

Abstract
Background
While 2–4% of lung cancers possess alterations in BRAF, little is known about the immune responsiveness of these tumours.
Methods
Clinical and genomic data were collected from 5945 patients with lung cancers whose tumours underwent next-generation sequencing between 2015 and 2018. Patients were followed through 2020.
Results
In total, 127 patients with metastatic BRAF-altered lung cancers were identified: 29 tumours had Class I mutations, 59 had Class II/III alterations, and 39 had variants of unknown significance (VUS). Tumour mutation burden was higher in Class II/III than Class I-altered tumours (8.8 mutations/Mb versus 4.9, P < 0.001), but this difference was diminished when stratified by smoking status. The overall response rate to immune checkpoint inhibitors (ICI) was 9% in Class I-altered tumours and 26% in Class II/III (P = 0.25), with median time on treatment of 1.9 months in both groups. Among patients with Class I–III-altered tumours, 36-month HR for death in those who ever versus never received ICI was 1.82 (1.17–6.11). Nine patients were on ICI for >2 years (two with Class I mutations, two with Class II/III alterations, and five with VUS).
Conclusions
A subset of patients with BRAF-altered lung cancers achieved durable disease control on ICI. However, collectively no significant clinical benefit was seen.
Subject terms: Lung cancer, Metastasis
Background
BRAF alterations have been identified in 2–4% of lung cancers [1–7]. Through the MAP kinase pathway, they can lead to cell proliferation and tumour growth [8, 9]. Three distinct categories of alterations in BRAF have been identified: Class I (V600) mutations are the most common BRAF alterations in lung cancers in most series [2–4, 8, 10–12], and operate as kinase monomers. Class II alterations, by contrast, promote kinase activity as dimers. Class III alterations have impaired kinase activity and drive signalling in a RAS-dependent, heterodimer-dependent fashion [8, 13–15].
Class I-mutant lung cancers (BRAFV600) are clinically distinct from other BRAF (non-V600) alterations in several respects [3, 16]. In some studies, BRAFV600 mutations have been associated with the female sex [4, 6, 17, 18] and with being a never-smoker [1, 17]. Patients with Class I-altered cancers also may benefit from targeted therapies, including BRAF inhibitors and combined BRAF and MEK inhibitors, which have been approved by the FDA and included in national guidelines [19–24]. By contrast, patients with non-V600 mutant lung cancers may have higher rates of brain metastasis and tobacco exposure [4, 16]. Although some patients with non-V600 alterations may respond to combined BRAF and MEK inhibition [25, 26], neither BRAF nor combined BRAF and MEK inhibitors are approved to treat non-V600 mutant lung cancers [20]. Furthermore, patients with non-V600 mutant tumours have shorter survival times [4, 16, 27, 28].
Because of the relative rarity of BRAF alterations in lung cancers and with investigations ongoing regarding the biology of different classes of BRAF-mutant disease, there is limited information on the immunogenicity of these tumours [27, 29, 30]. Thus, it remains unclear whether BRAF-altered lung cancers are similar to other oncogene-addicted lung cancers, including those with EGFR and ALK mutations, which are generally characterised by lower tumour mutation burden (TMB) and insensitivity to immune checkpoint inhibitors (ICI) [31–37]. The IMMUNOTARGET registry recorded a 24% response rate to ICI among 43 patients with BRAF-mutant lung cancers across classes, suggestive of benefit among a subset of patients with the mutation [38]. However, the extent to which responses differ by BRAF alteration class remains unknown [27, 28, 38]. To determine whether the benefit from ICI for patients differs among classes of BRAF alterations, we investigated immune biomarkers as well as response to treatment of a large cohort of patients with lung cancers, with the aim of defining optimal approaches to therapy for different BRAF classes.
Methods
We identified patients with lung cancers who underwent next-generation sequencing (NGS) at our institution using the MSK-IMPACT assay between January 2015 and January 2018 [39]. Data on treatment outcomes and survival were collected through April 2020. To allow sufficient time for follow-up of patients’ treatment course in the metastatic setting, patients were considered to have had metastatic disease if their tumours had been found to have recurred/metastasised before June of 2019, with survival defined from the date of metastatic diagnosis. The study was approved by the Institutional Review Board, performed in accordance with the United States Common Rule, and all patients provided written informed consent for data collection.
BRAF alterations were categorised as Class I (V600), Class II or Class III, or as variants of unknown significance (VUS) using standard criteria [13, 14]. For purposes of analysis, patients were grouped into a cohort having cancers with BRAFV600 mutant disease or as having either BRAF non-V600 alterations (Class II or III alterations) or VUS.
TMB was assessed using previously published methods and reported as mutations/megabase [34, 40–42]. PD-L1 was determined based on institutional standards using the verified E1L3N antibody (Cell Signaling Technology, Danvers, MA) [43].
The overall response rate (ORR) to ICI was assessed by RECIST v1.1 in all evaluable patients by a dedicated study radiologist who was blinded to treatment and mutational status. Some patients were treated with more than one line of ICI. For all patients, data from their first treatment with ICI was analysed.
Categorical variables, including clinicopathologic characteristics, were compared using Fisher’s exact test. Differences in TMB were compared using the Mann–Whitney U test. A stratified Wilcoxon (Van Elteren) test was used to assess TMB across BRAF classes, stratified by smoking status. Overall survival (OS) and time to treat discontinuation (TTD) were computed using Kaplan–Meier estimates, in the former case with adjustment for left truncation to account for the time of sequencing. Patients who were known to have died prior to having available sequencing data were excluded from the survival analyses. Patients who received ICI as part of non-approved experimental combination therapy on a trial were also excluded from survival and time-on-treatment analyses.
Comparisons of survival time with respect to BRAF mutation status were computed using the Cox proportional hazards model with left truncation. In addition, the relationship between survival time post-metastatic diagnosis with and without ICI treatment was depicted with smoothed hazard estimates and evaluated as a time-dependent covariate in a Cox proportional hazards model.
Because of the known association between EGFR activating mutations (L858R or exon 19 deletions) and lack of benefit from immunotherapy [31], we carried out sensitivity analyses of survival with and without receipt of immunotherapy both including and excluding patients with these tumour mutations.
All statistical analyses were performed using GraphPad Prism version 8 (San Diego, CA), STATA version 16 (College Station, TX), or R version 4.0 (R Project for Statistical Computing, Vienna, Austria)).
Results
Clinical characteristics and immunogenicity of BRAF-altered lung cancers
We identified 5945 patients who were diagnosed with lung cancers and underwent NGS during the study period. Of these, 177 patients had tumours harbouring alterations in BRAF, and 127 had the metastatic disease during the study period. Of the patients with metastatic disease, 29 patients had Class I mutant-tumors (22.8%), 36 had Class II-altered cancer (28.3%), 23 had Class III alterations (18.1%) and 39 had VUS (30.7%).
Baseline characteristics of all patients with Class I, II and III BRAF-altered lung cancers are shown in Table 1, and of patients with a VUS in Supplementary Table S-1. Across BRAF Classes I–III, the median age at diagnosis of metastatic disease was 59–70 years. While more patients with Class I tumours were female as compared to patients with Class II/III-altered cancers, a significant differential effect was not detected (69% versus 58%, P = 0.36). More patients with Class I-mutant cancers were never-smokers compared to patients with lung cancers harbouring Class II/III alterations (45% versus 10%, P = 0.002).
Table 1.
Baseline demographic characteristics of patients with BRAF-altered lung cancers.
| V600 | Non-V600 | |||
|---|---|---|---|---|
| Class I (n = 29) | Class II (n = 36) | Class III (n = 23) | P value* | |
| Median age at metastatic diagnosis, years (range) | 65 (43–93) | 70 (39–88) | 59 (46–84) | 0.70 |
| Sex | ||||
| Female | 20 (69%) | 20 (56%) | 14 (61%) | 0.36 |
| Smoking status | ||||
| Never | 13 (45%) | 4 (11%) | 2 (9%) | 0.002 |
| Ever (median pack-years) | 16 (20) | 32 (30) | 21 (30) | — |
| Histology | ||||
| Adenocarcinoma | 29 (100%) | 30 (83%) | 23 (100%) | 0.17 |
| Squamous cell | 0 | 3 | 0 | — |
| Neuroendocrine | 0 | 3 | 0 | — |
| Co-mutation | ||||
| NF1 | 0 | 3 (8%) | 4 (17%) | 0.09 |
| RAS | 0 | 4 (11%) | 2 (9%) | 0.17 |
| EGFR** | 1 (0.03%) | 5 (14%) | 2 (9%) | 0.26 |
| KEAP1 | 0 | 9 (25%) | 14 (39%) | <0.001 |
| STK11 | 0 | 9 (25%) | 13 (57%) | <0.001 |
| TMB (range) | 4.9 (1–19.3) | 8.9 (0–82.5) | 9.8 (2–32.5) | <0.001 |
| PD-L1 status known | 11 (38%) | 19 (53%) | 11 (48%) | 0.27 |
| PD-L1 status | 0.039 | |||
| 0% | 3 | 11 | 8 | |
| 1–49%*** | 4 | 6 | 3 | |
| ≥50% | 4 | 2 | 0 | |
| ICI for metastatic disease (median line) | 13 (2) | 21 (3) | 16 (2.5) | 0.17**** |
| Pembrolizumab | 4 | 4 | 2 | — |
| Nivolumab | 6 | 11 | 9 | — |
| Ipilimumab/nivolumab | 2 | 2 | 1 | — |
| Atezolizumab | 1 | 1 | 0 | — |
| Experimental***** | 0 | 3 | 4 | — |
ICI immune checkpoint inhibitors, TMB tumour mutation burden.
Bold entries indicate significant P values.
*P values represent comparisons between patients with V600 vs. other BRAF mutations.
**Sensitising mutations, including L858R and exon 19 deletion.
***Includes one case of PD-L1 staining called as “<5”.
****P value refers to the receipt of ICI ever.
*****Includes clinical trials of ICI in combination with another agent.
Fewer patients with BRAF Class I mutations than with Class II/III-altered lung cancers had co-mutations in NF1 (0 versus 7 in Class II/III, P = 0.09). Similarly, no patients with Class I mutations had co-mutations in RAS, while six with Class II/III alterations had a RAS co-mutation (P = 0.17). Of patients with RAS mutant tumours, three had KRAS G12V mutations, and one each had KRAS G12D, G13C and G13D-mutant tumours. Eight tumours had activating EGFR mutations (L858R or exon 19 deletions); one had a Class I alteration, five had Class II and two had Class III BRAF alterations. Seven (87.5%) of the eight patients with tumours harbouring EGFR co-mutations had received a prior EGFR-directed TKI, with the BRAF alteration found in the setting of resistance in all cases. Seven BRAF fusions were detected, with four of these patients having a concomitant sensitising EGFR mutation present. All four had received EGFR-directed therapy prior to BRAF fusion detection. None of the patients with BRAF Class I-mutant tumours had KEAP1 or STK11 mutations. Of patients with BRAF Class II-altered tumours, eight had mutations in STK11 and eight had mutations in KEAP1. Among patients with Class III-altered tumours, nine had mutations in STK11 and ten had mutations in KEAP1, with four patients with VUS having mutations in STK11 and six having mutations in KEAP1, respectively.
Median TMB was higher in tumours with BRAF Class II/III alterations than in those with Class I alterations (median: 8.8 mutations/Mb versus 4.9, P < 0.001). However, when differences in TMB between Class I versus II/III-altered tumours were stratified by smoking status (ever versus never-smokers), differences in TMB between Class I versus Class II/III-altered tumours were diminished (P = 0.09).
Data on PD-L1 status were available for 11 tumours in the Class I cohort (38%) and 30 tumours in the Class II/III cohort (51%, P = 0.27). The distribution of PD-L1 status (0%, 1–49%, or ≥50%) differed between Class I and Class II/III BRAF-altered cancers (P = 0.04). Among patients with Class I cancers, 27% (3/11) had PD-L1 negative tumours, compared to 63% (19/30) of patients with Class II/III tumours. Moderate (1–49%) PD-L1 expression was observed in 36% (4/11) of Class I tumours and 30% (9/30) of Class II/III tumours. Four patients with Class I-mutant tumours had high (≥50%) PD-L1 expression (36.4%) versus two (6.7%) with Class II/III-altered lung cancers (Table 1). In all, 13% of patients with VUS mutations had high PD-L1 expression (Supplementary Table S-1).
Patients were followed for a median of 47.9 months (4.0 years) from the date of sequencing (interquartile range: 34.4–55.7). The median OS of patients with Class I-mutant lung cancers was 26 months compared to 9.1 months overall in patients with BRAF Class II/III-altered lung cancers (HR = 1.9, P = 0.02) (Fig. 1). When patients with Class II and III alterations were compared to those with Class I-mutant lung cancers, a median OS of 8.9 months for Class II and 13 months for Class III alterations was observed (HR 2.27 and 1.52, respectively, P = 0.03) (Supplementary Fig. S-1A). Median survival for patients whose tumours had VUS was 16 months (Supplementary Fig. S-1B).
Fig. 1. Overall survival of patients with lung cancers according to the type of BRAF alteration.
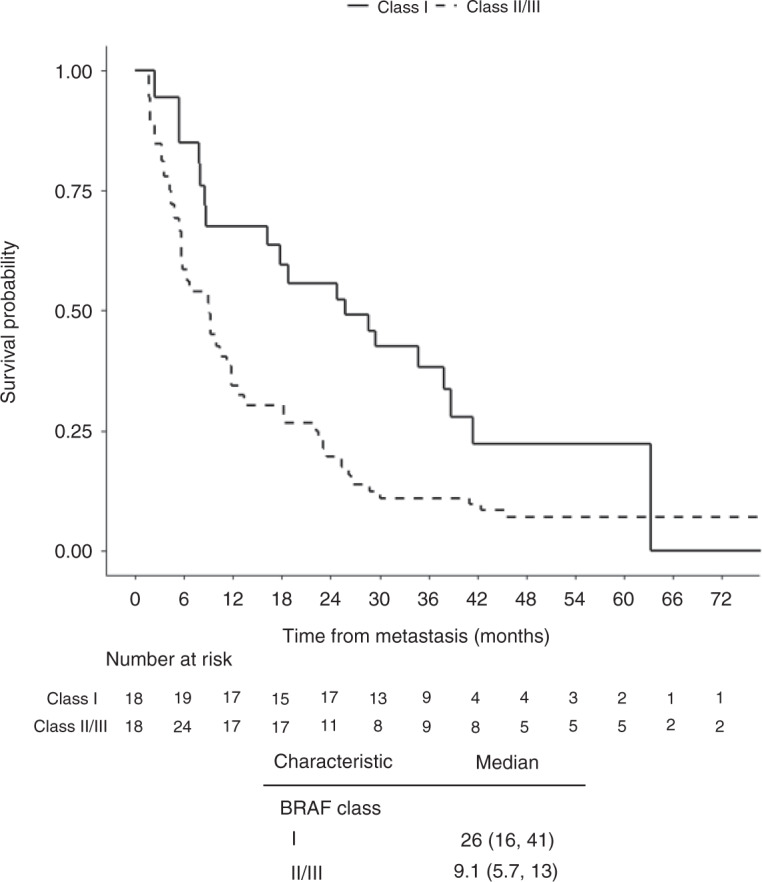
Overall survival in patients with Class I-mutant versus BRAF Class II/III lung cancers.
In order to exclude the possibility that concomitant sensitising EGFR alterations could be driving the clinical characteristics of those eight patients with Class I–III BRAF mutations and a concomitant sensitising EGFR alteration, we further assessed survival excluding patients with sensitising EGFR alterations. Median survival for patients with Class I-mutant disease was 29, while it remained 9.1 months among those patients with Class II/III-altered disease (HR 1.96, P = 0.02).
Response to ICI
Among patients with metastatic disease, 13 patients with Class I-mutant lung cancers and 37 patients with Class II/III BRAF-altered lung cancers received ICI. The median line of therapy at which patients received ICI was either the second or third line across all cohorts (Table 1). Eight of the patients who were treated with ICI had received prior BRAF-directed targeted therapy; of these, seven had Class I cancers and one patient had a Class II AGK-BRAF rearrangement.
Among patients with Class I–III alterations, nivolumab monotherapy was the most common ICI received (N = 26). Five patients received nivolumab with ipilimumab, two received atezolizumab and ten received pembrolizumab. Seven patients with Class I–III alterations were treated on a clinical trial in which ICI was given as part of an experimental combination with a different agent, including with kinase inhibitors, immune-stimulating agents, and angiogenesis modulators; these patients were excluded from further analysis. Demographic and clinical information for patients who received ICI either alone or as part of non-experimental therapies are presented in Table 2.
Table 2.
Demographic and clinical characteristics of patients who received non-experimental ICI.
| Histology | BRAF alteration | BRAF class | Concurrent alterations | TMB | PD-L1 (%) | ICI line of therapy | ICI | Time on ICI (months) | Best RECIST response |
|---|---|---|---|---|---|---|---|---|---|
| Adeno | V600E | I | 5 | n/a | 2 | Nivolumab | 1.3* | NA | |
| Adeno | p.V600E | I | 6 | 0 | 2 | Nivolumab | 0.5 | PD | |
| Adeno | p.V600E | I | 10 | n/a | 1 | Ipilimumab and Nivolumab | 5.5 | PD | |
| Adeno | p.V600E | I | 5 | n/a | 2 | Nivolumab | 27.9 | SD | |
| Adeno | p.V600E | I | 10 | n/a | 2 | Nivolumab | 0.4 | PD | |
| Adeno | p.V600E | I | 7 | 99 | 3 | Atezolizumab | 1.4 | SD | |
| Adeno | p.V600E | I | 3 | n/a | 5 | Nivolumab | 1.4 | SD | |
| Adeno | V600E | I | 1 | 90 | 3 | Pembrolizumab | 11.3 | NA | |
| Adeno | p.V600E | I | 13 | n/a | 4 | Pembrolizumab | 1.4 | PD | |
| Adeno | p.V600E | I | 6 | n/a | 2 | Nivolumab | 0.6 | PD | |
| Adeno | p.V600E | I | 19 | 70 | 3 | Pembrolizumab | 28.2* | PR | |
| Adeno | V600E | I | 2 | 10 | 1 | Ipilimumab and Nivolumab | 0.5 | SD | |
| Adeno | V600E | I | 2 | 10 to 20 | 4 | Pembrolizumab | 11.5* | SD | |
| Adeno | p.G469A | II | 7 | 20 | 4 | Nivolumab | 1.8 | PD | |
| Adeno | p.T599dup | II | 14 | n/a | 3 | Nivolumab | 0.04 | NA | |
| Adeno | p.G469A | II | 10 | 10 | 3 | Nivolumab | 1.4 | PD | |
| Squamous Cell | p.G469A | II | 1 | 0 | 3 | Nivolumab | 40.1 | PR | |
| Adeno | SND1 (NM_014390) - BRAF (NM_004333) | II | 4 | n/a | 4 | Nivolumab | 0.5 | PD | |
| Adeno | BRAF (NM_004333) - MRPS33 (NM_053035)Rearrangement | II | 3 | n/a | 6 | nivolumab | 0.5 | NA | |
| Adeno | p.G469A | II | KRAS G13C | 9 | n/a | 2 | Nivolumab | 0.06 | NA |
| Adeno | AGAP3 (NM_031946) - BRAF (NM_004333) rearrangement | II | 3 | 0 | 4 | Nivolumab | 2.3 | PD | |
| Adeno | p.G + AD2:AF121464R | II | KRAS G12V and NF1 | 10 | 0 | 1 | Ipilimumab and Nivolumab | 3.9 | PD |
| Adeno | AGK (NM_018238) - BRAF (NM_004333) rearrangement | II | 0 | n/a | 3 | Nivolumab | 0.06 | NA | |
| Adeno | p.G464V | II | 6 | n/a | 2 | Nivolumab | 2.2 | PD | |
| Neuroendocrine Tumour | p.G469A | II | 19 | n/a | 2 | Ipilimumab and Nivolumab | 1.4 | PD | |
| Adeno | p.G469A | II | 5 | 0 | 3 | Nivolumab | 4.4 | SD | |
| Adeno | p.G464V | II | KRAS G13D | 8 | 80 | 1 | Pembrolizumab | 0.06 | NA |
| Adeno | p.K601E | II | 11 | 0 | 2 | Pembrolizumab (with chemotherapy) | 5.6 | CR | |
| Adeno | p.G469S | II | NF1 | 6 | n/a | 1 | Pembrolizumab | 6.0 | PR |
| Large Cell Neuroendocrine | G469A | II | 8 | 1 | 2 | Atezolizumab | 1.4 | PD | |
| Adeno | p.V600_K601delinsE | II | 3 | 99 | 1 | Pembrolizumab (with chemotherapy) | 4.2 | PR | |
| Adeno | p.G596R | III | NF1 | 22 | 0 | 2 | Nivolumab | 34.8 | PD |
| Adeno | p.G596R | III | 11 | n/a | 3 | Nivolumab | 0.8 | PD | |
| Adeno | p.G466E | III | 20 | n/a | 1 | Ipilimumab and Nivolumab | 1.9 | PD | |
| Adeno | p.D594G | III | 6 | 5 | 4 | Nivolumab | 0.9 | PD | |
| Adeno | p.D594N | III | 7 | 0 | 2 | Nivolumab | 0.04 | NA | |
| Adeno | p.G469A | III | 14 | 0 | 2 | Nivolumab | 0.9 | NA | |
| Adeno | p.V471F | III | NF1 | 29 | n/a | 3 | Nivolumab | 5.8 | SD |
| Adeno | p.G466A | III | 4 | 10 | 2 | Pembrolizumab | 18.4 | PR | |
| Adeno | p.G596V | III | KRAS G12V | 10 | 5 | 1 | Pembrolizumab | 9.2 | CR |
| Adeno | p.S467L | III | 33 | 0 | 4 | Nivolumab | 9.7 | SD | |
| Adeno | p.N581S | III | 11 | n/a | 3 | Nivolumab | 1.9 | PD | |
| Adeno | p.N581I | III | 2 | 0 | 4 | Nivolumab | 1.3 | PD | |
| Adeno | p.V168L | U | 100 | n/a | 3 | Nivolumab | 44.2 | PD | |
| Adeno | p.I572F | U | KRAS G12V | 21 | 0 | 2 | Nivolumab | 39.8* | PD |
| Adeno | p.K483E | U | 2 | 0 | 6 | Nivolumab | 5.6 | PD | |
| Adeno | p.M620I | U | 23 | 5 | 2 | Nivolumab | 41.2 | SD | |
| Adeno | splicing variant | U | 20 | n/a | 3 | Atezolizumab | 0.7 | NA | |
| Adeno | p.M117L | U | 8 | 60 | 1 | Ipilimumab and Nivolumab | 3.7 | SD | |
| Small Cell Cancer | p.D40Y | U | 17 | n/a | 2 | Nivolumab | 0.06 | NA | |
| Adeno | p.N412S | U | 39 | 0 | 7 | Nivolumab | 7.1 | SD | |
| Adeno | E695Q | U | 17 | 0 | 3 | Nivolumab | 18.9 | PR | |
| Squamous Cell | p.S616Y | U | 21 | 20 | 2 | Nivolumab | 41.8* | CR | |
| Adeno | p.T274A | U | KRAS G12D and NF1 | 16 | 100 | 2 | Nivolumab | 0.9 | PD |
| Adeno | p.R239L | U | KRAS G12C | 18 | 0 | 2 | Nivolumab | 35.7* | SD |
| Adeno | splicing variant p.X380_splice | U | KRAS G12C | 14 | 0 | 2 | Atezolizumab | 1.4 | PD |
| Adeno | p.R266T | U | 48 | 0 | 2 | Nivolumab | 2.6 | PD | |
| Adeno | A404Cfs*9 | U | EGFR exon 20 insertion | 4 | 0 | 2 | Nivolumab | 0.9 | PD |
| Squamous Cell | p.I556F | U | 12 | 0 | 2 | Atezolizumab | 1.4 | NA | |
| Small Cell Cancer | p.P152S | U | 9 | n/a | 2 | Ipilimumab and Nivolumab | 0.06 | NA | |
| Adeno | p.A404Cfs*9 | U | 18 | n/a | 3 | Nivolumab | 0.8 | PD | |
| Adeno | p.V487L | U | NF1 | 18 | 1 | 2 | Pembrolizumab | 2.1 | PD |
Adeno adenocarcinoma, CR complete response, ICI immune checkpoint inhibitor, NA not available/not evaluable, PD progressive disease, PR partial response, SD stable disease, TMB tumour mutation burden, U variant of unknown significance. *Treatment ongoing.
Among the 34 patients treated with non-experimental ICI with available scans assessed by RECIST, the ORR of patients with Class I tumours was 9% (1/11) as compared to 26% (6/23) in patients with Class II/III-altered disease (P = 0.25) (Fig. 2a). The response rate among the 15 patients whose tumours had VUS was 13% (2/15) (Supplementary Fig. S-2). Among patients with an EGFR co-alteration, three received non-experimental ICI, two of whom were evaluable by RECIST and had progression of the disease as their best overall response.
Fig. 2. Response to ICI according to BRAF mutation class, TMB and PD-L1 status.

a Waterfall plot illustrating individual responses of patients treated with ICI with available scans assessed by RECIST. Boxes below indicate BRAF mutation Class (I–III indicates class), PD-L1 expression and tumour mutation burden (TMB). The best overall response was coded as progression of disease (PD), stable disease (SD), partial response (PR) or complete response (CR). b Overall survival of patients across all classes of BRAF-altered tumours in patients who received ICI versus those who never received ICI. c Overall survival of patients with Class I–III BRAF-altered tumours who received ICI versus those who never received ICI. d Overall survival from the time of first immunotherapy treatment by line of therapy at which ICI was received. e Overall survival of patients with Class I-mutant lung cancers who received ICI versus those who never received ICI. f Overall survival of patients whose cancers had Class II/III BRAF alterations who received ICI versus those who never received ICI.
Across all BRAF alterations, patients had a higher risk of death if they had received ICI (at 24 months, HR equalled 1.64, 95% CI: 1.04–3.15; at 36 months HR was 1.69, 95% CI: 1.00–4.60) (Fig. 2b). Worse survival was also seen across Class I–III-altered tumours (at 24 months, HR equalled 1.88, 95% CI (1.18–4.05); at 36 months HR was 1.82, 95% CI: 1.17–6.11) (Fig. 2c). There was no evidence of a difference in survival between patients who received ICI as part of first- or second-line therapy versus those who received ICI as part of later lines of therapy (first line: log-rank P = 0.40; second line: log-rank P = 0.70) (Fig. 2d).
Given that patients with sensitising EGFR mutations generally do not benefit from immunotherapy [31], we further analysed differences in survival excluding patients with these alterations who did and did not receive ICI. In the smaller cohort excluding these patients, a trend toward worse survival was seen in patients with any BRAF alteration who had received immunotherapy across classes (at 24 months, HR was 1.40, 95% CI 0.87–3.11; at 36 months, HR was 1.18, 95% CI 0.82–4.42). A similar trend was seen in those patients with Classes I–III alterations (at 24 months, HR was 2.29, 95% CI 0.98–3.78; at 36 months HR was 2.21, 95% CI 0.95–6.54).
A trend toward worse survival was also evident among patients with Class I alterations who received ICI versus those who never received ICI (HR at 24 months, 2.19, 95% CI: 0.46–9.61; HR at 36 months 1.56, 95% CI 0.39–8.08) (Fig. 2e). There was a trend toward worse survival in those with Class II/III-altered cancers who received ICI at 24 months of follow-up and this difference was significant at 36 months follow-up (HR at 24 months 2.67, 95% CI 0.72–9.63; HR at 36 months 4.83, 95% CI 1.35–34.24) (Fig. 2f).
Time on ICI therapy
The time on treatment of patients with Class I–III alterations ranged from 1 dose to 3.3 years (Fig. 3). The median time on treatment was 1.9 months in patients with Class I mutations and was also 1.9 months in patients with Class II/III alterations (P = 0.31). The median time on treatment of patients with BRAF VUS was longer than for patients with Class II/III alterations (2.6 versus 1.9 months, P = 0.05) (Supplementary Fig. S-3). One patient in the Class II/III cohort was treated with ICI for >3 years. Four patients were on ICI for >2 years, including two patients with Class I mutations and two patients with Class II/III alterations (one Class II BRAF p.G469A and one Class III BRAF p.G596R). Five patients with BRAF VUS were on therapy for >2 years, and four remained on treatment for >3 years.
Fig. 3. Time on ICI (in years) among patients with BRAF-altered lung cancer.
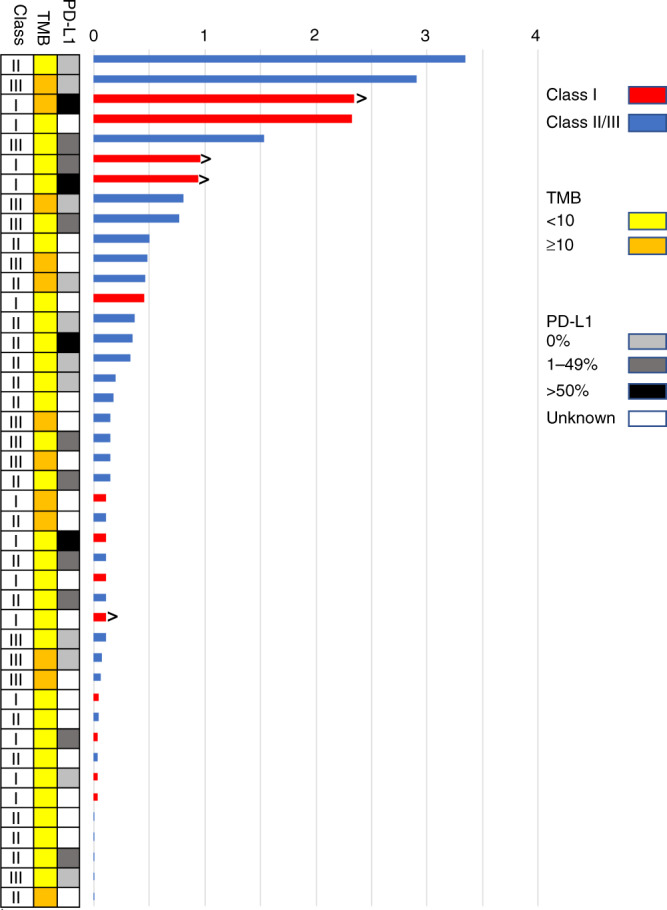
Patients still on treatment are denoted with arrows. Boxes at right indicate BRAF mutation Class (I–III indicates class), PD-L1 expression, and tumour mutation burden (TMB).
Using established cut-offs for TMB (≥10 mutations/Mb) [42], time on treatment did not differ among all patients with high TMB ≥ 10 mutations/Mb or low TMB <10 mutations/Mb (1.9 versus 1.8 months, P = 0.6).
Among the eight patients with Class I–III alterations who received BRAF-directed therapy before getting ICI for metastatic disease, time on ICI ranged from 1 day to 2.3 years. While one patient in this group had an OS from metastatic diagnosis of 4.9 months, five patients had an OS greater than 2 years, including three who were alive at the time of data collection.
Discussion
In this study, we analysed data from the largest cohort reported to date of patients with BRAF-altered lung cancers treated with ICI. Our study affirms that BRAF-altered lung cancers are clinically heterogeneous, with patients with Class I-mutant disease having longer survival than patients whose tumours had Class II/III alterations [4, 16, 27, 28]. While patients with Class I-mutant tumours have options for targeted therapy, our study found no significant clinical benefit from ICI in either the Class I or Class II/III groups. The median time on treatment of patients with either Class I-mutant or Class II/III-altered disease was just 1.9 months.
Moreover, long-term follow-up of our cohort enabled comparison of overall survival data from patients who had and had not received ICI. The hazard ratio for deaths per person-months comparing patients with Class I–III-altered tumours who ever received ICI versus those who never received ICI at 36 months was 1.82 (1.17–6.11). Our analysis of survival by receipt of ICI was able to account for several clinical factors that can influence treatment outcomes, including BRAF class and line of therapy. Notably, there was no difference in survival observed between patients who had received ICI as first- or second-line treatment versus those who had received ICI later on in their disease course. We further analysed differences in survival after receipt of ICI among patients who did not have activating EGFR alterations, given the known association of these alterations with lack of ICI benefit [31]. In this smaller cohort of patients who did not have a concomitant EGFR alteration, there was still a trend toward worse outcomes with ICI, although this narrowly missed the cut-off for statistical significance.
There are a variety of potential explanations for why we did not see a clear benefit from ICI in most patients with BRAF alterations in our cohort. We cannot entirely exclude the possibility that receipt of ICI directly caused harm, such as by delaying more effective treatment. Nonetheless, given the benefit of ICI for other lung cancers [44, 45], we believe this explanation is less likely, especially for those patients who received ICI as second-line or later therapy. We hypothesise that the patients treated with ICI may have been sicker prior to treatment and were therefore deemed unlikely to tolerate standard chemotherapy. These patients may have had poor baseline expected survival and may have been more likely to come off treatment faster.
The lack of a clear observed benefit with ICI for BRAF-altered tumours in our study differs from prior data presented by the Israel Lung Cancer group from Dudnik and colleagues. In this above-mentioned work, which included an investigation of 22 patients with BRAF-altered NSCLC, median overall survival was not reached among those patients who had received prior ICI, while it was just 21.1 months in those without prior ICI exposure [27]. Notably, Dudnik et al. did not explicitly differentiate between VUS and other non-V600 BRAF-altered tumours. Nonetheless, this difference in methodology alone likely does not account for the disparate results seen in our cohort, as our results were unchanged when patients with VUS were included in our supplementary survival analyses.
Differences in data analysis technique likely is a key factor in the disparate findings of our study and the Israel Lung Cancer investigation. In particular, our analysis took into consideration left truncation to account for differences in the timing at which patients underwent next-generation sequencing. This statistical adjustment can help to account for patients who may not have survived to the time of sequencing [46]. The importance of this adjustment for an aggressive molecular alteration was underscored by the experience of our patients, with at least five patients in the cohort passing away prior to receipt of a sequencing report showing a BRAF alteration.
While our results showing a signal of harm with ICI are novel, the lack of benefit from ICI observed does accord with previous data showing low progression-free survival (PFS) among patients with BRAF-mutant lung cancers [27, 28, 38]. In the Dudnik study, PFS on ICI was 3.7 months in patients with Class I mutations, as compared to 4.1 months in patients with all other BRAF alterations [27]. Similarly, the IMMUNOTARGET registry included 43 patients with BRAF-altered lung cancers, with the Class II/III cohort having a PFS of 4.1 months on ICI as compared to 1.8 months in the Class I cohort [38]. Guisier et al.’s study included 26 patients with Class I-mutant lung cancers and 18 with Class II/III alterations, with a recorded PFS in the former group of 5.3 months compared to 4.9 months in the latter group [28].
While collectively patients in our study did not benefit from ICI, our data did show durable responses in a select group of patients with BRAF alterations. These durable responses were observed across BRAF classes. In the case of Class I-mutant tumours, our identification of select patients with durable responses to ICI contrasts with the typical finding of a lack of response to ICI among oncogene-addicted tumours [31–37]. Response to ICI among patients with BRAF Class I-mutant tumours is not unique to lung cancer. In melanoma, both ICI and targeted therapies are considered first-line options for Class I disease based on robust overall response rates [47, 48]. Given the lack of survival benefit observed in patients with Class I-mutant tumours who received ICI in our study and the response rate of ~64% to targeted therapy in patients with Class I mutations [49], our results affirm a strategy of prioritising BRAF/MEK inhibition for Class I-mutant lung cancers. These data should not be interpreted as precluding immunotherapy in those patients whose tumours progress on BRAF-directed therapy, given the durable responses observed in a minority of patients across the BRAF-mutant class. Rather, our study may point to the benefits of using BRAF/MEK-directed targeted therapy before ICI in patients with a BRAF Class I mutation. Indeed, several patients in our cohort who had received BRAF-directed therapy prior to ICI achieved prolonged responses.
Our study also included patients who achieved durable responses with Class II/III-altered tumours. Prior research has shown a trend towards higher TMB, generally a marker of ICI benefit [40, 42], in Class II/III-altered tumours as compared to Class I-altered tumours [27]. In our study, TMB was significantly higher in patients with Class II/III alterations than in patients with Class I-mutant lung cancers. Based on our analyses, this difference could be accounted for by smoking status. Overall survival was ultimately worse in the Class II/III patients who received ICI than in those who had never received ICI despite the higher TMB, and time-on-treatment did not appear to differ by TMB in our study.
Durable responses to ICI were also seen in the cohort of patients with VUS. Interestingly, the largest subset of BRAF alterations in our cohort were categorised as VUS using an established, NGS assay [50]. Further multi-centre investigations in additional patients will enable a more granular assessment of whether individual Class II/III alterations and VUS have different sensitivity to ICI.
Our study has several limitations. First, while it represents the largest cohort to date of patients with BRAF-mutant disease who received ICI, it is a single-institution study from a tertiary cancer centre. Reassuringly, demographic features of the patients in our study, including higher rates of never-smokers among those with lung cancers possessing Class I alterations, are in agreement with prior investigations [1, 17].
Our analysis is retrospective, and we cannot rule out the possibility that providers may have chosen ICI over chemotherapy for patients with lower performance scores, who would be expected to have a worse prognosis regardless of therapeutic strategy. Our real-world data also includes patients who received different immunotherapy and chemo-immunotherapy regimens. In particular, it is notable that the median line of therapy at which patients in our cohort received ICI was between two and three depending on BRAF class. Due to evolving standards of care, current first-line therapy for most patients with lung cancers entails the use of ICI, often in combination with chemotherapy [44, 45]. While no difference in survival was observed between patients who received ICI as first- or second-line therapy and those who received ICI in later lines, further studies will be needed to fully investigate the role of ICI in the early-line setting for patients with BRAF alterations, particularly when used as part of combination chemo-immunotherapy.
Finally, not all patients who received ICI had available archival tissue for PD-L1 immunohistochemistry. Previous research on patients with BRAF-mutant lung cancers has shown that higher PD-L1 levels correlate with a higher likelihood of response to immunotherapy [27], and future investigations of the PD-L1 positive, BRAF-altered cohort are warranted.
Clinically, our study has several advantages, including its analysis of real-world data with long-term follow-up of patients with standardised RECIST measures. This longer-term follow-up enabled the investigation of differences in the durability of responses in both the Class I–III and VUS cohorts. Moreover, a large majority of patients in the study received ICI monotherapy, enabling interrogation of the efficacy of ICI, while minimising the confounding factor of combination therapies.
Overall, our data from a large cohort of patients do not demonstrate an overall survival benefit for ICI among patients with Class I–III disease. Nonetheless, a subset of patients with BRAF-altered disease may achieve durable cancer control on ICI. Further investigations into the immunogenicity of individual BRAF non-V600 lung cancers, including VUS, are warranted.
Supplementary information
Acknowledgements
YRMG gratefully acknowledges receipt of the Kristina M. Day Young Investigator Award from Conquer Cancer, the ASCO Foundation. She has received training as part of an institutional K30 grant from the National Cancer Institute (CTSA #UL1TR00457). The authors acknowledge funding for Memorial Sloan Kettering Cancer Center received through the NIH/NCI institutional P30 CA008748 grant. The authors appreciate the editorial and administrative contributions of Clare Wilhelm, Reeja Thomas and Jessica Moore.
Author contributions
MO and BTL conceived of/ designed the study. YRMG, TP, SM, DH, AJP, DL and MO acquired the data. All authors were involved in data analysis and/or interpretation. YRMG and MO drafted the manuscript. All authors approved the final version of the manuscript.
Funding
This research was supported in part by the National Cancer Institute of the National Institutes of Health P30 CA008748.
Data availability
All genomic data from sequenced tumours are included in the cBioportal for Cancer Genomics repository (http://cbioportal.org/msk-impact). Relevant clinical data are included in the manuscript.
Ethics approval and consent to participate
This study was approved by the institutional review board of Memorial Sloan Kettering Cancer Center.
Consent to publish
Not applicable.
Competing interests
MSK was awarded grants from the National Institutes of Health/National Cancer Institute during the conduct of the study. YRMG acknowledges receipt of travel, accommodation, and expenses from AstraZeneca. MGK reports personal fees from AstraZeneca, Pfizer, Regeneron, Daiichi Sankyo, outside the submitted work; he has received honoraria for participation in educational programs from WebMD, OncLive, Physicians Education Resources, Prime Oncology, Intellisphere, Creative Educational Concepts, Peerview, i3 Health, Paradigm Medical Communications, AXIS, Carvive Systems, AstraZeneca, and Research to Practice. Funds for travel and lodging, and food and beverage have been provided by AstraZeneca, Pfizer, Regeneron, and Genentech. Dr. Kris is an employee of Memorial Sloan Kettering. Memorial Sloan Kettering has received research funding from The National Cancer Institute (USA), The Lung Cancer Research Foundation, Genentech Roche, and PUMA Biotechnology for research conducted by Dr. Kris. Memorial Sloan Kettering has an institutional agreement with IBM for Watson For Oncology and receives royalties from IBM. MSK has licensed testing for EGFR T790M to MolecularMD. PKP reports grants and personal fees from Celgene; personal fees from Takeda, Abbvie, Lilly, Boehringer Ingelheim, EMD Serono, Calithera, Bicara, Xencor, GlaxoSmithKline, AstraZeneca, outside the submitted work. GJR reports grants from Novartis, Roche, Genentech, Millennium, GlaxoSmithKline, Pfizer, Infinity Pharmaceuticals, ARIAD; non-financial support from Merck Sharp & Dohme, outside the submitted work. In addition, Dr. Riely has a patent US20170273982A1 pending, and a patent WO2017164887A8 pending. HAY reports grants and personal fees from AstraZeneca and Daiichi; grants and non-financial support from Lilly, grants from Novartis, Pfizer, and Cullinan; personal fees from Blueprint Medicine, Janssen, outside the submitted work. In addition, Dr. Yu has a patent US20170273982A1 pending, and a patent WO2017164887A1 pending. CMR reports personal fees from AbbVie, Amgen, Ascentage, AstraZeneca, Bicycle, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Jansen, Jazz, Lilly/Loxo, Pfizer, PharmaMar, Syros, Vavotek, Bridge Medicines, Harpoon Therapeutics, and Earli outside the submitted work. M.D.H. reports personal fees from AstraZeneca/MedImmune; grants and personal fees from Bristol-Myers Squibb; personal fees from Merck, Genentech/Roche, Novartis, Janssen Pharmaceuticals, Nektar, Syndax Pharmaceuticals, Mirati Therapeutics, Shattuck Labs, outside the submitted work. In addition, Dr. Hellmann has a patent PCT/US2015/062208 pending and Stock and Other Ownership Interests: Shattuck Labs; Dr. Hellman is a Damon Runyon Clinical Investigator supported (in part) by the Damon Runyon Cancer Research Foundation (CI-98-18) and is a member of the Parker Institute for Cancer Immunotherapy. LWB reports personal fees from Amgen, Jazz Pharmaceuticals, Heron, Pfizer, outside the submitted work. PL reports research support to MSK from Mirati, Revolution Medicines, Amgen and Strategia during the conduct of the study. Dr Lito is listed as an inventor on patent applications filed by MSKCC describing therapeutic interventions for BRAF or KRAS mutant tumours. Dr. Lito is supported in part by the NIH/NCI (1R01CA230745-01 and 1R01CA230267-01A1), the Pew Charitable Trusts and the Damon Runyon Cancer Research Foundation. AD reports personal fees from Ignyta/Genentech/Roche, Loxo/Bayer/Lilly, Takeda/Ariad/Millenium, TP Therapeutics, AstraZeneca, Blueprint Medicines, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Tyra Biosciences, Verastem, MORE Health, Abbvie, 14ner/Elevation Oncology, Remedica Ltd., ArcherDX, Monopteros, Elevation Oncology, Novartis, EMD Serono, Melendi, Faculty RTP, Repare RX, Pfizer, Liberum, outside the submitted work; and Associated research paid to MSK from Pfizer, Exelixis, GlaxoSmithKline, Teva, Taiho, PharmaMar; research from Foundation Medicine; royalties from Wolters Kluwer; other support from Merck, Puma, Merus, Boehringer Ingelheim; Únd CME honoraria from Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis, Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences, Med Learning, Imedex, Answers in CME, Medscape, Clinical Care Options. DL reports personal fees from Pfizer, Heron Therapeutics, outside the submitted work. BTL reports non-financial support from Amgen, Genentech, and Boehringer Ingelheim; grants and non-financial support from Lilly, AstraZeneca, Daiichi Sankyo; grants and personal fees from Guardant Health, Hengrui Therapeutics, MORE Health; grants from Amgen, Illumina, GRAIL, Bolt Biotherapeutics; personal fees from Resolution Bioscience, Jiangsu Hengrui Medicine, outside the submitted work; Dr. Li is an inventor on two institutional patents at MSK and has intellectual property rights as a book author at Karger Publishers. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yonina R. Murciano-Goroff, Terry Pak.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01679-1.
References
- 1.Chen D, Zhang LQ, Huang JF, Liu K, Chuai ZR, Yang Z, et al. BRAF mutations in patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS ONE. 2014;9:e101354. doi: 10.1371/journal.pone.0101354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–51. doi: 10.1200/JCO.2010.33.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. 2013;19:4532–40. doi: 10.1158/1078-0432.CCR-13-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29:3574–9. doi: 10.1200/JCO.2011.35.9638. [DOI] [PubMed] [Google Scholar]
- 5.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 6.Lin Q, Zhang H, Ding H, Qian J, Lizaso A, Lin J, et al. The association between BRAF mutation class and clinical features in BRAF-mutant Chinese non-small cell lung cancer patients. J Transl Med. 2019;17:298. doi: 10.1186/s12967-019-2036-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183–99.. doi: 10.1038/s41388-018-0171-x. [DOI] [PubMed] [Google Scholar]
- 9.Baik CS, Myall NJ, Wakelee HA. Targeting BRAF-mutant non-small cell lung cancer: from molecular profiling to rationally designed therapy. Oncologist. 2017;22:786–96.. doi: 10.1634/theoncologist.2016-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvak AM, Paik PK, Woo KM, Sima CS, Hellmann MD, Arcila ME, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol. 2014;9:1669–74. doi: 10.1097/JTO.0000000000000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villaruz LC, Socinski MA, Abberbock S, Berry LD, Johnson BE, Kwiatkowski DJ, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer. 2015;121:448–56. doi: 10.1002/cncr.29042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinno T, Tsuta K, Shiraishi K, Mizukami T, Suzuki M, Yoshida A, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol. 2014;25:138–42. doi: 10.1093/annonc/mdt495. [DOI] [PubMed] [Google Scholar]
- 13.Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–8. doi: 10.1038/nature23291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–83. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontana E, Valeri N. Class(y) dissection of BRAF heterogeneity: beyond non-V600. Clin Cancer Res. 2019;25:6896–8. doi: 10.1158/1078-0432.CCR-19-2732. [DOI] [PubMed] [Google Scholar]
- 16.Dagogo-Jack I, Martinez P, Yeap BY, Ambrogio C, Ferris LA, Lydon C, et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin Cancer Res. 2019;25:158–65.. doi: 10.1158/1078-0432.CCR-18-2062. [DOI] [PubMed] [Google Scholar]
- 17.Tissot C, Couraud S, Tanguy R, Bringuier PP, Girard N, Souquet PJ. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer. 2016;91:23–8. doi: 10.1016/j.lungcan.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 18.Cui G, Liu D, Li W, Fu X, Liang Y, Li Y, et al. A meta-analysis of the association between BRAF mutation and nonsmall cell lung cancer. Medicine. 2017;96:e6552. doi: 10.1097/MD.0000000000006552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Planchard D, Besse B, Groen HJM, Souquet PJ, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17:984–93.. doi: 10.1016/S1470-2045(16)30146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazieres J, Cropet C, Montané L, Barlesi F, Souquet PJ, Quantin X, et al. Vemurafenib in non-small-cell lung cancer patients with BRAF(V600) and BRAF(nonV600) mutations. Ann Oncol. 2020;31:289–94.. doi: 10.1016/j.annonc.2019.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:642–50. doi: 10.1016/S1470-2045(16)00077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18:1307–16.. doi: 10.1016/S1470-2045(17)30679-4. [DOI] [PubMed] [Google Scholar]
- 23.Subbiah V, Puzanov I, Blay JY, Chau I, Lockhart AC, Raje NS, et al. Pan-cancer efficacy of vemurafenib in BRAF (V600)-mutant non-melanoma cancers. Cancer Discov. 2020;10:657–63.. doi: 10.1158/2159-8290.CD-19-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. Non-small cell lung cancer (version 2.2021) 2020. Available from: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. [DOI] [PubMed]
- 25.Dagogo-Jack I. Durable response to dabrafenib combined with trametinib in a patient with NSCLC harboring a BRAF G469A mutation. J Thorac Oncol. 2020;15:e174–e6.. doi: 10.1016/j.jtho.2020.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Negrao MV, Raymond VM, Lanman RB, Robichaux JP, He J, Nilsson MB, et al. Molecular landscape of BRAF-mutant NSCLC reveals an association between clonality and driver mutations and identifies targetable non-V600 driver mutations. J Thorac Oncol. 2020;15:1611–23. doi: 10.1016/j.jtho.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dudnik E, Peled N, Nechushtan H, Wollner M, Onn A, Agbarya A, et al. BRAF mutant lung cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol. 2018;13:1128–37.. doi: 10.1016/j.jtho.2018.04.024. [DOI] [PubMed] [Google Scholar]
- 28.Guisier F, Dubos-Arvis C, Viñas F, Doubre H, Ricordel C, Ropert S, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol. 2020;15:628–36.. doi: 10.1016/j.jtho.2019.12.129. [DOI] [PubMed] [Google Scholar]
- 29.Mansuet-Lupo A, Alifano M, Pécuchet N, Biton J, Becht E, Goc J, et al. Intratumoral immune cell densities are associated with lung adenocarcinoma gene alterations. Am J Respir Crit Care Med. 2016;194:1403–12.. doi: 10.1164/rccm.201510-2031OC. [DOI] [PubMed] [Google Scholar]
- 30.Rihawi K, Giannarelli D, Galetta D, Delmonte A, Giavarra M, Turci D, et al. BRAF mutant NSCLC and immune checkpoint inhibitors: results from a real-world experience. J Thorac Oncol. 2019;14:e57–e9.. doi: 10.1016/j.jtho.2018.11.036. [DOI] [PubMed] [Google Scholar]
- 31.Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585–93. doi: 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berghoff AS, Bellosillo B, Caux C, de Langen A, Mazieres J, Normanno N, et al. Immune checkpoint inhibitor treatment in patients with oncogene- addicted non-small cell lung cancer (NSCLC): summary of a multidisciplinary round-table discussion. ESMO Open. 2019;4:e000498. doi: 10.1136/esmoopen-2019-000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CK, Man J, Lord S, Links M, Gebski V, Mok T, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer—a meta-analysis. J Thorac Oncol. 2017;12:403–7. doi: 10.1016/j.jtho.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–41.. doi: 10.1200/JCO.2017.75.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spigel DR, Schrock AB, Fabrizio D, Frampton GM, Sun J, He J, et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J Clin Oncol. 2016;34:9017–9017. [Google Scholar]
- 36.Nagahashi M, Sato S, Yuza K, Shimada Y, Ichikawa H, Watanabe S, et al. Common driver mutations and smoking history affect tumor mutation burden in lung adenocarcinoma. J Surg Res. 2018;230:181–5. doi: 10.1016/j.jss.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. 2019;25:1063–9. doi: 10.1158/1078-0432.CCR-18-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–8. doi: 10.1093/annonc/mdz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–64. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–6. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vokes NI, Liu D, Ricciuti B, Jimenez-Aguilar E, Rizvi H, Dietlein F, et al. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non-small-cell lung cancer. JCO Precis Oncol. 2019;3:1–12. [DOI] [PMC free article] [PubMed]
- 42.Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl J Med. 2018;378:2093–104.. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaule P, Smithy JW, Toki M, Rehman J, Patell-Socha F, Cougot D, et al. A quantitative comparison of antibodies to programmed cell death 1 ligand 1. JAMA Oncol. 2017;3:256–9. doi: 10.1001/jamaoncol.2016.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl J Med. 2018;378:2078–92.. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 45.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl J Med. 2016;375:1823–33.. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 46.Shen R, Martin A, Ni A, Hellmann M, Arbour KC, Jordan E, et al. Harnessing clinical sequencing data for survival stratification of patients with metastatic lung adenocarcinomas. JCO Precis Oncol. 2019;3:1–9. doi: 10.1200/PO.18.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl J Med. 2019;381:1535–46. doi: 10.1056/NEJMoa1910836. [DOI] [PubMed] [Google Scholar]
- 48.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl J Med. 2015;372:2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 49.Planchard D, Besse B, Groen HJM, Souquet P-J, Quoix E, Baik CS, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17:984–93.. doi: 10.1016/S1470-2045(16)30146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13.. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic data from sequenced tumours are included in the cBioportal for Cancer Genomics repository (http://cbioportal.org/msk-impact). Relevant clinical data are included in the manuscript.