

Abstract

Aberrant gene-silencing through dysregulation of polycomb protein activity has emerged as an important oncogenic mechanism in cancer, implicating polycomb proteins as important therapeutic targets. Recently, an inhibitor targeting EZH2, the methyltransferase component of PRC2, received U.S. Food and Drug Administration approval following promising clinical responses in cancer patients. However, the current array of EZH2 inhibitors have poor brain penetrance, limiting their use in patients with central nervous system malignancies, a number of which have been shown to be sensitive to EZH2 inhibition. To address this need, we have identified a chemical strategy, based on computational modeling of pyridone-containing EZH2 inhibitor scaffolds, to minimize P-glycoprotein activity, and here we report the first brain-penetrant EZH2 inhibitor, TDI-6118 (compound 5). Additionally, in the course of our attempts to optimize this compound, we discovered TDI-11904 (compound 21), a novel, highly potent, and peripherally active EZH2 inhibitor based on a 7 member ring structure.
Keywords: EZH2 inhibitor, cancer, epigenetics, BBB (blood brain barrier), central nervous system
The characterization of somatic genomes in cancer through efforts such as TCGA (The Cancer Genome Atlas)1 has shown that chromatin factors are highly mutated in cancer, with up to 40% of cancers bearing at least one mutation in an epigenetic regulator.2 This work has generated much interest in targeting epigenetic regulators therapeutically in cancer. One of the most well-characterized epigenetic regulators is enhancer of zeste homologue 2 (EZH2), the methyltransferase (“writer”) component of the polycomb repressive complex 2 (PRC2).3 EZH2 catalyzes the generation of histone-3 lysine 27 methylation (H3K27me),4 an epigenetic mark which is critical for mediating gene repression in many contexts including development.5 A wealth of studies have implicated an oncogenic function for EZH2 through its ability to repress tumor suppressor genes6 stimulating many EZH2 drug discovery programs.7−9 For example, certain cancers such as lymphoma harbor recurrent gain-of-function mutations in EZH2, leading to aberrant gene repression through enhanced production of H3K27 trimethylation (H3K27me3).10,11 Additionally, tumors which bear mutations in the SWI-SNF (SWItch/Sucrose Non-Fermentable) complex, including sarcomas, are frequently sensitive to EZH2 inhibition,12−14 due to an evolutionary conserved synthetic lethal relationship between SWI-SNF and polycomb.15,16
Validation of EZH2 as a bona fide therapeutic target in cancer was recently achieved with encouraging clinical responses to tazemetostat in patient trials leading to U.S. Food and Drug Administration approval in epithelioid sarcoma and follicular lymphoma.17,18 Although this and other drug discovery programs have been effective in generating EZH2 inhibitors for human clinical trials, there remains a need for novel chemical matter to address other specific contexts in oncology and beyond. One important area of unmet need is central nervous system malignancies.19 Twenty percent of all patients with cancer will develop brain metastases,20,21 and several of the cancer types which have a propensity to metastasize to the brain have been shown to be effectively treated with EZH2 inhibition in preclinical cancer models, including small cell lung cancer,22 specific breast adenocarcinoma subtypes, and melanoma.23,24 Furthermore, malignant brain tumors are the leading cause of cancer-related mortality in children.25 Notably, several pediatric brain tumors are characterized by a reprogrammed epigenomic landscape in which EZH2 is essential for tumor maintenance including midline glioma,26,27 ependymoma,28 and atypical rhabdoid tumors of the brain,29,30 all of which lack effective therapies. Thus, a brain-penetrant EZH2 inhibitor could be of great utility in oncology.
Many successful case studies have been reported in the literature where brain penetration is enhanced by minimizing P-glycoprotein (P-gp) efflux transport and increasing passive permeability.32 Hydrogen bond donor count is one key physicochemical property which influences P-gp substrate activity,33,34 and reducing the number of hydrogen bond donors has been shown as an effective strategy to lower efflux transport.32,35−37 While EZH2 inhibitors reported in the literature possess an overall broad diversity of structures, most share a common, conserved pyridone which is critical for engaging and inhibiting EZH2 enzymatic activity [Tazemetostat (1),7 PF-06726304 (2),38 PF-06821497 (3),39 CPI-1205 (4),8 (Figure 1)]. It has been shown that pyridone containing EZH2 inhibitors are substrates of efflux transporters which severely restrict their distribution into the brain.40 We hypothesized that the common pyridone motif represents a good substrate for efflux transporters due to the presence of a hydrogen bond donor NH group. We envisioned that masking the hydrogen bond donor NH group in the pyridone could produce compounds that maintain a favorable potency profile and possess a reduced efflux ratio leading to small molecule inhibitors with improved brain-penetration properties.
Figure 1.

EZH2 inhibitors reported in the literature.31
We initiated our medicinal chemistry campaign with an analysis of the crystal structure of polycomb repressive complex 2 (PRC2) bound to a pyridone-containing inhibitor (PDB: 5IJ7).41 The pyridone moiety forms two hydrogen-bond interactions with the backbone of Trp624 from the conserved GXG motif of the SET domain and a water-mediated interaction with the side chain of Asn688. Calculation of hydration site thermodynamics using WaterMap42 suggested that the bridging water is only weakly bound (ΔG = 3.09 kcal/mol; ΔH = −1.36; −TΔS = 4.45), indicating that it could be displaced without a large desolvation penalty (Figure 2, left). Methylation of the pyridone N–H would remove one of the two hydrogen-bond interactions with Trp624 and displace the bridging water, as predicted by the docking model of a methylated analogue (Figure 2, right). Our analysis and a literature report43 suggested that methylation of the free NH in the pyridone would be tolerated. However, we also reasoned that the binding energy could be recovered through modification on the other parts of the molecule such as the 4-position of the pyridone, as shown by others.44
Figure 2.

(Left) WaterMap calculation showed destabilizing energy at the bridging water hydration site. (Right) Docking model of methylated pyridone compound 5 in EZH2 (PDB: 5IJ7).
Encouraged by this analysis, we selected a set of disclosed pyridone series EZH2 inhibitors and methylated the NH group in the conserved dimethylpyridone substituent, reducing the number of hydrogen bond donors and aiming to increase brain penetration. While methylation on the pyridone resulted in an expected reduction in biochemical potency, we were still able to identify compounds with low nanomolar activity against EZH2. More importantly, several of these modified EZH2 inhibitors exhibited improved multidrug resistance (MDR) efflux ratios compared to the parent N–H pyridone, supporting our strategy of N-methylation on the pyridone. One of these compounds, 5, exhibited an IC50 of 14 nM in our biochemical assay, a modest drop in potency relative to the original compound 2. Compound 5 retained moderate potency in the cellular EZH2 inhibition assay (flow cytometric quantitation of H3K27me3) (Table 1) while displaying a very favorable MDR ratio of 0.95 and good permeability, representing a significant improvement relative to compound 2. Furthermore, the concentration ratio of unbound drug in brain to blood (Kp,uu = 0.29) obtained from in vivo analysis in rats (see the Supporting Information, SI) demonstrated that compound 5 is brain-penetrant in vivo and provided proof-of principle that N-methylation of the pyridone group was sufficient to confer brain penetrance. Unfortunately, the instability toward both human and mouse liver microsomes as well as a modest in vitro antiproliferation effect compared to parent compound 2 limited the utility of 5 in follow up studies. To utilize this novel class of brain-penetrant EZH2 inhibitor for in vivo studies, we sought to further improve the compound’s cell potency and liver microsomal stability.
Table 1. Methylation of Dimethylpyridone to Improve Blood-Brain Barrier (BBB) Penetrance.

| 2 | 5a | |
|---|---|---|
| EZH2 IC50 (nM)b | 1.0 | 14 |
| cellular IC50 (H3K27me3) (nM)b | 16 | 580 |
| Karpas-422 proliferation (nM)b | 122 | 4680 |
| HLM/MLM (μL/min/mg) | 210/253 | 449/>768 |
| Cltotal (μL/min/g) | 35 | 100 |
| AUCpo (h·ng/mL)/bioavailability | 67.1/13.7% | 5.8/3.5% |
| MDCK/MDR1 permeability(nm/s) AB, efflux ratio | 37, 4.8 | 105, 0.95 |
| Kp,uu | <0.003 | 0.29 |
Preparation of compound 5 is summarized in Scheme S1 in the SI.
Mean of two determinations.
Our approach to improve potency and metabolic stability was initially guided by literature reports exploring substitution at the 4-position of the pyridone.44 These showed that the 4-position of the pyridone ring can accommodate a larger hydrophobic group, which is involved with van der Waals contacts with hydrophobic residues on the protein. Changes at this position can have a pronounced effect on EZH2 enzyme activity45 and we reasoned that the potency loss resulting from N-methylation could be recovered by introducing favorable substituents at the 4-position of the pyridone in our brain-penetrant molecules. We then prepared several analogues related to 5 in an effort to improve EZH2 inhibition activity (Table 2). When we incorporated increasingly more hydrophobic substituents (compounds 6a–c), we found that the MDR ratios were maintained at a desirable level. However, their biochemical potency dropped dramatically. We did observe moderate improvement in human microsome stability; however, stability against mouse liver microsome remained very low. A methoxy group was incorporated to reduce lipophilicity, anticipating improved microsomal stability predicted by lower AlogP value. Compound 6d displayed a moderate human liver microsome (HLM) increase, however mouse liver microsomes (MLM), biochemical and cellular potency were similar to compound 5. Surprisingly, compound 6d exhibited a greatly increased MDR ratio suggesting a very narrow SAR space to maintain desirable permeability characteristics. A similar observation was made by incorporation of a 4-thiomethyl pyridone, recently reported to enhance EZH2 potency of compound 4 (Figure 1) with increased residence time,45 to provide compound 6e which exhibited biochemical and cellular potency similar to parent compound 2. Therefore, while the 4-SMe group was able to restore the potency lost due to N-methylation in compound 5, the desirable brain-penetrating properties gained through N-methylation were lost.
Table 2. SAR on the 4-Position of the Pyridone.

| compda | R4 | EZH2 IC50 (nM)b | Karpas-422 proliferation (nM)b | HLM/MLM (μL/min/mg) | AlogP | MDCK/MDR1 permeability (nm/s) AB, BA/AB |
|---|---|---|---|---|---|---|
| 5 | Me | 14 | 4680 | 449/>768 | 4.4 | 105, 0.95 |
| 6a | Et | 82 | >768/>768 | 4.9 | 110, 0.72 | |
| 6b | c-Pr | 302 | 490/>768 | 4.7 | 52, 1.5 | |
| 6c | CF3 | >1000 | 287/592 | 4.9 | 49, 2.4 | |
| 6d | OMe | 11.6 | 2950 | 117/656 | 3.9 | 10, 23 |
| 6e | SMe | 1.3 | 632 | 235/796 | 4.5 | 3, 110 |
Preparation of compounds 6a–e summarized in Schemes S2–S6 in the SI.
Mean of two determinations.
Further efforts to improve the potency and metabolic stability of N-methyl pyridone-containing EZH2 inhibitors led us to initiate a medicinal chemistry effort to identify a new class of scaffold. Kung et al. showed that a 7-membered ring analogue related to compound 2 exhibited comparable potency and metabolic stability.38 Our modeling studies based on crystal data (Figure 3) showed that there is overall good overlay between 6- and 7-membered ring structures. The expanded 7-membered ring occupied a similar space with the potential for additional interactions with the protein. Guided by our computational docking model, we then prepared several analogues related to compound 2 (Table 3) with progressively larger substituents R1 at the aniline nitrogen. We chose to use the 4-OMe pyridone because it imparted higher microsomal stability (likely due to lower lipophilicity), as well as slightly more potent EZH2 biochemical activity. Our first set of compounds included 7 (R1 = H) and 8 (R1 = Me), which both exhibited subnanomolar potency in the biochemical assay and were as potent as the parent 6-membered ring compound 2. The enhanced biochemical potency translated into similar or better potency in cellular H3K27me3 assays and antiproliferation assays (Table 3). Compounds 7 and 8 also displayed improvements in HLM/MLM stability parameters relative to the analogous 6-membered-ring compound 2, presumably attributed to the decrease in AlogP values by incorporating the additional nitrogen in the 7-membered ring. Substitution of the aniline nitrogen with a larger ethyl group, compound 9, displayed dramatic attenuation of biochemical potency. It is unclear whether the potency drop was caused by the conformation change in the 7-membered ring due to steric repulsion between the ethyl group and the Cl group, or simply steric hindrance in the binding pocket to accommodate the ethyl group that is not predicted by our docking model. By removing the Cl group, we anticipated that a less restricted conformation would be populated due to the relief of steric hindrance, and at the same time, further reduction of AlogP will bring the HLM/MLM stability to a favorable level. We then prepared analogous des-Cl compounds 10 (R1 = H) and 11 (R1 = Me). Compound 10 displayed a modest loss in biochemical potency and antiproliferation efficacy, which is consistent with the effect of Cl substituent in the 6-membered ring reported in the literature.38 Interestingly, the decrease in AlogP resulted in significant improvement in both HLM and MLM stability. Unexpectedly, the ethyl substituted compound 11 exhibited more significant potency loss and we decided not to pursue more extensive SAR at this position.
Figure 3.
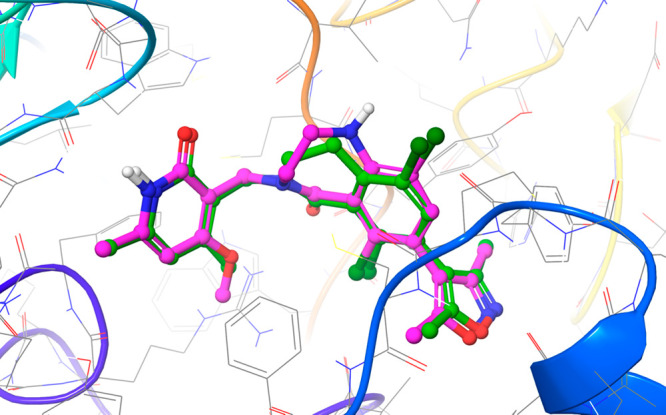
Predicted binding mode between EZH2 and compound 7, overlaid with the crystallographic binding mode of compound 2 from PDBID 6B3W.39 The protein is displayed as cartoon ribbons with atoms in gray wire, and the crystallographic binding mode of compound 2 is shown as green balls and sticks. The predicted binding mode of compound 7 is shown as magenta balls and sticks.
Table 3. SAR on a 7-Membered Ring Scaffold.

| compda | R1 | R2 | EZH2 IC50 (nM)b | Cell IC50 (H3K27me3) (nM)b | Karpas-422 proliferation (nM)b | HLM/MLM (μL/min/mg) | AlogP | MDCK/MDR1 permeability(nm/s) AB, BA/AB |
|---|---|---|---|---|---|---|---|---|
| 2 | 1.0 | 16 | 122 | 210/253 | 3.9 | 37, 4.8 | ||
| 7 | H | Cl | 0.35 | 17 | 65 | 79/123 | 3.0 | <1, >51 |
| 8 | Me | Cl | 0.87 | 208 | 245 | 80/112 | 3.3 | <1, >49 |
| 9 | Et | Cl | 33.4 | 3.7 | ||||
| 10 | Me | H | 2.9 | 1350 | 9/–7 | 2.6 | <1, >19 | |
| 11 | Et | H | 489 | 3.0 |
Preparation of compounds 7–11 is summarized in Schemes S7–S11 in the SI.
Mean of two determinations.
The potency and metabolic stability profile of compound 8 led us to explore this substitution pattern in conjunction with SAR at the R3 position (Table 4). More importantly, we chose compound 8 over the slightly more potent compound 7 as the starting point because it has one less hydrogen bond donor which would have reduced efflux potential. The substitution explored at the R3 position included moieties previously demonstrated by us and others in the 6-membered ring lactam scaffold to display potent biochemical inhibition of EZH2 and/or good metabolic stability.40 We were curious to find out if the SAR previously established was transferable to the new 7-membered ring lactam scaffold. It was shown that introduction of the sp3-carbon atom at this position was tolerated. Substitution of the R3 position with an isopropyl group (12) in compound 8 resulted in significant loss of potency, suggesting that dimethylisoxazole made similar hydrophobic interactions with the protein as the 6-membered ring lactam scaffold as predicted in the docking model. We then extended the isopropyl group with a branched tetrahydropyran (13, 15) to fill the two hydrophobic binding pockets occupied by the dimethylisoxazole, either through a carbon or nitrogen linkage. Both compounds 13 and 15 indeed displayed similar potency to compound 8 but with much lower HLM/MLM stability. Reduction of AlogP through a hydroxyl group (14) increased HLM/MLM stability with modest loss of potency. Replacement of dimethylisoxazole with aromatic moieties (16–18) that we identified in the 6-membered ring lactam series maintained the microsomal stability but displayed significant loss of potency in the new series.
Table 4. SAR for Dimethylisoxazole Replacement.
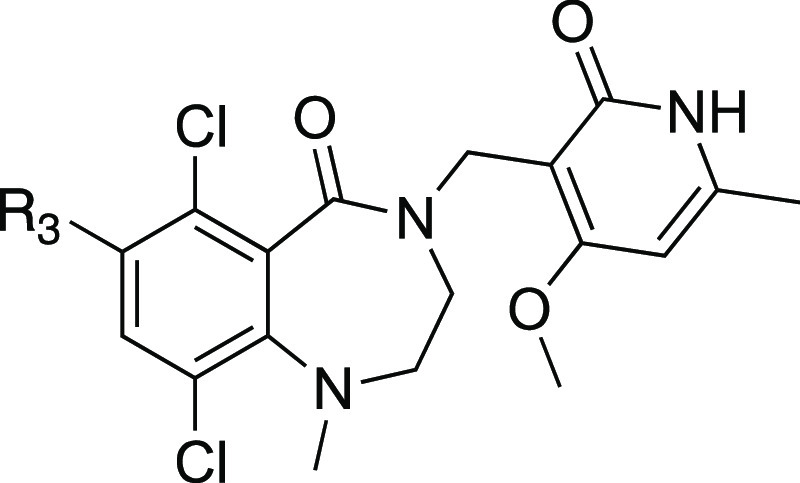

Preparation of compounds 12–18 is summarized in Schemes S12–S18 in the SI.
Mean of 2 determinations.
To access potent EZH2 inhibitors with favorable brain-penetrating properties, we reasoned that the potency profile of the 6,7-scaffold in combination with substitution to either increase lipophilicity or mask hydrogen bond donor functionality would provide a balanced profile to test this drug concept. To this end, we explored replacement of the 4-OMe group with the more lipophilic 4-ethyl substituted pyridone. Unfortunately the resulting compound 19 displayed has no obvious advantage in permeability relative to compound 8 and also appeared to have a similar potency. This was surprising since this substitution has been associated with enhanced EZH2 potency through increasing residence time.45 However, the SAR analysis for high affinity compounds is complicated by the active enzyme concentration of 2 nM resulting in a floor of the assay of 1 nM. For example, incorporation of the N-methyl pyridone into compound 20 displayed a greater than 500-fold loss of potency relative to compound 8 but the exact magnitude of the loss is unclear.
During the course of our program, Khanna et. al disclosed that incorporating a 4-thiomethyl pyridone in CPI-1205 (4) as a key modification significantly increased biochemical and cellular potency due to prolonged residence time, leading to the development of the second generation EZH2 inhibitor CPI-0209 with extended target coverage.45 We incorporated this finding into our new 7-membered ring structure of EZH2 inhibitor (Table 5). The resulting compound, 21, exhibited similar biochemical potency relative to compound 8 but superior cellular efficacy in the H3K27me3 assay (>30-fold improvement), consistent with the literature finding. Importantly, this improvement translated to markedly enhanced potency in antiproliferation assays in Karpas-422 cells, comparable to compound 2 and only 3-fold less active than the clinical compound PF-06821497 (compound 3) in the same assay. Despite the desirable potency, this compound proved to be a significant MDR substrate and therefore was not suitable for advancement as a brain-penetrant analogue. Nevertheless, the efficacy improvement from the thiomethyl pyridone was very encouraging. Accordingly, we prepared the analogous N-methyl pyridone compound 22. Although we did observe a significant improvement in biochemical potency for compound 22 versus 20, this compound still suffered from poor cellular efficacy. Removal of the R1 methyl group (23) provided a further improvement in both biochemical and cellular efficacy. Unfortunately, these compounds still displayed a high value of the MDCK/MDR1 ratio, suggesting that they are good substrates for efflux transporters and would not likely achieve significant brain penetration. The higher MDR1 ratio may be reflective of increased PSA of this scaffold due to incorporation of the additional atoms (e.g., compound 5 = 68 A2 vs compound 22 = 97 A2 and compound 23 = 106 A2).46
Table 5. Thiomethyl Pyridone Analogues with 7-Membered Ring Structure.
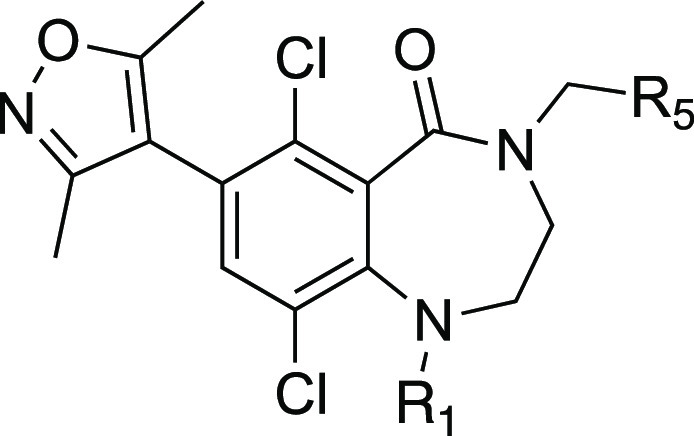

Preparation of compounds 19–23 is summarized in Schemes S19–S22 in the SI.
Mean of two determinations.
The synthesis of compounds 5 and 6a–6e (Scheme 1) started with the lactam bromide 24 intermediate and benzyloxy-protected chloromethylpyridine intermediate 26d, which were prepared from commercially available 2-hydroxy-4-methoxy-6-methylpyridine-3-carbonitrile according to literature procedure.36 The synthesis of benzyloxy-protected chloromethylpyridine intermediates 26a–c and 26e are described in further details in the SI.
Scheme 1. Synthesis of compounds 5 and 6a–6e.
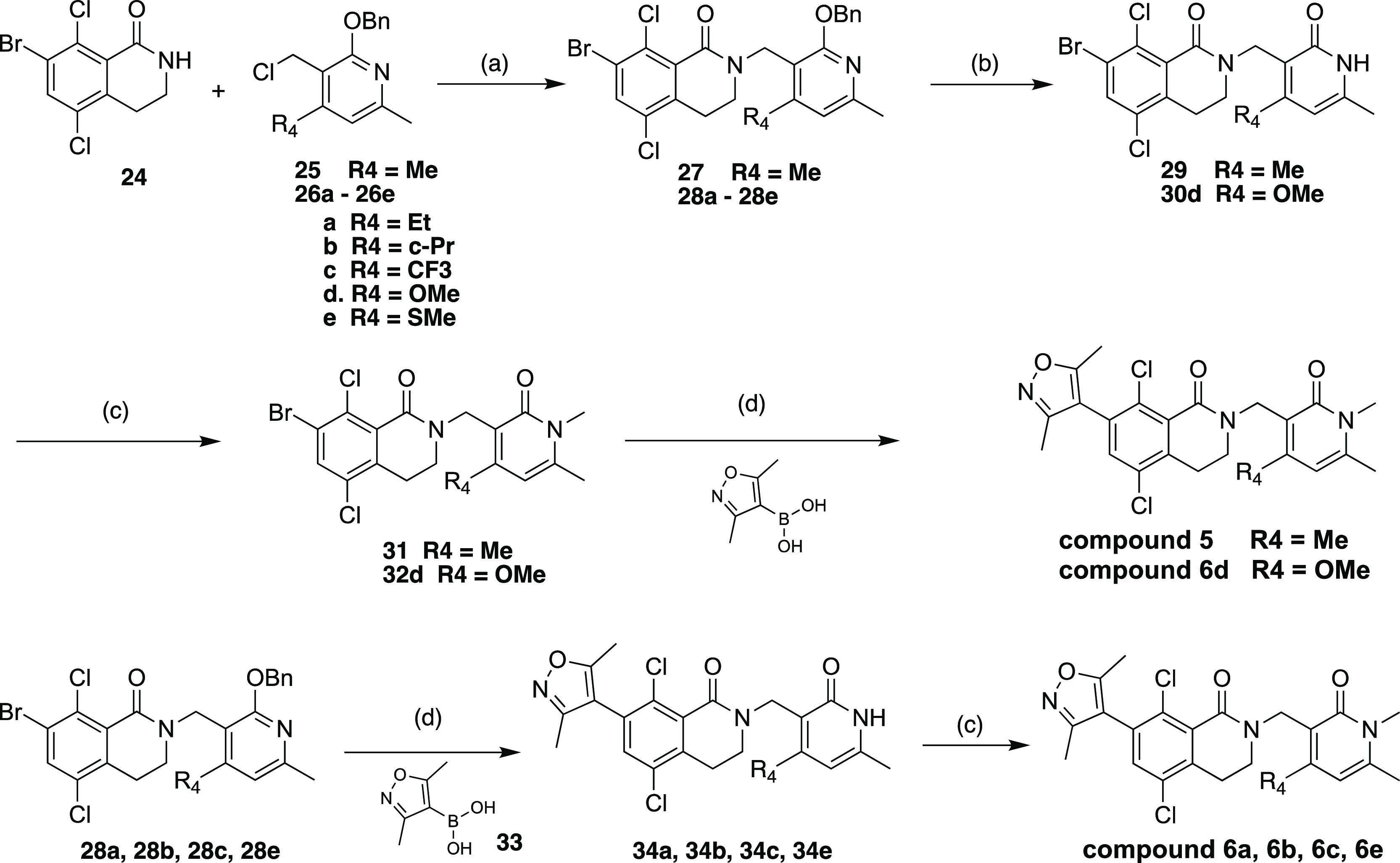
Reagents and conditions: (a) t-BuOK, DMF, 25°C, 12 h, 32–72%; (b) TFA, DCM, RT, 16 h, 72-97% (c) NaH, DMF for 30, 40°C, 30 min, 30–77%; (d) Pd(dppf)Cl2, K3PO4, dioxane, H2O, 90°C, 2 h, 32–67%.
Synthesis of R1 methyl-substituted 7-membered lactam was accomplished starting from 2-amino-3,6-dichlorobenzoic acid 35 (Scheme 2). Bromination of compound 35 followed by reductive amination of compound 36 with N-Boc-2-aminoaldehyde 37 afforded bromide carboxylic acid 38. Treatment of 38 with HCl deprotected the Boc group from the amine moiety, and subsequent cyclization under amide coupling condition generated the lactam intermediate 40, which upon methylation afforded intermediate 41.
Scheme 2. Synthesis of 7-Membered Ring Core Bromides 40 and 41.
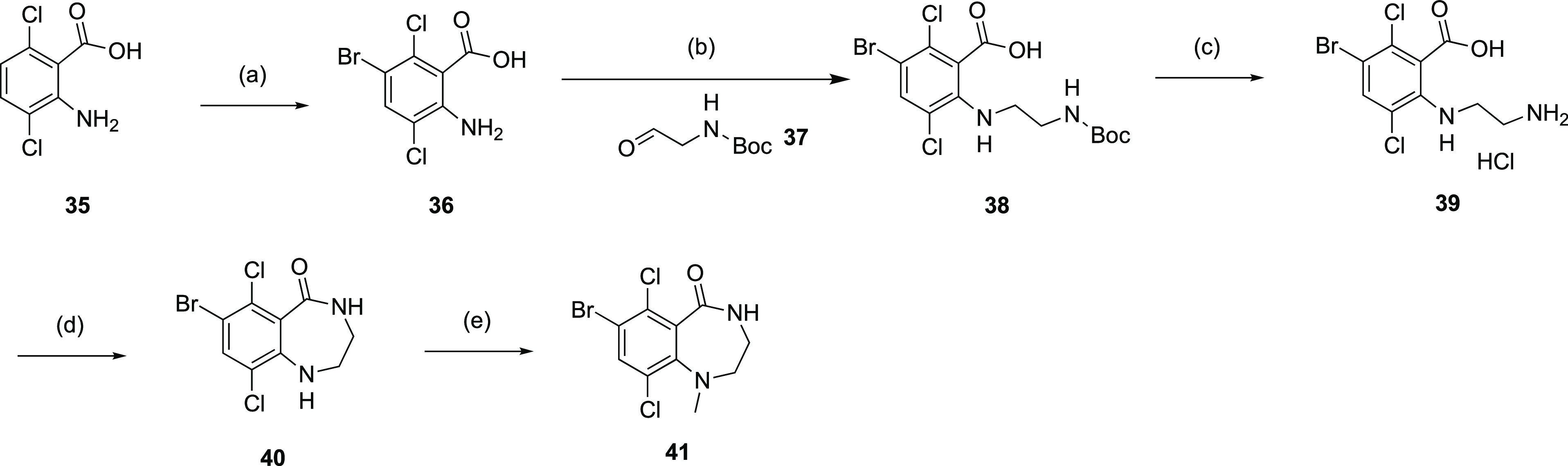
Reagents and conditions: (a) NBS, DMF, RT, 80%; (b) NaBH4, AcOH, DCE, 60°C, 12 h, 44%; (c) 4M HCl in dioxane, RT, 97%; (d) HOBt, BOP, NMM, DMF, RT, 16 h, 52%; (e) TEA, MeI, DMF, 70°C, 16 h, 81%.
Alkylation of the lactam nitrogen in intermediates 42 and 43 with the chloride intermediate 26d under basic conditions afforded the common intermediates 44 and 45 (Scheme 2). Suzuki coupling with dimethylisoxazole boronic acid 33 followed by the removal of benzyl protecting group with TFA afforded the final compounds 7 and 8 (Scheme 3).
Scheme 3. Synthesis of Compounds 7 and 8.
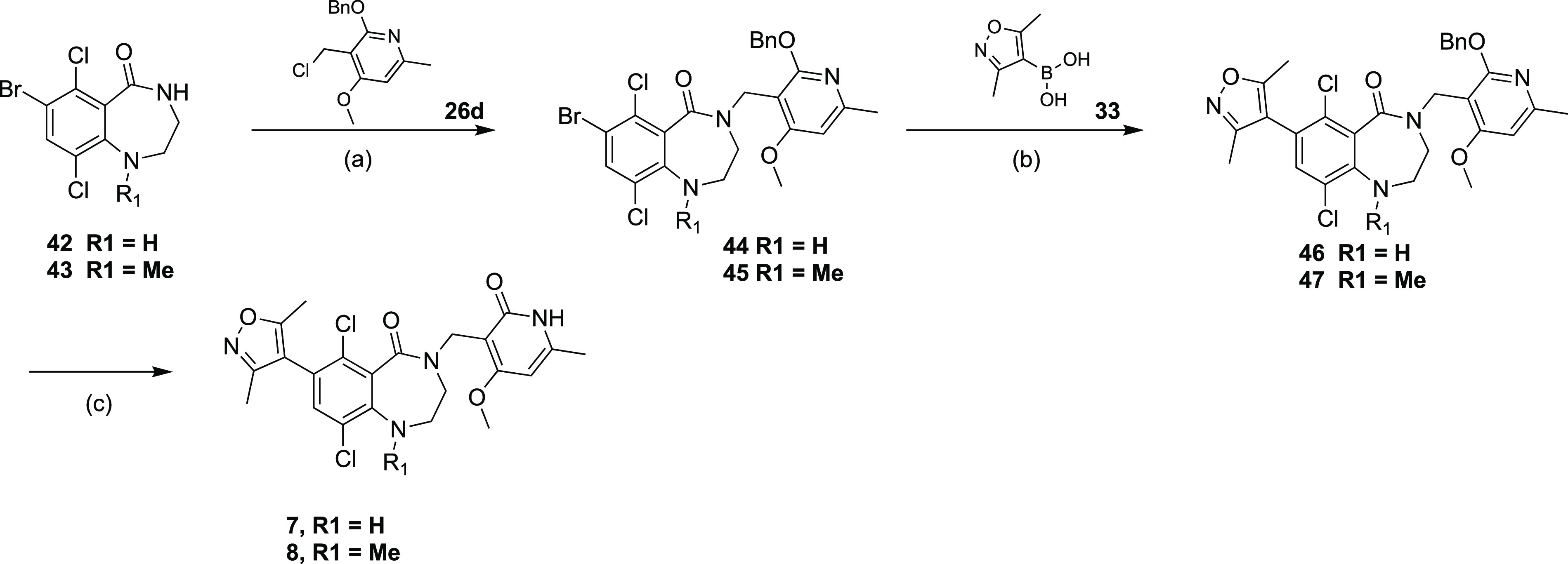
Reagents and conditions: (a) t-BuOK, DMF for 42, NaH, DMF for 43, 40°C, 30 min, 41-64%; (b) Pd(dppf)Cl2, K3PO4, dioxane, H2O, 90°C, 2 h, 29–48%; (c) TFA, DCM, RT, 16 h, 51–57%.
Compound 12 was prepared through two sequential alkylations (Scheme 4), one with 2-iodopropane on hydroxyl group of phenol intermediate 49, the other with chloride intermediate 26d on NH group of amide intermediate 50. Compounds 13 was accessed through two Grinard reactions (Scheme 5) starting from bromide intermediate 45. The first Grignard reaction with 4-formyl tetrahydropyran gave secondary alcohol intermediate 52, which was subjected to either acidic conditions to provide compound 14 or oxidation conditions to ketone intermediate 53. The second Grignard reaction generated tertiary alcohol intermediate 54 which was converted to compound 13 by elimination under acidic conditions.
Scheme 4. Synthesis of Compound 12.

Reagents and conditions: (a) Bis(pinacolato)diborane, Pd(dppf)Cl2, KOAc, dioxane, 90°C, 3 h, 42%; (b) H2O2, NaOH, THF, water, 25°C, 2 h, 97%; (c) 2-iodopropane, DMF, K2CO3, 40°C, 2 h, 95%; (d) 26d, NaH, DMF, 40°C, 1 h, 56%; (e) TFA, DCM, 45°C, 1.5 h, 35%.
Scheme 5. Synthesis of Compounds 13 and 14.
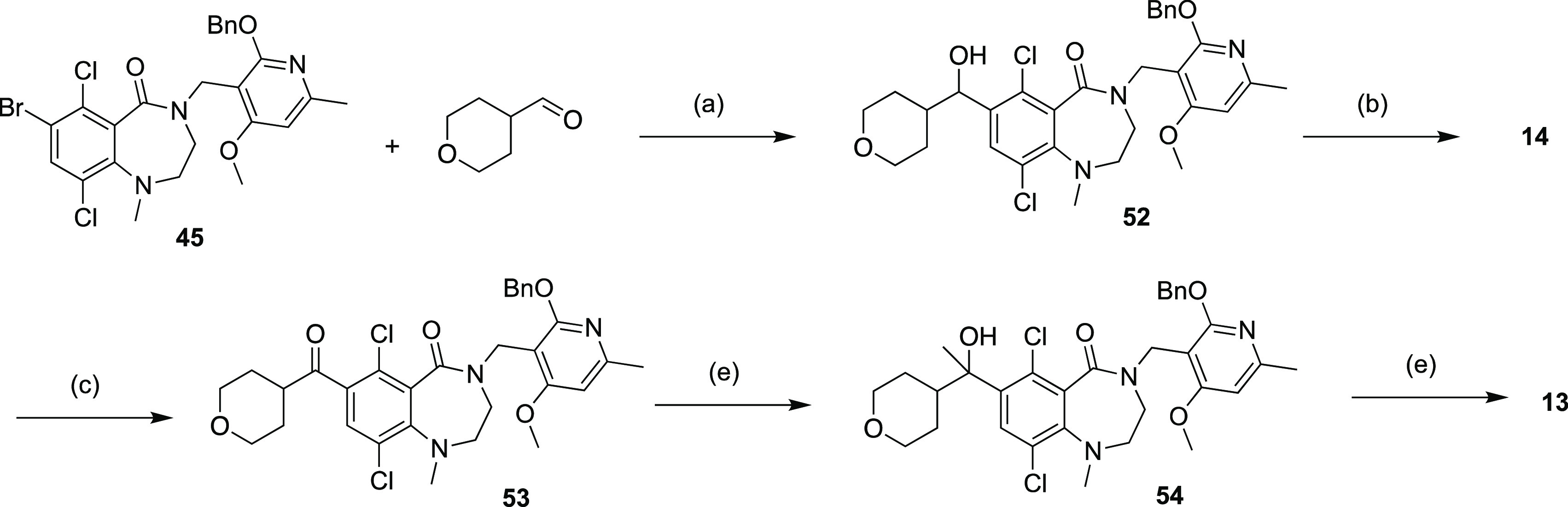
Reagents and conditions: (a) i-PrMgCl, THF, −65 to 10°C, 1.5 h, 2.3%; (b) TFA, DCM, 45°C, 1.5 h, 41%; (c) Dess–Martin, DCM, 15°C, 12 h, 34%; (d) MeMgBr, THF, 0°C, 2 h, 78%; (e) Et3SiH, TFA, 0–45°C, 4 h, 32%.
Compound 15 was prepared through reductive amination using amine intermediate 55 (Scheme 6), which was generated by amination of the bromide 45. Compounds 16–18 with aromatic R3 substitutions (Scheme 7) were synthesized via Suzuki reaction using the corresponding commercial boronic acids 58–60. Methylation of NH group in pyridone followed the general procedure (Scheme 8).
Scheme 6. Synthesis of Compounds 15.

Reagents and conditions: (a) Pd(dba)2, xantphos, Cs2CO3, dioxane, 110°C, 2 h, 89%; (b) HCl/EtOAC, 15°C, 2 h; (c) tetrahydropyran-4-one, tetramethylammonium, triacetoxyboranuide, TFA, DCM, 15°C, 2 h; (d) acetaldehyde, tetramethylammonium, triacetoxyboranuide, TFA, DCM, 15°C, 2 h; (e) TFA, DCM, 45°C, 2 h, 14% (3 steps).
Scheme 7. Synthesis of Compounds 16–18.

Reagents and conditions: (a) Pd(dppf)Cl2, K3PO4 or CsF, dioxane, H2O, 90°C, 2 h, 46-61%; (b) TFA, DCM, RT, 16 h, 22-62%.
Scheme 8. General Procedure for N-Methylation of Pyridone.

Reagents and conditions: NaH, MeI, DMF, 0–20°C for 12 h.
To summarize, we show here a new strategy to create brain-penetrant EZH2 inhibitors through N-methylation of the conserved pyridone found in many EZH2 inhibitors. N-Methylation of compound 2 identified compound 5 (TDI-6118), which displayed brain-penetrating properties both in vitro and in vivo (as determined by Kp,uu), and this marks the first report of a brain-penetrant EZH2 inhibitor. The reduction in cell-based activity accompanying this modification required the exploration of novel scaffolds in an attempt to identify a suitable candidate for further in vivo characterization.
To this end, a scaffold hopping approach was undertaken utilizing a structure-based approach to design a novel and highly potent class of 6,7-fused EZH2 inhibitors. This series provided compounds with subnanomolar enzymatic activity and potent antiproliferative effects. Enhanced inhibition of cellular trimethylation could be achieved by compound 21 (TDI-11904), derived from the incorporation of thiomethyl substitution on the key pyridone moiety (8 vs 21). Although not determined, this may be consistent with reports of improved on target residence time with this substitution on a different scaffold.45,46 Our results demonstrate that this is a general effect that can be applied to different chemical series of EZH2 inhibitors. The improved efflux ratio observed with compound 5 could not be maintained with either further modification on the pyridone or with the change to the 7-membered ring lactam structure. However, the enzymatic potency seen by combining the N-methylation with pyridone substitution for increased residence time (e.g., 23) suggests that additional structural modifications or alternate cores may provide compounds that balance high potency (and antiproliferative effects) with brain penetrance.
Acknowledgments
The authors gratefully acknowledge the support to the project generously provided by the Tri-Institutional Therapeutics Discovery Institute (TDI), a 501(c)(3) organization. TDI receives financial support from Takeda Pharmaceutical Company, TDI’s parent institutes (Memorial Sloan Kettering Cancer Center, The Rockefeller University, and Weill Cornell Medicine) and from a generous contribution from Mr. Lewis Sanders and other philanthropic sources. R.E.P. is supported by the MSK IDEA Award and the Center for Experimental Therapeutics, The Cure Starts Now, and Team Jack Foundation.
Glossary
Abbreviations
- EZH2
enhancer of zeste homologue 2
- BBB
blood brain barrier
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00448.
Synthetic experimental details, characterization data, and biological assay protocols (PDF)
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455 (7216), 1061–1068. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A. M.; Kadoch C. Chromatin Regulatory Mechanisms and Therapeutic Opportunities in Cancer. Nat. Cell Biol. 2019, 21, 152. 10.1038/s41556-018-0258-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B.; Chourrout D.; Vervoort M.; Leblanc B.; Cavalli G. Genome Regulation by Polycomb and Trithorax Proteins. Cell 2007, 128 (4), 735–745. 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Cao R.; Wang L.; Wang H.; Xia L.; Erdjument-Bromage H.; Tempst P.; Jones R. S.; Zhang Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298 (5595), 1039–1043. 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B.; Bourbon H.-M.; Di Croce L.; Cavalli G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171 (1), 34–57. 10.1016/j.cell.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Kim K. H.; Roberts C. W. M. Targeting EZH2 in Cancer. Nat. Med. 2016, 22 (2), 128–134. 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Scott M. P.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M.; Kuntz K. W.; Keilhack H. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (19), 7922–7927. 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani R. G.; Gehling V. S.; Dakin L. A.; Cook A. S.; Nasveschuk C. G.; Duplessis M.; Iyer P.; Balasubramanian S.; Zhao F.; Good A. C.; Campbell R.; Lee C.; Cantone N.; Cummings R. T.; Normant E.; Bellon S. F.; Albrecht B. K.; Harmange J.-C.; Trojer P.; Audia J. E.; Zhang Y.; Justin N.; Chen S.; Wilson J. R.; Gamblin S. J. Identification of (R)-N-((4-Methoxy-6-Methyl-2-Oxo-1,2-Dihydropyridin-3-Yl)Methyl)-2-Methyl-1-(1-(1-(2,2,2-Trifluoroethyl)Piperidin-4-Yl)Ethyl)-1H-Indole-3-Carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016, 59 (21), 9928–9941. 10.1021/acs.jmedchem.6b01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma D.; Kanno O.; Watanabe J.; Kinoshita J.; Hirasawa M.; Nosaka E.; Shiroishi M.; Takizawa T.; Yasumatsu I.; Horiuchi T.; Nakao A.; Suzuki K.; Yamasaki T.; Nakajima K.; Hayakawa M.; Yamazaki T.; Yadav A. S.; Adachi N. Novel Orally Bioavailable EZH1/2 Dual Inhibitors with Greater Antitumor Efficacy than an EZH2 Selective Inhibitor. Cancer Sci. 2017, 108 (10), 2069–2078. 10.1111/cas.13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer C. J.; Scott M. P.; Kuntz K. W.; Knutson S. K.; Pollock R. M.; Richon V. M.; Copeland R. A. Coordinated Activities of Wild-Type plus Mutant EZH2 Drive Tumor-Associated Hypertrimethylation of Lysine 27 on Histone H3 (H3K27) in Human B-Cell Lymphomas. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (49), 20980–20985. 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle T. J.; Knutson S. K.; Jin L.; Kuntz K. W.; Pollock R. M.; Richon V. M.; Copeland R. A.; Scott M. P. The Y641C Mutation of EZH2 Alters Substrate Specificity for Histone H3 Lysine 27 Methylation States. FEBS Lett. 2011, 585 (19), 3011–3014. 10.1016/j.febslet.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Bitler B. G.; Aird K. M.; Garipov A.; Li H.; Amatangelo M.; Kossenkov A. V.; Schultz D. C.; Liu Q.; Shih I.-M.; Conejo-Garcia J. R.; Speicher D. W.; Zhang R. Synthetic Lethality by Targeting EZH2Methyltransferase Activity in ARID1A-Mutated Cancers. Nat. Med. 2015, 21, 231. 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming K. C.; Wang X.; Roberts C. W. M. Vulnerabilities of Mutant SWI/SNF Complexes in Cancer. Cancer Cell 2014, 26 (3), 309–317. 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. G.; Wang X.; Shen X.; McKenna E. S.; Lemieux M. E.; Cho Y.-J.; Koellhoffer E. C.; Pomeroy S. L.; Orkin S. H.; Roberts C. W. M. Epigenetic Antagonism between Polycomb and SWI/SNF Complexes during Oncogenic Transformation. Cancer Cell 2010, 18 (4), 316–328. 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennison J. A.; Tamkun J. W. Dosage-Dependent Modifiers of Polycomb and Antennapedia Mutations in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 1988, 85 (21), 8136–8140. 10.1073/pnas.85.21.8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamkun J. W.; Deuring R.; Scott M. P.; Kissinger M.; Pattatucci A. M.; Kaufman T. C.; Kennison J. A. Brahma: A Regulator of Drosophila Homeotic Genes Structurally Related to the Yeast Transcriptional Activator SNF2/SWI2. Cell 1992, 68 (3), 561–572. 10.1016/0092-8674(92)90191-E. [DOI] [PubMed] [Google Scholar]
- Groisberg R.; Subbiah V. EZH2 Inhibition for Epithelioid Sarcoma and Follicular Lymphoma. Lancet Oncology 2020, 21 (11), 1388–1390. 10.1016/S1470-2045(20)30530-1. [DOI] [PubMed] [Google Scholar]
- Stacchiotti S.; Schoffski P.; Jones R.; Agulnik M.; Villalobos V. M.; Jahan T. M.; Chen T. W.-W.; Italiano A.; Demetri G. D.; Cote G. M.; Chugh R.; Attia S.; Gupta A. A.; Loggers E. T.; Van Tine B.; Sierra L.; Yang J.; Rajarethinam A.; Gounder M. M. Safety and Efficacy of Tazemetostat, a First-in-Class EZH2 Inhibitor, in Patients (Pts) with Epithelioid Sarcoma (ES) (NCT02601950). JCO 2019, 37 (15_suppl), 11003–11003. 10.1200/JCO.2019.37.15_suppl.11003. [DOI] [Google Scholar]
- Lin X.; DeAngelis L. M. Treatment of Brain Metastases. J. Clin Oncol 2015, 33 (30), 3475–3484. 10.1200/JCO.2015.60.9503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall W. A.; Djalilian H. R.; Nussbaum E. S.; Cho K. H. Long-Term Survival with Metastatic Cancer to the Brain. Med. Oncol 2000, 17 (4), 279–286. 10.1007/BF02782192. [DOI] [PubMed] [Google Scholar]
- Tabouret E.; Chinot O.; Metellus P.; Tallet A.; Viens P.; Gonçalves A. Recent Trends in Epidemiology of Brain Metastases: An Overview. Anticancer Res. 2012, 32 (11), 4655–4662. [PubMed] [Google Scholar]
- Gardner E. E.; Lok B. H.; Schneeberger V. E.; Desmeules P.; Miles L. A.; Arnold P. K.; Ni A.; Khodos I.; de Stanchina E.; Nguyen T.; Sage J.; Campbell J. E.; Ribich S.; Rekhtman N.; Dowlati A.; Massion P. P.; Rudin C. M.; Poirier J. T. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31 (2), 286–299. 10.1016/j.ccell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffen J. C.; Gunatilake D.; Gallagher S. J.; Gowrishankar K.; Heinemann A.; Cullinane C.; Dutton-Regester K.; Pupo G. M.; Strbenac D.; Yang J. Y.; Madore J.; Mann G. J.; Hayward N. K.; McArthur G. A.; Filipp F. V.; Hersey P. Targeting Activating Mutations of EZH2 Leads to Potent Cell Growth Inhibition in Human Melanoma by Derepression of Tumor Suppressor Genes. Oncotarget 2015, 6 (29), 27023–27036. 10.18632/oncotarget.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingg D.; Debbache J.; Schaefer S. M.; Tuncer E.; Frommel S. C.; Cheng P.; Arenas-Ramirez N.; Haeusel J.; Zhang Y.; Bonalli M.; McCabe M. T.; Creasy C. L.; Levesque M. P.; Boyman O.; Santoro R.; Shakhova O.; Dummer R.; Sommer L. The Epigenetic Modifier EZH2 Controls Melanoma Growth and Metastasis through Silencing of Distinct Tumour Suppressors. Nat. Commun. 2015, 6, ncomms7051 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- QuickStats: Brain Cancer Death Rates Among Children and Teens Aged 1–19 Years, by Sex and Age Group — United States, 2013–2015. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 461. 10.15585/mmwr.mm6617a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F.; Weissmann S.; Leblanc B.; Pandey D. P.; Højfeldt J. W.; Comet I.; Zheng C.; Johansen J. V.; Rapin N.; Porse B. T.; Tvardovskiy A.; Jensen O. N.; Olaciregui N. G.; Lavarino C.; Suñol M.; de Torres C.; Mora J.; Carcaboso A. M.; Helin K. EZH2 Is a Potential Therapeutic Target for H3K27M-Mutant Pediatric Gliomas. Nat. Med. 2017, 23, 483. 10.1038/nm.4293. [DOI] [PubMed] [Google Scholar]
- Piunti A.; Hashizume R.; Morgan M. A.; Bartom E. T.; Horbinski C. M.; Marshall S. A.; Rendleman E. J.; Ma Q.; Takahashi Y.-H.; Woodfin A. R.; Misharin A. V.; Abshiru N. A.; Lulla R. R.; Saratsis A. M.; Kelleher N. L.; James C. D.; Shilatifard A. Therapeutic Targeting of Polycomb and BET Bromodomain Proteins in Diffuse Intrinsic Pontine Gliomas. Nat. Med. 2017, 23, 493. 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michealraj K. A.; Kumar S. A.; Kim L. J. Y.; Cavalli F. M. G.; Przelicki D.; Wojcik J. B.; Delaidelli A.; Bajic A.; Saulnier O.; MacLeod G.; Vellanki R. N.; Vladoiu M. C.; Guilhamon P.; Ong W.; Lee J. J. Y.; Jiang Y.; Holgado B. L.; Rasnitsyn A.; Malik A. A.; Tsai R.; Richman C. M.; Juraschka K.; Haapasalo J.; Wang E. Y.; De Antonellis P.; Suzuki H.; Farooq H.; Balin P.; Kharas K.; Van Ommeren R.; Sirbu O.; Rastan A.; Krumholtz S. L.; Ly M.; Ahmadi M.; Deblois G.; Srikanthan D.; Luu B.; Loukides J.; Wu X.; Garzia L.; Ramaswamy V.; Kanshin E.; Sánchez-Osuna M.; El-Hamamy I.; Coutinho F. J.; Prinos P.; Singh S.; Donovan L. K.; Daniels C.; Schramek D.; Tyers M.; Weiss S.; Stein L. D.; Lupien M.; Wouters B. G.; Garcia B. A.; Arrowsmith C. H.; Sorensen P. H.; Angers S.; Jabado N.; Dirks P. B.; Mack S. C.; Agnihotri S.; Rich J. N.; Taylor M. D. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 2020, 181 (6), 1329–1345. 10.1016/j.cell.2020.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay H.; Kogiso M.; Qi L.; Murray J.; Perlaky L.; Su J.; Baxter P.; Adesina A.; Parsons D.; Chintagumpala M.; Li X.-N. AT-01THERAPEUTIC TARGETING OF INI1 DEFICIENCY IN PEDIATRIC ATRT: A PRE-CLINICAL STUDY UTILIZING PATIENT DERIVED ORTHOTOPIC XENOGRAFT (PDOX) MODELS. Neuro-Oncology 2015, 17 (Suppl 3), iii1 10.1093/neuonc/nov061.1. [DOI] [Google Scholar]
- Li X.-N.; Qi L.; Kogiso M.; Lindsay H. B.; Erickson S. W.; Guo Y.; Smith M. A.; Teicher B. A. Pediatric Preclinical Testing Consortium Evaluation of the EZH2 Inhibitor Tazemetostat in Orthotopic PDX Models of Pediatric Brain Tumors. JCO 2018, 36 (15_suppl), 10551–10551. 10.1200/JCO.2018.36.15_suppl.10551. [DOI] [Google Scholar]
- Lue J. K.; Amengual J. E. Emerging EZH2 Inhibitors and Their Application in Lymphoma. Curr. Hematol Malig Rep 2018, 13 (5), 369–382. 10.1007/s11899-018-0466-6. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58 (6), 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- Desai P. V.; Raub T. J.; Blanco M.-J. How Hydrogen Bonds Impact P-Glycoprotein Transport and Permeability. Bioorg. Med. Chem. Lett. 2012, 22 (21), 6540–6548. 10.1016/j.bmcl.2012.08.059. [DOI] [PubMed] [Google Scholar]
- Di L.; Artursson P.; Avdeef A.; Benet L. Z.; Houston J. B.; Kansy M.; Kerns E. H.; Lennernäs H.; Smith D. A.; Sugano K. The Critical Role of Passive Permeability in Designing Successful Drugs. ChemMedChem. 2020, 15 (20), 1862–1874. 10.1002/cmdc.202000419. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Carter G. T. Strategies to Assess Blood-Brain Barrier Penetration. Expert Opin Drug Discov 2008, 3 (6), 677–687. 10.1517/17460441.3.6.677. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A. Structural Modifications That Alter the P-Glycoprotein Efflux Properties of Compounds. J. Med. Chem. 2012, 55 (11), 4877–4895. 10.1021/jm201136z. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying Brain Penetration in Central Nervous System Drug Discovery. Miniperspective. J. Med. Chem. 2013, 56 (1), 2–12. 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- Kung P.-P.; Rui E.; Bergqvist S.; Bingham P.; Braganza J.; Collins M.; Cui M.; Diehl W.; Dinh D.; Fan C.; Fantin V. R.; Gukasyan H. J.; Hu W.; Huang B.; Kephart S.; Krivacic C.; Kumpf R. A.; Li G.; Maegley K. A.; McAlpine I.; Nguyen L.; Ninkovic S.; Ornelas M.; Ryskin M.; Scales S.; Sutton S.; Tatlock J.; Verhelle D.; Wang F.; Wells P.; Wythes M.; Yamazaki S.; Yip B.; Yu X.; Zehnder L.; Zhang W.-G.; Rollins R. A.; Edwards M. Design and Synthesis of Pyridone-Containing 3,4-Dihydroisoquinoline-1(2H)-Ones as a Novel Class of Enhancer of Zeste Homolog 2 (EZH2) Inhibitors. J. Med. Chem. 2016, 59 (18), 8306–8325. 10.1021/acs.jmedchem.6b00515. [DOI] [PubMed] [Google Scholar]
- Kung P.-P.; Bingham P.; Brooun A.; Collins M.; Deng Y.-L.; Dinh D.; Fan C.; Gajiwala K. S.; Grantner R.; Gukasyan H. J.; Hu W.; Huang B.; Kania R.; Kephart S. E.; Krivacic C.; Kumpf R. A.; Khamphavong P.; Kraus M.; Liu W.; Maegley K. A.; Nguyen L.; Ren S.; Richter D.; Rollins R. A.; Sach N.; Sharma S.; Sherrill J.; Spangler J.; Stewart A. E.; Sutton S.; Uryu S.; Verhelle D.; Wang H.; Wang S.; Wythes M.; Xin S.; Yamazaki S.; Zhu H.; Zhu J.; Zehnder L.; Edwards M. Optimization of Orally Bioavailable Enhancer of Zeste Homolog 2 (EZH2) Inhibitors Using Ligand and Property-Based Design Strategies: Identification of Development Candidate (R)-5,8-Dichloro-7-(Methoxy(Oxetan-3-Yl)Methyl)-2-((4-Methoxy-6-Methyl-2-Oxo-1,2-Dihydropyridin-3-Yl)Methyl)-3,4-Dihydroisoquinolin-1(2H)-One (PF-06821497). J. Med. Chem. 2018, 61 (3), 650–665. 10.1021/acs.jmedchem.7b01375. [DOI] [PubMed] [Google Scholar]
- Zhang P.; de Gooijer M. C.; Buil L. C. M.; Beijnen J. H.; Li G.; van Tellingen O. ABCB1 and ABCG2 Restrict the Brain Penetration of a Panel of Novel EZH2-Inhibitors. Int. J. Cancer 2015, 137, 2007–2018. 10.1002/ijc.29566. [DOI] [PubMed] [Google Scholar]
- Brooun A.; Gajiwala K. S.; Deng Y.-L.; Liu W.; Bolaños B.; Bingham P.; He Y.-A.; Diehl W.; Grable N.; Kung P.-P.; Sutton S.; Maegley K. A.; Yu X.; Stewart A. E. Polycomb Repressive Complex 2 Structure with Inhibitor Reveals a Mechanism of Activation and Drug Resistance. Nat. Commun. 2016, 7, 11384. 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for Molecular Recognition Exploiting Hydrophobic Enclosure in Protein-Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (3), 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz K. W.; Campbell J. E.; Keilhack H.; Pollock R. M.; Knutson S. K.; Porter-Scott M.; Richon V. M.; Sneeringer C. J.; Wigle T. J.; Allain C. J.; Majer C. R.; Moyer M. P.; Copeland R. A.; Chesworth R. The Importance of Being Me: Magic Methyls, Methyltransferase Inhibitors, and the Discovery of Tazemetostat. J. Med. Chem. 2016, 59 (4), 1556–1564. 10.1021/acs.jmedchem.5b01501. [DOI] [PubMed] [Google Scholar]
- Yang X.; Li F.; Konze K. D.; Meslamani J.; Ma A.; Brown P. J.; Zhou M.-M.; Arrowsmith C. H.; Kaniskan H. Ü.; Vedadi M.; Jin J. Structure-Activity Relationship Studies for Enhancer of Zeste Homologue 2 (EZH2) and Enhancer of Zeste Homologue 1 (EZH1) Inhibitors. J. Med. Chem. 2016, 59 (16), 7617–7633. 10.1021/acs.jmedchem.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuckey J. I.; Cantone N. R.; Côté A.; Arora S.; Vivat V.; Ramakrishnan A.; Mertz J. A.; Khanna A.; Brenneman J.; Gehling V. S.; Moine L.; Sims R. J.; Audia J. E.; Trojer P.; Levell J. R.; Cummings R. T. Identification and Characterization of Second-Generation EZH2 Inhibitors with Extended Residence Times and Improved Biological Activity. J. Biol. Chem. 2021, 296, 100349. 10.1016/j.jbc.2021.100349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong B.; Wang Y.; Chen Y.; Xing S.; Liao Q.; Chen Y.; Li Q.; Li W.; Sun H. Strategies for Structural Modification of Small Molecules to Improve Blood-Brain Barrier Penetration: A Recent Perspective. J. Med. Chem. 2021, 64 (18), 13152–13173. 10.1021/acs.jmedchem.1c00910. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.