

Abstract
We recently reported a potent, selective, and in vivo efficacious AKT degrader, MS21, which is a von Hippel–Lindau (VHL)-recruiting proteolysis targeting chimera (PROTAC) based on the AKT inhibitor AZD5363. However, no structure–activity relationship (SAR) studies that resulted in this discovery have been reported. Herein, we present our SAR studies that led to the discovery of MS21, another VHL-recruiting AKT degrader, MS143 (compound 20) with similar potency as MS21, and a novel cereblon (CRBN)-recruiting PROTAC, MS5033 (compound 35). Compounds 20 and 35 induced rapid and robust AKT degradation in a concentration- and time-dependent manner via hijacking the ubiquitin-proteasome system. Compound 20 suppressed cell growth more effectively than AZD5363 in multiple cancer cell lines. Furthermore, 20 and 35 displayed good plasma exposure levels in mice and are suitable for in vivo efficacy studies. Lastly, compound 20 effectively suppressed tumor growth in vivo in a xenograft model without apparent toxicity.
Graphical Abstract
INTRODUCTION
AKT [also known as protein kinase B (PKB)] has three isoforms (AKT1, AKT2, and AKT3) that share a high degree of sequence identity in their catalytic domains.1–4 AKT functions as a central element of the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway, which is frequently hyperactivated in human cancers and other human diseases.5 The activation mechanisms include amplification and/or mutation of three AKT genes, somatic mutation of PI3K (encoded by the PIK3CA gene) at the p110α catalytic subunit, loss-of-function mutations or downregulation of tumor suppressor PTEN, and alterations of receptor tyrosine kinase (RTK) signaling.4,6 Apart from the dysregulated PI3K/AKT/mTOR signaling pathway that plays a major role in tumor growth, survival, metabolism, and resistance to anticancer therapy,7–10 the overexpression of AKT has also been implicated in cancer progression and development.11 Therefore, AKT has been considered as an important therapeutic target for the prevention and treatment of human cancers.12–14
A number of AKT inhibitors including adenosine triphosphate (ATP)-competitive inhibitors (such as GSK690693,15 GDC-0068,16 and AZD536317) and allosteric inhibitors (such as MK220618 and ARQ-09219) have entered clinical investigations. However, to date, none of these inhibitors have demonstrated robust clinical efficacy, under-scoring the urgent need for novel therapeutic strategies for the treatment of AKT-mediated cancers and other diseases.13,20–23 Extensive studies have established that specific knockdown/knockout of AKT isoforms through genetic technologies significantly inhibited cancer cell proliferation and suppressed tumor progression in many AKT-driven human malignancies.24–27 For example, depletion of AKT1 and AKT2 substantially inhibited the proliferation of prostate cancer cells,28,29 and knockdown of AKT2 or AKT3 by RNA interference remarkably reduced the growth of glioma cells.30 In addition, the downregulation of AKT3 attenuated the growth of triple-negative breast cancer (TNBC) cells.31 Taken together, these studies highlighted that the downregulation or depletion of AKT protein levels can be a potential therapeutic strategy for treating AKT-dependent cancers.
Proteolysis targeting chimeras (PROTACs) are promising new therapeutic modalities with a unique mechanism of action (MOA), which induces the degradation of the target protein through hijacking the endogenous ubiquitin proteasome system (UPS).32–38 PROTACs, which chemically down-regulate target protein levels, could recapitulate biological effects of genetic knockout/knockdown and provide a potentially more effective therapeutic strategy than pharmacological inhibition of AKT kinase activity.
A few AKT PROTAC degraders have been reported by us and others. You et al. reported INY-03–041 as the first GDC-0068-derived cereblon (CRBN)-recruiting AKT degrader.39 We recently reported an extensive SAR study using GDC-0068 as an AKT binding moiety and the discovery of two additional AKT degraders that recruit CRBN (MS170) and von Hippel–Lindau (VHL) (MS98), respectively.40 We also reported extensive in vitro and in vivo characterization of a potent, selective, and in vivo efficacious AKT degrader, MS21, which is VHL-recruiting and derived from the AKT inhibitor AZD5363.41 However, SAR studies that resulted in the discovery of MS21 have not been reported. In this paper, we report the design, synthesis, and biological evaluation of a variety of AZD5363-derived AKT degraders. Through this extensive SAR campaign, we show how MS2141 was discovered as a potent and selective AKT degrader. In addition, we report another potent VHL-recruiting AKT degrader 20 (MS143) and a novel CRBN-recruiting AKT degrader 35 (MS5033). Both 20 and 35 induced rapid and efficient AKT degradation in cancer cells and displayed good bioavailability in mice via intraperitoneal (IP) administration. Moreover, 20 effectively inhibited tumor growth in vivo in a PC-3 cell line xenograft mouse model. Compounds 20 and 35 could be useful chemical tools for further studying AKT functions in health and disease. Further optimization of these degraders could provide potential AKT degradation therapies for AKT-dependent cancers and other diseases.
RESULTS AND DISCUSSION
Design and Biological Evaluation of AZD5363-Based AKT PROTAC Degraders.
AZD5363 is a highly potent and selective ATP-competitive pan-AKT inhibitor, which has been advanced into clinical trials for the treatment of solid tumors.17,21 The co-crystal structure of AKT1 in complex with AZD5363 revealed that the hydroxyl group of AZD5363 is solvent-exposed (Figure 1A, PDB: 4GV1).17 Previous SAR studies indicated that the hydroxyl group can be replaced with a morpholinyl group without a significant decrease in potency against AKT isomers (Figure 1B).17 To facilitate the linker installation, precursor 2 (AZD5363*), bearing a piperazine ring, was designed (Figure 1B).
Figure 1.
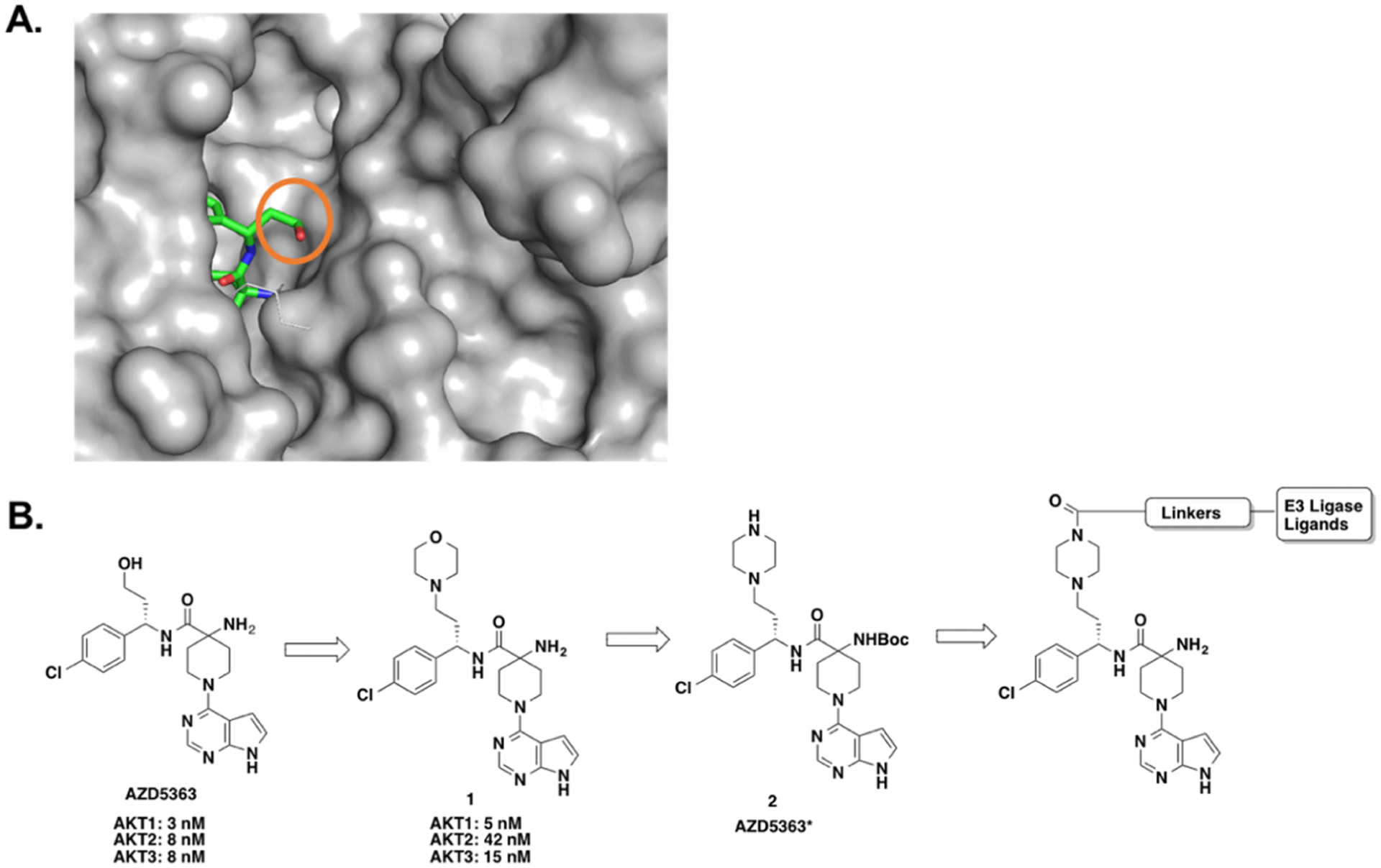
Design of putative AKT degraders. (A) Co-crystal structure of AKT1 (in gray) in complex with AZD5363 (in green, blue, and red) (PDB: 4GV1). The hydroxyl group of AZD5363 (marked by the orange cycle) is solvent-exposed. (B) Schematic design of AKT putative degraders based upon the AKT inhibitor AZD5363.
VHL/cullin 2 and CRBN/cullin 4A E3 ligase complexes have been widely harnessed by PROTACs to induce the degradation of targeted proteins, mainly because well-characterized small-molecule ligands of VHL and CRBN are readily available.42,43 VHL-1 and its analogues display good binding affinities to the VHL E3 ligase,44–46 and immunomodulatory drugs (IMiDs), such as pomalidomide (POM), lenalidomide, and thalidomide, are well characterized CRBN ligands.47,48 Therefore, we designed an initial set of AKT putative degraders by conjugating compound 2 to VHL-1 or POM through a variety of polyethylene glycol (PEG) or alkyl linkers (Figure 2).
Figure 2.
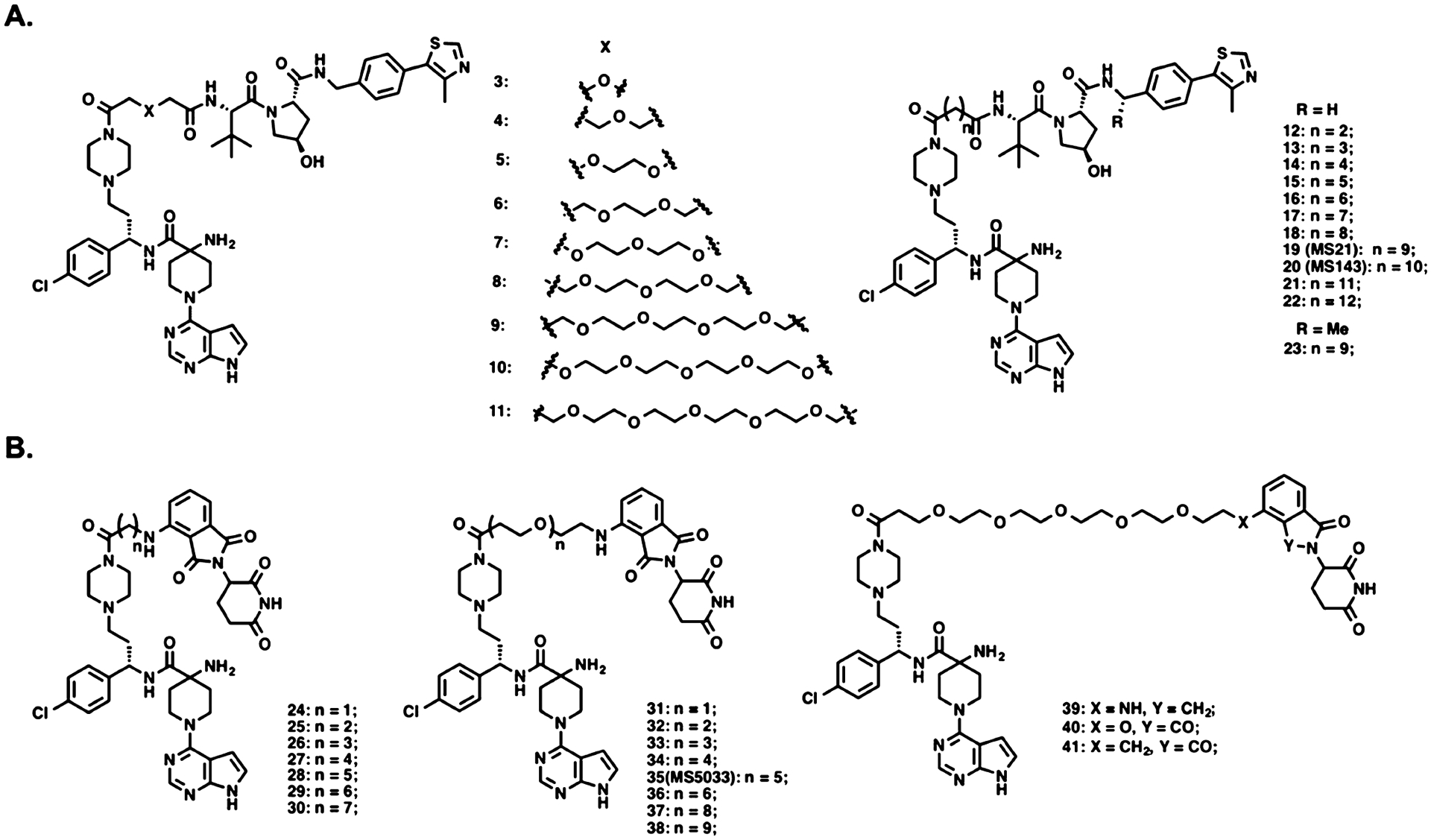
Chemical structures of designed AKT degraders based on AZD5363. (A) VHL-recruiting AKT degraders. (B) CRBN-recruiting AKT degraders.
These putative AKT degraders were evaluated using an immunoblotting assay through assessing the protein levels of total AKT (T-AKT), phosphorylated AKT (P-AKT), downstream signaling P-PRAS40, and biomarker P-S6 in BT474 cells, a breast cancer cell line (Figures 3 and 4). The cells were treated with the putative degraders at 1, 5, or 10 μM for 24 h using AZD5363 and the corresponding E3 ligase ligand (VHL-1 or POM) as controls. As shown in Figure 3, among the VHL-recruiting compounds with a PEG linker (3–11), compound 3, with the shortest PEG linker, induced the most significant T-AKT degradation, and displayed the most effective inhibition of the downstream signaling. VHL-1 as a control had no effect on the T-AKT protein level and downstream signaling. Interestingly, while the AKT inhibitor AZD5363 effectively inhibited the phosphorylation of downstream PRAS40 and ribosomal protein S6 and paradoxically upregulated the phosphorylated AKT level as expected,17,49 it also downregulated the T-AKT protein level. However, the effect of AZD5363 on the T-AKT protein level was less profound than that of 3. Increasing the linker length by two methylene groups (4) decreased the AKT degradation effectiveness. Longer PEG linkers (5–11) further diminished the degradation effectiveness. This linker length related SAR trend for compounds with a PEG linker is opposite to that of compounds with an alkyl linker. Compounds with a short alkyl linker, such as 12 (ethylene) and 13 (propylene), were inactive at reducing the T-AKT protein level. Compounds with a medium length linker, such as 14 (butylene) and 15 (pentylene), showed improved AKT degradation and significant downstream signaling inhibition at 5 μM. Compounds with an alkyl linker longer than pentylene (16–19) are the most potent AKT degraders and induced near-complete depletion of the T-AKT protein at concentrations as low as 1 μM. Among these compounds, compound 19 (MS21) exhibited the most profound AKT degradation and downstream signaling inhibition. Based on previous reports that a methylated VHL-1 analogue, (S,R,S)-AHPC-Me (VHL-2), can improve degradation potency,50–52 we designed compound 23, which incorporated this VHL ligand, as a direct analogue of 19. However, 23 did not show significant difference from 19 in degrading AKT and inhibiting the downstream signaling and colony formation (Figures 3 and S1). We further extended the alkyl linker by 1–3 methylene units to obtain compounds 20–22. Among these three analogues, compounds 20 and 21 displayed similar AKT protein degradation potency to 19 but slightly enhanced downstream signaling inhibition compared to 19 (Figure 3). Compounds 20 and 21 were also highly effective in inhibiting colony formation in BT474 cells, similar to compound 19 (Figure S1). Because compound 20 has a slightly shorter linker than compound 21, we selected compound 20 for further studies. Taken together, our SAR results of VHL-recruiting AKT degraders demonstrated that compounds with an alkyl linker induced AKT degradation more effectively than the compounds with a PEG linker, and compounds with a relatively long alkyl linker are more potent than the ones with a short alkyl linker.
Figure 3.
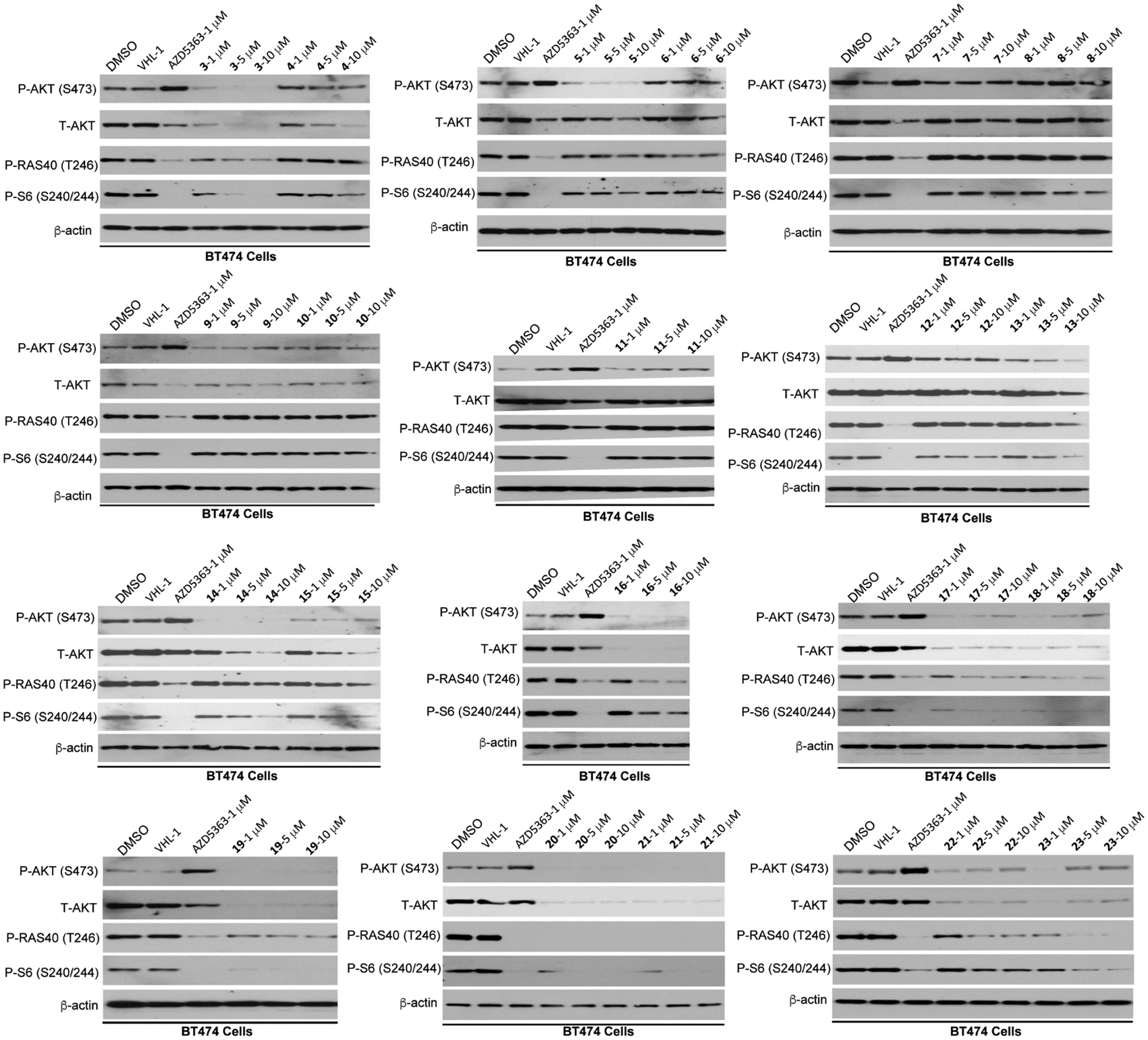
Effects of compounds 3–23 on reducing the AKT protein level and inhibiting the downstream signaling. BT474 cells were treated with AZD5363 (1 μM), VHL-1 (1 μM), or the indicated compound at 1, 5, and 10 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of T-AKT, P-AKT (S473), P-PRAS40 (T246), and P-S6 (S240/244). β-Actin was used as the loading control.
Figure 4.
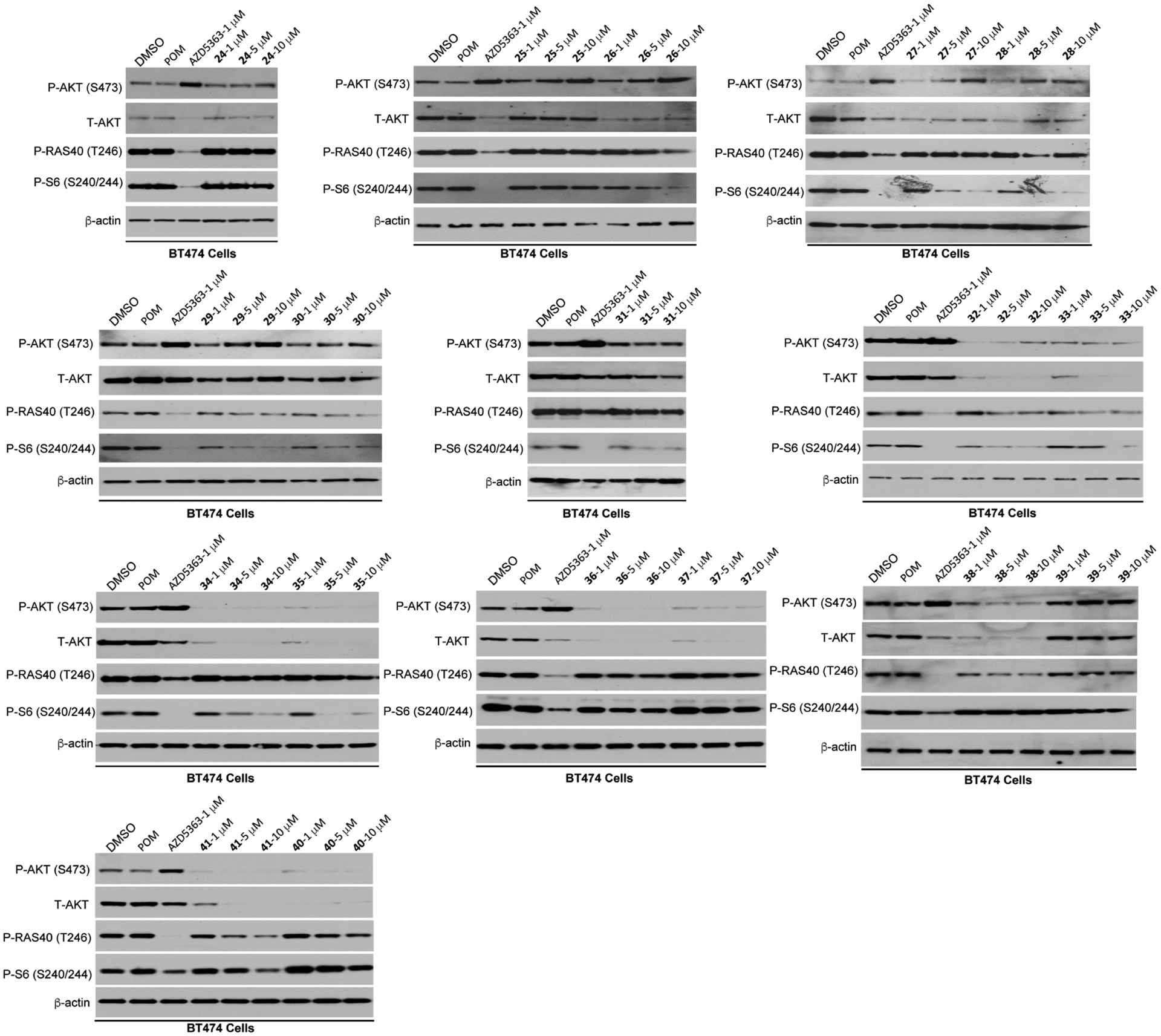
Effects of compounds 24–41 on reducing the AKT protein level and inhibiting the downstream signaling. BT474 cells were treated with AZD5363 (1 μM), POM (1 μM), or indicated compound at 1, 5, and 10 μM for 24 h. The cell lysates were analyzed by western blotting to examine the protein levels of T-AKT, P-AKT (S473), P-PRAS40 (T246), and P-S6 (S240/244). β-Actin was used as the loading control.
Compared with the VHL-recruiting degraders, the linker of the CRBN-recruiting compounds has different effects (Figure 4). Compounds with a medium length alkyl linker, such as 26, 27, and 28, displayed moderate effectiveness in AKT degradation and downstream signaling inhibition. Compounds with a short (24 and 25) or long (29 and 30) alkyl linker induced negligible AKT degradation at 1–10 μM concentrations. Switching from alkyl linkers to PEG linkers resulted in much more potent CRBN-recruiting AKT degraders (32–38). While compound 31 with the shortest PEG linker induced only moderate AKT degradation at 10 μM, compounds with a longer PEG linker (32–38) significantly reduced the AKT protein level at 1 μM. Among them, compound 35 was most effective in inhibiting the phosphorylation of ribosomal protein S6. Next, we designed three analogues of 35 by modifying the CRBN binder moiety (Figure 2B). Removal of one carbonyl group of the phthalimide moiety (39) significantly decreased effectiveness in AKT degradation (Figure 4). Replacing the amino group in compound 35 with either oxygen (40) or methylene group (41) did not improve either the AKT degradation or inhibition of colony formation in BT474 cells (Figures 4 and S2). Based on these SAR results, we selected compound 35 for additional characterization. Overall, these studies revealed the following SAR trends for CRBN-recruiting AKT degraders: (1) compounds with a PEG linker showed better AKT degradation potency than compounds with an alkyl linker, and (2) compounds with a long PEG linker are more potent than compounds with a short PEG linker.
Through the extensive SAR studies of AZD5363-based AKT degraders, we identified compounds 20 and 35 as promising VHL- and CRBR-recruiting AKT degraders, respectively. We thus developed two structurally similar negative control compounds: compound 42 (MS143N) for degrader 20, and compound 43 (MS5033N) for degrader 35 (Figure 5). Compound 42 incorporates a benzyl-protected hydroxyl proline group at the VHL binding region and preserves the same AKT binding motif and linker as compound 20 (Figure 5). The addition of the benzyl group to the hydroxyl proline moiety of VHL-1 (VHL-1-Bn) was designed to abrogate the key hydrogen bond interaction between the hydroxyl group and VHL.53 Using isothermal titration calorimetry (ITC), we confirmed that benzyl-protected VHL-1 indeed did not bind VHL (Figure S3). Compound 43 contains a methylated glutarimide ring, which abrogates its binding to CRBN54 and retains the identical AKT binding moiety and linker as compound 35 (Figure 5).
Figure 5.
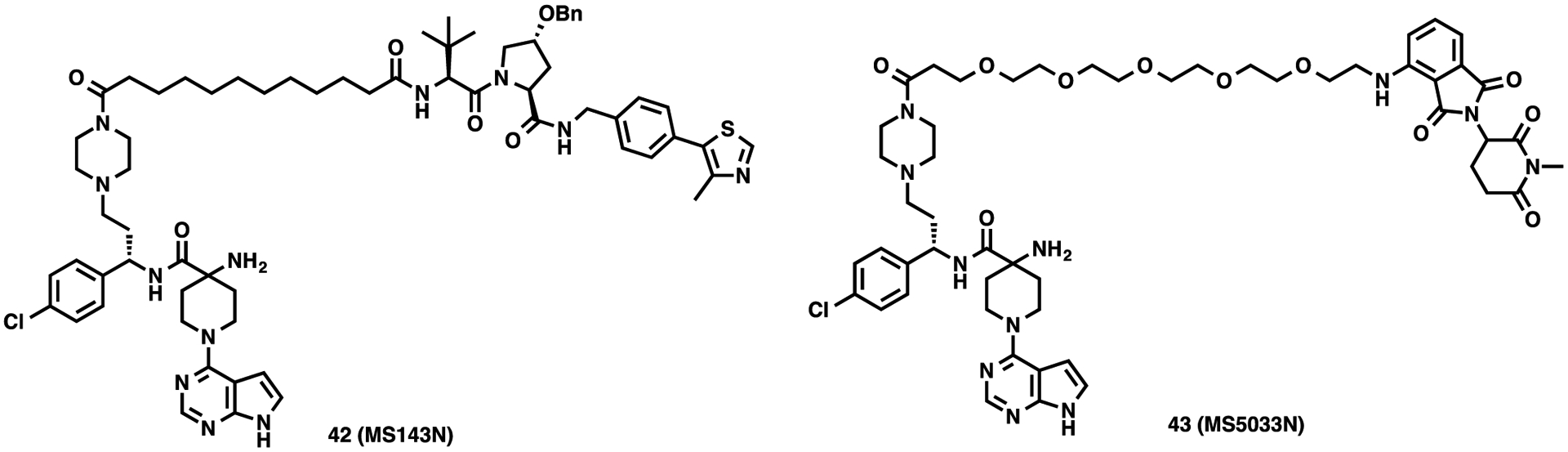
Chemical structures of negative control compounds 42 and 43.
Binding of Compounds 20, 35, 42, and 43 to AKT.
We next assessed binding affinities of degraders 20 and 35, and their negative compounds 42 and 43 to three AKT isoforms using a biochemical competition assay (Figure 6). The VHL-based degrader 20 showed good binding affinity to AKT1 (Kd = 26 ± 6.3 nM), AKT2 (Kd = 400 ± 200 nM), and AKT3 (Kd = 88 ± 140 nM), whereas the corresponding control compound 42 displayed weaker binding affinities to AKT1 (Kd = 150 ± 130 nM), AKT2 (Kd > 10 μM), and AKT3 (Kd = 2.9 ± 2.7 μM). The CRBN-based degrader 35 exhibited strong binding to AKT1 (Kd = 4.8 ± 5.6 nM), AKT2 (Kd = 160 ± 120 nM), and AKT3 (Kd = 22 ± 8.8 nM). Compared to 35, the N-methyl glutarimide negative control 43 retained similar binding affinities to AKT1 and AKT3 and displayed two fold decreased affinity to AKT2. We previously reported binding affinities of compound 19 and AZD5363 to the AKT isoforms in the same assay.41 The binding affinities of compound 20 to AKT1/2/3 are similar to those of compound 19.41 Compared to the parent inhibitor AZD5363,41 these degraders and their corresponding controls displayed weaker binding affinities to all three AKT isoforms. While it was unexpected that the binding affinities of compound 42 to AKT isoforms were lower than that of 20, compound 42 still retained good binding affinity to AKT1 and moderate binding affinity to AKT3. Because compounds 20 and 35 exhibited better affinity to AKT1 and AKT3 over AKT2, we sought to determine whether the observed binding selectivity for AKT1 and AKT3 over AKT2 could lead to selective degradation of AKT1 and AKT3 over AKT2. As illustrated in Figure S4, compound 20 at 100 nM reduced AKT1 and AKT3 protein levels by about 90% and the AKT2 protein level by about 50% in PC3 cells. To achieve approximately 90% of AKT2 degradation, a higher compound concentration (such as 300 nM–1 μM) was required. Therefore, degrader 20 indeed showed moderate degradation selectivity for AKT1 and AKT3 over AKT2.
Figure 6.
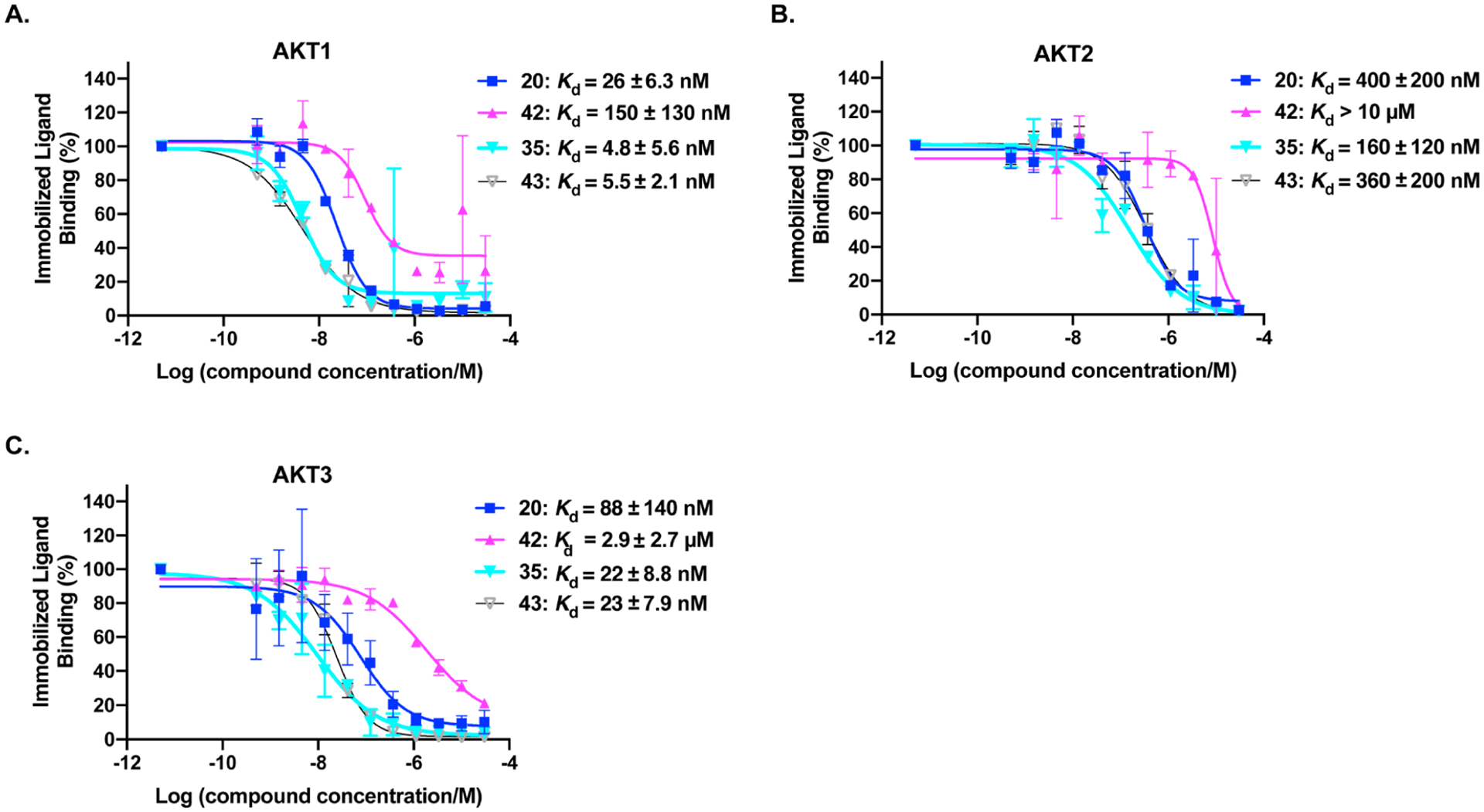
Binding affinities of degraders 20 and 35, and negative controls 42 and 43 to AKT1 (A), AKT2 (B), and AKT3 (C). The Kd determination experiments were performed in a competitive binding assay (KINOMEscan assay by Eurofins DiscoverX) in duplicate. The lowest concentration points represent data from the DMSO samples. Error bars represent ± SEM in duplicate independent experiments.
Compounds 20 and 35 Potently Induce AKT Degradation in PC3 Cells.
We further assessed the AKT degradation potency of 20 and 35 in PC3 cells (Figure 7A–B). Both 20 and 35 effectively induced T-AKT degradation in a concentration-dependent manner. The VHL-recruiting degrader 20 (DC50 = 46 ± 21 nM) was significantly more potent than the CRBN-recruiting degrader 35 (DC50 = 430 ± 300 nM) at degrading T-AKT. The complete T-AKT depletion was achieved when PC3 cells were treated with 20 at 300 nM or higher concentrations. Importantly, the “hooker effect” was not observed for either compound at concentrations up to 10 μM. To demonstrate that the observed AKT degradation is due to recruiting the corresponding E3 ligase, we evaluated the AKT degradation effect of negative controls 42 and 43. As expected, both 42 and 43 did not reduce the T-AKT protein level in PC3 cells at concentrations up to 10 μM (Figure 7C,D), suggesting that the VHL and CRBN E3 ligases were involved in the AKT degradation induced by degraders 20 and 35, respectively.
Figure 7.
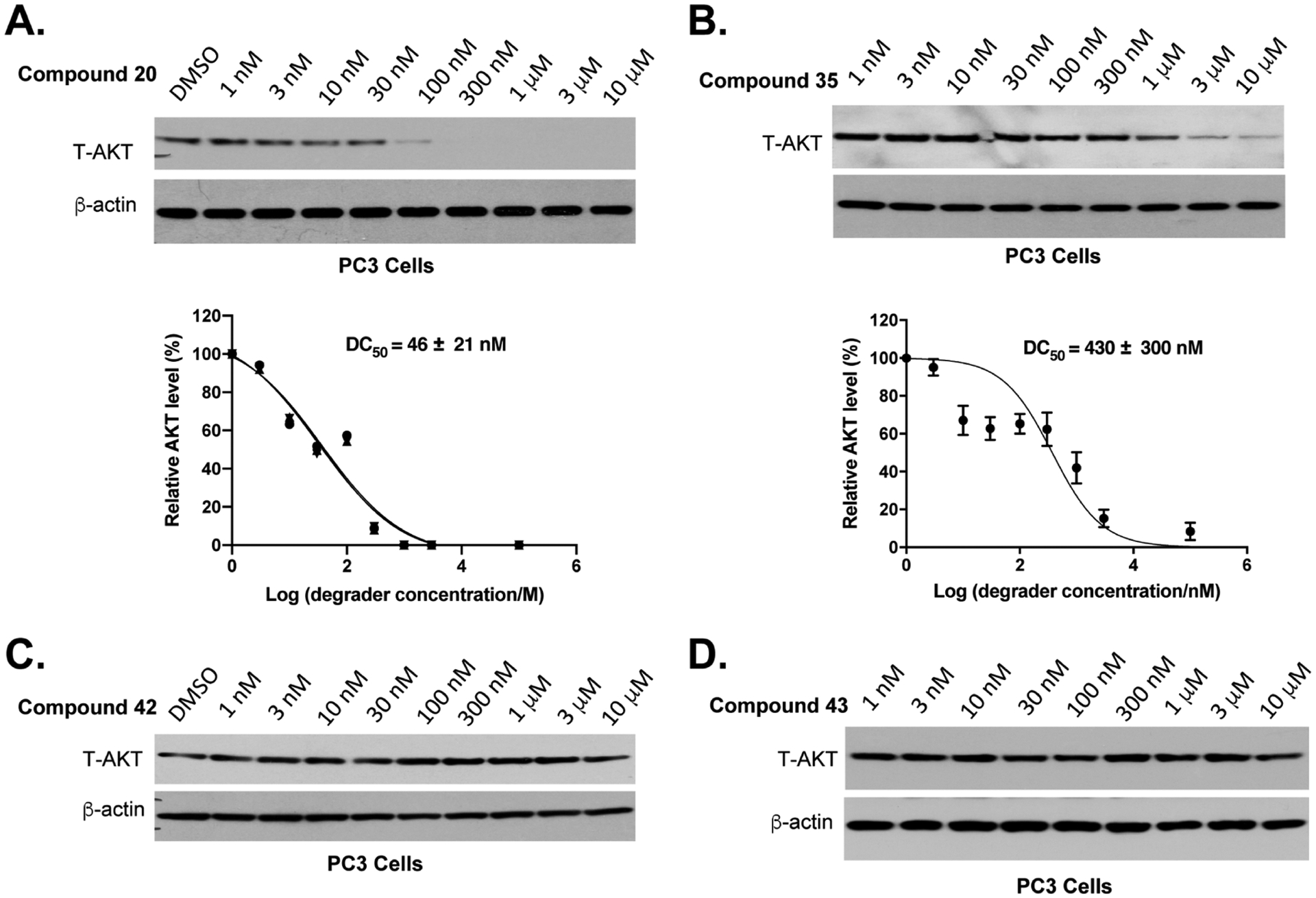
Effects of compounds 20, 35, 42, and 43 on reducing the AKT protein level in PC3 cells. PC3 cells were treated with DMSO or compound 20 (A), 35 (B), 42 (C), or 43 (D) at the indicated concentrations for 24 h. The T-AKT protein level was determined using western blots. The DC50 values of 20 (A) and 35 (B) were quantified from three independent biological experiments.
We next assessed kinetics of the AKT degradation and downstream signaling inhibition in PC3 cells treated with 20 or 35 at 1 μM (Figure 8). As a control, the parent inhibitor AZD5363 did not significantly reduce the AKT protein level at 1 μM for 24 h, but effectively inhibited PRAS40 and S6 phosphorylation. On the other hand, compound 20 induced substantial AKT degradation and inhibited PRAS40 and S6 phosphorylation within 4 h (Figure 8A). Complete depletion of T-AKT was achieved within 8 h. Compared to the VHL-recruiting degrader 20, the CRBN-recruiting degrader 35 displayed slower kinetics in degrading AKT. T-AKT was significantly downregulated at 8 h, and the maximum degradation was achieved at around 24 h (Figure 8B). We also performed washout studies for both 20 and 35. Interestingly, treatment with degrader 20 led to profound and persistent degradation of both T-AKT and P-AKT. The T-AKT level and downstream signaling inhibition were partially recovered after 48 h post-treatment (Figure 8C). On the other hand, treatment with degrader 35, the AKT protein level rebounded substantially at 24 h and was completely recovered at 48 h (Figure 8D). We also observed that downstream signaling inhibition was near fully recovered at 24 h after the treatment of 35 or the parent inhibitor AZD5363. Taken together, these results suggest that the VHL-recruiting degrader 20 mediates rapid and sustained AKT degradation and the CRBN-recruiting degrader 35 is less effective than the VHL-recruiting degrader 20.
Figure 8.
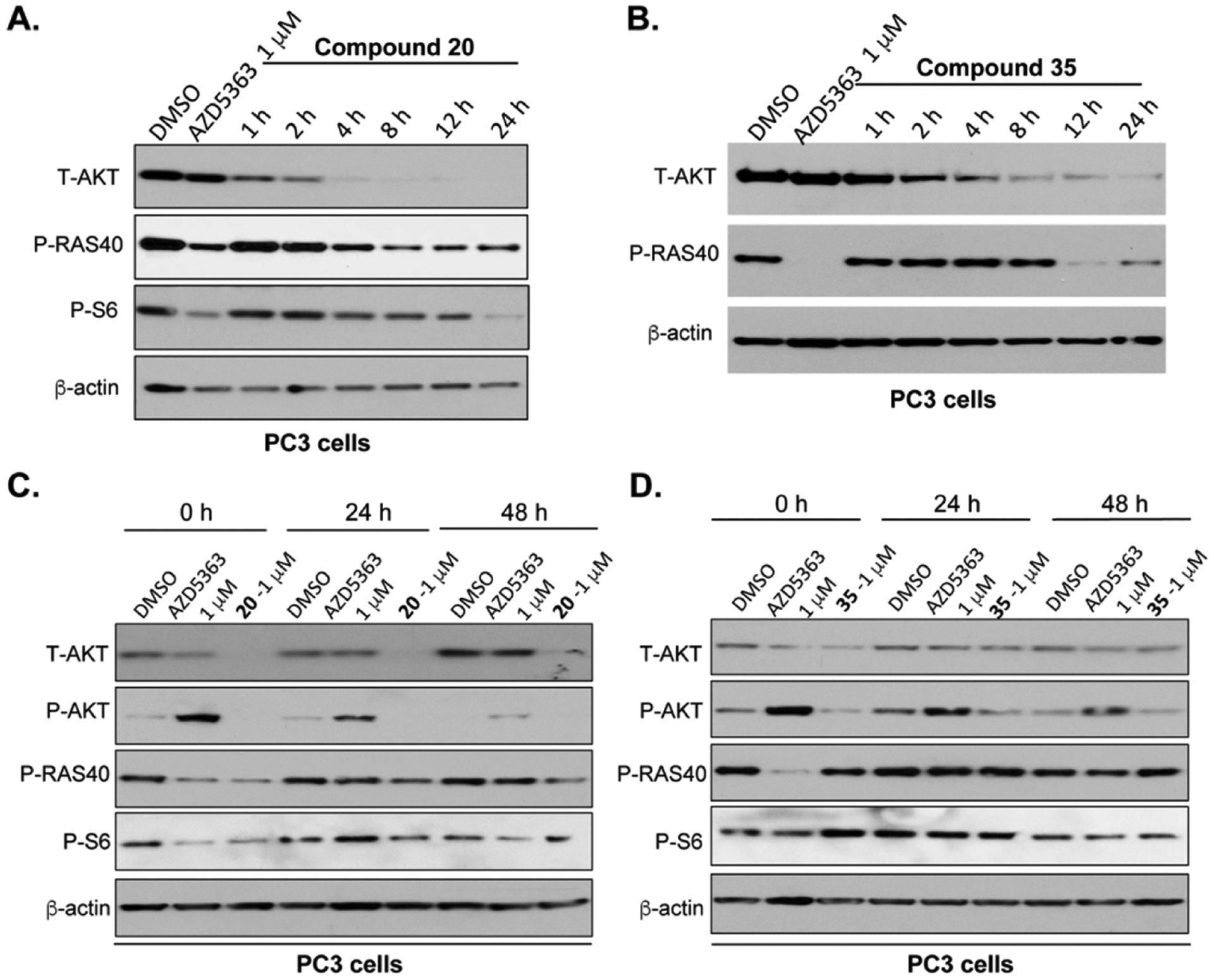
Compounds 20 and 35 induced AKT degradation and inhibited the downstream signaling in a time-dependent and reversible manner. (A,B) PC3 cells treated with DMSO (24 h), AZD5363 (24 h), 20, or 35 at 1 μM were collected at the indicated time points for western blotting analysis. (C,D) Western blots of T-AKT, P-AKT, P-PRAS40, and P-S6 and β-actin post-treatment of PC3 cells with DMSO, AZD5363, 20, or 35 at 1 μM for 24 h at the indicated time. The compounds were washed out with fresh medium after the 24 h treatment.
Compounds 20 and 35 Induce AKT Degradation through Hijacking the UPS.
To determine whether the AKT protein degradation induced by degraders 20 and 35 is mediated through the corresponding E3 ligase and UPS, we conducted a series of competition experiments in PC3 cells. As shown in Figure 9A, an excess amount of VHL ligand (S,R,S)-AHPC-Me (VHL-2), which competes with 20 at the VHL E3 ligase binding site, restored the AKT protein level effectively, suggesting that the AKT degradation induced by 20 is dependent on the VHL E3 ligase. Pretreatment with the NEDD8-activating enzyme (NAE) inhibitor MLN4924 or proteasome inhibitor MG-132 also significantly diminished the AKT degradation effect of 20, suggesting that the AKT degradation mediated by 20 is dependent on the cullin E3 ligase complex and proteasome. Furthermore, saturating the AKT binding site with an excess amount of AZD5363 also rescued the AKT protein level, indicating that AKT binding is required for AKT degradation. Similar results were obtained for the CRBN-recruiting degrader 35. Pretreatment with POM (1 μM), MLN4924 (1 μM), MG-132 (20 μM), or AZD5363 (1 μM) diminished 35’s ability in reducing the AKT protein in PC3 cells (Figure 9B). Moreover, pretreatment with these compounds, except AZD5363, not only rescued the AKT protein level but also attenuated the downstream signaling inhibition. These MOA study results, together with our observation that negative controls 42 and 43 were unable to induce T-AKT degradation (Figure 7C,D), demonstrated that 20 and 35 were bona fide AKT degraders, promoting AKT degradation in an E3 ligase- and UPS-dependent manner.
Figure 9.
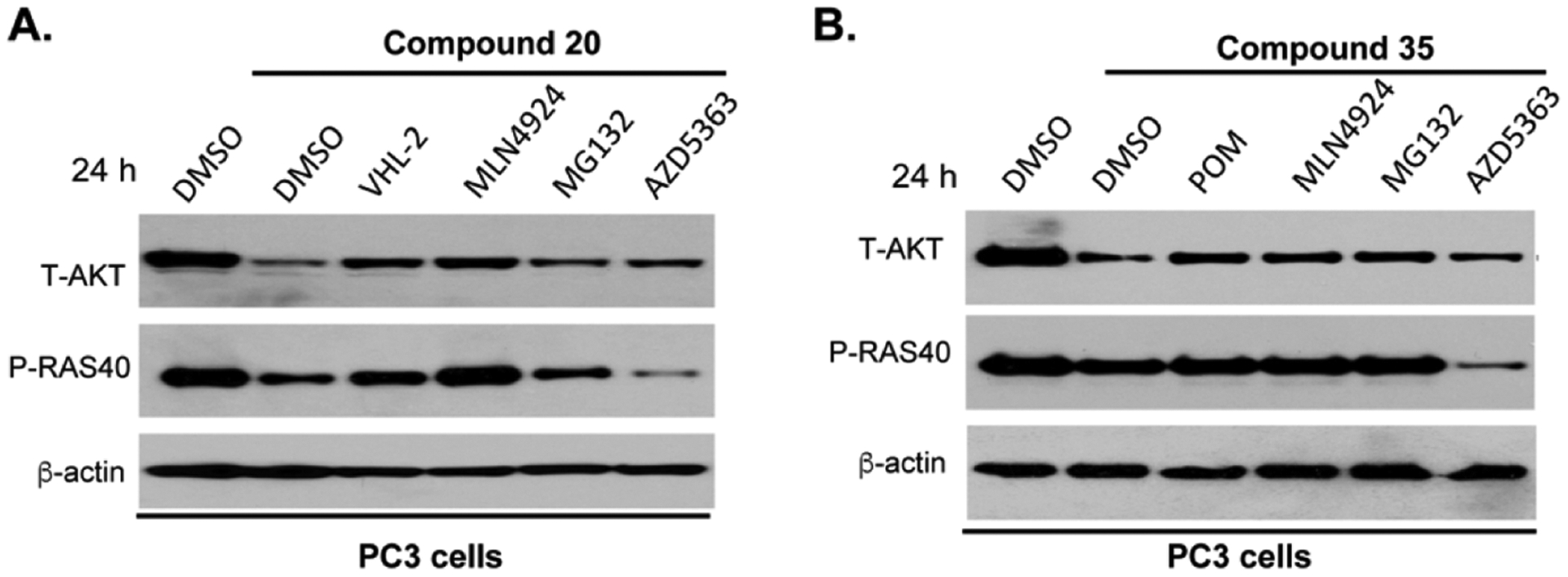
AKT degradation induced by compound 20 (A) or compound 35 (B) is mediated through the UPS. PC3 cells were treated with DMSO, AZD5363 (1 μM), MLN4924 (1 μM), MG132 (10 μM), VHL-2 (1 μM), or POM (1 μM) for 2 h, before the cells were treated with 20 or 35 for additional 24 h. The T-AKT and P-PRAS40 protein levels were determined by western blots.
Compounds 20 and 35 Potently Inhibit the Proliferation of Cancer Cells.
As described earlier, degrader 20 effectively inhibited colony formation in BT474 cells (Figure S1). We next investigated the antiproliferation activity of 20 and 35 in prostate cancer PC3 cells and TNBC MDA-MB-468 cells using AZD5363, 42, and 43 as controls (Figure 10). The VHL-recruiting compound 20 (GI50 = 0.8 ± 0.3 μM) was approximately 8-fold more potent than AZD5363 (GI50 = 6.5 ± 2.9 μM) in inhibiting the cell growth of PC3 cells (Figure 10A). The CRBN-recruiting degrader 35 (GI50 = 10.8 ± 5.6 μM) was about 14-fold less potent than 20, which is consistent to the weaker degradation potency of 35 (DC50 = 430 ± 300 nM) than that of 20 (DC50 = 46 ± 21 nM, Figure 7A,B). In MDA-MB-468 cells, compound 20 (GI50 = 1.0 ± 0.3 μM) was also more potent than AZD5363 (GI50 = 3.8 ± 1.6 μM) and 35 (GI50 = 4.8 ± 1.9 μM) in inhibiting cell growth. Negative controls 42 and 43 were inactive in these antiproliferation assays, suggesting that compound 20’s improved potency in cell growth inhibition is at least partially due to its AKT degradation activity (Figure 10A,B). Furthermore, as described earlier, degrader 20 effectively inhibited colony formation in BT474 cells (Figure S1). Overall, these results indicate that AKT degraders 20 and 35 were more potent in suppressing cancer cell proliferation than their corresponding negative control compounds 42 and 43. Importantly, the VHL-recruiting degrader 20 was more potent than AZD5363 and the CRBN-recruiting degrader 35 in inhibiting the growth in both PC3 and MDA-MB-468 cells.
Figure 10.
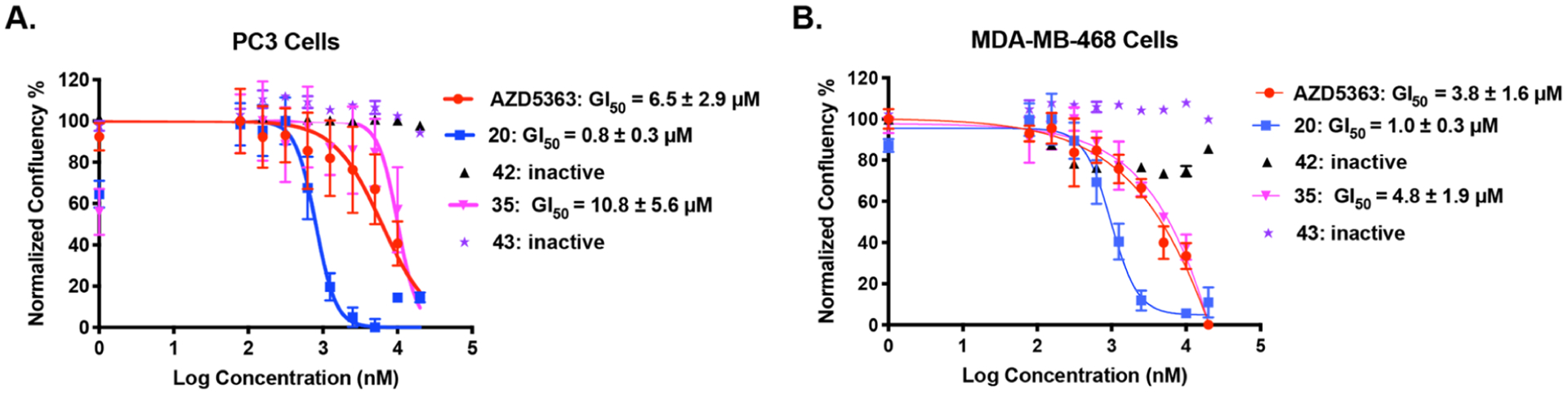
Compounds 20 and 35 inhibit cancer cell proliferation. PC3 (A) and MDA-MB-468 (B) cells were treated with serial dilutions of AZD5363, 20, 42, 35, or 43 for 5 days. Cell growth was determined using calculations derived from phase-contrast images in IncuCyte. Error bars indicate the standard errors in three independent experiments.
Compounds 20 and 35 are Bioavailable in Mice.
We next assessed the pharmacokinetic (PK) properties of compounds 20 and 35 in mice (Figure 11). A single dose of compound 20 at 75 mg/kg by IP injection achieved the maximum plasma concentration (Cmax) of 7 μM at 2 h (Tmax). The exposure level of approximate 3.7 μM was maintained over the 12 h period post-injection with an area under the curve (AUC) value of 63 600 h·ng/mL. Furthermore, the CRBN-recruiting compound 35 showed comparable Cmax (8 μM) at 1 h (Tmax) to 20 at a higher dose (150 mg/kg) via IP injection with an AUC value of 32 500 h·ng/mL. Compared to their determined DC50 and GI50 values in cellular assays, these high plasma concentrations and exposure suggest that these degraders have the potential to degrade ATK in vivo. A limitation in these PK studies is that we did not determine free drug concentrations. Future studies to determine this parameter are warranted. It is worth noting that these degraders were well tolerated by the tested mice, suggesting that they could be suitable for in vivo efficacy studies.
Figure 11.
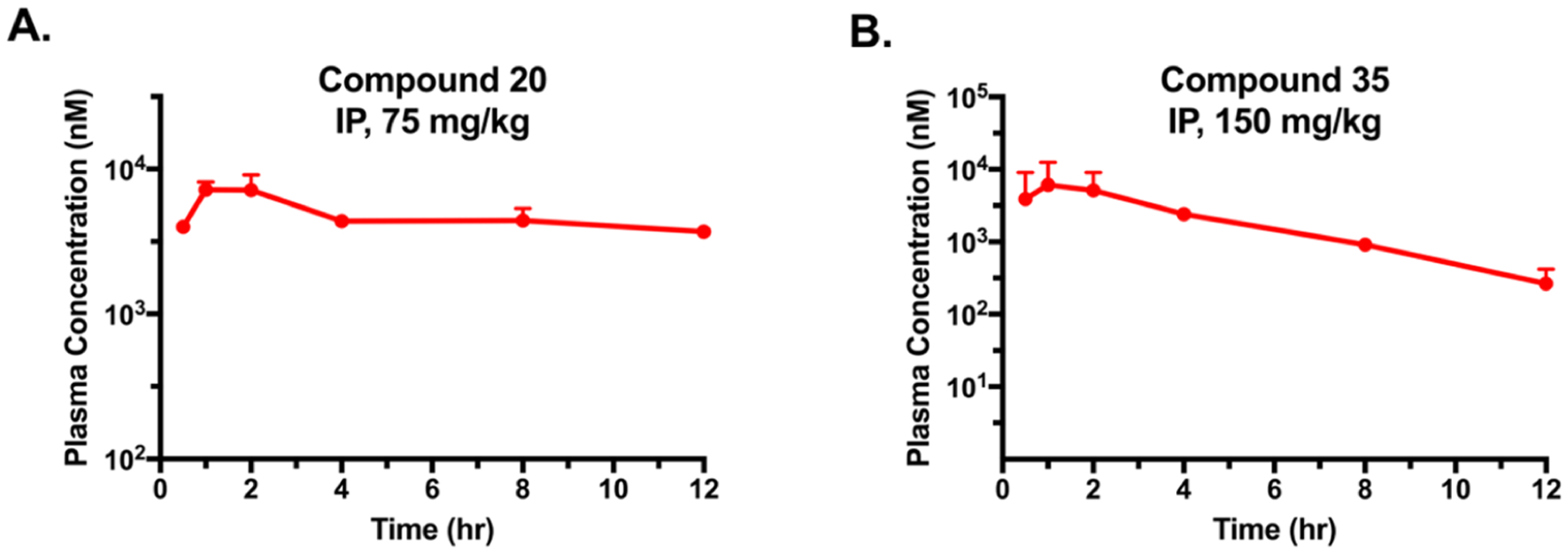
AKT degraders 20 and 35 are bioavailable in mice. Plasma concentrations of 20 (A) following a single 75 mg/kg IP injection over 12 h and 35 (B) following a single 150 mg/kg IP injection over 12 h in male Swiss Albino mice. Experiments were carried out in biological triplicate per time point, with the values representing mean concentrations ±SEM.
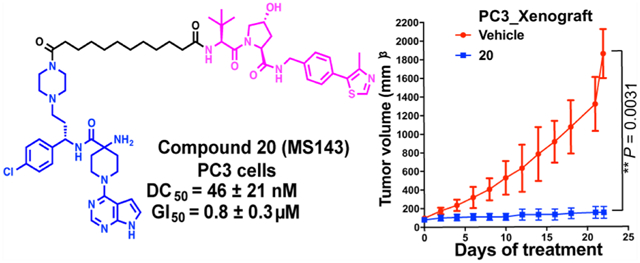
Compound 20 Effectively Suppresses Tumor Growth In Vivo in a Xenograft Model.
Because compound 20 exhibited more potent cell antiproliferation activity and displayed higher exposure in mouse plasma than compound 35, we selected compound 20 for testing its in vivo efficacy in a PC3 cell line xenograft model (Figure 12). We treated mice bearing PC3 xenografts with vehicle or compound 20 at a dose of 75 mg/kg once daily through IP injection. After the 22-day treatment, compound 20 drastically inhibited the tumor growth by 92% compared to the vehicle treatment (P = 0.0031, Figure 12A). In addition, compound 20 was well tolerated by the mice treated at this dose through the entire in vivo experiment, without significant body weight loss (Figure 12B) and other clinical signs. We previously reported that AZD5363 via the same dose regimen (75 mg/kg once daily IP injection) achieved only 75% tumor growth inhibition in the same xenograft model.41 Lastly, western blotting analysis of the tumor samples collected from mice treated with 20 or vehicle at the end of the study showed that compound 20 substantially degraded T-AKT and P-AKT and effectively inhibited the downstream signaling (PRAS40 phosphorylation) in vivo (Figure 12C).
Figure 12.
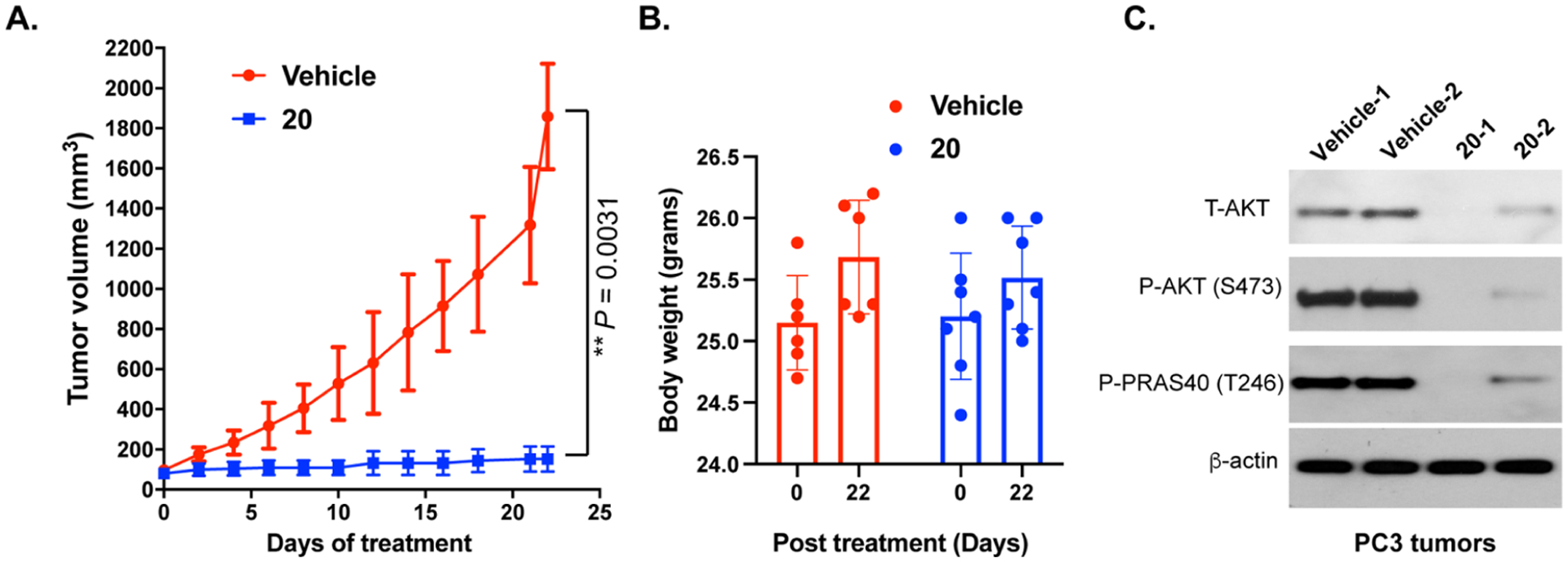
AKT degrader 20 suppresses the tumor growth in vivo in a PC3 cell line xenograft mouse model. (A) Nude mice (Foxn1nu, athymic nude) bearing established PC-3 xenografts were treated with vehicle (n = 6) or 20 (n = 7, 75 mg/kg, i.p., once daily) for 22 days. Data points represent mean tumor volume ±SE. (B) Body weights of mice at the beginning and the end of the in vivo studies. (C) Western blotting analysis of P-AKT and T-AKT protein levels, and downstream signaling inhibition (P-PRAS40) in the tumor samples isolated from the nude mice treated with vehicle or 20.
Compound Synthesis.
The synthetic route for preparing key intermediate 2 is described in Scheme 1. The synthetic effort commenced with Boc-protection of commercially available (S)-3-amino-3-(4-chlorophenyl)propan-1-ol (44) and subsequent halogenation reaction, yielding iodide compound 45. N-Alkylation of benzyl piperazine-1-carboxylate (46) with compound 45, followed by the deprotection of the Boc group provided compound 47. Finally, the amide coupling reaction between 47 and 4-[(tert-butoxycarbonyl)amino]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxylic acid (48),17 followed by the removal of the Cbz protecting group afforded the common intermediate 2.
Scheme 1. Synthesis of Key Intermediate 2a.
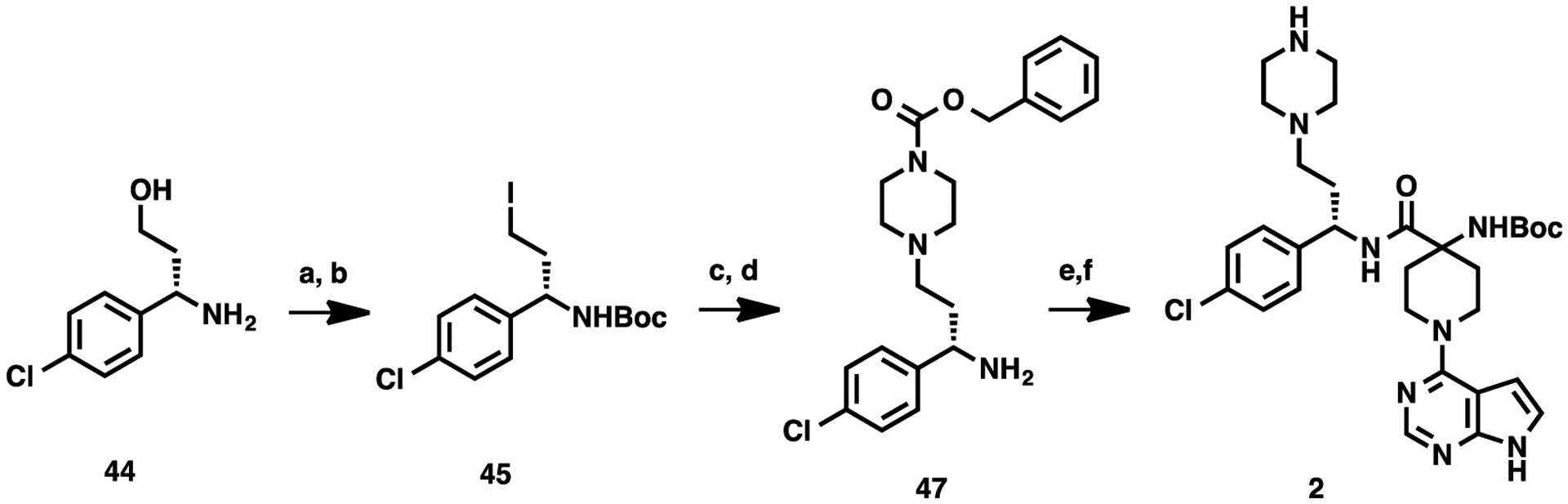
aReaction conditions: (a) Boc2O, NEt3, rt, 2 h; (b) PPh3, I2, imidazole, DCM, 4 h; (c) Benzyl piperazine-1-carboxylate (46), K2CO3, CH3CN, 80 °C, overnight; (d) TFA/DCM, rt, 30 min; (e) 4-[(tert-butoxycarbonyl)amino]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxylic acid (48), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-hydroxy-7-azabenzo-triazole (HOAt), N-methylmorpholine (NMM), dimethyl sulfoxide (DMSO), rt; and (f) Pd/C, H2, MeOH, rt.
As illustrated in Scheme 2A, AZD5363-based VHL-recruiting AKT degraders 3–23 were synthesized from intermediate 2 and various linker attached VHL-1 derivatives (49–69) through amide coupling and subsequent Boc-deprotecting. Following the same reaction sequence, CRBN-recruiting compounds 24–41 were synthesized from intermediate 2 and various linker attached POM derivatives (70–87) (Scheme 2B). Compounds 49–65 and 70–81 were synthesized following the reported procedures.55
Scheme 2. Syntheses of Compounds 3–41a.
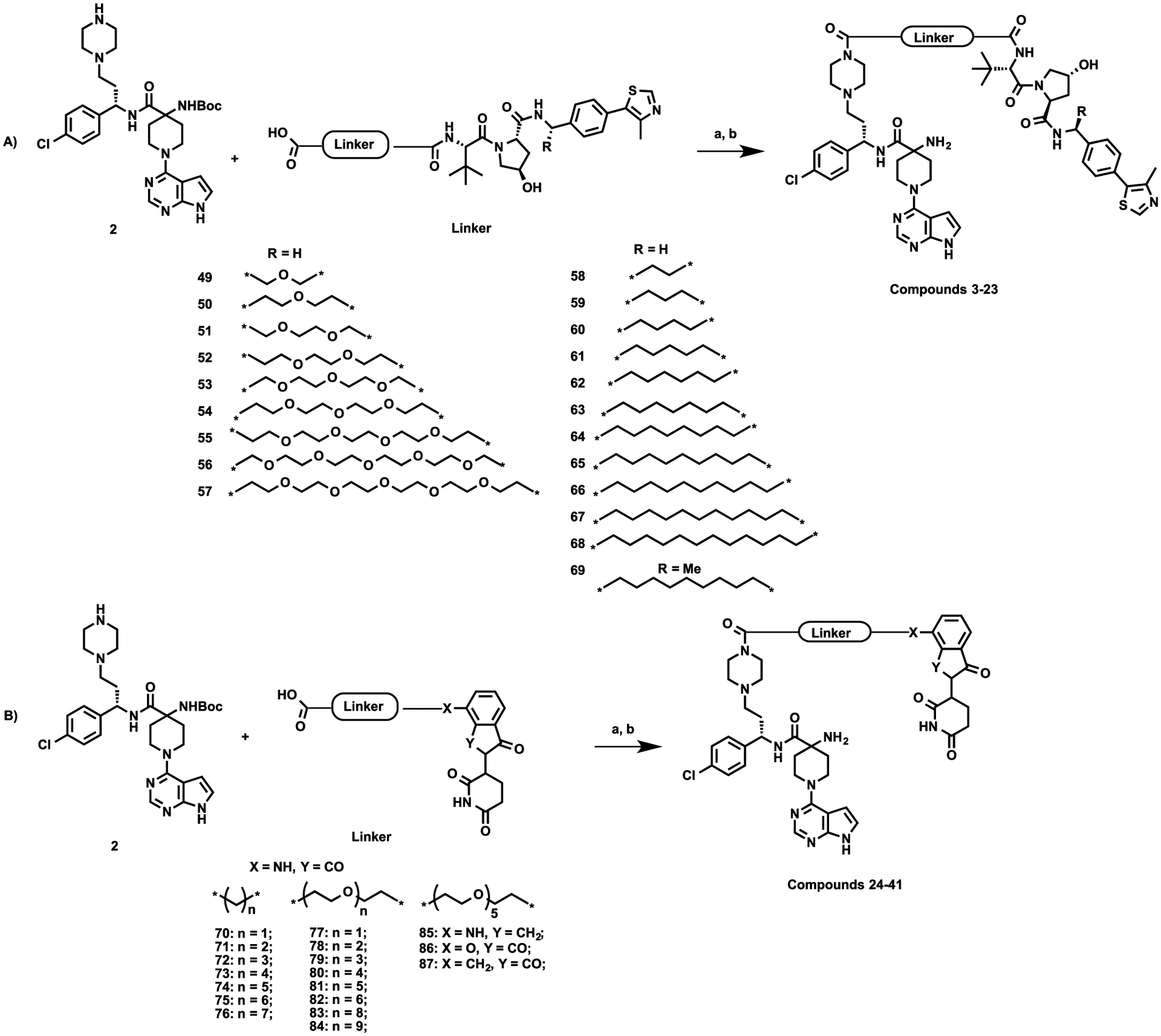
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) TFA, DCM, rt, 30 min.
The synthetic routes for compounds 42 and 43 are described in Scheme 3. Intermediate 90 was synthesized using the amide coupling reaction between commercially available compounds 88 and 89, followed by Boc-deprotection. Amide coupling between 90 and (S)-2-[(tert-butoxycarbonyl)-amino]-3,3-dimethylbutanoic acid (91) and subsequent Boc-deprotection led to intermediate 92, which was then coupled with undecanedioic acid (93) to yield 94. Negative control compound 42 was synthesized from intermediates 2 and 94 by amide coupling and Boc deprotection reactions (Scheme 3A). Intermediate 97 was obtained via a nucleophilic aromatic substitution reaction of the commercially available tert-butyl 1-amino-3,6,9,12,15-pentaoxaoctadecan-18-oate (95) and 4-fluoro-2-(1-methyl-2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (96),56 and subsequent hydrolysis of the t-butyl ester. Amide coupling between 2 and 97, followed by the removal of the Boc-protecting group provided compound 43 (Scheme 3B).
Scheme 3. Syntheses of Negative Control Compounds 42–43a.
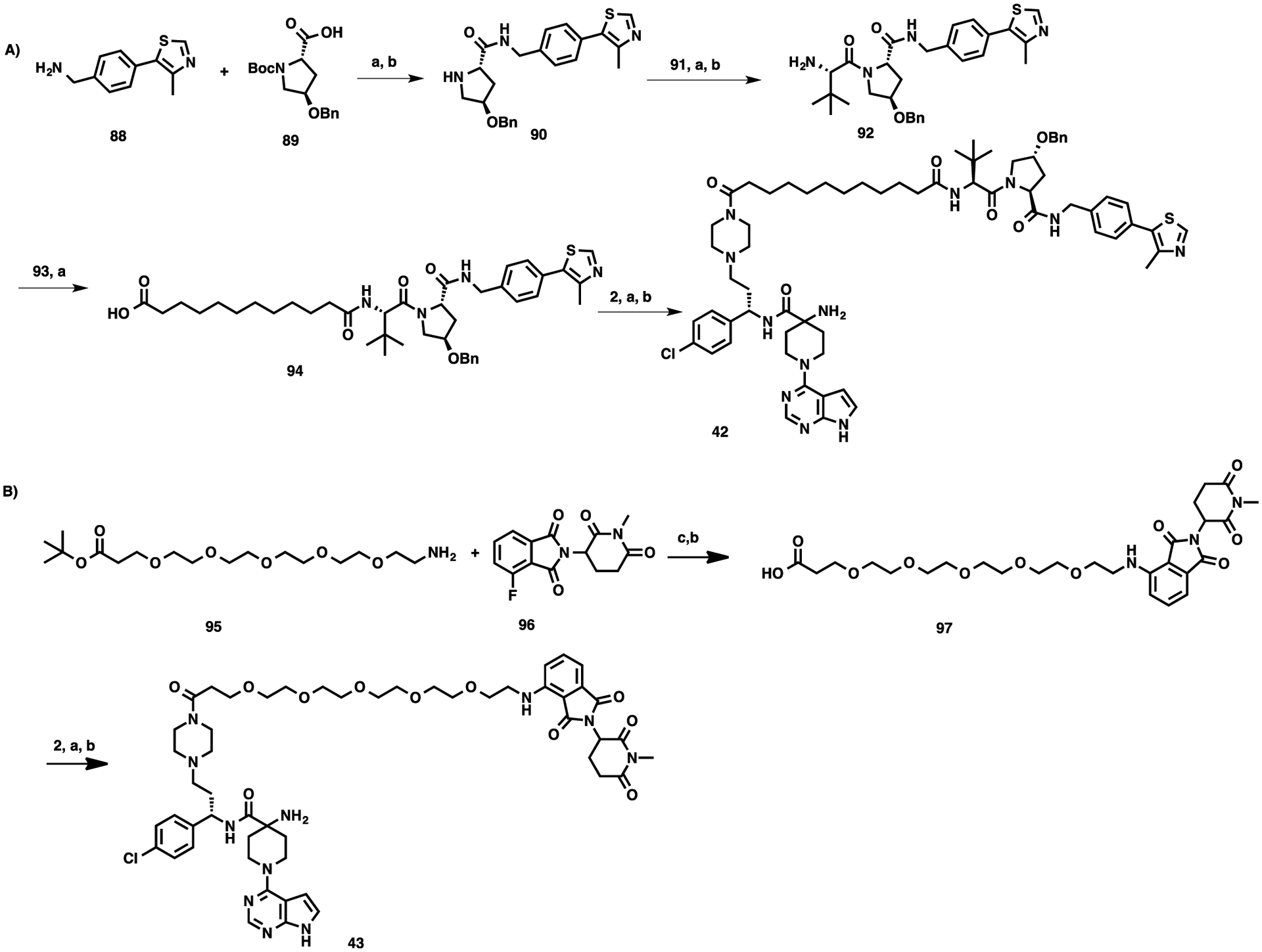
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) TFA, DCM, rt, 30 min; and (c) N-methyl-2-pyrrolidone (NMP), diisopropylethylamine (DIPEA), MW, 100 °C, 1 h.
The syntheses of linker attached E3 ligase ligands 66–69 and 82–87 are outlined in Scheme 4. Compounds 66–68 were prepared using amide coupling between commercially available di-acids 93, 98–99, and VHL-1 (101). Compound 69 was synthesized using the same amide coupling reaction between undecanedioic acid (100) and VHL-2 (102) (Scheme 4A). Compounds 82–84 were synthesized through nucleophilic aromatic substitution of 106 and commercially available 103–105, and subsequent Boc-protecting group removal (Scheme 4B). Compounds 85 and 86 were prepared through nucleophilic substitution of 108 and 109 with tert-butyl 1-bromo-3,6,9,12,15-pentaoxaoctadecan-18-oate (107) under mild basic conditions, followed by t-butyl ester hydrolysis (Scheme 4C). Compound 87 was synthesized via a three-step reaction sequence: Sonogashira coupling of commercially available tert-butyl 4,7,10,13,16-pentaoxanonadec-18-ynoate (110) with 4-bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (111), reduction of the alkynyl moiety by hydrogenation, and hydrolysis of the tert-butyl ester group (Scheme 4D).
Scheme 4. Syntheses of Linkers 66–69 and 82–87a.
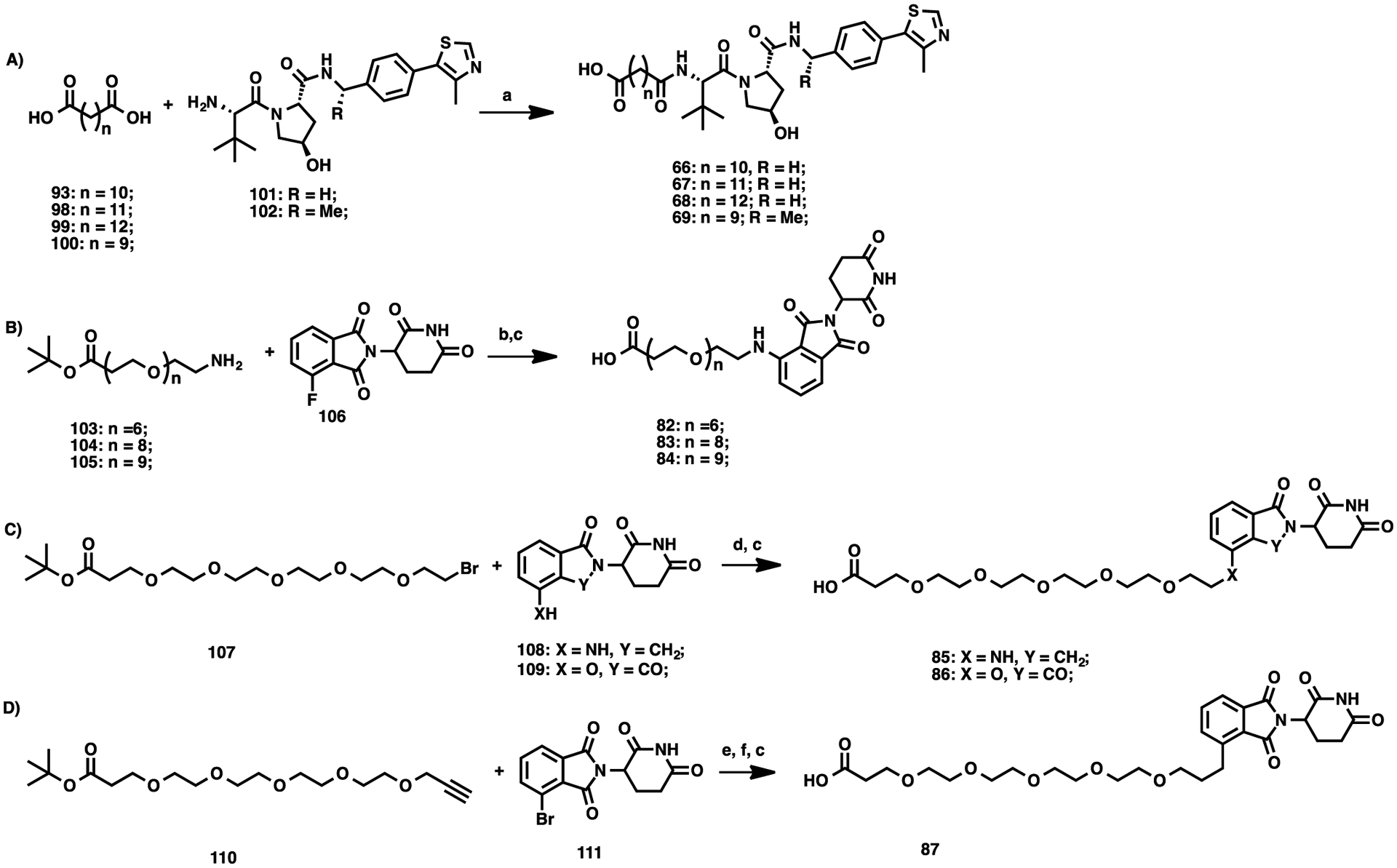
aReaction conditions: (a) EDCI, HOAt, NMM, DMSO, rt, 12 h; (b) NMP, DIPEA, MW, 100 °C, 1 h; (c) TFA, DCM, rt, 30 min; (d) NaHCO3, KI, DMF, 60 °C, overnight; (e) Pd(PPh3)2Cl2, CuI, NEt3, DMF, 70 °C, 2 h; and (f) Pd/C, H2, MeOH, rt, 1 h.
CONCLUSIONS
Starting from the ATP-competitive AKT inhibitor AZD5363, we designed and synthesized a series of putative AKT degraders by conjugating a solvent-exposed site of AZD5363 to two types of E3 ligase (VHL and CRBN) ligands via various linkers. Through extensive SAR studies on the composition and length of linkers and different VHL- and CRBN-recruiting ligands, we identified compounds 20 and 35 as potent VHL-and CRBN-recruiting AKT degraders, respectively. We also developed two structurally similar analogues, 42 and 43, as the negative controls of degraders 20 and 35, respectively. These two degraders, but not their negative controls, achieved rapid and effective AKT degradation and downstream signaling inhibition in PC3 cells in a concentration- and time-dependent manner. Washout experiments further demonstrated that the AKT degradation induced by compound 20 can be sustained for 1–2 days post treatment. Our MOA studies indicated that the AKT degradation induced by 20 and 35 is dependent on the corresponding E3 ligase (VHL/CRBN) and the UPS. In cell proliferation assays, degraders 20 and 35 were more potent than their corresponding negative controls in inhibiting the growth of cancer cells. Moreover, degrader 20 was more potent than AZD5363 and degrader 35 in suppressing cell proliferation in PC3 and MDA-MB-468 cells. In addition, both 20 and 35 displayed good plasma exposure levels in mouse PK studies, suggesting that they are potentially suitable for in vivo efficacy studies. Furthermore, degrader 20 effectively suppressed the tumor growth and reduced the AKT protein level in vivo in a PC3 cell line xenograft mouse model. Overall, we present a comprehensive SAR study to the targeted protein degradation research community, which resulted in the discovery of two in vivo efficacious VHL-recruiting AKT degraders (1941 and 20) and a novel CRBN-recruiting AKT degrader (35).
EXPERIMENTAL SECTION
Chemistry General Procedures.
All commercially available solvents and reagents were used as received without further purification. Microwave-heated reactions were carried out with a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, UK). Normal and reverse-phase flash chromatographies were performed using a Teledyne ISCO CombiFlash instrument and HP C18 RediSep Rf reverse-phase silica columns, respectively. Final compounds for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254 or 220 nm with solvent A (0.1% of TFA in water) and solvent B (methanol or acetonitrile) as eluents with a flow rate of 40 mL/min at rt. Purities of the final compounds were determined by HPLC and were >95%. HPLC conditions to assess purity were as follows: Agilent 1200 series system, 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column 1–99% gradient of 0.1% trifluoroacetic acid in water, and 0.1% trifluoroacetic acid in acetonitrile; flow rate, 0.4 mL/min; acquisition time, 8 min; and high-resolution mass spectra (HRMS) data were acquired on an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were recorded on either Bruker DXI 600 MHz (or 800 MHz) or AVANCE NEO 600 MHz NMR spectrometer. Proton nuclear magnetic resonance spectra are reported in parts per million (ppm) on the δ scale and are referenced from the residual protium in the NMR solvent (CD3OD, δ 3.31). C13 nuclear magnetic resonance spectra are reported in parts per million (ppm) on the δ scale and are referenced from the carbon resonances of the solvent (CD3OD, δ 49.15). Data are reported in the following format: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet), coupling constant, and integration.
tert-Butyl(S)-(4-((1-(4-chlorophenyl)-3-(piperazin-1-yl)propyl)-carbamoyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidin-4-yl)-carbamate (2).
To the solution of (S)-3-amino-3-(4-chlorophenyl)-propan-1-ol (44, 1 g, 5.4 mmol) in DCM was added di-tert-butyl dicarbonate (1.42 g, 6.5 mmol). After the reaction was stirred for 2 h at rt, the solvent was removed. The resulting residue was purified by flash chromatography (MeOH/DCM = 1/9) to provide tert-butyl (S)-[1-(4-chlorophenyl)-3-hydroxypropyl]carbamate as a white solid (1.31 g, yield 85%). ESI-MS (m/z) [M + H]+: 286.2; To a solution of triphenylphosphine (1.84 g, 7 mmol) and iodine (1.80 g, 7 mmol) in DCM (50 mL) was added imidazole (0.89 g, 14 mmol). The reaction was stirred at rt for 30 min before tert-butyl (S)-[1-(4-chlorophenyl)-3-hydroxypropyl]carbamate (1.0 g, 3.5 mmol) in 30 mL of DCM was added. After the resulting mixture was stirred for 3 h, saturated NaHCO3 and 10% of Na2S2O3 aqueous solution were added. The mixture was extracted with DCM (20 mL × 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel flash chromatography (Hexane/EtOAc = 1/1) to provide compound 45 as a white solid (2.3 g, yield 65%). 1H NMR (600 MHz, CDCl3): δ 7.34−7.30 (m, 2H), 7.22 (d, J = 8.2 Hz, 2H), 4.85−4.61 (m, 1H), 3.11 (d, J = 8.5 Hz, 1H), 3.01 (ddd, J = 10.0, 8.0, 6.8 Hz, 1H), 2.37−2.14 (m, 2H), 1.41 (s, 9H); ESI-MS (m/z): [M + H]+ 396.1.
To a solution of compound 45 (1.48 g, 3.7 mmol) in CH3CN (30 mL) were added K2CO3 (2.1 g, 14.8 mmol) and benzyl piperazine-1-carboxylate (46, 1.65 g, 7.4 mmol). After stirring at 80 °C overnight, the mixture was purified by silica gel flash chromatography (Hexane/EtOAc = 1/1) to provide an intermediate. The obtained intermediate was dissolved in DCM (20 mL) and TFA (20 mL). After the mixture was stirred for 30 min, solvents were evaporated to afford (S)-4-[3-amino-3-(4-chlorophenyl)propyl]piperazine-1-carboxylate 47 as a white foam solid (1.35 g, yield 95%). ESI-MS (m/z): [M + H]+ 388.3.
To a solution of benzyl (S)-4-[3-amino-3-(4-chlorophenyl)propyl]-piperazine-1-carboxylate (47, 1.3 g, 3.4 mmol) and 4-[(tert-butoxycarbonyl)amino]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxylic acid17 (48, 1.2 g, 3.4 mmol) in DMSO (15 mL) were added EDCI (0.98 g, 5.1 mmol), HOAt (0.69 g, 5.1 mmol), and NMM (1.0 g, 10.2 mmol). After stirring overnight at rt, the resulting mixture was purified by reverse-phase ISCO (10–100% methanol/0.1% TFA in H2O) to afford a white solid (1.71 g, 69% yield). This product was dissolved in methanol (30 mL) and 10% palladium on carbon was added. After this reaction was stirred under H2 for 4 h, it was filtered through celite. The filtrate was concentrated, and the resulting mixture was purified by a reverse-phase column (0–100% methanol/0.1% TFA in H2O) to afford the title compound 2 as a white solid (1.1 g, yield 78%). 1H NMR (600 MHz, CD3OD): δ 8.43 (d, J = 8.3 Hz, 1H), 8.31 (s, 1H), 7.42−7.33 (m, 4H), 6.95 (d, J = 3.7 Hz, 1H), 5.09 (q, J = 7.2 Hz, 1H), 4.46−4.29 (m, 2H), 3.93−3.80 (m, 2H), 3.57 (t, J = 5.3 Hz, 4H), 3.52−3.38 (m, 4H), 3.24−3.09 (m, 2H), 2.42−2.14 (m, 6H), 1.45 (s, 9H); 13C NMR (151 MHz, CD3OD): δ 174.66, 155.30, 142.13, 139.81, 133.10, 128.62, 128.39 (2C), 128.19 (2C), 128.14, 123.58, 103.83, 101.96, 79.76, 56.68 (2C), 54.29 (2C), 50.34, 48.73 (2C), 43.01, 41.22 (2C), 31.55, 29.71, 27.34 (3C); HRMS (m/z): for C30H42ClN8O3+ [M + H]+, calcd 597.3063; found, 597.3079.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)acetyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (3).
To a solution of intermediate 2 (15.9 mg, 0.022 mmol) in DMSO (1 mL) were added intermediate 49 (12.2 mg, 0.022 mmol, 1.0 equiv), EDCI (6.5 mg, 0.033 mmol, 1.5 equiv), HOAt (4.5 mg, 0.033 mmol, 1.5 equiv), and NMM (6.7 mg, 0.066 mmol, 3.0 equiv). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10%−100% methanol/0.1% TFA in H2O) to afford a white solid (18.2 mg, yield 91%). The solid was dissolved in DCM (1 mL) and TFA (1 mL) and the reaction was stirred at rt for 30 min. Then, the solvent was evaporated, and the resulting mixture was purified by preparative HPLC (10%–100% methanol/0.1% TFA in H2O) to afford compound 3 as a white solid (19.4 mg, yield 86%). 1H NMR (600 MHz, CD3OD): δ 8.98 (s, 1H), 8.39 (s, 1H), 7.47−7.43 (m, 2H), 7.41 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 3.7 Hz, 1H), 7.37−7.31 (m, 4H), 6.94 (d, J = 3.7 Hz, 1H), 5.00 (dd, J = 9.3, 5.8 Hz, 1H), 4.67 (s, 1H), 4.66−4.61 (m, 2H), 4.58−4.53 (m, 1H), 4.53−4.46 (m, 2H), 4.42−4.34 (m, 4H), 4.17−4.03 (m, 3H), 3.92−3.78 (m, 5H), 3.27 (dd, J = 12.2, 4.4 Hz, 2H), 3.14 (td, J = 12.1, 5.3 Hz, 2H), 2.72−2.58 (m, 3H), 2.47 (s, 3H), 2.39−2.14 (m, 5H), 2.12−2.00 (m, 3H), 1.04 (s, 9H). HRMS (m/z): for C51H66ClN12O7S+ [M + H]+, calcd 1025.4581; found, 1025.4590.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(3-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)-propanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (4).
Compound 4 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 50 (12.7 mg, 0.022 mmol, 1.0 equiv). Compound 4 was obtained as a white solid (11.9 mg, yield 52%). 1H NMR (600 MHz, CD3OD): δ 8.95 (s, 1H), 8.39 (s, 1H), 7.47−7.30 (m, 9H), 6.94 (d, J = 3.8 Hz, 1H), 5.01 (dd, J = 9.2, 5.9 Hz, 1H), 4.70−4.60 (m, 4H), 4.53 (dd, J = 9.4, 7.5 Hz, 1H), 4.49−4.36 (m, 4H), 3.90−3.76 (m, 5H), 3.75−3.66 (m, 5H), 3.26 (dd, J = 12.2, 4.5 Hz, 2H), 3.18−3.09 (m, 2H), 2.71−2.57 (m, 4H), 2.50 (t, J = 5.9 Hz, 2H), 2.47 (s, 3H), 2.39−2.13 (m, 5H), 2.11−1.98 (m, 3H), 1.02 (s, 9H). HRMS (m/z): for C55H70ClN12O7S+ [M + H]+, calcd 1053.4894; found, 1053.4895.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(2-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)-pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)ethoxy)acetyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (5).
Compound 5 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 51 (13 mg, 0.022 mmol, 1.0 equiv). Compound 5 was obtained as a white solid (21.1 mg, yield 89%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.40 (d, J = 2.8 Hz, 1H), 7.48−7.29 (m, 9H), 6.94 (d, J = 3.6 Hz, 1H), 5.01 (dd, J = 9.2, 5.8 Hz, 1H), 4.69 (d, J = 2.7 Hz, 1H), 4.66−4.61 (m, 2H), 4.56−4.51 (m, 1H), 4.47 (d, J = 17.9 Hz, 2H), 4.41−4.23 (m, 4H), 4.04 (t, J = 2.9 Hz, 2H), 3.90−3.78 (m, 5H), 3.75−3.68 (m, 5H), 3.26 (dd, J = 12.1, 4.3 Hz, 2H), 3.19−3.10 (m, 2H), 2.72−2.57 (m, 3H), 2.48 (s, 3H), 2.39−2.14 (m, 5H), 2.12−1.97 (m, 3H), 1.02 (s, 9H). HRMS (m/z): for C53H70ClN12O8S+ [M + H]+, calcd 1069.4843; found, 1069.4850.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(3-(2-(3-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)-pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-3-oxopropoxy)ethoxy)propanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (6).
Compound 6 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 52 (13.6 mg, 0.022 mmol, 1.0 equiv). Compound 6 was obtained as a white solid (18.3 mg, yield 76%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.39 (s, 1H), 7.49−7.31 (m, 9H), 6.94 (d, J = 3.7 Hz, 1H), 5.02 (dd, J = 9.2, 5.8 Hz, 1H), 4.64 (s, 4H), 4.59−4.45 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.91−3.77 (m, 5H), 3.75−3.66 (m, 5H), 3.59−3.55 (m, 4H), 3.26 (dd, J = 12.3, 4.5 Hz, 2H), 3.13 (td, J = 12.1, 5.2 Hz, 2H), 2.74−2.58 (m, 5H), 2.54 (dt, J = 15.0, 5.9 Hz, 1H), 2.49−2.42 (m, 4H), 2.40−2.15 (m, 5H), 2.11−2.01 (m, 2H), 1.03 (s, 9H). HRMS (m/z): for C55H74ClN12O8S+ [M + H]+, calcd 1097.5156; found, 1097.5160.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-((S)-13-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbony l) - 14, 14 - dimethyl - 11 - oxo - 3, 6, 9 - trioxa - 12 - azapentadecanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (7).
Compound 7 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 53 (14 mg, 0.022 mmol, 1.0 equiv). Compound 7 was obtained as a white solid (19.2 mg, yield 78%). 1H NMR (600 MHz, CD3OD): δ 9.01 (d, J = 3.0 Hz, 1H), 8.39 (d, J = 3.0 Hz, 1H), 7.48−7.30 (m, 9H), 6.95 (d, J = 3.7 Hz, 1H), 5.01 (dd, J = 9.2, 5.9 Hz, 1H), 4.66 (brs, 4H), 4.57−4.48 (m, 3H), 4.37 (d, J = 15.5 Hz, 1H), 4.30−4.17 (m, 2H), 4.04 (s, 2H), 3.93−3.77 (m, 5H), 3.72−3.62 (m, 9H), 3.26 (dd, J = 12.2, 4.5 Hz, 2H), 3.12 (td, J = 12.1, 5.2 Hz, 2H), 2.73−2.57 (m, 3H), 2.48 (d, J = 2.9 Hz, 3H), 2.39−2.15 (m, 5H), 2.13−1.98 (m, 3H), 1.03 (s, 9H). HRMS (m/z): for C55H74ClN12O9S+ [M + H]+, calcd 1113.5105; found, 1113.5120.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-((S)-15-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-16, 16-dimethyl −13-oxo - 4, 7, 10- trioxa - 14-azaheptadecanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (8).
Compound 8 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 54 (14 mg, 0.022 mmol, 1.0 equiv). Compound 8 was obtained as a white solid (13.2 mg, yield 53%). 1H NMR (600 MHz, CD3OD): δ 8.99 (s, 1H), 8.40 (s, 1H), 7.51−7.28 (m, 9H), 6.94 (d, J = 3.6 Hz, 1H), 5.01 (dd, J = 8.9, 6.1 Hz, 1H), 4.74−4.61 (m, 4H), 4.60−4.46 (m, 4H), 4.37 (d, J = 15.5 Hz, 1H), 3.91−3.75 (m, 5H), 3.73−3.67 (m, 5H), 3.60−3.54 (m, 8H), 3.25 (dd, J = 12.2, 4.5 Hz, 2H), 3.13−3.08 (m, 2H), 2.73−2.52 (m, 6H), 2.48 (d, J = 7.7 Hz, 4H), 2.41−2.14 (m, 5H), 2.12−2.00 (m, 2H), 1.03 (s, 9H). HRMS (m/z): for C57H78ClN12O9S+ [M + H]+, calcd 1141.5418; found, 1141.5438.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-((S)-18-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-19,19-dimethyl-16-oxo-4,7,10,13-tetraoxa-17-azaicosanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (9).
Compound 9 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 55 (15.6 mg, 0.022 mmol, 1.0 equiv). Compound 9 was obtained as a white solid (14.0 mg, yield 54%). 1H NMR (600 MHz, CD3OD): δ 8.95 (s, 1H), 8.39 (s, 1H), 7.51−7.26 (m, 9H), 6.93 (d, J = 3.7 Hz, 1H), 5.00 (dd, J = 8.8, 6.3 Hz, 1H), 4.70−4.60 (m, 4H), 4.60−4.45 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.90−3.76 (m, 5H), 3.75−3.66 (m, 5H), 3.62−3.51 (m, 12H), 3.26 (td, J = 12.4, 4.7 Hz, 2H), 3.10 (td, J = 12.1, 5.3 Hz, 2H), 2.72−2.55 (m, 6H), 2.51−2.42 (m, 4H), 2.39−2.14 (m, 5H), 2.11−1.99 (m, 2H), 1.02 (s, 9H). HRMS (m/z): for C59H82ClN12O10S+ [M + H]+, calcd 1185.5681; found, 1185.5665.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-((S)-19-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (10).
Compound 10 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 56 (15.9 mg, 0.022 mmol, 1.0 equiv). Compound 10 was obtained as a white solid (16.3 mg, yield 62%). 1H NMR (600 MHz, CD3OD): δ 8.96 (s, 1H), 8.39 (s, 1H), 7.55−7.17 (m, 9H), 6.93 (d, J = 3.7 Hz, 1H), 5.01 (t, J = 7.7 Hz, 1H), 4.68−4.46 (m, 8H), 4.36 (d, J = 15.5 Hz, 2H), 4.16−4.02 (m, 3H), 3.93−3.82 (m, 4H), 3.78 (dd, J = 10.9, 3.8 Hz, 1H), 3.68−3.54 (m, 16H), 3.29−3.20 (m, 2H), 3.17−3.09 (m, 2H), 2.74−2.57 (m, 3H), 2.47 (s, 3H), 2.41−2.13 (m, 5H), 2.12−1.99 (m, 3H), 1.04 (s, 9H). HRMS (m/z): for C59H82ClN12O11S+ [M + H]+, calcd 1201.5630; found, 1201.5651.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-((S)-21-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-22,22-dimethyl-19-oxo-4,7,10,13,16-pentaoxa-20-azatricosanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (11).
Compound 11 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 57 (16.5 mg, 0.022 mmol, 1.0 equiv). Compound 11 was obtained as a white solid (17.3 mg, yield 64%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.40 (s, 1H), 7.53−7.28 (m, 9H), 6.94 (d, J = 3.7 Hz, 1H), 5.00 (dd, J = 8.9, 6.3 Hz, 1H), 4.70−4.60 (m, 4H), 4.60−4.46 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.91−3.76 (m, 5H), 3.76−3.67 (m, 5H), 3.65−3.49 (m, 16H), 3.26 (dd, J = 12.3, 4.5 Hz, 2H), 3.11 (td, J = 12.2, 5.4 Hz, 2H), 2.72−2.54 (m, 6H), 2.52−2.43 (m, 4H), 2.40−2.13 (m, 5H), 2.12−1.99 (m, 2H), 1.02 (s, 9H). HRMS (m/z): for C61H86ClN12O11S+ [M + H]+, calcd 1229.5943; found, 1229.5950.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(4-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-4-oxobutanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (12).
Compound 12 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 58 (11.7 mg, 0.022 mmol, 1.0 equiv). Compound 12 was obtained as a white solid (19.9 mg, yield 90%). 1H NMR (600 MHz, CD3OD): δ 8.99 (s, 1H), 8.40 (s, 1H), 7.52−7.25 (m, 9H), 6.95 (d, J = 3.8 Hz, 1H), 5.01 (dd, J = 9.2, 5.9 Hz, 1H), 4.71−4.60 (m, 3H), 4.60−4.45 (m, 5H), 4.37 (d, J = 15.6 Hz, 1H), 3.92−3.74 (m, 5H), 3.27 (dd, J = 12.3, 4.4 Hz, 2H), 3.13 (td, J = 12.2, 5.2 Hz, 2H), 2.74−2.52 (m, 7H), 2.48 (s, 3H), 2.41−2.13 (m, 5H), 2.13−1.98 (m, 3H), 1.02 (s, 9H). HRMS (m/z): for C51H66ClN12O6S+ [M + H]+, calcd 1009.4632; found, 1009.4638.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(5-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-5-oxopentanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (13).
Compound 13 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 59 (12 mg, 0.022 mmol, 1.0 equiv). Compound 13 was obtained as a white solid (13.9 mg, yield 62%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.40 (s, 1H), 7.51−7.26 (m, 9H), 6.95 (dd, J = 3.7, 1.7 Hz, 1H), 5.00 (dd, J = 9.2, 5.9 Hz, 1H), 4.70−4.61 (m, 3H), 4.60−4.45 (m, 5H), 4.37 (d, J = 15.4 Hz, 1H), 3.94−3.75 (m, 5H), 3.26 (dd, J = 12.2, 4.3 Hz, 2H), 3.12 (td, J = 12.1, 5.2 Hz, 2H), 2.72−2.57 (m, 3H), 2.48 (d, J = 1.5 Hz, 3H), 2.45−2.38 (m, 2H), 2.38−2.12 (m, 8H), 2.12−1.98 (m, 2H), 1.88 (p, J = 7.3 Hz, 2H), 1.03 (d, J = 1.7 Hz, 9H). HRMS (m/z): for C52H68ClN12O6S+ [M + H]+, calcd 1023.4789; found, 1023.4794.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(6-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (14).
Compound 14 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 60 (12.3 mg, 0.022 mmol, 1.0 equiv). Compound 14 was obtained as a white solid (14 mg, yield 61%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.39 (s, 1H), 7.53−7.22 (m, 9H), 6.95 (d, J = 3.7 Hz, 1H), 5.00 (dd, J = 9.2, 5.9 Hz, 1H), 4.69−4.59 (m, 4H), 4.59−4.44 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.91−3.76 (m, 5H), 3.26 (dd, J = 12.3, 4.4 Hz, 2H), 3.13 (td, J = 12.1, 5.2 Hz, 2H), 2.73−2.57 (m, 3H), 2.48 (s, 3H), 2.43 (td, J = 7.1, 4.8 Hz, 2H), 2.40−2.13 (m, 8H), 2.11−2.01 (m, 2H), 1.68−1.57 (m, 4H), 1.03 (s, 9H). HRMS (m/z): for C53H70ClN12O6S+ [M + H]+, calcd 1037.4945; found, 1037.4923.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(7-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-7-oxoheptanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (15).
Compound 15 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 61 (12.6 mg, 0.022 mmol, 1.0 equiv). Compound 15 was obtained as a white solid (16.3 mg, yield 70%). 1H NMR (600 MHz, CD3OD): δ 9.04 (s, 1H), 8.40 (s, 1H), 7.55−7.25 (m, 9H), 6.95 (d, J = 3.7 Hz, 1H), 5.01 (dd, J = 9.3, 5.8 Hz, 1H), 4.64 (d, J = 18.7 Hz, 4H), 4.58−4.45 (m, 4H), 4.37 (d, J = 15.5 Hz, 1H), 3.93−3.77 (m, 5H), 3.27 (dd, J = 12.3, 4.5 Hz, 2H), 3.14 (td, J = 12.2, 5.2 Hz, 2H), 2.74−2.55 (m, 3H), 2.48 (s, 3H), 2.41 (t, J = 7.5 Hz, 2H), 2.39−2.14 (m, 8H), 2.12−2.00 (m, 2H), 1.66−1.56 (m, 4H), 1.41−1.29 (m, 2H), 1.02 (s, 9H). HRMS (m/z): for C54H72ClN12O6S+ [M + H]+, calcd 1051.5102; found, 1051.5094.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(8-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-8-oxooctanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (16).
Compound 16 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 62 (12.9 mg, 0.022 mmol, 1.0 equiv). Compound 16 was obtained as a white solid (11.9 mg, yield 51%). 1H NMR (600 MHz, CD3OD): δ 8.97 (s, 1H), 8.39 (s, 1H), 7.53−7.19 (m, 9H), 6.94 (d, J = 3.8 Hz, 1H), 5.01 (dd, J = 9.2, 5.9 Hz, 1H), 4.71−4.61 (m, 4H), 4.59−4.45 (m, 4H), 4.36 (d, J = 15.4 Hz, 1H), 3.92−3.76 (m, 5H), 3.26 (dd, J = 12.3, 4.5 Hz, 2H), 3.13 (td, J = 12.2, 5.3 Hz, 2H), 2.73−2.55 (m, 3H), 2.48 (s, 3H), 2.40 (t, J = 7.6 Hz, 3H), 2.37−2.14 (m, 7H), 2.11−2.00 (m, 2H), 1.64−1.55 (m, 4H), 1.35 (dd, J = 7.5, 4.0 Hz, 4H), 1.03 (s, 9H). HRMS (m/z): for C55H74ClN12O6S+ [M + H]+, calcd 1065.5258; found, 1065.5272.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(9-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-9-oxononanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (17).
Compound 17 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 63 (13.2 mg, 0.022 mmol, 1.0 equiv). Compound 17 was obtained as a white solid (10.4 mg, yield 44%). 1H NMR (600 MHz, CD3OD): δ 8.98 (s, 1H), 8.40 (d, J = 1.9 Hz, 1H), 7.56−7.25 (m, 9H), 6.95 (dd, J = 3.7, 1.9 Hz, 1H), 5.01 (dd, J = 9.3, 5.8 Hz, 1H), 4.70−4.61 (m, 4H), 4.59−4.47 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.92−3.78 (m, 5H), 3.26 (dd, J = 12.3, 4.3 Hz, 2H), 3.13 (td, J = 12.0, 5.0 Hz, 2H), 2.73−2.58 (m, 3H), 2.48 (s, 3H), 2.43−2.37 (m, 3H), 2.37−2.14 (m, 7H), 2.11−2.00 (m, 2H), 1.65−1.54 (m, 4H), 1.34 (s, 6H), 1.03 (d, J = 1.9 Hz, 9H). HRMS (m/z): for C56H76ClN12O6S+ [M + H]+, calcd 1079.5415; found, 1079.5414.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(10-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-10-oxodecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (18).
Compound 18 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 64 (13.5 mg, 0.022 mmol, 1.0 equiv). Compound 18 was obtained as a white solid (13.6 mg, yield 60%). 1H NMR (600 MHz, CD3OD): δ 9.00 (s, 1H), 8.40 (s, 1H), 7.53−7.22 (m, 9H), 6.95 (d, J = 3.8 Hz, 1H), 5.01 (dd, J = 9.3, 5.8 Hz, 1H), 4.71−4.61 (m, 4H), 4.59−4.46 (m, 4H), 4.36 (d, J = 15.5 Hz, 1H), 3.93−3.76 (m, 5H), 3.26 (dd, J = 12.3, 4.4 Hz, 2H), 3.13 (td, J = 12.2, 5.1 Hz, 2H), 2.73−2.57 (m, 3H), 2.48 (s, 3H), 2.40 (t, J = 7.6 Hz, 3H), 2.37−2.14 (m, 7H), 2.11–2.00 (m, 2H), 1.63−1.53 (m, 4H), 1.32 (s, 8H), 1.03 (s, 9H). HRMS (m/z): for C57H78ClN12O6S+ [M + H]+, calcd 1093.5571; found, 1093.5561.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(11-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (19).
Compound 19 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 65 (13.5 mg, 0.022 mmol, 1.0 equiv). Compound 19 was obtained as a white solid (16.0 mg, yield 66%). 1H NMR (800 MHz, CD3OD): δ 8.96 (s, 1H), 8.41 (s, 1H), 7.51−7.35 (m, 9H), 6.97 (d, J = 3.8 Hz, 1H), 5.05 (dd, J = 9.6, 5.7 Hz, 1H), 4.65 (d, J = 11.3 Hz, 3H), 4.62−4.50 (m, 3H), 4.39 (d, J = 15.4 Hz, 1H), 3.95−3.85 (m, 3H), 3.83 (dd, J = 10.9, 3.9 Hz, 1H), 3.31 (dt, J = 12.0, 6.3 Hz, 1H), 3.18 (td, J = 12.2, 5.1 Hz, 1H), 2.77−2.70 (m, 1H), 2.68 (s, 1H), 2.50 (s, 3H), 2.45−2.37 (m, 4H), 2.35−2.19 (m, 6H), 2.13−2.06 (m, 2H), 1.67−1.54 (m, 6H), 1.34 (d, J = 8.5 Hz, 14H), 1.06 (s, 9H). HRMS (m/z): for C58H80ClN12O6S+ [M + H]+, calcd 1107.5728; found, 1107.5751.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(12-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (20).
Compound 20 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 66 (12.8 mg, 0.02 mmol, 1.0 equiv). Compound 20 was obtained as a white solid (13.5 mg, yield 60%). 1H NMR (600 MHz, CD3OD): δ 9.03 (s, 1H), 8.40 (s, 1H), 7.53−7.27 (m, 9H), 6.95 (d, J = 3.7 Hz, 1H), 5.03 (dd, J = 9.5, 5.7 Hz, 1H), 4.69−4.45 (m, 6H), 4.37 (d, J = 15.4 Hz, 1H), 3.99−3.85 (m, 3H), 3.81 (dd, J = 11.0, 3.9 Hz, 1H), 3.69−3.38 (m, 8H), 3.36−3.23 (m, 3H), 3.23−3.10 (m, 1H), 2.78−2.55 (m, 2H), 2.49 (s, 3H), 2.43−2.14 (m, 6H), 2.14−2.00 (m, 2H), 1.71−1.47 (m, 4H), 1.43−1.18 (m, 12H), 1.04 (s, 9H). 13C NMR (151 MHz, CD3OD): δ 174.66, 173.08, 172.75, 170.97, 169.25, 154.17, 151.87, 146.99, 142.67, 139.59, 139.10, 133.39, 129.73, 129.09, 128.95 (2C), 128.56 (2C), 128.06 (2C), 128.02, 127.61 (2C), 123.61, 103.43, 102.33, 69.68, 59.44, 58.13, 57.58, 56.60, 53.97, 51.61, 51.50, 51.38, 42.29, 42.17, 41.89, 38.20, 37.53, 35.27, 35.17, 32.17, 30.68, 30.60, 29.24, 29.18, 29.13 (2C), 29.06, 29.00, 28.94, 28.90, 25.64 (3C), 25.60, 24.74, 14.14. HRMS (m/z): for C59H82ClN12O6S+ [M + H]+, calcd 1121.5884; found, 1121.5868.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(13-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-13-oxotridecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (21).
Compound 21 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 67 (13.1 mg, 0.02 mmol, 1.0 equiv). Compound 21 was obtained as a white solid (15 mg, yield 66%). 1H NMR (600 MHz, CD3OD): δ 9.06−8.86 (m, 1H), 8.41−8.34 (m, 1H), 7.57−7.27 (m, 9H), 6.94 (q, J = 3.9 Hz, 1H), 5.00 (td, J = 8.9, 5.1 Hz, 1H), 4.71−4.64 (m, 3H), 4.63−4.46 (m, 3H), 4.35 (dd, J = 15.5, 7.0 Hz, 1H), 3.95−3.76 (m, 4H), 3.61−2.89 (m, 10H), 2.65−1.95 (m, 15H), 1.58 (dh, J = 15.0, 7.3 Hz, 4H), 1.30 (dd, J = 13.9, 6.9 Hz, 14H), 1.09−0.89 (m, 9H). HRMS (m/z): for C60H84ClN12O6S+ [M + H]+, calcd 1135.6041; found, 1135.6034.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(14-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-14-oxotetradecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (22).
Compound 22 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 68 (13.4 mg, 0.02 mmol, 1.0 equiv). Compound 22 was obtained as a white solid (20.2 mg, yield 88%). 1H NMR (600 MHz, CD3OD): δ 9.04 (s, 1H), 8.40 (s, 1H), 7.55−7.24 (m, 9H), 7.07−6.84 (m, 1H), 5.01 (dd, J = 9.3, 5.8 Hz, 1H), 4.71−4.63 (m, 3H), 4.63−4.43 (m, 3H), 4.36 (d, J = 15.5 Hz, 1H), 3.95−3.77 (m, 4H), 3.61−2.95 (m, 10H), 2.68−2.61 (m, 2H), 2.49 (s, 3H), 2.45−2.14 (m, 8H), 2.12−1.97 (m, 2H), 1.63−1.53 (m, 4H), 1.39−1.19 (m, 16H), 1.03 (s, 9H). HRMS (m/z): for C61H86ClN12O6S+ [M + H]+, calcd 1149.6197; found, 1149.6176.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(11-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)-pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (23).
Compound 23 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (23.5 mg, 0.022 mmol) and 69 (21.2 mg, 0.022 mmol, 1.0 equiv). Compound 23 was obtained as a white solid (22.4 mg, yield 61%). 1H NMR (600 MHz, CD3OD): δ 8.95 (d, J = 6.3 Hz, 1H), 8.39 (s, 1H), 7.47−7.40 (m, 4H), 7.40−7.30 (m, 5H), 6.94 (d, J = 3.8 Hz, 1H), 5.08−4.95 (m, 1H), 4.69−4.63 (m, 3H), 4.61−4.52 (m, 1H), 4.46−4.35 (m, 1H), 3.98−3.81 (m, 4H), 3.74 (dd, J = 11.0, 4.0 Hz, 1H), 3.64−2.91 (m, 10H), 2.73−2.67 (m, 1H), 2.66−2.60 (m, 1H), 2.48 (s, 3H), 2.44−2.13 (m, 8H), 2.07−2.01 (m, 1H), 1.98−1.93 (m, 1H), 1.66−1.53 (m, 4H), 1.50 (d, J = 7.0 Hz, 3H), 1.41−1.23 (m, 10H), 1.04 (s, 9H). HRMS (m/z): for C59H82ClN12O6S+ [M + H]+, calcd 1121.5884; found, 1121.5869.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)glycyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (24).
Compound 24 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 70 (7.3 mg, 0.022 mmol, 1.0 equiv). Compound 24 was obtained as a yellow solid (2.9 mg, yield 16%). 1H NMR (600 MHz, CD3OD): δ 8.36 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.44 (dd, J = 8.5, 7.0 Hz, 1H), 7.36 (dd, J = 12.3, 2.7 Hz, 3H), 7.10 (d, J = 7.2 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 6.99 (dd, J = 8.4, 3.6 Hz, 1H), 6.89 (d, J = 3.7 Hz, 1H), 5.10−4.99 (m, 2H), 4.75−4.64 (m, 2H), 4.24 (s, 2H), 3.76 (q, J = 12.1 Hz, 4H), 3.44−3.32 (m, 2H), 3.28−3.22 (m, 2H), 3.12 (td, J = 12.2, 5.2 Hz, 2H), 2.90−2.80 (m, 2H), 2.78−2.51 (m, 5H), 2.40−2.24 (m, 2H), 2.17−2.06 (m, 2H), 2.04−1.96 (m, 1H). HRMS (m/z): for C40H45ClN11O6 +[M + H]+, calcd 810.3237; found, 810.3246.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(3-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)propanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (25).
Compound 25 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 71 (7.6 mg, 0.022 mmol, 1.0 equiv). Compound 25 was obtained as a yellow solid (16.2 mg, yield 89%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.56 (dd, J = 8.5, 7.2 Hz, 1H), 7.39−7.31 (m, 5H), 7.11 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 6.93 (d, J = 3.7 Hz, 1H), 5.07−4.97 (m, 2H), 4.64 (s, 2H), 3.84 (q, J = 12.2 Hz, 4H), 3.68−3.63 (m, 4H), 3.26−3.15 (m, 2H), 3.08 (td, J = 11.9, 4.9 Hz, 2H), 2.90−2.79 (m, 2H), 2.77−2.71 (m, 3H), 2.71−2.58 (m, 3H), 2.37−1.98 (m, 6H). HRMS (m/z): for C41H47ClN11O6+ [M + H]+, calcd 824.3394; found, 824.3396.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(4-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)butanoyl)piperazin-1-yl)-propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (26).
Compound 26 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 72 (7.9 mg, 0.022 mmol, 1.0 equiv). Compound 26 was obtained as a yellow solid (12.5 mg, yield 68%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.54 (dd, J = 8.5, 7.2 Hz, 1H), 7.37 (dd, J = 17.5, 3.5 Hz, 5H), 7.10 (d, J = 8.6 Hz, 1H), 7.04 (d, J = 7.1 Hz, 1H), 6.94 (d, J = 3.7 Hz, 1H), 5.05 (dd, J = 12.8, 5.5 Hz, 1H), 5.00 (dd, J = 9.2, 5.9 Hz, 1H), 4.70−4.60 (m, 2H), 3.84 (dd, J = 13.8, 10.5 Hz, 4H), 3.39 (t, J = 6.7 Hz, 4H), 3.23 (t, J = 12.2 Hz, 2H), 3.11−3.05 (m, J 2H), 2.93−2.79 (m, 2H), 2.78−2.57 (m, 5H), 2.53 (t, J = 6.9 Hz, 2H), 2.34 (d, J = 11.8 Hz, 1H), 2.26 (dd, J = 12.3, 6.2 Hz, 1H), 2.17 (d, J = 14.9 Hz, 1H), 2.12−2.08 (m, 1H), 2.04 (d, J = 14.7 Hz, 1H), 1.99−1.94 (m, 2H). HRMS (m/z): for C42H49ClN11O6+[M + H]+, calcd 838.3550; found, 838.3552.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(5-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)pentanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (27).
Compound 27 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 73 (8.2 mg, 0.022 mmol, 1.0 equiv). Compound 27 was obtained as a yellow solid (9.6 mg, yield 51%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.54 (dd, J = 8.4, 7.2 Hz, 1H), 7.36 (dd, J = 6.5, 3.5 Hz, 5H), 7.07−7.00 (m, 2H), 6.91 (d, J = 3.7 Hz, 1H), 5.07−5.03 (m, 1H), 5.00 (dd, J = 9.2, 5.9 Hz, 1H), 4.71−4.61 (m, 2H), 3.82 (q, J = 12.0 Hz, 4H), 3.36 (d, J = 5.8 Hz, 4H), 3.28−3.20 (m, 2H), 3.14−3.09 (m, 2H), 2.88−2.82 (m, 2H), 2.78−2.55 (m, 5H), 2.48 (s, 2H), 2.38−2.29 (m, 1H), 2.28−2.23 (m, 1H), 2.19−2.06 (m, 2H), 2.02 (d, J = 14.6 Hz, 1H), 1.75−1.67 (m, 4H). HRMS (m/z): for C43H51ClN11O6+ [M + H]+, calcd 852.3707; found, 852.3710.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(6-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)hexanoyl)piperazin-1-yl)-propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (28).
Compound 28 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 74 (8.5 mg, 0.022 mmol, 1.0 equiv). Compound 28 was obtained as a yellow solid (13.9 mg, yield 73%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.59−7.48 (m, 1H), 7.43−7.24 (m, 5H), 7.03 (dd, J = 7.8, 5.8 Hz, 2H), 6.93 (d, J = 3.7 Hz, 1H), 5.09−4.96 (m, 2H), 4.70−4.57 (m, 2H), 3.85 (q, J = 12.1 Hz, 4H), 3.33 (t, J = 6.8 Hz, 4H), 3.29−3.22 (m, 2H), 3.14−3.08 (m, 2H), 2.92−2.79 (m, 2H), 2.78−2.57 (m, 5H), 2.43 (t, J = 7.4 Hz, 2H), 2.38−2.30 (m, 1H), 2.28−2.20 (m, 1H), 2.17 (d, J = 14.7 Hz, 1H), 2.13−2.07 (m, 1H), 2.07−1.99 (m, 1H), 1.73−1.61 (m, 4H), 1.48−1.44 (m, 2H). HRMS (m/z): for C44H53ClN11O6+ [M + H]+, calcd 866.3863; found, 866.3859.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(7-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)heptanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (29).
Compound 29 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 75 (8.9 mg, 0.022 mmol, 1.0 equiv). Compound 29 was obtained as a yellow solid (14.5 mg, yield 75%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.54 (dd, J = 8.6, 7.1 Hz, 1H), 7.40−7.30 (m, 5H), 7.03 (t, J = 7.3 Hz, 2H), 6.94 (d, J = 3.7 Hz, 1H), 5.08−4.96 (m, 2H), 4.65 (d, J = 13.8 Hz, 2H), 3.85 (q, J = 12.1 Hz, 4H), 3.36−3.30 (m, 4H), 3.29−3.25 (m, 2H), 3.15−3.10 (m, 2H), 2.91−2.78 (m, 2H), 2.77−2.59 (m, 5H), 2.41 (d, J = 1.6 Hz, 2H), 2.37−2.31 (m, 1H), 2.26 (d, 1H), 2.17 (d, J = 15.0 Hz, 1H), 2.14−2.07 (m, 1H), 2.07−1.98 (m, 1H), 1.68−1.63 (m, 4H), 1.49−1.35 (m, 4H). HRMS (m/z): for C45H55ClN11O6+ [M + H]+, calcd 880.4020; found, 880.4026.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)octanoyl)piperazin-1-yl)-propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (30).
Compound 30 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 76 (9.1 mg, 0.022 mmol, 1.0 equiv). Compound 30 was obtained as a yellow solid (15.5 mg, yield 78%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.54 (dd, J = 8.6, 7.1 Hz, 1H), 7.39−7.32 (m, 5H), 7.03 (dd, J = 7.8, 4.3 Hz, 2H), 6.94 (d, J = 3.7 Hz, 1H), 5.08−4.97 (m, 2H), 4.68−4.62 (m, 2H), 3.84 (q, J = 12.2 Hz, 4H), 3.35−3.31 (m, 4H), 3.28−3.24 (m, 2H), 3.16−3.11 (m, 2H), 2.89−2.81 (m, 2H), 2.78−2.56 (m, 5H), 2.39 (t, J = 7.5 Hz, 2H), 2.34 (t, J = 10.4 Hz, 1H), 2.29−2.22 (m, 1H), 2.17 (d, J = 14.8 Hz, 1H), 2.14−2.10 (m, 1H), 2.03 (d, J = 14.6 Hz, 1H), 1.69−1.64 (m, 2H), 1.62−1.56 (m, 2H), 1.47−1.33 (m, 6H). HRMS (m/z): for C46H57ClN11O6+ [M + H]+, calcd 894.4176; found, 894.4192.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(3-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)propanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (31).
Compound 31 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 77 (8.6 mg, 0.022 mmol, 1.0 equiv). Compound 31 was obtained as a yellow solid (16.7 mg, yield 87%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.59−7.52 (m, 1H), 7.37 (dd, J = 3.9, 1.1 Hz, 1H), 7.34−7.28 (m, 4H), 7.10−7.03 (m, 2H), 6.93 (dd, J = 3.7, 1.5 Hz, 1H), 5.06−5.03 (m, 1H), 4.68−4.59 (m, 3H), 3.91−3.74 (m, 6H), 3.72−3.67 (m, 3H), 3.51−3.45 (m, 3H), 3.19−3.11 (m, 2H), 2.98−2.93 (m, 2H), 2.88−2.78 (m, 2H), 2.77−2.54 (m, 6H), 2.32−2.06 (m, 5H), 2.02 (d, J = 14.6 Hz, 1H). HRMS (m/z): for C43H51ClN11O7+ [M + H]+, calcd 868.3656; found, 868.3661.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(3-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)-propanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (32).
Compound 32 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 78 (9.5 mg, 0.022 mmol, 1.0 equiv). Compound 32 was obtained as a yellow solid (15.8 mg, yield 79%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.54 (m, 1H), 7.37 (dd, J = 3.8, 1.3 Hz, 1H), 7.34−7.28 (m, 4H), 7.08 (d, J = 8.5 Hz, 1H), 7.04 (dd, J = 7.0, 2.7 Hz, 1H), 6.92 (dd, J = 3.8, 1.6 Hz, 1H), 5.08−5.03 (m, 1H), 4.99−4.90 (m, 1H), 4.68−4.54 (m, 3H), 3.89−3.80 (m, 3H), 3.78−3.72 (m, 2H), 3.69−3.66 (m, 2H), 3.65−3.54 (m, 5H), 3.46 (t, J = 5.1 Hz, 3H), 3.28−3.22 (m, 2H), 3.10−3.01 (m, 2H), 2.88−2.81 (m, 2H), 2.77−2.57 (m, 6H), 2.35−2.07 (m, 5H), 2.02 (d, J = 14.7 Hz, 1H). HRMS (m/z): for C45H55ClN11O8 + [M + H]+, calcd 912.3918; found, 912.3902.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(3-(2-(2-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)-ethoxy)propanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (33).
Compound 33 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 79 (10.5 mg, 0.022 mmol, 1.0 equiv). Compound 33 was obtained as a yellow solid (16.2 mg, yield 77%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.55−7.53 (m, 1H), 7.39−7.28 (m, 5H), 7.07 (d, J = 8.6 Hz, 1H), 7.04 (s, 1H), 6.92 (dd, J = 3.7, 1.6 Hz, 1H), 5.07−5.04 (m, 1H), 5.01−4.98 (m, 1H), 4.67−4.53 (m, 3H), 3.91−3.80 (m, 3H), 3.73−3.67 (m, 4H), 3.64−3.53 (m, 8H), 3.53−3.44 (m, 4H), 3.25 (t, J = 12.8 Hz, 2H), 3.11−3.07 (m, 2H), 2.91−2.80 (m, 2H), 2.77−2.57 (m, 7H), 2.37−2.23 (m, 2H), 2.20−2.07 (m, 2H), 2.03 (d, J = 14.7 Hz, 1H). HRMS (m/z): for C47H59ClN11O9+ [M + H]+, calcd 956.4180; found, 956.4183.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12-tetraoxapentadecan-15-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (34).
Compound 34 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 80 (11.5 mg, 0.022 mmol, 1.0 equiv). Compound 34 was obtained as a yellow solid (12.8 mg, yield 58%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.56−7.53 (m, 1H), 7.38−7.31 (m, 5H), 7.08 (d, J = 8.5 Hz, 1H), 7.04 (dd, J = 7.1, 2.1 Hz, 1H), 6.92 (dd, J = 3.8, 1.3 Hz, 1H), 5.05 (dd, J = 12.8, 5.5 Hz, 1H), 5.02−4.96 (m, 1H), 4.68−4.57 (m, 3H), 3.87−3.84 (m, 3H), 3.73−3.68 (m, 4H), 3.65−3.60 (m, 4H), 3.59−3.53 (m, 9H), 3.49 (t, J = 5.2 Hz, 3H), 3.28−3.24 (m, 2H), 3.12−3.07 (m, 2H), 2.89−2.80 (m, 2H), 2.77−2.56 (m, 7H), 2.38−2.24 (m, 2H), 2.19−2.08 (m, 2H), 2.03 (d, J = 14.7 Hz, 1H). HRMS (m/z): for C49H63ClN11O10+ [M + H]+, calcd 1000.4442; found, 1000.4462.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaoctadecan-18-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (35, MS5033).
Compound 35 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15.9 mg, 0.022 mmol) and 81 (9.5 mg, 0.022 mmol, 1.0 equiv). Compound 35 was obtained as a yellow solid (5.0 mg, yield 22%). 1H NMR (600 MHz, CD3OD) 8.41 (s, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.40 (d, J = 7.6 Hz, 3H), 7.36 (d, J = 8.0 Hz, 2H), 7.09 (d, J = 8.5 Hz, 1H), 7.06 (d, J = 7.1 Hz, 1H), 6.95 (s, 1H), 5.07 (dd, J = 12.9, 5.5 Hz, 1H), 5.04 (t, J = 7.7 Hz, 1H), 4.66−4.53 (m, 2H), 3.99−3.90 (m, 2H), 3.74 (t, J = 5.8 Hz, 4H), 3.70−3.53 (m, 23H), 3.53−3.46 (m, 2H), 3.38 (s, 2H), 3.34 (s, 1H), 3.19−3.10 (m, 1H), 2.92−2.83 (m, 1H), 2.81−2.61 (m, 5H), 2.43−2.27 (m, 2H), 2.24−2.19 (m, 1H), 2.16−2.05 (m, 2H); 13C NMR (201 MHz, CD3OD): δ 173.27, 171.13, 170.29, 169.30, 169.25, 167.90, 154.31, 146.81, 143.35, 142.95, 139.52, 135.89, 133.43, 132.43, 128.61 (2C), 128.18 (2C), 123.56, 116.95, 110.71, 109.83, 103.36, 102.37, 70.22 (2C), 70.11, 70.07 (2C), 69.90 (2C), 69.85, 69.17, 67.14, 58.15, 53.94, 51.78, 51.67, 51.45, 48.84 (2C), 48.47, 42.75, 41.86, 38.36, 32.80, 30.83, 30.75, 30.62, 29.25, 22.42. HRMS (m/z): for C51H67ClN11O11+ [M + H]+, calcd 1044.4705; found, 1044.4708.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15,18-hexaoxahenicosan-21-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]-pyrimidin-4-yl)piperidine-4-carboxamide (36).
Compound 36 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 82(12.2 mg, 0.02 mmol, 1.0 equiv). Compound 36 was obtained as a yellow solid (10.3 mg, yield 47%). 1H NMR (600 MHz, CD3OD): δ 8.38 (s, 1H), 7.69 (d, J = 7.3 Hz, 1H), 7.61−7.51 (m, 1H), 7.44−7.28 (m, 5H), 7.07 (dd, J = 20.2, 7.8 Hz, 1H), 6.92 (d, J = 3.6 Hz, 1H), 5.09−4.98 (m, 2H), 4.65 (d, J = 12.4 Hz, 2H), 3.91−3.79 (m, 2H), 3.79−3.69 (m, 4H), 3.69−3.48 (m, 24H), 3.41−3.07 (m, 9H), 2.87 (t, J = 14.8 Hz, 1H), 2.80−2.53 (m, 5H), 2.41−2.24 (m, 2H), 2.18−2.11 (m, 2H), 2.04 (d, J = 14.7 Hz, 1H). HRMS (m/z): for C53H71ClN11O12+ [M + H]+, calcd 1088.4967; found, 1088.4974.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (37).
Compound 37 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 83 (14.1 mg, 0.02 mmol, 1.0 equiv). Compound 37 was obtained as a yellow solid (16.1 mg, yield 68%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.41−7.26 (m, 5H), 7.06 (dd, J = 21.5, 7.8 Hz, 2H), 6.93 (d, J = 3.7 Hz, 1H), 5.13−5.01 (m, 2H), 4.61 (d, J = 14.1 Hz, 2H), 3.94−3.79 (m, 2H), 3.80−3.37 (m, 38H), 3.36−3.00 (m, 8H), 2.91−2.83 (m, 1H), 2.79−2.53 (m, 4H), 2.40−2.26 (m, 2H), 2.22−2.00 (m, 3H). HRMS (m/z): for C57H79ClN11O14+ [M + H]+, calcd 1176.5491; found, 1176.5475.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15,18,21,24,27-nonaoxatriacontan-30-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo-[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (38).
Compound 38 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 84 (14.8 mg, 0.02 mmol, 1.0 equiv). Compound 38 was obtained as a yellow solid (15.9 mg, yield 65%). 1H NMR (600 MHz, CD3OD): δ 8.40 (s, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.46−7.30 (m, 5H), 7.10−7.07 (m, 2H), 6.94 (d, J = 3.6 Hz, 1H), 5.15−4.98 (m, 2H), 4.64 (d, J = 14.3 Hz, 2H), 3.87 (q, J = 10.9 Hz, 2H), 3.81−3.39 (m, 40H), 3.39−2.96 (m, 10H), 2.89−2.85 (m, 1H), 2.81−2.57 (m, 4H), 2.41−2.24 (m, 2H), 2.24−1.97 (m, 3H). HRMS (m/z): for C59H83ClN11O15 +[M + H]+, calcd 1220.5753; found, 1220.5767.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaoctadecan-18-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (39).
Compound 39 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 85 (11 mg, 0.02 mmol, 1.0 equiv). Compound 39 was obtained as a yellow solid (16.1 mg, yield 78%). 1H NMR (600 MHz, CD3OD): δ 8.42 (s, 1H), 7.58−7.37 (m, 6H), 7.08 (d, J = 8.5 Hz, 1H), 7.05 (d, J = 7.1 Hz, 1H), 6.96 (s, 1H), 5.07 (dd, J = 12.9, 5.5 Hz, 1H), 5.04 (t, J = 7.7 Hz, 1H), 4.66−4.53 (m, 2H), 3.99−3.90 (m, 2H), 3.75 (d, J = 5.8 Hz, 4H), 3.69−3.54 (m, 23H), 3.53−3.47 (m, 2H), 3.38 (s, 2H), 3.35 (s, 1H), 3.19−3.10 (m, 1H), 2.92−2.83 (m, 1H), 2.83−2.62 (m, 7H), 2.42−2.24 (m, 2H), 2.23−2.18 (m, 1H), 2.15−2.05 (m, 2H); HRMS (m/z): for C51H69ClN11O10+ [M + H]+, calcd 1030.4912; found, 1030.4945.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)-3,6,9,12,15-pentaoxaoctadecan-18-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (40).
Compound 40 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 86 (11.2 mg, 0.02 mmol, 1.0 equiv). Compound 40 was obtained as a white solid (13.4 mg, 64%). 1H NMR (600 MHz, CD3OD): δ 8.39 (s, 1H), 7.78 (t, J = 7.5 Hz, 1H), 7.47 (d, J = 8.2 Hz, 2H), 7.42−7.31 (m, 5H), 6.92 (d, J = 3.6 Hz, 1H), 5.11 (dd, J = 12.8, 5.4 Hz, 1H), 5.01 (d, J = 8.2 Hz, 1H), 4.79−4.60 (m, 3H), 4.38 (s, 1H), 3.92 (d, J = 4.5 Hz, 2H), 3.88−3.78 (m, 3H), 3.78−3.69 (m, 3H), 3.64−3.58 (m, 16H), 3.37−3.02 (m, 12H), 2.88 (t, J = 15.1 Hz, 1H), 2.81−2.54 (m, 3H), 2.43−2.25 (m, 2H), 2.22−2.10 (m, 1H), 2.04 (d, J = 14.6 Hz, 1H). HRMS (m/z): for C51H66ClN10O12+ [M + H]+, calcd 1045.4545; found, 1045.4534.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(19-(2-(2,6-dioxopiperidin-3-yl) - 1, 3-dioxoisoindolin-4 - yl) - 4, 7, 10, 13, 16-pentaoxanonadecanoyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (41).
Compound 41 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (14.2 mg, 0.02 mmol) and 87 (11.4 mg, 0.02 mmol, 1.0 equiv). Compound 41 was obtained as a white solid (12.1 mg, yield 58%). 1H NMR (600 MHz, CD3OD): δ 8.40 (s, 1H), 7.80−7.71 (m, 2H), 7.66 (dd, J = 6.1, 2.8 Hz, 1H), 7.45−7.26 (m, 5H), 6.95 (d, J = 3.6 Hz, 1H), 5.13 (dd, J = 12.8, 5.4 Hz, 1H), 5.02 (t, J = 7.7 Hz, 1H), 4.66 (s, 2H), 3.86 (q, J = 11.8 Hz, 2H), 3.73 (t, J = 6.1 Hz, 2H), 3.63−3.57 (m, 17H), 3.51 (t, J = 6.4 Hz, 2H), 3.36−3.24 (m, 9H), 3.23−3.06 (m, 3H), 2.94−2.84 (m, 1H), 2.81−2.55 (m, 5H), 2.42−2.25 (m, 2H), 2.16 (t, J = 17.3 Hz, 2H), 2.04 (d, J = 14.8 Hz, 1H), 1.99−1.91 (m, 2H). HRMS (m/z): for C52H68ClN10O11+ [M + H]+, calcd 1043.4752; found, 1043.4746.
4-Amino-N-((S)-1-(4-chlorophenyl)-3-(4-(11-(((S)-1-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecanoyl)-piperazin-1-yl)propyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-piperidine-4-carboxamide (42, MS143N).
Compound 42 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (15 mg, 0.025 mmol) and 12-(((S)-1-((2S,4R)-4-(benzyloxy)-2-((4-(4-methylthiazol-5-yl)benzyl)-carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (94, 18.4 mg, 0.025 mmol, 1.0 equiv). Compound 42 was obtained as a white solid (15.8 mg, yield 52%). 1H NMR (600 MHz, CD3OD): δ 8.80 (s, 1H), 8.26 (s, 1H), 7.41−7.07 (m, 14H), 6.78 (d, J = 3.7 Hz, 1H), 4.92 (dd, J = 9.2, 5.9 Hz, 1H), 4.61 (s, 1H), 4.58−4.47 (m, 3H), 4.47−4.35 (m, 3H), 4.26 (d, J = 15.4 Hz, 1H), 4.22−4.14 (m, 2H), 3.76−3.60 (m, 3H), 3.28−3.19 (m, 8H), 3.18−3.07 (m, 1H), 3.07−2.97 (m, 1H), 2.58−2.43 (m, 2H), 2.37 (s, 3H), 2.33−2.22 (m, 4H), 2.22−1.95 (m, 5H), 1.92 (dd, J = 14.9, 4.1 Hz, 1H), 1.55−1.41 (m, 4H), 1.28−1.12 (m, 12H), 0.94 (s, 9H). 13C NMR (151 MHz, CD3OD): δ 174.69, 172.96, 172.73, 171.07, 169.44, 154.95, 151.52, 147.60, 144.60, 139.53, 138.88, 138.02, 133.46, 130.09, 129.10, 128.97 (2C), 128.60 (2C), 128.09 (2C), 127.98, 127.96 (2C), 127.60 (2C), 127.58 (2C), 127.43, 127.30, 123.12, 102.71, 77.13, 75.29, 70.26, 59.44, 58.34, 57.42, 54.00, 53.11, 51.59, 51.43, 42.30, 42.22, 41.52, 38.25, 36.08, 35.34, 35.27, 35.02, 32.17, 30.77, 30.65, 29.22, 29.15 (2C), 29.09, 29.01, 28.96, 28.90, 25.65, 25.60 (3C), 24.75, 14.38. HRMS (m/z): for C66H88ClN12O6S+ [M + H]+, calcd 1211.6354; found, 1211.6365.
4-Amino-N-((1S)-1-(4-chlorophenyl)-3-(4-(1-((2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)-3,6,9,12,15-pentaoxaoctadecan-18-oyl)piperazin-1-yl)propyl)-1-(7H-pyrrolo-[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (43).
Compound 43 was synthesized following the standard procedure for preparing compound 3 from intermediates 2 (57.3 mg, 0.096 mmol) and 1-{[2-(1-methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}−3,6,9,12,15-pentaoxaoctadecan-18-oic acid (97, 55.4 mg, 0.096 mmol, 1.0 equiv). Compound 43 was obtained as a yellow solid (63.7 mg, yield 63%). 1H NMR (600 MHz, CD3OD): δ 8.42 (s, 1H), 7.57 (t, J = 7.8 Hz, 1H), 7.44−7.34 (m, 5H), 7.09 (dd, J = 28.6, 7.8 Hz, 2H), 6.97 (d, J = 3.5 Hz, 1H), 5.10 (dd, J = 13.0, 5.4 Hz, 1H), 5.05 (t, J = 7.6 Hz, 1H), 4.61 (d, J = 14.4 Hz, 2H), 4.00−3.90 (m, 2H), 3.79−3.70 (m, 4H), 3.70−3.66 (m, 4H), 3.66−3.46 (m, 20H), 3.39 (s, 2H), 3.36−3.33 (m, 2H), 3.16 (s, 3H), 2.94−2.85 (m, 2H), 2.77−2.54 (m, 5H), 2.44−2.28 (m, 2H), 2.23 (d, J = 14.7 Hz, 1H), 2.16−2.05 (m, 2H); 13C NMR (201 MHz, CD3OD): δ 172.30, 171.10, 170.07, 169.33, 169.24, 167.92, 154.31, 146.84, 143.33, 142.91, 139.56, 135.90, 133.44, 132.44, 128.62 (2C), 128.19 (2C), 123.59, 116.96, 110.72, 109.85, 103.39, 102.38, 70.22 (2C), 70.14 (2C), 70.09 (2C), 69.93 (2C), 69.86, 69.22, 67.16, 58.15, 53.95, 51.79, 51.68, 51.46, 49.50, 48.51, 42.76, 41.88, 38.38, 32.81, 31.14, 30.77, 30.64, 29.28, 26.05, 21.68; HRMS (m/z): for C52H69ClN11O11+ [M + H]+, calcd 1058.4861; found, 1058.4864.
12-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-12-oxododecanoic Acid (66).
To the solution of commercially available (2S,4R)-1-[(S)-2-amino-3,3-dimethylbutanoyl]-4-hydroxy-N-[4-(4-methylthiazol-5-yl)benzyl]pyrrolidine-2-carboxamide (101, 215 mg, 0.5 mmol) in DMSO (1.5 mL) were added dodecanedioic acid (93, 138 mg, 0.6 mmol, 1.5 equiv), EDCI (215 mg, 0.75 mmol, 1.5 equiv), HOAt (147 mg, 0.75 mmol, 1.5 equiv), and NMM (102 mg, 1.5 mmol, 3.0 equiv). After stirring overnight at rt, the resulting mixture was purified by reverse-phase ISCO (methanol/0.1% TFA in H2O) to afford compound 66 as a white solid (154 mg, yield 48%). 1H NMR (600 MHz, CD3OD): δ 8.91 (s, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 4.69−4.63 (m, 1H), 4.61−4.49 (m, 3H), 4.38 (dd, J = 15.4, 5.3 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 2.50 (s, 3H), 2.36−2.19 (m, 5H), 2.14−2.06 (m, 1H), 1.65−1.58 (m, 4H), 1.34 (t, J = 4.9 Hz, 12H), 1.06 (s, 9H). HRMS (m/z): for C34H51N4O6S+ [M + H]+, calcd 643.3524; found, 643.3523.
13-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-13-oxotridecanoic Acid (67).
Compound 67 was synthesized following the standard procedure for preparing compound 66 from (2S,4R)-1-[(S)-2-amino-3,3-dimethylbutanoyl]-4-hydroxy-N-[4-(4-methylthiazol-5-yl)benzyl]pyrrolidine-2-carboxamide (101, 215 mg, 0.5 mmol) and tridecanedioic acid (98, 147 mg, 0.6 mmol, 1.0 equiv). Compound 67 was obtained as a white solid (158 mg, yield 48%). 1H NMR (600 MHz, CD3OD): δ 8.91 (s, 1H), 7.49 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 4.68−4.64 (m, 1H), 4.61−4.49 (m, 3H), 4.38 (dd, J = 15.4, 5.1 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.82 (dd, J = 11.0, 3.9 Hz, 1H), 2.50 (s, 3H), 2.38−2.20 (m, 5H), 2.15−2.05 (m, 1H), 1.65−1.58 (m, 4H), 1.42−1.29 (m, 14H), 1.06 (s, 9H). HRMS (m/z): for C35H53N4O6S+ [M + H]+, calcd 657.3680; found, 657.3683.
14-(((S)-1-((2S,4R)-4-Hydroxy-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-14-oxotetradecanoic Acid (68).
Compound 68 was synthesized following the standard procedure for preparing compound 66 from (2S,4R)-1-[(S)-2-amino-3,3-dimethylbutanoyl]-4-hydroxy-N-[4-(4-methylthiazol-5-yl)benzyl]pyrrolidine-2-carboxamide (101, 215 mg, 0.5 mmol) and tetradecanedioic acid (99, 155 mg, 0.6 mmol, 1.0 equiv). Compound 68 was obtained as a white solid (145 mg, yield 43%). 1H NMR (600 MHz, CD3OD): δ 8.92 (s, 1H), 7.49 (d, J = 8.1 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 4.68−4.63 (m, 1H), 4.62−4.50 (m, 3H), 4.39 (dd, J = 15.4, 5.1 Hz, 1H), 3.92 (d, J = 11.0 Hz, 1H), 3.83 (dd, J = 11.0, 3.9 Hz, 1H), 2.50 (s, 3H), 2.36−2.20 (m, 5H), 2.17−2.03 (m, 1H), 1.67−1.59 (m, 4H), 1.43−1.29 (m, 16H), 1.06 (s, 9H). HRMS (m/z): for C36H55N4O6S+ [M + H]+, calcd 671.3837; found, 671.3875.
11-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)-phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecanoic Acid (69).
To a solution of (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide51 (102, 150 mg, 0.34 mmol) in DMSO (1.5 mL) were added undecanedioic acid (100, 88 mg, 0.41 mmol, 1.5 equiv), EDCI (98 mg, 0.51 mmol, 1.5 equiv), HOAt (70 mg, 0.51 mmol, 1.5 equiv), and NMM (103 mg, 1.02 mmol, 3.0 equiv). After stirring overnight at rt, the resulting mixture was purified by preparative HPLC (10%−100% methanol/0.1% TFA in H2O) to afford compound 69 as a white solid (134.8 mg, yield 62%). 1H NMR (600 MHz, CD3OD): δ 9.01 (s, 1H), 7.47−7.40 (m, 4H), 5.00 (q, J = 7.0 Hz, 1H), 4.62 (s, 1H), 4.59−4.54 (m, 1H), 4.46−4.42 (m, 1H), 3.89−3.87 (m, 1H), 3.74 (dd, J = 11.0, 4.0 Hz, 1H), 2.49 (s, 3H), 2.34−2.15 (m, 5H), 1.96−1.92 (m, 1H), 1.63−1.55 (m, 4H), 1.50 (d, J = 7.0 Hz, 3H), 1.32 (t, J = 4.4 Hz, 10H), 1.04 (s, 9H). HRMS (m/z): for C34H51N4O6S+ [M + H]+, calcd 643.3524; found, 643.3527.
1-{[2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}−3,6,9,12,15,18-hexaoxahenicosan-21-oic Acid (82).
To a solution of 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (106, 169 mg, 0.61 mmol) in NMP (2 mL) were added commercially available tert-butyl 1-amino-3,6,9,12,15,18-hexaoxahenicosan-21-oate (103, 250 mg, 0.61 mmol, 1.0 equiv) and DIPEA (266 μL, 1.53 mmol, 2.5 equiv). The reaction was heated at 100 °C for 1 h under microwave. The reaction mixture was purified by reverse-phase ISCO (acetonitrile/0.1% TFA in H2O) to afford the intermediate as a yellow solid. This intermediate was dissolved in DCM (3 mL) and TFA (3 mL). After the reaction was stirred for 30 min, solvents were removed. The resulting residue was purified by reverse-phase ISCO (acetonitrile/0.1% TFA in H2O) to afford the title compound 82 as a yellow solid (124 mg, yield 33%). 1H NMR (600 MHz, CD3OD): δ 7.83 (s, 1H), 7.57−7.52 (m, 2H), 5.05 (dd, J = 12.6, 5.5 Hz, 1H), 3.85 (t, J = 5.8 Hz, 1H), 3.78−3.63 (m, 20H), 3.57−3.46 (m, 4H), 2.95−2.81 (m, 1H), 2.73 (t, J = 16.1 Hz, 1H), 2.63−2.45 (m, 4H), 2.11 (s, 1H). HRMS (m/z): for C28H40N3O12+ [M + H]+, calcd 610.2607; found, 610.2612.
1-{[2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}−3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oic Acid (83).
Compound 83 was synthesized following the standard procedure for preparing compound 82 from 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (106, 139 mg, 0.5 mmol) and tert-butyl 1-amino-3,6,9,12,15,18,21,24-octaoxaheptacosan-27-oate (104, 250 mg, 0.5 mmol, 1.0 equiv). Compound 83 was obtained as a yellow solid (188 mg, yield 54%). 1H NMR (600 MHz, CD3OD): δ 7.56 (t, J = 7.8 Hz, 1H), 7.10−7.08 (m, 2H), 5.05 (dd, J = 12.9, 5.4 Hz, 1H), 3.84−3.47 (m, 34H), 2.94−2.81 (m, 1H), 2.73 (t, J = 15.5 Hz, 2H), 2.54 (t, J = 6.3 Hz, 2H), 2.11 (d, J = 12.4 Hz, 1H). HRMS (m/z): for C32H48N3O14+ [M + H]+, calcd 698.3131; found, 698.3138.
1-{[2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}−3,6,9,12,15,18,21,24,27-nonaoxatriacontan-30-oic Acid (84).
Compound 84 was synthesized following the standard procedure for preparing compound 82 from 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (106, 51 mg, 0.18 mmol) and tert-butyl 1-amino-3,6,9,12,15,18,21,24,27-nonaoxatriacontan-30-oate (105, 100 mg, 0.18 mmol, 1.0 equiv). Compound 84 was obtained as a yellow solid (107 mg, yield 81%). 1H NMR (600 MHz, CD3OD): δ 7.56 (t, J = 7.9 Hz, 1H), 7.09−7.07 (dd, 2H), 5.05 (dd, J = 12.9, 5.4 Hz, 1H), 3.81−3.48 (m, 38H), 2.85 (d, J = 15.4 Hz, 1H), 2.73 (t, J = 15.6 Hz, 2H), 2.54 (t, J = 6.2 Hz, 2H), 2.16−2.05 (m, 1H). HRMS (m/z): for C34H52N3O15+ [M + H]+, calcd 742.3393; found, 742.3397.
1-{[2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl]amino}−3,6,9,12,15-pentaoxaoctadecan-18-oic Acid (85).
To a solution of 3-(4-amino-1-oxoisoindolin-2-yl)piperidine-2,6-dione (108, 50 mg, 0.19 mmol) in DMF (2 mL) were added tert-butyl 1-bromo-3,6,9,12,15-pentaoxaoctadecan-18-oate (107, 100 mg, 1.2 mmol, 1.2 equiv), NaHCO3 (37 mg, 0.44 mmol, 2.3 equiv), and NaI (6.3 mg, 0.038 mmol, 0.2 equiv). After the reaction was stirred at 60 °C overnight, it was quenched by water. The reaction was extracted with EtOAc (3 × 10 mL), dried over Na2SO4, filtered, and concentrated. The resulting residue was dissolved in DCM (2 mL) and TFA (2 mL). After the reaction was stirred for 30 min, solvents were removed. The residue was purified by preparative HPLC (10%–100% methanol/0.1% TFA in H2O) to afford compound 85 as a yellow solid (84.8 mg, yield 81%). 1H NMR (600 MHz, CD3OD): δ 7.44−7.33 (m, 2H), 7.15 (d, J = 7.8 Hz, 1H), 5.22 (d, J = 12.4 Hz, 1H), 4.02 (s, 2H), 3.78−3.56 (m, 22H), 3.05−2.84 (m, 2H), 2.61−2.39 (m, 3H), 2.25−2.14 (m, 1H). HRMS (m/z): for C26H38N3O10+ [M + H]+, calcd 552.2552; found, 552.2556.
1-{[2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]oxy}−3,6,9,12,15-pentaoxaoctadecan-18-oic Acid (86).
Compound 86 was synthesized following the standard procedure for preparing compound 85 from 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione (109, 79 mg, 0.29 mmol) and 107 (150 mg, 0.35 mmol, 1.2 equiv). Compound 86 was obtained as a white solid (113 mg, yield 69%). 1H NMR (600 MHz, CD3OD): δ 7.77 (t, J = 8.0 Hz, 1H), 7.47 (dd, J = 11.5, 7.8 Hz, 2H), 5.09 (dd, J = 12.9, 5.4 Hz, 1H), 4.38 (t, J = 4.6 Hz, 2H), 3.92 (t, J = 4.6 Hz, 2H), 3.83−3.55 (m, 18H), 2.95−2.83 (m, 1H), 2.79−2.66 (m, 2H), 2.60−2.50 (m, 2H), 2.19−2.03 (m, 1H). HRMS (m/z): for C26H35N2O12+ [M + H]+, calcd 567.2185; found, 567.2197.
19-[2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]-4,7,10,13,16-pentaoxanonadecanoic Acid (87).
To a solution of 4-bromo-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (111, 33.7 mg, 0.1 mmol) and tert-butyl 4,7,10,13,16-pentaoxanonadec-18-ynoate (110, 72 mg, 0.2 mmol, 2 equiv) in DMA (1.5 mL) were added tetrakis(triphenylphosphine)palladium (0) (2.3 mg, 2 mol %), copper iodide (1 mg, 4 mol %), and triethylamine (22 mg, 0.3 mmol, 3.0 equiv). After the reaction was stirred at 100 °C for 3 h, the mixture was purified by preparative HPLC (10%−100% acetonitrile/0.1% TFA in H2O) to afford the desired product (49.1 mg, yield 80%). This intermediate was dissolved in MeOH, and palladium on carbon (5 mg, 10%) was added. The reaction was stirred at rt under hydrogen for 1 h, before it was filtered. The filtrate was concentrated. The resulting residue was dissolved in DCM (1 mL) and TFA (1 mL). After the reaction was stirred for 30 min, solvents were removed. The resulting residue was purified by preparative HPLC (10%−100% acetonitrile/0.1% TFA in H2O) to afford the title compound 87 as a white solid (24 mg, yield 57%). 1H NMR (600 MHz, CD3OD): δ 7.74−7.63 (m, 2H), 7.58−7.47 (m, 1H), 5.12 (dd, J = 12.7, 5.2 Hz, 1H), 3.71 (t, J = 6.1 Hz, 2H), 3.67−3.55 (m, 16H), 3.50 (t, J = 6.1 Hz, 2H), 3.17 (t, J = 7.8 Hz, 2H), 2.95−2.81 (m, 1H), 2.74 (t, J = 14.8 Hz, 2H), 2.53 (t, J = 6.2 Hz, 2H), 2.13 (t, J = 8.9 Hz, 1H), 1.94 (t, J = 7.5 Hz, 2H). HRMS (m/z): for C27H37N2O11+ [M + H]+, calcd 565.2392; found, 565.2388.
12-(((S)-1-((2S,4R)-4-(Benzyloxy)-2-((4-(4-methylthiazol-5-yl)-benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)-amino)-12-oxododecanoic Acid (94).
To a solution of [4-(4-methylthiazol-5-yl)phenyl]methanamine (88, 1 mmol, 204 mg) in DMSO (5 mL) were added commercially available (2S,4R)-4-(benzyloxy)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (89, 1 mmol, 321 mg), EDCI (288 mg, 1.5 mmol), HOAt (204 mg, 1.5 mmol), and NMM (303 mg, 3 mmol). After stirring for 2 h at rt, the resulting mixture was purified by reverse-phase ISCO (methanol/0.1% TFA in H2O) to afford an intermediate, which was dissolved in TFA (5 mL) and DCM (5 mL). After the resulting mixture was stirred for 30 min, solvents were removed. The resulting residue was purified by reverse-phase ISCO (methanol/0.1% TFA in H2O) to afford (2S,4R)-4-(benzyloxy)-N-[4-(4-methylthiazol-5-yl)-benzyl]pyrrolidine-2-carboxamide (90, 276 mg, yield 68%). To a solution of 90 (84 mg, 0.2 mmol) in DMSO (1 mL) was added commercially available (S)-2-[(tert-butoxycarbonyl)amino]-3,3-dimethylbutanoic acid (91, 48.5 mg, 0.2 mmol), EDCI (11.6 mg, 0.06 mmol), HOAt (8.2 mg, 0.06 mmol), and NMM (12.7 mg, 0.12 mmol), the reaction was stirred at rt for 3 h, and the mixture was purified by preparative HPLC (10%−100% acetonitrile/0.1% TFA in H2O) to afford the intermediate. This intermediate was dissolved in DCM (1 mL) and TFA (1 mL) and the resulting mixture was stirred at rt for 30 min. The solvent was evaporated and the resulting mixture was purified by preparative HPLC (10%−100% acetonitrile/0.1% TFA in H2O) to afford (2S,4R)-1-[(S)-2-amino-3,3-dimethylbutano-yl]-4-(benzyloxy)-N-[4-(4-methylthiazol-5-yl)benzyl]pyrrolidine-2-carboxamide (92, 39.5 mg, yield 38% for two steps) as a white solid. ESI (m/z): [M + H+]: 521.3; To a solution of 92 (21 mg, 0.04 mmol) in DMSO (1 mL) were added dodecanedioic acid (93, 18.5 mg, 0.08 mmol), EDCI (11.6 mg, 0.06 mmol), HOAt (8.2 mg, 0.06 mmol), and NMM (12.7 mg, 0.12 mmol). After stirring for 2 h at rt, the resulting mixture was purified by preparative HPLC (10%−100% acetonitrile/0.1% TFA in H2O) to afford the title compound 94 as a white solid (18.4 mg, yield 63%). 1H NMR (600 MHz, CD3OD): δ 8.94 (s, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 7.38−7.31 (m, 4H), 7.31−7.25 (m, 1H), 4.73 (s, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.58−4.49 (m, 3H), 4.38 (d, J = 15.4 Hz, 1H), 4.35−4.28 (m, 2H), 3.76 (dd, J = 11.3, 3.6 Hz, 1H), 2.50 (s, 3H), 2.46–2.38 (m, 1H), 2.32−2.20 (m, 4H), 2.15−2.08 (m, 1H), 1.66−1.54 (m, 4H), 1.38−1.25 (m, 12H), 1.06 (s, 9H). HRMS (m/z): for C41H57N4O6S+ [M + H]+, calcd 733.3993; found, 733.3987.
1-{[2-(1-Methyl-2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl]amino}−3,6,9,12,15-pentaoxaoctadecan-18-oic Acid (97).
Compound 97 was synthesized following the standard procedure for preparing compound 82 from commercially available tert-butyl 1-amino-3,6,9,12,15-pentaoxaoctadecan-18-oate (95, 74 mg, 0.2 mmol) and 4-fluoro-2-(1-methyl-2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione56 (96, 58 mg, 0.2 mmol). Compound 97 was obtained as a yellow solid (56.4 mg, yield 49%). 1H NMR (600 MHz, CD3OD): δ 7.54 (dd, J = 8.5, 7.1 Hz, 1H), 7.09 (d, J = 8.5 Hz, 1H), 7.04 (d, J = 7.0 Hz, 1H), 5.07 (dd, J = 12.9, 5.4 Hz, 1H), 3.74−3.68 (m, 4H), 3.67−3.55 (m, 16H), 3.50 (t, J = 5.3 Hz, 2H), 3.14 (s, 3H), 2.90−2.85 (m, 2H), 2.74−2.62 (m, 1H), 2.53 (t, J = 6.2 Hz, 2H), 2.12−2.05 (m, 1H); HRMS (m/z): for C27H38N3O11+ [M + H]+, calcd 580.2501; found, 580.2513.
Cell Culture.
All cell lines were purchased from ATCC and authenticated. Cell lines were regularly tested in the lab for mycoplasma. All cells were cultured at 37 °C under 5% CO2. Cell lines were cultured in 1× DMEM (Corning, 10-013-CV) with 10% fetal bovine serum (Atlanta Biologicals S11150) and 1X Penicillin/Streptomycin. Cells were split using 0.05 or 0.25% trypsin (Corning 25-051-Cl or 25-053-Cl, respectively) before they reached full confluence and media were changed every 3–4 days.
Western Blotting Assay.
Cells were lysed in a 2× sample buffer (125 mM Tris–HCl at pH 6.8, 10% βME, 2% sodium dodecyl sulfate, 20% glycerol, 0.05% Bromophenol Blue, 8 M urea). Protein lysates were loaded into 4–12% Bis-Tris gels and resolved by electrophoresis. Samples were then blotted on a poly(vinylidene difluoride) membrane (Millipore IPVH00010) using the wet transfer technique (Invitrogen). Membranes were blocked in 5% milk-TBST for 30 min, and incubated in primary antibody in 5% milk-TBST or 5% bovine serum albumin-TBST at 4 °C for 16 h. Membranes were rinsed (3 × 10 min) in TBST and incubated in horseradish peroxidase-conjugated secondary antibodies in 5% milk-TBST for at least 2 h and rinsed again in TBST (3 × 6 min). Membranes were visualized using the chemiluminescence system (Thermo 34080, 37075) on an auto-radiography film (Denville E3018). Primary antibodies: β-actin (Sigma A5316), p-AKT (Ser473 CST-9721), total AKT (CST-9272), AKT1 (2H10, CST-2967), AKT2 (CST-3063), AKT3 (CST-8018), p-S6 (Ser240/244, CST-5364), and p-PRAS40 (Thr246, CST-2997). Secondary antibodies: Mouse (Thermo 31432) and Rabbit (Thermo 31460).
AKT Binding Assay.
Binding affinities of 20, 35, 42, and 43 to three AKT isoforms (AKT1, AKT2, and AKT3) were determined by DiscoverX using a competition binding assay (KINOMEscan assay) to quantitatively measure the ability of a compound to compete with an immobilized, active-site directed ligand. The assay comprises three components: DNA-tagged AKT, immobilized ligand, and a test compound. The ability of the test compound to compete with the immobilized ligand was measured by quantitative polymerase chain reaction of the DNA tag. The Kd values were determined by using an 11-point threefold compound dilution (the top concentration of 30 μM) with three DMSO control points in duplicates.
ITC Assay.
VHL (Elongin C, Elongin B, and Von Hippel–Lindau) protein was prepared for ITC by desalting and equilibrating using ITC buffer (25 mM Tris pH 7.5 RT, 500 mM NaCl) in a GE healthcare PD-20 Sephadex column by gravity. Protein was then concentrated to stock 650 μM and final sample concentrations prepared (40 μM) with DMSO added from 1% (v/v) to minimize buffer mismatch between the cell and syringe. Compounds were diluted to a final concentration of 400 μM in a buffer with additional 1% DMSO (v/v). ITC was performed on a MicroCal ITC 200 device (Malvern, Worcestershire, UK) at 25 °C with an initial injection of 0.4 μL and 19 injections of 2.0 μL (180 s spacing, reference power 11) at a stirring rate of 750 rpm. Data were analyzed with Origin MicroCal Analysis software using a chi-squared fit model.
Cell Proliferation Assay.
Experiments were carried out in 96-well plates in triplicates (Corning 720089). A total of 1–3 × 103 cells per well were grown in the presence of indicated compounds, and cells were then monitored for 3–5 days using the IncuCyte live cell imaging system (Essen BioScience, Ann Arbor, MI, USA), which was placed in a cell culture incubator operated at 37 °C under 5% CO2. Cell confluence was determined using calculations derived from phase-contrast image readings on an IncuCyte ZOOM (Essen Biosciences) on live cells over time.
Cell Colony Formation Assay.
Cells were cultured for 14 days in the presence of different compounds. Media with compound were replenished every 2 days. At the end of the experiment, media were aspirated and viable cells were stained with 0.5% crystal violet dye.
Mouse Pharmacokinetic Study.
Compounds 20 and 35 in HCl salt form were dissolved in a formulation of 5% DMA, 5% Solutol HS-15, and 90% normal saline. Six male Swiss Albino mice were administered with a solution formulation of each compound at an indicated dose via IP injection. All samples were maintained below −70 ± 10 °C until performing bioanalysis. Approximately 60 μL of plasma samples were collected from three mice at 0.5, 1, 2, 4, 8, and 12 h. Pharmacokinetic analysis was conducted using the NCA module of Phoenix WinNonlin (Version 8.0) and plasma concentrations were quantified by fit-for-purpose LC–MS/MS method (LLOQ: 10.18 ng/mL). Compound concentrations in plasma at each time point are average values from three tested mice. Error bars represent ±SEM. Experiments involving mice were performed according to the Institutional Animal Care and Use Committee (IACUC)-approved protocol.
Mice Xenograft Studies.
Male immunocompromised NU/J mice (6 weeks old) (The Jackson Laboratory) were engrafted with PC-3 human prostate cancer cells. After tumor volumes reach ~100 mm3, mice were randomized into different treatment arms and administrated with vehicle control or 75 mg/kg compound 20 daily for 22 days via IP injections. The experimenters were not blinded. Tumor volume was calculated as follows: tumor size (mm3) = (longer measurement × shorter measurement2) × 0.5. Tumor sizes were recorded every other day over the course of the studies. All procedures involving mice and experimental protocols (LA13–00024) were approved by the Institutional Animal Care and Use Committee (IACUC) of Icahn School of Medicine at Mount Sinai (ISMMS).
Statistical Analysis.
No statistical methods were used to determine the sample size. Student t-tests (parametric) were used to compare two data sets. GraphPad Prism was used to make these simple predetermined statistical comparisons.
Supplementary Material
ACKNOWLEDGMENTS
J.J. acknowledges the support by an endowed professorship by the Icahn School of Medicine at Mount Sinai. This research was supported by the grant R35CA220491 (to R.P.) from the National Cancer Institute (NCI) at the National Institutes of Health (NIH). R.P. and J.J. were also supported by P30 CA196521 grant from the NCI at the NIH. J.X. is supported by T32 postdoc fellow training grant (T32CA078207) and Leo and Julia Forchheimer Foundation Postdoc Fellowship. This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
The authors declare the following competing financial interest(s): J.J., R.P., J.L. J.X., and X.Y. are inventors of a patent application filed by the Icahn School of Medicine at Mount Sinai. J.J. is a cofounder, scientific advisory board member, and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. R.P. is a shareholder and advisor of Therapten Bioscience, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Inc., Cullgen, Inc., and Cullinan Oncology, Inc.
ABBREVIATIONS USED
- ATP
adenosine triphosphate
- AUC
area under the curve
- DIPEA
diisopropylethylamine
- DMSO
dimethyl sulfoxide
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOAt
1-hydroxy-7-azabenzo-triazole
- IMiDs
immunomodulatory drugs
- IP
intraperitoneal
- ITC
isothermal titration calorimetry
- MOA
mechanism of action
- m-TOR
mammalian target of rapamycin
- NAE
NEDD8-activating enzyme
- NMM
N-methylmorpholine
- NMP
N-methyl-2-pyrrolidone
- P-AKT
phosphorylated AKT
- PEG
polyethylene glycol
- PI3K
phosphatidylinositol 3-kinase
- PK
pharmacokinetic
- POI
protein of interest
- POM
pomalidomide
- PROTACs
proteolysis targeting chimeras
- RTKs
receptor tyrosine kinases
- SAR
structure–activity relationship
- T-AKT
total AKT
- TNBC
triple-negative breast cancer
- UPS
ubiquitin proteasome system
- VHL
von Hippel–Lindau
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c02165
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02165.
Effects of compounds 19–23 on colony formation inhibition in BT474 cells; effects of compounds 35–41 on colony formation inhibition in BT474 cells; binding of benzylated VHL-1 (VHL-1-Bn) to the E3 ligase VHL assessed via ITC; effects of compound 20 on degradation of AKT1, AKT2, and AKT3 isoforms in PC3 cells; and 1H NMR, 13C NMR, and HPLC spectra of compounds 20, 35, 42, and 43 (PDF)
Molecular formula strings for all compounds (CSV)
Contributor Information
Xufen Yu, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jia Xu, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Kaitlyn M. Cahuzac, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States
Kwang-su Park, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Brandon Dale, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;.
Ramon E. Parsons, Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Department of Pharmacological Sciences and Department of Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States;.
REFERENCES
- (1).Scheid MP; Woodgett JR PKB/AKT: functional insights from genetic models. Nat. Rev. Mol. Cell Biol 2001, 2, 760–768. [DOI] [PubMed] [Google Scholar]
- (2).Cheng JQ; Lindsley CW; Cheng GZ; Yang H; Nicosia SV The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [DOI] [PubMed] [Google Scholar]
- (3).Manning BD; Cantley LC AKT/PKB signaling: navigating downstream. Cell 2007, 129, 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Manning BD; Toker A AKT/PKB signaling: navigating the network. Cell 2017, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mayer IA; Arteaga CL The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med 2016, 67, 11–28. [DOI] [PubMed] [Google Scholar]
- (6).Cheung M; Testa R, Diverse J mechanisms of AKT pathway activation in human malignancy. Curr. Cancer Drug Targets 2013, 13, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Berns K; Horlings HM; Hennessy BT; Madiredjo M; Hijmans EM; Beelen K; Linn SC; Gonzalez-Angulo AM; Stemke-Hale K; Hauptmann M; Beijersbergen RL; Mills GB; van de Vijver MJ; Bernards R A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [DOI] [PubMed] [Google Scholar]
- (8).Eichhorn PJA; Gili M; Scaltriti M; Serra V; Guzman M; Nijkamp W; Beijersbergen RL; Valero V; Seoane J; Bernards R; Baselga J Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68, 9221–9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Miller TW; Rexer BN; Garrett JT; Arteaga CL Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wong K-K; Engelman JA; Cantley LC Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev 2010, 20, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mundi PS; Sachdev J; McCourt C; Kalinsky K AKT in cancer: new molecular insights and advances in drug development. Br. J. Clin. Pharmacol 2016, 82, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nitulescu GM; Van De Venter M; Nitulescu G; Ungurianu A; Juzenas P; Peng Q; Olaru OT; Grădinaru D; Tsatsakis A; Tsoukalas D; Spandidos DA; Margina D The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol 2018, 53, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shariati M; Meric-Bernstam F Targeting AKT for cancer therapy. Expert Opin. Invest. Drugs 2019, 28, 977–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Landel I; Quambusch L; Depta L; Rauh D Spotlight on AKT: current therapeutic challenges. ACS Med. Chem. Lett 2020, 11, 225–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Heerding DA; Rhodes N; Leber JD; Clark TJ; Keenan RM; Lafrance LV; Li M; Safonov IG; Takata DT; Venslavsky JW; Yamashita DS; Choudhry AE; Copeland RA; Lai Z; Schaber MD; Tummino PJ; Strum SL; Wood ER; Duckett DR; Eberwein D; Knick VB; Lansing TJ; McConnell RT; Zhang S; Minthorn EA; Concha NO; Warren GL; Kumar R Identification of 4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}−1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a Novel Inhibitor of AKT Kinase. J. Med. Chem 2008, 51, 5663–5679. [DOI] [PubMed] [Google Scholar]
- (16).Blake JF; Xu R; Bencsik JR; Xiao D; Kallan NC; Schlachter S; Mitchell IS; Spencer KL; Banka AL; Wallace EM; Gloor SL; Martinson M; Woessner RD; Vigers GPA; Brandhuber BJ; Liang J; Safina BS; Li J; Zhang B; Chabot C; Do S; Lee L; Oeh J; Sampath D; Lee BB; Lin K; Liederer BM; Skelton NJ Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J. Med. Chem 2012, 55, 8110–8127. [DOI] [PubMed] [Google Scholar]
- (17).Addie M; Ballard P; Buttar D; Crafter C; Currie G; Davies BR; Debreczeni J; Dry H; Dudley P; Greenwood R; Johnson PD; Kettle JG; Lane C; Lamont G; Leach A; Luke RWA; Morris J; Ogilvie D; Page K; Pass M; Pearson S; Ruston L Discovery of 4-Amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. J. Med. Chem 2013, 56, 2059–2073. [DOI] [PubMed] [Google Scholar]
- (18).Hirai H; Sootome H; Nakatsuru Y; Miyama K; Taguchi S; Tsujioka K; Ueno Y; Hatch H; Majumder PK; Pan B-S; Kotani H MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther 2010, 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- (19).Lapierre J-M; Eathiraj S; Vensel D; Liu Y; Bull CO; Cornell-Kennon S; Iimura S; Kelleher EW; Kizer DE; Koerner S; Makhija S; Matsuda A; Moussa M; Namdev N; Savage RE; Szwaya J; Volckova E; Westlund N; Wu H; Schwartz B Discovery of 3-(3-(4-(1-Aminocyclobutyl)phenyl)-5-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amine (ARQ 092): An Orally Bioavailable, Selective, and Potent Allosteric AKT Inhibitor. J. Med. Chem 2016, 59, 6455–6469. [DOI] [PubMed] [Google Scholar]
- (20).Nitulescu GM; Margina D; Juzenas P; Peng Q; Olaru OT; Saloustros E; Fenga C; Spandidos DA; Libra M; Tsatsakis AM Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol 2016, 48, 869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Brown JS; Banerji U Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther 2017, 172, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pal SK; Reckamp K; Yu H; Figlin RA Akt inhibitors in clinical development for the treatment of cancer. Expert Opin. Invest. Drugs 2010, 19, 1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Huck BR; Mochalkin I Recent progress towards clinically relevant ATP-competitive Akt inhibitors. Bioorg. Med. Chem. Lett 2017, 27, 2838–2848. [DOI] [PubMed] [Google Scholar]
- (24).Koseoglu S; Lu Z; Kumar C; Kirschmeier P; Zou J AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol. Ther 2007, 6, 755–762. [DOI] [PubMed] [Google Scholar]
- (25).Roy N; Bordoloi D; Monisha J; Padmavathi G; Kotoky J; Golla R; Kunnumakkara A Specific targeting of Akt kinase isoforms: taking the precise path for prevention and treatment of cancer. Curr. Drug Targets 2017, 18, 421–435. [DOI] [PubMed] [Google Scholar]
- (26).Hinz N; Jücker M Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Commun. Signal 2019, 17, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Skeen JE; Bhaskar PT; Chen C-C; Chen WS; Peng X.-d.; Nogueira V; Hahn-Windgassen A; Kiyokawa H; Hay N Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 2006, 10, 269–280. [DOI] [PubMed] [Google Scholar]
- (28).Sasaki T; Nakashiro K.-i.; Tanaka H; Azuma K; Goda H; Hara S; Onodera J; Fujimoto I; Tanji N; Yokoyama M; Hamakawa H Knockdown of Akt isoforms by RNA silencing suppresses the growth of human prostate cancer cells in vitro and in vivo. Biochem. Biophys. Res. Commun 2010, 399, 79–83. [DOI] [PubMed] [Google Scholar]
- (29).Nogueira V; Patra KC; Hay N Selective eradication of cancer displaying hyperactive Akt by exploiting the metabolic consequences of Akt activation. Elife 2018, 7, No. e32213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mure H; Matsuzaki K; Kitazato KT; Mizobuchi Y; Kuwayama K; Kageji T; Nagahiro S Akt2 and Akt3 play a pivotal role in malignant gliomas. Neuro Oncol. 2010, 12, 221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chin YR; Yoshida T; Marusyk A; Beck AH; Polyak K; Toker A Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014, 74, 964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Deshaies RJ Prime time for PROTACs. Nat. Chem. Biol 2015, 11, 634–635. [DOI] [PubMed] [Google Scholar]
- (33).Toure M; Crews CM Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem., Int. Ed. Engl 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- (34).Burslem GM; Crews CM Small-molecule modulation of protein homeostasis. Chem. Rev 2017, 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- (35).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (37).Chamberlain PP; Hamann LG Development of targeted protein degradation therapeutics. Nat. Chem. Biol 2019, 15, 937–944. [DOI] [PubMed] [Google Scholar]
- (38).Dale B; Cheng M; Park K-S; Kaniskan HÜ; Xiong Y; Jin J Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).You I; Erickson EC; Donovan KA; Eleuteri NA; Fischer ES; Gray NS; Toker A Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem. Biol 2020, 27, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Yu X; Xu J; Xie L; Wang L; Shen Y; Cahuzac KM; Chen X; Liu J; Parsons RE; Jin J Design, synthesis, and evaluation of potent, selective, and bioavailable AKT kinase degraders. J. Med. Chem 2021, 64, 18054–18081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xu J; Yu X; Martin TC; Bansal A; Cheung K; Lubin A; Stratikopoulos E; Cahuzac KM; Wang L; Xie L; Zhou R; Shen Y; Wu X; Yao S; Qiao R; Poulikakos PI; Chen X; Liu J; Jin J; Parsons R AKT Degradation Selectively Inhibits the Growth of PI3K/PTEN Pathway-Mutant Cancers with Wild-Type KRAS and BRAF by Destabilizing Aurora Kinase B. Cancer Discovery 2021, 11, 3064–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Lucas X; Ciulli A Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr. Opin. Struct. Biol 2017, 44, 101–110. [DOI] [PubMed] [Google Scholar]
- (43).Ottis P; Toure M; Cromm PM; Ko E; Gustafson JL; Crews CM Assessing Different E3 ligases for small molecule induced protein ubiquitination and degradation. ACS Chem. Biol 2017, 12, 2570–2578. [DOI] [PubMed] [Google Scholar]
- (44).Frost J; Galdeano C; Soares P; Gadd MS; Grzes KM; Ellis L; Epemolu O; Shimamura S; Bantscheff M; Grandi P; Read KD; Cantrell DA; Rocha S; Ciulli A Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun 2016, 7, 13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Han X; Wang C; Qin C; Xiang W; Fernandez-Salas E; Yang C-Y; Wang M; Zhao L; Xu T; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem 2019, 62, 941–964. [DOI] [PubMed] [Google Scholar]
- (46).Hu J; Hu B; Wang M; Xu F; Miao B; Yang C-Y; Wang M; Liu Z; Hayes DF; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem 2019, 62, 1420–1442. [DOI] [PubMed] [Google Scholar]
- (47).Krönke J; Fink EC; Hollenbach PW; MacBeth KJ; Hurst SN; Udeshi ND; Chamberlain PP; Mani DR; Man HW; Gandhi AK; Svinkina T; Schneider RK; McConkey M; Järås M; Griffiths E; Wetzler M; Bullinger L; Cathers BE; Carr SA; Chopra R; Ebert BL Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE DRUG DEVELOPMENT Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Okuzumi T; Fiedler D; Zhang C; Gray DC; Aizenstein B; Hoffman R; Shokat KM Inhibitor hijacking of Akt activation. Nat. Chem. Biol 2009, 5, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wei J; Hu J; Wang L; Xie L; Jin MS; Chen X; Liu J; Jin J Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J. Med. Chem 2019, 62, 10897–10911. [DOI] [PubMed] [Google Scholar]
- (52).Shen Y; Gao G; Yu X; Kim H; Wang L; Xie L; Schwarz M; Chen X; Guccione E; Liu J; Bedford MT; Jin J Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J. Med. Chem 2020, 63, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Galdeano C; Gadd MS; Soares P; Scaffidi S; Van Molle I; Birced I; Hewitt S; Dias DM; Ciulli A Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem 2014, 57, 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Cheng M; Yu X; Lu K; Xie L; Wang L; Meng F; Han X; Chen X; Liu J; Xiong Y; Jin J Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem 2020, 63, 1216–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jin J; Yang X; Liu J; Xiong Y; Poulikakos P; Karoulia Z; Wu X; Ahmed T Compositions and methods for treating CDK4/6-mediated cancer. International Patent Application WO 2018106870 A1, 2018. [Google Scholar]
- (56).Zhang C; Han X-R; Yang X; Jiang B; Liu J; Xiong Y; Jin J Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem 2018, 151, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.