool.
Pharmaceutical Interventions
Pharmacological interventions have been used for many years to reprogram tumor metabolism. This section will briefly discuss the use of pharmaceutical interventions in rodent models of cancer while focusing on metabolically relevant compounds [PI3K/AKT/mTOR inhibitors, insulin-like growth factor 1 (IGF1)-signaling inhibitors, nonsteroidal anti-inflammatory drugs (NSAIDs), and statins/bisphosphonates]. Chemotherapeutic agents have targeted various aspects of cancer metabolism in humans and rodent models for decades and are expertly reviewed elsewhere (118).
Inhibitors of growth factor signaling.
PI3K/AKT/mTOR signaling—frequently activated in cancer—regulates cancer growth, survival, immune evasion, and metabolism. Much work has been conducted to determine how oncogenic mutations alter sensitivity to PI3K/mTOR inhibition. In addition to cancer cell targeting, PI3K/AKT/mTOR inhibitors in combination with immune checkpoint inhibitors are of growing interest given their immunomodulatory effects (144).
Insulin and IGF1 integrate host nutritional status with cellular response in part via activation of PI3K/AKT/mTOR signaling (Figure 2). While IGF1 receptor (IGF1R)-based therapies did not prove effective in phase III trials, early clinical and preclinical work was highly promising. Such therapies include monoclonal antibodies targeting the IGF1 ligand or IGF1R and small-molecule inhibitors of IGF1R/insulin receptor kinase activity (11). Pharmacologic inhibitors of mTOR, including rapamycin analogs (rapalogs) and active site inhibitors (TORKinibs), have also shown promise in clinical studies (147).
Metformin.
Numerous clinical trials have investigated the antitumoral effects of metformin in patients with and without diabetes (193). Metformin mediates antitumor activity via several distinct pathways, including 5′ adenosine monophosphate protein kinase (AMPK), mTOR, and TCA cycle modulation (193). Doses used in rodent models to elicit beneficial antitumor effects typically reflect high metformin levels in humans and thus may only model the effects of high metformin doses (48). Accordingly, metformin dosing requires particular attention when attempting to align rodent models and human studies.
Mevalonate and cholesterol metabolism.
Mevalonate metabolism is critical for the production of cholesterol, vitamin D, lipoproteins, ubiquinone, farnesyl pyrophosphate, and geranylgeranyl pyrophosphate. Disruption of mevalonate metabolism by statins (to target 3-hydroxy-3-methylglutaryl coenzyme A (HMG-coA) reductase) or nitrogen-containing bisphosphonates (to target farnesyl pyrophosphate synthase) reduces tumor growth and metastasis (70). Notably, bisphosphonates are rapidly sequestered and accumulate in bone (41). While this may be desirable for skeletal tumor models or studies examining bone metastasis, this pharmacokinetic profile may pose a significant limitation for most studies. To circumvent this issue, nanoparticle bisphosphonate formulations have been manufactured to promote extraskeletal availability and should be considered when administering a bisphosphonate in rodent cancer studies (41).
Nonsteroidal anti-inflammatory drugs.
Chronic inflammation is a hallmark of both cancer (77) and obesity (42) and promotes tumor growth and progression (42). NSAIDs are a broad family of drugs that inhibit cyclooxygenase (COX)1 and/or 2 activity and effectively reduce cancer incidence progression and metastasis via suppression of proinflammatory signaling (35). Two of the most commonly studied NSAIDs in both humans and rodents are aspirin, which inhibits COX1 and blocks prostacyclin production by COX2 at higher doses, and celecoxib, which selectively inhibits COX2 (195). Investigators examining the interactions between acute interventions and NSAIDs will likely need to consider the timing of each intervention. For example, promotion of immune resolution by NSAIDs prior to, but not following, either surgery or chemotherapy protects against tumor escape and metastasis (150). Chronic oral NSAID use is associated with bleeding events and intestinal damage, with concurrent COX1/2 inhibition imparting the greatest risk (199). Thus, using a COX-selective NSAID or administering low aspirin doses to selectively inhibit COX1 may reduce the rate of such complications.
General recommendations for pharmaceutical interventions.
Investigators wishing to apply human data to rodent designs or to translate rodent data to humans should be aware of several general considerations. Pharmacokinetics of any drug can differ significantly between humans and rodents, which limits the predictive power of preclinical work. For example, statin efficacy in rodents is poorly correlated with efficacy in humans, and this observation has been partially explained by differences in hepatic statin transport (192).
Other considerations include dosages used, timing of dosage, drug distribution, and microbiome interactions with drugs. Scaling doses between humans and rodents is complex and requires consideration of several factors. Differences in physiology (e.g., larger animals have greater muscle mass), pharmacokinetics (e.g., smaller animals distribute a drug more quickly), and pharmacodynamics (e.g., a drug response can differ widely among species) are important considerations (172). Allometric scaling, which involves dose extrapolation based on normalization of dose-to-body surface area, is particularly useful in preclinical studies as it requires limited a priori knowledge of drug metabolism and accounts for differences in animal size (136).
Circadian regulation of cancer development progression and therapy response is a growing area of interest. Given that DNA repair and the cell cycle display circadian oscillation, several clinical trials have synchronized chemotherapy with circadian rhythm and found significantly improved response rates and reduced treatment toxicity (183). While human and mouse circadian rhythms share some similarities, they are not the same. For example, human and mouse leukocytes in humanized mice both regulate chemokine receptor expression cyclically over a 24-h period in a reactive oxygen species–dependent fashion but are approximately 12 h out of phase with one another (219).
Drug distribution determines what tissues—namely healthy versus tumor in cancer—are exposed to the drug (72). For many drugs, the tumor response may be informed by both cancer cell and noncancerous cell effects, either of which can underlie therapy resistance (72). The regional pH or O2 variation (163) and vascular composition within the tumor microenvironment (110) further complicate this issue.
The intestinal microbiome, which is sensitive to dietary manipulation (123), is also significantly altered by many common drugs (197), particularly those that are orally administered. Interestingly, the protective effects of aspirin in models of colorectal cancer are antagonized by intestinal microbial metabolism of aspirin (218). Thus, effective dosages of NSAIDs in germ-free (i.e., microbially depleted) rodents may differ from animals with microbial communities.
Surgical Interventions for Obesity and Type 2 Diabetes
Bariatric surgery is the only clinical treatment currently available that achieves and sustains clinically meaningful weight loss in patients with morbid obesity. Large epidemiological studies have observed that people who lose weight via bariatric surgery have reduced rates of mortality and overall cancer risk compared with people who remain obese (2, 167). This inverse association becomes stronger when considering only cancers with obesity as an established risk factor (167). Despite the notable benefits of surgery, surgery is not a large-scale public health solution and carries inherent, nonnegligible risks. Thus, there is significant interest in understanding the underlying mechanisms by which bariatric surgery imparts these long-term health benefits to eventually target the essential pathways with pharmaceuticals.
Roux-en-Y gastric bypass and vertical sleeve gastrectomy are the two major forms of bariatric surgery. There is some evidence that Roux-en-Y gastric bypass may produce higher remission rates of type 2 diabetes (86), while vertical sleeve gastrectomy is now the more frequently performed procedure as it is associated with fewer surgical and postoperative complications (6). Vertical sleeve gastrectomy is also the preferred procedure in rodents because it takes less time to perform and is technically easier than Roux-en-Y gastric bypass (21). Bruinsma et al. (21) have published a comprehensive protocol for performing both Roux-en-Y and vertical sleeve gastrectomy surgeries in rodents. Importantly, they also discuss necessary controls to help interpret outcomes. While some control groups will depend on the study question, Bruinsma et al. highlight the importance of performing sham surgeries on all control groups as well as including a diet group that is isocalorically matched to the bariatric surgery rodents. One recent study demonstrated that the development of spontaneous pancreatic acinar cell carcinomas was prevented in nonobese mice receiving Roux-en-Y gastric bypass (82). Further studies using obese mice are therefore warranted to elucidate potential mechanisms that are either unique to bariatric surgery or common to both surgical and dietary weight loss interventions.
CONCLUDING REMARKS
Cancer remains a leading cause of death worldwide. Rodent models of cancer have been and remain a leading tool in disentangling complex cellular interactions, determining treatment responses, and elucidating metabolic regulatory networks within the tumor. While numerous therapies have been developed using preclinical rodent models, considerable work remains to be done in mitigating the public health impacts of cancer. Development of appropriate models to translate fundamental biochemistry and cell biology findings into tractable treatments is critical to triage such discoveries. Moreover, dietary modulation of cancer outcomes is understudied and requires very careful consideration in design and execution. This review highlights some of the key variables and relevant considerations for designing and conducting rodent studies to enhance the rigor and pace of translational research on nutrition, obesity, and cancer.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Acosta-Rodriguez VA, de Groot MHM, Rijo-Ferreira F, Green CB, Takahashi JS. 2017. Mice under caloric restriction self-impose a temporal restriction of food intake as revealed by an automated feeder system. Cell Metab 26:267–77.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams TD, Stroup AM, Gress RE, Adams KF, Calle EE, et al. 2009. Cancer incidence and mortality after gastric bypass surgery. Obesity 17:796–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal U, Didelija IC, Yuan Y, Wang X, Marini JC. 2017. Supplemental citrulline is more efficient than arginine in increasing systemic arginine availability in mice. J. Nutr 147:596–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen BG, Bhatia SK, Anderson CM, Eichenberger-Gilmore JM, Sibenaller ZA, et al. 2014. Ketogenic diets as an adjuvant cancer therapy: history and potential mechanism. Redox Biol 2:963–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altman BJ, Stine ZE, Dang CV. 2016. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16:619–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aminian A 2018. Sleeve gastrectomy: metabolic surgical procedure of choice? Trends Endocrinol. Metab 29:531–34 [DOI] [PubMed] [Google Scholar]
- 7.Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, et al. 2003. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. PNAS 100:6216–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aune D, Chan DS, Lau R, Vieira R, Greenwood DC, et al. 2011. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ 343:d6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, et al. 2020. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368:85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banan B, Beckstead JA, Dunavant LE, Sohn Y, Adcock JM, et al. 2020. Development of a novel murine model of lymphatic metastasis. Clin. Exp. Metastasis 37:247–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckwith H, Yee D. 2015. Minireview: Were the IGF signaling inhibitors all bad? Mol. Endocrinol 29:1549–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, et al. 2017. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet 49:1567–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berrigan D, Perkins SN, Haines DC, Hursting SD. 2002. Adult-onset calorie restriction and fasting delay spontaneous tumorigenesis in p53-deficient mice. Carcinogenesis 23:817–22 [DOI] [PubMed] [Google Scholar]
- 14.Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, et al. 2011. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1:508–23 [DOI] [PubMed] [Google Scholar]
- 15.Bonnet D, Dick JE. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 3:730–37 [DOI] [PubMed] [Google Scholar]
- 16.Bott AJ, Maimouni S, Zong W-X. 2019. The pleiotropic effects of glutamine metabolism in cancer. Cancers 11:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouchlaka MN, Sckisel GD, Chen M, Mirsoian A, Zamora AE, et al. 2013. Aging predisposes to acute inflammatory induced pathology after tumor immunotherapy. J. Exp. Med 210:2223–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bousquenaud M, Fico F, Solinas G, Rüegg C, Santamaria-Martínez A. 2018. Obesity promotes the expansion of metastasis-initiating cells in breast cancer. Breast Cancer Res 20:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowers LW, Rossi EL, McDonell SB, Doerstling SS, Khatib SA, et al. 2018. Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol. Cancer Res 16:869–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, et al. 2015. A periodic diet that mimics fasting promotes multi-system regeneration, enhanced cognitive performance, and healthspan. Cell Metab 22:86–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruinsma BG, Uygun K, Yarmush ML, Saeidi N. 2015. Surgical models of Roux-en-Y gastric bypass surgery and sleeve gastrectomy in rats and mice. Nat. Protoc 10:495–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caffa I, Spagnolo V, Vernieri C, Valdemarin F, Becherini P, et al. 2020. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583:620–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med 348:1625–38 [DOI] [PubMed] [Google Scholar]
- 24.Chaix A, Zarrinpar A, Miu P, Panda S. 2014. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab 20:991–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chansky K, Benedetti J, Macdonald JS. 2005. Differences in toxicity between men and women treated with 5-fluorouracil therapy for colorectal carcinoma. Cancer 103:1165–71 [DOI] [PubMed] [Google Scholar]
- 26.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, et al. 2016. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165:153–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chassaing B, Miles-Brown J, Pellizzon M, Ulman E, Ricci M, et al. 2015. Lack of soluble fiber drives diet-induced adiposity in mice. Am. J. Physiol. Gastrointest. Liver Physiol 309:G528–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S, Chen Y, Ma S, Zheng R, Zhao P, et al. 2016. Dietary fibre intake and risk of breast cancer: a systematic review and meta-analysis of epidemiological studies. Oncotarget 7:80980–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi B-H, Coloff JL. 2019. The diverse functions of non-essential amino acids in cancer. Cancers 11:675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coleman DL. 1973. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 9:294–98 [DOI] [PubMed] [Google Scholar]
- 31.Coleman DL, Hummel KP. 1969. Effects of parabiosis of normal with genetically diabetic mice. Am. J. Physiol 217:1298–304 [DOI] [PubMed] [Google Scholar]
- 32.Combs JA, DeNicola GM. 2019. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers 11:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eur. Comm. 2019. 2019 report on the statistics on the use of animals for scientific purposes in the Member States of the European Union in 2015–2017. Rep., Eur. Comm., Brussels [Google Scholar]
- 34.Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, et al. 2017. Systemic depletion of l-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med 23:120–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crusz SM, Balkwill FR. 2015. Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol 12:584–96 [DOI] [PubMed] [Google Scholar]
- 36.Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. 2018. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients 10:1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day CP, Merlino G, Van Dyke T. 2015. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163:39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Cabo R, Mattson MP. 2019. Effects of intermittent fasting on health, aging, and disease. N. Engl. J. Med 381:2541–51 [DOI] [PubMed] [Google Scholar]
- 39.de Groot S, Lugtenberg RT, Cohen D, Welters MJP, Ehsan I, et al. 2020. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun 11:3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de la Ballina LR, Cano-Crespo S, González-Muñoz E, Bial S, Estrach S, et al. 2016. Amino acid transport associated to cluster of differentiation 98 heavy chain (CD98hc) is at the cross-road of oxidative stress and amino acid availability. J. Biol. Chem 291:9700–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Rosa G, Misso G, Salzano G, Caraglia M. 2013. Bisphosphonates and cancer: what opportunities from nanotechnology? J. Drug Deliv 2013:637976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. 2016. Obesity, inflammation, and cancer. Annu. Rev. Pathol. Mech. Dis 11:421–49 [DOI] [PubMed] [Google Scholar]
- 43.Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, et al. 2016. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell 30:136–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobbs NA, Twelves CJ, Gillies H, James CA, Harper PG, Rubens RD. 1995. Gender affects doxorubicin pharmacokinetics in patients with normal liver biochemistry. Cancer Chemother. Pharmacol 36:473–76 [DOI] [PubMed] [Google Scholar]
- 45.Doetschman T, Gregg RG, Maeda N, Hooper ML, Melton DW, et al. 1987. Targetted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330:576–78 [DOI] [PubMed] [Google Scholar]
- 46.Dorff TB, Groshen S, Garcia A, Shah M, Tsao-Wei D, et al. 2016. Safety and feasibility of fasting in combination with platinum-based chemotherapy. BMC Cancer 16:360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorling JL, Das SK, Racette SB, Apolzan JW, Zhang D, et al. 2020. Changes in body weight, adherence, and appetite during 2 years of calorie restriction: the CALERIE 2 randomized clinical trial. Eur. J. Clin. Nutr 74:1210–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dowling RJO, Lam S, Bassi C, Mouaaz S, Aman A, et al. 2016. Metformin pharmacokinetics in mouse tumors: implications for human therapy. Cell Metab 23:567–68 [DOI] [PubMed] [Google Scholar]
- 49.Ducker GS, Chen L, Morscher RJ, Ghergurovich JM, Esposito M, et al. 2016. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab 23:1140–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ducker GS, Ghergurovich JM, Mainolfi N, Suri V, Jeong SK, et al. 2017. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. PNAS 114:11404–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ducker GS, Rabinowitz JD. 2017. One-carbon metabolism in health and disease. Cell Metab 25:27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunlap SM, Chiao LJ, Nogueira L, Usary J, Perou CM, et al. 2012. Dietary energy balance modulates epithelial-to-mesenchymal transition and tumor progression in murine claudin-low and basal-like mammary tumor models. Cancer Prev. Res 5:930–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, et al. 2007. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–94 [DOI] [PubMed] [Google Scholar]
- 54.Erickson N, Boscheri A, Linke B, Huebner J. 2017. Systematic review: isocaloric ketogenic dietary regimes for cancer patients. Med. Oncol 34:72. [DOI] [PubMed] [Google Scholar]
- 55.Fane M, Weeraratna AT. 2020. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20:89–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farooqi IS, O’Rahilly S. 2008. Mutations in ligands and receptors of the leptin–melanocortin pathway that lead to obesity. Nat. Clin. Pract. Endocrinol. Metab 4:569–77 [DOI] [PubMed] [Google Scholar]
- 57.Fischer AW, Cannon B, Nedergaard J. 2019. The answer to the question “What is the best housing temperature to translate mouse experiments to humans?” is: thermoneutrality. Mol. Metab 26:1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, et al. 2015. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res 75:275–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fogh J, Fogh JM, Orfeo T. 1977. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst 59:221–26 [DOI] [PubMed] [Google Scholar]
- 60.Fontana L, Klein S. 2007. Aging, adiposity, and calorie restriction. JAMA 297:986–94 [DOI] [PubMed] [Google Scholar]
- 61.Fontana L, Partridge L. 2015. Promoting health and longevity through diet: from model organisms to humans. Cell 161:106–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fox JG. 2007. The Mouse in Biomedical Research Amsterdam/Boston: Elsevier [Google Scholar]
- 63.Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. 2018. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 14:576–90 [DOI] [PubMed] [Google Scholar]
- 64.Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, et al. 2019. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 79:3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, et al. 2019. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572:397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gast CE, Shaw AK, Wong MH, Coussens LM. 2017. Surgical procedures and methodology for a preclinical murine model of de novo mammary cancer metastasis. J. Vis. Exp 125:e54852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gibson JT, Orlandella RM, Turbitt WJ, Behring M, Manne U, et al. 2020. Obesity-associated myeloid-derived suppressor cells promote apoptosis of tumor-infiltrating CD8 T cells and immunotherapy resistance in breast cancer. Front. Immunol 11:590794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Giles ED, Jackman MR, MacLean PS. 2016. Modeling diet-induced obesity with obesity-prone rats: implications for studies in females. Front. Nutr 3:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Giles ED, Wellberg EA. 2020. Preclinical models to study obesity and breast cancer in females: considerations, caveats, and tools. J. Mammary Gland Biol. Neoplasia In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Göbel A, Rauner M, Hofbauer LC, Rachner TD. 2020. Cholesterol and beyond—The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta 1873:188351. [DOI] [PubMed] [Google Scholar]
- 71.Golovko D, Kedrin D, Yilmaz OH, Roper J. 2015. Colorectal cancer models for novel drug discovery. Expert Opin. Drug Discov 10:1217–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gottesman MM, Lavi O, Hall MD, Gillet J-P. 2016. Toward a better understanding of the complexity of cancer drug resistance. Annu. Rev. Pharmacol. Toxicol 56:85–102 [DOI] [PubMed] [Google Scholar]
- 73.Gravel SP, Hulea L, Toban N, Birman E, Blouin MJ, et al. 2014. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res 74:7521–33 [DOI] [PubMed] [Google Scholar]
- 74.Grzywa TM, Sosnowska A, Matryba P, Rydzynska Z, Jasinski M, et al. 2020. Myeloid cell-derived arginase in cancer immune response. Front. Immunol 11:938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guerin MV, Finisguerra V, Van den Eynde BJ, Bercovici N, Trautmann A. 2020. Preclinical murine tumor models: a structural and functional perspective. eLife 9:e50740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hanahan D 1985. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315:115–22 [DOI] [PubMed] [Google Scholar]
- 77.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–74 [DOI] [PubMed] [Google Scholar]
- 78.Hansen MF, Jensen SO, Fuchtbauer EM, Martensen PM. 2017. High folic acid diet enhances tumour growth in PyMT-induced breast cancer. Br. J. Cancer 116:752–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harvie MN, Pegington M, Mattson MP, Frystyk J, Dillon B, et al. 2011. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int. J. Obes 35:714–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hatchwell L, Harney DJ, Cielesh M, Young K, Koay YC, et al. 2020. Multi-omics analysis of the intermittent fasting response in mice identifies an unexpected role for HNF4α. Cell Rep 30:3566–82.e4 [DOI] [PubMed] [Google Scholar]
- 81.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, et al. 2012. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab 15:848–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He R, Yin Y, Yin W, Li Y, Zhao J, Zhang W. 2018. Prevention of pancreatic acinar cell carcinoma by Roux-en-Y gastric bypass surgery. Nat. Commun 9:4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hervey GR. 1959. The effects of lesions in the hypothalamus in parabiotic rats. J. Physiol 145:336–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, et al. 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res 67:9721–30 [DOI] [PubMed] [Google Scholar]
- 85.Hirai K, Watanabe S, Nishijima N, Shibata K, Hase A, et al. 2020. Molecular and functional analysis of choline transporters and antitumor effects of choline transporter-like protein 1 inhibitors in human pancreatic cancer cells. Int. J. Mol. Sci 21:5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hofso D, Fatima F, Borgeraas H, Birkeland KI, Gulseth HL, et al. 2019. Gastric bypass versus sleeve gastrectomy in patients with type 2 diabetes (Oseberg): a single-centre, triple-blind, randomised controlled trial. Lancet Diabetes Endocrinol 7:912–24 [DOI] [PubMed] [Google Scholar]
- 87.Hong J, Stubbins RE, Smith RR, Harvey AE, Nunez NP. 2009. Differential susceptibility to obesity between male, female and ovariectomized female mice. Nutr. J 8:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hopkins BD, Pauli C, Du X, Wang DG, Li X, et al. 2018. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560:499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Howlader N, Noone AM, Krapcho M, Miller D, Brest A, et al. 2019. SEER Cancer Statistics Review 1975–2017 Bethesda, MD: Natl. Cancer Inst. [Google Scholar]
- 90.Hsieh MH, Choe JH, Gadhvi J, Kim YJ, Arguez MA, et al. 2019. p63 and SOX2 dictate glucose reliance and metabolic vulnerabilities in squamous cell carcinomas. Cell Rep 28:1860–78.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hylander BL, Punt N, Tang H, Hillman J, Vaughan M, et al. 2013. Origin of the vasculature supporting growth of primary patient tumor xenografts. J. Transl. Med 11:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, et al. 2005. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chainnull mice. Blood 106:1565–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jackson SJ, Andrews N, Ball D, Bellantuono I, Gray J, et al. 2017. Does age matter? The impact of rodent age on study outcomes. Lab. Anim 51:160–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, et al. 2012. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336:1040–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang W, Zhu Z, Thompson HJ. 2008. Dietary energy restriction modulates the activity of AMP-activated protein kinase, Akt, and mammalian target of rapamycin in mammary carcinomas, mammary gland, and liver. Cancer Res 68:5492–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kanarek N, Petrova B, Sabatini DM. 2020. Dietary modifications for enhanced cancer therapy. Nature 579:507–17 [DOI] [PubMed] [Google Scholar]
- 97.Kang J-S. 2020. Dietary restriction of amino acids for cancer therapy. Nutr. Metab 17:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kawaguchi K, Igarashi K, Li S, Han Q, Tan Y, et al. 2018. Recombinant methioninase (rMETase) is an effective therapeutic for BRAF-V600E-negative as well as -positive melanoma in patient-derived orthotopic xenograft (PDOX) mouse models. Oncotarget 9:915–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Keshet R, Szlosarek P, Carracedo A, Erez A. 2018. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 18:634–45 [DOI] [PubMed] [Google Scholar]
- 100.Kim J, Hu Z, Cai L, Li K, Choi E, et al. 2017. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546:168–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, et al. 2018. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol 14:140–62 [DOI] [PubMed] [Google Scholar]
- 102.Kok DE, Steegenga WT, Smid EJ, Zoetendal EG, Ulrich CM, Kampman E. 2020. Bacterial folate biosynthesis and colorectal cancer risk: more than just a gut feeling. Crit. Rev. Food Sci. Nutr 60:244–56 [DOI] [PubMed] [Google Scholar]
- 103.Kugel CH 3rd, Douglass SM, Webster MR, Kaur A, Liu Q, et al. 2018. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin. Cancer Res 24:5347–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kuijk E, Jager M, van der Roest B, Locati MD, Van Hoeck A, et al. 2020. The mutational impact of culturing human pluripotent and adult stem cells. Nat. Commun 11:2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kurmi K, Haigis MC. 2020. Nitrogen metabolism in cancer and immunity. Trends Cell Biol 30:408–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. 2014. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep 7:1248–58 [DOI] [PubMed] [Google Scholar]
- 107.Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, et al. 2016. Body fatness and cancer—viewpoint of the IARC working group. N. Engl. J. Med 375:794–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.LeBoeuf SE, Wu WL, Karakousi TR, Karadal B, Jackson SR, et al. 2020. Activation of oxidative stress response in cancer generates a druggable dependency on exogenous non-essential amino acids. Cell Metab 31:339–50.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, et al. 2018. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174:1559–70.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee SS-Y, Bindokas VP, Kron SJ. 2019. Multiplex three-dimensional mapping of macromolecular drug distribution in the tumor microenvironment. Mol. Cancer Ther 18:213–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Levin N, Nelson C, Gurney A, Vandlen R, de Sauvage F. 1996. Decreased food intake does not completely account for adiposity reduction after ob protein infusion. PNAS 93:1726–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, et al. 2013. The cystine/glutamate antiporter system xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 18:522–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li AM, Ye J. 2020. The PHGDH enigma: Do cancer cells only need serine or also a redox modulator? Cancer Lett 476:97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lieu EL, Nguyen T, Rhyne S, Kim J. 2020. Amino acids in cancer. Exp. Mol. Med 52:15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim JKM, Delaidelli A, Minaker SW, Zhang H-F, Colovic M, et al. 2019. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. PNAS 116:9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Longo VD, Mattson MP. 2014. Fasting: molecular mechanisms and clinical applications. Cell Metab 19:181–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu W-C, Saha A, Yan W, Garrison K, Lamb C, et al. 2020. Enzyme-mediated depletion of serum l-Met abrogates prostate cancer growth via multiple mechanisms without evidence of systemic toxicity. PNAS 117:13000–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Luengo A, Gui DY, Vander Heiden MG. 2017. Targeting metabolism for cancer therapy. Cell Chem. Biol 24:1161–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lukey MJ, Katt WP, Cerione RA. 2017. Targeting amino acid metabolism for cancer therapy. Drug Discov. Today 22:796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lutz TA. 2018. Considering our methods: methodological issues with rodent models of appetite and obesity research. Physiol. Behav 192:182–87 [DOI] [PubMed] [Google Scholar]
- 121.MacLean PS, Giles ED, Johnson GC, McDaniel SM, Fleming-Elder BK, et al. 2010. A surprising link between the energetics of ovariectomy-induced weight gain and mammary tumor progression in obese rats. Obesity 18:696–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, et al. 2017. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544:372–76 [DOI] [PubMed] [Google Scholar]
- 123.Mai V, Colbert LH, Perkins SN, Schatzkin A, Hursting SD. 2007. Intestinal microbiota: a potential diet-responsive prevention target in ApcMin mice. Mol. Carcinog 46:42–48 [DOI] [PubMed] [Google Scholar]
- 124.Marinac CR, Nelson SH, Breen CI, Hartman SJ, Natarajan L, et al. 2016. Prolonged nightly fasting and breast cancer prognosis. JAMA Oncol 2:1049–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mariotto E, Viola G, Ronca R, Persano L, Aveic S, et al. 2018. Choline kinase alpha inhibition by EB-3D triggers cellular senescence, reduces tumor growth and metastatic dissemination in breast cancer. Cancers 10:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, et al. 2017. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun 8:14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mesnage R, Defarge N, Rocque L-M, Spiroux de Vendômois J, Séralini G-E. 2015. Laboratory rodent diets contain toxic levels of environmental contaminants: implications for regulatory tests. PLOS ONE 10:e0128429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meynet O, Ricci JE. 2014. Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends Mol. Med 20:419–27 [DOI] [PubMed] [Google Scholar]
- 129.Minton DR, Nam M, McLaughlin DJ, Shin J, Bayraktar EC, et al. 2018. Serine catabolism by SHMT2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl-tRNAs. Mol. Cell 69:610–21.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mittelman SD. 2020. The role of diet in cancer prevention and chemotherapy efficacy. Annu. Rev. Nutr 40:273–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, et al. 2013. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56:1129–39 [DOI] [PubMed] [Google Scholar]
- 132.Morscher RJ, Ducker GS, Li SH, Mayer JA, Gitai Z, et al. 2018. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554:128–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Moser AR, Pitot HC, Dove WF. 1990. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247:322–24 [DOI] [PubMed] [Google Scholar]
- 134.Murakami T, Li S, Han Q, Tan Y, Kiyuna T, et al. 2017. Recombinant methioninase effectively targets a Ewing’s sarcoma in a patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 8:35630–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Muthusamy T, Cordes T, Handzlik MK, You L, Lim EW, et al. 2020. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature 586:790–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nair AB, Jacob S. 2016. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm 7:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nakamura M, Suetsugu A, Hasegawa K, Matsumoto T, Aoki H, et al. 2017. Choline-deficient-diet-induced fatty liver is a metastasis-resistant microenvironment. Anticancer Res 37:3429–34 [DOI] [PubMed] [Google Scholar]
- 138.Nambiar PR, Girnun G, Lillo NA, Guda K, Whiteley HE, Rosenberg DW. 2003. Preliminary analysis of azoxymethane induced colon tumors in inbred mice commonly used as transgenic/knockout progenitors. Int. J. Oncol 22:145–50 [PubMed] [Google Scholar]
- 139.NIH (Natl. Inst. Health). 2015. Consideration of sex as a biological variable in NIH-funded research Notice NOT-OD-15–102, NIH, Bethesda, MA. https://grants.nih.gov/grants/guide/notice-files/not-od-15-102.html [Google Scholar]
- 140.Nencioni A, Caffa I, Cortellino S, Longo VD. 2018. Fasting and cancer: molecular mechanisms and clinical application. Nat. Rev. Cancer 18:707–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, et al. 2009. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136:521–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Niklison-Chirou MV, Erngren I, Engskog M, Haglöf J, Picard D, et al. 2017. TAp73 is a marker of glutamine addiction in medulloblastoma. Genes Dev 31:1738–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nunez NP, Perkins SN, Smith NC, Berrigan D, Berendes DM, et al. 2008. Obesity accelerates mouse mammary tumor growth in the absence of ovarian hormones. Nutr. Cancer 60:534–41 [DOI] [PubMed] [Google Scholar]
- 144.O’Donnell JS, Massi D, Teng MWL, Mandala M. 2018. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol 48:91–103 [DOI] [PubMed] [Google Scholar]
- 145.Oleinik NV, Helke KL, Kistner-Griffin E, Krupenko NI, Krupenko SA. 2014. Rho GTPases RhoA and Rac1 mediate effects of dietary folate on metastatic potential of A549 cancer cells through the control of cofilin phosphorylation. J. Biol. Chem 289:26383–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Orillion A, Damayanti NP, Shen L, Adelaiye-Ogala R, Affronti H, et al. 2018. Dietary protein restriction reprograms tumor-associated macrophages and enhances immunotherapy. Clin. Cancer Res 24:6383–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Owonikoko TK. 2015. Inhibitors of mTOR pathway for cancer therapy, moving on from rapalogs to TORKinibs. Cancer 121:3390–92 [DOI] [PubMed] [Google Scholar]
- 148.Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, et al. 2016. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol 12:452–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Panda S 2016. Circadian physiology of metabolism. Science 354:1008–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Panigrahy D, Gartung A, Yang J, Yang H, Gilligan MM, et al. 2019. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. J. Clin. Investig 129:2964–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. 2014. Obesity and cancer—mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol 10:455–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parzych K, Saavedra-García P, Valbuena GN, Al-Sadah HA, Robinson ME, et al. 2019. The coordinated action of VCP/p97 and GCN2 regulates cancer cell metabolism and proteostasis during nutrient limitation. Oncogene 38:3216–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, et al. 2018. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab 27:428–38.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pellizzon MA, Ricci MR. 2018. The common use of improper control diets in diet-induced metabolic disease research confounds data interpretation: the fiber factor. Nutr. Metab 15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Phillips MS, Liu Q, Hammond HA, Dugan V, Hey PJ, et al. 1996. Leptin receptor missense mutation in the fatty Zucker rat. Nat. Genet 13:18–19 [DOI] [PubMed] [Google Scholar]
- 156.Pieroth R, Paver S, Day S, Lammersfeld C. 2018. Folate and its impact on cancer risk. Curr. Nutr. Rep 7:70–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Prendergast BJ, Onishi KG, Zucker I. 2014. Female mice liberated for inclusion in neuroscience and biomedical research. Neurosci. Biobehav. Rev 40:1–5 [DOI] [PubMed] [Google Scholar]
- 158.Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, et al. 2017. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol 19:974–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ribeiro AM, Andrade S, Pinho F, Monteiro JD, Costa M, et al. 2010. Prostate cancer cell proliferation and angiogenesis in different obese mice models. Int. J. Exp. Pathol 91:374–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Riess C, Shokraie F, Classen CF, Kreikemeyer B, Fiedler T, et al. 2018. Arginine-depleting enzymes—an increasingly recognized treatment strategy for therapy-refractory malignancies. Cell. Physiol. Biochem 51:854–70 [DOI] [PubMed] [Google Scholar]
- 161.Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, et al. 2010. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis 31:946–52 [DOI] [PubMed] [Google Scholar]
- 162.Roper J, Tammela T, Akkad A, Almeqdadi M, Santos SB, et al. 2018. Colonoscopy-based colorectal cancer modeling in mice with CRISPR-Cas9 genome editing and organoid transplantation. Nat. Protoc 13:217–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Saggar J, Yu M, Tan Q, Tannock I. 2013. The tumor microenvironment and strategies to improve drug distribution. Front. Oncol 3:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sanderson SM, Gao X, Dai Z, Locasale JW. 2019. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19:625–37 [DOI] [PubMed] [Google Scholar]
- 165.Saxena M, Christofori G. 2013. Rebuilding cancer metastasis in the mouse. Mol. Oncol 7:283–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, et al. 2017. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 6:e28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schauer DP, Feigelson HS, Koebnick C, Caan B, Weinmann S, et al. 2019. Bariatric surgery and the risk of cancer in a large multisite cohort. Ann. Surg 269:95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Schemmel R, Mickelsen O, Gill JL. 1970. Dietary obesity in rats: body weight and body fat accretion in seven strains of rats. J. Nutr 100:1041–48 [DOI] [PubMed] [Google Scholar]
- 169.Sengupta P 2013. The laboratory rat: relating its age with human’s. Int. J. Prev. Med 4:624–30 [PMC free article] [PubMed] [Google Scholar]
- 170.Serra M, Marongiu F, Pisu MG, Serra M, Laconi E. 2019. Time-restricted feeding delays the emergence of the age-associated, neoplastic-prone tissue landscape. Aging 11:3851–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Seyhan AA. 2019. Lost in translation: the valley of death across preclinical and clinical divide—identification of problems and overcoming obstacles. Transl. Med. Commun 4:18 [Google Scholar]
- 172.Sharma V, McNeill JH. 2009. To scale or not to scale: the principles of dose extrapolation. Br. J. Pharmacol 157:907–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Shin C-S, Mishra P, Watrous JD, Carelli V, D’Aurelio M, et al. 2017. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun 8:15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. 2014. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harb. Protoc 2014:694–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, et al. 2019. Antigen receptor control of methionine metabolism in T cells. eLife 8:e44210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Siolas D, Hannon GJ. 2013. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res 73:5315–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Smallwood TL, Gatti DM, Quizon P, Weinstock GM, Jung KC, et al. 2014. High-resolution genetic mapping in the diversity outbred mouse population identifies Apobec1 as a candidate gene for atherosclerosis. G3 4:2353–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Speakman JR. 2019. Use of high-fat diets to study rodent obesity as a model of human obesity. Int. J. Obes 43:1491–92 [DOI] [PubMed] [Google Scholar]
- 179.Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, et al. 2017. Genomes of the mouse collaborative cross. Genetics 206:537–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Steeg PS. 2016. Targeting metastasis. Nat. Rev. Cancer 16:201–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stemmer K, Kotzbeck P, Zani F, Bauer M, Neff C, et al. 2015. Thermoneutral housing is a critical factor for immune function and diet-induced obesity in C57BL/6 nude mice. Int. J. Obes 39:791–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stewart TA, Pattengale PK, Leder P. 1984. Spontaneous mammary adenocarcinomas in transgenic mice that carry and express MTV/myc fusion genes. Cell 38:627–37 [DOI] [PubMed] [Google Scholar]
- 183.Sulli G, Lam MTY, Panda S. 2019. Interplay between circadian clock and cancer: new frontiers for cancer treatment. Trends Cancer 5:475–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, et al. 2012. High-resolution genetic mapping using the mouse diversity outbred population. Genetics 190:437–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Swindell WR. 2012. Dietary restriction in rats and mice: a meta-analysis and review of the evidence for genotype-dependent effects on lifespan. Ageing Res. Rev 11:254–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Thomas KR, Capecchi MR. 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51:503–12 [DOI] [PubMed] [Google Scholar]
- 187.Thomas RM, Van Dyke T, Merlino G, Day CP. 2016. Concepts in cancer modeling: a brief history. Cancer Res 76:5921–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Threadgill DW, Miller DR, Churchill GA, de Villena FP. 2011. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J 52:24–31 [DOI] [PubMed] [Google Scholar]
- 189.Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, et al. 2020. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585:113–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Agric. Res. Serv. 2018. What we eat in America, NHANES 2015–2016. Rep., Agric. Res. Serv, US Dep. Agric., Washington, DC. https://www.ars.usda.gov/ARSUserFiles/80400530/pdf/1516/tables_1-36_2015-2016.pdf [Google Scholar]
- 191.US Dep. Agric., US Dep. Health Hum. Serv. 2020. Dietary guidelines for Americans, 2020–2025 Guidel., US Dep. Agric., US Dep. Health Hum. Serv., Washington, DC. https://www.dietaryguidelines.gov/sites/default/files/2020-12/Dietary_Guidelines_for_Americans_2020-2025.pdf [Google Scholar]
- 192.van de Steeg E, Kleemann R, Jansen HT, van Duyvenvoorde W, Offerman EH, et al. 2013. Combined analysis of pharmacokinetic and efficacy data of preclinical studies with statins markedly improves translation of drug efficacy to human trials. J. Pharmacol. Exp. Ther 347:635–44 [DOI] [PubMed] [Google Scholar]
- 193.Vancura A, Bu P, Bhagwat M, Zeng J, Vancurova I. 2018. Metformin as an anticancer agent. Trends Pharmacol. Sci 39:867–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Varady KA. 2011. Intermittent versus daily calorie restriction: which diet regimen is more effective for weight loss? Obes. Rev 12:e593–601 [DOI] [PubMed] [Google Scholar]
- 195.Veettil SK, Lim KG, Ching SM, Saokaew S, Phisalprapa P, Chaiyakunapruk N. 2017. Effects of aspirin and non-aspirin nonsteroidal anti-inflammatory drugs on the incidence of recurrent colorectal adenomas: a systematic review with meta-analysis and trial sequential analysis of randomized clinical trials. BMC Cancer 17:763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vettore L, Westbrook RL, Tennant DA. 2020. New aspects of amino acid metabolism in cancer. Br. J. Cancer 122:150–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vich Vila A, Collij V, Sanna S, Sinha T, Imhann F, et al. 2020. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun 11:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Visentin M, Zhao R, Goldman ID. 2012. The antifolates. Hematol. Oncol. Clin. North Am 26:629–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wallace JL, McKnight W, Reuter BK, Vergnolle N. 2000. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 119:706–14 [DOI] [PubMed] [Google Scholar]
- 200.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, et al. 2017. Humanized mouse models of clinical disease. Annu. Rev. Pathol. Mech. Dis 12:187–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wanders D, Stone KP, Forney LA, Cortez CC, Dille KN, et al. 2016. Role of GCN2-independent signaling through a noncanonical PERK/NRF2 pathway in the physiological responses to dietary methionine restriction. Diabetes 65:1499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang P, Wang Y, Langley SA, Zhou YX, Jen KY, et al. 2019. Diverse tumour susceptibility in Collaborative Cross mice: identification of a new mouse model for human gastric tumourigenesis. Gut 68:1942–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Watanabe S, Nishijima N, Hirai K, Shibata K, Hase A, et al. 2020. Anticancer activity of Amb4269951, a choline transporter-like protein 1 inhibitor, in human glioma cells. Pharmaceuticals 13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wege AK, Ernst W, Eckl J, Frankenberger B, Vollmann-Zwerenz A, et al. 2011. Humanized tumor mice—a new model to study and manipulate the immune response in advanced cancer therapy. Int. J. Cancer 129:2194–206 [DOI] [PubMed] [Google Scholar]
- 205.Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, et al. 2017. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med 9:eaai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wei Z, Song J, Wang G, Cui X, Zheng J, et al. 2018. Deacetylation of serine hydroxymethyl-transferase 2 by SIRT3 promotes colorectal carcinogenesis. Nat. Commun 9:4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wilkinson MJ, Manoogian ENC, Zadourian A, Lo H, Fakhouri S, et al. 2020. Ten-hour time-restricted eating reduces weight, blood pressure, and atherogenic lipids in patients with metabolic syndrome. Cell Metab 31:92–104.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Winter JM, Gildea DE, Andreas JP, Gatti DM, Williams KA, et al. 2017. Mapping complex traits in a diversity outbred F1 mouse population identifies germline modifiers of metastasis in human prostate cancer. Cell Syst 4:31–45.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, et al. 2014. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26:549–64 [DOI] [PubMed] [Google Scholar]
- 210.World Cancer Res. Fund, Am. Inst. Cancer Res. 2018. Diet, nutrition, physical activity and cancer: a global perspective: the third expert report Rep., World Cancer Res. Fund, London [Google Scholar]
- 211.Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, et al. 2017. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171:642–54.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yang L, Garcia Canaveras JC, Chen Z, Wang L, Liang L, et al. 2020. Serine catabolism feeds NADH when respiration is impaired. Cell Metab 31:809–21.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yang X, Wang Z, Li X, Liu B, Liu M, et al. 2018. SHMT2 desuccinylation by SIRT5 drives cancer cell proliferation. Cancer Res 78:372. [DOI] [PubMed] [Google Scholar]
- 214.Ye J, Fan J, Venneti S, Wan YW, Pawel BR, et al. 2014. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov 4:1406–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yoon DY, Mansukhani NA, Stubbs VC, Helenowski IB, Woodruff TK, Kibbe MR. 2014. Sex bias exists in basic science and translational surgical research. Surgery 156:508–16 [DOI] [PubMed] [Google Scholar]
- 216.Zeisel S 2017. Choline, other methyl-donors and epigenetics. Nutrients 9:445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, et al. 2019. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol 26:623–33.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhao R, Coker OO, Wu J, Zhou Y, Zhao L, et al. 2020. Aspirin reduces colorectal tumor development in mice and gut microbes reduce its bioavailability and chemopreventive effects. Gastroenterology 159:969–83.e4 [DOI] [PubMed] [Google Scholar]
- 219.Zhao Y, Liu M, Chan XY, Tan SY, Subramaniam S, et al. 2017. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 130:1995–2005 [DOI] [PubMed] [Google Scholar]