

Abstract
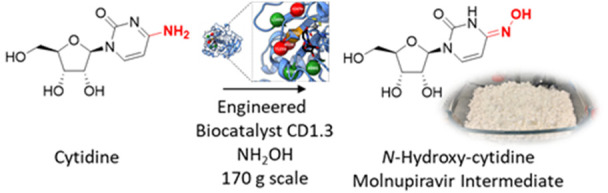
The Covid-19 pandemic highlights the urgent need for cost-effective processes to rapidly manufacture antiviral drugs at scale. Here we report a concise biocatalytic process for Molnupiravir, a nucleoside analogue recently approved as an orally available treatment for SARS-CoV-2. Key to the success of this process was the development of an efficient biocatalyst for the production of N-hydroxy-cytidine through evolutionary adaption of the hydrolytic enzyme cytidine deaminase. This engineered biocatalyst performs >85 000 turnovers in less than 3 h, operates at 180 g/L substrate loading, and benefits from in situ crystallization of the N-hydroxy-cytidine product (85% yield), which can be converted to Molnupiravir by a selective 5′-acylation using Novozym 435.
Molnupiravir 1 is a nucleoside analogue that was recently approved as an antiviral therapy to treat adult patients with mild to moderate Covid-19 who are at risk of progressing to severity or hospitalization.1 The structural simplicity of Molnupiravir compared to alternative therapies (e.g., Remdesivir), combined with its oral availability and broad-spectrum antiviral activities, makes it an attractive candidate for the global treatment of viral infections. In view of the urgent need to develop therapies for treating the Covid-19 pandemic, there has been considerable interest in developing synthetic routes to this active pharmaceutical ingredient (API). The original synthesis required 10 steps and proceeded with <10% overall yield.2 More streamlined approaches have subsequently been reported; however, these routes rely on chemical methods to install the N-hydroxy unit, which require elevated temperatures, prolonged reaction times, and/or additional steps for activation and protection/deprotection chemistry.3−7 These factors compromise reaction yields and process productivity, ultimately leading to increased costs.
We sought to develop a more efficient and sustainable route to 1 that could be implemented on scale, where modification of the base to install the N-hydroxy unit is achieved catalytically using an engineered enzyme. Since N-hydroxy-cytidine 4 has previously been shown to undergo lipase-catalyzed acylation at C-5-OH,3,4 this approach would provide an integrated biocatalytic process to synthesize Molnupiravir 1 from readily available starting materials. Our desired route requires an engineered cytidine deaminase (CD) capable of selectively installing the N-hydroxy unit from NH2OH in a bulk water phase (Figure 1). To meet the demands of a robust industrial biocatalyst, we set ourselves the target of developing an engineered enzyme capable of operating at [cytidine] > 100 g/L and achieving >90% conversion within 12 h using low catalyst loadings to drive down the enzyme cost contribution to the overall process. At the outset of the project there was no known enzyme for this transformation. However, we were intrigued by a report which demonstrated that N-hydroxy-cytidine 4 could be hydrolyzed to uridine 3 by wild-type CD, albeit at a very low rate (ca. 2% of activity compared to cytidine 2).8 Cytidine deaminase (CD; EC 3.5.4.5) is a zinc-containing hydrolytic enzyme which catalyzes the conversion of cytidine 2 to uridine 3.9−13 The enzyme is widely distributed among organisms and is involved in the salvage of cytidine for uridine monophosphate synthesis. Interestingly wild-type CD from Escherichia coli (E. coli) has been used on scale for hydrolysis of the nucleoside analogue 2′-deoxy-3′-thiacytidine, and hence it appeared to be an attractive choice as a potential industrial biocatalyst.14 On the basis of these observations, we initiated experiments aimed at converting cytidine 2 to N-hydroxy-cytidine 4 catalyzed by CD.
Figure 1.

Proposed biocatalytic route to Molnupiravir. (a) Wild-type cytidine deaminase (CD) catalyzes the hydrolysis of 2 to 3. (b) The active site of CD with uridine bound (PDB code: 1AF2). The Zn2+ ion is shown in gray. His102, Cys129, Cys132, and catalytic Glu104 are shown as atom-colored sticks with blue carbons. Uridine ligand is shown as atom-colored sticks with black carbons. (c) Proposed synthetic route to Molnupiravir 1. 2 is converted to 4 by an engineered CD followed by acylation using Novozym 435.3,4
CD from E. coli was expressed in BL21(DE3) cells and purified to homogeneity via nickel affinity chromatography. To investigate whether this enzyme could serve as a biocatalyst for the preparation of N-hydroxy-cytidine 4, we initially established direct spectrophotometric assays to monitor the interconversion of cytidine 2, uridine 3, and N-hydroxy-cytidine 4, based on diagnostic differences between the UV–vis spectra of these species. Such assays are valuable to allow high-throughput and real-time analysis of biotransformations. In particular, the N-hydroxy-cytidine 4 spectrum shows a diagnostic feature at >310 nm that is not present in either cytidine 2 or uridine 3, thus allowing N-hydroxy-cytidine 4 formation/decay to be easily monitored.
We initially employed this assay to investigate the conversion of N-hydroxy-cytidine 4 to uridine 3. Consistent with previous reports, CD promotes hydrolysis of N-hydroxy-cytidine 4 under ambient conditions, with complete conversion of 100 μM substrate achieved in 60 min (0.1% enzyme loading, Figure S2). Encouraged by these results, we next turned our attention to the more challenging synthesis of N-hydroxy-cytidine 4 using either uridine 3 or cytidine 2 as a starting material. Pleasingly, biotransformations carried out in the presence of 1% NH2OH (∼300 mM) in water led to the accumulation of a product with spectral features consistent with N-hydroxy-cytidine 4. Similar final concentrations of 4 are formed using either 2 or 3 as a starting material, or in reactions starting from 4, suggesting that the enzyme establishes an equilibrium distribution of products (Figure 2b). As anticipated, the ratio of 4:3 formed is dependent on NH2OH concentration, and 1:6 and 3:1 mixtures were achieved using 10% (∼3 M) and 50% (∼15 M) NH2OH solutions, respectively (Figures 2c and S3). Analysis of biotransformations over time reveals that cytidine hydrolysis followed by uridine amination (pathway B) outcompetes direct amine transfer (pathway A) with the wild-type enzyme (Figure 2a), irrespective of the NH2OH concentration used.
Figure 2.
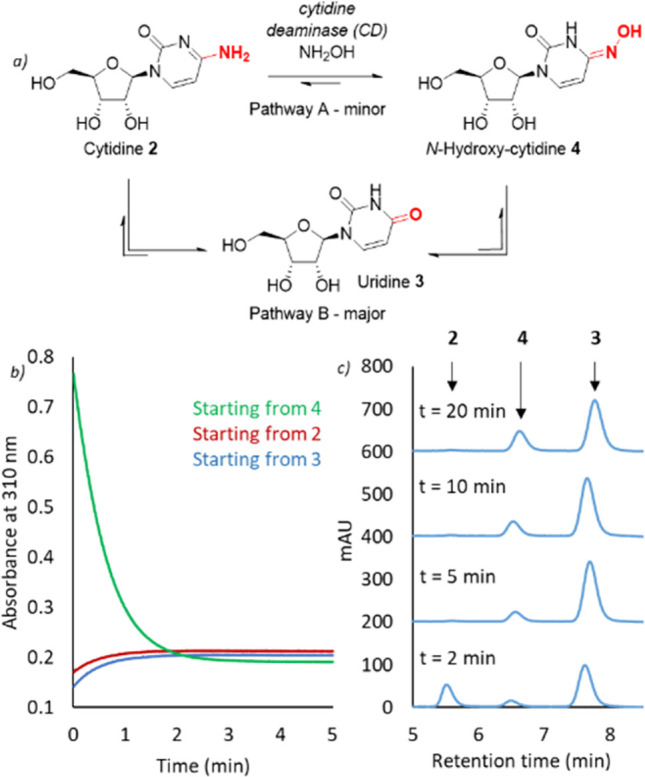
Characterization of wild-type cytidine deaminase (CD). (a) Pathways for conversion of 2 to 4 catalyzed by CD. Pathway A involves direct conversion of 2 to 4 using NH2OH as a nucleophile. Pathway B involves initial hydrolysis of 2 to uridine 3, which is then transformed to 4 through condensation with NH2OH. Pathway B is the dominant pathway when using the wild-type enzyme, leading to an equilibrium distribution of 3 and 4. (b) The conversion of 2 (1 mM) and 3 (1 mM) to 4 by CD (5 μM) in the presence of 1% NH2OH (∼300 mM, pH 7) is monitored by increasing absorbance at 310 nm. Similar final concentrations of 4 are formed using either 2 (red) or 3 (blue) as a starting material, or in reactions starting from 4 (green). (c) The conversion of 2 (750 mM) by CD (25 μM) to 3 and 4 is monitored by HPLC analysis in the presence of 10% NH2OH (∼3 M, pH 7). The time course of the reaction indicates CD operates via pathway B.
These results highlight biotransformations with CD as a promising strategy for the production of N-hydroxy-cytidine 4. Unfortunately, under thermodynamic control significant quantities of uridine byproduct are formed, even at elevated NH2OH concentrations. Furthermore, the use of such high NH2OH concentrations is undesirable for large-scale applications and leads to the formation of impurities over extended reaction times. To address these shortcomings, we elected to engineer CD via directed evolution to optimize pathway A while minimizing pathway B. Similar strategies have been employed to control the partitioning of transglycosylation/hydrolysis by glycoside hydrolases.15,16 Accelerating the direct amination of cytidine in this way should allow accumulation of 4 under kinetic control at reduced NH2OH concentrations.
To this end, iterative rounds of site saturation mutagenesis were performed using NNK degenerate codons, targeting residues within close proximity to the Zn2+ cofactor and the substrate binding pocket. Beneficial mutations identified during rounds of evolution were combined by overlap extension PCR (Table S1). The aforementioned spectrophotometric assay was used to evaluate individual variants as crude cell lysates arrayed in 96-well plates, using 50 mM 2 as the substrate and 1% NH2OH (∼300 mM, pH 7) as the reaction medium. Throughout evolution, we identified improved variants with kinetic profiles consistent with rapid initial accumulation of 4 beyond the equilibrium position, followed by slower redistribution of products to equilibrium (Figure 3a–c). The apparent lag phase observed with the evolved variant between 0 and 4 min can be attributed to pH changes in biotransformations carried out at low (1%, ∼300 mM) NH2OH concentrations (note that the extinction coefficient of N-hydroxy-cytidine at 340 nm is sensitive to changes in pH). At 10% (∼3 M) NH2OH, the pH is essentially unchanged throughout and no “lag phase” is observed under these conditions (Figure S4).
Figure 3.
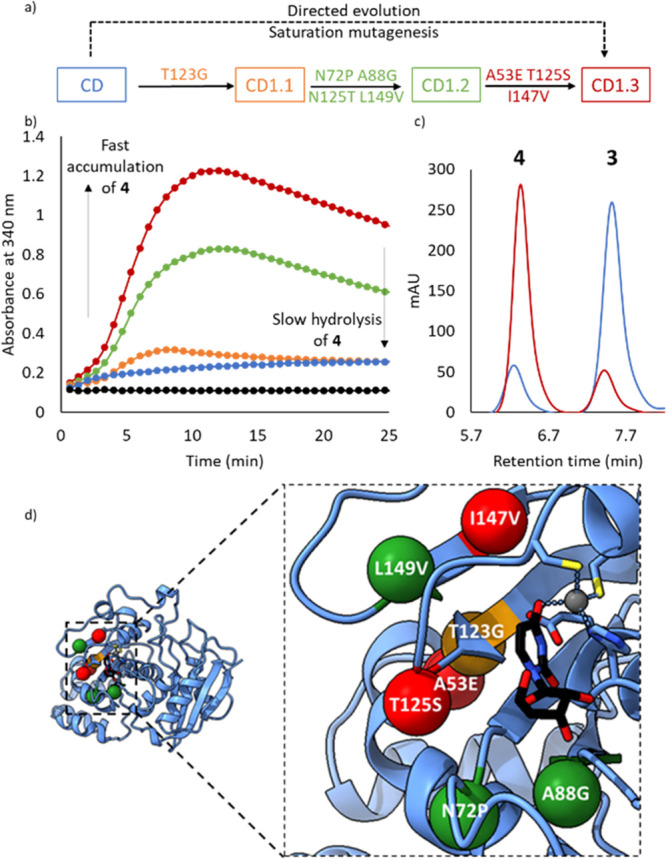
Directed evolution of a cytidine deaminase (CD). (a) Directed evolution of CD to CD1.3 showing mutations installed during each round. (b) Time course for the formation of 4 from 2 (50 mM) catalyzed by CD1.3 (2.5 μM, red), CD1.2 (2.5 μM, green), CD1.1 (2.5 μM, orange), wild-type CD (2.5 μM, blue), and no enzyme (black) in the presence of 1% (∼300 mM) NH2OH, pH 7. (c) HPLC traces showing 2 (500 mM) conversion to 4 and 3 catalyzed by CD1.3 (25 μM, red) and wild-type CD (25 μM, blue) in the presence of 10% (∼3 M) NH2OH, pH 7. (d) The active site of CD with uridine bound (PDB code: 1AF2). Mutations installed in rounds 1, 2, and 3 are shown as orange, green, and red spheres, respectively. The uridine ligand is shown as atom-colored sticks with black carbons, and the Zn2+ cofactor is shown in gray. His102, Cys129, Cys132, and catalytic Glu104 are shown as atom-colored sticks with blue carbons.
The most promising variant to emerge following three rounds of evolution (CD1.3) contains seven mutations clustered around the active site (Figure 3d). As intended, CD1.3 operates efficiently, promoting the conversion of 2 to 4 even at low NH2OH concentrations and high substrate loadings. For example, at 500 mM 2 and 10% NH2OH (∼3 M, pH 7), CD1.3 produces an 8:1 ratio of 4:3. In contrast, the wild-type enzyme produces a 1:6 ratio of 4:3 under identical conditions at equilibrium. The improved performance of CD1.3 can, in part, be attributed to substantially reduced rates of cytidine and N-hydroxy-cytidine hydrolysis, ∼4.5-fold and ∼2.5-fold, respectively, when compared with the wild-type enzyme CD (Figure S5). Significantly, CD1.3 is also able to promote hydroxyaminolysis of cytidine analogues 5-fluorocytidine and 2′-deoxycytidine and can promote the formation of N-methyl-cytidine in biotransformations using methylamine as a nucleophile (Figure S6).
With a promising biocatalyst in hand, we turned our attention to reaction intensification and product isolation. We observed that N-hydroxy-cytidine is substantially less soluble than either cytidine or uridine in aqueous solutions, presenting an opportunity for dynamic in situ crystallization of 4 to allow facile product isolation and further favor the distribution of 4:3. Upon increasing the substrate loading (750 mM) and reducing the reaction temperature (4 °C), in situ crystallization of 4 was observed. Following optimization of reaction conditions, 4 was isolated in 85% yield (4.9 g from 5 g of cytidine) and >98% purity in only 3 h using 0.001 mol% of purified CD1.3 (Figure 4). To circumvent costs associated with enzyme purification, a biotransformation was subsequently performed on a 200 mL scale (38 g of cytidine) using lyophilized cell-free extract, affording N-hydroxy-cytidine in 89% isolated yield and >96% purity. Preliminary efforts toward scale-up of this process by our external collaborators, Sterling Pharma Solutions, generated 137 g of N-hydroxy-cytidine 4 in 71% isolated yield and >95% purity from a 900 mL (169 g of cytidine) reaction, further highlighting the potential of this approach for large scale N-hydroxy-cytidine production (Figure S8). We anticipate that careful optimization of process parameters will lead to further improvements in product yields and purity.
Figure 4.
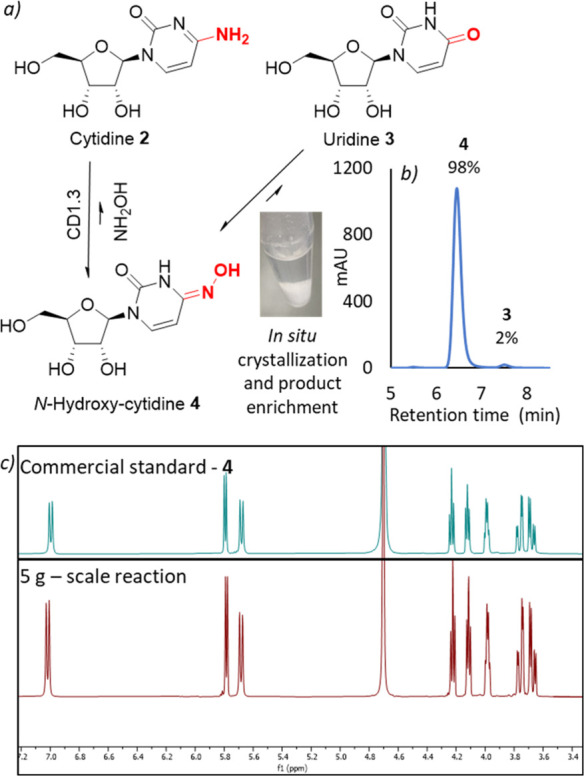
Biocatalytic process for N-hydroxy-cytidine synthesis. (a) In situ crystallization of 4 in reactions catalyzed by CD1.3 leads to product enrichment. Reaction conditions: 750 mM 2, 7.5 μM CD1.3, 10% NH2OH (∼3 M, pH 7), 4 °C. (b) HPLC trace of the product isolated from the biotransformation described in (a). (c) Stacked 1H NMR traces of 4, commercial standard (top), and the product isolated from the biotransformation described in (a) (bottom).
In summary, we have developed an efficient biocatalytic route to N-hydroxy-cytidine 4, a key intermediate for the production of Molnupiravir 1. The process takes advantage of an engineered cytidine deaminase and benefits from dynamic product crystallization to provide a scalable and sustainable manufacturing route to an important molecule in the fight against Covid-19. Through rounds of directed evolution, we were able to achieve the target metrics initially set, namely to operate at [cytidine] > 100 g/L with 90% conversion in 24 h. Remarkably, this engineered biocatalyst is able to achieve TON > 85 000, which places it within the sphere of well-established industrial biocatalysts of proven utility.17−20 Given that selective 5′-acylation of N-hydroxy-cytidine can be achieved with lipases,3,4 this work establishes an integrated biocatalytic strategy for Molnupiravir synthesis using cytidine as an inexpensive and readily available starting material.
Acknowledgments
We would like to thank the Bill & Melinda Gates Foundation for funding and are particularly grateful to Dr. John Dillon, Dr. Trevor Laird, and Dr. Silpa Sundaram for their helpful comments and advice during this project. We also acknowledge Dr. Aisling Ní Cheallaigh of the Future Biomanufacturing Research Hub for helpful discussions on product isolation. We are grateful to Manchester SYNBIOCHEM Centre (BB/M017702/1) and the Henry Royce Institute for Advanced Materials (funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1) for access to their facilities. We acknowledge the Biotechnology and Biological Sciences Research Council (David Phillips Fellowship BB/M027023/1 to A.P.G.), the European Research Council (ERC Starter Grant, no. 757991 to A.P.G. and ERC Advanced Grant no. 742987 to N.J.T) and the UK Research and Innovation Council (Future Leader Fellowship MR/T041722/1 to S.L.L.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c11048.
Experimental details, supplementary Figures S1–S8 and Tables S1 and S2, and DNA and protein sequences (PDF). Source data are available from the corresponding author upon reasonable request.
Author Contributions
‡ W.R.B., Y.Z., and T.W.T. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Cox R. M.; Wolf J. D.; Plemper R. K. Therapeutically administered ribonucleoside analogue MK-4482/EIDD-2801 blocks SARS-CoV-2 transmission in ferrets. Nat. Microbiol. 2021, 6, 11–18. 10.1038/s41564-020-00835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter G. R.; Bluemling G. R.; Natchus M. G.; Guthrie D.. N4-hydroxycytidine and derivatives and anti-viral uses related thereto. WO2019113462, 2018.
- Vasudevan N.; Ahlqvist G. P.; McGeough C. P.; Paymode D. J.; Cardoso F. S. P.; Lucas T.; Dietz J.-P.; Opatz T.; Jamison T. F.; Gupton F. B.; Snead D. R. A concise route to MK-4482 (EIDD-2801) from cytidine. Chem. Commun. 2020, 56, 13363–13364. 10.1039/D0CC05944G. [DOI] [PubMed] [Google Scholar]
- Ahlqvist G. P.; McGeough C. P.; Senanayake C.; Armstrong J. D.; Yadaw A.; Roy S.; Ahmad S.; Snead D. R.; Jamison T. F. Progress toward a large-scale synthesis of Molnupiravir (MK-4482, EIDD-2801) from cytidine. ACS Omega 2021, 6, 10396–10402. 10.1021/acsomega.1c00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner A.; Znidar D.; Ötvös S. B.; Snead D. R.; Dallinger D.; Kappe C. O. A high-yielding synthesis of EIDD-2801 from uridine. Eur. J. Org. Chem. 2020, 2020, 6736–6739. 10.1002/ejoc.202001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J. A.; Benkovics T.; Silverman S. M.; Huffman M. A.; Kong J.; Maligres P.; Itoh T.; Yang H.; Verma D.; Pan W.; Ho H.; Vroom J.; Knight A. M.; Hurtak J. A.; Klapars A.; Fryszkowska A.; Morris W. J.; Strotman N.; Murphy G.; Maloney K.; Fier P. Engineered Ribosyl-1-Kinase Enables Concise Synthesis of Molnupiravir, an Antiviral for COVID-19. ACS Cent. Sci. 2021, 10.1021/acscentsci.1c00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paymode D. J.; Vasudevan N.; Ahmad S.; Kadam A. L.; Cardoso F. S. P.; Burns J. M.; Cook D. W.; Stringham R. W.; Snead D. R. Toward a practical, two-step process for Molnupiravir: direct hydroxamination of cytidine followed by selective esterification. Org. Process Res. Dev. 2021, 25, 1822–1830. 10.1021/acs.oprd.1c00033. [DOI] [Google Scholar]
- Trimble R. B.; Maley F. Metabolism of 4-N-hydroxy-cytidine in Escherichia coli. J. Bacteriol. 1971, 108, 145–153. 10.1128/jb.108.1.145-153.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick L.; Mac Neela J. P.; Wolfenden R. Transition State Stabilization by Deaminases: Rates of Nonenzymatic Hydrolysis of Adenosine and Cytidine. Bioorg. Chem. 1987, 15, 100–108. 10.1016/0045-2068(87)90011-3. [DOI] [Google Scholar]
- Cohen R. M.; Wolfenden R. Cytidine Deaminase from Escherichia coli: purification, properties, and inhibition by the potential transition state analog 3,4,5,6-tetrahydrouridine. J. Biol. Chem. 1971, 246, 7561–7565. 10.1016/S0021-9258(19)45812-2. [DOI] [PubMed] [Google Scholar]
- Cohen R. M.; Wolfenden R. The equilibrium of hydrolytic deamination of cytidine and N4-methylcytidine. J. Biol. Chem. 1971, 246, 7566–7568. 10.1016/S0021-9258(19)45813-4. [DOI] [PubMed] [Google Scholar]
- Xiang S.; Short S. A.; Wolfenden R.; Carter C. W. Jr. Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry 1995, 34, 4516–4523. 10.1021/bi00014a003. [DOI] [PubMed] [Google Scholar]
- Xiang S.; Short S. A.; Wolfenden R.; Carter C. W. Jr. The structure of the cytidine deaminase-product complex provides evidence for efficient proton transfer and ground-state destabilization. Biochemistry 1997, 36, 4768–4774. 10.1021/bi963091e. [DOI] [PubMed] [Google Scholar]
- Ferrero M.; Gotor V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides, and C-nucleosides. Chem. Rev. 2000, 100, 4319–4348. 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]
- Romero-Téllez S.; Lluch J. M.; González-Lafont À.; Masgrau L. Comparing hydrolysis and transglycosylation reactions catalyzed by Thermus thermophilus β-glycosidase. A combined MD and QM/MM study. Front. Chem. 2019, 7, 200. 10.3389/fchem.2019.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulis C.; Guieysse D.; Morel S.; Séverac E.; Remaud-Siméon M. Natural and engineered transglycosylases: Green tools for the enzyme-based synthesis of glycoproducts. Curr. Opin. Chem. Biol. 2021, 61, 96–106. 10.1016/j.cbpa.2020.11.004. [DOI] [PubMed] [Google Scholar]
- Rogers T. A.; Bommarius A. S. Utilizing simple biochemical measurements to predict lifetime output of biocatalysts in continuous isothermal processes. Chem. Eng. Sci. 2010, 65, 2118–2124. 10.1016/j.ces.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton S. G.; Cowan D. A.; Woodley J. M. The search for the ideal biocatalyst. Nat. Biotechnol. 2002, 20, 37–45. 10.1038/nbt0102-37. [DOI] [PubMed] [Google Scholar]
- Woodley J. M. Accelerating the implementation of biocatalysis in industry. Appl. Microbiol. Biotechnol. 2019, 103, 4733–4739. 10.1007/s00253-019-09796-x. [DOI] [PubMed] [Google Scholar]
- Birmingham W. R.; Toftgaard Pedersen A.; Dias Gomes M.; Bøje Madsen M.; Breuer M.; Woodley J. M.; Turner N. J. Toward scalable biocatalytic conversion of 5-hydroxymethylfurfural by galactose oxidase using coordinated reaction and enzyme engineering. Nat. Commun. 2021, 12, 4946. 10.1038/s41467-021-25034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.