

Summary
Prokaryotic organisms have developed multiple defense systems against phages, however little is known about whether and how these interact with each other. Here we studied the connection between two of the most prominent prokaryotic immune systems: restriction-modification and CRISPR. While both systems employ enzymes that cleave a specific DNA sequence of the invader, CRISPR nucleases are programmed with phage-derived spacer sequences, which are integrated into the CRISPR locus upon infection. We found that restriction endonucleases provide a short-term defense that is rapidly overcome through methylation of the phage genome. In a small fraction of the cells, however, restriction results in acquisition of spacer sequences from the cleavage site, which mediates a robust type II-A CRISPR-Cas immune response against the methylated phage. This mechanism is reminiscent of eukaryotic immunity, where the innate response offers a first temporary line of defense, and also activates a second and more robust adaptive response.
Graphical Abstract
Blurb
Restriction and CRISPR-Cas nucleases cleave phage DNA. Maguin et al. show how these are coordinated to defend bacteria from infection. Restriction provides initial protection and also generates free dsDNA ends for the acquisition of spacers by CRISPR-Cas systems. Spacers guide CRISPR nucleases to destroy methylated phage DNA that bypasses restriction.
Introduction
Archaea and bacteria have evolved many mechanisms to resist the constant assault of their viruses (phages) (Bernheim and Sorek, 2020; Samson et al., 2013). One such strategy is provided by restriction-modification (RM) systems, present in over 90% of sequenced prokaryote genomes (Roberts et al., 2015). Typically, RM systems encode endonuclease and methyltransferase activities that “restrict” and “modify”, respectively, the same short DNA sequence (Bickle and Kruger, 1993). The methylation of chromosomal target sites inhibits cleavage by the restriction enzyme to prevent the attack of “self” DNA. On the other hand, unmodified sites on the phage, or “foreign”, genome are recognized and cleaved by the restriction endonuclease to provide antiviral defense. This discrimination strategy, however, can easily be overcome by phages, as methyltransferases can erroneously modify incoming invaders before restriction occurs (Bickle and Kruger, 1993). This allows the completion of the phage’s lytic cycle and the release of viral progeny with modified genomes, which will go on to infect and kill the rest of the bacterial population. RM systems are divided into four types according to their subunit organizations, recognition sequences, and co-factor requirements (Tock and Dryden, 2005). Type I RM systems are composed of three different subunits: S and M form a complex in which the S subunit recognizes the target DNA sequence, and M carries out methylation of adenosine residues using S-adenosylmethionine as the methyl group donor. The R subunit, when associated with the SM complex, can translocate DNA in an ATP-dependent process and cleaves unmethylated DNA at a distance of several thousand base-pairs from the recognition site (Loenen et al., 2014). By contrast, in the majority of type II RM systems, the most prevalent, the methyltransferase and endonuclease act independently (Pingoud et al., 2005). While the methyltransferase acts as a monomer, modifying one strand of the target DNA at a time, the endonuclease acts as a homodimer, cleaving both strands at a defined position close to or within the recognition sequence. While Type III systems also recognize and cleave unmodified DNA, type IV typically lack methyltransferase activity and cleave modified DNA instead (Tock and Dryden, 2005).
Present in ~40% of bacterial and ~80% of archaeal genomes (Makarova et al., 2011), clustered regularly interspaced short palindromic repeats (CRISPR) loci and their associated genes (cas) constitute another defense system that cleaves foreign DNA. The CRISPR locus contains short DNA repeats (30–40 nucleotides), separated by equally short unique sequences of viral or plasmid origin, called spacers (Bolotin et al., 2005; Mojica et al., 2005; Pourcel et al., 2005), which are acquired during infection (Barrangou et al., 2007). These are transcribed and processed into short RNA guides, the CRISPR RNAs (crRNAs) that mediate, through base pair complementarity, target recognition and cleavage by Cas nucleases (Brouns et al., 2008; Hale et al., 2009; Jore et al., 2011). Depending on the cas gene content, CRISPR-Cas systems can be classified into six types, with types I, II, and III being the most abundant and studied (Makarova et al., 2020). Type II-A CRISPR systems are thought to use free double-stranded DNA (dsDNA) ends as substrates for new spacers (Modell et al., 2017; Nussenzweig et al., 2019), which are recognized and incorporated into the CRISPR locus by the Cas9-Cas1-Cas2-Csn2 supercomplex (Heler et al., 2015; Jakhanwal et al., 2021; Xiao et al., 2017). The number of dsDNA ends for spacer acquisition can be amplified through degradation of these ends by the AddAB nuclease (in Gram-positive bacteria; RecBCD in Gram-negative) (Levy et al., 2015; Modell et al., 2017). Finally, immunity is achieved by the introduction of dsDNA breaks (DSBs) on the phage genome by the RNA-guided nuclease Cas9 (Garneau et al., 2010). Although individual phages harboring target mutations can avoid Cas9 cleavage (Barrangou et al., 2007; Deveau et al., 2008; Gasiunas et al., 2012; Jiang et al., 2013; Jinek et al., 2012), the host population as a whole harbors many different spacer sequences, ensuring the neutralization of the escaper phages and the survival of the bacterial community (Pyenson and Marraffini, 2020; van Houte et al., 2016).
Despite being two of the most well-studied defense systems, that often cohabit in the same host (Oliveira et al., 2014), only two studies explored the possibility of interactions between RM and CRISPR-Cas systems. The first found that in Streptococcus thermophilus with a type II-A CRISPR-Cas locus carrying spacers against phage Φ2972, the propagation of unmodified viruses was further reduced by three orders of magnitude during heterologous expression of the Lactococcus lactis type II RM system LlaDCHI (Dupuis et al., 2013). Southern blots showed cleavage of the viral genome by both systems, further demonstrating that these defense mechanisms can work together to increase immunity. Using the same experimental system, the second study revealed that infection of naïve bacteria; i.e., without targeting spacers, with a mixed population of methylated and unmethylated phage increased the numbers of CRISPR-resistant colonies, with a proportional correlation to the amount of unmodified Φ2972 (Hynes et al., 2014). This result suggested that the inactivation of the viral lytic cycle through restriction prevents the irreversible damage of the host cell and at the same time enables the acquisition of new spacers from the inactivated phage.
Using Staphylococcus aureus as hosts for the Streptococcus pyogenes type II-A CRISPR-Cas locus, we show that both the native SauI (type I) and the heterologous BglII (type II, from Bacillus globigii) RM systems provide a short-lived defense against the unmodified staphylococcal phages ΦNM4γ4 and Φ12ρ1. Although temporary and weak, restriction stimulates a robust type II-A CRISPR-Cas immune response through the acquisition of new spacers from the cleavage site on the phage genome. We also show that infection with a mutant phage incapable of replication is not sufficient to promote robust spacer acquisition. Our results reveal the molecular mechanisms connecting RM and CRISPR systems to show that the manner in which restriction attacks the phage is as important as the inactivation of the viral lytic cycle to spare the host.
Results
The SauI RM system provides temporary defense against phage
Our lab has characterized spacer acquisition by the Streptococcus pyogenes SF370 type II-A system through plasmid expression (pCRISPR) in Staphylococcus aureus RN4220 (Heler et al., 2015). This strain harbors the SauI (type I) RM system encoding hsdM (methyltransferase), hsdS (sequence specificity factor), and hsdR (restriction endonuclease) genes (Waldron and Lindsay, 2006). Two complexes with different subunit stoichiometries and activities are formed: M2S1, which acts as a methyltransferase capable of modifying a specific sequence defined by its S subunit (5’-CCAYN6TGT-3’ and 5’-ATCN5CCT-3’ (Roberts et al., 2013)), and R2M2S1, which initiates DNA translocation upon binding to unmethylated sites (Loenen et al., 2014), cleaving the DNA at a variable distance from the recognition site. However, S. aureus RN4220 contains a single nucleotide mutation in the hsdR gene that results in a premature stop codon (Nair et al., 2011), thus preventing type I DNA restriction in this strain.
To investigate the relationship between RM and CRISPR at the molecular level, we restored type I restriction by cloning an intact copy of the hsdR gene into the plasmid pLZ12 (Perez-Casal et al., 1991), and introduced the resulting plasmid, pSauI, into RN4220 cells. We tested restriction of the staphylococcal phage ΦNM4γ4 (Goldberg et al., 2014) (which contains 26 SauI recognition sites) after propagating it on strain sPM02, a hsdS1/hsdM1 and hsdS2/hsdM2 double mutant incapable of methylating the phage DNA (Fig. S1A,B). Enumeration of plaque-forming units (PFUs) revealed that phage propagation was reduced by two orders of magnitude on RN4220/pSauI cells when compared to propagation on RN4220/pLZ12 staphylococci or to the propagation of the methylated phage. (Fig. 1A). This relatively limited defense is usually attributed to the rise of phages with modified genomes that can avoid DNA cleavage and lyse the host (Bickle and Kruger, 1993). To test whether this is the case for the SauI RM system, we treated S. aureus RN4220/pSauI and RN4220/pLZ12 with unmodified ΦNM4γ4 at a multiplicity of infection (MOI) equal to 10 and measured the OD600 of the cultures to monitor their growth. Staphylococci lacking restriction (RN4220/pLZ12, with functional methylation) succumbed to the virus and were unable to grow. In contrast, cells equipped with SauI (RN4220/pSauI) were initially resistant to infection and displayed similar growth to uninfected cultures, but eventually lysed at about four hours after the addition of phage (Fig. 1B). To determine how phages escaped restriction, we collected the supernatants and confirmed that they contained viral particles fully resistant to restriction (Fig. 1C). However, when these phages were grown in strain sPM02, incapable of modifying DNA, they became fully sensitive to restriction again (Fig. 1C), a result that implicates DNA modification (an epigenetic but not genetic change) in the rapid escape of ΦNM4γ4 from restriction. We observed growth at the end of the experiment for the pSauI cultures and therefore decided to test whether the cells carried mutations that confer phage resistance, such as receptor mutations (Bae et al., 2006). We plated the culture, selected 10 colonies, and tested their susceptibility to infection with unmethylated phage (Fig. S1C). ΦNM4γ4 was able to form plaques on lawns of all ten cultures derived from the colonies, with very similar efficiency of plaquing to that observed on a lawn of RN4220/pSauI. These results demonstrate that the recovery is not due to receptor or any other inheritable mutations. Moreover, no regrowth was observed at MOI 250 (Figs S1D,E), a result that suggests the presence of a small fraction of uninfected cells at MOI 10 that were able to regrow at the end of the experiment. Altogether, our experiments show that the SauI RM system provides only temporary protection to staphylococci due to the rise of modified phage progeny that can overcome restriction and lyse the cultures.
Figure 1. SauI restriction of ΦNM4γ4 promotes the type II CRISPR-Cas response in staphylococci.
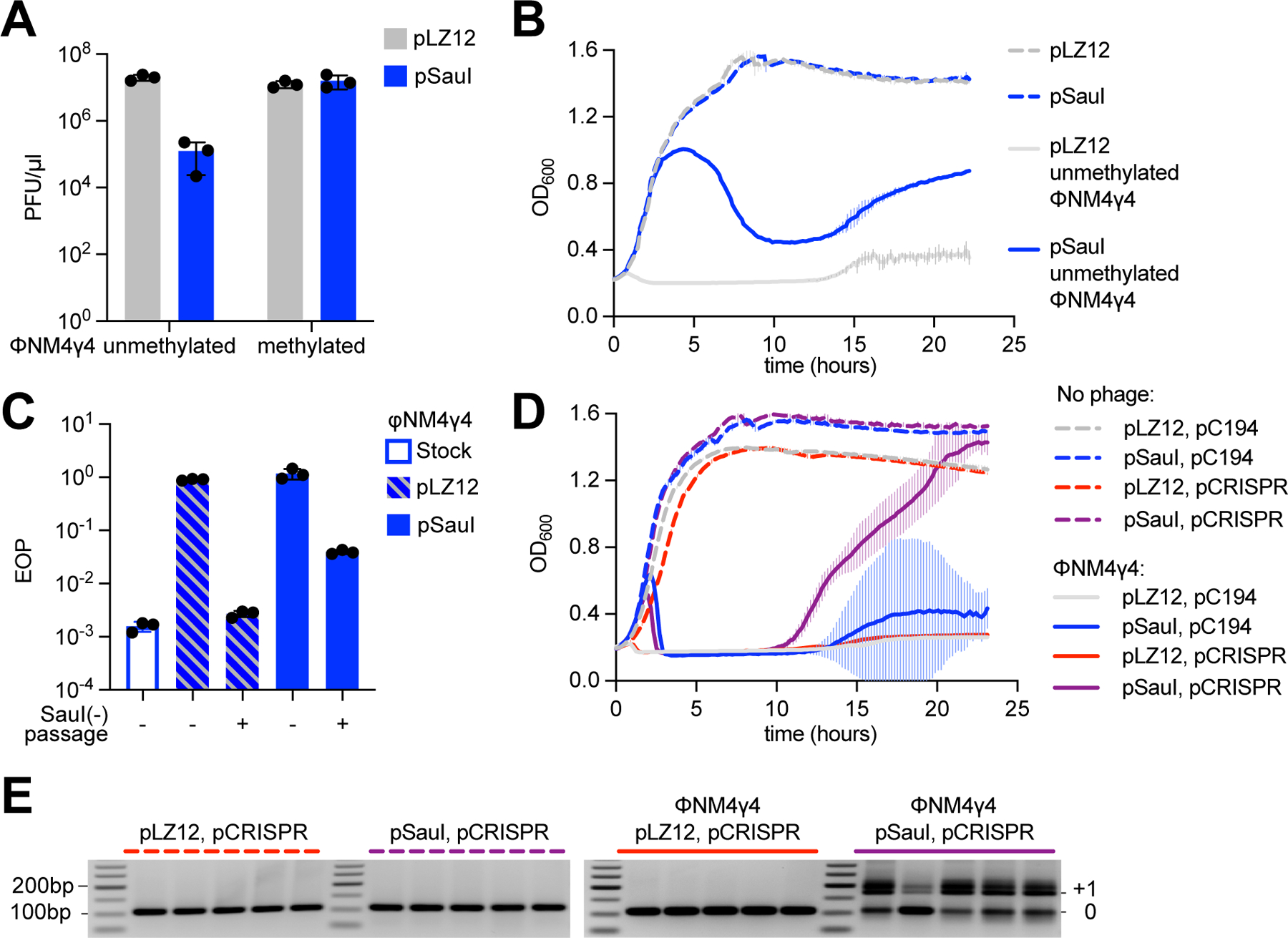
(A) Enumeration of ΦNM4γ4 PFU on lawns of staphylococci expressing SauI or carrying a vector control. Mean of three biological replicates ± SD are reported. (B) Growth of staphylococci expressing SauI or carrying an empty vector control in the presence or absence of ΦNM4γ4 infection, measured as the OD600 of the cultures over time. MOI ~10. Mean of three biological replicates ± SD are reported. (C) EOP of a phage stock, or of phages obtained at the end of the growth curve shown in (B), amplified or not through the non-methylating strain sPM02, after plating on lawns of staphylococci expressing SauI, relative to PFUs obtained after plating on lawns of cells carrying an empty vector control. Mean of three biological replicates ± SD are reported. (D) Same as (B), but using strains carrying an additional pCRISPR plasmid. MOI ~250. Mean of five biological replicates ± SD are reported. (E) Agarose gel electrophoresis of PCR products obtained after amplification of the CRISPR array using DNA obtained from the cultures shown in (D). See also Figure S1.
SauI restriction increases spacer acquisition during the type II-A CRISPR-Cas response
Previous work showed that phage restriction increased the number of colonies that survive infection via type II-A CRISPR immunity in S. thermophilus (Hynes et al., 2014). We decided to determine whether this is also the case in our S. aureus system. To do this, we eliminated all sequences but a single repeat of the type II-A locus CRISPR array in the pCRISPR plasmid (Fig. S1F), for two reasons. First, we wanted to prevent any confounding effects of the phenomenon known as priming, in which previously acquired spacers enhance the acquisition of new ones (Nussenzweig et al., 2019). Second, the lack of pre-existing spacers produces a stronger transcription repression of the type II locus (Workman et al., 2021), leading to a low rate of acquisition that would make more evident any enhancement of the process. We infected S. aureus RN4220/pCRISPR/pSauI as well as S. aureus RN4220/pCRISPR/pLZ12 cultures (five independent replicates) with unmodified ΦNM4γ4, at high MOI (see below) and we monitored their growth over time (Fig. 1D). Neither type II-A CRISPR immunity nor SauI alone were sufficient to enable survival and the cultures rapidly succumbed to infection. In contrast, the combination of CRISPR and SauI restriction allowed an initial growth that was followed by the collapse of the cultures and the eventual recovery of the infected staphylococci. To test whether CRISPR immunity mediated this recovery, we amplified the CRISPR array of each replicate culture (Fig. 1E). All the cultures harboring pSauI, but not those carrying the pLZ12 control, showed integration of one or two new spacers into the CRISPR locus. Amplification of pCRISPR isolated from 50 individual colonies followed by Sanger sequencing of the PCR products showed that 48/50 harbored an expanded array (Fig. S1G), and that the new spacer sequences matched the ΦNM4γ4 genome (Data S1). Similar results were obtained after infection at a lower MOI, 10 (Figs. S1H–J and Data S1). Our data not only corroborate previous results in S. thermophilus, but also suggest a dynamic in which restriction provides a first, short-lived, line of defense that is quickly bypassed by the modification of phage DNA and that also stimulates spacer acquisition by the type II-A CRISPR-Cas immune response to enable the survival of the infected population.
SauI inactivation of the ΦNM4γ4 lytic cycle is not sufficient to enhance spacer acquisition
The previous work in S. thermophilus suggested that defective phages could drive spacer acquisition by type II-A CRISPR systems (Hynes et al., 2014). It was hypothesized that restriction of the phage genome would prevent the completion of the lytic cycle and the death of the host, allowing for the process of spacer acquisition to occur and thus leading to the observed increase in phage-resistant colonies. We decided to test this hypothesis using our experimental system through the engineering of a phage that, similar to the conditions of infection in the presence of restriction, could inject its genome but fail at mounting a lytic cycle. To do this, we deleted the dnaC gene from phage ΦNM4γ4, which was shown previously to be essential for DNA replication and lysis in the related phage 80α (Neamah et al., 2017) (Fig. S2A,B). ΦNM4γ4-ΔdnaC was unfit to propagate in S. aureus, but otherwise able to form plaques on (Fig. 2A) and limit the growth of (Fig. 2B) cultures expressing dnaC from a plasmid, pDnaC. To quantify viral replication directly, we performed quantitative PCR (qPCR) to measure the relative amounts of phage DNA at 10 and 30 minutes after infection of RN4220/pLZ12 cells with unmethylated ΦNM4γ4 or ΦNM4γ4-ΔdnaC (Fig. 2C). While the qPCR value for the wild-type genome showed a 15-fold increase from 10 to 30 minutes, the value for the mutant genome remained low, confirming its inability to carry DNA replication. To determine if the effect of the ΔdnaC mutation is comparable to that of restriction, we performed the same experiment using RN4220/pSauI cells. We found that in the presence of SauI activity, the levels of phage DNA were equivalent to those for the ΦNM4γ4-ΔdnaC phage in the absence of restriction (Fig. 2D, compare to Fig. 2C), suggesting that the replication of the mutant virus is similar to that of the wild-type phage in the presence of restriction. Finally, we tested whether infection with the defective ΦNM4γ4-ΔdnaC phage could lead to an increase in spacer acquisition like the one observed for SauI restriction of the wild-type phage. To do this we infected RN4220/pCRISPR/pSauI and RN4220/pCRISPR/pLZ12 cultures with unmethylated ΦNM4γ4 or ΦNM4γ4-ΔdnaC and collected cells after 30 minutes to extract plasmid DNA. CRISPR loci within the pCRISPR plasmids were amplified and the PCR products were subjected to next-generation sequencing (NGS) to obtain the sequences and relative abundance of the acquired spacers (Fig. 2E and Data S1). Since the lytic cycle of ΦNM4γ4 takes 40–50 minutes (Fig. S2C), sample collection at 30 minutes prevents both the depletion of cells that acquire spacers that mediate poor CRISPR immunity and succumb to phage infection, as well as the positive selection of cells containing spacers with high efficiency of targeting. As expected from our previous results, in the presence of SauI activity >70% of the new spacers matched the viral genome, after infection with both phages (Fig. 2E, Fig. S2D, and Data S1). In the absence of restriction, this fraction decreased to ~1%. In this case, the great majority were derived from the pCRISPR plasmid (however these spacers do not significantly affect plasmid stability over time (Heler et al., 2017)), not only after infection with the wild-type phage, but also during infection with the non-replicating ΦNM4γ4-ΔdnaC. This result demonstrates that not any defective phage can drive spacer acquisition and suggests that specific events that occur during SauI restriction, beyond halting the progression of the viral lytic cycle, are important for the generation of new spacers.
Figure 2. Inactivation of the viral lytic cycle is not sufficient to promote CRISPR immunity.
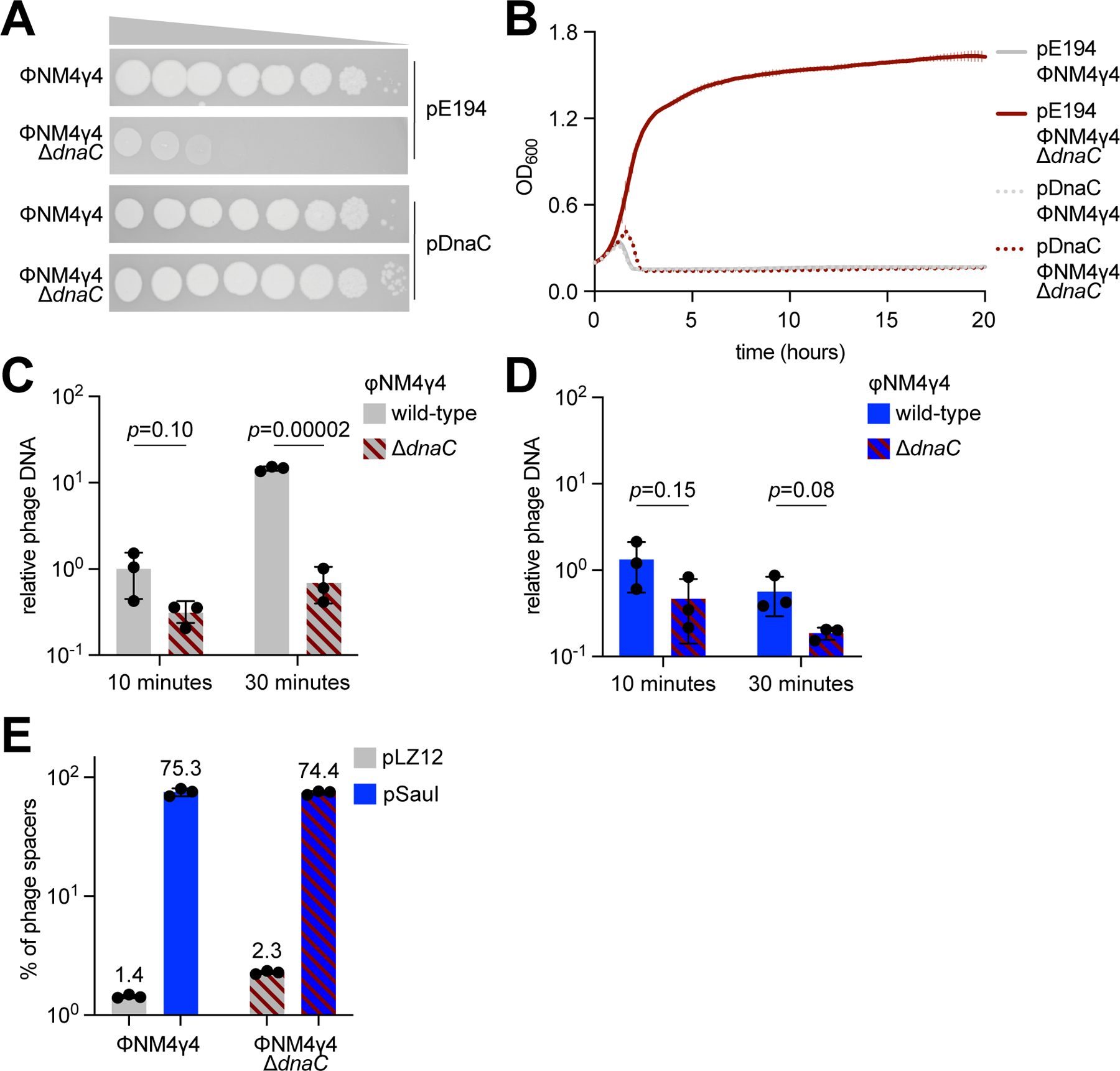
(A) Detection of plaque formation after spotting 10-fold serial dilutions of ΦNM4γ4 or ΦNM4γ4-ΔdnaC phages on top agar plates seeded with S. aureus expressing DnaC or carrying an empty vector control. (B) Growth of staphylococci expressing DnaC or carrying a vector control after infection with ΦNM4γ4 or ΦNM4γ4-ΔdnaC phages, measured as the OD600 of the cultures over time. MOI ~1. Mean of three biological replicates ± SD are reported. (C) Phage DNA quantification via qPCR, relative to host DNA, 10 and 30 minutes post-infection of S. aureus harboring pCRISPR in the absence of restriction with ΦNM4γ4 and ΦNM4γ4-ΔdnaC. MOI ~0.1. Mean of three biological replicates ± SD are reported. (D) Same as (C) but following infection of staphylococci expressing SauI. (E) Quantification of phage-derived spacers, relative to total new spacers, acquired 30 minutes after infection of staphylococci harboring pCRISPR and expressing SauI or carrying a vector control with ΦNM4γ4 or ΦNM4γ4-ΔdnaC phages, via NGS of the CRISPR locus. MOI ~250. Mean of three biological replicates ± SD are reported. See also Figure S2.
BglII restriction sites are hotspots of spacer acquisition
A unique characteristic of restriction is the generation of free DNA ends in the phage genome (Tock and Dryden, 2005), which we previously reported are substrates for the new spacers acquired by type II-A systems. As a consequence of this, both cos sites (Modell et al., 2017) and Cas9 cleavage targets (Nussenzweig et al., 2019) become hotspots of spacer acquisition within the viral genome. To determine if new spacers originate from SauI cleavage sites, we mapped the spacer sequences obtained from the NGS experiments of Figure 2E to the ΦNM4γ4 genome (Fig. S2E and Data S1). However, even after infection at a very high MOI of 250, which should markedly increase the frequency of spacer acquisition (Modell et al, 2017), the graphs did not reveal any notable correlation between the new spacer sequences and the SauI recognition sites. We hypothesized that the difficulty to interpret these data was due to both the large number of SauI sites (twenty-six, Fig. S2F) as well as the random cleavage of this type I restriction enzyme (Loenen et al., 2014).
Therefore, we decided to use a type II RM, which would cleave the viral genome at a few and defined sequences. We cloned the BglII RM system from B. globigii on a modified pLZ12 vector (carrying mutations that eliminate a BglII site, see Methods), obtaining pBglII. This system is composed of three genes, bglIIC, bglIIM, and bglIIR, coding for a controller protein (C.BglII), a methyltransferase (M.BglII), and a restriction enzyme (R.BglII), respectively, and specifically recognizes and cleaves 5’-A↓GATCT-3’ sequences that contain non-methylated cytosines (Duncan et al., 1978). The ΦNM4γ4 genome contains a single BglII site (Fig. 3A), which we named site “A”, and it is susceptible to cleavage in vitro (Fig. S3A,B). In vivo, we looked at the propagation of ΦNM4γ4 in lawns of RN4220/pBglII or RN4220/pLZ12, and found that BglII restriction reduced the PFU count by approximately one order of magnitude (Fig. S3C). In liquid cultures treated with ΦNM4γ4, cells expressing BglII continued growing for ~ 30 minutes longer than those harboring the vector control, but ultimately succumbed to infection (Fig. S3D), presumably due to the rise of methylated phage. As a control, we engineered a mutant virus in which the “A” site was eliminated through the introduction of silent mutations (to TGACTT), ΦNM4γ4-ΔBglII (Fig. 3A). The genomic DNA of this phage was not cleaved in vitro (Fig. S3A,B), the PFU count was not reduced in lawns of RN4220/pBglII when compared to RN4220/pLZ12 (Fig. S3C), and the expression of BglII did not prolong the growth of liquid cultures (Fig. S3D).
Figure 3. BglII promotes CRISPR spacer acquisition at the restriction site.
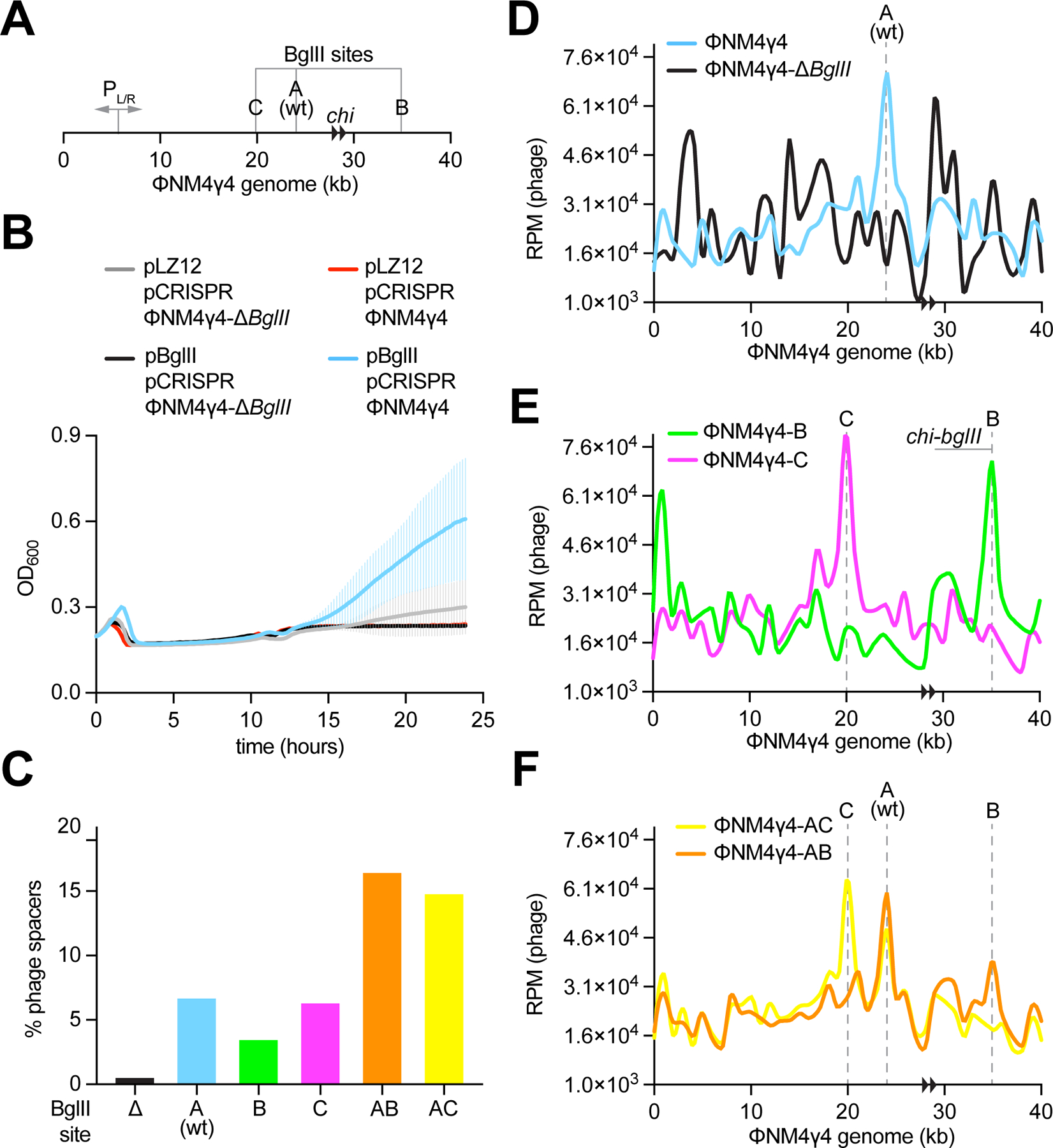
(A) Schematic representation of the ΦNM4γ4 genome showing the BglII sites analyzed in this study, its two chi sites (black arrowheads) and the PL/R bidirectional promoter. (B) Growth of staphylococci harboring pCRISPR and expressing BglII or carrying a vector control after infection with ΦNM4γ4 or ΦNM4γ4-ΔBglII phages, measured as the OD600 of the cultures over time. MOI ~10. Mean of five biological replicates ± SD are reported. (C) Quantification of phage-derived spacers, relative to total new spacers, acquired 30 minutes after infection of staphylococci harboring pCRISPR and expressing BglII or carrying a vector control with ΦNM4γ4 phages containing different BglII sites, via NGS of the CRISPR locus. MOI ~25. (D) Distribution of spacer abundance (measured as RPM of phage-matching reads) obtained in (C) across the ΦNM4γ4 genome, using data from wild-type and ΔBglII infections. (E) Same as (D) using data from ΦNM4γ4-B and ΦNM4γ4-C infections. (F) Same as (D) using data from ΦNM4γ4-AB and ΦNM4γ4-AC infections. See also Figure S3.
After establishing a functional BglII RM system in staphylococci, we tested whether it could synergize with the type II-A CRISPR-Cas response as was the case for SauI. First, we infected RN4220/pCRISPR/pBglII or RN4220/pCRISPR/pLZ12 cultures with ΦNM4γ4 or ΦNM4γ4-ΔBglII and followed their growth over time (Fig. 3B). CRISPR immunity was able to promote the regrowth of cells infected with wild-type phage, but not ΦNM4γ4-ΔBglII, only when they expressed BglII. PCR amplification of the CRISPR array at the end of the experiment detected acquisition of new spacers for all the replicates of the cultures carrying pBglII but not for those harboring the pLZ12 control or infected with the unrestricted ΦNM4γ4-ΔBglII phage (Fig. S3E). These results confirmed that, similar to SauI restriction, the heterologous expression of the BglII RM system can promote the type II-A CRISPR-Cas immune response in staphylococci.
Next, we used the heterologous BglII RM system to test our hypothesis that the free DNA ends generated by restriction are substrates for spacer acquisition. To do this, we infected RN4220/pCRISPR/pBglII cells with ΦNM4γ4 or ΦNM4γ4-ΔBglII and extracted plasmid DNA for the amplification of the CRISPR array, 30 minutes post-infection. The PCR products were subjected to NGS to capture the sequence and relative abundance of the new spacers (all NGS data presented is compiled in Data S1). Using this data, we first determined the fraction of reads corresponding to spacers acquired from the viral DNA (Fig. 3C). Consistent with our previous results, BglII restriction increased this value by ~ 10-fold, from 0.5 % during ΦNM4γ4-ΔBglII infections, to 6.7 % in experiments using wild-type ΦNM4γ4. We also mapped these reads along the phage genome to obtain the pattern of spacer acquisition (Fig. 3D). We detected a hotspot of spacer acquisition at the BglII cleavage site A in ΦNM4γ4, which was absent for ΦNM4γ4-ΔBglII. To determine if this result was specific to this BglII site, we engineered phages lacking site A but having sites B and C (Fig. 3A). Both engineered sites were cleaved in vitro by the commercial restriction enzyme (Fig. S3A,B), reduced plaque formation to a similar extent as site A in the wild-type phage (Fig. S3C), and increased the fraction of spacers acquired from the viral genome during the type II-A CRISPR response by approximately an order of magnitude (Fig. 3C). Most importantly, analysis of the spacer acquisition map of these phages showed hotspots centered at the new sites, as well as the elimination of the peak in site A (Fig. 3E). Finally, we wanted to investigate the effect of multiple BglII sites and therefore we created phages with sites A and B, or A and C (Fig. 3A). Both viral genomes were restricted in vitro at both sites (Fig. S3A,B). In vivo, the additional BglII cleavage did not reduce plaque formation further than any of the single sites (Fig. S3C). In contrast, the fraction of spacers acquired from the viral genome during type II-A CRISPR immunity was significantly increased to ~15% (Fig. 3C). Note that this value is still considerably lower than what we detected for the SauI RM system (~70%, Fig. 2E) and is most likely due to the presence of a much larger number of SauI target sites (twenty-six, Fig. S2F) in the ΦNM4γ4 genome. More important, the spacer acquisition pattern of the AB and AC phages showed hotspots centered at both BglII sites (Fig. 3F). Altogether, these experiments demonstrate that restriction of ΦNM4γ4 by BglII promotes the acquisition of new spacers at the cleavage site, and suggest that the dsDNA ends generated by this RM system are substrates for the spacer integration complex of type II-A CRISPR-Cas systems.
AddAB amplifies spacer acquisition from the BglII restriction sites
Previously, we showed that the DNA repair complex AddAB, which is the main nuclease that processes free dsDNA ends in Gram-positive bacteria (Wigley, 2013), is responsible for the amplification of spacer acquisition from the cos site of the staphylococcal phage Φ12γ3 (Modell et al., 2017). Cos phages translocate a specific free dsDNA end during every infection (Hershey et al., 1963), which is thought to be processed by AddAB to generate the DNA substrates for the acquired spacers. This processing is limited by chi sites, a 7 base pair sequence (5’-GAAGCGG-3’ for S. aureus) that stops DNA degradation by AddAB (Halpern et al., 2007), and therefore it creates a hotspot of spacer acquisition between the cos site and the first upstream chi site (Modell et al., 2017). In contrast to Φ12γ3, ΦNM4γ4 is a pac phage that, due to its headful packaging mechanism (Casjens and Gilcrease, 2009), injects a variable dsDNA end of its genome during each infection event. Therefore, a cos-chi hotspot is not detected for this phage (Modell et al., 2017). To determine whether and how AddAB affects spacer acquisition from the restriction site, we decided to analyze the spacers acquired by the type II-A CRISPR-Cas system upon BglII restriction of the cos phage Φ12γ3, and take advantage of the chi-cos hotspot of spacer acquisition observed after infection with this phage as an internal control. To do this, we removed from Φ12γ3 (through the introduction of silent mutations) three BglII recognition sequences residing within the chi-cos hotspot, and three chi sites located in close proximity upstream of the remaining BglII restriction sequence, generating phage Φ12ρ1 (Fig. 4A). BglII was able to cleave the genomic DNA of this phage in vitro (Fig. S4A,B) and also mediated a reduction in plaque formation (Fig. S4C). To determine the pattern of spacer acquisition, we infected RN4220/pCRISPR or RN4220/pCRISPR/pBglII cultures with Φ12ρ1 and performed NGS experiments. As was the case for infections with ΦNM4γ4, restriction increased the fraction of viral-derived spacers approximately 4-fold (Fig. 4B). After mapping the reads obtained from the control cells (without BglII restriction) to the Φ12ρ1 genome, we observed the pattern that we described previously for Φ12γ3 (Modell et al., 2017), with the cos-chi spacer acquisition hotspot (Fig. 4C). As reported before (Modell et al., 2017), a strong peak around 10 kb, immediately downstream of the chi site, was observed. Although we do not understand the factors that lead to the generation of this peak, given that it is the result of one spacer with an unusually high number of reads (5’-TCGCCGTATGTGTAGTGCGC-3’, 10 to 100 fold higher than the rest of the spacer reads in that bin; Data S1), we suspect that this sequence is preferred for binding and/or integration by the Cas1/Cas2 integrase complex (Xiao et al., 2017). In the presence of BglII, a new hotspot of spacer acquisition appeared, encompassing the restriction site and the region immediately upstream limited by the nearest chi sequence (Fig. 4D). The marked reduction of acquisition beyond (upstream of) the chi site suggests the involvement of AddAB in this process; i.e., degrading the free dsDNA ends generated after BglII restriction. This was confirmed after repeating the experiment with S. aureus JW418 hosts, a derivative of RN4220 carrying a mutation in the addA gene that inactivates the nuclease activity of the AddAB complex (Modell et al., 2017). In this genetic background, the pattern of spacer acquisition displayed sharp peaks; i.e., not extended, at the cos and BglII sites (Fig. 4E). In addition, the distribution of new spacers displayed a very low level of acquisition from the rest of the genome outside of the main peaks, as well as peaks at ~4, 8, and 42 kb of unknown origin. Interestingly, during infection of wild-type hosts, spacer acquisition from the chi-cos area seemed to be diminished at the expense of the chi-bglII hotspot. We believe this to be the consequence of the rapid circularization of the Φ12ρ1 genome at the cos site, which eliminates the dsDNA ends for the spacer acquisition machinery, making this process infrequent. In contrast, approximately 90 % of the phages are cut by BglII, an estimation based on the decrease in PFUs caused by this restriction nuclease (Fig. S4C), greatly elevating the chances of spacer acquisition from the cleavage site.
Figure 4. AddAB nuclease activity amplifies the region of spacer acquisition.
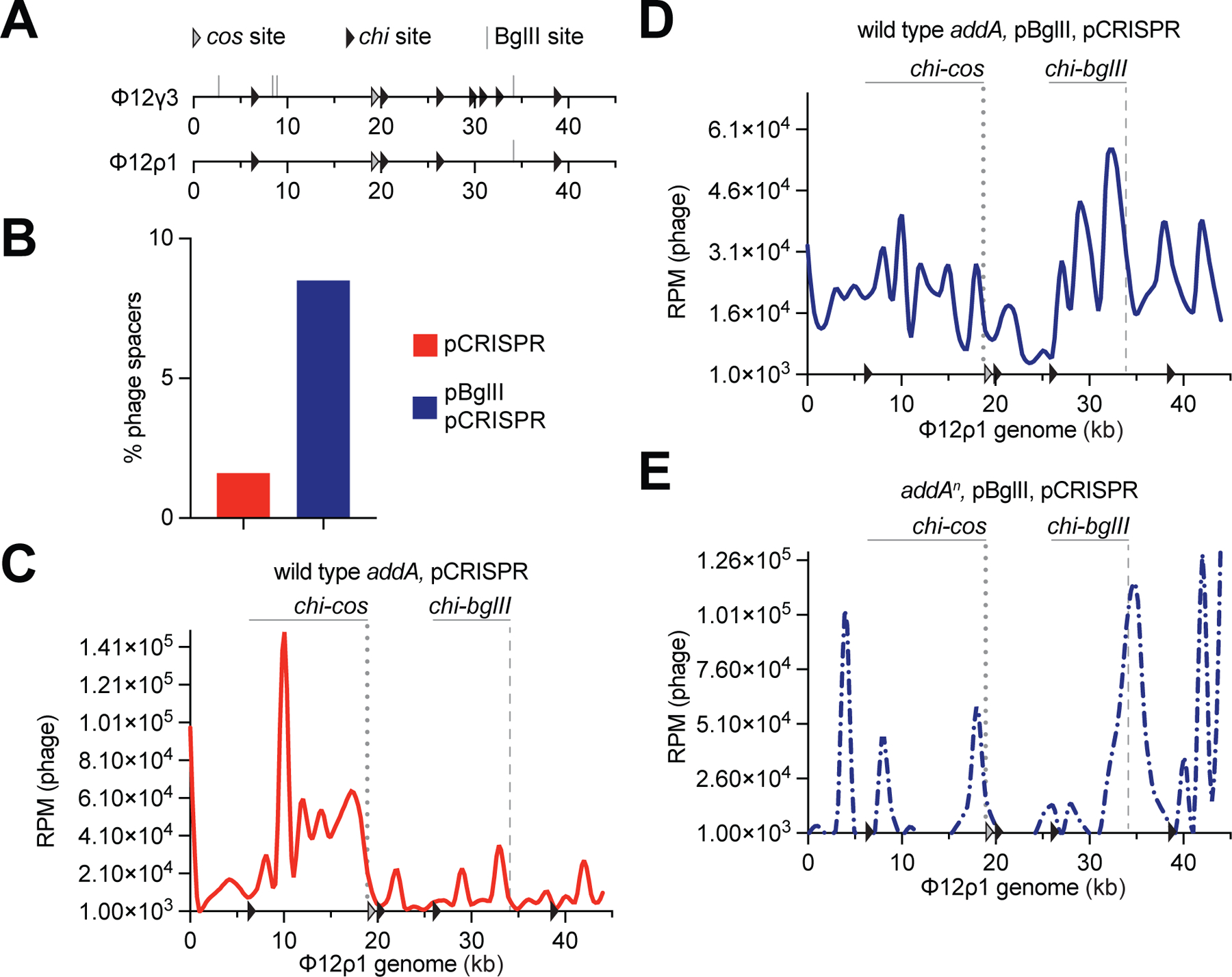
(A) Schematic representation of the Φ12γ3 and Φ12ρ1 genomes showing the BglII sites analyzed in this study (grey lines), their chi sites (black arrowheads) and the cos site (white arrowhead). (B) Quantification of phage-derived spacers, relative to total new spacers, acquired 30 minutes after infection of staphylococci harboring pCRISPR in the presence or absence of BglII expression with Φ12ρ1, via NGS of the CRISPR locus. MOI ~25. (C) Distribution of spacer abundance (measured as RPM of phage-matching reads) obtained in (B) across the Φ12ρ1 genome, using data from infection of cells not expressing BglII. (D) Same as (C) using data from infection of cells carrying pBglII. (E) Same as (D) using data from infection of addAn mutant staphylococci. See also Figure S4.
While ΦNM4γ4 does not have a cos sequence, it contains two contiguous chi sites (Fig. 3A). Both of them are downstream of BglII sites A and C, and in an orientation that will be recognized by an AddAB nuclease complex starting degradation at the upstream restriction site, approximately 35 kb away (once the phage’s genome circularize). We believe that because of this distance, we cannot discern whether we have amplification of acquisition from site A and C extending to the chi sites. BglII site B, on the other hand, is located only ~10 kb downstream of the chi sites, which should limit the AddAB upstream activity that originates at the DSB generated at this restriction site. This short distance creates a hotspot of spacer acquisition in the ΦNM4γ4-B genome delimited by the BglII-B site and these two chi sites (Fig. 3E). AddAB degradation of the DNA downstream of the DSB generated by restriction is not antagonized by the chi sites and therefore can produce only a general increase of spacer acquisition, but not generate hotspots. To directly test the involvement of AddAB in spacer acquisition, we repeated the experiment using the addA mutant host S. aureus JW418. We found that the pattern of spacer acquisition after infection of JW418/pCRISPR/pBglII cells with ΦNM4γ4-B contained a single peak at the BglII recognition site, which was narrow and not as extended to the upstream chi sites (Fig. S4D), with low levels of acquisition from the rest of the viral DNA. A similar result was obtained for the BglII site A when we infected AddA mutant cells with wild-type ΦNM4γ4 (Fig. S4E). Therefore, absence of AddAB activity eliminated both background as well as the minor hotspots of spacer acquisition and revealed only one strong and sharp hotspot at the BglII cut site. Altogether, these experiments demonstrate that AddAB is involved in the amplification of spacer acquisition that starts at the BglII restriction site, allowing it to expand to other regions of the viral genome.
BglII defense is rapidly overcome by phage DNA methylation
Our results suggest that restriction is a first, vulnerable, line of defense against phage infection that can stimulate CRISPR immunity as a second, more reliable, response. Interestingly, cultures that combine restriction (either SauI or BglII) and type II-A CRISPR immunity to fight against phage infection at a high multiplicity of infection (MOI), collapse 2–5 hours after infection and recover at 10–15 hours (Figs. 1D and 3B), integrating new spacers as early as 30 minutes post-infection (Figs. 2E and 3C). To investigate the dynamics of the interplay between restriction and spacer acquisition by the type II-A CRISPR system, we infected RN4220/pCRISPR/pBglII or RN4220/pCRISPR/pLZ12 cultures with ΦNM4γ4 at an MOI of 1. We decreased the MOI to induce a smoother transition in the timing of phage modification and of the expansion of the CRISPR array, in order to avoid abrupt changes caused by a very high MOI that could prevent us from observing the dynamics of the process we want to study. We took 16 samples for a period of 24 hours and measured the CFU, PFU, DNA methylation, and spacer acquisition. The CFU count reflected the growth curves we previously observed, upon infection at a higher MOI (Fig. S5A), with a rapid collapse and later recovery in the absence of restriction, and delayed lysis and earlier regrowth of cells expressing BglII. Expectedly, the total number of phage in the culture supernatants followed the opposite trend (Fig. S5B), with a decrease in PFU over time, followed by a steady accumulation of phage. Viral titers are approximately two orders of magnitude higher in cultures carrying BglII, most likely a result of the partial survival of staphylococci due to restriction earlier during infection, which increased the number of hosts for the subsequent infection by methylated phages. As noted before, we hypothesized that this higher PFU number could be attributed to the evasion of BglII restriction through methylation of the phage DNA. To test this, we plated the culture supernatants on lawns of staphylococci expressing the BglII RM system (Fig. S5C) and calculated the fraction of total phage resistant to restriction (Fig. 5A). We observed that approximately 90 % of the phage from RN4220/pCRISPR/pLZ12 cultures remained sensitive to BglII restriction over the 24-hour period. In contrast, ΦNM4γ4 taken from RN4220/pCRISPR/pBglII cultures gained resistance to restriction within an hour of infection. To determine if this is due to DNA methylation, we performed bisulfite sequencing, a method that uses this chemical to convert unmodified, but not methylated, cytosines into uracil residues, which are recognized as thymine in subsequent PCR amplification and NGS (Frommer et al., 1992). We found that whereas the phage DNA isolated from RN4220/pCRISPR/pLZ12 supernatants contained background levels of converted uracil residues, the phage refractory to BglII restriction contained maximum levels of genomic modification after one hour of infection (Fig. 5B). These results demonstrate that ΦNM4γ4 can rapidly evade RM systems through DNA methylation.
Figure 5. Phage DNA methylation and spacer acquisition occur shortly after phage infection.
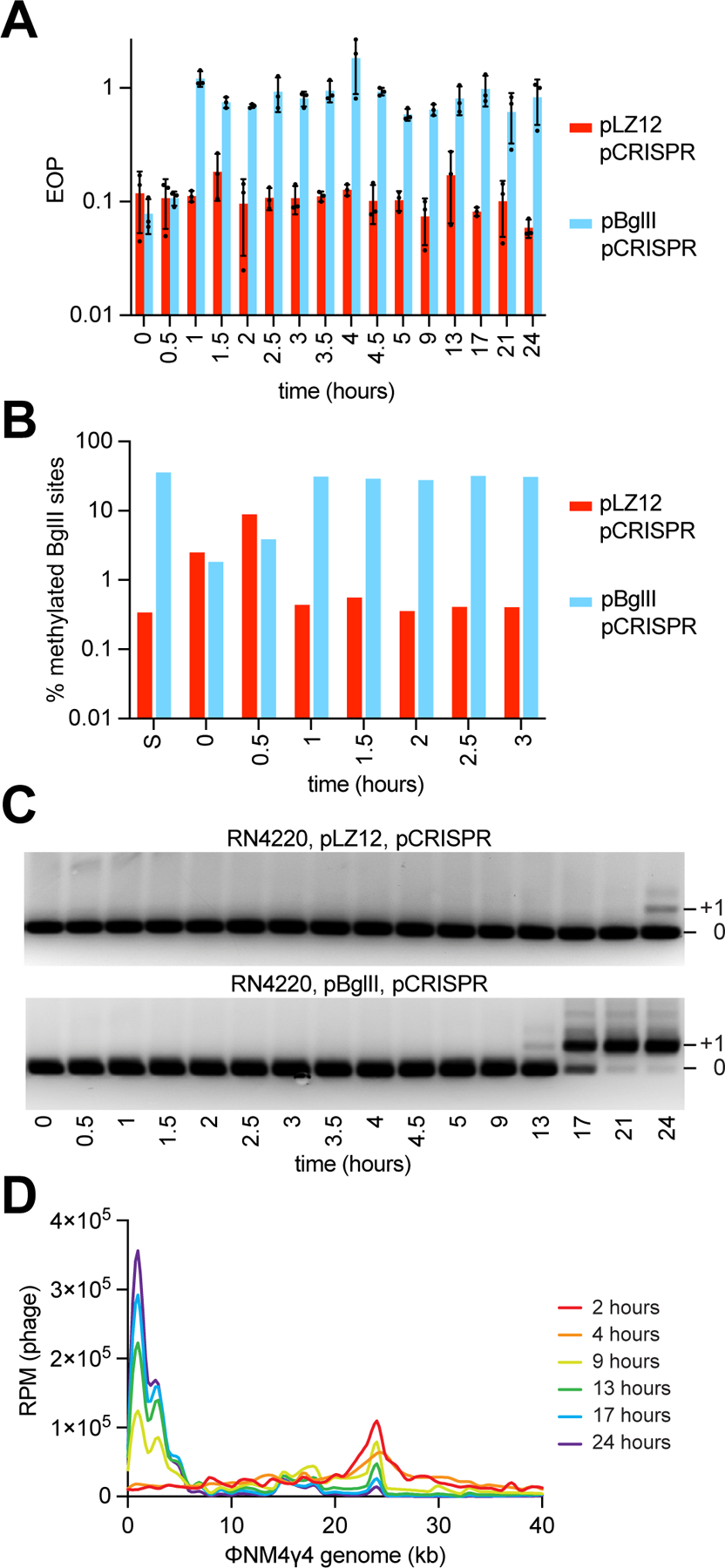
(A) EOP of ΦNM4γ4 phages obtained at different time points following infection (MOI ~1) of S. aureus cells harboring pCRISPR and expressing BglII or carrying a vector control, after plating on lawns of staphylococci expressing BglII, relative to PFUs obtained with cells carrying a vector control. Mean of three technical replicates ± SD are reported. (B) Percent of methylated BglII sites on a unmethylated or methylated ΦNM4γ4 stocks (“S”) and on phages obtained in (A), measured using bisulfite NGS. (C) Agarose gel electrophoresis of PCR products obtained after amplification of the CRISPR array using DNA obtained from the cultures used in (A). (D) Distribution of spacer abundance (measured as RPM of phage-matching reads) obtained after NGS of the CRISPR locus present in the cultures used in (A) across the ΦNM4γ4 genome, using data from the indicated time points. See also Figure S5.
The type II-A CRISPR immune response initially targets the BglII recognition site and expands to attack other regions of the viral genome
Next, we investigated the details of the type II-A CRISPR-Cas immune response during restriction. Amplification of the CRISPR array showed that, in the presence of BglII activity, by 13 hours after infection with ΦNM4γ4, a small fraction of the population harboring new spacers was detected by PCR (Fig. 5C, compare the intensity of the PCR products with and without a new spacer). Accordingly, this is also the time when we observed a rebound in CFU in the culture (Fig. S5A). This fraction increased over the next 8 hours, and at 21 hours post-infection, the great majority of the cells had expanded CRISPR arrays. In contrast, in the absence of restriction, the first sign of spacer acquisition appeared at our last data point, 24 hours post-infection, for only a small proportion of the staphylococci in the RN4220/pCRISPR/pLZ12 culture. This result shows a much less efficient CRISPR-Cas immune response in the absence of restriction, which leads to a limited recovery of the cells in this culture (Fig. S5A). Next, we performed NGS analysis to determine more accurately the timing and levels of spacer acquisition (Fig. S5D). As mentioned above, as early as 30–60 minutes after infection with ΦNM4γ4, the levels of viral spacers acquired in the presence of BglII restriction were an order of magnitude higher than those acquired in the absence of the nuclease. Given that the duration of the ΦNM4γ4 lytic cycle is 40–50 minutes, the NGS data after the first two time points reflects not only spacer acquisition, but also the enrichment of staphylococci harboring anti-phage spacers. Therefore, the fraction of ΦNM4γ4-derived spacers increases steadily, and at 17 hours post-infection all spacers in both populations matched the phage genome. However, the CFU count at this time point is too low for the pLZ12/pCRISPR sample (Fig. S5A) and therefore the expanded CRISPR array cannot be amplified via PCR. We also mapped the new spacers to obtain their distribution across the phage genome over time. Compared to the previous experiment performed at a much higher MOI (250), at MOI 1 the number of spacers acquired 30 and 60 minutes after infection were not sufficient to generate a map (Fig. S5E). At two hours, however, a distinct peak was detected at the BglII site (Fig. 5D). Interestingly, this peak decreased with time, seemingly at the expense of a growing area of spacer acquisition at the 5’ end of the viral genome (Fig. 3A), a region which has been shown before to be a preferred source of new spacers against ΦNM4γ4 (Heler et al., 2019; Modell et al., 2017). This result suggests that, after the initial stimulation of the type II-A CRISPR-Cas response by BglII restriction, the subsequent interplay between additional spacer acquisition and phage selection of the most efficient crRNA guides further shape the distribution of spacer sequences in the bacterial population.
Discussion
Incorporation of our results in a model for CRISPR spacer acquisition
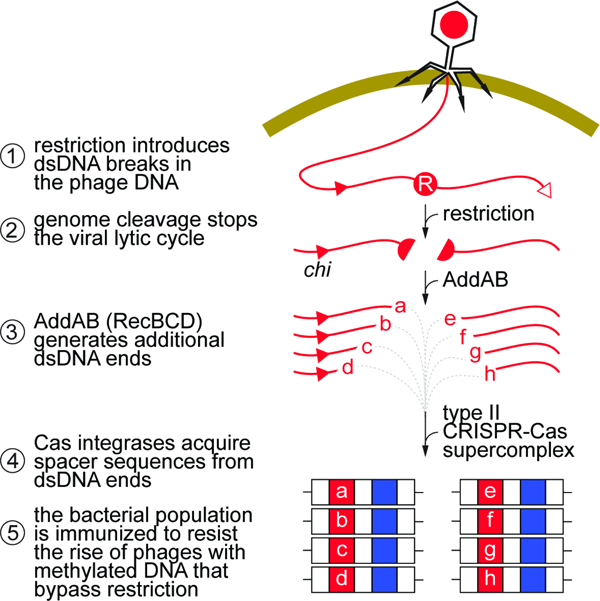
Previous work that studied the relationship between type II-A CRISPR immunity and RM systems found that infections with a mix of modified and unmodified phage, resistant and susceptible to restriction, respectively, increased the levels of CRISPR-resistant survivors in a manner proportional to the levels of restriction-sensitive virus (Hynes et al., 2014). It was hypothesized that the inactivation of the phage would prevent it from damaging the host cell beyond recovery and allow time for the process of spacer acquisition to occur. Here we describe the molecular mechanisms that explain and expand these results. We found that upon infection with unmodified phage, the cleavage of the viral DNA by restriction nucleases provides only a temporary protection that is rapidly overcome by DNA methylation, causing the death of most bacteria in the culture (Fig. 6). Although not efficient enough to ensure survival, RM activity stimulates the type II-A CRISPR-Cas immune response. As early as 30 minutes after infection, restriction promotes spacer acquisition in a small fraction of the cells of the bacterial population. New spacers are extracted predominantly from both ends of the cleavage site, likely due to the creation of the dsDNA ends used by the type II supercomplex as substrates for integration into the CRISPR array (Jakhanwal et al., 2021; Modell et al., 2017; Nussenzweig et al., 2019). The host’s DNA repair machinery (RecBCD in E. coli; AddAB in S. aureus) can further degrade the restricted DNA at the cleavage site (Simmon and Lederberg, 1972). Assuming that these complexes fall off the substrate DNA at some rate, their activity would generate additional free dsDNA ends. As a result, a hotspot of spacer acquisition is generated in the region spanning the restriction site and the first chi site with the appropriate orientation to stop AddAB activity. In turn, the newly acquired spacers direct Cas9 to cleave the viral DNA, an event that not only provides immunity but also generates more free dsDNA ends in other areas of the viral genome that are also used by the spacer acquisition machinery, a process known as priming (Nussenzweig et al., 2019). We believe that over time, priming expands the repertoire of new spacers from an original set targeting the vicinity of the restriction site to a final spacer population matching many other zones of the phage DNA. The fraction of CRISPR-immunized cells (each containing a different spacer) is able to grow in the presence of modified phage, allowing the recovery and survival of the infected culture. Ultimately, escaper phages with target mutations that abrogate Cas9 recognition and/or cleavage will rise. However, these phages do not take over the bacterial population for two reasons. First, the frequency of escape mutations is low, ~10–5 in our experimental system (Pyenson et al., 2017) (much lower than the frequency of escape from RM systems). Second, the population is immunized with many different spacers, ensuring the neutralization of phages that can escape the defense provided by a single spacer (Pyenson and Marraffini, 2020; van Houte et al., 2016). As a result of this, the type II-A CRISPR-Cas immune response provides a more stable defense than restriction. The synergy between RM and CRISPR systems is reminiscent of mammalian immunity, where the activation of pattern recognition receptors (PRRs) during the innate immune response triggers an immediate defense that also activates a second, more robust, highly specific, but temporally delayed, adaptive immunity (Palm and Medzhitov, 2009).
Figure 6. Restriction prevents the death of the host and at the same time provides the substrates for new spacers.
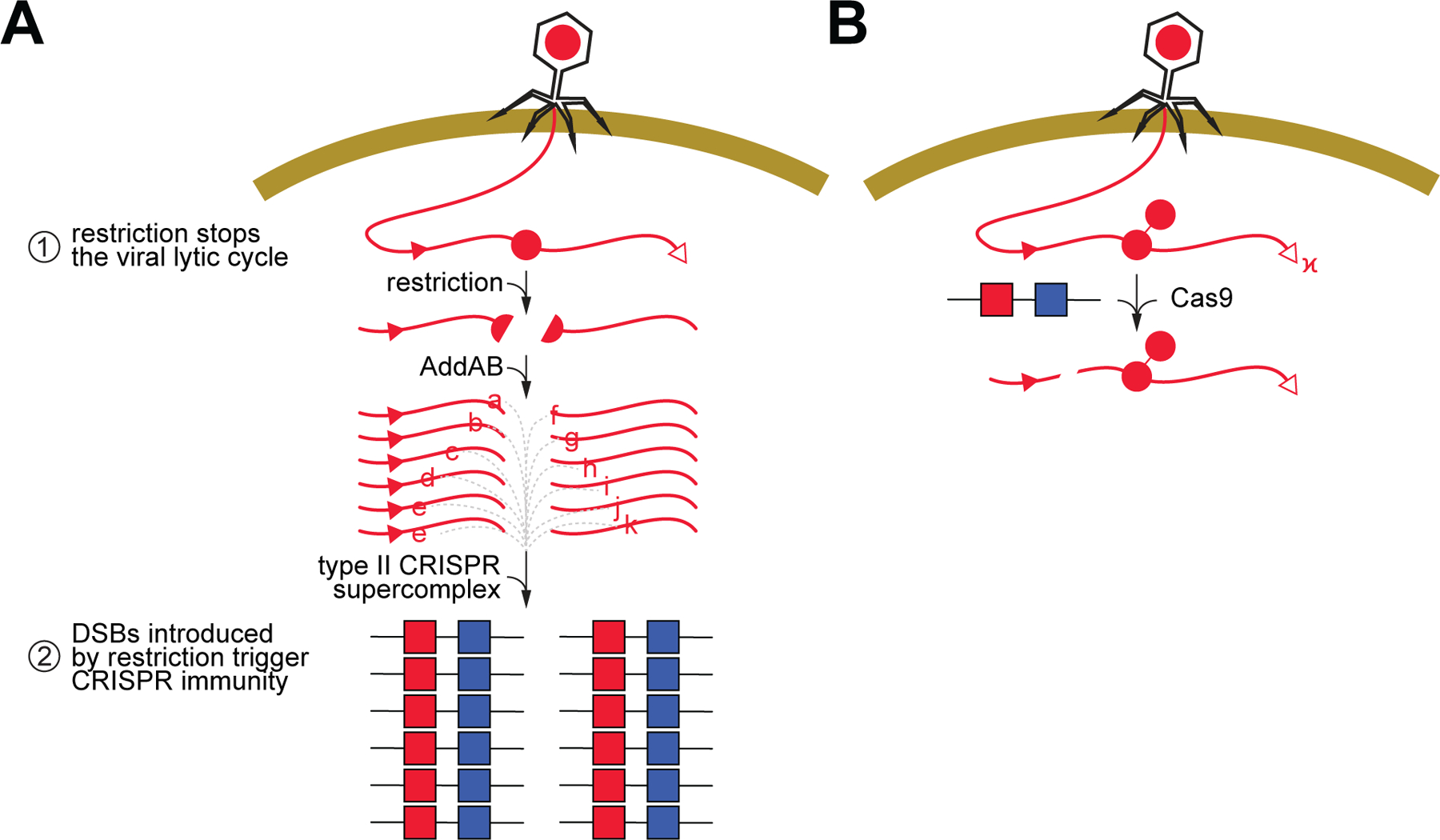
(A) In our model for the synergistic effect of RM and type II-A CRISPR systems, cleavage of the viral DNA at the restriction site (red circle, “R”) shortly after infection has a dual effect: (i) it prevents the completion of the lytic cycle and death of the host, and (ii) generates free dsDNA ends that are processed by AddAB and then used by the type II supercomplex to acquire new spacers (red squares, a, b,…, k). Chi sites (red triangles), if positioned in the correct direction to inhibit AddAB, limit the region of spacer acquisition to a hotspot between the restriction and the chi site. Acquisition from the free dsDNA at the injected cos site is less frequent. (B) When methylation of the restriction site (red circle, “m”) prevents this first line of defense, a subpopulation of bacterial hosts are already immunized with the spacers acquired during restriction, enabling the Cas9 endonuclease to cleave the viral DNA and prevent infection. See also Figure S6.
While, as previously hypothesized (Hynes et al., 2014), the inactivation of the phage via restriction is probably required to prevent its lytic cycle and allow the events required for spacer acquisition to occur, using a non-replicating phage we demonstrated that this inactivation is not sufficient for the increased levels of CRISPR immunization. Instead, how restriction inactivates the phage; i.e., generating free dsDNA ends that are the substrates for the spacer acquisition machinery, is critical for the synergistic relationship between RM and CRISPR systems. Therefore, our results add to a body of work that highlights the requirement of a two-pronged attack on the viral genome, made by different nucleases, for efficient spacer acquisition during the type II CRISPR-Cas immune response. First, nucleases have to halt the phage lytic cycle. This can be mediated by AddAB degradation of the injected dsDNA end of the viral genome (Modell et al., 2017), by Cas9 cleavage during priming (Nussenzweig et al., 2019), or by restriction endonucleases (this work). Second, the product of nuclease activity has to generate free DNA ends, the preferred substrates for the Cas9-Cas1-Cas2-Csn2 spacer integration supercomplex (Jakhanwal et al., 2021). In the case of AddAB, while degrading the injected viral DNA end, the complex could occasionally disengage from its substrate before reaching a chi site, leaving free DNA ends for the spacer acquisition machinery (Modell et al., 2017). In the case of Cas9 cleavage, it generates blunt DNA ends (Gasiunas et al., 2012; Jinek et al., 2012) that also serve as spacer substrates (Nussenzweig et al., 2019). Finally, in the case of restriction endonucleases, DNA ends generated after cleavage, including those with 3’ and 5’ ssDNA overhangs, are not only used as sources of spacers, but also further degraded by AddAB to expand the region of the phage genome from which spacers are acquired. All three mechanisms also contribute to the discrimination against acquisition of “self” spacers, from the host genome, that would lead to lethal cleavage of the bacterial chromosome (Jiang et al., 2013), a form of type II-A autoimmunity. Injection of free DNA ends is an obligate step of the infection cycle of most dsDNA phages, Cas9 is usually programmed with crRNAs that target invaders, and restriction only cleaves foreign, unmodified, DNA. Therefore, all these strategies will bias spacer acquisition towards DNA invaders and therefore contribute to the fundamental goal of all immune systems: to recognize and eliminate invading pathogens with maximal efficacy and minimal damage to self.
Limitations of this study
Due to the extensive distribution of both CRISPR-Cas and RM systems, present in 40% and 90% of bacterial genomes, respectively, there is a wide range of organisms that contain both systems (Oliveira et al., 2014). However, there are many different types of RM and CRISPR systems, the latter with diverse molecular mechanisms (Makarova et al., 2020). Our work explored the effect of type I and II restriction nucleases on type II CRISPR spacer acquisition. Although it is not known what type of DNA ends are generated by SauI, BglII cleavage results in 4-base 5’ overhangs (Duncan et al., 1978). Many other type II restriction nucleases, however, generate 3’ overhangs. Previously, we showed that chromosomal cleavage by the yeast homing endonuclease I-SceI, which produces in 4-base 3’ overhangs (Colleaux et al., 1988), also leads to AddAB-enhanced hotspots of type II-A CRISPR spacer acquisition (Modell et al., 2017). We corroborated that this is also the case during ΦNM4γ4 infection, when the I-SceI recognition site is on the phage (Fig. S6). Therefore, it appears that the type II-A spacer acquisition machinery can utilize both types of overhangs usually generated by restriction enzymes. Some type III RM systems have also been shown to produce DNA ends with small overhangs (Tock and Dryden, 2005) suggesting that they could also facilitate the type II-A CRISPR-Cas response. Additional experimental work will be required to determine whether these RM systems, as well as the least studied type IV, can generate substrates for spacer acquisition.
With regards to the different CRISPR types, both the previous work in S. themophilus (Hynes et al., 2014) and ours studied the interaction between RM and a type II-A CRISPR-Cas system; therefore whether restriction can enhance spacer acquisition in other CRISPR types is unknown. In addition to type II, the type I-E system of Escherichia coli (Levy et al., 2015) and type III-A system of Staphylococcus epidermidis (Aviram et al., 2022) can use dsDNA ends as preferred substrates for new spacers, with the RecBCD (E. coli) and AddAB (S. epidermidis) complexes being also involved in the expansion of the hotspot of spacer acquisition at the dsDNA break. These similarities with the spacer acquisition mechanism of type II-A systems suggest that restriction would also enhance the type I and III CRISPR-Cas responses through the incorporation of new spacers from restricted phage DNA. In contrast, the importance of DSBs for spacer acquisition in the other CRISPR types (IV, V, and VI) is not known, and therefore we cannot speculate whether and how restriction of viral DNA would affect these CRISPR systems. Interestingly, recent work revealed that the prokaryotic Argonaute, a DNA-guided nuclease involved in anti-phage defense, also acquires new guides at DSB sites (Kuzmenko et al., 2020). Our results suggest the possibility of a dynamic between RM and pAgo similar to the RM-CRISPR interplay, where restriction creates the substrates for guide acquisition, triggering a pAgo defense against methylated phages that raise later during infection.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luciano A. Marraffini (marraffini@rockefeller.edu).
Materials availability
All materials generated for this study are available upon request from the Lead Contact, Luciano A. Marraffini, with a completed Materials Transfer Agreement.
Data and code availability
• All the datasets generated during this study are all available. Next-generation sequencing data is deposited under the BioProject accession number PRJNA782062. The unprocessed version of the images included in this study can be found at http://dx.doi.org/10.17632/gnhrzd5sny.1
• Codes for all the analyses performed are publicly available at
Github: https://github.com/Marraffini-Lab/Maguin_etal_2022. Version v1.0 of the code can be found at http://dx.doi.org/10.5281/zenodo.5831938
• Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and growth conditions
Growth of S. aureus was carried out in brain-heart infusion broth (BHI) at 37 °C with agitation at 220 RPM. Whenever needed, the media was supplemented with chloramphenicol at 10 µg/ml, erythromycin at 10 µg/ml, or spectinomycin at 250 µg/ml for maintenance of pC194 (Horinouchi and Weisblum, 1982a), pE194 (Horinouchi and Weisblum, 1982b), and pLZ12-derived (Perez-Casal et al., 1991) plasmids, respectively. For bacteriophage infection, the media was supplemented with 5 mM CaCl2 to facilitate adsorption. The bacterial strains and phage used in this study can be found in Methods S1.
METHOD DETAILS
Bacteriophage propagation
Overnight cultures of S. aureus were diluted 1:100 in fresh BHI supplemented with 5 mM CaCl2 and the appropriate antibiotic, if needed, and grown for 1 h 15 min at 37 °C. A small volume of bacteriophage was added, and the cultures were grown for an additional 4 hours. Then, the cultures were spun down for 5 minutes at 4300 RPM and the lysates were filtered through 0.45 µm syringe filters (Acrodisc). Plaque formation assays were conducted to assess the number of infectious particles in the resulting stocks.
Plasmid construction
All the plasmids used in this study can be found in Methods S1 along with their cloning strategies and the oligos used to construct them. All the constructed plasmids were electroporated in S. aureus as previously described elsewhere (Goldberg et al., 2014).
Strain construction
To make the S. aureus RN4220 hsdM1/hsdS1 and hsdM2/hsdS2 double knockout, sPM02, we used a method previously described ((Modell et al., 2017). Briefly, RN4220 was electroporated with pPM48 and integrants were checked by PCR (Phusion High-Fidelity, ThermoFischer) using the primer PM96/PM37 and then isolated. Selection for plasmid excision was performed using a temperature-sensitive cat targeting Cas9 phagemid, pJW326. Deletion of hsdM1/hsdS1 was confirmed by PCR using primers PM37/PM174. The resulting strain was electroporated with pPM49 and integrants were isolated and confirmed by PCR with the primer pair PM181/PM185. Selection for plasmid excision was performed using a temperature-sensitive cat targeting Cas9 phagemid, pJW326. Deletion of hsdM2/hsdS2 was confirmed by PCR using the primer pair PM181/PM184 resulting in strain sPM02.
Phage construction
To make phage ΦNM4γ4-BglIIAB and ΦNM4γ4-BglIIAC, ΦNM4γ4 (Heler et al., 2015) was spotted on a layer of S. aureus harboring pPM96 or pPM98, respectively. pPM96 and pPM98 harbor the S. pyogenes type II-A CRISPR system with a unique spacer targeting ΦNM4γ4. Each spacer was chosen carefully to have its PAM sequence (5′-NGG-3′) in a region of DNA that resembles a BglII recognition site except for one bp. CRISPR Phage escapers for pPM96 and pPM98 were selected on soft agar and checked by PCR (Phusion High-Fidelity, ThermoFischer) using primers PM438/PM439 and PM454/PM455, respectively. Phage escapers that acquired a mutation in the PAM sequence creating a new BglII recognition site were spotted again on a lawn of RN4220 harboring either pPM96 or pPM98 to further purify the mutant phage from wild type phage. Then, a single plaque for each was isolated and propagated on S. aureus sPM02.
To make ΦNM4γ4-ΔBglII, ΦNM4γ4-BglIIB and ΦNM4γ4-BglIIC, phage ΦNM4γ4, ΦNM4γ4-BglIIAB and ΦNM4γ4-BglIIAC were propagated in liquid cultures of S. aureus RN4220 harboring pPM117, a plasmid with roughly a 1000 bp of homology to the ΦNM4γ4 genome centered on a mutated BglII recognition sequence (site A). To select for phage with a mutated BglII site A, each resulting lysate was used in a plaque formation assay with S. aureus RN4220 harboring a plasmid with the S. pyogenes type II-A CRISPR and a spacer targeting wild type ΦNM4γ4 (pPM116) but not the recombined phage. Correct editing was confirmed by PCR using the primer pair PM493/PM472. A single plaque for each was propagated again on pPM117 to further purify edited phage from wild type. Then, a single plaque for each was isolated and propagated on S. aureus sPM02.
To make ΦNM4γ4-ΔdnaC, ΦNM4γ4 was propagated in a liquid culture of S. aureus RN4220 harboring a plasmid containing 700 bp arms with downstream and upstream homology to the dnaC gene of ΦNM4γ4, pPM135. The resulting lysate was spotted on S. aureus harboring pPM235 and pPM134. pPM134 provided the dnaC gene on a plasmid to allow phage with the deleted dnaC gene to form plaques and pPM235 allowed for selection of the recombinant phage by having a spacer targeting ΦNM4γ4 but not the dnaC deletion mutant. Correct deletion of dnaC was verified by PCR amplification with the primer pair PM592/PM593. A single plaque was propagated again on pPM235/pPM134, and one of the resulting plaques was used to lyse a liquid culture of pPM134 to obtain a final stock of ΦNM4γ4-ΔdnaC.
To make Φ12ρ1, Φ12γ3 (Modell et al., 2017) was propagated on S. aureus RN4220 with pPM166 to mutate one of its BglII recognition sites through silent mutations. A plaquing assay with S. aureus harboring pPM167 was performed with the resulting lysate to select for recombinant phage. PCR amplification with PM824/PM825 was performed to check for the mutated BglII recognition site. One plaque was purified further on pPM167 and then propagated on RN4220. The resulting lysate was propagated on S. aureus RN4220 with pPM168 to mutate a second BglII site through silent mutations. A plaquing assay with S. aureus harboring pPM169 was performed to select for recombinant phage. PCR amplification with PM822/PM823 was performed to verify that the second BglII recognition site was mutated. One plaque was purified further on pPM169 and then propagated on RN4220. The resulting lysate was propagated on S. aureus with pPM170 to mutate a third BglII site through silent mutations. A plaquing assay with S. aureus RN4220 harboring pPM171 was performed to select for recombinant phage. PCR amplification with PM826/PM827 was performed to verify that the third BglII recognition site was mutated. One plaque was purified further on pPM171 and then propagated on RN4220. The resulting phage was propagated on S. aureus pPM176 to mutate two chi sites through silent mutations. A plaquing assay with a 1:1 mixed culture of S. aureus harboring pPM177 or pPM178 was performed to select for phage with both chi sites mutated. Correct editing was verified by PCR amplification with PM902/PM903. Then, a single plaque was purified further on the mixed culture and then propagated on RN4220. The resulting phage stock was propagated on S. aureus harboring pPM179 to mutate a third chi site. The resulting lysate was used in a plaque assay with S. aureus harboring pPM180 to select for recombinant phage. Plaques were PCR amplified with the primer pair PM904/PM905 to check for mutation of the chi site. A single plaque was then spotted on a lawn of pPM180 to further purify the phage and a single plaque was then amplified on sPM02. This resulted in phage Φ12ρ1. The final stock of Φ12ρ1was again amplified by PCR to check that all the mutations created successively were still present.
To make ΦNM4γ4I-sceI, ΦNM4γ4 was propagated in a liquid culture of S. aureus harboring a plasmid, pJW241, containing about 2000 bp of homology to the ΦNM4γ4 genome with an I-sceI recognition sequence in the middle. The resulting lysate was spotted on a lawn of S. aureus harboring a plasmid with the S. pyogenes type II-A CRISPR system with a spacer targeting ΦNM4γ4 but not the edited phage, pJW237. Plaques were PCR amplified to check for proper editing and a single edited plaque was amplified on sPM02 to create a stock of ΦNM4γ4I-sceI.
Colony formation assay
Ten-fold dilutions of S. aureus were spotted on BHI agar plate supplemented with the appropriate antibiotic, if needed. The plates were incubated overnight at 37 °C and colony-forming units (CFU) were enumerated the next day.
Plaque formation assay
Ten-fold dilutions of bacteriophage were spotted on a layer of S. aureus cells suspended in 50 % heart infusion agar (HIA) supplemented with 5 mM CaCl2 and the appropriate antibiotic, if needed. The plates were incubated overnight at 37 °C and plaque-forming units (PFU) were enumerated the next day.
ΦNM4γ4 growth curve
Overnight cultures of S. aureus RN4220 were diluted 1:100 in 50 ml of fresh BHI supplemented with 5 mM CaCl2 and grown for 1 h 15 min at 37 °C. Following incubation, the cultures were infected with ΦNM4γ4 at MOI = 0.1. Immediately, 1 ml was removed from each culture and filtered through a 0.45 µm syringe filter (Acrodisc) to remove the bacteria. Then every 10 minutes, 1 ml was removed and filtered from each culture. All the filtered lysates were used in plaque formation assays to enumerate the number of phage particles at each time point.
SauI escaper assay
Overnight cultures of S. aureus RN4220 harboring either pLZ12 or pSauI (pPM61) were launched from single colonies. The following day, the cultures were diluted 1:100 in fresh BHI supplemented with spectinomycin at 250 µg/mL and 5 mM CaCl2 and outgrown for about 1 hour 15min and normalized for optical density. The cultures were then aliquoted to a 96-well plate (Cellstar) and half were infected with unmodified ΦNM4γ4 (obtained by lysing S. aureus sPM02) at an MOI of 10. Absorbance at 600 nm was measured every 10 minutes for 22 hours using a microplate reader (TECAN Infinite 200 PRO). At the end of the experiment, the bacteriophage obtained from the infected wells were collected by briefly centrifuging the cultures at high speed and collecting the lysates. The lysates were then propagated on S. aureus sPM02. The original lysates and the ones passaged on sPM02 were used to perform plaque formation assays with RN4220/pLZ12 and RN4220/pPM61 to assess the sensitivity of each phage stock to the SauI R-M system.
CRISPR and RM synergy growth curves
Overnight cultures were launched from single colonies. The next day, the cultures were diluted 1:100 in fresh BHI and 5 mM CaCl2. The cultures were outgrown for about 1 hour 15 minutes and then normalized for optical density and 150uL of cultures were seeded in a flat-bottom 96-well plate (Cellstar). Half of the cultures were infected with phage and the absorbance at 600 nm was recorded every 10 minutes for 24 hours in a microplate reader (TECAN Infinite 200 PRO). After 24 hours, 2 µl from the uninfected and infected pCRISPR cultures were resuspended in 30 µl of colony lysis buffer (250 mM KCl, 5 mM MgCl2, 50 mM Tris-HCl at pH 9.0 and 0.5% Triton X-100) supplemented with 200 ng/µl of lysostaphin. The reactions were incubated for 20 minutes at 37° C and then for 10 minutes at 98° C in a thermocycler. 0.5 µl of each reaction was used to PCR amplify the CRISPR locus using the primer pair PM223/PM225 with TopTaq@ master mix (Qiagen). The PCR products were analyzed on a 2 % agarose gel stained with ethidium bromide and imaged with FluorChem HD2 (Protein simple). Additionally, one well for pSauI-pCRISPR cultures infected with ΦNM4γ4 was streaked on a BHI agar plate at the end of the experiment and incubated overnight at 37 °C. The next day, 50 single colonies were resuspended in 30 µl of colony lysis buffer (see above) supplemented 200 ng/µl of lysostaphin. 0.5 µl of each reaction was PCR amplified as described just above with the primer pair PM223/PM225. The PCR products were analyzed on a 2 % agarose gel stained with ethidium bromide and imaged with FluorChem HD2 (Protein simple). Also, each PCR product was sent for Sanger sequencing to obtain the sequences of the newly acquired spacers (Data S1).
ΦNM4γ4-ΔdnaC infectivity assay
Plaque formation assays (see above) were performed using ΦNM4γ4 and ΦNM4γ4-ΔdnaC on lawns of S. aureus RN4220 harboring either an empty pE194 plasmid or pE194 with a copy of the dnaC gene, pDnaC. Liquid infection was also tested by recording the absorbance of infected culture over time. Briefly, overnight of S. aureus RN4220 harboring either pE194 or pDnaC were launched from single colonies. The next day, the cultures were diluted 1:100 in fresh BHI supplemented with 10 µg/ml erythromycin and 5 mM CaCl2, outgrown for 1 hour 15 minutes and normalized for optical density. Cultures were seeded to wells of a flat bottom 96-well plate (Cellstar) and each culture was either uninfected or infected with ΦNM4γ4 or ΦNM4γ4-ΔdnaC at an MOI equal to 1. Growth curves were obtained by measuring the absorbance at 600 nm every 10 minutes for 20 hours in a microplate reader (TECAN Infinite 200 PRO).
DNA extraction and qPCR for phage DNA replication assay
Overnight cultures of pLZ12 and pPM61 (pSauI) were launched from single colonies. The next day, the cultures were diluted 1:100 in 20 ml of fresh BHI supplemented with 250 µg/ml spectinomycin and 5 µM CaCl2 and outgrown until OD600 reached 0.4. The cultures were then infected with either ΦNM4γ4 and ΦNM4γ4-ΔdnaC at an MOI equal to 0.1. At 10 minutes and 30 minutes post-infection, 10 ml from each culture was collected and spun down at 10000 RPM for 3 minutes and the pellets were flash-frozen in liquid nitrogen and stored at −80 °C until DNA extraction. For DNA extraction, the pellets were resuspended in 500uL of P1 buffer (Qiagen) supplemented with 2 µg/µl lysozyme (Ambi Products) and 200 ng/µL−1 of lysostaphin (Sigma-Aldrich) and incubated for 10 minutes at 37° C. Then, 60 µl of 10 % N-lauroylsarcosine (Sigma-Aldrich) was mixed-in and 600 µl of phenol/chloroform/isoamyl alcohol (Fischer Scientific) was added. The samples were vortexed at high force until the mixtures were white and opaque and then spun down at 13000 RPM for 5 minutes. 550 µl of the top layer was collected and 60 µl of 3M sodium acetate (Fisher Scientific) was added and briefly vortexed. 1 ml of 100% ethanol was added to precipitate the DNA and then spun down at 13000 RPM for 1 minute. The DNA pellets were washed with 200 µl of 70 % ethanol and spun down again for one minute. The ethanol was removed, and the DNA pellets were air-dried at room temperature. Once dried, they were resuspended in 500 µl of water. 100 ng of DNA was used from each sample for qPCR using Fast SYBR Green Master Mix (Life Technologies) and QuantStudio® 3 Real-Time PCR System (Applied Biosystems). The relative phage DNA content was calculated by the ΔΔCt method. Ct values were measured for a phage-specific primer set (PM1250/PM1251), then normalized to Ct values for a host-specific primer set (PM1248/1249) to control for total DNA content. Then phage DNA content values were normalized to the 10 min pLZ12 sample infected with ΦNM4γ4, which was set to 1.
30 minutes post-infection spacer acquisition assay
Spacer acquisition was performed as previously described (Nussenzweig et al., 2019) with slight modifications. Briefly, overnight cultures launched from single colonies were diluted 1:100 in 10 ml of BHI with 5mM CaCl2 and outgrown for about 1 hour 15 minutes at 37° C with agitation. For the addA nuclease mutant, strain JW418 was used instead of RN4220. The optical densities (OD600) were measured for all the cultures and each was diluted to OD600 = 0.3. Cultures were infected with the appropriate bacteriophage at MOI = 250 for experiments with pSauI and MOI= 25 for pBlII. 30 minutes post-infection, the cells were spun down at 10000 RPM for 3 minutes and flash-frozen in liquid nitrogen. The frozen pellets were stored at −80 °C until plasmid extraction for next-generation sequencing.
Spacer acquisition time course experiment
Overnight cultures of S. aureus RN4220/pLZ12/pGG32 and RN4220/pPM120/pPM118 were launched from single colonies. The next day, the cultures were diluted 1:100 in 1 liter of fresh BHI supplemented with 5mM CaCl2, outgrown for 1 hour 15 minutes. Each culture was infected with ΦNM4γ4 at an MOI equal to 1. From each culture, 25 ml was retrieved at each time point (every 0.5 hours for the first 5 hours post-infection and then every 4 hours). From the 25ml sample, 1 ml aliquot was removed, spun down, and resuspended in 1ml of fresh BHI and used in colony formation assays. The rest of the 25 ml was spun down at 10000 RPM for 3 minutes at 4 °C. The supernatant was collected and filtered through a 0.45 µm syringe filter (Acrodisc) and used to perform plaque formation assays on both S. aureus RN4220/pLZ12 and RN4220/pPM120. The pellet was flash-frozen in liquid nitrogen and stored at −80° c until plasmid extraction for next-generation sequencing.
Phage DNA extraction and bisulfite sequencing on the time course samples
Phage DNA was extracted from the spacer acquisition time course samples collected during the first 3 hours post-infection. Additionally, DNA was also extracted from control stocks of unmethylated ΦNM4γ4 (propagated on S. aureus sPM02) and BglII methylated ΦNM4γ4 (propagated on S. aureus sPM02/pPM212). The phage supernatants were concentrated using Ultra-4 100K centrifugal 50-ml spin columns (Amicon) to about 500 µl. The concentrates were resuspended in 15 ml of DNase I buffer (DNase I buffer, 20 mM Tris-HCl, pH 8.0, and 2 mM MgCl2) and concentrated again. This was done three times to buffer exchange the lysate with DNAse I buffer. 450 µL of each concentrate was treated with 25 units of DNase I (Sigma) for 1 h at 37° C. DNAse I was inactivated by adding 25 µl of stop solution (Sigma) and by heating the reactions for 10 min at 70 °C. The samples were then treated with 8 units of proteinase K (NEB) and 12 µl of 20 % SDS for 1 h at 37 °C. Finally, the DNA was extracted using phenol /chloroform/isoamyl alcohol extraction (Fisher) and resuspended in 100 µl of water. 500 ng of each DNA sample was bisulfite treated using the epiTect kit (Qiagen).
Following bisulfite treatment, the region of DNA encompassing the single BglII site of ΦNM4γ4 was PCR amplified using pyroMARK PCR kit (Qiagen) and a set of top strand-specific primers (PM1033/PM1035). Correct PCR amplification was checked on a 2 % agarose gel stained with ethidium bromide. The amplicons were cleaned up using a QIAquick PCR purification kit (Qiagen) and prepared for high-throughput sequencing exactly as previously described (Nussenzweig et al., 2019). Then, the samples were sequenced on a MiSeq instrument (Illumina). A custom python script was used to compute the number of reads containing an intact Bglll site 5’-AGATCT-3’ and the number of reads containing a bisulfite modified Bglll site 5’-AGATTT-3’.
Spacer acquisition with I-sceI cleavage
Overnight cultures launched from single colonies of RN4220/pC194/pWJ259/ and RN4220/pWJ250/pWJ259 were diluted 1:100 in 10 ml of BHI supplemented 5mM CaCl2 and 1 mM IPTG. The cultures were outgrown for about 1 hour and 15 minutes at 37° C with agitation and then normalized for optical density. Following the outgrowth, the cultures were infected with ΦNM4γ4I-sceI at an MOI equal to 1. The cultures were collected 5 hours post-infection and quickly spun down at 10000 RPM for 3 minutes. The resulting pellets were flash-frozen in liquid nitrogen and store at −80 °C until plasmid extraction for next-generation sequencing.
CRISPR plasmid extraction and amplification for next-generation sequencing
For all the deep sequencing spacer acquisition experiments, the CRISPR plasmids from the frozen pellets were extracted using a modified QIAprep Spin Miniprep Kit (Qiagen) protocol described previously (Modell et al., 2017). The CRISPR loci were amplified using 250 ng of plasmid DNA with Phusion High-Fidelity enzyme (Thermo Fischer). For the acquisition time course experiment, each PCR reaction was performed using modified PM900 and PM901 primers containing 5 random bp and a unique 3–6bp barcode at their 5’ ends to keep track of each sample in the downstream NGS data. Successful amplification was checked on a 2 % agarose gel stained with ethidium bromide. Then, the PCR products were cleaned using a MinElute PCR purification kit (Qiagen). The PCR amplicons corresponding to expanded CRISPR loci were extracted using the PippinHT instrument (Sage Science) set on the range mode (136bp to 450bp) with a 2 % agarose gel cassette. For all the other acquisition experiments, PCR amplifications of the CRISPR loci were performed as described elsewhere with some modifications (Jakhanwal et al., 2021). In short, the plasmids were PCR amplified with a cocktail of three reverse primers mixed 1:1 (PM375, PM376, and PM377) and one forward primer PM168 to preferentially amplify expanded CRISPR loci (Heler et al., 2015). For each sample, a modified pPM168 primer with 5 random nucleotides and a unique 3–6 bp barcode at its 5’ end was used to track each sample in the resulting next-generation sequencing data. The PCR products were analyzed on a 2 % agarose gel stained with ethidium bromide and locations corresponding to expanded CRISPR loci were gel extracted using QIAquick gel extraction kit (Qiagen). For all the acquisition experiments, the PCR amplicons were prepared for high-throughput sequencing exactly as previously described (Nussenzweig et al., 2019). Samples from the acquisition time course were sequenced on a NovaSeq instrument (Illumina) and all the other experimental samples were sequenced on a MiSeq instrument (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
High Throughput Sequencing Data Analysis
Using python, the spacer sequences were extracted and the number of reads for each spacer was recorded from the Illumina raw FASTQ files. For all the deep sequencing experiments (except for the time course acquisition experiment), the number of reads for each spacer was corrected to account for the PCR biased introduced by the reverse primer cocktail (PM375, PM376, PM377), as previously described (Modell et al., 2017). Each spacer sequence was aligned to the phage, plasmids, and bacterial genome. If an exact match was found (except for Figure 2E), the origin of the spacer and its PAM sequence was recorded. Additionally, for spacer sequences matching the phage genome, the position of the spacer sequence on the genome was recorded. For the quantification of spacer acquisition in the presence of the SauI RM system (Figure 2E), the spacers were matched to each possible DNA source using Bowtie2 (Langmead and Salzberg, 2012; Langmead et al., 2009) on usegalaxy.org (Afgan et al., 2016) to allow for mismatches. To create the pattern of acquisition for each phage, the number of reads for each phage spacer sequence with a correct PAM (5′-NGG-3′) was binned in roughly 1 kb bins along the phage genome (985 bp for ΦNM4γ4 and its mutants and 993 bp for Φ12ρ1). Following binning, the number of reads in each bin was normalized to the number of PAM sequences in the phage genome within that bin. Finally, reads per million were calculated as RPMphage, as previously described (Modell et al., 2017). The sequence 5’-AGACAAAAATAGTCTACGAG-3’ was removed from our NovaSeq data as it corresponds to the leader sequence but is sometimes perceived by our script as a spacer in some reads representing a DNA recombination event.
The error bars in the quantification of spacer acquisition in Figure 2E represent the SD of 3 biological replicates. Statistical analysis was carried using Prism 9.2.0 (GraphPad).
Growth curves
The error bars for growth curves in Figures 1D, 3B, S1H, S3D represent the SD of 5 biological replicates. In Figures 1B, 2B, and S1D, the error bars represent the SD of 3 biological replicates. Statistical analysis was carried using Prism 9.2.0 (GraphPad).
Plaque assays
The error bars for the quantification of plaque assays in Figures 1A, 1C, S1E, and S2C represent the SD of 3 biological replicates. In Figure S3C the error bars represent the SD of 4 biological replicates. The error bars in Figures 5A, S5B, and S5C represent the SD of 3 technical replicates. Statistical analysis was carried using Prism 9.2.0 (GraphPad).
Colony formation assay
The error bars in Figure S5A represent the SD of 3 technical replicates. Statistical analysis was carried using Prism 9.2.0 (GraphPad).
qPCR quantification
The error bars in Figures 2C and 2D represent the SD of 3 biological replicates. Statistical analysis was carried using Prism 9.2.0 (GraphPad).
Supplementary Material
Data S1. Related to Figures 2, 3, 4, 5, S1, S2, S4, S5, and S6. Spacer sequences obtained in this study.
Methods S1. Related to STAR Methods. Tables of bacterial strains, plasmids, bacteriophages, and oligonucleotides used in this study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| ΦN4220 | Kreiswirth et al., 1983 | N/A |
| sPM02 | This paper | N/A |
| TB4 | Bae et al., 2006 | N/A |
| ΦNM4γ4 | Goldberg et al., 2014 | N/A |
| ΦFNM4γ4-ΔdnaC | This paper N/A | |
| ΦFNM4γ4-ΔBglII | This paper | N/A |
| ΦFNM4γ 4-BglIIAB | This paper | N/A |
| ΦFNM4γ 4-BglIIAC | This paper | N/A |
| ΦFNM4γ 4-BglIIB | This paper | N/A |
| ΦFNM4γ 4-BglIIC | This paper | N/A |
| ΦFNM4γ4-I-sceI | This paper | N/A |
| ΦF12γ3 | Modell et al., 2017 | N/A |
| ΦF12ρ1 | This paper | N/A |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Brain-Heart Infusion (BHI) Broth | Becton Dickson | Catalog No. 238400 |
| BglII Restriction Enzyme | New England Biolabs | Catalog No. R0144 |
| BsrBI Restriction Enzyme | New England Biolabs | Catalog No. R0102 |
| BssHII Restriction Enzyme | New England Biolabs | Catalog No. R0199 |
| DNase I | Sigma | Catalog No. AMPD1-1KT |
| Heart Infusion (HI) Agar | Becton Dickson | Catalog No. 244400 |
| Lysostaphin | Ambi Products | Catalog No. LSPN-50 |
| Lysozyme | Sigma-Aldrich | Catalog No. L6876 |
| N-Lauroylsarcosine Sodium Salt | Sigma-Aldrich | Catalog No. L9150 |
| Phenol/Chloroform/Isoamyl Alcohol | Fisher Scientific | Catalog No. BP1752I-400 |
| Sodium Acetate Anhydrous | Fisher Scientific | Catalog No. S210-500 |
|
Critical Commercial Assays | ||
| 2 % Agarose Gel Cassette, Pippin HT | Sage Science | Catalog No. HTC2010 |
| Amicon®Ultra-15 Centrifugal Filter Unit | Millipore | Catalog No. UFC9100 |
| EpiTect Bisulfite Kit | Qiagen | Catalog No. 59104 |
| Fast SYBR™ Green Master Mix | Applied Biosystems | Catalog No. 4385612 |
| Minelute PCR Purification Kit | Qiagen | Catalog No. 28004 |
| MiSeq reagent Kit v3 (150-cycle) | Illumina | Catalog No. MS-102-3001 |
| NovaSeq SP Reagent kit (500-cycle) | Illumina | Catalog No. 20029137 |
| Phusion High-Fidelity PCR Kit | Thermo Fisher Scientific | Catalog No. F530 |
| QIAprep Spin Miniprep Kit | Qiagen | Catalog No. 27104 |
| QIAquick Gel Extraction Kit | Qiagen | Catalog No. 28115 |
| TopTaq PCR Kit | Qiagen | Catalog No. 200403 |
| TruSeq Nano DNA LT Library Prep Kit | Illumina | Catalog No. 20015964 |
|
Deposited Data | ||
| Next-generation Sequencing Files | This paper | PNRJNA782062 |
| Unprocessed images | This paper; Mendeley Data | https://doi.org/10.17632/gnhrzd5sny.1 |
|
Oligonucleotides | ||
| See Methods S1 | This paper | N/A |
|
Recombinant DNA | ||
| pAV268 | This paper | N/A |
| pC194 | Horinouchi and Weisblum, 1982a | N/A |
| pDB114 | Bikard et al., 2014 | N/A |
| pE194 | Horinouchi and Weisblum, 1982b | N/A |
| pGG32 | Heler et al., 2015 | N/A |
| pJW215 | Modell et al., 2017 | N/A |
| pJW237 | This paper | N/A |
| pJW241 | This paper | N/A |
| pJW250 | This paper | N/A |
| pJW259 | Modell et al., 2017 | N/A |
| pLM9B | Jiang et al., 2013b | N/A |
| pLZ12 | Perez-Casal et al., 1991 | N/A |
| pPM48 | This paper | N/A |
| pPM49 | This paper | N/A |
| pPM61 | This paper | N/A |
| pPM82 | This paper | N/A |
| pPM96 | This paper | N/A |
| pPM98 | This paper | N/A |
| pPM116 | This paper | N/A |
| pPM117 | This paper | N/A |
| pPM118 | This paper | N/A |
| pPM120 | This paper | N/A |
| pPM134 | This paper | N/A |
| pPM135 | This paper | N/A |
| pPM166 | This paper | N/A |
| pPM167 | This paper | N/A |
| pPM168 | This paper | N/A |
| pPM169 | This paper | N/A |
| pPM170 | This paper | N/A |
| pPM171 | This paper | N/A |
| pPM176 | This paper | N/A |
| pPM177 | This paper | N/A |
| pPM179 | This paper | N/A |
| pPM180 | This paper | N/A |
| pPM212 | This paper | N/A |
| pPM235 | This paper | N/A |
| pT181 | Khan and Novick, 1983 | N/A |
| pWJ244 | Modell et al., 2017 | N/A |
| pWJ326 | Modell et al., 2017 | N/A |
|
Software and Algorithms | ||
| Illustrator | Adobe | N/A |
| Prism 9 | GraphPad | N/A |
| Python scripts for NGS data (Maguin_etal_2022-v1.0) |
This paper | https://doi.org/10.5281/zenodo.5831938 |
Highlights.
Restriction nucleases generate free DNA ends and stop the viral lytic cycle
DNA repair nucleases degrade DNA ends to generate new ones
Type II CRISPR-Cas integrases acquire new spacers from free DNA ends
Bacteria is immunized to neutralize restriction escapers with methylated genomes
Acknowledgements.
We would like to thank J. T. Rostøl for helpful discussions. We also thank the Rockefeller University Genomics Resource Center for its assistance with the NGS experiments. Support for this work comes from the National Institute of Health Director’s Pioneer Award 1DP1GM128184-01 and the Burroughs Wellcome Fund PATH Award to LAM. LAM is an Investigator of the Howard Hughes Medical Institute. AV was supported by the Arnold and Mabel Beckman Postdoctoral Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests. L.A.M. is a founder and advisor of Intellia Therapeutics, CRISPR Biotechnologies, and Eligo Biosciences.
References
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44, W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram N, Thornal AN, Zeevi D, and Marraffini LA (2022). Different modes of spacer acquisition by the Staphylococcus epidermidis type III-A CRISPR-Cas system. Nucleic Acids Res In press. [DOI] [PMC free article] [PubMed]
- Bae T, Baba T, Hiramatsu K, and Schneewind O (2006). Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol 62, 1035–1047. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bernheim A, and Sorek R (2020). The pan-immune system of bacteria: antiviral defence as a community resource. Nat. Rev. Microbiol 18, 113–119. [DOI] [PubMed] [Google Scholar]
- Bickle TA, and Kruger DH (1993). Biology of DNA restriction. Microbiol Rev 57, 434–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, and Marraffini LA (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol 32, 1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, and Ehrlich SD (2005). Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens SR, and Gilcrease EB (2009). Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol Biol 502, 91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleaux L, D’Auriol L, Galibert F, and Dujon B (1988). Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci U S A 85, 6022–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, and Moineau S (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CH, Wilson GA, and Young FE (1978). Biochemical and genetic properties of site-specific restriction endonucleases in Bacillus globigii. J. Bacteriol 134, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis ME, Villion M, Magadan AH, and Moineau S (2013). CRISPR-Cas and restriction-modification systems are compatible and increase phage resistance. Nat Commun 4, 2087. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, and Paul CL (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GW, Jiang W, Bikard D, and Marraffini LA (2014). Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 514, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, and Terns MP (2009). RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern D, Chiapello H, Schbath S, Robin S, Hennequet-Antier C, Gruss A, and El Karoui M (2007). Identification of DNA motifs implicated in maintenance of bacterial core genomes by predictive modeling. PLoS Genet 3, 1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, and Marraffini LA (2015). Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519, 199–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Wright AV, Vucelja M, Bikard D, Doudna JA, and Marraffini LA (2017). Mutations in Cas9 Enhance the Rate of Acquisition of Viral Spacer Sequences during the CRISPR-Cas Immune Response. Mol. Cell 65, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heler R, Wright AV, Vucelja M, Doudna JA, and Marraffini LA (2019). Spacer Acquisition Rates Determine the Immunological Diversity of the Type II CRISPR-Cas Immune Response. Cell Host Microbe 25, 242–249 e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey AD, Burgi E, and Ingraham L (1963). Cohesion of DNA molecules isolated from phage lambda. Proc Natl Acad Sci U S A 49, 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi S, and Weisblum B (1982a). Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol 150, 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi S, and Weisblum B (1982b). Nucleotide sequence and functional map of pE194, a plasmid that specifies inducible resistance to macrolide, lincosamide, and streptogramin type B antibodies. J. Bacteriol 150, 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Villion M, and Moineau S (2014). Adaptation in bacterial CRISPR-Cas immunity can be driven by defective phages. Nat Commun 5, 4399. [DOI] [PubMed] [Google Scholar]
- Jakhanwal S, Cress BF, Maguin P, Lobba MJ, Marraffini LA, and Doudna JA (2021). A CRISPR-Cas9-integrase complex generates precise DNA fragments for genome integration. Nucleic Acids Res 49, 3546–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, and Marraffini LA (2013a). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol 31, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Maniv I, Arain F, Wang Y, Levin BR, and Marraffini LA (2013b). Dealing with the evolutionary downside of CRISPR immunity: bacteria and beneficial plasmids. PLoS Genet 9, e1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jore MM, Lundgren M, van Duijn E, Bultema JB, Westra ER, Waghmare SP, Wiedenheft B, Pul U, Wurm R, Wagner R, et al. (2011). Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol 18, 529–536. [DOI] [PubMed] [Google Scholar]
- Khan SA, and Novick RP (1983). Complete nucleotide sequence of pT181, a tetracycline-resistance plasmid from Staphylococcus aureus. Plasmid 10, 251–259. [DOI] [PubMed] [Google Scholar]
- Kreiswirth BN, Lofdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, and Novick RP (1983). The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305, 709–712. [DOI] [PubMed] [Google Scholar]
- Kuzmenko A, Oguienko A, Esyunina D, Yudin D, Petrova M, Kudinova A, Maslova O, Ninova M, Ryazansky S, Leach D, et al. (2020). DNA targeting and interference by a bacterial Argonaute nuclease. Nature 587, 632–637. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, and Sorek R (2015). CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loenen WA, Dryden DT, Raleigh EA, and Wilson GG (2014). Type I restriction enzymes and their relatives. Nucleic Acids Res 42, 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol 9, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Jiang W, and Marraffini LA (2017). CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 544, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, and Soria E (2005). Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Nair D, Memmi G, Hernandez D, Bard J, Beaume M, Gill S, Francois P, and Cheung AL (2011). Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol 193, 2332–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neamah MM, Mir-Sanchis I, Lopez-Sanz M, Acosta S, Baquedano I, Haag AF, Marina A, Ayora S, and Penades JR (2017). Sak and Sak4 recombinases are required for bacteriophage replication in Staphylococcus aureus. Nucleic Acids Res 45, 6507–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig PM, McGinn J, and Marraffini LA (2019). Cas9 Cleavage of Viral Genomes Primes the Acquisition of New Immunological Memories. Cell Host Microbe 26, 515–526 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira PH, Touchon M, and Rocha EP (2014). The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res 42, 10618–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, and Medzhitov R (2009). Pattern recognition receptors and control of adaptive immunity. Immunol. Rev 227, 221–233. [DOI] [PubMed] [Google Scholar]
- Perez-Casal J, Caparon MG, and Scott JR (1991). Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J. Bacteriol 173, 2617–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud V, and Wende W (2005). Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci 62, 685–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, and Vergnaud G (2005). CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151, 653–663. [DOI] [PubMed] [Google Scholar]
- Pyenson NC, Gayvert K, Varble A, Elemento O, and Marraffini LA (2017). Broad Targeting Specificity during Bacterial Type III CRISPR-Cas Immunity Constrains Viral Escape. Cell Host Microbe 22, 343–353 e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenson NC, and Marraffini LA (2020). Co-evolution within structured bacterial communities results in multiple expansion of CRISPR loci and enhanced immunity. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts GA, Houston PJ, White JH, Chen K, Stephanou AS, Cooper LP, Dryden DT, and Lindsay JA (2013). Impact of target site distribution for Type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res 41, 7472–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, and Macelis D (2015). REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43, D298–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson JE, Magadan AH, Sabri M, and Moineau S (2013). Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol 11, 675–687. [DOI] [PubMed] [Google Scholar]
- Simmon VF, and Lederberg S (1972). Degradation of bacteriophage lambda deoxyribonucleic acid after restriction by Escherichia coli K-12. J. Bacteriol 112, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tock MR, and Dryden DT (2005). The biology of restriction and anti-restriction. Curr. Opin. Microbiol 8, 466–472. [DOI] [PubMed] [Google Scholar]
- van Houte S, Ekroth AK, Broniewski JM, Chabas H, Ashby B, Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, and Westra ER (2016). The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron DE, and Lindsay JA (2006). Sau1: a novel lineage-specific type I restriction-modification system that blocks horizontal gene transfer into Staphylococcus aureus and between S. aureus isolates of different lineages. J. Bacteriol 188, 5578–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley DB (2013). Bacterial DNA repair: recent insights into the mechanism of RecBCD, AddAB and AdnAB. Nat. Rev. Microbiol 11, 9–13. [DOI] [PubMed] [Google Scholar]
- Workman RE, Pammi T, Nguyen BTK, Graeff LW, Smith E, Sebald SM, Stoltzfus MJ, Euler CW, and Modell JW (2021). A natural single-guide RNA repurposes Cas9 to autoregulate CRISPR-Cas expression. Cell 184, 675–688 e619. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Ng S, Nam KH, and Ke A (2017). How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature 550, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Related to Figures 2, 3, 4, 5, S1, S2, S4, S5, and S6. Spacer sequences obtained in this study.
Methods S1. Related to STAR Methods. Tables of bacterial strains, plasmids, bacteriophages, and oligonucleotides used in this study.
Data Availability Statement
• All the datasets generated during this study are all available. Next-generation sequencing data is deposited under the BioProject accession number PRJNA782062. The unprocessed version of the images included in this study can be found at http://dx.doi.org/10.17632/gnhrzd5sny.1
• Codes for all the analyses performed are publicly available at
Github: https://github.com/Marraffini-Lab/Maguin_etal_2022. Version v1.0 of the code can be found at http://dx.doi.org/10.5281/zenodo.5831938
• Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.