

Abstract
Site-specific labeling and conjugation of antibodies are highly desirable for fundamental research and for developing more efficient diagnostic and therapeutic methods. We report here a general and robust chemoenzymatic method that permits a one-pot site-specific functionalization of antibodies. A series of selectively modified disaccharide oxazoline derivatives were designed, synthesized, and evaluated as donor substrates of different endoglycosidases for antibody Fc glycan remodeling. We found that among several endoglycosidases tested, wild-type endoglycosidase from Streptococcus pyogenes of serotype M49 (Endo-S2) exhibited remarkable activity in transferring the functionalized disaccharides carrying site-selectively modified azide, biotin, or fluorescent tags to antibodies without hydrolyzing the resulting transglycosylation products. This discovery, together with the excellent Fc deglycosylation activity of Endo-S2 on recombinant antibodies, allowed direct labeling and functionalization of antibodies in a one-pot manner without the need of intermediate and enzyme separation. The site-specific introduction of varied numbers of azide groups enabled a highly efficient synthesis of homogeneous antibody–drug conjugates (ADCs) with a precise control of the drug-to-antibody ratio (DAR) ranging from 2 to 12 via a copper-free strain-promoted click reaction. Cell viability assays showed that ADCs with higher DARs were more potent in killing antigen-overexpressed cells than the ADCs with lower DARs. This new method is expected to find applications not only for antibody–drug conjugation but also for cell labeling, imaging, and diagnosis.
Graphical Abstract
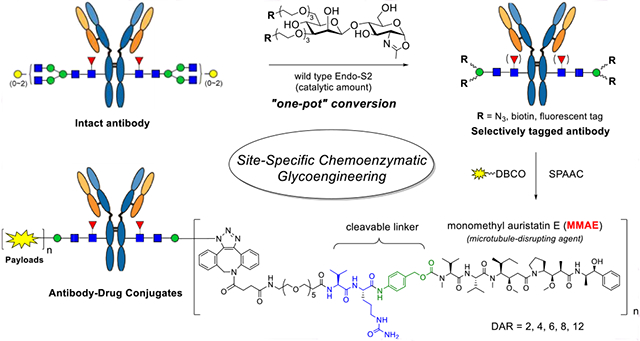
INTRODUCTION
Antibodies are an important class of biologics that have been used for the treatment of many challenging diseases, such as cancer,1 autoimmune disorders,2 and inflammatory diseases.3 Featured by their high specificity and affinity to antigens, antibodies are also widely used for specific labeling of cells and for targeted delivery of small-molecule drugs in the form of antibody–drug conjugates (ADCs). In all these pursuits, site-specific labeling and conjugation of antibodies are highly desirable.4-6 ADCs that combine the specificity of antibodies and the high potency of drugs hold great promise for targeted cell killing.7 So far, 11 ADCs have been approved by the US FDA for the treatment of cancers.5,8,9 The first-generation ADCs in clinical use or preclinical development are generated through nonspecific conjugation,10 resulting in heterogeneous mixtures with varied DARs and distinct pharmacological properties. Recent studies have shown that ADCs obtained by site-specific conjugations give more homogeneous components, which offer improved stability, pharmacokinetics, and safety profiles.11-13 Significant progress has been made in the development of site-specific antibody–drug conjugation strategies,5 which include incorporation of unpaired cysteines or unnatural amino acids,11,14-17 disulfide rebridging,18,19 selective C-/N-terminal modifications,20,21 and enzyme-mediated bioconjugations.22-25 Although homogeneous products can be obtained, most of these strategies require re-engineering of each antibody sequence and/or partially denaturing the antibodies with subsequent site-selective conjugations.
Another approach is to perform site-specific conjugation at the conserved glycans attached to the Asn-297 of the Fc domain, which are spatially distant from the antigen-binding region.5 Early methods have focused on the oxidation of 1,2-diols of terminal monosaccharides, which, however, produces a mixture of conjugates due to the heterogeneity of the glycoforms and the possibility of oxidation at different sugar units.26-28 The application of the galactosyltransferase mutants capable of accommodating modified galactose nucleotide as the donor substrates has made it possible to incorporate a handle selectively at the antibody’s glycans for subsequent bioorthogonal reactions with modified cytotoxic agents.6 A variety of tags including azide,29,30 thiol,31 and keto32 derivatives have been introduced. However, this method requires trimming of the N-glycans to the terminal GlcNAc-glycan forms, and an excess of modified sugar nucleotide and enzymes and long incubation time are usually needed to drive the reaction, which often leads to incomplete reaction and thus heterogeneity of the products. On the other hand, the endoglycosidase-catalyzed glycan remodeling strategy,33,34 particularly the discovery of the Endo-S and Endo-S2 glycosynthase mutants,35-38 has gained attention in recent years for generating various homogeneous antibody glycoforms for functional studies, including antibody labeling and the development of ADCs.6,37,39-43 Despite these successful applications, however, most of the current studies rely on the extraction of natural N-glycans, and for ADC preparation, the N-glycans have to be functionalized for drug conjugation. Thus, the exploitation of simpler donor substrates with a tailored number of tags is greatly demanded for site-specific antibody labeling and conjugation.
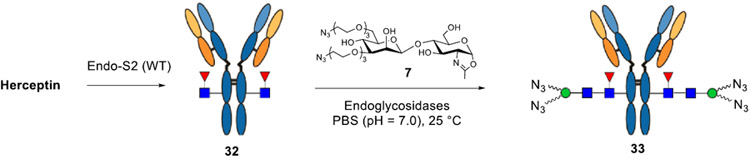
As an alternate, we have previously shown that a synthetic azido-functionalized tetrasaccharide oxazoline can serve as a donor substrate of the Endo-S mutant (D233A or D233Q) for glycosylating the deglycosylated antibody to introduce azide tags at the Fc domain without product hydrolysis.35 Previous studies have shown that wild-type Endo-S could perform glycosylation with the core tetrasaccharide (Man3GlcNAc) oxazoline or the azide-tagged tetrasaccharide oxazoline, but the yield was relatively low, as the wild-type enzyme could hydrolyze the tetrasaccharide oxazoline and the product relatively quickly.44,45 We report in this paper the design, synthesis, and evaluation of a series of simple disaccharide oxazoline derivatives modified with different functional groups and the evaluation of them as donor substrates for antibody glycoengineering. We examined the substrate specificity of different ENGases (Endo-S, Endo-S2, Endo-F3, Endo-A, Endo-CC, and their mutants) toward these synthetic substrates in glycan remodeling of therapeutic antibodies, using trastuzumab (Herceptin) as a model antibody. We found that wild-type Endo-S2 exhibited the best activity in transferring the functionalized disaccharides with site-selectively modified azide, biotin, or fluorescent tags to antibodies, but the transglycosylation products, once formed, were resistant to hydrolysis by the wild-type enzyme, due to the truncated modifications. Combining with the excellent Fc deglycosylation activity of Endo-S2, we devised a simple one-pot deglycosylation and transglycosylation method for direct labeling and functionalization to give structurally well-defined and homogeneously tagged antibodies (Figure 1). The site-specific introduction of varied numbers of azide groups enabled a highly efficient synthesis of homogeneous ADCs with a precise control of the DARs via a copper-free strain-promoted click reaction. Cytotoxicity assays showed that ADCs with higher DARs were more potent in killing antigen-overexpressed cells than those with lower DARs.
Figure 1.

One-pot and site-specific labeling and conjugation of antibodies enabled by simultaneous deglycosylation and transglycosylation with wild-type Endo-S2.
RESULTS AND DISCUSSION
Chemical Synthesis of the Azido-Tagged Disaccharides.
While we and others have shown that smaller synthetic disaccharide oxazolines could serve as substrates of endoglycosidases Endo-A and Endo-M for transglycosylation,46,47 it is not clear whether these antibody-specific ENGases and their mutants can recognize the smaller substrates for transglycosylation using antibodies as acceptors. To test this hypothesis, we sought to synthesize a series of azido-tagged disaccharide oxazolines. Considering the flexibility and solubility of PEGs, we decided to introduce azido groups with PEG-derived scaffolds. Recently, Mizuno and co-workers have shown that PEGylated sugar oxazolines could serve as donor substrates of Endo-M for transglycosylation.47 Thus, we designed a Manβ1,4GlcNAc oxazoline carrying two azide groups as our first target, in which the PEG-linkers resembled the natural glycan branches, so that it might be recognized favorably by the endoglycosidases (Scheme 1). The synthesis was started with the known disaccharide 148 through selective removal of the PMB group with DDQ, giving 2 in 90% yield. Regioselective reductive ring-opening reaction on the benzylidene acetal of 2 provided the diol 3. After conversion of the azido group to an acetamido group with AcSH to afford 4, two azide-tagged polyethylene chains were introduced at the 3′ and 6′ positions of the disaccharide by treatment of 4 with NaH and N3(CH2CH2O)3Ts, giving 5 in 78% yield. To selectively remove the benzyl groups in the presence of azides, a biphasic oxidative condition (NaBrO3/Na2S2O4) was adopted,49 which furnished the desired free disaccharide derivative 6 in 65% yield. Finally, oxazoline formation was achieved in a single step by treatment of 6 with an excess amount of 2-chloro-1,3-dimethylimidazolinium chloride (DMC) and TEA in water,50 affording the desired disaccharide oxazoline 7 carrying two azide groups in 90% isolated yield.
Scheme 1. Synthesis of Disaccharide Oxazoline 7 Carrying Two Azide Tagsa.
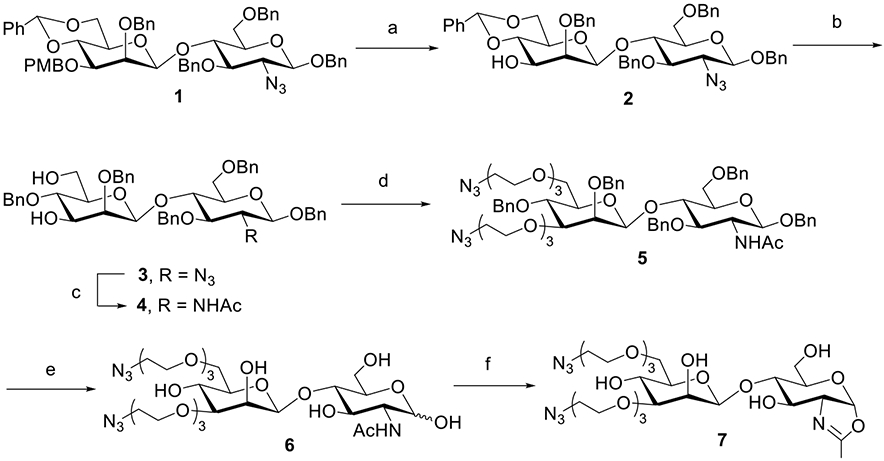
aReagents and conditions: (a)DDQ, CH2Cl2/H2O, 0 °C ~ RT, and 90%; (b) BH3·THF, Bu2BOTf, CH2Cl2, 0 °C, and 94%; (c) AcSH, pyridine/CHCl3, RT, and 82%; (d) N3(CH2CH2O)3Ts, NaH, DMF, 0 °C ~ RT, and 78%; (e) NaBrO3, NaS2O4, EtOAc/H2O, RT, and 65%; and (f) DMC, Et3N, H2O, 0 °C, and 90%.
Next, we synthesized the disaccharides with one or three azido groups (Scheme 2). The 6-position of the mannose residue was selected for the introduction of a single azido-ended PEG linker. Regioselective ring-opening of 3 smoothly exposed the hydroxyl group to obtain disaccharide 8. After the reduction of the azido group to an acetamido group, the PEG-spaced azide group was introduced in high yield to afford 10. Selective debenzylation under oxidative conditions proved to be efficient, giving the free disaccharide 11 in 90% yield, which was converted to oxazoline 12 after treatment with DMC/TEA in water. In parallel, the synthesis of a three azido-tagged disaccharide also started from 3. Upon the reduction of the azido group to the acetamido group, the benzylidene and PMB groups were simultaneously removed under acidic conditions,48 yielding 14 with a free hydroxyl group at the 3′, 4′, and 6′ positions. Next, the azide-terminated PEG chains were introduced at the three free hydroxyl groups to yield 15. Following the same debenzylation conditions, we found that the reaction proceeded very fast, but the desired product was isolated in low yield along with some decomposed byproducts as monitored by ESI–MS. We speculated that the increase in the PEG component resulted in higher water solubility; thus, the partially deprotected intermediate migrated to the aqueous phase and directly interacted with the free radical,51 which led to a rapid reaction but undesired products. To address this issue, we attempted a two-step deprotection method.48 First, the benzyl groups in 15 were removed by catalytic hydrogenation. Under these conditions, the azido groups were simultaneously reduced to amino groups, which were then transformed back to azido functionality by the copper-catalyzed diazo transfer reaction,52 giving 16 in 66% yield in two steps. Finally, standard oxazoline formation by treatment of 16 with DMC/TEA in water gave the disaccharide 17 carrying three azide groups in excellent yield (Scheme 2).
Scheme 2. Synthesis of Disaccharide Oxazolines 12 and 17 Carrying One or Three Azide Tagsa.
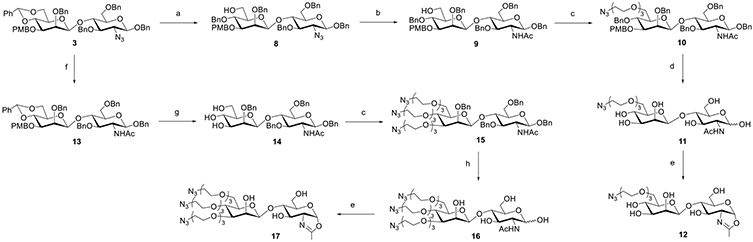
aReagents and conditions: (a) BH3·THF, Bu2BOTf, CH2Cl2 0 °C, and 91%; (b) AcSH, pyridine/CHCl3, RT, and 84%; (c) N3(CH2CH2O)3Ts, NaH, DMF, 0 °C ~ RT, 10, 89%, 15, and 73%; (d) NaBrO3, NaS2O4, EtOAc/H2O, RT, and 90%; (e) DMC, Et3N, H2O, 0 °C, 12, 90%, 17, and 85%; (f) AcSH, pyridine/CHCl3, 60 °C, and 85%; (g) TFA, CH2Cl2, −20–0 °C, and 82%; (h) Pd/C, H2, HCl (aq), THF/H2O, then, TfN3, K2CO3, CuSO4, CH2Cl2/MeOH/H2O, RT, and 66%.
Chemical Synthesis of the Disaccharide Derivatives Carrying a Cluster of Azide Groups.
Previous studies have demonstrated that ADCs with higher DARs tend to provide increased target cell killing potency.13,53 In a handful of cases, a DAR as high as 8 has been achieved through the use of hydrophilic linker payloads, as exemplified with clinically approved Enhertu and Trodelvy.53-55 To equip antibodies with more biorthogonal tags, we designed disaccharides carrying four or six azido groups. The synthesis commenced with the branched scaffolds (Scheme 3). First, the diol 1856 and triol 1957 were extended by an azido-ended linker, which, respectively, furnished 22 and 23 after the removal of the trityl group. The exposed OH in 22 and 23 was further extended with a bis-tosyl linker to give 24 and 25, respectively, which were ready for conjugation. Next, disaccharide 4, which carries free hydroxyl groups at the 4′ and 6′ positions, was reacted with the tosyl derivatives (24 and 25) in the presence of NaH, giving 26 and 27, respectively. Finally, the two-step deprotection followed by oxazoline formation gave disaccharide oxazolines 30 and 31 carrying four and six azido groups, respectively (Scheme 3).
Scheme 3. Synthesis of Disaccharide Oxazolines 30 and 31 Carrying Four or Six Azide Tagsa.
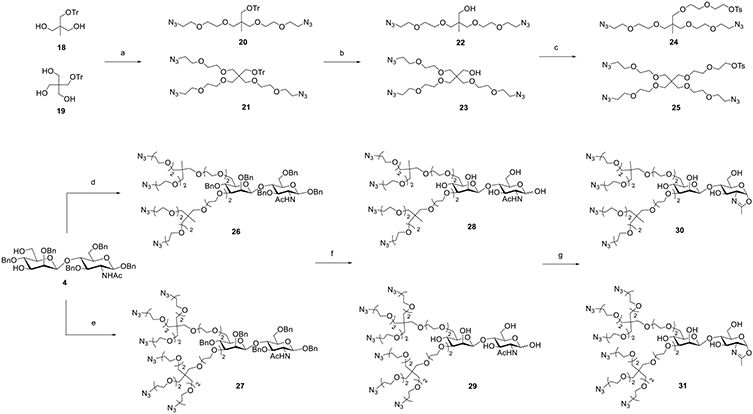
a Reagents and conditions: (a) N3(CH2CH2O)2Ts, NaH, DMF, and 0 °C ~ RT; (b) TsOH, MeOH, 60 °C, 20, 72% for two steps, 21, and 54% for two steps; (c) TsO(CH2CH2O)2Ts, NaH, DMF, 0 °C ~ RT, 22, 85%, 23, and 72%; (d) 24, NaH, DMF, 0 °C ~ RT, and 78%; (e) 25, NaH, DMF, 0 °C ~ RT, and 74%; (f) Pd/C, H2, HCl (aq), THF/H2O, then TfN3, K2CO3, CuSO4, CH2Cl2/MeOH/H2O, RT, 28, 67%, 29, and 59%; and (g) DMC, Et3N, H2O, 0 °C, 30, 89%, 31, and 84%.
Evaluation of the Synthetic Disaccharide Oxazolines as Donor Substrates for Enzymatic Antibody Glycan Remodeling.
With the disaccharide oxazolines in hand, we tested their suitability as donor substrates for antibody–glycan remodeling by the catalysis of different endoglycosidases. For the purpose, we chose trastuzumab, an anti-Her2 antibody (Herceptin), as a typical monoclonal antibody and the synthetic disaccharide oxazoline (7) carrying two PEG-spaced azide groups as the donor substrate to examine the enzymatic reactions. The results are summarized in Table 1. Trastuzumab (Herceptin) was first deglycosylated with wild-type Endo-S2 to provide the Fucα1,6GlcNAc glycoform of Herceptin as the acceptor (32).38
Table 1.
Screening of the Transglycosylation Conditions
| |||||
|---|---|---|---|---|---|
| ENGases | transglycosylation |
product hydrolysis | |||
| oxazoline (equiv)a | enzyme (w/w) (%) | time (h) | conversion yield (%) | ||
| Endo-S2 | 20 | 0.1 | 1 | 98 | <1%c |
| Endo-S2 D184M | 20 | 1 | 2 | 70 | no |
| Endo-S | 20 + 20 | 1 | 2 | 65 | no |
| Endo-S D233Q | 20 | 10 | 3 | 15 | no |
| Endo-F3 | 20 | 10 | 3 | <5 | d |
| Endo-F3 D165A | 20 | 10 | 3 | <5 | d |
| Endo-A | 20 | 10 | 3 | 0 (<5%b) | d |
| Endo-D | 20 | 10 | 3 | 0 (10%b) | no |
| Endo-CC | 20 | 10 | 3 | 0 (<5%b) | d |
Based on each glycosylation site.
Nonfucosylated antibody (GlcNAc-Herceptin) was used as the acceptor, as these three enzymes do not recognize the Fuca1,6GlcNAc-antibody as the acceptor.
After 6 h at room temperature.
Not determined.
Among the endoglycosidases tested, we found that Endo-S2,58 an endoglycosidase from Streptococcus pyogenes of serotype M49 (Endo-S2) with relaxed substrate specificity, exhibited remarkable activity toward the azido-tagged disaccharide oxazoline 7 for transglycosylation. With only a catalytic amount of enzyme (0.1%, w/w, enzyme/antibody) and 20 mol equivalents of the azido-disaccharide oxazoline, the reaction could reach completion within 1 h under mild conditions [rt, phosphate-buffered saline (PBS) buffer, and pH 7.0]. We found that the transglycosylation product (33), once formed, was largely resistant to hydrolysis by the wild-type enzyme, mainly due to truncated modifications. We also tested the Endo-S2 mutant (D184M), a glycosynthase with broad substrate specificity and diminished hydrolytic activity,38 and found that the D184M mutant could also use the azide-disaccharide oxazoline for transglycosylation, but the activity was lower than that of the wild-type enzyme, and a relatively large amount of the mutant enzyme (1%, w/w) was required to drive the reaction. Again, the transglycosylation product was not hydrolyzed by the mutant, allowing accumulation of the product. Next, we tested wild-type Endo-S, the first endoglycosidase from Streptococcus pyogenes that shows Fc-specific deglycosylation activity.44,59 Endo-S could transfer the modified disaccharide oxazoline (7), but, in contrast to wild-type Endo-S2, at least 10-fold more enzyme was required to drive the reaction. Similarly, the Endo-S D233Q mutant, which was a glycosynthase that could act efficiently on complex type N-glycan oxazoline and azido-Man3GlcNAc oxazoline corresponding to the N-glycan core for Fc glycan remodeling,35 just showed very low activity on 7, probably because the minimal structure and the modification made it a poor substrate for the enzyme.
In addition to Endo-S2 and Endo-S, we also tested several other endoglycosidases. Endo-F3 is an endoglycosidase from Elizabethkingia meningoseptica that efficiently hydrolyzes core-fucosylated complex-type N-glycans.60,61 We have previously reported that wild-type Endo-F3 and its mutant Endo-F3 D165A can transfer with complex-type glycan oxazoline and core Man3GlcNAc oxazoline for N-glycopeptide synthesis and antibody glycan remodeling.62-64 However, we found that neither wild-type Endo-F3 nor its glycosynthase mutant (D165A) could efficiently act on the azido-tagged disaccharide oxazoline for transglycosylation. As for endoglycosidases Endo-A (from Arthrobacter protophormiae),48,65 Endo-D,66 and Endo-CC,67 we found that they were not able to transfer the azide-disaccharide oxazoline (7) to the Fucα1,6GlcNAc-Herceptin. This is expected, as these three endoglycosidases cannot accept core-fucosylated GlcNAc as an acceptor. To test if nonfucosylated GlcNAc-Herceptin could serve as an acceptor, we removed the core-fucose from the Fucα1,6Glc-NAc-Herceptin (32) by treatment with an α-fucosidase (BfFucH) to produce the nonfucosylated Fc glycoform. However, Endo-A, Endo-D, and Endo-CC were not able to show the transglycosylation product, as monitored by LC–ESI–MS analysis, when even a large amount of enzyme (10%, w/w) was used (Table 1). These studies established wild-type Endo-S2 as a remarkably efficient endoglycosidase to act on the azido-tagged disaccharide oxazoline for transglycosylation without product hydrolysis.
One-Pot Chemoenzymatic Fc Glycan Remodeling with Different Azido-Tagged Disaccharide Oxazolines using Wild-Type Endo-S2.
Given the observation that wild-type Endo-S2 could act on the minimal azido-disaccharide oxazoline for efficient transglycosylation without product hydrolysis and the fact that Endo-S2 is highly active for antibody Fc deglycosylation, we envisioned that a “one-pot” and site-specific Fc glycan remodeling of antibodies should be possible to produce homogeneous azido-tagged antibody glycoforms. Indeed, when commercial Herceptin was treated with Endo-S2 together with the azido-disaccharide oxazoline (7) at rt in a PBS buffer (pH 7.0), LC–ESI–MS monitoring indicated that the Herceptin deglycosylation was complete within 10 min, followed by the appearance and accumulation of the transglycosylation product (33). The transglycosylation was complete within 1 h under the described conditions (Scheme 4) to give essentially quantitative transformation without the need of isolating the intermediates. We next examined the one-pot reactions with other synthetic disaccharide oxazolines carrying varied numbers of azide functional groups. Although these azide-functionalized disaccharide derivatives showed different activities toward wild-type Endo-S2, with the heavily functionalized disaccharide being less active, we found that all of them could serve as donor substrates for Endo-S2-catalyzed transgylcosylation, and excellent yields were obtained in a one-pot manner within 1–3 h when the enzyme amount was adjusted (0.1–1.0%, w/w) (Scheme 4). Notably, one portion of oxazoline was enough to achieve complete conversion for the disaccharides carrying one–three azido groups, because 7 and 12 were such good substrates that the transglycosylation could be finished within 1 h, whereas 17 was resistant to hydrolysis, and as a result, a relatively larger amount of enzyme but no additional oxazoline was needed. However, for the disaccharide carrying four (30) or six (31) azido groups, an additional portion of oxazoline was necessary to drive the reaction to completion due to the moderate hydrolytic activity of Endo-S2 toward these oxazoline donors. The final product was isolated by the protein A affinity column, and their identity and homogeneity were confirmed by LC–ESI–MS analysis of the intact whole antibody and the Fc domain glycoforms after IdeS treatment to disconnect the Fab and Fc domains (Supporting Information). It should be mentioned that in the case when a large excess of glycan oxazolines were used to drive the reaction, the excess glycan oxazoline could be recovered in the form of free oligosaccharide during protein A purification, which could be further purified by RP-HPLC and then converted into the glycan oxazoline in a single step with DMC/TEA, thus permitting the recycling of glycan oxazoline for transglycosylation.
Scheme 4. One-Pot Transglycosylation with Different Azido-Tagged Disaccharide Oxazolinesa.

aReagents and conditions: the reactions were conducted with 20 equiv oxazolines in PBS buffer at 25 °C; (a) one additional portion of oxazoline (10 equiv) was added after 1 h; (b) three more portions of oxazoline (10 equiv each) were added every 30 min.
One-Pot Chemoenzymatic Fc Glycan Remodeling with Biotin- and Fluorophore-tagged Disaccharide Oxazolines.
Encouraged by the one-pot introduction of the azido functionality, we investigated whether we could directly transfer a more complex structure, such as biotin- or fluorophore-tagged disaccharide, to an intact antibody with Endo-S2; in this way, the antibody could be labeled in one step, which would be attractive for diagnostics and in vivo imaging and as tools for molecular biology.68-70 To this end, biotin or TAMRA was introduced to the disaccharide, either via amine coupling reaction (39) or click chemistry (40), and we found that the modified disaccharide oxazolines could still serve as good substrates of Endo-S2. The biotinylated disaccharide was transferred to the intact antibody to give 41 in 95% yield within 1.5 h with the catalytic amount of enzyme (0.1%, w/w), and the TAMRA-tagged disaccharide oxazoline could afford about 85% of product (42) if additional portions of oxazoline were added (Scheme 5). Taken together, the chemoenzymatic remodeling method described here showed great potential for one-step labeling of intact antibodies.
Scheme 5a.

aReagents and conditions: the reactions were conducted with 20 equiv oxazolines in PBS buffer; (a) in the case of the biotin tag, one additional portion (10 equiv) of oxazoline 39 was added after 60 min; (b) in the case of the fluorescent tag, four more portions (10 equiv each) of oxazoline 40 were added every 60 min to push the reaction to completion.
Synthesis of Structurally Well-Defined, Homogeneous ADCs by Copper-free Strain-Promoted Click Reactions.
With these azido-tagged antibodies in hand, next, we tried click chemistry to make ADCs, using monomethyl auristatin E (MMAE) as a model warhead, a microtubule-disrupting agent that has been used for making the FDA-approved ADCs (Scheme 6).5,8 The dibenzoazacyclooctyne (DBCO)-ended MMAE derivative with a cleavable dipeptide linker was incubated with the azido-tagged antibodies, and the reactions were monitored by LC–ESI–MS. We found that the click chemistry gradually afforded the desired compounds (43–47). Take the reaction between 8N3-modified Herceptin (36) and DBCO-PEG5-VC-PAB-MMAE as an example. All the eight sites were conjugated with drugs within 20 h as indicated by LC–ESI–MS (Figure 2). To further verify that the drugs were specifically conjugated to the Fc domain, the product was digested with the protease IdeS followed by LC–ESI–MS analysis.71 The results showed that the shift of molecular weight was consistent with the attached payloads (calculated for the Fc domain glycoform, M = 32,140; observed, 32,143, deconvoluted data), thus confirming the structure of the product. Notably, ion fragments (asterisked peaks in Figure 2) derived from the whole antibody or Fc domain were detected in ESI–MS analysis, which corresponded to a loss of the drug fragment (~762 Da) that has been reported in previous studies.24,72 Finally, the Man3-derived antibody carrying four azido groups was also conjugated with the payloads, affording another DAR4-ADC (48) that allowed us to investigate the influence of different components (Scheme 6).
Scheme 6. Synthesis of Structurally Defined ADCs with Different DARs by Click Chemistrya.

aReagents and conditions: the azido-tagged antibodies (final concentration 2 mg/mL) were incubated with DBCO-PEG5-VC-PAB-MMAE (3.3–5.0 equiv/per azido group) in DMSO/50 mM PB (3:7, v/v) at room temperature for 8–24 h.
Figure 2.

LC–ESI–MS analysis of ADC 46. (a) Whole antibody analysis of compound 36; (b) whole antibody analysis of compound 46; (c) Fc domain of compound 36; and (d) Fc domain of compound 46. Asterisked peaks indicate the ion fragments derived from the intact antibody (b) or the Fc domain (d). Shift of molecular weight for the whole antibody: 161,880 – 148,262 = 13,618 ≈ 1702 × 8 (equaling to attachment of eight payloads) and for the Fc fragment: 32,143 – 25,332 = 6811 ≈ 1702 × 4 (equaling to attachment of four payloads).
Comparative Study of the Cancer Cell Killing Potency of the ADCs with Different DARs.
To demonstrate the potency of ADCs with different DARs, we conducted cytotoxicity assay in breast cancer cell lines expressing high or low levels of HER2. The results showed that all the ADCs achieved significant cell killing of the high target (HER2)—expressing cell line BT474—and ADCs with higher DARs were more potent, as indicated by the half-maximal effective concentration (EC50) values (Figure 3). It was found that the potency of the ADCs for cell killing was proportional to the DAR of the ADCs. For example, the EC50 of 44 (0.104 nM) was reduced to about a half of that of 43 (0.170 nM) when the DAR increased from 2 to 4; the EC50 of 46 (0.080 nM) was reduced to about threefold of that of 43 when the DAR was increased from 2 to 8 (Figure 3). These results are consistent with the previously reported observation, where site-specifically conjugated ADCs with varied DARs (2–8) are compared.13 In addition, the site-specific high-loaded ADC (e.g., DAR8) is substantially more efficacious than the low-loaded ADC (e.g., DAR2) in a mouse xenograft model.13 On the other hand, no substantial killing was observed in the T47D cell line that expresses a low level of HER2 by ADCs with DAR 2–6 under the tested concentrations, indicating the high selectivity of the ADCs for the target cells. However, for ADCs with higher DARs, nonspecific cell killing was observed under high concentrations especially for the ADC (47) with DAR12, which was not surprising since ADCs carrying multiple hydrophobic linkers tended to attach to the cell surface nonspecifically, resulting in cytotoxicity to normal cells independent of specific antigen expression. Swapping with hydrophilic payloads might be a solution to overcome the nonspecific cell killing. Taken together, the results suggest that ADCs made by this method carrying 6–8 payloads per antibody might result in an optimal potency and safety profile. In addition to ADCs, the azido-tagged antibodies described here can be also used for other antibody conjugations, such as site-selective construction of antibody–enzyme conjugates,73,74 antibody–cell conjugates,75,76 antibody–antibiotic conjugates,77 and lysosome-targeting chimeras for targeted protein degradation.78-81
Figure 3.
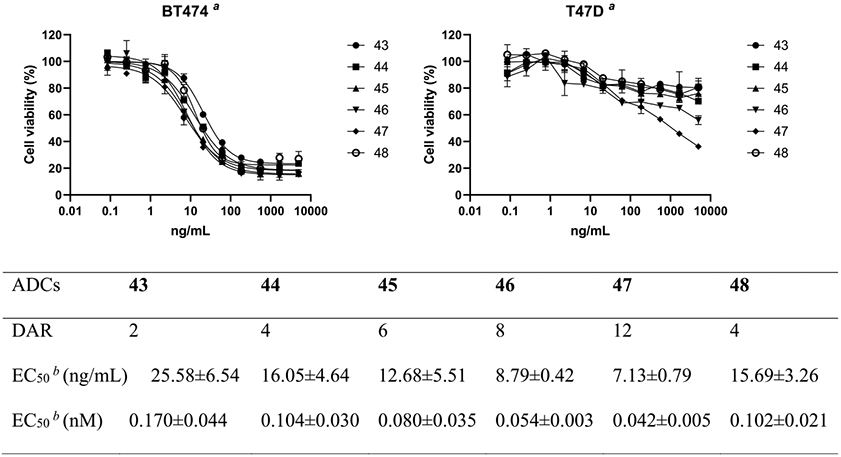
Cell killing studies in the breast cancer cell line BT474 (HER2 overexpression) and T47D (HER2 low expression). All assays were performed in triplicate. aRepresentative results of two independent assays. bAverage of two independent assays for the BT474 cell line.
CONCLUSIONS
A general and robust chemoenzymatic method for Fc glycan-mediated antibody labeling and conjugation is established. This method is enabled by the design, synthesis, and evaluation of various functionalized disaccharide oxazolines as donor substrates for Fc-specific endoglycosidase-catalyzed transglycosylation. The discovery that wild-type Endo-S2 exhibited excellent activity toward various selectively modified disaccharide oxazolines for transglycosylation, yet the resulting modified antibodies were resistant to enzymatic hydrolysis due to the modifications has led to a general platform for site-specific antibody labeling and conjugation. In particular, the relaxed substrate specificity of Endo-S2 allows direct labeling of antibodies with azide-, biotin-, or fluorescent tags, making it possible to achieve single-step labeling of intact antibodies in a site-selective manner. The flexibility to introduce varied numbers of azide functional groups provides a general and robust strategy to produce homogeneous ADCs with well-defined DARs ranging from 2 to 12. Since all the IgG antibodies carry highly conserved Fc-N-glycans, it is expected that this general Fc-glycan-mediated labeling and conjugation method will find wide applications not only for antibody–drug conjugation but also for cell labeling, imaging, and diagnosis.
METHODS
Chemical Synthesis of Glycan Substrates.
General.
All chemicals, reagents, and solvents were purchased from Sigma-Aldrich and TCI and unless specially noted applied in the reaction without further purification. Thin-layer chromatography was performed using silica gel on glass plates (Sigma-Aldrich), and spots were detected under UV light (254 nm) and then charred with 5% (v/v) sulfuric acid in EtOH or cerium molybdate stain followed by heating at 150 °C. Silica gel (200–425 mesh) for flash chromatography was purchased from Sigma-Aldrich. NMR spectra were recorded on a 400 MHz spectrometer (Bruker, Tokyo, Japan) with CDCl3 or D2O as the solvent. The chemical shifts were assigned in ppm, and multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Coupling constants (J) are reported in Hertz. MALDI-TOF was performed on a Bruker Autoflex speed mass spectrometer in the positive reflectron mode with DHB (ACN/H2O = 1:1) as the matrix. HRMS was performed on an Exactive Plus Orbitrap mass spectrometer (Thermo Scientific) equipped with a C18 column. Preparative HPLC was performed with a Waters 600 HPLC instrument and Waters C18 columns (7.0 μm 19 × 300 mm). The column was eluted with a suitable gradient of MeCN–H2O containing 0.1% FA at a flow rate of 10 mL/min. The detailed procedures for the chemical synthesis of the disaccharide derivatives and other small-molecule compounds (1—31, 39, and 40) are provided in the Supporting Information. The syntheses of antibody conjugates are described below.
Preparation of Azido-, Biotin-, or TAMRA-Functionalized Antibodies.
General.
LC–MS analysis was performed on an Ultimate 3000 HPLC system coupled to an Exactive Plus Orbitrap mass spectrometer (Thermo Fischer Scientific) with a C4 (whole antibody, gradient, 5–95% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) or C8 (IdeS digestion, gradient, 25–35% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min, or 5–95% aq MeCN containing 0.1% FA for 6 min, 0.4 mL/min) column. Deconvolution data were transformed using MagTran software.
Synthesis of 33.
A solution of commercial Herceptin (1.0 mg) and oxazoline 7 (186 μm, 20 eq per reaction site) was incubated with wild-type Endo-S2 (1.0 μg) at 25 °C in 40 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. Within 1 h, LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%; the product was purified using protein A chromatography to give 33 (920 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 147,224 Da; found (m/z), 147,224 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 24,813 Da; found (m/z), 24,814 (deconvolution data).
Synthesis of 34.
A solution of commercial Herceptin (1.0 mg) and oxazoline 12 (143 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (1.0 μg) at 25 °C in 40 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. Within 30 min, LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%; the product was purified using protein A chromatography to give 34 (900 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 146,909 Da; found (m/z), 146,912 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 24,656 Da; found (m/z), 24,656 (deconvolution data).
Synthesis of 35.
A solution of commercial Herceptin (1.0 mg) and oxazoline 17 (230 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (10 μg) at 25 °C in 40 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. After 2 h, LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%; the product was purified using protein A chromatography to give 35 (900 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 147,538 Da; found (m/z), 147,539 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 24,970 Da; found (m/z), 24,972 (deconvolution data).
Synthesis of 36.
A solution of commercial Herceptin (1.0 mg) and oxazoline 30 (328 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (2.0 μg) at 25 °C in 40 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. After 1 h, another portion of oxazoline (164 μg, 10 equiv per reaction site) was added, and the reaction was incubated for another 30 min. When LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%, the product was purified using protein A chromatography to give 36 (900 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 148,260 Da; found (m/z), 148,262 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 25,332 Da; found (m/z), 25,332 (deconvolution data).
Synthesis of 37.
A solution of commercial Herceptin (1.0 mg) and oxazoline 31 (400 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (10 μg) at 25 °C in 40 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. After 30 min, another portion of oxazoline (200 μg, 10 equiv per reaction site) was added, and the reaction was incubated for another 30 min. This procedure was repeated 2–3 times until LC–MS analysis indicated the conversion yield >90%, and the product was purified using protein A chromatography to give 37 (900 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 148,776 Da; found (m/z), 148,778 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 25,590 Da; found (m/z), 25,590 (deconvolution data).
Synthesis of 38.
A solution of deglycosylated Herceptin 32 (1.0 mg) and Man3GlcNAc tetrasaccharide oxazoline35 (200 μg, 20 equiv per reaction site) was incubated with Endo-S2 D184M (10 μg) at 25 °C in 100 μL of 150 mM PBS buffer (pH = 7.4), and the reaction was monitored by LC–MS of aliquots. Within 30 min, LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%, and the product was purified using protein A chromatography to give 38 (910 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 147,344 Da; found (m/z), 147,346 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 24,873 Da; found (m/z), 24,874 (deconvolution data).
Synthesis of 41.
A solution of commercial Herceptin (100 μg) and Biotin-tagged oxazoline 39 (38.4 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (0.1 μg) at 25 °C in 5 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. After 1 h, another portion of oxazoline (19.2 μg, 10 equiv per reaction site) was added, and the reaction was incubated for another 30 min. LC–MS analysis indicated the completion of the transglycosylation with conversion yield >95%. LC–MS: calculated for the whole antibody, M = 148,660 Da; found (m/z), 148,663 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 25,532 Da; found (m/z), 25,533 (deconvolution data).
Synthesis of 42.
A solution of commercial Herceptin (100 μg) and TAMRA-tagged oxazoline 40 (70 μg, 20 equiv per reaction site) was incubated with wild-type Endo-S2 (1.0 μg) at 25 °C in 5 μL of 150 mM PBS buffer (pH = 7.0), and the reaction was monitored by LC–MS of aliquots. After 60 min, another portion of oxazoline (35 μg, 10 equiv per reaction site) was added, and the reaction was incubated for another 60 min. This procedure was repeated 4–5 times, and LC–MS analysis indicated that the conversion yield was ca. 85%. LC–MS: calculated for the whole antibody, M = 150,968 Da; found (m/z), 150,967 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 26,687 Da; found (m/z), 26,685 (deconvolution data).
Synthesis of 43.
To a solution of 34 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/25 μL) was added the drug payload (DBCO-PEG5-VC-PAB-MMAE)43 (5.0 mg/mL, 4.6 μL, 10 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 43 (150 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 150,313 Da; found (m/z), 150,314 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 26,358 Da; found (m/z), 26,359 (deconvolution data).
Synthesis of 44.
To a solution of 33 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/20 μL) was added the drug payload (5.0 mg/mL, 9.2 μL, 20 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 44 (142 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 154,032 Da; found (m/z), 154,034 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 28,217 Da; found (m/z), 28,219 (deconvolution data).
Synthesis of 45.
To a solution of 35 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/19 μL) was added the drug payload (5.0 mg/mL, 11.3 μL, 25 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 45 (120 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 157,750 Da; found (m/z), 157,753 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 30,076 Da; found (m/z), 30,079 (deconvolution data).
Synthesis of 46.
To a solution of 36 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/15 μL) was added the drug payload (5.0 mg/mL, 13.6 μL, 30 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 46 (120 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 161,876 Da; found (m/z), 161,880 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 32,140 Da; found (m/z), 32,143 (deconvolution data).
Synthesis of 47.
To a solution of 37 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/10 μL) was added the drug payload (5.0 mg/mL, 18.5 μL, 40 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 47 (100 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 169,200 Da; found (m/z), 169,205 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 35,802 Da; found (m/z), 35,805 (deconvolution data).
Synthesis of 48.
To a solution of 38 (200 μg) in a mixture of 50 mM PB/DMSO (70 μL/20 μL) was added the drug payload (5.0 mg/mL, 9.2 μL, 20 equiv), and the reaction was incubated at room temperature. After LC–MS analysis indicated the completion of the reaction, the mixture was diluted with 50 mM PB (3 mL) and filtered using a 0.22 μm filter to remove most of the hydrophobic payload, and the residue was purified using protein A chromatography to give 48 (130 μg as measured using Nanodrop). LC–MS: calculated for the whole antibody, M = 154,151 Da; found (m/z), 154,154 (deconvolution data); after IdeS digestion, LC–MS calculated for the Fc part, M = 28,277 Da; found (m/z), 28,279 (deconvolution data).
Cell Killing Studies with Breast Cancer Cell Lines.
BT474 cells (ATCC HTB-20) were maintained in a suspension in Hybri-Care medium (ATCC 46-X) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT). T47D cells (ATCC HTB-133) were maintained in a suspension in RPMI-1640 medium (ATCC 30-2001) containing FBS, 4 mg/L insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT). For the cytotoxicity assays, cells were planted into 96-well plates with 10,000 cells per well. These plates were incubated overnight at 37 °C and 5% CO2. Serial threefold dilution was applied to the ADC samples with the corresponding medium from 5000 to 0.085 ng/mL. The samples were added to three wells (150 μL per well) with every single concentration, and the cells were cultured at 37 °C and 5% CO2 for three days before the addition of cell counting kit-8 (Sigma). The absorbance of formazan released by viable cells was measured at 450 nm using a spectrophotometer after incubation for 2–3 h at 37 °C and 5% CO2. Finally, the EC50 values and the cell viability curve were calculated using GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH Grants R01AI155716 and R01GM096973 to L.-X.W.)
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00597.
Synthetic procedures and characterization of compounds 1–31, 39, and 40; LC–ESI–MS analysis of transglycosylation and click reactions; and copies of NMR spectra (PDF)
The authors declare the following competing financial interest(s): LXW is the founder and a major shareholder of GlycoT Therapeutics LLC. Other authors declare no conflict of interest.
REFERENCES
- (1).Scott AM; Wolchok JD; Old LJ Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [DOI] [PubMed] [Google Scholar]
- (2).Chan AC; Carter PJ Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol 2010, 10, 301–316. [DOI] [PubMed] [Google Scholar]
- (3).Samuelsson A; Towers TL; Ravetch JV Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 2001, 291, 484–486. [DOI] [PubMed] [Google Scholar]
- (4).Sadiki A; Vaidya SR; Abdollahi M; Bhardwaj G; Dolan ME; Turna H; Arora V; Sanjeev A; Robinson TD; Koid A; Amin A; Zhou ZS Site-specific conjugation of native antibody. Antibiot. Ther 2020, 3, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Walsh SJ; Bargh JD; Dannheim FM; Hanby AR; Seki H; Counsell AJ; Ou X; Fowler E; Ashman N; Takada Y; Isidro-Llobet A; Parker JS; Carroll JS; Spring DR Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev 2021, 50, 1305–1353. [DOI] [PubMed] [Google Scholar]
- (6).Wang L-X; Tong X; Li C; Giddens JP; Li T Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem 2019, 88, 433–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chari RVJ; Miller ML; Widdison WC Antibody–drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed 2014, 53, 3796–3827. [DOI] [PubMed] [Google Scholar]
- (8).do Pazo C; Nawaz K; Webster RM The oncology market for antibody–drug conjugates. Nat. Rev. Drug Discovery 2021, 20, 583–584. [DOI] [PubMed] [Google Scholar]
- (9).Drago JZ; Modi S; Chandarlapaty S Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol 2021, 18, 327–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (11).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; Lu Y; Meng YG; Ng C; Yang J; Lee CC; Duenas E; Gorrell J; Katta V; Kim A; McDorman K; Flagella K; Venook R; Ross S; Spencer SD; Lee Wong W; Lowman HB; Vandlen R; Sliwkowski MX; Scheller RH; Polakis P; Mallet W Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol 2008, 26, 925–932. [DOI] [PubMed] [Google Scholar]
- (12).Shen B-Q; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu S-F; Mai E; Li D; Tibbitts J; Baudys J; Saad OM; Scales SJ; McDonald PJ; Hass PE; Eigenbrot C; Nguyen T; Solis WA; Fuji RN; Flagella KM; Patel D; Spencer SD; Khawli LA; Ebens A; Wong WL; Vandlen R; Kaur S; Sliwkowski MX; Scheller RH; Polakis P; Junutula JR Conjugation site modulates the in vivo stability and therapeutic activity of antibody–drug conjugates. Nat. Biotechnol 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- (13).Strop P; Delaria K; Foletti D; Witt JM; Hasa-Moreno A; Poulsen K; Casas MG; Dorywalska M; Farias S; Pios A; Lui V; Dushin R; Zhou D; Navaratnam T; Tran T-T; Sutton J; Lindquist KC; Han B; Liu S-H; Shelton DL; Pons J; Rajpal A Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol 2015, 33, 694–696. [DOI] [PubMed] [Google Scholar]
- (14).Shinmi D; Taguchi E; Iwano J; Yamaguchi T; Masuda K; Enokizono J; Shiraishi Y One-Step Conjugation Method for Site-Specific Antibody–Drug Conjugates through Reactive Cysteine-Engineered Antibodies. Bioconjugate Chem. 2016, 27, 1324–1331. [DOI] [PubMed] [Google Scholar]
- (15).Axup JY; Bajjuri KM; Ritland M; Hutchins BM; Kim CH; Kazane SA; Halder R; Forsyth JS; Santidrian AF; Stafin K; Lu Y; Tran H; Seller AJ; Biroc SL; Szydlik A; Pinkstaff JK; Tian F; Sinha SC; Felding-Habermann B; Smider VV; Schultz PG Synthesis of site-specific antibody–drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).VanBrunt MP; Shanebeck K; Caldwell Z; Johnson J; Thompson P; Martin T; Dong H; Li G; Xu H; D’Hooge F; Masterson L; Bariola P; Tiberghien A; Ezeadi E; Williams DG; Hartley JA; Howard PW; Grabstein KH; Bowen MA; Marelli M Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody–Drug Conjugates Using Click Cycloaddition Chemistry. Bioconjugate Chem. 2015, 26, 2249–2260. [DOI] [PubMed] [Google Scholar]
- (17).Nilchan N; Li X; Pedzisa L; Nanna AR; Roush WR; Rader C Dual-mechanistic antibody–drug conjugate via site-specific selenocysteine/cysteine conjugation. Antibiot. Ther 2019, 2, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Forte N; Chudasama V; Baker JR Homogeneous antibody–drug conjugates via site-selective disulfide bridging. Drug Discovery Today: Technol. 2018, 30, 11–20. [DOI] [PubMed] [Google Scholar]
- (19).Badescu G; Bryant P; Bird M; Henseleit K; Swierkosz J; Parekh V; Tommasi R; Pawlisz E; Jurlewicz K; Farys M; Camper N; Sheng X; Fisher M; Grygorash R; Kyle A; Abhilash A; Frigerio M; Edwards J; Godwin A Bridging disulfides for stable and defined antibody drug conjugates. Bioconjugate Chem. 2014, 25, 1124–1136. [DOI] [PubMed] [Google Scholar]
- (20).Casi G; Huguenin-Dezot N; Zuberbühler K; Scheuermann J; Neri D Site-Specific Traceless Coupling of Potent Cytotoxic Drugs to Recombinant Antibodies for Pharmacodelivery. J. Am. Chem. Soc 2012, 134, 5887–5892. [DOI] [PubMed] [Google Scholar]
- (21).Zhang C; Dai P; Vinogradov AA; Gates ZP; Pentelute BL Site-Selective Cysteine-Cyclooctyne Conjugation. Angew. Chem., Int. Ed 2018, 57, 6459–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schneider H; Deweid L; Avrutina O; Kolmar H Recent progress in transglutaminase-mediated assembly of antibody–drug conjugates. Anal. Biochem 2020, 595, 113615. [DOI] [PubMed] [Google Scholar]
- (23).Dennler P; Chiotellis A; Fischer E; Brégeon D; Belmant C; Gauthier L; Lhospice F; Romagne F; Schibli R Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjugate Chem. 2014, 25, 569–578. [DOI] [PubMed] [Google Scholar]
- (24).Anami Y; Xiong W; Gui X; Deng M; Zhang CC; Zhang N; An Z; Tsuchikama K Enzymatic conjugation using branched linkers for constructing homogeneous antibody–drug conjugates with high potency. Org. Biomol. Chem 2017, 15, 5635–5642. [DOI] [PubMed] [Google Scholar]
- (25).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stan AC; Radu DL; Casares S; Bona CA; Brumeanu T-D Antineoplastic Efficacy of Doxorubicin Enzymatically Assembled on Galactose Residues of a Monoclonal Antibody Specific for the Carcinoembryonic Antigen. Cancer Res. 1999, 59, 115–121. [PubMed] [Google Scholar]
- (27).Zhou Q; Stefano JE; Manning C; Kyazike J; Chen B; Gianolio DA; Park A; Busch M; Bird J; Zheng X; Simonds-Mannes H; Kim J; Gregory RC; Miller RJ; Brondyk WH; Dhal PK; Pan CQ Site-specific antibody–drug conjugation through glycoengineering. Bioconjugate Chem. 2014, 25, 510–520. [DOI] [PubMed] [Google Scholar]
- (28).Faridoon F; Shi W; Qin K; Tang Y; Li M; Guan D; Tian X; Jiang B; Dong J; Tang F; Huang W New linker structures applied in glycosite-specific antibody drug conjugates. Org. Chem. Front 2019, 6, 3144–3149. [Google Scholar]
- (29).van Geel R; Wijdeven MA; Heesbeen R; Verkade JMM; Wasiel AA; van Berkel SS; van Delft FL Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconjugate Chem. 2015, 26, 2233–2242. [DOI] [PubMed] [Google Scholar]
- (30).Li X; Fang T; Boons GJ Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem., Int. Ed 2014, 53, 7179–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Okeley NM; Toki BE; Zhang X; Jeffrey SC; Burke PJ; Alley SC; Senter PD Metabolic engineering of monoclonal antibody carbohydrates for antibody–drug conjugation. Bioconjugate Chem. 2013, 24, 1650–1655. [DOI] [PubMed] [Google Scholar]
- (32).Zhu Z; Ramakrishnan B; Li J; Wang Y; Feng Y; Prabakaran P; Colantonio S; Dyba MA; Qasba PK; Dimitrov DS Site-specific antibody–drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs 2014, 6, 1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li C; Wang L-X Chemoenzymatic Methods for the Synthesis of Glycoproteins. Chem. Rev 2018, 118, 8359–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Fairbanks AJ The ENGases: versatile biocatalysts for the production of homogeneous N-linked glycopeptides and glycoproteins. Chem. Soc. Rev 2017, 46, 5128–5146. [DOI] [PubMed] [Google Scholar]
- (35).Huang W; Giddens J; Fan S-Q; Toonstra C; Wang L-X Chemoenzymatic glycoengineering of intact IgG antibodies for gain of functions. J. Am. Chem. Soc 2012, 134, 12308–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Li T; Li C; Quan DN; Bentley WE; Wang L-X Site-specific immobilization of endoglycosidases for streamlined chemoenzymatic glycan remodeling of antibodies. Carbohydr. Res 2018, 458–459, 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Parsons TB; Struwe WB; Gault J; Yamamoto K; Taylor TA; Raj R; Wals K; Mohammed S; Robinson CV; Benesch JLP; Davis BG Optimal Synthetic Glycosylation of a Therapeutic Antibody. Angew. Chem., Int. Ed 2016, 55, 2361–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Li T; Tong X; Yang Q; Giddens JP; Wang L-X Glycosynthase Mutants of Endoglycosidase S2 Show Potent Transglycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem 2016, 291, 16508–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Manabe S; Yamaguchi Y Antibody Glycoengineering and Homogeneous Antibody–Drug Conjugate Preparation. Chem. Rec 2021, DOI: 10.1002/tcr.202100054. [DOI] [PubMed] [Google Scholar]
- (40).Tang F; Wang L-X; Huang W Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody–drug conjugates. Nat. Protoc 2017, 12, 1702–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Guo W; Tang F; Qin K; Zhou M; Le Z; Huang W Glycoengineering and glycosite-specific labeling of serum IgGs from various species. Carbohydr. Res 2017, 446–447, 32–39. [DOI] [PubMed] [Google Scholar]
- (42).Tang F; Yang Y; Tang Y; Tang S; Yang L; Sun B; Jiang B; Dong J; Liu H; Huang M; Geng M-Y; Huang W One-pot N-glycosylation remodeling of IgG with non-natural sialylglycopeptides enables glycosite-specific and dual-payload antibody–drug conjugates. Org. Biomol. Chem 2016, 14, 9501–9518. [DOI] [PubMed] [Google Scholar]
- (43).Ou C; Li C; Zhang R; Yang Q; Zong G; Dai Y; Francis RL; Bournazos S; Ravetch JV; Wang L-X One-Pot Conversion of Free Sialoglycans to Functionalized Glycan Oxazolines and Efficient Synthesis of Homogeneous Antibody–Drug Conjugates through Site-Specific Chemoenzymatic Glycan Remodeling. Bioconjugate Chem. 2021, 32, 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Goodfellow JJ; Baruah K; Yamamoto K; Bonomelli C; Krishna B; Harvey DJ; Crispin M; Scanlan CN; Davis BG An endoglycosidase with alternative glycan specificity allows broadened glycoprotein remodelling. J. Am. Chem. Soc 2012, 134, 8030–8033. [DOI] [PubMed] [Google Scholar]
- (45).Tong X; Li T; Orwenyo J; Toonstra C; Wang L-X One-pot enzymatic glycan remodeling of a therapeutic monoclonal antibody by endoglycosidase S (Endo-S) from Streptococcus pyogenes. Bioorg. Med. Chem 2018, 26, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zeng Y; Wang J; Li B; Hauser S; Li H; Wang L-X Glycopeptide synthesis through endo-glycosidase-catalyzed oligosaccharide transfer of sugar oxazolines: probing substrate structural requirement. Chem.—Eur. J 2006, 12, 3355–3364. [DOI] [PubMed] [Google Scholar]
- (47).Goto K; Kurogochi M; Takashima S; Mori M; Matsuda A; Mizuno M Site-specific protein PEGylation catalyzed by endo-β-N-acetylglucosaminidase. Tetrahedron Lett. 2020, 61, 151475. [Google Scholar]
- (48).Ochiai H; Huang W; Wang L-X Expeditious chemo-enzymatic synthesis of homogeneous N-glycoproteins carrying defined oligosaccharide ligands. J. Am. Chem. Soc 2008, 130, 13790–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Niemietz M; Perkams L; Hoffman J; Eller S; Unverzagt C Selective oxidative debenzylation of mono- and oligosaccharides in the presence of azides. Chem. Commun 2011, 47, 10485–10487. [DOI] [PubMed] [Google Scholar]
- (50).Noguchi M; Tanaka T; Gyakushi H; Kobayashi A; Shoda S.-i. Efficient synthesis of sugar oxazolines from unprotected N-acetyl-2-amino sugars by using chloroformamidinium reagent in water. J. Org. Chem 2009, 74, 2210–2212. [DOI] [PubMed] [Google Scholar]
- (51).Adinolfi M; Barone G; Guariniello L; Iadonisi A Facile cleavage of carbohydrate benzyl ethers and benzylidene acetals using the NaBrO3Na2S2O4 reagent under two-phase conditions. Tetrahedron Lett. 1999, 40, 8439–8441. [Google Scholar]
- (52).Nyffeler PT; Liang C-H; Koeller KM; Wong C-H The chemistry of amine-azide interconversion: catalytic diazotransfer and regioselective azide reduction. J. Am. Chem. Soc 2002, 124, 10773–10778. [DOI] [PubMed] [Google Scholar]
- (53).Lyon RP; Bovee TD; Doronina SO; Burke PJ; Hunter JH; Neff-LaFord HD; Jonas M; Anderson ME; Setter JR; Senter PD Reducing hydrophobicity of homogeneous antibody–drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol 2015, 33, 733–735. [DOI] [PubMed] [Google Scholar]
- (54).Bardia A; Mayer IA; Vahdat LT; Tolaney SM; Isakoff SJ; Diamond JR; O’Shaughnessy J; Moroose RL; Santin AD; Abramson VG; Shah NC; Rugo HS; Goldenberg DM; Sweidan AM; Iannone R; Washkowitz S; Sharkey RM; Wegener WA; Kalinsky K Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med 2019, 380, 741–751. [DOI] [PubMed] [Google Scholar]
- (55).Viricel W; Fournet G; Beaumel S; Perrial E; Papot S; Dumontet C; Joseph B Monodisperse polysarcosine-based highly-loaded antibody–drug conjugates. Chem. Sci 2019, 10, 4048–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Linclau B; Cini E; Oakes CS; Josse S; Light M; Ironmonger V Stereoarrays with an all-carbon quaternary center: diastereoselective desymmetrization of prochiral malonaldehydes. Angew. Chem., Int. Ed 2012, 51, 1232–1235. [DOI] [PubMed] [Google Scholar]
- (57).Tanaka K; Tanaka T; Hasegawa T; Shionoya M A close-packed, highly insulating organic thin monolayer on Si(111). Chem. Lett 2008, 37, 440–441. [Google Scholar]
- (58).Sjögren J; Struwe WB; Cosgrave EFJ; Rudd PM; Stervander M; Allhorn M; Hollands A; Nizet V; Collin M EndoS2 is a unique and conserved enzyme of serotype M49 group A Streptococcus that hydrolyses N-linked glycans on IgG and alpha1-acid glycoprotein. Biochem. J 2013, 455, 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Collin M; Olsen A EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J. 2001, 20, 3046–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Tarentino AL; Quinones G; Plummer TH Jr. Overexpression and purification of non-glycosylated recombinant endo-beta-N-acetylglucosaminidase F3. Glycobiology 1995, 5, 599–601. [DOI] [PubMed] [Google Scholar]
- (61).Plummer TH; Phelan AW; Tarentino AL Porcine fibrinogen glycopeptides: substrates for detecting endo-beta-N-acetylglucosaminidases F2 and F3. Anal. Biochem 1996, 235, 98–101. [DOI] [PubMed] [Google Scholar]
- (62).Huang W; Li J; Wang L-X Unusual transglycosylation activity of Flavobacterium meningosepticum Endoglycosidases enables convergent chemoenzymatic synthesis of core fucosylated complex N-glycopeptides. ChemBioChem 2011, 12, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Giddens JP; Lomino JV; Amin MN; Wang L-X Endo-F3 Glycosynthase Mutants Enable Chemoenzymatic Synthesis of Core-fucosylated Triantennary Complex Type Glycopeptides and Glycoproteins. J. Biol. Chem 2016, 291, 9356–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Giddens JP; Lomino JV; DiLillo DJ; Ravetch JV; Wang L-X Site-selective chemoenzymatic glycoengineering of Fab and Fc glycans of a therapeutic antibody. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 12023–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Takegawa K; Yamabe K; Fujita K; Tabuchi M; Mita M; Izu H; Watanabe A; Asada Y; Sano M; Kondo A; Kato I; Iwahara S Cloning, sequencing, and expression of Arthrobacter protophormiae endo- beta-N-acetylglucosaminidase in Escherichia coli. Arch. Biochem. Biophys 1997, 338, 22–28. [DOI] [PubMed] [Google Scholar]
- (66).Fan S-Q; Huang W; Wang L-X Remarkable transglycosylation activity of glycosynthase mutants of endo-D, an endo-beta-N-acetylglucosaminidase from Streptococcus pneumoniae. J. Biol. Chem 2012, 287, 11272–11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Eshima Y; Higuchi Y; Kinoshita T; Nakakita S.-i.; Takegawa K Transglycosylation Activity of Glycosynthase Mutants of Endo-beta-N-Acetylglucosaminidase from Coprinopsis cinerea. PLoS One 2015, 10, No. e0132859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Freise AC; Wu AM In vivo imaging with antibodies and engineered fragments. Mol. Immunol 2015, 67, 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Zhou Q Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Boeggeman E; Ramakrishnan B; Pasek M; Manzoni M; Puri A; Loomis KH; Waybright TJ; Qasba PK Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: application for cell surface antigen detection. Bioconjugate Chem. 2009, 20, 1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Chevreux G; Tilly N; Bihoreau N Fast analysis of recombinant monoclonal antibodies using IdeS proteolytic digestion and electrospray mass spectrometry. Anal. Biochem 2011, 415, 212–214. [DOI] [PubMed] [Google Scholar]
- (72).Anami Y; Yamazaki CM; Xiong W; Gui X; Zhang N; An Z; Tsuchikama K Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun 2018, 9, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Gray MA; Stanczak MA; Mantuano NR; Xiao H Pijnenborg JFA; Malaker SA; Miller CL; Weidenbacher PA; Tanzo JT; Ahn G; Woods EC; Läubli H; Bertozzi CR Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol 2020, 16, 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Xiao H; Woods EC; Vukojicic P; Bertozzi CR Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 10304–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Wang X; Luo X; Tian Y; Wu T; Weng J; Li Z; Ye F; Huang X Equipping Natural Killer Cells with Cetuximab through Metabolic Glycoengineering and Bioorthogonal Reaction for Targeted Treatment of KRAS Mutant Colorectal Cancer. ACS Chem. Biol 2021, 16, 724–730. [DOI] [PubMed] [Google Scholar]
- (76).Li J; Chen M; Liu Z; Zhang L; Felding BH; Moremen KW; Lauvau G; Abadier M; Ley K; Wu P A Single-Step Chemoenzymatic Reaction for the Construction of Antibody–Cell Conjugates. ACS Cent. Sci 2018, 4, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lehar SM; Pillow T; Xu M; Staben L; Kajihara KK; Vandlen R; DePalatis L; Raab H; Hazenbos WL; Hiroshi Morisaki J; Kim J; Park S; Darwish M; Lee B-C; Hernandez H; Loyet KM; Lupardus P; Fong R; Yan D; Chalouni C; Luis E; Khalfin Y; Plise E; Cheong J; Lyssikatos JP; Strandh M; Koefoed K; Andersen PS; Flygare JA; Wah Tan M; Brown EJ; Mariathasan S Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [DOI] [PubMed] [Google Scholar]
- (78).Ahn G; Banik SM; Miller CL; Riley NM; Cochran JR; Bertozzi CR LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol 2021, 17, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Banik SM; Pedram K; Wisnovsky S; Ahn G; Riley NM; Bertozzi CR Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Zhou Y; Teng P; Montgomery NT; Li X; Tang W Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci 2021, 7, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Powell M; Blaskovich MAT; Hansford KA Targeted Protein Degradation: The New Frontier of Antimicrobial Discovery? ACS Infect. Dis 2021, 7, 2050–2067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.