

Abstract
Over the past decade, tremendous progress has been made in defining autism spectrum disorder (ASD) as a disorder of brain connectivity. Indeed, whole-brain imaging studies revealed altered connectivity in the brains of individuals with ASD, and genetic studies identified rare ASD-associated mutations in genes that regulate synaptic development and function. However, it remains unclear how specific mutations alter the development of neuronal connections in different brain regions and whether altered connections can be restored therapeutically. The main challenge is the lack of preclinical models that recapitulate important aspects of human development for studying connectivity. Through recent technological innovations, it is now possible to generate patient- or mutation-specific human neurons or organoids from induced pluripotent stem cells (iPSCs) and to study altered connectivity in vitro or in vivo upon xenotransplantation into an intact rodent brain. Here, we discuss how deficits in neurodevelopmental processes may lead to abnormal brain connectivity and how iPSC-based models can be used to identify abnormal connections and to gain insights into underlying cellular and molecular mechanisms to develop novel therapeutics.
Introduction
Autism spectrum disorder (ASD) represents a group of neurodevelopmental disorders characterized by impaired social communication [1]. ASD has an estimated prevalence of 1 in 59 children in the USA [2] and affects approximately four times more males than females [3, 4]. Phenotypic heterogeneity significantly complicates the study of ASD, and the neurobiological abnormalities responsible for ASD remain largely unknown [5].
Clinical observations of complex impairments in ASD patients suggested the involvement of multiple brain regions, leading to a prevailing hypothesis that the neurobiological abnormalities in ASD are caused by altered brain connectivity [6–11]. Consistently, whole-brain imaging studies have detected abnormally decreased or increased connectivity in local and distal brain networks in the autistic brain [12]. More imaging studies are underway to identify altered connections [13, 14]. Imaging studies have the strengths of being performed in individuals with ASD and the potential to identify new diagnostic biomarkers, but an important weakness of providing no or very limited insight into the underlying pathology.
Anatomical studies of postmortem brain tissue from individuals with ASD have revealed potential causes of disrupted brain connectivity, including altered cell numbers [15–18]; a thinned corpus callosum [19]; disorganized neurons [20]; deficient axons, dendrites, and/or dendritic spines [21–23]; and molecular changes in specific cortical neuron and glial subtypes [24]. Thus, multiple neurodevelopmental processes, including proliferation (altered cell numbers), migration (focal disorganization), neurite outgrowth (corpus callosum abnormalities), morphogenesis (altered dendrites and dendritic spines), and synaptogenesis and gliogenesis (altered connections), may cause disrupted connectivity in the autistic brain.
ASD mouse models, with modifications in ASD genes, have allowed mechanistic experiments to identify cellular and molecular abnormalities associated with ASD. Interestingly, multiple mouse studies detected connectivity-related synaptic abnormalities [25, 26] and demonstrated that both synaptic and ASD-related behavioral deficits can be compensated by targeted drug therapies [27–31]. These encouraging results led to clinical trials of mGluR5 inhibitor for Fragile X syndrome (FXS) and insulin-like growth factor 1 (IGF1) for Rett syndrome, which, unfortunately, were terminated due to the lack of clinical improvements [32–34]. These failures suggest that effective translation of results obtained in mice to humans requires a thorough understanding of the differences between the human and mouse brains [35, 36].
Patient and genetically engineered induced pluripotent stem cell (iPSC)-derived neurons offer an in vitro human model for studying the cellular and molecular deficits in ASD (reviewed in [5]). The key advantage of patient iPSC-derived neurons is that they carry patient-specific mutations and genetic background, while the main limitation is that phenotypes detected in vitro may deviate substantially from those present in vivo. Thus, more advanced stem cell-based platforms that mimic the diversity and organization of brain cells are also needed.
In this review, we discuss when and how connectivity deficits may arise during human brain development (Fig. 1) and how human iPSC-derived neurons and organoids can be used to investigate the underlying mechanisms (Fig. 2) as well as potentially develop novel gene-, cell-, and network-based therapies for patients.
Figure 1: How ASD-associated connectivity deficits may arise during brain development.
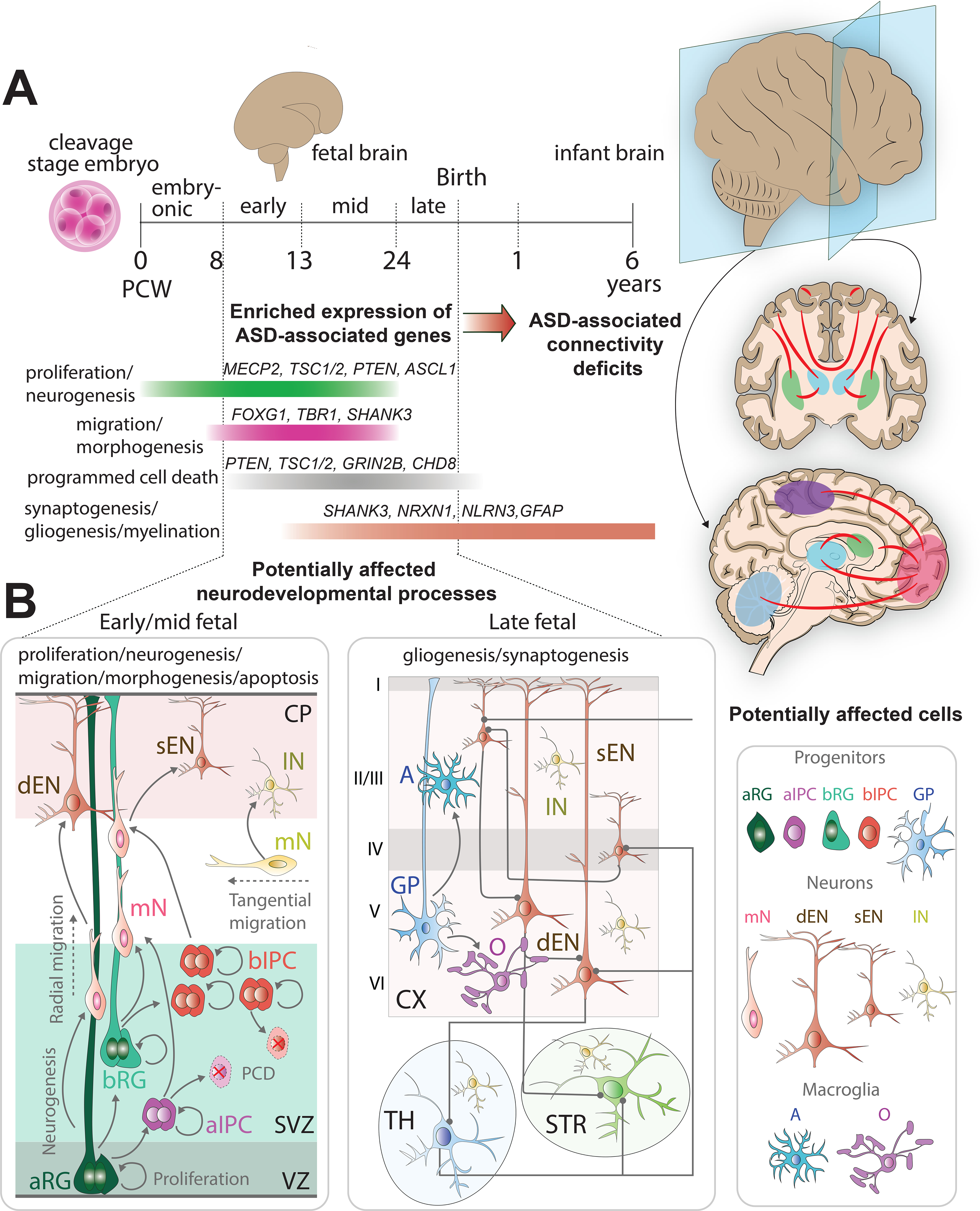
A: Timeline of human brain development. Human cortical development begins early in the embryo, in the first weeks post-conception (PCW 0–8), with proliferation and neurogenesis. The time following the embryonic stage until birth can be divided into three fetal developmental timeframes, early fetal (PCW 8–13), mid-fetal (PCW 13–24), and late fetal (PCW 24-birth) stages marked by specific neurodevelopmental processes. These fetal neurodevelopmental stages show enriched expression of genes implicated in ASD [46, 151]. Neurogenesis and proliferation, during which NPs divide to expand the pool and produce different subtypes of neurons, continue until the late fetal stage. Neuronal migration, the process by which newly born neurons migrate to their destinations in the brain and establish anatomical connections with other brain regions, begins at the end of the embryonic stage and continues through the early and mid-fetal stages. Apoptosis, required for regulation of brain cell type number, distribution, identity, and connectivity, occurs post embryonic stage until birth. Lastly, gliogenesis, the process of glial cell formation, and synaptogenesis, when neurons form synapses and establish interneuronal connectivity, initiate in the mid-fetal stage and continue until after birth.
B: Neurodevelopmental processes and associated cells during early/mid and late stages of fetal development. During the early/mid fetal stages of human cortical development, apical radial glia (aRG) cells differentiate to give rise to apical intermediate progenitors (aIPC), migratory neurons (mN) and basal radial glia (bRG). bRG and aIPC proliferate, differentiate into migratory neurons and undergo apoptosis. mN migrate from the subventricular zone (SVZ) through the intermediate zone to the cortical plate (CP) where they form cortical layers in an inside-out fashion with deep land superficial layer excitatory neurons (dEN and sEN, respectively). Inhibitory neurons (IN) tangentially migrate into the cortex from the ganglionic eminence. During the late fetal stages of human cortical development, RG differentiate in glial progenitors (GPs) that form microglial cells, such as astrocytes (A) and oligodendrocytes (O). Neurons establish intra- and inter-regional synaptic connections with other brain regions such as the striatum (STR) and thalamus (TH). Cellular abnormalities that emerge during fetal neurodevelopment could cause connectivity deficits. The potentially affected cortico-cortical, cortico-thalamic, and cortico-striatal connections are labels in red. These connections can be studied using organoids and transplantation models (Fig. 2).
Figure 2: Application of iPSC-derived neurons and organoids to study connectivity deficits in ASD.
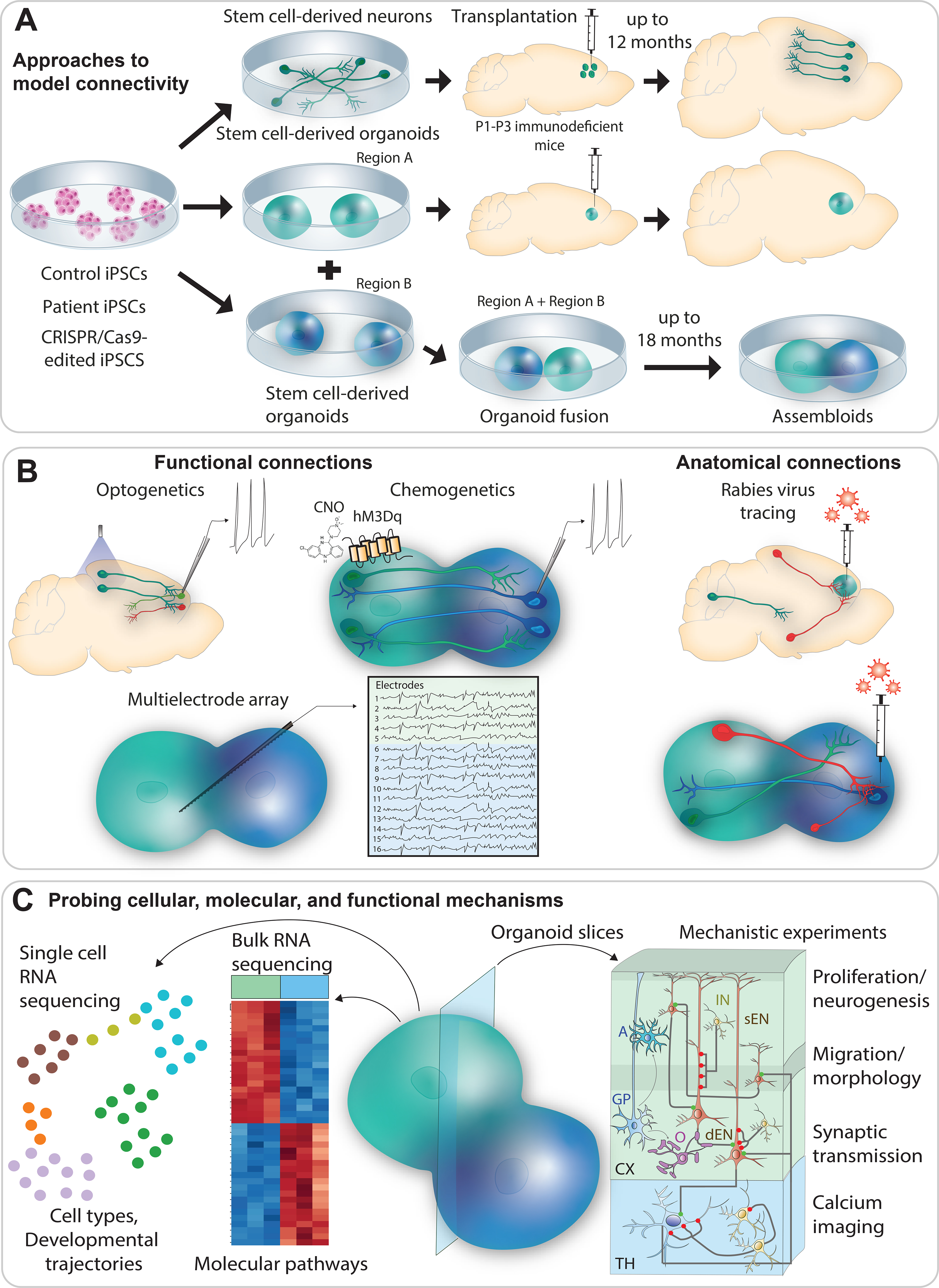
A: Use of iPSC-derived neurons and organoids to model connectivity. iPSC-derived neurons and organoids can be produced from patients with ASD, control individuals, and CRISPR/Cas9-engineered stem cells with ASD-associated mutations. Connectivity can be studied by transplanting iPSC-derived neurons or organoids into immunodeficient mouse brains and tracking their connectivity with the host brain. iPSCs can also be differentiated into brain organoids of selective nervous system regions, such as the striatum, thalamus, spinal cord or cortex. Fusion of these organoids can be applied to form assembloids for studying inter-organoid neural connections.
B: Methods for studying functional and anatomical connections. iPSC-derived neurons and organoids can be engineered to express light- or chemical-activated receptors for studying functional connectivity. Optical or chemical stimulations of iPSC-derived or host neurons can reveal functional connections between human and mouse neurons in different brain regions. Microelectrode arrays can be used to measure neuronal activity. Anatomical connections can be probed using monosynaptic RV tracing. This approach can visualize anatomical connections in both iPSC-derived organoids transplanted in the mouse brain and fusion organoids.
C: Methods for probing cellular, molecular and functional mechanisms underlying connectivity deficits. Single-cell RNA sequencing (scRNAseq) can provide unbiased information about the abundance of different cell types, developmental stage and developmental trajectories in organoids. Bulk RNAseq can identify molecular pathways potentially disrupted in patient iPSC-derived organoids. Lastly, slices of organoids can be used for time-lapse imaging of neuronal migration, electrophysiology measurement of intrinsic and synaptic excitability, and measurement of calcium fluctuations using calcium imaging to gain mechanistic insights into processes affected by ASD mutations.
From disrupted neurodevelopment to altered connectivity in ASD
Human brain development involves several dynamically-regulated stages: proliferation and neurogenesis; migration and morphogenesis; apoptosis; and synaptogenesis, gliogenesis, and myelination (Fig. 1). In the earliest stages, proliferation and neurogenesis, neural stem cells (NSCs), or neural progenitors (NPs), divide symmetrically and asymmetrically to expand the pool of NPs and produce different neuronal subtypes [37]. Multiple high-confidence ASD genes, including MECP2, FMR1, TSC1, TSC2, PTEN, CDH2, DYRK1A, KMT2B, ARID1B, MYT1L, ASCL1, and PAX6, are implicated in cell-cycle and differentiation regulation, suggesting both proliferation and neurogenesis may be altered in ASD. Some genes may influence brain connectivity through region- or progenitor type-specific patterns of expression. For example, PAX6 is predominantly expressed in dorsal telencephalic progenitors [38]; ASCL1 (a.k.a. MASH1) regulates neurogenesis in the ventral telencephalon [39]; and other genes, including the mTOR pathway genes, are enriched in outer radial glia (oRG) [40]. Thus, mutations in these genes may cause imbalanced production of dorsal or ventral telencephalic neurons, disrupting brain connectivity in the cortex or between the cortex and striatum.
Newly-born neurons in the ventricular or subventricular zone migrate to the cortical plate and extend processes, axons and dendrites to establish anatomical connections with neurons in other regions. Many ASD-associated genes, including FOXG1, TBR1, SATB2, FOXP1, DSCAM, ANK2, MAP1A, CACNA1C, CNTNAP2, and SHANK3, play roles in neuronal migration and morphogenesis. Given the cell type-specific expression patterns of some genes, including FOXG1 (telencephalic progenitors and neurons), TBR1 (deep-layer cortical neurons), SATB2 (superficial-layer cortical neurons), and FOXP1 (striatal projection neurons), it is conceivable that disrupted region- or cell type-specific migration and/or morphogenesis could alter brain connectivity. Consistently, Satb2 deletion in mice alters neuronal migration and identity, disrupting cortico-cortical connectivity [41], and disorganized cortical lamination and corpus callosum abnormalities are observed in the postmortem ASD brain [19, 20].
Cell numbers within brain regions are regulated via programmed cell death (PCD) [42], which may be affected in ASD, as many high-confidence ASD genes, including PTEN, TSC1, TSC2, GRIN1, GRIN2A, GRIN2B, and CHD8, regulate this process [43, 44]. However, because most of these genes regulate multiple neurodevelopmental processes, the specific contribution of PCD to ASD and altered brain connectivity is unclear. One possibility is that over- or under-production of specific cell types could influence brain wiring, an idea supported by the observation of neuron overproduction in the prefrontal cortex of the postmortem ASD brain [45].
During synaptogenesis, gliogenesis, and myelination stage, differentiated neurons establish functional synaptic connections and glial cells provide support synapse development and fast signal propagation. Disturbances in these later neurodevelopmental processes are likely in ASD, as mutations in synaptic and glial genes, including SHANK3, SYNGAP1, GRIN2B, and GFAP, are observed in individuals with ASD [46]. Additionally, neuroimaging studies have identified aberrant development of white matter tracks, particularly the corpus callosum, in ASD [47, 48]. Finally, ASD-related behavioral deficits in mice can be compensated by small molecules or trophic factors that increase synaptic transmission [25, 26]. Synaptic genes including SHANK3 and GRIN2B exhibit developmental stage- and cell type-specific expression patterns [49, 50]; therefore, their disruption may diminish specific functional connections in the developing brain. Indeed, mouse and primate models of SHANK3 deficiency exhibit functional connectivity deficits [51–55].
In summary, connectivity deficits in ASD may arise from impairments in multiple neurodevelopmental processes (Fig. 1), and further research is needed to characterize the relationships between specific impairments and connectivity deficits.
Connectivity-related deficits in human iPSC-derived neurons
Human iPSC-derived neurons produced from patients, control individuals, and engineered stem cell lines offer several advantages for studying ASD-associated neurodevelopmental deficits, including: 1) patient-specific mutations and genetic background, 2) human-specific cells, and 3) opportunities for mechanistic experiments and drug discovery. Here, we summarize connectivity-related neurodevelopmental deficits identified using iPSC-derived neurons from patients with rare genetic abnormalities.
MECP2 (Rett syndrome):
The transcription regulator MECP2 (methyl CpG-binding protein 2) controls the expression of multiple genes important for normal brain development [56]. MECP2 mutation is detected in >95% of Rett syndrome patients [56]. Several studies have employed telencephalic iPSC-derived neurons from patients and/or engineered lines [57–62]. Consistent with disrupted connectivity, MECP2-deficient neurons show abnormalities in excitatory synaptic transmission, somatic and dendritic morphologies, dendritic spines, and excitability. Additionally, deficits in MECP2-deficient neurons can be reversed by MECP2 overexpression or treatment with IGF1 [62], choline [57], and chemicals that modulate cholinergic responses [61]. Further research is needed to understand the effects of the identified neurodevelopmental abnormalities and pharmacological treatments on brain connectivity.
SHANK3 (Phelan-McDermid syndrome [PMDS]):
PMDS is a genetic neurodevelopmental disorder associated with severe intellectual disability and ASD [63]. Hemizygous SHANK3 deletions and mutations are considered primarily responsible for these deficits [64], but loss of other genes in the 22q13 region may also contribute [65]. SHANK3 is a postsynaptic protein that regulates important signaling cascades at excitatory synapses [66]. Telencephalic iPSC-derived excitatory neurons produced from PMDS patients show elevated intrinsic excitability and excitatory synaptic deficits [67]. Additionally, SHANK3-deficient neurons with cortical-like identities have fewer dendrites and dendritic spines [68–70]. Interestingly, the excitatory synaptic deficits in SHANK3-deficient human neurons are at least partially attributable to elevated intrinsic excitability caused by reduced expression of hyperpolarization-activated Ih channels [70]. IGF1 treatment compensates excitatory synaptic transmission deficits in patient neurons [67], but the underlying mechanisms and side effects remain to be determined [71].
CACNA1C (Timothy syndrome [TS]):
TS is a congenital disorder involving cardiac arrhythmia and ASD. It is caused by de novo missense mutations in CACNA1C [72]. Telencephalic iPSC-derived neurons from TS patients exhibit elevated depolarization-induced calcium influxes, altered neuronal identities, and defective dendritic arbors [73, 74]. This example illustrates that disruption of a single gene can cause multiple neurodevelopmental abnormalities in ASD.
FMR1 (Fragile X syndrome [FXS]):
FXS is a genetic neurodevelopmental disorder involving severe intellectual disability and ASD [75]. It is caused by CGG-repeat expansions in FMR1 that completely silence or significantly reduce expression of fragile X mental retardation protein, an important regulator of protein translation [76]. FMR1-deficient human stem cell-derived excitatory neurons with cortical-like identities have deficits in differentiation, neurite outgrowth, intrinsic excitability, and homeostatic synaptic plasticity [77–79]. Importantly, removal of the pathogenic CGG repeats rescues the homeostatic synaptic deficits in FMR1-deficient patient neurons. Homeostatic plasticity is considered important for the establishment of stable functional neuronal connections during brain development [80].
In summary, studies of patient and engineered iPSC-derived neurons have identified several cellular deficits that may lead to altered brain connectivity in ASD.
Xenotransplantation and organoid models for studying connectivity
To overcome the limitations of iPSC-derived neurons, xenotransplantation and organoid models have been developed [81–84], with the key advantages of modeling long-term development under more physiological conditions and possessing relevant cells and neural networks.
Xenotransplantation:
Transplantation of human stem cell-derived neurons into the mouse brain allows the study of anatomical and functional connections between human and mouse neurons in different brain regions (Fig. 2A–B). Thus, human iPSC-derived cortical excitatory neurons transplanted into the mouse frontal cortex acquire proper identities, establish expected efferent connections with mouse neurons in different brain regions, and receive synaptic inputs [81]. Human iPSC-derived neurons with visual-like cortical identities efficiently integrate into the lesioned mouse visual cortex, but not the motor cortex, suggesting that regional identity match is important for successful integration of transplanted human neurons [85]. An optimization of cell dissociation and delivery into the lateral ventricle of neonatal mice further improves human neuron integration in the mouse cortex [86]; such that the xenotransplanted neurons develop elaborated dendrites, spines, and synapses capable of undergoing plasticity. Remarkably, the xenotransplanted neurons also integrate into physiological circuits in the mouse visual cortex and respond to visual stimuli similarly to host mouse neurons. This research provided a blueprint for using xenotransplanted human neurons to study connectivity.
Xenotransplantation of human stem cell-derived neurons has been used most often to study neurodegenerative disorders and develop cell-based therapies for Parkinson’s disease [87], Alzheimer’s disease [88], stroke [89], epilepsy [90], and spinal cord injury [91]. Fewer studies have used xenotransplantation to investigate connectivity in neurodevelopmental disorders [92, 93]. Upon transplantation of iPSC-derived cortical excitatory neurons from Down syndrome (DS) patients into the somatosensory cortex of adult mice, longitudinal in vivo GCaMP6 imaging showed that DS neurons develop more stable dendritic spines and significantly increased spine density than isogenic control neurons [92]. Although excitatory synaptic transmission remains unchanged, DS neurons exhibit increased somatic calcium oscillations. Overall, this research identified cellular deficits that could lead to altered brain connectivity in DS [94].
Transplantation of SHANK3-deficient human cortical neurons into the mouse prefrontal cortex revealed that SHANK3 deficiency causes different synaptic deficits at different developmental stages [93]. Initially, the strength of excitatory AMPA receptor-mediated synaptic currents is reduced. Later (13–18 months post-transplantation), reduced numbers of excitatory synapses and spines and underdeveloped dendritic arbors are observed. Further research is needed to determine how SHANK3 deficiency affects different types of neuronal connections in the brain.
Organoids:
Human stem cell-derived brain organoids are self-organized, three-dimensional tissues produced from PSCs or multipotent NSCs. Cortical organoids recapitulate early stages of human cortical development, including oRG production in the subventricular zone, sequential generation of cortical neurons, synaptogenesis, and gliogenesis [95–98]. Importantly, organoids can be cultured for extended periods (up to 2 years) [99–101], develop functional neuronal connections [102, 103], and exhibit spontaneously oscillating neural networks responsive to external stimuli [104].
To study interregional neurodevelopmental processes, such as migration and connectivity, brain organoids with different regional identities can be fused for co-development (assembloids) (Fig. 2). For example, cortical inhibitory interneuron migration was studied in pallial-subpallial assembloids [105–107], and cortico-thalamic connections in cortico-thalamic assembloids [108]. More recently, unilateral cortico-striatal connectivity was recapitulated using cortico-striatal assembloids [109].
Furthermore, studies of iPSC-derived cortical organoids co-cultured with mouse spinal cord explants containing intact peripheral nerves and muscles [102] or with human iPSC-derived spinal cord organoids and skeletal muscle cells [110] provided proof-of-concept that fusion of central nervous system components enables the formation of neural circuits that can be manipulated to understand their development under normal and pathological conditions.
Neural oscillations have also been measured in organoids [103, 104]. Cerebral organoids consisting of cells with diverse regional identities, including forebrain, midbrain, hindbrain, and retina, exhibited organized patterns of spontaneous electrical activity [104]. Remarkably, some neurons showed attenuated firing rates upon light exposure, suggesting the formation of functional networks responsive to physiological sensory stimuli. In another study, bursts of synchronous activity appeared in organoids already after 2 months post-plating on two-dimensional multi-electrode arrays [103]. This activity became increasingly regular and transitioned into robust oscillations by 4–6 months. Notably, 6-month-old organoids displayed coupling between oscillations at different frequency bands (cross-frequency coupling), possibly representing functional neural network communication. Although plated cerebral organoids lack other brain regions important for oscillatory rhythm generation and the unique organization present in three-dimensional tissue, this proof-of-principle study showed that stem cell-derived human neural tissue can generate a wide range of electrical activities.
More recently, cortico-subcortical assembloids were shown to develop functionally active neural networks with robust interneuron-dependent oscillatory rhythms of varying frequency [111]. Using this system, hypersynchronous and hyperexcitable network activity was detected in organoids from Rett syndrome patients, and Pifithrin-α, a putative inhibitor of TP53, reduced spike frequency and suppressed high-frequency oscillations in these patient organoids. These results demonstrate the feasibility of using human organoids to study oscillatory rhythms and disease pathophysiology.
Several studies have used brain organoids to study cellular and molecular deficits in human neurodevelopmental disorders associated with ASD [84, 107, 109, 112–114]. Telencephalic organoids produced from individuals with idiopathic ASD showed increased production of GABAergic inhibitory NPs and neurons compared with organoids produced from unaffected parents [112]. Although no ASD-related genetic abnormalities were identified in patient cells, inhibitory deficits were attributed to elevated FOXG1 expression. In pallial-subpallial assembloids produced from TS patients, impaired migration of MGE-derived inhibitory interneurons was detected and rescued by an L-type calcium channel inhibitor, nimodipine [107]. Together, these studies suggest that abnormal development of inhibitory neurons could be responsible for some ASD-related deficits.
Connectivity deficits in PMDS were investigated using iPSC-derived cortico-striatal fusion organoids produced from patients and unrelated controls [109]. The patient-derived organoids exhibited increased frequency of spontaneous calcium spikes in the striatal region but dramatically reduced network synchronization, a phenotype consistent with the elevated intrinsic excitability and reduced excitatory synaptic transmission detected in cultured PMDS neurons [67].
The molecular mechanisms disrupted by PMDS-related hemizygous SHANK3 deletion were investigated in cortico-striatal organoids generated from stem cell-derived single neural rosettes (SNRs) [113]. This research confirmed the presence of synaptic and intrinsic excitability deficits in SHANK3-deficient organoids and identified impaired expression of several clustered protocadherins as potentially responsible. Further research is needed to understand how SHANK3 regulates these genes and how their altered expression contributes to the observed deficits.
The cellular and molecular deficits caused by CDKL5 deficiency were investigated using iPSC-derived neurons and organoids from patients and engineered lines [115]. CDKL5-deficient neurons exhibit increased dendrites, intrinsic excitability, and excitatory synaptic transmission. Consistent with these phenotypes, CDKL5-deficient organoids show increased electrical activity and overly synchronized networks at early developmental stages. A high-throughput drug-screening assay targeting excitability deficits identified the inhibitors of Ih channels (Ivabradine), muscarinic receptors (Solifenacin) and GSK3 (AZD1080) as promising therapeutic compounds. The same group found that treatments with chemicals that modulate cholinergic signaling, Nefiracetam and PHA543613, rescued morphological and synaptic deficits in iPSC-derived neurons with MECP2 mutation and improved functional activity in mutant organoids [61]. Collectively, these results suggest that cortical organoids can be used as a drug screening platform.
To investigate organoid development under more physiological conditions, iPSC-derived cortical organoids were transplanted into the cortex of immunodeficient mice [116]. Remarkably, these organoids became functionally and anatomically integrated into the mouse brain and developed a functional vasculature system. Moreover, neuronal activity in the transplanted organoids was influenced by environmental stimuli.
In summary, xenotransplanted iPSC-derived neurons and organoids can integrate into the mouse brain and form proper connections with mouse neurons in different brain regions. Fused organoids can recapitulate inter-regional migration and connectivity. Both systems can be used to study disrupted connections in ASD and the underlying neurodevelopmental mechanisms (Fig. 2).
Quality control considerations
Sample size and isogenic lines:
Most ASD patients carry genetic variants of low penetrance with unclear etiopathogenesis [26]. To account for heterogeneity in ASD, iPSCs and iPSC-derived neurons should be generated from male and female patients as well as unaffected siblings or age- and sex-matched control individuals. The sample size required for studying idiopathic ASD using iPSC-derived neurons or organoids is prohibitively large, reaching hundreds of lines from unrelated individuals [117]; therefore, most studies have focused on syndromic ASDs or genetic abnormalities in “high-confidence” ASD genes [46]. The state-of-the-art approach for modeling syndromic ASD uses 2–4 pairs of isogenic lines with mutations introduced on different genetic backgrounds of both sexes with differentiation and transplantation repeated 3–5 times. New collaborative consortiums that can handle large sample collations and big data acquisition and analysis are needed identify the cellular and network mechanisms disrupted in idiopathic ASD using iPSC-based models.
Neuronal maturation and identity:
Although multiple differentiation protocols have been developed for producing human neurons with cortical identities from PSCs, none generates a pure neuron population with a well-defined and physiologically-relevant identity. Cultures of human iPSC-derived neurons typically contain multiple cell types, including NPs, neurons, and glial cells, at different developmental stages. Despite the importance of knowing the developmental stage and identity of differentiated cells, few studies have assessed these properties [118].
For neuron enrichment, cytosine arabinoside, which inhibits DNA synthesis, can be used to eliminate proliferative cells [119]. Additionally, neurons can be labeled using lenti- or AAV-based reporters carrying a fluorescent protein under control of a neuronal promoter [62, 67]. Also, a combination of small-molecule inhibitors can be applied during neural induction to accelerate the production of functionally mature cortical neurons [120]. Alternatively, directed reprogramming of stem cells into neurons using neurogenic transcription factors can produce a relatively uniform population of functionally mature neurons [121, 122]. However, several caveats of directed reprogramming include the unclear identities of generated neurons that co-express a mix of neuronal and stem cell-related genes [121, 123], low-to-no expression of NMRA receptor subunits [123], and omission of early developmental stages potentially critical for ASD-associated deficits [124]. A common limitation of differentiation and reprogramming approaches is that they do not allow efficient production of later-specified neurons, such as superficial-layer cortical excitatory neurons and fast-spiking parvalbumin-expressing inhibitory interneurons, which could be disrupted in ASD [24, 46, 112].
Dual-color genotype labeling:
Transplantation of human cells into a specific brain region of neonatal mice is technically challenging. Injection precision can be improved by using a stereotactic apparatus and well-defined coordinates, but is still limited by experimenter skill and inter-animal brain size variation. To overcome these challenges, a dual-color fluorescent labeling scheme can be used [67, 125], in which control and patient iPSC-derived neurons labeled with different fluorescent proteins are co-injected [93]. This approach is only suitable for studying cell-autonomous deficits; the study of non-cell autonomous deficits requires separate transplantations or organoids.
SNR-derived organoids:
One limitation of organoids for connectivity studies is the unpredictable organization of cells and germinal zones, which may create conflicting gradients of secreted morphogens, affecting neuronal migration and maturation [126]. To overcome this limitation, we developed a method for generating telencephalic organoids from SNRs [113]. Each SNR is a neural tube-like structure containing neuroepithelial cells radially organized around the lumen [127]. To a certain extent, this mimics neural tissue development from a singular neural tube. Indeed, NPs and neurons in SNR-derived organoids are predictably organized around the lumen [113]. However, the potential advantages for studying connectivity remain unknown.
Other considerations:
Control experiments using fetal human brain tissue are needed to validate results obtained using organoids. Most studies have used bulk and/or single-cell RNA-sequencing for validation [107, 128, 129], but this approach is limited by transcript degradation and loss of cell in dissociation. In addition, similarities in cellular compositions and/or gene expression profiles do not guarantee similarities in functional network activity. Network activity in organoids can be evaluated using extracellular field potential recording and compared to electroencephalographic recording in the human brain [103].
Finally, questions remain regarding the ethics of research with patient iPSC-derived neurons and organoids, including the measure of consciousness in organoids, the use of human-animal chimeras, and informed consent and ownership [130–133].
Future applications
Xenografted human iPSC-derived neurons or organoids in vivo and brain region-specific organoids assembled in vitro can be used to study the disrupted connections and underlying mechanisms (Fig. 2).
In xenotransplantation models, iPSC-derived neurons or organoids can be delivered to a specific location in the mouse brain for integration into developing brain networks, and anatomical and functional inputs/outputs on/from transplanted human cells can be evaluated using optogenetics, chemogenetics, implanted multielectrode arrays, and rabies virus-based tracing approaches. The main advantage of this platform is the ability to assess multiple connections under physiological conditions. A disadvantage is that interactions between human neurons and mouse cells in the immunodeficient mouse brain could differ substantially from those in the human brain. In addition, the absence of human brain-specific connections in the mouse brain could further complicate the interpretation of obtained results.
Fused organoids or assembloids consisting of two or more brain region-specific organoids are another innovative platform for studying connectivity deficits in ASD. Assembloids can be maintained in culture for long periods to allow establishment of enter- and inter-regional functional neural networks and investigation of specific connections using anatomical and functional assays (Fig. 2B). The main advantage of this approach is its modularity. Assembloids can be generated using different brain region-specific organoids with and without ASD mutations. Such experiments should facilitate discovery of the origins of disrupted connections and the underlying cellular and molecular deficits. The main disadvantages of this system are the absences of other non-ectodermal cell types, brain-like organization, and inputs from sensory organs known to regulate proper anatomical and functional connections. Thus, both xenotransplantation and assembloid platforms are likely needed to identify connections disrupted in ASD.
Understanding connectivity deficits in ASD may support novel therapeutics. For example, activity in specific brain circuits can be modulated using electrical, magnetic, or ultrasound stimuli [134], as tested for treatment of Parkinson’s disease, major depression, certain migraine types, and obsessive-compulsive disorder [135, 136]. Interestingly, high-frequency stimulation of the subthalamic nucleus alleviates excessive grooming deficits in Mecp2- and Shank3-deficient mice [137], and optogenetic stimulation of dopaminergic neurons in the ventral tegmental area enhances social preference in Shank3-deficient mice [138].
Cell- or gene-based therapies represent additional avenues for correcting disrupted connectivity. For example, transplantation of inhibitory neurons into the cortex alleviated behavioral deficits in animal ASD models [139–141]. Furthermore, gene-based therapies, including AAV-based gene replacement [142], gene activation or inhibition using the dead Cas9 system (CRISPRa/i) [143], and synthetic antisense oligonucleotide use to control gene expression [144], have shown encouraging results in mouse models of Rett, Fragile X, Angelman and Dravet syndromes [145–150]. Organoid and xenotransplantation models may facilitate the optimization of gene-based therapies for use in humans.
Overall, there is hope that delivery of proper network stimuli, cells, or genes into specific brain regions at defined developmental stages may correct connectivity abnormalities and alleviate certain ASD-associated behavioral deficits.
Footnotes
The authors declare no competing interests
References:
- 1.APA. Diagnostic and statistical manual of mental disorders (5th ed.). 2013. [Google Scholar]
- 2.Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, et al. Prevalence of autism spectrum disorder among children aged 8 Years - Autism and developmental disabilities monitoring network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018. 2018. 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werling DM, Parikshak NN, Geschwind DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hull L, Mandy W, Petrides KV. Behavioural and cognitive sex/gender differences in autism spectrum condition and typically developing males and females. Autism. 2017;21:706–727. [DOI] [PubMed] [Google Scholar]
- 5.Yang G, Shcheglovitov A. Probing disrupted neurodevelopment in autism using human stem cell‐derived neurons and organoids: An outlook into future diagnostics and drug development. Dev Dyn. 2019:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belmonte MK, Allen G, Beckel-Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J. Neurosci, vol. 24, 2004. p. 9228–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courchesne E, Pierce K. Why the frontal cortex in autism might be talking only to itself: Local over-connectivity but long-distance disconnection. Curr Opin Neurobiol. 2005;15:225–230. [DOI] [PubMed] [Google Scholar]
- 9.Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. [DOI] [PubMed] [Google Scholar]
- 10.Just MA, Cherkassky VL, Keller TA, Kana RK, Minshew NJ. Functional and anatomical cortical underconnectivity in autism: Evidence from an fmri study of an executive function task and corpus callosum morphometry. Cereb Cortex. 2007;17:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Just MA, Keller TA, Malave VL, Kana RK, Varma S. Autism as a neural systems disorder: A theory of frontal-posterior underconnectivity. Neurosci Biobehav Rev. 2012;36:1292–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uddin LQ, Supekar K, Menon V. Reconceptualizing functional brain connectivity in autism from a developmental perspective. Front Hum Neurosci. 2013;7:458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Martino A, O’Connor D, Chen B, Alaerts K, Anderson JS, Assaf M, et al. Enhancing studies of the connectome in autism using the autism brain imaging data exchange II. Sci Data. 2017;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holiga Š, Hipp JF, Chatham CH, Garces P, Spooren W, D’Ardhuy XL, et al. Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci Transl Med. 2019;11. [DOI] [PubMed] [Google Scholar]
- 15.Hanson KL, Lew CH, Hrvoj-Mihic B, Cuevas D, Greiner DMZ, Groeniger KM, et al. Decreased density of cholinergic interneurons in striatal territories in Williams syndrome. Brain Struct Funct. 2020;225:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanson KL, Cuevas DL, Groeniger KM, Lew CH, Hrvoj-Mihic BL, Raghanti MA, et al. Decreased Density of Cholinergic Interneurons in the Medial Caudate Nucleus in Humans with Williams Syndrome. FASEB J. 2018;225:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashemi E, Ariza J, Rogers H, Noctor SC, Martínez-Cerdeño V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb Cortex. 2017;27:1931–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lew CH, Brown C, Bellugi U, Semendeferi K. Neuron density is decreased in the prefrontal cortex in Williams syndrome. Autism Res. 2017;10:99–112. [DOI] [PubMed] [Google Scholar]
- 19.Wegiel J, Flory M, Kaczmarski W, Brown WT, Chadman K, Wisniewski T, et al. Partial agenesis and hypoplasia of the corpus callosum in idiopathic autism. J Neuropathol Exp Neurol. 2017;76:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weir RK, Bauman MD, Jacobs B, Schumann CM. Protracted dendritic growth in the typically developing human amygdala and increased spine density in young ASD brains. J Comp Neurol. 2018;526:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. 2018;19:215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Cerdeño V. Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol. 2017;77:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. Single-cell genomics identifies cell type–specific molecular changes in autism. Science. 2019;364:685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4:a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–563. [DOI] [PubMed] [Google Scholar]
- 27.Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30:5303–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicidomini C, Ponzoni L, Lim D, Schmeisser M, Reim D, Morello N, et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol Psychiatry. 2017;22:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan ZF, Mao SS, Shen J, Jiang LH, Xu L, Xu JL, et al. Insulin-Like Growth Factor-1 Down-Regulates the Phosphorylation of FXYD1 and Rescues Behavioral Deficits in a Mouse Model of Rett Syndrome. Front Neurosci. 2020;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dölen G, Osterweil E, Rao BSS, Smith GB, Auerbach BD, Chattarji S, et al. Correction of Fragile X Syndrome in Mice. Neuron. 2007;56:955–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erickson CA, Davenport MH, Schaefer TL, Wink LK, Pedapati E V, Sweeney JA, et al. Fragile X targeted pharmacotherapy: Lessons learned and future directions. J Neurodev Disord. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Leary HM, Kaufmann WE, Barnes KV., Rakesh K, Kapur K, Tarquinio DC, et al. Placebo-controlled crossover assessment of mecasermin for the treatment of Rett syndrome. Ann Clin Transl Neurol. 2018;5:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeste SS, Geschwind DH. Clinical trials for neurodevelopmental disorders: At a therapeutic frontier. Sci Transl Med. 2016;8:1–4. [DOI] [PubMed] [Google Scholar]
- 35.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krienen F, Goldman M, Zhang Q, del Rosario R, Florio M, Machold R, et al. Innovations in Primate Interneuron Repertoire. Nature. 2019;586:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. [DOI] [PubMed] [Google Scholar]
- 38.Shen T, Ji F, Yuan Z, Jiao J. CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells. 2015;33:1794–1806. [DOI] [PubMed] [Google Scholar]
- 39.Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. [DOI] [PubMed] [Google Scholar]
- 40.Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, et al. Satb2 Regulates Callosal Projection Neuron Identity in the Developing Cerebral Cortex. Neuron. 2008;57:364–377. [DOI] [PubMed] [Google Scholar]
- 42.Yamaguchi Y, Miura M. Programmed cell death in neurodevelopment. Dev Cell. 2015;32:478–490. [DOI] [PubMed] [Google Scholar]
- 43.Nishiyama M, Oshikawa K, Tsukada YI, Nakagawa T, Iemura SI, Natsume T, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009;11:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4:e560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ, et al. Neuron number and size in prefrontal cortex of children with autism. JAMA - J Am Med Assoc. 2011;306:2001–2010. [DOI] [PubMed] [Google Scholar]
- 46.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180:568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolff JJ, Gu H, Gerig G, Elison JT, Styner M, Gouttard S, et al. Differences in white matter fiber tract development present from 6 to 24 months in infants with autism. Am J Psychiatry. 2012;169:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pagnozzi AM, Conti E, Calderoni S, Fripp J, Rose SE. A systematic review of structural MRI biomarkers in autism spectrum disorder: A machine learning perspective. Int J Dev Neurosci. 2018;71:68–82. [DOI] [PubMed] [Google Scholar]
- 49.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:1988–1991. [DOI] [PubMed] [Google Scholar]
- 50.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, Sabatini BL, Peixoto R, Wang W, Croney D, Kozorovitskiy Y, Sabatini B. Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B−/− mice. Nat Neurosci. 2016;19:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peixoto RT, Chantranupong L, Hakim R, Levasseur J, Wang W, Merchant T, et al. Abnormal Striatal Development Underlies the Early Onset of Behavioral Deficits in Shank3B−/− Mice. Cell Rep. 2019;29:2016–2027.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pagani M, Bertero A, Liska A, Galbusera A, Sabbioni M, Barsotti N, et al. Deletion of autism risk gene shank3 disrupts prefrontal connectivity. J Neurosci. 2019;39:5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Y, Sharma J, Ke Q, Landman R, Yuan J, Chen H, et al. Atypical behaviour and connectivity in SHANK3-mutant macaques. Nature. 2019;570:326–331. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, et al. Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun. 2016;7:11459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ip JPK, Mellios N, Sur M. Rett syndrome: Insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018;19:368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chin EWM, Marcy G, Yoon SI, Ma D, Rosales FJ, Augustine GJ, et al. Choline Ameliorates Disease Phenotypes in Human iPSC Models of Rett Syndrome. NeuroMolecular Med. 2016;18:364–377. [DOI] [PubMed] [Google Scholar]
- 58.Kim KY, Hysolli E, Park IH. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011;108:14169–14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, et al. Global transcriptional and translational repression in human-embryonic- stem-cell-derived rett syndrome neurons. Cell Stem Cell. 2013;13:446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Djuric U, Cheung AYL, Zhang W, Mok RS, Lai W, Piekna A, et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol Dis. 2015;76:37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trujillo CA, Adams JW, Negraes PD, Carromeu C, Tejwani L, Acab A, et al. Pharmacological reversal of synaptic and network pathology in human MECP2 ‐KO neurons and cortical organoids. EMBO Mol Med. 2021;13:e12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol. 2012;2:186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harony-Nicolas H, De Rubeis S, Kolevzon A, Buxbaum JD. Phelan McDermid Syndrome: From Genetic Discoveries to Animal Models and Treatment. J Child Neurol. 2015;30:1861–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mitz AR, Philyaw TJ, Boccuto L, Shcheglovitov A, Sarasua SM, Kaufmann WE, et al. Identification of 22q13 genes most likely to contribute to Phelan McDermid syndrome. Eur J Hum Genet. 2018;26:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol Psychiatry. 2018;23:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gouder L, Vitrac A, Goubran-Botros H, Danckaert A, Tinevez JY, André-Leroux G, et al. Altered spinogenesis in iPSC-derived cortical neurons from patients with autism carrying de novo SHANK3 mutations. Sci Rep. 2019;9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;2669:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ranke MB, Wölfle J, Schnabel D, Bettendorf M. Treatment of Dwarfism With Recombinant Human Insulin-Like Growth Factor-1. Dtsch Arztebl Int. 2009;106:703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. [DOI] [PubMed] [Google Scholar]
- 73.Krey JF, Paşca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122:4314–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Verkerk AJMH, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DPA, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med. 2018;10:eaar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, et al. IPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23:1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Telias M, Kuznitsov-Yanovsky L, Sega M, Ben-Yosef D. Functional deficiencies in fragile X neurons derived from human embryonic stem cells. J Neurosci. 2015;35:15295–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tien NW, Kerschensteiner D. Homeostatic plasticity in neural development. Neural Dev. 2018;13:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J, et al. Pyramidal Neurons Derived from Human Pluripotent Stem Cells Integrate Efficiently into Mouse Brain Circuits In Vivo. Neuron. 2013;77:440–456. [DOI] [PubMed] [Google Scholar]
- 82.Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, et al. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell. 2008;3:519–532. [DOI] [PubMed] [Google Scholar]
- 83.Mariani J, Simonini MV, Palejev D, Tomasini L, Coppola G, Szekely AM, et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2012;109:12770–12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Espuny-Camacho I, Michelsen KA, Linaro D, Bilheu A, Acosta-Verdugo S, Herpoel A, et al. Human Pluripotent Stem-Cell-Derived Cortical Neurons Integrate Functionally into the Lesioned Adult Murine Visual Cortex in an Area-Specific Way. Cell Rep. 2018;23:2732–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Linaro D, Vermaercke B, Iwata R, Ramaswamy A, Libé-Philippot B, Boubakar L, et al. Xenotransplanted Human Cortical Neurons Reveal Species-Specific Development and Functional Integration into Mouse Visual Circuits. Neuron. 2019;104:972–986.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Barker RA, Parmar M, Studer L, Takahashi J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell. 2017;21:569–573. [DOI] [PubMed] [Google Scholar]
- 88.Kwak KA, Lee SP, Yang JY, Park YS. Current perspectives regarding stem cell-based therapy for Alzheimer’s disease. Stem Cells Int. 2018;6392986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tornero D, Wattananit S, Madsen MG, Koch P, Wood J, Tatarishvili J, et al. Human induced pluripotent stem cell-derived cortical neurons integrate in stroke-injured cortex and improve functional recovery. Brain. 2013;136:3561–3577. [DOI] [PubMed] [Google Scholar]
- 90.Cunningham M, Cho JH, Leung A, Savvidis G, Ahn S, Moon M, et al. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. 2014;15:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antonic A, Sena ES, Lees JS, Wills TE, Skeers P, Batchelor PE, et al. Stem Cell Transplantation in Traumatic Spinal Cord Injury: A Systematic Review and Meta-Analysis of Animal Studies. PLoS Biol. 2013;11:e1001738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Real R, Peter M, Trabalza A, Khan S, Smith MA, Dopp J, et al. In vivo modeling of human neuron dynamics and down syndrome. Science. 2018;362:eaau1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chiola S, Napan K, Wang Y, Lazarenko R, Armstrong CJ, Cui J, et al. Defective AMPA-mediated synaptic transmission and morphology in human neurons with hemizygous SHANK3 deletion engrafted in mouse prefrontal cortex. Mol Psychiatry. 2021;Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu SY, Lu FM, Wang MY, Hu ZS, Zhang J, Chen ZY, et al. Altered Functional Connectivity in the Motor and Prefrontal Cortex for Children With Down’s Syndrome: An fNIRS Study. Front Hum Neurosci. 2020;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell–derived neocortex. Proc Natl Acad Sci. 2013;110:20284–20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watanabe M, Buth JE, Vishlaghi N, de la Torre-Ubieta L, Taxidis J, Khakh BS, et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep. 2017;21:517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 2016;165:1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon SJ, Huber N, Pritchard JK, et al. Chromatin accessibility dynamics in a model of human forebrain development. Science. 2020;367:eaay1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sloan SA, Darmanis S, Huber N, Khan TA, Birey F, Caneda C, et al. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron. 2017;95:779–790.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gordon A, Yoon SJ, Tran SS, Makinson CD, Park JY, Andersen J, et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat Neurosci. 2021;24:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat Neurosci. 2019;22:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell. 2019;25:558–569.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Yang SM, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell. 2017;21:383–398.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bagley JA, Reumann D, Bian S, Lévi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14:743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiang Y, Tanaka Y, Cakir B, Patterson B, Kim KY, Sun P, et al. hESC-Derived Thalamic Organoids Form Reciprocal Projections When Fused with Cortical Organoids. Cell Stem Cell. 2019;24:487–497.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miura Y, Li M, Birey F, Ikeda K, Revah O, Thete MV, et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat Biotechnol. 2020;38:1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Andersen J, Revah O, Miura Y, Thom N, Amin ND, Kelley KW, et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell. 2020;183:1913–1929.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Samarasinghe RA, Miranda OA, Mitchell S, Ferando I, Watanabe M, Buth JE, et al. Identification of neural oscillations and epileptiform changes in human brain organoids. BioRxiv. 2021: 10.1101/820183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang Y, Chiola S, Yang G, Russell C, Armstrong CJ, Wu Y, et al. Modeling autism-associated SHANK3 deficiency using human cortico-striatal organoids generated from single neural rosettes. Cold Spring Harbor Laboratory; 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun AX, Yuan Q, Fukuda M, Yu W, Yan H, Lim GGY, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019;366:1486–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Negraes PD, Trujillo CA, Yu N-K, Wu W, Yao H, Liang N, et al. Altered network and rescue of human neurons derived from individuals with early-onset genetic epilepsy. Mol Psychiatry. 2021;Online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mansour AA, Gonçalves JT, Bloyd CW, Li H, Fernandes S, Quang D, et al. An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol. 2018;36:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nehme R, Barrett LE. Using human pluripotent stem cell models to study autism in the era of big data. Mol Autism. 2020;11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stein JL, de la Torre-Ubieta L, Tian Y, Parikshak NN, Hernández IA, Marchetto MC, et al. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron. 2014;83:69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Frega M, van Gestel SHC, Linda K, van der Raadt J, Keller J, Van Rhijn J-R, et al. Rapid Neuronal Differentiation of Induced Pluripotent Stem Cells for Measuring Network Activity on Micro-electrode Arrays. J Vis Exp. 2017:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qi Y, Zhang X-J, Renier N, Wu Z, Atkin T, Sun Z, et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat Biotechnol. 2017;35:154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Y, Pak CH, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jo J, Xiao Y, Sun AX, Cukuroglu E, Tran HD, Göke J, et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell. 2016;19:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 2018;23:2509–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci. 2019;22:243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zaslavsky K, Zhang WB, McCready FP, Rodrigues DC, Deneault E, Loo C, et al. SHANK2 mutations associated with autism spectrum disorder cause hyperconnectivity of human neurons. Nat Neurosci. 2019;22:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ladewig J, Koch P, Brüstle O. Auto-attraction of neural precursors and their neuronal progeny impairs neuronal migration. Nat Neurosci. 2014;17:24–26. [DOI] [PubMed] [Google Scholar]
- 127.Elkabetz Y, Studer L. Human ESC-derived neural rosettes and neural stem cell progression. Cold Spring Harb Symp Quant Biol. 2008;73:377–387. [DOI] [PubMed] [Google Scholar]
- 128.Yoon SJ, Elahi LS, Pașca AM, Marton RM, Gordon A, Revah O, et al. Reliability of human cortical organoid generation. Nat Methods. 2019;16:75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 2020;578:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Farahany NA, Greely HT, Hyman S, Koch C, Grady C, Pasca SP, et al. The ethics of experimenting with human brain tissue comment. Nature. 2018;556:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sawai T, Sakaguchi H, Thomas E, Takahashi J, Fujita M. The Ethics of Cerebral Organoid Research: Being Conscious of Consciousness. Stem Cell Reports. 2019;13:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lovell-Badge R Stem-cell guidelines: why it was time for an update. Nature. 2021;593:479–479. [DOI] [PubMed] [Google Scholar]
- 133.Lavazza A, Massimini M. Cerebral organoids: Ethical issues and consciousness assessment. J Med Ethics. 2018;44:606–610. [DOI] [PubMed] [Google Scholar]
- 134.Johnson MD, Lim HH, Netoff TI, Connolly AT, Johnson N, Roy A, et al. Neuromodulation for brain disorders: Challenges and opportunities. IEEE Trans Biomed Eng. 2013;60:610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bourdillon P, Hermann B, Sitt JD, Naccache L. Electromagnetic Brain Stimulation in Patients With Disorders of Consciousness. Front Neurosci. 2019;13:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fang JY, Tolleson C. The role of deep brain stimulation in parkinson’s disease: An overview and update on new developments. Neuropsychiatr Dis Treat. 2017;13:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chang AD, Berges VA, Chung SJ, Fridman GY, Baraban JM, Reti IM. High-Frequency Stimulation at the Subthalamic Nucleus Suppresses Excessive Self-Grooming in Autism-Like Mouse Models. Neuropsychopharmacology. 2016;41:1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bariselli S, Tzanoulinou S, Glangetas C, Prévost-Solié C, Pucci L, Viguié J, et al. SHANK3 controls maturation of social reward circuits in the VTA. Nat Neurosci. 2016;19:926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Donegan JJ, Boley AM, Lodge DJ. Embryonic stem cell transplants as a therapeutic strategy in a rodent model of autism. Neuropsychopharmacology. 2018;43:1789–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tyson JA, Anderson SA. GABAergic interneuron transplants to study development and treat disease. Trends Neurosci. 2014;37:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Southwell DG, Seifikar H, Malik R, Lavi K, Vogt D, Rubenstein JL, et al. Interneuron transplantation rescues social behavior deficits without restoring wild-type physiology in a mouse model of autism with excessive synaptic inhibition. J Neurosci. 2020;40:2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu X, Qi LS. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J Mol Biol. 2018;431:34–47. [DOI] [PubMed] [Google Scholar]
- 144.Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Garg SK, Lioy DT, Cheval H, McGann JC, Bissonnette JM, Murtha MJ, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33:13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tillotson R, Selfridge J, Koerner MV., Gadalla KKE, Guy J, De Sousa D, et al. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature. 2017;550:398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hampson DR, Hooper AWM, Niibori Y. The application of adeno-associated viral vector gene therapy to the treatment of fragile X syndrome. Brain Sci. 2019;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Colasante G, Lignani G, Brusco S, Di Berardino C, Carpenter J, Giannelli S, et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol Ther. 2020;28:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bailus BJ, Pyles B, Mcalister MM, O’Geen H, Lockwood SH, Adams AN, et al. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an angelman syndrome mouse brain. Mol Ther. 2016;24:548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014. 2014. 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]