

Abstract
Background
Thyroid-associated ophthalmopathy (TAO), one of the most common orbital diseases in adults, seriously reduces patients’ quality of life. Although human tear proteomics identified many abnormal expressed proteins and proposed several pathogeneses of TAO, most of these studies focused on the active stage or mixed types in TAO. In this study we identified significantly changed proteins and preliminary revealed the potential signalling pathways and mechanisms of TAO with the late, inactive stage.
Patients and Methods.
Tears from TAO patients (n=6) with a CAS score < 3 and 6 control healthy subject were collected. The pooled tears were further fractionated using high pH reversed-phase chromatography, then submitted to LC-MS/MS and subsequent bioinformatic analysis.
Results.
Proteomic profiling identified 107 significantly changed proteins between the inactive stage of TAO patients and healthy cases. Among these proteins, 62 were upregulated, and 45 were downregulated in TAO cases compared to healthy individuals. Enrichment analysis revealed that the immune system, cell cycle, metabolism (carbohydrate metabolism and metabolism of cofactors and vitamins), protein synthesis and degradation might play a vital role in the progress of inactive TAO. The present investigation represents the first proteomic tear study of TAO patients in the inactive stage.
Conclusion.
The results shed light on the differences between inactive TAO patients and healthy cases, thus enabling us to understand better the molecular mechanisms and potential targets for the treatment of inactive TAO.
Keywords: Tear proteomics, Thyroid-associated ophthalmopathy, Inactive stage, Bioinformatics
Introduction
As one of the autoimmune diseases, thyroid-associated ophthalmopathy (TAO), also termed Graves’ orbitopathy or thyroid eye disease, is one of the most common orbital diseases in adults. About 60% of patients will develop TAO within 18 months of diagnosing Graves’ disease (1). Common symptoms of TAO include exophthalmos, eyelid retraction, strabismus, and even comppressive optic neuropathy, which results in a severe decline in the quality of life of these patients. Previous clinical trials indicated that high-dose glucocorticoids alone or with radiotherapy could attenuate inflammation-related symptoms in active TAO patients (2, 3). However, glucocorticoids and orbital radiotherapy are inadequate for the treatment of exophthalmos (2). Interestingly, teprotumumab, a monoclonal antibody against insulin-like growth factor I (IGF-I) receptor (IGF-IR), significantly reduced proptosis in active patients with TAO may result from attenuation of signalling pathway through both IGF-IR and the thyrotropin receptor (4). During the active phase, inflammation of orbital tissues was active, while in the inactive phase, the inflammation of orbital tissues was relatively stable. These previous studies suggested that inflammation-based antibody therapy may be less effective for inactive TAO patients. Therefore, revealing and elucidating the mechanism of TAO in the inactive phase is urgent.
As a non-invasive method, tear test plays an increasingly significant role in the diagnosis and management of ocular surface diseases (5). Previous studies revealed that abnormal proteins could be found in tears of TAO patients (6). Proteins with a molecular weight between 3 and 20k Da changed significantly (7). In patients with active TAO, a number of proteins changed significantly compared with healthy individuals (8, 9). For example, alpha-1-antichymotrypsin, cystatin-C, NADPH dehydrogenase [quinone] 1, 8-hydroxy-2 deoxyguanosine, MDA, IL-1β, IL-6, etc. were upregulated, while phospholipase A2, signal transducer and activator of transcription 1-alpha/beta, protein ABHD14B and S100A4, etc. were downregulated in TAO patients (8-12). In addition, the levels of some significantly changed proteins were different in the active and inactive stages. For example, a low level of IL-7 was found in active TAO patients’ tears, while the level of IL-7 was higher in inactive TAO patients’ tears than in healthy cases’ tears (13). These suggested that among the differentially expressed proteins (DEPs), the expression of protein profiles is diverse in the different stages of TAO patients. Furthermore, previous investigations on tear proteomics of TAO mainly focus on active or mixed patients. Tear proteome analysis in TAO cases with an inactive stage is still confused. Therefore, the present study identified the abnormally changed tear proteomes and explored the potential enrichment signalling pathways and their interactions in inactive TAO cases using HPLC-MS/MS and bioinformatics analyses.
Materials and methods
Patients
A total of 6 inactive TAO patients (3 males and 3 females) with a CAS score < 3 were collected from the department of ophthalmology, Zhabei central hospital during November 2017 till August 2018. Tears collected from 6 age-matched healthy cases were chosen as control (CTL). The degree of TAO was measured by clinical activity with a clinical activity score (CAS), which consists of 7 orbital sign and symptom variables (14). The CAS < 3 was selected as an inactive stage (15). The determination of thyroid hormones and related antibodies was carried out in the laboratory of Zhabei central hospital. In this investigation, all participants gave their written informed consent. All protocols were approved by the local ethical standard committee in accordance with the ethical standards laid down in the Declaration of Helsinki.
Chemicals
Trypsin was purchased from Promega Corporation (Madison, WI, USA). Dithiothreitol, formic acid, iodoacetamide, NH4HCO3, and trifluoroacetic acid were obtained from Sinopharm Chemical Reagent (Shanghai, China). Acetonitrile (HPLC-grade) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). SiRT peptides were acquired from Biognosys (Schlieren, Switzerland). The other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise indicated.
Tear collection and preparation
Tear collection and preparation were according to our previous work (16). During the processing, tear samples were collected by not touching the ocular surface and without application of any local drug. All tears were stored in a –80 °C freezer prior to analysis. The pretreatment of tear liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed as described in our recent study (17). False discovery rate (FDR) was set to 1% for protein and peptide spectrum matches.
LC-MS/MS determination and analysis
LC-MS/MS was carried out according to our previous study (17). Briefly, the pooled tears were further fractionated using high pH reversed-phase chromatography (Dionex UHPLC, Thermo Scientific, Waltham, MA, USA) for preparing the digested peptides. Then the peptides were resuspended in 1% formic acid 5% acetonitrile for DDA analysis by Orbitrap Fusion LUMOS Tribrid mass spectrometer (Thermo Fisher Scientific) connected to an Easy-nLC 1200 via an Easy Spray (Thermo Fisher Scientific). During the spectral library generation, the false discovery rate (FDR) was set to 1% for protein and peptide spectrum matches. DIA data analysis was analyzed in Spectronaut X (Biognosys, Schlieren, Switzerland), and the results were filtered by a Q value < 0.01.
Bioinformatics analysis
Bioinformatics analysis was performed according to our recent study (17). Briefly, g:Profiler analysis was carried out to our previous work (17) at https://biit.cs.ut.ee/gprofiler/gost. The version was e95_eg42_p13_f6e58b9, and the database was updated on 25/05/2019. The significantly changed terms were enriched in the GO, KEGG, and Reactome (REAC) databases with an adjusted p-value <0.05. NetworkAnalyst analysis was performed according to our previous study (17) at https://www.networkanalyst.ca/NetworkAnalyst/faces/home.xhtml. The website was upgraded and maintained until 8/05/2019. Reactome (REAC) enrichment analysis was performed against Reactome version 66 on 25/05/2019 at https://reactome.org/PathwayBrowser.
Statistics
FDR < 1% was set to protein and peptide spectrum matches. Significance threshold in g:Profiler analysis was g:SCS threshold. The adjusted p value method was used in Benjamini-Hochberg (BH) procedure, and the adjusted p value was transformed to negative log10 (-log10(adjp)). All significantly changed pathways and interactions were used with an adjusted p-value < 0.05.
Results
Clinical and serological data
The clinical and serological data were shown in Table 1. Six TAO patients (3 males and three females) with a CAS score < 3 were chosen as inactive cases. The indexes of thyroid hormone function in these cases were relatively normal. OSDI score indicated these patients had mild symptoms of dry eye. Six healthy age-matched cases (2 males and four females) were adopted as controls in this investigation.
Table 1.
Clinical characteristics of TAO patients and healthy controls
| CTL | TAO | Normal ranges | |
|---|---|---|---|
| Cases (M/F) | 2/4 | 3/3 | |
| Ages | 46.7±6.3 | 50.2±5.4 | |
| CAS Score | N.D. | 1.7±0.5 | |
| OSDI Score | 6.2±2.3 | 18.0±1.7 ** | |
| TSH (mIU/L) | N.D. | 1.9±1.3 | 0.55~4.73 |
| FT3 (pmol/L) | N.D. | 4.5±0.4 | 3.5~6.5 |
| FT4 (pmol/L) | N.D. | 15.2±2.3 | 11.5~22.7 |
| T3 (ng/mL) | N.D. | 1.09±0.13 | 0.60~1.81 |
| T4 (μg/dL) | N.D. | 7.9±1.5 | 4.5~10.9 |
| rT3 (ng/mL) | N.D. | 0.57±0.089 | 0.31~0.95 |
| A TPO (IU/mL) | N.D. | 6.2±4.3 | 1~16 |
| TGAb (%) | N.D. | 5.4±6.3 | 0~30 |
| TMAb (%) | N.D. | 4.7±3.3 | 0~20 |
| TRAb (IU/L) | N.D. | 5.7±4.8 | 0 - 1.75 UI/L |
| Tg (ng/mL) | N.D. | 20.8±22.8 | 0.2~70.0 |
N.D. Not determined. ** p<0.01 compared with CTL group.
Proteomics changes between TAO and healthy individuals
MS/MS analysis was carried out to identify proteins following to tear processing as described in the Methods section. The test analysis process was shown in Figure 1A. PLS-DA of proteomics data suggested that the difference in the same group of tears was slight, while the difference between different groups was prominent (Fig. 1B). A total of 671 proteins were identified by MS. Among them, 611 proteins were found in healthy individuals, and 666 were found in TAO cases (For more information, refer to Supplementary materials: Differentially_expressed_proteins.xlsx). Through the statistical analysis, we find that 107 proteins changed significantly between TAO and health cases with a |Fold Change|>2, a q-value < 0.01, and a p-value < 0.05 (Supplementary Table 1). Volcano showed the distribution of these proteins (Fig. 1C). Please note that 60 extremely upregulated and five downregulated proteins in TAO cases compared with healthy’s were not mapped in the volcano due to these proteins being expressed only in TAO or healthy group (Fig. 1C). Heatmap of differentially expressed proteins (DEPs) indicated the clustering relationship of these proteins (Fig. 1D). Additionally, the chord diagram and Venn of DEPs intuitively showed the distribution of these differential proteins (Fig. 1E/F). There were 62 upregulated and 45 downregulated proteins in the TAO group compared with the healthy group (Fig. 1F). Besides, E5RK69, F2Z2Y4, F6TLX2, Q14435, and Q92520 were only expressed in healthy individuals. However, A0A087WSY5, A0A087WW43, A0A087X1X7, A0A0A0MRM8, A0A0D9SF54, etc. were only expressed in TAO cases.
Figure 1.
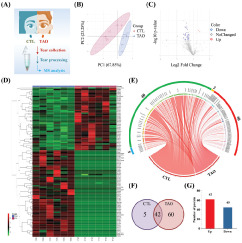
Results of tear proteomics in inactive TAO by HPLC-MS/MS. The proteomics method is described in the Methods section. (A) A schematic diagram of tear proteomics. (B) PLS-DA of proteomics data. (C) Volcano of DEPs from proteomics data. (D) Heatmap of DEPs. (E) Chord diagram of DEPs. (F) Venn of DEPs. (G) Number of DEPs.
Results of enrichment analysis by g:Profiler and Reactome
To visualize the enrichment information of these DEPs, g: Profiler and Reactome were applied to perform bioinformatics analysis. A large number of pathways were enriched by g:Profiler in GO, KEGG, and REAC databases (Fig. 2A). A large number of signalling pathways were further enriched by Reactome (Fig. 2B). Additionally, there are extensive interactions among these signalling pathways (Fig. 2B). For example, signal transduction interacts with the immune system, metabolism, neuronal system, hemostasis, or gene expression.
Figure 2.
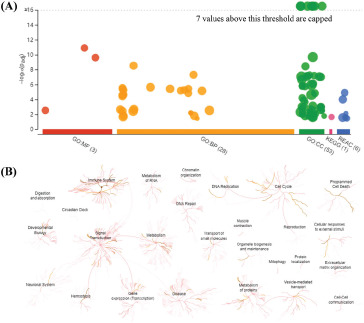
Overall results of bioinformatics analysis using g:Profiler and Reactome. The bioinformatics method is described in the Methods section. (A) The significantly changed terms enriched by GO, KEGG, and Reactome (REAC) databases. (B) The enrichment pathways and their interactions were enriched by REAC.
To clarify the categories of these pathways, we summarized these pathways enriched by REAC. The immune system, cell cycle, metabolism, and signal transduction were dominant, and their percentage was as high as 68% (Fig. 3A/B). Additionally, programmed cell death, cell response to external stimulus, and molecule transport were also preponderant (Fig. 3B). We further explored the interactions among these significantly changed pathways and found that extensive interactions existed in these pathways (Fig. 3C). For example, cell-cell communication interacted with DSCAM interactions, Nef and signal transduction, Robo receptor signalling and inactivation of Cdc42 and Rac.
Figure 3.
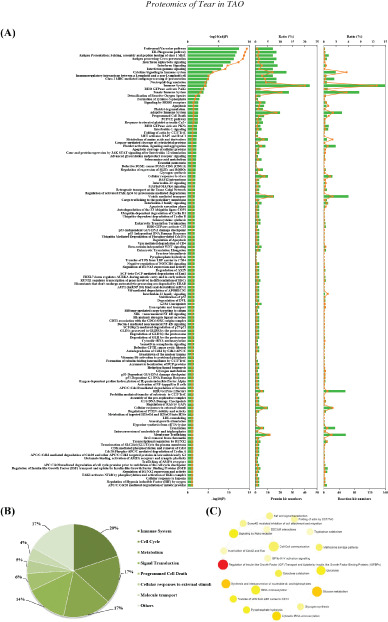
Classification statistics of significantly enriched pathways by REAC were shown in Figure 2B. Enrichment analysis methods were described in the Methods section. (A) Enrichment pathways by REAC analysis. Left: -log(adjP) and -log(P) were shown as line chart and column. Middle: numbers and ratio of DEPs involved in enrichment pathways were shown as column and line chart. Right: numbers and ratio of reactions were shown as column and line charts. (B) Statistical results of classification shown in A. (C) Interactions among these significantly changed pathways by NetworkAnalyst.
Results of GO enrichment analysis
GO enrichment analysis was further applied to explore the BP, MF and CC terms induced by these DEPs. A total of 28, 3, and 53 GO terms were significantly enriched by g:Profiler in BP, MF, and CC with an adjusted p-value < 0.05, respectively (Fig. 4A-C). In the BP terms, immune or inflammatory response, modulation of growth of symbiont involved in interaction with host, exocytosis or secretion, vesicle-mediated transport were significant terms (Fig. 4A). These enriched terms were also proved by Blast2GO. Additionally, chemotaxis, monosaccharide catabolic process, and regulation of protein polymerization were also found by Blast2GO (Fig. 4D). In the MF terms, cadherin binding, cell cadherin binding and actin binding were enriched by g:Profiler (Fig. 4B). In addition, actin binding, protein binding, insulin-like growth factor binding, RNA binding, antioxidant activity, D-xylulose reductase activity, glutathione-disulfide reductase activity, and IL-1 receptor antagonist activity were also enriched by Blast2GO (Fig. 4E). In CC terms, a large number of extracellular and intracellular components were enriched by g:Profiler and Blast2GO (Fig. 4C/F), such as exosome, vesicle, and organelle, cytosol, cytoskeleton and actin filament. According to the order of the ratio of the participated proteins, the terms mentioned above were also the top (Fig. 4G).
Figure 4.

Gene Ontology (GO) enrichment analysis by g:Profiler and Blast2GO. The bioinformatics method is described in the Methods section. (A) BP, (B) MF, and (C) CC terms by g:Profiler. (D) BP, (E) MF and (F) CC terms by Blast2GO. (G) Top GO terms by Blast2GO. (H) Interactions of GO (H) BP, (I) MF, and (J) CC terms by NetworkAnalyst.
We further analyze interactions of these GO terms using NetworkAnalyst. A large number of interactions existed in the GO terms (Fig. 4H-J). For instance, cell activation interacted with cell defense response, immune response, leukocyte migration/chemotaxis, hemostasis, cell adhesion, or actin filament organization in BP terms (Fig. 4H). Moreover, actin binding interacted with GTPase binding and cytoskeletal protein binding in MF terms (Fig. 4I). Additionally, there were also extensive interactions between intracellular and extracellular components (Fig. 4J).
Results of KEGG enrichment analysis
We further analyze KEGG enrichment pathways and their interactions using KOBAS and NetworkAnalyst. There were 13 and 16 significantly enriched pathways using KOBAS and NetworkAnalyst, respectively (Fig. 5A/B). Among them, salmonella infection, fructose and mannose metabolism, shigellosis, glycolysis/gluconeogenesis, and carbohydrate metabolism were both enriched by KOBAS and NetworkAnalyst. Classification of these pathways indicated that metabolism pathways, carbohydrate metabolism, infectious diseases, metabolism of cofactors and vitamins, and protein synthesis and degradation were dominant (Fig. 5D/E). Additionally, extensive interactions among carbohydrate metabolism, protein metabolism, riboflavin metabolism, adherens junction, and infectious diseases were identified by NetworkAnalyst (Fig. 5C).
Figure 5.
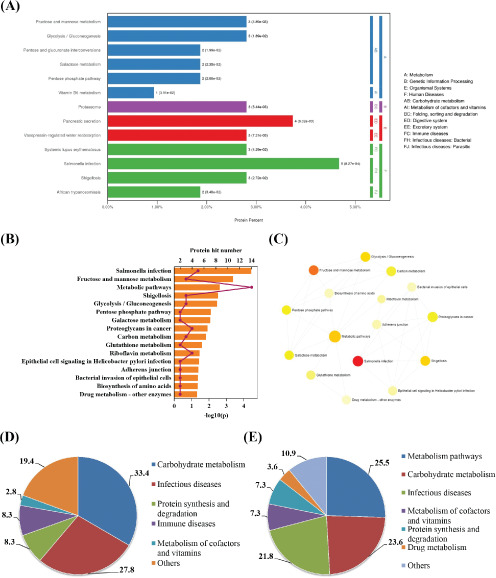
KEGG enrichment pathways and their interactions using KOBAS and NetworkAnalys. The bioinformatics method is described in the Methods section. (A) Enrichment pathways by KOBAS. (B) Enrichment pathways by NetworkAnalys. DEP hit numbers and –log10(P) were shown as columns and line charts. (C) Interactions of KEGG enrichment pathways. Classified statistical results from NetworkAnalys (D) and KOBAS (E).
Results of KOG enrichment analysis
KOG enrichment analysis indicated that cell functions were focused on cytoskeleton, general function prediction, posttranslational modification, protein turnover, chaperones, amino acid transport and metabolism, and signal transduction mechanisms (Fig. 6A/B). In addition, proteins involved in functions of defense mechanisms, secondary metabolites biosynthesis, transport and catabolism, nuclear structure, transcription and inorganic ion transport and metabolism were only upregulated (Fig. 6B). However, proteins involved in functions of chromatin structure and dynamics, coenzyme transport and metabolism, and nucleotide transport and metabolism were only downregulated (Fig. 6B). Further classification analysis revealed that DEPs were focused on protein synthesis and degradation, cytoskeleton, general function prediction and metabolism and the participated proteins in these functions accounted for 68.4% of the total (Fig. 6C).
Figure 6.
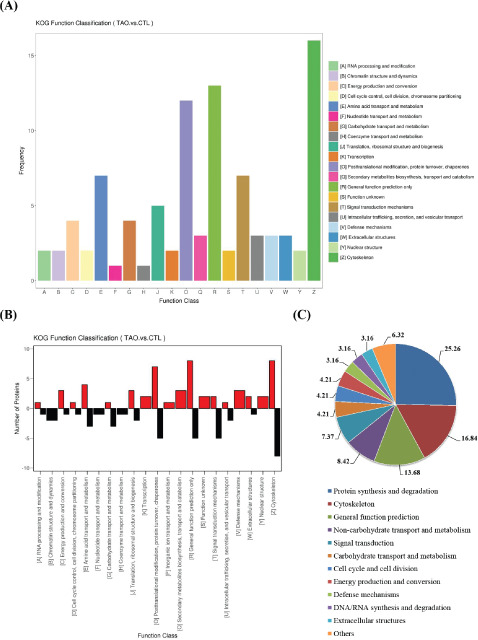
Results of KOG enrichment analysis. The bioinformatics method is described in the Methods section. (A) Top KOG enrichment pathways. (B) KOG function classification. (C) Classified statistical results of KOG enrichment pathways.
PPI and GRN results of DEPs
To explore the protein-protein interactions and gene regulatory networks, we performed NetworkAnalyst and STRING analysis, respectively. A large number of interactions are enriched in these databases. CAND1, RAC1, RPS8, YWHAH, and FLNA frequently interacted with other proteins in PPI (Fig. 7A). A large number of miRNAs, such as miR-92a-3p and miR-16-5p, interacted with these DEPs in gene-miRNA interactions (Fig. 7B). Moreover, transcription factors, such as SMAD5 and TFDP1, regulated these protein expressions in TF-gene interactions (Fig. 7C). Additionally, transcription factors EGR1, MAX, MYC, PDLIM1, JUN, SP1, USF1, etc. interacted with miRNAs, such as miR-15a, miR-16, miR-195, miR-424, and miR-497, coregulated protein expression in TF-miRNA coregulatory interactions (Fig. 7D). We further conducted a statistical analysis of these interactions. In the PPI, CAND1, RAC1, RPS8, YWHAH, and FLNA were top proteins (Fig. 7F). SERBP1, GSR, FAM3C, CAND1, and RAC1 were top proteins in Gene-miRNA interactions (Fig. 7F). APOM, HIST1H2BK, LASP1, PCBP1, and RPS8 were top in TF-gene interactions (Fig. 7F). While SERBP1, YWHAH, VAT1, PCBP1, PLEC, and PFN1 were top in TF-miRNA coregulatory interactions (Fig. 7F). We found that CAND1, PCBP1, PFN1, SERBP1 and YWHAH were the top proteins in the four interactions (Fig. 7G). Using another functional protein association networks STRING, core protein TPI1, CCT4, RPS8, PCBP, and PLEC that were proved by the four interactions mentioned above were at the center of the PPI network (Fig. 7E). Besides, RPL18, RPS3, HP, RPDX6, and CCT3 were also at the center of the network (Fig. 7E).
Figure 7.
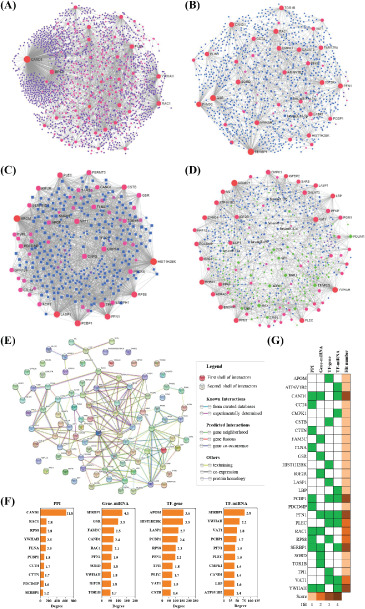
Protein-protein interactions (PPI) and gene regulatory networks (GRN) analysis of these DEPs. The bioinformatics method is described in the Methods section. PPI (A), gene-miRNA interactions (B), TF-gene interactions (C), and TF-miRNA coregulatory interactions (D) by NetworkAnalyst. (E) PPI by STRING. (F) Top DEPs in PPI and GRN. Hit numbers were shown as a column. The number was the ratio of PPI and GRN.
Discussion
Proteomic profiling identified 107 significantly changed proteins between the inactive stage of TAO patients and healthy cases. Among these proteins, 62 proteins were upregulated, and 45 proteins were downregulated in TAO cases compared to healthy individuals. In these inactive TAO patients, 60 proteins were uniquely expressed, and five proteins were not expressed compared with healthy controls. These significantly changed DEPs were focused on the progress of protein synthesis and degradation, cytoskeleton, general function prediction and metabolism. Enrichment analysis revealed that signalling pathways of the immune system, cell cycle, metabolism (carbohydrate metabolism and metabolism of cofactors and vitamins), as well as protein synthesis and degradation might play a significant role in the progress of inactive TAO.
In the active stage of TAO, autoimmune inflammation is usually active. Furthermore, large numbers of T-cell clones directed against varieties of orbital antigens are involved during these stages. While in an inactive stage of TAO, lymphocytes may produce predominantly cytokines, and leukocyte infiltration was less evident (18, 19). Autoimmune inflammation seems to be self-limiting in the majority of patients with inactive TAO. However, the immune system was the top enrichment pathway by Reactome analysis which indicated that inflammation is continuously activated in inactive TAO patients. These results were further verified by GO enrichment analysis. A total of 44 DEPs were involved in these signalling pathways. For example, HLA class I histocompatibility antigen and complement factor H-related protein 1 changed significantly. The results of the present study supported the idea that many molecules, such as lipopolysaccharide-binding protein, HLA class I histocompatibility antigen, and proteasome subunit beta type-8, appear central to inflammation (20). Although direct evidence should be supplied, the results of Reactome enrichment analysis indicated that immune system-related pathways, such as interferon signalling, interleukin-1 signalling, ER-phagosome pathway, cytokine signalling, antigen processing-cross presentation, and class I MHC mediated antigen processing & presentation, may play a significant role in activating of inflammation in inactive TAO patients.
Vesicle-mediated transport of membrane and proteins plays a significant role in molecular transport, signal transfer, and clearance of heterogeneous substances (21). Both Reactome and GO enrichment analyses confirmed that vesicle-mediated transport was critical for the pathogenic mechanism of TAO. Fifteen proteins were identified as membrane trafficking associated proteins using Reactome analysis. Such as perilipin-3, unconventional myosin-VI, capping protein, torsin-1B, and 14-3-3 eta protein. In the ciliary body, unconventional myosins are related to the ciliary muscle that controls the shape of the lens during accommodation (22). These suggested that some proteins expressed in the eyes may be released into tears during inflammation in TAO patients. These membrane trafficking proteins released into tears may be as the result of sustained autoimmune inflammation or innate immune system against xenobiotics.
A previous work found that Fas-mediated apoptosis was involved in extraocular muscle tissue from patients with the late stage of TAO (23). Many molecules, such as the proteasome, polyubiquitin-B, plectin, lamin B1, histone, and 14-3-3 eta protein, were identified through proteomics analysis in inactive TAO tear and may activate apoptosis and cell cycle signalling pathways. These data further demonstrated the idea that cell cycle, apoptosis regulators and proteasomes play a key role in TAO development (24).
KEGG enrichment analysis indicated that metabolism signalling, carbohydrate metabolism, infectious diseases, metabolism of cofactors and vitamins, and protein synthesis and degradation were top pathways. Previous work has proved that thyroid hormones play key roles in the processes of growth, development, and metabolism (25). Thyroid hormones also play a significant role in regulating lipid, cholesterol, glucose, and protein metabolism (26. 27). The thyroid hormone signalling pathway regulates energy expenditure through both central and peripheral pathways (28). Although thyroid hormones were relatively normal in the late, inactive stage of TAO, thyroid hormones altered significantly in the active stage of TAO. During the active stage, thyroid hormone regulates the basal metabolic rate and energy intake and thus affects metabolism homeostasis (29). The enriched results of the present investigation further suggested that even if thyroid function returns to a relatively normal level in the stages of inactive TAO patients, metabolic abnormalities may persist for a long time. These suggested that metabolic abnormalities should not be neglected in the prevention and treatment of inactive TAO.
We further performed KOG enrichment analysis with these DEPs. We found that these proteins involved in functions of defense mechanisms, secondary metabolites biosynthesis, transport and catabolism, nuclear structure, transcription and inorganic ion transport and metabolism were upregulated. At the same time, DEPs involved in functions of chromatin structure and dynamics, coenzyme transport and metabolism, and nucleotide transport and metabolism were downregulated. KOG enrichment results further indicated that protein synthesis and degradation, cytoskeleton, general function prediction, metabolism, cell cycle, energy production and conversion, and the immune system play essential roles in inactive TAO. The results of KOG analysis directly proved the pathways of the ribosome, protein synthesis and degradation, and retinol metabolism play significant roles in patients with inactive TAO (24).
In conclusion, a total of 107 proteins changed significantly between the inactive stage of TAO patients and healthy cases. The signalling pathways of the immune system, apoptosis, cell cycle, cytoskeleton, metabolism of carbohydrates, cofactors and vitamins, protein synthesis and degradation, vesicle-mediated transport of membrane and proteins may play key roles in patients with inactive TAO.
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
List of Abbreviations: DEPs: differentially expressed proteins; GO: BP: GO biological process GO: CC: GO cellular component; GO: MF: GO molecular function; IGF-I: insulin-like growth factor I; IGF-IR: IGF-I receptor; REAC: Reactome; TAO: thyroid-associated ophthalmopathy; CTL: healthy control.
Ethics approval and consent to participate: All protocols were approved by Tongji Hospital ethical standard committee in accordance with the ethical standards laid down in the Declaration of Helsinki. The clinical trial record number was 2019000067.
Funding: The current study was supported by the National Natural Science Foundation of China (No. 81770959) and the Shanghai Municipal Commission of Health and Family Planning (No. 201640215).
References
- 1.Garrity JA, Bahn RS. Pathogenesis of Graves ophthalmopathy: Implications for prediction, prevention, and treatment. Am J Ophthalmol. 2006;142:147–153. doi: 10.1016/j.ajo.2006.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sisti E, Coco B, Menconi F, Leo M, Rocchi R, Latrofa F, Profilo MA, Mazzi B, Albano E, Vitti P, Marcocci C, Brunetto M, Marinò M. Intravenous glucocorticoid therapy for Graves’ ophthalmopathy and acute liver damage: an epidemiological study. Eur J Endocrinol. 2015;172:269–276. doi: 10.1530/EJE-14-0712. [DOI] [PubMed] [Google Scholar]
- 3.Tanda ML, Bartalena L. Efficacy and safety of orbital radiotherapy for Graves’ orbitopathy. J Clin Endocrinol Metab. 2012;97:3857–3865. doi: 10.1210/jc.2012-2758. [DOI] [PubMed] [Google Scholar]
- 4.Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, Tang RA, Harris GJ, Antonelli A, Salvi M, Goldberg RA, Gigantelli JW, Couch SM, Shriver EM, Hayek BR, Hink EM, Woodward RM, Gabriel K, Magni G, Douglas RS. Teprotumumab for thyroid-associated ophthalmopathy. N Engl J Med. 2017;376(18):1748–1761. doi: 10.1056/NEJMoa1614949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herbaut A, Liang H, Denoyer A, Baudouin C, Labbé A. Tear film analysis and evaluation of optical quality: A review of the literature. J Fr Ophtalmol. 2019;42(2):e21–e35. doi: 10.1016/j.jfo.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhou L, Beuerman RW. The power of tears: how tear proteomics research could revolutionize the clinic. Expert Rev Proteomics. 2017;14(3):189–191. doi: 10.1080/14789450.2017.1285703. [DOI] [PubMed] [Google Scholar]
- 7.Okrojek R, Grus F H, Matheis N, Kahaly GJ. Proteomics in autoimmune thyroid eye disease. Horm Metab Res. 2009;41(06):465–470. doi: 10.1055/s-0029-1214413. [DOI] [PubMed] [Google Scholar]
- 8.Kishazi E, Dor M, Eperon S, Hamedani M, Turck N. Thyroid-associated orbitopathy and tears: a proteomics study. J Proteomics. 2018;170:110–116. doi: 10.1016/j.jprot.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Aass C, Norheim I, Eriksen EF, Børnick EC, Thorsby PM, Pepaj M. Comparative proteomic analysis of tear fluid in Graves’s disease with and without orbitopathy. Clin Endocrinol. 2016;85(5):805–812. doi: 10.1111/cen.13122. [DOI] [PubMed] [Google Scholar]
- 10.Choi W, Li Y, Ji Y S, Yoon KC. Oxidative stress markers in tears of patients with Graves’ orbitopathy and their correlation with clinical activity score. BMC Ophthalmol. 2018;18(1):303. doi: 10.1186/s12886-018-0969-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edina K, Marianne D, Simone E, Oberic A, Turck N, Hamedani M. Differential profiling of lacrimal cytokines in patients suffering from thyroid-associated orbitopathy. Sci Rep. 2018;8(1):10792. doi: 10.1038/s41598-018-29113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matheis N, Grus F H, Breitenfeld M, Knych I, Funke S, Pitz S, Ponto KA, Pfeiffer N, Kahaly GJ. Proteomics differentiate between thyroid-associated orbitopathy and dry eye syndrome. Invest Ophthalmol Vis Sci. 2015;56(4):2649–2456. doi: 10.1167/iovs.15-16699. [DOI] [PubMed] [Google Scholar]
- 13.Yang M, Chung Y, Lang S, Yawata N, Seah LL, Looi A. The tear cytokine profile in patients with active Graves’ orbitopathy. Endocrine. 2017;59(2):402–409. doi: 10.1007/s12020-017-1467-2. [DOI] [PubMed] [Google Scholar]
- 14.Kim JW, Woo YJ, Yoon JS. Is modified clinical activity score an accurate indicator of diplopia progression in Graves’ orbitopathy patients? Endoc J. 2016;63(12):1133–1140. doi: 10.1507/endocrj.EJ16-0165. [DOI] [PubMed] [Google Scholar]
- 15.Sun B, Zhang Z, Dong C, Zhang Y, Yan C, Li S Medscape. 99Tcm-octreotide scintigraphy and serum eye muscle antibodies in evaluation of active thyroid-associated ophthalmopathy. Eye. 2017;31(5):668–676. doi: 10.1038/eye.2017.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang L, Wei R. Analysis of Graves’ ophthalmopathy patients’ tear protein spectrum. Chin Med J. 2013;126(23):4493–4498. [PubMed] [Google Scholar]
- 17.Jiang L, Rong A, Wei R, Diao J, Ding H, Wang W. Tear proteomics of orbital decompression for disfiguring exophthalmos in inactive thyroid-associated ophthalmopathy. Exp Ther Med. 2020;20(6):253. doi: 10.3892/etm.2020.9383. 10.3892/etm.2020.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bednarczuk T, Hiromatsu Y, Inoue Y, Yamamoto K, Wall JR, Nauman J. T-cell-mediated immunity in thyroid-associated ophthalmopathy. Thyroid. 2002;12(3):209–215. doi: 10.1089/105072502753600151. [DOI] [PubMed] [Google Scholar]
- 19.Garrity JA, Bahn RS. Pathogenesis of Graves ophthalmopathy: Implications for prediction, prevention, and treatment. Am J Ophthalmol. 2006;142:147–153. doi: 10.1016/j.ajo.2006.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naik V, Khadavi N, Naik MN, Hwang C, Goldberg RA, Tsirbas A, Smith TJ, Douglas RS. Biologic therapeutics in thyroid-associated ophthalmopathy: translating disease mechanism into therapy. Thyroid. 2008;18(9):967–971. doi: 10.1089/thy.2007.0403. [DOI] [PubMed] [Google Scholar]
- 21.Graham T R. Flippases and vesicle-mediated protein transport. Trends Cell Biol. 2004;14(12):670–677. doi: 10.1016/j.tcb.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Kirby D, Dufresne C, Chen Y, Turner R, Ferri S, Edward DP, Van Eyk JE, Semba RD. Defining the proteome of human iris, ciliary body, retinal pigment epithelium, and choroid. Proteomics. 2016;16:1146–1153. doi: 10.1002/pmic.201500188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koga M, Hiromatsu Y, Jimi A, Inoue Y, Nonaka K. Possible involvement of Fas-Mediated apoptosis in eye muscle tissue from patients with thyroid-associated ophthalmopathy. Thyroid. 1998;8(4):311–318. doi: 10.1089/thy.1998.8.311. [DOI] [PubMed] [Google Scholar]
- 24.Zhao P, Yin H, Tao C, Chen P, Song Y, Yang W, Liu L. Latent pathways identification by microarray expression profiles in thyroid-associated ophthalmopathy patients. Endocr Pathol. 2015;26(3):200–210. doi: 10.1007/s12022-015-9373-8. [DOI] [PubMed] [Google Scholar]
- 25.Hollenberg AN. The role of the thyrotropin-releasing hormone (TRH) neuron as a metabolic sensor. Thyroid. 2008;18:131–139. doi: 10.1089/thy.2007.0251. [DOI] [PubMed] [Google Scholar]
- 26.Potenza M, Via M, Yanagisawa R. Excess thyroid hormone and carbohydrate metabolism. Endocr Pract. 2009;15(3):254–262. doi: 10.4158/EP.15.3.254. [DOI] [PubMed] [Google Scholar]
- 27.Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol Metab. 2014;25(10):538–545. doi: 10.1016/j.tem.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Mcaninch EA, Bianco AC. Thyroid hormone signalling in energy homeostasis and energy metabolism. Ann NY Acad Sci. 2014;1311(1):77–87. doi: 10.1111/nyas.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tohma Y, Aktürk M, Altınova A, Yassibas E, Cerit ET, Gulbahar O, Arslan M, Sanlier N, Toruner F. Circulating levels of orexin-A, nesfatin-1, agouti related peptide, and neuropeptide Y in patients with hyperthyroidism. Thyroid. 2015;25(7):776–783. doi: 10.1089/thy.2014.0515. [DOI] [PubMed] [Google Scholar]