

Abstract
Large-scale molecular profiling and genotyping provide a unique opportunity to systematically compare the genetically predicted effects of therapeutic targets on the human metabolome. We firstly constructed genetic risk scores for 8 drug targets on the basis that they primarily modify low-density lipoprotein (LDL) cholesterol (HMGCR, PCKS9, and NPC1L1), high-density lipoprotein (HDL) cholesterol (CETP), or triglycerides (APOC3, ANGPTL3, ANGPTL4, and LPL). Conducting mendelian randomisation (MR) provided strong evidence of an effect of drug-based genetic scores on coronary artery disease (CAD) risk with the exception of ANGPTL3. We then systematically estimated the effects of each score on 249 metabolic traits derived using blood samples from an unprecedented sample size of up to 115,082 UK Biobank participants. Genetically predicted effects were generally consistent among drug targets, which were intended to modify the same lipoprotein lipid trait. For example, the linear fit for the MR estimates on all 249 metabolic traits for genetically predicted inhibition of LDL cholesterol lowering targets HMGCR and PCSK9 was r2 = 0.91. In contrast, comparisons between drug classes that were designed to modify discrete lipoprotein traits typically had very different effects on metabolic signatures (for instance, HMGCR versus each of the 4 triglyceride targets all had r2 < 0.02). Furthermore, we highlight this discrepancy for specific metabolic traits, for example, finding that LDL cholesterol lowering therapies typically had a weak effect on glycoprotein acetyls, a marker of inflammation, whereas triglyceride modifying therapies assessed provided evidence of a strong effect on lowering levels of this inflammatory biomarker. Our findings indicate that genetically predicted perturbations of these drug targets on the blood metabolome can drastically differ, despite largely consistent effects on risk of CAD, with potential implications for biomarkers in clinical development and measuring treatment response.
Introduction
Cardiovascular disease (CVD), including coronary artery disease (CAD) and ischaemic stroke, is the leading cause of death worldwide [1]. Circulating lipoprotein lipid concentrations are of central importance to the aetiology of CAD [2,3]. For example, clinical trials [4] and studies of human genetics [5–7] converge to support a causal role of apolipoprotein B (apoB) and low-density lipoprotein (LDL) cholesterol concentrations in the initial development and subsequent progression of CAD.
Pharmacological therapies that target the metabolism of blood lipids are routinely used for the prevention and treatment of CVD and are among the most widely prescribed medicines in the world [8]. Interestingly, drug targets that modify concentrations of LDL cholesterol (for instance, statins, acting on HMG-CoA reductase [HMGCR]) and those designed to modify high-density lipoprotein (HDL) cholesterol (for instance, cholesteryl ester transfer protein [CETP] inhibitors) and triglycerides (for instance, angiopoietin-like protein 3 [ANGPLT3] inhibitors) act on discrete pathways involved in lipid metabolism. Therefore, while each of these drug classes has proven [9–11] or emerging [12–15] efficacy for CVD risk reduction, their effects on the blood lipidome and metabolome are likely to vary considerably. This has implications on understanding which biomarkers to measure (for instance, during clinical development in randomised controlled trials) and on gauging markers of treatment response [16].
In this study, we sought to estimate the effects of lipid-modifying therapeutic targets on the blood metabolome to better characterise their impact on biomarkers related to CVD risk reduction. We constructed genetic instruments for drug targets that are either currently licenced or under development and grouped them according to their primary lipid of pharmacological focus: LDL cholesterol, HDL cholesterol, or triglycerides. We then compared the genetically predicted effects of therapeutic targets on CAD risk, before evaluating their effects on circulating lipoprotein lipid concentrations newly measured at large scale in the UK Biobank (UKB) study through the application of drug-target mendelian randomisation (MR) [17–19] (Fig 1).
Fig 1. A schematic representation of the drug-target MR approach undertaken in this study using, for instance, HMGCR variants to proxy for HMG-CoA reductase inhibition (the mechanism of action of statin therapy) to estimate its genetically predicted effect on CAD.

Genetic variants robustly associated with a lipoprotein lipid trait (for instance, LDL cholesterol) based on P < 1 × 10−6 within 100 kbs of encoding genes were identified as genetic proxies for perturbing therapeutic targets. A sensitivity analysis restricted to 50 kbs on either side of encoding genes was also undertaken in this study. CAD, coronary artery disease; kbs, kilobases; LDL, low-density lipoprotein; MR, mendelian randomisation.
Results
Genetic instrumentation of lipid-modifying drug targets to estimate their therapeutic effects on coronary artery disease and type 2 diabetes risk
We conducted genome-wide association studies (GWAS) on measures of LDL cholesterol (n = 328,111), HDL cholesterol (n = 300,528), and triglycerides (n = 328,498) in the UKB using biochemistry measures of these traits. Sample sizes were determined based on standard exclusion criteria (see Materials and methods), as well as excluding participants with measures of metabolic traits derived from a newly available nuclear magnetic resonance (NMR) platform in UKB. This was to avoid overlapping samples in our planned MR analyses of metabolic traits, which has been reported to potentially lead to overfitting in estimates [20]. Instrumental variables for 8 drug targets were identified using results of these GWAS for planned MR analyses. These were PCSK9, HMGCR, and NPC1L1 [4] (using LDL cholesterol results), CETP (using HDL cholesterol results), and APOC3, ANGPTL3, ANGPTL4, and LPL [15,21] (using triglyceride results). In total, we identified 137 instruments based on P < 1 × 10−6, r2 < 0.1, and a window size of 100 kbs either side of the 8 genetic loci responsible for encoding the lipid-modifying targets evaluated in this study (S1 Table).
Two-sample MR analyses were undertaken using the inverse variance weighted (IVW) method while accounting for the correlation between instruments [22,23] (Fig 1). Using results on 60,801 CAD cases and 123,504 control from the CARDIoGRAMplusC4D consortium, we found strong evidence of a genetically predicted effect for each therapeutic target on CAD risk (based on false discovery rate (FDR) < 5%) with the exception of ANGPTL3 (S2 Table and Fig 2), in keeping with prior findings [5,24–28]. Likewise, analyses on type 2 diabetes (T2D) risk using results from a GWAS of 74,124 cases and 824,006 controls from the DIAMANTE consortium supported previous findings (S2 Table). For instance, this included a genetically predicted adverse effect for the HMGCR score with T2D risk (OR = 1.64, 95% CI = 1.22 to 2.20, P = 0.001), whereas a protective effect was found for the LPL score (OR = 0.73, 95% CI = 0.66 to 0.80, P = 6.05 × 10−10). There was additionally strong evidence of a genetically predicted effect on T2D risk for the ANGPTL4 score (OR = 0.62, 95% CI = 0.50 to 0.76, P = 2.65 × 10−6). F-statistics did not indicate that drug target scores were prone to weak instrument bias (F = 58.3 to 297.1) (S2 Table). Genetically predicted effects on LDL cholesterol, HDL cholesterol, and triglycerides based on UKB biochemistry measures in the participants with NMR metabolites data (i.e., nonoverlapping with the partitioned sample, which instruments were selected in) can be found in S3 Table.
Fig 2. A forest plot visualising the genetically predicted effects of lipid-modifying drug targets on risk of CAD and T2D using MR.

Estimates are colour coded based on the lipoprotein lipid trait estimates used to derive genetic scores. Each genetic score was oriented to mimic the putative effects of drug targets on lipoprotein traits, meaning that effect estimates correspond to relative odds of disease per 1 SD change in either lower LDL cholesterol, higher HDL cholesterol, or lower triglyceride levels via each specific drug target. Note that in the case of CETP, we are not ascribing causal effects to HDL cholesterol—rather, we are orientating CAD/T2D effect estimates corresponding to a genetically predicted increase in HDL cholesterol arising from pharmacological inhibition of CETP. The data underlying this figure can be found in S2 Table. CAD, coronary artery disease; CETP, cholesteryl ester transfer protein; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MR, mendelian randomisation; SD, standard deviation; T2D, type 2 diabetes.
Systematic evaluation of genetically predicted therapeutic target effects on metabolic traits
Next, we applied our GWAS pipeline to all 249 metabolic traits measured by targeted high-throughput NMR metabolomics from Nightingale Health (biomarker quantification version 2020) in the separate subset of UKB participants with these measures. Sample sizes after QC ranged between n = 110,051 to n = 115,082 (S4 Table). In total, there were 2,814 genetic variants robustly associated with at least one measure (based on the conventional GWAS cutoff P < 5 × 10−8) across 721 independent genetic loci (S5 Table). All of the 249 metabolic traits were represented among these findings (i.e., each trait quantified by the NMR platform had at least one SNP association at GWAS levels of significance) with the majority having dozens of independent variants associated with their levels (median: 74 variants, interquartile range: 27 variants) (S6 Table).
Systematically estimating genetically predicted effects of each lipid-modifying target in turn on each of the 249 metabolic traits using MR identified a total of 1,588 effects robust to FDR < 5% corrections (S7–S14 Tables). Investigating the robustness of our results to a more stringent instrument selection criteria (i.e., 50 kbs either side of encoding genes as compared to 100 kbs in our main analyses) provided strong evidence of homogeneity between genetically predicted drug-target effects in the original analysis (S1–S8 Figs).
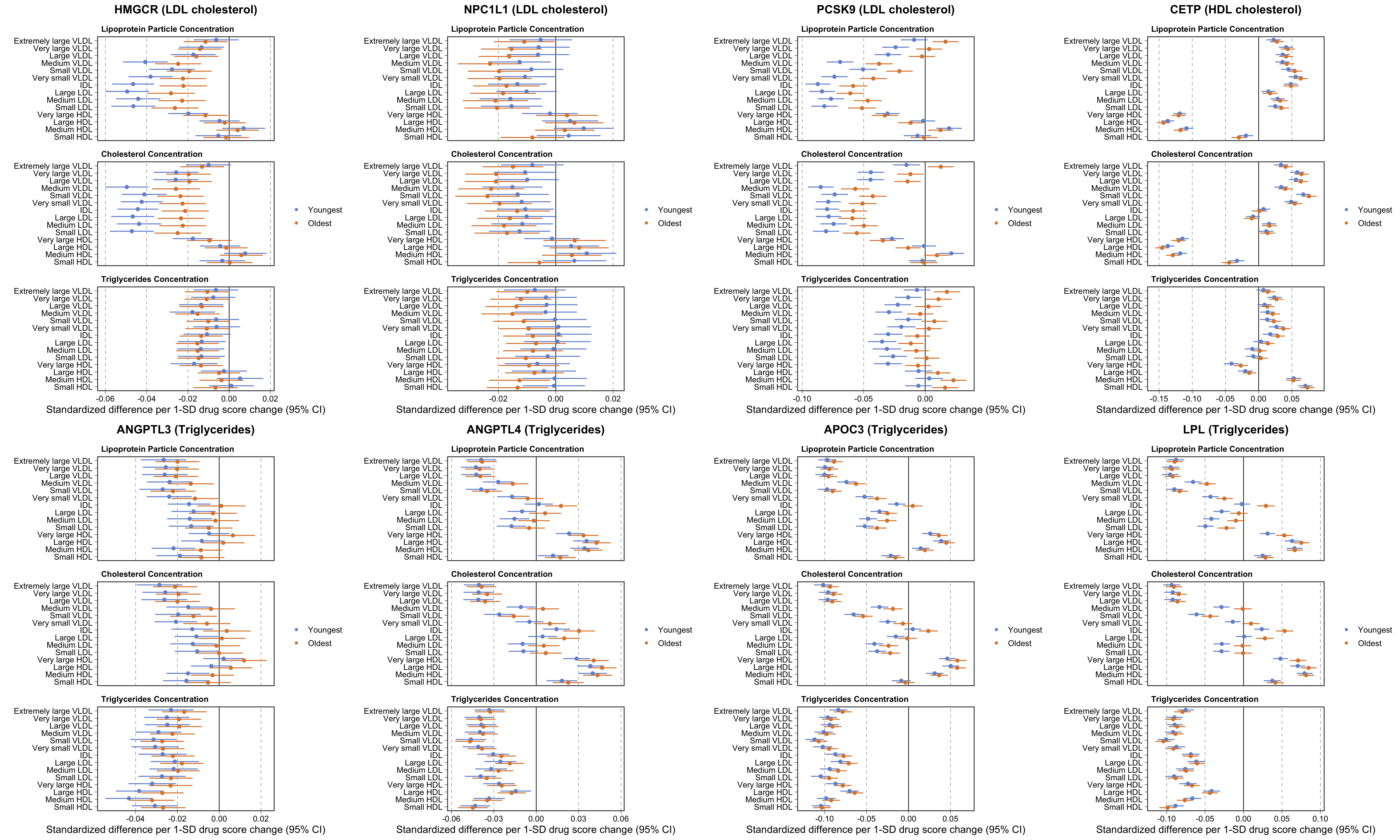
A subset of these estimates related to lipoprotein particle, cholesterol, and triglyceride concentrations across the 8 drug targets have been highlighted in Fig 3. Broadly, the LDL cholesterol modifying targets (HMGCR, PCSK9, and NPC1L1) provided evidence of genetically predicted effects on lower levels of very low-density lipoprotein (VLDL), intermediate density lipoprotein (IDL), and LDL-related particle concentrations, but typically weak evidence on HDL-related markers (with the exception of very large HDL particles, for which genetic instruments for both HMGCR and PCSK9 showed strong evidence of lowering). For example, the strongest evidence identified using the PCSK9 score was on large LDL particle concentrations (Beta = 0.96 SD reduced per 1-SD lowering in LDL cholesterol, 95% CI = 0.87 to 1.04, P = 3 × 10-113). The concentration of cholesterol within lipoprotein particle subclasses tended to mimic the associations identified for lipoprotein particle concentration. In contrast, generally weaker effects of genetic instruments for HMGCR, PCSK9, and NPC1L1 were identified for triglyceride concentrations within the same lipoprotein particle subclasses.
Fig 3. Forest plots illustrating the genetically predicted effects of lipid-modifying drug targets on measures of circulating metabolite concentrations using NMR in the UKB study.

Effect estimates are based on an SD change in the genetically predicted drug target scores oriented to reflect therapeutic intervention (i.e., lower LDL cholesterol, lower triglycerides, and higher HDL cholesterol). Scores were derived using genetic variants robustly associated which lipoprotein lipid traits (as indicated in each target’s legend) at each encoding gene’s region. The data underlying this figure can be found in S7–S14 Tables. CETP, cholesteryl ester transfer protein; HDL, high-density lipoprotein; IDL, intermediate density lipoprotein; LDL, low-density lipoprotein; NMR, nuclear magnetic resonance; SD, standard deviation; UKB, UK Biobank; VLDL, very low-density lipoprotein.
Orientated to a lowering of CAD risk, the HDL cholesterol modifying target CETP provided evidence of lower genetically predicted effects on VLDL and LDL circulating concentrations. Notably, comparatively larger effects were identified on lipoprotein particle concentration and cholesterol concentrations within HDL subclasses with positive associations identified for these HDL-related traits. In contrast, marked heterogeneity was found in relation to triglycerides concentrations, with genetically predicted estimates suggesting an effect on higher very large and large HDL-C concentrations and on lower levels of medium and small HDL concentrations.
Genetically predicted triglyceride modifying targets (APOC3, ANGPTL3, ANGPTL4, and LPL) markedly lowered triglyceride concentrations across the spectrum of lipoprotein subclasses—this was in contrast to genetic instruments for HMGCR, PCSK9, NPC1L1, and CETP where effect estimates were weaker and tended to be on both sides of the null. For lipoprotein particle and cholesterol concentrations, lowering effects of triglyceride modifying targets were typically found for the VLDL-related traits, with an inflection point at IDL seen for ANGTPL4, APOC3, and LPL but not for ANGPTL3.
A comparison of these analyses repeated in the youngest and oldest subgroups using individual-level data from unrelated individuals within UKB (both n = 30,000) can be found in S9 Fig. Overall metabolic signatures did not appear to drastically differ between these strata defined by age, suggesting that treatment with statins was unlikely to lead to major perturbations in the effect estimates we present. While overall trends did not typically vary from those identified in the full sample, these findings suggest that analyses, which directly adjust for contingent factors within UKB, such as statin medications, are likely to introduce collider bias into their findings (as proposed previously [29]).
We also identified differing signatures between drug target classes for non-lipoprotein lipid–related traits. For instance, LDL lowering targets typically provided weak evidence of a genetically predicted effect on glycoprotein acetyls (GlycAs), a marker of inflammation (for instance, PCSK9: Beta = 0.01, 95 CI% = −0.06 to 0.08, P = 0.78). In contrast, all triglyceride lowering targets (i.e., ANGPTL3, ANGPTL4, APOC3, and LPL) provided strong evidence of a genetically predicted effect on lowering GlycA (for instance, LPL: Beta = −0.43, 95 CI% = −0.37 to −0.48, P = 9 × 10−50), as well as CETP. All drug target estimates on GlycA have been collated in S15 Table. Although GlycA is an adjunct of inflammation, we also provide genetically predicted effects of each target on C-reactive protein (CRP), measured using the biochemistry in the same participants with measures of NMR metabolites, given that it is a more widely and clinically used biomarker of inflammation (S16 Table). Similar directions of effect for targets on GlycA were found on CRP, although the only target to provide evidence of an effect robust to multiple comparisons was ANGPTL3 (Beta = −0.22, 95 CI% = −0.39 to −0.04, P = 0.02).
Genetically predicted metabolic effects for drug targets in comparison to statin medication
We systematically compared the genetically predicted effects of each drug target on all 249 metabolic traits with HMGCR acting as a proxy for statin therapy. For comparative purposes, estimates were scaled in accordance with their respective effect estimates on CAD as reported in S2 Table. In general, we identified strong evidence of concordance between the other LDL cholesterol lowering therapies (PCKS9 and NPC1L1) with the HMGCR score (r2 = 0.91 and r2 = 0.79, respectively). Fig 4A illustrates the linear trend identified between genetically predicted effects of PCSK9 and HMGCR on metabolic markers. S10 Fig contains the corresponding plot for NPC1L1.
Fig 4.
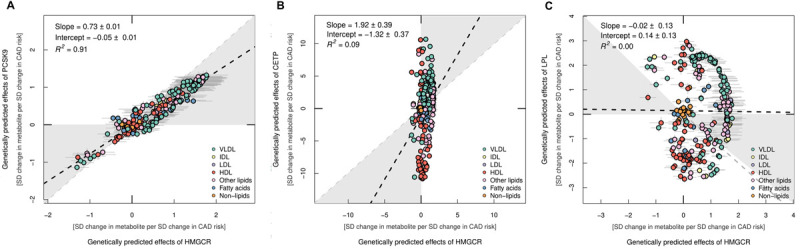
A comparison of distributions between genetically predicted drug target effects (A) PCSK9, (B) CETP, and (C) LPL on metabolic traits using NMR in the UKB study. In each figure estimates are compared with the results from the HMGCR score. Effect estimates were scaled in accordance with the corresponding effects of these genetically predicted drugs targets on risk of CAD, which is why axes vary between plots. Points are coloured based on subcategories of metabolic traits indicated in the figure legends. The data underlying this figure can be found in S7, S8, S10, and S14 Tables. CAD, coronary artery disease; CETP, cholesteryl ester transfer protein; HDL, high-density lipoprotein; IDL, intermediate density lipoprotein; LDL, low-density lipoprotein; NMR, nuclear magnetic resonance; SD, standard deviation; UKB, UK Biobank; VLDL, very low-density lipoprotein.
In contrast, comparisons with the HDL cholesterol raising (CETP) (r2 = 0.09) (Fig 4B) and triglyceride lowering (APOC3, ANGPTL3, ANGPTL4, and LPL) (all r2 ≤ 0.02) targets provided weak evidence of concordance with the HMGCR score. Fig 4C visualises this general lack of concordance using the LPL and HMGCR score comparison as an exemplar. Broadly, both the LPL and HMGCR scores provided evidence of genetically predicted effects on higher levels of various triglyceride-rich VLDL-related traits (highlighted in green). However, conversely, the LPL score typically provided stronger evidence of an effect on HDL-related traits (highlighted in red), which was generally not the case for the HMGCR score. As expected, both scores provided weak evidence of an effect on non-lipid-related traits (highlighted in orange). All other figures generated from this analysis for the other targets in comparison to the HMGCR score can be found in S11–S13 Figs. There was also typically good concordance among metabolomic profiles derived from drug targets within the same lipoprotein lipid class, for instance, when comparing all pairwise combinations of triglyceride lowering drug targets (S14–S19 Figs). These findings were similar to those reported previously by a study conducted by Wang and colleagues [28], for example, with very strong concordance between ANGPTL4 and LPL profiles identified (r2 = 0.96 by Wang and colleagues compared with r2 = 0.99 in this study). This is likely attributed to the large sample size harnessed in the study by Wang and colleagues (n = 61,240), which is also the case in this current study. APOC3, which was not evaluated by this previous study, likewise had a similar metabolomic profile as the other triglyceride lowering targets assessed (for instance, LPL and APOC3 r2 = 0.94).
Discussion
In this study, we explored the genetically predicted metabolic effects of modifying LDL, HDL, and triglycerides via drug targets that are either well established, recently licenced, and/or under development [30–33]. Our findings demonstrate that drug targets that principally act to modify LDL cholesterol (for instance, statins, PCSK9 inhibitors, and ezetimibe) have broadly similar effects on the blood metabolome. In contrast, effects of drugs designed to modify HDL cholesterol and triglycerides had very different effects on metabolic biomarkers, even when scaled to the same difference in risk of CAD. These findings provide a catalogue of genetically predicted pharmacological effects on the blood metabolome, which serves to illustrate the heterogeneity between different lipid-modifying therapies, highlighting the need for rich phenotyping of lipoprotein lipids in developing assays that gauge treatment response.
Our findings illustrate the tapestry of metabolic biomarker associations that are predicted to be downstream consequences of pharmacological modification of a therapeutic target. While these findings do not provide evidence of causation of these metabolic biomarkers, rather, they employ drug target MR as a means of characterising therapeutic effects on the metabolome [34,35]. Such diverse effects can then potentially be triangulated [36] to explore patterns of metabolomics where signatures are consistent with cardiovascular risk reduction. In-depth investigations into the independent causal role of specific metabolic traits at a granular level can then be explored using approaches such as multivariable MR [37]. For example, one might construct genetic instruments for biomarkers that are downstream consequences of HMGCR inhibition and conduct de novo multivariable MR analyses of these traits in order to identify the mediating mechanisms beyond apoB or LDL cholesterol. While previous studies, including those that we conducted, identified apoB as the fundamental driver of lipid-mediated CVD [7], a greater understanding of the causal components should facilitate new avenues of investigation and resultant pharmacological development. We identified important differences in the genetically predicted effects of some therapeutic targets on risks of CAD and T2D. HMGCR, NPC1L1 and PCSK9 all lowered risk of CAD, yet HMGCR, and to a lesser extent NPC1L1 and PCSK9, increased risk of T2D (as presented in Fig 2). In contrast, ANGPTL4 and LPL were genetically predicted to lower risks of both CAD and T2D. Comparisons of these therapeutic targets on detailed measurements using omics approaches such as those employed in this study may clarify the underlying aetiological mechanisms driving these differences and aid in the development of medicines that are protective for both vascular and metabolic diseases. For example, by exploring disease-specific effects of genetically instrumented drug targets and partitioning metabolic biomarkers according to such, it may be possible, through approaches such as multivariable MR, to identify in finer detail which metabolic biomarkers are causally implicated in CVD and metabolic disease. Additional approaches including “reverse-MR” [38,39], where genetic instruments for liability to disease are explored for their metabolomic signatures, may reveal biomarkers on the causal pathway to disease. Integration of these types of genetic epidemiological avenues of investigation are likely to resolve disease-specific roles of these metabolic biomarkers and, consequently, facilitate new therapeutic targets for clinical development. Additionally, our results provide granular insight into the genetically predicted effects on the plethora of circulating metabolic traits investigated in this study. For instance, the triglyceride lowering targets evaluated in this study typically provided strong evidence of an effect on GlycAs, a marker of inflammation, whereas in contrast, the LDL cholesterol lowering targets provided weak evidence of an effect on this circulating metabolic trait. This suggests that, although triglyceride lowering medications may not provide the same magnitude of effect towards lowering CAD risk as LDL lowering therapies, they may yield additional benefit towards reducing inflammation. Given that the role of inflammation in CVD is gaining traction as an orthogonal avenue of therapeutic potential, such effects of TG-modifying therapies on inflammation biomarkers offer potential therapeutic indications, which may have roles beyond CVD.
One of the striking findings is the general consistency of associations between drug targets and particle concentration and cholesterol concentration (likely owing to the high correlation between these phenotypic traits) and the divergence between particle and cholesterol concentration and triglycerides concentration. This was most notable when comparing drugs across their primary lipid indication—i.e., drugs that were developed on the basis of LDL cholesterol lowering tended to have modest associations with a general reduction in triglyceride concentrations across lipoprotein particles. In contrast, HDL cholesterol raising variants in CETP were identified to have effects on lower triglycerides in apoB containing lipoproteins, whereas for HDL particles, triglycerides were increased in very large and large HDL particles and reduced in medium and small HDL particles. Our CETP genetic score also had a genetically predicted effect on lower IDL and LDL lipoprotein particle and cholesterol concentrations, which has not been reported by previous MR evaluations of this target [19,40]. Possible explanations for this include the much larger number of genetic instruments leveraged in this study (n = 57), in comparison to previous studies that harnessed n ≤ 3 instruments, as well as performing analyses on a much larger sample size of individuals with NMR metabolites data in this work (n = 115,082).
For the drug targets where triglycerides metabolism was the primary lipid of pharmacological focus for development, triglycerides concentrations were lower across the lipoprotein particle spectrum. Since most of these drug targets demonstrated genetic evidence of CAD lowering, one might draw conclusions from such heterogeneity of triglycerides effects across these lipid-lowering therapies indicative that perhaps triglycerides were less important and that it was cholesterol or lipoprotein particle concentration (indexed, for instance, by apoB concentrations) that mediated these causal effects. However, previous multivariable MR analyses that included triglycerides, apoB, and LDL-C in the model demonstrated a direct effect of triglycerides consistent with a potential causal role of triglycerides in CAD [7,41] using the same dataset from the CARDIoGRAMplusC4D consortium as analysed in this study [42]. Thus, while our findings illustrate pronounced heterogeneity in cholesterol and triglyceride lipoprotein lipid concentrations arising from genetically predicted pharmacological inhibition of lipid modifying drug targets, drawing causal conclusions from such perturbations is nontrivial and requires MR of the individual phenotypes, as described previously.
The findings presented here have been made available by large-scale phenotyping using NMR-targeted metabolomics in UKB in combination with GWAS genotyping. Such data provide resolution of lipoprotein lipids at scale and enable genetic analyses of the type we present. The value of metabolomics may be to offer signatures of treatment response, which can then be used to guide pharmacological treatment. Such may be of utility from an early stage—for instance, during Phase I, II, and III clinical trials, where biomarkers are often used as a means of measuring treatment response across different concentrations of drugs [17], and postmarketing, when assessing interindividual response to treatment. Equally, our study has noteworthy limitations. For example, although previous studies have used similar criteria for instrument selection for the gene-based drug scores used in this study, we are unable to rule out genetic confounding as a potential source of bias in our analyses. Our analyses were also based on the European subset of the UKB study, and, therefore, evaluations in individuals of non-European ancestry would be valuable to investigate how representative our findings in diverse populations. Furthermore, we have used common genetic variants associated with lipoprotein lipid traits as a source of genetic instruments in this work. Future endeavours harnessing genetic effects on molecular traits (for instance, circulating proteins) or rare (and potentially highly penetrant) genetic variants may yield alternate strands of evidence to complement (or contradict) our results. In particular, these alternative approaches to genetic instrument selection for may identify a more powerful proxy for targets such as ANGPTL3 [43].
In summary, our study characterises the repertoire of genetically predicted lipid-modifying therapies on the blood metabolome. These findings demonstrate the widespread metabolic perturbance that arises from genetically evaluated modifications of therapeutic targets and heterogeneity between discrete classes of drugs, especially when their primary lipid trait differs. Such findings may be useful to illustrate the utility of drug target MR in gauging the predicted effects of drugs on omics traits to guide dose-ranging studies during clinical development and as a marker of treatment response.
Materials and methods
Instrument identification
Genetic instruments for each lipid-modifying drug target were selected by undertaking GWAS of lipoprotein lipid traits measured using a conventional biochemistry assay in UKB [44]. These included HDL cholesterol (field 30760), LDL cholesterol (field 30780), and triglycerides (field 30870). Details on genotyping quality control, phasing, and imputation in UKB have been described previously [45]. Briefly, GWAS were undertaken after excluding individuals with sex-mismatch (derived by comparing genetic sex and reported sex) or individuals with sex-chromosome aneuploidy were excluded from the analysis (n = 814). Next, a K-mean clustering algorithm was applied to remove UKB participants of non-European descent (based on K = 4) and also those with withdrawn consent leaving a maximum sample size of n = 463,005. Individuals who had measures of metabolic traits derived from a newly available NMR platform in UKB were also excluded from these GWAS (up to n = 121,727 participants) to avoid overlap with outcome samples (a potential source of bias in MR due to overfitting [20]). LDL cholesterol, HDL cholesterol, and triglycerides were normalised using inverse rank-normalisation such that their mean was 0 and their standard deviation was 1. We used the BOLT-LMM (linear mixed model) software with adjustment for age, sex, fasting status (i.e., the interval between consumption of food or drink and blood samples being taken), and a binary variable denoting the genotyping chip used in individuals (the UKBB Axiom array or the UK BiLEVE array) [46]. BOLT-LMM uses a linear mixed effect model to account for the population structure within UKB, which is why principal components were not included as covariates in the model. All analyses were conducted under UKB application #15825.
Final instrument selection for all 8 drug targets was based on results obtained from the GWAS of HDL cholesterol (to instrument CETP), LDL cholesterol (to instrument PCSK9, HMGCR, and NPC1L1 [4]), and triglycerides (to instrument APOC3, ANGPTL3, ANGPTL4, and LPL [15,21]). A selection criteria of genetic variants with P < 1 × 10−6, which were located within a 100-kbs region either side of encoding genes, was applied to select instruments. This window size was selected to reduce the likelihood of including instruments proximal to other genetic targets, which may influence these lipoprotein lipid traits via alternate biological pathways (i.e., horizontal pleiotropy). We conducted linkage disequilibrium (LD) pruning for variants such that they had r2 < 0.1 using a reference panel of 503 European individuals enrolled in the 1,000 Genomes Project phase 3 (version 5) [47]. We additionally set out to identify genetic instruments for PPARA as a proxy for triglyceride modification through fibrates, although our GWAS only identified a single variant associated with triglyceride levels at this gene’s locus. We did not carry this target forward into downstream analyses given the challenges for genetic confounding of conducting gene-centric MR analyses with a single genetic instrument, particularly in gene dense regions where the function of each gene is not well understood, which may hinder inference [48].
Genome-wide association studies of metabolic traits, coronary artery disease, and type 2 diabetes
We applied the same GWAS pipeline described above to all 249 metabolic traits measured by targeted high-throughput NMR metabolomics from Nightingale Health (biomarker quantification version 2020) in UKB. These analyses were conducted under UKB project #15825. Measures were taken using nonfasting EDTA plasma samples (aliquot) obtained from a random subsample of 121,584 UKB participants. Sample sizes on the 249 metabolic traits for GWAS after QC ranged between n = 110,051 to n = 115,082 UKB participants. A full summary of sample sizes can be found in S3 Table. Each metabolic trait was normalised to have a mean of 0 and standard deviation of 1 using inverse rank-normalisation as above allowing comparisons to be made between derived effect estimates. As before, all GWAS were adjusted for age, sex, fasting time, and genotyping chip.
Among these biomarkers were various lipoprotein lipids and their concentrations within 14 subclasses, fatty acids, ketone bodies, glycolysis metabolites, and amino acids (see S3 Table). Further details have been described previously [49]. Additionally, we conducted a GWAS of CRP measured using the biochemistry assay in UKB on the subset of participants with NMR metabolic traits. Ethical approval for this study was obtained from the Research Ethics Committee (REC; approval number: 11/NW/0382), and informed consent was collected from all participants enrolled in UKB.
Genome-wide summary-level estimates on CAD risk were obtained from a previously conducted GWAS from the CARDIoGRAMplusC4D consortium [42]. CAD cases in this consortium were defined as myocardial infarction, acute coronary syndrome, chronic stable angina, or coronary stenosis >50%. In total, there were 60,801 cases and 123,504 control assembled by CARDIoGRAMplusC4D across 48 studies, which did not include the UKB study. Genetic estimates on T2D were extracted from a previous GWAS from the DIAMANTE consortium consisting of 74,124 cases and 824,006 controls of European ancestry [50].
Statistical analysis
Drug-target mendelian randomisation
Univariable MR analyses were firstly undertaken to estimate the genetically predicted effects of each therapeutic target on risk on CAD. Estimates were derived by applying the IVW method while accounting for the correlation between instruments using the same reference panel as above [22,23]. Further details on this approach have been described previously [22]. Briefly, we calculated the pairwise correlations between all variants included in genetic scores. These were then incorporated into the standard error terms of test statistics for the summary-level weighted generalised linear regression MR models.
This approach was then applied systematically to estimate the genetically predicted effects of each target on each of the 249 metabolic traits in turn. Analyses were conducted in a two-sample data setting to ensure that our sample of UKB participants from which instruments were identified (i.e., the non-NMR subset of UKB) did not overlap with individuals analysed in the GWAS of metabolic traits in UKB. To account for multiple testing, FDR corrections were applied for each drug target analysed as a heuristic to highlight the most noteworthy findings with strong statistical support based on current sample sizes, although all results are reported in the Supporting information (S1–S19 Figs, S1–S16 Tables). We repeated this analysis restricting our instrument selection criteria to a 50-kbs window around encoding genes for targets to assess the robustness of our findings to genetic confounding (i.e., variants influencing metabolic traits via neighbouring genes).
Comparison between different drug target effects across metabolome-wide traits
We initially compared effect estimates for a subset of lipid concentrations across drug targets using forest plots. More comprehensive comparisons on metabolome-wide results (i.e., on all 249 traits) were illustrated using scatter plots proposed previously to compare pairwise estimates between 2 targets with metabolic traits coloured based on their subcategories [28]. For comparative purposes, we scaled all metabolite estimates using a scaling factor based on each target’s corresponding genetically predicted effect on CAD risk. We used HMGCR estimates as our baseline comparison for each of the other 7 drug targets given the widespread adoption of statin therapy to treat individuals at elevated risk of CVD. Comparisons between HMGCR estimates and those for each of the other scores were evaluated using generated R2 values as applied previously [28]. R2 values are the coefficients of determination estimating the quotient of the variances of the fitted values and observed values of the dependent variable using a linear regression model. Here, it describes the linear fit for the estimates on metabolic traits between the 2 drug targets assessed.
As a sensitivity analysis, we used individual-level data from UKB to investigate whether the genetically predicted effects of drug targets on lipoprotein lipid concentrations varied among participant subgroups stratified by their age. As described previously [29], this approach permits the investigation of whether putative contingent factors in UKB may influence conclusions without directly conditioning of them. For example, in this study, we might anticipate that the influence of statin medications on metabolic markers may distort effect estimates. However, adjusting for this factor either as a covariate or by stratifying participants on it is likely to induce collider bias into analyses, which is recognised to potentially undermine causal inference [51]. As such, we partitioned the unrelated European sample from UKB into the youngest (range from 40 to 54 years) and oldest (range from 61 to 71 years) subgroups (both n = 30,000), where the number of reported participants taking statin medications was 5.6% and 27.7%, respectively. Instruments for drug targets were then constructed as genetic risk scores using individual-level data from UKB and analysed against each measure of lipoprotein lipid concentrations in turn using linear regression adjusted for age, sex, and the top 10 principal components.
All plots in this study were generated using the R package “ggplot2” [52]. MR analyses were conducted using the R package “MendelianRandomization” [53]. All analyses were undertaken using R (version 3.5.1).
Supporting information
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PNG)
{kind=link}
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Acknowledgments
We would like to thank the CARDIoGRAMplusC4D consortium for making their summary statistics available for the benefit of this work and to the participants of the UK Biobank study. Additionally, the authors are grateful to UK Biobank for access to data to undertake this study (Project #15825 and #30418). MVH works in a unit that receives funding from the UK Medical Research Council and is supported by a British Heart Foundation Intermediate Clinical Research Fellowship (FS/18/23/33512) and the National Institute for Health Research Oxford Biomedical Research Centre. George Davey Smith and Joshua Bell work in the Medical Research Council Integrative Epidemiology Unit at the University of Bristol (MC_UU_00011/1).
Abbreviations
- ANGPLT3
angiopoietin-like protein 3
- apoB
apolipoprotein B
- CAD
coronary artery disease
- CETP
cholesteryl ester transfer protein
- CRP
C-reactive protein
- CVD
cardiovascular disease
- FDR
false discovery rate
- GlycA
glycoprotein acetyl
- GWAS
genome-wide association studies
- HDL
high-density lipoprotein
- IDL
intermediate density lipoprotein
- IVW
inverse variance weighted
- LD
linkage disequilibrium
- LDL
low-density lipoprotein
- MR
mendelian randomization
- NMR
nuclear magnetic resonance
- T2D
type 2 diabetes
- UKB
UK Biobank
- VLDL
very low-density lipoprotein
Data Availability
Data supporting this paper can be found within the Supporting Information files. Full GWAS summary statistics for all 249 circulating metabolites generated in this work are accessible via the GWAS catalog (https://www.ebi.ac.uk/gwas/) under accession IDs GCST90092803 to GCST90093051.
Funding Statement
MVH works in a unit that receives funding from the UK Medical Research Council and is supported by a British Heart Foundation Intermediate Clinical Research Fellowship (FS/18/23/33512) and the National Institute for Health Research Oxford Biomedical Research Centre. GDS works at the Medical Research Council Integrative Epidemiology Unit at the University of Bristol (MC_UU_00011/1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.World Health Organization. Cardiovasc Dis. 2020. Available from: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1.
- 2.Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388(10059):2532–61. Epub 2016/09/13. doi: 10.1016/S0140-6736(16)31357-5 . [DOI] [PubMed] [Google Scholar]
- 3.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459–72. Epub 2017/04/27. doi: 10.1093/eurheartj/ehx144 ; PubMed Central PMCID: PMC5837225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, et al. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. JAMA. 2016;316(12):1289–97. Epub 2016/09/28. doi: 10.1001/jama.2016.13985 . [DOI] [PubMed] [Google Scholar]
- 5.Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–61. Epub 2015/03/17. doi: 10.1016/j.jacc.2015.02.020 ; PubMed Central PMCID: PMC6101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmes MV, Asselbergs FW, Palmer TM, Drenos F, Lanktree MB, Nelson CP, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36(9):539–50. Epub 2014/01/30. doi: 10.1093/eurheartj/eht571 ; PubMed Central PMCID: PMC4344957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson TG, Sanderson E, Palmer TM, Ala-Korpela M, Ference BA, Davey Smith G, et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17(3):e1003062. Epub 2020/03/24. doi: 10.1371/journal.pmed.1003062 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuentes AV, Pineda MD, Venkata KCN. Comprehension of Top 200 Prescribed Drugs in the US as a Resource for Pharmacy Teaching, Training and Practice. Pharmacy (Basel). 2018;6(2). Epub 2018/05/15. doi: 10.3390/pharmacy6020043 ; PubMed Central PMCID: PMC6025009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cholesterol Treatment Trialists C. Efficacy and safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet. 2019;393(10170):407–15. Epub 2019/02/05. doi: 10.1016/S0140-6736(18)31942-1 ; PubMed Central PMCID: PMC6429627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med. 2015;372(25):2387–97. Epub 2015/06/04. doi: 10.1056/NEJMoa1410489 . [DOI] [PubMed] [Google Scholar]
- 11.Group HTRC, Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N Engl J Med. 2017;377(13):1217–27. Epub 2017/08/30. doi: 10.1056/NEJMoa1706444 . [DOI] [PubMed] [Google Scholar]
- 12.Schmitz J, Gouni-Berthold I. APOC-III Antisense Oligonucleotides: A New Option for the Treatment of Hypertriglyceridemia. Curr Med Chem. 2018;25(13):1567–76. Epub 2017/06/10. doi: 10.2174/0929867324666170609081612 . [DOI] [PubMed] [Google Scholar]
- 13.Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N Engl J Med. 2015;373(5):438–47. Epub 2015/07/30. doi: 10.1056/NEJMoa1400283 . [DOI] [PubMed] [Google Scholar]
- 14.Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. 2017;377(3):211–21. Epub 2017/05/26. doi: 10.1056/NEJMoa1612790 ; PubMed Central PMCID: PMC5800308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laufs U, Parhofer KG, Ginsberg HN, Hegele RA. Clinical review on triglycerides. Eur Heart J. 2020;41(1):99–109c. Epub 2019/11/26. doi: 10.1093/eurheartj/ehz785 ; PubMed Central PMCID: PMC6938588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holmes MV, Ala-Korpela M. What is ’LDL cholesterol’? Nat Rev Cardiol. 2019;16(4):197–8. Epub 2019/02/01. doi: 10.1038/s41569-019-0157-6 . [DOI] [PubMed] [Google Scholar]
- 17.Holmes MV, Richardson TG, Ference BA, Davies NM, Davey Smith G. Integrating genomics with biomarkers and therapeutic targets to invigorate cardiovascular drug development. Nat Rev Cardiol. 2021. Epub 2021/03/13. doi: 10.1038/s41569-020-00493-1 [DOI] [PubMed] [Google Scholar]
- 18.Ference BA, Holmes MV, Davey Smith G. Using Mendelian Randomization to Improve the Design of Randomized Trials. Cold Spring Harb Perspect Med. 2021. Epub 2021/01/13. doi: 10.1101/cshperspect.a040980 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kettunen J, Holmes MV, Allara E, Anufrieva O, Ohukainen P, Oliver-Williams C, et al. Lipoprotein signatures of cholesteryl ester transfer protein and HMG-CoA reductase inhibition. PLoS Biol. 2019;17(12):e3000572. Epub 2019/12/21. doi: 10.1371/journal.pbio.3000572 ; PubMed Central PMCID: PMC6944381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Zhao Q, Bowden J, Hemani G, Davey Smith G, Small DS, et al. Causal inference for heritable phenotypic risk factors using heterogeneous genetic instruments. PLoS Genet. 2021;17(6):e1009575. Epub 2021/06/23. doi: 10.1371/journal.pgen.1009575 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med. 2016;374(12):1123–33. Epub 2016/03/05. doi: 10.1056/NEJMoa1510926 ; PubMed Central PMCID: PMC4900689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906. Epub 2015/12/15. doi: 10.1002/sim.6835 ; PubMed Central PMCID: PMC4832315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65. Epub 2013/10/12. doi: 10.1002/gepi.21758 ; PubMed Central PMCID: PMC4377079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, et al. Variation in PCSK9 and HMGCR and Risk of Cardiovascular Disease and Diabetes. N Engl J Med. 2016;375(22):2144–53. Epub 2016/12/14. doi: 10.1056/NEJMoa1604304 . [DOI] [PubMed] [Google Scholar]
- 25.Ference BA, Kastelein JJP, Ray KK, Ginsberg HN, Chapman MJ, Packard CJ, et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA. 2019;321(4):364–73. Epub 2019/01/30. doi: 10.1001/jama.2018.20045 ; PubMed Central PMCID: PMC6439767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ference BA, Kastelein JJP, Ginsberg HN, Chapman MJ, Nicholls SJ, Ray KK, et al. Association of Genetic Variants Related to CETP Inhibitors and Statins With Lipoprotein Levels and Cardiovascular Risk. JAMA. 2017;318(10):947–56. Epub 2017/08/29. doi: 10.1001/jama.2017.11467 ; PubMed Central PMCID: PMC5710502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wulff AB, Nordestgaard BG, Tybjaerg-Hansen A. APOC3 Loss-of-Function Mutations, Remnant Cholesterol, Low-Density Lipoprotein Cholesterol, and Cardiovascular Risk: Mediation- and Meta-Analyses of 137 895 Individuals. Arterioscler Thromb Vasc Biol. 2018;38(3):660–8. Epub 2018/01/20. doi: 10.1161/ATVBAHA.117.310473 . [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Oliver-Williams C, Raitakari OT, Viikari J, Lehtimaki T, Kahonen M, et al. Metabolic profiling of angiopoietin-like protein 3 and 4 inhibition: a drug-target Mendelian randomization analysis. Eur Heart J. 2021;42(12):1160–9. Epub 2020/12/23. doi: 10.1093/eurheartj/ehaa972 ; PubMed Central PMCID: PMC7982288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell JA, Richardson TG, Wang Q, Sanderson E, Palmer T, Walker V, et al. Dominant role of abdominal adiposity in circulating lipoprotein, lipid, and metabolite levels in UK Biobank: Mendelian randomization study. medRxiv. 2021:2021.05.29.21258044. doi: 10.1101/2021.05.29.21258044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moura JMSP Mendes PFAPP, Reigota CP Gemas VJV. New drugs coming up in the field of lipid control. e-Journal of Cardiology. Practice. 2020;19. [Google Scholar]
- 31.Reeskamp LF, Tromp TR, Stroes ESG. The next generation of triglyceride-lowering drugs: will reducing apolipoprotein C-III or angiopoietin like protein 3 reduce cardiovascular disease? Curr Opin Lipidol. 2020;31(3):140–6. Epub 2020/04/24. doi: 10.1097/MOL.0000000000000679 . [DOI] [PubMed] [Google Scholar]
- 32.Mullard A. FDA approves first anti-ANGPTL3 antibody, for rare cardiovascular indication. Nat Rev Drug Discov. 2021;20(4):251. Epub 2021/03/13. doi: 10.1038/d41573-021-00047-1 . [DOI] [PubMed] [Google Scholar]
- 33.Geldenhuys WJ, Lin L, Darvesh AS, Sadana P. Emerging strategies of targeting lipoprotein lipase for metabolic and cardiovascular diseases. Drug Discov Today. 2017;22(2):352–65. Epub 2016/10/30. doi: 10.1016/j.drudis.2016.10.007 ; PubMed Central PMCID: PMC5482225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wurtz P, Wang Q, Soininen P, Kangas AJ, Fatemifar G, Tynkkynen T, et al. Metabolomic Profiling of Statin Use and Genetic Inhibition of HMG-CoA Reductase. J Am Coll Cardiol. 2016;67(10):1200–10. Epub 2016/03/12. doi: 10.1016/j.jacc.2015.12.060 ; PubMed Central PMCID: PMC4783625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sliz E, Kettunen J, Holmes MV, Williams CO, Boachie C, Wang Q, et al. Metabolomic consequences of genetic inhibition of PCSK9 compared with statin treatment. Circulation. 2018;138(22):2499–512. Epub 2018/12/14. doi: 10.1161/CIRCULATIONAHA.118.034942 ; PubMed Central PMCID: PMC6254781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munafo MR, Higgins JPT, Davey Smith G. Triangulating Evidence through the Inclusion of Genetically Informed Designs. Cold Spring Harb Perspect Med; 2020. Epub 2020/12/24. doi: 10.1101/cshperspect.a040659 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol. 2019;48(3):713–27. Epub 2018/12/12. doi: 10.1093/ije/dyy262 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohammadi-Shemirani P, Sjaarda J, Gerstein HC, Treleaven DJ, Walsh M, Mann JF, et al. A Mendelian Randomization-Based Approach to Identify Early and Sensitive Diagnostic Biomarkers of Disease. Clin Chem. 2019;65(3):427–36. Epub 2018/10/20. doi: 10.1373/clinchem.2018.291104 . [DOI] [PubMed] [Google Scholar]
- 39.Holmes MV, Davey Smith G. Can Mendelian Randomization Shift into Reverse Gear? Clin Chem. 2019;65(3):363–6. Epub 2019/01/30. doi: 10.1373/clinchem.2018.296806 . [DOI] [PubMed] [Google Scholar]
- 40.Blauw LL, Noordam R, Soidinsalo S, Blauw CA, Li-Gao R, de Mutsert R, et al. Mendelian randomization reveals unexpected effects of CETP on the lipoprotein profile. Eur J Hum Genet. 2019;27(3):422–31. Epub 2018/11/14. doi: 10.1038/s41431-018-0301-5 ; PubMed Central PMCID: PMC6460571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richardson TG, Wang Q, Sanderson E, Mahajan A, McCarthy MI, Frayling TM, et al. Effects of apolipoprotein B on lifespan and risks of major diseases including type 2 diabetes: a mendelian randomisation analysis using outcomes in first-degree relatives. Lancet Healthy Longev. 2021;2(6):e317–e26. Epub 2021/06/01. doi: 10.1016/S2666-7568(21)00086-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30. Epub 2015/09/08. doi: 10.1038/ng.3396 ; PubMed Central PMCID: PMC4589895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raal FJ, Rosenson RS, Reeskamp LF, Hovingh GK, Kastelein JJP, Rubba P, et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N Engl J Med. 2020;383(8):711–20. Epub 2020/08/20. doi: 10.1056/NEJMoa2004215 . [DOI] [PubMed] [Google Scholar]
- 44.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. Epub 2015/04/01. doi: 10.1371/journal.pmed.1001779 ; PubMed Central PMCID: PMC4380465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9. Epub 2018/10/12. doi: 10.1038/s41586-018-0579-z . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjalmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284–90. Epub 2015/02/03. doi: 10.1038/ng.3190 ; PubMed Central PMCID: PMC4342297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. Epub 2012/11/07. doi: 10.1038/nature11632 ; PubMed Central PMCID: PMC3498066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson TG, Hemani G, Gaunt TR, Relton CL, Davey Smith G. A transcriptome-wide Mendelian randomization study to uncover tissue-dependent regulatory mechanisms across the human phenome. Nat Commun. 2020;11(1):185. Epub 2020/01/12. doi: 10.1038/s41467-019-13921-9 ; PubMed Central PMCID: PMC6954187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nightingale Health UKBI, Julkunen H, Cichonska A, Slagboom PE, Wurtz P. Metabolic biomarker profiling for identification of susceptibility to severe pneumonia and COVID-19 in the general population. elife. 2021;(10). Epub 2021/05/05. doi: 10.7554/eLife.63033 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–13. Epub 2018/10/10. doi: 10.1038/s41588-018-0241-6 ; PubMed Central PMCID: PMC6287706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffith GJ, Morris TT, Tudball MJ, Herbert A, Mancano G, Pike L, et al. Collider bias undermines our understanding of COVID-19 disease risk and severity. Nat Commun. 2020;11(1):5749. Epub 2020/11/14. doi: 10.1038/s41467-020-19478-2 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ginestet C. ggplot2: Elegant Graphics for Data Analysis. J R Stat Soc Ser A Stat Soc. 2011;174:245. doi: 10.1111/j.1467-985X.2010.00676_9.x WOS:000285969600026. [DOI] [Google Scholar]
- 53.Broadbent JR, Foley CN, Grant AJ, Mason AM, Staley JR, Burgess S. MendelianRandomization v0.5.0: updates to an R package for performing Mendelian randomization analyses using summarized data. Wellcome Open Res. 2020;5:252. Epub 2021/01/02. doi: 10.12688/wellcomeopenres.16374.2 ; PubMed Central PMCID: PMC7745186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PNG)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
(PDF)
Data Availability Statement
Data supporting this paper can be found within the Supporting Information files. Full GWAS summary statistics for all 249 circulating metabolites generated in this work are accessible via the GWAS catalog (https://www.ebi.ac.uk/gwas/) under accession IDs GCST90092803 to GCST90093051.