

Abstract
We report a series of palladium(ii)-catalyzed, intramolecular alkene hydrofunctionalization reactions with carbon, nitrogen, and oxygen nucleophiles to form five- and six-membered carbo- and heterocycles. In these reactions, the presence of a proximal bidentate directing group controls the cyclization pathway, dictating the ring size that is generated, even in cases that are disfavored based on Baldwin's rules and in cases where there is an inherent preference for an alternative pathway. DFT studies shed light on the origins of pathway selectivity in these processes.
We report a series of palladium(ii)-catalyzed, intramolecular alkene hydrofunctionalization reactions with carbon, nitrogen, and oxygen nucleophiles to form five- and six-membered carbo- and heterocycles.
Introduction
Catalytic alkene hydrofunctionalization reactions are of growing importance within the modern synthetic repertoire.1 Among established methods for alkene hydrofunctionalization, palladium(ii) catalysis involving Wacker-type nucleopalladation2 followed by protodepalladation3 of the resulting alkylpalladium(ii) intermediate is an attractive approach because such reactions proceed in a redox-neutral manner and with ideal atom-economy.4–7 Early intermolecular versions of such reactions were restricted to conjugated alkenes, such as enones and acrylates.4 Pioneering work by Widenhoefer and Michael in the 2000s then established that non-conjugated alkenes could engage in palladium(ii)-catalyzed intramolecular hydroalkylation (6-endo-trig)5 and hydroamination (5/6-exo-trig)6 with tethered 1,3-dicarbonyl and amide/carbamate nucleophiles, respectively (Scheme 1A). Building on these important precedents, our lab developed a substrate-directed approach to intermolecular hydrofunctionalization of non-conjugated alkenes with N–H and C–H (pro)nucleophiles that is highly regioselective, even with challenging 1,2-disubstituted alkene substrates (Scheme 1B).7–9
Scheme 1. Background and synopsis of current work.

Results and discussion
Based on these precedents, we questioned whether it would be possible to control the regioselectivity of intramolecular hydrofunctionalization through substrate directivity,10,11 whereby the directing group would override any inherent pathway bias and enhance reactivity with more highly substituted alkenes (Scheme 1C). In this way, we envisioned that this approach could grant access to product structures that are not readily accessible using existing hydrofunctionalization methods, thereby complementing other approaches. At the outset, we anticipated several potential challenges that would need to be overcome to reduce this idea to practice, including the attenuated reactivity of more substituted nucleophiles (e.g., protected alkyl amine compounds), the diminished reactivity of internal alkenes, and the fact that inherent ring-closing tendencies may compete (or even predominate) against the directed pathway. Additionally, our previous attempts to integrate oxygen-based nucleophiles in directed intermolecular nucleopalladation systems had been unsuccessful, presumably due to the endergonic and reversible nature of C–O bond formation.12
To initiate our investigation, we elected to use alkenyl amine 1c as our pilot substrate, which we hypothesized would react via 6-exo-trig cyclization facilitated by the 8-aminoquinoline-derived amide directing group (AQ) (Table 1). Using Pd(OAc)2 as precatalyst and MeCN as solvent, we evaluated a series of carboxylic acid additives (entries 1–6) and found that sterically bulky aliphatic acids led to slightly better yields, with 1-AdaCO2H being the best among those tested (entry 1). Another key finding was that polar aprotic solvents, such as MeCN and PhCN, outperformed nonpolar solvents, and that acidic protic solvents, such as HFIP and TFE, led to diminished yields (entries 7–11). When the reaction temperature was reduced to 80 °C, slightly lower yield was observed, even when the reaction time was extended (entry 12). Raising the temperature to 120 °C led to partial decomposition of the substrate, with a concomitant reduction in yield (78% yield, entry 13). Interestingly, unlike many other intramolecular cyclization reactions, this reaction gives high yields at higher concentration (entries 14 and 15). This suggests that intermolecular oligomerization is not a competitive process. Directing groups that are similar in structure to AQ but less rigid (DG1 and DG2) only gave 44% and 16% yield, respectively (entries 16 and 17). The corresponding free carboxylic acid substrate decomposed under the reaction condition, and in this case, the desired product could not be detected. These results illustrate the importance of the AQ group for promoting the desired reaction pathway.
Optimization of reaction conditions for intramolecular hydroamination of 1ca.
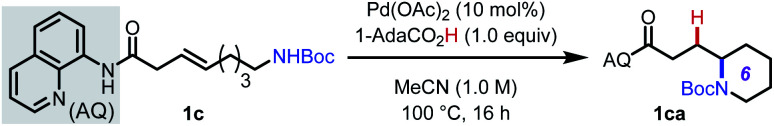
| ||
|---|---|---|
| Entrya | Variation from standard conditions | Yieldb |
| 1 | (None) | 97% (92%) |
| 2 | AcOH in place of 1-AdaCO2H | 89% |
| 3 | PivOH in place of 1-AdaCO2H | 92% |
| 4 | PhCO2H in place of 1-AdaCO2H | 87% |
| 5 | A1 in place of 1-AdaCO2H | 90% |
| 6 | A2 in place of 1-AdaCO2H | 88% |
| 7 | PhMe as solvent | 82% |
| 8 | PhCN as solvent | 88% |
| 9 | t-AmylOH as solvent | 77% |
| 10 | TFE as solvent | 77% |
| 11 | HFIP as solvent | 65% |
| 12c | 80 °C | 91% |
| 13 | 120 °C | 78% |
| 14 | 0.5 M | 96% |
| 15 | 2.0 M | 90% |
| 16d | DG1 in place of AQ | 44% |
| 17d | DG2 in place of AQ | 16% |
| 18d | OH in place of AQ | n.d. |
| ||
1c (0.1 mmol).
1H NMR yield with CH2Br2 as internal standard; isolated yield given in parentheses.
40 h.
1c (0.05 mmol).
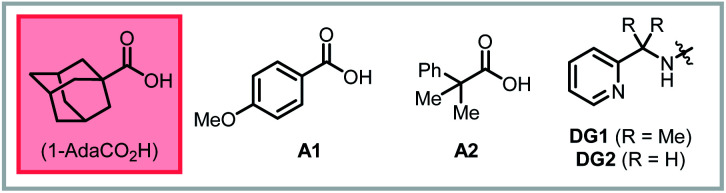
After identifying optimal reaction conditions, we turned our attention to examining the scope and limitations of this protocol. First, ring size effects were tested with substrates bearing different chain length. Based on Baldwin's rules, 4-, 5-, 6- and 7-exo-trig ring closure are all predicted to be allowed.13,14 However, only product 2ba and 2ca, corresponding to the 5-exo-trig and 6-exo-trig pathways, respectively, were formed in high yields. In the other two cases, only unreacted starting material could be recovered. Based on the results above, we next investigated the N-substituent compatibility of the reaction. For the 5-exo-trig cyclization, changing the R group to Cbz, Ms, or Ts gave similar yields of around 90% (2bb–2bd), while a more electron-withdrawing group (Ns) only gave 73% yield (2be). Similar trends were observed with the 6-exo-trig pathway, where the Ns-protected substrate furnished only 32% yield (2ce). Other sulfonamide substituents provided good to excellent yield (2cf–2cj). Gratifyingly, heterocyclic substituents were competent, with 3-pyridyl (2cg) and 2-thiofuranyl sulfonamides (2ch) giving 67% and 88% yields, respectively. We examined the effect of substitution at the α-position and found that 6-exo-trig cyclization with a Boc-protected nitrogen nucleophile proceeded in 88% yield and 1.5 : 1 d.r. (2e).
We next questioned whether directed cyclization was possible with carbon- and oxygen-based nucleophiles. Not only could this expand the utility of this approach, but it would also allow us to test whether substrate directivity could enforce otherwise disfavored ring closures across diverse substrate classes. To this end, we tested a series of substrates bearing potentially reactive nucleophiles at various positions. Indeed, we found that a substrate bearing tethered malonate nucleophile, reacted in a 5-exo-trig mode, rather than the 6-endo-trig mode documented by Widenhoefer,5 to give the product 3a in 79% yield with HFIP as the optimal solvent. This example underscores the complementary nature of directed and non-directed metal-catalyzed cyclizations. Though oxygen-based nucleophiles have not been extensively studied in palladium(ii)-catalyzed hydrofunctionalization reactions with non-conjugated alkenes, we were pleased to find that alkenyl alcohols were competent substrates under these conditions. A tethered phenol nucleophile reacted in a 5-exo-trig mode to give product 3b in 71% yield. Likewise, with a tethered naphthol nucleophile, 6-exo-trig cyclization onto a trisubstituted alkene proceeded in 86% yield (3c). We have previously shown that product 3c can alternatively be prepared by a cascade process from 2-naphthol and the corresponding 1,3-diene, where this trisubstituted alkenyl alcohol is generated in situ (see ESI†).10 Additionally, we found that oxaspiro compounds 3d and 3e could be synthesized in nearly quantitative yields through a mechanism involving a Baldwin's-disallowed 5-endo-trig cyclization.15
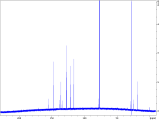
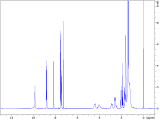
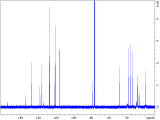
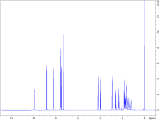
To interrogate the mechanistic underpinnings and origins of regioselectivity in these reactions, we next performed a series of mechanistic experiments. We first prepared π-alkene palladium complex 4 by combining Pd(OAc)2 (1 equiv.) with a representative alkenyl amine derivative and characterized it by NMR and X-ray crystallography. In the X-ray structure, the substrate binds analogously to related examples with alkenyl AQ-amide substrates;7a,7b however in this case the pendant nucleophile occupies the fourth coordination site as an X-type ligand, such that the substrate adopts an overall tetradentate coordination mode. We next probed for the possibility of E/Z isomerization16 during the course of catalysis, which is important in establishing what cyclization pathways are potentially operative in the reaction. To this end, we subjected substrate 1c to the reaction conditions, and halted the reaction prematurely. In this case, we were able to observe a mixture of product 2ca, small amounts of other alkene isomers, and recovered starting material 1c (E/Z ratio = 8 : 1). The presence of Z-isomer, albeit in small amounts, establishes that reaction through either isomer is potentially possible in this system,16b,16c which complicates experimental interrogation of the mechanism.
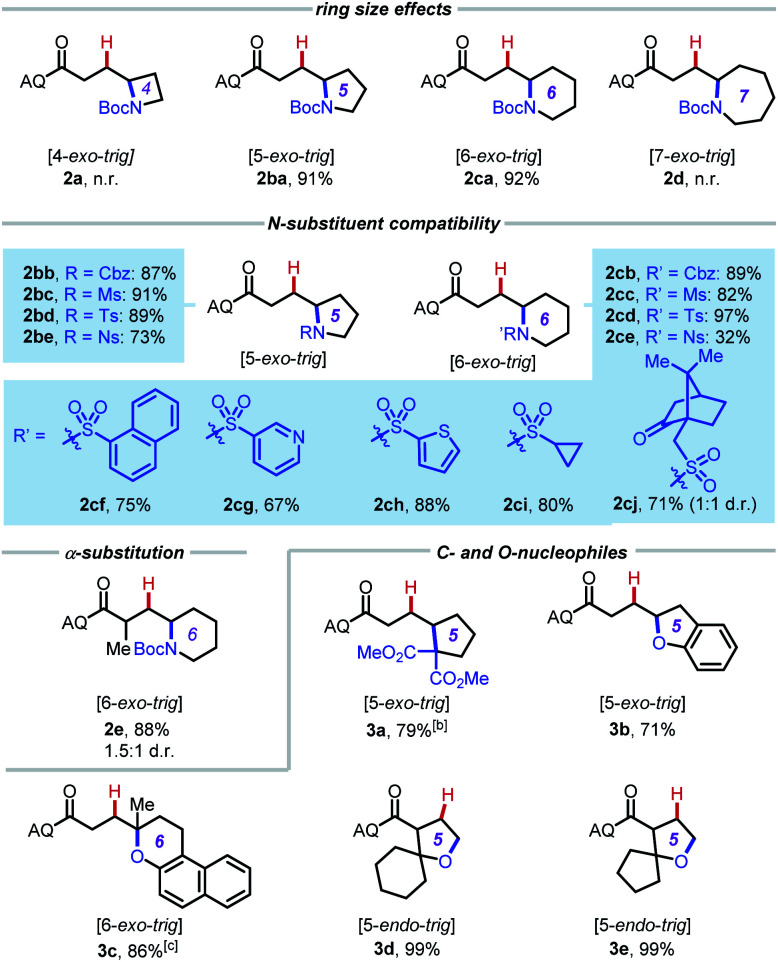
A general mechanism of this palladium-catalyzed intramolecular hydrofunctionalization process is shown in Scheme 3.7 Pd(OAc)2 coordinates to the aminoquinoline directing group and the alkene moiety on the substrate to form a π-alkene complex, which then undergoes nucleopalladation, protodepalladation, and ligand exchange to generate the hydroamination product. Several pertinent mechanistic questions remained outstanding: (1) What is the mechanism of nucleopalladation? (2) How does tether length affect the reactivity? (3) Which factors control the regioselectivity of the hydroamination? To seek a better understanding of these questions, we turned our efforts to density functional theory (DFT) calculations.17
Scheme 3. General depiction of a plausible reaction mechanism.

The computational studies were initiated by elucidating the reaction mechanisms of the intramolecular cyclization of alkenyl amine 1b, which forms the 5-exo-trig product 2ba with high regioselectivity (Table 2).18 Several potential syn- and anti-nucleopalladation processes to access the 5-exo-trig and 6-endo-trig cyclization products were considered. The most favorable pathway, as shown in Fig. 1, involves tetradentate square-planar complex IM2b formed via the acetate-assisted concerted metalation/N–H deprotonation viaTS1b. In IM2b, the pendent nucleophile occupies the coordination site cis to the alkene, a geometry akin to the X-ray crystal structure of 4 (Scheme 2). From IM2b, intramolecular syn-nucleopalladation takes place viaTS2b. This 5-exo-trig cyclization process leads to palladacycle intermediate IM3b, which then undergoes protodepalladation viaTS3b to form product complex IM4b. The β-hydride elimination (TS4b) requires a 2.2 kcal mol−1 higher barrier than that of protodepalladation and forms a thermodynamically less stable palladium–hydride complex IM6b. These results agree with the absence of β-hydride elimination products in the crude reaction mixture. Finally, ligand exchange with another reactant molecule releases the hydroamination product 2ba and regenerates the π-alkene complex IM1b. The computed activation free energies of several less favorable nucleopalladation pathways are summarized in Table 3 (entry 2). Transition state TS5b involves syn-nucleopalladation from the π-alkene complex IM1b. Here, the deprotonation of the Boc-protected amine and the nucleopalladation take place in a concerted fashion. Although TS5b is slightly less stable by 2.1 kcal mol−1, the relatively small energy difference between TS2b and TS5b suggests that both processes may be operative depending on the substrate (vide infra). anti-Nucleopalladation pathways to access the 5-exo-trig and 6-endo-trig cyclization products (TS6b and TS7b) are both much less favorable than TS2b. Furthermore, the syn-nucleopalladation transition state leading to the 6-endo-trig product is highly distorted and is 13.7 kcal mol−1 less favorable than TS2b (see Fig. S8†). Here, syn-nucleopalladation pathways for 5-exo-trig cyclization (TS2b and TS5b) are the most favorable due to either facile deprotonation of the Boc-protected amine to form IM2b or a concerted N–H deprotonation during nucleopalladation. Both scenarios provide stronger nucleophiles relative to the protonated state, which facilitate the ring formation. By contrast, N–H deprotonation in anti-nucleopalladation pathways is disfavored due to the low acidity of the (alkyl)NHBoc group.19 The less nucleophilic protonated state is involved in the anti-nucleopalladation, leading to their higher activation energies. Because the anti-nucleopalladation pathways are less regioselective (i.e. the activation energies of TS6b and TS7b are comparable), the lower activation energy of syn-nucleopalladation in 5-exo-trig cyclization plays an important role in controlling the regioselectivity of the cyclization product.
Electrophile and nucleophile scopea.
|
Reactions performed on 0.1 mmol scale using standard conditions from Table 1 unless otherwise specified. Percentages represent isolated yields.
HFIP was used as solvent.
Reaction was performed on 0.012 mmol scale and at 0.5 M concentration. The reduced scale in this case reflects difficulties in preparing pure starting material (see ESI).
Fig. 1. Computed energy profile of the 5-exo-trig cyclization of 1b (see ESI† for energy profiles of 4-, 6-, and 7-exo-trig cyclizations of 1a, 1c, and 1d).

Scheme 2. Mechanistic experiments.

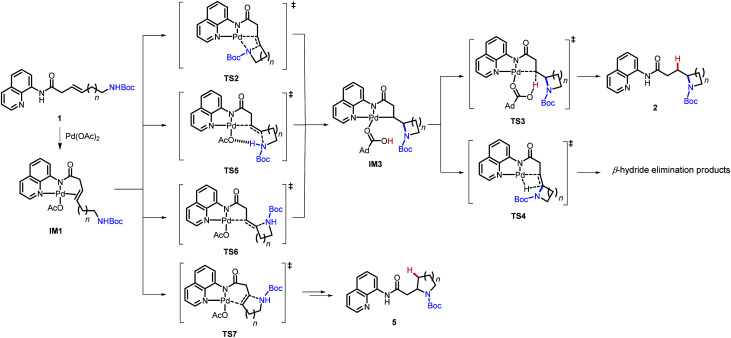
Gibbs free energies of stationary points in the Pd-catalyzed intramolecular hydroamination of 1a–d. All energy values (in kcal mol−1) are relative to IM1. Bold numbers indicate the most favorable mechanisms in the nucleopalladation step.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Alkenyl amine | (n + 3)-Exo-trig | (n + 4)-Endo-trig | (n + 3)-Exo-trig | ||||
| syn-Nucleo-palladation TS | anti-Nucleo-palladation TS | anti-Nucleo-palladation TS | Pallada-cycle | Protode-palladation TS | β-Hydride elimination TS | |||
| TS2 | TS5 | TS6 | TS7 | IM3 | TS3 | TS4 | ||
| 1 | 1a (n = 1) | 31.0 | 29.3 | Cannot locate | 28.0 | 21.8a | 39.6a | 37.6 |
| 2 | 1b (n = 2) | 19.7 | 21.8 | 24.0 | 24.6 | 5.8 | 22.0 | 24.2 |
| 3 | 1c (n = 3) | 23.3 | 21.7 | 24.9 | 32.2 | 5.7 | 22.7 | 23.9 |
| 4 | 1d (n = 4) | 27.1 | 29.1 | 31.3 | Cannot locate | 8.1 | 24.5 | 26.9 |
In the reaction with 1a, the 5-endo-trig regioisomeric pathway is more favorable. The Gibbs free energies of the palladacycle and the protodepalladation TS in the 5-endo-trig pathway with 1a are 9.2 and 29.4 kcal mol−1, respectively.
We then evaluated the effects of ring size on the mechanisms of nucleopalladation. Computed activation barriers of (n + 3)-exo-trig and (n + 4)-endo-trig pathways (n = 1–4) of alkenyl amines 1a–d are summarized in Table 3. The most favorable cyclization mechanism depends upon the ring-size. Computationally, reaction with 1a favors the 5-endo-trig cyclization due to the high ring-strain of the four-membered azetidine ring20 in the 4-exo-trig pathway (entry 1), but experimentally neither putative products are observed. In reactions with 1b–d, the syn-nucleopalladation leading to the 5-, 6-, and 7-exo-trig products are always favored, with either the N–H-deprotonated amide or the neutral Boc-protected amine as the active nucleophile (TS2 and TS5, respectively). Although both 6- and 7-endo-trig ring closures are allowed based on Baldwin's rules, these pathways with 1b and 1c require 4.9 and 10.5 kcal mol−1 higher barriers than corresponding 5- and 6-exo-trig pathways to form 2ba and 2ca.
Ring size also plays a significant role in the activation barriers of the nucleopalladation. syn-Nucleopalladation with 1a and 1dvia 4- and 7-exo-trig cyclization require much higher barriers (29.3 and 27.1 kcal mol−1, respectively) than the 5- and 6-exo-trig cyclizations with 1b and 1c (19.7 and 21.7 kcal mol−1, respectively). This trend agrees with the fact that four- and seven-membered ring products were not observed in experiments. It should be noted that the reactivity trend in cyclization does not completely follow the trend of the strain energies20 of the heterocycle products (Fig. 2). For example, although the strain energies of pyrrolidine (n = 2) and azepane (n = 4) are comparable, the cyclization to form the latter requires a substantially higher activation energy. In addition, formation of the less-strained piperidine (n = 3) requires a higher energy barrier than that of pyrrolidine (n = 2). Our results suggest that besides the ring strain of the cyclization product, steric effects also contribute to the activation energies of syn-nucleopalladation. Greater steric repulsions between the nitrogen nucleophile and the AQ directing group are observed in the syn-nucleopalladation transition states to form larger rings (e.g. seven-membered. See TS2d in Fig. 3).
Fig. 2. Activation free energies of syn-nucleopalladation transition states relative to IM1 and strain energies of the N-heterocycles.20b.

Fig. 3. Greater steric repulsions in the syn-nucleopalladation transition state TS2d to form a seven-membered ring.

Next, we evaluated the barriers of protodepalladation to investigate whether nucleopalladation or protodepalladation is rate- and selectivity-determining. The stabilities of palladacycle IM3 and the subsequent protodepalladation transition state (TS3) both have good correlation with the ring strain energy of the formed N-heterocycle (see ESI† for details).20b This indicates steric effects of the N-heterocycle play a less important role in the protodepalladation step. In reactions with 1a–1c, the protodepalladation requires a higher barrier than the nucleopalladation, and is thus rate-determining.21 While with 1d, nucleopalladation was calculated to be the highest-energy step due to the unfavorable steric repulsions in TS2d.
Furthermore, we computed the reaction energy profile of the Baldwin-disallowed 5-endo-trig cyclization giving oxaspiro compound 3d (see Fig. S6 in the ESI†). The cyclization occurs via an anti-nucleopalladation TS that only requires 26.0 kcal mol−1 relative to the π-alkene complex. The syn-nucleopalladation TS with this substrate is less favorable due to steric repulsions between the spiro center and the Pd. The relatively low barrier in the anti-nucleopalladation is in accordance with the effective formation of 5-endo-trig cyclization products experimentally. One likely contributor for preference for this mode of closure, in this case, is the fact that 4-exo-trig cyclization is disfavored because of the high ring-strain of the four-membered oxetane ring.20
Conclusions
In conclusion, we developed a palladium catalyzed intramolecular hydrofunctionalization system with 8-aminoquinoline as directing group. This method can tolerate a variety of nucleophiles including nitrogen-, oxygen- and carbon-based nucleophiles. Moreover, a Baldwin's rule disallowed 5-endo-trig cyclization was achieved for the preparation of a unique spirocycle. A detailed mechanistic study was performed. For the 5-exo-trig and 6-exo-trig cyclization, the reaction proceeds through a syn-addition, protodepalladation pathway. For the Baldwin's-disallowed 5-endo-trig cyclization, on the other hand, the computational results favor an anti-nucleopalladation pathway. Further studies on achieving the asymmetric version of this transformation are in progress in our lab.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was financially supported by the NIH (R35GM125052-03, R35GM128779). We further acknowledge the China Scholarship Council for visiting student scholarship (X. W.), the NSF for a Graduate Research Fellowship (DGE-1346837, J. A. G.), the Life Science Summer Institute for an internship (J. E. X.), and Bristol-Myers Squibb for a Graduate Fellowship (Z. L.). We thank Prof. Arnold L. Rheingold (UCSD) for X-ray crystallographic analysis. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation grant number ACI-1548562. Zibo Bai and Prof. Gong Chen (Nankai University) are acknowledged for helpful discussion.
Electronic supplementary information (ESI) available. CCDC 1995204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03409f
Notes and references
- For representative reviews, see: ; (a) Foley N. A. Lee J. P. Ke Z. Gunnoe T. B. Cundari T. R. Acc. Chem. Res. 2009;42:585–597. doi: 10.1021/ar800183j. [DOI] [PubMed] [Google Scholar]; (b) Huang L. Arndt M. Gooßen K. Heydt H. Gooßen L. J. Chem. Rev. 2015;115:2596–2697. doi: 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]; (c) Hesp K. D. Stradiotto M. ChemCatChem. 2010;2:1192–1207. doi: 10.1002/cctc.201000102. [DOI] [Google Scholar]; (d) Yamamoto Y. Radhakrishnan U. Chem. Soc. Rev. 1999;28:199–207. doi: 10.1039/A806581K. [DOI] [Google Scholar]
- For reviews, see: ; (a) Jensen K. H. Sigman M. S. Org. Biomol. Chem. 2008;6:4083–4088. doi: 10.1039/B813246A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McDonald R. I. Liu G. Stahl S. S. Chem. Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kočovský P. Bäckvall J.-E. Chem.–Eur. J. 2015;21:36–56. doi: 10.1002/chem.201404070. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yin G. Mu X. Liu G. Acc. Chem. Res. 2016;49:2413–2423. doi: 10.1021/acs.accounts.6b00328. [DOI] [PubMed] [Google Scholar]
- O'Duill M. L. Engle K. M. Synthesis. 2018;50:4699–4714. doi: 10.1055/s-0037-1611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Stark M. A. Richards C. J. Tetrahedron Lett. 1997;38:5881–5884. doi: 10.1016/S0040-4039(97)01309-9. [DOI] [Google Scholar]; (b) Stark M. A. Jones G. Richards C. J. Organometallics. 2000;19:1282–1291. doi: 10.1021/om990710h. [DOI] [Google Scholar]; (c) Gaunt M. J. Spencer J. B. Org. Lett. 2001;3:25–28. doi: 10.1021/ol0066882. [DOI] [PubMed] [Google Scholar]; (d) Takenaka K. Uozumi Y. Org. Lett. 2004;6:1833–1835. doi: 10.1021/ol0494515. [DOI] [PubMed] [Google Scholar]; . For related processes involving Heck-type migratory insertion, see: ; (e) Yamamura K. J. Org. Chem. 1978;43:724–727. doi: 10.1021/jo00398a044. [DOI] [Google Scholar]; (f) Cacchi S. La Torre F. Misiti D. Tetrahedron Lett. 1979;20:4591–4594. doi: 10.1016/S0040-4039(01)86658-2. [DOI] [Google Scholar]; (g) Lu X. Lin S. J. Org. Chem. 2005;70:9651–9653. doi: 10.1021/jo051561h. [DOI] [PubMed] [Google Scholar]
- (a) Pei T. Widenhoefer R. A. J. Am. Chem. Soc. 2001;123:11290–11291. doi: 10.1021/ja011764x. [DOI] [PubMed] [Google Scholar]; (b) Qian H. Widenhoefer R. A. J. Am. Chem. Soc. 2003;125:2056–2057. doi: 10.1021/ja0293002. [DOI] [PubMed] [Google Scholar]
- (a) Michael F. E. Cochran B. M. J. Am. Chem. Soc. 2006;128:4246–4247. doi: 10.1021/ja060126h. [DOI] [PubMed] [Google Scholar]; (b) Cochran B. M. Michael F. E. J. Am. Chem. Soc. 2008;130:2786–2792. doi: 10.1021/ja0734997. [DOI] [PubMed] [Google Scholar]
- (a) Gurak Jr J. A. Yang K. S. Liu Z. Engle K. M. J. Am. Chem. Soc. 2016;138:5805–5808. doi: 10.1021/jacs.6b02718. [DOI] [PubMed] [Google Scholar]; (b) Yang K. S. Gurak Jr J. A. Liu Z. Engle K. M. J. Am. Chem. Soc. 2016;138:14705–14712. doi: 10.1021/jacs.6b08850. [DOI] [PubMed] [Google Scholar]; (c) Gurak Jr J. A. Tran V. T. Sroda M. M. Engle K. M. Tetrahedron. 2017;73:3636–3642. doi: 10.1016/j.tet.2017.03.091. [DOI] [Google Scholar]
- We and others have shown that such processes can be rendered enantioselective in the presence of an appropriate chiral promoter: ; (a) Wang H. Bai Z. Jiao T. Deng Z. Tong H. He G. Peng Q. Chen G. J. Am. Chem. Soc. 2018;140:3542–3546. doi: 10.1021/jacs.8b00641. [DOI] [PubMed] [Google Scholar]; (b) Shen H.-C. Zhang L. Chen S.-S. Feng J. Zhang B.-W. Zhang Y. Zhang X. Wu Y.-D. Gong L.-Z. ACS Catal. 2019;9:791–797. doi: 10.1021/acscatal.8b04654. [DOI] [Google Scholar]; (c) Nimmagadda S. K. Liu M. Karunananda M. K. Gao D.-W. Apolinar O. Chen J. S. Liu P. Engle K. M. Angew. Chem., Int. Ed. 2019;58:3923–3927. doi: 10.1002/anie.201814272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wei C. Ye X. Xing Q. Hu Y. Xie Y. Shi X. Org. Biomol. Chem. 2019;17:6607–6611. doi: 10.1039/C9OB01165J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related reactions involving Heck-type organopalladium migratory insertion followed by protodepalladation, see: ; (a) Matsuura R. Jankins T. C. Hill D. E. Yang K. S. Gallego G. M. Yang S. He M. Wang F. Marsters R. P. McAlpine I. Engle K. M. Chem. Sci. 2018;9:8363–8368. doi: 10.1039/C8SC03081B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bai Z. Bai Z. Song F. Wang H. Chen G. He G. ACS Catal. 2020;10:933–940. doi: 10.1021/acscatal.9b04285. [DOI] [Google Scholar]
- In ref. 7b our lab described a cascade reaction of 1,3-dienes in which intramolecular alkene hydrofunctionalization was proposed as the second step. Prior to the present study, this cyclization process had not been studied systematically in isolation
- For a related 1,1-heterodifunctionalization process involving intramolecular trapping, see: ; Jeon J. Ryu H. Lee C. Cho D. Baik M.-H. Hong S. J. Am. Chem. Soc. 2019;141:10048–10059. doi: 10.1021/jacs.9b04142. [DOI] [PubMed] [Google Scholar]
- (a) Tran V. T. Gurak Jr J. A. Yang K. S. Engle K. M. Nat. Chem. 2018;10:1126–1133. doi: 10.1038/s41557-018-0110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ni H.-Q. Kevlishvili I. Bedekar P. G. Barber J. S. Yang S. Tran-Dubé M. Lu H.-X. McAlpine I. Liu P. Engle K. M. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12510038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J. E. J. Chem. Soc., Chem. Commun. 1976:734–736. doi: 10.1039/C39760000734. [DOI] [Google Scholar]
- It should be noted that transition-metal-mediated cyclizations are mechanistically distinct processes from those included in Baldwin's original empirical models and thus exhibit distinct reactivity trends in some cases. For a computational analysis of intramolecular migratory insertion pathways, see: ; Fiser B. Cuerva J. M. Gómez-Bengoa E. Organometallics. 2018;37:390–395. doi: 10.1021/acs.organomet.7b00812. [DOI] [Google Scholar]
- While this work was in progress, this O-cyclization method was used to prepare one starting material in ref. 12a
- (a) Liu Z. Zeng T. Yang K. S. Engle K. M. J. Am. Chem. Soc. 2016;138:15122–15125. doi: 10.1021/jacs.6b09170. [DOI] [PubMed] [Google Scholar]; (b) Liu Z. Li X. Zeng T. Engle K. M. ACS Catal. 2019;9:3260–3265. doi: 10.1021/acscatal.9b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bai Z. Zheng S. Bai Z. Song F. Wang H. Peng Q. Chen G. He G. ACS Catal. 2019;9:6502–6509. doi: 10.1021/acscatal.9b01350. [DOI] [Google Scholar]
- DFT calculations were performed at the M06/6-311+G(d,p)-SDD/SMD(MeCN)//M06/6-31G(d)-SDD level of theory. See the ESI† for computational details
- Because E/Z isomerization of the alkene was observed experimentally, we also calculated the cyclization of the Z-isomers of 1a–d. The calculations indicate that the reactions with the Z-isomers are less favorable (see ESI†)
- In the reaction with 1c, the anti-nucleopalladation transition state with the deprotonated N-nucleophile is 2.9 kcal mol−1 higher in energy than TS6c. This transition state is disfavored due to the high energy required (21.1 kcal mol−1) to deprotonate the (alkyl)NHBoc with OAc− as the base. It should be noted that reactions with a more acidic nucleophile (e.g., succinimide) may occur with the nucleophile in its deprotonated state and that this may be a contributing factor for the experimentally observed preference for anti-nucleopalladation in previously reported intermolecular systems (see ref. 7)
- (a) Benson S. W. Cruickshank F. R. Golden D. M. Haugen G. R. O'Neal H. E. Rodgers A. S. Shaw R. Walsh R. Chem. Rev. 1969;69:279–324. doi: 10.1021/cr60259a002. [DOI] [Google Scholar]; (b) Dudev T. Lim C. J. Am. Chem. Soc. 1998;120:4450–4458. doi: 10.1021/ja973895x. [DOI] [Google Scholar]
- Although protodepalladation is turnover-limiting in reactions with 1b and 1c, the regioselectivity in these reactions is still controlled in the nucleopalladation step, because the 6- and 7-endo-trig nucleopalladation transition states (TS7b and TS7c) have higher barriers than the protodepalladation transition states in the 5- and 6-exo-trig pathways (TS3b and TS3c)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.