

Abstract
Polymorphism drives survival under stress and provides adaptability. Genetic polymorphism of ribosomal RNA (rRNA) genes derives from internal repeat variation of this multicopy gene, and from interindividual variation. A considerable amount of rRNA sequence heterogeneity has been proposed but has been challenging to estimate given the scarcity of accurate reference sequences. We identified four rDNA copies on chromosome 21 (GRCh38) with 99% similarity to recently introduced reference sequence KY962518.1. We customized a GATK bioinformatics pipeline using the four rDNA loci, spanning a total 145 kb, for variant calling and used high-coverage whole-genome sequencing (WGS) data from the 1000 Genomes Project to analyze variants in 2504 individuals from 26 populations. We identified a total of 3791 variant positions. The variants positioned nonrandomly on the rRNA gene. Invariant regions included the promoter, early 5′ ETS, most of 18S, 5.8S, ITS1, and large areas of the intragenic spacer. A total of 470 variant positions were observed on 28S rRNA. The majority of the 28S rRNA variants were located on highly flexible human-expanded rRNA helical folds ES7L and ES27L, suggesting that these represent positions of diversity and are potentially under continuous evolution. Several variants were validated based on RNA-seq analyses. Population analyses showed remarkable ancestry-linked genetic variance and the presence of both high penetrance and frequent variants in the 5′ ETS, ITS2, and 28S regions segregating according to the continental populations. These findings provide a genetic view of rRNA gene array heterogeneity and raise the need to functionally assess how the 28S rRNA variants affect ribosome functions.
Keywords: rRNA, ribosome, rDNA array, 1000 Genomes Project
INTRODUCTION
The ribosomal RNA (rRNA) genes encode for the main RNA component of the ribosome and are essential for ribosome synthesis and hence protein translation. Ribosome biogenesis is dependent on transcription of the rRNAs involving a coordinated action by three polymerases and synthesis of hundreds of proteins. RNA polymerase I (Pol I) transcribes a 13 kb polycistronic 47S rRNA precursor that is cotranscriptionally processed and cleaved into the mature 5.8S, 18S, and 28S rRNAs (Moss et al. 2007). Transcription terminates at a series of nonconserved termination repeat elements, followed by a long ∼30 kb intragenic spacer (IGS) (Németh et al. 2013). The mature rRNAs are flanked by noncoding spacers called 5′ and 3′ external transcribed spacers (5′ and 3′ ETSs) and are separated by internal transcribed spacers 1 and 2 (ITS1 and ITS2) (Henras et al. 2015). Precise, sequential cleavage occurs at the processing sites and is assisted by a multitude of rRNA biogenesis proteins and small nucleolar noncoding RNAs (Lafontaine 2015). The processing, folding and maturation requires also extensive post-translational modification of the rRNAs (Sloan et al. 2017). Ultimately, the rRNAs are assembled into ribosomes, large protein–RNA complexes composed in the human of a large 60S subunit consisting of 28S, 5S, and 5.8S rRNAs and 47 proteins, and a small 40S subunit with 18S rRNA and 33 proteins (Anger et al. 2013; Khatter et al. 2015).
The human multicopy rRNA genes are organized in repeat arrays on five acrocentric chromosomes: 13, 14, 15, 21, and 22 (Henderson et al. 1972). Copy numbers are estimated to vary between 100–600 per diploid genome, are unevenly distributed on the acrocentric chromosomes, and vary between individuals (Gibbons et al. 2015; McStay 2016; Xu et al. 2017; van Sluis et al. 2020). The rRNA gene loci contain not only the gene arrays, but are also highly repetitive due to the presence of satellite repeats and other repetitive elements rendering them challenging to study and assess their genetic variability (McStay 2016). The Telomere-to-Telomere (T2T) Consortium very recently provided in-depth mapping of these previously unannotated gene arrays covering a total of 10 Mb rDNA sequence derived from a functionally haploid CHM13 hydatidiform mole cell line (Nurk et al. 2021). This analysis, using a combination of PacBio HiFi, Oxford Nanopore and PCR-free sequencing and bioinformatic methods, indicated chromosome-dependent variation of the rDNA copies (Nurk et al. 2021).
While substantial progress has been made in identifying the proximal and distal sequences of the arrays, consensus or reference sequences for the human rRNA gene have been lacking (McStay 2016). Only a few reference sequences have existed until recently (U13369, AL353644.34) (Gonzalez and Sylvester 1995). The reference sequence U13369.1 was deposited in 1994 (Gonzalez and Sylvester 1995) and has been used in a vast majority of studies since then. Yet, reports recognizing rRNA sequence variation have been published since the 1980s (Gonzalez et al. 1985, 1988; Worton et al. 1988; Kuo et al. 1996). Recent transformation-associated recombination (TAR) cloning and long-read coding efforts, using a single chromosome 21 mouse/human hybrid as a source, enabled the accurate assembly of a new 45 kb rDNA sequence, identified numerous discrepancies to the earlier reference and substantially refined it, and introduced a reference sequence (KY962518.1) which was 1.8 kb longer than the previous (Kim et al. 2018). These sequencing efforts also identified single-nucleotide variants (SNV) and insertion/deletions (INDEL) in the rRNA coding and noncoding IGS sequences based on multiple sequenced clones of chromosome 21 (Kim et al. 2018). Several earlier studies have conducted variant calling approaches of the rRNA genes, but may be compromised by calling variants representing sequencing or alignment errors due to the use of the earlier reference U13369 (Babaian 2017; Xu et al. 2017; Parks et al. 2018). The identification of variants still requires refinement and their implications are yet to be determined.
Here we identified three full and one partial rRNA gene copies on chromosome 21 (GRCh38) with 99% similarity to the most recent rDNA reference KY962518.1. We used whole-genome sequencing (WGS) new high-coverage data from the 1000 Genomes Project, including 2504 individuals from 26 populations (1000 Genomes Project Consortium et al. 2015; Byrska-Bishop et al. 2021) and performed variant calling against the identified rDNA sequences in GRCh38 to generate a comprehensive set of SNVs and INDEL variant positions (SNV/INDEL) of the rDNA arrays. We discovered a total of 3791 variant positions on the rRNA genes. Moreover, the variants distinctly clustered on the rRNA gene coding and IGS regions, while regions for core ribosomal protein interactions and rRNA modification sites and large areas in the IGS were near invariant. A high number of variant positions, 380 SNVs and 90 INDELs, were detected on the mature 28S rRNA. The majority of the 28S rRNA variants were located on the highly flexible human-expanded rRNA helical folds ES7L and ES27L. We further validated several variant positions using high-coverage RNA-seq data from the 1000 Genomes Project, and annotated these variants across the human population. These analyses identified shared and population-stratified variants. We infer that the rRNA array mosaicism substantially contributes to the number of detected variants, some of which have evolved in a population-specific manner. Collectively, our findings provide a genetic view for rRNA heterogeneity and suggest potential links between variants and ribosome functions.
RESULTS
Genomic variation of rDNA copies in GRCh38 chromosome 21
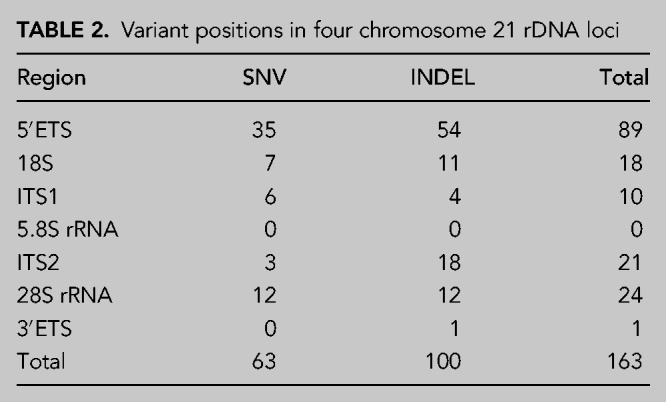
To identify rDNA loci in the GRCh38 human genome reference, we performed pairwise alignment of the human rDNA reference sequence assembled by using TAR cloning and long-read sequencing (GenBank: KY962518.1) against current human reference genome GRCh38. We identified three full and one partial rDNA copies on chromosome 21 (GRCh38) with 99% similarity to the reference KY962518 (Fig. 1A; Supplemental Table S1). The 13.3 kb rRNA coding sequence of these copies varied in length by up to 42 nt due to length variation of all but the 5.8S rRNA domain (Table 1; Supplemental Table S1). Next, we performed multiple sequence alignment of rDNA reference sequence KY962518 and the four chromosome 21 rDNA copies (Fig. 1B). We identified a total of 163 SNV/INDELs among these rDNA coding regions. Over half of the variants were detected in the 5′ ETS region, 15% in the 28S rRNA, and 11% in 18S rRNA, whereas there were no variants in the 5.8S rRNA coding region (Table 2). Among the variants in the 28S rRNA, two 4–6 nt INDELs were found, which were present in two rDNA copies and absent in the other two (Fig. 1C,D). The two short INDELs consisted of GC-repeats (Fig. 1D) and were located in the ES7L and ES15L expansion segments (not shown). The observed variation between the repeats in one chromosome suggests that diversity between different copies is likely to exist. This is in accordance with the mosaic pattern of rDNA array copies detailed in the study by Nurk et al. (2021).
FIGURE 1.

Multiple sequence alignment of human rRNA coding region in chromosome 21. (A) Schematic representation of four rDNA copies in human acrocentric chromosome 21 (GRCh38). rDNA copies are labeled by different colors. (B) Multisequence alignment of the coding regions of the chromosome 21 rDNA copies and KY962518 reference sequence. The differences are colored by red. rRNA coding regions are shown on the top. (C) Multisequence alignment of 28S rRNA. Asterisks indicate the position of small INDELs. (D) Multisequence alignment of two INDELs in 28S rRNA. Numbering on the left refers to 28S rRNA positions.
TABLE 1.
Coding region length (nt) variation of rDNA loci on chromosome 21
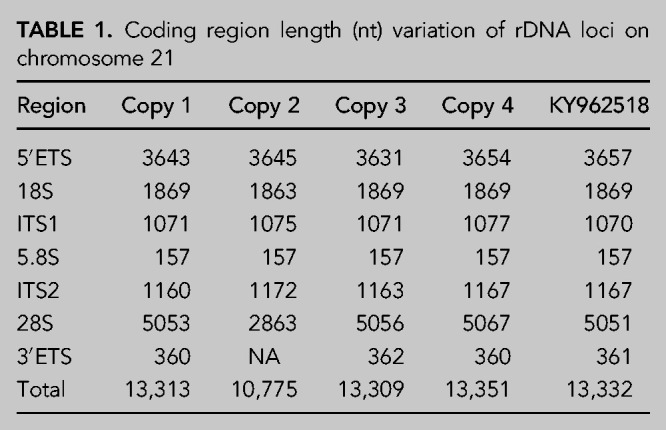
TABLE 2.
Variant positions in four chromosome 21 rDNA loci
High rDNA GC content anticorrelates with WGS read depth
The GC content of rDNA is high and reaches up to 90%. High genomic GC content poses a challenge to PCR-based sequencing technologies (Meienberg et al. 2015). To assess the impact of GC content on the sequencing depth throughout the rRNA coding region, we used high coverage WGS data from the Cancer Cell Line Encyclopedia (CCLE) project (Barretina et al. 2012). For this purpose, we randomly selected 70 cancer cell lines, conducted read alignment to the reference KY962518 using Sentieon DNASeq tools and calculated the read depth across the rDNA sequence. We normalized the read depth per position against the total rDNA reads in the 13.3 kb coding region for each cancer cell line. The GC content was calculated in 70 bp bins and correlated with the normalized read depth. This analysis showed that the GC content strongly negatively correlated with the read depth (Fig. 2A). We then plotted the read depth and GC content per 70 bp bins. Notably, the majority of the rDNA coding region read depth of the 70 cancer cell lines was high. Areas with very low read depths were evident and largely correlated with areas of over 80% GC content (Fig. 2B).
FIGURE 2.

WGS sequence read depth anticorrelates with rDNA GC content. (A) Scatter plot showing GC content as compared to the normalized mean read depth in rDNA coding region. High coverage WGS data were obtained from 70 cancer cell lines in the CCLE database. P was determined by two-sided Pearson's correlation test. (B) Normalized read depth and GC content are plotted across the rDNA coding region. (Top) rRNA coding regions.
Variant discovery in rRNA genes across human 2504 genomes
To explore rRNA gene variation in human genomes, we used the most recent iteration of the 1000 Genomes Project high-coverage WGS resource that has enabled variant discovery across populations (1000 Genomes Project Consortium et al. 2015; Byrska-Bishop et al. 2021). We first examined the sequence coverage of rDNA and WGS data on 2504 individuals using Samtools. Consistent with the description of the WGS data, we observed 30-fold or higher whole genome coverage of the 2504 samples (Fig. 3A). The coverage for rDNA was on average 5000-fold in the human WGS data set (Fig. 3A). This estimation indicates that despite the existence of some highly challenging read areas, the coverage for the rDNA sequences is deep and suitable for variant calling.
FIGURE 3.

rRNA gene variant discovery in the human genome. (A) Coverage of rDNA and whole genome reads are shown for each individual from the 1000 Genomes Project (n = 2504). (B) Number of variant positions in the rRNA gene domains per kilobase (kb). (C) Manhattan plot of variant distribution in a full-length 45 kb rDNA copy. Colors depict rRNA regions, which are indicated on the top. INDELs are shown in solid dots, and SNVs in circles. Gray horizontal line represents the variant quality score significance (QUAL) threshold set at 30. (D) Density plot of the variants in the 13 kb rDNA coding region. Coding region domains are color-labeled as indicated by the legend. (E) Manhattan plot of variant distribution in the rRNA coding region. Color-coding as in C.
To perform large scale variant calling of WGS data, the standard software package for variant calling, GATK (Genome Analysis Toolkit) pipeline, is computing-intense for the analysis of single WGS samples even using multiple computing nodes and after optimization (Van der Auwera et al. 2013). To offer further computing speed over GATK, several ultrafast options have been developed (Raczy et al. 2013; Weber et al. 2016; Pluss et al. 2017). Given that the DNASeq pipeline of Sentieon provides faster variant calling without compromising accuracy, we used it for the analysis of the genomic variation in human rDNA. The workflow was applied on WGS data of 2504 individuals to perform variant calling against the four chromosome 21 rDNA copies, including the coding and IGS sequences, totaling 145 kb. The cut-off for variant calling was defined as variant quality score (QUAL) > 30 (<0.1% error), and rare variants observed in <3 alleles were excluded.
The final variant call set across the 2504 samples identified 2659 SNV and 1132 INDEL positions (Table 3; Supplemental Table S2). The mean variant density across the rDNA coding region was 86 variants/kb. The density was highest on the terminator (over 500 variants/kb), whereas overall was low on the IGS (54 variants/kb) (Fig. 3B). The data showed that the terminator domain and IGS region had the largest number of variant positions (2691 positions). Notably, the ITS2 and 3′ ETS domains had very high variant densities (over 200 variants/kb) (Fig. 3B). The variants clustered around peaks with very high QUAL scores (>106) and positioned nonrandomly on the rDNA (Table 3; Fig. 3C).
TABLE 3.
Variant position calls of rDNA copies in 2504 human genomes

We further visualized the variant density in the coding region (Fig. 3D). In addition to the ITS2 and 3′ ETS domains, the 5′ ETS domain had a high density of variants clustered around 2100 to 3000 bp from transcription start site (Fig. 3D). None of these variants position to the known human rRNA processing sites (Henras et al. 2015). Of note, 18S and 5.8S rRNA had only a few variants, whereas 470 variant positions and a total of 592 variants were observed in the 28S rRNA (Fig. 3D,E). This indicates that 18S and 5.8S rRNAs are highly conserved compared to 28S rRNA. Overall, these results, based on diverse populations and thousands of individuals, reveal highly conserved and variable regions throughout the rDNA sequence.
Annotation of variants on the 28S rRNA structure
Given the abundance of variants in 28S rRNA and their clustering to distinct high- and low-density regions, and significance of the mature 28S rRNA for the 60S subunit function, we focused on annotation of the variants on 28S rRNA. We mapped the variant density profile on the human 28S rRNA secondary structure derived from Ribovision (Bernier et al. 2014). Interestingly, variants with the highest QUAL scores were located in the highly flexible expansion segment helical folds ES27L and ES7L (Fig. 4A). To position the variants to the three-dimensional environment in the rRNA structure, we mapped the variant density profile to the 28S rRNA in the 80S ribosome cryo-EM structure (Anger et al. 2013). The regions with highest density of variants were located on the surface of the ribosome (Fig. 4B). These regions represent expansion segments forming A-form helices that are under species-specific evolution (Anger et al. 2013; Fujii et al. 2018).
FIGURE 4.

Annotation of variants on 28S rRNA structure. (A) Variant density on the secondary structure diagram of 28S rRNA. Nucleotides are color-labeled by the variant density value. (B) Variants of 28S rRNA (shown in blue) are shown on the 3D structure model (PDB 4V6X). Nucleotides are color-labeled by the variant density value as in A. High variant density expansion segments (ES) ES7L and ES27L are indicated. Images were generated with RiboVision.
Chemical modifications of human rRNA are introduced during ribosome biogenesis and required for the rRNA folding and stability. More than 130 individual rRNA modifications are indicated in the 3D structure of the human ribosome (Natchiar et al. 2017). We further compared the human genome variant positions to the recorded modification sites in 28S rRNA. Only one modification site (C2861 methylation) had a SNV in the human genomes, but had a very low allele count and QUAL score (7 and 99, respectively). The combined results suggest that the rRNA core ribosomal protein interaction sites and chemical modifications on rRNA are critical for functioning ribosomes and are rarely altered. The high number of variants in the 28S rRNA flexible expansion segments suggest that these may supply ribosome heterogeneity.
Population-stratified rRNA variants
To validate whether the genomic variants are expressed as RNA, high-coverage RNA-seq data available on five individuals in the 1000 Genomes Project were downloaded, and the variant calling pipeline was used to analyze for presence of rRNA variants compared to the 145 kb rRNA gene reference. This resulted in identification of 172 unique variant positions of which close to 30% matched with those of the DNA variants in the coding region. We considered these 50 variant positions as high-confidence positions for further population analyses (Supplemental Table S3). We refined this data set by excluding data on positions where it was available in <30% of all analyzed individuals or <2% variant events were recorded, reasoning that these have a low level of population impact. This retained 38 variant positions for which the variant allele frequencies were calculated as the number of genomic variant reads/number of total reads for each position. This showed large, position-dependent variation in the allele frequencies (0.4%–76%) (Fig. 5A; Supplemental Table S4). Positions with common, high variant frequencies (>10%) were observed in the ITS2, 28S, and 3′ ETS domains. There were no private variants in any of the 26 populations, and the averaged variant frequency distribution of the 38 variants among the populations was very similar (Supplemental Table S4; Supplemental Fig. S1). These findings indicated extensive sharing of the variants among the populations.
FIGURE 5.

Population-wide analyses of the RNA-validated genomic variants. Variant allele frequency was calculated as the number of variant reads/number of total reads for each position and is expressed as %. (A) Average allele frequency (%) per position. (B) Supervised heat map organized by population (y-axis) and position (x-axis). Superpopulations are indicated by color codes, and populations are abbreviated using their 1000 Genomes Project acronyms. Heatmap is depicted as light blue to maroon gradient. White cells, no data.
To visualize individual variation and potential clustering of some of the variants in a population-dependent manner, the variant frequencies were displayed as a supervised ancestry-organized heatmap. A large degree of interindividual variation was observed across all populations, as well as population clustering of certain variants (Fig. 5B). To further assess this, we conducted unsupervised analyses on eight variant positions. We first calculated the average variant frequency for each population per position and then conducted Euclidian distance matrix analysis. This showed organization of the variants according to their superpopulations (African, European, American, South Asian, East Asian) (Fig. 6A). Two populations were outliers of their superpopulations, Finnish (FIN) (present in the American instead of European) and Peruvian (PEL) (present in the East Asian instead of American). We then computed the variance in the 26 populations for each position and conducted K-means clustering analyses. As shown in Figure 6B, this led to the clustering of 23 out of 26 populations according to their superpopulations. In this analysis, all except the FIN and PEL populations were found in clusters expected of their superpopulations, and the Bengali population (BEB) clustered at the edge of the South Asian superpopulation. To assess the population variance in detail, the variant frequencies of all 38 positions were plotted according to their superpopulations using violin plots (Supplemental Fig. S2). These showed that in a subset of 5′ ETS, ITS2, and 28S rRNA positions, there was a strong tendency for a superpopulation-dependent variation frequency.
FIGURE 6.

Multidimensional scaling of variants. (A) Euclidian distance matrix analysis. Variant positions (2422, 7196, 7222, 7234, 7443, 7460, 7760, 11390) were used to calculate the average variant frequency for each population per position. Below, superpopulations are indicated by color codes. (B) K-means clustering analysis was conducted based on the variance in each population. Clusters of superpopulations are indicated. Dashed lines indicate outlier populations in the cluster.
To further illustrate the ancestry of selected variants, we plotted their variation frequency in each population (Fig. 7). For example, 5′ ETS C2422A and C2443G were very rare or absent (<2%) in the African populations, whereas were present in all others. ITS2 G7196GT was present at a much higher frequency in the South Asian (>20%) and European (>14%) populations than the African, American or East Asian (10% and less) populations, whereas ITS2 A7222C was high in European (>20%) and rare in the East Asian populations (less than 1%). The highest variant frequency of ITS2 C7760G was observed in the South Asian populations (>24%), whereas was present in <17% in East Asian, European and American populations. On the other hand, G11390A, locating to the 28S rRNA ES27L position 3490, had a higher frequency in the African (>20%) and East Asian (>18%) populations, whereas was present on average at <10% in others. 28S rRNA G11050GGA in ES27L position 3126 was highest in the East Asian (>13%) populations, but rare or absent in the African (<1%). On the other hand, 5′ ETS T2177C and CA11266C in 28S position 3342 are examples of positions in which no marked distribution differences were detected (Fig. 7). Hence, distinct ancestry-linked variants were observed in several positions in the rRNA coding region.
FIGURE 7.

Population stratification of variants. Population distribution of intra-individual variant frequencies of selected 5′ ETS, ITS2, and 28S rRNA positions are shown as violin plots. Solid lines, median; dashed lines, quartiles. Populations are color-coded according to their superpopulations. Top, rRNA coding regions and variant positions.
DISCUSSION
The current knowledge of the variation in human rDNA sequences is incomplete. This study shows the heterogeneity of human rRNA genes in a comprehensive study of individuals from the 1000 Genomes Project populations. We show that diverse lengths and considerable sequence variation exists within the established human rDNA GRCh38 chromosome 21 sequences. We annotated the human rDNA variants (SNV/INDEL) using the high coverage WGS data including 2504 individuals from 26 ancestry-diverse populations. We performed SNV/INDEL calling on the GRCh38 reference rRNA gene copies and generated a comprehensive rDNA variant map. In total, 3791 SNV/INDEL variant positions were identified. Density distribution analysis of variants showed noticeable clustering throughout the gene, including in the rRNA coding region, providing a perspective of both invariant and highly diverse regions. Several of the variants were validated by their presence in RNA-seq data sets. Furthermore, the frequency of some of the 5′ ETS, ITS2, and 28S rRNA variants were ancestry-linked, suggesting that they represent sites with genetic drift. Also, many highly penetrant variants were identified, suggesting that they represent rDNA array mosaicism. We mapped the 28S rRNA high density variants on the ribosome structure, which showed their location in the 28S rRNA expansion segments. This suggests that the variation in the 28S rRNA can potentially contribute to heterogeneity in ribosome function. The advantage of analysis of this large data set, instead of single-genome sample comparisons, is that it not only affirms confidence of the reference genotype, but provides a resource to assess the impact of rRNA diversity in subsequent functional studies and population-based analyses.
Genomic GC content affects sequencing depth due to PCR bias during the sequencing process (Dohm et al. 2008; Meienberg et al. 2015; Laursen et al. 2017). Reduction of GC bias is critical in improving the assembly of the genomes and increasing the accuracy of biomedical or clinical application. Given that the rDNA copies have high GC content regions in eukaryotes (Escobar et al. 2011; Parks et al. 2018) and that PCR bias affects the accuracy of rDNA gene sequencing, we calculated the sequencing depth of rDNA using cancer cell line WGS data from the CCLE resource. The CCLE WGS data set was generated using PCR amplification (Ghandi et al. 2019). Relatively low read depth was observed in the high GC content regions, showing a significant negative correlation with the GC-rich areas. High GC content will also impact other PCR-based methods such as chromatin and RNA immunoprecipitation and sequencing analyses, and has been observed in studies involving rRNA genes, such as low read coverage areas. Computational methods and PCR-free technology in WGS have been developed to decrease this bias and to obtain better coverage of biologically important loci with high GC content (Benjamini and Speed 2012; Ross et al. 2013). Benefiting from the high coverage of 1000 genomes WGS data sequenced using PCR-free technology, we report a mean 5000-fold coverage on the rRNA gene, enabling robust variant calling analysis. Furthermore, the first complete human genome sequence that detailed the acrocentric rDNA arrays in the haploid CHM13 cells using PacBio HiFi and Oxford Nanopore sequencing was released by the T2T consortium (Nurk et al. 2021). These advanced techniques and resources help remove obstacles in the identification of human variants both in discovery research and precision medicine.
Few studies have evaluated the heterogeneity of rRNA genes across human populations. This is partly due to the difficulty of assembling a reference for the highly variable rDNA loci. Unfortunately, assembly continues to be a computationally challenging problem for rDNA tandem repeat units using shotgun sequencing. A refined reference for a rDNA copy unit, assembled applying TAR cloning and long sequencing technologies, has enabled further assessment of individual rDNA units on chromosomes 21 and 22 (Kim et al. 2018, 2021). Here, based on the refined human rDNA reference sequence (KY962518), we identified four copies of rDNA in the current human reference genome GRCh38 suitable for rDNA SNV/INDEL calling. Though they were 99% identical to the reference KY962518, each copy presented with both length and sequence variation, and a total of 163 variant positions were identified. This copy variation is consistent with the observation that chromosome 21 rRNA gene copies are mosaic (Nurk et al. 2021). Based on the T2T consortium data on CHM13 cell line HiFi reads, the degree of mosaicism varies between the acrocentric arrays, being high on chromosomes 13, 15, and 21 and low on 14 and 22. The mosaicism in the copies used here is hence present as a natural variation in our variant calling pipeline. The variant calling pipeline was designed to identify both intra-array and interindividual heterogeneity in the rRNA arrays compared to the reference. We took advantage of the 1000 Genomes Project, the largest fully open resource of whole-genome sequencing data, making this resource an ideal starting point to identify human rDNA variants. Our analysis was based on the new high coverage WGS resource and generated a comprehensive set of rDNA SNV/INDELs.
In contrast to previous published studies, based on the earlier reference sequence U13369 (Babaian 2017; Xu et al. 2017; Parks et al. 2018), this study demonstrates distinct clustering of the variants to specific rRNA regions. The highest density of variants was observed in the rRNA transcription termination sites containing repetitive CT-sequences, as well as in the 5′ ETS and ITS2 domains flanking 18S and 28S rRNA coding regions. However, we did not detect any variants in the rRNA processing sites present in these regions. Also, the 18S and 5.8S rRNA coding sequences, in agreement with Xu et al. (2017), but in contrast to Parks et al. (2018), were near invariant in this deep data set, suggesting a low tolerance for adaptation of these mature rRNA coding sequences. Also, in contrast to the study by Parks et al. (2018), we did not detect variants in the 28S rRNA modification sites or high-confidence, frequent variants in the ribosome bridge intersubunit sites. Overall, these findings suggest that rRNA gene domains and sites essential for the processing, maturation and assembly into ribosomes are selected against for further variation.
Notably, and surprisingly, large areas of the IGS were devoid of variants. This is in contrast to the earlier notion that IGS is highly variable (Gonzalez and Sylvester 1995, 2001). The conservation of IGS may be driven by as of yet unidentified functional relevance of these sequences. Stress-inducible noncoding IGS-derived RNAs have been identified, and IGS contains regions with transcriptionally active chromatin states, with both sense and antisense transcription by Pol I and Pol II being reported (Audas et al. 2012; Agrawal and Ganley 2018; Abraham et al. 2020). On the other hand, high variant densities with high QUAL scores were observed in the IGS at 16, 23–24, 28, 31, 33, and 39 kb that frequently were represented by CT and TG repeat sequences. Overall, these results suggest a high degree of conservation of not only the rDNA arrays between chromosomes, but also low variance between individuals. Given that the rDNA copies are considered to undergo frequent genomic alterations, this conservation is remarkable.
Based on the population analyses of the RNA-validated variants, a majority displayed high (>5%) frequencies. The average variant frequency of all positions was similar among the populations. However, when inspected by position, remarkable, population-linked differences were observed that consistently segregated with their continental superpopulations. Given the rRNA gene array mosaicism, the likeliest explanation is that these represent variation in the arrays and their copy numbers in individual chromosomes. As an example, 5% variant frequency corresponds to its presence in 20 copies in an average 400 copy genome. Copy number variation of an array carrying a particular variant(s) will result in the findings described here. Hence, variants that display a highly population-stratified frequency represent these selective changes in the rRNA arrays. Interestingly, no population private variants were detected, that is, all variants were observed in multiple populations albeit at variable frequencies. Once further long-read rRNA array assemblies representing humans of diverse ancestry origins are compiled, these variants will be useful in identification and quantification of the rRNA arrays. Nevertheless, these findings are reflective of a remarkable variance of the rRNA genes and that this genetic variance is also ancestry-linked. This suggests that these variant positions may have undergone adaptive selection or otherwise remarkable genetic drift potentially at times of population genetic bottlenecks.
Genomic variants of rRNA genes may provide a molecular basis for heterogeneous ribosomes, potentially leading to functional consequences. rDNA unit diversity has been observed in various species (Tseng et al. 2008; Matyasek et al. 2012; Agrawal and Ganley 2018). In mouse, the rDNA array consists of genetically distinct variants, and a subset of rDNA genes are regulated in a cell type-specific manner (Tseng et al. 2008). Mouse rRNA variants are differentially expressed in organs and the epigenetic state of rDNA copies is influenced by in utero nutrition (Holland et al. 2016; Parks et al. 2018). Hence, variation arising from the rRNA sequence can lead to ribosome heterogeneity (Sauert et al. 2015; Shi and Barna 2015; Emmott et al. 2019; Ferretti and Karbstein 2019). Ribosome-associated proteins further functionally diversify mammalian ribosomes (Simsek et al. 2017). Heterogeneous ribosomes, containing diverse post-translational modifications of the ribosomal proteins or distinct ribosome binding factors, can preferentially translate different subsets of mRNAs (Emmott et al. 2019; Li and Wang 2020). However, whether variation in the rRNA coding sequences have an impact on ribosomal functional heterogeneity remains poorly understood. The expansion segments, especially ES27L, are dynamic and assist in connecting the 40S mRNA exit and 60S tunnel exit sites on the ribosome, and interact with export factors and other regulators (Anger et al. 2013). For example, the human ES27L acts as an RNA scaffold to facilitate binding of the methionine amino peptidase to control translation fidelity (Fujii et al. 2018). We observed that positions relevant for the rRNA-ribosomal protein interaction are much less variant compared to the highly flexible human-expanded rRNA ES7L and ES27L helical folds on the surface of the complex. Some of the 28S rRNA variant sites, such as ES27L 3342, showed very high variant frequencies (>40%), suggesting highly penetrating heterogeneity. Also, 28S ES27L 3490 had a remarkable population distribution, the variant allele being most frequent in the African populations (>20%), whereas was present on average at 5% in the American and European populations. Hence, variation in the 28S ES27L may provide a genetic basis for heterogeneous translation fidelity in human individuals. On the other hand, our data show that rRNA modification sites are highly conserved. This is consistent with the notion that rRNA-ribosomal protein interaction sites and chemical modifications on rRNA are critical for ribosome assembly and function and are under strict preservation. Yet, the high number of 28S rRNA variants and the frequencies of the validated sites implicate that they potentially contribute to translation control or mediate selective mRNA translation. However, this study does not directly explore whether the variant rRNAs are incorporated into ribosomes. Also, we do not account for putative tissue-specific heterogeneity or somatic alterations of the rDNA genes. These important aspects await validation in future studies.
In addition to the challenge of the sequence identity of the gene copies, the rDNA repeats are highly dynamic due to recombination events during meiosis, DNA repair and in diseases such as cancer. Identification of disease-linked variants will require comprehensive assessment of the rDNA variation in the normal human genome. Our study of genetic variation on the rRNA genes across human populations, aided by high coverage WGS data, narrows down the previously presumed variation and provides a resource of rRNA variant detection and basis for the understanding of the impact of rRNA variants potentially affecting physiology and disease.
MATERIALS AND METHODS
Identification of rDNA regions in the human reference genome GRCh38
rDNA reference KY962518 (GenBank: KY962518.1) was aligned to the human genome reference genome (Homo sapiens genome assembly GRCh38.p13) by using the Basic Local Alignment Search Tool (BLAST). Highly similar sequences (Megablast) with 100% query coverage and ≥99% identity were considered as high confidence rDNA copies. An unlocalized genomic scaffold and a PATCHES sequence were excluded from blast results. The rDNA locus on chromosome 21 was visualized using RIdeogram R package (Hao et al. 2020).
Multiple sequence alignments
Multiple sequence alignment of the rDNA reference KY962518 and rDNA copies identified on chromosome 21 was performed with Clustal Omega Multiple Sequence Alignment (MSA) to detect variation across copies. The alignment results were visualized by NCBI MSA viewer. The number of SNVs and INDELs were counted by Jvarkit and VCFtools (Danecek et al. 2011). INDELs from multiple sequence alignment were visualized using the ggmsa R package (Yu 2020).
Comparison of GC content and read depth
Genomic GC content percentage was calculated as Count(G + C)/Count(A + T + G + C) × 100%. A GC% value was computed for each 70 nt interval in the rDNA reference KY962518. We randomly selected 70 cancer cell lines from the CCLE project, downloaded related WGS data and performed read alignment to KY962518 using Sentieon DNASeq tools. Read depth at each position or region was computed by samtools depth according to the Samtools manual. For each sample, we normalized the read depth per position as depth per position/mean depth in the rDNA coding region. Mean normalized read depth values for 70 cancer cell lines are presented. We then calculated Pearson correlation between the GC content and mean normalized read depth of the 70 samples.
Coverage for rDNA and WGS sequence reads
The average rDNA read coverage was computed by samtools flagstat according to protocol in the Samtools manual for WGS read analysis. GRCh38 was used as reference, and reads mapping to the identified rDNA copies were calculated. Mapped reads for each sample were extracted and rDNA coverage was computed as (mapped read count × read length)/rDNA copy size. Genome sequence read coverage was computed as (mapped read count × read length)/total genome size.
Variant calling and variant density measurements
High coverage (mean 34×) WGS data for 2504 individuals were obtained from the 1000 Genomes Project, and read alignment to the human reference genome GRCh38, duplicate marking, and Base Quality Score Recalibration (BQSR) was performed (Byrska-Bishop et al. 2021). The sequencing was conducted using NovaSeq 6000 using PCR-free technology (Byrska-Bishop et al. 2021). The full GRCh38 reference, including ALT contigs, decoy, EBV and HLA sequences, was included to avoid mismapping. Alignment files in CRAM format were used for variant calling. Germline SNVs and INDEL variants were called with the DNAseq pipeline (Sentieon, Release 201911). The DNAseq pipeline has demonstrated strong performance for variant calling and suitability of use with the NovaSeq 6000 technology (Kendig et al. 2019; Pei et al. 2021). We used the following variant call criteria: QUAL score >30 PHRED (P < 0.001) and presence in three or more alleles. Computing was conducted using Maryland Advanced Research Computing Center (MARCC) high performance cluster computing nodes. The Integrative Genomics Viewer (IGV, version 2.8.6) was used to visualize read alignments. SNVs and INDELs were summarized from gVCF files derived by Sentieon DNAseq pipeline and visualized by the ggplot2 R package. The density of variants located in the rDNA region was calculated and visualized by the ggridges and ggplot2 R packages. The bandwidth used for density calculation was provided as 50.
RNA variant analyses and data sets
High-coverage RNA-seq reads from five individuals (NA19650, NA19240, HG03732, HG03371, HG00096) included in the 1000 Genomes Project were downloaded from the International Genome Sample Resource (www.internationalgenome.org). FASTQ files were used for variant calling against the GRCh38 reference containing four rRNA gene copies on chromosome 21. SNVs and INDEL variants were called using the RNA Variant Calling pipeline (Sentieon) similarly to the DNA variant calling using a threshold for QUAL > 20. RNA and DNA variants were aligned according to their genomic positions resulting in validation of 50 variants overlapping in the coding region (Supplemental Table S3). The variant frequencies were calculated for each position as the number of variant reads/number of total reads for each position, and were plotted using Prism9 supervised heatmaps and violin plots. Multidimensional scaling was conducted in R using Euclidian distance matrix and K-means cluster analysis using k = 5. The population nomenclature according to the 1000 Genomes Project Consortium et al. (2015) was adhered to and is provided in Supplemental Table S5.
Annotation of variants on 28S rRNA 2D and 3D structure
Ribosome Visualization Suite RiboVision2 (http://apollo.chemistry.gatech.edu/RiboVision2/) was used to visualize the density of variants on 28S rRNA (Bernier et al. 2014). Variant density was mapped on the secondary (2D) structure of 28S rRNA and portrayed by color gradient. All proteins in the tool were checked for visualizing the protein contacts on 28S rRNA. The variant densities were mapped to the three-dimensional (3D) structures of ribosomes and the density intensity is indicated by a color gradient. Images were generated using RiboVision2 based on PDB 4V6X (Anger et al. 2013).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
M.L. holds patents on RNA polymerase I inhibitors, which are managed by the Johns Hopkins University. These are unrelated to the work described here. The other authors declare no competing interests.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by National Institute of General Medical Sciences (NIGMS) R01GM121404 and National Cancer Institute (NCI) P30 CA006973. We thank Dr. Steven Salzberg for helpful discussions and the Maryland Advanced Research Computing Center (MARCC) for access to the high computing resource.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078925.121.
MEET THE FIRST AUTHOR
Wenjun Fan.

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Wenjun Fan is the first author of this paper, “Widespread genetic heterogeneity of human ribosomal RNA genes.” Wenjun is a post-doctoral fellow working with Marikki Laiho at the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine. His research has focused on developing and optimizing small molecules targeting Pol I transcription in cancers and exploring the mechanisms of drug resistance by using biochemistry and bioinformatic methods.
What are the major results described in your paper and how do they impact this branch of the field?
Our study shows the heterogeneity of human rRNA genes across 2504 individuals from 26 ancestry-diverse populations. rRNAs are key components of ribosomes, and we identified conserved regions in 5.8S, 18S, and 28S rRNAs that are essential for ribosome assembly. Interestingly, we also revealed highly dynamic regions in expansion segments of 28S. The highly flexible human-expanded rRNA ES7L and ES27L helical folds locate on the surface of the ribosome complex and control translation fidelity. Genetic variation in 28S ES27L may lead to heterogeneous translation fidelity in cells. Our work provides evidence for variation of rRNAs and may provide a molecular basis for heterogeneity of protein synthesis.
What led you to study RNA or this aspect of RNA science?
Pol I is a large transcriptional holocomplex that synthesizes ribosomal RNAs. Pol I activity is frequently deregulated in cancers by oncogene activation and/or tumor suppressor inactivation. Despite the key importance of Pol I activity to cancer cells, only limited efforts have been directed to its therapeutic targeting. Our laboratory has identified small molecules targeting Pol I transcription. However, potential alterations or variation of rRNA genes in cancer are poorly understood. We decided to start exploring this question by assessing the genetic variation of rRNA genes in human populations.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
The great success of Imatinib in treatment of BCR-ABL positive CML made me interested in cancer therapy and to believe that scientists who are involved in the biomarker discovery and drug development are heroes. Special thanks to Dr. Laiho for providing me with an opportunity to engage my skills in molecular biology and bioinformatics to study the mechanisms of the RNA polymerase I inhibitors and expand on the aspects of drug development.
What are your subsequent near- or long-term career plans?
I will continue my biomedical research training and will finish the training in a few years. Going forward, my goal is to seek independent research funding and find a faculty position to continue my career in cancer research.
REFERENCES
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham KJ, Khosraviani N, Chan JNY, Gorthi A, Samman A, Zhao DY, Wang M, Bokros M, Vidya E, Ostrowski LA, et al. 2020. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 585: 298–302. 10.1038/s41586-020-2497-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Ganley ARD. 2018. The conservation landscape of the human ribosomal RNA gene repeats. PLoS ONE 13: e0207531. 10.1371/journal.pone.0207531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. 2013. Structures of the human and Drosophila 80S ribosome. Nature 497: 80–85. 10.1038/nature12104 [DOI] [PubMed] [Google Scholar]
- Audas TE, Jacob MD, Lee S. 2012. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell 45: 147–157. 10.1016/j.molcel.2011.12.012 [DOI] [PubMed] [Google Scholar]
- Babaian A. 2017. Intra- and inter-individual genetic variation in human ribosomal RNAs. bioRxiv 10.1101/118760 [DOI] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP. 2012. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40: e72. 10.1093/nar/gks001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier CR, Petrov AS, Waterbury CC, Jett J, Li F, Freil LE, Xiong X, Wang L, Migliozzi BL, Hershkovits E. 2014. RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss 169: 195–207. 10.1039/C3FD00126A [DOI] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. 2021. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 10.1101/2021.02.06.430068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm JC, Lottaz C, Borodina T, Himmelbauer H. 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36: e105. 10.1093/nar/gkn425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmott E, Jovanovic M, Slavov N. 2019. Ribosome stoichiometry: from form to function. Trends Biochem Sci 44: 95–109. 10.1016/j.tibs.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar JS, Glemin S, Galtier N. 2011. GC-biased gene conversion impacts ribosomal DNA evolution in vertebrates, angiosperms, and other eukaryotes. Mol Biol Evol 28: 2561–2575. 10.1093/molbev/msr079 [DOI] [PubMed] [Google Scholar]
- Ferretti MB, Karbstein K. 2019. Does functional specialization of ribosomes really exist? RNA 25: 521–538. 10.1261/rna.069823.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii K, Susanto TT, Saurabh S, Barna M. 2018. Decoding the function of expansion segments in ribosomes. Mol Cell 72: 1013–1020.e1016. 10.1016/j.molcel.2018.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER III, Barretina J, Gelfand ET, Bielski CM, Li H. 2019. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569: 503–508. 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Godinho SA, Yu S, Lemos B. 2015. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc Natl Acad Sci 112: 2485–2490. 10.1073/pnas.1416878112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE. 1995. Complete sequence of the 43-kb human ribosomal DNA repeat: analysis of the intergenic spacer. Genomics 27: 320–328. 10.1006/geno.1995.1049 [DOI] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE. 2001. Human rDNA: evolutionary patterns within the genes and tandem arrays derived from multiple chromosomes. Genomics 73: 255–263. 10.1006/geno.2001.6540 [DOI] [PubMed] [Google Scholar]
- Gonzalez IL, Gorski JL, Campen TJ, Dorney D, Erickson JM, Sylvester JE, Schmickel RD. 1985. Variation among human 28S ribosomal RNA genes. Proc Natl Acad Sci 82: 7666–7670. 10.1073/pnas.82.22.7666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE, Schmickel RD. 1988. Human 28S ribosomal RNA sequence heterogeneity. Nucleic Acids Res 16: 10213–10224. 10.1093/nar/16.21.10213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z, Lv D, Ge Y, Shi J, Weijers D, Yu G, Chen J. 2020. RIdeogram: drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput Sci 6: e251. 10.7717/peerj-cs.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson A, Warburton D, Atwood K. 1972. Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci 69: 3394–3398. 10.1073/pnas.69.11.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras AK, Plisson-Chastang C, O'Donohue MF, Chakraborty A, Gleizes PE. 2015. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA 6: 225–242. 10.1002/wrna.1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Lowe R, Caton PW, Gemma C, Carbajosa G, Danson AF, Carpenter AA, Loche E, Ozanne SE, Rakyan VK. 2016. Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 353: 495–498. 10.1126/science.aaf7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendig KI, Baheti S, Bockol MA, Drucker TM, Hart SN, Heldenbrand JR, Hernaez M, Hudson ME, Kalmbach MT, Klee EW, et al. 2019. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front Genet 10: 736. 10.3389/fgene.2019.00736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatter H, Myasnikov AG, Natchiar SK, Klaholz BP. 2015. Structure of the human 80S ribosome. Nature 520: 640–645. 10.1038/nature14427 [DOI] [PubMed] [Google Scholar]
- Kim JH, Dilthey AT, Nagaraja R, Lee HS, Koren S, Dudekula D, Wood WH Iii, Piao Y, Ogurtsov AY, Utani K, et al. 2018. Variation in human chromosome 21 ribosomal RNA genes characterized by TAR cloning and long-read sequencing. Nucleic Acids Res 46: 6712–6725. 10.1093/nar/gky442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Noskov VN, Ogurtsov AY, Nagaraja R, Petrov N, Liskovykh M, Walenz BP, Lee HS, Kouprina N, Phillippy AM, et al. 2021. The genomic structure of a human chromosome 22 nucleolar organizer region determined by TAR cloning. Sci Rep 11: 2997. 10.1038/s41598-021-82565-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo BA, Gonzalez IL, Gillespie DA, Sylvester JE. 1996. Human ribosomal RNA variants from a single individual and their expression in different tissues. Nucleic Acids Res 24: 4817–4824. 10.1093/nar/24.23.4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DL. 2015. Noncoding RNAs in eukaryotic ribosome biogenesis and function. Nat Struct Mol Biol 22: 11–19. 10.1038/nsmb.2939 [DOI] [PubMed] [Google Scholar]
- Laursen MF, Dalgaard MD, Bahl MI. 2017. Genomic GC-content affects the accuracy of 16S rRNA gene sequencing based microbial profiling due to PCR bias. Front Microbiol 8: 1934. 10.3389/fmicb.2017.01934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Wang J. 2020. Ribosome heterogeneity in stem cells and development. J Cell Biol 219: e202001108. 10.1083/jcb.202001108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyasek R, Renny-Byfield S, Fulnecek J, Macas J, Grandbastien MA, Nichols R, Leitch A, Kovarik A. 2012. Next generation sequencing analysis reveals a relationship between rDNA unit diversity and locus number in Nicotiana diploids. BMC Genomics 13: 722. 10.1186/1471-2164-13-722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B. 2016. Nucleolar organizer regions: genomic ‘dark matter’ requiring illumination. Genes Dev 30: 1598–1610. 10.1101/gad.283838.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, Xu Z, Steinmann B, Carrel T, Rothlisberger B, et al. 2015. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res 43: e76. 10.1093/nar/gkv216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss Langlois T, Gagnon-Kugler F, Stefanovsky TV. 2007. A housekeeper with power of attorney: the rRNA genes in ribosome biogenesis. Cell Mol Life Sci 64: 29–49. 10.1007/s00018-006-6278-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP. 2017. Visualization of chemical modifications in the human 80S ribosome structure. Nature 551: 472–477. 10.1038/nature24482 [DOI] [PubMed] [Google Scholar]
- Németh A, Perez-Fernandez J, Merkl P, Hamperl S, Gerber J, Griesenbeck J, Tschochner H. 2013. RNA polymerase I termination: Where is the end? Biochim Biophys Acta 1829: 306–317. 10.1016/j.bbagrm.2012.10.007 [DOI] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. 2021. The complete sequence of a human genome. bioRxiv 10.1101/2021.2005.2026.445798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D, Vincent CT, Blanchard SC. 2018. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci Adv 4: eaao0665. 10.1126/sciadv.aao0665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei S, Liu T, Ren X, Li W, Chen C, Xie Z. 2021. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief Bioinform 22: bbaa148. 10.1093/bib/bbaa148 [DOI] [PubMed] [Google Scholar]
- Pluss M, Kopps AM, Keller I, Meienberg J, Caspar SM, Dubacher N, Bruggmann R, Vogel M, Matyas G. 2017. Need for speed in accurate whole-genome data analysis: GENALICE MAP challenges BWA/GATK more than PEMapper/PECaller and Isaac. Proc Natl Acad Sci 114: E8320–E8322. 10.1073/pnas.1713830114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczy C, Petrovski R, Saunders CT, Chorny I, Kruglyak S, Margulies EH, Chuang HY, Kallberg M, Kumar SA, Liao A, et al. 2013. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 29: 2041–2043. 10.1093/bioinformatics/btt314 [DOI] [PubMed] [Google Scholar]
- Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, Nusbaum C, Jaffe DB. 2013. Characterizing and measuring bias in sequence data. Genome Biol 14: R51. 10.1186/gb-2013-14-5-r51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauert M, Temmel H, Moll I. 2015. Heterogeneity of the translational machinery: variations on a common theme. Biochimie 114: 39–47. 10.1016/j.biochi.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Barna M. 2015. Translating the genome in time and space: specialized ribosomes, RNA regulons, and RNA-binding proteins. Annu Rev Cell Dev Biol 31: 31–54. 10.1146/annurev-cellbio-100814-125346 [DOI] [PubMed] [Google Scholar]
- Simsek D, Tiu GC, Flynn RA, Byeon GW, Leppek K, Xu AF, Chang HY, Barna M. 2017. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 169: 1051–1065.e18. 10.1016/j.cell.2017.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Warda AS, Sharma S, Entian KD, Lafontaine DLJ, Bohnsack MT. 2017. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol 14: 1138–1152. 10.1080/15476286.2016.1259781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng H, Chou W, Wang J, Zhang X, Zhang S, Schultz RM. 2008. Mouse ribosomal RNA genes contain multiple differentially regulated variants. PLoS ONE 3: e1843. 10.1371/journal.pone.0001843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J. 2013. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Sluis M, van Vuuren C, Mangan H, McStay B. 2020. NORs on human acrocentric chromosome p-arms are active by default and can associate with nucleoli independently of rDNA. Proc Natl Acad Sci 117: 10368–10377. 10.1073/pnas.2001812117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JA, Aldana R, Gallagher BD, Edwards JS. 2016. Sentieon DNA pipeline for variant detection-Software-only solution, over 20× faster than GATK 3.3 with identical results. PeerJ Preprints 4: e1672v2. 10.7287/peerj.preprints.1672v1 [DOI] [Google Scholar]
- Worton RG, Sutherland J, Sylvester JE, Willard HF, Bodrug S, Dube I, Duff C, Kean V, Ray PN, Schmickel RD. 1988. Human ribosomal RNA genes: orientation of the tandem array and conservation of the 5′ end. Science 239: 64–68. 10.1126/science.3336775 [DOI] [PubMed] [Google Scholar]
- Xu B, Li H, Perry JM, Singh VP, Unruh J, Yu Z, Zakari M, McDowell W, Li L, Gerton JL. 2017. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet 13: e1006771. 10.1371/journal.pgen.1006771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. 2020. Using ggtree to visualize data on tree-like structures. Curr Protoc Bioinformatics 69: e96. 10.1002/cpbi.96 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.