

Abstract
Background
Cancer cells selectively promote the translation of oncogenic transcripts to stimulate cancer progression. Although growing evidence has revealed that tRNA modifications and related genes participate in this process, their roles in head and neck squamous cell carcinoma (HNSCC) remain largely uncharacterized. Here, we sought to investigate the function and mechanisms of the transfer RNA (tRNA) N7‐methylguanosine (m7G) modification in regulating the occurrence and development of HNSCC.
Methods
Cell lost‐of‐function and gain‐of‐function assays, xenograft models, conditional knockout and knockin mouse models were used to study the physiological functions of tRNA m7G modification in HNSCC tumorigenesis. tRNA modification and expression profiling, mRNA translation profiling and rescue assays were performed to uncover the underlying molecular mechanisms. Single‐cell RNA sequencing (scRNA‐seq) was conducted to explore the tumor microenvironment changes.
Results
The tRNA m7G methyltransferase complex components Methyltransferase‐like 1 (METTL1)/WD repeat domain 4 (WDR4) were upregulated in HNSCC and associated with a poor prognosis. Functionally, METTL1/WDR4 promoted HNSCC progression and metastasis in cell‐based and transgenic mouse models. Mechanistically, ablation of METTL1 reduced the m7G levels of 16 tRNAs, inhibiting the translation of a subset of oncogenic transcripts, including genes related to the phosphatidylinositol‐3‐kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway. In addition, chemical modulators of the PI3K/Akt/mTOR signaling pathway reversed the effects of Mettl1 in mouse HNSCC. Furthermore, scRNA‐seq results revealed that Mettl1 knockout in mouse tumor cells altered the immune landscape and cell‐cell interaction between the tumor and stromal compartment.
Conclusions
The tRNA m7G methyltransferase METTL1 was found to promote the development and malignancy of HNSCC through regulating global mRNA translation, including the PI3K/AKT/mTOR signaling pathway, and found to alter immune landscape. METTL1 could be a promising treatment target for HNSCC patients.
Keywords: head and neck squamous cell carcinoma, m7G modification, metastasis, METTL1, microenvironment, PI3K/AKT/mTOR signaling, scRNA‐seq, tRNA, WDR4
Abbreviations
- 4NQO
4‐nitroquinoline N‐oxide
- Adrb2
adrenoceptor beta 2
- AlkB
alpha‐ketoglutarate‐dependent dioxygenase
- BMI1
B cell‐specific Moloney murine leukemia virus insertion site 1
- CCK‐8
Cell Counting Kit‐8
- CCLE
Cancer Cell Line Encyclopedia
- CSC
cancer stem cell
- Cx3cr1
C‐X3‐C motif chemokine receptor 1
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- FDR
false discovery rate
- GO
gene ontology
- GSEA
gene set enrichment analysis
- GSVA
gene set variation analysis
- HNSCC
head and neck squamous cell carcinoma
- HOK
human oral keratinocytes
- Il1b
interleukin 1 beta
- Il1r2
interleukin 1 receptor type 2
- IP
immunoprecipitation
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- m6A
N6‐methyladenosine
- m7G
N7‐methylguanosine
- Macro‐3
macrophage‐3
- mESCs
embryonic stem cells
- METTL1
methyltransferase‐like 1
- Mrc1
mannose receptor C type 1
- NES
normalized enrichment score
- NKT
natural killer cells
- ORFs
open reading frames
- PCA
principal component analysis
- p‐EMT
partial epithelial‐to‐mesenchymal transition
- PI3K/AKT/mTOR
phosphatidylinositol‐3‐kinase/ protein kinase B/ mammalian target of rapamycin
- PTEN
phosphatase and tensin homolog
- qRT‐PCR
quantitative real‐time PCR
- Ribo‐seq
ribosome sequencing
- RNC‐seq
ribosome nascent‐chain complex‐bound mRNA sequencing
- RPL22
ribosomal protein L22
- scRNA‐seq
single‐cell RNA sequencing
- siRNAs
small interfering RNAs
- snoRNA
small nucleolar RNA
- TE
translation efficiency
- Th17
T helper 17
- TRAC‐seq
m7G site‐specific tRNA reduction and cleavage‐sequencing
- Treg
regulatory T
- tRNA
transfer RNA
- TRs
translation ratios
- UMAP
uniform manifold approximation and projection
- UTR
untranslated region
- WDR4
WD repeat domain 4
1. BACKGROUND
Growing evidence supports the idea that perturbations in RNA post‐transcriptional modification and related translational control contribute greatly to tumor oncogenesis [1, 2, 3]. We recently reported that the RNA modification enzyme methyltransferase‐like 3 (METTL3) can promote head and neck squamous cell carcinoma (HNSCC) progression by regulating the N6‐methyladenosine (m6A) modification of B cell‐specific Moloney murine leukemia virus insertion site 1 (BMI1) mRNA and enhancing its translational activity [4]. Furthermore, defects in transfer RNA (tRNA) modifications are frequently involved in cancer etiology [5, 6, 7]. However, it is unclear how alterations in tRNA modifications are involved in HNSCC development.
It is estimated that approximately 15% of tRNA nucleotides are covalently modified [8]. To date, over 80 tRNA modifications have been characterized [9]. These tRNA modifications can govern a wide variety of processes, including regulating the stability of tRNA fragments [10], maintaining the structural flexibility and rigidity [11], and regulating the translation [12] and decoding [13] of tRNAs. As one of the most common tRNA modifications, N7‐methylguanosine (m7G) is catalyzed and installed by the Methyltransferase‐like 1 (METTL1)/WD repeat domain 4 (WDR4) complex in the tRNA variable loop in humans. In this methyltransferase complex, METTL1 serves as the m7G catalytic enzyme while WDR4 plays a stabilizing role [14, 15, 16]. METTL1 has been shown to play a role in the self‐renewal and differentiation of embryonic stem cells and cancer [16, 17, 18, 19], and a mutation in WDR4 has been proposed to cause primordial dwarfism [20]. Previous reports have shown that various tRNA modifications can affect the efficacy and fidelity of translation and participate in determining cellular behaviors in cancer [2, 21, 22]. Abnormal tRNA m7G modified by METTL1/WDR4 was found to be closely related to the occurrence and progression in some tumors, such as lung cancer [21] and intrahepatic cholangiocarcinoma [22].
HNSCC is the sixth most common cancer globally, and its incidence is expected to increase 30% by 2030 (1.08 million new cases per year) [23, 24]. The principal treatment modalities for HNSCC are surgical resection, adjuvant irradiation, chemoradiation, and treatment with immune checkpoint inhibitors according to the disease stage. However, tumors frequently become refractory to these treatments. Hence, there is a clinical need to develop effective therapeutic regimens for these patients; these could include molecular targeted therapy, which is promising as an alternative or combination treatment for HNSCC.
In this study, we investigated the function and molecular mechanism of METTL1/WDR4‐mediated m7G tRNA modification in the development of HNSCC and explored its possible application in the diagnosis, molecular targeted therapy, and prognosis of HNSCC.
2. MATERIALS AND METHODS
2.1. Clinical tissue specimens, the Cancer Genome Atlas (TCGA) database analysis, and Cancer Cell Line Encyclopedia (CCLE) database analysis
A total of 140 HNSCC specimens and 69 adjacent normal tissue samples (1 cm away from HNSCC tissues) were collected from HNSCC patients diagnosed at the Department of Oral and Maxillofacial Surgery, First Affiliated Hospital, Sun Yat‐sen University (Guangzhou, Guangdong, China) between November 2004 and September 2014. None of the patients had received preoperative irradiation or chemotherapy. The overall survival (OS) time of these patients was calculated from the date of diagnosis to September 2020 (or death). METTL1/WDR4 expression levels were compared between 502 HNSCC tissues and 44 adjacent normal tissue samples using TCGA database. METTL1 mutation signature was analyzed with CCLE database (https://sites.broadinstitute.org/ccle) and 1478 HNSCC samples from 6 data sets: head and neck squamous cell carcinoma (TCGA, Nature 2015; TCGA, Firehose Legacy; Broad, Science 2011; TCGA, PanCancer Atlas; Johns Hopkins, Science 2011) and oral squamous cell carcinoma (MD Anderson, Cancer Discov 2013).
2.2. Immunohistochemistry (IHC) staining
All specimens collected were routinely embedded in paraffin, and the tissue slides were deparaffinized and rehydrated. After incubation with 3% H2O2 and antigen retrieval, the slides were blocked in 5% bovine serum albumin followed by incubation with primary anti‐METTL1 (1:100, 14994, Proteintech, Rosemont, IL, USA), anti‐phosphatidylinositol‐3‐kinase (PI3K) (1:100, AF5112, Affinity, Changzhou, Jiangsu, China), anti‐AKT (1:100, AF6261, Affinity), anti‐pAKT (1:100, AF0016, Affinity), anti‐mammalian target of rapamycin (mTOR) (1:100, AF6308, Affinity), anti‐pmTOR (1:100, AF3309, Affinity), anti‐pan‐cytokeratin (anti‐PCK, 1:100, ab9377, Abcam, Cambridge, UK), or anti‐Ki67 antibody (1:100, ab15580, Abcam) overnight at 4°C. After washing, secondary antibodies and strept avidin‐biotin complex (SABC) were applied with a SABC‐POD (rabbit IgG) kit (BOSTER, Wuhan, Hubei, China). Then, the slides were stained with a 3,3’‐diaminobenzidine (DAB) kit (BOSTER) and counterstained with hematoxylin. The IHC score is a continuous scale of 0‐300 according to the fraction of cells stained at each intensity multiplying the staining intensity (intensity score 0‐3) [25]. For Ki67, the proportion of positively stained cells was calculated. The anti‐PCK antibody was used for IHC staining of metastatic cells in cervical lymph nodes. The diagnosis was made by two pathologists, blinding to the patients’ clinical data, according to 8th edition of the Union for International Cancer Control/ American Joint Committee on Cancer (UICC/AJCC) TNM Staging Manual.
2.3. Western blotting
Cells were treated in radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China) with proteinase inhibitor cocktail (Roche, Shanghai, China) and conditionally adding phosphatase inhibitors (Sangon Biotech, Shanghai, China). The aliquots of protein were subjected to 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and electrotransferred to the polyvinylidene fluoride membrane. After blocking with 5% skimmed milk and incubating with primary anti‐METTL1 (1:1500), anti‐WDR4 (1:1500, ab169526, Abcam), anti‐PI3K (1:1000), anti‐AKT (1:1000), anti‐pAKT (1:1000), anti‐mTOR (1:1000), anti‐pmTOR (1:1000), anti‐GAPDH (1:2000, 10494, Proteintech), anti‐β‐Tubulin (1:2000, 10094, Proteintech), anti‐Cyclin D1 (1:1000, ab134175, Abcam), anti‐Vimentin (1:1000, ab92547, Abcam), anti‐matrix metalloprotein 9 (MMP9) (1:1000, ab38898, Abcam), anti‐B‐cell lymphoma‐2 (Bcl‐2) (1:1000, 12789‐1‐AP, Proteintech) , anti‐Bcl‐2‐associated X protein (BAX) (1:1000, ab32503, Abcam) or anti‐phosphorylation of S6 kinase (P‐S6K) antibody (1:1000, 14485‐1‐AP, Proteintech), the membrane was then incubated with anti‐rabbit secondary antibodies (1:5000, SA00001‐2, Proteintech). Protein levels were detected using Tanon 5200 Multi intelligent imaging system (Tianneng, Shanghai, China).
2.4. Northern blotting and northwestern blotting
Northern blotting of tRNAs or U6 small nucleolar RNA (snoRNA) was performed as previously described [16]. Briefly, 2 μg total RNA samples were mixed with 2× RNA loading buffer and denatured at 95°C for 5 min. The samples were subjected to 15% urea‐polyacrylamide (UREA‐PAGE) electrophoresis in 1× Tris‐borate‐ethylene diamine tetraacetic acid (EDTA) (TBE) buffer (Solarbio, Beijing, China). After electrophoresis, the RNAs were transferred onto a positively charged nylon membrane, crosslinked with ultraviolet, and then blotted with digoxigenin‐labeled probes against tRNAs or U6 snoRNA (Sangon Biotech). For Northwestern blotting, the RNA‐containing membranes were blotted with anti‐m7G antibody (1:1000, RN017M, Medical Biological Laboratories, Nagoya, Japan) overnight. Finally, the digoxigenin or anti‐m7G antibody signals were detected following the Western blotting protocol described above.
2.5. Cell culture and transfection
Human oral keratinocytes (HOK) were purchased from ScienCell (Carlsbad, California, USA). HNSCC cell lines UM1, SCC9, and SCC25 were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA), and HNSCC cell lines SCC4 and SCC15 were provided by Cheng Wang from Department of Maxillofacial Surgery, Guanghua Stomatological Hospital, Sun Yat‐sen University, with the cell line STR authentication report.
To knockout METTL1 via the CRISPR technique, small guide RNA (sgRNA; Supplementary Table S1) targeting the METTL1 gene was cloned into LentiCRISPRv2 (Addgene, Cambridge, MA, USA). To knockdown WDR4, short hairpin RNA (shRNA; Supplementary Table S1) targeting the WDR4 gene was cloned into the pLKO.1 vector (Addgene). For METTL1 and phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha (PIK3CA) overexpression plasmids, the full‐length open reading frames (ORFs) of the human METTL1 gene (NM_005371.6) and the PIK3CA gene (NM_006218) were separately cloned into the pEZ‐Lv201 vector (GeneCopoeia, Rockville, MD, USA) and the pLV vector (Addgene). The METTL1 catalytic inactive mutant (aa160‐163, LFPD to AFPA) was generated using the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA). For the rescue assay, SCC9 and SCC15 cells were given SC79 (5 μg/mL; Beyotime) to activate AKT.
2.6. Puromycin uptake assay
Small interfering RNAs (siRNAs; Supplementary Table S1) (RiboBio, Guangzhou, Guangdong, China) targeting the 3’‐untranslated region (UTR) of METTL1 were designed and used in these experiments to avoid the degradation of exogenous METTL1 by siRNAs. Cells were incubated with puromycin (1 μmol/L final concentration) for 30 min, followed by protein extraction and Western blotting using anti‐puromycin antibody (Abcam).
2.7. Cell proliferation assay, colony formation assay, cell migration and invasion assay
The cell proliferation, colony formation, cell migration and invasion assays were performed as described in our previous study [4]. Cell proliferation was measured at scheduled time points using Cell Counting Kit‐8 (CCK‐8) (Dojindo, Tokyo, Japan) following the manufacturer's protocol. For the colony formation experiments, cells were plated into 6‐well plates (500 cells/well) in DMEM/F12 with 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA) and cultured at 37°C with 5% CO2 in a humidified atmosphere for 14 days. The colonies were fixed with 4% paraformaldehyde and stained with crystal violet, and then the visible colonies were counted. For migration assay, cells (5 × 104 cells/well) were suspended in 200 μL FBS‐free DMEM/F12 culture medium and added into a transwell insert (Corning, Shanghai, China), and 500 μL of DMEM/F12 containing 1% FBS was added to the lower compartment. For invasion assay, the chambers were pre‐coated with 5% Matrigel (BioCoat, Guangzhou, Guangdong, China) with DMEM/F12 at 37°C for 1 hour. Then the cells were added as described in migration assay. After 48 or 72 hours, the chambers were collected and stained by 0.1% crystal violet. Images of the stained lower surface were captured, and the cells were quantified under a microscope in five random fields.
2.8. Cell apoptosis and cell cycle assay
Cell cycle assays were performed using a cell cycle staining kit (MultiSciences, Hangzhou, Zhejiang, China) following the manufacturer's protocol. Cell apoptosis assays were conducted with the Annexin V‐FITC/PI Apoptosis Assay Kit (MultiSciences) according to the manufacturer's protocol. The fixed and stained cells were analyzed by flow cytometry using CytoFLEX (Beckman, Brea, CA, USA). Apoptotic cells consist of Q2 and Q3 cells. The FlowJo software version 7.6 (FlowJo LLC, Ashland, KY, USA) was employed for data analysis.
2.9. Tumorigenesis and metastasis assay in vivo
Our animal experiments were approved by the Laboratory Animal Center of Sun Yat‐sen University. Female BALB/c nude mice (5 weeks old) purchased from the Model Animal Research Center (Nanjing, Jiangsu, China) were used to investigate the effect of METTL1 on tumor growth and metastasis in vivo. The mice were kept in the Laboratory Animal Center of Sun Yat‐sen University under specific pathogen‐free conditions. To construct the orthotopic transplantation model, 1 × 106 SCC9 cells were suspended in 50 μL PBS and injected into the middle‐posterior dorsal tongue of the mice. The mice were euthanized when the tumor volumes reached 100 mm3 or body weight loss of higher than 15% from baseline. After 2 weeks, the mice were euthanized by carbon dioxide inhalation, and the tumor volumes were calculated using the following formula: volume = (length × width2) / 2. The tongue and cervical lymph node were embedded, sectioned, and stained for H&E and IHC analysis.
2.10. Mettl1‐knockout (KO) or ‐knockin (KI) mouse model
K14Cre, K14CreER, and Ribotag mouse strains were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mettl1flox and Mettl1cKI mice were kindly provided by Dr. Shuibin Lin from The First Affiliated Hospital of Sun Yat‐sen University. Mice were treated with 50 μg/mL 4‐nitroquinoline N‐oxide (4NQO) (Sigma‐Aldrich, St. Louis, MO, USA) in drinking water as previously described [26]. Four‐week‐old K14CreER;Mettl1wt/wt (Mettl1cKO‐Ctrl) and K14CreER;Mettl1fl/fl (Mettl1cKO) mice were injected with tamoxifen (Sigma‐Aldrich) at a dose of 4.44 mg/g body weight for 5 consecutive days and then treated with 4NQO in their drinking water for 16 weeks followed by another 12 weeks with normal drinking water. For the Mettl1 overexpression study, we crossed male and female K14Cre;Mettl1cKI/wt mice to obtain K14Cre;Mettl1wt/wt (Mettl1cKI‐Ctl) and K14Cre;Mettl1cKI/cKI (Mettl1cKI) mice. For the mouse rescue assay, SC79 (0.04 mg/g), an AKT activator [27], was intraperitoneally injected for 4 weeks after 4NQO treatment and fed for 20 weeks in Mettl1cKO‐Ctrl and Mettl1cKO mice. Mettl1cKI‐Ctrl and Mettl1cKI mice were treated with BKM120 (MedChemExpress, Shanghai, China), a PI3K inhibitor [28], by oral gavage (35 mg/kg) for 4 weeks after 4NQO treatment and feeding for 20 weeks.
2.11. m7G site‐specific tRNA reduction and cleavage‐sequencing (TRAC‐seq) and data analysis
TRAC‐seq was performed as previously described [16, 29]. After extraction of total RNA from METTL1‐KO and METTL1‐wild type (WT) cells, small RNAs were isolated using the mirVana miRNA Isolation Kit (Invitrogen, Carlsbad, CA, USA). Then recombinant wild‐type and D135S Alpha‐ketoglutarate‐dependent dioxygenase (AlkB) proteins provided by Dr. Shuibin Lin were used to demethylate the dominant methylation sites on small RNAs [30, 31]. Half of the AlkB‐demethylated RNAs were removed as input for small RNA sequencing library construction. The remaining AlkB‐treated RNAs were treated with 0.1 mol/L NaBH4 (Sigma‐Aldrich) for 30 min on ice at dark in the presence of 1 mmol/L free 7‐methylguanosine 5’‐triphosphate sodium salt (Sigma‐Aldrich) as methylation carrier. The remaining NaBH4‐treated RNAs were subsequently treated with aniline‐acetate solution (at a H2O:glacial acetate acid:aniline ratio of 7:3:1) at 25°C in the dark for 2 hours to induce site‐specific cleavage of m7G sites. Then, the NaBH4/aniline‐treated RNA together with input demethylated RNA was processed with small RNA sequencing library construction using the Multiplex Small RNA Library Prep set for Illumina Kit (New England Biolabs) and subjected to high‐throughput sequencing with Illumina NextSeq 500.
The analysis of tRNA expression and m7G modification was conducted as previously described [16, 29]. To analyze the expression of RNA, the reads per million mapped reads (RPM) values of individual tRNAs were calculated with the sequencing data of AlkB‐demethylated input samples. To analyze the m7G tRNA modifications, the clean reads from the chemically processed sequencing data and the control input sequencing data were first mapped to the human mature tRNA sequences. NaBH4/aniline treatment induced cleavage at m7G sites, causing the sequencing reads to start after the m7G sites. Then, Bedtools (http://bedtools.readthedocs.io/en/latest) was used to calculate the read depth (tRNA abundance) of each locus and the reading from that position (cleaved reads). For example, for a specific m7G‐modified site i, the cleavage score of site i was calculated as the ratio between the number of reads starting at site i and the read depth of site i [32]. Then, the cleavage score of site i was calculated as follows:
Positions 46‐48 with cleavage scores >4 and coverage >100 were applied to identify the candidate m7G sites.
2.12. Ribosome sequencing (Ribo‐seq) and data analysis
Ribo‐seq was performed as previously described [33]. SCC15 cells were first incubated with harringtonine (Solarbio) at a concentration of 2 μg/mL for 2 min, and then 100 μg/mL cycloheximide (Sigma‐Aldrich) was used to block translation. To prepare ribosome footprints, 300 μL lysate was mixed with 7.5 μL RNase I and 5 μL DNase I and incubated at 25°C for 45 min later. Nuclease digestion was stopped by adding 10 μL RNase inhibitor (Vazyme). Then, 100 μL of digested ribosome footprints was added to the column and centrifuged at 600 ×g for 2 min. Ribo‐seq libraries were constructed using NEBNext® Multiple Small RNA Library Prep Set for Illumina® (New England Biolabs).
The Ribo‐seq data were analyzed using the RiboTool kit (https://bioinformatics.sc.cn/RiboToolkit) [34]. The translation efficiency (TE) was calculated as the ratio between the numbers of ribosome fragments aligned to the sequence coding for amino acids in protein (CDS) region and mRNA abundance for individual genes. Codon frequency analysis was performed after eliminating the genes with mRNA abundances lower than 5, and TEs with a 1‐fold decrease were considered TE‐downregulated genes.
2.13. Ribosome nascent‐chain complex‐bound mRNA sequencing (RNC‐seq) and data analysis
RNC‐seq was performed as previously described [35]. Briefly, cells were pretreated with 100 μg/mL cycloheximide for 15 min at 37°C followed by immediate cold PBS wash. Then, cells were incubated with 1 mL cell lysis buffer. Cell lysates were centrifuged at 16,200 ×g for 10 min at 4°C, and 10% of the extract was used as an input control. The remaining extraction was layered onto 35 mL sucrose buffer (30% sucrose in polysome cell extraction buffer) and then subjected to ultracentrifugation at 174,900 ×g for 5 hours at 4°C in a SW32 rotor (Beckman) to collect the RNC pellets containing the polysome fractions. Next, RNA samples were isolated from the input and RNC samples for sequencing and quantitative real‐time PCR (qRT‐PCR; Supplementary Table S1).
cDNA library construction and sequencing were performed by Beijing Genomics Institute (Beijing, China) using the BGISEQ‐500 platform (BGI, Shenzhen, Guangdong, China). High‐quality reads were mapped to the University of California Santa Cruz (UCSC) human reference genome (GRCH38/hg38) using the alignment tool Hisat2. The gene expression level was normalized by using the fragments per kilobase of transcript per million mapped reads (FPKM) method. Translation ratios (TRs) were calculated using the following formula: TR = (FPKM in RNC‐seq) / (FPKM in input RNA‐seq). Genes with FPKM > 0.01 and 0.6‐fold change TR were considered differentially translated genes.
2.14. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and gene set enrichment analysis (GSEA)
KEGG analysis of mRNAs with decreased TRs identified by RNC‐seq data was conducted using the KEGG Pathway Database (https://www.kegg.jp/kegg/pathway.html). The statistical significance of genes with decreased TRs was evaluated according to the P value, and P < 0.05 was considered to indicate statistical significance.
GSEA software downloaded from the GSEA website (https://www.gsea‐msigdb.org/gsea/downloads.jsp) was used to determine significantly enriched signaling pathways after METTL1 knockout. The following parameters were used in GSEA version 4.0: the annotated gene sets of h.all.v7.2.symbols.gmt were performed; the number of permutations was 1,000; the phenotype label was ‘‘ WT vs. KO”; the collapse/remap to gene symbols was ‘‘collapse’’; and the normalized enrichment score (NES) served as the GSEA main statistic. The conditions for gene sets selecting were as below: nominal P < 0.05, false discovery rate (FDR) < 0.25, and NES > 1.0 [36, 37].
2.15. Polysome profiling
Polysome profiling was performed as described previously [16]. METTL1‐KO or control HNSCC cells were treated with 100 μg/mL cycloheximide for 15 min at 37°C, followed by immediate precooled PBS washing. Then, the cells were lysed with polysome cell extraction buffer for 10 min on ice and centrifuged at 13,000 ×g for 10 min at 4°C. Then 1 mL supernatant was slowly layered onto 11 mL of a preprepared 10%‐50% sucrose gradient and subsequently centrifuged at 36,000 rpm at 4°C for 3 hours in an SW41 rotor (Beckman). The stratified samples were separated by a BR‐188 Density Gradient Fractionation System (Brandel, Gaithersburg, MD, USA) at 0.75 mL/min, and the absorbance was assessed at 254 nm. The area under the receiver operating characteristic curve (AUC) was calculated to quantify the polysome level with GraphPad (GraphPad, San Diego, CA, USA).
2.16. Immunoprecipitation (IP) assays
IP assays of HNSCC cells from K14CreER;RiboTag;Mettl1fl/fl or K14CreER;RiboTag;Mettl1wt/wt mice were performed as described previously [38, 39]. Briefly, HNSCC tissues were mashed by a multisample tissue grinding machine (Jingxin, Xinchang, Zhejiang, China) in supplemental homogenization buffer. Parts of the supernatant were removed as input to analyze the expression level of total RNA. The remaining cleared homogenates were added anti‐HA tag antibody (1:125, ab9110, Abcam) and rotated for 4 hours at 4°C. Then protein A/G magnetic beads (Thermo Fisher Scientific, Waltham, MA, USA) were added to homogenization buffer and incubated with rotation for 10 min at 4°C, followed by adsorption of the magnetic beads with a magnetic frame and removal of the supernatant. Then, homogenates containing anti‐HA tag antibody were added to tubes containing protein A/G magnetic beads and rotated overnight at 4°C. After incubation, the beads were washed three times for 10 min in high‐salt buffer. After that, input and IP samples were added to proteinase reaction buffer. Then, the RNA samples were purified with the RNA Clean & Concentrator kit (Zymo Research, Irvine, CA, USA).
2.17. HNSCC tissue collection and single‐cell preparation of mouse samples
To prepare a single cell suspension, Mettl1cKO and Mettl1cKO‐Ctrl HNSCC samples were first dissected from the mouse oral cavity and minced with scissors. Tissues were then processed using a tumor dissociation kit inmice (Miltenyi, Berlin, Germany) and enzymatically digested in a gentle MACS Octo Dissociator (Miltenyi) following the manufacturer's instructions. After dissociation, cells were passed through a 40 mm cell strainer (Corning) and rinsed with RPMI‐1640 containing 10% FBS. The single‐cell suspension was centrifuged at 1,500 rpm for 5 min and resuspended in DMEM containing 10% FBS. Cells were stained with trypan blue and counted using Countess II Automated Cell Counter Invitrogen (Thermo Fisher). A total of 10,000‐15,000 cells were then loaded onto a 10X Chromium microfluidics platform (10X Genomics, San Francisco, CA, USA) according to the manufacturer's guidelines. scRNA‐seq libraries were prepared using Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 (10X Genomics) according to the manufacturer's guidance. The library was sequenced with an Illumina NovaSeq 6000 System using 75 base pair (bp) paired‐end (PE) reads.
2.18. Preprocessing of scRNA‐seq data using Cell Ranger
For scRNA‐seq data analysis, we first used Cell Ranger software (https://support.10xgenomics.com/single‐cell‐gene‐expression/software/overview/ welcome) to perform sample demultiplexing, barcode processing, and single‐cell 3’ counting. CellRanger mkfastq was then used to demultiplex raw base call files from the Illumina NovaSeq 6000 sequencer into fastq files. We then used CellRanger count to align fastq files to the mm10 genome, filter, and quantify.
2.19. Processing analysis with the seurat package
Seurat version 3.2.2 was used to integrate datasets [40]. Genes that were expressed in fewer than 3 cells, cells that expressed fewer than 200 and more than 5000 genes, and cells with mitochondrial content greater than 12.5% were filtered out. Doublets were identified by DoubletFinder in each sample, and cells co‐expressing markers of different cell types were also excluded from further analysis. We used the sctransform function in Seurat to normalize the data. Principal component analysis (PCA) was performed using the top 3000 variable genes. For clustering, we used the FindClusters function. Uniform manifold approximation and projection (UMAP) was used to project cells into two dimensions using the top 20 principal components. For reclustering of immune cells, we selected clusters that were classified as myeloid/lymphoid cells. The same strategy was then applied to these cells by performing PCA, UMAP, and clustering as described above. For gene ontology (GO) enrichment analyses of epithelial cells, we used the gene set variation analysis (GSVA) package with default parameters. The C2 collection of curated gene sets used in this analysis was documented by the molecular signature database and implemented in the GSEABase package. For cell‐cell communication analysis, we used the iTALK R package to identify and map the differentially expressed ligands/receptors [41].
2.20. Flow cytometry for separation of Mrc1+ macrophages and langerhans cells
Three pairs of tumor samples from Mettl1cKO‐Ctrl and Mettl1cKO mice were lysed into single‐cell suspension by the above method. The cells from mouse tumor were stained with the antibodies against CD45 (1:200, 11‐0451‐81, Thermo Fisher), CD11b (1:200, 48‐0112‐82, Thermo Fisher), CD11c (1:200, 25‐0114‐81, Thermo Fisher), F4/80 (1:200, 45‐4801‐80, Thermo Fisher), and CD326 (1:200, 17‐5791‐80, Thermo Fisher) for 20 min at 25°C, followed by cleaning with staining buffer. Intracellular staining (CD206, 1:200, 12‐2061‐80, Thermo Fisher) was performed using Intracellular FIX/PERM kit (Thermo Fisher). Data acquisition was performed on Attune NxT4 (Thermo Fisher) and analyzed via FlowJo.
2.21. Statistical analysis
Significant tests for difference analysis were conducted by means of unpaired parametric two‐tailed Student's t‐test between two groups or chi‐square for tumor grade analysis. Survival analysis was performed with a Kaplan‐Meier curve and log‐rank test. The correlations between METTL1 and WDR4 expression levels were analyzed using IHC scores with Pearson's correlation test. P < 0.05 was defined as the threshold for statistical significance. Data management and statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA) and SPSS 20.0 software (IBM SPSS, Armonk, NY, USA).
3. RESULTS
3.1. METTL1 and WDR4 were upregulated and associated with poor prognosis of HNSCC patients
We analyzed the expression levels of the m7G tRNA methyltransferase complex components METTL1 and WDR4 and found that METTL1 and WDR4 were significantly upregulated and positively correlated with each other in HNSCC (r = 0.53) (Figure 1A and B). METTL1 was significantly associated with clinical stage, tumor stage, and differentiation grade, while WDR4 was only significantly associated with tumor stage in HNSCC (Supplementary Figure S1). Patients with high METTL1 or WDR4 expression had shorter OS (Figure 1C).
FIGURE 1.
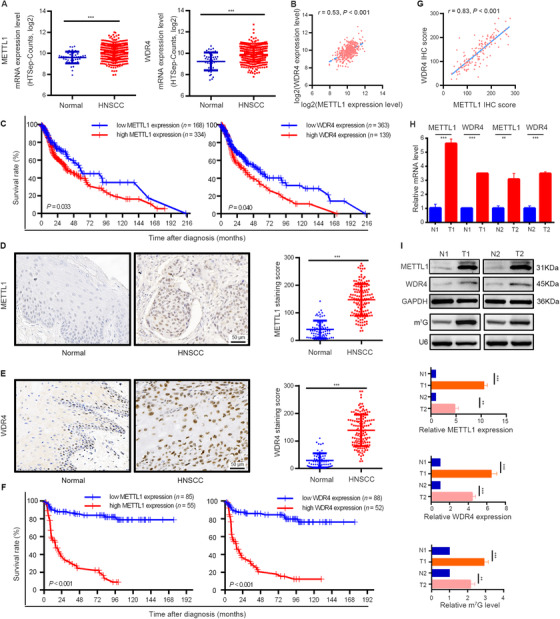
METTL1 and WDR4 are upregulated and associated with poor prognosis of HNSCC patients. (A) TCGA data showed that METTL1 (left) and WDR4 (right) were significantly upregulated in HNSCC tissues (n = 502) as compared with normal tissues (n = 44). (B) The correlation between METTL1 and WDR4 levels in HNSCC tissues using TCGA data. (C) Kaplan‐Meier curves of overall survival based on METTL1 (left) and WDR4 (right) levels in HNSCC tissues from TCGA data. (D) Representative images of IHC staining for METTL1 (left) and quantification (right) between 140 HNSCC tissue samples and 69 normal tissue samples. (E) Representative images of IHC staining for WDR4 (left) and quantification (right) between 140 HNSCC tissue samples and 69 normal tissue samples. (F) Kaplan‐Meier curves of overall survival based on METTL1 (left) and WDR4 (right) levels in HNSCC patients from our cohort. (G) The correlation between METTL1 and WDR4 levels in HNSCC patients from our cohort. (H,I) The mRNA levels and protein levels of METTL1 and WDR4 and m7G tRNA modification were upregulated in human HNSCC specimens examined by qRT‐PCR (H), Western blotting and Northwestern blotting (I). T1/T2 and N1/N2 represented tumor tissues and surrounding normal mucosal tissues from two HNSCC patients, respectively. Data are presented as the mean ± SD and analyzed by Student's t‐test, log rank test, or Pearson's correlation test. **, P < 0.01, ***, P < 0.001. Abbreviations: METTL1: Methyltransferase‐like 1; WDR4: WD repeat domain 4; HNSCC: head and neck squamous cell carcinoma; TCGA: the Cancer Genome Atlas; IHC: immunohistochemistry; m7G: N7‐methylguanosine; qRT‐PCR: quantitative real‐time PCR
To further validate this finding, IHC staining of 140 HNSCC tissues and 69 normal tissues obtained from the First Affiliated Hospital of Sun Yat‐sen University was carried out. We found that METTL1 and WDR4 were predominately expressed in the nucleus (Figure 1D and E). Quantification analysis demonstrated that METTL1 and WDR4 expression levels in HNSCC tissues were significantly higher than those in normal tissues (Figure 1D and E). METTL1, but not WDR4, was significantly associated with clinical stage and lymph node metastasis (Supplementary Figure S2). Consistently, Kaplan‐Meier analysis showed that patients with high METTL1 or WDR4 protein expression had shorter OS than patients with low METTL1 or WDR4 protein expression (Figure 1F). A significant correlation between METTL1 and WDR4 expression levels was also found in these HNSCC tissues (Figure 1G). Moreover, both univariate and multivariate analyses indicated that METTL1 and WDR4 expression levels were prognostic factors for patients with HNSCC (Supplementary Table S2).
Furthermore, we verified that both METTL1 and WDR4 were significantly more highly expressed in HNSCC tissues by Western blotting and qRT‐PCR, and the m7G modification levels in HNSCC tissues were correspondingly higher than those in adjacent normal tissues according to anti‐m7G Northwestern blotting (Figure 1H and I). Thus, we presumed that METTL1 and WDR4 may be related to HNSCC progression and associated with poor prognosis of HNSCC patients.
3.2. METTL1/WDR4 was required for HNSCC progression both in vitro and in vivo
First, we detected the METTL1 expression level in five HNSCC cell lines and found that all HNSCC cell lines expressed higher METTL1 protein levels than HOK cells (Figure 2A). To investigate whether high expression level of METTL1 was caused by mutation, we explored METTL1 mutational signature in upper aerodigestive squamous cell lines with CCLE database and 1478 HNSCC samples from 6 data sets. The results showed that there were no cell lines with METTL1 mutation, 0.6% samples with METTL1 amplification, and 0.9% samples with METTL1 mutation(Supplementary Figure S3A and S3B), suggesting that the high expression of METTL1 in HNSCC may not be caused by mutations in METTL1 itself. We then knocked out the METTL1 gene using sgRNA in SCC9 and SCC15 cell lines (Figure 2B), which expressed the highest levels of METTL1 protein. We found that knockout of METTL1 in SCC9 and SCC15 cells inhibited cell proliferation, migration, and invasion, suppressed colony formation, and promoted cell apoptosis (Figure 2C‐G and Supplementary Figure S3C‐F). Cell cycle analysis showed that the percentage of cells in the G1 phase was significantly increased in the METTL1‐KO group compared to that in the METTL1‐WT group (Figure 2H and Supplementary Figure S3G). We further investigated the oncogenic function of METTL1 in vivo using an orthotopic transplantation model. The tumors formed by METTL1‐KO cells grew slower, and the tumor volume was significantly smaller than those formed by METTL1‐WT cells (Figure 2I and J). Anti‐PCK staining revealed that METTL1 knockout resulted in a significant reduction in lymph node metastasis (Figure 2K).
FIGURE 2.
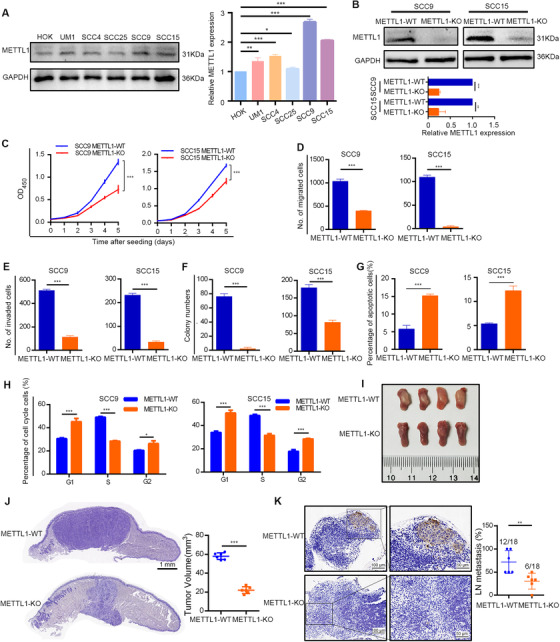
METTL1 is required for HNSCC progression both in vitro and in vivo. (A) Western blotting of METTL1 protein expression in HNSCC cell lines and HOK cells. (B) The effects of METTL1 knockout in SCC9 and SCC15 cells were verified by Western blotting. (C‐H) METTL1 knockout reduced the proliferation ability (C), restrained the migration (D), invasion (E) and colony formation (F), and promoted the apoptosis (G) and G1‐phase retention (H) of HNSCC cells. I. Representative images of mouse tongues from the orthotopic transplantation model. J. Representative H&E staining (left) and tumor volume (right) of METTL1‐WT and METTL1‐KO cell xenografts (n = 6). (K) Representative IHC staining (left) for PCK and LN metastasis percentage (right; the number of metastasis LN of the total LN) of METTL1‐WT and METTL1‐KO cells in xenografts. Data are presented as the mean ± SD and analyzed by Student's t‐test. *, P < 0.05, **, P < 0.01, ***, P < 0.001. Abbreviations: METTL1: Methyltransferase‐like 1 (METTL1); HOK: human oral keratinocytes; WT: wild‐type; KO: knockout; PCK: pan‐cytokeratin; LN: lymph node; IHC: immunohistochemistry; SD: standard deviation
In addition, we used WDR4 shRNA to knockdown WDR4 expression in SCC9 and SCC15 cell lines. Consistent with the results from METTL1‐KO cell lines, knockdown of WDR4 in SCC9 and SCC15 cells had similar functional roles in vitro and in a xenograft mouse model (Supplementary Figures S4 and S5).
Complementarily, we overexpressed METTL1 and its catalytically inactive mutant (without m7G tRNA methyltransferase activity) in the SCC25 cell line, which displayed the lowest expression of METTL1 (Figure 2A). The overexpression of wild‐type METTL1, but not of its catalytically inactive mutant, increased the m7G tRNA modification levels and significantly promoted SCC25 cell proliferation, migration, and invasion (Supplementary Figure S6). Overall, our data indicated that METTL1 and WDR4 acted as oncogenes in the progression and metastasis of HNSCC.
3.3. METTL1‐mediated m7G tRNA modification regulated mRNA translation and the PI3K/AKT/mTOR signaling pathway in HNSCC
To probe the mechanism of METTL1 in regulating HNSCC progression, we first examined tRNA m7G signals in METTL1‐KO or WDR4‐knockdown cells. We found that either METTL1 knockout or WDR4 knockdown led to reduced levels of tRNA m7G modification (Figure 3A and Supplementary Figure S7A). We then utilized TRAC‐seq to profile the single‐nucleotide resolution mapping of global m7G tRNA modification in SCC15 cells. A total of 16 tRNAs containing m7G modification in HNSCC and the motif “RRGGYYS” were identified within the V‐loop of tRNA (Figure 3B and C). METTL1 knockout significantly decreased the m7G tRNA modification levels of the identified tRNAs compared with the METTL1‐WT group (Figure 3D and E). While the non‐m7G‐modified tRNA levels were not substantially altered, the majority of m7G‐modified tRNAs showed decreased expression levels (Figure 3F and G). Northern blotting confirmed that the expression levels of m7G‐modified tRNAs such as ValACC and ThrTGT were decreased, while the expression level of the non‐modified tRNA ProAGG remained unchanged (Figure 3H).
FIGURE 3.
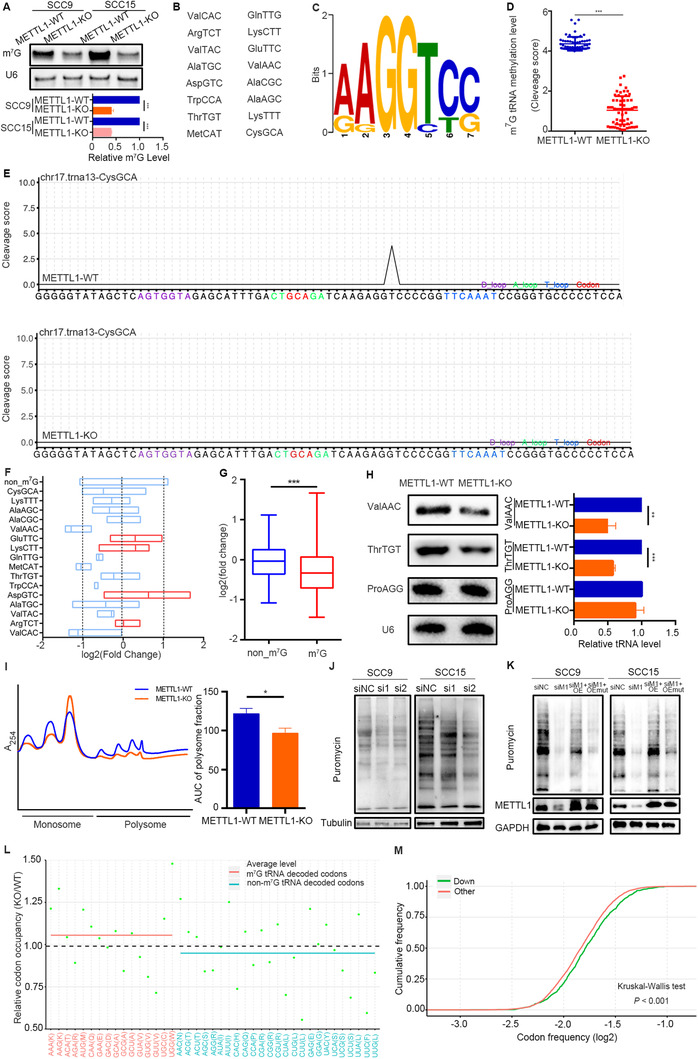
METTL1‐mediated m7G tRNA modification regulates tRNA expression and mRNA translation in HNSCC. (A) Northwestern blotting of m7G modification in tRNAs from METTL1‐WT and METTL1‐KO SCC9 and SCC15 cells. (B) List of m7G‐modified tRNAs identified by TRAC‐seq in SCC15 cells. (C) Sequence motif in the m7G sites identified by TRAC‐seq in SCC15 cells. (D) Quantitative comparison of cleavage scores between METTL1‐WT and METTL1‐KO SCC15 cells. (E) Representative images showing the cleavage score of identified tRNAs at the motif site from different groups (METTL1‐WT, top; METTL1‐KO, bottom). (F) Expression profile of the 16 m7G‐modified tRNAs. Each boxplot shows the expression of a tRNA type that was calculated from the combined expression of all tRNA genes for the same tRNA type. (G) Quantitative comparison of the fold change in expression between m7G‐modified and non‐m7G‐modified tRNAs. (H) Validation of the expression of representative m7G‐modified tRNAs by Northern blotting using the indicated tRNA probes. ProAGG (non‐m7G‐modified tRNA) and U6 snoRNA were used as loading controls. (I) Polysome profiling of METTL1‐WT and METTL1‐KO SCC15 cells. (J) Puromycin uptake assay of METTL1 knockout using two independent siRNAs. Total protein samples were examined by Western blotting using an anti‐puromycin antibody. (K) Puromycin uptake assay of METTL1‐KO cells transfected with wild‐type overexpression or catalytic inactive METTL1 plasmids. (L) Change in ribosome occupancy at A sites in METTL1‐KO cells compared to METTL1‐WT SCC15 cells. Only codons detected in both TRAC‐seq and Ribo‐Seq are shown. The average relative occupancy ratio of codons decoded by m7G tRNAs (pink) and codons decoded by non‐m7G tRNAs (blue) are presented. (M) Codon frequency in the CDS region of genes with decreased TE (down) and other genes. Data are presented as the mean ± SD and analyzed by Student's t‐test or Kruskal‐Wallis test. *, P < 0.05, **, P < 0.01, ***, P < 0.001. Abbreviations: m7G: N7‐methylguanosine; WT: wild‐type; KO: knockout; METTL1: Methyltransferase‐like 1; TRAC‐seq: m7G site‐specific tRNA reduction and cleavage‐sequencing; snoRNA: small nucleolar RNA; siRNAs: small interfering RNAs; siNC, negative control siRNA; si1, siMETTL1‐1; si2, siMETTL1‐2; OE, wild‐type METTL1 overexpression; OEmut, catalytically inactive METTL1; Ribo‐Seq: ribosome sequencing; TE: translation efficiency; CDS: sequence coding for amino acids in protein; SD: standard deviation
Furthermore, polysome profiling results demonstrated that METTL1 knockout led to reduced polyribosome peaks, indicating that global translation was inhibited in METTL1‐KO cells (Figure 3I and Supplementary Figure S7B). Moreover, a puromycin uptake assay showed that the incorporation of puromycin was significantly decreased in METTL1‐KO cells (Figure 3J). We then performed a rescue assay and found that overexpression of wild‐type METTL1 (but not a catalytically inactive mutant) rescued impaired mRNA translation in METTL1‐KO cells (Figure 3K). We conducted ribosome foot printing and found that the absence of METTL1 increased the codon‐dependent ribosome pause of m7G tRNA (Figure 3L), indicating that METTL1‐mediated m7G tRNA modification was necessary for efficient codon recognition during the ribosomal transition of m7G tRNA decoding codons. Codon frequency analysis revealed that the mRNAs with decreased TE had a significantly higher frequency of m7G‐modified tRNA‐decoding codons (Figure 3M). Thus, genes with a higher frequency of m7G tRNA‐decoded codons had decreased TE in METTL1‐KO cells, which may be related to ribosome pausing at m7G‐tRNA‐dependent codons.
To determine the downstream mRNA targets of METTL1‐mediated m7G tRNA modification, we conducted RNC‐seq to investigate actively translated mRNAs and identified 3945 mRNAs with decreased TRs and 2172 mRNAs with increased TRs (Figure 4A). KEGG enrichment analysis and GSEA revealed that the PI3K/AKT/mTOR signaling pathway was consistently enriched in genes regulated by METTL1 (Figure 4B and C, Supplementary Figure S7C). Consistently, Western blotting and IHC staining assays demonstrated that the level of PI3K protein and the phosphorylation level of AKT and mTOR were significantly reduced in METTL1‐KO cells (Figure 4D and Supplementary Figure S8). We further examined expression of downstream proteins and found that in METTL1‐KO cells, the expression of Cyclin D1, Vimentin, MMP9, Bcl‐2, and P‐S6K were significantly decreased, while the expression of BAX was significantly upregulated, compared with METTL1‐WT cells (Figure 4D). Furthermore, RNC‐qPCR assays showed that METTL1 knockout barely influenced the transcription of PIK3CA, but significantly decreased its translation (Figure 4E). We performed a rescue assay through PIK3CA overexpression or treatment with the AKT activator SC79 in METTL1‐KO cells and found that both approaches partially reversed the effects of METTL1 knockout on HNSCC cells (Figure 4F‐I).
FIGURE 4.
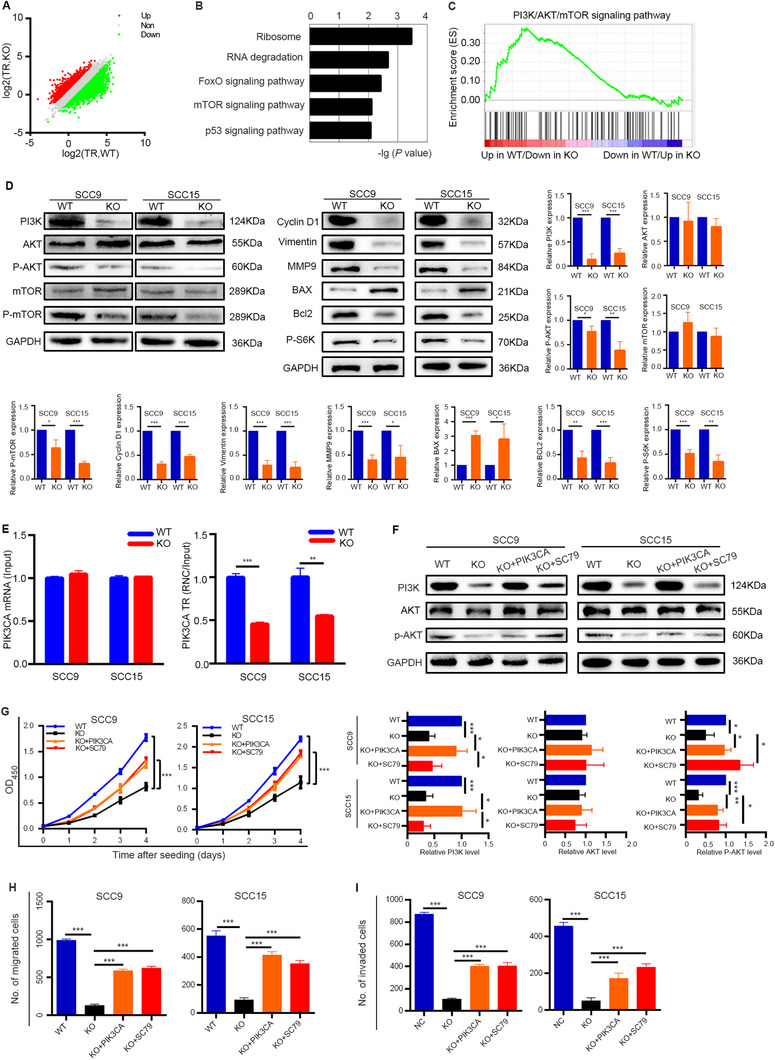
METTL1‐mediated m7G tRNA modification regulates the activity of the PI3K/AKT/mTOR signaling pathway. (A) Scatterplot of the TRs in METTL1‐WT and METTL1‐KO SCC15 cells. TRs were calculated by dividing the ribosome‐binding transcript signals by input RNA‐seq signals. (B) KEGG pathway analysis of the genes with decreased TRs upon METTL1 knockout. (C) The PI3K/AKT/mTOR signaling pathway was enriched in RNC‐seq datasets by GSEA (NES = 1.64, FDR = 0.165, P < 0.001). (D) Western blotting of PI3K/AKT/mTOR signaling pathway proteins and downstream proteins using the indicated antibodies. (E) qRT‐PCR analysis of PIK3CA with RNC and input samples in SCC9 and SCC15 cells. (F) The protein levels of PI3K, AKT, and p‐AKT in METTL1‐WT, METTL1‐KO, PI3K‐transfected METTL1‐KO cells (KO + PIK3CA) and 5 μg/mL SC79‐treated METTL1‐KO cells cultured with (KO + SC79). (G‐I) The proliferation (G), migration (H) and invasion abilities (I) were partially restored after transfecting METTL1‐KO cells with the PI3K plasmid or activating AKT. Data are presented as the mean ± SD and analyzed by Student's t‐test. *, P < 0.05, **, P < 0.01, ***, P < 0.001. Abbreviations: PI3K/AKT/mTOR: phosphatidylinositol‐3‐kinase/protein kinase B/mammalian target of rapamycin; METTL1: Methyltransferase‐like 1; WT: wild‐type; KO: knockout; TRs: translation ratios; KEGG: Koto Encyclopedia of Genes and Genomes; GSEA: gene set enrichment analysis; NES: normalized enrichment score; FDR: false discovery rate; qRT‐PCR: quantitative real‐time PCR; RNC: Ribosome nascent‐chain complex‐bound; MMP9: matrix metalloprotein 9; Bcl‐2: B‐cell lymphoma‐2; P‐S6K: phosphorylation of S6 kinase; BAX: Bcl‐2‐associated X protein; PIK3CA: phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha; SD: standard deviation
3.4. A transgenic mouse model verified that Mettl1‐PI3K/Akt/mTOR was required for HNSCC carcinogenesis and metastasis
To explore the role of Mettl1 in spontaneous HNSCC, we generated Mettl1cKO‐Ctrl and Mettl1cKO mice, which allowed us to specifically delete Mettl1 expression in the mouse oral epithelium. Mice were treated with 4NQO for 16 weeks to develop HNSCC and then euthanized to collect their tongues and cervical lymph nodes 12 weeks later. We first validated the knockout efficiency of Mettl1 in the oral epithelium by IHC staining (Figure 5A). We found that Mettl1cKO mice developed fewer and smaller clinically visible lesions than Mettl1cKO‐Ctrl mice (Figure 5B). The numbers of visible lesions in Mettl1cKO‐Ctrl and Mettl1cKO mice were 3.7 ± 0.3, and 2.8 ± 0.2, respectively (Figure 5C). In addition, Mettl1cKO mice manifested a lower histopathological grade than Mettl1cKO‐Ctrl mice (Figure 5D). Most lesions of Mettl1cKO mice were carcinoma in situ (37.0%) and epithelial dysplasia (37.5%); however, Mettl1cKO‐Ctrl mice developed more invasive carcinoma (50.0%) and carcinoma in situ (37.0%). Furthermore, Mettl1cKO mice displayed more suppressive proliferation ability (Ki67) in lesions and lower rate of lymph node metastasis (PCK) (Figure 5E and F).
FIGURE 5.
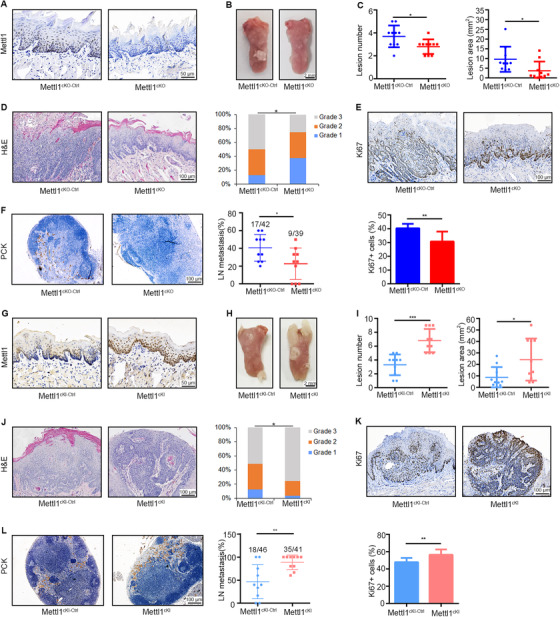
A transgenic mouse model verifies that Mettl1 is required for HNSCC carcinogenesis and metastasis. (A) IHC staining of Mettl1+ cells in tongue tissues from Mettl1cKO‐Ctrl and Mettl1cKO mice. (B) Representative images of tongue lesions 28 weeks after 4NQO treatment in Mettl1cKO‐Ctrl and Mettl1cKO mice. (C) The number of lesions (left) and quantification of lesion areas (right) visible in the tongues of Mettl1cKO‐Ctrl and Mettl1cKO mice (n = 10). (D) Representative H&E staining of lesions (left) from Mettl1cKO‐Ctrl and Mettl1cKO mice. Quantification of histological grade of lesions in different groups (right). Grade 1, epithelium dysplasia; grade 2, carcinoma in situ; grade 3, invasive carcinoma. (E) Representative IHC staining (up) and quantification (bottom) of Ki67 in Mettl1cKO‐Ctrl and Mettl1cKO mice. (F) Representative IHC staining (left) and quantification (right) of lymph node metastasis rate (the proportion of metastasis lymph nodes in all lymph nodes) in Mettl1cKO‐Ctrl and Mettl1cKO mice. (G) IHC staining of Mettl1+ cells in tongue tissues from Mettl1cKI‐Ctrl and Mettl1cKI mice. (H) Representative images of tongue lesions from Mettl1cKI‐Ctrl and Mettl1cKI mice 28 weeks after 4NQO treatment. (I) The number of lesions (left) and quantification of lesion areas (right) visible in the tongue from Mettl1cKI‐Ctrl and Mettl1cKI mice (n = 10). (J) Representative H&E staining (left) of lesions from Mettl1cKI‐Ctrl and Mettl1cKI mice and quantification of histological grade of lesions (right) in different groups. (K) Representative IHC staining (up) and quantification (bottom) of Ki67 in Mettl1cKI‐Ctrl and Mettl1cKI mice. (L) Representative IHC staining (left) and quantification (right) of lymph node metastasis rate in Mettl1cKI‐Ctrl and Mettl1cKI mice. Data are presented as the mean ± SD and analyzed by Student's t test or Chi‐squared Test. *, P < 0.05, **, P < 0.01, ***, P < 0.001. Abbreviations: Mettl1: Methyltransferase‐like 1; Mettl1cKO‐Ctrl: K14CreER;Mettl1wt/wt mice; Mettl1cKO: K14CreER;Mettl1fl/fl mice; Mettl1cKI‐Ctrl: K14Cre;Mettl1wt/wt mice; Mettl1cKI: K14Cre;Mettl1KI/KI mice; IHC: immunohistochemistry; 4NQO: 4‐nitroquinoline N‐oxide; PCK, pan‐cytokeratin; LN, lymph node; SD: standard deviation
Mettl1cKI and Mettl1cKI‐Ctrl mice were treated with 4NQO to facilitate HNSCC development. IHC staining showed that the epithelium of Mettl1cKI mice exhibited a more strongly positive Mettl1 signal than Mettl1cKI‐Ctrl mice (Figure 5G). Quantitative analysis revealed that Mettl1cKI mice developed more lesions with larger lesion areas (Figure 5H and I). Tumors from Mettl1cKI mice had higher histopathological grades and higher proliferation (Ki67) and metastasis abilities than those from Mettl1cKI‐Ctrl mice (Figure 5J‐L).
To further understand how Mettl1 regulates HNSCC progression and metastasis in vivo, we collected HNSCC tissues from two pairs of Mettl1cKO and Mettl1cKO‐Ctrl mice and performed scRNA‐seq using the 10X Genomics Chromium platform. After quality control and filtering, we obtained 18,665 transcriptomes of single cells in total. Further unsupervised clustering identified HNSCC epithelial cells as well as stromal subtypes, including fibroblasts, endothelial cells, muscle cells, myeloid cells, B cells, and T cells (Figure 6A and Supplementary Figure S9A), which were in accordance with previous studies [42, 43]. We reclustered epithelial cells and found that the Mettl1 gene was downregulated in Mettl1cKO mice compared with Mettl1cKO‐Ctrl mice (Figure 6B and C). Moreover, the results from GSVA of epithelial cells revealed that PI3K/Akt activation was significantly decreased in Mettl1cKO HNSCC samples (Figure 6D). We next explored how Mettl1/Wdr4 expression states varied among different subsets of malignant cells. Mettl1/Wdr4 expression varied within and between tumors in their expression of signatures related to cell cycle, epithelial differentiation, partial epithelial‐to‐mesenchymal transition (p‐EMT), stress, and undefined subsets (Supplementary Figure S9B). The Mettl1 expression levels were higher in cell cycle, epithelial differentiation, p‐EMT, and stress subsets than in undefined subsets. Meanwhile, p‐EMT subset expressed the highest Wdr4 levels, followed by cell cycle subset (Supplementary Figure S9B). Based on the cancer stem cell (CSC) gene signatures, we found that the CSC scores of cells with Mettl1/Wdr4 expression were significantly higher than those of cells without Mettl1/Wdr4 expression (Supplementary Figure S9C).
FIGURE 6.
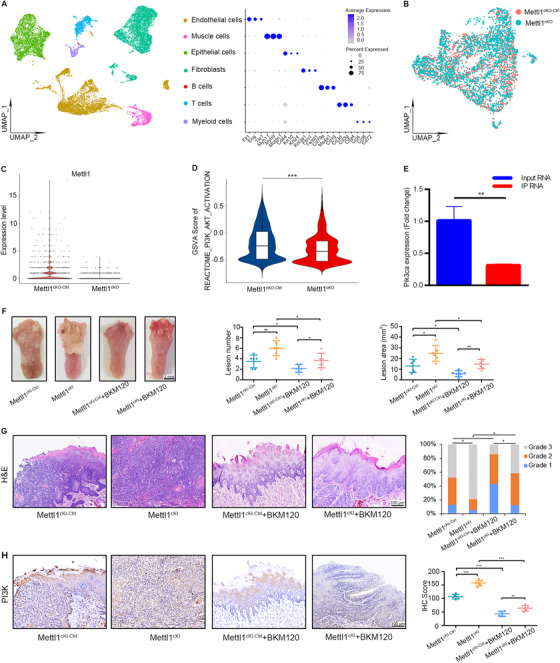
A transgenic mouse model verifies that Mettl1 regulates PI3K/Akt/mTOR activity in HNSCC. (A) UMAP of scRNA‐seq cells recovered from both Mettl1cKO‐Ctrl and Mettl1cKO mice labeled by cell type (left). Dot plot of selected subset‐associated genes across cell types from scRNA‐seq data (right). (B) UMAP of reclustered epithelial cells from Mettl1cKO‐Ctrl and Mettl1cKO mice. (C) Violin plots of the expression of the Mettl1 gene in epithelial cells from Mettl1cKO‐Ctrl and Mettl1cKO mice. (D) Violin plots of the GSVA results for epithelial cells revealed that PI3K/Akt activation was significantly decreased in Mettl1cKO HNSCC samples. (E) qRT‐PCR of Pik3ca expression (K14CreER;RiboTag;Mettl1fl/fl /K14CreER;RiboTag;Mettl1wt/wt ) in input RNA was normalized to that of Gapdh and the ratio of IP RNA. (F) Representative images (left) of tongue lesions from Mettl1cKI‐Ctrl and Mettl1cKI mice by oral gavage of BKM120 (Mettl1cKI‐Ctrl + BKM120 and Mettl1cKI + BKM120) compared with Mettl1cKI‐Ctrl and Mettl1cKI mice by oral gavage of buffer agent (Mettl1cKI‐Ctrl and Mettl1cKI). Quantification of the number of lesions (middle) and the lesion areas (right) visible in the tongue (n = 6). (G) Representative H&E staining of lesions (left) from Mettl1cKI‐Ctrl, Mettl1cKI, Mettl1cKI‐Ctrl + BKM120 and Mettl1cKI + BKM120 mice. Quantification of histological grade of lesions in different groups (right). (H) Representative IHC staining (left) and quantification (right) of PI3K in Mettl1cKI‐Ctrl, Mettl1cKI, Mettl1cKI‐Ctrl + BKM120 and Mettl1cKI + BKM120 mice. Data are presented as the mean ± SD and analyzed by Student's t‐test or Chi‐squared Test. *, P < 0.05, **, P < 0.01, ***, P < 0.001. Abbreviations: PI3K/Akt/mTOR: phosphatidylinositol‐3‐kinase/protein kinase B/mammalian target of rapamycin; Pik3ca: phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha; Mettl1: Methyltransferase‐like 1; IHC: immunohistochemistry; IP: immunoprecipitation; UMAP: Uniform manifold approximation and projection; GSVA: gene set variation analysis; Mettl1cKO‐Ctrl: K14CreER;Mettl1wt/wt mice; Mettl1cKO: K14CreER;Mettl1fl/fl mice; Mettl1cKI‐Ctrl: K14Cre;Mettl1wt/wt mice; Mettl1cKI: K14Cre;Mettl1KI/KI mice; SD: standard deviation
To validate the effects of Mettl1 knockout on the PI3K/Akt signaling pathway in mouse HNSCC, we used RiboTag mice and generated K14CreER;RiboTag;Mettl1fl/fl and K14CreER;RiboTag;Mettl1wt/wt mice. The immunofluorescent staining results verified that HA‐tagged ribosomal protein ribosomal protein L22 (RPL22) was specifically expressed in the epithelial tissues of mouse HNSCC (Supplementary Figure S10A). We then immunoprecipitated mRNA being translated from the tongue lesions with anti‐HA antibody and found that the expression of the Pik3ca gene in K14CreER;RiboTag;Mettl1fl/fl mice was decreased compared with that in K14CreER;RiboTag;Mettl1wt/wt mice according to the qRT‐PCR assay (Figure 6E). Moreover, the IHC staining results showed that the expression levels of members of the PI3K/Akt/mTOR signaling pathway were decreased after Mettl1 knockout in transgenic mouse models (Supplementary Figure S10B).
Interestingly, treatment with SC79 for 4 weeks after 4NQO treatment led to an increase in the lesion number, lesion area, malignancy, proliferation, and metastasis of HNSCC in Mettl1cKO mice (Supplementary Figure S11). On the other hand, we used an anti‐PI3K agent (BKM120) to inhibit the PI3K/Akt/mTOR signaling pathway in Mettl1cKI mice. We found that the phospho‐Akt level was significantly decreased after BKM120 treatment (Supplementary Figure S12A). Mettl1cKI mice treated with BKM120 developed fewer lesions with reduced lesion area, decreased invasive carcinomas, and reduced lymph node metastasis (Figure 6F‐H and Supplementary Figure S12B‐C).
3.5. scRNA‐seq analysis of microenvironment alterations in HNSCC after Mettl1 knockout
Tumor‐infiltrating myeloid cells are essential parts of the HNSCC microenvironment [44]. We then analyzed scRNA‐seq data to further examine the landscape of immune cells in HNSCC after Mettl1 knockout. In total, we found 471 myeloid cells, which were classified into seven subclusters: neutrophils, C‐X3‐C motif chemokine receptor 1 (Cx3cr1+) macrophages, mannose receptor C type 1 (Mrc1+) macrophages, macrophage‐3 (Macro‐3) cells, dendritic cells, Langerhans cells, and migrating cells. Notably, the proportions of Mrc1+ macrophages, Macro‐3 cells, and Langerhans cells in Mettl1cKO HNSCC were elevated compared with those in Mettl1cKO‐Ctrl samples (Figure 7A‐C). Then, we conducted flow cytometry to explore the proportion changes of Mrc1+ macrophages and Langerhans cells. The results showed that the proportion of Mrc1+ macrophages and Langerhans cells were significantly elevated in Mettl1cKO HNSCC compared with those in Mettl1cKO‐Ctrl samples (Supplementary Figure S13), which were consistent with scRNA‐seq data. In addition, we identified a total of 1,013 tumor‐infiltrating lymphocytes (TILs), which were clustered into seven different subsets as CD4 memory T, CD4+ naïve T, CD4+ T exhausted, CD8+ naïve T, natural killer cells (NKT), T helper 17 (Th17), and regulatory T (Treg) cells according to their gene signatures (Figure 7D‐F). We quantified the cell number and proportion of each subcluster and found a substantial decrease in CD4+ T exhaustion and Tregs in Mettl1cKO HNSCC samples (Figure 7E).
FIGURE 7.
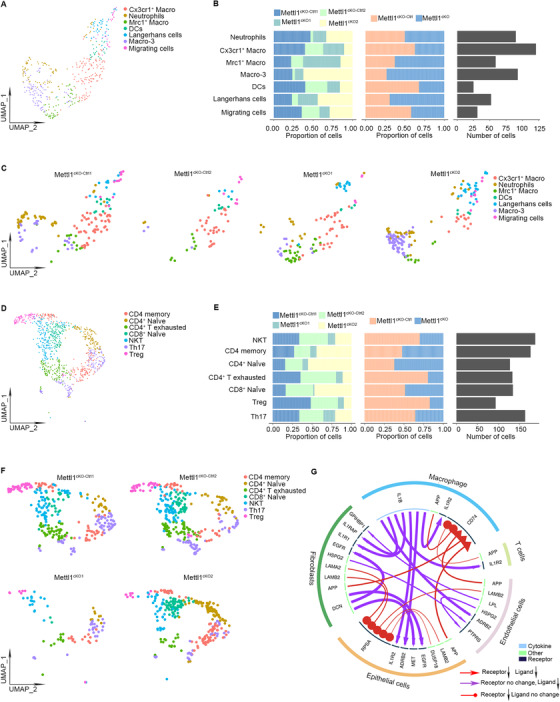
scRNA‐seq analysis of tumor cell‐microenvironment crosstalk in HNSCC tumors after Mettl1 knockout. (A) UMAP of myeloid subsets, including neutrophils, Cx3cr1+ macrophages, Mrc1+ macrophages, Macro‐3, dendritic cells, Langerhans cells, and migrating cells, in Mettl1cKO‐Ctrl and Mettl1cKO mice. (B) Bar plots of the proportions of cell types and total cell number in Mettl1cKO‐Ctrl and Mettl1cKO mice. (C) UMAP of myeloid subsets of each sample. (D) UMAP of TIL subsets, including memory CD4 T cells, naïve CD4+ T cells, exhausted CD4+ T cells, naïve CD8+ T cells, NKT cells, Th17 cells, and Tregs, in Mettl1cKO‐Ctrl and Mettl1cKO mice. (E) Bar plots of the proportions of cell types and total cell number in Mettl1cKO‐Ctrl and Mettl1cKO mice. (F) UMAP of TIL subsets of each sample. (G) Circos plot showing significant crosstalk alterations between stromal cells and epithelial cell subclusters (Mettl1cKO vs. Mettl1cKO‐Ctrl). Abbreviations: Mettl1: Methyltransferase‐like 1; KO: knockout; TIL: tumor‐infiltrating lymphocytes; Mettl1cKO‐Ctrl: K14CreER;Mettl1wt/wt mice; Mettl1cKO: K14CreER;Mettl1fl/fl mice; Macro‐3: macrophage‐3; DCs: dendritic cells; UMAP: Uniform manifold approximation and projection
Because stromal cells may affect HNSCC via direct interactions with epithelial cells or vice versa, we investigated stromal and epithelial cell receptor–ligand pairing in both Mettl1cKO and Mettl1cKO‐Ctrl HNSCC samples. We found alterations in crosstalk between 1,739 receptor‐ligand pairs in stromal cells versus paired epithelial cell subclusters. As shown in the chord diagrams (Figure 7G), we found inhibition of multiple receptor‐ligand pairs, including interleukin 1 beta (Il1b)‐interleukin 1 receptor type 2 (Il1r2) and Il1b‐adrenoceptor beta 2 (Adrb2), in Mettl1cKO HNSCC samples. These data indicated that Mettl1 knockout in tumor cells led to alterations in the communication between cancer cells and their microenvironment.
4. DISCUSSION
In the current study, we detected elevated levels of m7G tRNA modifications and upregulation of the m7G tRNA methyltransferase complex components METTL1 and WDR4 in human HNSCC. In addition, we found that high METTL1 expression was associated with a poor prognosis in our clinical cohort and the TCGA database, indicating that METTL1 is a promising prognostic factor for HNSCC patients. The difference between TCGA data and patients’ sample data would attribute to the following reasons. Firstly, TCGA data sources are mostly Caucasian, while our patients are all East Asian. Racial differences may affect METTL1 expression. Secondly, TCGA data analysis was based on mRNA levels, while IHC analysis of our patients’ sample was based on protein levels. However, the abundance of protein in cells is reported to be much higher than the abundance of their own transcript [45]. Although there were differences between TCGA data and our patients’ sample data, their trend is consistent, showing higher METTL1 levels in HNSCC than in normal epithelium. Functionally, METTL1 knockout or WDR4 knockdown resulted in decreased cell proliferation, migration and invasion abilities and survival in HNSCC cells. An orthotopic transplantation model confirmed that METTL1 knockout and WDR4 knockdown greatly impaired HNSCC tumorigenesis and lymph node metastasis in vivo. Our results indicate a broad impact of METTL1 and m7G tRNA methylation on HNSCC development and targeted therapy.
Fundamentally, METTL1 and WDR4 are required for the introduction of the m7G at position 46 of tRNA and for managing appropriate downstream translation [46]. However, the repertoire of tRNAs regulated by METTL1 and WDR4 may vary among different contexts, hence driving the translation of different subsets of mRNAs. For example, Lin et al. [16] reported 22 species of mouse m7G‐modified tRNA by Mettl1 in mouse embryonic stem cells (mESCs), which are responsible for the translation of genes related to the self‐renewal and neural differentiation of mESCs. In human HNSCC, our TRAC‐seq results revealed 16 different types of m7G‐modified tRNAs, 13 of which overlap with those reported in mESCs. This distinction in m7G‐modified tRNA species might lead to the biased use of codons correlated with different gene expression levels. Indeed, we found that ribosomes were suspended in m7G tRNA‐dependent codons and that genes with a higher frequency of m7G tRNA codons had reduced TE after the knockout of METTL1.
We found that METTL1/WDR4‐mediated m7G tRNA modification led to inhibition of the PI3K/AKT/mTOR signaling pathway in HNSCC via the integration of bioinformatics and histological data. Importantly, the effects of Mettl1 overexpression in promoting mouse HNSCC progression and metastasis were rescued by treatment with a PI3K/Akt/mTOR signaling pathway inhibitor, further supporting this pathway as a functional mediator of Mettl1. The PI3K/AKT/mTOR signaling pathway is active in over 90% of HNSCC and is linked to the failure of current therapy for HNSCC [47]. Previous reports have shown that activation of the PI3K/AKT/mTOR signaling pathway is mainly due to genetic alterations in key factors involved in this pathway, including epidermal growth factor receptor (EGFR), PIK3CA, and phosphatase and tensin homolog (PTEN) [47]. Our study indicated that tRNA modification‐mediated translational regulation also played a critical role in determining the activity of the PI3K/AKT/mTOR signaling pathway in HNSCC cells. Interestingly, the PI3K/AKT/mTOR signaling pathway has also been shown to play a pivotal role in mRNA translation through ribosomal S6 kinase (S6K) and eukaryotic translation initiation factor 4E‐binding protein (4E‐BP1) [48, 49]. However, whether METTL1/WDR4‐mediated m7G tRNA modifications and S6K or 4E‐BP1 can coordinately control gene expression at the level of translation remains to be further investigated.
The tumor microenvironment and tumor cells are closely related and interact perpetually [44]. In the present study, we found disruption of cell‐cell communication between stromal cells and tumor cells after Mettl1 knockout. We noticed the downregulation of Il1b‐Il1r2 and Il1b‐Adrb2 interactions between epithelial cells and macrophages. Intriguingly, a recent scRNA‐seq study reported that IL1B‐IL1R2 interactions between macrophages and cancer cells exert tumor‐promoting functions in gastric cancer [50]. In addition, blockade of β2‐adrenergic receptor, which is encoded by the Adrb2 gene, was shown to regulate the immune status of the tumor microenvironment and enhance the response to immunotherapy in patients [51]. However, clinical application of METTL1‐targeting therapy is hampered by lack of METTL1 small molecule inhibitors. Together, our findings provide fundamental insight into how METTL1/WDR4‐mediated m7G tRNA modification controls multifaceted HNSCC progression.
5. CONCLUSIONS
The tRNA m7G methyltransferase METTL1 was found to promote the development and malignancy of HNSCC through regulating global mRNA translation, including the PI3K/AKT/mTOR signaling pathway, and found to alter immune landscape. METTL1 could be a promising treatment target for HNSCC patients.
DECLARATIONS
CONTRIBUTIONS
DMC, AXW and QMC conceived of, designed, and supervised the study; JC performed most of the experiments and wrote the manuscript; KL, and JWC, XCW performed some of the experiments; RSL performed scRNA‐seq assay; MSC and ZC fed the transgenic mice and performed part of the experiments; FFC, QTH, SL, CHZ, and YZJ, provided technical assistance with the experiments; All co‐authors have reviewed and approved this version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Human data and samples were obtained with informed consent and the use was approved by the Institutional Review Board of the First Affiliated Hospital of Sun Yat‐Sen University (2016074). The nude mouse experiments performed were approved by the Laboratory Animal Center of Sun Yat‐Sen University (SYSU‐IACUC‐2021‐000092). The transgenic mouse experiments performed were approved by the Laboratory Animal Center of Sun Yat‐Sen University (SYSU‐IACUC‐2020‐000569).
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This study was supported by National Natural Science Foundation of China (81872409, 82173362), Natural Science Foundation of Guangdong Province (2018A030313610, 2019A1515110110 and 2020A1515010291), The Open Funding of the State Key Laboratory of Oral Diseases (SKLOD2021OF02). We are indebted to Yan Zhu for help with some of the experiments and for help discussion.
Chen J, Li K, Chen J, Wang X, Ling R, Cheng M, et al. Aberrant translation regulated by METTL1/WDR4‐mediated tRNA N7‐methylguanosine modification drives head and neck squamous cell carcinoma progression. Cancer Commun. 2022;42:223–244. 10.1002/cac2.12273
Contributor Information
Qianming Chen, Email: qmchen@zju.edu.cn.
Anxun Wang, Email: wanganx@mail.sysu.edu.cn.
Demeng Chen, Email: chendm29@mail.sysu.edu.cn.
DATA AVAILABILITY STATEMENT
We have deposited all sequencing datasets in this study on GEO (GSE172146, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172146).
REFERENCES
- 1. Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA‐mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun (Lond). 2021;41(2):109‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janin M, Coll‐SanMartin L, Esteller M. Disruption of the RNA modifications that target the ribosome translation machinery in human cancer. Mol Cancer. 2020;19(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ju H, Hu Z, Wei D, Huang J, Zhang X, Rui M, et al. A novel intronic circular RNA, circGNG7, inhibits head and neck squamous cell carcinoma progression by blocking the phosphorylation of heat shock protein 27 at Ser78 and Ser82. Cancer Commun (Lond). 2021;41(11):1152‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu L, Wu Y, Li Q, Liang J, He Q, Zhao L, et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m6A Methylation in Oral Squamous Cell Carcinoma. Mol Ther. 2020;28(10):2177‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Santos M, Fidalgo A, Varanda AS, Oliveira C, Santos MAS. tRNA Deregulation and Its Consequences in Cancer. Trends Mol Med. 2019;25(10):853‐65. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol. 2021;22(6):375‐92. [DOI] [PubMed] [Google Scholar]
- 7. Pan Y, Yan TM, Wang JR, Jiang ZH. The nature of the modification at position 37 of tRNAPhe correlates with acquired taxol resistance. Nucleic Acids Res. 2021;49(1):38‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chou HJ, Donnard E, Gustafsson HT, Garber M, Rando OJ. Transcriptome‐wide Analysis of Roles for tRNA Modifications in Translational Regulation. Mol Cell. 2017;68(5):978‐92.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He C, Bozler J, Janssen KA, Wilusz JE, Garcia BA, Schorn AJ, et al. TET2 chemically modifies tRNAs and regulates tRNA fragment levels. Nat Struct Mol Biol. 2021;28(1):62‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49(24):4934‐44. [DOI] [PubMed] [Google Scholar]
- 12. Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, et al. ALKBH1‐Mediated tRNA Demethylation Regulates Translation. Cell. 2016;167(3):816‐28.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nedialkova DD, Leidel SA. Optimization of Codon Translation Rates via tRNA Modifications Maintains Proteome Integrity. Cell. 2015;161(7):1606‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alexandrov A. tRNA m7G methyltransferase Trm8p/Trm82p: Evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA. 2005;11(5):821‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boulias K, Greer EL. Put the Pedal to the METTL1: Adding Internal m7G Increases mRNA Translation Efficiency and Augments miRNA Processing. Mol Cell. 2019;74(6):1105‐7. [DOI] [PubMed] [Google Scholar]
- 16. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4‐Mediated m7G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self‐Renewal and Differentiation. Mol Cell. 2018;71(2):244‐55.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu Y, Yang C, Zhao Y, Chi Q, Wang Z, Sun B. Overexpressed methyltransferase‐like 1 (METTL1) increased chemosensitivity of colon cancer cells to cisplatin by regulating miR‐149‐3p/S100A4/p53 axis. Aging (Albany NY). 2019;11(24):12328‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Na W, Fu L, Luu N, Shi YB. Direct activation of tRNA methyltransferase‐like 1 (Mettl1) gene by thyroid hormone receptor implicates a role in adult intestinal stem cell development and proliferation during Xenopus tropicalis metamorphosis. Cell Biosci. 2020;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng Y, Zhou Z, Ji W, Lin S, Wang M. METTL1‐mediated m7G methylation maintains pluripotency in human stem cells and limits mesoderm differentiation and vascular development. Stem Cell Res Ther. 2020;11(1):306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Filonava L, Torres AG, Ribas de Pouplana L. A novel cause for primordial dwarfism revealed: defective tRNA modification. Genome Biol. 2015;16:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma J, Han H, Huang Y, Yang C, Zheng S, Cai T, et al. METTL1/WDR4‐mediated m7G tRNA modifications and m7G codon usage promote mRNA translation and lung cancer progression. Mol Ther. 2021:S1525‐0016(21):00399‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dai Z, Liu H, Liao J, Huang C, Ren X, Zhu W, et al. N7‐Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol Cell. 2021;81(16):3339‐55.e8. [DOI] [PubMed] [Google Scholar]
- 23. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, et al. EGFR expression as a predictor of survival for first‐line chemotherapy plus cetuximab in patients with advanced non‐small‐cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13(1):33‐42. [DOI] [PubMed] [Google Scholar]
- 26. Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan‐Salvo M, et al. Targeting BMI1+ Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell. 2017;20(5):621‐34.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, et al. Small molecule‐induced cytosolic activation of protein kinase Akt rescues ischemia‐elicited neuronal death. Proc Natl Acad Sci USA. 2012;109(26):10581‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang HN, Kim JH, Park AY, Choi JW, Lim SM, Kim J, et al. Establishment and characterization of patient‐derived xenografts as paraclinical models for head and neck cancer. BMC Cancer. 2020;20(1):316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin S, Liu Q, Jiang YZ, Gregory RI. Nucleotide resolution profiling of m7G tRNA modification by TRAC‐Seq. Nat Protoc. 2019;14(11):3220‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cozen AE, Quartley E, Holmes AD, Hrabeta‐Robinson E, Phizicky EM, Lowe TM. ARM‐seq: AlkB‐facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods. 2015;12(9):879‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, et al. Efficient and quantitative high‐throughput tRNA sequencing. Nat Methods. 2015;12(9):835‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León‐Ricardo BX, et al. Transcriptome‐wide mapping reveals widespread dynamic‐regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159(1):148‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ingolia NT, Brar, G.A. , Rouskin, S. , McGeachy, A.M. , Weissman, J.S . The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome‐protected mRNA fragments. Nat Protoc. 2012;7(8):1534‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Q, Shvarts T, Sliz P, Gregory RI. RiboToolkit: an integrated platform for analysis and annotation of ribosome profiling data to decode mRNA translation at codon resolution. Nucleic Acids Res. 2020;48(W1):W218‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang T, Cui Y, Jin J, Guo J, Wang G, Yin X, et al. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013;41(9):4743‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vickers AJ, Cronin AM, Elkin EB, Gonen M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med Inform Decis Mak. 2008;8:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reimand J, Isserlin R, Voisin V, Kucera M, Tannus‐Lopes C, Rostamianfar A, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat Protoc. 2019;14(2):482‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell‐type‐specific isolation of ribosome‐associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang K, Li J, Ito M, Takeda JI, Ohkawara B, Ogi T, et al. Gene Expression Profile at the Motor Endplate of the Neuromuscular Junction of Fast‐Twitch Muscle. Front Mol Neurosci. 2020;13:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Y, Wang R, Zhang S, Song S, Jiang C, Han G, et al. iTALK: an R Package to Characterize and Illustrate Intercellular Communication. bioRxiv. 2019. [Google Scholar]
- 42. Cillo AR, Kürten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. Immune Landscape of Viral‐ and Carcinogen‐Driven Head and Neck Cancer. Immunity. 2020;52(1):183‐99.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single‐Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171(7):1611‐24.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang G, Zhang M, Cheng M, Wang X, Li K, Chen J, et al. Tumor microenvironment in head and neck squamous cell carcinoma: Functions and regulatory mechanisms. Cancer Lett. 2021;507:55‐69. [DOI] [PubMed] [Google Scholar]
- 45. Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Corrigendum: Global quantification of mammalian gene expression control. Nature. 2013;495(7439):126‐7. [DOI] [PubMed] [Google Scholar]
- 46. Bahr A, Hankeln T, Fiedler T, Hegemann J, Schmidt ER. Molecular Analysis of METTL1,a Novel Human Methyltransferase‐like Gene with a High Degree of Phylogenetic Conservation. Genomics. 1999;57(3):424‐8. [DOI] [PubMed] [Google Scholar]
- 47. Marquard FE, Jücker M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem Pharmacol. 2020;172:113729. [DOI] [PubMed] [Google Scholar]
- 48. Ma XM, Yoon SO, Richardson CJ, Jülich K, Blenis J. SKAR links pre‐mRNA splicing to mTOR/S6K1‐mediated enhanced translation efficiency of spliced mRNAs. Cell. 2008;133(2):303‐13. [DOI] [PubMed] [Google Scholar]
- 49. She QB, Halilovic E, Ye Q, Zhen W, Shirasawa S, Sasazuki T, et al. 4E‐BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18(1):39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eum HH, Kwon M, Ryu D, Jo A, Chung W, Kim N, et al. Tumor‐promoting macrophages prevail in malignant ascites of advanced gastric cancer. Exp Mol Med. 2020;52(12):1976‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, et al. β‐Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8+ T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017;77(20):5639‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
We have deposited all sequencing datasets in this study on GEO (GSE172146, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE172146).