

Abstract
Intercalated cells (IC) make up about a third of all cells within the connecting tubule and the collecting duct and are subclassified as type A, type B and Non-A, non-B based on the subcellular distribution of the H+-ATPase, which dictates whether it secretes H+ or HCO3−. Type B intercalated cells mediate Cl− absorption and HCO3− secretion, which occurs largely through the anion exchanger, pendrin. Pendrin is stimulated by angiotensin II via the angiotensin type 1a receptor and by aldosterone through the mineralocorticoid receptor. Aldosterone stimulates pendrin expression and function, in part through the alkalosis it generates. Pendrin-mediated HCO3− secretion increases in models of metabolic alkalosis, which attenuates the alkalosis. However, pendrin positive ICs also regulate blood pressure, at least partly, through pendrin-mediated Cl− absorption, and through their indirect effect on the epithelial Na+ channel, ENaC. This aldosterone-induced increase in pendrin secondarily stimulates ENaC, thereby contributing to the aldosterone pressor response. This review describes the contribution of pendrin positive ICs to Na+, K+, Cl− and acid-base balance.
Keywords: Slc26a4, pendrin, Cl−/HCO3− exchange, ENaC, intercalated cells, blood pressure
1. INTRODUCTION
The connecting segment (CNT) and the cortical collecting duct (CCD) consist of principal cells, connecting tubule cells and intercalated cells (ICs, Figure 1). Within these segments, ICs represent ~30–40% of all cells 1, although they represent only ~1% of kidney volume 2 and only ~2 to 2.5% of kidney mass 3. Nevertheless, they play an important role in kidney function. Type A intercalated cells secrete H+s through the apical plasma membrane H+-ATPase, which is stimulated during metabolic acidosis, which helps correct the acidosis 4–7. The apical plasma membrane of type B intercalated cells expresses the Cl−/HCO3− exchanger, pendrin, encoded by Slc26a4 8–10. This anion exchanger acts in series with the H+-ATPase on the basolateral plasma membrane to mediate HCO3− secretion and Cl− absorption, which increases in models of metabolic alkalosis, thereby promoting HCO3− secretion, which helps correct the alkalosis 9, 11, 12. Because ICs mediate the secretion of H+ or HCO3−, early reports studied primarily the role of these cells in acid-base balance.
Figure 1: Intercalated and principal cells transporters and channels within the cortical collecting duct:
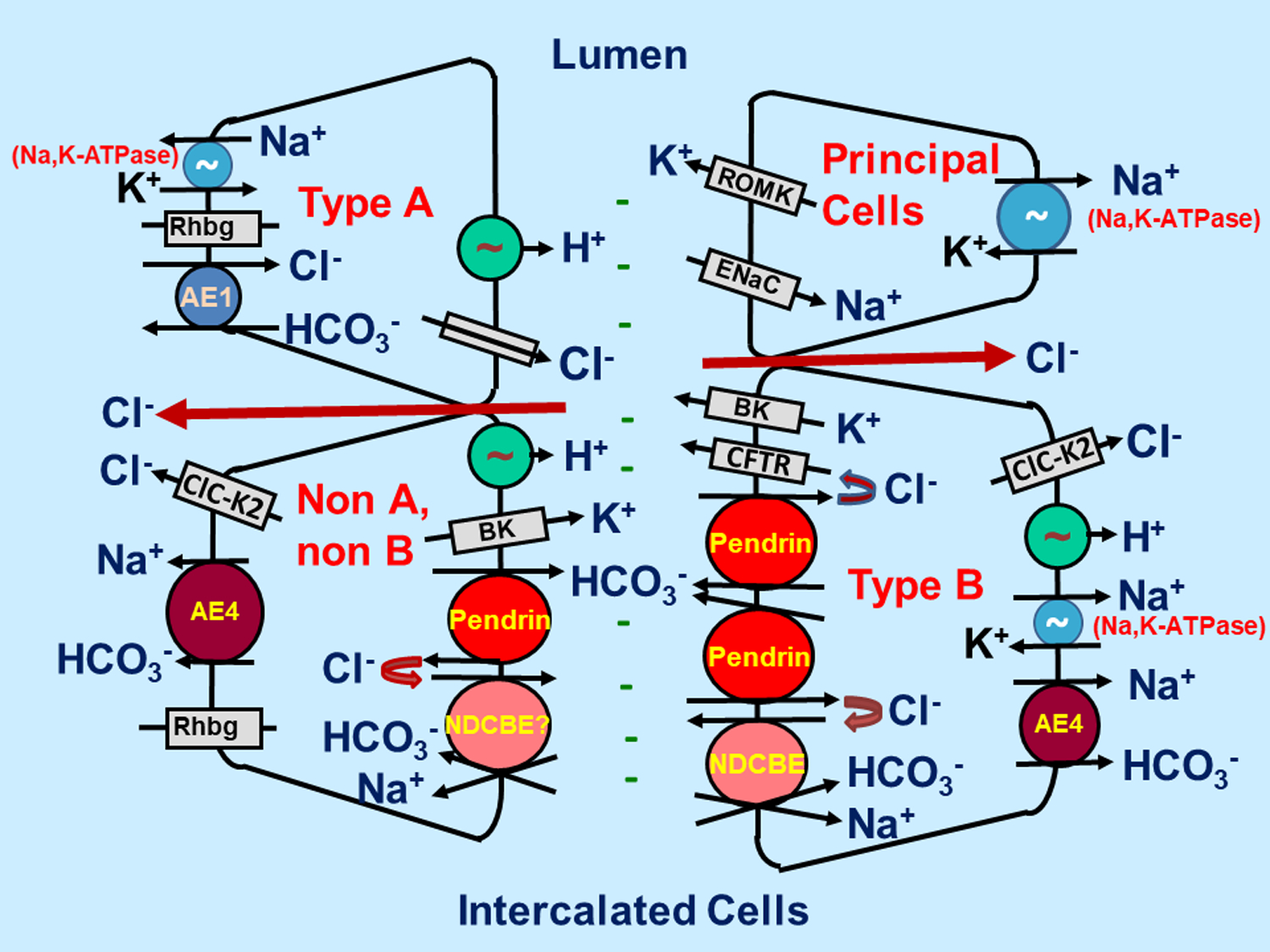
Type B ICs absorb NaCl through pendrin-mediated apical Cl− /HCO3− and the Na+-dependent Cl−/HCO3− exchanger, NDCBE, acting in tandem. Pendrin and the CFTR Cl− channel mediate HCO3− secretion, while they recycle Cl− across the apical plasma membrane. H+ and Cl− exit the cell through the basolateral plasma membrane ClC-K2 Cl− channel and the H+-ATPase, while the basolateral membrane Na+-HCO3− cotransporter, AE4, and the Na,K-ATPase mediate Na+ exit. In type A ICs the apical membrane H+-ATPase and the basolateral membrane Cl−/HCO3− exchanger, AE1, act in series to mediate H+ secretion. Principal cells absorb Na+ through the apical membrane epithelial Na+ channel, ENaC, which generates the lumen-negative transepithelial voltage, providing the driving force for K+ secretion through K+ channels such as ROMK and Maxi K+ channels.
More recent studies have shown that ICs play an important role in Na+ 13, 14, K+ 15, 16 and Cl− 17, 18 balance, as well as in blood pressure regulation11, 14. Pendrin regulates NaCl balance and hence blood pressure by mediating Cl− absorption 17, and by modulating ENaC-dependent Na+ absorption 14, 19, 20. Therefore, with ablation of the gene encoding pendrin (Slc26a4), a natriuresis, a chloriuresis, and reduced blood pressure are observed in both people and rodents 11, 14, 21, 22. As such, there has been interest in developing pendrin inhibitors for use as diuretics or antihypertensives in clinical practice 23, 24.
Pendrin-positive ICs also modulate blood pressure through the action of other transporters within these cells. For example, Slc4a8 encodes a Na+-dependent Cl−/HCO3− exchanger (NDCBE), which operates in tandem with pendrin-mediated Cl−/HCO3− exchange to mediate net NaCl absorption by the CCD (Figure 1) 13.
Pendrin is greatly upregulated by aldosterone and angiotensin II, which increases renal NaCl absorption. In so doing, this anion exchanger contributes to the rise in blood pressure that occurs in response to these hormones 11, 17, 25, 26. Whether or not angiotensin II stimulates pendrin independently of its effect on aldosterone release is debated 26, 27. Aldosterone has a unique interaction with the intercalated cell mineralocorticoid receptor (MR) due to a novel MR phosphorylation site found only in these cells, giving the receptor novel properties 28. Dephosphorylation of the MR at S843, which is enhanced with either angiotensin II or reduced serum K+ concentration, promotes aldosterone binding to the receptor.
Electroneutral, pendrin-dependent transport provides a mechanism for renal NaCl absorption when renal K+ conservation is needed, such as during vascular volume contraction 16. At least under some conditions, serum K+ is lower in pendrin null mice as well as in people with SLC26A4 inactivation sequence variants, relative to controls 15, 29–31. However, the conditions that unmask hypokalemia in the mutant mice is unresolved.
This review summarizes the role of pendrin-positive intercalated cells in acid-base, as well as in Na+, K+ and Cl− balance.
II. THE ROLE OF PENDRIN-MEDIATED HCO3− SECRETION IN ACID-BASE BALANCE.
A. The Role of Intercalated Cells in Acid-Base balance:
Because the ability of intercalated cells in the CCD to secrete or absorb HCO3− correlates with the subcellular distribution of the H+-ATPase in the cell 32, 33, IC subtype is determined by H+-ATPase subcellular distribution and whether or not it expresses the Cl−/HCO3− exchangers, AE1 1, 34 or pendrin 8–10 (Figure 1). In type A ICs, the H+-ATPase localizes to the apical plasma membrane, whereas AE1 is found on the basolateral membrane. This subtype mediates H+ secretion, which is stimulated in models of metabolic acidosis 35 through increased apical plasma membrane H+-ATPase abundance and activity 36, 37.
The CCD secretes HCO3− through an apical, electroneutral, stilbene-insensitive Cl−/HCO3− exchanger known as pendrin, which localizes to type B intercalated cells (Figure 1) 38–42 and is markedly stimulated during metabolic alkalosis 11, 18, 43–46. In type B ICs, the basolateral H+-ATPase 47 acts in series with apical plasma membrane pendrin to mediate HCO3− secretion 25, 48. AE1 immunoreactivity is not detected in this cell type 8, 10.
The third IC subtype is the Non-A, non-B IC 47, which in mouse localizes primarily to the CNT 10. Their transport properties are less understood than type B intercalated cells, which localize to the CCD in mouse, since the mouse CCD, but not the CNT, has been perfused in vitro. Non-A, non-B ICs likely mediate either HCO3− or H+ secretion, depending on the experimental condition, since both pendrin and the H+-ATPase localize to the apical membrane 49 47.
B. Acid-Base balance in people and mice with Slc26a4 gene ablation
Inactivation SLC26A4 sequence variants results in Pendred Syndrome, which is characterized by deafness and goiter 50. While initial reports observed no kidney involvement in these individuals, after the gene responsible (slc26a4) was cloned 51 and mouse models were developed 52, slc26a4 was found to encode the protein (pendrin) that mediates the apical anion exchange of the type B IC 9, 17. Experiments in our laboratory showed that the Cl− absorption and HCO3− secretion seen in CCDs from aldosterone-treated wild type mice is markedly reduced in pendrin null mice 9, 17. Within kidney, pendrin localizes to the apical regions of human, rat and mouse type B and non-A, non-B intercalated cells of the CNT and the CCD 8–10 and within the distal portion of the distal convoluted tubule 10.
Since apical anion exchange increases in rodent models of metabolic alkalosis, our laboratory and others asked if pendrin abundance and/or subcellular distribution are regulated by changes in acid-base balance. In models of metabolic alkalosis, such as aldosterone or NaHCO3 administration, total and apical plasma membrane pendrin abundance increase 11, 18. This increase in apical plasma membrane pendrin abundance occurs more through pendrin subcellular redistribution than through changes in protein abundance 11, 18 44. Conversely, pendrin protein abundance falls in models of metabolic acidosis, as seen with either AE1 53 or the H+-ATPase B1 subunit (ATP6V1B1) 54 gene ablation or with the administration of NH4+ 44, 45, 55, 56 or a carbonic anhydrase inhibitor 56.
Since pendrin mediates HCO3− secretion, we hypothesized that in the absence of pendrin, arterial pH and HCO3− concentration will increase. To test this hypothesis, we measured arterial blood gases in aldosterone and NaHCO3-treated wild type and pendrin null mice. With either treatment, urinary pH was lower, while arterial pH and HCO3− concentration were higher in pendrin null than in wild type mice 11, 18. Therefore, the pendrin null kidney has an impaired ability to secrete HCO3−, which limits its capacity to correct an alkalosis.
While there is limited data supporting a role of pendrin in human acid-base balance, case reports have observed severe metabolic alkalosis in individuals with Pendred Syndrome following thiazide treatment 57 or vomiting 30. Moreover, pendrin immunolabel intensity is lower in kidney biopsy samples from people with distal renal tubular acidosis relative to normal controls 58, 59. Finally, human pendrin urinary exosome abundance, an index of pendrin abundance in kidney, was lower following administration of NH4Cl than NaHCO3 60, consistent with rodent observations.
III. THE ROLE OF PENDRIN IN Na+ AND K+ BALANCE
A. The mechanism of NaCl absorption by the rodent CCD:
Figure 1 shows the transporters and channels that localize to principal cells and intercalated cells. Principal cells of the CCD absorb Na+ through ENaC 20, 61. While ICs do not express ENaC 62, ENaC generates a lumen-negative transepithelial voltage that increases the driving force for electrogenic Cl− absorption either through paracellular Cl− transport or electrogenic, transcellular transport through a Cl− channel or a Cl− exchanger 20, 63–65 66. Conversely, either ENaC or Na+, K+-ATPase inhibitors reduce paracellular of electrogenic, transcellular Cl− absorption in the CCD 25.
NaCl absorption by the CCD occurs through this electrogenic, ENaC-dependent mechanism and through a thiazide-sensitive, electroneutral mechanism 13, 67. The magnitude of the amiloride- and the thiazide- sensitive components of NaCl absorption by the CCD has differed among published reports and likely depends on the species being studied and the experimental conditions 13, 63, 67.
While thiazides inhibit electroneutral, transcellular NaCl absorption in rat and mouse CCD 13, 67, the collecting duct does not express the NaCl cotransporter of the DCT (NCC, encoded by Slc12a3) 68, which is the classic target of thiazides 69. Instead, Leviel et al. observed that Slc4a8 encodes a Na+-dependent Cl−/HCO3− exchanger (NDCBE) that mediates thiazide-sensitive Na+ absorption in mouse CCD 13. In this segment, pendrin-mediated Cl−/HCO3− exchange acts in tandem with NDCBE-mediated Na+-dependent Cl−/HCO3− exchange to mediate electroneutral NaCl absorption, although other groups have not observed NDCBE transcript expression within ICs 70. Na+ absorbed across the apical plasma membrane of type B ICs exits the cell across the basolateral plasma membrane through the Na+-HCO3− cotransporter, AE4 71, and most probably the Na,K-ATPase, while Cl− exits through ClC-K2/barttin Cl− channels 72, 73.
Pendrin-mediated Cl−/HCO3− exchange also acts in parallel with CFTR-mediated Cl− secretion (Figure 1) 74 to mediate HCO3− secretion, while Cl− is recycled across the apical membrane 74. Through a mechanism analogous to its action in the exocrine pancreas, secretin acts on type B IC basolateral plasma membrane secretin receptors to stimulate HCO3− secretion through this mechanism 74. As such, following a HCO3− load, a profound metabolic alkalosis is observed in both CFTR null mice and patients with inactivation sequence variants in CFTR (cystic fibrosis) 74, 75.
While Na,K-ATPase subunit abundance is lower in intercalated than in principal cells 76–80, the Na+ pump augments IC-mediated transport. Na+ pump inhibitor application to the bath reduces Cl− absorption across intercalated cells of CCDs perfused in vitro 66, 25 and increases intercalated cell intracellular Na+ concentration 78. The basolateral plasma membrane H+-ATPase acts independently of the Na+, K+-ATPase, to further enhance the driving force for Cl− absorption by intercalated cells in mouse CCD 25, 71. Thus, the H+-ATPase and most probably the Na,K-ATPase provide the active steps for Cl− absorption by ICs.
B. Compensation by other electroneutral NaCl transporters
Pendrin null mice have a very mild renal phenotype under basal conditions. Following a Na+, K+ and Cl− -replete diet, NaCl excretion, serum electrolytes and arterial blood gases are similar in wild type and pendrin null mice 14, 17, 18, although blood pressure is slightly lower in the mutant mice 14, 81. The absence of a phenotype is thought to be due to compensation by other mechanisms of electroneutral NaCl absorption, particularly the thiazide-sensitive NaCl cotransporter, encoded by Slc12a3. Under basal conditions, NCC abundance is higher in kidneys from pendrin null than wild type mice 14, which should at least partially offset the NaCl loss that follows pendrin gene ablation. As such, the natriuresis and chloriuresis that follow thiazide administration is enhanced in pendrin null mice 82, which is consistent with greater NCC abundance in the mutant mice. How pendrin regulates NCC is, however, unresolved.
Type B intercalated cell transporters, such as pendrin, are upregulated in NCC null mice and in mice lacking active, phosphorylated NCC, such as in SPAK null mice 16, 83, 84. The increase in pendrin abundance observed when NCC is reduced or absent occurs, in part, through increased α ketoglutarate (αKG) production by the proximal tubule 83. αKG is then secreted into the luminal fluid of this segment, which stimulates pendrin abundance and transport downstream 83, 85.
Whereas pendrin null mice have a very mild phenotype under basal conditions, a very prominent renal phenotype is observed with ablation of the genes encoding both pendrin (Slc26a4) and NCC (Slc12a3) 86. Mice lacking both NCC and pendrin have more profound apparent intravascular volume contraction than those that are pendrin null alone.
C. Regulation of pendrin by aldosterone
Angiotensin II and aldosterone play an important role in the regulation of NaCl balance and blood pressure. Since apical anion exchange increases in response to these hormones 12, 38,87, we explored whether these hormones change renal pendrin abundance, subcellular distribution or function.
In the mouse, apical plasma membrane pendrin abundance is low under basal conditions, but increases ~6-fold in response to aldosterone 11. This increase in apical plasma membrane pendrin abundance occurs mainly through subcellular redistribution with a smaller contribution from increased pendrin protein abundance per cell 11. Ochiai-Homma et al quantified pendrin abundance in human urinary extracellular vesicles as an index of renal pendrin abundance and showed that aldosterone upregulates pendrin in human kidney 88, consistent with observations in mouse. Moreover, they showed a strong correlation between pendrin abundance in these human urinary extracellular vesicles and serum aldosterone concentration 88. Following either adrenalectomy or mineralocorticoid receptor antagonist administration, pendrin abundance in urinary vesicles fell substantially 88. Therefore, in rodents and in humans, aldosterone greatly stimulates pendrin abundance in the kidney, which occurs, at least partly, through the alkalosis it generates 89. Aldosterone increases apical plasma membrane pendrin expression in tandem with increased pendrin-mediated Cl− absorption and HCO3− secretion 9, 11, 12, 17, 90.
D. Role of the mineralocorticoid receptor in intercalated cell aldosterone signaling.
Upon aldosterone binding, the mineralocorticoid receptor (MR) undergoes a conformational change and is then translocated to the nucleus, which stimulates gene transcription 91. While both aldosterone and cortisol/corticosterone are MR ligands, circulating corticosterone/cortisol is much higher than aldosterone under most physiological conditions 92. Therefore the ability of aldosterone to bind to the MR is predicated on 11β-hydroxysteroid dehydrogenase type II expression within the cell, which oxidizes and therefore inactivates these glucocorticoids, making aldosterone the preferred ligand 92.
The MR is highly expressed in ICs 28, 93, 94 where it regulates the abundance of transporters such as pendrin and the H+-ATPase 28, 89, 94, and modulates the rate of Cl− absorption by the CCD 94. However, the mechanism of MR activation differs in principal and in ICs. Although aldosterone is an IC MR ligand in heterologous expression systems, whether it acts as a receptor ligand in vivo is controversial. Whereas 11β-hydroxysteroid dehydrogenase type II (11 βHSD2) is highly expressed in principal cells, its expression is very low or absent in intercalated cells 93, 95. Thus, the fall in intercalated cell transporter expression seen with MR inhibitors, such as spironolactone 28, might occur from an indirect effect of MR blockade, such as through changes in serum K+ concentration or acid-base balance 96.
The IC MR is phosphorylated at S843, which represents a phosphorylation site unique to this cell type 28. When phosphorylated at this site, aldosterone binds to the receptor poorly, and therefore activates the IC MR very little (Figure 2). Conversely, IC MR S843 dephosphorylation, which occurs in response to either angiotensin II or aldosterone-induced hypokalemia, enhances aldosterone binding, which activates the receptor. Therefore, during dietary NaCl restriction, where angiotensin II production increases, IC MR S843 dephosphorylation increases, which enhances aldosterone binding to the receptor and hence its activation, thereby increasing pendrin and H+-ATPase total protein abundance.
Figure 2: Mechanism of MR activation within ICs.
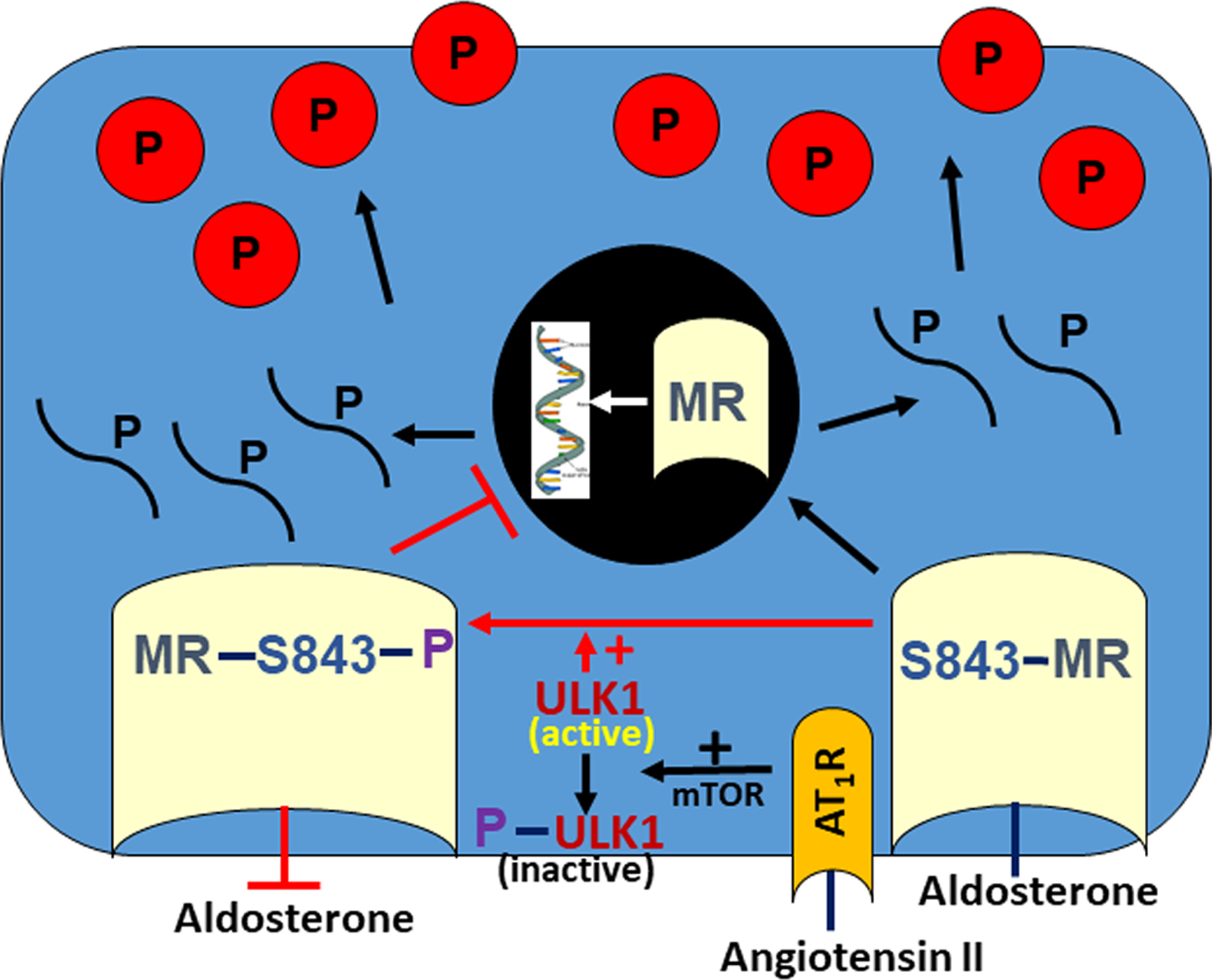
The kinase, ULK1, is highly expressed in ICs and mediates the phosphorylation of the IC MR at S843. Upon angiotensin II binding to the angiotensin type 1 receptor (AT1R), mTOR phosphorylates, and thus inactivates, ULK1. With this ULK1 inactivation, the MR is dephosphorylated at S843, which enhances aldosterone binding to the MR. The IC MR is therefore activated and thus translocates to the nucleus, which stimulates gene transcription.
Using high throughput screening, Shibata et al determined that this MR dephosphorylation event is catalyzed by the kinase, unc-51-like kinase 1 (ULK1) 97, which is highly expressed in ICs 97. They also showed that angiotensin II acts through the angiotensin type 1 receptor to activate mTOR, which phosphorylates, and thus inactivates, ULK1 (Figure 2). Upon angiotensin II binding to the angiotensin type 1 receptor, ULK1 is inactivated, which dephosphorylates the IC MR at S843, thereby activating the receptor 97.
The effect of K+ on IC MR activation, however, has been questioned by our laboratory and others 89, 94. To explore the effect of K+ on IC activation, we compared pendrin label per cell as well as pendrin’s relative label in the most apical region of ICs from the same mouse that either express or do not express the MR 94. Experiments were conducted in a variety of treatment models, such as in aldosterone-treated mice, where serum K+ is 3.1 mEq and in mice given aldosterone and amiloride, where serum K+ is 5.1 mEq. In both models, pendrin label per cell, as well as pendrin’s relative abundance in the most apical 10% of the cell were significantly higher in ICs that express the MR than in cells that did not. We conclude that the IC MR regulates pendrin directly over a very wide range in serum K+, consistent with observations by others 89.
E. Regulation of pendrin by angiotensin II
Since angiotensin II acts through an angiotensin type 1 receptor to increase apical Cl−/HCO3− exchange in rabbit CCDs perfused in vitro 87, we explored how this occurs 25. In CCDs perfused in vitro from furosemide-treated wild type mice, adding angiotensin II to the bath increased Cl− absorption 2-fold, although transepithelial voltage, VT, was unchanged. However, in CCDs from pendrin null mice, angiotensin II changed neither Cl− absorption nor VT 25. These data suggest that this peptide hormone increases Cl− absorption through a transcellular rather than paracellular transport process 25.
To determine if angiotensin II alters pendrin or H+-ATPase apical plasma membrane abundance, we quantified transporter subcellular distribution using immunogold cytochemistry with morphometric analysis of CCDs perfused in vitro. While angiotensin II application changed neither pendrin nor H+-ATPase subcellular distribution in type B ICs, within type A ICs, it increased both apical plasma membrane H+-ATPase abundance and HCO3− absorption, which reflects increased H+ secretion 98 99.
In CCDs perfused in vitro, angiotensin II increases ENaC-mediated Na+ absorption 100, which should increase the lumen-negative voltage, VT. However, since angiotensin II application did not change transepithelial voltage, VT, 25, 98, movement of a counter ion may shunt this ENaC-mediated current. The increased H+ secretion that follows angiotensin II application may provide such a counter-ion 98, 101. In CCDs perfused in vitro, this lumen-negative VT increases with the application of H+-ATPase inhibitors to the perfusate 101, but falls with ENaC inhibitors 14, 19. Since angiotensin II increases both H+ secretion and Na+ absorption in mouse CCDs, movement of these 2 ions may shunt the voltage generated by transport of the other.
Because angiotensin II application in vitro does not change pendrin or H+-ATPase subcellular distribution within the type B IC, how it stimulates pendrin-dependent Cl− absorption remains unexplained. This peptide hormone might activate a plasma membrane Cl− channel, which generates a more favorable ion gradient for anion exchange. However, other mechanisms are also possible.
In contrast to in vitro studies, we observed that angiotensin II given in vivo acts through the angiotensin type 1 receptor, independently of aldosterone, to increase pendrin abundance in the most apical region of Non-A, non-B ICs of the CNT through subcellular redistribution 26. However, other studies observed that angiotensin II upregulates pendrin total protein abundance by increasing aldosterone production 27. If so, aldosterone and angiotensin II should produce similar changes in pendrin abundance and subcellular distribution within the various IC subtypes. Instead, angiotensin II increases pendrin label intensity in type B IC, but not Non-A, non-B ICs 26, whereas aldosterone increases pendrin label in Non-A, non-B ICs, but not in type B cells 11. Moreover, angiotensin II and aldosterone did not produce the same changes in pendrin subcellular distribution within the various pendrin positive cell types 11, 26. These data suggest that within ICs, angiotensin II and aldosterone do not have identical downstream signaling mechanisms.
F. Role of pendrin in NaCl balance and blood pressure regulation.
Because pendrin null mice lose body weight with NaCl restriction 17, we compared Na+ and Cl− balance in NaCl-restricted wild type and pendrin knockout mice 14, 18. Urinary NaCl excretion was similar in wild type and in global, pendrin null mice that consumed a standard, NaCl-replete rodent diet 17, 18. However, with dietary NaCl restriction, pendrin null mice excreted more NaCl than wild type mice 14, 17, 18, which led to a higher BUN, a lower blood pressure and greater weight loss in the former 14, 17–19. Therefore, the pendrin null kidney cannot fully conserve NaCl, which leads to intravascular volume contraction and lower blood pressure 14, 17, 18. This fall in blood pressure occurs whether pendrin gene ablation is induced in utero or during adulthood 11, 14, 19, 22. Conversely, when given a high NaCl diet, mice that overexpress pendrin have much higher blood pressure than wild type controls 102.
Other groups have examined blood pressure and salt balance in people with Pendred Syndrome and in normal controls. The first of these did a retrospective chart review, which examined the incidence of hypertension in persons homozygous for inactivation sequence variants of Slc26a4 and in their unaffected family members 103. While inadequately powered, it suggested that pendrin gene ablation is protective against hypertension. A later study examined blood pressure and NaCl excretion in a larger cohort of people with biallelic, inactivating sequence variants of SLC26a4 and in unaffected controls 21 and showed that blood pressure is lower, while and NaCl excretion is higher, in persons with Pendred Syndrome, consistent with previous rodent studies.
Since blood pressure is the product of systemic vascular resistance and cardiac output, we examined vascular tone in wild type and pendrin knockout mice 104. Although pendrin expression is either low or absent in vascular tissue, pendrin knockout mice have greater thoracic aorta contractile force in response to either α adrenergic agonists (phentolamine) or angiotensin II administration in vitro than do wild type mice 104, which may blunt the fall in blood pressure that follows pendrin gene ablation. Because pendrin expression in vascular tissue is very low or absent, pendrin must affect vascular tone through an indirect mechanism, such as through changes angiotensin II and/or catecholamine release 104.
G. The interaction of pendrin and ENaC and its physiological significance
Because pendrin is not a Na+ transporter, we explored why pendrin null mice excrete more Na+ than do wild type mice 14. To do so, we used “targeted proteomics” to quantify renal Na+ transporter abundance in pendrin null and wild type mice. The abundance of NHE3, NKCC2, α1 Na,K-ATPase, and ENaC were similar in wild type and pendrin null mice following a NaCl-replete diet, where renin and aldosterone are suppressed 14. However, following either an aldosterone infusion or dietary NaCl restriction, where circulating aldosterone concentration is increased, α, β and γ ENaC subunit abundance is lower in kidneys from pendrin knockout than wild type mice 14, 19, 20.
While aldosterone administration in vivo greatly increases ENaC-mediated Na+ absorption in the CCD of rats and mice 105, ENaC inhibitor (benzamil) application to the luminal fluid eliminates this response 20, 61, 106. Therefore, aldosterone stimulates Na+ absorption in the CCD primarily through ENaC. Further studies by our laboratory explored the effect of pendrin gene ablation on ENaC function and found that benzamil-sensitive Na+ absorption is much lower in CCDs from aldosterone-treated pendrin knockout than wild type mice 20. Therefore, pendrin modulates ENaC-mediated Na+ absorption.
Aldosterone amplifies ENaC channel activity (NPo) by increasing the frequency at which the channel is open (open probability, Po) 107 and by increasing channel surface density (N), either through changes in ENaC subunit abundance or subcellular distribution 62. To determine if pendrin changes one or more of these ENaC properties, we made single channel recordings of principal cells in split open mouse CCDs taken from aldosterone-treated wild type and pendrin null mice. We observed that pendrin gene ablation lowers channel activity by reducing both channel surface density 14, 19, 20 and channel open probability 20. This fall in ENaC activity contributes to the lower blood pressure seen in the pendrin null mice.
The mineralocorticoid receptor modulates ENaC, in part, through its effect on ICs. While ENaC is highly regulated by the principal cell MR, we and others observed that γ ENaC abundance is paradoxically higher in MR negative than MR positive principal cells taken from the same mouse 94, 108. In contrast, γ ENaC abundance as well as ENaC channel activity are lower in principal cells taken from intercalated cell MR knockout mice than from wild type mice, even though the MR is expressed in ENaC positive principal cells from both groups of mice 94. Therefore, the MR regulates ENaC, in large part, through its effect on intercalated cells, which secondarily regulates principal cell function.
Our laboratory and others have explored how pendrin changes ENaC abundance and function 19, 48. Because they localize to different cell types 8–10, 109 (Figure 1), these proteins cannot associate directly. Moreover, pendrin does not modulate ENaC through changes in the circulating concentration of a hormone that regulates the channel, e.g. vasopressin, thyroid hormone, renin, aldosterone or corticosterone 14, 110. Finally, while pendrin gene ablation reduces ENaC abundance and activity in kidney, it is unchanged in thyroid or colon 14. Paradoxically, pendrin gene ablation increases ENaC subunit mRNA in the inner ear 111. Therefore, the reduced ENaC subunit abundance and function seen in pendrin null mice appears limited to the kidney.
Since acid-base balance modulates ENaC subunit abundance and function 19, 112–114, we posited that pendrin stimulates ENaC by increasing luminal HCO3− concentration and/or pH. To test this hypothesis, we give mice NaHCO3 and aldosterone to stimulate pendrin-mediated HCO3− secretion in the CNT and CCD 19, 115. Other mice received aldosterone and NaHCO3 plus a carbonic anhydrase inhibitor (acetazolamide) to increase distal delivery of HCO3− from upstream segments. While pendrin is upregulated in the first protocol, it is downregulated in the second 19, 116. Following aldosterone and NaHCO3, pendrin null mice had a more severe metabolic alkalosis and reduced renal ENaC subunit abundance and function relative to wild type mice 19. However, acetazolamide addition eliminated these differences 19. Therefore, increasing distal HCO3− delivery attenuates the alkalosis and the fall in renal ENaC abundance and function observed in pendrin knockout mice.
We used a mouse principal cell model (mpkCCD) to examine changes in ENaC abundance and function when extracellular HCO3− concentration was varied 19. Raising HCO3− on the apical side of the monolayer increased ENaC abundance and function, independently of substituting anion. Therefore, pendrin modulates ENaC, at least partly, through changes in luminal pH or HCO3− concentration.
Prostaglandin E2 is a well-known ENaC inhibitor. Since B1-H+-ATPase gene ablation increases urinary prostaglandin E2 (PGE2) excretion, Gueutin et al. posited that ENaC abundance and function is reduced in B1-H+-ATPase null mice 48 (Figure 3). He observed greater ATP secretion by CCDs from B1 ATPase null than from wild type mice. This luminal ATP acts on the principal cell apical plasma membrane purinergic receptors to stimulate principal cell Ca2+ release, which increases prostaglandin E2 production, thereby reducing ENaC abundance and function 48. Since H+-ATPase gene ablation greatly reduces pendrin abundance in type B intercalated cells, these H+-ATPase null rodents represent a pendrin knockdown model. As such, pendrin gene ablation may reduce ENaC through a similar mechanism. If so, pendrin gene ablation should reduce type B IC H+-ATPase abundance, which increases ATP secretion and therefore prostaglandin E2 production thereby reducing ENaC abundance and activity. Therefore, pendrin gene ablation reduces ENaC abundance and function by changing luminal HCO3− and ATP concentration, although other pathways may also contribute to this interaction.
Figure 3: Pendrin may regulate ENaC through changes in ATP secretion.
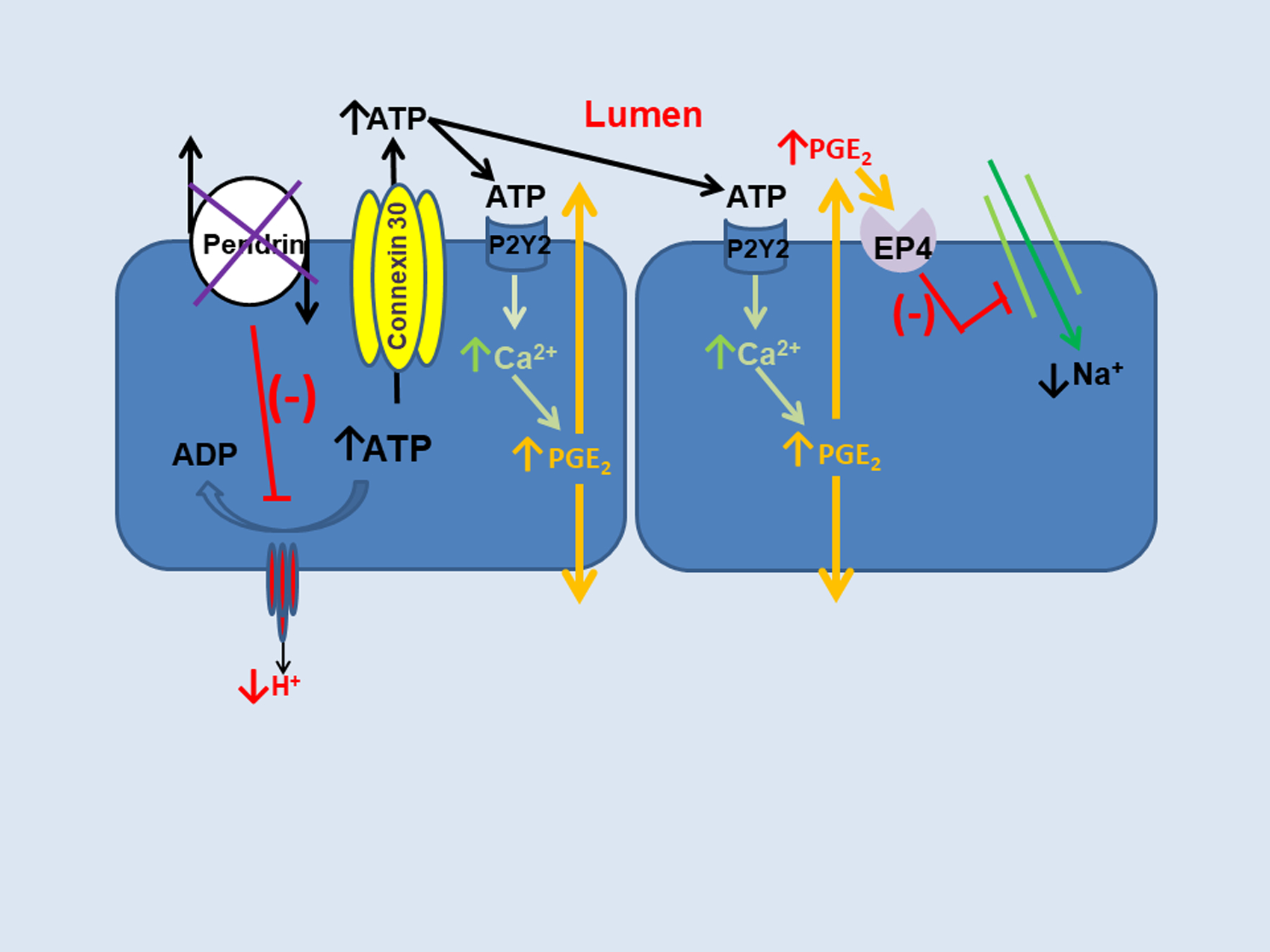
Pendrin gene ablation reduces H+-ATPase abundance in type B intercalated cells, which increases intracellular ATP content and connexin 30-mediated ATP secretion. Luminal ATP acts on principal cell apical membrane purinergic receptors to stimulate calcium release, which increases prostaglandin E2 production (PGE2) and thereby reduces ENaC abundance and function.
ENaC is upregulated with NaCl restriction, which increases renal Na+ absorption, thereby helping to maintain intravascular volume and blood pressure. Because NaCl-restricted pendrin null mice are hypotensive and volume contracted, why ENaC is downregulated in these mutant mice has therefore been puzzling. Consistent with its presumed role as a K+-sparing mechanism of NaCl absorption 13, 16, 117, serum K+ is slightly lower in pendrin knockout than wild type mice, under at least some conditions 15. Downregulating ENaC may enable pendrin null mice to maintain K+ homeostasis.
CONCLUSIONS:
Pendrin is an electroneutral, Cl−/HCO3− exchanger found in the thyroid, inner ear, kidney, and adrenal gland. In kidney, pendrin mediates Cl− absorption and HCO3− secretion in subsets of intercalated cells and is stimulated in rodent models of metabolic alkalosis. In so doing, HCO3− excretion increases, which helps correct the alkalosis. Pendrin-positive ICs also play an important role in Na+ and Cl− absorption by the kidney and hence in blood pressure regulation. NaCl restriction increases apical plasma membrane pendrin abundance, which increases renal NaCl absorption and hence blood pressure. Aldosterone upregulates pendrin, at least in part, by activating the IC MR, which has properties unique to this cell type. Aldosterone stimulates pendrin, which secondarily increases ENaC abundance and function. Therefore, aldosterone stimulates ENaC directly through the principal cell MR and stimulates the channel indirectly vis-à-vis pendrin-positive ICs. Angiotensin II acts via the angiotensin type 1a receptor to stimulate pendrin in vivo and in vitro. Whether pendrin changes blood pressure, at least partly, through its action outside kidney, remains to be determined.
Disclosures:
This work was supported by DK 110375 (to S.M. Wall).
Footnotes
DISCLOSURES:
Dr. Wall owns stock in Johnson & Johnson.
REFERENCES
- 1.Kim J, Kim Y-H, Cha J-H, Tisher CC and Madsen KM. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. JAmSocNephrol. 1999;10:1–12. [DOI] [PubMed] [Google Scholar]
- 2.Pfaller W Structure Function Correlation on Rat Kidney. New York: Springer-Verlag; 1982. [PubMed] [Google Scholar]
- 3.Clark JZ, Chen L, Chou CL, Jung HJ, Lee JW and Knepper MA. Representation and relative abundance of cell-type selective markers in whole-kidney RNA-Seq data. Kidney Int. 2019;95:787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown D, Hirsch S and Gluck S. An H+-ATPase in opposite plasma membrane domains in kidney epithelial cell subpopulations. Nature. 1988;331:622–624. [DOI] [PubMed] [Google Scholar]
- 5.Gluck S, Kelly S and Al-Awqati Q. The proton translocating ATPase responsible for urinary acidification. JBiolChem. 1982;257:9230–9233. [PubMed] [Google Scholar]
- 6.Sabolic I, Brown D, Gluck SL and Alper SL. Regulation of AE1 anion exchanger and H+-ATPase in rat cortex by acute metabolic acidois and alkalosis. Kidney Int. 1997;51:125–137. [DOI] [PubMed] [Google Scholar]
- 7.Verlander JW, Madsen KM, Cannon JK and Tisher CC. Activation of acid-secreting intercalated cells in rabbit collecting duct with ammonium chloride loading. Am J Physiol. 1994;266:F633–45. [DOI] [PubMed] [Google Scholar]
- 8.Kim Y-H, Kwon T-H, Frische S, Kim J, Tisher CC, Madsen KM and Nielsen S. Immunocytochemical localization of pendrin in intercalated cell subtypes in rat and mouse kidney. AmJPhysiol. 2002;283:F744–F754. [DOI] [PubMed] [Google Scholar]
- 9.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA and Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci U S A. 2001;98:4221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, Shipley GL and Verlander JW. Localization of pendrin in mouse kidney. AmJPhysiol. 2003;284:F229–F241. [DOI] [PubMed] [Google Scholar]
- 11.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED and Wall SM. Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension. 2003;42:356–62. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Austt J, Good DW, Burg MB and Knepper MA. Deoxycorticosterone-stimulated bicarbonate secretion in rabbit cortical collecting duct: effects of luminal chloride removal and in vivo acid loading. AmJPhysiol. 1985;249:F205–F212. [DOI] [PubMed] [Google Scholar]
- 13.Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hatim H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R and Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. JClinInvest. 2010;120:1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y-H, Pech V, Spencer KB, Beierwaltes WH, Everett LA, Green ED, Shin WK, Verlander JW, Sutliff RL and Wall SM. Reduced ENaC expression contributes to the lower blood pressure observed in pendrin null mice. AmJPhysiol. 2007;293:F1314–F1324. [DOI] [PubMed] [Google Scholar]
- 15.Xu N, Hirohama D, Ishizawa K, Chang WX, Shimosawa T, Fujita T, Uchida S and Shibata S. Hypokalemia and Pendrin Induction by Aldosterone. Hypertension. 2017;69:855–862. [DOI] [PubMed] [Google Scholar]
- 16.Sinning A, Radionov N, Trepiccione F, Lopez-Cayuqueo KI, Jayat M, Baron S, Corniere N, Alexander RT, Hadchouel J, Eladari D, Hubner CA and Chambrey R. Double Knockout of the Na+-Driven Cl-/HCO3- Exchanger and Na+/Cl- Cotransporter Induces Hypokalemia and Volume Depletion. J Am Soc Nephrol. 2017;28:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED and Verlander JW. NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension. 2004;44:982–7. [DOI] [PubMed] [Google Scholar]
- 18.Verlander JW, Kim YH, Shin W, Pham TD, Hassell KA, Beierwaltes WH, Green ED, Everett L, Matthews SW and Wall SM. Dietary Cl(−) restriction upregulates pendrin expression within the apical plasma membrane of type B intercalated cells. Am J Physiol Renal Physiol. 2006;291:F833–9. [DOI] [PubMed] [Google Scholar]
- 19.Pech V, Pham TD, Hong S, Weinstein AM, Spencer KB, Duke BJ, Walp E, Kim Y-H, Sutliff RL, Bao H-F, Eaton DC and Wall SM. Pendrin modulates ENaC function by changing luminal HCO3−. JAmSocNephrol. 2010;21:1928–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pech V, Wall SM, Nanami M, Bao HF, Kim YH, Lazo-Fernandez Y, Yue Q, Pham TD, Eaton DC and Verlander JW. Pendrin gene ablation alters ENaC subcellular distribution and open probability. Am J Physiol Renal Physiol. 2015;309:F154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim BG, Yoo TH, Yoo JE, Seo YJ, Jung J and Choi JY. Resistance to hypertension and high Cl- excretion in humans with SLC26A4 mutations. Clin Genet. 2016;91:448–452. [DOI] [PubMed] [Google Scholar]
- 22.Trepiccione F, Soukaseum C, Baudrie V, Kumai Y, Teulon J, Villoutreix B, Corniere N, Wangemann P, Griffith AJ, Byung Choi Y, Hadchouel J, Chambrey R and Eladari D. Acute genetic ablation of pendrin lowers blood pressure in mice. Nephrol Dial Transplant. 2017;32:1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haggie PM, Phuan PW, Tan JA, Zlock L, Finkbeiner WE and Verkman AS. Inhibitors of pendrin anion exchange identified in a small molecule screen increase airway surface liquid volume in cystic fibrosis. FASEB J. 2016;30:2187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cil O, Haggie PM, Phuan PW, Tan JA and Verkman AS. Small-Molecule Inhibitors of Pendrin Potentiate the Diuretic Action of Furosemide. J Am Soc Nephrol. 2016;27:3706–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pech V, Kim Y-H, Weinstein AM, Everett LA, Pham TD and Wall SM. Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. AmJPhysiol. 2007;292:F914–F920. [DOI] [PubMed] [Google Scholar]
- 26.Verlander JW, Hong S, Pech V, Bailey JL, Agazatian D, Matthews SW, Coffman TM, Le T, Inagami T, Whitehill FM, Weiner ID, Farley DB, Kim YH and Wall SM. Angiotensin II acts through the angiotensin 1a receptor to upregulate pendrin. AmJPhysiol. 2011;301:F1314–F1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirohama D, Ayuzawa N, Ueda K, Nishimoto M, Kawarazaki W, Watanabe A, Shimosawa T, Marumo T, Shibata S and Fujita T. Aldosterone Is Essential for Angiotensin II-Induced Upregulation of Pendrin. J Am Soc Nephrol. 2018;29:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata S, Rinehart J, Zhang J, Moeckel G, Castaneda-Bueno M, Stiegler AL, Boggon TJ, Gamba G and Lifton RP. Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab. 2013;18:660–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemoine S, Eladari D, Juillard L, Bonnefond A, Froguel P and Dubourg L. The Case | Hypokalemia and severe renal loss of sodium. Kidney Int. 2020;97:1305–1306. [DOI] [PubMed] [Google Scholar]
- 30.Kandasamy N, Fugazzola L, Evans M, Chatterjee K and Karet F. Life-threatening metabolic alkalosis in Pendred syndrome. Eur J Endocrinol. 2011;165:167–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanei-Moghaddam A, Wilson T, Kumar S and Gray R. An unfortunate case of Pendred syndrome. J Laryngol Otol. 2011;125:965–7. [DOI] [PubMed] [Google Scholar]
- 32.Emmons C New subtypes of rabbit CCD intercalated cells as functionally defined by anion exchange and H+-ATPase activity. Paper presented at: JAmSocNephrol; 1993. [Google Scholar]
- 33.Schuster VL. Function and regulation of collecting duct intercalated cells. AnnuRevPhysiol. 1993;55:267–288. [DOI] [PubMed] [Google Scholar]
- 34.Alper SL, Natale J, Gluck S, Lodish HF and Brown D. Subtypes of intercalated cells in rat kidney collecting duct defined by antibodies against erythroid band 3 and renal vacuolar H+-ATPase. ProcNatlAcadSciUSA. 1989;86:5429–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuruoka S and Schwartz GJ. Metabolic acidosis stimulates H+ Secretion in the rabbit outer medullary collecting duct (inner stripe) of the kidney. JClinInvest. 1997;99:1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verlander JW, Madsen KM, Cannon JK and Tisher CC. Activation of acid-secreting intercalated cells in rabbit collecting duct with ammonium chloride loading. AmJPhysiol. 1994;266:F633–F645. [DOI] [PubMed] [Google Scholar]
- 37.Bastani B, Purcell H, Hemken P, Trigg D and Gluck S. Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. JClinInvest. 1991;88:126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Star RA, Burg MB and Knepper MA. Bicarbonate secretion and chloride absorption by rabbit cortical collecting ducts: role of chloride/bicarbonate exchange. JClinInvest. 1985;76:1123–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiner ID, Weill AE and New AR. Distribution of Cl-/HCO3- exchange and intercalated cells in rabbit cortical collecting duct. AmJPhysiol. 1994;267:F952–F964. [DOI] [PubMed] [Google Scholar]
- 40.Weiner ID and Hamm LL. Regulation of intracellular pH in the rabbit cortical collecting tubule. JClinInvest. 1990;85:274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emmons C Transport characteristics of the apical anion exchanger of rabbit cortical collecting duct β-cells. AmJPhysiol. 1999;276:F635–F643. [DOI] [PubMed] [Google Scholar]
- 42.Emmons C and Kurtz I. Functional characterization of three intercalated cell subtypes in the rabbit outer cortical collecting duct. JClinInvest. 1994;93:417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohebbi N, Perna A, van der Wijst J, Becker HM, Capasso G and Wagner CA. Regulation of two renal chloride transporters, AE1 and pendrin, by electrolytes and aldosterone. Plos One. 2013;8:e55286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frische S, Kwon T-H, Frokiaer J, Madsen KM and Nielsen S. Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. AmJPhysiol. 2003;284:F584–F593. [DOI] [PubMed] [Google Scholar]
- 45.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS and Geibel JP. Regulation of the expression of the Cl-/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 2002;62:2109–2117. [DOI] [PubMed] [Google Scholar]
- 46.Atkins JL and Burg MB. Bicarbonate transport by isolated perfused rat collecting ducts. Am J Physiol. 1985;249:F485–9. [DOI] [PubMed] [Google Scholar]
- 47.Teng-ummnuay P, Verlander JW, Yuan W, Tisher CC and Madsen KM. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. JAmSocNephrol. 1996;7:260–274. [DOI] [PubMed] [Google Scholar]
- 48.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Corniere N, Leviel F, Sohet F, Wagner CA, Eladari D and Chambrey R. Renal beta-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest. 2013;123:4219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuruoka S and Schwartz GJ. Mechanisms of HCO3- secretion in the rabbit connecting segment. AmJPhysiol. 1999;277:F567–F574. [DOI] [PubMed] [Google Scholar]
- 50.Pendred V Deaf-mutism and goitre. Lancet. 1896;2:532. [Google Scholar]
- 51.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC and Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Genetics. 1997;17:411–422. [DOI] [PubMed] [Google Scholar]
- 52.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK and Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Human Molecular Genetics. 2001;10:153–161. [DOI] [PubMed] [Google Scholar]
- 53.Stehberger PA, Shmukler BE, Stuart-Tilley AK, Peters LL, Alper SL and Wagner CA. Distal renal tubular acidosis in mice lacking the AE1 (band 3) Cl/HCO3 exchanger (Slc4a1). JAmSocNephrol. 2007;18:1408–1418. [DOI] [PubMed] [Google Scholar]
- 54.Bourgeois S, Bettoni C, Baron S and Wagner CA. Haploinsufficiency of the Mouse Atp6v1b1 Gene Leads to a Mild Acid-Base Disturbance with Implications for Kidney Stone Disease. Cell Physiol Biochem. 2018;47:1095–1107. [DOI] [PubMed] [Google Scholar]
- 55.Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, Paillard M, Aronson PS and Eladari D. The Cl-/HCO3- exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. AmJPhysiol. 2004;287:F1179–F1188. [DOI] [PubMed] [Google Scholar]
- 56.Hafner P, Grimaldi R, Capuano P, Capasso G and Wagner CA. Pendrin in the mouse kidney is primarily regulated by Cl- excretion but also by systemic metabolic acidosis. Am J Physiol Cell Physiol. 2008;295:C1658–67. [DOI] [PubMed] [Google Scholar]
- 57.Pela I, Bigozzi M and Bianchi B. Profound hypokalemia and hypochloremic metabolic alkalosis during thiazide therapy in a child with Pendred Syndrome. ClinNephrol. 2008;69:450–453. [DOI] [PubMed] [Google Scholar]
- 58.Kim HY, Kim SS, Bae EH, Ma SK and Kim SW. Decreased Renal Expression of H(+)-ATPase and Pendrin in a Patient with Distal Renal Tubular Acidosis Associated with Sjogren’s Syndrome. Intern Med. 2015;54:2899–904. [DOI] [PubMed] [Google Scholar]
- 59.van den Wildenberg MJ, Hoorn EJ, Mohebbi N, Wagner CA, Woittiez AJ, de Vries PA and Laverman GD. Distal renal tubular acidosis with multiorgan autoimmunity: a case report. Am J Kidney Dis. 2015;65:607–10. [DOI] [PubMed] [Google Scholar]
- 60.Pathare G, Dhayat N, Mohebbi N, Wagner CA, Cheval L, Neuhaus TJ and Fuster DG. Acute regulated expression of pendrin in human urinary exosomes. Pflugers Arch. 2018;470:427–438. [DOI] [PubMed] [Google Scholar]
- 61.Stoos BA, Garcia NH and Garvin JL. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J Am Soc Nephrol. 1995;6:89–94. [DOI] [PubMed] [Google Scholar]
- 62.Masilamani S, Kim G-H, Mitchell C, Wade JB and Knepper MA. Aldosterone-mediated regulation of ENaC a,b and g subunit proteins in rat kidney. JClinInvest. 1999;104:R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pech V, Thumova M, Kim Y-H, Agazatian D, Hummler E, Rossier BC, Weinstein AM, Nanami M and Wall SM. ENaC inhibition stimulates Cl- secretion in the mouse cortical collecting duct through an NKCC1-dependent mechanism. AmJPhysiol. 2012;303:F45–F55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nanami M, Lazo-Fernandez Y, Pech V, Verlander JW, Agazatian D, Weinstein AM, Bao HF, Eaton DC and Wall SM. ENaC inhibition stimulates HCl secretion in the mouse cortical collecting duct I: stilbene-sensitive Cl- secretion. Am J Physiol Renal Physiol. 2015;309:F251–F258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pech V, Thumova M, Dikalov S, Hummler E, Rossier BC, Harrison DG and Wall SM. Nitric oxide reduces Cl- absorption in the mouse cortical collecting duct through an ENaC-dependent mechanism. Am J Physiol Renal Physiol. 2013;304:F1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schlatter E, Greger R and Schafer JA. Principal cells of the cortical collecting ducts of the rat are not a route of transepithelial Cl− transport. Pflugers Arch. 1990;417:317–323. [DOI] [PubMed] [Google Scholar]
- 67.Terada Y and Knepper MA. Thiazide-sensitive NaCl absorption in rat cortical collecting duct. AmJPhysiol. 1990;259:F519–F528. [DOI] [PubMed] [Google Scholar]
- 68.Kim G-H, Masilamani S, Turner R, Mitchell C, Wade JB and Knepper MA. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. ProcNatlAcadSciUSA. 1998;95:14552–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ellison DH, Velazquez H and Wright FS. Thiazide-sensitive sodium chloride cotranport in early distal tubule. AmJPhysiol. 1987;253:F546–F554. [DOI] [PubMed] [Google Scholar]
- 70.Xu J, Barone S, Zahedi K, Brooks M and Soleimani M. Slc4a8 in the Kidney: Expression, Subcellular Localization and Role in Salt Reabsorption. Cell Physiol Biochem. 2018;50:1361–1375. [DOI] [PubMed] [Google Scholar]
- 71.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hubner CA and Eladari D. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci U S A. 2013;110:7928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Corniere N, Bohm D, Jentsch TJ, Chambrey R, Teulon J, Hubner CA and Eladari D. The ClC-K2 Chloride Channel Is Critical for Salt Handling in the Distal Nephron. J Am Soc Nephrol. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pinelli L, Nissant A, Edwards A, Lourdel S, Teulon J and Paulais M. Dual regulation of the native ClC-K2 chloride channel in the distal nephron by voltage and pH. J Gen Physiol. 2016;148:213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Berg P, Svendsen SL, Sorensen MV, Larsen CK, Andersen JF, Jensen-Fangel S, Jeppesen M, Schreiber R, Cabrita I, Kunzelmann K and Leipziger J. Impaired Renal HCO3 (−) Excretion in Cystic Fibrosis. J Am Soc Nephrol. 2020;31:1711–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Varasteh Kia M, Barone S, McDonough AA, Zahedi K, Xu J and Soleimani M. Downregulation of the Cl-/HCO3-Exchanger Pendrin in Kidneys of Mice with Cystic Fibrosis: Role in the Pathogenesis of Metabolic Alkalosis. Cell Physiol Biochem. 2018;45:1551–1565. [DOI] [PubMed] [Google Scholar]
- 76.Kashgarian M, Biemesderfer D, Caplan M and Forbush B III. Monoclonal antibody to Na,K-ATPase: Immunocytochemical localization along nephron segments. Kidney Int. 1985;28:899–913. [DOI] [PubMed] [Google Scholar]
- 77.Fejes-Toth G and Naray-Fejes-Toth A. Isolated principal and intercalated cells: hormone responsiveness and Na+-K+-ATPase activity. Am J Physiol. 1989;256:F742–50. [DOI] [PubMed] [Google Scholar]
- 78.Sauer M, Flemmer A, Thurau K and Beck F-X. Sodium entry routes in principal and intercalated cells of the isolated perfused cortical collecting duct. Pflugers Arch. 1990;416:88–93. [DOI] [PubMed] [Google Scholar]
- 79.Takada T, Yamamoto A, Omori K and Yashiro Y. Quantitative immunogold localization of Na,K-ATPase along rat nephron. Histochemistry. 1992;98:183–197. [DOI] [PubMed] [Google Scholar]
- 80.Sabolic I, Herak-Kramberger CM, Breton S and Brown D. Na/K-ATPase in intercalated cells along the rat nephron revealed by antigen retrieval. J Am Soc Nephrol. 1999;10:913–22. [DOI] [PubMed] [Google Scholar]
- 81.Lazo-Fernandez Y, Aguilera G, Pham TD, Park AY, Beierwaltes WH, Sutliff RL, Verlander JW, Pacak K, Osunkoya AO, Ellis CL, Kim YH, Shipley GL, Wynne BM, Hoover RS, Sen SK, Plotsky PM and Wall SM. Pendrin localizes to the adrenal medulla and modulates catecholamine release. Am J Physiol Endocrinol Metab. 2015;309:E534–E545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alshahrani S and Soleimani M. Ablation of the Cl-/HCO3- Exchanger Pendrin Enhances Hydrochlorothiazide-Induced Diuresis. Kidney Blood Press Res. 2017;42:444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB and Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest. 2015;125:2136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschenes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R and Eladari D. Pendrin regulation in mouse kidney primarily is chloride-dependent. JAmSocNephrol. 2006;17:2153–2163. [DOI] [PubMed] [Google Scholar]
- 85.Lazo-Fernandez Y, Welling PA and Wall SM. alpha-Ketoglutarate stimulates pendrin-dependent Cl(−) absorption in the mouse CCD through protein kinase C. Am J Physiol Renal Physiol. 2018;315:F7–F15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K and Amlal H. Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci U S A. 2012;109:13368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weiner ID, New AR, Milton AE and Tisher CC. Regulation of luminal alkaliniation and acidification in the cortical collecting duct by angiotensin II. AmJPhysiol. 1995;269:F730–F738. [DOI] [PubMed] [Google Scholar]
- 88.Ochiai-Homma F, Kuribayashi-Okuma E, Tsurutani Y, Ishizawa K, Fujii W, Odajima K, Kawagoe M, Tomomitsu Y, Murakawa M, Asakawa S, Hirohama D, Nagura M, Arai S, Yamazaki O, Tamura Y, Fujigaki Y, Nishikawa T and Shibata S. Characterization of pendrin in urinary extracellular vesicles in a rat model of aldosterone excess and in human primary aldosteronism. Hypertens Res. 2021;44:1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ayuzawa N, Nishimoto M, Ueda K, Hirohama D, Kawarazaki W, Shimosawa T, Marumo T and Fujita T. Two Mineralocorticoid Receptor-Mediated Mechanisms of Pendrin Activation in Distal Nephrons. J Am Soc Nephrol. 2020;31:748–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stokes JB, Ingram MJ, Williams AD and Ingram D. Heterogeneity of rabbit collecting tubule: localization of mineralocorticoid hormone action to the cortical portion. Kidney Int. 1981;20:340–347. [DOI] [PubMed] [Google Scholar]
- 91.Soundararajan R, Lu M and Pearce D. Organization of the ENaC-regulatory machinery. Crit Rev Biochem Mol Biol. 2012;47:349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kyossev Z, Walker PD and Reeves WB. Immunolocalization of NAD-dependent 11 beta-hydroxysteroid dehydrogenase in human kidney and colon. Kidney Int. 1996;49:271–81. [DOI] [PubMed] [Google Scholar]
- 93.Ackermann D, Gresko N, Carrel M, Loffing-Cueni D, Habermehl D, Gomez-Sanchez C, Rossier BC and Loffing J. In vivo nuclear translocation of mineralocorticoid and glucocorticoid receptors in rat kidney: differential effect of corticosteroids along the distal tubule. Am J Physiol Renal Physiol. 2010;299:F1473–85. [DOI] [PubMed] [Google Scholar]
- 94.Pham TD, Verlander JW, Wang Y, Romero CA, Yue Q, Chen C, Thumova M, Eaton DC, Lazo-Fernandez Y and Wall SM. Aldosterone regulates pendrin and ENaC through intercalated cell mineralocorticoid receptor-dependent and independent mechanisms over a wide range in serum K+. J Am Soc Nephrol. 2020;31:483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Paunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB and Knepper MA. Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc Natl Acad Sci U S A. 2017;114:E9989–E9998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL and Ellison DH. Direct and Indirect Mineralocorticoid Effects Determine Distal Salt Transport. J Am Soc Nephrol. 2015;27:2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shibata S, Ishizawa K, Wang Q, Xu N, Fujita T, Uchida S and Lifton RP. ULK1 Phosphorylates and Regulates Mineralocorticoid Receptor. Cell Rep. 2018;24:569–576. [DOI] [PubMed] [Google Scholar]
- 98.Pech V, Zheng W, Pham TD, Verlander JW and Wall SM. Angiotensin II activates H+-ATPase in type A intercalated cells in mouse cortical collecting duct. JAmSocNephrol. 2008;19:84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rothenberger F, Velic A, Stehberger PA, Kovacikova J and Wagner CA. Angiotensin II stimulates vacuolar H+-ATPase activity in renal acid-secretory intercalated cells rom the outer medullary collecting duct. JAmSocNephrol. 2007;18:2085–2093. [DOI] [PubMed] [Google Scholar]
- 100.Peti-Peterdi J, Warnock DG and Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT1 receptors. JAmSocNephrol. 2002;13:1131–1135. [DOI] [PubMed] [Google Scholar]
- 101.Nanami M, Pech V, Lazo-Fernandez Y, Weinstein AM and Wall SM. ENaC inhibition stimulates HCl secretion in the mouse cortical collecting duct II: Bafilomycin-sensitive H+ secretion. Am J Physiol Renal Physiol. 2015;309:F259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jacques T, Picard N, Miller RL, Riemondy KA, Houillier P, Sohet F, Ramakrishnan SK, Busst CJ, Jayat M, Corniere N, Hassan H, Aronson PS, Hennings JC, Hubner CA, Nelson RD, Chambrey R and Eladari D. Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol. 2013;24:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Madeo AC, Manichaikul A, Pryor SP and Griffith AJ. Do mutations of the Pendred syndrome gene, SLC26A4, confer resistance to asthma and hypertension? JMedGenet. 2009;46:405–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sutliff RL, Walp ER, Kim YH, Walker LA, El-Ali AM, Ma J, Bonsall R, Ramosevac S, Eaton DC, Verlander JW, Hansen L, Gleason RL, Pham TD, Hong S, Pech V and Wall SM. Contractile Force Is Enhanced in Aortas from Pendrin Null Mice Due to Stimulation of Angiotensin II-Dependent Signaling. Plos One. 2014;9:E105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pacha J, Frindt G, Antonian L, Silver RB and Palmer LG. Regulation of Na channels of the rat cortical collecting tubule by aldosterone. JGenPhysiol. 1993;102:25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stoner LC, Burg MB and Orloff J. Ion transport in cortical collecting tubule; effect of amiloride. AmJPhysiol. 1974;227:453–459. [DOI] [PubMed] [Google Scholar]
- 107.Garty H and Palmer LG. Epithelial sodium channels: function, structure, and regulation. PhysiolRev. 1997;77:359–395. [DOI] [PubMed] [Google Scholar]
- 108.Czogalla J, Vohra T, Penton D, Kirschmann M, Craigie E and Loffing J. The mineralocorticoid receptor (MR) regulates ENaC but not NCC in mice with random MR deletion. Pflugers Arch. 2016. [DOI] [PubMed] [Google Scholar]
- 109.Hager H, Kwon T-H, Vinnikova AK, Masilamani S, Brooks HL, Frokiaer J, Knepper MA and Nielsen S. Immunocytochemical and immunoelectron microscopic localization of a-, b- and g-ENaC in rat kidney. AmJPhysiol. 2001;280:F1093–F1106. [DOI] [PubMed] [Google Scholar]
- 110.Kim Y-H, Pham TD, Zheng W, Hong S, Baylis C, Pech V, Beierwaltes WH, Farley DB, Braverman LE, Verlander JW and Wall. SM Role of pendrin in iodide balance: going with the flow. AmJPhysiol. 2009;297:1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim BG, Kim JY, Kim HN, Bok J, Namkung W, Choi JY and Kim SH. Developmental Changes of ENaC Expression and Function in the Inner Ear of Pendrin Knock-Out Mice as a Perspective on the Development of Endolymphatic Hydrops. Plos One. 2014;9:e95730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim G-H, Martin SW, Fernandez-Llama P, Masilamani S, Packer RK and Knepper MA. Long-term regulation of renal Na-dependent cotransporters and ENaC: response to altered acid-base intake. AmJPhysiol. 2000;279:F459–F467. [DOI] [PubMed] [Google Scholar]
- 113.Palmer LG and Frindt G. Effects of cell Ca and pH on Na channels from rat cortical collecting tubule. AmJPhysiol. 1987;253:F333–F339. [DOI] [PubMed] [Google Scholar]
- 114.Shipway A, Danahay H, Williams JA, Tully DC, Backes BJ and Harris JL. Biochemical characterization of prostasin, a channel activating protease. BiochemBiophysResCommun. 2004;324:953–963. [DOI] [PubMed] [Google Scholar]
- 115.Knepper MA, Good DW and Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. AmJPhysiol. 1985;249:F870–F877. [DOI] [PubMed] [Google Scholar]
- 116.Milton AE and Weiner ID. Regulation of B-type intercalated cell apical anion exchange activity by CO2/HCO3−. AmJPhysiol. 1998;274:F1086–F1094. [DOI] [PubMed] [Google Scholar]
- 117.Lopez-Cayuqueo KI, Chavez-Canales M, Pillot A, Houillier P, Jayat M, Baraka-Vidot J, Trepiccione F, Baudrie V, Busst C, Soukaseum C, Kumai Y, Jeunemaitre X, Hadchouel J, Eladari D and Chambrey R. A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int. 2018;94:514–523. [DOI] [PubMed] [Google Scholar]