

Abstract
Cell therapies present an entirely new paradigm in drug development. Within this class, immune cell therapies are among the most advanced, having already demonstrated definitive evidence for clinical benefit in cancer and infectious disease. Numerous features distinguish these “living therapies” from traditional medicines, including their ability to expand and contract in proportion to need and to mediate therapeutic benefits for months or years following a single application. Continued advances in fundamental immunology, genetic engineering, gene editing, and synthetic biology exponentially expand opportunities to enhance the sophistication of T cell therapies, thereby increasing potency and safety and broadening their potential for treatment of disease. This perspective will summarize the current status of immune cell therapies for cancer, infectious disease, and autoimmunity, and discuss advances in cellular engineering currently under study to overcome barriers to progress.
Mackall eTOC blurb
Mackall, Maus & Weber describe the continuum of T cell therapeutics, including engineered and non-engineered approaches, and explore the progress and challenges in applying them to cancer, infectious disease, and autoimmunity.
Introduction
The transformative potential of cells as therapeutics was first realized in the mid 20th century, when widespread availability of red blood cell transfusions dramatically improved outcomes following trauma, surgery and some medical conditions. Subsequently, platelet transfusion and bone marrow transplantation enhanced survival of patients with hematologic diseases. In the modern era, fundamental advances in immunology, molecular biology, and virology alongside technological advances in cell manufacturing and genetic engineering have led to exciting progress in the development of immune cell therapies, with T cell therapies emerging as the most advanced within this therapeutic class. Adoptive transfer of tumor-infiltrating T cells and T cells engineered to express recombinant T cell receptors recognizing tumor antigens mediate impressive response rates in some solid cancers, chimeric antigen receptor modified T cells demonstrate impressive responses in B cell malignancies resistant to all standard agents, and viral-specific cytotoxic T lymphocytes (CTL) potently control some viral infections in immunocompromised hosts (Figure 1). This success has catapulted immune cell therapies from treatments studied on a small scale at research institutions to a global commercial enterprise. The convergence of continuing progress in immunobiology and synthetic biology, rapid advances in technologies for genetic engineering and gene editing at clinical scale, and private sector investment has positioned immune cell therapies for substantially greater impact on human health in the decades to come. In this perspective, we summarize the current status and future prospects for immune cell therapies for cancer, infectious disease, autoimmunity and other conditions.
Figure 1. Immune cell therapies for the treatment of human disease.
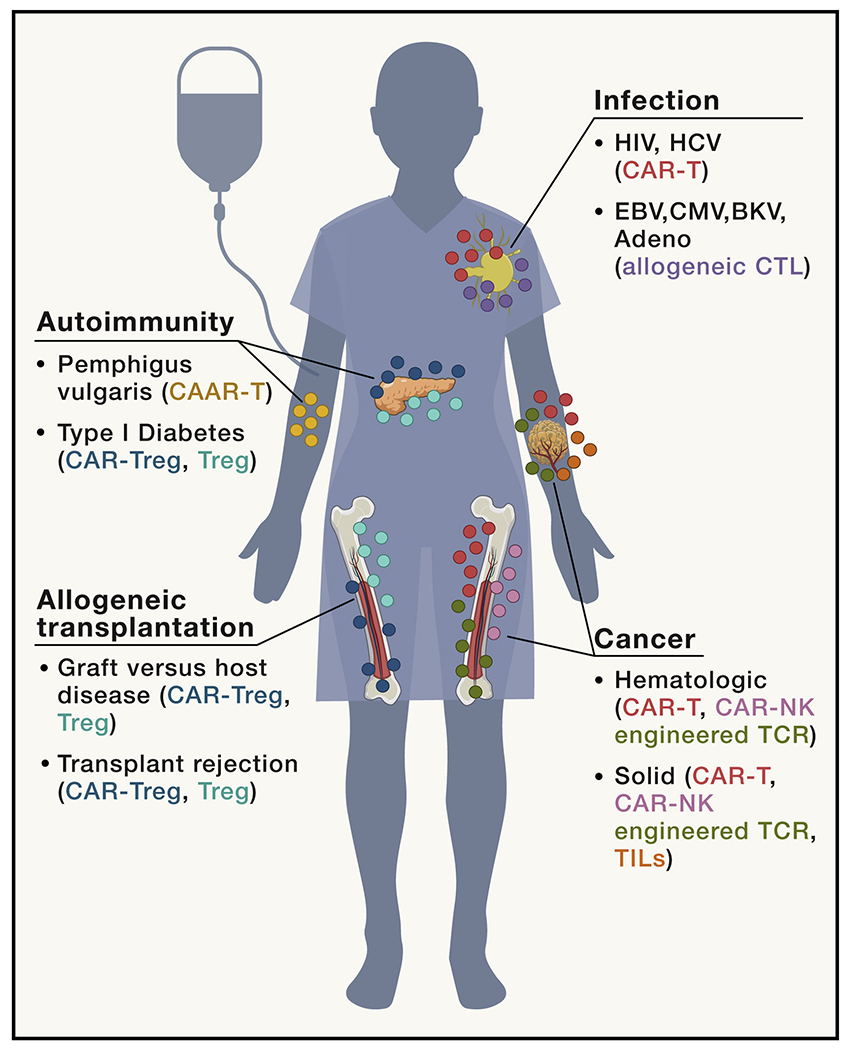
Recent advances in synthetic biology and bioengineering have broadened the applicability of immune cell therapies to include cancer, infection, allogeneic transplantation, and autoimmunity. CAR-T or NK cells, engineered TCRs, and TIL therapy have been and continue to be tested in hematologic and solid cancers. Tregs, CAR-Tregs, and CAAR-T cells are being developed to treat various autoimmune diseases and prevent rejection of transplanted tissues.
T Cell Therapies for Cancer
Adoptive Transfer of Tumor Infiltrating Lymphocytes
Studies conducted in the late 1980s and early 1990s demonstrated that 25-50% of patients with metastatic malignant melanoma treated with infusions of ex vivo expanded autologous tumor infiltrating T cells (TILs) plus recombinant IL-2 (rhIL-2) experienced long-lasting complete remission (Rosenberg et al., 1988; Rosenberg et al., 1994). These impressive results were seminal since they provided irrefutable evidence for tumor specific T cell-mediated immunity in humans and led to the molecular definition of self- and neo-antigens that can serve as the basis for T cell recognition of cancer (Brichard et al., 1993; Coulie et al., 1994; Kawakami et al., 1994a; Kawakami et al., 1994b; Kawakami et al., 1994c). Additional studies demonstrating a critical role for lymphopenia induced elevations in homeostatic cytokines in supporting expansion of adoptively transferred T cells (Fry et al., 2001; Fry and Mackall, 2001; Gattinoni et al., 2005), led to the demonstration that lymphodepleting therapies are critical components of effective adoptive T cell regimens for cancer (Dudley et al., 2008; Kochenderfer et al., 2017; Turtle et al., 2016a; Turtle et al., 2016b). While the overall clinical impact of TIL therapy in oncology has been limited in part because adequate numbers of bioactive TILs have not been reliably generated from patients with non-melanoma cancers, promising results have also been observed in individual patients with metastatic colorectal or breast cancer following infusion of TILs enriched for patient-specific neoantigens (Tran et al., 2015; Zacharakis et al., 2018).
T Cells Expressing Engineered TCRs
Advances in genetic engineering enabled investigators to clone tumor reactive T cell receptors (TCR) from TILs in responding patients and express the TCR in T cells expanded from the peripheral blood of other patients with cancer, providing a potentially limitless quantity of cells for therapeutic application (Figure 2). While generating T cells expressing such “engineered TCRs” was technologically feasible, early experience in translating this approach into the clinic faced several challenges. One problem was the transgenic, tumor-specific, TCR alpha and beta chains paired with endogenous receptors, which led to low levels of transgenic TCR expression and risks for off-target toxicity (Sarukhan et al., 1998). This challenge has been largely overcome by optimizing vector design (Jones et al., 2009) and incorporating cysteines (Cohen et al., 2007; Kuball et al., 2007) and/or mouse elements (Cohen et al., 2006; Hart et al., 2008; Voss et al., 2006) into the transgenic alpha and beta chains to induce preferential pairing of the transgenic proteins.
Figure 2. The continuum of immune cell therapies.
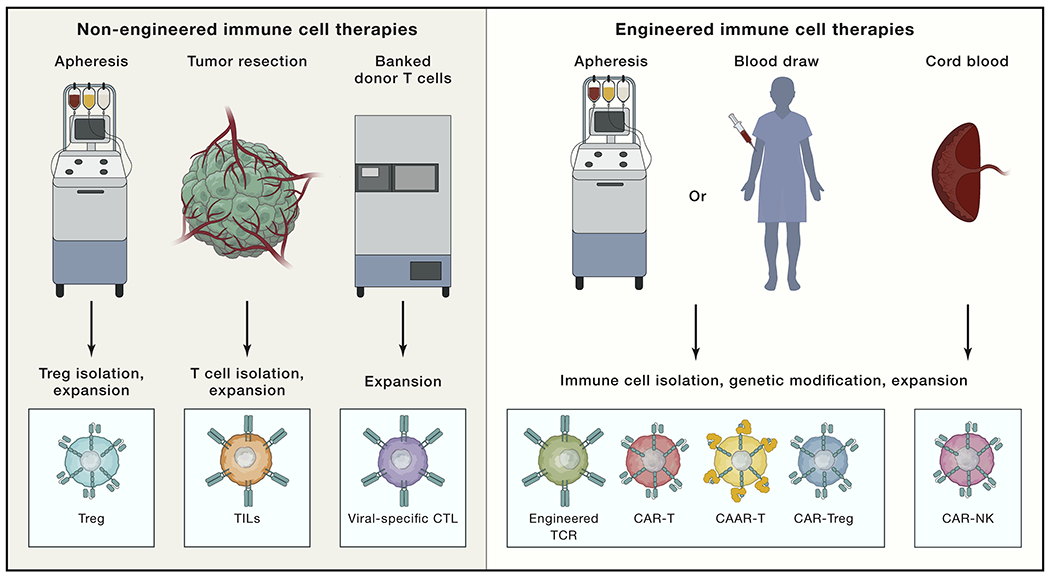
Non-engineered immune cell therapies (left) have also exhibited clinical efficacy in various disease contexts, including peripheral Tregs isolated from patient apheresis that are expanded and reinfused into the patient to treat autoimmune disease, GVHD, or organ rejection. Tumor-infiltrating lymphocytes (TILs) are isolated from resected tumors (usually from melanoma patients), expanded ex vivo, and reinfused into the patient. Banked donor virus-specific CTLs are thawed, expanded, and reinfused into HLA-matched recipients for treatment of chronic infections. Engineered immune cell therapies (right) are generated by first apheresing or drawing blood from the patient, isolating T cells, and using viral or non-viral approaches to insert a transgene encoding a synthetic receptor. Examples of engineered T cells include (1) T cells expressing an engineered TCR consisting of TCR alpha and beta subunits; (2) CAR-expressing T cells (CAR-Ts) or NK cells (CAR-NKs), which consist of an extracellular antigen-binding domain fused to intracellular domains involved in TCR signaling; (3) CAAR T cells (CAAR-Ts), where the chimeric receptor is comprised of an antigen-binding domain that targets autoreactive B cells; and (4) CAR-Tregs, where Tregs are isolated from peripheral blood and engineered to express a CAR that redirects them to tissue affected by autoimmune disease. All engineered T cell types are further expanded ex vivo prior to re-infusion into the patient. Of note, T cells modified to express a CAR or TCR mainly comprise cytotoxic effectors since Tregs are not substantially enriched by current culture methods.
A second challenge was identifying the safe and effective T cell receptors contained within the TIL population to be used for genetic transfer. A significant fraction of TILs present in melanoma tumors can recognize self-antigens that are expressed at low levels on healthy tissues (Yee et al., 2000), underscoring the potential for engineered TCRs to elicit significant on-target, off-tumor toxicity (Table 1). Compared to TCRs recognizing foreign antigens, TCRs recognizing self-antigens generally manifest low potency, related in part to low affinity as a result of thymic selection (Bowerman et al., 2009; Chervin et al., 2009; Stone et al., 2009). To improve the potency of TCRs recognizing tumor associated self-antigens, investigators enhanced their affinity using yeast or bacteriophage display or by immunizing mice with human antigens (Robbins et al., 2008; Schmitt et al., 2013). Affinity-enhanced, tumor-reactive TCRs generally demonstrated increased potency, but in clinical trials posed an increased risk of on-target off-tumor toxicity due to recognition of low levels of antigen on normal tissues (Johnson et al., 2009; Yee et al., 2000). Affinity-enhanced TCRs are also prone to cross-reactivity (Zhao et al., 2007), as illustrated by two deaths occurring in one study wherein T cells engineered to express a high affinity TCR targeting melanoma antigen gene (MAGE)-A3-derived peptide in the context of HLA-A*01, cross-reacted with titin expressed on cardiac tissue (Cameron et al., 2013; Linette et al., 2013). In a related instance, a high affinity TCR designed to target MAGE-A3 induced lethal neurotoxicity, presumably due to cross-reactivity with a MAGE-12-derived peptide expressed in brain tissue (Morgan et al., 2013).
Table 1.
Challenges and Opportunities for Immune Cell Therapies of Cancer
| Targeting Receptor | Opportunities | Challenges |
|---|---|---|
| TCR targeting overexpressed cancer associated self-antigens | • Can target a wide range of intracellular or cell surface molecules overexpressed in cancer • Antigens are prevalent in a wide range of cancer histologies • Early evidence for a therapeutic window enabling safe targeting (e.g. NY-ESO-1/LAGE TCR, WT1 TCR) |
•MHC restriction limits clinical applicability of any specific TCR • Requirement for affinity maturation is labor intensive and increases risk of off-target toxicity • Risk of toxicity due to low level antigen expression on normal tissues • Most antigens in this class do not contribute to oncogenic fitness, increasing the risk of selection of antigen neg variants |
| TCR targeting individualized neoantigens | •TCRs do not require affinity maturation • Likely to be safe since the antigen is not expressed in normal tissue |
• Labor and cost intensive process to generate unique products for individual patients limits availability • Most neoantigens are passenger mutations, increasing the risk of selection of antigen neg variants |
| TCR targeting public neoantigens in “hot spot” regions of oncogenes | • TCRs do not require affinity maturation • Mutations likely contribute to oncogenic fitness diminishing the risk of antigen neg variants • Modern oncology sequencing platforms can identify subsets of patients with targetable mutations |
• MHC restriction limits clinical applicability of any specific TCR • Limited immunogenicity of most tumor-specific mutations renders it likely that TCRs availability will be restricted to unique MHC alleles |
| CAR targeting overexpressed cell surface molecules | • Lack of MHC restriction enables applicability to all patients regardless of MHC allele expression • MHC downregulation does not confer resistance • Requirement for high antigen density for optimal CAR activation provides the potential for a therapeutic window even when targeting molecules expressed on normal tissues • scFvs and other binders can be generated to specifically recognize a vast array of molecules, including post-translational modifications |
• Risk of toxicity due to low level expression on normal tissues • Heterogeneous antigen expression increases the risk of antigen neg escape • Requirement for high antigen density for optimal CAR activation increases the risk of antigen neg and antigen lo escape |
Approaches to predict off-target toxicity of engineered TCRs have now been developed and some high affinity TCRs demonstrate both safety and significant clinical activity. Most notable is a high affinity TCR (c259) that recognizes a peptide derived from NY-ESO-1/LAGE-1 expressed on HLA-A2 (Zhao et al., 2007). Approximately 50% of patients treated with a lymphodepleting preparative regimen followed by 1-10 x 109 c259 expressing T cells with or without rhIL-2 experienced sustained antitumor effects (D’Angelo et al., 2018; Robbins et al., 2015; Robbins et al., 2011). Despite the favorable response rate, complete tumor eradication was not observed in most patients and work is underway to better understand the basis for resistance to this therapy. Favorable clinical outcomes were also observed in patients with NY-ESO-1/LAGE expressing multiple myeloma treated with adoptive transfer of c259 expressing T cells following autologous hematopoietic stem cell transplantation (HSCT) (Rapoport et al., 2015). Similarly, T cells expressing an engineered TCR targeting the tumor-associated antigen WT-1 (Xue et al., 2005) demonstrate a favorable safety profile and have shown impressive results in preventing relapse of acute myelogenous leukemia when administered following HSCT (Chapuis et al., 2019; Chapuis et al., 2013).
To overcome the challenges associated with targeting tumor-associated self-antigens, several groups have sought to identify TCRs that mediate recognition of tumor-specific neoantigens (Cohen et al., 2015; Yossef et al., 2018)(Table 1). Immune responses to neoantigens appear to play a major role in the antitumor response following treatment with immune checkpoint inhibition (Ribas and Wolchok, 2018) and TCRs recognizing neoantigens do not require affinity maturation and would be expected to demonstrate a safe profile. Autologous T cells expressing engineered patient-specific, neoantigen-targeting T cells have been generated on a small scale (Deniger et al., 2016), but the costs associated with such an individualized approach may be prohibitive for large scale application. One approach to overcome this challenge is to focus development on engineered TCR therapies that target shared neoantigens (Klebanoff and Wolchok, 2018) (Table 1). Several recurring “hotspot” mutations have been identified in common oncogenes, such as PI3K, Ras and p53 (Deniger et al., 2018; Lo et al., 2019), and some peptides derived from them are expressed on some HLA alleles, as was observed with the KRAS G12D mutation in colorectal cancer (Tran et al., 2016) and the H3K27M mutation in brain tumors (Chheda et al., 2018). Additionally, there is renewed interest in identifying TCRs that target breakpoint regions of oncogenic fusion proteins (Zamora et al., 2019). It may be possible to generate a bank of TCRs recognizing peptides expressed on a range of HLA alleles and use next generation sequencing and MHC typing to identify appropriate patients for such therapies. However, the laborious nature involved in identifying TCRs that exhibit potency and specificity, as well as the relatively small fraction of patients who match both HLA-type and tumor hotspot mutation limit the wide applicability of this strategy.
Avenues for Future Progress with Engineered TCR Therapeutics
Continued progress with T cell therapeutics incorporating engineered TCRs will require technologies to enhance potency, specificity, and safety. Thus far, the substantial effort required to develop potent and safe TCRs have limited the availability of such therapeutics to a small set of alleles, such as HLA-A2, resulting in significant barriers to access for testing in patients with less common HLA alleles. Progress in this arena is ongoing, and includes a recent approach to modify the framework regions of the TCR variable domain to increase expression levels and enhance potency while limiting the risk of cross-reactivity (Thomas et al., 2019), as well as new techniques to generate de novo TCRs (Sharma et al., 2019) and to identify the antigenic targets of tumor-reactive T cells (Gee et al., 2018). Furthermore, a multitude of engineering improvements are under development to enhance the potency of adoptively transferred T cells for cancer (Figure 3) and integrating such improvements into cells expressing tumor-reactive TCRs could improve efficacy. Examples already in clinical trials include a trial testing NY-ESO-1/LAGE-1 specific-TCR T cells in which PD-1, the inhibitory co-receptor, was eliminated using CRISPR/Cas9 (NCT03399448) (Stadtmauer et al., 2020) and trials administering engineered T cells with immune checkpoint inhibitors (NCT02775292, NCT03709706).
Figure 3. Engineering strategies to enhance adoptive Immune cell therapy.
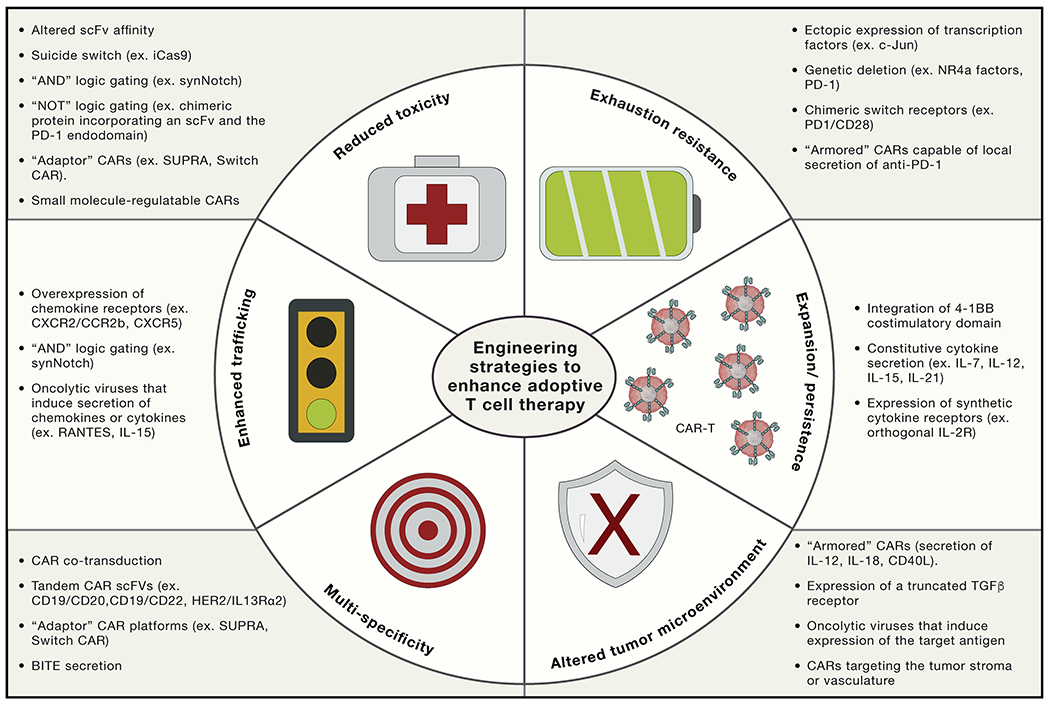
Sophisticated bioengineering approaches are under development to enhance the potency, specificity, and safety of T cell therapies. Suicide switches, AND gating, and adaptor CAR platforms are being developed to mitigate CAR-mediated toxicity. Ectopic expression of c-Jun or genetic deletion of NR4a factors endows CAR T cells with exhaustion resistance, potentially improving efficacy in solid tumors and enhancing persistence. CAR T cells can also be engineered to secrete specific factors to augment expansion or persistence (e.g., IL-7, IL-12, IL-15, or IL-21), diminish the need for a lymphodepleting regimen, resist the suppressive tumor microenvironment (e.g., secretion of IL-18, expression of a truncated transforming growth factor β [TGF-β] receptor), or act as tumor-specific drug delivery vehicles (e.g., local secretion of anti-PD-1).
Chimeric Antigen Receptor (CAR) T cells for B Cell Malignancies
CARs combine an extracellular antigen-targeting domain, which usually comprise a single chain variable fragment (scFv) but may utilize any antigen-specific binder, with intracellular signaling domains typically derived from the TCR. T cells expressing CAR receptors are not restricted by MHC, enabling applicability across patients regardless of HLA type and also preventing resistance due to MHC downregulation, which occurs commonly in cancer. Technologies are now readily available to generate scFvs and other binders to essentially any cell surface molecule (e.g. modified proteins, lipids, sugars, MHC-restricted peptides) and to engineer binders with a broad range of biochemical properties.
Early studies utilized first-generation CARs, which incorporate the TCR zeta signaling domain without any co-stimulatory domain (Eshhar et al., 1993; Gross et al., 1989a; Gross et al., 1989b). Some first-generation CAR T cells showed long-term persistence (Louis et al., 2011; Pule et al., 2008; Scholler et al., 2012), but they generally underwent limited expansion and failed to induce meaningful anti-tumor effects (Kershaw et al., 2006; Lamers et al., 2006; Park et al., 2007; Till et al., 2008). A major breakthrough ensued with the development of second-generation CARs, which integrate a co-stimulatory endodomain (usually CD28 or 4-1BB) upstream of CD3z (Brentjens et al., 2007; Imai et al., 2004). Beginning in 2010, second-generation CD19-CARs emerged as the most efficacious adoptive T cell therapy to date, mediating potent and long-lasting responses in high-grade B cell lymphoma in adults (Abramson et al., 2017; Kochenderfer et al., 2012; Kochenderfer et al., 2010; Neelapu et al., 2017; Schuster et al., 2017; Turtle et al., 2016b) and in B cell acute lymphoblastic leukemia in both children and adults who are refractory to all standard therapies (Brentjens et al., 2011; Gardner et al., 2017; Grupp et al., 2013; Lee et al., 2015; Maude et al., 2018; Maude et al., 2015; Turtle et al., 2016a).
CAR T cells incorporating a CD28 or 4-1BB endodomain both demonstrate significant clinical activity; however these products differ in the rate of expansion (CD28 expands more quickly), peak expansion level (CD28 expands to a higher degree), and the propensity for persistence (4-1BB demonstrates greater persistence) (Majzner and Mackall, 2019). Differences in expansion kinetics and persistence are associated with clinical outcomes, since CD19.28.z-CARs exhibit the highest response rates in lymphoma along with higher rates of CAR-mediated toxicity while post-CAR relapse appears to be lower in CD19.BB.z-CARs for B-ALL, likely because CAR persistence for at least 3-6 months is important for long-term control of this disease (Finney et al., 2019; Majzner and Mackall, 2019). These observations underscore the potential benefits of tailoring CAR T cell constructs based on the unique characteristics of the tumor being targeted. Although unproven, it is possible that more aggressive tumors may require CD28 co-stimulation due to rapid expansion kinetics, while those that progress more slowly could be controlled more safely with CARs T cells that integrate 4-1BB co-stimulation. Further, scFv affinity and/or CAR density could be optimized to target tumors that express heterogeneous levels of antigen, or to limit CAR-mediated toxicity (as discussed in more detail below).
Significant toxicities have been observed in patients treated with T cells expressing CD19 CARs, including cytokine release syndrome (CRS), whereby CAR T cell anti-tumor activity results in high levels of secreted IL-6, IL-1, and sepsis-like symptoms (Giavridis et al., 2018; Neelapu et al., 2017; Norelli et al., 2018), as well as immune-effector cell associated neurotoxicity syndrome (ICANS), which is associated with endothelial cell dysfunction at the blood-brain barrier induced by a hyperinflammatory milieu (Gust et al., 2017). While these toxicities have resulted in serious or even fatal complications, lowering CAR T cell dose and treatment with steroid therapy or antibodies blocking IL-6R (i.e. tocilizumab) has been highly effective and treatment-related mortality in large multicenter trials is currently <5% (Locke et al., 2019), which is not dissimilar from other standard treatment regimens for these refractory diseases. Additional pharmacologic strategies like anti-IL1 receptor blockade (Giavridis et al., 2018) or the tyrosinekinase inhibitory dasatinib, which potently and reversibly inhibits CAR T cell function (Mestermann et al., 2019; Weber et al., 2019), have been tested in preclinical models and may provide readily available, US Food and Drug Administration (FDA) -approved alternatives for patients who are refractory to standard CRS management therapies. Engineered safety switches, such as an inducible Caspase 9 (Di Stasi et al., 2011; Diaconu et al., 2017), enable CAR T cell depletion in the case of severe toxicities, but are irreversible by nature and have yet to be tested clinically. An alternative suicide switch that incorporates a thymidine kinase derived from herpes simplex virus initiated an immunogenic response leading to diminished survival of adoptively transferred T cells, thus illustrating the potential risk of enhanced immunogenicity following engineered expression of foreign proteins (Riddell et al., 1996).
The clinical success of CD19-targeting CARs has resulted in their approval by the US Food and Drug Administration and the European Medicine Agency, and has sparked widespread academic and private sector investment in CAR T cell research, including development of CAR T cells against other targets on B cell malignancies. CD22-targeting CARs also demonstrate remarkable success in patients with ALL, with over 80% of patients treated at the highest dose level achieving complete remission (Fry et al., 2018), and B cell maturation antigen (BCMA)-targeting CARs induce remissions in a high fraction of patients with multiple myeloma (Ali et al., 2016; Brudno et al., 2018; Cohen et al., 2019; Raje et al., 2019). The high response rates observed for CAR T cells targeting B cell malignancies are unprecedented, especially considering that patients treated thus far with these agents are refractory to all other therapies.
CAR T cells for Non-hematologic Solid Tumors
In stark contrast to the success observed in B cell malignancies, CAR T cells have not demonstrated convincing evidence of activity in patients with solid tumors. Current concepts hold that this likely represents a convergence of several barriers. A major challenge is the dearth of identified cell membrane targets with high level, homogenous expression on solid tumors and limited expression on normal tissue. However, as investigators have focused more intently on cataloguing the surfaceome of cancer, molecules with significant differential expression have been identified, including the Tn glycoform of MUC1 on adenocarcinomas (Posey et al., 2016), GD2 ganglioside on diffuse intrinsic pontine glioma (Mount et al., 2018), GPC2 on neuroblastoma (Bosse et al., 2017), and PAPP-A on Ewing sarcoma (Heitzeneder et al., 2019). These results, combined with an emerging understanding that CARs require high levels of antigen density for optimal activation (Harris et al., 2018; Majzner and Mackall, 2018; Majzner et al., 2019; Walker et al., 2017) and that engineering approaches can tune the antigen density threshold (Liu et al., 2015) (Figure 3) provide optimism that a therapeutic window for CAR T cell targeting of cell surface molecules overexpressed on solid tumors can be identified.
A related issue is heterogeneity of antigen expression on cancer in general, and solid tumors in particular, which has fueled interest in developing multi-specific CARs as discussed below (Figure 3). Effective treatment of solid tumors with cell therapies is also potentially hampered by limited trafficking. Recent work has demonstrated that regional delivery may mitigate this challenge in CNS tumors and other cancers such a mesothelioma which primarily demonstrate regional spread (Adusumilli et al., 2014; Brown et al., 2016; Priceman et al., 2018). Finally, solid cancers are well known to harbor a suppressive microenvironment, which inhibits CAR T cell functionality through multiple routes, including expression of checkpoint receptor ligands (ex. PD-L1), hypoxia and nutrient depletion, and suppressive immune cells (ex. Tregs, MDSCs) (Chong et al., 2017; Juillerat et al., 2017; Long et al., 2016). As discussed below, numerous engineering approaches are in development to address this challenge to enhance the potency of CAR T cells for solid tumors (Figure 3).
T Cell Therapies for Infectious Disease
Viral-Specific T Cell Therapy
Clinical investigators in the early 1990s observed that some patients with leukemia relapse following allogeneic HSCT could be rendered into remission following the infusion of T cells from the donor (Bonini et al., 1997; Cullis et al., 1992; Drobyski et al., 1992; Kolb et al., 1990). These data ultimately provided convincing evidence for T cell mediated antileukemic effects, as well as the potential for such cells to treat uncontrolled viral infection, which occurred not uncommonly in this setting (Papadopoulos et al., 1994). In patients with severe immunosuppression, allogeneic viral-specific CTL induce very high response rates against Epstein-Barr virus (EBV) infection and EBV-associated lymphomas, cytomegalovius, adenovirus, BK virus and human herpesvirus-6 infection, with limited evidence for graft-versus-host disease (GVHD) despite the use of minimally MHC matched products (Heslop et al., 1996; Heslop et al., 2010; McLaughlin et al., 2018; Melenhorst et al., 2010). Efficacy is dependent upon limited rejection of the allogeneic cells due to recipient immunoincompetence, while the absence of GVHD presumably is due to the limited alloreactivity in CTL products that are devoid of naïve T cells. “Off-the-shelf” banks of viral-specific CTL are now available to treat multiple viruses covering the vast majority of HLA-alleles such that an appropriate product is likely to be available for the majority of patients (O’Reilly et al., 2016; Tzannou et al., 2017) (Figure 2), and private investment is seeking to commercialize this therapy. The success of these products has also raised the prospect of using viral-specific CTL to target viral-associated cancers, including those driven by EBV (Bollard et al., 2014; Bollard et al., 2018; McLaughlin et al., 2018), hepatitis B (Bertoletti et al., 2015), and others._However, because patients with viral-associated cancer retain sufficient immunity to reject allogeneic cell products, these efforts largely focus on adoptive transfer of autologous viral-specific CTL or genetically-engineered viral-reactive TCRs, and thus are not dissimilar to the approaches discussed earlier that utilize non-viral directed T cells for cancer therapy.
Viral-specific CAR T cells for HIV Infection
Beginning in 1998, clinical trials of CAR T cells were launched to target HIV infection. T cells were engineered to express a first-generation CAR receptor that utilized the CD4 exodomain recognizing the gp120 subunit of the HIV Env protein as its antigen-binding domain, enabling recognition of HIV infected cells (Deeks et al., 2002; Scholler et al., 2012). These trials did not demonstrate efficacy in controlling HIV infection, but many patients exhibited long-term survival as a result of contemporaneous advances in antiretroviral therapeutics. Remarkably 98% of samples tested more than 10 years following infusion demonstrated evidence for persistent CAR T cells, with no evidence for persistent clonal expansion or enrichment for cells in which the vector had integrated near oncogenes (Scholler et al., 2012), nor any evidence for the emergence of replication-competent retrovirus/lentivirus (Marcucci et al., 2018). With more than 500 patient-years of follow-up, this experience is the strongest evidence to date that adoptive transfer of retrovirally-engineered T cells is safe and that such cells or their progeny are capable of persisting for more than a decade.
Numerous efforts are ongoing to improve the efficacy of HIV-specific CAR T cells (reviewed in Maldini et al., 2018), many of which overlap with efforts to enhance the functionality of CARs for cancer, including efforts to enhance CAR T cell persistence and engineering multi-specificity to overcome viral heterogeneity (Hale et al., 2017). However, some challenges are uniquely relevant to CARs for HIV infection, including the need to engineer resistance to viral infection in the engineered T cells themselves, which investigators are attempting via gene editing of CCR5 (Hale et al., 2017; Perez et al., 2008; Tebas et al., 2014) and overexpression of proteins that interfere with viral machinery (Maldini et al., 2018). Because the toxicity associated with lymphodepleting regimens is not likely to be acceptable in this clinical setting and because CAR bioactivity must be sustained for many years, engineering cells to ensure long-term persistence represents a major focus. In one primate model, SIV-reactive CAR T cells generated from HSCs compared to those generated from blood showed enhanced persistence and protection from rebound viremia upon cessation of antiretroviral therapy, raising the prospect that HSC-derived CAR expressing T cells could be preferred in this setting (Zhen et al., 2017). Work is also underway to target the latent HIV reservoir by co-expressing CXCR5 (Ayala et al., 2017) to home HIV-specific CAR T cells to CD4+ T follicular helper cells (Tfh) in the B cell follicle of lymphoid tissues (Figure 3).
T Cell Therapies for Autoimmunity and Other Diseases
Despite the introduction of improved therapeutics for autoimmunity over the past several decades, more progress is needed. Small molecule tyrosine kinase inhibitors and cytokine-targeting antibodies demonstrate remarkable clinical efficacy, but are broadly immunosuppressive and are not applicable to the full spectrum of autoimmune diseases. These challenges are driving efforts to develop more targeted approaches, such as adoptive transfer of regulatory T cells (Tregs) (Figure 2), which play a major role in preserving self-tolerance, maintaining immune homeostasis, and preventing autoimmunity (Sakaguchi et al., 2008). Adoptive transfer of non-engineered Tregs mediates impressive results in a variety of murine models of autoimmunity and GVHD (Perdigoto et al., 2015), and adoptive transfer of human Tregs in clinical settings of GVHD, organ transplantation, and type I diabetes (T1D) have proven to be safe, feasible, and have demonstrated persistence for up to 1-year post-infusion (Figure 1) (Bluestone et al., 2015; Brunstein et al., 2011). However, the near- and long-term efficacy of these therapies remains unproven.
CARs are being expressed in Tregs as a strategy to improve the potency and specificity of Treg therapies. In a landmark study, investigators engineered CAR Tregs specific for 2,4,6-trinitrobenzenesulfonic acid (TNBS) in murine model of colitis (Elinav et al., 2009). CAR Tregs secreted suppressive factors, proliferated, and ameliorated disease symptoms in an antigen-specific manner. Similar results were observed in murine models of multiple sclerosis and transplant rejection (Boroughs et al., 2019; Fransson et al., 2012; MacDonald et al., 2016). CAR Tregs exhibited therapeutic effects at doses that were suboptimal for non-engineered Tregs, providing evidence that CAR expression improves the potency of Treg therapies in addition to enhancing specificity. Collectively, these studies provide a strong rationale for clinical testing of CAR Treg therapy.
A potential advantage of CAR Treg therapy over effector CAR T cell therapy is maintenance of the target population, which may result in sustained Treg expansion, persistence, and durable immunosuppression in the target tissue. Further, whereas off-target toxicity of effector CAR T cell therapies could result in substantial tissue damage, off-target effects of CAR Tregs are expected to be less severe, but could include prolonged immunosuppression at non-diseased tissues, opportunistic infections, or suppression of local tumor immunity. Similar side effects could also manifest if Tregs were to express a CAR that exhibits antigen-independent tonic signaling (Long et al., 2015), resulting in constitutive and non-specific immunosuppression.
A distinct safety risk associated with CAR Tregs relates to the potential plasticity of the Treg lineage. In a murine model of Type 1 diabetes (T1D), Tregs exposed to inflammatory conditions in situ lost expression of FOXP3 and converted to an effector-like T cell (Zhou et al., 2009). Further, CAR Tregs incorporating a 4-1BB co-stimulatory domain converted to cytotoxic CAR T cells that lacked immunosuppressive capacity (Boroughs et al., 2019), raising the prospect that conversion of engineered Tregs into effector cells could potentiate autoimmunity rather than suppress it. A similar phenomenon could manifest in the event of effector CAR T cell contamination in the CAR Treg manufactured product. Given that effector T cells expand much more quickly and robustly upon activation compared to Tregs, a minor population of effector CAR T cells could quickly outcompete CAR Tregs and mediate devastating autoimmunity.
Cell engineering and synthetic biology provide potential opportunities to mitigate such toxicities (Figure 3). In addition to drug-inducible suicide switches like those used in effector CAR T cells (Diaconu et al., 2017), a cell intrinsic suicide switch that activates autonomously upon loss of FOXP3 expression or in response to transcription of inflammatory cytokines could help protect against CAR Treg conversion (Maldini et al., 2018). Alternatively, ectopic expression of FOXP3 in CAR Tregs could mitigate conversion by maintaining FOXP3 expression and promoting suppressive function (Maldini et al., 2018). To prevent long-term immunosuppression of tumor immunity or opportunistic infections at target tissue sites, one could employ tunable platforms wherein CAR activity is dependent on a protein therapeutic infused into the patient (Cho et al., 2018; Leung et al., 2019; Pishali Bejestani et al., 2017; Rodgers et al., 2016; Wu et al., 2015). In autoimmune diseases that manifest periods of sudden and severe symptoms (i.e. flare ups), like rheumatoid arthritis or relapsing remitting multiple sclerosis, such platforms could enable the induction of CAR Treg activity only during these periods while keeping CAR Tregs dormant when disease symptoms are minimal.
Recently, investigators developed chimeric autoantibody receptors (CAARs) by using a creative strategy to exploit effector CAR T cells for treatment of autoimmune disease (Figure 1). CAARs are similar to typical CARs, except the extracellular antigen-binding domain targets the B cell receptor (BCR) of self-reactive B cells. Investigators utilized a murine model of pemphigus vulgaris, wherein self-reactive B cells that target desmogleins mediate skin damage. T cells expressing a CAAR with a desmoglein 3 extracellular domain specifically targeted pathogenic B cells, resulting in cure (Ellebrecht et al., 2016). This study provides important proof-of-concept that CAAR T cells could be utilized in B cell-mediated autoimmune diseases in which specific autoantigens are well-defined, such as rheumatoid arthritis and lupus erythematosus.
A provocative report recently utilized CAR T cells targeting fibroblast activating protein (FAP) to prevent fibrosis in a murine model of fibrosis-induced cardiomyopathy (Aghajanian et al., 2019). Fibrosis was induced over the span of 4 weeks and CAR T cells were administered one week following the inducing stimulus. The CAR T cells infiltrated the heart, induced killing of the reactive fibroblasts, diminished fibrosis, and led to improved cardiac function. No apparent toxicity was observed over 12 weeks, presumably reflecting a therapeutic window between high levels of FAP expressed on fibroblasts within the inflamed cardiac tissue and low levels of FAP expressed on fibroblasts in normal tissues. Whether such time-dependent and antigen-dependent therapeutic windows can be identified in the context of human fibrotic diseases remains to be seen, but this study demonstrates the versatility of CAR T cells to treat a variety of human diseases, and will surely be an area of future study.
Next Generation Engineering to Address Major Barriers to Progress
Advances in genetic engineering, gene editing, cellular reprogramming and synthetic biology are providing an increasingly robust toolbox with which to engineer solutions to the problems of resistance and toxicity that currently limit the field. Many of these solutions are modular and can potentially be integrated in a multiplex fashion in individual cells and cell products, thus dramatically increasing the sophistication of immune cell therapeutics.
Antigen Negative and Antigen Low Escape Following Treatment with Monospecific CAR T cells
Similar to other targeted therapeutics in oncology or in infectious diseases like tuberculosis or HIV infection, selective pressure on any one target often leads to emergence of escape variants. Not surprisingly, antigen loss represents a major form of resistance to CAR T cell therapy (reviewed in Majzner and Mackall, 2018; Maude et al., 2018; Sotillo et al., 2015). A related issue is the increasing recognition that, in contrast to TCR therapeutics which can recognize very low levels of antigen, high levels of antigen are required for optimal CAR T cell activation (Harris et al., 2018; Majzner and Mackall, 2018; Majzner et al., 2019; Walker et al., 2017). While this property can provide a therapeutic window for targeting antigens with low expression in normal tissues, such as the GD2 ganglioside or mesothelin, resistance to CAR therapeutics can also occur by selection of variants with subthreshold levels of the targeted antigen as observed with clinical trials of CD22-CAR (Fry et al., 2018). To address these issues, efforts are underway to engineer effective multi-specific CAR T cells (discussed below, Figure 3), modulate the antigen density threshold for CAR T cell activation by altering scFv affinity (Ahmed et al., 2017; Ahmed et al., 2015; Drent et al., 2016; Liu et al., 2015), upregulate the antigen density on targeted cells (Pont et al., 2019; Ramakrishna et al., 2019), target tissue stroma to prevent the escape of variant tumor cells (Spiotto et al., 2004), or engineer approaches to enhance the induction of natural immunity and thereby broaden the CAR-induced immune response to include bystander, antigen-loss variants (Beatty et al., 2014; Slaney et al., 2017).
Endowing Multi-specificity to Enhance Efficacy (OR gates)
Engineering combinatorial antigen recognition could enhance CAR T cell efficacy by overcoming antigen escape and/or by increasing the repertoire of targetable antigens (Figure 3). The administration of multiple CAR T cell products represents one strategy for multi-antigen targeting. However, this method imposes significantly increased cost and labor. Further, in preclinical models this approach was less effective than engineering multispecific recognition into a single cell (Fry et al., 2018; Hegde et al., 2016), and in the only reported clinical trial to date, response rates were similar to patients treated with a single CAR T cell product (Yan et al., 2019). Several approaches are under development to engineer individual cells capable of targeting two antigens, where binding of EITHER antigen would trigger CAR T cell activation (an “OR” gate, in terms of Boolean logic). One approach is to co-transduce a single population of T cells with vectors encoding two CARs (Ruella et al., 2016), while a related approach incorporates a bicistronic vector to express two separate chimeric receptors on every cell (Majzner and Mackall, 2018). An alternative approach is to create a bivalent or “tandem” construct, where recognition of antigen by either one of two binding domains on the extracellular portion of the CAR can trigger effector function. Hedge and colleagues reported on a HER2/IL13Rα2 tandem CAR for the treatment of glioblastoma, and observed protection against antigen escape as well as a synergistic effect on CAR T cell activation when both antigens were present (Hegde et al., 2016). Numerous tandem CARs have been developed and studied in preclinical models (Ormhoj et al., 2019; Scarfo et al., 2018; Schmidts et al., 2019; Schneider et al., 2017; Zah et al., 2016) and clinical trials of CD19/20 and CD19/22 CARs for lymphoma (Shah et al., 2019) and CD19/22 CARs for leukemia (Schultz et al., 2018) are underway. Interestingly, all of the tandem CAR designs have required systematic testing of various configurations to determine the optimal design for each antigen. For example, the CD19/20 CARs required different testing of different lengths of linkers between specificities, where only short linkers were proved to preserve function (Zah et al., 2016), and the optimal CD19/22 CAR required a looped structure, where the two variable regions of the CD19 component were interspersed with the variable regions of the CD22 binder component (Qin et al., 2018).
Dual-or multi-targeted “OR” CARs can also be generated using so-called “adaptor” CARs, which have an extracellular domain that can bind a variety of binders (Cho et al., 2018; Kudo et al., 2014; Rodgers et al., 2016; Tamada et al., 2012; Urbanska et al., 2012) (Figure 3). A soluble adaptor must be administered for the CAR T cell to be activated and “OR” gating can be achieved if multiple binders are administered simultaneously. Such platforms theoretically provide a safety switch since CAR T cell function is ablated by clearance of the soluble adaptor, though the ability to regulate the activity of the CAR is dependent not only on the kinetics of T cell expansion and persistence, but also on the half-life and stability of the adaptor protein that confers antigen specificity. An alternative approach involves engineering CAR T cells to secrete a bispecific antibody-like molecule that triggers T cell activation on one end and binds a second antigen on the tumor with the other end (Bonifant et al., 2016; Iwahori et al., 2015; Velasquez et al., 2016). This approach was recently pioneered in the setting of glioblastoma, where CAR T cells directed to the oncogenic tumor antigen EGFR variant III also secreted bispecific molecules targeting EGFR (Choi et al., 2019).
“AND” and “NOT” Gating Strategies to Enhance Safety
CAR T cells can also be engineered to activate only in response to target cells expressing two antigens concurrently, thus enabling discriminating between tumor cells expressing antigen pairs versus healthy tissue expressing only one of the targets (Figure 3). In one strategy, one receptor incorporates a CD3 zeta endodomain and a second incorporates a co-stimulatory domain. In order to prevent an “OR” gate, the CAR incorporating CD3 zeta must be engineered to have a very low affinity such that it induces only subpar activation upon antigen binding (Kloss et al., 2013). A different approach involves the use of synthetic Notch (synNotch) receptors (Roybal et al., 2016b) wherein sensing of antigen 1 by the synNotch receptor induces transcription of a CAR with specificity for antigen 2 (Roybal et al., 2016a). This strategy was efficacious in the context of a pre-clinical model of anatomically separated solid tumors, where one tumor expressing both antigens was injected into one flank while a second (control) tumor expressing only one antigen was on the second flank. However, in a liquid tumor model, where normal stroma expressing antigen 1 (ROR1) was co-mingled anatomically with tumor cells expressing antigen 1 (ROR1) and antigen 2 (EpCAM or B7-H3), the synNotch logic gate failed to spare the antigen 1-only expressing healthy cells (Srivastava et al., 2019). Some investigators have also sought to develop “NOT” gates whereby antigen 1 is targeted only in the absence of antigen 2, by incorporating the intracellular domain of either CTLA-4, or more effectively, PD1, on the CAR targeting antigen 2 (Fedorov et al., 2013). This has been demonstrated to be efficacious in a pre-clinical model of allogeneic rejection of fibroblasts, but has not yet entered clinical trials.
Targeting T Cell Exhaustion and the Tumor Microenvironment to Enhance Potency
Chronic antigen stimulation leads to a state of T cell exhaustion characterized by functional impairment (Wherry et al., 2003), surface expression of multiple inhibitory receptors, including PD-1, TIM-3, and LAG-3 among others, and a distinct transcriptional and epigenetic profile (Bengsch et al., 2018; Blackburn et al., 2009; Blank et al., 2019; Pauken et al., 2016; Quigley et al., 2010; Sen et al., 2016). There is ample pre-clinical and clinical evidence that CAR T cells are predisposed to exhaustion and that this limits efficacy. Canonical exhaustion markers on tumor-infiltrating CD19 CAR T cells were higher in non-responders versus those who exhibited a complete response (CR)(Schuster et al., 2017), and high exhaustion marker expression on the CAR T manufactured product was found to be predictive of non-response (Finney et al., 2019). The phenomena of tonic signaling due to antigen-independent aggregation of the CAR receptors in the cell membrane and exposure to high tumor burdens can also lead to exhaustion (Frigault et al., 2015; Long et al., 2015). In cases where exhausted T cells are present prior to engineering, selection of T cell subsets with greater proliferative capacity prior to genetic manipulation could improve outcomes (Busch et al., 2016; Sabatino et al., 2016; Sommermeyer et al., 2016; Turtle et al., 2016a; Wang et al., 2016). Interestingly, pre-clinical and clinical studies have shown that small molecule drugs such as dasatinib or ibrutinib may prevent or reverse T cell exhaustion (Fraietta et al., 2016; Long et al., 2017; Sagiv-Barfi et al., 2015; Weber et al., 2020). Finally, transient disruption of tonic CAR signaling via regulation of CAR protein can epigenetically reprogram exhausted CAR T cells and augment efficacy in pre-clinical models (Weber et al., 2020).
A recent case study reported the enrichment of a single T cell clone in which the CAR transgene integrated into the TET2 locus, resulting in a loss of function mutation. This mutation endowed CAR T cells with increased potency, expansion, persistence, and a memory-like phenotype ultimately leading to a 5-year complete remission at the time of the report (Fraietta et al., 2018), raising the prospect that CAR T cells can be engineered to avoid or resist exhaustion (Figure 3). This has emerged as an active and promising area of research in the field. Insertion of the CAR transgene into the TRAC locus and endogenous control of CAR expression prevented exhaustion in pre-clinical leukemia models (Eyquem et al., 2017). Overexpression of the transcription factor c-Jun (Lynn et al., 2019) has been shown to protect T cells from exhaustion from even the most exhausting CAR designs. In murine models, expression of the nuclear receptor transcription factors NR4A1, NR4A2, and NR4A3 was also associated with CAR T cell exhaustion, but without available pharmacologic inhibitors of these, it is a less clear how this strategy could be applied in human T cells, where 3 separate genes would require knockout (Chen et al., 2019).
Numerous approaches to modify the tumor microenvironment or confer resistance to it in the CAR T cells are also under investigation. Some groups have focused on engineering blockade of the PD-1 axis by engineered secretion of anti-PD1 nanobodies (Rafiq et al., 2018), gene-editing to delete the PD-1 protein altogether (Ren et al., 2017), or engineering a “switch” receptor composed of extracellular PD-1, that would bind to PD-L1 expressed in the tumor microenvironment, fused to the intracellular domain of a costimulatory molecule like CD28 (Liu et al., 2016). Similarly, to overcome death signaling imposed by tumor overexpression of Fas ligand, investigators expressed a dominant negative Fas receptor, which conferred increased expansion and persistence in CD19-targeting CAR T cells (Yamamoto et al., 2019). Brentjens has a series of publications on “armored” CAR T cells, where a second transgene is meant to modify the tumor environment: this includes secretion of the inflammatory cytokines IL-12 (Koneru et al., 2015; Pegram et al., 2012) or IL-18 (Avanzi et al., 2018), or CD40L (Curran et al., 2015) to enhance antigen cross-presentation and promote epitope spreading. Other groups have also used a non-signaling form the TGF beta receptor which outcompetes the endogenous receptor due to its constitutive and high expression. This transgene was first applied to human EBV-specific T cells (Foster et al., 2008) and then included as a second transgene in human CAR T cells targeting prostate cancer in preclinical models (Kloss et al., 2018) and in clinical trials (NCT03089203). Finally, preclinical models have indicated that targeting the tumor stroma and/or vasculature can enhance CAR-T efficacy and potentially limit the escape of antigen-negative variants (Kakarla et al., 2013; Seaman et al., 2017; Spiotto et al., 2004; Wang et al., 2014).
Alternative Immune Cells (NK cells, γδ cells, NKT cells, iPSC derived immune effectors) and Allogeneic Immune Cell Therapies
While this review has primarily focused on therapies that administer and engineer αβ T cells, there is an emerging body of work demonstrating progress in using similar techniques to engineer other immune effector cells, which can could confer certain advantages. The principal advantage of natural killer (NK), gamma-delta (γδ) T cells, natural killer T (NKT) cells is that they all possess cytotoxic capacity, but none express an endogenous TCR and therefore do not mediate GvHD if administered to MHC mismatched hosts. However, adult peripheral blood NK cells are relatively resistant to retroviral and lentiviral transduction, and exhibit poor persistence in the absence of high levels of IL-2 or IL-15. To circumvent this, Rezvani and colleagues pioneered an approach whereby NK cells contained in unrelated cord blood are transduced to express both a CD19-targeting CAR and a transgene coding for IL-15, which in a recent report mediated complete responses in 7 out of 11 patients treated with this therapy (Liu et al., 2020; Liu et al., 2018). Such an approach could provide an “off-the-shelf” CAR-NK cell product, thus enabling unprecedented scaling of CAR-NK therapy. γδ T cells are rare populations in the peripheral blood and thus require substantial enrichment and bisphosphonates during ex vivo culture (Xiao et al., 2018). Nonetheless, investigators have successfully transduced γδ T cells with CARs, which have demonstrated activity in pre-clinical models (Capsomidis et al., 2018; Harrer et al., 2017). Similarly, invariant NKT cells are very rare populations, but demonstrate pre-clinical efficacy in solid tumors (Heczey et al., 2014) and show enhanced persistence when they are engineered to secrete IL-15 (Xu et al., 2019).
Improved approaches are emerging to generate immune effector cells from induced pluripotent stem cells (iPSC) (Li et al., 2018; Themeli et al., 2013), further opening the potential for scalability of engineered immune cell populations. It is not clear yet how such cells compare in cytotoxicity and persistence to cell products engineered from mature alpha-beta T cells and whether clinically relevant cell numbers can be produced using artificial cell culture systems; however successful approaches to generate and deliver effective iPS-derived engineered immune effector cells offer the tantalizing possibility of manufacturing hundreds of doses of therapeutic cells from an inexhaustible source.
A major barrier to the success of this approach is rejection of the allogeneic product, although progress is being made in this arena. Recent preliminary reports have demonstrated feasibility and some clinical activity of allogeneic CAR T cells engineered to delete the TCR as well as CD52 (Qasim et al., 2017; Torikai et al., 2012), enabling selective depletion of lymphocytes in the host to prevent rejection using a CD52 directed mAb. Recent progress has also been reported in diminishing rejection by deletion of HLA class I and II combined with overexpression of CD47 (Deuse et al., 2019). If the dual challenges of GVHD and rejection could be overcome, the availability of banks of immune effector generated from healthy donors could transform the field of immune cell therapies, by enabling more cost-effective therapies, reducing the time needed to provide such therapies to ill patients, providing a platform for more sophisticated multi-engineering and resulting in standardization of quality across products beyond that which can be accomplished using the autologous platform.
Conclusions
Immune cell therapies are a rapidly emerging class of therapeutics that have already demonstrated transformative impact in some B cell malignancies, and are well positioned for increasing impact in cancer and beyond in the coming years. Seminal work conducted in the 1980s and 1990s established the foundational principles of immunotherapy and genetic engineering that are now being leveraged to engineer human immune cells into “living” drugs. Remarkable progress has culminated in FDA-approval of CAR T cell therapy for the treatment of B cell malignancies; yet, translation to solid tumors has proven immensely challenging and these therapeutics have had limited impact in other diseases. Sophisticated bioengineering approaches utilizing genetic deletion, ectopic overexpression of transcription factors, multi-specific binders, Boolean gating, and other synthetic systems will ultimately determine the extent to which next-generation immune cell therapies emerge as efficacious alternatives to traditional medicines.
Acknowledgments
This work was supported by a St Baldrick’s/Stand Up 2 Cancer Pediatric Dream Team Translational Cancer Research Grant (C.L.M.) and SU2C innovator award (MVM). Stand Up 2 Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. C.L.M is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. The work was also supported by the Virginia and D.K. Ludwig Fund for Cancer Research (C.L.M.) and NIH grants 2P01CA049605-29A1, P01CA217959-01 and 5P30CA124435 (C.L.M.) and U54 CA232568-01 (E.W.W. and C.L.M.).
Declaration of Interests
C.L.M. is an inventor on numerous patent applications in the area of CAR T cell immunotherapy and has received royalties for the CD22-CAR from NIH following licensure to Opus Bio and Juno Therapeutics. CLM is a founder of, holds equity in, and receives consulting fees from Lyell Immunopharma, which develops cellular therapies for cancer. She is also a consultant for Nektar, Neoimmune Tech, and Apricity and holds equity in Apricity and Allogene. M.V.M. is an inventor on numerous patent applications in the area of CAR T cell immunotherapy and has received royalties; MVM is a consultant or advisory board member for multiple companies developing cellular therapies, and holds equity in TCR2 and Century therapeutics. E.W.W. is an inventor on numerous patent applications in the area of CAR T cell immunotherapy, holds equity in, and receives consulting fees from Lyell Immunopharma.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramson JS, Palomba ML, Gordon LI, Lunning MA, Arnason JE, Wang M, Forero A, Maloney DG, Albertson T, Garcia J, et al. (2017). High Durable CR Rates in Relapsed/Refractory (R/R) Aggressive B-NHL Treated with the CD19-Directed CAR T Cell Product JCAR017 (TRANSCEND NHL 001): Defined Composition Allows for Dose-Finding and Definition of Pivotal Cohort. Blood 130, 581–581.28584136 [Google Scholar]
- Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, and Sadelain M (2014). Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 6, 261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, et al. (2019). Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, et al. (2017). HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 3, 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. (2015). Human Epidermal Growth Factor Receptor 2 (HER2) - Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33, 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, Brudno JN, Stetler-Stevenson M, Feldman SA, Hansen BG, et al. (2016). T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 128, 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, Park H, Purdon TJ, Daniyan AF, Spitzer MH, et al. (2018). Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep 23, 2130–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala VI, Deleage C, Trivett MT, Jain S, Coren LV, Breed MW, Kramer JA, Thomas JA, Estes JD, Lifson JD, et al. (2017). CXCR5-Dependent Entry of CD8 T Cells into Rhesus Macaque B-Cell Follicles Achieved through T-Cell Engineering. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al. (2014). Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer immunology research 2, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O’Brien S, Gherardini PF, Herati RS, Huang AC, Chang KM, et al. (2018). Epigenomic-Guided Mass Cytometry Profiling Reveals Disease-Specific Features of Exhausted CD8 T Cells. Immunity 48, 1029–1045 e1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoletti A, Brunetto M, Maini MK, Bonino F, Qasim W, and Stauss H (2015). T cell receptor-therapy in HBV-related hepatocellularcarcinoma. Oncoimmunology 4, e1008354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, and Wherry EJ (2009). Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, et al. (2019). Defining ‘T cell exhaustion’. Nat Rev Immunol 19, 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, et al. (2015). Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7, 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Gottschalk S, Torrano V, Diouf O, Ku S, Hazrat Y, Carrum G, Ramos C, Fayad L, Shpall EJ, et al. (2014). Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol 32, 798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, Dakhova O, Carrum G, Ramos CA, Liu H, et al. (2018). Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol 36, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifant CL, Szoor A, Torres D, Joseph N, Velasquez MP, Iwahori K, Gaikwad A, Nguyen P, Arber C, Song XT, et al. (2016). CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol Ther 24, 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F, Traversari C, et al. (1997). HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 276, 1719–1724. [DOI] [PubMed] [Google Scholar]
- Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, Kulkarni AS, Cetrulo CL, Ting D, Blazar BR, et al. (2019). Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, Rathi KS, Kendsersky NM, Randall M, Donovan L, et al. (2017). Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell 32, 295–309 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman NA, Crofts TS, Chlewicki L, Do P, Baker BM, Christopher Garcia K, and Kranz DM (2009). Engineering the binding properties of the T cell receptor:peptide:MHC ternary complex that governs T cell activity. Mol Immunol 46, 3000–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. (2011). Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118, 4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, La Perle K, Quintas-Cardama A, Larson SM, and Sadelain M (2007). Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res 13, 5426–5435. [DOI] [PubMed] [Google Scholar]
- Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, Coulie P, and Boon T (1993). The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 178, 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. (2016). Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, Lam N, Stetler-Stevenson M, Salem D, Yuan C, Pavletic S, et al. (2018). T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J Clin Oncol 36, 2267–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, Defor T, Levine BL, June CH, Rubinstein P, et al. (2011). Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, and Riddell SR (2016). Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol 28, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. (2013). Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 5, 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, Majani Y, Gileadi T, Wallace R, Gustafsson K, et al. (2018). Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther 26, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, Voillet V, Gottardo R, Ragnarsson GB, Bleakley M, et al. (2019). T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med 25, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, et al. (2013). Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 5, 174ra127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, Yoshimura A, Scott-Browne JP, and Rao A (2019). NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervin AS, Stone JD, Holler PD, Bai A, Chen J, Eisen HN, and Kranz DM (2009). The impact of TCR-binding properties and antigen presentation format on T cell responsiveness. J Immunol 183, 1166–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, Yang X, Carrera DA, Downey KM, Shrivastav S, et al. (2018). Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med 215, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Collins JJ, and Wong WW (2018). Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173, 1426–1438 e1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, Bailey SR, Boroughs AC, Frigault MJ, and Leick MB (2019). CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nature biotechnology 37, 1049–1058. [DOI] [PubMed] [Google Scholar]
- Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, and Schuster SJ (2017). PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129, 1039–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, Vogl DT, Weiss BM, Dengel K, Nelson A, et al. (2019). B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 129, 2210–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS, et al. (2015). Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 125, 3981–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, and Morgan RA (2007). Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67, 3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, and Morgan RA (2006). Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66, 8878–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, Mattei S, De Plaen E, Lurquin C, Szikora JP, et al. (1994). A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 180, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis JO, Jiang YZ, Schwarer AP, Hughes TP, Barrett AJ, and Goldman JM (1992). Donor leukocyte infusions for chronic myeloid leukemia in relapse after allogeneic bone marrow transplantation. Blood 79, 1379–1381. [PubMed] [Google Scholar]
- Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, Purdon T, Pegram HJ, and Brentjens RJ (2015). Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther 23, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, Grupp S, Tap WD, Chagin K, Binder GK, et al. (2018). Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov 8, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C, June CH, et al. (2002). A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther 5, 788–797. [DOI] [PubMed] [Google Scholar]
- Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, Malekzadeh P, Jia L, Yossef R, Langhan MM, et al. (2018). T-cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin Cancer Res 24, 5562–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniger DC, Pasetto A, Tran E, Parkhurst MR, Cohen CJ, Robbins PF, Cooper LJ, and Rosenberg SA (2016). Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol Ther 24, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, Thayer WO, Wahl A, Garcia JV, Reichenspurner H, et al. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol 37, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, et al. (2011). Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365, 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, and Savoldo B (2017). Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol Ther 25, 580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drent E, Themeli M, Poels R, van de Donk NWCJ, Lokhorst HM, and Mutis T (2016). Reducing on-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors By Affinity Optimization. Blood 128, 2170–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobyski WR, Roth MS, Thibodeau SN, and Gottschall JL (1992). Molecular remission occurring after donor leukocyte infusions for the treatment of relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation. Bone Marrow Transplant 10, 301–304. [PubMed] [Google Scholar]
- Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. (2008). Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol 26, 5233–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Adam N, Waks T, and Eshhar Z (2009). Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 136, 1721–1731. [DOI] [PubMed] [Google Scholar]
- Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, et al. (2016). Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, Gross G, and Schindler DG (1993). Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 90, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, and Sadelain M (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov VD, Themeli M, and Sadelain M (2013). PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 5, 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, Futrell RB, Orentas RJ, Li D, Gardner RA, et al. (2019). CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest 129, 2123–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, Rooney CM, and Bollard CM (2008). Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother 31, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, Lacey SF, Melenhorst JJ, McGettigan SE, Cook DR, et al. (2016). Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 127, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, Cogdill AP, Morrissette JJD, DeNizio JE, Reddy S, et al. (2018). Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, and Loskog AS (2012). CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD Jr., et al. (2015). Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer immunology research 3, 356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, Steinberg SM, Wood LV, Yarchoan R, Zuckerman J, et al. (2001). A potential role for interleukin-7 in T-cell homeostasis. Blood 97, 2983–2990. [DOI] [PubMed] [Google Scholar]
- Fry TJ, and Mackall CL (2001). Interleukin-7: master regulator of peripheral T-cell homeostasis? Trends Immunol 22, 564–571. [DOI] [PubMed] [Google Scholar]
- Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, Wolters P, Martin S, Delbrook C, Yates B, et al. (2018). CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 24, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, Bleakley M, Brown C, Mgebroff S, Kelly-Spratt KS, et al. (2017). Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129, 3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, et al. (2005). Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee MH, Han A, Lofgren SM, Beausang JF, Mendoza JL, Birnbaum ME, Bethune MT, Fischer S, Yang X, Gomez-Eerland R, et al. (2018). Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 172, 549–563 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M (2018). CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 24, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross G, Gorochov G, Waks T, and Eshhar Z (1989a). Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc 21, 127–130. [PubMed] [Google Scholar]
- Gross G, Waks T, and Eshhar Z (1989b). Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 86, 10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. (2013). Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368, 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, et al. (2017). Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov 7, 1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, Scharenberg AM, Rawlings DJ, and Wagner TA (2017). Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol Ther 25, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrer DC, Simon B, Fujii SI, Shimizu K, Uslu U, Schuler G, Gerer KF, Hoyer S, Dorrie J, and Schaft N (2017). RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 17, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, Lever M, Dushek O, Schmitt TM, Greenberg PD, et al. (2018). Comparison of T Cell Activities Mediated by Human TCRs and CARs That Use the Same Recognition Domains. J Immunol 200, 1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart DP, Xue SA, Thomas S, Cesco-Gaspere M, Tranter A, Willcox B, Lee SP, Steven N, Morris EC, and Stauss HJ (2008). Retroviral transfer of a dominant TCR prevents surface expression of a large proportion of the endogenous TCR repertoire in human T cells. Gene Ther 15, 625–631. [DOI] [PubMed] [Google Scholar]
- Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, Gao X, Guo L, Yvon E, Hicks J, et al. (2014). Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 124, 2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK, et al. (2016). Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 126, 3036–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzeneder S, Sotillo E, Shern JF, Sindiri S, Xu P, Jones R, Pollak M, Noer PR, Lorette J, Fazli L, et al. (2019). Pregnancy-Associated Plasma Protein-A (PAPP-A) in Ewing Sarcoma: Role in Tumor Growth and Immune Evasion. J Natl Cancer Inst 111, 970–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, and Rooney CM (1996). Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med 2, 551–555. [DOI] [PubMed] [Google Scholar]
- Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, et al. (2010). Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 115, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, and Campana D (2004). Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18, 676–684. [DOI] [PubMed] [Google Scholar]
- Iwahori K, Kakarla S, Velasquez MP, Yu F, Yi Z, Gerken C, Song XT, and Gottschalk S (2015). Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther 23, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. (2009). Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Peng PD, Yang S, Hsu C, Cohen CJ, Zhao Y, Abad J, Zheng Z, Rosenberg SA, and Morgan RA (2009). Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther 20, 630–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, Duchateau P, and Poirot L (2017). An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep 7, 39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K, et al. (2013). Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther 21, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, and Rosenberg SA (1994a). Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci U S A 91, 3515–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, and Rosenberg SA (1994b). Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci U S A 91, 6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, and Rosenberg SA (1994c). Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med 180, 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. (2006). A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12, 6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, and Wolchok JD (2018). Shared cancer neoantigens: Making private matters public. J Exp Med 215, 5–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, and Sadelain M (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, and June CH (2018). Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther 26, 1855–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. (2012). B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119, 2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, et al. (2017). Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol 35, 1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. (2010). Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb HJ, Mittermuller J, Clemm C, Holler E, Ledderose G, Brehm G, Heim M, and Wilmanns W (1990). Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 76, 2462–2465. [PubMed] [Google Scholar]
- Koneru M, Purdon TJ, Spriggs D, Koneru S, and Brentjens RJ (2015). IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 4, e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, and Greenberg PD (2007). Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 109, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo K, Imai C, Lorenzini P, Kamiya T, Kono K, Davidoff AM, Chng WJ, and Campana D (2014). T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res 74, 93–103. [DOI] [PubMed] [Google Scholar]
- Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, and Oosterwijk E (2006). Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, e20–22. [DOI] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung WH, Gay J, Martin U, Garrett TE, Horton HM, Certo MT, Blazar BR, Morgan RA, Gregory PD, Jarjour J, et al. (2019). Sensitive and adaptable pharmacological control of CAR T cells through extracellular receptor dimerization. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hermanson DL, Moriarity BS, and Kaufman DS (2018). Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 23, 181–192 e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. (2013). Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, et al. (2020). Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 382, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, et al. (2018). Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 32, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, et al. (2015). Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 75, 3596–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, Newick K, Lo A, June CH, Zhao Y, et al. (2016). A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res 76, 1578–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W, Parkhurst M, Robbins PF, Tran E, Lu YC, Jia L, Gartner JJ, Pasetto A, Deniger D, Malekzadeh P, et al. (2019). Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer immunology research 7, 534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, Lin Y, Braunschweig I, Hill BT, Timmerman JM, et al. (2019). Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al. (2015). 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 21, 581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, El-Etriby R, Galli S, Tsokos MG, Orentas RJ, et al. (2016). Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer immunology research 4, 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, Maddocks KJ, Cheney C, Jones JA, Flynn JM, et al. (2017). Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest 127, 3052–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, Anbunathan H, Lattin J, Jones R, Tieu V, et al. (2019). c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, and Levings MK (2016). Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest 126, 1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2018). Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov 8, 1219–1226. [DOI] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2019). Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355. [DOI] [PubMed] [Google Scholar]
- Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, Rietberg SP, Linde MH, Xu P, Rota C, et al. (2019). CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin Cancer Res 25, 2560–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldini CR, Ellis GI, and Riley JL (2018). CAR T cells for infection, autoimmunity and allotransplantation. Nat Rev Immunol 18, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci KT, Jadlowsky JK, Hwang WT, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, Gupta M, Lacey SF, Plesa G, Chew A, et al. (2018). Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol Ther 26, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Teachey DT, Porter DL, and Grupp SA (2015). CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125, 4017–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin LP, Rouce R, Gottschalk S, Torrano V, Carrum G, Wu MF, Hoq F, Grilley B, Marcogliese AM, Hanley PJ, et al. (2018). EBV/LMP-specific T cells maintain remissions of T- and B-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood 132, 2351–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, Brenner MK, Barrett AJ, and Heslop HE (2010). Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood 116, 4700–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, Mades A, Sadelain M, Einsele H, and Hudecek M (2019). The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. (2013). Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, Labanieh L, Hulleman E, Woo PJ, Rietberg SP, et al. (2018). Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat Med 24, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. (2018). Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24, 739–748. [DOI] [PubMed] [Google Scholar]
- O’Reilly RJ, Prockop S, Hasan AN, Koehne G, and Doubrovina E (2016). Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transplant 51, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormhoj M, Scarfo I, Cabral ML, Bailey SR, Lorrey SJ, Bouffard AA, Castano AP, Larson RC, Riley LS, Schmidts A, et al. (2019). Chimeric Antigen Receptor T Cells Targeting CD79b Show Efficacy in Lymphoma with or without Cotargeting CD19. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, et al. (1994). Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med 330, 1185–1191. [DOI] [PubMed] [Google Scholar]
- Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, et al. (2007). Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 15, 825–833. [DOI] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, and Brentjens RJ (2012). Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdigoto AL, Chatenoud L, Bluestone JA, and Herold KC (2015). Inducing and Administering Tregs to Treat Human Disease. Front Immunol 6, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. (2008). Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pishali Bejestani E, Cartellieri M, Bergmann R, Ehninger A, Loff S, Kramer M, Spehr J, Dietrich A, Feldmann A, Albert S, et al. (2017). Characterization of a switchable chimeric antigen receptor platform in a pre-clinical solid tumor model. Oncoimmunology 6, e1342909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, Hudecek M, Comstock ML, Rajan A, Patel BKR, et al. (2019). gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 134, 1585–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey AD Jr., Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, et al. (2016). Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 44, 1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, Chang WC, Ostberg JR, Neman J, Jandial R, et al. (2018). Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res 24, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. (2008). Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, et al. (2017). Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med 9. [DOI] [PubMed] [Google Scholar]
- Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, Yuan CM, Haso W, Shern JF, Shah NN, et al. (2018). Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol Ther Oncolytics 11, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S, et al. (2010). Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med 16, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al. (2018). Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol 36, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, Liedtke M, Rosenblatt J, Maus MV, Turka A, et al. (2019). Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med 380, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, Qin H, Kraft IL, Stetler-Stevenson M, Yuan CM, et al. (2019). Modulation of Target Antigen Density Improves CAR T-cell Functionality and Persistence. Clin Cancer Res 25, 5329–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, et al. (2015). NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21, 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu X, Fang C, Jiang S, June CH, and Zhao Y (2017). Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 23, 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, and Wolchok JD (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, Lupton SD, Overell RW, Reynolds TC, Corey L, et al. (1996). T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med 2, 216–223. [DOI] [PubMed] [Google Scholar]
- Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, et al. (2015). A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, et al. (2008). Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 180, 6116–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O, et al. (2016). Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A 113, E459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. (1988). Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 319, 1676–1680. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, and White DE (1994). Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 86, 1159–1166. [DOI] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016a). Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016b). Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432 e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, and Kozlowski M (2016). Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. The Journal of clinical investigation 126, 3814–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, Dougherty S, Qin H, Klebanoff CA, Fry TJ, et al. (2016). Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 128, 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, and Levy R (2015). Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A 112, E966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Sarukhan A, Garcia C, Lanoue A, and von Boehmer H (1998). Allelic inclusion of T cell receptor alpha genes poses an autoimmune hazard due to low-level expression of autospecific receptors. Immunity 8, 563–570. [DOI] [PubMed] [Google Scholar]
- Scarfo I, Ormhoj M, Frigault MJ, Castano AP, Lorrey S, Bouffard AA, van Scoyk A, Rodig SJ, Shay AJ, Aster JC, et al. (2018). Anti-CD37 chimeric antigen receptor T cells are active against B and T cell lymphomas. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts A, Ormhøj M, Choi BD, Taylor AO, Bouffard AA, Scarfò I, Larson RC, Frigault MJ, Gallagher K, and Castano AP (2019). Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Advances 3, 3248–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, Aggen DH, Stromnes IM, Dossett ML, Richman SA, Kranz DM, and Greenberg PD (2013). Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 122, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D, Xiong Y, Wu D, Nlle V, Schmitz S, Haso W, Kaiser A, Dropulic B, and Orentas RJ (2017). A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, et al. (2012). Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4, 132ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LM, Davis KL, Baggott C, Chaudry C, Marcy AC, Mavroukakis S, Sahaf B, Kong KA, Muffly LS, and Kim S (2018). Phase 1 study of CD19/CD22 bispecific chimeric antigen receptor (CAR) therapy in children and young adults with B cell acute lymphoblastic leukemia (ALL) (Am Soc Hematology). [Google Scholar]
- Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, et al. (2017). Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 377, 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA, et al. (2017). Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 31, 501–515 e508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NN, Zhu F, Schneider D, Taylor C, Krueger W, Worden A, Longo WL, Hamadani M, Fenske T, and Johnson B (2019). Results of a phase I study of bispecific anti-CD19, anti-CD20 chimeric antigen receptor (CAR) modified T cells for relapsed, refractory, non-Hodgkin lymphoma (American Society of Clinical Oncology). [Google Scholar]
- Sharma P, Harris DT, Stone JD, and Kranz DM (2019). T-cell Receptors Engineered De Novo for Peptide Specificity Can Mediate Optimal T-cell Activity without Self Cross-Reactivity. Cancer immunology research 7, 2025–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaney CY, von Scheidt B, Davenport AJ, Beavis PA, Westwood JA, Mardiana S, Tscharke DC, Ellis S, Prince HM, Trapani JA, et al. (2017). Dual-specific Chimeric Antigen Receptor T Cells and an Indirect Vaccine Eradicate a Variety of Large Solid Tumors in an Immunocompetent, Self-antigen Setting. Clin Cancer Res 23, 2478–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, and Riddell SR (2016). Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. (2015). Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5, 1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiotto MT, Rowley DA, and Schreiber H (2004). Bystander elimination of antigen loss variants in established tumors. Nat Med 10, 294–298. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, et al. (2019). Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 35, 489–503 e488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, et al. (2020). CRISPR-engineered T cells in patients with refractory cancer. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JD, Chervin AS, and Kranz DM (2009). T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology 126, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, and Davila E (2012). Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res 18, 6436–6445. [DOI] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. (2014). Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, and Sadelain M (2013). Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol 31, 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Mohammed F, Reijmers RM, Woolston A, Stauss T, Kennedy A, Stirling D, Holler A, Green L, Jones D, et al. (2019). Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat Commun 10, 4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ, et al. (2008). Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 112, 2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovich B, et al. (2012). A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, et al. (2015). Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, et al. (2016a). CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126, 2123–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, et al. (2016b). Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8, 355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, Carrum G, Sasa G, Lulla P, Watanabe A, et al. (2017). Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol 35, 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, and Powell DJ Jr. (2012). A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res 72, 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez MP, Torres D, Iwahori K, Kakarla S, Arber C, Rodriguez-Cruz T, Szoor A, Bonifant CL, Gerken C, Cooper LJ, et al. (2016). T cells expressing CD19-specific Engager Molecules for the Immunotherapy of CD19-positive Malignancies. Sci Rep 6, 27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss RH, Kuball J, Engel R, Guillaume P, Romero P, Huber C, and Theobald M (2006). Redirection of T cells by delivering a transgenic mouse-derived MDM2 tumor antigen-specific TCR and its humanized derivative is governed by the CD8 coreceptor and affects natural human TCR expression. Immunol Res 34, 67–87. [DOI] [PubMed] [Google Scholar]
- Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, Lopomo P, Vigny M, Fry TJ, Orentas RJ, et al. (2017). Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol Ther 25, 2189–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer immunology research 2, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, Norris AP, Wong CW, Urak RZ, Chang WC, et al. (2016). Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 127, 2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Lynn RC, Parker KR, Anbunathan H, Lattin J, Sotillo E, Good Z, Malipatlolla M, Xu P, Vandris P, et al. (2020). Transient “rest” induces functional reinvigoration and epigenetic remodeling in exhausted CAR-T cells. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, and Mackall CL (2019). Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv 3, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77, 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Roybal KT, Puchner EM, Onuffer J, and Lim WA (2015). Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Chen C, Li Z, Zhu S, Tay JC, Zhang X, Zha S, Zeng J, Tan WK, Liu X, et al. (2018). Large-scale expansion of Vgamma9Vdelta2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy 20, 420–435. [DOI] [PubMed] [Google Scholar]
- Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, Jin J, Courtney AN, Liu B, Di Pierro EJ, et al. (2019). NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin Cancer Res 25, 7126–7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue SA, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A, Apperley J, Engels B, Uckert W, Morris E, et al. (2005). Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood 106, 3062–3067. [DOI] [PubMed] [Google Scholar]
- Yamamoto TN, Lee PH, Vodnala SK, Gurusamy D, Kishton RJ, Yu Z, Eidizadeh A, Eil R, Fioravanti J, Gattinoni L, et al. (2019). T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J Clin Invest 129, 1551–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y, Shi M, Lan J, Fei X, Jin L, et al. (2019). A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol 6, e521–e529. [DOI] [PubMed] [Google Scholar]
- Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, Kenyon K, Davis MM, Riddell SR, and Greenberg PD (2000). Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo. J Exp Med 192, 1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, Gartner JJ, Prickett TD, Cafri G, Robbins PF, et al. (2018). Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zah E, Lin MY, Silva-Benedict A, Jensen MC, and Chen YY (2016). T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer immunology research 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora AE, Crawford JC, Allen EK, Guo XJ, Bakke J, Carter RA, Abdelsamed HA, Moustaki A, Li Y, Chang TC, et al. (2019). Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, et al. (2007). High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol 179, 5845–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen A, Peterson CW, Carrillo MA, Reddy SS, Youn CS, Lam BB, Chang NY, Martin HA, Rick JW, Kim J, et al. (2017). Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog 13, e1006753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, and Bluestone JA (2009). Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol 10, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]