

Abstract
Proteomics has revealed that the ~20,000 human genes engender a far greater number of proteins, or proteoforms, that are diversified in large part by post-translational modifications (PTMs). How such PTMs affect protein structure and function is an active area of research but remains technically challenging to assess on a proteome-wide scale. Here, we describe a chemical proteomic method to quantitatively relate serine/threonine phosphorylation to changes in the reactivity of cysteine residues, a parameter that can affect the potential for cysteines to be post-translationally modified or engaged by covalent drugs. Leveraging the extensive high-stoichiometry phosphorylation occurring in mitotic cells, we discover numerous cysteines that exhibit phosphorylation-dependent changes in reactivity on diverse proteins enriched in cell cycle regulatory pathways. The discovery of bidirectional changes in cysteine reactivity often occurring in proximity to serine/threonine phosphorylation events points to the broad impact of phosphorylation on the chemical reactivity of proteins and the future potential to create small-molecule probes that differentially target PTM-modified proteoforms.
Introduction
Large-scale genomic and transcriptomic profiling methods have transformed our understanding of the molecular composition of biological systems and their regulation by physiological and pathological stimuli1–4, as well as by pharmacological perturbation5. Much of the biochemistry of human cells is dedicated, however, to shaping the proteome in a contextual and temporal manner through post-translational events that are poorly captured by genomic and transcriptomic profiling. Indeed, it has been estimated that the ~20,000 human genes in the human genome produce a much larger number of unique proteins (~10X), or proteoforms6, due to splice variations and post-translational modifications (PTMs)7.
Understanding PTMs and their impact on protein function is an intense area of research, but also a daunting task because, as the number of proteoforms originating from a single gene increases, so does their potential for combinatorial representation in a given cellular state, and methods to resolve each proteoform remain limited. One effective strategy is top-down proteomics, which enables independent analysis of individual proteoforms; but, despite recent advances, this method is still challenged to analyze proteins of larger size (> 50 kDa), especially in complex biological systems8, 9. Bottom-up proteomics, on the other hand, where cell lysates are digested by a protease such as trypsin before analysis by liquid chromatography-mass spectrometry (LC-MS), typically assesses all proteoforms of a given protein as a single integrated signal10. Various strategies have been introduced to enrich specific PTM forms of proteins, such as phosphorylated11, glycosylated12, or lipidated proteins13, and recent studies have shown how such PTMs can be related to the thermal stability of proteins in cells14. However, the impact of PTMs on many other features of protein structure and function remains unknown. Addressing this important topic, especially on a global scale, would facilitate identification of PTM events that create proteoforms with distinct chemical properties, biochemical activities, and druggability, thereby refining our understanding of, and potential to pharmacologically control, cellular processes.
Mitosis is the biological process responsible for faithfully segregating the duplicated genome to two daughter cells for proper cell division15, 16. Failure to regulate cell division has severe consequences, including the uncontrolled proliferation state that is a hallmark of cancer17–19. Proper cell cycle progression is temporally controlled through a complex network of PTMs that influence protein activity, stability, localization, and biomolecular interactions20. Phosphorylation plays a major role in regulating cell cycle, and phosphoproteomic studies have revealed a massive burst of protein phosphorylation during mitosis involving over 30,000 distinct mitotic phosphosites21–24, with 20% of these phosphosites being exclusively found in mitosis21. These phosphorylation events mainly occur on serine and threonine residues (referred to hereafter as S/T phosphorylation events) and conform to a consensus sequence where the phosphorylated residue is followed by proline ([S/T]P motif), reflecting the robust activation of cyclin-dependent kinases (CDKs) during mitosis24. Notably, quantitative proteomic methods have also revealed that many mitotic phosphorylation events occur with high stoichiometry, meaning that the majority of a given protein undergoing mitotic phosphorylation has been converted into its phosphorylated proteoform21, 22.
Because mitosis is rich in unique proteoform content, in part through PTMs like phosphorylation, it has been intensively investigated by large-scale methods aimed at globally dissecting protein state and activity relationships. These approaches include not only phosphoproteomics,21–24 but also complementary proteogenomic methods, such as cellular thermal shift assays (MS-CETSA) that give insight into biophysical changes in proteins that may arise, for instance, from cell cycle-specific PTMs or protein-protein/metabolite interactions25, 26, size exclusion chromatography (MS-SEC) that identifies mitotic protein complex reorganization27, as well as single-cell immunofluorescence microscopy to map proteins with temporal or spatial changes throughout the cell cycle16.
While the aforementioned proteomic approaches have revealed striking changes in protein modification state, structure, localization, and function during mitosis, it remains unclear how unique mitotic proteoforms, or phosphorylated proteoforms more generally, may differ in their chemical properties compared to non-mitotic/unphosphorylated states of proteins. The chemical reactivity of proteins, for instance, is a dynamic property that can influence the potential for covalent modification by endogenous or exogenous small molecules28–32. Cysteine is the most intrinsically nucleophilic amino acid in the proteome and plays important roles in enzyme catalysis and metal coordination, as well as also being subject to diverse post-translational forms of oxidative and chemical modification33–35. Because the cysteine thiol pKa is close to physiological pH, slight perturbations in the microenvironment surrounding cysteines can strongly influence their reactivity and potential for modification28, 36. These characteristics enable cysteine residues to serve as sensors of changes in cell and protein state, as well as primary targets for covalent chemical probe and drug development37, 38. We have previously described an activity-based protein profiling (ABPP) platform that leverages broad, cysteine-directed iodoacetamide (IA) chemical probes coupled with multiplexed (tandem mass tagging, or TMT) proteomic analysis to quantitatively monitor changes in the reactivity of many thousands of cysteines in native human proteomes32, 39–41. This TMT-ABPP platform has been used to assess changes in cysteine reactivity that occur, for instance, in stimulated human T cells39, as well as to assess electrophilic small-molecule interactions (or the covalent ligandability) of cysteines on a global scale39–42. Nonetheless, how cysteine reactivity may be modulated by PTMs occurring on other proteinaceous amino acids, such as S/T phosphorylation, remains unexplored. Here, we describe a method that integrates TMT-ABPP with protocols for altering the phosphoproteome to enable the quantification of phosphorylation-dependent changes in cysteine reactivity on a global scale. We discover numerous cysteines on structurally and functionally diverse proteins that undergo phosphorylation-dependent increases or decreases in reactivity in mitotic cells, underscoring the widespread potential for phosphorylation pathways to shape the nucleophilic character of cysteines in the human proteome.
Results
Cysteine reactivity profiling of mitotic cells
We first set out to establish a baseline understanding of changes in cysteine reactivity in mitotic cells. HeLa cells were synchronized in early mitosis by a thymidine-nocodazole block, and corresponding asynchronous HeLa cells were seeded and harvested in parallel (Extended Data Fig. 1a). Proteomic lysates from each cell state were then analyzed by TMT-ABPP, specifically being treated with the cysteine-reactive probe iodoacetamide-desthiobiotin (IA-DTB,100 μM, 1 h), trypsinized, and biotinylated cysteine-containing peptides isolated and comparatively quantified by streptavidin enrichment, TMT labeling, and liquid chromatography-mass spectrometry (MS) using an Orbitrap Fusion instrument. Across ~17,000 quantified cysteines from ~5,800 proteins, we identified >3,000 cysteines displaying a two-fold or greater change in reactivity between mitotic and asynchronous HeLa cells (Fig. 1a and Supplementary Dataset 1). Though there have been reported increases in reactive oxygen species in mitosis43, we did not observe changes in highly oxidation-sensitive cysteines in mitotic HeLa cells (e.g., GAPDH C152 and PARK7 C10644; Extended Data Fig. 1b), which may indicate that such oxidation events occur with too low of a stoichiometry to alter the relative IA-DTB reactivity of cysteines in mitotic and asynchronous cell states.
Figure 1. Cysteine reactivity profiling of mitotic and asynchronous cells.
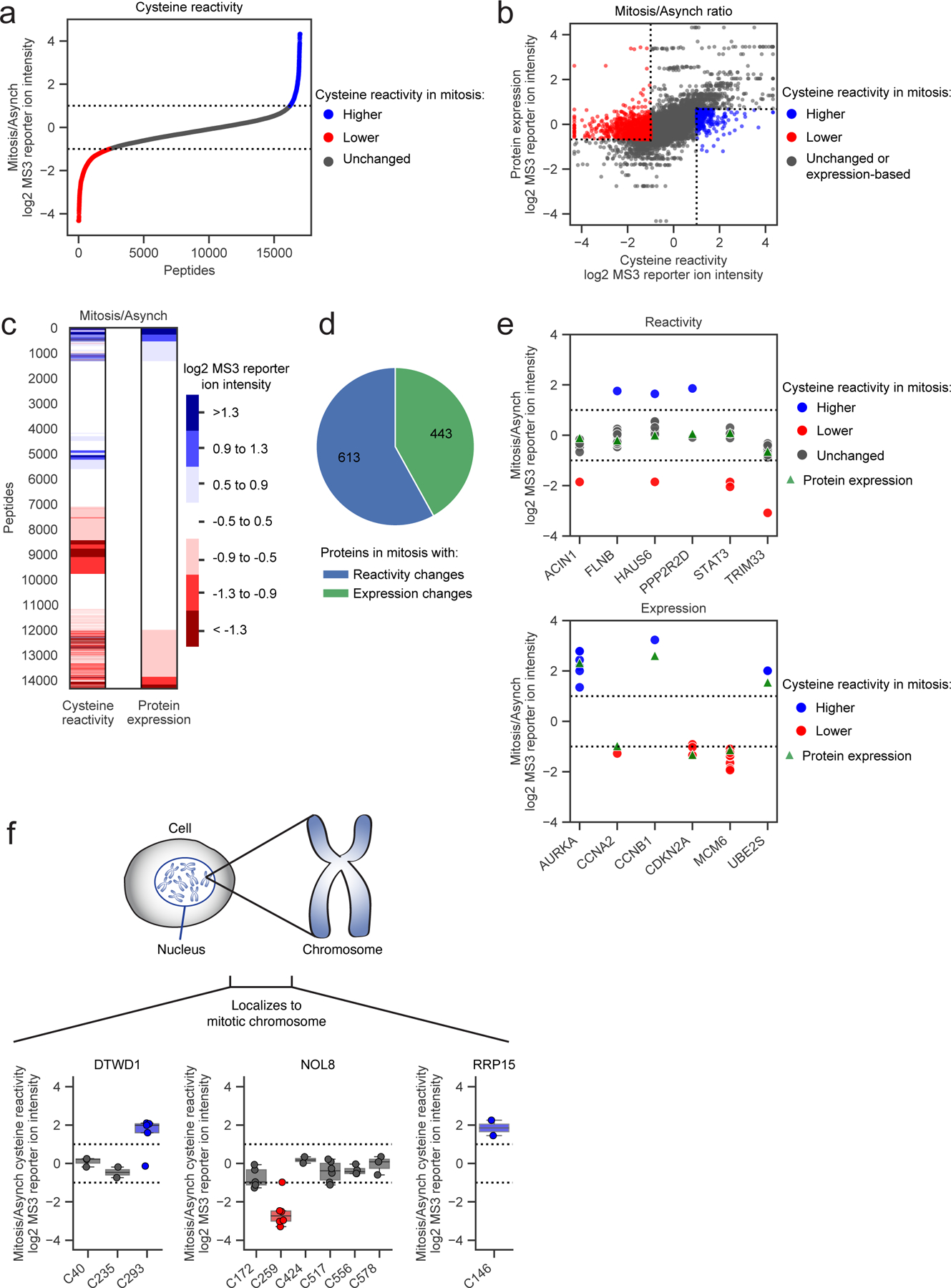
a, Cysteine reactivity ratio values of mitotic vs asynchronous (Mitosis/Asynch) HeLa cell proteomes. Blue and red data points mark cysteine reactivity values that are ≥ two-fold higher (dotted lines) in mitotic or asynchronous proteomes, respectively. Each data point is median value for n = 2 (or more) independent experiments. b, c, Comparison of cysteine reactivity values to the expression values for corresponding proteins harboring these cysteines in scatter plot (b) or heat map (c) format. Ratio values are presented as Mitosis/Asynch (log2 transformed). For b, blue and red mark cysteine reactivity values ≥ two-fold higher in mitotic or asynchronous proteomes, respectively, where the corresponding protein expression value is unchanged (< 1.6x-fold different Mitosis/Asynch). For c, blue and red mark cysteine reactivity or protein expression values that are ≥ two-fold higher in mitotic or asynchronous cell proteomes, respectively. Data shown are median values for n = 2 (or more) independent TMT-ABPP experiments and n = 1 (or more) independent unenriched proteomics experiments. d, Distribution of proteins with cysteine changes assigned as reactivity-based (blue) or expression-based (green) in the mitotic proteome. Proteins with unassigned cysteine changes are not shown. e, Representative reactivity-based (top) and expression-based (bottom) cysteine changes with corresponding protein expression values (Mitosis/Asynch). Dotted lines designate ≥ two-fold changes. Data shown are median values for n = 2 (or more) independent TMT-ABPP experiments and n = 1 (or more) independent unenriched proteomics experiments f, Cysteine reactivity changes in proteins DTWD1 (left), NOL8 (middle), and RRP15 (right) shown to localize to mitotic chromosomes16 (top). Bottom, box plots showing cysteine reactivity profiles for each protein, where blue and red colors mark cysteines with greater reactivity in the mitotic and asynchronous proteomes, respectively. Horizontal black line for each cysteine marks median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range. Dotted lines designate boundaries for ≥ two-fold changes for n = 2 (or more) independent experiments (circles).
In parallel with our TMT-ABPP approach, we also employed unenriched proteomics (Extended Data Fig. 1c), allowing us to distinguish cysteines with authentic changes in reactivity versus those showing signal differences caused by alterations in protein abundance. More specifically, cysteines with authentic reactivity changes were expected to show substantial difference in IA-DTB reactivity (≥ two-fold change) in mitotic cells against a backdrop of unaltered protein expression (<1.5-fold change). While we observed a generally good overlap in proteins quantified by unenriched proteomics versus TMT-ABPP (Extended Data Fig. 1d), each approach quantified ~20–30% of proteins not detected by the other method. We therefore included an additional filter, where cysteine changes were assigned as reactivity-based if additional cysteines quantified in the same protein were unaltered between mitotic and asynchronous cell states (<1.6-fold change). These analyses identified many instances of cysteines that showed striking apparent increases or decreases in reactivity in mitotic cells (Fig. 1b, c). In total, ~1800 proteins were found to display cysteine changes, and of the >1000 proteins confidently assigned to reactivity or expression change groups, ~60% reflected reactivity changes (Fig. 1d and Supplementary Dataset 1). Representative proteins with cysteine reactivity or expression changes are shown in Fig. 1e (upper and lower graphs, respectively). Proteins with reactivity-based cysteine changes were enriched in cellular processes involved in the cell cycle as determined by GO analysis45, 46 (Extended Data Fig. 1e).
In comparing our cysteine reactivity changes to proteomic alterations mapped in mitotic cells by complementary methods, we noted interesting examples of convergent findings. For instance, two cysteines in thymidylate kinase (DTYMK) – C117 and C163 – showed substantial reductions in reactivity in mitotic cells (Extended Data Fig. 1f). DTYMK was also shown to exhibit impaired thermal stability in S and prometaphase states by MS-CETSA analysis25, a feature that was interpreted to reflect the removal of the thymidine block as part of the protocol to induce mitosis. Interestingly, we found that gel filtration of mitotic cell lysates restored DTYMK C117 reactivity, while no such change in the reactivity of this cysteine was observed in gel-filtered asynchronous cell lysates (Extended Data Fig. 1g, h). Considering that C117 is distal (> 10 angstroms away) to the ATP and TMP binding sites of DTYMK47 (Extended Data Fig. 1i), we interpret the change in reactivity of this cysteine to potentially reflect an allosteric effect caused by altered metabolite interactions with DTYMK in the mitotic cell state. Changes in cysteine reactivity in mitotic cells might also reflect alterations in protein-protein interactions, which may in turn affect protein localization. Consistent with this hypothesis, a multitude of proteins exhibiting cysteine reactivity changes, including the tRNA aminocarboxypropyltransferase DTWD1, the nucleolar protein NOL8, and the ribosomal RNA processing protein RRP15, were recently shown to redistribute to chromosomes during mitosis by single-cell immunofluorescence microscopy16 (Fig. 1f and Extended Data Fig. 1j). These findings indicate that integrating TMT-ABPP with other large-scale proteomic analyses can reveal how cell state-dependent biomolecular interactions may promote site-specific changes in the chemical reactivity of proteins.
Mapping phosphorylation-dependent cysteine reactivity changes
We next set out to determine what fraction of cysteine reactivity changes in the mitotic cell proteome may be caused by S/T phosphorylation, which has been shown to be globally increased in the mitotic cell state to produce many high stoichiometry phosphorylation sites21–24. We first showed that the majority of S/T phosphorylation events in the HeLa mitotic proteome could be removed by treatment with lambda phosphatase (LPP) by both immunonoblot analysis (Fig. 2a) and MS-based phosphoproteomic analysis (Fig. 2b and Supplementary Dataset 1) without affecting protein abundances (Supplementary Dataset 1). We quantified over 11,000 phosphorylation sites, which had good overlap with previously quantified mitotic phosphorylation sites21 (Extended Data Fig. 2a). LPP treatment of the mitotic proteome generally did not affect protein abundance (Supplementary Dataset 1). The effective in vitro dephosphorylation by LPP enabled a robust and convenient protocol for a pairwise comparison of conditions – high phosphorylation (LPP-untreated (LPP(−)) mitotic proteome) and low phosphorylation (LPP-treated (LPP(+)) mitotic proteome) – for cysteine reactivity profiling (Fig. 2c). With this method, we comparatively quantified >16,000 cysteines on ~5,600 proteins between LPP(−) and LPP(+) mitotic proteomes, an extent of coverage that compares favorably to previous TMT-ABPP studies, indicating that the incorporation of an LPP treatment step did not substantially alter sensitivity. Across this global landscape of quantified cysteines, we found ~1,000 cysteines showing substantial (> two-fold) differences in reactivity in the LPP(+) proteome (Fig. 2d and Supplementary Dataset 1)39.
Figure 2. Proteomic mapping of phosphorylation-dependent changes in cysteine reactivity.
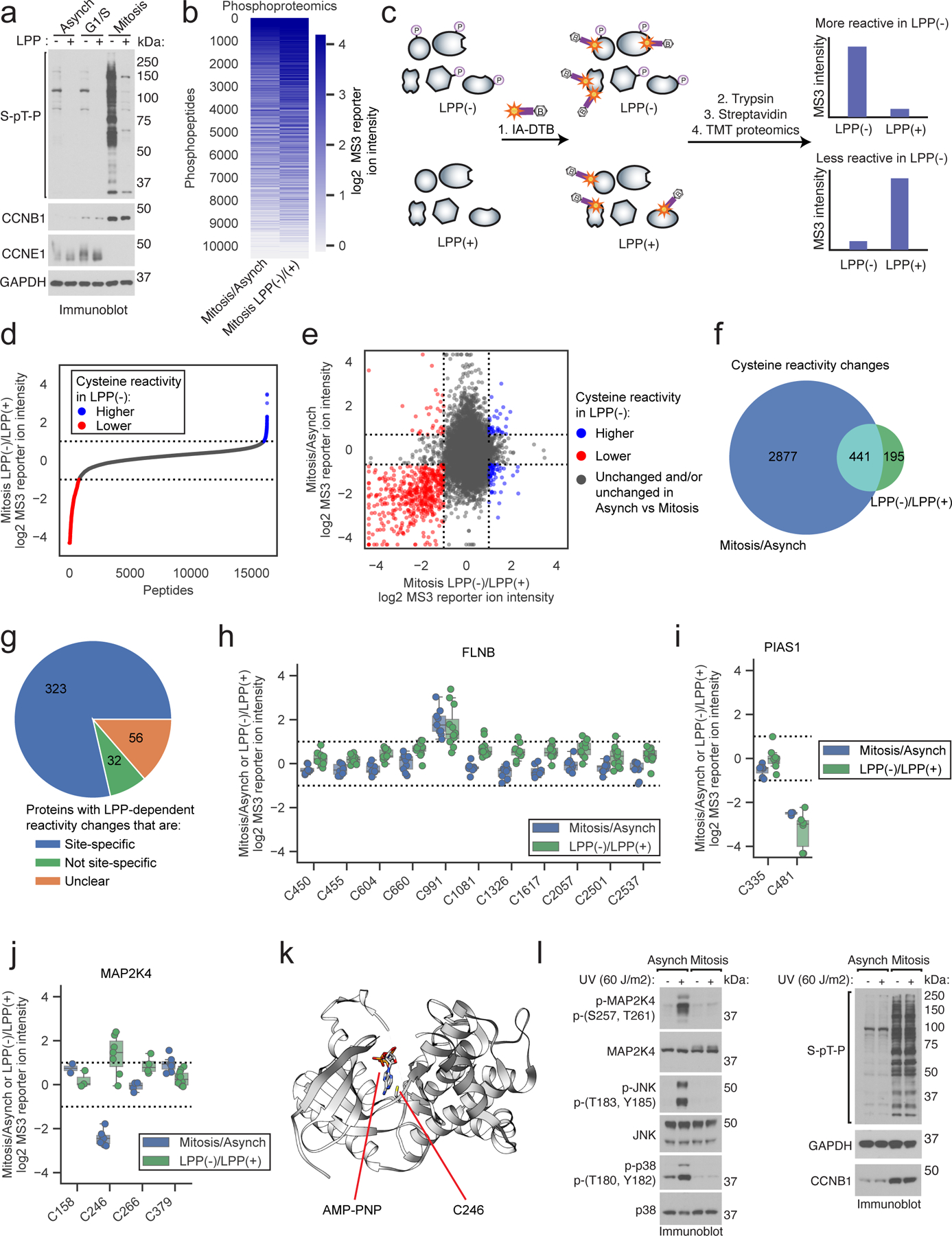
a, Immunoblot of proteomes from synchronized cell states treated with lambda protein phosphatase (LPP). Data are from a single experiment representative of two independent experiments. b, Heatmap showing relative S/T-phosphopeptide enrichment in mitotic versus asynchronous proteomes (Mitosis/Asynch, left) and mitotic proteomes +/− LPP treatment (Mitosis LPP(−)/(+), right). Data are median ratio values for n = 1 (or more) independent experiments. c, TMT-ABPP workflow for measuring LPP-dependent changes in cysteine reactivity. d, Cysteine reactivity ratio values for mitotic proteomes treated with or without LPP (LPP(−)/LPP(+)). Blue and red mark cysteine reactivity values ≥ two-fold higher (boundaries marked by dotted lines) in LPP(−) and LPP(+) proteomes, respectively. Each data point is median value for n = 2 (or more) independent experiments. e, Comparison of cysteine reactivity values from Mitosis/Asynch (y-axis) and Mitosis LPP(−)/(+) (x-axis) proteomes. Blue and red mark cysteine reactivity values ≥ two-fold higher in LPP(−) and LPP(+) proteomes, respectively, that are also changing ≥ 1.6-fold in Mitosis/Asynch (boundaries marked by dotted lines). Data are median values for n = 2 (or more) independent LPP(−)/LPP(+) experiments and n = 1 (or more) Mitosis/Asynch experiments. f, Venn diagram comparing cysteine reactivity changes in LPP(−)/LPP(+) (≥ two-fold, green) and Mitosis/Asynch (≥ 1.6-fold, blue) proteomes. Cysteine reactivity changes calculated from median values from n = 2 (or more) independent experiments. Cysteines from proteins with Mitosis/Asynch expression changes were excluded from the analysis. g, Pie chart showing fraction of site-specific cysteine reactivity changes in the LPP(+) mitotic proteome (i.e., other cysteines in parent proteins were unchanged by LPP (blue)). Unclear proteins had only one total quantified cysteine (orange). h-j, Mitosis/Asynch (blue) and Mitosis LPP(−)/LPP(+) (green) cysteine reactivity values for FLNB, PIAS1, and MAP2K4. Horizontal black line marks median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments. Dotted lines designate boundaries for ≥ two-fold changes. k, Structure of MAP2K4 kinase domain (PDB: 3ALO)49 with C246 highlighted in yellow. l, Immunoblot of UV-induced stress-activated pathway in mitotic vs asynchronous cells. Data are from a single experiment representative of two independent experiments.
To verify that the observed phosphorylation-dependent cysteine reactivity changes were not indirectly reflective of protein misfolding potentially caused by the LPP protocol, we also evaluated cysteine reactivity profiles in mitotic cell proteomes denatured with high molarity urea. Consistent with past studies, which have shown that protein misfolding radically alters cysteine reactivity profiles42, we found that ~75% of the ~15,500 quantified cysteines exhibited reactivity changes upon denaturation (Extended Data Fig. 2b and Supplementary Dataset 1). Importantly however, these denaturation-induced changes in cysteine reactivity did not correlate with LPP-mediated cysteine reactivity changes (Extended Data Fig. 2c–f). On the contrary, we interestingly observed a population of LPP-induced increases in cysteine reactivity that were anti-correlated with the effects of denaturation (Extended Data Fig. 2c). Such cysteines showing decreased reactivity in the denatured proteome were also more likely than unchanging cysteines to reside in predicted disordered regions (blue, Extended Data Fig. 2g), which might indicate that solvent-exposed cysteines in disordered regions become aggregated or solvated by urea and less accessible to reaction with IA-DTB upon denaturation. In contrast, cysteines with increased reactivity in the denatured mitotic proteome were much less likely than unchanging cysteines to reside in predicted disordered regions48 (orange, Extended Data Fig. 2g), possibly indicating that these residues are buried in structured regions of proteins and consequently shielded from IA-DTB labeling until denaturation, which exposes them to reaction with the IA-DTB probe. We believe that these data, taken together, support that our LPP treatment protocol achieves global dephosphorylation while preserving most proteins in their folded state, allowing us to profile the effects of S/T-phosphorylation on cysteine reactivity in native proteomes.
Most of the cysteine reactivity changes caused by LPP treatment were also observed as reactivity differences between mitotic and asynchronous cells (Fig. 2e, f), consistent with the much greater overall S/T phosphorylation in the mitotic cell state (Fig. 2a, b). The vast majority of LPP-induced changes in cysteine reactivity were site-specific, in that other quantified cysteines within the same protein were unchanged (Fig. 2g). As one example, we show the cysteine reactivity profile for the actin-binding protein filamin B (FLNB), in which a single cysteine C991 was substantially increased in reactivity in the mitotic proteome compared to the asynchronous proteome, as well as in the LPP(−) versus LPP(+) mitotic proteomes, whereas the many other quantified cysteines in this protein were unaltered across the comparison groups (Fig. 2h). As a converse case study, a site-specific reduction in reactivity was observed for C481 in the E3 SUMO-ligase PIAS1 in the mitotic/LPP(−) proteome compared to the asynchronous or LPP(+) proteomes (Fig. 2i). The directionality of change for these examples is interpreted to reflect phosphorylation-dependent increases (e.g., FLNB C991) or decreases (PIAS1 C481) in cysteine reactivity in the mitotic cell proteome.
While many of the differences in cysteine reactivity between LPP(−) and LPP(+) proteomes directionally aligned with the changes observed in mitotic versus asynchronous cells (Fig. 2e), there were instances of anti-correlated cysteines reactivity changes, such as C246 in the stress-activated protein kinase MAP2K4, which displayed elevated reactivity in the asynchronous compared to mitotic proteome, but decreased cysteine reactivity in the LPP(+) versus LPP(−) mitotic proteome (Fig. 2j). Other cysteines in MAP2K4 were generally unchanged across the comparison groups (Fig. 2j), indicating bidirectional, site-specific effects of phosphorylation and the mitotic cell state on C246 reactivity. In contemplating potential mechanisms for the observed reactivity profile of MAP2K4 C246, we noted that this residue is located in the ATP-binding pocket of the kinase49 (Fig. 2k) and that early mitotic cells have been reported to display elevated ATP content50, 51. We furthermore found that gel filtration to remove small-molecule metabolites resulted in a site-selective increase in MAP2K4 C246 reactivity in mitotic, but not asynchronous cell proteomes (Extended Data Fig. 2h, i). From these data, we propose a model in which MAP2K4 C246 reactivity is suppressed in mitotic cells by greater occupancy of the kinase active site by ATP (or ADP), and mitotic S/T-phosphorylation may then serve to counteract ATP/ADP binding to MAP2K4 (by, for instance, lowering the affinity of this interaction) such that LPP-treatment leads to even greater ATP/ADP engagement, further decreasing MAP2K4 C246 reactivity. We also note that MAP2K4 has been shown to display an unusually high affinity for ATP among protein kinases (Kd = 1.5 μM)52, indicating that it could be particularly susceptible to high-occupancy nucleotide binding based on cell state-dependent changes in ATP/ADP concentration, though we cannot rule out that other metabolite interactions may also contribute to the altered MAP2K4 C246 reactivity in mitotic proteomes. Interestingly, the high-stoichiometry mitotic phosphorylation sites found in MAP2K4 – S26, S90 and S394 – differ from the canonical stress-activated phosphorylation sites (S257 and T261)21, 53. Consistent with a model where mitosis produces functionally distinct proteoforms of MAP2K4, we found that UV irradiation-induced phosphorylation and activation of MAP2K4 were disrupted in mitotic, but not asynchronous HeLa cells (Fig. 2l). Finally, we noted that active-site proximal cysteines in other MAP2K proteins were also altered in mitotic and/or LPP-treated cell proteomes (Extended Data Fig. 2i), suggesting a more generalized impact of S/T-phosphorylation on altering the reactivity of ATP pocket cysteines in this kinase family.
Interpreting proximal phosphorylation-cysteine interactions
In analyzing the cysteine reactivity changes caused by LPP treatment in mitotic proteomes, we noted a strong enrichment in S/T-P sequences on tryptic peptides containing cysteines with apparent increases in reactivity following exposure to LPP (Fig. 3a, b). This outcome pointed to a potential technical problem with the original protocol for cysteine reactivity profiling because high-stoichiometry phosphorylation of a S/T residue on the same tryptic peptide as the quantified cysteine would produce a loss in MS-based proteomic signal that may be unrelated to a change in cysteine reactivity (Fig. 3c, top). We therefore sought to adapt the TMT-ABPP method to distinguish authentic versus artifactual changes in cysteine reactivity caused by proximal S/T phosphorylation events (Fig. 3c). We initially considered directly quantifying the phosphorylated peptides in our TMT-ABPP data sets, but very few peptides bearing both differential modifications (S/T phosphorylation and IA-DTB-labeled cysteines) were quantified in the search outputs (Supplementary Dataset 1), consistent with the low overall coverage of phosphorylated peptides in proteomic experiments that lack a specific enrichment step54. We therefore instead established an adapted TMT-ABPP protocol that identifies artifactual differences in cysteine reactivity by removing proximal S/T phosphorylation after IA-DTB probe labeling (Fig. 3c, bottom). Importantly, treatment with LPP after probe labeling does not affect IA-DTB-cysteine interactions, since they represent irreversible adducts. Specifically, mitotic cell proteomes were exposed to LPP or control buffer (LPP(+) or LPP(−)), followed by IA-DTB treatment and streptavidin-mediated enrichment of IA-DTB-labeled proteins; then, both samples were once again dephosphorylated with LPP (Fig. 3c, d), and proteins digested with trypsin and processed as normal for MS analysis (Fig. 3c). We reasoned that, in this adapted protocol, authentic cysteine reactivity changes would be preserved following the second LPP treatment (Fig. 3c, red) and thus distinguished from artifactual cysteine reactivity changes (Fig. 3c, yellow), which would be removed by the second LPP treatment. We refer hereafter to the four different proteomic groups based on their LPP treatment (− or +) relative to IA-DTB treatment (before, after) as: LPP(−, −), LPP(+, −), LPP(−, +), LPP(+, +), with the second position discerning the original protocol (LPP(−, −) and LPP(+, −)) from the adapted protocol (LPP(−, +) and LPP(+, +)).
Figure 3. Adapted protocol for interpreting proximal phosphorylation-cysteine interactions.
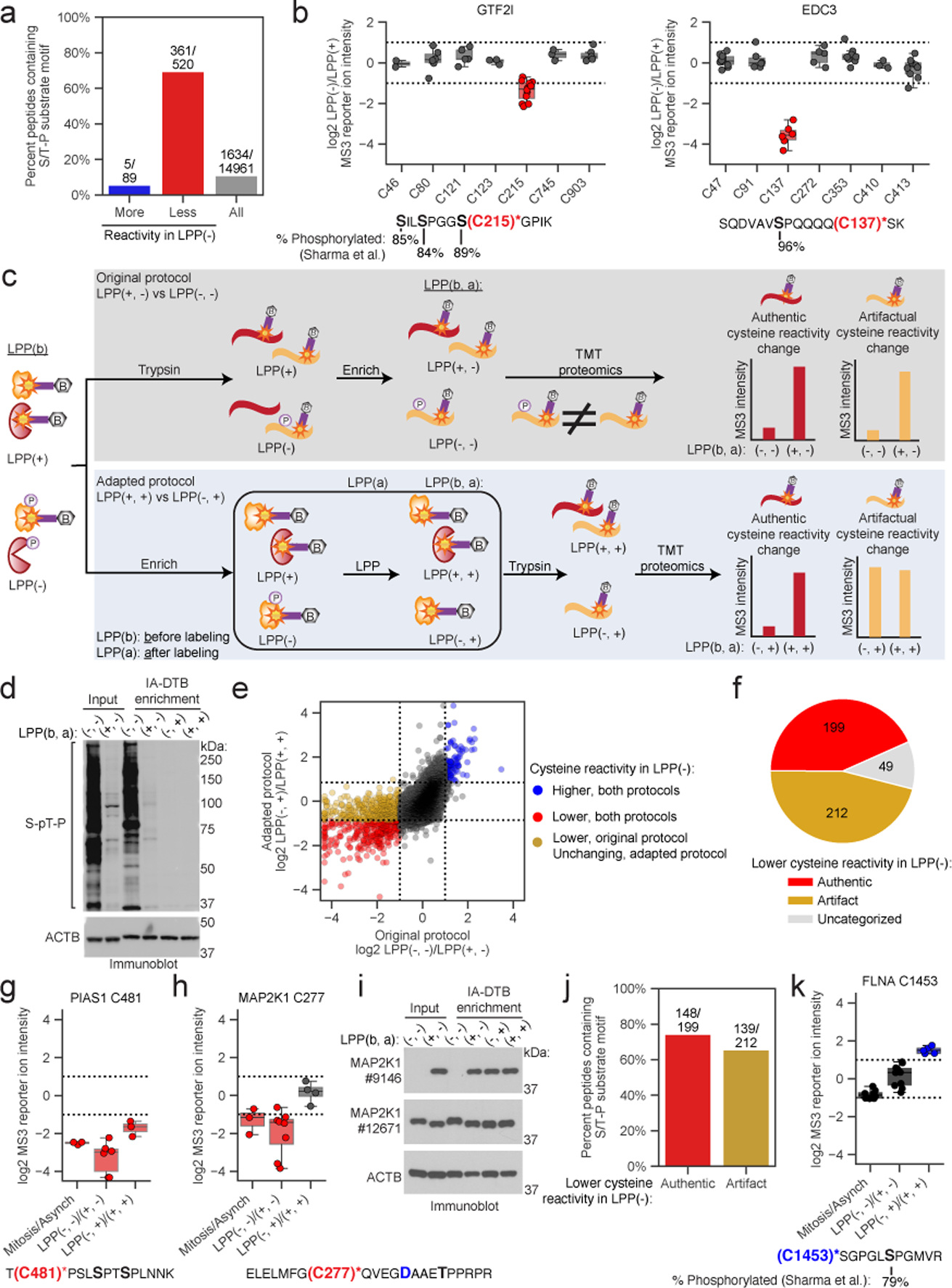
a, Fraction of tryptic peptides with a S/T-P motif for cysteines showing ≥ two-fold reductions (red) and increases (blue) in reactivity, and all quantified cysteines (gray) in Mitotic LPP(−)/LPP(+) proteomes. b, Top: cysteine reactivity values of Mitotic LPP(−)/LPP(+)proteomes for GTF2I (left) and EDC3 (right). Horizontal black lines mark median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments. Dotted lines designate boundaries for ≥ two-fold changes. Bottom: tryptic peptides containing the LPP-dependent cysteine reactivity changes (red) and high occupancy phosphorylation sites (black, bold)21. c, Original (top, gray) and adapted (bottom, blue) TMT-ABPP workflow for measuring phosphorylation-dependent changes in cysteine reactivity. d, Immunoblot confirming LPP-mediated dephosphorylation of the mitotic proteome after IA-DTB treatment and streptavidin enrichment. The data are from a single experiment representative of three independent experiments. e, Comparison of cysteine reactivity values for original (x-axis, LPP(−, −)/LPP(+, −)) and adapted (y-axis, LPP(−, +)/LPP(+, +)) TMT-ABPP protocols. Blue and red data points mark cysteine reactivity values that are higher or lower, respectively, in the LPP(−, ) groups using either protocol (≥ two-fold in the original protocol and ≥ 1.8-fold in adapted protocol, boundaries marked by dotted lines). Yellow data points mark cysteine reactivity values that are ≥ two-fold lower in the LPP(−, −) group of the original protocol, but unchanged in LPP(−, +) group of the adapted protocol. Each data point is the median value for n = 2 (or more) independent LPP(−, −)/LPP(+, −) experiments (original) and n = 1 (or more) LPP(−, +)/LPP(+, +) experiments (adapted). f, Pie chart showing distribution of LPP-dependent authentic and artifactual cysteine reactivity changes. g, h, Top, cysteine reactivity values for indicated comparison groups for PIAS1 C481 (g) and MAP2K1 C277 (h). Horizontal black lines mark median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 3 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. Bottom, tryptic peptides containing PIAS1 C481 (g) and MAP2K1 C277 (h) (red) and potential proximal S/T-P sites (black, bold). i, Immunoblot with antibody #9146 (Cell Signaling), which recognizes D282 in MAP2K1 (panel h; blue, bold). The data are from a single experiment representative of two independent experiments. j, Fraction of parent tryptic peptides with S/T-P motifs for cysteines showing authentic (red) and artifactual (yellow) reactivity changes. k, Top, reactivity values for FLNA C1453. Horizontal black lines mark median values, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 5 (or more) independent experiments. Dotted lines designate boundaries for ≥ two-fold changes. Bottom, tryptic peptide containing FLNA C1453 (blue) and FLNA S1459 (black, bold).
The cysteine reactivity profiles were generally similar between the original and adapted TMT-ABPP protocols, as reflected in the reactivity ratios for LPP(−, −)/(+, −) (original) versus LPP(−, +)/(+, +) (adapted) treatment groups (Fig. 3e). The most striking pattern of change was found for cysteines exhibiting LPP-induced increases in apparent reactivity using the original protocol (i.e., high LPP(−, −)/(+, −) ratios), which split into two nearly equal subsets displaying either preservation (red dots, Fig. 3e) or loss (yellow dots, Fig. 3e) of this increased reactivity as reflected in their LPP(−, +)/(+, +) ratios using the adapted protocol (Fig. 3f). We interpret the preservation of LPP-dependent increases in reactivity using the adapted protocol, as exemplified by PIAS1 C481 (Fig. 3g), as indicative of authentic decreases in cysteine reactivity caused by S/T phosphorylation. In contrast, cysteines that no longer showed a change in reactivity using the adapted protocol, as exemplified by MAP2K1 C277, were considered artifactual events presumed to be associated with high stoichiometry S/T-phosphorylation sites on the same tryptic peptide as the quantified (and unchanging) cysteine reactivity (e.g., MAP2K1 T286, Fig. 3h; also see Extended Data Fig. 3a, b for other examples where the proximal S/T-phosphorylation sites were directly mapped by phosphoproteomics). We found that immunoblotting of IA-DTB-enriched proteins with a monoclonal antibody #9146 centered on MAP2K1 D282 mirrored our chemical proteomic results, showing suppression of MAP2K1 signal in the LPP(−, −) group, that is recovered in the other three groups exposed to LPP (Fig. 3i), consistent with loss of epitope binding due to T286 phosphorylation during mitosis (also see Extended Data Fig. 3c). Strikingly, immunoblotting with monoclonal antibody #12671, which centers on a different region of MAP2K1, confirmed expression of MAP2K1 in LPP(−, −) groups and even supported high stoichiometry phosphorylation through a band shift that is lost upon LPP treatment (Fig. 3i, and Extended Data Fig. 3c).
Importantly, we found that not only artifactual, but also authentic changes in reactivity were frequently associated with cysteines on the same tryptic peptide as S/T phosphorylation sites (Fig. 3j), pointing to the potential for proximal phosphorylation events to perturb the nucleophilicity of neighboring cysteines. PIAS1 C481, for instance, is found on a tryptic peptide with two S/P sequences, the phosphorylation of which may account for the decreased reactivity of C481 in mitotic cells (Fig. 3g). Even more surprising, using the adapted protocol we also discovered masked phosphorylation-induced increases in cysteine reactivity, such as FLNA C1453, which resides on a tryptic peptide along with S1459, a high stoichiometry mitotic phosphorylation site (Fig. 3k and Extended Data Fig. 3d). The profile of FLNA C1453 across different conditions underscored how its reactivity change would have been overlooked without the adapted protocol. For instance, C1453 shows an apparent decrease in reactivity in mitotic versus asynchronous cells and unchanged reactivity using the original LPP protocol. The adapted protocol clarifies this type of unexpected profile as reflecting phosphorylation-induced increases in cysteine reactivity (C1453 in FLNA; or see Extended Data Fig. 3e, f for SLAIN2 C152 as another example) counterbalanced by a proximal mitotic phosphorylation event on the same tryptic peptide (S1459 in FLNA; see Supplementary Dataset 1) that prevents visualization of heightened cysteine reactivity until the proteome is exposed to LPP both before and after treatment with IA-DTB.
Features of proteins with cysteine reactivity changes
Phosphorylation-dependent cysteine reactivity changes were found to be enriched on proteins with high stoichiometry phosphorylation events mapped previously in phosphoproteomic studies of mitotic cells21 (Fig. 4a and Extended Data Fig. 4a), as well as on proteins with LPP-sensitive phosphorylation sites identified in this study (Extended Data Fig. 4b). These relationships support a model where the observed cysteine reactivity changes are often caused by phosphorylation events on the same parent protein (versus being indirectly modulated by phosphorylation events on other proteins). Cysteines with phosphorylation-dependent changes in reactivity, as well as those changing in reactivity in mitotic versus asynchronous cells, were found on a diverse array of cell cycle-related proteins55 (Fig. 4b, and Extended Data Fig. 4c), and GO analysis45, 46 revealed that these proteins were also enriched in cellular processes related to mitosis, such as microtubule formation and chromosome condensation, even after removing artifactual cysteine reactivity changes caused by proximal phosphorylation events on the same tryptic peptide (Fig. 4c).
Figure 4. Features of proteins with mitotic phosphorylation-dependent changes in cysteine reactivity.
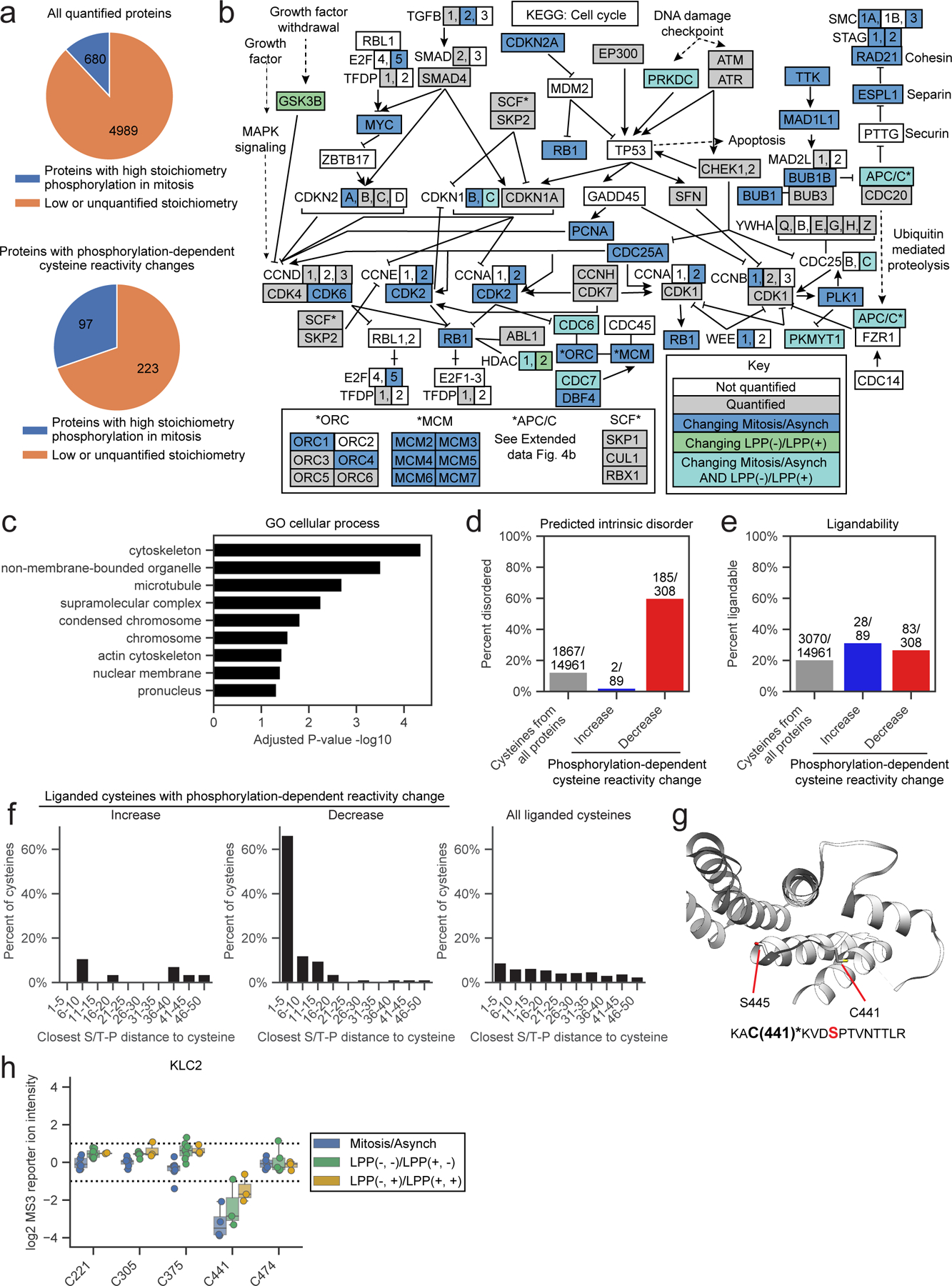
a, Proteins with phosphorylation-dependent cysteine reactivity changes (bottom) are enriched for high stoichiometry mitotic phosphorylation sites21 (> 50% occupancy; blue) compared to all quantified proteins (top). Proteins that only contained artifactual phosphorylation-dependent cysteine reactivity changes were excluded from the bottom pie chart. Proteins lacking sufficient data for phosphorylation stoichiometry calculation or exhibiting low stoichiometry (< 50% occupancy) sites in phosphoproteomics datasets21 were grouped and labeled as ‘Low or unquantified stoichiometry’ (orange). b, KEGG cell cycle pathway (HSA04110) diagram55 marking proteins with i) cell state- and phosphorylation-dependent cysteine reactivity changes in light blue; ii) only cell state-dependent cysteine reactivity changes in dark blue, iii) only phosphorylation-dependent cysteine reactivity changes, and iv) unchanging cysteine reactivities in gray. Proteins not quantified in our proteomic experiments are in white. c, GO cellular enrichment analysis of proteins with cell state- and phosphorylation-dependent cysteine reactivity changes.45, 46 Proteins with only artifactual phosphorylation-dependent cysteine reactivity changes were omitted from analysis. d, Percentage of cysteines from the indicated categories that reside in predicted disordered domains (IUPreD score > 0.5) based on IUPreD2A analysis.48 e, Percentage of cysteines from the indicated categories that are liganded by cysteine-reactive electrophilic small molecules; >80% engagement, as determined in references.39, 40, 42 f, Fraction of ligandable cysteines from the indicated categories showing phosphorylation-dependent reactivity changes within the specified amino acid distances from an S/T-P site. For d-f, artifactual phosphorylation-dependent cysteine reactivity changes were omitted from analysis. g, X-ray crystal structure of KLC2 (PDB: 3EDT)57 with the phosphorylation-dependent cysteine reactivity change – C441 – and the proximal serine in an S/T-P site – S445 – highlighted. Bottom, tryptic peptide from KLC2 containing C441 (asterisks, bold) and S445 (red, bold). h, Box plot showing cysteine reactivity values across indicated comparison groups for quantified cysteines in KLC2. Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes.
High stoichiometry phosphorylation events have been shown to frequently occur at intrinsically disordered regions of proteins21, 56. Consistent with this past work, we observed that >50% of cysteines with authentic phosphorylation-dependent decreases in reactivity were located in predicted disordered regions48, which was a much greater fraction than all cysteines quantified in our chemical proteomic experiments (< 20% in disordered regions; Fig. 4d). This finding also helps to contextualize the previously noted anti-correlation between LPP- and denaturation-dependent cysteine reactivity changes (Extended Data Fig. 2c), indicating that cysteines in predicted disordered regions are more likely to show both phosphorylation- and denaturation-induced decreases in reactivity (Extended Data Fig. 2g). Interestingly, only ~5% of cysteines showing phosphorylation-dependent increases in reactivity were found in disordered regions (Fig. 4d), possibly suggesting that phosphorylation is more likely to enhance the nucleophilicity of cysteines that are within structured regions of proteins (e.g., see aforementioned active site-proximal cysteines in MAP2Ks as examples).
To set the stage for future studies where the phosphorylation-dependent changes in cysteine reactivity discovered herein may serve as starting points for proteoform-restricted chemical probe development, we note that a substantial fraction of cysteines with reactivity changes have shown evidence of ligandability as determined in previous chemical proteomic studies mapping cysteine interactions with electrophilic small molecules39, 40, 42 (Fig. 4e). Cysteines were defined as ligandable in these past studies based on substantial engagement (> 80%) by fragment or elaborated electrophilic small molecules, and we observed a similar fraction of ligandable cysteines across all three categories of phosphorylation-dependent effects (Fig. 4e). Interestingly, a strong enrichment of S/T-P sequences was found to surround ligandable cysteines that showed phosphorylation-dependent decreases in reactivity (Fig. 4f), suggesting that many of these effects are mediated by local changes in structure or formation of protein-protein interactions that are caused by proximal phosphorylation events. No such enrichment was observed for ligandable cysteines showing either phosphorylation-dependent increases in reactivity or phosphorylation-independent reactivity in the mitotic proteome (Fig. 4f). A similar profile was also observed for all cysteines showing authentic phosphorylation-dependent changes in reactivity (Extended Data Fig. 4d). Many other cysteines, however, were unaffected in reactivity by proximal phosphorylation events, as exemplified by the artifactual category uncovered using the adapted protocol (Extended Data Fig. 4e), indicating that the potential impact of phosphorylation events on cysteine reactivity cannot be simply predicted by sequence proximity relationships.
Very few of the phosphorylation-dependent decreases in reactivity for ligandable cysteines occurred in proteins (or regions of proteins) with three-dimensional structures. One exception was C441 in the kinesin light chain KLC2, which has been found to react with electrophilic fragments in human T cell proteomes39 and resides in close proximity to S-P site S445-P446 (Fig 4g). This CXXXSP sequence is conserved in other KLCs (e.g., KLC1; see Extended Data Fig. 4f) and is located at a junction region of the C-terminal TRP domain57 involved in binding cellular cargo for transport along microtubules58. Interestingly, ERK-mediated phosphorylation of S460 of KLC1 (corresponding to S445 of KLC2) increases affinity for the cargo protein calsytenin-159, which has been suggested to reflect a phosphorylation-induced change in TRP conformation57. In mitotic cells, we only observed phosphorylation-dependent decreases in reactivity for KLC2 C441, while KLC1 C456 was unchanged (Fig. 4h, and Extended Data Fig. 4g). These finding suggest that the C-terminal domains of KLC1 and KLC2 may be regulated by distinct kinases, and, consistent with a possible role for KLC2 in the cell cycle, this protein has been shown to bind to SMAD2 through its C-terminal domain during mitosis60. We also note the presence of additional phosphorylation-dependent changes in the reactivity of ligandable cysteines that were not obviously associated with proximal mitotic phosphorylation events, such as the decreases observed for the shared cysteine in the ATP-binding pocket of MAP2Ks (Extended Data Fig. 2j). Taken together, our findings point to cysteines in proteins that, due to mitotic phosphorylation-dependent alterations in their reactivity, might show distinct susceptibility to covalent modification in actively dividing cells.
Finally, while we focused our efforts on mapping phosphorylation-dependent changes in cysteine reactivity that occurred specifically in mitotic cells, we noted rare examples of LPP-sensitive cysteines showing equivalent reactivity in asynchronous and mitotic cell proteomes. One such instance was C270 on the RNA-binding protein FXR2, which interestingly also possessed an LPP-sensitive phosphorylation event on S450 that was similar in magnitude between asynchronous and mitotic cell proteomes (Extended Data Fig. 4h, i). Considering that asynchronous cells exhibit a much less extensively phosphorylated proteome compared to mitotic cells, these findings indicate the potential of our method to discover phosphorylation-dependent cysteine reactivity changes across diverse landscapes of global protein phosphorylation.
Discussion
The molecular characterization of proteoforms presents a major challenge for the field of proteomics, as these protein variants are often dynamically regulated and produced at low concentrations in cells6. Enrichment strategies to isolate specific post-translationally modified states of proteins (e.g., phosphopeptide enrichment) have proven effective at addressing, at least in part, this challenge14, but are still typically performed with bottom-up protocols, which are not well-suited to readout the effect of PTMs on many aspects of protein structure/function. Top-down proteomics, while generally limited at present to proteins of smaller size (< 50 kDa)8, 9, 61, has underscored the importance of PTM crosstalk in regulating protein structure and function62, 63. More generally, how individual proteoforms may differ in their chemical reactivity, a feature that impacts the potential for post-translational regulation32 and covalent engagement by small molecules42, remains poorly understood. Here, we have addressed this question by developing a chemical proteomic method to globally assess the impact of S/T phosphorylation on cysteine reactivity in biological systems.
Our method leverages the broad substrate scope of lambda protein phosphatase (LPP), which we found to globally strip proteomes of S/T phosphorylation events while maintaining proteins in a folded state, providing a convenient compare-and-contrast system for mapping phosphorylation-dependent changes in cysteine reactivity. Furthermore, by studying mitosis – a cell state rich in high stoichiometry S/T phosphorylation events – we increased the probability of identifying such cysteine reactivity changes. When considering extending our method to additional biological systems where fewer high stoichiometry phosphorylation events may exist, we note that phosphorylation-induced increases in cysteine reactivity should be less dependent on stoichiometry due to the technical ease in detecting gain-of-signal changes. Future studies could also map tyrosine phosphorylation effects on cysteine reactivity by enriching proteins with anti-phosphotyrosine antibodies prior to cysteine reactivity profiling or study the effect of other PTMs (e.g., N-linked/O-linked glycosylation, K-acetylation, K/R-methylation) on cysteine reactivity. Finally, LPP should find broader use as a tool to study phosphorylation effects on other properties of proteins, such as protein complexation state.64
One limitation of our method is that the LPP treatment occurs in vitro, and therefore some phosphorylation-dependent changes in cysteine reactivity may have been lost after cell lysis. To the extent that the phosphorylated proteome can be experimentally controlled in cells (e.g., by treatment with kinase or phosphatase inhibitors), in situ cysteine reactivity profiling using, for instance, caged electrophilic probes65, may provide a means to identify phosphorylation-dependent cysteine reactivity changes that are caused, for instance, by dynamic interactions or localization in the cell. As a bottom-up approach, our strategy also does not specifically assign the phosphorylation events responsible for causing changes in cysteine reactivity. Nonetheless, integrating our data with phosphoproteomic information suggests that the observed cysteine reactivity changes are frequently caused by phosphorylation events occurring on the same proteoform of a protein (versus indirectly caused by phosphorylation events on other interacting proteins). Future studies where candidate S/T sites are mutated to prevent (e.g., alanine mutants) or mimic (e.g., glutamate mutants) phosphorylation should allow for more conclusive assignment of phosphorylation events that cause cysteine reactivity changes. We additionally note the importance of relating phosphorylation-dependent changes in cysteine reactivity to potential effects on protein function, which could be explored in the future using cysteine-directed ligands and/or cysteine mutagenesis.
Some of the most striking phosphorylation-dependent changes in cysteine reactivity occurred on proteins with important roles in cell division. Filamin A (FLNA), for instance, which possesses two cysteines showing phosphorylation-dependent increases in reactivity (C1260, C1453; Extended Data Fig. 3d) – one of which (C1453) was only revealed using the adapted TMT-ABPP protocol – forms a complex with CDC25C to stimulate the activity of this phosphatase during mitosis66, and mutation of mitotic phosphorylation sites in FLNA, including S1459, impairs daughter cell separation67. SLAIN2, another protein found to possess a phosphorylation-induced increase in reactivity (C152) regulates processive microtubule depolymerization in interphase cells68. Other cysteine reactivity changes may point to alternative functions for proteins in mitosis, such as for MAP2K4, where high-stoichiometry mitotic phosphorylation events at sites different from those regulated by canonical stress-activation pathways may create proteoforms with altered cysteine reactivity (C246) and sensitivity to stressors (e.g., UV irradiation; see Fig. 2l).
Projecting forward, we believe that proteomic methods relating the dynamic PTM-modified states of proteins to changes in cysteine reactivity have the potential to identify opportunities for proteoform-selective chemical probe development. Considering that the reactivity of cysteines can influence their covalent ligandability42, an increase or decrease in cysteine reactivity associated with a specific phosphorylated proteoform of a protein may point to greater or lesser potential for targeting this cysteine with electrophilic small molecules. Phosphorylation may also affect reversible ligand binding to proteins, as recently shown for the HPK1 kinase69. We further speculate that the cysteine reactivity changes discovered herein may facilitate PTM crosstalk in cells, by, for instance, enabling phosphorylation events to influence the introduction of cysteine-related PTMs (e.g., nitrosylation, sulfenylation, electrophilic lipidation). Finally, we should point out that the IA-DTB probe used herein to map cysteine reactivity changes is a structurally simple compound and may overlook phosphorylation-dependent changes in the environment surrounding cysteines that could positively or negatively influence modification by more elaborated endogenous or exogenous small molecules. This concept could be explored in the future by using different types of cysteine-reactive probes or even probes targeting modified forms of cysteine (e.g., sulfenylation probes70). In this way, the impact of dynamic phosphorylation events on cysteine modification and ligandability can be fully realized to understand and control proteoform function.
Methods
Cell culture and cell synchronization
HeLa cells were obtained from ATCC and were maintained at 37 ˚C with 5% CO2. HeLa cells were cultured and synchronized in DMEM (Corning) supplemented with 10% fetal bovine serum (FBS, Omega Scientific), penicillin (100 U/mL), streptomycin (100 μg/mL) and L-glutamine (2 mM). Cells were synchronized by a thymidine-nocodazole block. 2.2–2.4e6 cells were plated in a 15-cm dish. The next day, the media was replaced with fresh media with or without thymidine (2 mM thymidine, 15–16 mL) for mitotic and asynchronous cells, respectively. After 24 hours, the cells were rinsed twice with warm DPBS and resuspended in fresh media (14 mL). After 3.5–4.5 hours, nocodazole (100 ng/mL) or DMSO was added to the cells (1:1000 v/v). After 13 hours, asynchronous cells were rinsed 1x with warm DPBS and resuspended in 13 mL of warm media. Mitotic cells were loosely attached after 13 hours. Media and floating cells were separated by centrifugation (400 x g, 3 min), and cells were rinsed 1x with warm DPBS, separated by centrifugation (400 x g, 3 min) and resuspended in 13 mL of warm media. After 30 minutes at 37 ˚C, cells were scraped on ice, rinsed 1x with ice cold DPBS with phosphatase inhibitors (1 mM beta-glycerophosphate, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate), then two plates of cells were transferred in 1 mL ice cold DPBS with phosphatase inhibitors to an Eppendorf tube. Cells were centrifuged (800 x g, 5 min), DPBS was aspirated and cell pellets were flash frozen in liquid nitrogen until further use.
Proteomics: Asynchronous vs. Mitosis whole proteome tandem mass tagging (TMT-exp)
TMT-exp – sample preparation
All comparison groups were run in either duplicate or triplicate. Cell pellets from 2 × 15-cm were resuspended in 700 μL of DPBS with EDTA-free protease inhibitors (Sigma) and PhosStop (Sigma). All samples were lysed using a Branson Ultrasonics Sonifier S-250A cell disruptor (~2 × 8 pulses, 35% duty cycle, output setting = 3.5). Following lysis, cell lysates were clarified by centrifugation by 9,000 x g for 10 min. Protein concentration was determined with the DC Protein Assay (Bio-Rad) and the absorbance was measured using a CLARIOstar microplate reader following manufacturer’s instructions. Protein concentration was diluted to 2 mg/mL in 100 μL. 400 μL of ice cold 4:1 MeOH:CHCl3 was added to each sample followed by an 200 μL of water. The mixture was vortexed and centrifuged (9,000 x g, 3 min) to afford a protein disc at the interface of CHCl3 and aqueous layers. Both layers were aspirated without perturbing the disk, which was gently rinsed 1x with cold methanol (300 μL) and then re-suspended in cold methanol (300 μL) by brief sonication. The proteins were pelleted (9,000 g, 5 min, 4°C), and the resulting pellets were allowed air dry.
TMT-exp – trypsin digestion and TMT labeling
Dried protein pellets were resuspended in 100 μL EPPS buffer (200 mM, pH 8) containing proteomics-grade urea (8M) and DTT (10 mM) by either vortexing or bath sonication (5 min). Samples were heated at 65°C for 15 min. Sample was cooled to room temp, iodoacetamide (10 μL, 500 mM solution in H2O) was added, and the samples were incubated at 37°C for 30 min with shaking. Samples were diluted with EPPS buffer (250 μL, 200 mM, pH 8) for a final urea concentration of 2M. Trypsin (8 μL, 0.25 μg/μL in trypsin buffer containing 25 mM CaCl2) was then added and the samples were incubated at 37°C with shaking overnight. Peptide concentrations from digests were determined by microBCA assay (Thermo Scientific).
TMT labeling and mass spectrometry were performed as previously described with the following changes.39 In brief, for each sample, 25 μg of peptides (in 35 μL of EPPS buffer) was transferred to a new Eppendorf tube, and diluted with 9 μL acetonitrile. TMT tags (Thermo Scientific) 5 μL/channel in dry CH3CN, 20 μg/μL) were added to the corresponding tubes and the reaction was allowed to proceed at room temperature for 1 h. Labeling reaction was quenched with 5 μL of 5% hydroxylamine for 15 minutes, followed by acidification by 5 μL formic acid. Samples were vortexed, spun down and combined in a low binding 1.5 mL Eppendorf tube, and vacuumed-centrifuged to dryness and the samples were stored at −80°C until further analysis.
TMT-exp – liquid chromatography-mass spectrometry (LC-MS) analysis
Samples were desalted and fractionated as previously described39. In brief, samples were re-suspended in buffer A (1 mL, 95% H2O, 5% CH3CN, 0.1% formic acid) by bath sonication (~5 min) and desalted by passing through Sep-Pak C18 cartridges (55–105 μm, Waters). After eluted peptides were dried by vacuum centrifugation, peptides were resuspended in buffer A (510 μL) by bath sonication and fractionated into a 96 deep-well plate using HPLC (Agilent) using a gradient between 10 mM sodium bicarbonate in water and 100% acetonitrile detailed in Vinogradova et al.39 Samples were combined and analyzed by liquid chromatography tandem mass-spectrometry using an Xcalibur v4.0 software on an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex), using the same capillary column, flow rate, and gradient as previously detailed.39 Methods of data acquisition are as previously described.39 In brief, data were acquired using an MS3-based TMT method with dynamic exclusion enabled, choosing the top ten precursors for MS2/MS3 analysis. MS2 analysis consisted of: quadrupole isolation of precursor ion followed by collision-induced dissociation (CID) in the ion trap. Following the acquisition of each MS2 spectrum, synchronous precursor selection (SPS) enabled the selection of up to 10 MS2 fragment ions for MS3 analysis. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap using charge state–dependent isolation windows. Raw files were uploaded to Integrated Proteomics Pipeline (IP2), whereupon MS2 and MS3 spectra were extracted and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release-2016_07).
Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146 Da). N-termini and lysine residues were also searched with a static modification corresponding to the TMT tag (+229.1629 Da). All other relevant settings are detailed in Vinogradova et al.39
TMT-exp – protein ratios calculation
At the individual TMT experiment level, filters were applied to remove low-quality peptides as previously performed with the following differences:39 removal of peptides with more than one internal missed cleavage sites with allowing for missed tryptic sites if they are immediately followed/preceded by another tryptic site (e.g. R.RXXRXR.X is not removed) and removal of peptides with low (5,000 x number of channels) sum of reporter ion intensities (3 channels/group), and peptides with high variation between both of the replicate channels for asynchronous or mitotic cells (coefficient of variance > 0.5). Ratios for (mitotic over asynchronous) for each peptide entry were calculated using the median reporter ion intensity of mitosis and asynchronous channels. The final protein ratio was the median of the ratios of all quantified peptides for a given protein. All ratios were log2 transformed for presentation. Proteins were required to have at least 2 unique quantified peptides in an independent experiment for quantification. Quantification in at least one experimental replicate was required for interpretation.
Proteomics: Asynchronous vs. Mitosis Cysteine Reactivity Profiling (TMT-ABPP)
TMT-ABPP – sample preparation
All comparison groups were run in either duplicate or triplicate. Cell pellets from 2× 15-cm were resuspended in 700 μL of DPBS with EDTA-free protease inhibitors (Sigma) and PhosStop (Sigma). Samples were lysed, clarified, and protein concentration measured as described in TMT-exp – sample preparation. Protein concentration was diluted to 2 mg/mL in 500 μL, and samples were then treated with iodoacetamide polyethyleneoxide desthiobiotin (IA-DTB (Santa Cruz), 5 μL of 10 mM stock in DMSO, final concentration: 100 μM) or iodoacetamide alkyne (IA-alkyne, synthesized in house as previously described,31 5 μL of 10 mM stock in DMSO, final concentration: 100 μM), for 1 h at ambient temperature with end-over-end rotation. For samples treated with IA-alkyne, alkynylated proteins were then conjugated to Desthiobiotin-PEG3-Azide (N3-DTB BroadPharm) using copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC). Reagents for the CuAAC reaction were pre-mixed prior to their addition to the proteome samples: 100 μM N3-DTB (5 μL of 10 mM in DMSO), 100 μM tris(benzyltriazolylmethyl)amine ligand (TBTA; 30 μL of 1.7 mM stock in DMSO:t-butanol 1:4), 1 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP; 10 μL of fresh 50 mM stock in water), and 1 mM CuSO4 (10 μL of 50 mM stock in water) were combined, vortexed and added to the proteomes (55 μL/sample), vortexed, and incubated at RT for 1h. For all samples, 600 μL of ice-cold 4:1 MeOH:CHCl3 were then added, the mixture was vortexed and centrifuged (9,000 x g, 3 min) to afford a protein disc at the interface of CHCl3 and aqueous layers. Both layers were aspirated without perturbing the disk, which was gently rinsed 1x with cold methanol (1 mL) and aspirated. Pellets were frozen at −80 ˚C until the following day.
TMT-ABPP – trypsin digestion and streptavidin enrichment
Samples were re-suspended in cold methanol (300 μL) by sonication, centrifuged (10,000 x g, 10 min, 4°C), and the resulting pellets were allowed to air dry. Samples were then re-suspended in 90 μL of buffer containing 9M urea, 10 mM DTT in triethylammonium bicarbonate (TEAB) buffer (50 mM, pH 8.5) and cysteines were alkylated with iodoacetamide (10 μL, 500 mM solution in H2O) as previously described.39 Following alkylation, samples were diluted with 300 μL of triethylammonium bicarbonate buffer (50 mM, 1/20 dilution of 1.0 M stock, pH 8.5) and vortexed. If precipitate was present in samples, samples were probe sonicated until minimal precipitation (typically 10 pulses, 40% and output 4). Trypsin (8 μL of 0.25 μg/μL trypsin in trypsin buffer, containing 25 mM CaCl2) was then added and the digestion proceeded at 37°C overnight. The following day, samples were diluted with wash buffer (350 μL, 50 mM TEAB, 150 mM NaCl, 0.2% NP-40) containing streptavidin-agarose beads (40 μL/compact bead per sample) and the bead mixture was rotated for 2 h at RT or overnight at 4 ˚C. Beads were transferred to a BioSpin column affixed to a vacuum, and washed (2 × 1 mL wash buffer, 5 × 1 mL PBS, 5 × 1 mL H2O). Peptides were eluted into a Protein LoBind Eppendorf tube (VWR) by the addition of 300 μL of 50% aqueous acetonitrile containing 0.1% formic acid. The eluate was then evaporated to dryness using SpeedVac vacuum concentrator.
TMT-ABPP– TMT labeling
TMT labeling and mass spectrometry were performed as previously described.39 Briefly, peptides were re-suspended in 100 μL EPPS/CH3CN buffer (200 mM, pH 8.0, 30% CH3CN), TMT labeled (3 μL/channel in dry CH3CN, 20 μg/μL) for 1 h at room temperature. Reactions were quenched with 5% hydroxylamine (3 μL per sample), acidified with formic acid (5 μL), then combined and dried in a Protein LoBind Eppendorf tube (VWR). Samples were stored at −80°C until the high pH fractionation step. Samples were fractionated as described in TMT-exp – high pH offline fractionation.
TMT-ABPP – liquid chromatography-mass spectrometry (LC-MS) analysis
Peptides were analyzed by mass spectrometry and assigned as described in TMT-exp – liquid chromatography-mass-spectrometry (LC-MS) analysis, in addition to searching cysteine residues with a static modification for carboxyamidomethylation (+57.02146 Da) and N-termini and lysine residues with a static modification corresponding to the TMT tag (+229.1629 Da), cysteine residues were also searched with up to one differential modification for either the IA-DTB tag (+398.2529 Da) or IA-alkyne conjugated to N3-DTB (494.3217), depending on the probe used. If searching for phosphorylated peptides dually modified with probe, serine and threonine residues were also searched with a differential modification for phosphorylation (+79.9663 Da), with a total of two differential modifications allowed for quantification. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a spectrum false-positive rate below 1%. MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
TMT-ABPP – cysteine reactivity ratio calculation
At the individual TMT experiment level, the following filters were applied to remove low-quality peptides: removal of half-tryptic peptides with the exception those at the beginning and end of protein sequences, removal of peptides with more than one internal missed cleavage sites with allowing for missed tryptic sites if they are immediately followed/preceded by another tryptic site (i.e. R.RXXRXR.X is not removed), removal of peptides with low (< 5,000 x number of channels) sum of reporter ion intensities for channels from both groups, removal of peptides with high variation (coefficient of variance > 0.5) between the channels for both group (asynchronous or mitosis). Ratios (mitosis over asynchronous) for each peptide were calculated using the average reporter ion intensities of mitotic and asynchronous TMT channels and were capped at 20 or 0.05. Once the Mitosis/Asynch cysteine ratio values were calculated, overlapping peptides with the same modified cysteine (such as different charge states, tryptic termini, high pH fractionation fractions,) were grouped together, and the median ratio value was reported for a given TMT experiment. Representative median ratio values for a given peptide from each TMT experiment were grouped. If the median value was less than 1, all ratios for that given peptide were inverted for standard deviation and mean calculation only. The median value was reported unless the standard deviation was greater than 60% of the mean. For peptides quantified in fewer than 4 replicates, peptides that had standard deviations greater than 60% of the mean were not interpreted unless all ratios were considered changing (>1.5-fold difference). For peptides quantified in 4 or more replicates, peptides with standard deviations greater than 60% of the mean were not interpreted unless at least 80% of ratios were changing 1.5-fold. The overall median of all TMT replicates was reported alongside the shortest quantified tryptic peptide sequence. All ratios were log2 transformed for presentation. For interpretation in Fig. 1 and Extended Data Fig. 1, cysteine-containing peptides were required to be quantified in at least two replicates.
TMT-ABPP – categorization of cysteine reactivity changes
For Fig. 1d, categorization of cysteine changes were determined as previously described with minor changes.39 For proteins with 5 or more quantified peptides, cysteine changes were considered reactivity-based if i) its peptide ratio (Mitosis/Asynch) value differed more than two-fold from both the median ratio value of all quantified cysteines on the same protein and, if available, the protein expression ratio (Mitosis/Asynch) measured in TMT-exp experiments; and ii) the protein had at least one unchanging cysteine (<1.5-fold). Unchanging cysteines (<1.5-fold change) were considered to have a reactivity change if: i) it differed two-fold both the median ratio value of all quantified cysteines on the same proteins or protein expression level measured in TMT-exp experiments (if available) and ii) the median ratio value of all quantified cysteines was also changing at least two-fold. For proteins with 3–4 quantified peptides, cysteine changes were considered reactivity-based if i) its peptide ratio (Mitosis/Asynch) value differed more than two-fold from both the median ratio value of all quantified cysteines on the same protein or from the protein expression level measured in unenriched proteomics (if available), and ii) the parent protein had at least one unchanging cysteine (<1.5-fold). For proteins with 2 quantified peptides, a cysteine was considered to have a change in reactivity if its peptide ratio value differed more than two-fold from the protein expression level measured in unenriched proteomics or was 2x greater from the other peptide that was also required to be unchanging or changing in the opposite direction. If there was only 1 peptide quantified, it was required to be two-fold greater than the protein expression.
For proteins with 5 or more quantified peptides, a cysteine was considered to have a change in expression if the median ratio value (Mitosis/Asynch) of all quantified cysteines on the same protein was changing at least two-fold or if the protein expression level measured in TMT-exp experiments was also changing (if available). For proteins with 3–4 peptides, a cysteine was considered to have a change in expression if at least one cysteine was changing two-fold and either the median value of all its peptides was also 1.5-fold changing or the protein expression was changing 1.5-fold in the same direction. For proteins with 1–2 quantified peptides, a cysteine was considered to have a change in expression if its peptide ratio value changed more than two-fold and the corresponding protein expression changed at least 1.5-fold.
Proteomics: LPP(+) vs. LPP(−) (original LPP TMT-ABPP protocol)
Original LPP TMT-ABPP – sample preparation
All comparison groups were run in either duplicate or triplicate. Cell pellets from 2 × 15-cm mitotic and asynchronous cells were resuspended in 700 μL of lysis buffer (DPBS with EDTA-free protease inhibitors (Sigma)). Cell pellets were split in half, 10x PhosStop in DPBS (Sigma) was added to a final concentration 1x to all LPP(−) samples and the same volume of DPBS was added to all LPP(+) samples. Samples were lysed, clarified, and protein concentration measured as described in TMT-exp – sample preparation. Protein concentration was diluted to 2.2 mg/mL in 450 μL with lysis buffer with 1x PhosStop or lysis buffer for LPP(−) and LPP(+) samples, respectively. 50 μL of MnCl2 (10 mM, NEB) was added to a final concentration of 1 mM. Lambda phosphatase (NEB) or vehicle (50 mM HEPES pH 7.5, 100 mM NaCl, 2 mM DTT, 0.01 Brig 35, 0.1 mM EGTA, 0.1 mM MnCl2, 50% glycerol) was added to LPP(+) and LPP(−) samples, respectively. Samples were lightly flicked to mixed, then incubated prone at 30 ˚C for 45 minutes. Samples were allowed to cool to room temperature, then were treated with IA-DTB, followed by protein precipitation as described in TMT-ABPP – sample preparation.
Original LPP TMT-ABPP – liquid chromatography-mass spectrometry (LC-MS) analysis
Samples were processed according to the protocol described in Asynchronous vs. Mitosis TMT-ABPP trypsin digestion and streptavidin enrichment and Asynchronous vs. Mitosis TMT-ABPP tag labeling and TMT high pH offline fractionation. Data were processed according to the protocol described in TMT-ABPP cysteine reactivity ratio calculation for cell state-dependent reactivity dataset. Peptides with two-fold differences between LPP(+) and LPP(−) groups were considered as cell state and phosphorylation-dependent cysteine reactivity changes if they also had at least 1.6-fold change in reactivity in asynchronous vs mitotic groups.
Original LPP TMT-ABPP – categorization of cysteine reactivity changes
For proteins with 5 or more quantified peptides, a cysteine was considered to have a change in reactivity if its peptide ratio (LPP(−)/LPP(+)) value differed more than two-fold from the median ratio value of all quantified cysteines on the same protein and had at least one unchanging cysteine (<1.6-fold) or cysteine changing in the opposite direction. For proteins with 3–4 quantified peptides, a cysteine was considered to have a change in reactivity if its peptide ratio (LPP(−)/LPP(+)) value differed more than two-fold from the median ratio value of all quantified cysteines on the same protein and had at least one unchanging cysteine (<1.6-fold) or cysteine changing in the opposite direction. For proteins with 2 quantified peptides, a cysteine was considered to have a change in reactivity if its peptide ratio (LPP(−)/LPP(+)) value was two-fold greater from the other peptide that was also required to be unchanging or changing in the opposite direction.
Proteomics: Asynchronous, Mitosis LPP(+), and Mitosis LPP(−) TMT phosphopeptide enrichment
TMT phosphoproteomics – sample preparation
All comparison groups were run in duplicate. Cells were lysed, protein concentrations were determined, and appropriate samples were dephosphorylated as described in Original LPP TMT-ABPP – sample preparation. Proteins were then precipitated as described in Original LPP TMT-ABPP – sample preparation.
Whole protein expression TMT-exp was performed for all samples in parallel as described in TMT-exp – sample preparation, TMT-exp – trypsin digestion and TMT labeling, TMT-exp – high pH offline fractionation, TMT-exp – liquid chromatography-mass-spectrometry (LC-MS) analysis, and TMT-exp – protein ratios calculation.
TMT phosphoproteomics –phosphopeptide enrichment
Samples were processed and digested with trypsin according to the protocol described in Asynchronous vs. Mitosis TMT-ABPP trypsin digestion and streptavidin enrichment with the following changes. Instead of proceeding to streptavidin enrichment, samples were diluted with 400 μL of buffer A (1 mL, 95% H2O, 5% acetonitrile, 0.1% formic acid) by bath sonication (~5 min). Formic acid was added until a final pH of < 3. Some precipitate was present upon acidification. Samples were centrifuged and supernatant was desalted by passing through Sep-Pak C18 cartridges (55–105 μm, Waters), eluted and dried into a a Protein LoBind Eppendorf tube (VWR) as described in TMT-exp – high pH offline fractionation and liquid chromatography-mass spectrometry (LC-MS) analysis
Phosphopeptides were enriched using High-Select Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific) using manufacturer’s instructions. In brief, dried peptides were resuspended in Binding/Wash buffer (200 μL) by bath sonication for 5 min. Columns were equilibrated twice with Binding/Wash buffer (1,000 x g, 30 s). Columns were capped and suspended peptides were added to the column and gently flicked to mix. For a total of 30 min of incubation at RT, every 10 min, samples were gently mixed. Columns were washed (3x with wash buffer, 1x with water, 1,000 x g, 30 s) and samples were eluted into fresh eppindorf tubes with Elution buffer. Samples were immediately dried thoroughly by speed vacuum. Note that the elution buffer contains ammonium hydroxide, so samples were dried thoroughly.
TMT phosphoproteomics – TMT labeling and high pH offline fractionation
Peptides were TMT-labeled as described in TMT-exp – trypsin digestion and TMT labeling with the following exceptions. Peptides were re-suspended in 100 μL EPPS/CH3CN buffer (200 mM, pH 8.0, 30% CH3CN) by bath sonication, and TMT tags (5 μL/channel in dry CH3CN, 20 μg/μL) were added to the corresponding tubes and the reaction was allowed to proceed at room temperature for 60 min. The reaction was quenched by the addition of 5% hydroxylamine (5 μL per sample), acidified with formic acid (5 μL), then samples were combined and dried in a SpeedVac vacuum concentrator and kept at −80°C until the high pH fractionation step. Samples were fractionated as described in TMT-exp – high pH offline fractionation.
TMT phosphoproteomics – liquid chromatography-mass spectrometry (LC-MS) analysis
Peptides were analyzed by mass spectrometry and assigned as described in TMT-exp – liquid chromatography-mass-spectrometry (LC-MS) analysis, in addition to searching cysteine residues with a static modification for carboxyamidomethylation (+57.02146 Da) and N-termini and lysine residues with a static modification corresponding to the TMT tag (+229.1629 Da), serine and threonine residues were also searched with a differential modification for phosphorylation (+79.9663 Da), with a total of two differential modifications allowed for quantification. Peptides were required to be at least 6 amino acids long, and to have at least one tryptic terminus. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a spectrum false-positive rate below 1%. The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
Peptide ratios were calculated for Mitosis/Asynch and LPP(−)/LPP(+) as described in TMT-ABPP – cysteine reactivity ratio calculation. For interpretation in Fig. 2, Extended Data Figs. 3, 4, phosphosites were required to be quantified in one replicate. For bar plots in Extended Data Figs. 3, 4, ratios are normalized to Mitosis LPP(−).
Proteomics: Mitotic Native vs. Denatured Cysteine Reactivity Profiling (TMT-ABPP)
Denatured TMT-ABPP – sample preparation
All comparison groups were run in triplicate. Cell pellets from 1 × 15-cm mitotic and asynchronous cells were resuspended in 300 μL of lysis buffer (DPBS with EDTA-free protease inhibitors (Sigma)). Samples were lysed, clarified, and protein concentration was determined as described above. 1 mg of proteome was brought up in 320 μL of lysate. 240 mg of urea were added to samples to be denatured, and 180 μL of DPBS was added to samples preserved in native state (final volume = 500 μL). Denatured samples were vortexed to dissolve urea, then heated at 65 ˚C for 15 min. Native samples were kept on ice. All samples were then allowed to come to room temperature, then were treated with IA-DTB, followed by protein precipitation as described in TMT-ABPP – sample preparation. Denatured samples had fragile protein disks, or protein pellets at the bottom of the tube. Solvent was aspirated carefully, resuspended in 1 mL MeOH and vortexed. Proteins were pelleted (16,000 x g for 10 min at 4 ˚C) and MeOH was aspirated. Protein pellets were allowed to air dry.
Denatured TMT-ABPP – liquid chromatrograph-mass spectrometry (LC-MS) analysis
Samples were processed according to the protocol described in Asynchronous vs. Mitosis TMT-ABPP trypsin digestion and streptavidin enrichment and Asynchronous vs. Mitosis TMT-ABPP tag labeling and TMT high pH offline fractionation. Data were processed according to the protocol described in TMT-ABPP – cysteine reactivity ratio calculation. For interpretation in Figure S2, cysteine-containing peptides were required to be quantified in one replicate.
Proteomics: LPP(+, +), LPP(−, +), LPP(+, −), and LPP(−, −) (adapted LPP TMT-ABPP protocol)
Adapted LPP TMT-ABPP – sample preparation
Samples were processed as described in Original LPP TMT-ABPP – sample preparation.
Samples were then resolubilized as described in Original LPP TMT-ABPP – sample preparation, but also included the addition 25 μL SDS (10% in water). Samples were reduced and alkylated as described in Original LPP TMT-ABPP – sample preparation . Following the labeling with iodoacetamide, samples were diluted with 350 μL of wash buffer (50 mM TEAB, 150 mM NaCl, 0.1% NP-40). If precipitate was present in samples, samples were probe sonicated until minimal precipitation (typically 10 pulses, 35% and output 3). Samples were diluted with wash buffer (300 μL, 50 mM TEAB, 150 mM NaCl, 0.1% NP-40) containing streptavidin-agarose beads (20 μL/compact bead per sample) and the bead mixture was rotated for 2 h at RT or overnight at 4 ˚C. After incubation, the beads were pelleted by centrifugation (2,000 g, 1 min), and washed with wash buffer (2 × 0.75 mL 50 mM TEAB containing 1% NP40, 150 mM NaCl and 0.5% Triton X) and DPBS (2 × 0.75 mL). Samples were rinsed 1x with 1x PMP buffer (NEB, 100 μL) and resuspended in 100 μL of 1x PMP buffer containing 1 mM MnCl2. 2 μL of LPP or vehicle was added to LPP( , +) and LPP( , −) samples, respectively. Samples were flicked gently, taking care to ensure beads did not get stuck on the Eppendorf tube walls. Samples were laid flat at 30 ˚C for 1 hour, then washed with DPBS (2 × 1 mL) and resuspended in 150 μL of TEAB (50 mM). Trypsin (4 μL of 0.25 μg/μL trypsin in trypsin buffer, containing 25 mM CaCl2) was then added and the proteins were digested at 37°C overnight. The following day, samples were diluted with wash buffer (500 μL, 50 mM TEAB, 150 mM NaCl, 0.2% NP-40) transferred to a BioSpin column affixed to a vacuum. Streptavidin enrichments were washed and peptides eluted as described in TMT-ABPP – trypsin digestion and streptavidin enrichment. Samples were processed according to the protocol described in TMT-ABPP tag labeling, TMT liquid chromatography-mass spectrometry (LC-MS) analysis and TMT-ABPP – cysteine reactivity ratio calculation.
Adapted LPP TMT-ABPP – data filtering and processing
The adapted protocol processed 4x groups simultaneously, LPP(−, −), LPP(−, +), LPP(+, −) and LPP(+, +). While the LPP(−, +)/(+, +) ratio (LPP after IA-DTB labeling) can differ legitimately from the LPP(−)/(+) ratio of the original protocol (no LPP after IA-DTB labeling) (detailed above in Proteomics: LPP(+) and LPP(−) TMT-ABPP), the LPP(−, −)/(+, −) ratio (no LPP after IA-DTB labeling) should, in principle, be equivalent to the LPP(−)/LPP(+) ratios of the original protocol, with any differences pointing to technical or biological variation. If the LPP(−, −)/(+, −) ratio was not quantified or it varied 2x from original LPP(−)/(+) ratio, the LPP(−, +)/(+, +) ratio (LPP after IA-DTB labeling) was flagged for manual review. If a flagged peptide had an original LPP(−)/(+) ratio that was changing 2x but the LPP(−, −)/(+, −) ratio was not changing (<1.6x), or vice versa, the LPP(−, +)/(+, +) ratio was not interpreted. If there was only 1 TMT replicate for the LPP(−, +)/(+, +) ratio, the corresponding replicate for the LPP(−, −)/(+, −) ratio was required to be quantified and not vary more than 2x from the combined median of all LPP(−, −)/(+, −) and LPP(−)/LPP(+) replicates. All other flagged peptides were manually inspected.
Adapted LPP TMT-ABPP – categorization of cysteine reactivity changes
For cysteines that exhibited phosphorylation-dependent reduction in cysteine reactivity (two-fold), reactivity changes were categorized as follows: 1) authentic changes were defined as those that maintained at least a 1.8-fold decrease in reactivity in LPP(−, +)/(+, +) and 2) artifactual changes were defined as those that changed less than 1.6-fold change in reactivity in LPP(−, +)/(+, +).
Proteomics: Asynchronous vs. Mitosis gel filtration isoTOP-ABPP (isoTOP-ABPP)
isoTOP-ABPP – sample preparation
Cell pellets from 2 × 15-cm were resuspended in 500 μL of DPBS with EDTA-free protease inhibitors (Sigma). Samples were lysed and clarified as described in TMT-exp – sample preparation. Zeba Spin Desalting Column (7 K molecular weight cut-off, 2 mL, Thermo Scientific) were equilibrated 3x times DPBS according to manufacturer’s instruction (1,000 x g, 2 min). Samples were split in half and brought up to 400 μL each, and half were gel-filtered by passing through equilibrated Zeba columns by centrifugation (1,000 x g, 2 min). 10x PhosStop (Sigma) was added to both unfiltered and filtered samples to a final concentration of 1x. Sample protein concentration was determined followed by treatment with iodoacetamide alkyne as described in Original LPP TMT-ABPP – sample preparation. Modified proteins were then conjugated to isotopically labeled, TEV-cleavable biotin tags (“heavy” or “light” TEV-tags) using copper-catalyzed azide-alkyne cycloaddition reaction (CuAAC), precipitated with ice-cold methanol and chloroform, then resolubilized in 1.2% SDS in PBS (1 mL) in a 15-mL falcoln tube as detailed in Vinogradova et al.39 Streptavidin enrichment, tryptic and TEV digestion was performed as previously described.39 Samples were stored at −80°C prior to analysis.
isoTOP-ABPP – liquid-chromatography-mass spectrometry (LC-MS) analysis
Samples were analyzed by LC-MS/MS analysis and peptides were identified as previously described with minor changes.39 The MS2 spectra data were searched with Integrated Proteomics Pipeline (IP2), and searched using the ProLuCID algorithm (publicly available at http://fields.scripps.edu/downloads.php) using a reverse concatenated, non-redundant variant of the Human UniProt database (release-2016_07). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146) and up to one differential modification for either the light or heavy TEV tags (+464.28595 or +470.29976 respectively). ProLuCID data was filtered through DTASelect (version 2.0) to achieve a spectrum false-positive rate below 1%.
isoTOP-ABPP ratio value calculation and data processing
Ratios were calculated as described in Vinogradova et al. with minor changes. In brief, the heavy/light isoTOP-ABPP ratios for each unique peptide (Gel-filtered/Unfiltered) were quantified with in-house CIMAGE software32 with a maximal ratio of 20. At the individual isoTOP experiment level, the following filters were applied to remove low-quality peptides: removal of half-tryptic peptides with the exception those at the beginning and end of protein sequences, removal of peptides with more than one internal missed cleavage sites with allowing for missed tryptic sites if they are immediately followed/preceded by another tryptic site (i.e. R.RXXRXR.X is not removed), removal of peptides with R = 20 and only a single MS2 event triggered during the elution of the parent ion, removal of non-unique peptides. Overlapping peptides with the same modified cysteine were grouped together and the median ratio was reported as the final ratio. When aggregating data across experimental replicates, the mean of each experimental median ratio was reported. Peptides that had standard deviations greater than 60% of the mean were not interpreted unless all ratios were considered changing (>1.6-fold difference).
Gene Ontology (GO) Enrichment Analysis
For Fig. 4, cysteines reactivity changing two-fold in LPP(−, −) vs LPP(+, −) and 1.6-fold in mitosis versus asynchronous cell lysates were grouped. Of this group, cysteines with artifactual phosphorylation-dependent changes (two-fold reduction reactivity in LPP(−, −) over LPP(+, −) but less than 1.6-fold reduction in LPP(−, +) vs LPP(+, +)) were removed from analysis. The proteins containing cysteines that passed these filters were used for enrichment of GO cellular process terms. The background list represented proteins that had at least one cysteine that was quantified in at least two LPP(−)/(+) TMT-ABPP replicates (from either the original or adapted protocol).
For Extended Data Fig. 1, proteins with cysteine reactivity changes in mitotic versus asynchronous cell proteomes that were reactivity-based (as opposed to expression-based, as defined in TMT-ABPP – categorization of cysteine reactivity changes. The background list represented proteins that had at least one cysteine that was quantified in at least two Mitosis/Asynch TMT-ABPP replicates.
All GO analyses were performed with WebGestalt 2019 (http://www.webgestalt.org/) using the significance level (FDR < 0.05)45. The minimum and maximum number of genes for a given category was set at 5 and 2,000, respectively. Benjamini-Hochberg was used for q-value adjustment. Enriched terms were then passed through REVIGO (http://http://revigo.irb.hr/), which identifies redundant terms and chooses representative terms for each group46. The following settings were used with REVIGO: allowed similarity of 0.5, searched against the human Uniprot database, using the SimRel semantic similarity measure71.
Immunoblot analysis
Sample preparation for immunoblot analysis of cell states with LPP treatment
1.1 e6 HeLa cells were seeded in 10-cm dishes for asynchronous, G1/S and mitosis groups (Fig 2). G1/S groups were synchronized using a double thymidine block. 24 h after seeding, media was replaced with new median containing thymidine (2 mM). After 18 h, cells were washed 2x with warm DPBS and released into fresh media. After 9 h, media was replaced with media containing thymidine (2 mM) was reintroduced. 18 h later, cells were harvested and flash frozen in liquid nitrogen. Mitosis groups were synchronized using a thymidine-nocodazole block and asynchronous groups underwent washing and media changes at the same time as mitosis groups. 24 h after seeding, media was replaced with new media for asynchronous groups and media containing thymidine (2 mM) for mitosis groups. After 24 hours, cells were washed 2x with warm DPBS and released into fresh media. After 3.5 h, nocodazole (100 ng/uL) or DMSO was added to mitosis groups and asynchronous groups, respectively. 13 h later, cells were rinsed 1x with DPBS and released into fresh media. 30 min later, cells were harvested and flash frozen in liquid nitrogen.
Cell pellets were resuspended in 250 μL lysis buffer (DPBS with EDTA-free protease inhibitors (Sigma)). Cell pellets were split in half, 10x PhosStop in DPBS (Sigma) was added to a final concentration 1x to all LPP(−) samples and the same volume of DPBS was added to all LPP(+) samples. Samples were lysed and clarified as described in TMT-exp – sample preparation. Protein concentration was diluted to 1.3 mg/mL in 100 μL with lysis buffer with 1x PhosStop or lysis buffer for LPP(−) and LPP(+) samples, respectively. 10 μL of MnCl2 (10 mM, NEB) was added to all samples to a final concentration of 1 mM. 2 μL of LPP (NEB) or vehicle (50 mM HEPES pH 7.5, 100 mM NaCl, 2 mM DTT, 0.01 Brig 35, 0.1 mM EGTA, 0.1 mM MnCl2, 50% glycerol) was added to LPP(+) and LPP(−) samples, respectively. Samples were lightly flicked to mix, then incubated prone at 30 ˚C. After 30 min, 4x loading dye (Laemmli buffer: 250 mM Tris-HCl, pH 6.8, 10% glycerol, 8% SDS, ~5 mM bromophenol blue, 20% 2-mercapto-ethanol) was added to samples.
Sample preparation for immunoblot analysis of UV treatment in asynchronous vs mitotic cells
Cells were synchronized in mitosis as described in Sample preparation for immunoblot analysis of cell states with LPP treatment but directly after release from nocodazole, cells were then UV irradiated (60 J/m2) and incubated for 30 min. Cells were then harvested and flash frozen in liquid nitrogen. Cell pellets were resuspended in 250 μL lysis buffer with 1x PhosStop. Samples were processed for immunoblot analysis as described in Sample preparation for immunoblot analysis of cell states with LPP treatment.
Sample preparation for immunoblot validation of adapted LPP TMT-ABPP protocol
Cells were synchronized, lysed, and labeled with IA-DTB. Modified proteins were enriched and desphosphorylated as described in Adapted LPP TMT-ABPP – sample preparation, but sample preparation diverged after LPP dephosphorylation. After the second LPP treatment, samples were washed twice with DPBS and 60 μL of 4x loading dye was added to beads. Samples were briefly boiled and then stored at −20 ˚C.
Immunoblot analysis
Proteins were resolved by SDS-PAGE, transferred to 0.45 μM PVDF membranes (GE Healthcare) which were blocked with 5% milk in TBST buffer (20 mM Tris-HCl 7.6, 150 mM NaCl with 0.1% tween 20). Primary antibodies were used at the following concentrations: 1:10,000 anti-GAPDH (Santa Cruz, sc-47724), 1:10,000 anti-ACTB-HRP (Santa Cruz, sc-47778 HRP), 1:2,000 anti-phospho-JNK (Cell Signaling, #4668), 1:2,000 anti-JNK (Cell Signaling, #9252), 1:2,000 anti-phospho-p38 (Cell Signaling, #4511), 1:2,000 p38 (Cell signaling, #8690), 1:500 CCNB1 (Santa Cruz, sc-245), 1:500 CCNE1 (Santa Cruz, sc-247), 1:1,000 anti-SpTP (Cell Signaling, #5243), 1:1,000 anti-MAP2K4 (Cell Signaling, #9152), 1:1,000 anti-phospho-MAP2K4 (Cell Signaling, #9156), 1:500 anti-MAP2K1 (Cell Signaling, #9146), 1:500 anti-MAP2K1 (Cell Signaling, #12671). Blots were incubated with primary antibodies (with the exception of anti ACTB-HRP) in either 5% BSA in TBST or 5% milk in TBST at 4°C overnight. Following another TBST wash (3 times), membranes were incubated with secondary antibody (1:5000 in 5% milk in TBST) at 4°C overnight. Membranes were washed with TBST (3x times, 5 min), developed with ECL western blotting detection reagent kit (Thermo Scientific) and recorded on CL-XPosure film (Thermo Scientific). Blots were then stripped with stripping buffer (Thermo Scientific, PI21059) for 20 minutes, blocked in 5% milk, then incubated with anti-ACTB-HRP antibody for at least 1 h at RT. Membranes were washed with TBST (3x times, 5 min) and developed with ECL western blotting detection reagent kit (Thermo Scientific) and detected with Bio-Rad ChemiDoc XRS, or recorded on CL-XPosure film (Thermo Scientific) and detected with Konica Minolta SRX 101 x-ray film processor.
KEGG cell cycle pathway containing proteins with cysteines quantified by TMT-ABPP
The KEGG cell cycle pathway (hsa04110) in Fig. 4 was regenerated using gene names used in TMT-ABPP datasets and color coding based on cysteine reactivity55. Members of the KEGG cell cycle pathway that had quantified cysteines by TMT-ABPP in two replicates of either the mitosis vs asynchronous dataset or the LPP(−) vs LPP(+) dataset, were marked in gray, proteins with cysteine reactivity changes in LPP(−) vs LPP(+) (two-fold) were marked in green, in mitosis vs. asynchronous (two-fold) were marked in blue, and proteins with cysteine reactivity changes in both LPP(−) vs LPP(+) (two-fold) and mitosis vs. asynchronous (1.6-fold) were marked in light blue. Proteins not quantified were in white.
Analysis of high occupancy phospho-proteins
Proteins containing cysteines with cysteine reactivity changes in both LPP(−) vs LPP(+) (two-fold) and in mitosis vs. asynchronous (>1.6-fold) were cross-referenced to a list of proteins with high occupancy mitotic phosphorylation sites (≥50%) were taken from Sharma et al.21
Analysis of predicted disordered regions
For Fig. 4d, “Cysteines from all proteins” refers to all cysteines that were quantified in at least two replicates of the LPP(−) vs LPP(+) TMT-ABPP dataset. “Phosphorylation-dependent cysteine reactivity change” refers to cysteines changing at least two-fold with LPP treatment and changing at least 1.6-fold in mitosis vs. asynchronous with those that no longer had phosphorylation-dependent changes with LPP after IADTB labeling) removed. Cysteines labeled as “Increase” have two-fold more reactivity in LPP(−) and cysteines that are “Decrease” have two-fold more reactivity in LPP(+).
For Extended Data Fig. 2g, “Lower” and “Higher” refer to cysteines with two-fold more or less reactivity in native mitotic proteome relative to mitotic proteome denatured by high molarity urea. “Unchanging” refers to cysteines that are changing less than two-fold upon denaturation.
For all analyses, protein sequences for proteins containing said cysteines were obtained from the same FASTA database used for peptide assignment, namely a reverse concatenated, non-redundant variant of the Human UniProt database (release-2016_07). Protein disorder for the protein sequences was predicted using the tool IUPred2A (downloaded March 2020), which outputs a value from 0 to 1 for each residue, with higher values indicating higher likelihood of residing in a disordered region48. For these analyses, a cutoff of ≥0.5 was used to categorize likely disordered regions.
Analysis of ligandability
“Cysteines from all proteins,” “Increase” and “Decrease” in phosphorylation-dependent cysteine reactivity change are as described in Analysis of predicted disordered regions. Ligandability data were taken from Vinogradova et al. 2020, Bar-Peled et al 2017 and Backus et al. 201639, 40, 42. Cysteines were considered liganded if they were engaged at least 80% by the small molecule ligands listed in the indicated papers. Cysteines changing at least two-fold with LPP treatment and changing at least 1.6-fold in mitosis vs. asynchronous were grouped and those that no longer had phosphorylation-dependent changes with LPP after IADTB labeling were removed.
Analysis of [S/T]P phosphorylation
For Fig. 4f, “All liganded cysteines” are as described in Analysis of ligandability. Of these cysteines, cysteines were defined as “Increase” or “Decrease” in phosphorylation-dependent cysteine reactivity change based on the same criteria as for Fig. 4d in Analysis of predicted disordered.
For Extended Data Fig. 4d, categories are as defined in Analysis of predicted disordered regions. For Extended Data Fig. 4e, categories are as defined in Adapted LPP TMT-ABPP – Categorization of TMT-ABPP cysteine reactivity changes. In brief, authentic changes were defined as cysteines with at least two-fold decrease in LPP(−, −)/(+, −) reactivity that still had at least 1.8-fold decrease in reactivity in LPP(−, +)/(+, +). Artifactual changes were defined as those that had less than 1.6-fold decrease in reactivity in LPP(−, +)/(+, +). Protein sequences for proteins containing said cysteines were obtained from the same FASTA database used for peptide assignment, namely a reverse concatenated, non-redundant variant of the Human UniProt database (release-2016_07). Only the closest distance [S/T]P motif was quantified for each cysteine.
Phosphoproteomic comparison to literature resources
Phosphoproteomic data were taken from Sharma et al21. Only phosphoserines and phosphothreonines were considered for analysis, and phosphosites from Sharma et al. were required to have intensities in either mitotic or asynchronous samples. Uniprot accession IDs from Sharma et. al were mapped to IDs from a reverse concatenated, non-redundant variant of the Human UniProt database (release-2016_07) used for proteomic analysis in this paper. Sequence windows reported in Sharma et al. were mapped onto the respective protein sequence from the 2016 database. Phosphosites were considered if they were within a sequence of at least 15 consecutive amino acids (of the 31 amino acid window) that matched the 2016 database sequence. Phosphosite positions from Sharma et al. were remapped to the 2016 database and compared to phosphosites quantified in at least one replicate of either LPP(−)/LPP(+) or Mitosis/Asynch in this study.
Analysis of phospho-proteins with LPP-sensitive phosphosites
For Extended Data Fig. 4b, “All proteins with phosphorylation-dependent cysteine reactivity changes” refers to proteins containing cysteines with cysteine reactivity changes in both LPP(−) vs LPP(+) (two-fold, at least two replicates) and in mitosis vs. asynchronous (>1.6-fold, at least one replicate). Proteins with only artifactual cysteine reactivity changes were removed from analysis (two-fold reduced reactivity in LPP(−, −) relative to LPP(+, −) but changing less than 1.6x-fold in LPP(−, +) vs LPP(+, +)). “All proteins” are any proteins with at least one cysteine quantified in at least two replicates of LPP(−) vs LPP(+). Proteins were considered to have LPP-sensitive phosphosites if a given phosphosite had a two-fold change in enrichment between LPP(−) and LPP(+). Proteins with only LPP-insensitive (less than a two-fold change) or no quantified phosphosites were grouped and labeled as “Proteins with phosphosites not quantified or unchanging”.
Code availability
TMT-based data output from Integrated Proteomics Pipeline (IP2) and isoTOP data output from CIMAGE was further analyzed with custom scripts, available on Zenodo at https://zenodo.org/badge/latestdoi/419072418.
Data availability
All mass spectrometry data are available via PRIDE with identifier PXD026730.
Source data, in addition to Supplementary Dataset 1, is available for all figure panels.
Extended Data
Extended Data Fig. 1. Cysteine reactivity profiling of mitotic and asynchronous cells.
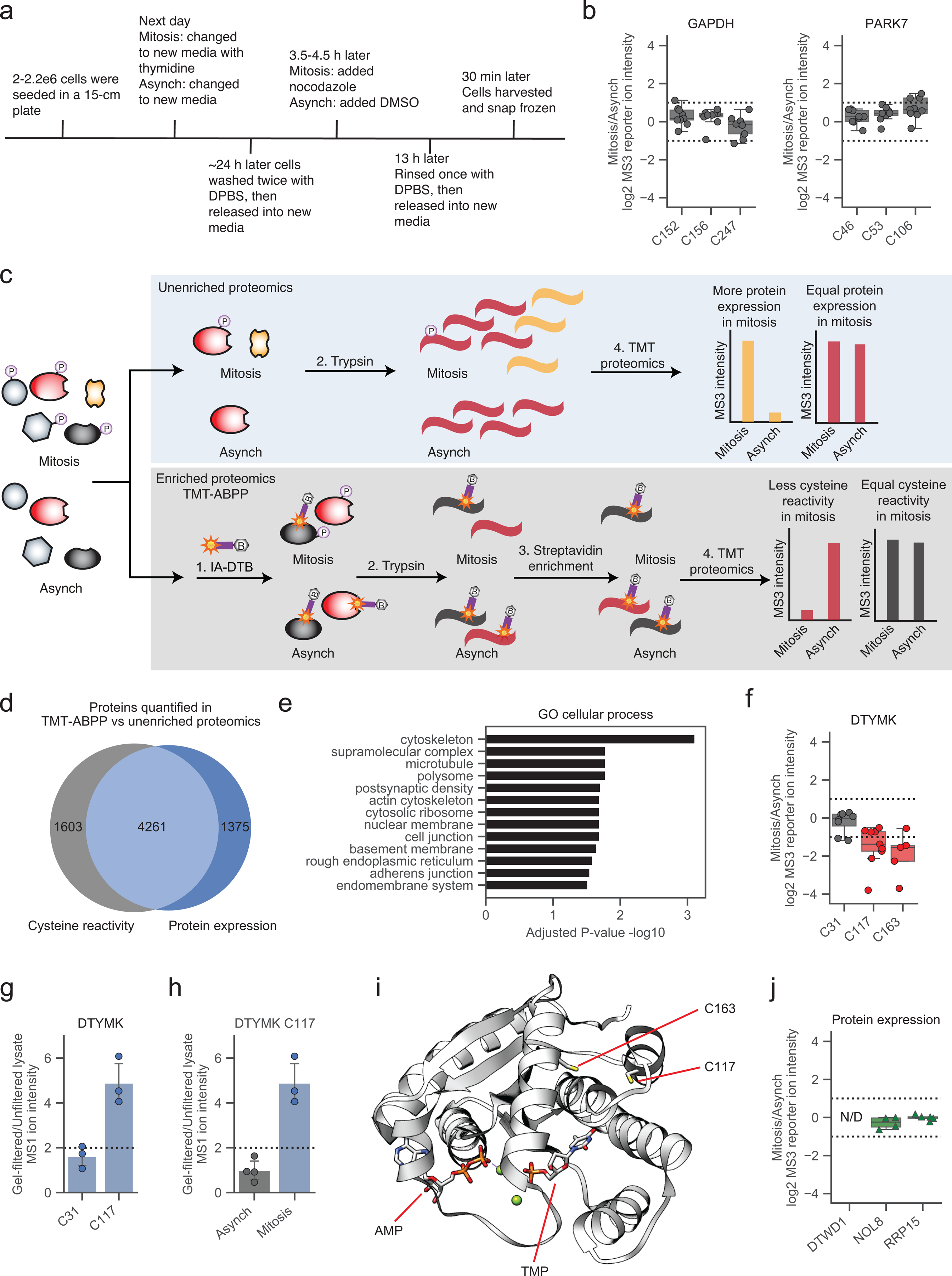
a, Timeline for mitotic HeLa cell proteome generation. b, Cysteine reactivity values for GAPDH and PARK7 (Mitosis/Asynch) cell proteomes. Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 9 independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. c, TMT-ABPP workflow for measuring protein expression (top, blue) and cysteine reactivity (bottom, gray) in the mitotic and asynchronous HeLa cell proteome. d, Venn diagram showing overlap (light blue) in proteins quantified by TMT-ABPP (gray) and unenriched proteomics (dark blue). For inclusion, proteins had at least one quantified cysteine in at least two replicate experiments of TMT-ABPP and/or two unique quantified peptides quantified from at least one replicate of unenriched proteomics. e, GO cellular analysis of proteins with reactivity-based cysteine changes in mitotic vs asynchronous cell proteomes45, 46. Proteins with reactivity-based cysteine changes correspond to those defined in Fig. 1d. f, Box plot showing DTYMK cysteine reactivity values (Mitosis/Asynch). Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 5 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. g, Cysteine reactivity values for all quantified DTYMK cysteines following gel filtration of the mitotic cell proteome. Data represent average values +/− standard deviation for n = 3 independent experiments (circles). h, DTYMK C117 reactivity following gel filtration of asynchronous (gray) vs mitotic (blue) cell proteomes. Data represent average values +/− standard deviation for n = 3 (or more) independent experiments (circles). i, X-ray crystal structure of DTYMK in complex with AMP and TMP with C163 and C117 highlighted in yellow (PDB: 1E2D)47. j, Protein expression values for DTWD1 (not detected), NOL8, and RRP15 (Mitosis/Asynch). Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 9 independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes.
Extended Data Fig. 2. A proteomic method to map phosphorylation-dependent changes in cysteine reactivity.
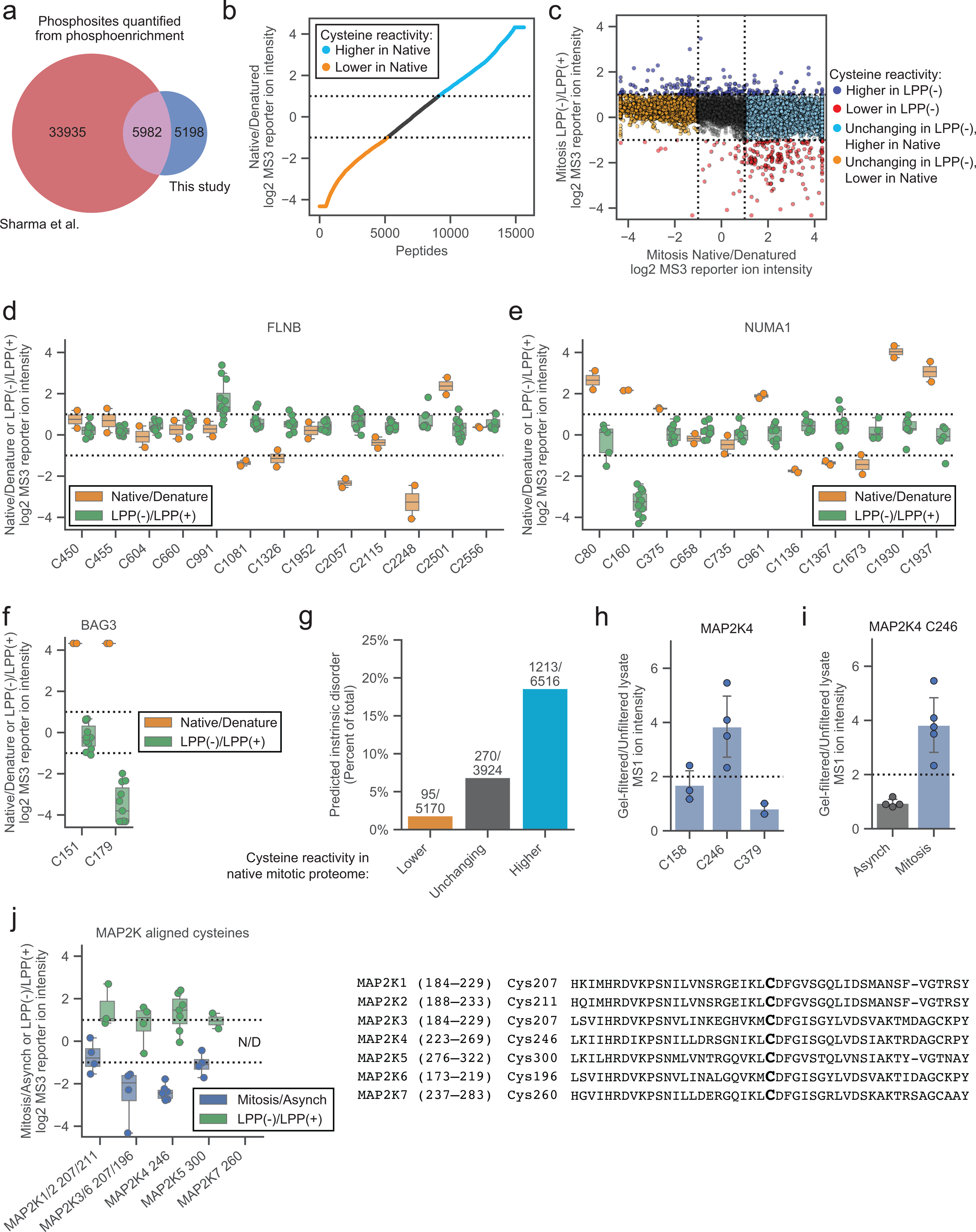
a, Venn diagram of phosphorylated S/T residues quantified by Sharma et al.21 (red) and this study (dark blue) in asynchronous and mitotic cell proteomes. b, Cysteine reactivity ratio values in Native/Denatured mitotic proteome. Light blue and orange data mark cysteine reactivity values that are ≥ two-fold higher (boundary marked by dotted lines) in native and denatured cell proteome, respectively. Data are the median value for n = 1 (or more) independent experiments. c, Comparison of cysteine reactivity values from LPP(−)/LPP(+) (y-axis) and Native/Denatured (x-axis) proteomes. Blue and red data mark cysteine reactivity values that are ≥ two-fold higher in LPP(−) and LPP(+) cell proteomes, respectively. Light blue and orange data mark cysteines that are unchanging in LPP(−)/LPP(+), but changing two-fold in Native/Denatured mitotic proteomes. Dotted lines mark boundaries for cysteines that change ≥ two-fold in reactivity in LPP(−)/LPP(+) and Native/Denatured. Data are the median value for n = 2 (or more) independent LPP(−)/LPP(+) experiments and n = 1 (or more) Native/Denatured experiments. d-f, Cysteine reactivity values across Native/Denatured proteome (orange) and LPP(−)/LPP(+) (green) mitotic proteome for cysteines in d) FLNB, e) NUMA1, and f) BAG3. Horizontal black lines mark median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. g, Percentage of cysteines in predicted disordered domains (IUPreD ≥ 0.5)48. h, Cysteine reactivity values for MAP2K4 in gel-filtred mitotic cell proteome. Data represent the average values +/− standard deviation for n = 2 (or more) independent experiments (circles). i, MAP2K4 C246 reactivity in gel-filtered asynchronous (gray) vs mitotic (blue) cell proteomes. Data represent the average values +/− standard deviation for n = 4 (or more) independent experiments (circles). j, Left, MAP2K ATP-binding pocket cysteine reactivity. Nonunique peptides are assigned to both MAP2Ks. Horizontal black lines mark median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. Right, sequence alignment of MAP2K proteins centered on MAP2K4 ATP-binding pocket C246.
Extended Data Figure 3. Adapted protocol for interpreting proximal phosphorylation-cysteine interactions.
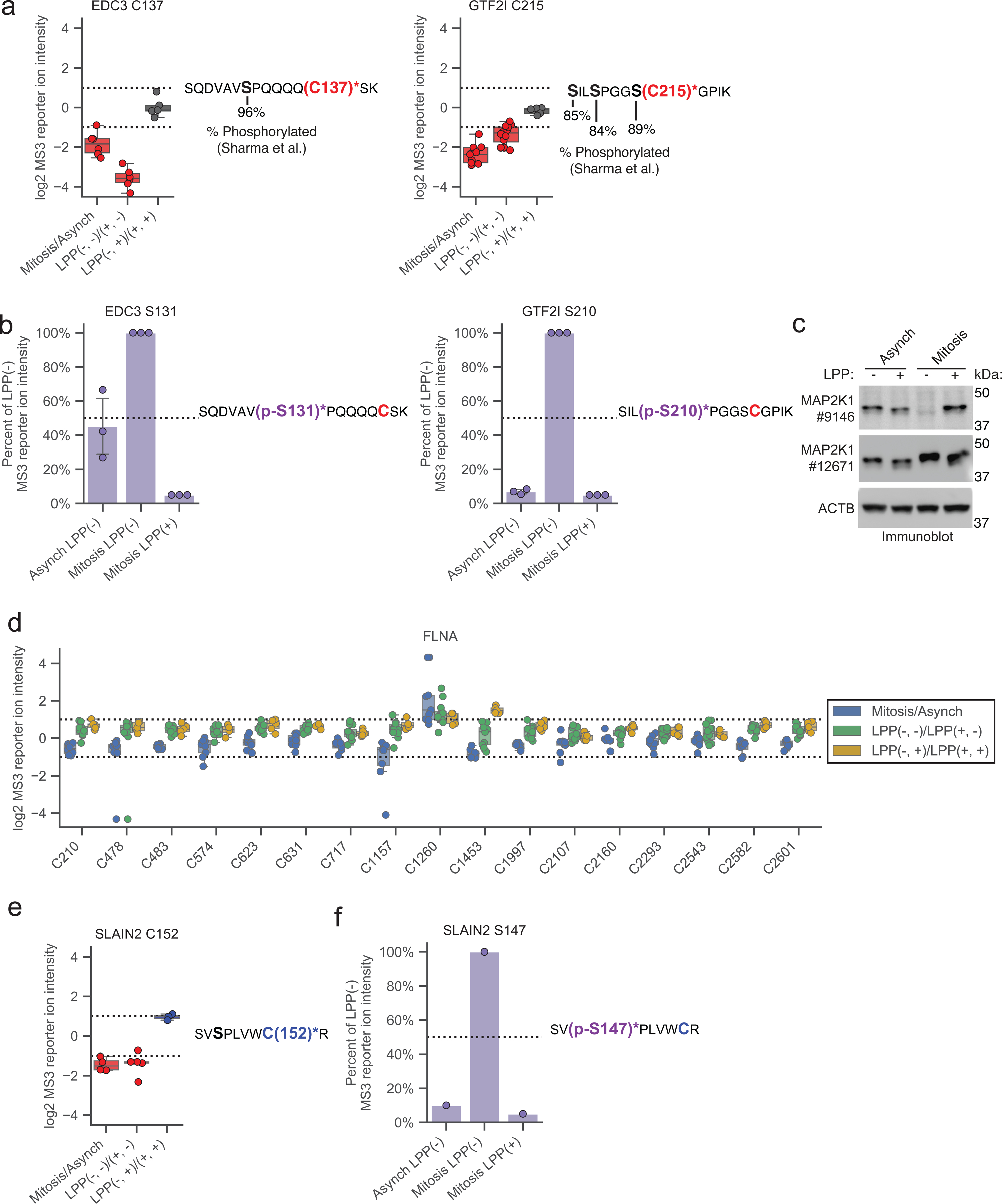
a, Left, cysteine reactivity values for indicated comparison groups for quantified cysteines from ECD3 (C137, top) and GTF2I (C215, bottom). Horizontal black line for each cysteine marks median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 5 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. Right, tryptic peptides containing EDC3 C137 (asterisks, red, bold; top) and GTF2I C215 (asterisks, red, bold; bottom) and high occupancy phosphorylation sites (black, bold)21 b, Left, bar graph showing phosphopeptide enrichment of EDC3 p-S131 (top) and GTF2I p-S210 (bottom). Data were normalized to mitotic proteome without LPP treatment (Mitosis LPP(−)) and represent the median values ± standard deviation for n = 3 independent experiments (circles). Right, tryptic peptides containing phosphorylated (p-, purple, bold) EDC p-S131 (asterisks, purple, bold; top) and GTF2I p-S210 (asterisks, purple, bold; bottom) and cysteines from Extended Data Fig. 3a marked (red, bold). c, Immunoblot analysis of MAP2K1 with antibody #9146 in mitosis. Data are from a single experiment representative of two independent experiments. d, FLNA cysteine reactivity values across the indicated comparison groups. Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 5 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. e, Left, SLAIN2 C152 reactivity values across the indicated comparison groups. Horizontal black line for each cysteine marks median value, boxes mark the upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 3 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. Right, tryptic peptide containing SLAIN2 C152 (asterisks, blue, bold) and a potential S/T-P phosphorylation site (black, bold). f, Left, bar graph showing phosphopeptide enrichment of SLAIN2 p-S147. Data were normalized to mitotic proteome without LPP treatment (Mitosis LPP(−)) and are from n = 1 experiment. Right, tryptic peptide containing phosphorylated (p-, purple, bold) SLAIN2 S147 (asterisks, purple, bold) with cysteine from Extended Data Fig. 3e marked (blue, bold).
Extended Data Figure 4. Features of proteins with mitotic phosphorylation-dependent changes in cysteine reactivity.
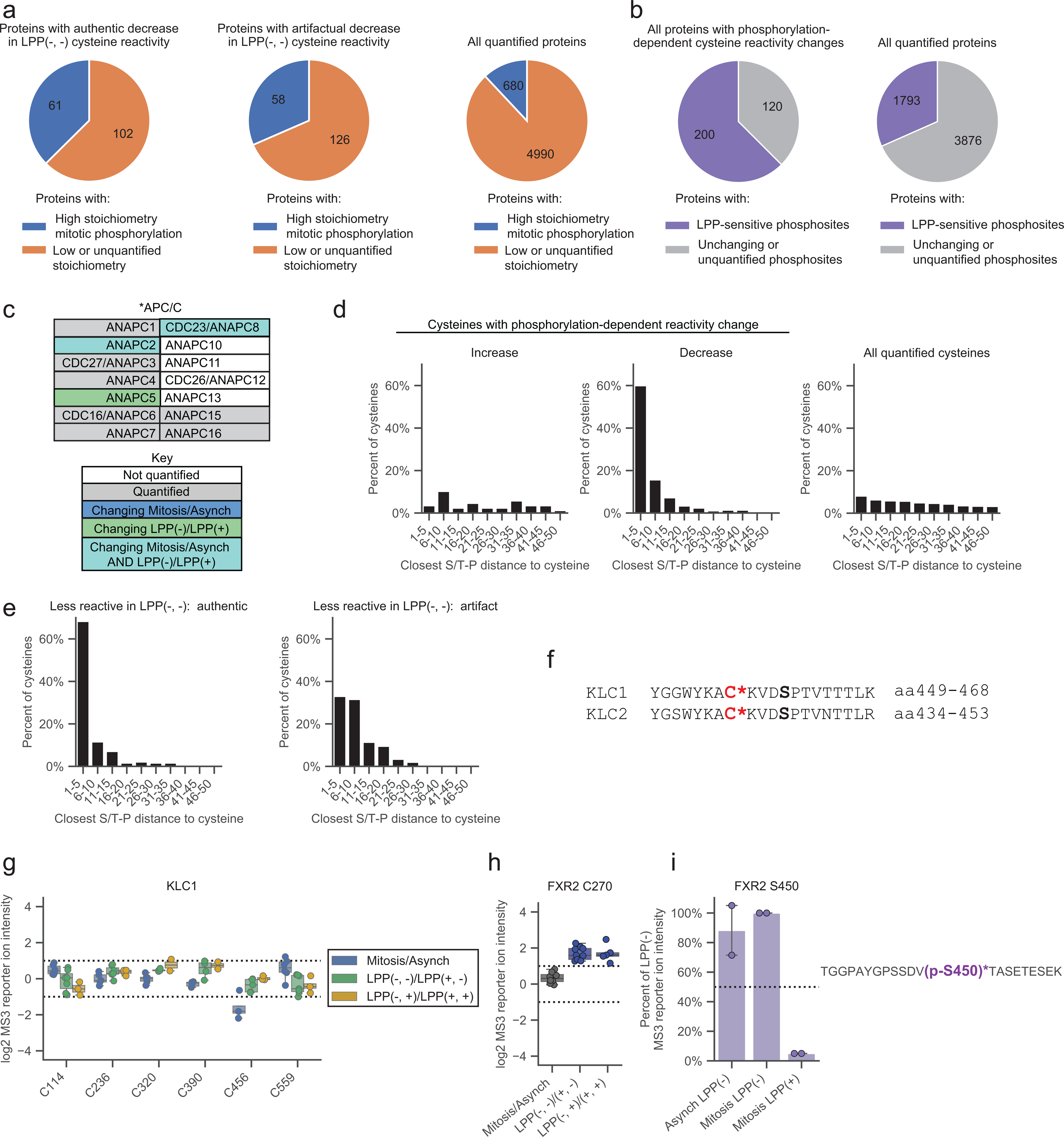
a, Proteins with authentic (left) and artifactual (middle) phosphorylation-dependent cysteine reactivity changes are enriched for high stoichiometry mitotic phosphorylation sites21 (blue) compared to all quantified proteins (right). Proteins lacking sufficient data for phosphorylation stoichiometry calculation or exhibiting only low stoichiometry (< 50% occupancy) sites21 were labeled as ‘Low or unquantified stoichiometry’ (orange). b, Proteins with phosphorylation-dependent cysteine reactivity changes (left) are enriched for LPP-sensitive mitotic phosphorylation sites (purple) compared to all quantified proteins (right). Proteins with only artifactual cysteine reactivity changes were removed from analysis. Proteins with LPP-insensitive or no quantified phosphorylation sites were labeled as “Unchanging or unquantified phosphosites” (gray). c, Members of the anaphase-promoting complex (APC/C) of the KEGG cell cycle pathway (HSA04110).55 Proteins are as described in Fig. 4b. d, Fraction of cysteines showing phosphorylation-dependent reactivity changes within the specified amino acid distances from an S/T-P site. Artifactual phosphorylation-dependent cysteine reactivity changes were omitted from analysis. e, Fraction of cysteines showing authentic (left) versus artifactual (right) phosphorylation-dependent reactivity changes within the specified amino acid distances from an S/T-P site. Authentic and artifactual changes were determined as described in Figure 3f. f, Sequence alignment of the KLC1 and KLC2 proteins centered on C456 and C441, respectively (asterisks, red, bold). Known (KLC1) and predicted (KLC2) S-P phosphorylation motifs are marked (black, bold). g, KLC1 cysteine reactivity values across the indicated comparison groups. Horizontal black lines mark median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 2 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. h, FXR2 C270 reactivity values for indicated comparison groups. Horizontal black lines mark median value, boxes mark upper and lower quartiles, and whiskers mark 1.5x interquartile range for n = 5 (or more) independent experiments (circles). Dotted lines designate boundaries for ≥ two-fold changes. i, Left, phosphopeptide enrichment of FXR2 p-S450. Data were normalized to Mitosis LPP(−) and represent the median values +/− standard deviation for n = 2 independent experiments (circles). Right, tryptic peptides containing phosphorylated (p-, purple, bold) FXR2 p-S450 (asterisks, purple, bold).
Supplementary Material
Acknowledgements
We thank Vincent Vartabedian from the Teijaro lab for technical assistance and Gabe Simon (Vividion) and Sherry Niessen and Matthew Hayward (Pfizer) for valuable feedback throughout this project. This work was supported by NIH (CA231991 to B.F.C), NIH-NCI (CA239556 to E.K.K.), and Vividion Therapeutics.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Tsherniak A; Vazquez F; Montgomery PG; Weir BA; Kryukov G; Cowley GS; Gill S; Harrington WF; Pantel S; Krill-Burger JM; Meyers RM; Ali L; Goodale A; Lee Y; Jiang G; Hsiao J; Gerath WFJ; Howell S; Merkel E; Ghandi M; Garraway LA; Root DE; Golub TR; Boehm JS; Hahn WC, Defining a Cancer Dependency Map. Cell 2017, 170 (3), 564–576 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forbes SA; Beare D; Gunasekaran P; Leung K; Bindal N; Boutselakis H; Ding M; Bamford S; Cole C; Ward S; Kok CY; Jia M; De T; Teague JW; Stratton MR; McDermott U; Campbell PJ, COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015, 43 (Database issue), D805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consortium, E. P., An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489 (7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research, N., Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455 (7216), 1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spreafico R; Soriaga LB; Grosse J; Virgin HW; Telenti A, Advances in Genomics for Drug Development. Genes (Basel) 2020, 11 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schluter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlen M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B, How many human proteoforms are there? Nat Chem Biol 2018, 14 (3), 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen ON, Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 2004, 8 (1), 33–41. [DOI] [PubMed] [Google Scholar]
- 8.Cai W; Tucholski T; Chen B; Alpert AJ; McIlwain S; Kohmoto T; Jin S; Ge Y, Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal Chem 2017, 89 (10), 5467–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen B; Brown KA; Lin Z; Ge Y, Top-Down Proteomics: Ready for Prime Time? Anal Chem 2018, 90 (1), 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y; Fonslow BR; Shan B; Baek MC; Yates JR 3rd, Protein analysis by shotgun/bottom-up proteomics. Chem Rev 2013, 113 (4), 2343–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Low TY; Mohtar MA; Lee PY; Omar N; Zhou H; Ye M, Widening the Bottleneck of Phosphoproteomics: Evolving Strategies for Phosphopeptide Enrichment. Mass Spectrom Rev 2020. [DOI] [PubMed] [Google Scholar]
- 12.Riley NM; Bertozzi CR; Pitteri SJ, A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry-Based Glycoproteomics. Mol Cell Proteomics 2020, 20, 100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin BR; Cravatt BF, Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods 2009, 6 (2), 135–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang JX; Lee G; Cavanaugh KE; Chang JW; Gardel ML; Moellering RE, High throughput discovery of functional protein modifications by Hotspot Thermal Profiling. Nat Methods 2019, 16 (9), 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batty P; Gerlich DW, Mitotic Chromosome Mechanics: How Cells Segregate Their Genome. Trends Cell Biol 2019, 29 (9), 717–726. [DOI] [PubMed] [Google Scholar]
- 16.Mahdessian D; Cesnik AJ; Gnann C; Danielsson F; Stenstrom L; Arif M; Zhang C; Le T; Johansson F; Shutten R; Backstrom A; Axelsson U; Thul P; Cho NH; Carja O; Uhlen M; Mardinoglu A; Stadler C; Lindskog C; Ayoglu B; Leonetti MD; Ponten F; Sullivan DP; Lundberg E, Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 2021, 590 (7847), 649–654. [DOI] [PubMed] [Google Scholar]
- 17.Levine MS; Holland AJ, The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev 2018, 32 (9–10), 620–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins K; Jacks T; Pavletich NP, The cell cycle and cancer. Proc Natl Acad Sci U S A 1997, 94 (7), 2776–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanahan D; Weinberg RA, Hallmarks of cancer: the next generation. Cell 2011, 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 20.Cuijpers SAG; Vertegaal ACO, Guiding Mitotic Progression by Crosstalk between Post-translational Modifications. Trends Biochem Sci 2018, 43 (4), 251–268. [DOI] [PubMed] [Google Scholar]
- 21.Sharma K; D’Souza RC; Tyanova S; Schaab C; Wisniewski JR; Cox J; Mann M, Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 2014, 8 (5), 1583–94. [DOI] [PubMed] [Google Scholar]
- 22.Olsen JV; Vermeulen M; Santamaria A; Kumar C; Miller ML; Jensen LJ; Gnad F; Cox J; Jensen TS; Nigg EA; Brunak S; Mann M, Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 2010, 3 (104), ra3. [DOI] [PubMed] [Google Scholar]
- 23.Kettenbach AN; Schweppe DK; Faherty BK; Pechenick D; Pletnev AA; Gerber SA, Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal 2011, 4 (179), rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dephoure N; Zhou C; Villen J; Beausoleil SA; Bakalarski CE; Elledge SJ; Gygi SP, A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A 2008, 105 (31), 10762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai L; Zhao T; Bisteau X; Sun W; Prabhu N; Lim YT; Sobota RM; Kaldis P; Nordlund P, Modulation of Protein-Interaction States through the Cell Cycle. Cell 2018, 173 (6), 1481–1494 e13. [DOI] [PubMed] [Google Scholar]
- 26.Becher I; Andres-Pons A; Romanov N; Stein F; Schramm M; Baudin F; Helm D; Kurzawa N; Mateus A; Mackmull MT; Typas A; Muller CW; Bork P; Beck M; Savitski MM, Pervasive Protein Thermal Stability Variation during the Cell Cycle. Cell 2018, 173 (6), 1495–1507 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heusel M; Frank M; Kohler M; Amon S; Frommelt F; Rosenberger G; Bludau I; Aulakh S; Linder MI; Liu Y; Collins BC; Gstaiger M; Kutay U; Aebersold R, A Global Screen for Assembly State Changes of the Mitotic Proteome by SEC-SWATH-MS. Cell Syst 2020, 10 (2), 133–155 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wani R; Nagata A; Murray BW, Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front Pharmacol 2014, 5, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung HS; Wang SB; Venkatraman V; Murray CI; Van Eyk JE, Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res 2013, 112 (2), 382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backus KM, Applications of Reactive Cysteine Profiling. Curr Top Microbiol Immunol 2019, 420, 375–417. [DOI] [PubMed] [Google Scholar]
- 31.Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF, Global profiling of lysine reactivity and ligandability in the human proteome. Nat Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MB; Bachovchin DA; Mowen K; Baker D; Cravatt BF, Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468 (7325), 790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan J; Carroll KS, Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers 2014, 101 (2), 165–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Couvertier SM; Zhou Y; Weerapana E, Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim Biophys Acta 2014, 1844 (12), 2315–30. [DOI] [PubMed] [Google Scholar]
- 35.Gould NS; Evans P; Martinez-Acedo P; Marino SM; Gladyshev VN; Carroll KS; Ischiropoulos H, Site-Specific Proteomic Mapping Identifies Selectively Modified Regulatory Cysteine Residues in Functionally Distinct Protein Networks. Chem Biol 2015, 22 (7), 965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulaj G; Kortemme T; Goldenberg DP, Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry 1998, 37 (25), 8965–72. [DOI] [PubMed] [Google Scholar]
- 37.Long MJC; Aye Y, Privileged Electrophile Sensors: A Resource for Covalent Drug Development. Cell Chem Biol 2017, 24 (7), 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurais AJ; Weerapana E, Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 2019, 50, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vinogradova EV; Zhang X; Remillard D; Lazar DC; Suciu RM; Wang Y; Bianco G; Yamashita Y; Crowley VM; Schafroth MA; Yokoyama M; Konrad DB; Lum KM; Simon GM; Kemper EK; Lazear MR; Yin S; Blewett MM; Dix MM; Nguyen N; Shokhirev MN; Chin EN; Lairson LL; Melillo B; Schreiber SL; Forli S; Teijaro JR; Cravatt BF, An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182 (4), 1009–1026 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bar-Peled L; Kemper EK; Suciu RM; Vinogradova EV; Backus KM; Horning BD; Paul TA; Ichu TA; Svensson RU; Olucha J; Chang MW; Kok BP; Zhu Z; Ihle NT; Dix MM; Jiang P; Hayward MM; Saez E; Shaw RJ; Cravatt BF, Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171 (3), 696–709 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C; Weerapana E; Blewett MM; Cravatt BF, A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 2014, 11 (1), 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Backus KM; Correia BE; Lum KM; Forli S; Horning BD; Gonzalez-Paez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF, Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534 (7608), 570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson JC; Joughin BA; van de Kooij B; Lim DC; Lauffenburger DA; Yaffe MB, ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Syst 2019, 8 (2), 163–167 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Reest J; Lilla S; Zheng L; Zanivan S; Gottlieb E, Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat Commun 2018, 9 (1), 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y; Wang J; Jaehnig EJ; Shi Z; Zhang B, WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 2019, 47 (W1), W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Supek F; Bosnjak M; Skunca N; Smuc T, REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011, 6 (7), e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ostermann N; Schlichting I; Brundiers R; Konrad M; Reinstein J; Veit T; Goody RS; Lavie A, Insights into the phosphoryltransfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate. Structure 2000, 8 (6), 629–42. [DOI] [PubMed] [Google Scholar]
- 48.Meszaros B; Erdos G; Dosztanyi Z, IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 2018, 46 (W1), W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsumoto T; Kinoshita T; Kirii Y; Yokota K; Hamada K; Tada T, Crystal structures of MKK4 kinase domain reveal that substrate peptide binds to an allosteric site and induces an auto-inhibition state. Biochem Biophys Res Commun 2010, 400 (3), 369–73. [DOI] [PubMed] [Google Scholar]
- 50.Zhao H; Li T; Wang K; Zhao F; Chen J; Xu G; Zhao J; Li T; Chen L; Li L; Xia Q; Zhou T; Li HY; Li AL; Finkel T; Zhang XM; Pan X, AMPK-mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol 2019, 21 (4), 476–486. [DOI] [PubMed] [Google Scholar]
- 51.Marcussen M; Larsen PJ, Cell cycle-dependent regulation of cellular ATP concentration, and depolymerization of the interphase microtubular network induced by elevated cellular ATP concentration in whole fibroblasts. Cell Motil Cytoskeleton 1996, 35 (2), 94–9. [DOI] [PubMed] [Google Scholar]
- 52.Becher I; Savitski MM; Savitski MF; Hopf C; Bantscheff M; Drewes G, Affinity profiling of the cellular kinome for the nucleotide cofactors ATP, ADP, and GTP. ACS Chem Biol 2013, 8 (3), 599–607. [DOI] [PubMed] [Google Scholar]
- 53.Tibbles LA; Ing YL; Kiefer F; Chan J; Iscove N; Woodgett JR; Lassam NJ, MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J 1996, 15 (24), 7026–35. [PMC free article] [PubMed] [Google Scholar]
- 54.Posewitz MC; Tempst P, Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal Chem 1999, 71 (14), 2883–92. [DOI] [PubMed] [Google Scholar]
- 55.Kanehisa M; Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000, 28 (1), 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iakoucheva LM; Radivojac P; Brown CJ; O’Connor TR; Sikes JG; Obradovic Z; Dunker AK, The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 2004, 32 (3), 1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu H; Lee HY; Tong Y; Hong BS; Kim KP; Shen Y; Lim KJ; Mackenzie F; Tempel W; Park HW, Crystal structures of the tetratricopeptide repeat domains of kinesin light chains: insight into cargo recognition mechanisms. PLoS One 2012, 7 (3), e33943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirokawa N; Noda Y, Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev 2008, 88 (3), 1089–118. [DOI] [PubMed] [Google Scholar]
- 59.Vagnoni A; Rodriguez L; Manser C; De Vos KJ; Miller CC, Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J Cell Sci 2011, 124 (Pt 7), 1032–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Batut J; Howell M; Hill CS, Kinesin-mediated transport of Smad2 is required for signaling in response to TGF-beta ligands. Dev Cell 2007, 12 (2), 261–74. [DOI] [PubMed] [Google Scholar]
- 61.Savaryn JP; Catherman AD; Thomas PM; Abecassis MM; Kelleher NL, The emergence of top-down proteomics in clinical research. Genome Med 2013, 5 (6), 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng Y; Fornelli L; Compton PD; Sharma S; Canterbury J; Mullen C; Zabrouskov V; Fellers RT; Thomas PM; Licht JD; Senko MW; Kelleher NL, Unabridged Analysis of Human Histone H3 by Differential Top-Down Mass Spectrometry Reveals Hypermethylated Proteoforms from MMSET/NSD2 Overexpression. Mol Cell Proteomics 2016, 15 (3), 776–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ntai I; Fornelli L; DeHart CJ; Hutton JE; Doubleday PF; LeDuc RD; van Nispen AJ; Fellers RT; Whiteley G; Boja ES; Rodriguez H; Kelleher NL, Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc Natl Acad Sci U S A 2018, 115 (16), 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Floyd BM; Drew K; Marcotte EM, Systematic Identification of Protein Phosphorylation-Mediated Interactions. J Proteome Res 2021, 20 (2), 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abo M; Weerapana E, A Caged Electrophilic Probe for Global Analysis of Cysteine Reactivity in Living Cells. J Am Chem Soc 2015, 137 (22), 7087–90. [DOI] [PubMed] [Google Scholar]
- 66.Telles E; Gurjar M; Ganti K; Gupta D; Dalal SN, Filamin A stimulates cdc25C function and promotes entry into mitosis. Cell Cycle 2011, 10 (5), 776–82. [DOI] [PubMed] [Google Scholar]
- 67.Szeto SGY; Williams EC; Rudner AD; Lee JM, Phosphorylation of filamin A by Cdk1 regulates filamin A localization and daughter cell separation. Exp Cell Res 2015, 330 (2), 248–266. [DOI] [PubMed] [Google Scholar]
- 68.van der Vaart B; Manatschal C; Grigoriev I; Olieric V; Gouveia SM; Bjelic S; Demmers J; Vorobjev I; Hoogenraad CC; Steinmetz MO; Akhmanova A, SLAIN2 links microtubule plus end-tracking proteins and controls microtubule growth in interphase. J Cell Biol 2011, 193 (6), 1083–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang W; Mevellec L; Liu A; Struble G; Miller R; Allen SJ; Federowicz K; Wroblowski B; Vialard J; Ahn K; Krosky D, Discovery of an Allosteric, Inactive Conformation-Selective Inhibitor of Full-Length HPK1 Utilizing a Kinase Cascade Assay. Biochemistry 2021. [DOI] [PubMed] [Google Scholar]
- 70.Paulsen CE; Truong TH; Garcia FJ; Homann A; Gupta V; Leonard SE; Carroll KS, Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol 2011, 8 (1), 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schlicker A; Domingues FS; Rahnenfuhrer J; Lengauer T, A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics 2006, 7, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry data are available via PRIDE with identifier PXD026730.
Source data, in addition to Supplementary Dataset 1, is available for all figure panels.