

Abstract
Intra-host dynamics are a core component of virus evolution but most intra-host data come from a narrow range of hosts or experimental infections. Gaining broader information on the intra-host diversity and dynamics of naturally occurring virus infections is essential to our understanding of evolution across the virosphere. Here we used PacBio long-read HiFi sequencing to characterize the intra-host populations of natural infections of the RNA mycovirus Cryphonectria hypovirus 1 (CHV1). CHV1 is a biocontrol agent for the chestnut blight fungus (Cryphonectria parasitica), which co-invaded Europe alongside the fungus. We characterized the mutational and haplotypic intra-host virus diversity of thirty-eight natural CHV1 infections spread across four locations in Croatia and Switzerland. Intra-host CHV1 diversity values were shaped by purifying selection and accumulation of mutations over time as well as epistatic interactions within the host genome at defense loci. Geographical landscape features impacted CHV1 inter-host relationships through restricting dispersal and causing founder effects. Interestingly, a small number of intra-host viral haplotypes showed high sequence similarity across large geographical distances unlikely to be linked by dispersal.
Keywords: RNA virus, virus evolution, natural infection, PacBio HiFi, intra-host diversity, epistasis, chestnut blight
1. Introduction
Viruses are some of the most diverse life forms on Earth. Yet, very little of the virosphere has been explored, leaving many open evolutionary questions (Zhang, Shi, and Holmes 2018; Zhang et al. 2019). Fewer still are the viruses where the intra-host evolutionary dynamics have been characterized. Almost all are human pathogens, often subjected to medical interventions, which can alter viral intra-host genetic diversity and the selective landscape (e.g. Feder, Pennings, and Petrov 2021). The few non-human pathogens explored (e.g. Zucchini yellow mosaic virus) are often experimental infections of crop pathogens, where it has been shown that human interventions and artificial conditions can impact the virus evolutionary process (Simmons, Holmes, and Stephenson 2011; Dunham et al. 2014). Natural virus infections, those not subject to medical or human interventions, are therefore likely to display different intra-host dynamics and it is essential that we begin to study them. This is particularly pertinent as an incomplete picture of virus evolution is likely concealing interesting evolutionary insights and leaving many questions on natural intra-host virus dynamics unanswered.
Historically, characterizing intra-host virus populations was limited by sequencing capabilities that constrained researchers to host-level virus consensus sequences (often at small amplicons e.g.< 600 base pairs ‘bp’, Kinoti et al. 2017; Ježić et al. 2021) or a small number of intra-host haplotypes obtained through cloning and Sanger sequencing (e.g. Redd et al. 2012). This limited picture is often unrepresentative of genome-wide intra-host diversity and virus evolution. Consequently, high-throughput sequencing was quickly applied to viruses after its development (Wang et al. 2007).
High-throughput sequencing of virus infections has yielded many important insights into the evolutionary dynamics of, predominantly human pathogenic, viruses (Lauring 2020). Intra-host sequencing of virus populations has shown that genetic drift and purifying selection are often the dominant evolutionary forces (e.g. Kennedy and Dwyer 2018; Xue and Bloom 2020). Strong bottlenecks can arise during transmission and spread through host tissues (e.g. Simmons, Holmes, and Stephenson 2011; Dunham et al. 2014). Intra-host variants that are rare in the early stage of an infection have also been shown to increase in frequency over time and have a strong impact on treatment failure in HIV infection (Simen et al. 2009). Recently, intra-host viral structural variants have also begun to be classified, opening up a new component of virus diversity to be explored (e.g. hepatitis C virus, Yamashita et al. 2020). Yet, how these findings apply across the virosphere, to natural infections, and across different types of hosts (e.g. non-mammal or sessile hosts) remains unclear.
As for any parasite, a large component of the selective landscape of viruses is likely to be the host (Simmonds, Aiewsakun, and Katzourakis 2019). Host effects classically arise from immune and defense genes that can create hostile conditions leading to strong directional selection against the parasites, which must be overcome to maintain infection. This drives an antagonistic evolutionary host–parasite arms race leading to cycles of adaptation and counter adaptation (Brockhurst et al. 2014; Papkou et al. 2019). These cycles are coupled with frequency-dependent selection in both the host and parasite, the most widely known examples of which are at host resistance genes (Tellier and Brown 2007). Host–parasite interactions are further complicated by epistatic gene interactions. Epistatic interactions are where the combination of alleles at multiple loci have non-additive synergistic effects on phenotypes (Phillips 2008). These have been found at host resistance genes (Metzger et al. 2016) as well as in mutations favoring drug resistance or immune escape success in parasites (including viruses Barton et al. 2016; Ferretti et al. 2020). Due to the rapid nature of their evolutionary cycles, the host–parasite arms race can create population-specific ‘local’ adaptations (Ebert 1994). Spatial genetic structure in hosts or parasites may lead to localized differences in host–parasite interactions that impact evolution (Rousseau et al. 2009). Yet host genotype effects on parasites, including viruses, have rarely been tested in natural systems (Sallinen et al. 2020).
In this study, we addressed these knowledge gaps in virus evolution by characterizing the intra-host populations of natural RNA virus infections. To expand the explored fraction of the virosphere beyond human, mammalian, or crop pathogens, we focused on the mycovirus Cryphonectria hypovirus 1 (CHV1). CHV1 is a viral parasite of Cryphonectria parasitica, an ascomycete fungus that causes chestnut blight (Nuss 1992; Rigling and Prospero 2018). CHV1 is a well-characterized unencapsidated single-stranded RNA mycovirus, consisting of a 12.7 kilobase (kb) genome with two large open reading frames that are read in a single direction. It has no polymerase proofreading abilities (Dawe and Nuss 2001).
Chestnuts are an important food in Europe (Castanea sativa) (Conedera et al. 2004) and were a historically important food in North America (Castanea dentata). Chestnut wood also once underpinned an immensely valuable lumber industry in North America (Davis 2004). However, this rapidly changed after the accidental introduction of the Asian fungal pathogen C. parasitica in the early 1900s (Rigling and Prospero 2018). This bark pathogen drove the C. dentata to the brink of extinction, destroyed the American chestnut lumber industry, and permanently changed forest diversity and structure (Elliott and Swank 2008).
In Europe the chestnut blight epidemic was initially destructive, leading to rural food shortages and depopulation (Heiniger and Rigling 1994; Diamandis 2018). However, in the decade after its introduction into Europe, a subset of C. parasitica infections (hereafter ‘cankers’) began to partially heal and the trees survived. This was attributed to the presence of the mycovirus CHV1 (Heiniger and Rigling 1994). Fortuitously co-introduced with C. parasitica into Europe, CHV1 results in a chronic multi-year infection that eventually restricts the growth of the host and reduces its sporulation. Over time CHV1 infection in the fungus drives a transition from an actively expanding canker (hereafter ‘active’ canker) to a non-growing canker with a healed appearance (hereafter ‘passive’ canker, Rigling and Prospero 2018). CHV1 is now used as a biocontrol agent for chestnut blight in Europe (Nuss 1992; Rigling and Prospero 2018).
CHV1 spreads naturally in Europe through asexual C. parasitica spores (vertical transmission) and by hyphal anastomosis (i.e. fusion) between fungal hosts genetically compatible at self-recognition loci (horizontal transmission), referred to as vegetative incompatibility (vic) loci. Vegetative incompatibility is a key host defense mechanism that reduces the transmission rate of viruses between fungi through programmed cell death upon hyphal contact (Cortesi et al. 2001; Zhang et al. 2014). Six diallelic vic genes have been identified in C. parasitica, and polymorphisms at these vic loci constitute over 80 per cent of the fungus’s total genome-wide diversity in European populations (Stauber et al. 2020).
To study the natural evolutionary dynamics of CHV1, we characterized the intra-host CHV1 populations of naturally infected cankers. Intra-host samples from four geographic populations (Switzerland: Contone and Orselina; Croatia: Kast and Ozalj) were sequenced with PacBio’s long-read HiFi technology (Pacific Biosciences, California). We targeted two amplicons (5 and 4.6 kb) that both span the 5ʹ-end of CHV1’s genome (∼40 per cent of CHV1’s 12.7 kb genome). With the exception of a PacBio study of the HIV envelope (Laird et al. 2016; Kumar et al. 2019) and Hepatitis C virus structural variants (Yamashita et al. 2020), this promising new sequencing technology has not been widely utilized to study virus intra-host diversity and evolution.
2. Results
A total of thirty-eight samples were sequenced. Two samples and an additional single amplicon from a third sample failed to produce sufficient reads after demultiplexing and were removed (see Table S1 for the sample information).
A mean of 40.2 intra-host mutations per sample were found in the overlapping 4.4 kb region (overlap of mutation callers Freebayes, Garrison and Marth 2012, and Deepvariant; Poplin et al. 2018), with an average difference of only 1.3 ± 2.9 mutations across the two amplicons from each sample (only three pairs of replicate amplicons differed by >2 mutations). The additional mutations were more common in the longer 5 kb amplicon libraries. Consequently, it is likely that they are caused by the putative association between the sequencing error and length in PacBio HiFi reads (Kumar et al. 2019). The additional mutations are spread evenly across the reads (Fig. S1); therefore, they should not generate false biological trends. Mutation frequencies were highly similar across amplicons, differing by a mean of 0.01 ± 0.02 (90 per cent percentile of the cross-amplicon frequency differences was ≤0.028). The mean denoised intra-host haplotype number across all samples and amplicons was 12.2 ± 8.7 (ranging from 1 to 35), and the average frequency of the most common haplotype was 0.6 ± 0.2. Haplotype number was more variable across amplicons than single nucleotide variants (SNVs) differing by an average of 4.8 ± 5.66 between amplicon pairs. However, the variability in the haplotype number may be driven by biologically meaningful differences in the non-overlapping regions of our amplicons. Overall, the similarity across our amplicons indicates that a highly repeatable picture of intra-host viral populations is captured with our sequencing method.
2.1. Inter- and intra-host CHV1 population structure
Inter-host relationships of CHV1 viral populations were first assessed across Croatia and Switzerland using both a phylogeny (RAxML-NG, Kozlov et al. 2019) and a network analysis (PopNetD3, Zhang and Parkinson 2019). To ensure a reliable and representative characterization of inter-host relationships, we used host-specific consensus sequences that were generated by mapping intra-host mutations found in both replicate amplicons to the reference CHV1 sequence (CHV1-EP721, Lin et al. 2007). The two Croatian geographic populations were genetically differentiated from each other and from both Swiss populations, but the two Swiss geographic populations showed no genetic differentiation (Fig. 1A). The network-based visualization of inter-host relationships found similar groupings. In both analyses, one well-linked genetic cluster was found in the Croatian population Kast. This suggests that a single founder established the entire Kast population (Fig. 1B). In contrast, there were distinct clusters within the geographical populations Ozalj (n = 3) and Contone (n = 2) that overlap with different clades within our RAxML-NG tree, suggesting that multiple genetically diverse founders established these populations. PopNetD3 also implemented a chromosome painting sliding window analysis that looks for shared ancestry and colors the genomic window accordingly. Small sections of shared ancestry were visible across all our clusters.
Figure 1.
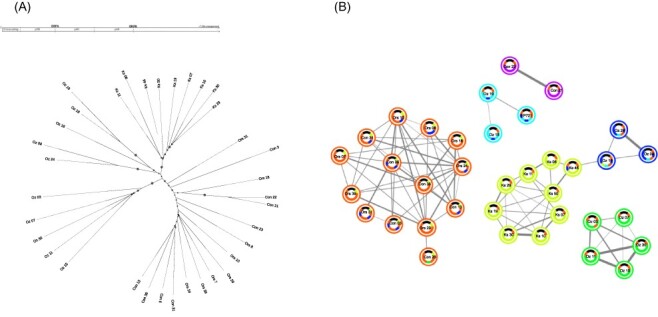
CHV1 inter-host relationships.
The consensus derived inter-host relationships as inferred by (A) a RAxML-NG unrooted phylogentic tree and (B) a PopNetD3 network analysis. Swiss geographical populations are denoted with Con (Contone) and Ors (Orselina), Croatian geographical populations are Oz (Ozalj) and Ks (Kast). The bootstrap values for the RAxML-NG tree are shown by node size, and larger nodes have higher bootstrap support (ranging from 0.3 to 0.99). The thickness of connecting lines in the network analysis shows the degree of sequence similarity across individuals with a cut-off value of >0.5 for edges to be shown. The outer circle color indicates cluster membership and the inner circle colors represent regions of shared ancestry across clusters. Black indicates the non-overlapping regions of our amplicons that were excluded from the analyses.
PacBio sequences can act as haplotypes, allowing for a more detailed phylogenetic comparison of viral populations between and within hosts. However, due to the expectation that some sequencing errors and PCR duplicates will be present, it is necessary to denoise PacBio sequencing reads and collapse them into biologically meaningful haplotypes (Kumar et al. 2019). Denoised haplotypes were generated for each intra-host population using Robust Amplicon Denoising (Kumar et al. 2019). These were then used to build an intra- and inter-host phylogenetic tree with Phyloscanner (Wymant et al. 2018). This phylogeny supports our results of a single well-connected Swiss meta-population and two distinct Croatian populations (Fig. 2). However, the intra-host phylogenies offer a much more detailed picture into CHV1 relationships. For example, there were a small number of hosts with many distinct haplotype groups, which could be caused by multiple infection or highly diverse founders (e.g. Contone 31 and Orselina 29 have multiple haplotype groups across all four phylogenetic trees). We also identified a small number of closely related intra-host viral haplotypes originating from highly geographically distant hosts (e.g. Kast 07 and Contone 13; Ozalj 24 and Orselina 18; Contone 36 and Ozalj 11, which all appear closely related across all four phylogenetic trees). There was also a pattern of long inter-host and short intra-host branches on the phylogeny, which is expected when strong genetic drift is experienced during transmission (Wymant et al. 2018). A similar phylogeny was obtained regardless of the amplicon used. The intra- and inter-host tree was also used to estimate the intra-host recombination rate, which on average was 0.001 ± 0.002 (estimates ranged from 0 to 0.008 by the metric defined in Wymant et al. 2018).
Figure 2.
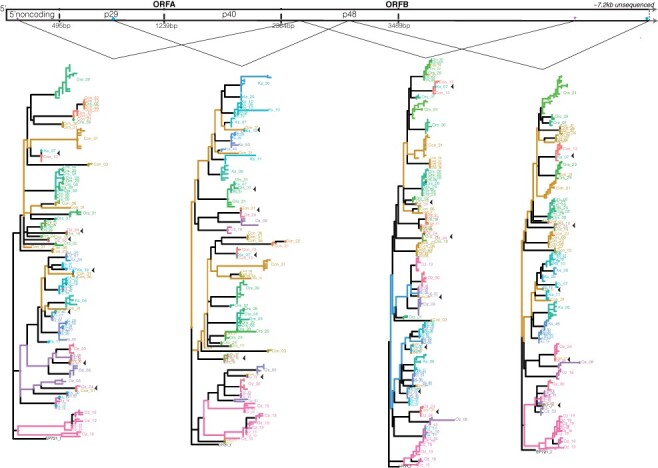
Haplotype-based inter- and intra-host relationship.
The relationship between intra-host haplotypes within each host and across hosts. Included is a representation of CHV1’s genome structure, and primer positions are shown by the stars (5 kb) and blue circles (4.6 kb). Different sections of the genome may contain different evolutionary information; therefore, to ensure our trends were robust each amplicon was divided into two sections. This roughly corresponds to the two ORFs but the complete annotated region they encompass is denoted by the lines joining each phylogeny to the CHV1 genome picture. Phylogenetic trees are shown in order of their starting position across the genome and thus originate from alternating amplicons starting with the 5 kb amplicon. Each intra-host population has a unique color, the same color separated across the phylogeny indicated a diverse intra-host viral population. To increase readability adjacent labels from the same host have been collapsed into one per node. Shown by the black triangle are closely related haplotypes sampled from unlikely dispersal distances (i.e. different geographic countries). Swiss sampling locations are abbreviated to Con (Contone) and Ors (Orselina), Croatian populations to Oz (Ozalj) and Ks (Kast).
2.2. Intra-host diversity and accumulation of mutations over time
Intra-host diversity was measured using intra-host mutations and denoised haplotypes. This was done separately for replicate amplicons from the same host and included the non-overlapping portion of the genome. Variation in sequencing depth can impact the detection of rare intra-host mutations and mutational diversity metrics. Consequently, intra-host mutational diversity was measured using nucleotide diversity, i.e. π (estimated in SNPGenie Nelson, Moncla, and Hughes 2015), because it is robust to large variation in sequencing depth (Zhao and Illingworth 2019). Across all samples and amplicons, the mean π was 3.9 × 10−4 ± 6.2 × 10−4 (range 9.3 × 10−6 to 3.1 × 10−3). Haplotype number was also correlated with sequencing depth (R2 = 0.24; e = 0.001 ± 0.0002, t = 4.659, P = 1.5 × 10−5; Fig. S2A, n = 71); therefore Nei’s H was calculated to characterize intra-host haplotype diversity (Nei and Tajima 1980). Nei’s H did not correlate with sequencing depth (Fig. S2B, e = 1.1 × 10−5 ±7.6 × 10−6, t = 1.447, P = 0.152, n = 71) and was consistent across replicate amplicons (mean difference in Nei’s H of only 0.11 ± 0.13). Nei’s H ranged from 0 to 0.99 across our samples (Figs S3 and S4), with a mean of 0.58 ± 0.27 across both amplicons.
Passive cankers had significantly higher intra-host π than active cankers (Fig. 3; GLMM e = 0.97 ± 0.41, t= 2.311, P = 2.08 × 10−2, n = 71). Mutational enrichment was visible in passive cankers at the 5ʹ-UTR genomic region (covered only by our 5 kb amplicon) and at the beginning of the p40 domain (1150–1950 bp, covered by both amplicons). Nei’s H was also significantly higher in passive cankers (active 0.53 ± 0.24, passive 0.60 ± 0.28, Nei’s H GLMM, e = 0.904 ± 0.451, t = 2.004, P = 4.5 × 10−2, n = 71). Both Nei’s H and π differed significantly across populations. However, post hoc pairwise comparisons between populations were all non-significant (P > 0.05), suggesting that this trend is weak and likely reflects host or virus population structure.
Figure 3.
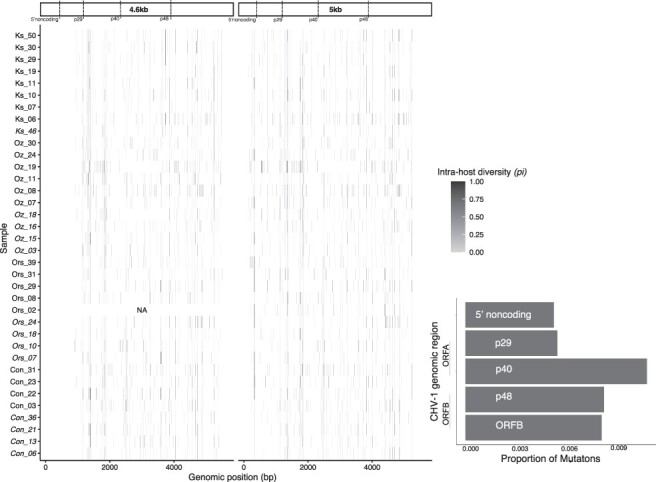
Intra-host diversity heatmap.
The intra-host diversity (measured as π) for each sequenced position across the genome. Darker colors indicate higher diversity. For this figure only, those positions fixed for differences from the reference were given a value of 1 as π cannot measure fixed differences. This shows regions with a high rate of mutation fixation.
A subgraph is a connected region of an inter-/intra-host phylogeny (both connected tips and internal nodes) that originates from the same infection (i.e. canker) (Wymant et al. 2018). The subgraph number can be used as an estimate of the number of infecting founder viruses (Wymant et al. 2018) (Fig. 2, mean of 1.99 ± 1.51). This can help determine if repeated infections are driving the higher CHV1 diversity in passive cankers. The factors affecting mean subgraphs number were assessed using the same model as π and Nei’s H. The subgraph number did not differ between active and passive cankers nor between geographical populations. However, it did differ significantly across replicate amplicons from the same sample (GLMM, e = 0.347 ± 0.0755, t = 4.603, P = 4.2 × 10−6, n = 69), which is unlikely to be biologically meaningful as the difference was <1 on average (Fig. S5). This analysis indicates that the number of infecting viruses is not higher in passive cankers, i.e. repeated infections are not likely to occur and do not drive the higher diversity seen in older passive cankers.
2.3. Epistatic effects of host vic genes on viral intra-host diversity and infection founders
Epistatic host vic interactions significantly impacted CHV1. Both metrics of CHV1 intra-host diversity were impacted by a significant interaction between the host alleles at vic7 and vic4 (π GLMM, e = 4.89 ± 1.57, t = 3.11, P = 1.8 × 10−3, n = 71; Nei’s H GLMM, e = 9.12 ± 1.695, t = 5.427, P = 5.7 × 10−8, n = 71), as well as at vic7 and vic6 (π GLMM, e = −3.91 ± 1.64, t = −2.38, P = 1.7 × 10−2, n = 71; Nei’s H GLMM, e = −8.273 ± 1.766, t = −4.684, P = 2.8 × 10−6, n = 71). An additional significant interaction between alleles at vic4 and vic2 also impacted haplotypic diversity (Nei’s H GLMM, e = 2.93 ± 1.376, t = 2.13, P = 3.3 × 10−2, n = 71). The epistasis (interaction) plots show the predicted directionality of these vic interactions (Fig. 4 and S6). There was also a significant interaction affecting subgraph number at loci vic7 and vic6 (GLMM, e = −2.365 ± 0.564, t = −4.191, P = 2.8 × 10−5, n = 69) and at vic7 and vic2 (GLMM, e = −2.164 ± 0.631, t = −3.429, P = 6.1 × 10−4, n = 69). This means that the combination of host vic alleles impacted both the number of viruses infecting the host and the intra-host diversity of the virus population.
Figure 4.
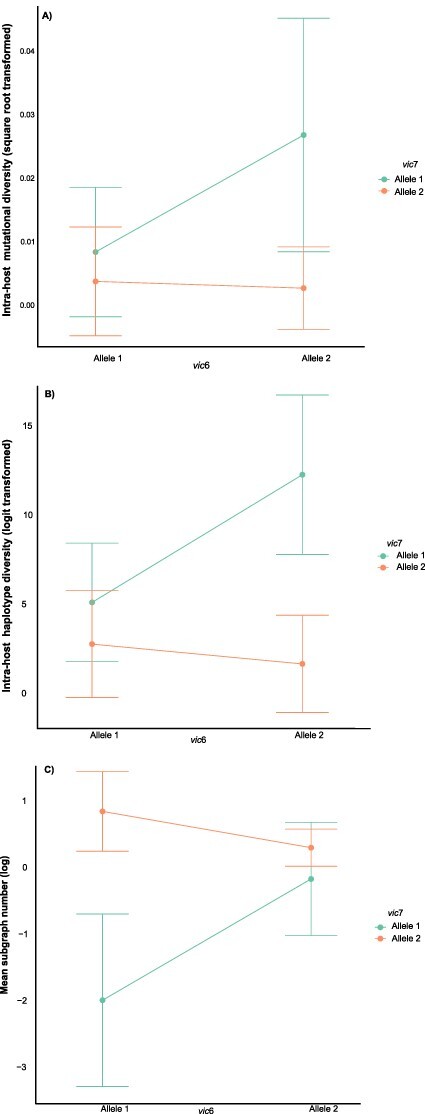
Epistatic sign plots for host vic alleles.
The directionality of statistically significant epistatic host vic allele interactions and their effect on intra-host virus populations as characterized by interaction plots. Intra-host viral diversity is estimated by (A) nucleotide diversity (π) at mutations (B) gene diversity (Nei’s H) across haplotypes and (C) the predicted number of founders (Phyloscanner subgraph number). Displayed is the only vic combination that significantly affects all three intra-host metrics.
2.4. Intra-host patterns of selection
To characterize the amplicon-wide signals of selection, π was compared between synonymous (hereafter πS) and non-synonymous (hereafter πN) mutations. πS was significantly higher than πN across all samples (πS = 3.3 × 10−4 ± 5.3 × 10−4, πN = 1.1 × 10−4 ± 1.8 × 10−4, paired t-test, t = −4.9, df = 70, mean of the differences = −2.2 × 10−4, P = 6.7 × 10−6, consistent when divided by amplicon). Dividing the genome into CHV1’s two ORFs, πS values commonly exceeded πN in at least one ORF per sample (n = 34/36 samples). This signified widespread purifying selection. Signs of positive selection (πN > πS) (Nelson, Moncla, and Hughes 2015) were rare and always limited to one ORF from each sample (ORFA n = 2 samples or ORFB n = 5).
At the codon level the sliding window analyses showed few genomic windows with robust signals of selection. All codons with significant πN/πS ratios (marked by a star in Fig. 5) showed signs of purifying selection (πN/πS < 1). These signals were largely sample specific, although codons showing signs of purifying selection in more than one sample were found at 1266–1269 bp in Ozalj 7 and 30, at 2414–2423 bp in Contone 21 and 22, and at ∼3340 bp in Ozalj 8 and 24.
Figure 5.
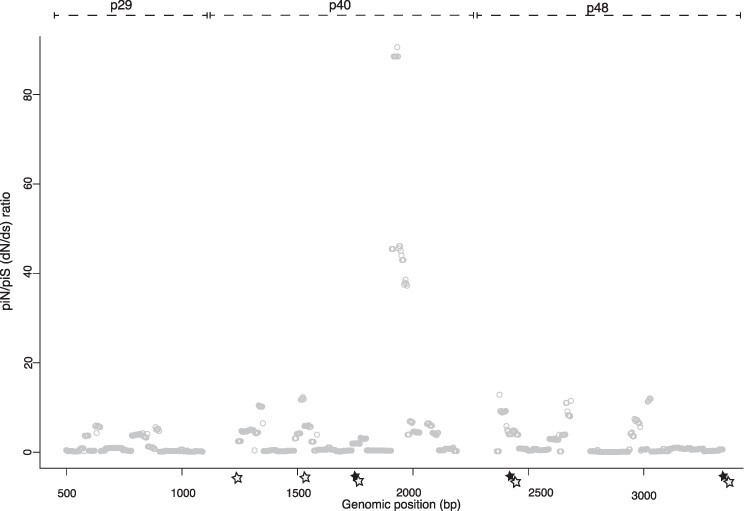
Patterns of selection across the genome.
The average intra-host πN/πS values across codons calculated in 50 bp windows with a 10 bp step. Codons where the πN/πS ratio is significant are indicated with stars. This was evaluated using a permutation two-sided test where the null hypothesis was that the ratio was not equal across a window (closed stars) and a one-sided permutation test where the null hypothesis was that πN > πS (open stars). The other tests performed by SNPGenie were not significant across any windows. Windows where either measured πN or πS was equal to 0 are excluded.
In four samples, there was a mismatched πN/πS at the ORF level. The 5 kb amplicon often showed πN < πS, while the 4.6 kb amplicon showed trends of πN > πS. These different signals of selection are likely to reflect the incomplete sequencing of ORFA and different information present across the genome.
2.5. Deleterious mutations arising in intra-host populations
Mutations in intra-host CHV1 viral populations were most often predicted by SNPGenie (Nelson, Moncla, and Hughes 2015) and SNPEff (Cingolani et al. 2012) to be either synonymous (n = 296 mutations detected in either amplicon) or non-synonymous amino acid changing (n = 202). Following this, variants in the 5ʹ-non-coding region were seen moderately often (n = 24). Severely deleterious mutations, such as disruptive deletions (n = 1) or frameshift mutations (n = 8), were rare.
Considering only severely deleterious mutations identified across both amplicons, thereby excluding sequencing or processing errors, only five frameshift or disruptive mutations were detected. A frameshift at 4660 bp (1 bp deletion) was the most common putative severe mutation. This mutation was detected in 24 samples but was always at a low intra-host frequency (<0.16). Two severe mutations were seen at a low intra-host frequency of <0.16 in only a single sample, at 4659 bp in Contone 13 (1 bp deletion) and 1799 bp in Kast 11 (1 bp insertion). Two were seen at a high intra-host frequency of ∼0.8 and present in the most common denoised haplotype, each was only found in one sample, at 3466 bp in Ozalj 07 (1 bp deletion) and 2374 bp in Ozalj 19. The latter is a large 1.2 kb deletion visible on an agarose gel that virtually removes the entire p48 domain. This large deletion makes the virus defective. Four of the five samples with severe deleterious mutations visible in both amplicons were derived from passive cankers. None of the samples showed signals of positive selection at the ORF level that could indicate genetic hitchhiking of the deleterious mutation.
3. Discussion
Intra-host diversity of CHV1 was moderately low relative to that estimated for the West Nile virus (>1000 mutations per sample, genome size ∼11 kb, Ehrbar et al. 2017). However, the mean π values of CHV1 fell within the lower range of reported values for human RNA virus pathogens during the acute infection stage (e.g. HIV, RSV Gelbart et al. 2020). Haplotype numbers were similar to those identified through PacBio HiFi sequencing of hepatitis C virus infections (Yamashita et al. 2020).
3.1. Characterizing inter-/intra-host CHV1 populations
CHV1’s inter-host relationships based on consensus sequences showed broad geographical structuring with distinct separation of Swiss and Croatian meta-populations. The two Swiss geographic populations were also much less genetically distinct than those in Croatia. Although the geographic populations were physically equidistant in each country, the Swiss populations are on opposing sides of a valley, while those in Croatia are separated by dense mixed forest over hilly terrain. The lack of divergence between the Swiss populations is likely facilitated by a sufficient exchange of migrant viruses between sites. This could occur through fungal asexual spores being dispersed by wind or vectors (Rigling and Prospero 2018). In contrast, within Croatia fungal spore dispersal appears to be more restricted due to the dense forest. This has likely fueled a single genetic founder in Kast, which in turn drove population differentiation through founder effect–derived genetic drift and no subsequent migrant exchange. Reduced landscape connectivity between the sites in Croatia is supported by previous studies, which showed a reduced diversity of the fungal host in Kast relative to other Croatian sites (Ježić et al. 2018, 2021).
Founder effects are extreme genetic drift events, where the random introduction of only a small number of individuals from a source population when establishing a new population results in an unrepresentative sample of the source population’s genetic diversity (Sirkkomaa 1983). Such founding bottlenecks can have long-term impacts on the genetic structure and diversity of a population (Ventura et al. 2014). Founder effects and signs of genetic drift have previously been seen in clinical or experimental samples of pathogenic RNA viruses and even occur within a host. For example in HIV, intra-host founder effects have impacted the virus haplotype frequencies in the spleen (Frost et al. 2001). In plant viruses, such as the Cucumber mosaic virus and Zucchini yellow mosaic virus, intra-host founder bottlenecks have previously been shown in different host tissues (Dunham et al. 2014; Alí and Roossinck 2010). Excitingly, our results suggest that despite the rapid evolution and high mutation rate of RNA viruses (e.g. Lauring and Andino 2010), CHV1 shows similar landscape effects on the population structure to those common in more complex multicellular organisms (Sork and Waits 2010).
Landscape studies on pathogens rarely use genetic tools (only 16/51 studies collated by Kozakiewicz et al. 2018). Additionally, although viruses (particularly rabies) have long been a focus of pathogen landscape genetics studies, the large majority of previous studies use the host genetic structure as a proxy for the pathogen (Kozakiewicz et al. 2018). This approach likely masks a lot of important patterns in viruses because of the fundamental differences in the mutation rate and the life history. Importantly, signs of population differentiation and landscape effects on CHV1 are not visible across the smaller amplicons and consensus sequences used in previous studies (e.g. Ježić et al. 2021), highlighting the importance of also revisiting the few existing viral landscape studies with high-throughput sequencing data and longer target amplicons. Further intra-host landscape genomic studies directly examining viruses in natural habitats are now necessary and will likely offer interesting evolutionary insights.
The haplotype-based intra-host phylogeny revealed that intra-host viral populations are also more complex than is visible at the consensus level. The overall signals of genetic drift remained evident. This is because phylogenetic tree branch lengths were long between haplotypes identified in different hosts and short between haplotypes from the same hosts (Wymant et al. 2018). Broad geographic patterns were stable; however, a small number of fungal hosts harbored genetically distinct viral variants, which are likely a result of either repeated infections or genetically diverse initial founders. Multiple virus infections have previously been observed in plant viruses, although this is a rare phenomenon (e.g. Predajňa et al. 2012). Accordingly, in our study signs of multiple infection were not common in CHV1.
In CHV1 a small number of closely related haplotypes were found in multiple hosts; most importantly this included hosts sampled across large inter-county distances. These long-distance haplotype pairs (n = 3) were consistently grouped together across all four replicate Phyloscanner trees, offering high support for their genetic similarity (Wymant et al. 2018). Recent migrant exchange does not appear to drive this sequence similarity, because such short-term dispersal is highly unrealistic between these locations (i.e. Croatia and Switzerland). Closely related haplotypes were isolated from hosts over 600 km apart and wind or vector-based dispersal has previous been shown to be limited to a few hundreds of meters in C. parasitica (Dutech et al. 2008). It is also not caused by artificial human-mediated dispersal of CHV1 for biocontrol, because country-specific virus strains have always been used. Furthermore, this is unlikely to be caused by sequencing technical artifacts (e.g. Leigh et al. 2018) as this phenomenon was seen across two of our sequencing libraries, all libraries contained samples of mixed origin and the relationship was visible in both (separately barcoded) amplicons.
It can be hypothesized that the rare high haplotype similarity seen across countries in CHV1 may be driven by limited genetic diversity and weak population structure of the host C. parasitica (Stauber et al. 2021). Parallel adaptive changes across vast geographical distances have previously been described in invasive rabbits that were exposed to the biocontrol Myxoma virus (i.e. in Europe and Australia). Such patterns are possible in invasive species because of the decoupling of evolutionary and geographic distance (Alves et al. 2019).
Alternatively, this may be a natural phenomenon of virus evolution. Sequence conservation has recently been proposed to be common in virus evolution. This is because highly similar viral haplotypes have been identified across substantial temporal scales: ∼3,000 years in the DNA virus Human Parvovirus B19 (Mühlemann et al. 2018) and ∼1,000 years in the double-stranded RNA virus Zea mays chrysovirus 1 (Peyambari et al. 2018). Although high intra-host diversity is observed in viruses, these results have suggested much of this diversity may be transient and leading to limited sequence change over time (Simmonds, Aiewsakun, and Katzourakis 2019). Under this scenario viral sequence conservation is driven by either the low fitness of new mutations, leading to their elevated loss or constraints on sequence evolution arising from strong host-derived evolutionary pressure (discussed in Simmonds, Aiewsakun, and Katzourakis 2019). Space-for-time proxies are common in landscape genetics (McGarigal and Cushman 2002), meaning that the temporal sequence conservation may also occur across space and drive the patterns observed here.
Finally, the high sequence similarity visible across space may also be because the evolutionary rate of natural virus infections is simply lower than previously estimated (Smith et al. 2014). Nevertheless, high sequence similarity between CHV1 haplotypes isolated in different countries was rare. These low levels of sequence conservation do not directly contradict our findings of landscape impacts on virus haplotype and allele frequencies but show virus population genetic patterns may be complex due to unexplored evolutionary forces.
3.2. Selection and intra-host mutations in CHV1
Within each intra-host population there were strong signs of purifying selection acting on CHV1. This result is in line with previous findings from other RNA viruses (e.g. influenza A virus, Xue and Bloom 2020; dengue virus, Lequime et al. 2016; West Nile virus, Jerzak et al. 2005; enterovirus C; Xiao et al. 2017). Purifying selection likely keeps a virus at the fitness peak for the host by preventing the accumulation of deleterious mutations (Simmonds, Aiewsakun, and Katzourakis 2019).
As expected in the presence of purifying selection, deleterious mutations were neither abundant nor often observed at high intra-host frequencies in CHV1. A notable exception was a defective virus haplotype present at an intra-host frequency of >0.8 in a single sample. Defective viruses have previously been described in some lab strains of CHV1 (Shapira et al. 1991). However, defective viruses in CHV1 were previously thought only to arise after the relaxation of selection and repeated bottleneck events endured during prolonged laboratory fungal culturing (Shapira et al. 1991). Defective viruses in natural populations are not known to be common, although a widely spread defective dengue virus haplotype caused by a stop codon in the surface envelope gene (E) has previously been described (Aaskov et al. 2006). Although seemingly extreme in the case of CHV1 (1.2 kb deletion that essentially removes the entire p48 domain), it is important to note that defective viruses can parasitize on correctly coded proteins produced by other complete viral genomes in the host cell, and thus may only be mildly deleterious (Aaskov et al. 2006). Furthermore, as they are shorter, defective viruses may rise to a high frequency due to faster replication (Tapia et al. 2013; Nelson and Hughes 2015).
Despite strong purifying selection, there were signs of mutation accumulation over the course of a CHV1 infection (using canker type as a proxy). This is because passive cankers, which are more likely to be older (Rigling and Prospero 2018), had significantly higher values of intra-host CHV1 diversity than active cankers. This was not due to repeated infections of the same canker (as seen in Daphnia magna, Ameline et al. 2020), because intra-host subgraph number is not higher in older passive cankers. Consequently the temporal increase in intra-host viral diversity appears to be driven by accumulation of de novo mutations over time. Accordingly, the non-coding 5ʹ-UTR where mutations may have a limited fitness effect was a hotspot for signs of mutation accumulation in isolates from passive cankers. HIV has also repeatedly been shown to accumulate intra-host mutations over time and this can be used to date infections (Carlisle et al. 2019). Whether these acquired mutations are transient or maintained and transmitted to new infections is unclear. Future studies involving serial temporal sampling of cankers are needed to address this and clarify if there is a decoupling of long and short-term CHV1 evolution.
3.3. Epistatic host–gene interactions and intra-host diversity
Epistatic interactions at host vic loci impacted CHV1 intra-host diversity as well as the number of founder viruses. Although vic interactions drove only small non-additive differences in the estimated number of founder viruses, even minute variations in founder number would easily leave lasting footprints on CHV1 population diversity because of the profound natural transmission bottlenecks observed. Intra-host populations of CHV1 in our study are often founded by less than two viruses. Discordance in alleles at vic loci have previously been shown to affect the probability of successful CHV1 horizontal virus transmission in laboratory cultures (Cortesi et al. 2001). This suggests that epistatic host effects may be common in this system and affect CHV1 beyond the well-characterized horizontal transmission effects visible in highly controlled laboratory conditions (Cortesi et al. 2001).
Unexpectedly, intra-host viral diversity was significantly impacted by epistatic interactions involving vic4. Individuals with discordant vic4 alleles do not trigger programmed cell death on hyphal contact and do not restrict virus transmission (Cortesi et al. 2001). The vic4 allele present did not affect subgraph number, suggesting that its effects on intra-host diversity must have arisen post-infection. Vic4 alleles encode for different proteins: allele 1 is a protein kinase c-like (PKC) gene while allele 2 is a NACHT-NTP/WD repeat-encoding gene that is considered a STAND protein (signal-transducing ATPase with numerous protein domains). PKC genes are often conserved and central to immune responses (shown in mammals and plants, Spitaler and Cantrell 2004). STAND proteins are rapidly diversifying in fungi and are involved in immune responses or programmed cell death (Dyrka et al. 2014). Vic2 and vic7 also encode for STAND proteins (vic2, Patatin-like protein; vic7, HET domain; Zhang et al. 2014). Epistatic interactions between either of these two loci and vic4 could thus be driven by disruption, or augmentation, of immune-related signal transduction cascades in the fungal host. This would alter the viral selective landscape and could lead to the observed differences in intra-host diversity. In a landmark experiment on Plantago plants on the Åland island system, the host genotype was shown to impact natural intra-host viral community diversity (i.e. number of species, Sallinen et al. 2020). An effect of host genotype on the intra-host viral diversity thus aligns with our expectations and reiterates that natural virus evolution is governed simultaneously by multiple factors.
3.4. Conclusions
Our results indicate that the evolutionary pressures on natural virus infections are multifaceted. In natural infections of CHV1 intra-host populations are predominately determined by the interplay between host effects, landscape impacts on virus dispersal, and standard population genetic processes (e.g. mutation accumulation over time). However, a small number of highly similar sequences were seen in different countries. This could support recent hypothesis of a potential decoupling between long- and short-term virus evolution due to sequence reversion but may also be a product of an invasive system and limited host genetic diversity. Further landscape or temporal genomic intra-host studies on natural infections from across the virome and a range of hosts are now necessary to fully characterize the evolutionary process of viruses and to connect long- and short-term virus evolution.
4. Methods
4.1. Sample collection
European chestnut trees (C. sativa) naturally infected with C. parasitica were sampled in two Swiss (Contone and Orselina) and two Croatian (Kast and Ozalj) forest sites in the summer of 2019. At each site, 40–50 cankers were sampled-each from different trees. Sampling consisted of extracting 0.5–1 cm of infected bark using a 2-mm bone marrow needle. To prevent contamination, the needle was dipped in 96 per cent ethanol and flamed after each sample. All samples were taken within a one-month period, and each site was sampled in a single day. There are several CHV1 viral subtypes found in Europe (Bryner, Rigling, and Brunner 2012), and the four focal populations are from the oldest and most widely spread European subtype, ‘subtype I’ (Bryner, Rigling, and Brunner 2012).
4.2. Culturing and confirming virus presence
All bark samples were cultured to confirm if C. parasitica was infected with CHV1. Culturing began the day after field sampling. First, bark samples were surface sterilized by dipping them in 70 per cent ethanol and drying them on sterile filter paper for 10 s. Second, the bark sample was placed on a 90 mm Petri dish containing potato dextrose agar (PDA, 39 g/l Difco BD Biosciences) using sterile tweezers and incubated in the dark for 2–3 days at 24 °C, 70 per cent humidity. Third, when the fungal colonies reached a diameter of ∼1.5 cm, a 2-mm square of each colony was transferred to a 60-mm PDA Petri dish. This was cultured in the dark for one week at 24 °C, 70 per cent humidity and then phenotyped to confirm virus presence. CHV1 infected C. parasitica isolates have a white appearance in culture, while virus-free isolates develop orange pigmentation and sporulate when exposed to light (Rigling and Prospero 2018). The original bark sample cultures were kept at 4 °C over this period to limit growth.
A subset of 9–10 virus-infected cultures were selected from each of the four sites for sequencing. Where possible, a balanced number of isolates from active and passive canker types were selected. To capture the entire intra-host CHV1 diversity present in a sample and produce enough material for sequencing, the original bark-derived colonies from the 90-mm plate were divided into four quarters. A small margin of mycelium that surrounded the initial bark sample was excluded to prevent any contamination or bark fragments in our samples. Each quarter of mycelium was transferred to a separate sterile 90-mm Petri dish with PDA overlaid with cellophane and cultured at 24 °C 70 per cent humidity for five to seven days until their diameter was 7 cm. After this period, the fungal mycelia from each plate were scrapped, lyophilized, the four samples merged, and stored at −80 °C until RNA extraction. All culture preparation and handling were conducted under a Biosafety Cabinet Class II.
4.3. PacBio sequencing library preparation
CHV1 is a single-stranded RNA virus with a double-stranded (dsRNA) replicative form (Nuss 2005). The dsRNA was extracted from 20 to 30 mg of lyophilized mycelium using the iNtRON dsRNA extraction mini kit following the manufacturer’s protocol. After the extraction, dsRNA was incubated at 100 °C for 2 min followed by a snap-chill on ice. Single-stranded cDNA was then synthesized using Maxima H Minus Reverse Transcriptase (Thermo Scientific) with Oligot(dt)12–18 primers. The fresh cDNA was immediately used as a template for long-range PCR. This reduced the risk of heteroduplex formation during the PCR (Waugh et al. 2015). Two overlapping amplicons of 4.6 kilobase (kb) and 5 kb were targeted. These spanned CHV1’s ORFA and the beginning of ORFB (reviewed in Nuss 2005). Primer combinations for the 5kb amplicon were as follows: forward: ATCYGGAGAARGTGATTTGC and reverse: YTTRTTGATGTAGCTGCGAGG. Primer combinations for the 4.6kb amplicon were as follows: forward: CCGATTCCTTCAGTTGGT and reverse: AGCGGAGCCATGTAGC. All primers were tagged with a 6-bp in-line barcode for sample identification (barcode sequences supplied by B. Murrell). PCR conditions were 95 °C for 1 min, 15 cycles of 95 °C for 30 s and 68 °C for 7 min, followed by a final 10-min extension at 68° C. The PCR protocol was optimized to reduce the risk of PCR duplicates and heteroduplex formation: we used multiple reactions for each amplicon (5–7 reactions), a low cycle number (15x), and included a substantial extension time (10 min) (Liu et al. 2014; Waugh et al. 2015). High-fidelity advantage 2 Taq polymerase (Takara, Japan) was used throughout (Laird Smith et al. 2016). After amplification, reactions from the same sample were pooled, bead cleaned with Ampure beads (Beckman Coulter, USA), and the final concentration measured using a Qubit (BR-DNA kit, ThermoFischer, USA).
Samples were equimolarly pooled onto four PacBio Sequel SMRTcell, each SMRTcell consisted of both amplicons derived from 9–10 of the focal samples. To prevent technical artifacts being confounded with biological trends (e.g. Leigh et al. 2018), populations were spread across the four SMRT cells. Further PacBio library preparation steps and sequencing were then performed at the Functional Genomics Centre Zurich (Switzerland). PacBio HiFi offers exciting potential to study virus evolution because it can produce long and accurate reads that allow us to directly observe intra-host virus haplotypes, thus circumventing the error-prone haplotype reconstruction step that remains necessary for short sequencing reads (Schirmer, Sloan, and Quince 2014). While MinION (Oxford Nanopore) sequencing can also sequence viral haplotypes, the error rate remains high (evaluated for CHV1 in Leigh, Schefer, and Cornejo 2020) and PacBio HiFi sequencing offers more accurate intra-host mutation calls.
4.4. PacBio long-read processing
PacBio polymerase reads were processed with the software pbccs (Pacific Biosciences, California) to generate consensus ‘ccs’ reads also known as ‘HiFi’ reads. These reads circumvent the moderate sequencing error expected with PacBio technology by resequencing (called ‘passes’) the same DNA molecule multiple times. This information is then merged to produce a HiFi read that is both long and has a high sequencing quality (Wenger et al. 2019). Reads were required to have a minimum length of 3 kb, 5 polymerase passes, and a predicted sequencing quality of 0.99. HiFi reads were then demultiplexed into samples using the in-line barcodes attached to our primers using lima (Pacific Biosciences, California). Lima was run with ‘peek-guess’ to remove spurious barcodes, barcodes were allowed a minimum quality score of 26, as well as different forward and reverse barcodes to match our read structure.
4.5. Mutation identification
The demultiplexed ‘bam’ files were converted to a ‘fasta’ format using samtools (v1.9, Li et al. 2009) and aligned to a CHV1 reference sequence (DQ861913 EP721, Lin et al. 2007) using minimap2 (v2.17, Li 2016) with the ‘map-pb’ option suitable for PacBio reads. The resulting files were sorted, indexed, and converted to a ‘bam’ format with samtools.
Intra-host mutations were called on the aligned HiFi reads using two programs and only the overlapping mutation calls found in both were used in downstream analysis. Specifically, mutations were called with Deepvariant (v.1.1.0, Poplin et al. 2018) using the PacBio option, as well as with Freebayes (v.1.3.1, Garrison and Marth 2012) assuming a ploidy of 1, and with the ‘pooled discrete’ and ‘pooled continuous’ options activated to allow for intra-host mutation calling. Both programs were run on each sample independently to reduce the memory required. Only mutation frequencies from Deepvariant were used, because the intra-host frequencies based on the observed sequencing depths reported in Freebayes appeared potentially inaccurate and were therefore ignored. Specifically, we had a defective virus sequencing in one sample that was visibly polymorphic on a gel but was estimated to be fixed using the read depths reported in Freebayes. Thus, we chose to exclude the frequencies Freebayes reported.
Based on the read depths and frequencies reported by Deepvariant, the average coverage across polymorphic sites was 1404 ± 239 reads and the average genotyping quality score was 49 ± 16. The minimum allele frequency across heterozygote or homozygous mutational calls for new mutations not seen in the reference sequence was 0.12 (average 0.97 ± 0.13). The minimum reference allele frequency across heterozygote and homozygous reference calls was 0.03 (average 0.48 ± 0.32), although all sites with a reference call frequency below 0.05 had a read depth of >40 for the reference allele.
Intra-host virus diversity as measured with mutations (π) was calculated for each amplicon from a sample using SNPGenie (Nelson, Moncla, and Hughes 2015) with a custom annotation of genomic structure developed for CHV1 using published descriptions (reviewed in, Nuss 2005). SNPGenie reports genome-wide values of π, thus values were corrected to the amplicon region by calculating the average sum of pairwise differences at all coding sites in the amplicon, divided by the total number of sites (Ndiffs + Sdiffs/NSites + Sites in the SNPGenie codon file). No minimum allele frequency or sliding window was used. We chose to measure viral intra-host diversity using π because it is robust to large variation in sequencing depth (Zhao and Illingworth 2019). Mutation effects were also annotated using SNPEff (v4.3, Cingolani et al. 2012) and a custom genome annotation for CHV1. Mutational effects were defined following SNPEff’s standard approach (Cingolani 2021). Specifically following the SNPEff manual: low-effect mutations were synonymous; moderate-effect mutations were almost entirely non-synonymous mutations causing an amino acid change; high-effect mutations were disruptive or frameshift mutations; and modifiers were mutations in non-coding regions.
4.6. Haplotype identification
Due to the sequencing error, PacBio HiFi reads have to be denoised and merged to generate accurate haplotypes. The unaligned demultiplexed and quality filtered HiFi reads were thus run through the Robust Amplicon Denoising pipeline that is designed for working with PacBio HiFi reads (Kumar et al. 2019). Quality filtered reads were additionally filtered to exclude those that were <3 kb and >6 kb in length. The denoising pipeline does not orientate reads and PacBio reads are not directional, thus identical haplotypes that are reverse complements may be present at this stage. To identify such haplotypes, denoised reads were first aligned to the CHV1 reference as described above and the Hamming distance calculated between every haplotype in an amplicon library in R using the package pegas (v0.14, Paradis 2010). The read count for those with a Hamming distance of zero were then merged. Haplotype diversity was measured using gene diversity i.e. Nei’s H (Nei and Tajima 1980).
4.7. Assessing sequencing accuracy with overlapping amplicons
The amplicons chosen (4.6 and 5 kb) balanced length with expected sequencing quality, which is determined for PacBio reads by the number of polymerase passes and sequence length (Wenger et al. 2019). As assessments of PacBio HiFi sequencing of viruses remain limited, the primer pairs were independent of each other and targeted an overlapping 4.4 kb of CHV1’s 5ʹ-end. This allowed us to assess the reproducibility of our analyses and the accuracy of our long-read sequencing method. To this end, we calculated the repeatability of mutation calls as well as the similarity in the estimated intra-host frequencies of mutations. Haplotypes were not compared as they cover separate genomic regions and could not be trimmed to only the overlapping region under the current pipeline. However, we did compare haplotype numbers and Nei’s H values across amplicons.
4.8. Relationship reconstruction
To characterize the broad-scale inter-host relationships within and between our four geographical populations, phylogenic relationships were inferred using host-specific consensus sequences and RAxML-NG (v0.9.0, Kozlov et al. 2019). Consensus sequences were constructed for each amplicon using bcftools (v1.9-259-gbd769ac, Li et al. 2009), which generates a consensus using intra-host mutations and the reference sequence as a backbone. To ensure an accurate relationship, only mutation calls found in the overlapping region of both amplicons (877–5269 bp) of a sample were considered for the consensus. Inter-host relationships were then reconstructed using RAxML-NG’s ‘all-in-one’ analysis option with GTR + G model and 1000 bootstrap replicates. Both ‘fbp’ and ‘tbe’ bootstrap metrics were calculated. These are shown by node size on our phylogenetic tree. A network-based inference of population structure was also constructed across the four populations using PopNetD3 (Zhang and Parkinson 2019). This only used mutation calls found in both amplicons of a sample. Due to program requirements, only SNV mutations were included and the reference sequence CHV1-EP721 was used as a backbone. The overlapping region of our two amplicons was divided into 800 bp windows for this analysis. The window size was chosen to balance the need for a sufficient number of polymorphic mutations to perform the chromosome painting analyses and small enough windows to capture different patterns across the genome. Shown are edges above the cut-off value of 0.5.
Fine-scale intra- and inter-host structures were then characterized using the intra-host haplotypes generated by Robust Amplicon Denoising and Phyloscanner (v.1.8, Wymant et al. 2018). As recommended, two phylogenetic trees were generated for each amplicon to capture the different evolutionary information present across a viral genome. We divided the genome into two equal parts that roughly separates the two ORFs. The 5-kb amplicon was divided from 101 to 2600 bp and 2600 to 5099 bp. The 4.6-kb amplicon was divided from 878 to 3177 bp and 3177 to 5476 bp. Trees were bootstrapped 100 times, rooted to the CHV1 Sanger reference sequence (EP721), and recombination was checked. The trees were then analyzed following the Phyloscanner pipeline without read blacklisting. The multifurcation threshold was estimated by the Phyloscanner. The ancestral state of each intra-host population was reconstructed using both the ‘s’ and ‘r’ settings and with k values of 0 and 12. Relationships and the subgraph number remained consistent across these settings. Shown are the values with ‘s’ and ‘k’ of 0. The recombination rates and predicated subgraph number were extracted for each individual. The Phyloscanner does not recommend reporting bootstrapped support values for relationships because this can unfairly penalize biologically meaningful relationships with similar haplotypes. The robustness of the relationships should instead be evaluated by the agreement across independent genomic windows, accordingly all four replicate windows are reported (Wymant et al. 2018).
4.9. Factors affecting intra-host diversity
To test for factors affecting π and Nei’s H, we ran each in a General Linear Mixed Effects Model (R version 4.0.2 using lme4 v1.1–25, Bates et al. 2015), with canker type (active/passive), geographic population, fungal host genotype at the five polymorphic vic loci and CHV1 amplicon (5 kb or 4.6 kb) as explanatory variables. An interaction was fit between the amplicon and host canker type. Canker type was used as a proxy for infection age, because passive cankers are more likely to be older infections. The change in appearance occurs over time due to the actions of the virus (Rigling and Prospero 2018). All possible pairwise interactions with the data were fit between vic loci. The sample (i.e. the sampled canker) was fit as a random variable. Values of π were log transformed and values of Nei’s H were logit transformed to improve model fit and meet model assumptions. Two samples had only one denoised haplotype giving Nei’s H value of 0. These could not be logit transformed at 0 and so were given a value of 0.01, which is equivalent to half the value of those that have two haplotypes. Model reduction was performed using backward stepwise deletion and variances were estimated using maximum likelihood. Term significance was assessed using the t-value and a type 3 ANOVA (R package Car v3.0–10, Fox and Weisberg 2019), as well as confirmed with a X2 model comparison. Interactions were visualized through the ‘interaction plot’ and ‘cat plot’ function in R. Post hoc pairwise comparisons across the levels of significant categorical variables with multiple levels (i.e. population) were assessed with the package emmeans (R package v1.5.2–1, Lenth et al. 2020). Final models are reported in Tables S2 and S3.
The subgraph number was analyzed using the identical parameters and model as both diversity metrics (described above). The subgraph number was averaged across the two replicate trees for each amplicon, making it a non-categorical variable. To meet model assumptions subgraph number was log transformed. The final model is reported in Table S4.
4.10. Intra-host patterns of selection
The mean values of π at non-synonymous (πN) and synonymous sites (πS) were calculated by SNPGenie (Nelson, Moncla, and Hughes 2015) and extracted for each replicate amplicon from each sample (i.e. each sampled canker). No location filtering was applied to our mutations. A two-sided paired t-test was used to test for significant differences in π across these site types, pairs consisted of the πN and πS values from each amplicon from a focal sample. The paired t-test was run separately for each amplicon, as well as for both amplicons combined. To examine fine-scale patterns of selection, a sliding window analyses was run on the intra-host mutation calls from each amplicon from a sample with SNPGenie. This calculated the πN/ πS values across 50 bp windows with a window step of 10 bp. Values were bootstrapped 1,000 times and a minimum of three codons were needed per window. The analysis was run for each p-domain separately, and only those entirely covered by an amplicon were analyzed. Significance assessed for those codons where both πN and πS were >0 using a permutated t-tests described in the SNPGenie manual.
4.11. Fungal host vegetative compatibility genotyping
Fungal host genotypes were obtained by standard protocols. DNA was extracted using the Kingfisher 96 Flex kit and vic loci genotyped using a published assay (Cornejo et al. 2019).
Supplementary Material
Acknowledgements
We would like to thank Benjamin Murrell for his help during the PCR design process, Javi Zhang for his guidance running PopNetD3, and Nobuhiro Suzuki for his feedback early on in this project. We would like to thank all members of WSL’s Phytopathology group, notably Hélèn Blauenstein, Sven Ulrich, Steffi Pfister, Lovro Ogresta, and Melanie Beck for their help in the lab. We would like to thank Lea Stauber and the teams at the GDC, FGCZ, and WSL’s Hyperion cluster for their support.
Contributor Information
Deborah M Leigh, Phytopathology, Swiss Federal Institute for Forest, Snow and Landscape Research WSL, Birmensdorf CH-8903, Switzerland.
Karla Peranić, Faculty of Science, University of Zagreb, Zagreb, Grad Zagreb 10000, Croatia.
Simone Prospero, Phytopathology, Swiss Federal Institute for Forest, Snow and Landscape Research WSL, Birmensdorf CH-8903, Switzerland.
Carolina Cornejo, Phytopathology, Swiss Federal Institute for Forest, Snow and Landscape Research WSL, Birmensdorf CH-8903, Switzerland.
Mirna Ćurković-Perica, Faculty of Science, University of Zagreb, Zagreb, Grad Zagreb 10000, Croatia.
Lucija Nuskern, Faculty of Science, University of Zagreb, Zagreb, Grad Zagreb 10000, Croatia.
Daniel Rigling, Phytopathology, Swiss Federal Institute for Forest, Snow and Landscape Research WSL, Birmensdorf CH-8903, Switzerland.
Marin Ježić, Faculty of Science, University of Zagreb, Zagreb, Grad Zagreb 10000, Croatia.
Data availability
Sample information including all intra-host diversity metrics are supplied in Table S1. All relevant raw sequencing data (including demultiplexing files) will be made accessible upon publication acceptance. All data underlying our figures are either in Table S1 or can be generated using our raw sequencing files. Processed sequencing data (SNVs, etc.) will be archived in the Phytopathology group at WSL and can be accessed or publicly archived upon request.
Supplementary data
Supplementary data is available at Virus Evolution online.
Funding
This work is supported by the Croatian-Swiss Research Program (Swiss National Science Foundation grant number IZHRZ0_180651, ‘Dynamics of virus infection in mycovirus-mediated biological control of a fungal pathogen’).
Conflict of interest:
The authors declare no known conflict or competing interests.
Author contributions
D.M.L. and D.R. collected the Swiss samples. K.P. and M.J. collected the Croatian samples. D.M.L., K.P., and L.N. conducted the fungal culture laboratory work. D.M.L., C.C., and Q.K. developed and optimized NGS library protocols. Q.K. prepared the NGS libraries. D.M.L. independently developed and ran all NGS processing, ran and interpreted all data analysis. D.M.L. wrote the manuscript and prepared the figures. C.C., S.P., D.R., Q.K., L.N., K.P., M.Ć.-P., and M.J. commented on the text. D.R., M.J., S.P., and M.Ć.-P. conceived, outlined, and secured funding for the overall project. D.M.L., D.R., and M.J. planned the experiments and oversaw the collaboration.
Code availability
Data were processed using the software and programs outlined in the methods. No new programs or algorithms were required for data analyses. An outline of the core commands used has been uploaded to GitHub https://github.com/deborahmleigh/CHV1_intra-host.git.
References
- Aaskov J. et al. (2006) ‘Long-Term Transmission of Defective RNA Viruses in Humans and Aedes Mosquitoes’, Science, 311: 236–8. [DOI] [PubMed] [Google Scholar]
- Ali A., and Roossinck M. J. (2010) ‘Genetic Bottlenecks during Systemic Movement of Cucumber Mosaic Virus Vary in Different Host plants’. Virology, 404: 279–83. [DOI] [PubMed] [Google Scholar]
- Alves J. M. et al. (2019) ‘Parallel Adaptation of Rabbit Populations to Myxoma Virus’, Science, 363: 1319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameline C. et al. (2020) ‘A Two-locus System with Strong Epistasis Underlies Rapid Parasite-mediated Evolution of Host Resistance’, MBE, 38: 1512–28.doi: 10.1093/molbev/msaa311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton J. P. et al. (2016) ‘Relative Rate and Location of Intra-host HIV Evolution to Evade Cellular Immunity are Predictable’, Nature Communications, 7: 11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D. et al. (2015) ‘Fitting Linear Mixed-Effects Models Using Lme4’, Journal of Statistical Software, 67: 1–48. [Google Scholar]
- Brockhurst M. A. et al. (2014) ‘Running with the Red Queen: The Role of Biotic Conflicts in Evolution’, Proceedings of the Royal Society B: Biological Sciences, 281: 20141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryner S. F., Rigling D., and Brunner P. C. (2012) ‘Invasion History and Demographic Pattern of Cryphonectria Hypovirus 1 across European Populations of the Chestnut Blight Fungus’, Ecology and Evolution, 2: 3227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle L. A. et al. (2019) ‘Viral Diversity Based on Next-Generation Sequencing of HIV-1 Provides Precise Estimates of Infection Recency and Time since Infection’, The Journal of Infectious Diseases, 220: 254–65. [DOI] [PubMed] [Google Scholar]
- Cingolani P. (2021), Input & Output Files—SnpEff & SnpSift Documentation. SNPeff Doc. <https://pcingola.github.io/SnpEff/se_inputoutput/#eff-field-vcf-output-files> accessed 19 Jan 2021.
- —— et al. (2012) ‘A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3’, Fly, 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conedera M. et al. (2004) ‘The Cultivation of Castanea sativa (Mill.) In Europe, from Its Origin to Its Diffusion on a Continental Scale’, Vegetation History and Archaeobotany, 13: 161–79. [Google Scholar]
- Cornejo C. et al. (2019) ‘A Multiplexed Genotyping Assay to Determine Vegetative Incompatibility and Mating Type in Cryphonectria Parasitica’, European Journal of Plant Pathology, 155: 81–91. [Google Scholar]
- Cortesi P. et al. (2001) ‘Genetic Control of Horizontal Virus Transmission in the Chestnut Blight Fungus, Cryphonectria Parasitica’, Genetics, 159: 107–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis D. E. (2004) ‘Historical Significance of American Chestnut to Appalachian Culture And Ecology’. In: Steiner, K. C., and Carlson, J. E. (eds) Restoration of American Chestnut to Forest Lands. The North Carolina Arboretum. Natural Resources Report NPS/NCR/CUE/NRR - 2006/001, National Park Service, Washington, DC, USA. [Google Scholar]
- Dawe A. L., and Nuss D. L. (2001) ‘Hypoviruses and Chestnut Blight: Exploiting Viruses to Understand and Modulate Fungal Pathogenesis’, Annual Review of Genetics, 35: 1–29. [DOI] [PubMed] [Google Scholar]
- Diamandis S. (2018) ‘Management of Chestnut Blight in Greece Using Hypovirulence and Silvicultural Interventions’, Forests, 9: 492. [Google Scholar]
- Dunham J. P. et al. (2014) ‘Analysis of Viral (Zucchini Yellow Mosaic Virus) Genetic Diversity during Systemic Movement through a Cucurbita Pepo Vine’, Virus Research, 191: 172–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutech C. et al. (2008) Geostatistical genetic analysis for inferring the dispersal pattern of a partially clonal species: example of the chestnut blight fungus. Mol Ecol, 17: 4597–607. [DOI] [PubMed] [Google Scholar]
- Dyrka W. et al. (2014) ‘Diversity and Variability of NOD-Like Receptors in Fungi’, Genome Biology and Evolution, 6: 3137–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D. (1994) ‘Virulence and Local Adaptation of a Horizontally Transmitted Parasite’, Science, 265: 1084–6. [DOI] [PubMed] [Google Scholar]
- Ehrbar D. J. et al. (2017) ‘High Levels of Local Inter- and Intra-host Genetic Variation of West Nile Virus and Evidence of Fine-scale Evolutionary Pressures’, Infection, Genetics and Evolution, 51: 219–26. [DOI] [PubMed] [Google Scholar]
- Elliott K. J., and Swank W. T. (2008) ‘Long-term Changes in Forest Composition and Diversity following Early Logging (1919–1923) and the Decline of American Chestnut (Castanea dentata)’, Plant Ecology, 197: 155–72. [Google Scholar]
- Feder A. F., Pennings P. S., and Petrov D. A. (2021) ‘The Clarifying Role of Time Series Data in the Population Genetics of HIV’, PLoS Genetics, 17: e1009050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti L. et al. (2020) ‘Pervasive Within-host Recombination and Epistasis as Major Determinants of the Molecular Evolution of the Foot-and-mouth Disease Virus Capsid’, PLoS Pathogens, 16: e1008235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J., and Weisberg S. (2019) An R Companion to Applied Regression. 3rd edn. Sage: Thousand Oaks, CA. [Google Scholar]
- Frost S. D. W. et al. (2001) ‘Genetic Drift and Within-host Metapopulation Dynamics of HIV-1 Infection’, Proceedings of the National Academy of Sciences, 98: 6975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E., and Marth G. (2012) ‘Haplotype-based Variant Detection from Short-read Sequencing’. 20 Jul. arXiv.:1207.3907. http://arxiv.org/abs/1207.3907. [Google Scholar]
- Gelbart M. et al. (2020) ‘Drivers of Within-host Genetic Diversity in Acute Infections of Viruses’, PLoS Pathogens, 16: e1009029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiniger U., and Rigling D. (1994) ‘Biological Control Of Chestnut Blight In Europe’, Annual Review of Phytopathology, 32: 581–99. [Google Scholar]
- Jerzak G. et al. (2005) ‘Genetic Variation in West Nile Virus from Naturally Infected Mosquitoes and Birds Suggests Quasispecies Structure and Strong Purifying Selection’, Journal of General Virology, 86: 2175–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ježić M. et al. (2018) ‘Changes in Cryphonectria Parasitica Populations Affect Natural Biological Control of Chestnut Blight’, Phytopathology, 108: 870–7. [DOI] [PubMed] [Google Scholar]
- —— et al. (2021) ‘Temporal and Spatial Genetic Population Structure of Cryphonectria Parasitica and Its Associated Hypovirus across an Invasive Range of Chestnut Blight in Europe’, Phytopathology, 111: 1327–37. [DOI] [PubMed] [Google Scholar]
- Kennedy D. A., and Dwyer G. (2018) ‘Effects of Multiple Sources of Genetic Drift on Pathogen Variation within Hosts’, PLoS Biology, 16: e2004444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoti W. M. et al. (2017) ‘Analysis of Intra-host Genetic Diversity of Prunus Necrotic Ringspot Virus (PNRSV) Using Amplicon Next Generation Sequencing’, PLoS One, 12: e0179284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakiewicz C. P. et al. (2018) ‘Pathogens in Space: Advancing Understanding of Pathogen Dynamics and Disease Ecology through Landscape Genetics’, Evolutionary Applications, 11: 1763–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A. M. et al. (2019) ‘RAxML-NG: A Fast, Scalable and User-friendly Tool for Maximum Likelihood Phylogenetic Inference’, Bioinformatics, 35: 4453–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V. et al. (2019) ‘Long-read Amplicon Denoising’, Nucleic Acids Research, 47: e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird Smith M. et al. (2016) ‘Rapid Sequencing of Complete Env Genes from Primary HIV-1 Samples’, Virus Evolution, 2: vew018.doi: 10.1093/ve/vew018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A. S. (2020) ‘Within-Host Viral Diversity: A Window into Viral Evolution’, Annual Review of Virology, 7: 63–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauring A. S., and Andino R. (2010) ‘Quasispecies Theory and the Behavior of RNA Viruses’, PLoS Pathogens, 6: e1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh D. M. et al. (2018) ‘Batch Effects in a Multiyear Sequencing Study: False Biological Trends Due to Changes in Read Lengths’, Molecular Ecology Resources, 18: 778–88. [DOI] [PubMed] [Google Scholar]
- Leigh D. M., Schefer C., and Cornejo C. (2020) ‘Determining the Suitability of MinION’s Direct RNA and DNA Amplicon Sequencing for Viral Subtype Identification’, Viruses, 12: 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth R. V. et al. (2020), Emmeans: Estimated Marginal Means, Aka Least-Squares Means. <https://CRAN.R-project.org/package=emmeans> accessed 19 Jan 2021.
- Lequime S. et al. (2016) ‘Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes’, PLoS Genetics, 12: e1006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2016) ‘Minimap and Miniasm: Fast Mapping and de Novo Assembly for Noisy Long Sequences’, Bioinformatics, 32: 2103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— et al. 1000 Genome Project Data Processing Subgroup . (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. et al. (2007) ‘Genome Sequence, Full-Length Infectious cDNA Clone, and Mapping of Viral Double-Stranded RNA Accumulation Determinant of Hypovirus CHV1-EP721’, Journal of Virology, 81: 1813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. (2014) ‘Extensive Recombination Due to Heteroduplexes Generates Large Amounts of Artificial Gene Fragments during PCR’, PLoS One, 9: e106658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarigal K., and Cushman S. A. (2002) ‘Comparative Evaluation of Experimental Approaches to the Study of Habitat Fragmentation Effects’, Ecological Applications, 12: 335–45. [Google Scholar]
- Metzger C. M. J. A. et al. (2016) ‘The Red Queen Lives: Epistasis between Linked Resistance Loci’, Evolution, 70: 480–7. [DOI] [PubMed] [Google Scholar]
- Mühlemann B. et al. (2018) ‘Ancient Hepatitis B Viruses from the Bronze Age to the Medieval Period’, Nature, 557: 418–23. [DOI] [PubMed] [Google Scholar]
- Nei M., and Tajima F. (1980) ‘DNA Polymorphism Detectable by Restriction Endonucleases’, Genetics, 97: 145–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. W., and Hughes A. L. (2015) ‘Within-host Nucleotide Diversity of Virus Populations: Insights from Next-generation Sequencing’, Infection, Genetics and Evolution, 30: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson C. W., Moncla L. H., and Hughes A. L. (2015) ‘SNPGenie: Estimating Evolutionary Parameters to Detect Natural Selection Using Pooled Next-generation Sequencing Data’, Bioinformatics (Oxford, England), 31: 3709–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss D. L. (1992) ‘Biological Control of Chestnut Blight: An Example of Virus-mediated Attenuation of Fungal Pathogenesis’, Microbiological Reviews, 56: 561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- —— (2005) ‘Hypovirulence: Mycoviruses at the Fungal–plant Interface’, Nature Reviews Microbiology, 3: 632–42. [DOI] [PubMed] [Google Scholar]
- Papkou A. et al. (2019) ‘The Genomic Basis of Red Queen Dynamics during Rapid Reciprocal Host–pathogen Coevolution’, Proceedings of the National Academy of Sciences, 116: 923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E. (2010) ‘Pegas: An R Package for Population Genetics with an Integrated–modular Approach’, Bioinformatics, 26: 419–20. [DOI] [PubMed] [Google Scholar]
- Peyambari M. et al. (2018) ‘A 1,000-Year-Old RNA Virus’, Journal of Virology, 93: e01188–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P. C. (2008) ‘Epistasis — the Essential Role of Gene Interactions in the Structure and Evolution of Genetic Systems’, Nature Reviews Genetics, 9: 855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R. et al. (2018) ‘A Universal SNP and Small-indel Variant Caller Using Deep Neural Networks’, Nature Biotechnology, 36: 983–7. [DOI] [PubMed] [Google Scholar]
- Predajňa L. et al. (2012) ‘Evaluation of the Genetic Diversity of Plum Pox Virus in a Single Plum Tree’, Virus Research, 167: 112–7. [DOI] [PubMed] [Google Scholar]
- Redd A. D. et al. (2012) ‘Previously Transmitted HIV-1 Strains are Preferentially Selected during Subsequent Sexual Transmissions’, The Journal of Infectious Diseases, 206: 1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigling D., and Prospero S. (2018) ‘Cryphonectria Parasitica, the Causal Agent of Chestnut Blight: Invasion History, Population Biology and Disease Control’, Molecular Plant Pathology, 19: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau C. M. et al. (2009) ‘Rare HLA Drive Additional HIV Evolution Compared to More Frequent Alleles’, AIDS Research and Human Retroviruses, 25: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallinen S. et al. (2020) ‘Intraspecific Host Variation Plays a Key Role in Virus Community Assembly’, Nature Communications, 11: 5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M., Sloan W. T., and Quince C. (2014) ‘Benchmarking of Viral Haplotype Reconstruction Programmes: An Overview of the Capacities and Limitations of Currently Available Programmes’, Briefings in Bioinformatics, 15: 431–42. [DOI] [PubMed] [Google Scholar]
- Shapira R. et al. (1991) ‘The Contribution of Defective RNAs to the Complexity of Viral-encoded Double-stranded RNA Populations Present in Hypovirulent Strains of the Chestnut Blight Fungus Cryphonectria Parasitica.’, The EMBO Journal, 10: 741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen B. B. et al. (2009) ‘Low-abundance Drug-resistant Viral Variants in Chronically HIV-infected, Antiretroviral Treatment-naive Patients Significantly Impact Treatment Outcomes’, Journal of Infectious Diseases, 199: 693–701. [DOI] [PubMed] [Google Scholar]
- Simmonds P., Aiewsakun P., and Katzourakis A. (2019) ‘Prisoners of War — Host Adaptation and Its Constraints on Virus Evolution’, Nature Reviews Microbiology, 17: 321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons H. E., Holmes E. C., and Stephenson A. G. (2011) ‘Rapid Turnover of Intra-host Genetic Diversity in Zucchini Yellow Mosaic Virus’, Virus Research, 155: 389–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirkkomaa S. (1983) ‘Calculations on the Decrease of Genetic Variation Due to the Founder Effect’, Hereditas, 99: 11–20. [DOI] [PubMed] [Google Scholar]
- Smith O. et al. (2014) ‘A Complete Ancient RNA Genome: Identification, Reconstruction and Evolutionary History of Archaeological Barley Stripe Mosaic Virus’, Scientific Reports, 4: 4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sork V. L., and Waits L. (2010) ‘Contributions of Landscape Genetics – Approaches, Insights, and Future Potential’, Molecular Ecology, 19: 3489–95. [DOI] [PubMed] [Google Scholar]
- Spitaler M., and Cantrell D. A. (2004) ‘Protein Kinase C and Beyond’, Nature Immunology, 5: 785–90. [DOI] [PubMed] [Google Scholar]
- Stauber L. et al. (2020) Emergence and population genomics of the highly invasive chestnut blight pathogen. Doctoral Thesis, University of Neuchatel, Switzerland. [Google Scholar]
- Stauber L. et al. (2021) ‘Emergence and Diversification of a Highly Invasive Chestnut Pathogen Lineage across South-eastern Europe’, Elife, 10: e56279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia K. et al. (2013) ‘Defective Viral Genomes Arising in Vivo Provide Critical Danger Signals for the Triggering of Lung Antiviral Immunity’, PLoS Pathogens, 9: e1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier A., and Brown J. K. M. (2007) ‘Polymorphism in Multilocus Host–Parasite Coevolutionary Interactions’, Genetics, 177: 1777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M. et al. (2014) ‘Local and Regional Founder Effects in Lake Zooplankton Persist after Thousands of Years despite High Dispersal Potential’, Molecular Ecology, 23: 1014–27. [DOI] [PubMed] [Google Scholar]
- Wang C. et al. (2007) ‘Characterization of Mutation Spectra with Ultra-deep Pyrosequencing: Application to HIV-1 Drug Resistance’, Genome Research, 17: 1195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh C. et al. (2015) ‘A General Method to Eliminate Laboratory Induced Recombinants during Massive, Parallel Sequencing of cDNA Library’, Virology Journal, 12: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger A. M. et al. (2019) ‘Accurate Circular Consensus Long-read Sequencing Improves Variant Detection and Assembly of a Human Genome’, Nature Biotechnology, 37: 1155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymant C. et al. STOP-HCV Consortium, the Maela Pneumococcal Collaboration, and the BEEHIVE Collaboration . (2018) ‘PHYLOSCANNER: Inferring Transmission from Within- and Between-Host Pathogen Genetic Diversity’, Molecular Biology and Evolution, 35: 719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y. et al. (2017) ‘Poliovirus Intrahost Evolution Is Required to Overcome Tissue-specific Innate Immune Responses’, Nature Communications, 8: 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue K. S., and Bloom J. D. (2020) ‘Linking Influenza Virus Evolution within and between Human Hosts’, Virus Evolution, 6: veaa010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T. et al. (2020) ‘Single-molecular Real-time Deep Sequencing Reveals the Dynamics of Multi-drug Resistant Haplotypes and Structural Variations in the Hepatitis C Virus Genome’, Scientific Reports, 10: 2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.-X. et al. (2014) ‘Vegetative Incompatibility Loci with Dedicated Roles in Allorecognition Restrict Mycovirus Transmission in Chestnut Blight Fungus’, Genetics, 197: 701–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., and Parkinson J. (2019) ‘PopNetD3—A Network-Based Web Resource for Exploring Population Structure’, Molecular Biology and Evolution, 11: 1730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-Z. et al. (2019) ‘Expanding the RNA Virosphere by Unbiased Metagenomics’, Annual Review of Virology, 6: 119–39. [DOI] [PubMed] [Google Scholar]
- Zhang Y.-Z., Shi M., and Holmes E. C. (2018) ‘Using Metagenomics to Characterize an Expanding Virosphere’, Cell, 172: 1168–72. [DOI] [PubMed] [Google Scholar]
- Zhao L., and Illingworth C. J. R. (2019) ‘Measurements of Intrahost Viral Diversity Require an Unbiased Diversity Metric’, Virus Evolution, 5: vey041.doi: 10.1093/ve/vey041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sample information including all intra-host diversity metrics are supplied in Table S1. All relevant raw sequencing data (including demultiplexing files) will be made accessible upon publication acceptance. All data underlying our figures are either in Table S1 or can be generated using our raw sequencing files. Processed sequencing data (SNVs, etc.) will be archived in the Phytopathology group at WSL and can be accessed or publicly archived upon request.