

Abstract
Allogeneic “off-the-shelf” (OTS) chimeric antigen receptor T cells (CAR-T cells) hold promise for more accessible CAR-T therapy. Here, we report a novel and simple way to make allogeneic OTS T cells targeting cancer. By engineering T cells with a bispecific T cell engager (BiTE), both TCRαβ and CD3ε expression on the T cell surface are dramatically reduced. BiTE-engineered T (BiTE-T) cells show reduced reaction to TCR stimulation in vitro and have low risk of graft-versus-host disease (GvHD) in vivo. BiTE-T cells down-regulated CD3ε/TCRαβ on bystander T cells by releasing BiTEs. BiTE-T cells produce much fewer cytokines and are comparable to CAR-T cells on anti-cancer efficacy in xenograft mouse models with pre-existing HLA-mismatched T cells. Co-expressing co-stimulatory factors or T cell-promoting cytokines enhanced BiTE-T cells. Our study suggests CD3ε engagement could be a new strategy for allogeneic T cell therapy worthy of further evaluation.
Keywords: bi-specific T cell engager, chimeric antigen receptor T cell, off-the-shelf, graft versus host disease
Graphical abstract
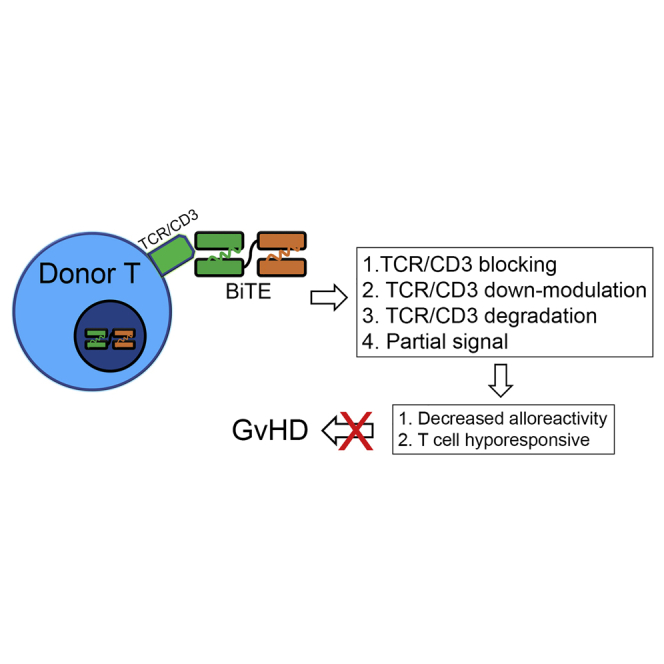
Allogeneic “off-the-shelf” cell therapies are extensively pursued. CD3 engagement has immune-suppressive effects on T cells. By engineering T cells with a bispecific T cell engager (BiTE), T cells show decreased alloreactivity due to CD3 binding while targeting tumor. Thus, T cells secreting BiTEs are potential allogeneic “off-the-shelf” T cells.
Introduction
Autologous chimeric antigen receptor T cells (CAR-T cells) have been a revolutionary therapy for B cell malignancies. Allogeneic “off-the-shelf” (OTS) T and NK cells have been pursued due to potential advantages over autologous CAR-T cells.1 Currently, there are a few promising OTS cell therapies, including allogeneic T cell receptor (TCR)αβ knockout CAR-T cells,2 CAR-NK cells,3 invariant NK T cells,4 gamma-delta (γδ) T cells,5 and virus-specific T cells.6 Compared with autologous CAR-T cells, allogeneic CAR-T cell therapy usually requires gene editing and TCR-negative cell purification to reduce risk of graft-versus-host disease (GvHD). In addition to GvHD, another challenge for OTS cell therapies is host-versus-graft (HvG) reaction, which decreases donor cell persistence and compromises anti-cancer efficacy. Current solutions include (1) deep lymphodepletion using alemtuzumab, fludarabine, and cyclophosphamide;7 (2) deleting HLA-I on donor T cells;8 and (3) co-expressing an alloimmune defense receptor to target alloreactive host T cells.9 Although effective, these strategies add more complex to allogeneic OTS CAR-T cell therapy. Meanwhile, like autologous CAR-T therapy, allogeneic OTS CAR-T cells have adverse effects such as cytokine release syndrome and neurotoxicity.10 Thus, current allogeneic OTS CAR-T therapy has challenges and there is a need for better options.
CD3 monoclonal antibodies (mAbs) are potent immunosuppressive drugs used in clinical transplantation.11, 12, 13 There are essentially two types of CD3 mAbs, mitogenic and nonmitogenic, depending on their capacity of Fc receptor (FcR) binding. Mitogenic CD3 mAbs, represented by OKT3, initiate T cell activation before the suppression of T cell responses. This mitogenic activity depends on TCR-CD3 cross-linking via Fc binding to FcR-expressing cells such as monocytes. Nonmitogenic CD3 mAbs have low or no FcR binding by altering binding to FcR or removing Fc region. Although both are immunosuppressive, mitogenic and nonmitogenic CD3 mAbs suppress T cell responses by distinct mechanisms. Mitogenic CD3 mAbs modulate TCR, induce internalization of the TCR complex, T cell apoptosis, and long-term T cell unresponsiveness. Nonmitogenic CD3 mAbs generally do not induce T cell activation, proliferation, or cytokine production. They tend to deliver a partial TCR signal and render T cell clones hyporesponsive.14,15 As a nonmitogenic CD3 mAb, CD3 single-chain variable fragments (scFvs) have been shown to modulate CD3 without causing T cell activation and cytokine release in vitro. They were able to suppress T cell activation and inflammatory cytokine release in mixed lymphocyte cultures.16 These data underscore immunosuppressive properties of CD3 scFv and their potential to mitigate alloreactions in vivo. Bispecific T cell engager (BiTE) is an emerging drug platform that has shown potent efficacy against hematological cancer.17 A typical BiTE consists of a CD3ε scFv and a target-binding scFv that are tandemly linked together. It enables T cells to kill tumor cells by engaging TCRαβ/CD3 complex to tumor target. Since half of BiTEs is an immunosuppressive drug, we hypothesize that BiTEs could have similar immunosuppressive impacts on T cells to those of CD3 Abs; thus, T cells secreting BiTEs (BiTE-T) are less likely to induce GvHD and are potential “natural” OTS T cells for cancer. The anti-cancer efficacy of BiTE-T cells has been demonstrated in previous pre-clinical studies.18, 19, 20
Results
CD3 engagement decreases TCR/CD3 detection by flow cytometry
To test our hypothesis, we first treated human T cells with increasing doses of human CD19 BiTEs in the presence of 3T3 cells expressing human CD19 or mouse CD19 for 24 h. CD3ε and TCRαβ expressions on the T cell surface were then measured by flow cytometry. CD19 BiTEs decreased CD3ε and TCRαβ detection in a dose-dependent manner (Figure 1A), suggesting that TCRαβ/CD3 complex was masked or modulated by BiTEs. Human CD19 target engagement appeared to further bring down CD3ε/TCRαβ (Figure 1A). Similar results were observed when we treated mouse T cells with mCD3ε antibody (Figure S1A).
Figure 1.

BiTE-T cells have decreased CD3/TCRαβ expression and alloreactivity
(A) CD3/TCRαβ expression on human T cells treated with human CD19 BiTE (blinatumomab) in the presence of 3T3.mCD19 (left) or 3T3.hCD19 cells (right). (B) CD3/TCRαβ expression on CD19 or Her2 BiTE-T cells. (C) T cell activation and cytokine production after TCRαβ stimulation. (D–G) GvHD risk evaluation of BiTE-T cells. Male NSG mice received cyclophosphamide (CTX) or whole-body irradiation (TBI) first and then 10 million T cells 1 day after. Mice were monitored for GvHD. (D) Study design. (E) Weight change over time. (F) NSG mice survival. (G) T cell persistence in peripheral blood. (H) CD3/TCRαβ expression on CD19 BiTE-T cells in peripheral blood of NSG mice without targets. (I) CD3/TCRαβ expression over time on Her2 BiTE-T cells in peripheral blood of NSG mice without targets. (J) CD3/TCRαβ expression on CD19 BiTE-T cells in serial-killing assay on CD19-expressing NALM6 cells. (K) CD3/TCRαβ expression on Her2 BiTE-T cells in serial killing assay on A375.Her2 cells. (L and M) CD3/TCRαβ expression on Her2-targeting T cells in vivo. NSG mice were subcutaneously injected with A375.Her2 cells and treated with Her2 BiTE-T or CAR-T cells 8–10 days later. (L) CD3/TCRαβ expression on donor T cells in peripheral blood 3 weeks after T cell injection. (M) CD3/TCRαβ expression of tumor infiltrating T cells 4 weeks after T cell injection in the A375.Her2 solid-tumor model. (N) CD3/TCRαβ expression on co-cultured T cells. BiTE-T or CAR-T cells from one healthy donor (as donor T cells) were co-cultured with activated T cells from another healthy donor (as recipient T cells) for 72 h and subjected to flow cytometry. DTC, donor T cells; RTC, recipient T cells. (O) CD3/TCRαβ expression over time on host and donor T cells in allogeneic MLR assay. CD19 BiTE-T or CAR-T cells were co-cultured with HLA-mismatched PBMCs at a 1:10 E:T ratio in 96-well plates, and CD3/TCRαβ expression on host and donor T cells were followed over time. Data are presented as means ± SD. ∗p<0.05; ∗∗p<0.01; ∗∗∗p<0.001; ∗∗∗∗p<0.0001; ns, not significant.
BiTE-T cells have reduced TCR/CD3 detection and alloreactivity in vitro
We next designed two BiTEs targeting CD19 and Her2, respectively (Figure S1B). Human T cells from healthy donor peripheral blood mononuclear cells (PBMCs) were transduced with BiTEs to produce BiTE-T cells. CD19 and Her2 BiTE-T cells showed minimal TCRαβ and CD3 detection by flow cytometry in contrast to untransduced T cells (UT) (Figures 1B and S1C). Consistent with this, CAR-T cells engineered with a soluble secreted or membrane-bound CD3ε scFv showed low CD3ε/TCRαβ and comparable cytotoxicity (Figures S1D and S1E) without TCRαβ depletion. BiTE-T cells showed comparable viability, proliferation, and immune phenotype to UT cells otherwise (Figures S1F–S1H). When stimulated with TCRαβ Abs, BiTE-T cells show much lower activation marker CD69 and less IFNγ or IL-2 production compared with untransduced T cells and CAR-T cells (Figure 1C), suggesting that BiTEs have a masking or modulation effect on the CD3/TCRαβ complex.
BiTE-T cells have low persistence and GvHD risk in immune-deficient mice
To evaluate BiTE-T cells' alloreactivity in vivo, we adopted a GvHD mouse model that was previously used for allogeneic OTS CAR-T cells.7,21 Immune-deficient NSG mice underwent full-body irradiation and then intravenously received a high dose of human T cells the following day (Figure 1D). Mice were monitored for weight loss, survival, and blood T cell persistence. The UT-treated group developed GvHD symptoms such as gradual weight loss in 2–3 weeks (Figure 1E), and eventually all died within 40 days (Figure 1F). Mice injected with CD19 or Her2 BiTE-T cells maintained or gained weight while appearing healthy throughout the monitoring period lasting over 120 days (Figures 1E and 1F). Both BiTE-T cells showed low persistence in peripheral blood in contrast to UT, that showed active expansion during the first month after T cell injection (Figure 1G). We also replaced irradiation with chemotherapy for this model and got similar results (Figures S1I and S1J). Despite low persistence in the blood, CD19 BiTE-T cells were able to offer some protection against CD19 + leukemia 4.5 months after injection (Figure S1K). The persisted few CD19 BiTE-T cells in peripheral blood had very low CD3ε and TCRαβ detection by flow cytometry (Figure 1H). Similarly, Her2 BiTE-T cells showed reduced CD3ε and TCRαβ detection in contrast to Her2 CAR-T cells over time in vivo (Figure 1I).
BiTE-T cells maintain low TCR/CD3 detection after target engaging
Next, we set up in vitro serial-killing assays to evaluate TCRαβ and CD3 on BiTE-T cells in the presence of target antigen. T cells were repeatedly challenged with target cells until they failed to kill. TCRαβ and CD3ε expressions were evaluated every 48 h during this period. For both CD19 and Her2 targets, BiTE-T cells maintained low TCRαβ/CD3 through the challenge assay in contrast to CAR-T and UT cells (Figures 1J and 1K). In vivo, we used a Her2-expressing solid-tumor model to evaluate Her2 BiTE-T cells. NSG mice were first inoculated with Her2-expressing human melanoma cell A375 (A375.Her2) and then received BiTE-T or UT treatment. Blood T cells and tumor-infiltrating T cells were evaluated by flow cytometry. In peripheral blood, Her2 BiTE-T cells showed limited presence with low CD3ε (Figure 1L). Tumor-infiltrated Her2 BiTE-T cells also showed lower TCRαβ/CD3 than CAR-T cells (Figure 1M). These results indicate that BiTE-T cells maintain low CD3ε and TCRαβ in the presence of target antigen.
BiTE-T cells decrease TCR/CD3 on bystander T cells in vitro and in vivo
Another challenge for allogeneic OTS T cells is the host-versus-graft (HvG) reaction due to the alloreactivity of host TCRs. We hypothesized that BiTEs could presumably mask or modulate TCRs on host/recipient T cells so they would less likely induce HvG reactions. To test this hypothesis, we co-cultured CD19 BiTE-T cells (donor) with HLA mismatched T cells (host or recipient) for 72 h, and CD3 and TCR of the mixed cells were measured by flow cytometry. As expected, the mixed population with BiTE-T cells had minimal CD3 and TCR detection (Figure 1N). We also set up an allo-MLR (mixed lymphocyte reaction) assay by co-culturing CD19 BiTE-T cells (donor) with HLA mismatched PBMCs (host). TCR/CD3 and cell counts of host and donor populations were measured at days 1, 3, 6, 9, and 12 after allo-MLR setup. Host T cells mixed with CD19 BiTE-T cells had decreased CD3/TCR compared with those mixed with CAR-T or untransduced T cells (Figure 1O, left). At day 1, host T cells mixed with CD19 BiTE-T cells were 4.66% CD3+TCR+, whereas host T cells mixed with either UT or CAR-T cells were over 70% CD3+TCR+ (Figure S2A). As expected, CD19 BiTE-T cells maintained minimal CD3/TCR levels throughout time (Figure 1O, right). Co-culturing Her2 BiTE-T cells with HLA mismatched PBMCs at different ratios also yielded a mix, with low TCR/CD3 ranging from 0.83 to 4.79% (Figure S2B). These data suggest that donor BiTE-T cells have masking or modulation effects on bystander T cells by secreting BiTEs in vitro. Next, we evaluated whether BiTE-T cells could secrete enough BiTEs in vivo so that sera from BiTE-T cell treated mice could mediate T cell killing and CD3/TCR masking or modulation. Her2 BiTE-T cells and UT cells were used to treat the A375.Her2 NSG mouse model. One week after T cell injection, sera were isolated and added to untransduced T cells and A375.Her2 tumor cells co-culture in vitro. After 72 h, IFNγ, IL-2, IL-6, and TNFα in the supernatant were measured. Supernatant containing sera from mice receiving Her2 BiTE-T cells showed significantly elevated IFNγ, TNFα, and IL-6 levels and lower IL-2 levels compared with the supernatant containing sera from mice that received UT cells (Figures S2C–S2F). We then measured the CD3/TCR on the T cells from co-culture. The CD3 levels on T cells treated with Her2 BiTE-T sera were lower (p = 0.0571) than those treated with UT sera (Figure S2G) while being comparable to T cells treated with supernatant from BiTE-T cell culture (positive control). These data demonstrated that BiTE-T cells can generate meaningful levels of BiTEs in vivo which potentially have impacts on TCR/CD3 on host T cells.
BiTE-T cells elicit curbed activation on targets and are less durable than second-gen CAR-T cells
The anti-cancer activities of BiTE-T cells have been demonstrated by other groups.18,19 Here, we did side-by-side comparisons between BiTE-T and the second-generation CAR-T cells on their anti-cancer activities. CD19 or Her2 BiTE-T and CAR-T cells sharing the same scFv were evaluated. For both targets, BiTE-T cells showed comparable, if not better, killing than CAR-T cells in vitro (Figures 2A and 2C). BiTE-T cells produced only a fraction of Th1 cytokines that CAR-T cell produced (Figures 2B and 2D), including IFNγ, IL-2, IL-6, and TNFα. Of note, IL-2 levels produced by BiTE-T cells are very minimal, suggesting that BiTE-T cells tend to elicit curbed activation and proliferation when targets are engaged. This is in contrast to the full-blown activation mediated by CAR-T cells and raises the possibility that BiTE-T cells may have less toxicity than CAR-T cells. Meanwhile, CAR-T cells outperformed BiTE-T cells in serial-killing assays (Figures S3A–S3D) and offered better survival, persistence, and efficacy in NSG models transplanted with NALM6 or A375.Her2 cells (Figures S3E–S3H). This is not surprising, considering that BiTE-T cells have curbed activation, whereas second-generation CAR-T cells have built-in co-stimulatory domains.
Figure 2.

BiTE-T cells produce fewer cytokines and are comparable to or better than CAR-T cells on efficacy in the presence of host T cells
(A and B) Cytotoxicity (A) and cytokine production (B) of CD19 BiTE-T or CAR-T cells against 3T3.hCD19 cells. (C and D) Cytotoxicity (C) and cytokine production (D) of Her2 BiTE-T or CAR-T cells against A375.Her2 cells. (E–H) a comparison study of CD19 BiTE-T and CAR-T cells in vivo with host T cell presence. (E) Study design. Male NSG mice received 1.2 Gy full-body irradiation and then were engrafted with 7 million HLA-A2+-activated T cells 1 day after. Nalm6GL cells were given at day 0. Mice were treated with HLA-A2– CD19 BiTE-T or CAR-T cells at day 3. Survival (F), Donor T cells (DTC; G), and recipient T cells (RTC; H) in peripheral blood were monitored over time. (I–L) a comparison study of Her2 BiTE-T and CAR-T cells in vivo with host T cell presence. (I) Study design. Male NSG mice received 1.2 Gy full-body irradiation and then were engrafted with 5 million HLA-A2+-activated T cells 1 day after. A375.Her2 cells were given at day 0. Mice were treated with HLA-A2– Her2 BiTE-T or CAR-T cells at day 8. Tumor growth (J), DTC (K), and RTC (L) in peripheral blood were monitored over time. Data are presented as means ± SD. ∗p<0.05; ∗∗p<0.01; ∗∗∗p<0.001; ∗∗∗∗p<0.0001; ns, not significant.
Donor BiTE-T cells show comparable efficacy to CAR-T cells in the presence of host T cells
One potential advantage of BiTE-T cells is their capability of mobilizing bystander or host T cells. Thus, we adopted a mouse model to mimic an environment with pre-existing host T cells.9 NSG mice were irradiated and then engrafted with activated HLA-A2+ human T cells. Mice were then injected with tumor cells and later treated with HLA-A2– BiTE-T or CAR-T cells (Figure 2E). In the NSG NALM6 model with pre-existing T cells, both CD19-targeting BiTE-T and CAR-T cells showed a comparable survival benefit over UT cells (Figure 2F). Donor CD19 CAR-T cell expansion was also curbed in this model, although host T cells showed expansion in the CAR-T-treated group (Figures 2G and 2H). In the Her2-overexpressing tumor model with pre-existing host T cells (Figure 2I), CAR-T cells were not able to inhibit tumor growth whereas BiTE-T cells still could (Figure 2J). In peripheral blood, both Her2-targeting BiTE-T and CAR-T cells showed very limited persistence (Figure 2K). However, Her2 BiTE-T cells induced greater host T cell expansion than CAR-T cells (Figure 2L).
Co-stimulation or T cell-promoting cytokines enhance BiTE-T cells while maintaining low CD3/TCR
To enhance efficacy, we added co-stimulation to BiTE-T cells. T cells were co-transduced with BiTE and a bicistronic construct of 4-1BBL and CD8022 to make enhanced BiTE-T cells. These enhanced Her2 BiTE-T cells showed better proliferation than Her2 CAR-T cells in the serial-killing assay while maintaining low levels of CD3/TCRαβ (Figures 3A–3C). In vivo (Figure 3D), enhanced Her2 BiTE-T cells showed comparable tumor suppression, blood persistence, and tumor infiltration and weights to those of CAR-T cells (Figures 3E–3H). Similarly, in the NALM6 model (Figure 3I), the enhanced CD19 BiTE-T cells had comparable efficacy and possibly better blood persistence than CAR-T cells (Figures 3J–3L and S4). Enhanced CD19 BiTE-T cells maintained low CD3/TCRαβ (Figure 3M) in vivo. We also enhanced BiTE-T cells by co-expressing a bicistronic construct of IL7 and IL15 genes. BiTE T cells engineered with cytokines out-performed CAR-T cells in the serial-killing assay in vitro (Figures 3N and 3O).
Figure 3.

Co-expressing co-stimulatory factors or cytokines enhances BiTE-T cells to match or overperform CAR-T cells
(A–C) a serial-killing assay on A375.Her2 cells. (A) T cell proliferation over time. (B) TCRαβ expression over time. (C) CD3 expression over time. (D–H) efficacy comparison of co-stimulation-enhanced Her2 BiTE-T and CAR-T cells. (D) Study design. (E) Tumor growth. (F) Percentages of tumor-infiltrating T cells. (G) Donor T cells in peripheral blood over time. (H) Weight change over time. (I–M) Data from a comparison study of co-stimulation-enhanced CD19 BiTE-T and CAR-T cells. (I) Study design. (J) Survival. Small arrows indicate death events unrelated to leukemia or GvHD. (K) Weight change. (L) Donor T persistence in peripheral blood. (M) CD3/TCRαβ expression on week 4 blood T cells. (N) Proliferation of Her2 targeting cytokine -enhanced BiTE-T cells in serial-killing assay in vitro. (O) Proliferation of CD19 targeting cytokine-enhanced BiTE-T cells in serial-killing assay in vitro. Data are presented as means ± SD. ∗p<0.05; ∗∗p<0.01; ∗∗∗p<0.001; ∗∗∗∗p<0.0001; ns, not significant.
Discussion
Our study for the first time brings attention to a neglected fact that one-half of a BiTE (i.e., CD3 scFv) is actually a potent immunosuppressive agent, suggesting that BiTEs could curb T cells' alloreactivity while enabling T cells to kill cancer cells. With this rationale, we propose BiTE-secreting T cells as a novel and simple strategy for allogeneic T cell therapy. BiTE-T cells potentially have a few benefits. First, BiTE-T cells are less complex to produce, with no need of gene editing or TCRαβ negative purification. Despite an exciting technology, gene editing adds more complexity, cost, and hurdles to OTS T cell production and could cause uncertain consequences.23 Second, BiTE-T cells may have low toxicity for producing fewer cytokines. Although manageable, cytokine release syndrome (CRS) is still a common adverse effect in allogeneic CAR-T therapy. Third, BiTE-T cells can secrete BiTEs to re-direct bystander T cells to kill tumor in vivo. Last, BiTE-T cells could potentially alleviate HvG reaction by secreting BiTEs, as they would have the same masking and modulation effects on host TCRs.
BiTEs are OTS anti-cancer agents that are readily available to patients, which raises the question whether allogeneic BiTE-T cells are superior to BiTEs. Regardless of efficacy, the two are different types of therapeutics, and each has its pros and cons. For BiTE-T cells, one advantage is that they do not require multi-weeks of daily infusions as BiTEs do. Another is that BiTE-T as a cell-based therapy can undergo further engineering to gain sophisticated functions and upgrades. Also, BiTEs rely on patient T cells to take effect, whereas BiTE-T cells do not. Finally, BiTE-T cells potentially have less toxicity than BiTEs, since BiTE-T cells elicit curbed activation when killing whereas BiTEs likely induce typical TCR activation on endogenous T cells.24
In our study, total T cells consisting of BiTE-expressing T cells and a significant amount of untransduced T cells were used for GvHD evaluation. Theoretically, these untransduced T cells risk developing GvHD. However, we found that high doses of human BiTE-T cells (including untransduced T cells) with 40–90% transduction efficiency did not induce GvHD in immune-deficient NSG mice. A possible explanation is that BiTE-treated T cells are prone to apoptosis if there are no further signals from co-stimulation, cytokines, or target engaging. Like BiTE-T cells, BiTE-treated untransduced T cells are potentially hyporesponsive, as discussed below.
The underlying mechanisms that BiTE-T cells have low risk of GvHD in the NSG mouse model could be multifaceted, and distinctive mechanisms may play a role before and after target engagement. Before engaging target, BiTEs potentially impact T cells like nonmitogenic CD3 mAbs, such as (1) they have direct masking and modulation effects that sterically block TCR recognizing alloantigens; and (2) they deliver constant partial TCR signals that render T cell hyporesponsive.13,14 Without other support (e.g., cytokines), these BiTE-T cells are prone to apoptosis, as evidenced by low persistence in NSG mice. After tumor cells are engaged, cross-linking of BiTEs induces TCR internalization and degradation and even apoptosis in T cells. Thus, BiTEs perform more like mitogenic CD3 mAbs, reducing alloreactivity at this stage. All these mechanisms and impacts of BiTEs could apply to host T cells as well, thus potentially mitigating HvG reactions. However, effects on host T cells require significant levels of BiTEs in circulation. Potential strategies include (1) using multiple and high doses of BiTE-T cells, (2) maximizing BiTE production and secretion by T cells, and (3) making T cells secrete BiTEs with extended half-life.
Since we have shown that BiTE-T cells are not as durable as second-generation CAR-T cells in terms of serial-killing capacity and persistence in vivo, the overarching challenge will be to increase the persistence of allogeneic BiTE-T cells while maintaining their OTS feature. There are multiple strategies such as adding co-stimulatory factors or pro-inflammatory cytokines. However, caution should be taken, as such genetic modifications might rescue BiTE-T cells from hyporesponsive mode.
In summary, BiTE-T cells have the unique characteristics to address both hurdles for OTS T cell therapy in a simple way, which warrants further investigation.
Materials and methods
Study design
The purpose of this study was to develop a novel allogeneic “off-the-shelf” T cell therapy. We evaluated CD19-and Her2-targeting BiTE-engineered T cells on their CD3/TCRαβ expression. GvHD risk of BiTE-T cells was evaluated using chemo-conditioned or whole-body-irradiated NSG mice. We also performed side-by-side comparisons between BiTE-T and CAR-T cells on cytotoxicity, cytokines, serial killing, and in vivo efficacy with or without host T cells. Two xenograft mouse models were used to compare the efficacy between BiTE-T and CAR-T cells, including hematological and solid tumor. A variety of healthy donors were used to produce engineered T cells. All experiments were independently repeated at least twice.
Cells and medium
The NALM6GL cell line expressing both GFP and firefly luciferase was purchased from ATCC and cultured in RPMI 1640 medium supplemented with 10% FBS, L-glutamine, and Pen/Strep (RPMI 10). The human melanoma A375 cell line was a gift from Dr. Cecilia Ramello and was cultured in DMEM supplemented with 10% FBS, L-glutamine, and Pen/Strep. Her2-expressing A375 (A375.Her2) cells were made by transducing A375 cells using retrovirus expressing a truncated Her2. Human T cells were cultured in RPMI 10 supplemented with 10 ng/mL IL-7 and 5 ng/mL IL-15.
Retroviral constructs, γ-retrovirus production, and T cell transduction
DNAs of human Her2 CAR, human CD19 BiTE, Her2 BiTE, 4-1BBL-CD80, and other constructs were synthesized and subcloned to an SFG retroviral vector by Genewiz (South Plainfield, NJ). Human CD19 CAR has been described previously.25 Her2 CAR and Her2 BiTE use the same scFv derived from Trastuzumab. CD19 CAR and CD19 BiTE use the same scFv derived from FMC63 clone. Recombinant γ-retrovirus production has been described previously.26 Briefly, retroviral constructs containing CAR or BiTE were transiently transfected to H29 cells. Supernatant from transfected H29 cells were used to transduce RD114 cells to make a stable producer cell line. Retroviral supernatant from RD114 cells was harvested, and 0.45 μm was filtered and cryopreserved for future use. For T cell transduction, healthy donor leukopaks were purchased from Stemcell Technologies (Vancouver, BC, Canada) and ALLCELLS (Alameda, CA). PBMCs were isolated using standard Ficoll method and cryopreserved for future use. Engineered-T cell production was described previously,26 with minor modification. At day 0, T cells were isolated from the cryopreserved PBMCs by using a human T cell isolation kit (Stemcell Technologies). T cells were then activated using human CD3/CD28 dynabeads (Thermo Fisher Scientific, Waltham, MA). At day 1 and day 2, T cells were transduced using fresh BiTE or CAR retrovirus in RetroNectin (Takara Bio, Mountain View, CA) -coated plates at 2,000 × g, 32°C for 1 h. At day 3, fresh medium with cytokines was added. Dynabeads were removed at day 6, and T cells were further expanded in complete medium with 10 ng/mL human IL-7 and 5 ng/mL human IL-15. Medium and cytokines were replenished every 2–3 days. Days 8–14 T cells were used for in vivo study.
Flow cytometry
The following antibody clones were used for flow cytometry: from BD: CD3 (Clone SK7), CD8 (Clone RPA-T8), CD45 (Clone HI30), TCRαβ (Clone T10B9.1A-31), CD45RA (HI100), PD1 (Clone EH12.2H7), CD69 (Clone FN50), and CD25 (Clone 2A3); from Biolegend: CD4 (Clone OKT4), TCRαβ (Clone IP26), CCR7 (Clone G043H7), nd aCD62L (Clone DREG-56). Fc receptor-binding inhibitor (Thermo Fisher) was routinely used for staining. Whole-blood samples were stained and lysed using BD FACS lysing solution. CountBright absolute counting beads (Thermo Fisher) were added for measuring cell numbers. Flow data were acquired on a BD LSRII or BD FACSymphony flow cytometer. Data were analyzed using FlowJo version 10.
ELISA
An Ella machine (ProteinSimple, San Jose, CA) was used for ELISA. Twenty-four- to 72-h supernatant from cytotoxicity or TCR stimulation assay was added to ELLA cartridges for cytokine measurements.
TCR/CD3 stimulation assay
For human T cell stimulation assay, non-tissue culture-treated 24-well plates were coated with 1 μg/mL human TCRαβ antibody (Clone IP26) overnight. T cells were added and cultured in the plates. For activation marker, T cells were subjected to flow analysis after 5-h stimulation. Supernatant was collected after 24-h stimulation, and cytokines were measured by ELISA. For BiTE stimulation, human CD19 BiTE (blinatumomab) was purchased from BPS Bioscience (San Diego, CA). T cells were co-cultured with 3T3.hCD19 or 3T3.mCD19 cells with increasing blinatumomab concentrations. For mouse T cell stimulation, T cells were isolated from spleen and added to 24-well plates coated with mouse CD3ε antibody (Clone 145-2C11). After 5 h, cells were analyzed by flow cytometry.
Cytotoxicity and serial-killing assay
An xCELLigence RTCA instrument (Agilent Technologies, Santa Clara, CA) was used for cytotoxicity assay. T cells were co-cultured with target cells at various effector-to-target (E:T) ratios in RTCA E-plates and real-time cell killing was recorded on the instrument. For serial-killing assays, T cells were co-cultured with target cells at a 5:1 E:T ratio in non-tissue culture-treated 24-well plates. Every 2 days or longer, a small fraction of T cells were analyzed by flow cytometry for phenotype and cell counts, while the remaining cells were transferred to a new plate with fresh medium and target cell challenge.
Mouse study
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the University of South Florida (USF). NSG mice were purchased from the Jackson Lab and bred in the USF animal facility. Eight- to twelve-week-old male NSG mice were used for in vivo studies. For GvHD evaluation, NSG mice underwent 2.5 Gy whole-body irradiation or 250 mg/kg cyclophosphamide (CTX) and then received T cells at a 10-million dose the following day. Mice were monitored for survival and any signs of GvHD (hunched posture, slow mobility, weight loss, etc.). Mice having more than 20% weight loss and other GvHD symptoms were euthanized. For efficacy study on the hematological tumor model, NSG mice were intravenously injected with 0.5 million NALM6GL cells. Three days later, mice were intravenously injected with 10 million CAR-T, BiTE-T, or UT cells. Survival was monitored twice a week. For the solid-tumor model, NSG mice were subcutaneously injected with 0.5 million A375.Her2 cells on the right flank. Seven to ten days later, mice were intravenously injected with 10 million CAR-T, BiTE-T, or UT cells. Blood was collected weekly, and tumor tissues were collected at end point. For mice with pre-existing T cells, NSG mice received 1.2 Gy whole-body irradiation and then were subsequently engrafted with activated T cells. Mice were injected with tumor cells and treated with HLA mismatched T cells. For details, see the timelines in the figures. Tumor tissues were processed on a gentleMACS Octo Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) and digested with Liberase and DNase I (Roche, Mannheim, Germany). Isolated tumor cells were subjected to flow analysis.
Statistical methods
GraphPad Prism 8 was used for statistical analysis, as indicated in figure legends. Data are presented as means ± SD. An unpaired parametric t test was used for two-group comparison. One-way ANOVA was used for multiple-group comparison. Survival was compared using log rank tests.
Acknowledgments
This study was funded by H. Lee Moffitt Cancer Center and Research Institute internal support grant (M.L.D.). The authors would like to acknowledge the animal care staff in the Department of Comparative Medicine at the University of South Florida for technical assistance and the flow cytometry core at Moffitt. We thank Dr. Emiliano Roselli for review of the manuscript.
Author contributions
G.L. conceived the idea, designed the study, performed experiments, acquired and analyzed data, wrote, reviewed, and edited the manuscript. M.L.D. funded and supervised the study and reviewed the manuscript. K.M.R., K.S., N.B., and J.B. performed experiments and acquired and analyzed data. K.M.R. and J.B. reviewed and edited the manuscript.
Declaration of interests
G.L. and M.L.D. are co-inventors of a patent related to the research. G.L. is the founder and holds equity in Immunebro Therapeutics, Inc., a company pursuing commercializing this technology.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.02.024.
Contributor Information
Gongbo Li, Email: gongbo.li@northwestern.edu.
Marco L. Davila, Email: marco.davila@moffitt.org.
Supplemental information
References
- 1.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 2.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K., et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 3.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y.R., Zhou Y., Kim Y.J., Zhu Y., Ma F., Yu J., Wang Y.C., Chen X., Li Z., Zeng S., et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021;2:100449. doi: 10.1016/j.xcrm.2021.100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makkouk A., Yang X.C., Barca T., Lucas A., Turkoz M., Wong J.T.S., Nishimoto K.P., Brodey M.M., Tabrizizad M., Gundurao S.R.Y., et al. Off-the-shelf Vdelta1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-003441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aftab B.T., Sasu B., Krishnamurthy J., Gschweng E., Alcazer V., Depil S. Toward “off-the-shelf” allogeneic CAR T cells. Advances Cell Gene Therapy. 2020;3:e86. doi: 10.1002/acg2.86. [DOI] [Google Scholar]
- 7.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R., et al. Multiplex genome-edited T-cell manufacturing platform for “Off-the-Shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 8.Wang D., Quan Y., Yan Q., Morales J.E., Wetsel R.A. Targeted disruption of the beta2-Microglobulin gene minimizes the immunogenicity of human embryonic stem cells. Stem Cells Transl. Med. 2015;4:1234–1245. doi: 10.5966/sctm.2015-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mo F., Watanabe N., McKenna M.K., Hicks M.J., Srinivasan M., Gomes-Silva D., Atilla E., Smith T., Ataca Atilla P., Ma R., et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol. 2021;39:56–63. doi: 10.1038/s41587-020-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neelapu S.S., Nath R., Munoz J., Tees M., Miklos D.B., Frank M.J., Malik S.A., Stevens D., Shin C.R., Balakumaran A., et al. ALPHA study: ALLO-501 produced deep and durable responses in patients with relapsed/refractory non-Hodgkin's lymphoma comparable to autologous CAR T. Blood. 2021;138:3878. doi: 10.1182/blood-2021-146038. [DOI] [Google Scholar]
- 11.Smith S.L. Ten years of Orthoclone OKT3 (muromonab-CD3): a review. J. Transpl. Coord. 1996;6:109–119. doi: 10.7182/prtr.1.6.3.8145l3u185493182. quiz 120-101. [DOI] [PubMed] [Google Scholar]
- 12.Ortho Multicenter Transplant Study G. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. N. Engl. J. Med. 1985;313:337–342. doi: 10.1056/NEJM198508083130601. [DOI] [PubMed] [Google Scholar]
- 13.Chatenoud L. CD3-specific antibody-induced active tolerance: from bench to bedside. Nat. Rev. Immunol. 2003;3:123–132. doi: 10.1038/nri1000. [DOI] [PubMed] [Google Scholar]
- 14.Smith J.A., Tso J.Y., Clark M.R., Cole M.S., Bluestone J.A. Nonmitogenic anti-CD3 monoclonal antibodies deliver a partial T cell receptor signal and induce clonal anergy. J. Exp. Med. 1997;185:1413–1422. doi: 10.1084/jem.185.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith J.A., Bluestone J.A. T cell inactivation and cytokine deviation promoted by anti-CD3 mAbs. Curr. Opin. Immunol. 1997;9:648–654. doi: 10.1016/s0952-7915(97)80044-1. [DOI] [PubMed] [Google Scholar]
- 16.Le Gall F., Reusch U., Moldenhauer G., Little M., Kipriyanov S.M. Immunosuppressive properties of anti-CD3 single-chain Fv and diabody. J. Immunol. Methods. 2004;285:111–127. doi: 10.1016/j.jim.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Goebeler M.E., Bargou R.C. T cell-engaging therapies - BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020;17:418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 18.Iwahori K., Kakarla S., Velasquez M.P., Yu F., Yi Z., Gerken C., Song X.T., Gottschalk S. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol. Ther. 2015;23:171–178. doi: 10.1038/mt.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi B.D., Yu X., Castano A.P., Bouffard A.A., Schmidts A., Larson R.C., Bailey S.R., Boroughs A.C., Frigault M.J., Leick M.B., et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019;37:1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 20.Liu X., Barrett D.M., Jiang S., Fang C., Kalos M., Grupp S.A., June C.H., Zhao Y. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood Cancer J. 2016;6:e430. doi: 10.1038/bcj.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamiya T., Wong D., Png Y.T., Campana D. A novel method to generate T-cell receptor-deficient chimeric antigen receptor T cells. Blood Adv. 2018;2:517–528. doi: 10.1182/bloodadvances.2017012823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velasquez M.P., Szoor A., Vaidya A., Thakkar A., Nguyen P., Wu M.F., Liu H., Gottschalk S. CD28 and 41BB costimulation enhances the effector function of CD19-specific engager T cells. Cancer Immunol. Res. 2017;5:860–870. doi: 10.1158/2326-6066.CIR-17-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020;183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 24.Slaney C.Y., Wang P., Darcy P.K., Kershaw M.H. CARs versus BiTEs: a comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 2018;8:924–934. doi: 10.1158/2159-8290.CD-18-0297. [DOI] [PubMed] [Google Scholar]
- 25.Li G., Boucher J.C., Kotani H., Park K., Zhang Y., Shrestha B., Wang X., Guan L., Beatty N., Abate-Daga D., Davila M.L. 4-1BB enhancement of CAR T function requires NF-kappaB and TRAFs. JCI Insight. 2018;3 doi: 10.1172/jci.insight.121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li G., Park K., Davila M.L. Gammaretroviral production and T cell transduction to genetically retarget primary T cells against cancer. Methods Mol. Biol. 2017;1514:111–118. doi: 10.1007/978-1-4939-6548-9_9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.