

Abstract
Primary myelofibrosis (PMF) is a clonal stem cell disorder characterized by myeloid dominant hematopoiesis and dysregulated proliferation of fibroblasts in the bone marrow. It has yet to be clarified by which mechanism aberrant myeloid cells and fibroblasts are produced. Here we examined in vivo engraftment kinetics of PMF patient-derived CD34+ cells in immune-compromised NOD/SCID/IL2rgKO (NSG) mice. Engrafted human cells were analyzed with flow cytometry, and proliferation of fibroblastic cells and bone marrow fibrosis were assessed with histo-pathological examination. Transplantation of PMF patient-derived circulating CD34+ fractions into NSG newborns recapitulates clinical features of human PMF. Engraftment of human CD45+ leukocytes resulted in anemia and myeloid hyperplasia accompanied by bone marrow fibrosis by six months post-transplantation. Fibrotic bone marrow contained CD45-vimentin+ cells of both human and mouse origin, suggesting that circulating malignant CD34+ subsets contribute to myelofibrotic changes in PMF through direct and indirect mechanisms. Patient-derived xenotransplantation (PDX) model of PMF allows in vivo examination of disease initiation and propagation originating from immature CD34+ cells and will support investigation of pathogenesis as well as therapeutic modalities of the disorder.
Introduction
Primary myelofibrosis (PMF) is a myeloproliferative neoplasm (MPN) which has been believed to originate from hematopoietic stem cells (HSCs). PMF is characterized by de novo bone marrow (BM) fibrosis with deposition of reticulin and collagen accompanied by myeloid metaplasia associated with progressive anemia and predominance of granulocytic and megakaryocytic elements in the BM [1]. Other typical features include circulating immature hematopoietic cells and extramedullary hematopoiesis [1].
The clonal basis of PMF pathogenesis has been intensively investigated. Based on G6-PD isozyme analysis, Fialkow and colleagues showed that erythroid cells, megakaryocytes, granulocytes and monocytes are clonal [2, 3]. B and T lymphocytes were also shown to be clonal by karyotype, FISH and RAS mutational analyses [4, 5], strongly suggesting that this disease originates from a single malignant HSC clone [6]. In a fraction of PMF patients, the presence of JAK2V617F, a somatic mutation of the JAK2 gene [7–10] also suggests that PMF is a clonal disorder, analogous to other Philadelphia chromosome-negative MPNs including polycythemia vera (PV) and essential thrombocytosis (ET). To date, mutations involving JAK2, MPL and CALR have been described as driver mutations responsible for the PMF phenotype[11]. The JAK2V617F mutation results in constitutively active tyrosine phosphorylation, and mice harboring a JAK2V617F transgene developed myeloproliferative conditions mimicking PV and ET [12]. In about 10% of PMF patients without JAK2 mutation, a somatic mutation of the MPL gene was identified [13, 14]. This gain-of-function mutation results in activation of downstream JAK-STAT signaling leading to cytokine-independent cell growth. In 20% to 25% of patients with ET and PMF, somatic mutations of CALR activate JAK-STAT signaling and lead to excessive myeloproliferation [15, 16]. While these mutations may be the basis for myeloproliferative phenotype common to the MPNs, the pathogenic mechanisms leading to distinct clinical features that characterize PV, ET and PMF are not defined.
The origin and mechanism of fibrotic changes in PMF, including cellular components of fibrotic lesions have been investigated. In one report, clonal analysis of ex vivo cultured PMF patient BM showed that hematopoietic cells were clonal while fibroblasts were polyclonal [2]. It has also been proposed that megakaryocyte proliferation plays a key role in development of BM fibrosis [17]. This is supported by the stimulation of myelofibroblast growth by locally-released cytokines such as TGF-β1 from abnormal megakaryocytes [18] and the development of BM fibrosis accompanied by elevation of serum TGF-β1 levels in TPO transgenic mice [19] as well as increased serum TGF-β1 levels in PMF patients [20]. These findings suggest that myelofibroblast proliferation might be a secondary event in PMF pathogenesis. On the other hand, single-cell transplantation of green fluorescence protein (GFP)-labeled murine HSCs resulted in the presence of GFP+ fibroblasts in multiple organs including the BM, suggesting that HSCs can differentiate and generate fibroblasts directly [21, 22]. CD34−CD14+ monocytes appear to be a key hematopoietic cellular component for initiation of PMF [23–25].
In this report, we created a patient-derived xenograft (PDX) model for human PMF using the newborn NOD/SCID/IL2rgKO (NSG) xenogeneic transplantation model [26], which supports high levels of normal and malignant human hematopoietic engraftment [27, 28]. In the xenograft, we found that a circulating CD34+ populations in PMF patients is enriched for in vivo PMF-initiating cells with long-term reconstitution potential and that PMF-engrafted NSG mice developed BM fibrosis with scattered megakaryocytes. Furthermore, we found co-existence of human chromosome+ fibroblast-like cells and mouse chromosome+ fibroblast-like cells in the BM of the recipients. These findings show that circulating CD34+ cell populations recapitulate in vivo key features of PMF, contributing to myeloid-dominant malignant hematopoiesis and to BM fibrosis through both direct and indirect mechanisms.
Materials and Methods
Mice
NOD.Cg-PrkdcscidIL2rgtmlWjl/Sz (NSG) mice were developed at the Jackson Laboratory [26]. Mice were bred and maintained with irradiated food and acidified water at the animal facility in Kyushu University. All experiments were performed according to the guidelines established by the Institutional Animal Committee and approved by the Institutional Review Board of Kyushu University.
Patient samples
Peripheral blood (PB) samples were obtained from PMF patients with written informed consent in accordance with the requirements of the Institutional Review Board of Kyushu University Hospital. Each patient met WHO classification criteria for PMF [29] and myelofibrosis grading was based on 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia (supplemental Table 1) [30]. Healthy donor cord blood cells were provided by Japan Red Cross Kyushu Block Blood Center.
Purification of PMF-initiating cells and xenotransplantation
Mononuclear cells (MNCs) were separated by gradient centrifugation using Lymphocyte Separation Medium (MP Biomedicals LLC). MNCs were depleted of human T cells using mouse anti-human CD3, CD4, and CD8 monoclonal antibodies (BD, San Jose, CA). Samples were enriched for CD34+ cells by using anti-CD34 microbeads (Miltenyi Biotec, Auburn, CA), labeled with anti-CD34 and CD38 antibodies (BD). CD3-CD4-CD8-CD34+CD38− and CD3-CD4-CD8-CD34+CD38+ cells were isolated by cell sorting using FACSAria (Becton Dickinson, San Jose, CA). Magnetic bead-isolated CD34+ cells (2–8×105 cells/mouse) or FACS-sorted CD34+CD38− cells (3.3×104 – 3.2×105 cells/mouse) or CD34+CD38+ cells (2.7×104 – 2.8×105 cells/mouse) were injected into sublethally-irradiated (100 cGy) NSG newborn mice within 48 hours of birth via the facial vein [27].
Flow cytometric analysis
Recipient mice were phlebotomized via retroorbital plexus every four weeks following transplantation, and BM cells were harvested from the recipients at 4–17 months post-transplantation. PB hemoglobin concentrations and leukocyte counts were evaluated by an automated cell analyzer (NIHON KOHDEN). Hematopoietic cells were analyzed using mouse anti-human CD3, CD11b, CD14, CD15, CD19, CD33, CD45 (eBioscience) by FACSCalibur or FACSAria (BD Biosciences).
Histological examination of NSG recipient bone sections
Paraformaldehyde-fixed, decalcified, paraffin-embedded sections were prepared from recipient femurs. Hematoxylin-eosin (H&E) staining and silver staining were performed according to standard procedures. BM fibrosis was graded based on the proposed WHO criteria. FISH analyses were performed using a FITC-conjugated mouse pan-centromeric probe (Cambio) and a Spectrum Orange-conjugated human X-chromosomal probe (Vysis) to distinguish between mouse cells and human cells. Immunofluorescence labeling of BM sections were performed using mouse anti-CD45 monoclonal antibody (cross-reacts with both human and mouse CD45; Dako), mouse anti-human CD45 monoclonal antibody (Dako), goat anti-vimentin polyclonal antibody (SIGMA), and rabbit anti-vimentin monoclonal antibody (Abcam) were used as primary antibodies (cross-reacts with both human and mouse vimentin), and visualized with Cy3–conjugated donkey anti-mouse IgG, AlexaFluor 549-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.), AlexaFluor 488-conjugated donkey anti-mouse IgG and AlexaFluor 647–conjugated donkey anti-goat IgG antibodies (Invitrogen). Nuclei were stained with 4’, 6-diamidino-2-phenylinodole (DAPI) (Vector Laboratories). Stained specimens were observed by light microscopy (Olympus and Zeiss). Specimens were photographed with a Binary Planner 4490 (Jenoptik) or Axiovert 200 (Zeiss).
Detection of JAK2V617F and CALR mutations
JAK2 gene, CALR gene, and MPL gene mutations were detected using nested polymerase chain reaction (PCR) and direct sequencing. Sequences of the oligonucleotide primers used are provided in supplemental Table 2. Sequencing products were analyzed using an ABI3130XL DNA sequencer (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Two-tailed paired and unpaired t-tests were performed using GraphPad Prism (GraphPad).
Results
PMF patient PB-derived SCID-repopulating cells preferentially differentiate into myeloid cells in vivo
Patient characteristics are summarized in Supplemental Table 1. All PMF patients studied met WHO criteria with biopsy-documented BM fibrosis. Of the four patients, Patient 2 had a type I CALR mutation and Patient 4 had the JAK2V617F mutation. Patient 3 had no mutations in JAK2, CALR or MPL. In the patients, CD34+ cells accounted for 1–4.9% of total nucleated blood cells (Figure 1A and supplemental Table 1). This CD34+ cell population contained a small fraction of CD38− cells, whose phenotype is similar to normal HSCs [31] with SCID mouse reconstitution potential [27] (Figure 1A).
Figure 1. Recipients of human primary PMF CD34+ and CD34+CD38− cells exhibit peripheral blood abnormalities similar to the PMF donor.
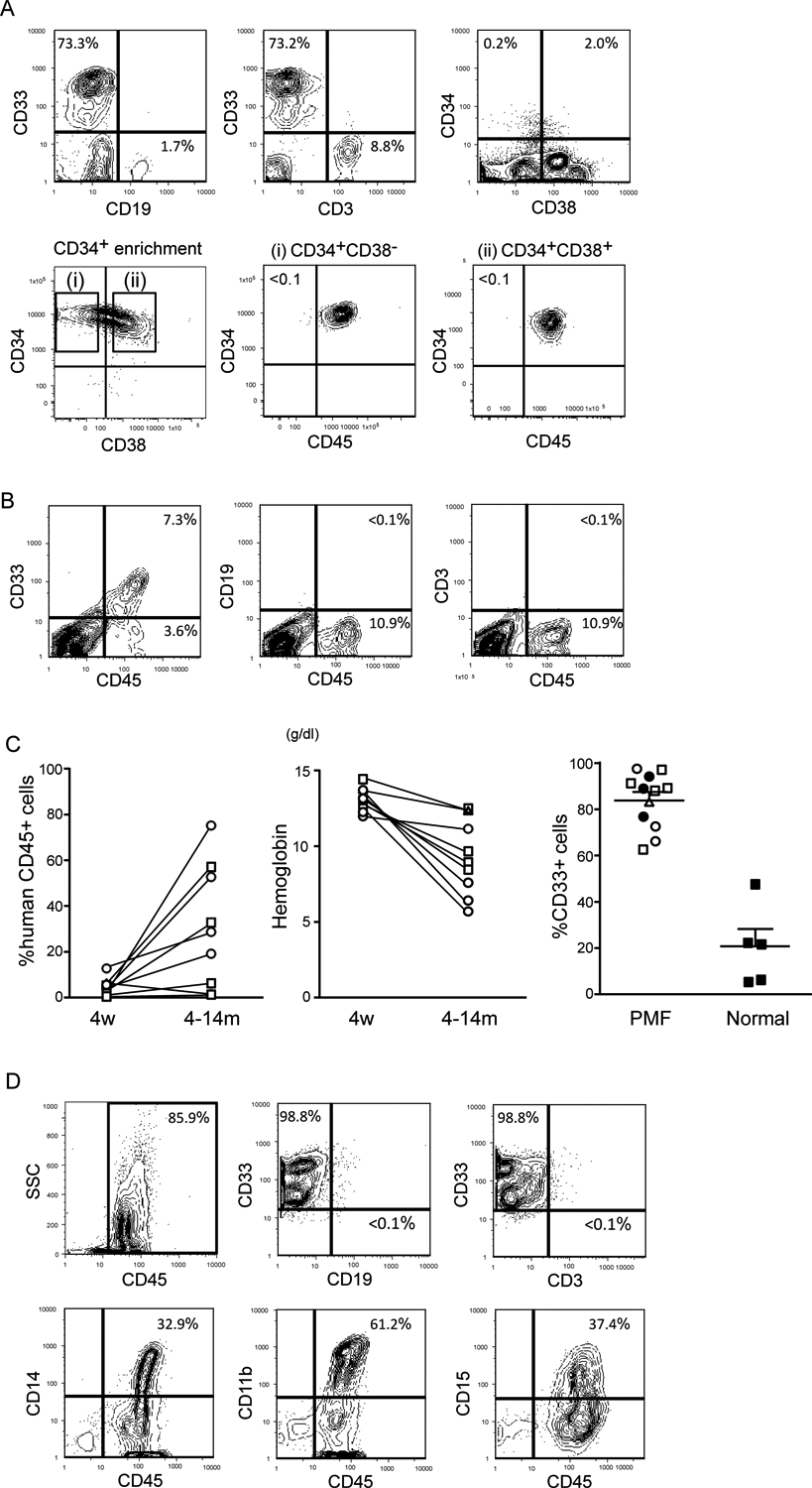
(A) Representative flow cytometric analysis of the blood in Patient 3. The majority of nucleated blood cells are CD33+ myeloid cells. The blood contained CD34+ cells with or without CD38 expression (Upper). CD45, CD34 and CD38 expression in blood cells after anti-CD34 microbeads enrichment is shown in the bottom panels. Purified CD34+ CD38− and CD34+CD38+ cells all expressed CD45. (B) Representative flow cytometric analyses of blood cells (recipient 3–3). (C) Engraftment levels of human hematopoietic cells in recipients’ peripheral blood at 4 weeks and 4–14 months post-transplantation (Left). Blood hemoglobin concentration of engrafted recipients at 4 weeks and 4–14 months post-transplantation. Open squares, open circles, and a triangle represent the recipients transplanted with circulating CD34+CD38− cells in Patients 2, 3 and 4, respectively (Middle). The percentages of CD33+ myeloid cells in the BM of recipients transplanted with circulating PMF CD34+CD38− cells or normal cord blood CD34+CD38− cells at 4–14 months post-transplantation (Right). Bars indicate mean + s.e.m. Filled circles, open squares, open circles, and a triangle represent the recipients transplanted with circulating CD34+ or CD34+CD38− cells of Patients 1, 2, 3 and 4, respectively. Filled squares represent recipients transplanted with normal cord blood CD34+CD38− cells. (D) Representative flow cytometric analysis of BM (recipient 3–3). CD33+ myeloid engraftment was predominant, and these cells co-expressed mature myeloid antigens including CD11b, CD14 and/or CD15.
We examined in vivo repopulation capacity of PB CD34+, CD34+CD38− and CD34+CD38+ cells obtained from these 4 patients by xenotransplantation into NSG newborns (representative flow cytometric analysis and cell sorting gates are shown for Patient 3 in Figure 1A). Long-term engraftment was observed in all mice transplanted with patient-derived CD34+ cells with 16–64% human hematopoietic chimerism (Table 1). CD38 expression correlated variably with long-term repopulation capacity: In Patients 2 and 4, in vivo repopulation capacity resided in CD34+CD38− but not CD34+CD38+ cells, while in Patient 3, both CD34+CD38− and CD34+CD38+ cells were capable of long-term hematopoietic reconstitution (Table 1).
Table 1.
Results of xenotransplantation expreiments
| UPN | Mouse | Transplanted cells | graft cells(×104) | Engraftment | Hb (g/dL) | % human CD45+ cells | % within human CD45+ cells | Survival (months) | Myelofibrosis grading | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Myeloid | T | B | |||||||||
| 1 | 1–1 | CD34+ | 20.0 | + | NE | 64.9 | 96.7 | 0.7 | 1.1 | 6 | MF-3 |
| 1–2 | CD34+ | 20.0 | + | NE | 16.8 | 88.7 | 5.3 | 5.7 | 6 | MF-3 | |
| 1–3 | CD34+ | 20.0 | + | NE | 16.2 | NE | NE | NE | 6 | MF-3 | |
| 2 | 2–1 | CD34+ | 80.0 | + | 12.4 | 41.1 | 66.1 | 0.2 | 31.4 | 5 | MF-2 |
| 2–2 | CD34+ | 80.0 | + | 9.6 | 56.0 | 82.0 | 3.2 | 9.9 | 6 | MF-2 | |
| 2–3 | CD34+CD38− | 17.4 | + | 8.5 | 15.8 | 62.8 | 27.1 | 10.1 | 13 | MF-2 | |
| 2–4 | CD34+CD38− | 8.7 | + | 8.9 | 79.5 | 97.0 | 0.0 | 2.7 | 14 | MF-2 | |
| 2–5 | CD34+CD38+ | 22.8 | − | 16.2 | <1.0 | NE | NE | NE | 5 | MF-0 | |
| 2–6 | CD34+CD38+ | 5.3 | − | 12.7 | <1.0 | NE | NE | NE | 13 | MF-0 | |
| 3 | 3–1 | CD34+CD38− | 4.7 | + | 11.1 | 82.0 | 72.4 | 0.1 | 19.9 | 4 | MF-2 |
| 3–2 | CD34+CD38− | 7.2 | + | 7.5 | 89.6 | 98.8 | <1.0 | <1.0 | 6 | MF-3 | |
| 3–3 | CD34+CD38− | 10.9 | + | 5.7 | 78.2 | 94.0 | 0.4 | 1.9 | 10 | MF-3 | |
| 3–4 | CD34+CD38− | 3.3 | + | 6.4 | 94.8 | 88.6 | 0.3 | 7.7 | 11 | MF-3 | |
| 3–5 | CD34+CD38+ | 10.0 | + | 8.6 | 80.6 | 94.6 | 0.0 | 0.3 | 16 | MF-3 | |
| 3–6 | CD34+CD38+ | 2.7 | + | 3.8 | 87.5 | 91.1 | 0.0 | 6.6 | 17 | MF-3 | |
| 3–7 | CD34+CD38+ | 4.4 | − | 11.8 | <1.0 | NE | NE | NE | 13 | MF-0 | |
| 4 | 4–1 | CD34+CD38− | 32.0 | + | 12.4 | 30.7 | 87.6 | 0.0 | 9.3 | 14 | MF-1 |
| 4–2 | CD34+CD38− | 10.0 | Transient | 12.8 | <1.0 | NE | NE | NE | 7 | MF-0 | |
| 4–3 | CD34+CD38− | 30.0 | Transient | 13.7 | <1.0 | NE | NE | NE | 8 | MF-0 | |
| 4–4 | CD34+CD38− | 30.0 | Transient | 14.0 | <1.0 | NE | NE | NE | 9 | MF-0 | |
| 4–5 | CD34+CD38− | 18.0 | Transient | 14.0 | <1.0 | NE | NE | NE | 14 | MF-0 | |
| 4–6 | CD34+CD38− | 12.0 | − | 14.0 | <1.0 | NE | NE | NE | 4 | MF-0 | |
| 4–7 | CD34+CD38− | 12.0 | − | 14.0 | <1.0 | NE | NE | NE | 9 | MF-0 | |
| 4–8 | CD34+CD38− | 9.6 | − | 13.0 | <1.0 | NE | NE | NE | 17 | MF-0 | |
| 4–9 | CD34+CD38+ | 15.5 | − | 13.7 | <1.0 | NE | NE | NE | 6 | MF-0 | |
| 4–10 | CD34+CD38+ | 15.5 | − | 14.0 | <1.0 | NE | NE | NE | 9 | MF-0 | |
| 4–11 | CD34+CD38+ | 28.0 | − | 12.4 | <1.0 | NE | NE | NE | 17 | MF-0 | |
NE, not evaluated
At four weeks post-transplantation, human CD45+ chimerism in the PB was less than 20%, and consisted mostly of CD33+ myeloid cells (Representative flow cytometry plots from recipient 3–3 shown in Figure 1B; Summary in Figure 1C). Percentages of PB human CD45+ cells increased over time (Figure 1C, left panel; p=0.0098 by paired two-tailed t test), accompanied by decrease in PB hemoglobin concentration (Figure 1C, middle panel; p=0.0003 by paired two-tailed t test). At 4 to 14 months post-transplantation, engrafted recipients of PMF patient-derived cells showed predominant CD33+ myeloid reconstitution, at a significantly higher frequency compared with mice transplanted with normal human HSCs (Figure 1C, right panel; p<0.0001 by two-tailed t test). In the recipient BM, human CD45+ cells co-expressed a variety of myeloid-associated antigens including CD11b, CD14 and CD15 (representative flow cytometry plots from recipient 3–2 shown in Figure 1D). These findings indicate that PMF patient PB contains long-term multilineage repopulating cells that favor myeloid lineage differentiation in vivo.
PMF patient-derived circulating CD34+ cells initiate BM fibrosis both directly and indirectly
Next, we examined roles of circulating CD34+ and CD34+CD38− long-term multilineage repopulating cells in BM fibrosis. We first assessed BM of mice transplanted with PMF patient-derived CD34+ and CD34+CD38− cells for fibrosis (Figure 2). In recipients transplanted with circulating CD34+ PMF cells from Patient 1 (1–1, 1–2 and 1–3), the BM was occupied by fibroblastic cells and silver staining-positive reticulin fibers, with scarcity of blood cells. In a recipient of Patient 3-derived circulating CD34+CD38− cells (3–3), fibrotic changes in the BM were less severe and the mouse BM contained sparse hematopoietic cells with increased number of abnormal megakaryocytes, as compared to control mice transplanted with normal human cord blood-derived HSCs. Patient 3-derived circulating CD34+CD38+ PMF cells also caused BM fibrosis in a recipient (3–6). These findings suggest that CD34+ cells are responsible for initiation and progression of BM fibrosis regardless of gene mutation types.
Figure 2. Development of BM fibrosis in mice engrafted with circulating PMF patient CD34+ cells.
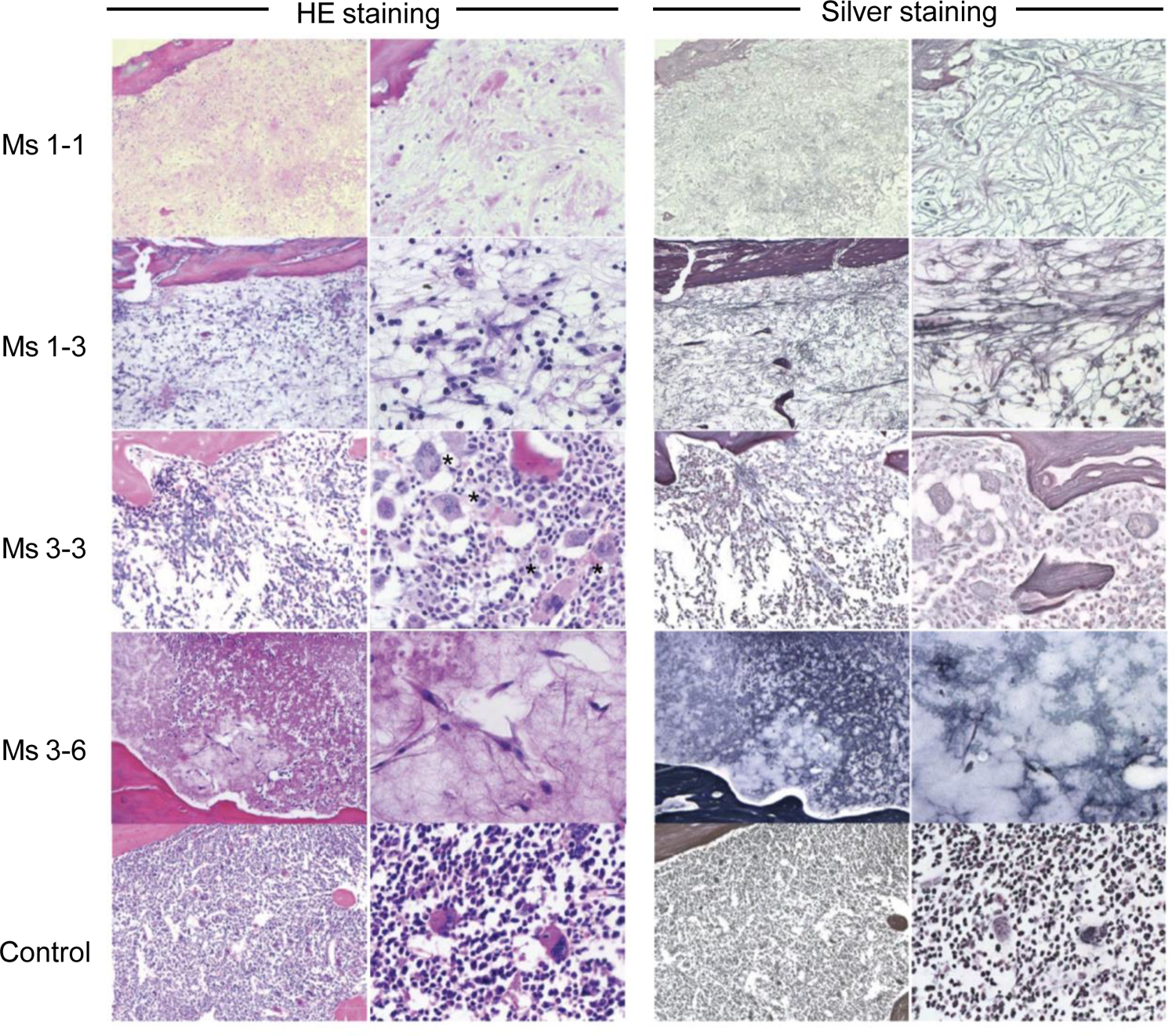
H&E and silver staining of femoral sections of two recipients transplanted with blood CD34+ cells (1–1 and 1–3), a recipient transplanted with blood CD34+CD38− cells (3–3), a recipient transplanted with blood CD34+CD38+ cells (3–6), and a recipient engrafted with normal cord blood HSCs (control) are shown. Multi-lineage hematopoiesis found in HSC recipient BM is replaced by fibroblasts, reticulin fibers, and abnormal megakaryocytes in the BM of PMF-initiating cell recipients. Asterisks indicate megakaryocytes.
We further examined mechanism for development of BM fibrosis following transplantation of PMF CD34+ cells by determining the origin of cells within fibrotic lesions through FISH analysis (representative images are shown in Figure 3 [recipient 1–1] and supplemental Figure 1 [recipient 3–3 and 3–6]). Anti-human X chromosome (red) and anti-mouse centromeric (green) probes were used to determine origin of the cells. Fibroblast-like cells were identified by spindle-shaped morphology and the presence of cytoplasmic vimentin, a marker for mesenchymal intermediate filaments [22, 32]. In recipients 1–1, 3–3 and 3–6 with BM fibrosis, 11 out of 43 vimentin+CD45− cells were positive for the anti-human X-chromosome probe, while the remaining 32 cells were positive for the anti-mouse centromeric probe. These findings indicate that circulating patient-derived CD34+ subpopulations contribute to fibrotic changes in the BM through two distinct mechanisms: Directly through differentiation into human fibroblast-like cells and indirectly through induction of proliferation of mouse fibroblast-like cells (Figure 4). Furthermore, we detected the small number of potential fibrocytes which were characterized by a spindle shape, anti-human X chromosome probe positive, vimentin+ and CD45+ (Supplemental Figure 2).
Figure 3. Co-existence of human fibroblast-like cells and mouse fibroblast-like cells in mice engrafted with circulating PMF patient CD34+ cells.
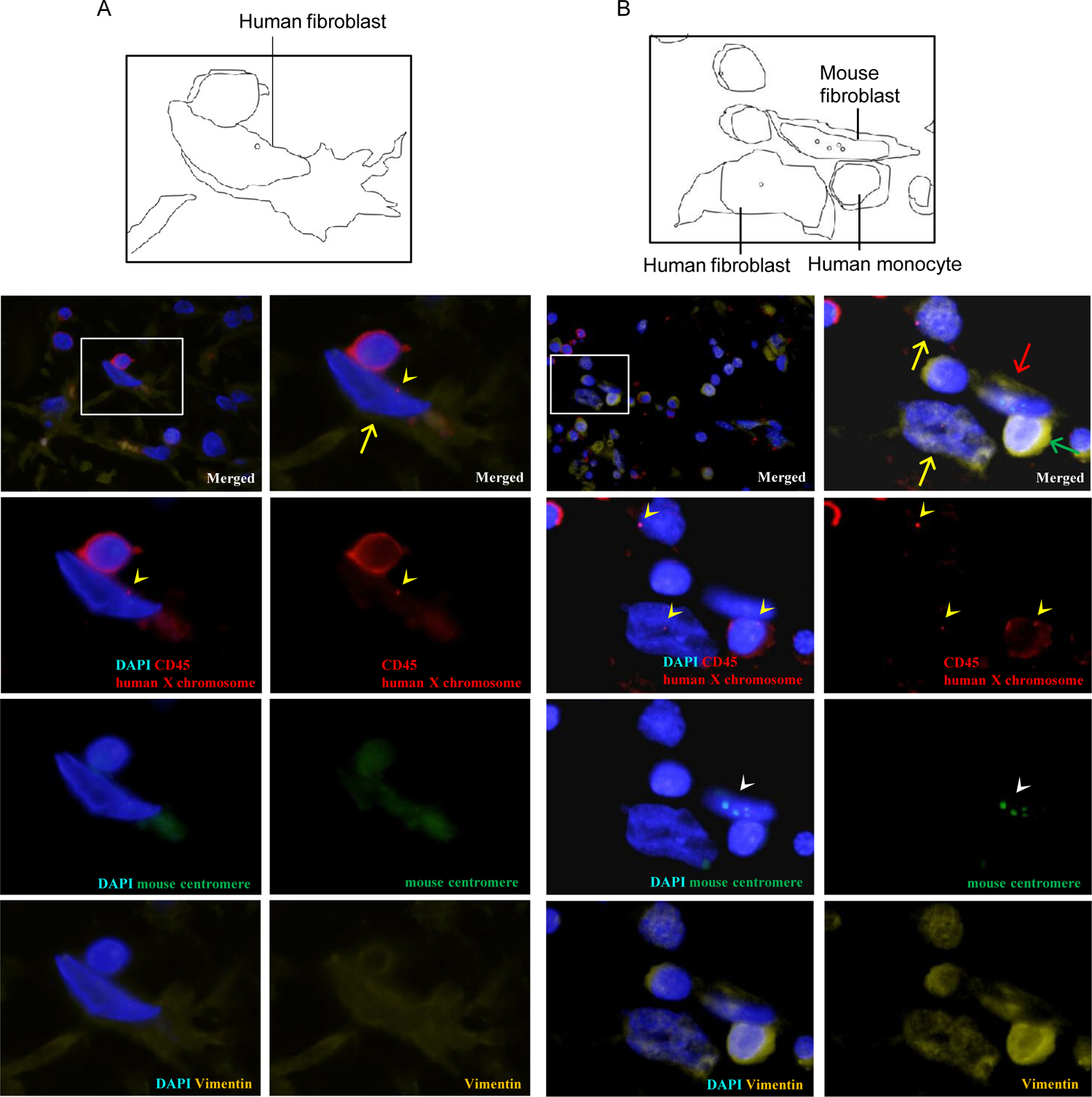
Femoral sections of recipients engrafted with (A) CD34+ PMF cells (recipient 1–1) and (B) CD34+CD38− PMF cells (recipient 3–3) were labeled with anti-human X chromosome probe (red), anti-mouse pan-centromeric probe (green), anti-CD45 antibody (red) and anti-vimentin antibody (yellow). Nuclei were stained with DAPI (blue). Schematic views are shown in left panels. Yellow arrowheads indicate the signals for human X chromosomes, and white arrowheads indicate those for mouse centromere. Fibroblast-like cells are characterized by CD45−vimentin+ phenotype, while monocytes and macrophages are characterized by CD45+vimentin+ phenotype. Both human X chromosome+CD45−vimentin+cells (yellow arrows) and mouse chromosome+CD45−vimentin+ cells (B, red arrow) were found in the fibrotic regions of recipient BM. Green arrow shows human X chromosome+CD45+vimentin+ monocyte.
Figure 4. Schematic diagram of potential pathogenesis of PMF.
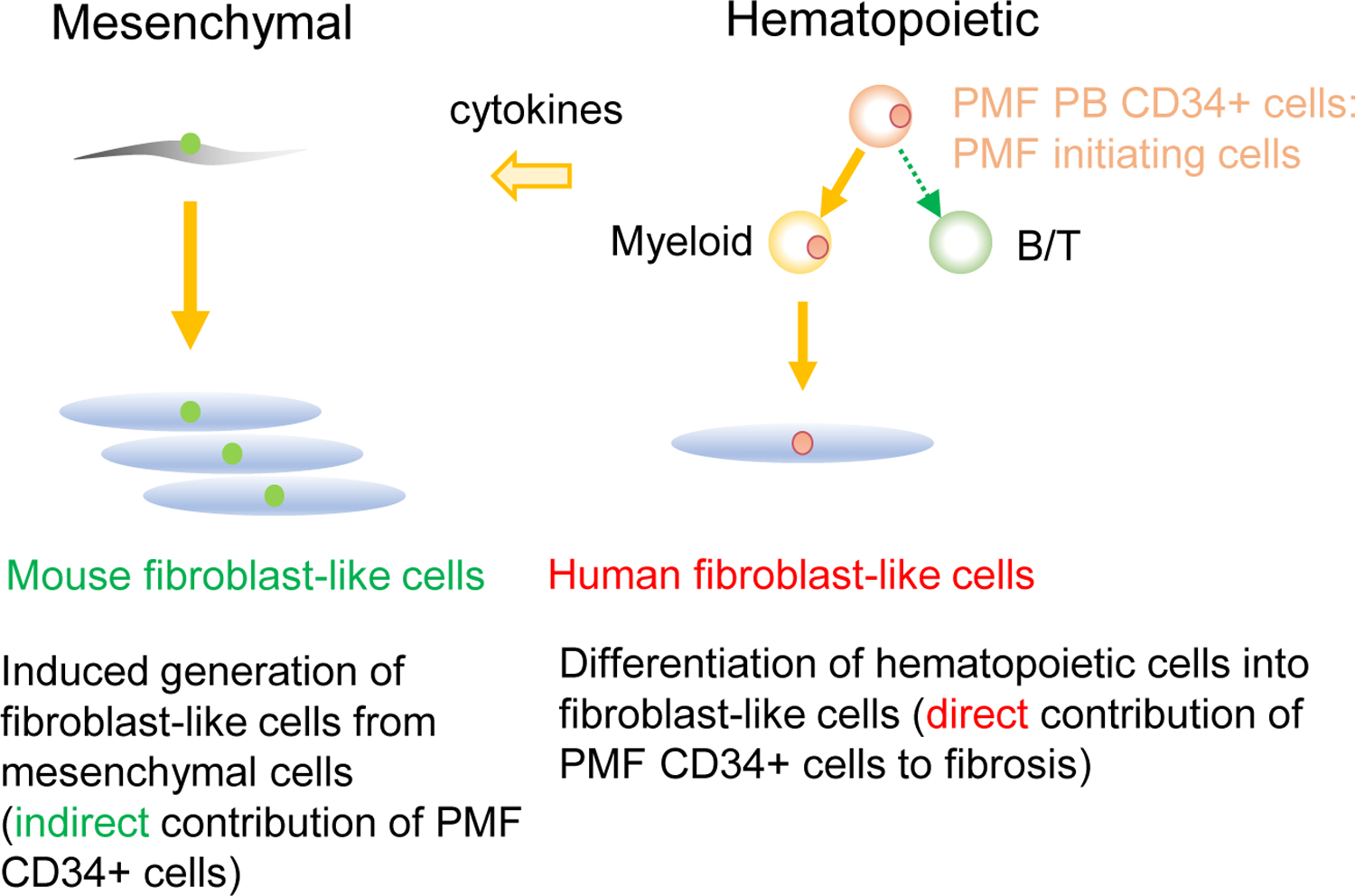
With an in vivo PMF xenograft model, we found both mouse chromosome+ fibroblast-like cells and human chromosome+ fibroblast-like cells in the recipient BM. The finding suggests that BM fibrosis occurs both through induction of fibroblasts from host mesenchymal stromal cells and partly through generation of fibroblast-like cells from malignant stem cells.
Discussion
PMF is categorized into MPN, a syndrome of chronic myeloid proliferation presumably due to “hematopoietic stem cell impairment” [33]. Previous studies showed engraftment of human CD45+ cells in immune-deficient mice by using PMF patient splenic CD34+ cells as donor cells [34]. Furthermore, transplantation of low density BM cells and CD34−CD14+ relatively mature monocytes of PMF patients resulted in recapitulation of BM fibrosis in immune-compromised mice [23, 24]. One of the unanswered questions regarding PMF pathogenesis is how circulating CD34+ cells contribute to fibrotic change in the BM. In the present paper, by transplanting PMF patient-derived circulating CD34+ subpopulations into NSG newborns, we achieved long-term (up to 17 months) myeloid-dominant engraftment with relatively high human cell chimerism. Furthermore, in mice which achieved long-term reconstitution, transplantation of CD34+ subpopulations resulted in generation of fibroblast-like cells in the BM, recapitulating BM fibrosis in vivo.
It has been shown that PMF patients have increased the number and frequencies of CD34+ cells in peripheral blood. The blood CD34+ cells in PMF patients could be functionally-normal HSCs and progenitors that escaped from the fibrotic BM into circulation. Consistent with a previous report [35], PMF patient-derived CD34+ or CD34+CD38− cells exhibited predominant myelopoiesis with relatively low levels of lymphocyte reconstitution in NSG recipients (Figure 1 and Table 1). This was in contrast to in vivo human hematopoietic repopulation by normal HSCs where B cell reconstitution is usually prominent in NSG and other immune-compromised mice [27]. Therefore, the phenotypically-identified HSC-like cells in PMF patients appear to have an intrinsic genetic programing that is biased for myeloid cell differentiation. In Patient 3, the blood CD34+CD38+ fraction as well as CD34+CD38− fraction engrafted mouse BM for a long-term, further suggesting the intrinsic abnormality of PMF HSCs in regulation of self-renewal or CD38 antigen expression. Collectively, the circulating CD34+ fractions in PMF patients contains malignant PMF-initiating cells that should be the target for PMF treatment.
It is important to note that circulating CD34+ PMF-initiating cells might belong to a single malignant PMF clone, because a number of previous studies have shown that all hematopoietic lineages in PMF patients were clonal [2–5]. Furthermore, previous findings showed that spleen endothelial cells from PMF patients were positive for the JAK2V617F mutation [36], but the origin of these mesenchymal lineage cells harboring JAK2V617F mutation is yet undefined. Our PDX model for PMF, via transplantation of purified PMF-initiating CD34+ subpopulations, results in predominantly hematopoietic not mesenchymal cell repopulation; However, human chromosome+ fibroblast-like cells were present in the recipient BM, albeit infrequently, suggesting that fibroblastic differentiation from PMF-initiating cells may occur at a low frequency. This potential of CD34+ cells generating fibroblast-like cells may reflect physiological differentiation into monocytes/macrophages, as shown by in vivo capacity of both fibroblastic and hematopoietic reconstitution from single murine HSCs [21, 22] and development of fibroblasts from macrophages through forced expression of transcription factors [37] and in the presence of vitamin D3 [25]. Recent studies reported that tumor stem cells could give rise to endothelial cells and fibroblasts in various cancers, possibly contributing to neovascularization and accelerated tumor growth [38–41]. Notably, monocyte/macrophage populations are well maintained in PMF patient BM, despite severe fibrosis. As recently reported, BM fibrosis in PMF may originate from malignant stem cells that differentiate into fibroblast-like cells and endothelial cells via monocytes/macrophages [23]. In addition, CD45−CD34− mesenchymal stem cells may also generate BM fibroblasts [22] [32]. In our in vivo PMF-initiation xenograft, the co-existence of human and mouse fibroblasts in the recipient BM were of mouse origin suggesting that BM fibrosis occurs through multiple distinct mechanisms: directly through generation of fibroblast-like cells from malignant stem cells carrying mutations such as JAK2V617F and indirectly through recruitment of resident mesenchymal lineage cells leading to pathological fibrotic changes in the BM (Figure 4).
As discussed above, we had focused on investigation of origins for hCD45-vimentin+ fibroblast-like mesenchymal cells. However, consistent with previous reports for neoplastic hematopoietic cell-derived fibrocytes [23, 42], we also found CD45+vimentin+ potential fibrocytes in the fibrotic lesions of NSG recipients. Roles of fibrocytes in PMF reported by Verstovsek et al. and Ozono et al. [23] [42] would further be investigated in our xenograft model in the future.
Delineation of mechanisms and possible points of therapeutic intervention await further clarification of genetic/cellular/autocrine/paracrine components that contribute to BM fibrosis in PMF. Furthermore, single-cell clonal analysis of PMF patient cell will help clarify not only its pathogenesis but also how an identical mutation, e.g. JAK2V617F results in multiple clinically distinct MPNs. Our human PMF xenograft system will enable future experiments to test therapeutic agents for PMF in vivo as well as to clarify the pathogenesis of PMF.
Supplementary Material
Supplemental Figure 1. Identification of human and mouse fibroblast like cells in NSG xenograft for PMF Femoral sections of NSG recipients engrafted with circulating PMF cells were labeled with anti-human X chromosome probe (red), anti-mouse pan centromeric probe (green), anti-human CD45 antibody (A C; green, D F; red), and anti-vimentin antibody (A C; red, D F; green). Nuclei were stained with DAPI (blue). Both human fibroblast like cells (white arrows) and mouse fibroblast like cells (yellow arrows) w ere present in the fibrotic region of the recipient bone marrow.
Supplemental Figure 2. Identification of potential human fibrocytes in NSG xenograft for PMF We detected potential fibrocytes (white arrows) which were characterized by a spindle shape, anti-human X chromosome probe positive, vimentin+ and CD45+. (Left) Images for every staining, (center) those with DAPI (blue) and human X chromosome probe and human CD45 (red), (right) and those with DAPI (blue) and vimentin (yellow) are shown. Arrow heads represent FISH signals.
Acknowledgments
The authors thank Drs. Takashi Okamura, Yoshinobu Asano, Tetsuya Eto, Yoshikiyo Ito for critical discussion.
Footnotes
Conflict of interest
The authors have no conflicts of interest to declare.
References
- 1.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson RJ, Salo A, Fialkow PJ. Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood. 1978;51:189–94. [PubMed] [Google Scholar]
- 3.Fialkow PJ. Use of glucose-6-phosphate dehydrogenase markers to study human myeloproliferative disorders. Haematol Blood Transfus. 1979;23:53–8. [DOI] [PubMed] [Google Scholar]
- 4.Buschle M, Janssen JW, Drexler H, Lyons J, Anger B, Bartram CR. Evidence for pluripotent stem cell origin of idiopathic myelofibrosis: clonal analysis of a case characterized by a N-ras gene mutation. Leukemia. 1988;2:658–60. [PubMed] [Google Scholar]
- 5.Reeder TL, Bailey RJ, Dewald GW, Tefferi A. Both B and T lymphocytes may be clonally involved in myelofibrosis with myeloid metaplasia. Blood. 2003;101:1981–3. [DOI] [PubMed] [Google Scholar]
- 6.Tefferi A Myelofibrosis with myeloid metaplasia. N Engl J Med. 2000;342:1255–65. [DOI] [PubMed] [Google Scholar]
- 7.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. [DOI] [PubMed] [Google Scholar]
- 8.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. [DOI] [PubMed] [Google Scholar]
- 9.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. [DOI] [PubMed] [Google Scholar]
- 10.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. [DOI] [PubMed] [Google Scholar]
- 11.Guglielmelli P, Lasho TL, Rotunno G, Mudireddy M, Mannarelli C, Nicolosi M, et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J Clin Oncol. 2018;36:310–8. [DOI] [PubMed] [Google Scholar]
- 12.Shide K, Shimoda HK, Kumano T, Karube K, Kameda T, Takenaka K, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. [DOI] [PubMed] [Google Scholar]
- 13.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–6. [DOI] [PubMed] [Google Scholar]
- 15.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90. [DOI] [PubMed] [Google Scholar]
- 16.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frey BM, Rafii S, Teterson M, Eaton D, Crystal RG, Moore MA. Adenovector-mediated expression of human thrombopoietin cDNA in immune-compromised mice: insights into the pathophysiology of osteomyelofibrosis. J Immunol. 1998;160:691–9. [PubMed] [Google Scholar]
- 18.Chagraoui H, Komura E, Tulliez M, Giraudier S, Vainchenker W, Wendling F. Prominent role of TGF-beta 1 in thrombopoietin-induced myelofibrosis in mice. Blood. 2002;100:3495–503. [DOI] [PubMed] [Google Scholar]
- 19.Kakumitsu H, Kamezaki K, Shimoda K, Karube K, Haro T, Numata A, et al. Transgenic mice overexpressing murine thrombopoietin develop myelofibrosis and osteosclerosis. Leuk Res. 2005;29:761–9. [DOI] [PubMed] [Google Scholar]
- 20.Rameshwar P, Chang VT, Thacker UF, Gascon P. Systemic transforming growth factor-beta in patients with bone marrow fibrosis--pathophysiological implications. Am J Hematol. 1998;59:133–42. [DOI] [PubMed] [Google Scholar]
- 21.Ebihara Y, Masuya M, Larue AC, Fleming PA, Visconti RP, Minamiguchi H, et al. Hematopoietic origins of fibroblasts: II. In vitro studies of fibroblasts, CFU-F, and fibrocytes. Exp Hematol. 2006;34:219–29. [DOI] [PubMed] [Google Scholar]
- 22.Ogawa M, LaRue AC, Drake CJ. Hematopoietic origin of fibroblasts/myofibroblasts: Its pathophysiologic implications. Blood. 2006;108:2893–6. [DOI] [PubMed] [Google Scholar]
- 23.Verstovsek S, Manshouri T, Pilling D, Bueso-Ramos CE, Newberry KJ, Prijic S, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. 2016;213:1723–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manshouri T, Verstovsek S, Harris DM, Veletic I, Zhang X, Post SM, et al. Primary myelofibrosis marrow-derived CD14+/CD34− monocytes induce myelofibrosis-like phenotype in immunodeficient mice and give rise to megakaryocytes. PLoS One. 2019;14:e0222912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wakahashi K, Minagawa K, Kawano Y, Kawano H, Suzuki T, Ishii S, et al. Vitamin D receptor-mediated skewed differentiation of macrophages initiates myelofibrosis and subsequent osteosclerosis. Blood. 2019;133:1619–29. [DOI] [PubMed] [Google Scholar]
- 26.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–89. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–21. [DOI] [PubMed] [Google Scholar]
- 29.Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009;115:3842–7. [DOI] [PubMed] [Google Scholar]
- 30.Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128–32. [PubMed] [Google Scholar]
- 31.Terstappen LW, Huang S, Safford M, Lansdorp PM, Loken MR. Sequential generations of hematopoietic colonies derived from single nonlineage-committed CD34+CD38− progenitor cells. Blood. 1991;77:1218–27. [PubMed] [Google Scholar]
- 32.Baertschiger RM, Serre-Beinier V, Morel P, Bosco D, Peyrou M, Clement S, et al. Fibrogenic potential of human multipotent mesenchymal stromal cells in injured liver. PLoS One. 2009;4:e6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112:2190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Prakash S, Lu M, Tripodi J, Ye F, Najfeld V, et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J Clin Invest. 2012;122:3888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M, Bruno E, Chao J, Ni H, Lindgren V, Nunez R, et al. The constitutive mobilization of bone marrow-repopulating cells into the peripheral blood in idiopathic myelofibrosis. Blood. 2005;105:1699–705. [DOI] [PubMed] [Google Scholar]
- 36.Rosti V, Villani L, Riboni R, Poletto V, Bonetti E, Tozzi L, et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood. 2013;121:360–8. [DOI] [PubMed] [Google Scholar]
- 37.Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, et al. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci U S A. 2008;105:6057–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McDonald LT, LaRue AC. Hematopoietic stem cell derived carcinoma-associated fibroblasts: a novel origin. Int J Clin Exp Pathol. 2012;5:863–73. [PMC free article] [PubMed] [Google Scholar]
- 40.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–8. [DOI] [PubMed] [Google Scholar]
- 41.Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–33. [DOI] [PubMed] [Google Scholar]
- 42.Ozono Y, Shide K, Kameda T, Kamiunten A, Tahira Y, Sekine M, et al. Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia. 2021;35:454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Identification of human and mouse fibroblast like cells in NSG xenograft for PMF Femoral sections of NSG recipients engrafted with circulating PMF cells were labeled with anti-human X chromosome probe (red), anti-mouse pan centromeric probe (green), anti-human CD45 antibody (A C; green, D F; red), and anti-vimentin antibody (A C; red, D F; green). Nuclei were stained with DAPI (blue). Both human fibroblast like cells (white arrows) and mouse fibroblast like cells (yellow arrows) w ere present in the fibrotic region of the recipient bone marrow.
Supplemental Figure 2. Identification of potential human fibrocytes in NSG xenograft for PMF We detected potential fibrocytes (white arrows) which were characterized by a spindle shape, anti-human X chromosome probe positive, vimentin+ and CD45+. (Left) Images for every staining, (center) those with DAPI (blue) and human X chromosome probe and human CD45 (red), (right) and those with DAPI (blue) and vimentin (yellow) are shown. Arrow heads represent FISH signals.