

Abstract
Emerging studies indicate that the immune system can regulate systemic metabolism. Here, we show that thymic stromal lymphopoietin (TSLP) stimulates T cells to induce selective white adipose loss, which protects against obesity, improves glucose metabolism, and mitigates nonalcoholic steatohepatitis. Surprisingly, adipose loss was not caused by alterations in food intake, absorption, or energy expenditure. Rather, it was induced by the excessive loss of lipids through the skin as sebum. TSLP and T cells regulated sebum release and sebum-associated antimicrobial peptide expression in the steady state. In human skin, TSLP expression correlated directly with sebum-associated gene expression. Thus, we establish a paradigm in which adipose loss can be achieved by sebum hypersecretion and uncover a role for adaptive immunity in skin barrier function through sebum secretion.
One-Sentence Summary:
TSLP-stimulated T cells promote sebum secretion, which can be pharmacologically manipulated to promote adipose loss.
Obesity is a serious public health concern. Globally, over 40% of adults are overweight or obese and face up to a sevenfold increased risk of associated chronic diseases, such as type II diabetes, fatty liver disease, heart disease, and certain cancers. Despite growing attention and significant public health initiatives, obesity rates continue to rise (1–3). Consequently, there is an urgent need to identify factors that can decrease adiposity.
There is emerging recognition that the immune system plays a key role in regulating adipose tissue and energy metabolism. Type 2 immune cells, such as type 2 innate lymphoid cells (ILC2s) and eosinophils, increase metabolic rate by promoting adipose beiging and the upregulation of thermogenic energy expenditure (4–8). Obesity also causes a state of chronic low-grade inflammation, which leads to the development of insulin resistance (type II diabetes). Regulatory T cells (Tregs) suppress obesity-induced inflammation and help maintain insulin sensitivity independently of weight, preventing type II diabetes (9, 10).
Thymic stromal lymphopoietin (TSLP) is a cytokine that is expressed by epithelial cells at barrier sites, such as the skin, lung, and gut. TSLP has been studied extensively as an activator of type 2 immune cells including ILC2s and eosinophils (11–17). More recently, TSLP has also been shown to increase systemic Treg numbers (18, 19). Thus, by expanding type 2 immune cells and Tregs, we hypothesized that TSLP could counteract obesity and its associated complications.
RESULTS
TSLP protects against obesity and obesity-related complications
We first tested the effect of TSLP on obesity by injecting a Tslp-expressing adeno-associated virus serotype 8 (TSLP-AAV) expressed in the liver to increase TSLP levels in mice fed a high-fat diet (HFD). Strikingly, HFD-fed mice given TSLP-AAV lost weight over 4 weeks compared to mice administered Control-AAV, which gained weight (Fig. 1A and fig. S1A). TSLP-AAV also caused weight loss in normal chow (NC)-fed hyperphagic ob/ob mice (fig. S1, B and C). Furthermore, TSLP not only prevented but also reversed obesity. Mice first fed HFD for 10 weeks (obese mice) and then given TSLP-AAV while continuing on HFD displayed weight loss, decreased visceral fat mass (epididymal white adipose tissue, eWAT), and markedly improved metabolic parameters, including glucose tolerance, fasting blood glucose levels, fasting insulin levels, homeostatic model assessment of insulin resistance (HOMA-IR), and hepatic triglyceride (TG) levels (Fig. 1, B to G, and fig. S1, D to G).
Fig. 1. TSLP protects against diet-induced obesity and glucose intolerance.
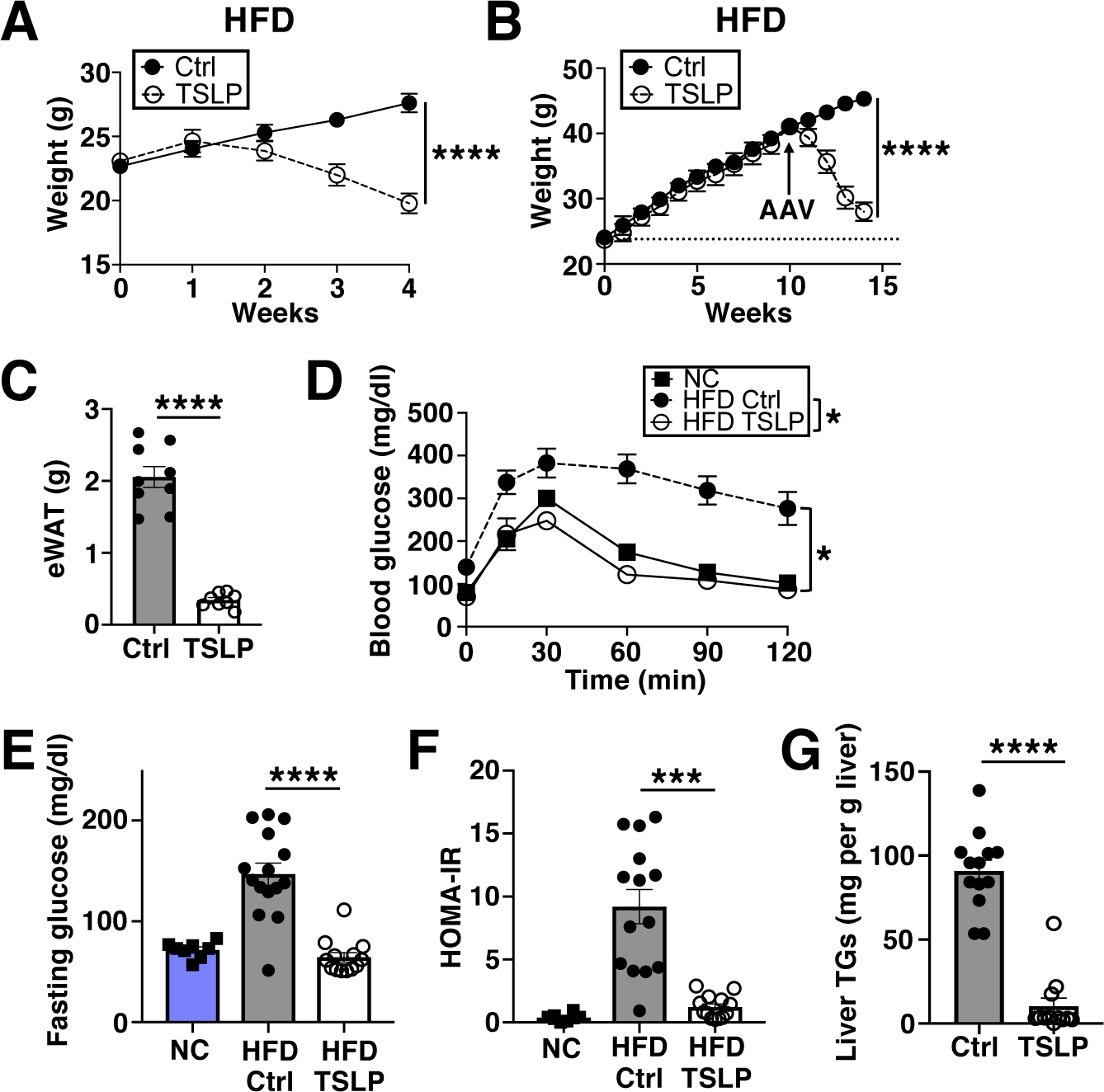
(A) Weights of HFD-fed mice post AAV (Control-AAV or TSLP-AAV). (n=13 mice/group, pooled from 3 independent experiments). F-test. (B to G) Mice fed HFD for 10 weeks prior to AAV and for another 4 weeks thereafter. (B) Weights (n=9 mice/group, pooled from 3 independent experiments). F-test. (C) eWAT masses (n=8–9 mice/group, pooled from 3 independent experiments). t-test. (D) Blood glucose following glucose tolerance test (GTT, n=8–10 mice/HFD group, pooled from 2 independent experiments, with n=5 NC mice shown for reference). F-test. (E) Fasting blood glucose (n=13–15 mice/HFD group, pooled from 3 independent experiments, with n=8 NC mice shown for reference). F-test. (F) HOMA-IR (n=13–14 mice/HFD group, pooled from 3 independent experiments, with n=8 NC mice shown for reference). t-test. (G) Liver TGs (n=12–13 mice/group, pooled from 3 independent experiments). t-test. N.S.=not significant, P≥0.05, *P<0.05, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
Given the reduction in hepatic steatosis, we further explored the effect of TSLP in a mouse model of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). To this end, mice were fed a methionine–choline–deficient diet (MCDD) for 4 weeks, and then administered either Control-AAV or TSLP-AAV for an additional 4 weeks. MCDD-fed mice given TSLP-AAV had lower liver TG levels, lower serum alanine aminotransferase (ALT) levels, and decreased hepatic fibrosis compared to controls (Fig. 2, A to D, and fig. S2A).
Fig. 2. TSLP ameliorates MCDD-driven liver damage and induces selective white adipose loss in NC-fed mice.
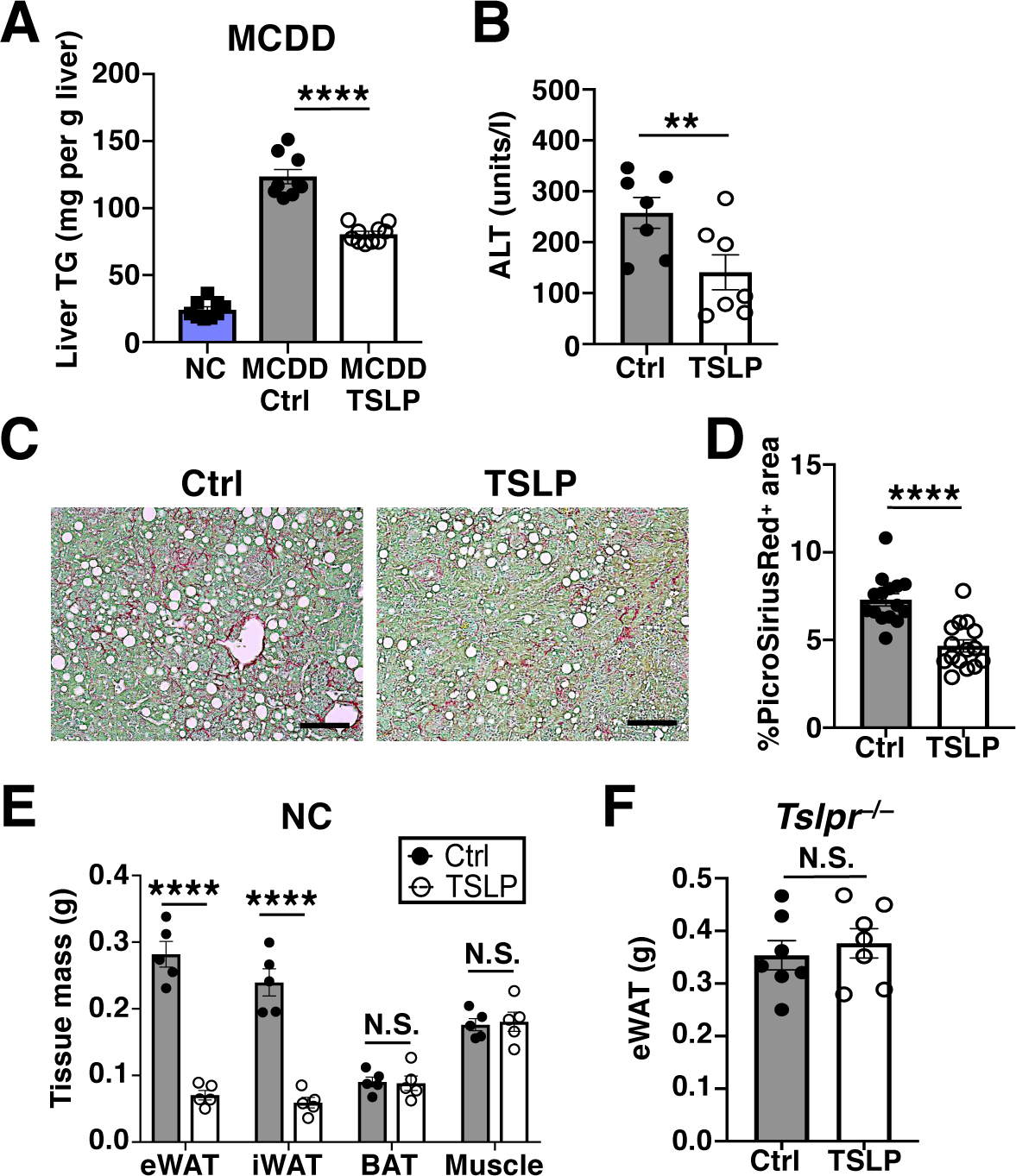
(A to D) Mice fed MCDD for 4 weeks prior to AAV and for another 4 weeks thereafter. (A) Liver TGs (n=9–10 mice/MCDD group, pooled from 2 independent experiments, with n=9 NC mice shown for reference). t-test. (B) Serum ALT levels (n=7 mice/group, pooled from 2 independent experiments). t-test. (C and D) Representative PicroSiriusRed liver staining and quantification (n=15 mice/group, pooled from 3 independent experiments). t-test. Scale bars=100 μm. (E and F) NC-fed mice. (E) eWAT, iWAT, BAT, and quadriceps muscle masses, 2 weeks post AAV (n=5 mice/group, 1 representative of at least 10 independent experiments shown). Student’s t test. (F) eWAT masses of Tslpr−/− mice, 2 weeks post AAV (n=7 mice/group, pooled from 2 independent experiments). t-test. N.S., P≥0.05, **P<0.01, ****P<0.0001. Data are mean ± SEM.
TSLP induces adipose loss in NC-fed mice
The effect of TSLP on adipose loss was also observed in NC-fed mice. Two weeks after TSLP-AAV, NC-fed mice displayed significantly decreased eWAT and subcutaneous inguinal white adipose tissue (iWAT) mass, without a corresponding decrease in brown adipose tissue (BAT) or muscle mass (Fig. 2E). The majority of the WAT loss occurred between 7–14 days after TSLP-AAV was administered (fig. S2, B to E). The loss of WAT correlated in a dose-dependent manner with the level of serum TSLP induced by TSLP-AAV (fig. S2, F to H). Body composition analysis also showed that TSLP-AAV decreased percentage of whole body fat mass (fig. S2I). Thus, mice given TSLP-AAV are not cachexic per se, as they exhibit a selective reduction in WAT.
TSLP is a well-known mediator of allergic inflammation and can activate numerous immune cell types across multiple organs (20–29). Thus, we examined various tissues for inflammatory damage in mice given TSLP-AAV. As expected, immune cell infiltration occurred, particularly in the lung and liver, but the histological architecture of each organ remained predominantly intact and no immune complex deposition in the kidney was observed (fig. S3A, and table S1). Importantly, the body-condition scoring (BCS) remained normal and mice only shed 5% of their total body weight, despite losing almost all of their WAT (fig. S3, B and C). The source and location of TSLP expression did not affect the adipose loss phenotype, because elevation of TSLP levels by subcutaneously injected TSLP-expressing lymphoma cells (RMA) also resulted in WAT loss (fig. S3, D and E). Finally, the effects of TSLP on adipose loss were dependent on signaling through the TSLP receptor (TSLPR), since Tslpr−/− mice did not lose WAT when administered TSLP-AAV (Fig. 2F). Thus, TSLP induces WAT loss through the TSLPR regardless of TSLP source and without causing severe systemic inflammation leading to anorexia or cachexia.
T cells mediate TSLP-driven adipose loss
To examine which cell type(s) might be responsible for mediating TSLP-induced WAT loss, we first tested whether TSLP causes adipose loss through TSLPR signaling in hematopoietic (radiosensitive) or nonhematopoietic (radioresistant) cells. Three types of bone marrow (BM) chimeric mice were generated (fig. S4A) and then administered Control-AAV or TSLP-AAV after BM reconstitution. WAT loss was observed in NC-fed Tslpr−/− recipient mice transplanted with wildtype (Wt) donor BM but not in Wt recipient mice transplanted with Tslpr−/− donor BM (fig. S4B). Similar results were seen in HFD-fed BM chimeras (fig. S4, C to E). Thus, this effect is likely mediated by TSLPR signaling in hematopoietic cells.
To identify the hematopoietic cell type required for TSLP-induced adipose loss, we administered Control-AAV or TSLP-AAV to a variety of NC-fed mouse strains lacking different immune cell types. TSLP-AAV caused significant WAT loss in mice lacking dendritic cells (DCs), eosinophils, or Tregs (fig. S5, A and B). However, Rag2−/−Il2rg−/− mice, which lack B, T, and innate lymphocytes, Rag2−/− mice, which lack B and T cells, and Ebeta−/− (which lack αβ T cells) mice did not lose WAT (fig. S5, C and D, and Fig. 3A). Thus, αβ T cells are necessary for TSLP-driven adipose loss. To distinguish whether CD4+ or CD8+ T cells were important for this effect, CD4+ and/or CD8+ T cells were depleted using antibodies. Surprisingly, we found that depletion of both CD4+ and CD8+ T cells was required to prevent TSLP-mediated WAT loss (Fig. 3B), signifying that either T cell subset was sufficient. Accordingly, reconstitution of Rag2−/− mice with either CD4+ or CD8+ T cells restored the ability of Rag2−/− mice to lose WAT in response to TSLP (Fig. 3C). Importantly, Rag2−/−Il2rg−/− mice with selective reconstitution of T cells became susceptible to TSLP-mediated WAT loss (fig. S5C). Thus, T cells but not B cells, NK cells, or ILCs are required for the effect of TSLP on adipose loss.
Fig. 3. TSLPR signaling in T cells is required for TSLP-driven adipose loss.
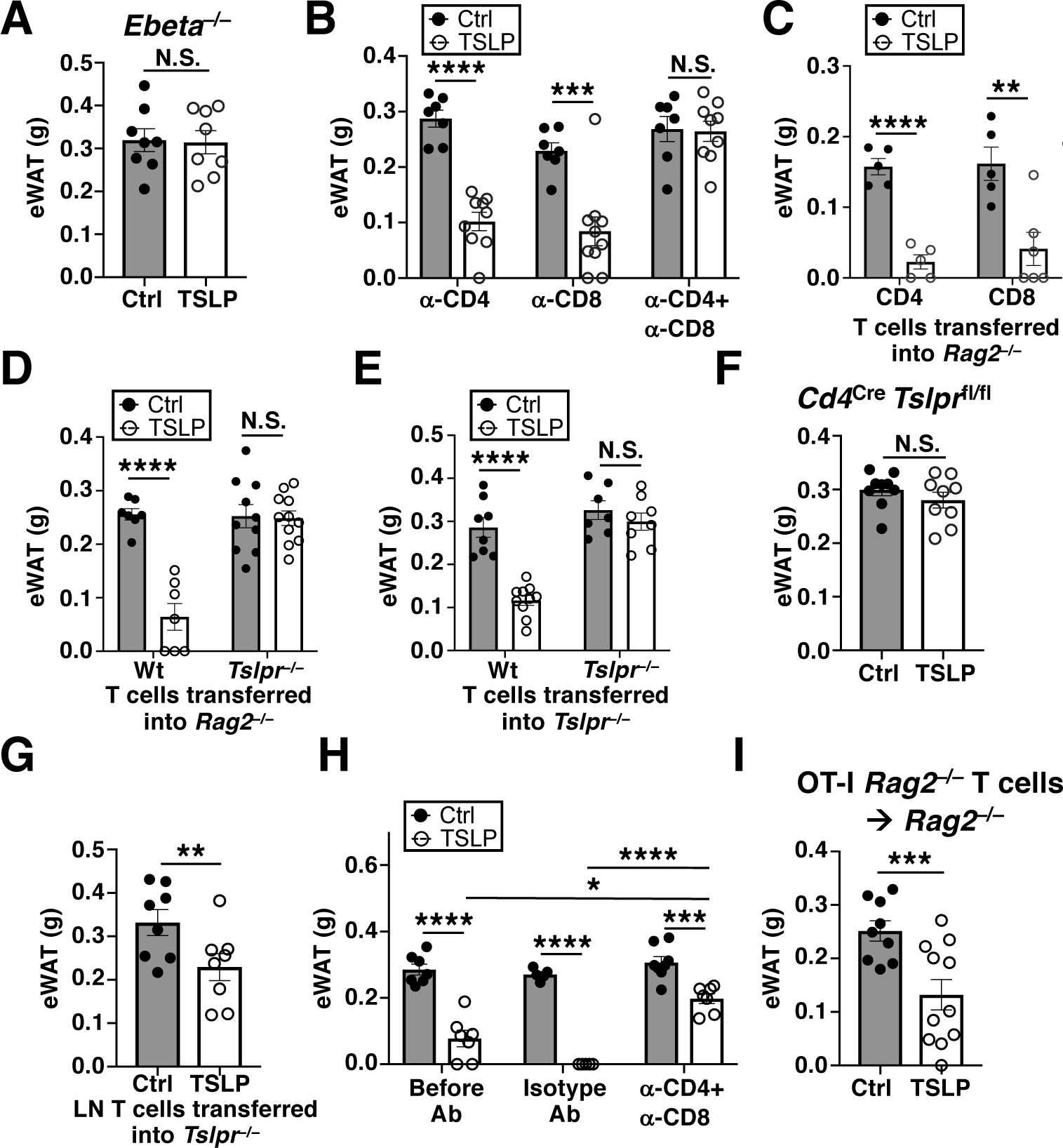
(A) eWAT masses of Ebeta−/− mice, 2 weeks post AAV (n=8 mice/group, pooled from 3 independent experiments). t-test. (B) eWAT masses of Wt mice injected with α-CD4±α-CD8, 2 weeks post AAV (n=7–10 mice/group, pooled from 3 independent experiments). t-test. (C) eWAT masses of Rag2−/− mice reconstituted with CD4+ or CD8+ T cells, 2 weeks post AAV (n=5–6 mice/group, pooled from 2 independent experiments). t-test. (D) eWAT masses of Rag2−/− mice reconstituted with Wt or Tslpr−/− T cells, 2 weeks post AAV (n=7–11 mice/group, pooled from 3 independent experiments). t-test. (E) eWAT masses of Tslpr−/− mice reconstituted with Wt or Tslpr−/− T cells, 2 weeks post AAV (n=7–10 mice/group, pooled from 3 independent experiments). t-test. (F) eWAT masses of Cd4Cre Tslprfl/fl mice, 2 weeks post AAV (n=9 mice/group, pooled from 3 independent experiments). t-test. (G) eWAT masses of Tslpr−/− mice transferred with skin-draining lymph node (LN) T cells from either Ctrl-AAV- or TSLP-AAV- injected Wt mice, 2 weeks post cell transfer (n=8 mice/group, pooled from 2 independent experiments). t-test. (H) eWAT masses of AAV-injected Wt mice before antibody injection (2 weeks post AAV, n=7 mice/group), 2 weeks after isotype antibody injection (4 weeks post AAV, n=5 mice/group), or 2 weeks after α-CD4+α-CD8 antibody injection (4 weeks post AAV, n=7–8 mice/group). Data pooled from 2 independent experiments. t-test. (I) eWAT masses of Rag2−/− mice reconstituted with T cells from OT-I Rag2−/− mice, 2 weeks post AAV (n=9–11 mice/group, pooled from 3 independent experiments). t-test. N.S., P≥0.05, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
To assess whether TSLP acts directly on T cells to promote adipose loss, we adoptively transferred Wt or Tslpr−/− T cells into Rag2−/− mice. TSLP-AAV-injected NC-fed Rag2−/− mice that were reconstituted with Wt but not Tslpr−/− T cells lost WAT (Fig. 3D). Furthermore, Tslpr−/− mice adoptively transferred with Wt T cells but not Tslpr−/− T cells also lost WAT with TSLP-AAV (Fig. 3E). We further verified these findings by generating Cd4Cre Tslprfl/fl mice, in which the TSLPR was specifically deleted in T cells (fig. S5E). Cd4Cre Tslprfl/fl mice remained insensitive to TSLP and did not lose any adipose upon TSLP-AAV injection (Fig. 3F). Thus, TSLPR signaling in T cells alone is sufficient to mediate TSLP-driven adipose loss. Remarkably, T cells that were previously exposed to TSLP appeared to retain their ability to induce adipose loss, since the adoptive transfer of lymph node T cells from TSLP-AAV-injected Wt mice into naïve NC-fed Tslpr−/− mice resulted in adipose loss of recipient mice (Fig. 3G). Moreover, T cells were continuously required to maintain the adipose loss in mice, as depleting T cells at 2 weeks post TSLP-AAV injection allowed for partial but significant restoration of WAT mass (Fig. 3H).
To determine whether antigen specificity of T cells was an important determinant for TSLP-induced adipose loss, we utilized TCR-transgenic OT-I Rag2−/− mice, which harbor only CD8+ T cells with a single fixed antigen specificity. Although OT-I Rag2−/− mice contain solely naïve T cells, adoptive transfer of OT-I Rag2−/− T cells into Rag2−/− mice generates “memory-like” CD44hi T cells with a single fixed antigen specificity (30). We found that NC-fed OT-I Rag2−/− mice were resistant to TSLP (fig. S5F), but Rag2−/− mice adoptively transferred with OT-I Rag2−/− T cells were susceptible (Fig. 3I), suggesting that TSLP-driven adipose loss is mediated by activated/memory T cells in an antigen-independent manner. Given that TSLP can stimulate T helper type 2 (Th2) cells, we examined whether IL-4, a major Th2 cell effector and differentiation cytokine, is important for the effect of TSLP on adipose loss. Mice injected with TSLP-AAV displayed increased serum IL-4 levels (fig. S5G). Despite this however, NC-fed Il4−/− mice were not resistant to TSLP-mediated adipose loss (fig. S5H), suggesting that IL-4 is dispensable for this process.
TSLP induces adipose loss by increasing sebum secretion
The loss of body weight and WAT establish that TSLP causes a net negative energy balance in mice, such that caloric output is greater than caloric intake. However, decreased caloric intake was not responsible for TSLP-induced adipose loss, as NC-fed mice administered TSLP-AAV consumed more food (Fig. 4A and fig. S6A), equally absorbed and cleared orally administered glucose and olive oil (fig. S6, B and C), and did not excrete more calories per day in their feces as measured by bomb calorimetry (Fig. 4B). Since TSLP is a known activator of type 2 responses, which can cause adipose beiging, we hypothesized that TSLP causes a net negative energy balance by increasing caloric output through increased thermogenic energy expenditure. Surprisingly however, we found no evidence of increased metabolic rate. Despite losing ~30% of total WAT over a 3-day metabolic cage measurement period, the locomotor activity and the rate of oxygen consumption, carbon dioxide production, energy expenditure, and respiratory exchange ratio were similar between NC-fed mice given Control-AAV and TSLP-AAV (Fig. 4C and fig. S6, D to H). Consistent with these results, NC-fed uncoupled protein 1–knockout (Ucp1−/−) mice, which have diminished adipose beiging capacity, were not resistant to TSLP-induced adipose loss (fig. S6I). Thus, TSLP does not increase thermogenic energy expenditure.
Fig. 4. TSLP induces adipose loss by promoting sebum secretion.
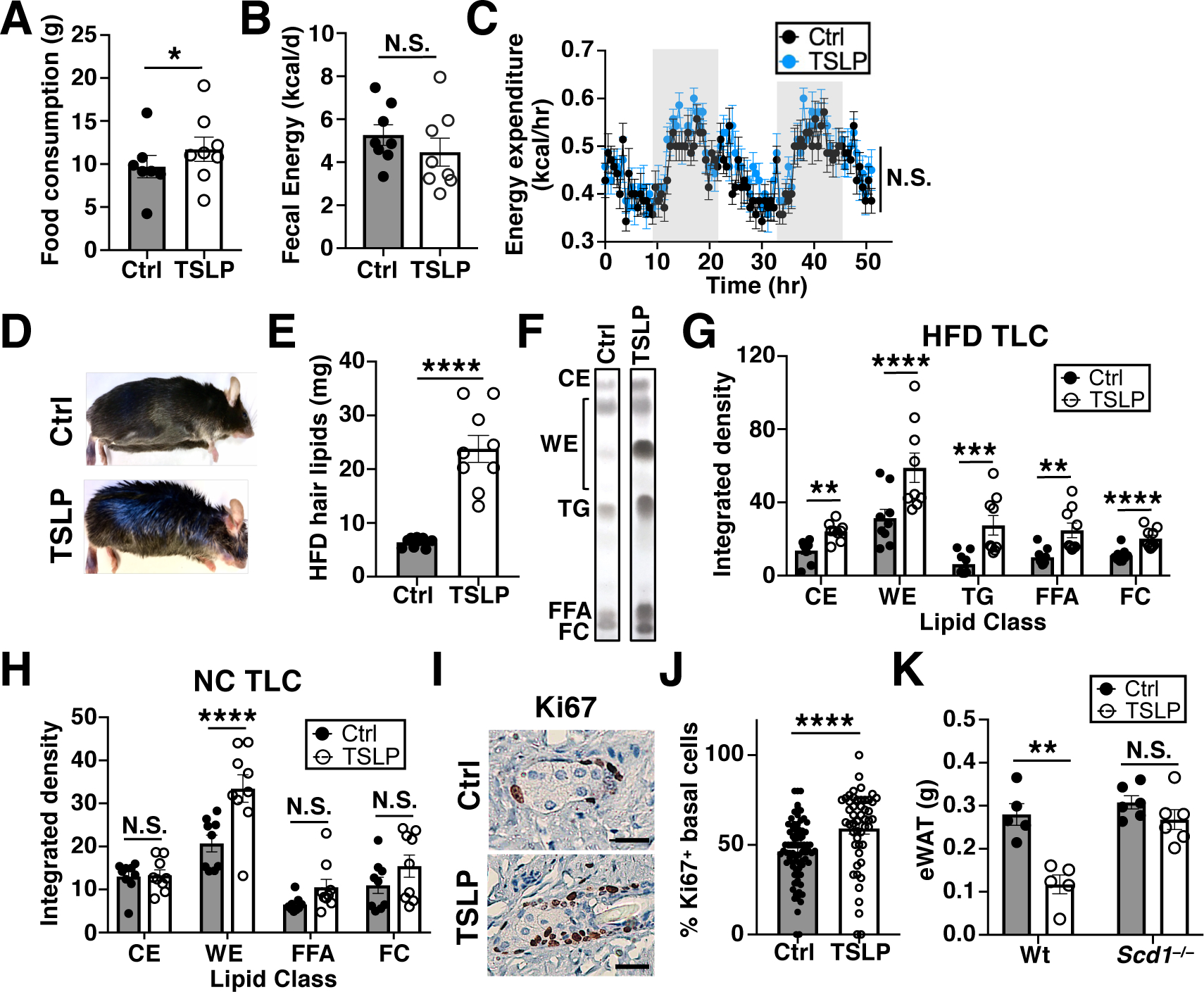
(A) Food consumption, 9–11 days post AAV (n=7–8 mice/group, 1 representative of 2 independent experiments shown). Student’s t test. (B) Fecal calories, 9–11 days post AAV (n=8 mice/group, pooled from 2 independent experiments). t-test. (C) Energy expenditure, 9–11 days post AAV (n=7–8 mice/group, 1 representative of 2 independent experiments shown). ANOVA. (D to G) Mice fed HFD for 10 weeks prior to AAV and for another 4 weeks thereafter. (D) Gross appearance. (E) Hair lipid mass (n=9 mice/group, pooled from 3 independent experiments). t-test. (F and G) TLC plot and quantification of hair lipids (CE=cholesterol esters, WE=wax esters, TG=triglycerides, FFA=free fatty acids, FC=free cholesterol, n=9 mice/group, pooled from 3 independent experiments). t-test. (H) TLC quantification of hair lipids from NC-fed mice, 10 days post AAV (n=9 mice/group, pooled from 3 independent experiments). t-test. (I and J) Sebaceous gland Ki67 staining and quantification, 10 days post AAV (n=78 Ctrl and 50 TSLP sebaceous glands from three mice/group, pooled from 2 independent experiments). t-test. Scale bars=20 μm. (K) eWAT masses of Wt or Scd1−/− mice, 2 weeks post AAV (n=5–6 mice/group, pooled from 2 independent experiments). t-test. N.S., P≥0.05, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
We next hypothesized that TSLP might cause energy loss through the excretion or secretion of calorie-containing metabolites. Fecal caloric content and urine glucose, ketones, protein, and free fatty acids were not increased in NC-fed mice given TSLP-AAV (Fig. 4B and fig. S6, J to M). However, mice administered TSLP-AAV began to develop strikingly greasy hair that was grossly visible at around 4 weeks post injection (Fig. 4D). This occurred in both HFD and NC-fed mice but was more prominent in HFD-fed mice. To identify this oleaginous substance on the hair of mice given TSLP-AAV, we performed hair lipid extraction and analyzed the composition by thin-layer chromatography (TLC). HFD-fed mice administered TSLP-AAV exhibited significantly increased hair lipid mass (Fig. 4E), which comprised a mixture of wax esters, cholesterol esters, triglycerides, free fatty acids, and free cholesterol (Fig. 4, F and G). The increase in wax esters, which are sebum-specific lipids, signified the presence of enhanced sebum secretion in mice given TSLP-AAV. Likewise, sebum secretion was also increased in TSLP-AAV-injected NC-fed mice at day 10, an early time-point when mice began to lose WAT but were not yet visibly greasy (Fig. 4H and fig. S7, A and B). Similar to TSLP-AAV, the topical application of MC903, a vitamin D analogue that stimulates skin keratinocytes to produce TSLP, also triggered an increase in hair wax esters in a TSLP-dependent manner (Fig. S7, C to E). Thus, endogenous skin-derived TSLP also promotes sebum secretion.
Skin histological analysis of NC-fed mice given TSLP-AAV revealed smaller sebaceous glands with similar total lipid content compared to control mice (fig. S8, A to D). Sebum release occurs via holocrine secretion, a process whereby sebocytes undergo programmed cell death and release their intracellular contents into the hair follicle (31). Although the smaller sebaceous glands seen in mice given TSLP-AAV seemed to be paradoxical, we reasoned that this was likely a result of increased holocrine secretion and turnover of mature sebocytes. The continual extrusion of sebaceous contents would not allow for prolonged sebaceous gland growth and accumulation of lipids within mature sebocytes. Rather, it would result in smaller sebaceous glands with an increased rate of mature sebocyte replenishment from sebocyte stem cells. Indeed, an increased fraction of the basal cells lining the sebaceous glands (proliferative sebocyte stem cells) were Ki67+ within each gland (Fig. 4, I and J, and fig. S8, E and F). Concordantly, delineation of sebaceous gland zones (32) by Krt5 (marking basal cells) and Krt79 (marking mature sebocytes) demonstrated that mice administered TSLP-AAV had an increased percentage of Krt5+ cells along with a decreased percentage of Krt79+ cells per gland (fig. S8, G and H). Further classification of Krt79+ sebocytes via nuclear staining showed an increased proportion of sebocytes without nuclei in mice given TSLP-AAV (fig. S8I), consistent with dead or dying mature sebocytes undergoing holocrine secretion. Thus, TSLP appears to promote sebum production by increasing the rate of holocrine secretion, which consequently enhances sebocyte turnover within sebaceous glands.
To determine whether sebum secretion was necessary for TSLP-mediated adipose loss, we utilized asebia mice, which lack the enzyme stearoyl-coA desaturase-1 (Scd1) and have marked sebaceous gland hypoplasia (33, 34). Compared to Wt mice, NC-fed asebia mice lost minimal amounts of WAT upon TSLP-AAV injection (Fig. 4K), indicating that sebum secretion is necessary for TSLP-driven adipose loss. To further establish the relationship between sebum secretion and adipose loss, we assessed the eWAT lipolysis rates and serum free fatty acid and TG levels of Wt mice administered Control- or TSLP-AAV. eWAT isolated ex vivo from mice administered TSLP-AAV released increased amounts of glycerol (an end-product readout of lipolysis, fig. S9A), demonstrating that TSLP increases lipolysis. If TSLP directly induced lipolysis, TSLP-AAV-injected mice would be expected to display increased circulating free fatty acid and TG levels. However, for both mice fed a NC and HFD, other than a subtle increase in TGs noted at week 2 in HFD-fed mice, TSLP-AAV injection did not increase serum free fatty acid or TG levels over time compared to Control-AAV injected mice. Rather, TSLP-AAV injection decreased serum free fatty acid levels at later timepoints in mice fed a HFD (fig. S9, B to E). Thus, adipose loss in TSLP-AAV-injected mice is not driven by direct induction of lipolysis causing an overflow “push” of lipids to the skin. Rather, the lipolysis is likely driven by a “pull” mechanism whereby the sebocytes actively consume lipids from the blood, which in turn induces lipolysis to replenish serum lipids. This increased rate of lipid consumption by sebaceous glands eventually outpaces the ability of adipose tissue to undergo lipolysis, such that serum lipids are ultimately decreased.
From a cellular perspective, the increased sebum secretion was dependent on TSLPR signaling in T cells. NC-fed Rag2−/− mice administered TSLP-AAV did not display increased hair wax esters (Fig. 5A and fig. S10A). However, NC-fed Rag2−/− mice reconstituted with Wt T cells but not Tslpr−/− T cells exhibited increased hair wax esters when administered TSLP-AAV (Fig. 5B, and fig. S10B). Examination of the skin by flow cytometry showed a significant increase in skin CD4+ and CD8+ T cells upon TSLP-AAV injection (Fig. 5, C to E, and fig. S11, A to E), with a slight increase in the skin CD8+ tissue resident memory (CD69+CD103+) T cell subset (fig. S12). Histological staining of the skin also showed a significant increase in skin CD4+ and CD8+ T cells, which were clustered in or around sebaceous glands (Fig. 5E). The increase in T cells was likely due to enhanced T cell trafficking to the skin, since the sphingosine-1-phosphate inhibitor FTY720, which blocks lymphocyte migration, inhibited the TSLP-driven increase in skin T cells (fig. S13A). Importantly, FTY720-treated NC-fed mice were also resistant to TSLP-induced adipose loss and sebum hypersecretion (fig. S13, B to D). Finally, TSLP-mediated recruitment of T cells to the skin and subsequent sebum hypersecretion was dependent on direct stimulation of T cells through the TSLPR, since Cd4Cre Tslprfl/fl mice administered TSLP-AAV did not exhibit increased skin CD4+ and CD8+ T cells or increased hair wax esters (fig. S13, E to L). Thus, TSLP directly stimulates T cells to migrate to skin sebaceous glands and this migration is required for both adipose loss and sebum hypersecretion.
Fig. 5. TSLP stimulates T cells to promote sebum production and increases T cells in the skin.
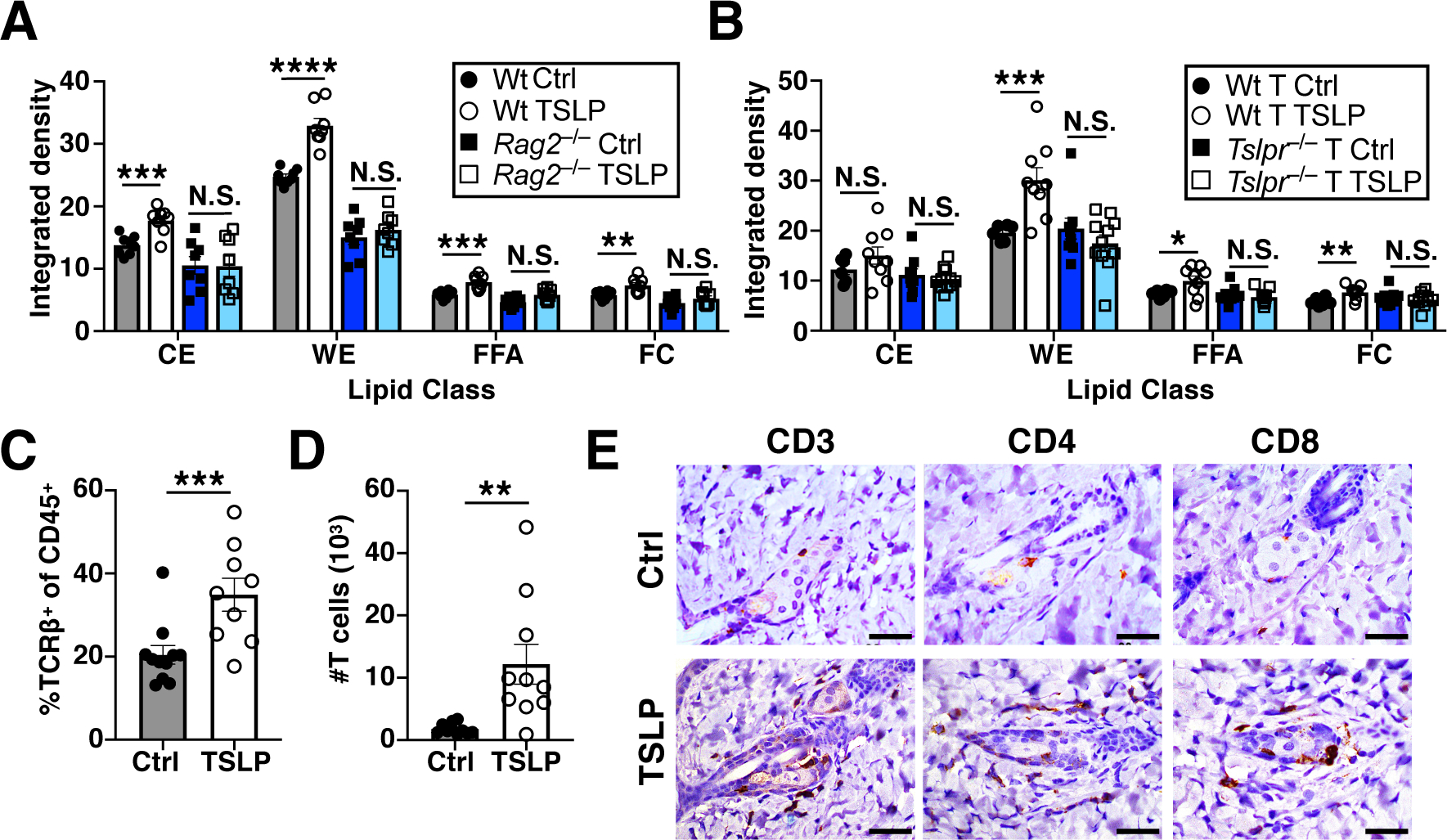
(A) TLC quantification of hair lipids from Wt and Rag2−/− mice, 2 weeks post AAV (n=8 mice/group, pooled from 3 independent experiments). t-test. (B) TLC quantification of hair lipids from Rag2−/− mice reconstituted with Wt or Tslpr−/− T cells, 2 weeks post AAV (n=6–9 mice/Wt T cell group, pooled from 2 independent experiments, and n=9–11 mice/Tslpr−/− T cell group, pooled from 3 independent experiments). t-test. (C and D) Percentage and number of skin T cells by flow cytometry, 10 days post AAV (n=9–11 mice/group, pooled from 3 independent experiments). t-test. (E) Skin CD3, CD4, and CD8 immunohistochemical staining, 10 days post AAV. Scale bars=40 μm. N.S., P≥0.05, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
TSLP regulates homeostatic sebum secretion
We next tested whether TSLP and T cells play a physiological role in the control of sebum secretion at homeostasis. Aside from calories, sebum also contains numerous antimicrobial fatty lipids, cathelicidins, β-defensins, and other antimicrobial peptides (AMPs), all of which form an important physical and immune-protective barrier for the skin (35–38). Since TSLP is a barrier cytokine whose expression is regulated by homeostatic and inflammatory triggers, we hypothesized that the physiologic role of endogenous TSLP may be to maintain skin barrier function by controlling sebum production. To this end, we examined the hair lipid composition of unmanipulated NC-fed Wt and Tslpr−/− mice. Compared to Wt mice, Tslpr−/− mice exhibited decreased hair wax esters and a lower fraction of Ki67+ basal cells without differences in sebaceous gland size or lipid content (Fig. 6, A and B, and fig. S14, A to F), signifying that TSLP regulates sebum secretion at baseline. Similarly, unmanipulated NC-fed Rag2−/− and Ebeta−/− mice also displayed decreased hair wax esters compared to Wt mice (Fig. 6C and fig. S14, G to I), indicating that T cells may play a homeostatic role in controlling sebum secretion. Furthermore, NC-fed Tslpr−/− mice expressed significantly lower levels of sebum-associated AMPs in back skin compared to Wt mice (Fig. 6D), consistent with the hypothesis that TSLP promotes barrier function through sebum.
Fig. 6. TSLP and T cells regulate sebum secretion at homeostasis.
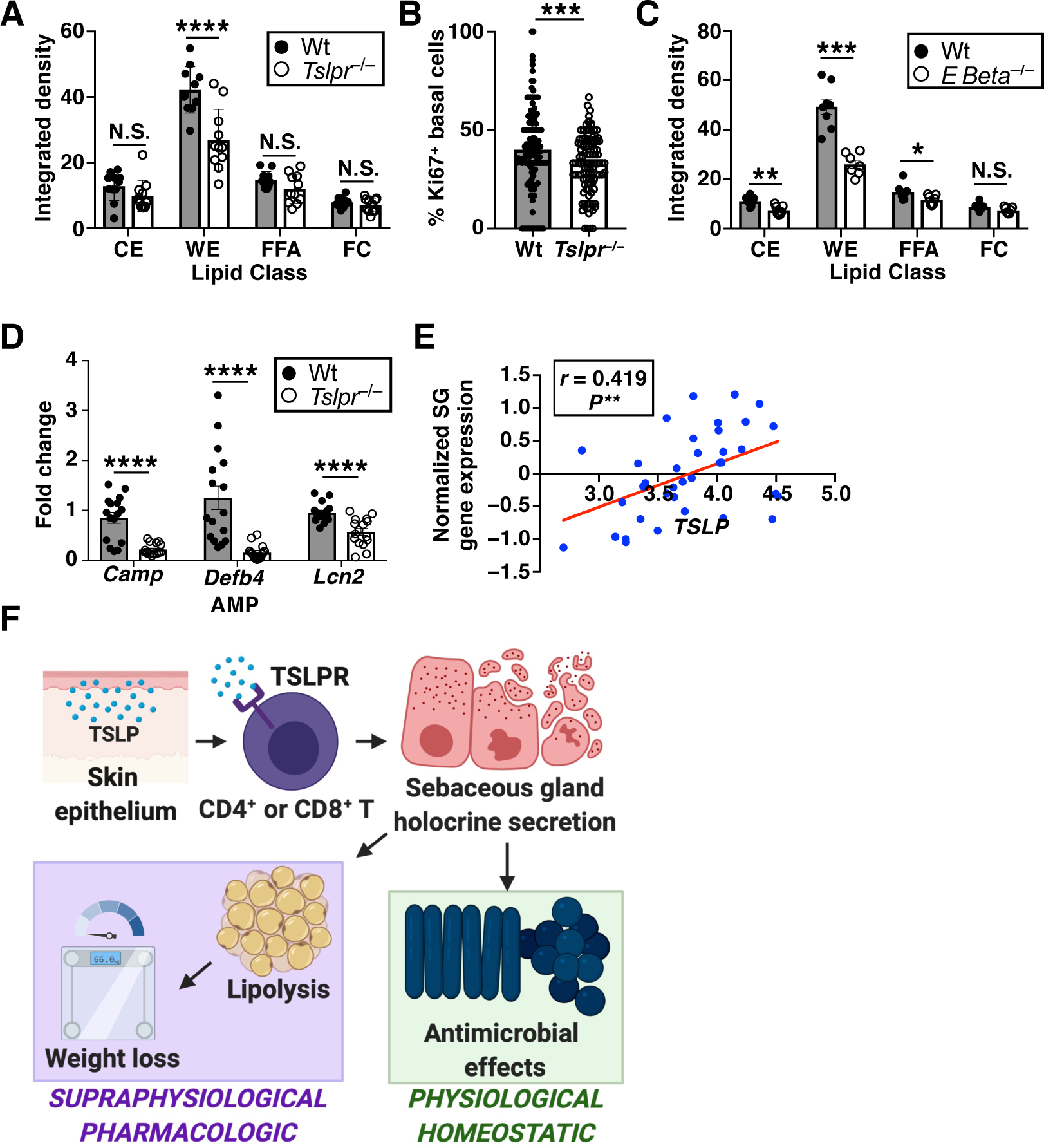
(A) TLC quantification of hair lipids from Wt versus Tslpr−/− mice at baseline (n=11 mice/group, pooled from 3 independent experiments). t-test. (B) Ki67 quantification of sebaceous glands from Wt versus Tslpr−/− mice (n=132 Wt and 109 Tslpr−/− sebaceous glands from three mice/group, pooled from 2 independent experiments). t-test. (C) TLC quantification of hair lipids from Wt versus Ebeta−/− mice at baseline (n=7–8 mice/group, pooled from 3 independent experiments). t-test. (D) Expression of sebum-associated AMPs in the skin (n=16 mice/group, qPCR normalized to Hprt expression, pooled from 3 independent experiments). t-test. (E) Correlation analysis of sebaceous gland (SG) gene expression versus TSLP expression in human skin (n=36 healthy subjects). Pearson correlation and linear regression slope test. (F) Model for pharmacologic and homeostatic roles of TSLP-driven sebum secretion. N.S., P≥0.05, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Data are mean ± SEM.
Finally, we asked whether this TSLP–sebum axis might be operational in humans as well. Examination of TSLP and a panel of 18 sebaceous gland-associated genes in a publicly available dataset (GSE98774) revealed that TSLP expression is significantly and positively correlated with sebaceous gland gene expression in healthy human skin (Fig. 6E and fig. S15, A to R). To ensure that this correlation was specific to the sebaceous gland gene set, we iteratively chose 10,000 random sets of 18 genes and plotted their correlation values versus TSLP expression. This resulted in a normal distribution of correlation values, where the sebaceous gland gene set lay in the top 4% of correlation values (fig. S15S). These findings are consistent with previous studies suggesting that TSLP expression is higher in seborrheic versus dry areas of healthy human skin (39). Thus, physiological levels of TSLP may control homeostatic sebum production in humans as well.
DISCUSSION
The results presented here support a model (Fig. 6F) in which therapeutic levels of TSLP induce selective white adipose loss by directly acting on T cells to induce sebum hypersecretion, whereas physiologic levels of TSLP regulate homeostatic sebum production and skin barrier function. Our data provide a therapeutic proof-of-concept that adipose loss can be achieved by secreting calories from the skin in the form of energy-rich sebum. Furthermore, we have demonstrated how the immune system plays an important role in sebum release.
Sebum production can be linked to systemic energy balance. Significant decreases in sebum production are seen within 1–2 weeks of limiting caloric intake (40) and changes in diet composition alter sebum composition (41). Thus, there is cross-regulation between skin energy expenditure and whole-body metabolism. In our model, we found that mice lose ~1.2 grams of adipose post TSLP-AAV injection, which is equivalent to loss of 1.5 kcal per day or ~15% of total daily caloric intake (42). At baseline, humans secrete ~130 kcal of sebum each day. A fourfold increase in the sebum production rate could result in an ~0.5 kg per week loss of WAT (43). Notably however, TSLP loss-of-function mutations may not necessarily cause adipose tissue gain in humans, since decreasing homeostatic sebum secretion by ~50% (based on Tslpr−/− mouse experiments) would only result in an ~65 kcal per day increase in caloric retention, which could easily be compensated for by other minute metabolic changes that decrease energy intake or increase energy expenditure. Thus, although weight is unlikely to be controlled by sebum release at homeostasis, it is feasible that the selective loss of WAT could be achieved via “sweating fat” in humans by therapeutically shifting sebum release into high gear. Diverting lipids towards the skin and away from the liver could also lead to improvement of obesity-associated NAFLD and NASH, as seen in our mouse models.
It is surprising that adaptive immunity regulates an innate aspect of skin barrier function such as sebum secretion. Although the control of sebum secretion by hormones is well-established, little is known about how the immune response is involved. Here, by uncovering the mechanism of therapeutic TSLP-driven adipose loss, we describe a role for TSLP and T cells in the upregulation of skin barrier function by sebum secretion. Recently, it was reported that ILCs in the skin negatively regulate sebaceous gland size and lipid content (44). Although we did not find a role for ILCs in our mouse model, it is possible that ILCs and T cells play opposing functions in sebum secretion.
Extensive studies in atopic dermatitis (AD) patients show that pathologic TSLP expression is associated with skin inflammation, skin barrier dysfunction, and decreased sebum production (22, 25, 27, 45–48). Although this may seem incongruent with our findings that TSLP promotes sebum secretion, the loss of skin barrier function and sebum production in AD is due to numerous factors, including the upregulation of other downstream type 2 cytokines and activation of proteases such as kallikreins and cathepsins (49–51). Furthermore, TSLP biology in humans is complex, since humans express two isoforms of TSLP—a long form (lfTSLP) and a short form (sfTSLP)—whereas mice and other mammals express only one isoform (52, 53). lfTSLP binds to the human TSLPR and is only induced and upregulated in inflammatory conditions such as allergic and atopic pathology. By contrast, sfTSLP binds to an unknown receptor, is expressed constitutively by normal healthy skin, is upregulated by homeostatic signals such as vitamin D, and exhibits AMP activity and anti-inflammatory functions (53–60). Thus, the homeostatic process of sebum secretion in humans may be regulated by sfTSLP rather than by lfTSLP. Our analysis of normal human skin showing a correlation between TSLP and sebum-related genes (Fig. 6E) is most likely to be sfTSLP, since lfTSLP is not generated in normal healthy skin. Furthermore, in AD patients, it has been shown that sfTSLP is decreased. By contrast, lfTSLP is increased in lesional compared to non-lesional skin (61). Thus, in AD, increased lfTSLP rather than sfTSLP generation may result in the observed skin barrier pathology, whereas decreased sfTSLP generation may explain why AD individuals have diminished sebum production.
Topological surveys comparing sebaceous gland-rich versus -poor regions of healthy human skin have shown that gland-rich areas have increased AMP expression, unique microbiomes, and a non-activated, tolerogenic, immune cell surveillance phenotype (39, 62). Thus, it is likely that TSLP and its isoforms play critical and unexplored roles that extend beyond pathologic inflammation by functioning in healthy skin barrier maintenance, sebum secretion, and immunometabolic regulation.
Materials and Methods
Mice.
C57BL/6 and B6-LY5.1/Cr mice were purchased from Charles River Laboratories (strain no. 556 and 564). Ob/ob, Scd1−/−, Rag2−/−Il2rg−/−, Rosa26DTA (DTA), and Cd11cCre mice were purchased from Jackson Laboratories (strain no. 000632, 005956, 014593, 009669, and 008068). OT-I (Tg(TcraTcrb)1100Mjb) Rag2−/− mice were purchased from Taconic (strain no. 2334-M). EoCre mice were created as previously described (63). Cd11cCre and Rosa26DTA mice were crossed to generate DC-deficient mice, and EoCre and Rosa26DTA mice were crossed to generate eosinophil-deficient mice. Tslpr−/− mice, as previously described (64), were a kind gift from W.J. Leonard (NIH) and were bred and maintained in our facility. Ebeta−/−, Rag2−/−, Foxp3GFP, Foxp3DTR-eGFP, Ucp1−/−, and Il4−/− mice were also bred and maintained in our facility. For CD4+ and CD8+ T cell depletion, antibodies against CD4 (BioXCell, BE0003–1, clone GK1.5) and CD8 (BioXCell, BE006, clone 2.43) were administered via intraperitoneal injection at a dose of 200 μg per mouse on days 0, 2, 4, 8, and 12 or on days 14, 16, 18, 22, and 26 following AAV injection. Foxp3GFP and Foxp3DTR-eGFP mice were administered diphtheria toxin (Santa Cruz Biotechnology, sc-391135) intraperitoneally at a dose of 10 μg per kg weight on days −2, 0, 2, 4, 6, 8, 10, and 12 after AAV injection. Tslprfl/+ mice were generated by the University of Pennsylvania CRISPR/Cas9 Mouse Targeting Core using the CRISPR/Cas9 system as previously described (65). Briefly, a long single-stranded DNA repair template (LONGssDNA) containing two LoxP sites in cis was designed and directed against intronic regions flanking the third exon of the Tslpr gene (sequence below). The Cas9-mediated double-stranded DNA breaks were resolved by homologous directed repair with ablation of the endogenous sequence and insertion of the LONGssDNA in C57BL/6 mouse zygotes. The resulting founder mice were Tslprfl/+, as confirmed by gene sequencing, and were outbred to C57BL/6 mice to ensure germline transmission and control for potential off-target effects. These mice were then crossed to Cd4Cre mice (Jackson Laboratories, 022071) to generate Cd4Cre Tslprfl/fl mice. For all NC-fed mouse experiments, mice were harvested 2 weeks after AAV injection and eWAT (bilateral), iWAT (bilateral), BAT (bilateral), and quadriceps muscle (left quadriceps) masses were measured. Ob/ob mice were injected with AAV for 5 weeks starting from 12 weeks of age. Unless otherwise specified, all mice were 8–10-week-old males at the time of use, were housed in pathogen-free conditions, and were treated in strict compliance with the Institutional Animal Care and Use Committee regulations at the University of Pennsylvania.
LONGssDNA template sequence (5′ to 3′ with LoxP sites in bold):
GGTACTTCCTTTCAGACTCTGATGAATGACAGGAAGGAGCGGTACTTCCTTTCTGCCCATCACTTTCTGTTTGAGGTGTTGAAATTCAGATGCTCGTGGGATAACTTCGTATAATGTATGCTATACGAAGTTATCGGGACTTCTGGTCCATCACTTCCCATCCAGCTGATCCTTCCCCCTTAATCCCCGACTCTGCAGTTATGGCACTGGCGCCCTGCAACCCTGCCCGCGATATTTCCTGTCCGGCGCTGGTGTCACTTCCGGGTGCATCCTCCCCGCGGCGAGGGCGGGGCTGCTGGAGCTGGCACTGCGCGACGGAGGCGGGGCCATGGTGTTTAAGGCTAGGCAGCGCGCGTCCGCCTGGCGTGAGTGGTGTGGGCGTGACATGGAGGGAGGGGGCGGGGCCTTGGGCGGAAGTACGCATAACTTCGTATAATGTATGCTATACGAAGTTATAGGAAGTTGCCTGATGATGTCTCTTCAACTCCCACTGAGTGGGGCAAACAGGAACTGGGCGGCGCTTAGACGGGAAGTTGGTCTTTGATACCGGAATTGG
HFD and MCDD models.
Mice were either fed a HFD consisting of 45 kcal% fat (Research Diets, D12451) or an MCDD consisting of 60 kcal% fat with 0.1% methionine and no added choline (Research Diets, A06071302). For HFD models, mice were either injected with AAV at day 0 or after 10 weeks of being on HFD while continuing on HFD for an additional 4 weeks (HFD for 14 weeks with AAV injection at week 10). Mice were weighed weekly. For MCDD models, mice were injected with AAV after 4 weeks of diet while continuing on MCDD for an additional 4 weeks (MCDD for 8 weeks with AAV injection at week 4).
AAV injections and cytokine levels.
Control-AAV (AAV8.TBG.PI.eGFP.WPRE.bGH) and TSLP-AAV (AAV8.TBG.PI.mTSLP.IRES.eGFP.WPRE.bGH) were generated by the Penn Vector Core. For all experiments besides the TSLP-AAV titrations, mice were injected intravenously with 5×1010 genome copies of AAV. For the TSLP-AAV titration curves, mice were injected intravenously with fivefold dilutions of 5×1010, 1×1010, 2×109, 4×108, and 8×107 genome copies of AAV per mouse. Mice were euthanized, blood was collected and centrifuged, and serum TSLP or IL-4 levels were measured using murine specific ELISAs (TSLP: R&D, MTLP00 and IL-4: BioLegend, 431104) according to the manufacturers’ protocols.
RMA tumor injections.
cDNA encoding TSLP was subcloned into the retroviral vector MigR to transduce RMA cells. GFP+ RMA cells were sorted and grown in RPMI media containing 10% FBS and 1% Pen-Strep. A total of 1×106 cells were injected subcutaneously into the flanks of Wt mice. Tumor size was measured to ensure equal tumor sizes between groups. Mice were euthanized at day 12 and eWAT mass was measured. Serum was collected for TSLP ELISA quantification.
BM chimeras.
Femurs and hip bones were isolated from donor mice and crushed with a mortar and pestle to obtain bone marrow. Red cells were lysed with ACK lysing buffer (ThermoFisher, A1049201) and bone marrow was filtered through a 70-μm filter (Sigma, CLS431751). Host mice were irradiated with 1000 cGy and injected intravenously with 2×106 donor bone marrow cells. Four weeks later, BM chimeras were injected with AAV.
Adoptive transfers.
Wt, Tslpr−/−, and OT-I Rag2−/− splenic T cells were isolated using a T cell negative selection kit (STEMCELL Technologies, 19851) and then sorted for CD19−B220−NK1.1−CD11c−CD11b− and CD90.2+CD4+ or CD8+ cells using a FACSAria cell sorter (BD Biosciences). Sorted cells (2×106) were transferred intravenously into Rag2−/−Il2rg−/−, Rag2−/−, or Tslpr−/− mice. Four weeks later, all adoptively transferred mice were injected with AAV. For lymph node T cell adoptive transfers, T cells from the skin-draining inguinal and cervical lymph nodes of Control-AAV or TSLP-AAV injected Wt mice were isolated 4 weeks post AAV and transferred intravenously into Tslpr−/− mice, which were lightly irradiated with 500 cGy prior to the cell transfers.
FTY720 treatment.
2.5% DMSO (Sigma, D2650) or 0.1% FTY720 (Tocris, 6176) + 2.5% DMSO were dissolved into PBS and injected intraperitoneally into mice daily at a dose of 10 μl per gram of body weight.
In vivo metabolic analysis.
Mice were individually housed and monitored during days 9–11 post AAV injection by the University of Pennsylvania’s Rodent Metabolic Phenotyping Core and by the Seale lab, using the OxyMax Comprehensive Laboratory Animal Monitoring System (CLAMS). Mice were fed NC and maintained on a standard 12:12 light–dark cycle at 24°C with ad libitum access to food and water. Accumulated feces were collected from singly housed mice on days 9–11 after AAV injection and bomb calorimetry of the feces was performed by the University of Michigan Mouse Phenotyping Core and by the University of Pennsylvania’s Rodent Metabolic Phenotyping Core. Fat mass and lean mass were measured by 1H-NMR spectroscopy by the University of Pennsylvania’s Rodent Metabolic Phenotyping Core on days 0 and 14 following AAV injection. For GTTs, mice were fasted overnight for 14–16 hours, and injected intraperitoneally with 10 μl per g body weight of a 20% w/v dextrose solution. Blood glucose levels were measured from blood collected from the tail vein at 0, 15, 30, 60, 90, and 120 minutes post dextrose challenge. Zero-minute time-point measurements were used to determine fasting glucose and insulin levels. Blood glucose measurements were performed using a handheld glucometer (Contour, 7151G) and plasma insulin levels were determined by ELISA (Crystal Chem, 90080). HOMA-IR index values were calculated as described previously (66). OGTT was performed in a similar manner as GTT, except that the dextrose solution was administered via oral gavage. OFTT was performed as previously described (67) and blood TG levels were measured via the Infinity TG kit (ThermoFisher, TR22421). Serum ALT assays were performed by the Translational Core Laboratory of the Children’s Hospital of Philadelphia (CHOP) Research Institute. For urinalysis, urine was collected from mice 10 days after AAV injection and 50 μl of urine was placed on urine dipsticks (Ketostix, 2881; Chemstrip 2GP, 11895397) for colorimetric analysis. Urine FFAs were measured using the FFA Fluorometric Assay Kit according to the manufacturer’s protocol (Cayman Chemicals, 700310).
Body condition scoring.
For BCS scoring, mice were scored on days 0, 3, 7, 10 and 14 after AAV injection with grading on a scale of 0–2 in five categories as follows: (1) Weight Loss: 2 points if >25%, 1 point if 10–25%, 0 points if <10%; (2) Hunching: 2 points if severe hunching that impairs movement, 1 point if hunched at rest, 0 points if normal posture; (3) Visible Lethargy: 2 points if stationary unless stimulated, 1 point if mild to moderately decreased movement, 0 points if normal; (4) Lethargy to touch: 2 points if stationary when stimulated, 1 point if mild to moderately to decreased movement upon stimulation, 0 points if normal; (5) Ruffled fur: 2 points if severe ruffling and poor grooming, 1 point if mild to moderate ruffling, 0 points if normal. Points from each category were summed to form the total score.
Liver TGs.
Liver tissue (20–100 mg) was homogenized with 10 μl of 5% NP-40 (Igepal CA630, Sigma-Aldrich, I8896) per mg liver tissue. The resulting homogenate was boiled at 80°C for 10 min, cooled to room temperature, boiled again at 80°C for 10 min and centrifuged at 10,000g for 10 min. The supernatant was then diluted 1:20 with dH2O. TG levels were quantified using the Infinity TG Kit.
Ex vivo glycerol release.
One hundred milligrams of eWAT from each sample was minced and transferred into a 24-well plate containing 300 μl of 3.7% BSA Krebs-Ringer Bicarbonate (KRB) buffer. Samples were incubated at 37°C for 30 min, and then the supernatant was collected and used for the Free Glycerol Reagent assay following the manufacturer’s protocol (Sigma, F6428). KRB buffer contained 1 liter of KRB powder (Sigma, K4002) with 10 mM NaHCO3 (Sigma, S5761) and 30 mM HEPES (ThermoFisher, 15630080), pH 7.4. The assay was performed in triplicates for each sample.
Serum FFAs and TGs.
For mice on NC, blood was collected from the tails of mice at 3, 7, 10, and 14 days post-AAV injection. For mice on HFD, mice were first fed HFD for 10 weeks, then injected with AAV, and blood was collected from the tail veins at 0, 1, 2, and 3 weeks post-AAV injection. Serum FFAs and TGs were measured using the FFA Fluorometric Assay Kit and the Infinity TG Kit, respectively.
Hair lipid isolation and TLC.
A 2.5 cm × 5 cm area of hair was shaved from back skin and immersed in 4 ml of chloroform–methanol (Sigma, 288306 and Sigma, 322415) (2:1 v/v) followed by 4 ml of acetone. Lipid extracts were pooled, syringe filtered, dried down overnight under a stream of N2 gas, and resuspended in equal volumes of chloroform–methanol (4:1, v/v) for loading onto TLC plates (Sigma, 100390). The TLC plates were developed three times using the following: (1) hexane (Sigma, 296090):isopropyl diether (Sigma, 673803):acetic acid (80:20:1) up to 50% of the plate height, (2) hexane:benzene (Sigma, 401765) (1:1) up to 80% of the plate height, and (3) hexane up to 90% of the plate height. Plates were allowed to dry between each developing solution. Plates were sprayed uniformly with 10% cupric sulfate (Sigma, 451657)/8% phosphoric acid (Sigma, P6560) solution and then baked at 120°C for 20 min to visualize lipid species. ImageJ (NIH) was used to quantify the intensity and area of the bands. The TLC non-polar lipid mixture A (Matreya, 1129) was used as a standard to identify lipid classes.
MC903 treatment.
MC903 (Tocris, 2700) was dissolved in 100% ethanol (Decon Labs, 2716) to 200 μM. Mice were treated on both ears with 2 nmol/10 μl of MC903 or ethanol vehicle on days 1–5 and 8–12. Serum and hair were collected for TSLP ELISA and TLC analysis respectively on day 14.
Flow cytometric analysis.
Dermal sheets of ear skin were separated, minced, and incubated in Hank’s balanced salt solution (ThermoFisher, 24020117) containing 0.25 mg/ml of Liberase TL (Roche, 5401020001), 0.1 mg/ml of DNase I (Roche, 10104159001), and 0.7 mg/ml of collagenase D (Roche, 11088882001) for 2 hours with shaking at 37°C. Contents were then strained though a 70-μm filter into a new tube containing 10 ml of PBS, centrifuged at 4°C for 5 min at 180g, and resuspended in PBS. Cells were stained with live–dead stain and cell surface stains at 4°C for 15 min in PBS. Flow cytometry was performed with an LSR II or LSR Fortessa (BD Biosciences). Data were analyzed using FlowJo software (TreeStar). Staining antibodies used are listed in table S2. Live/Dead Near-IR (ThermoFisher, L10119, 1:1000 dilution) and Live/Dead Aqua (ThermoFisher, L34957, 1:200 dilution) stains were used to exclude dead cells.
Histology.
Tissues were fixed in 10% formalin at 4°C overnight and embedded in paraffin before H&E or IHC staining. Liver sections were processed and stained by the University of Pennsylvania Cardiovascular Institute’s Histology and Gene Expression Core. PicroSiriusRed stains were performed as previously described (68). Skin samples were processed and stained (H&E, Ki67, ORO, CD3, CD4, CD8, Krt5, and Krt79) by the University of Pennsylvania’s Cutaneous Phenomics and Transcriptomics Core. Liver, lung, kidney, small intestine, and colon sections were processed and stained for H&E by the CHOP Research Institute’s Pathology core. Immunofluorescence was additionally performed on the formalin-fixed paraffin-embedded kidney tissue following protease digestion for IgG, IgA, IgM, C3, C1q, and kappa and lambda light chains by Arkana Labs. An independent pathologist then assessed and provided commentary on organ architecture and inflammation. For ORO staining of skin samples, frozen skin samples were embedded in cryosectioning medium prior to sectioning and staining. For quantification of sebocyte size, ORO lipid content, and PicroSiriusRed fibrosis, 8–10 sections at 20X magnification were captured per animal. For H&E skin sections, ImageJ was used to draw circumscribing ellipses around sebocytes to quantify sebocyte area. For ORO skin and PicroSiriusRed liver sections, ImageJ was used to split RGB channels, threshold positive staining, and measure the percentage of area stained and staining intensity. Integrated density was calculated as area × staining intensity. For quantification of cells in various SG zones, basal layer cells were identified as Krt5Bright, Krt79Dim, DAPI+ cells on the periphery of the SGs, differentiated sebocytes were identified as broad, flat Krt5Dim, Krt79Bright cells in the interior of the SGs, and were further classified as having a nucleus (mature sebocyte) or not (dying sebocyte) by DAPI staining. Although one single z-plane is shown, 4-μm stacks were acquired for quantitation to distinguish between cells without nuclei and those with nuclei in different z-planes. Samples were imaged using a 63X 1.4 NA objective on an Axiovert 200M (Zeiss) with a spinning disc confocal system (UltraVIEW ERS6, Perkin Elmer) equipped with an ORCA-Flash 4.0 CMOS camera (Hamamatsu). Images were acquired using Volocity v6.3 software (Perkin Elmer) and were prepared for publication using ImageJ (NIH). Immunohistochemistry antibodies used are listed in Table S3. A mouse and rabbit specific HRP IHC kit (Abcam, ab236466) was used to detect staining.
RNA isolation, cDNA synthesis, and qPCR.
Hair was shaved and back skin was isolated and stored in RNAlater (Qiagen, 76104) according to the manufacturer’s protocol. Skin was then transferred into TRIzol (ThermoFisher, 15596026) and minced in tissue lyser tubes (MP Biomedicals, 116910050) using a homogenizer (Qiagen, 69980). RNA was isolated according to the TRIzol manufacturer’s instructions. Glycogen (Roche, 10 901 393 001) was used as a carrier. Isolated total RNA was quantified using a Nanodrop 1000. cDNA was synthesized using Superscript Vilo (ThermoFisher, 11754050) according to the manufacturer’s protocol. qPCR was performed using the Taqman Fast gene expression assay (Taqman, 4444557) according to the manufacturer’s protocol, using the following primers from Taqman: Hprt (Mm03024075_m1), Camp (Mm00438285_m1), Defb4 (Mm00731768_m1), and Lcn2 (Mm01324470). All qPCR reactions were performed on a ViiA7 Real-Time PCR instrument (ThermoFisher).
Human gene expression analysis.
Publicly available human skin gene expression data was obtained from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98774. Gene expression values for sebum-production associated genes (SCD, FADS2, PPARG, FA2H, DGAT1, DGAT2, FABP4, FABP5, ACACA, FASN, AWAT1, ELOVL1, ELOVL3, ELOVL4, ELOVL5, MOGAT1, MOGAT2, MOGAT3) were plotted against corresponding TSLP expression values for each sample. For normalized SG gene expression, expression of each sebum-associated gene dataset was normalized to a mean of 0 and standard deviation of 1 and then all SG genes from each sample were averaged together and plotted against the corresponding TSLP expression. The correlation of the SG gene set (n=18 genes) was then compared to the correlation of 10,000 iterations of randomly selected gene sets (of n=18 genes/gene set, also normalized to a mean of 0 and standard deviation of 1). Pearson correlation and linear regression analysis were performed using R (https://www.r-project.org). The code is available upon request.
Statistical analysis and figure construction.
Data are reported as mean ± SEM. All measurements were made from distinct biological samples. Sample sizes were determined based on prior lab experiences on the number of mice needed for statistical significance, as well as the availability of mice. Samples were randomly allocated to experimental treatment groups. For pooled data, a mixed effect model was used with ANOVA F-tests and p-values using Satterthwaite’s or Kenward-Roger’s method for denominator degrees-of-freedom and F-statistic. Models were fitted using R with lmer from the lmerTestpackage. The model Y ~ (1|sample id) + time + TSLP + experiments+ TSLP*time was used when comparing differences across time, where a mixed effect regression model with random intercept was fitted, and then tested for whether the coefficient of TSLP*time = 0 or not term. The model Y ~ 1 + TSLP + experiments was used for comparing single time points, where a linear regression model was fitted, and then tested for whether the coefficient of TSLP term = 0 or not. For data that were normally distributed but were not pooled, statistical significance was determined by two-sided Student’s t test (Fig. 4A) or two-way ANOVA (Fig. 4C, and fig. S6A and D–H). Correlation analyses were conducted using Pearson correlation and linear regression slope test (Fig. 6E and fig. S15A–R). Statistical analyses were performed with Prism 8 and R. Data figures were created with Prism 8 and R. The Print Page Summary figure and Fig. 6F were created with BioRender (BioRender.com).
Supplementary Material
Acknowledgments:
We thank members of the T.K. lab (P. Lundgren, M. Kim, and M. Shimizu) for their technical assistance with experiments, as well as members of the Z.A. lab (B. Gosis and S. Jeong), and the P.S. lab (J. Ishibashi and A. Angueira) for their scientific and technical advice. We also thank L. Li of the Penn Cardiovascular Institute Histology and Gene Expression Co-Op and S. Prouty and T. Dentchev of the Cutaneous Phenomics and Transcriptomics Core in the Penn Skin Biology and Disease Resource-Based Center for their assistance with histological sample processing and staining. Lastly, we thank L. Joannas of the University of Pennsylvania CRISPR/Cas9 Mouse Targeting Core for generating the Tslprfl/+ mouse line.
Funding:
This research was supported by grants from the National Institutes of Health (R01-HL111501, R01-AI121250, R01-AR070116, T32-HL07439), the Doris Duke Charitable Foundation, and the University of Pennsylvania Medical Scientist Training Program. We also thank the Penn Vector Core, the Penn Mouse Phenotyping, Physiology & Metabolism Core at the Diabetes Research Center (DRC) of the Institute for Diabetes, Obesity & Metabolism (IDOM), the Penn Diabetes Endocrine Research Center Grant (P30-DK19525), the Cutaneous Phenomics and Transcriptomics Core in the Penn Skin Biology and Disease Resource-Based Center (P30-AR069589, Cotsarelis), the Penn Cardiovascular Institute Histology and Gene Expression Co-Op, the University of Michigan Mouse Metabolic Phenotyping Center (U2-CDK110768), the Penn CRISPR/Cas9 Mouse Targeting Core, and the Pathology Core and Translational Core Laboratory of the CHOP Research Institute.
Footnotes
Competing interests: U.S. Provisional Patent Application No. 62/972,462 was filed by the University of Pennsylvania. The inventors are Taku Kambayashi and Ruth Choa. The patent application is based on the finding that TSLP induces sebum secretion from the skin and that this can lead to treatment of skin conditions and obesity and its complications.
Data and materials availability:
The datasets analyzed during the current study are available in the NCBI Gene Expression Omnibus, GSE98774. All data are available in the main text or the supplementary materials.
References and Notes:
- 1.Hruby A, Hu FB, The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 33, 673–689 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell NS, Catenacci VA, Wyatt HR, Hill JO, Obesity: overview of an epidemic. Psychiatr Clin North Am 34, 717–732 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mokdad AH et al. , Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 289, 76–79 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Qiu Y et al. , Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157, 1292–1308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brestoff JR et al. , Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu D et al. , Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MW et al. , Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molofsky AB et al. , Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210, 535–549 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feuerer M et al. , Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolodin D et al. , Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab 21, 543–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong CK, Hu S, Cheung PF, Lam CW, Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: implications in allergic inflammation. Am J Respir Cell Mol Biol 43, 305–315 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Kabata H et al. , Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitajima M, Lee HC, Nakayama T, Ziegler SF, TSLP enhances the function of helper type 2 cells. Eur J Immunol 41, 1862–1871 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziegler SF et al. , The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol 66, 129–155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziegler SF, Artis D, Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol 11, 289–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhou B, Functions of thymic stromal lymphopoietin in immunity and disease. Immunol Res 52, 211–223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai T, TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int 61, 3–17 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Leichner TM et al. , Skin-derived TSLP systemically expands regulatory T cells. J Autoimmun 79, 39–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kashiwagi M et al. , Direct control of regulatory T cells by keratinocytes. Nat Immunol 18, 334–343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Astrakhan A et al. , Local increase in thymic stromal lymphopoietin induces systemic alterations in B cell development. Nat Immunol 8, 522–531 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Iseki M et al. , Thymic stromal lymphopoietin (TSLP)-induced polyclonal B-cell activation and autoimmunity are mediated by CD4+ T cells and IL-4. Int Immunol 24, 183–195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li M et al. , Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci U S A 103, 11736–11741 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessup HK et al. , Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol 181, 4311–4319 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z et al. , Thymic stromal lymphopoietin overproduced by keratinocytes in mouse skin aggravates experimental asthma. Proc Natl Acad Sci U S A 106, 1536–1541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demehri S, Morimoto M, Holtzman MJ, Kopan R, Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol 7, e1000067 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim BS et al. , TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med 5, 170ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim BS et al. , Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol 193, 3717–3725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noti M et al. , Thymic stromal lymphopoietin-elicited basophil responses promote eosinophilic esophagitis. Nat Med 19, 1005–1013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noti M et al. , Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J Allergy Clin Immunol 133, 1390–1399, 1399.e1391–1396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldrath AW, Bogatzki LY, Bevan MJ, Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med 192, 557–564 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer H et al. , Holocrine Secretion of Sebum Is a Unique DNase2-Dependent Mode of Programmed Cell Death. J Invest Dermatol 137, 587–594 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Veniaminova NA et al. , Niche-Specific Factors Dynamically Regulate Sebaceous Gland Stem Cells in the Skin. Dev Cell 51, 326–340.e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Y et al. , Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet 23, 268–270 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Sampath H et al. , Skin-specific deletion of stearoyl-CoA desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J Biol Chem 284, 19961–19973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lovaszi M, Szegedi A, Zouboulis CC, Torocsik D, Sebaceous-immunobiology is orchestrated by sebum lipids. Dermatoendocrinol 9, e1375636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee DY et al. , Sebocytes express functional cathelicidin antimicrobial peptides and can act to kill propionibacterium acnes. J Invest Dermatol 128, 1863–1866 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakatsuji T et al. , Sebum free fatty acids enhance the innate immune defense of human sebocytes by upregulating beta-defensin-2 expression. J Invest Dermatol 130, 985–994 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brogden NK, Mehalick L, Fischer CL, Wertz PW, Brogden KA, The emerging role of peptides and lipids as antimicrobial epidermal barriers and modulators of local inflammation. Skin Pharmacol Physiol 25, 167–181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dajnoki Z et al. , Sebaceous Gland-Rich Skin Is Characterized by TSLP Expression and Distinct Immune Surveillance Which Is Disturbed in Rosacea. J Invest Dermatol 137, 1114–1125 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Pochi PE, Downing DT, Strauss JS, Sebaceous gland response in man to prolonged total caloric deprivation. J Invest Dermatol 55, 303–309 (1970). [DOI] [PubMed] [Google Scholar]
- 41.MACDONALD I, CHANGES IN THE FATTY ACID COMPOSITION OF SEBUM ASSOCIATED WITH HIGH CARBOHYDRATE DIETS. Nature 203, 1067–1068 (1964). [DOI] [PubMed] [Google Scholar]
- 42.N. R. C. U. S. o. L. A. Nutrition. (1995). [Google Scholar]
- 43.Downing DT, Stewart ME, Strauss JS, Estimation of sebum production rates in man by measurement of the squalene content of skin biopsies. J Invest Dermatol 77, 358–360 (1981). [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi T et al. , Homeostatic Control of Sebaceous Glands by Innate Lymphoid Cells Regulates Commensal Bacteria Equilibrium. Cell 176, 982–997.e916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leyva-Castillo JM, Hener P, Jiang H, Li M, TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J Invest Dermatol 133, 154–163 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Cianferoni A, Spergel J, The importance of TSLP in allergic disease and its role as a potential therapeutic target. Expert Rev Clin Immunol 10, 1463–1474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han H, Roan F, Ziegler SF, The atopic march: current insights into skin barrier dysfunction and epithelial cell-derived cytokines. Immunol Rev 278, 116–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu YJ, Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med 203, 269–273 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cork MJ et al. , Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol 129, 1892–1908 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Rerknimitr P, Otsuka A, Nakashima C, Kabashima K, The etiopathogenesis of atopic dermatitis: barrier disruption, immunological derangement, and pruritus. Inflamm Regen 37, 14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SE, Jeong SK, Lee SH, Protease and protease-activated receptor-2 signaling in the pathogenesis of atopic dermatitis. Yonsei Med J 51, 808–822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varricchi G et al. , Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front Immunol 9, 1595 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsilingiri K, Fornasa G, Rescigno M, Thymic Stromal Lymphopoietin: To Cut a Long Story Short. Cell Mol Gastroenterol Hepatol 3, 174–182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bjerkan L et al. , The short form of TSLP is constitutively translated in human keratinocytes and has characteristics of an antimicrobial peptide. Mucosal Immunol 8, 49–56 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Martin Mena A et al. , The Expression of the Short Isoform of Thymic Stromal Lymphopoietin in the Colon Is Regulated by the Nuclear Receptor Peroxisome Proliferator Activated Receptor-Gamma and Is Impaired during Ulcerative Colitis. Front Immunol 8, 1052 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dong H et al. , Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite-induced asthmatic airway epithelial barrier disruption. Sci Rep 6, 39559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Landheer J et al. , TSLP is differentially regulated by vitamin D3 and cytokines in human skin. Immun Inflamm Dis 3, 32–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sonesson A et al. , Thymic stromal lymphopoietin exerts antimicrobial activities. Exp Dermatol 20, 1004–1010 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Harada M et al. , Functional analysis of the thymic stromal lymphopoietin variants in human bronchial epithelial cells. Am J Respir Cell Mol Biol 40, 368–374 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Xie Y, Takai T, Chen X, Okumura K, Ogawa H, Long TSLP transcript expression and release of TSLP induced by TLR ligands and cytokines in human keratinocytes. J Dermatol Sci 66, 233–237 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Fornasa G et al. , Dichotomy of short and long thymic stromal lymphopoietin isoforms in inflammatory disorders of the bowel and skin. J Allergy Clin Immunol 136, 413–422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Béke G et al. , Immunotopographical Differences of Human Skin. Front Immunol 9, 424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doyle AD et al. , Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol 94, 17–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Al-Shami A et al. , A role for thymic stromal lymphopoietin in CD4(+) T cell development. J Exp Med 200, 159–168 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henao-Mejia J et al. , Generation of Genetically Modified Mice Using the CRISPR-Cas9 Genome-Editing System. Cold Spring Harb Protoc 2016, pdb.prot090704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matthews DR et al. , Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985). [DOI] [PubMed] [Google Scholar]
- 67.Wada M, Tamura A, Takahashi N, Tsukita S, Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 144, 369–380 (2013). [DOI] [PubMed] [Google Scholar]
- 68.Jeong SH et al. , Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest 128, 1010–1025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Veniaminova NA et al. , Keratin 79 identifies a novel population of migratory epithelial cells that initiates hair canal morphogenesis and regeneration. Development 140, 4870–4880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available in the NCBI Gene Expression Omnibus, GSE98774. All data are available in the main text or the supplementary materials.