

Abstract
Prime editing enables the installation of virtually any combination of point mutations, small insertions, or small deletions in the DNA of living cells. A prime editing guide RNA (pegRNA) directs the prime editor protein to the targeted locus and also encodes the desired edit. Here we demonstrate that degradation of the 3′ region of the pegRNA that contains the reverse transcriptase template and the primer-binding site can poison the activity of prime editing systems, impeding editing efficiency. We incorporated structured RNA motifs to the 3′ terminus of pegRNAs that enhance their stability and prevent degradation of the 3′ extension. The resulting engineered pegRNAs (epegRNAs) improve prime editing efficiency 3 to 4-fold in HeLa, U2OS, and K562 cells and in primary human fibroblasts without increasing off-target editing activity. We optimized the choice of 3′ structural motif and developed pegLIT, a computational tool to identify non-interfering nucleotide linkers between pegRNAs and 3′ motifs. Finally, we demonstrated that epegRNAs enhance the efficiency of the installation or correction disease-relevant mutations.
Reporting Summary
Nelson Randolph Manuscript reporting summary revised.pdf
The ability to make targeted changes to the genome of living systems continues to advance the life sciences and medicine. Double-strand break (DSB)-mediated DNA editing strategies that use programmable nucleases such as ZFNs, TALENs, or CRISPR-Cas nucleases can efficiently disrupt genes by inducing insertions or deletions (indels) at the target site, but DSBs also result in outcomes that are often undesired, including uncontrolled mixtures of editing outcomes1,2, larger DNA rearrangements3–5, p53 activation6–8, and chromothrypsis9,10. Although targeted DSBs can stimulate precise gene correction through homology-directed repair, the process is inefficient in most therapeutically relevant cell types11. In contrast, base editors12,13 and prime editors14 can efficiently install precise changes in therapeutically relevant cells without requiring DSBs. Cytosine and adenosine base editors enable the conversion of C•G to T•A, and A•T to G•C, respectively, while prime editors enable the installation of virtually any local mutation, including the substitution, insertion, and/or deletion of up to dozens of base pairs at targeted DNA sites.
Prime editing (PE) systems minimally consist of two components: a protein containing a programmable DNA nickase fused to an engineered reverse transcriptase (RT), and a prime editing guide RNA, or pegRNA (Fig. 1a)14. The pegRNA contains a spacer that specifies the target site, an sgRNA scaffold, and a 3′ extension that encodes the desired edit. This extension contains a primer-binding site (PBS) that is complementary to a portion of the DNA protospacer, and an RT template that encodes the desired edit and downstream genomic sequence. After the PE ribonucleoprotein (RNP) binds the target site and nicks the PAM-containing DNA strand, the resulting nicked DNA strand base pairs to the PBS in the pegRNA, priming the reverse transcription of the RT template directly into the target DNA site14. The newly synthesized 3′ flap of edited DNA is then resolved by cellular DNA repair pathways, leading to installation of the desired edit at the target site.
Figure 1. Truncated pegRNAs limit prime editing efficiency.
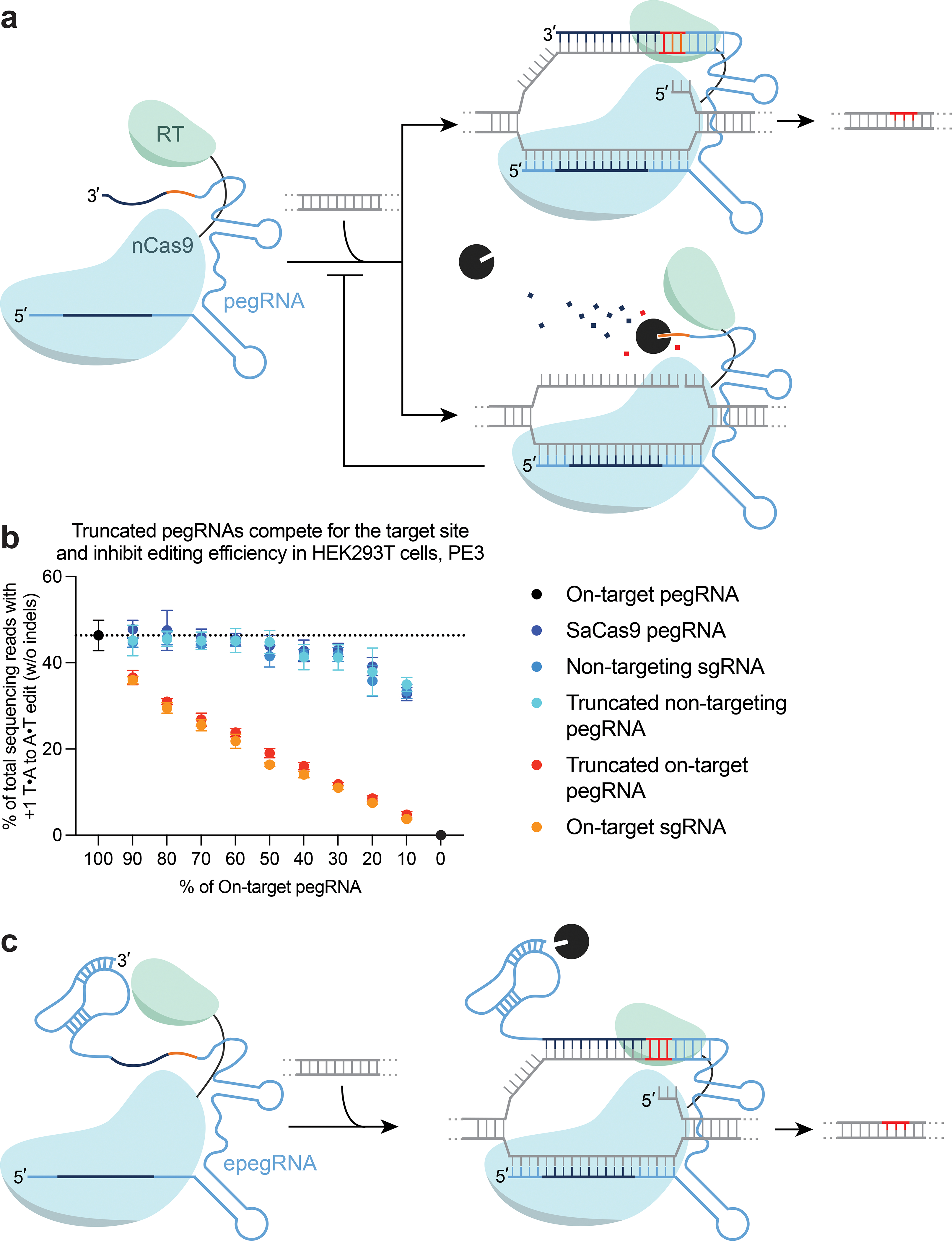
(a) (left) Schematic of a prime editing complex composed of a prime editor (PE) protein that consists of a Cas9 nickase (nCas9) fused to a modified reverse transcriptase via a flexible linker and a prime editing guide RNA (pegRNA). (right) Degradation of the 3′ extension of a pegRNA by exonucleases could impede editing efficiency through loss of the PBS. (b) PE3-mediated editing efficiencies with the addition of plasmids expressing sgRNAs, truncated pegRNAs that target the same genomic locus (HEK3), non-targeting pegRNA, or SaCas9 pegRNAs. All pegRNAs are expressed from a U6 promoter. Data and error bars reflect the mean and standard deviation of three independent biological replicates. (c) Design of engineered pegRNAs (epegRNAs) that contain a structured RNA pseudoknot, which protects the 3′ extension from degradation by exonucleases.
The versatility of prime editing arises from the ability of the 3′ extension of the pegRNA to encode a wide variety of edited sequences. Despite its versatility, the efficiency of current prime editors varies substantially among target sites and cell types14. In this study, we report that the putative degradation of the 3′ extension of pegRNAs can erode prime editing efficiency. Although the resulting truncated pegRNAs compete for target site engagement, they are incompetent for prime editing. To address this vulnerability, we identified RNA motifs that protect pegRNA integrity and broadly improve prime editing efficiencies at a variety of target sites in multiple cell lines and via multiple delivery modalities. The resulting engineered pegRNAs (epegRNAs) substantially advance the effectiveness and the application scope of prime editing.
Results
RNA stability limits pegRNA efficacy
Unprotected nuclear RNAs are susceptible to degradation from both the 5′ and 3′ termini by exonucleases15. In contrast to sgRNAs in which the entire guide RNA is protected by an associated Cas9 protein16, the 3′ extension of pegRNAs is likely to be exposed in cells and thus more susceptible to exonucleolytic degradation. We hypothesized that while partially degraded pegRNAs might retain their ability to bind Cas9 and engage the target DNA site, loss or truncation of the PBS might prevent their ability to install the desired edit, thereby occupying PE proteins and target sites with guide RNAs that cannot mediate prime editing.
To test this hypothesis, we transfected HEK293T cells with mixtures of two plasmids in varying ratios that generate either a full-length pegRNA containing an RT template encoding a T•A-to-A•T transversion, or a truncated pegRNA containing an RT template encoding a T•A-to-G•C transversion but lacking the PBS at the 3′ terminus. The two pegRNAs targeted either the same or different genomic loci in human cells. We also tested the effect of adding a plasmid that generated a non-interacting SaCas9 pegRNA that should compete for transcription with the SpCas9 pegRNA-encoding plasmids, but not interact with the prime editor protein. Increasing the production of truncated pegRNA resulted in inhibited PE activity when the full-length and truncated pegRNAs were targeted to the same site (Fig. 1b). In contrast, neither a truncated pegRNA targeted to a different genomic site nor a non-targeting SpCas9 sgRNA impeded PE activity any more than the SaCas9 pegRNA (Fig. 1b). These data suggest that degraded pegRNAs with truncated 3’ extensions inhibit PE activity by enabling editing-incompetent prime editor ribonucleoproteins (RNPs) to compete for the targeted genomic locus.
Design of engineered pegRNAs (epegRNAs) that improve prime editing efficiency
Having identified truncated pegRNAs as a potent inhibitor of prime editing, we next sought to minimize pegRNA degradation. We envisioned that structured RNA motifs at the 3′ end of the pegRNA (Fig. 1c) might improve pegRNA stability, consistent with the ability of RNA structures at the 5′ or 3′ termini to enhance mRNA stability in human cells and in yeast17,18. For instance, the long-noncoding RNA MALAT1 is stabilized by a triple helix that sequesters its poly(A) tail, limiting both degradation and nuclear export19.
We first tested whether prime editing efficiency could be improved by incorporating one of two stable pseudoknots at the 3′ end of the pegRNA: either a modified prequeosine1-1 riboswitch aptamer20,21, (evopreQ1) or the frameshifting pseudoknot from Moloney murine leukemia virus (MMLV)22, hereafter referred to as “mpknot” (Supplementary Fig. 1). We chose evopreQ1 because it is one of the smallest naturally derived RNA structural motifs with a defined tertiary structure (42 nucleotides, nt, in length)20,21. We reasoned that smaller motifs would minimize the formation of secondary structures that could interfere with pegRNA function. Furthermore, shorter pegRNAs can be more easily produced by chemical synthesis. We chose mpknot because of its tertiary structure and because it is an endogenous template for the MMLV RT from which the RT in canonical prime editors was engineered, raising the possibility that mpknot might help recruit the RT. We tested if these epegRNAs could insert a FLAG epitope tag sequence using PE3 at five genomic loci in HEK293T cells (Fig. 2a). To reduce the potential for the motif to interfere with pegRNA function during prime editing, we included an 8-nt linker to connect either evopreQ1 or mpknot to the 3′ end of the epegRNA PBS. Linker sequences were designed using ViennaRNA23 to avoid potential base pairing interactions between the linker and PBS, or between the linker and the pegRNA spacer14. We observed an average of 2.1-fold increased efficiency of FLAG tag insertion when using epegRNAs compared to canonical pegRNAs across all five genomic sites tested, with no apparent change in edit:indel ratios (Supplementary Fig. 2), suggesting that 3′ terminal pseudoknot motifs can improve PE efficacy.
Figure 2. PE editing efficiency is enhanced by the addition of structured RNA motifs to the 3′ terminus of pegRNAs.
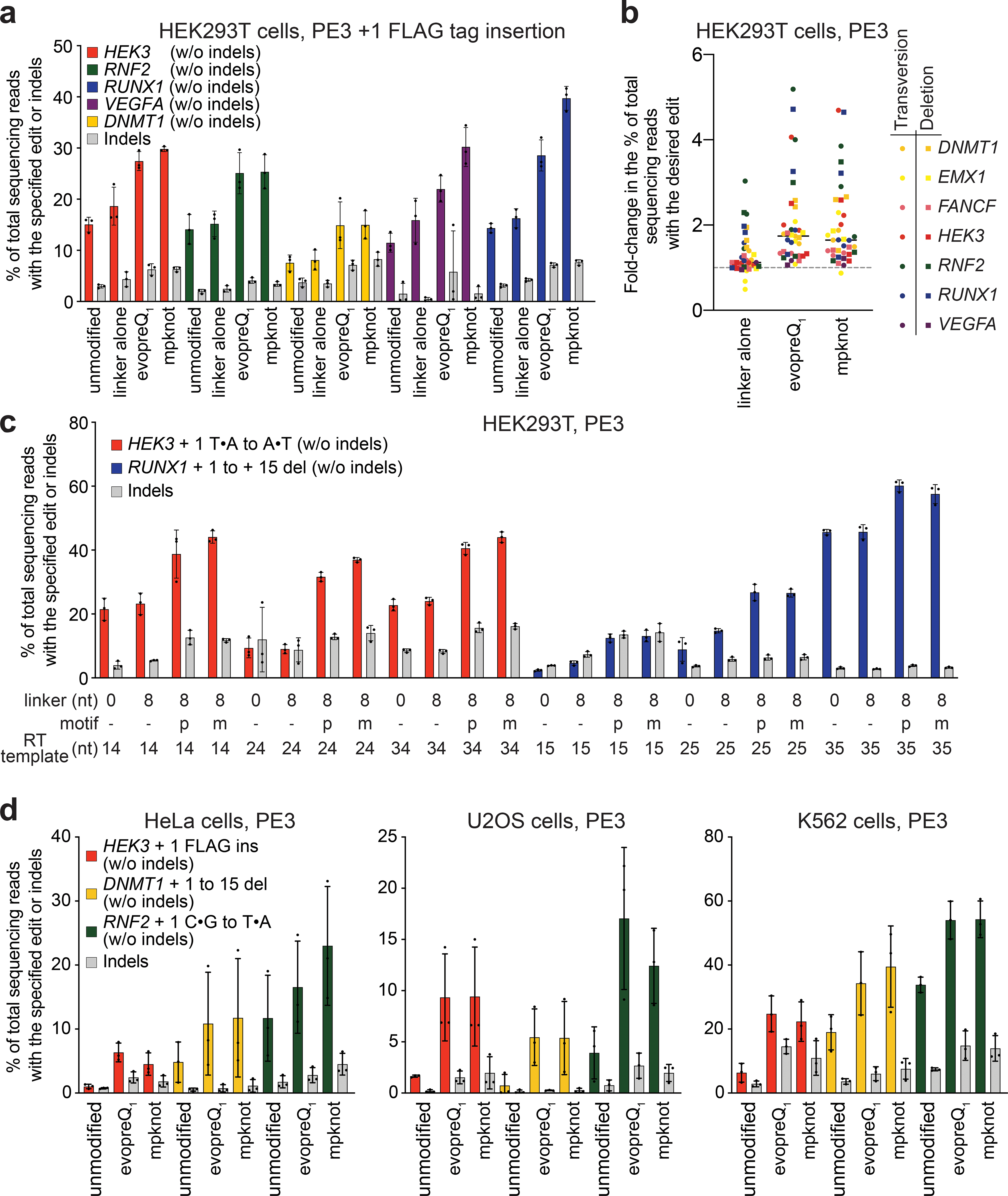
(a) Efficiency of PE3-mediated insertions of the FLAG epitope tag at the +1 editing position (insertion directly at the pegRNA-induced nick site) across multiple genomic loci in HEK293T cells using canonical pegRNAs (“unmodified”), pegRNAs with either evopreQ1 or mpknot appended to the 3′ end of the PBS via an 8-nt linker, or pegRNAs appended with only the 8-nt linker sequence. (b) Summary of the fold-change in PE editing efficiency relative to canonical pegRNAs of the indicated edit at various genomic loci upon addition of the indicated 3′ motif via an 8-nt linker, or the addition of the linker alone. “Transversion” denotes mutation of the +5 G•C to T•A at RUNX1, EMX1, VEGFA, and DNMT1, the +1 C•G to T•A at RNF2, and the +1 T•A to A•T at HEK3, where the positive integer indicates the distance from the Cas9 nick site. “Deletion” denotes a 15-bp deletion at the Cas9 nick site. Data summarized here are presented in (c) and Supplementary Fig. 2. The horizontal bars show the median values. (c) Representative improvements in PE editing efficiency from appending either evopreQ1 (p) or mpknot (m) via an 8-nt linker to pegRNAs with varying template lengths (in nucleotides, indicated). (d) Editing activities of canonical pegRNAs and modified pegRNAs across three genomic loci in HeLa cells, U2OS cells, and K562 cells. Data and error bars in a, c, and d reflect the mean and standard deviation of three independent biological replicates.
We characterized the necessity of the linker sequence by comparing the ability of epegRNAs with or without 8-nt linkers to mediate transversions or FLAG tag insertions. We observed a significant decrease in PE3 editing efficiency upon removing the linker for epegRNAs containing mpknot (p=0.022), but no significant difference for epegRNAs that contain evopreQ1 (Supplementary Fig. 3), perhaps because evopreQ1 is smaller than mpknot and is less prone to steric clashes with the RT. While the overall average editing efficiencies for epegRNAs with evopreQ1 were similar (with or without a linker) we noted occasional reduced performance for epegRNAs without a linker (Supplementary Fig. 3). We therefore opted to include an 8-nt linker unless otherwise noted for subsequent epegRNA designs.
To ensure that this improvement in PE efficacy was not limited to epegRNAs with longer extensions, we tested 148 additional epegRNAs that encoded a variety of point mutations or deletions with various RT template lengths at seven different genomic sites in HEK293T cells using PE3. Use of either motif resulted in a 1.5-fold average improvement in prime editing efficiency relative to that of canonical pegRNAs across all tested sites and pegRNAs in HEK293T cells, with no apparent change in edit:indel ratios (Figs. 2b–c and Supplementary Figs. 4 and 5). Together, these results establish that epegRNAs broadly improve PE efficacy in HEK293T cells.
Engineered pegRNAs improve prime editing in multiple mammalian cell lines
We previously observed that PE efficiency varies substantially between mammalian cell types14, highlighting the need to test improved PE systems in a variety of cells. We tested the ability of epegRNAs containing a 3′ evopreQ1 or mpknot motif to insert a 24-bp FLAG epitope tag at HEK3, delete 15 bp at DNMT1, or install a C•G-to-A•T transversion at RNF2 via PE3 in K562, U2OS, and HeLa cells. In each of these cell lines, epegRNAs resulted in large improvements in editing efficiency compared to pegRNAs, averaging 2.4-fold higher editing in K562 cells, 3.1-fold higher editing in HeLa cells, and 5.6-fold higher editing in U2OS cells across all tested edits (Fig. 2d) with no decrease in edit:indel ratios (Supplementary Fig. 2). These results indicate that epegRNAs can enhance prime editing in multiple mammalian cell lines. Additionally, epegRNAs improved editing efficiencies to a greater degree in non-HEK293T cells than in HEK293T cells, (Fig. 2a and Supplementary Fig. 4 compared to Fig. 2d), suggesting that epegRNAs are especially beneficial in cell lines that are less efficiently transfected or edited by the original PE systems.
Effect of engineered pegRNAs on off-target prime editing
Previous studies have demonstrated that prime editing results in substantially less off-target editing than other CRISPR gene editing strategies14,24–27. To determine if the addition of evopreQ1 or mpknot changed the extent of off-target editing, we treated HEK293T cells with pegRNAs or epegRNAs targeting HEK3, EMX1, or FANCF that template either a transversion (T•A-to-A•T at HEK3 or G•C-to-T•A at EMX1 and FANCF) or a 15-bp deletion using PE3. We measured the extent of indel generation and any nucleotide changes that could reasonably arise from prime editing at the top four experimentally confirmed off-target sites28 for each targeted locus and compared the extent of off-target editing between epegRNAs and unmodified pegRNAs following treatment with PE3. In all cases epegRNAs and pegRNAs exhibited ≤0.1% off-target prime editing and indels at the examined sites (Supplementary Fig. 6), suggesting that epegRNAs and pegRNAs exhibit similar levels of off-target editing.
Basis of enhanced prime editing with engineered pegRNAs
EpegRNAs may enhance prime editing outcomes through a variety of mechanisms, including resistance to degradation, higher expression levels, more efficient Cas9 binding, and/or target DNA engagement when complexed with Cas9; we probed each of these possibilities.
To determine whether evopreQ1 or mpknot impede degradation of the pegRNA 3′ extension, we compared the stability of epegRNAs and pegRNAs following in vitro incubation with HEK293T nuclear lysates containing endogenous exonucleases. We found that pegRNAs were degraded to a greater extent from this treatment compared to epegRNAs (1.9-fold compared to evopreQ1 and 1.8-fold compared to mpknot, p<0.005, Fig. 3a). Conversely, addition of Cas9, which binds the guide RNA scaffold and is likely to protect the core sgRNA from degradation, rescued pegRNA abundance compared to either epegRNA as determined by RT-qPCR quantification of the guide RNA scaffold (Fig. 3b).
Figure 3. Structural motifs increase RNA stability and efficiency of reverse transcription but reduce Cas9 binding affinity.
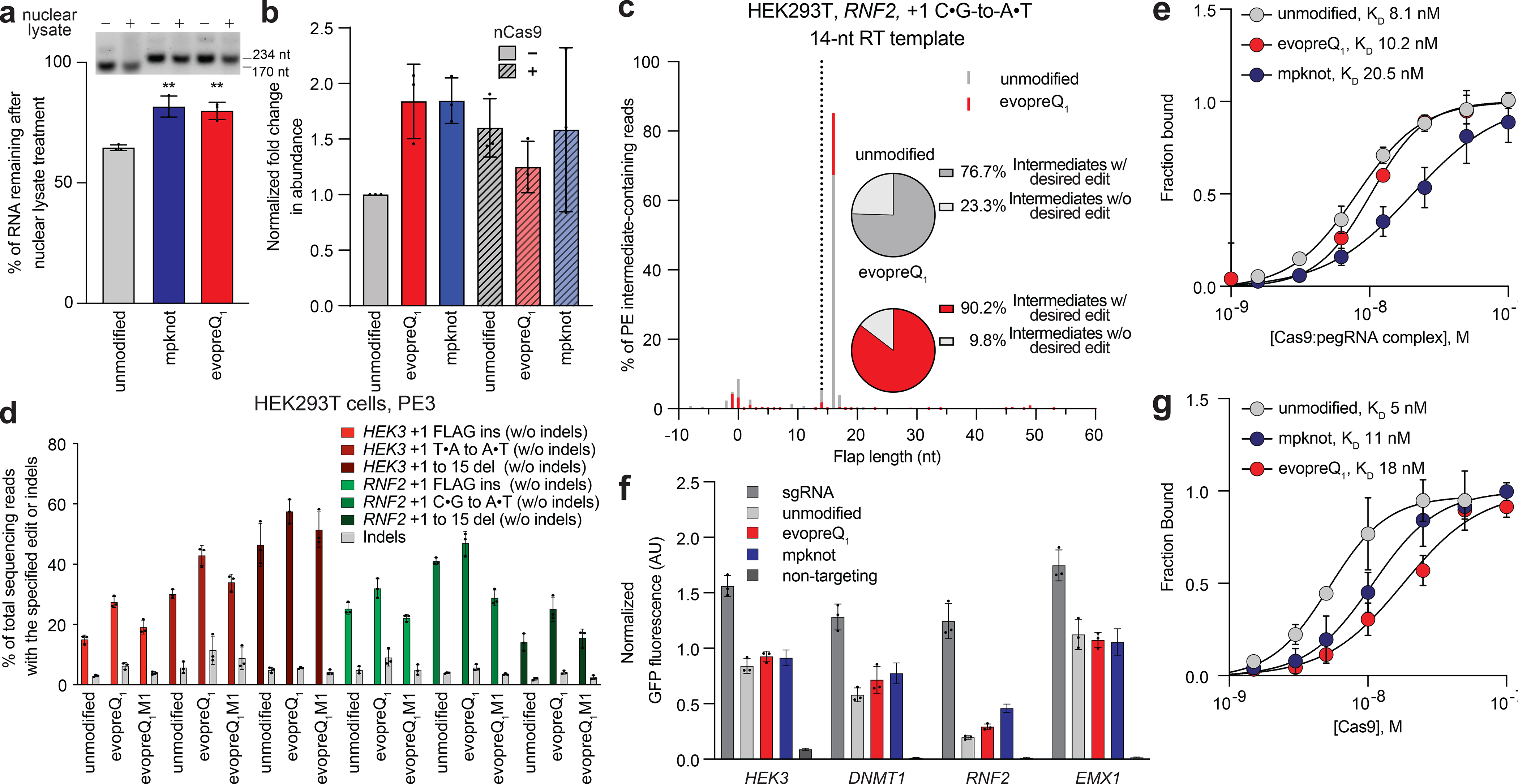
(a) Resistance of unmodified pegRNA or epegRNA containing evopreQ1 or mpknot to degradation upon exposure to HEK293T nuclear lysates. The agarose gel shown is representative of three experiments. Untreated in vitro transcribed pegRNAs or epegRNAs served as standards. Percent RNA remaining was calculated using densitometry. Significance was analyzed using a two-tailed unpaired Student’s t test (p=0.0028 for mpknot and 0.0022 for evopreQ1). (b) Fold change in abundance of the pegRNA scaffold relative to unmodified pegRNA upon exposure to HEK293T nuclear lysates in the absence and presence of nCas9 as determined by RT-qPCR of the sgRNA scaffold. (c) Comparison of prime-editing intermediates generated by PE2 with either pegRNAs or epegRNAs at RNF2. Dotted lines indicate the full-length reverse transcriptase product templated by the pegRNA or epegRNA tested at the indicated locus. X axis is relative to the position of the PE2-induced nick with the first base 3’ downstream represented as position +1. Histograms and pie charts are generated from the average of three independent biological replicates. (d) PE3 editing efficiencies in HEK293T cells using unmodified pegRNAs, pegRNAs containing the evopreQ1 motif, or pegRNAs containing a G15C point mutant of evopreQ1 (M1) that disrupts pseudoknot motif structure. (e) Fraction of Cas9 RNPs composed of dCas9 and either unmodified pegRNA or epegRNA containing either evopreQ1 or mpknot and templating a +1 FLAG tag insertion at HEK3 bound to dsDNA as determined by MST. (f) CRISPRa transcriptional activation by pegRNAs, epegRNAs, and sgRNAs. Reported GFP fluorescence is normalized to iRFP fluorescence expressed from a co-transfected plasmid. AU, arbitrary units. (g) Fraction of unmodified pegRNA or epegRNA (templating a +1 FLAG tag insertion at HEK3) containing either evopreQ1 or mpknot bound to H840A nCas9 as determined by microscale thermophoresis (MST). Data and error bars reflect the mean and standard deviation of three independent biological replicates.
The ability of 3′ structural motifs to increase the abundance of the upstream scaffold region (Fig. 3b) suggests that pegRNA degradation in the nucleus is dominated by 3′-directed degradation. This model is consistent with the characterized behavior of the nuclear exosome, the major source of RNA turnover in the nucleus29. However, partially degraded pegRNAs would generate editing-incompetent RNPs previously shown to inhibit prime editing (Fig. 1c). To detect partially degraded RNAs in cells, we analyzed lysates of HEK293T cells transfected with plasmids encoding PE2 and either pegRNAs or epegRNAs templating either a +1 FLAG tag insertion at HEK3 or a nucleotide transversion at EMX1 via northern blot. We observed RNA species containing the sgRNA scaffold and equivalent in size to the sgRNA, consistent with our previous finding (Fig. 3b) that Cas9 binding protects the scaffold from 3′-directed degradation (Supplementary Fig 7). However, lysates with different total levels of pegRNA or epegRNA had similar levels of sgRNA-like truncated species, which represented only a minority of the guide RNA content of the lysate (Supplementary Fig. 7). Since we observed robust degradation of pegRNAs exposed to nuclear lysate in vitro (Fig. 3a and 3b), and pegRNA is present in levels greater than PE2 in HEK293T cells (Fig. 1b), we suspect that partially degraded pegRNA species do not accumulate at levels amenable to northern blot detection.
Next, we examined genomic prime editing intermediates to better understand how epegRNAs might be mediating improved editing efficiency. In our current model, the 3′ flap intermediate generated by RT extension of the nicked targeted site is converted into a 5′ flap intermediate, replacing the original genomic sequence with the newly synthesized one14. This 5′ flap is then removed by 5′−3′ exonucleases and the resulting genomic nick undergoes ligation to install the prime edit14. While full-length pegRNAs would be expected to efficiently template RT extension of the nicked genomic strand, truncated pegRNAs without a PBS should be unable to do so, resulting instead in nicking of the targeted strand followed by chew-back or extension of the strand by DNA repair enzymes (lacking the templated edit in either case). If a greater fraction of RT-extended prime editing intermediates is observed with epegRNAs than with pegRNAs, this would suggest that addition of 3′ RNA motifs improve the integrity of the PBS.
To capture these intermediates, we transfected HEK293T cells with plasmids encoding PE2 and either unmodified pegRNAs or epegRNAs containing evopreQ1 or mpknot that template transversions at HEK3, DNMT1, EMX1, or RNF2. Next, we used terminal transferase to label with oligo-dG the 3’ termini of genomic DNA, which should include intermediates of prime editing that have not yet undergone ligation. In each case, epegRNAs reduced the extent of editing-incompetent intermediates at the targeted site by an average of 2.2-fold across the four sites (Fig. 3c and Supplementary Fig. 8). The dominant reverse transcription product contained the full sequence templated by the 3′ extension and two nucleotides templated by the last two nucleotides of the pegRNA scaffold, consistent with previous in vitro characterization of PE intermediates14. The scaffold-templated nucleotides are presumably removed during DNA repair of the targeted locus to produce the cleanly edited alleles that represent the dominant product of PE. These data are consistent with a model in which epegRNAs improve reverse transcription of the pegRNA extension into the target site by reducing the frequency of unproductive target-site nicking from prime editors bound to truncated pegRNAs.
Because single-stranded 3’ termini are a common feature of 3′ exonuclease substrates30, we next tested whether the degradation resistance conferred by these motifs could be explained by the more mechanically stable tertiary structures of pseudoknots. Notably, appending 15-bp (34-nt) hairpins to the 3′ terminus resulted in inconsistent improvements to PE efficiency compared to appending pseudoknots (Supplementary Fig. 9), suggesting that tertiary structure is indeed an important feature of epegRNAs.
To test if tertiary pseudoknot structure is required for epegRNA-mediated improvements in PE efficiency, we examined editing efficiency of epegRNAs containing the G15C point mutation within evopreQ1, a mutation known to disrupt pseudoknot formation (M1 in Supplementary Fig. 1)21. We used epegRNAs to install either a 24-bp FLAG epitope tag insertion, a 15-bp deletion, or transversions at HEK3 or RNF2 in HEK293T cells using PE3. Indeed, incorporation of the G15C mutation into evopreQ1 abolished the increases in editing efficiency (Fig. 3d). These results establish that the secondary or tertiary structure of the motifs are critical for epegRNA-mediated PE improvements, likely by stabilizing the 3’ extension.
Next, we tested whether the structured 3′ motifs in epegRNAs increase their expression level compared to pegRNAs. RT-qPCR quantification of the pegRNA scaffold revealed target-dependent differences in epegRNA expression levels relative to unmodified pegRNAs (Supplementary Fig. 7).
For a pegRNA that templates a +1 FLAG tag insertion at HEK3, we observed that addition of evopreQ1 or mpknot decreased pegRNA expression 9.2- to 9.6-fold, despite yielding a 1.9-fold improvement in the efficiency of FLAG tag epitope insertion at HEK3 (Fig. 2a). Similarly, epegRNAs that template a transversion at DNMT1 also exhibited reduced expression (1.6- to 2.1-fold). However, epegRNAs that template transversions at RNF2 or EMX1 were expressed to greater levels than those of unmodified pegRNA (2.2- to 2.4-fold and 1.4- to 3.7-fold, respectively, Supplementary Fig. 7). These data suggest that the 3′ motifs affect pegRNA expression inconsistently, concordant with our earlier finding (Fig. 1b) that PE efficiency under these transfection conditions is not limited by pegRNA expression in HEK293T cells. When epegRNA expression is more limiting, however, improving epegRNA expression might further improve editing efficiency.
Next, we tested if the addition of a 3′ RNA structural motif reduced engagement of the target DNA site by comparing the ability of epegRNAs and pegRNAs to support transcriptional activation by dCas9–VP64–p65–Rta (dCas9–VPR) fusions32,33. HEK293T cells were transfected with plasmids encoding dCas9-VPR, GFP downstream of either the HEK3, DNMT1, RNF2, or EMX1 target protospacer, and either pegRNAs, epegRNAs, or sgRNAs targeting the corresponding site. Transcriptional activation was measured via cellular GFP fluorescence. In contrast to their ability to enhance PE activity (Fig. 2a), epegRNAs showed similar Cas9-dependent transcriptional activation in HEK293T cells as pegRNAs (Fig. 3f). Both epegRNAs and pegRNAs resulted in lower transcriptional activation compared to an sgRNA targeting the same site (3.0-fold for pegRNA, 2.3-fold for evopreQ1 epegRNA, and 1.9-fold for mpknot epegRNA across four sites), suggesting that the 3′ extension in pegRNAs and epegRNAs modestly impedes target site engagement.
To deconvolute potential changes in target site engagement and differences in pegRNA and epegRNA expression, we performed microscale thermophoresis (MST) to measure the affinity of pre-incubated RNP complexes of catalytically inert Cas9 (dCas9) and pegRNAs or epegRNAs for a dsDNA substrate. We found that addition of mpknot or evopreQ1 resulted in comparable or modestly reduced binding affinity for dsDNA compared to unmodified pegRNA respectively (KD=10 nM for evopreQ1 epegRNA and 21 nM for mpknot pegRNA versus 8.1 nM for unmodified pegRNA, Fig. 3e). Affinity of pegRNAs for Cas9 H840A nickase was also modestly reduced by either motif (KD=18 nM for evopreQ1 epegRNA, 11 nM for mpknot pegRNA, and 5 nM for unmodified pegRNA; Fig. 3g). These findings suggest that increased PE efficiency from epegRNAs does not arise from improved binding of the pegRNA to Cas9, or of the PE RNP complex to the targeted site.
Taken together, these results suggest that epegRNAs are more resistant to cellular degradation than pegRNAs and thus generate fewer truncated pegRNA species that erode prime editing efficiency. Additional mechanisms behind improvements from epegRNAs cannot be excluded.
Optimization of engineered pegRNA 3′ motifs
Having established that epegRNAs improve editing efficiency by resisting exonucleolytic degradation, we speculated that more stable RNA motifs might further improve PE activity. We screened 25 additional structured RNA motifs for their ability to improve epegRNA editing efficiency across epegRNAs encoding either the installation of a 24-bp FLAG epitope tag insertion, a 15-bp deletion, or a transversion at HEK3 or RNF2 (Supplementary Figs. 9 and 10). These motifs included additional evolved prequeosine1-1 riboswitch aptamers21, mpknot variants with improved pseudoknot stability22, G-quadruplexes of increasingly stability34, 15-bp hairpins, an xrRNA35, and the P4-P6 domain of the group I intron36. While 123 of the 137 epegRNAs tested exhibited improved overall prime editing compared to the corresponding pegRNAs, none demonstrated consistent improvements over evopreQ1 or mpknot across the majority of edits tested (Supplementary Figs. 9 and 10).
Next, we hypothesized that trimming unnecessary sequence from the added evopreQ1 and mpknot motifs might further improve the epegRNA design because removing extraneous sequences within a structured RNA can reduce the propensity for misfolding37. We found that trimming 5 nt of excess sequence from evopreQ1 or mpknot resulted in marginal gains in average PE3-editing efficiency relative to the full-length epegRNAs (Supplementary Fig. 10). Since trimming these RNA motifs did not adversely affect editing efficiency and shorter epegRNAs are more readily prepared by chemical synthesis, we decided to use trimmed evopreQ1 (tevopreQ1) in epegRNAs when applying epegRNAs to install therapeutically relevant mutations (see below).
We also examined whether the “flip and extension” (F+E) sgRNA scaffold38 would further improve epegRNA editing efficiency. This guide RNA scaffold mutates the fourth base pair of the direct repeat from U•A to A•U to remove a potential pol III terminator and extends the direct repeat by five base pairs to improve Cas9 binding38. We transduced HEK293T cells with lentiviruses encoding either an unmodified (F+E) pegRNA, an (F+E) epegRNA containing tevopreQ1, or a tevopreQ1 epegRNA with the standard scaffold that templates a transversion at HEK3 or DNMT1, or a 3-nt insertion at HEK3. Use of tevopreQ1 substantially improved editing efficiency (3.8-fold for the nucleotide transversion and 2.6-fold for the 3-nt insertion at HEK3 and 6.8-fold at DNMT1) (Supplementary Fig. 11). Use of the (F+E) scaffold in a tevopreQ1 epegRNA further improved editing efficiency (1.1-fold for the nucleotide transversion, 1.5-fold for the 3-nt insertion at HEK3, and 2.5-fold at DNMT1). We also characterized sgRNA scaffold variants previously shown to increase Cas9-nuclease activity39 under transfection conditions with reduced amounts of plasmid and observed similar overall benefits, albeit with greater variability (Supplementary Discussion, Supplementary Fig. 12). These findings further suggest that epegRNAs mediate greater improvements in PE efficiency when expression is limited. Additionally, these data highlight the potential for modified scaffolds to improve PE efficiency in conjunction with epegRNAs, although a more in-depth exploration of this possibility is needed.
A computational tool to design epegRNA linkers
In contrast with protein linkers, RNA linkers more likely to be sequence-dependent, such that the same linker might function for one epegRNA but impede another. To minimize the possibility of interference from the epegRNA linker, we developed pegLIT (pegRNA Linker Identification Tool) (Supplementary Discussion, Supplementary Fig. 13), a computational tool that identifies linker sequences predicted to minimally base pair with the remainder of the epegRNA. For an initial validation, we tested two sets of 15 evopreQ1 epegRNAs with different linkers templating either a C•G-to-A•T transversion at RNF2 or a 15-bp deletion at DNMT1. Within each set, five linkers were recommended by pegLIT; five were predicted to base pair with the spacer, and five were predicted to base pair with the PBS. The use of pegLIT-designed linkers resulted in a modest increase in PE3 editing efficiency over the use of manually designed linkers (1.2-fold higher for RNF2 and 1.1-fold higher for DNMT1) (Supplementary Fig. 13). While spacer interactions did not significantly impact editing efficiency, linker-PBS interactions correlated with reduced PE3-editing efficiency, resulting in 1.3- and 1.1-fold lower editing efficiency compared to pegLIT linkers for RNF2 and DNMT1 respectively. The two worst-performing linkers, which resulted in 1.9- and 3.4-fold less efficient PE3 editing at RNF2 relative to optimal linker sequences, were correctly identified by pegLIT as scoring poorly for PBS interactions (Supplementary Fig. 13). The closer proximity of the linker to the PBS compared to the spacer may give linker:PBS interactions an entropic advantage compared to linker:spacer pairing. We then sought to determine whether pegLIT-designed linker sequences could improve the efficacy of two epegRNAs (templating a G•C-to-T•A transversion at EMX1 and a 15-bp deletion at VEGFA) which initially failed to exhibit improved editing (Supplementary Fig. 4). Indeed, using pegLIT-designed linkers increased PE3 editing efficiency by 1.3-fold and 1.4-fold, respectively, over that of pegRNAs for these two edits (Supplementary Fig. 13). Collectively, these findings demonstrate that pegLIT facilitates the use of epegRNAs to consistently improve prime editing outcomes.
We also examined whether pegLIT-designed linkers improved the activity of epegRNAs compared to epegRNAs without linkers. Compared to mpknot epegRNAs without a linker, adding a pegLIT-designed linker resulted in a significantly increased editing efficiency than when using manually designed linkers (Supplementary Figs. 3 and 13). In contrast, the use of pegLIT linkers with evopreQ1 or tevopreQ1 epegRNAs did not significantly increase editing relative to epegRNAs without a linker (Supplementary Fig. 13). We therefore recommend the use of pegLIT-designed linkers in epegRNAs using larger structured RNA motifs such as mpknot.
Improved editing efficiency with chemically modified epegRNAs
Chemically synthesized gRNAs are commonly used when transfecting cells with mRNA or RNPs40. Although synthetic gRNAs can incorporate chemical modifications that promote resistance to exonucleolytic-degradation16,40, we speculated that structural motifs might still mediate additional improvements in conjunction with such modifications.
To test this possibility, we compared prime editing efficiencies of synthetic tevopreQ1 epegRNAs with those of synthetic pegRNAs that install either a point mutation or 15-bp deletion at five genomic sites (HEK3, RNF2, DNMT1, RUNX1, and EMX1) in HEK293T cells. Both the epegRNAs and pegRNAs contained 2′-O-methyl modifications and phosphorothioate linkages between the first and last three nucleotides of the RNA. For six of the seven pegRNAs tested, the corresponding epegRNAs exhibited 1.1- to 3.1-fold higher editing with unchanged edit:indel ratios (Supplementary Fig. 14). These data suggest that epegRNAs also enhance PE outcomes compared to pegRNAs in applications that use chemically synthesized and modified pegRNAs.
Engineered pegRNAs improve prime editing of therapeutically relevant mutations
Having validated the use of epegRNAs as a strategy for broadly improving PE activity, we next compared the activity of epegRNAs containing tevopreQ1 with that of pegRNAs to install a variety of protective or therapeutic genetic mutations. We successfully used epegRNAs to install the PRNP G127V allele that protects against human prion disease41,42 in HEK293T cells with 1.4-fold higher efficiency over the canonical pegRNA (Fig. 4a). In addition, we used epegRNAs to correct the most common cause of Tay-Sachs disease (HEXA1278+TATC), both in previously constructed HEXA1278+TATC HEK293T cell lines14 via plasmid lipofection and in primary patient-derived fibroblasts via nucleofection of in vitro transcribed mRNA and synthetic pegRNA (Fig. 4b and c). In both cases, we observed improved editing efficiencies for tevopreQ1 epegRNAs containing pegLIT-designed 8-nt linkers over canonical pegRNAs (2.8-fold higher in HEK293T cells and 2.3-fold higher in patient-derived fibroblasts).
Figure 4. Prime editing-mediated editing efficiency of therapeutically relevant genome editing is improved by the use of epegRNAs.
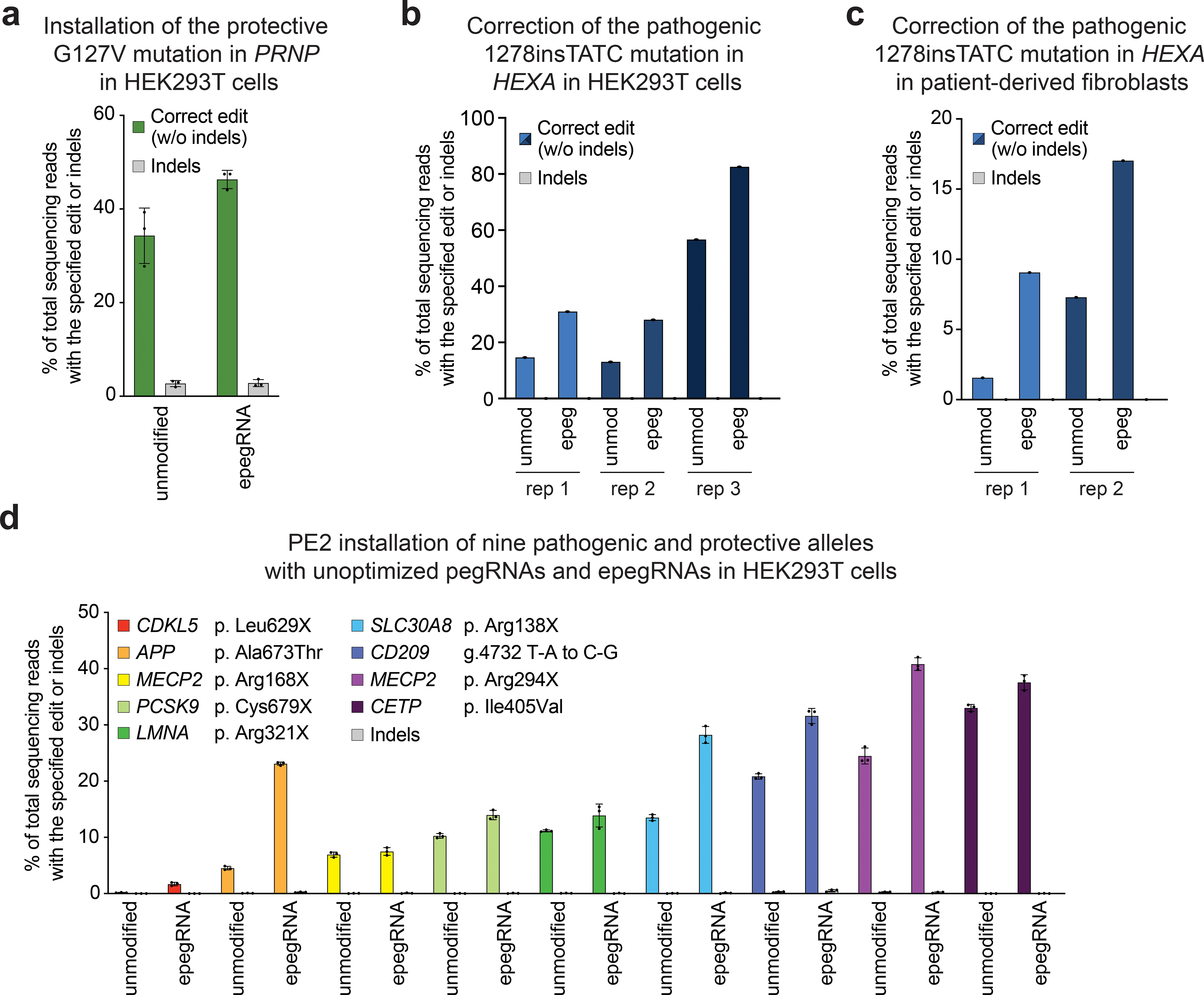
(a) PE3-mediated installation of the G127V mutation in PRNP that protects against human prion disease41,42, and (b) correction of the pathogenic c1278TATC insertion in HEXA that causes Tay Sachs disease in both HEK293T cells and (c) primary patient-derived fibroblasts. (d) Comparison of PE2-mediated installation of pathogenic and protective alleles using unoptimized epegRNAs or unoptimized pegRNAs at nine genomic sites. Reference SNP (rs) designations can be found for all mutations in Supplementary Table 6. Data and error bars in a, d, and e reflect the mean and standard deviation of three independent biological replicates.
Installation of therapeutically relevant edits using unoptimized epegRNAs
The design and screening of many pegRNAs with different PBS and RT templates is an important first step in the successful use of prime editing14. Although general rules to guide PBS and RT template length and composition have been described14,43, identifying optimal pegRNAs often requires extensive screening of pegRNA constructs. We speculated that epegRNAs might support more efficient installation of therapeutically relevant prime edits without extensive pegRNA optimization. We examined the ability of unoptimized pegRNAs and epegRNAs to template the installation of nine protective or pathogenic point mutations using PE2. In all cases, the pegRNAs and epegRNAs used in this experiment contained a 13-nt PBS and an RT template containing 10 nt of homology to the targeted site after the last edited nucleotide, except when the 3′ extension would begin with cytosine14, in which case it was extended to the nearest non-C nucleotide.
We examined pegRNAs that install therapeutically relevant mutations associated with Alzheimer’s disease44, coronary heart disease45,46, type-2 diabetes47, innate immunity48, CDKL5 deficiency disorder49, lamin A deficiency50, and Rett syndrome51,52. We compared the outcomes of prime editing with pegRNAs and corresponding tevopreQ1 epegRNAs with 8-nt pegLIT linkers in HEK293T cells (Fig. 4d). Only a single pegRNA or epegRNA design was tested per target. In every case, epegRNAs outperformed pegRNAs in editing efficiency. For five of the nine therapeutically relevant edits tested, epegRNAs resulted in ≥20% editing efficiency, which is typically sufficient to generate model cell lines. By comparison, only three of the nine pegRNAs achieved this level of editing efficiency. The higher editing efficiencies mediated by epegRNAs (2.8-fold higher than pegRNAs on average) should streamline the production of homozygous cell lines, an important consideration for modeling recessive mutations. Similarly, unoptimized epegRNAs mediated insertion of a 24-bp FLAG tag with ≥10% efficiency at 5 of 15 tested sites; the corresponding pegRNAs did not achieve ≥10% efficiency at any site tested (Supplementary Discussion and Supplementary Fig. 15). Taken together, these findings demonstrate that epegRNAs streamline the production of model cell lines with PE.
Discussion
Here we report the design, characterization, and validation of engineered pegRNAs to address a key bottleneck in prime editing. These epegRNAs contain a structured RNA motif 3′ of the PBS that prevents degradation of the pegRNA extension and the subsequent formation of editing-incompetent PE complexes that compete for access to the targeted genomic site. We found that epegRNAs broadly improve PE efficiency in all five cell lines and primary cell types tested, with larger improvements observed in cell lines that are more difficult to transfect. Additionally, we observed that the use of epegRNAs can enhance prime editing performance when using chemically modified pegRNAs, when installing therapeutically relevant edits in human cells, and when using unoptimized pegRNA designs. Finally, we describe a computational program that expedites epegRNA design by identifying linkers that minimize the risk of counterproductive secondary structure. In total, our findings establish that epegRNAs broadly improve prime editing outcomes at a wide variety of genomic loci, edit types (substitutions, insertions, and deletions), and cell types.
Improvements in prime editing enabled by epegRNAs are likely to depend on delivery strategy. Lower-expression delivery modalities such as some viral vectors might benefit more strongly from the use of epegRNAs when pegRNA concentration is limiting (Supplementary Fig. 12). Similarly, further improvements in the synthesis of chemically modified RNAs might decrease the benefits of epegRNAs by better mitigating degradation. Additionally, the longer length of epegRNAs (an additional 37 nt when using tevopreQ1) is an important consideration when using synthetic epegRNAs given current challenges of chemically synthesizing longer RNAs.
We recommend epegRNAs for all prime editing experiments that can support a modestly longer pegRNA. Importantly, researchers seeking to identify prime editing agents that install a desired edit with the highest possible efficiency should continue to test many epegRNAs that include a variety of PBS and RT template sequences and lengths, and a variety of nicking sgRNAs when using PE3. Incorporating guide RNA scaffold variants38,39 may also further improve PE efficiency on a site-dependent basis (Supplementary Figs. 11 and 12). As demonstrated in this study, however, extensive screening may not be needed when maximizing editing efficiency is not critical. In these cases, an epegRNA containing the trimmed evopreQ1 (tevopreQ1) motif with a PBS length of 13 and a template that includes either 10 nt of homology past the targeted edit for small insertions, deletions, and point mutations—or 25 nt of homology for larger insertions or deletions—provides a promising starting point for epegRNA designs. PBS, RT template length, scaffold sequence, and nicking sgRNA can then be optimized if observed editing efficiencies are insufficient.
Online Methods
General Methods.
Plasmids expressing pegRNAs and epegRNAs were cloned either by Gibson assembly, Golden Gate assembly using either a previously described custom acceptor plasmid14 or newly designed custom acceptor plasmids that contain trimmed evopreQ1 or mpknot (the use of which is described in Supplemental Note 1), or synthesized and cloned by Twist Biosciences. Plasmids expressing sgRNAs were cloned via Gibson or USER assembly. DNA amplification was accomplished by PCR with Phusion U or High Fidelity Phusion Green Hot Start II (New England Biolabs). Plasmids expressing pegRNAs were purified using PureYield plasmid miniprep kits (Promega) when transfecting HEK293T cells or Plasmid Plus Midiprep kits (Qiagen) when transfecting other cell types, while plasmids expressing prime editors were purified exclusively using Plasmid Plus Midiprep kits. Plasmids ordered from Twist Biosciences were resuspended in nuclease-free water and used directly. Primers and dsDNA fragments were ordered from Integrated DNA Technologies (IDT). Uncropped agarose and northern blot gels are provided in Supplementary Figs. 16 and 17.
Synthetic pegRNAs and in vitro transcribed mRNA generation.
Synthetic pegRNAs were ordered from IDT and contained 2′-O-methyl modifications at the first and last three nucleotides and phosphorothioate linkages between the three first and last nucleotides, and were used directly. Synthetic nicking sgRNAs were ordered from Synthego and contained 2′-O-methyl modifications at the three first and last nucleotides and phosphorothioate linkages between the first three and last two nucleotides. PE-encoded mRNA was transcribed in vitro using the protocol described previously53. Briefly, the PE2 cassette—consisting of a 5′ UTR, Kozak sequence, PE2 ORF and 3′ UTR—was cloned into a plasmid containing an inactive T7 (dT7) promoter. The mRNA transcription template was generated via PCR using a primer to install the correct T7 promoter sequence and a reverse primer which installed the poly-A tail. mRNA was generated using a HiScribe T7 High-Yield RNA Kit (New England Biolabs) according to the manufacturer’s instructions, with the exception that N1-methylpseudouridine triphosphate (Trilink) was substituted for uridine triphosphate and CleanCapAG (Trilink) was added to enable co-transcriptional capping. The resulting mRNA was purified via lithium chloride precipitation and reconstituted in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0 at 25 °C). Sequences of pegRNAs and sgRNAs used in this study can be found in Supplementary Table 1. A list of structured RNA motifs examined in this study can be found in Supplementary Table 2.
General mammalian cell culture conditions.
HEK293T (ATCC CRL-3216), U2OS (ATCC HTB-96), K562 (CCL-243), and HeLa (CCL-2) cells were purchased from ATCC and cultured and passaged in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with GlutaMax (Thermo Fisher Scientific), McCoy’s 5A Medium (Gibco), RPMI Medium 1640 plus GlutaMAX (Gibco), or Eagle’s Minimal Essential Medium (EMEM, ATCC), respectively, each supplemented with 10% (v/v) fetal bovine serum (Gibco, qualified). Primary Tay Sachs disease patient fibroblast cells were obtained from the Coriell Institute (Cat. ID GM00221) and grown in low-glucose DMEM (Sigma Aldrich) and 10% (v/v) FBS, supplemented with an additional 2 mM L-glutamine (Thermo Fisher Scientific). All cell types were incubated, maintained and cultured at 37 °C with 5% CO2. Each cell line was authenticated by their respective supplier and tested negative for mycoplasma.
Tissue culture transfection and nucleofection protocols and genomic DNA preparation.
For transfections, 10,000 HEK293T cells were seeded per well on 96-well plates (Corning). 16–24 hours post-seeding, cells were transfected at approximately 60% confluency with 0.5 μL of Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocols and 200 ng of PE plasmid, 40 ng of pegRNA plasmid, and 13 ng of sgRNA plasmid (for PE3). When transfecting reduced amounts of editor-encoded plasmids, 0.5 μL Lipofectamine 2000 was used to transfect 20 ng of PE plasmid, 4 ng of pegRNA plasmid, 1.3 ng of sgRNA plasmid (for PE3), and 228 ng pUC19.
For nucleofections, HEK293T cells were electroporated with in vitro transcribed mRNA and synthetic pegRNA using a Lonza 4D Nucleofector with an SF cell line kit (Lonza). 200,000 cells per electroporation were centrifuged for 8 min at 120 x g, then washed in 1 mL PBS (Thermo Fisher Scientific). After a second centrifugation, cells were resuspended in 5 μL reconstituted SF buffer per sample and added to microcuvettes.
For each cuvette, 17 μL of cargo mix (1 μg of PE2 mRNA in 0.5 μL, 90 pmol of pegRNA in 0.9 μL, and 60 pmol of nicking sgRNA in 0.6 μL, and 15 μL of reconstituted SF buffer) was added and pipetted up and down three times to mix. Cells were electroporated using program CM-130, then 80 μL of warm media was added and cells were incubated for 10 min at room temperature. The mixture was then pipetted to mix and 25 μL was added to the well of a 48-well plate, with a final culture volume of 250 μL per well. For experiments in HeLa, U2OS, and K562 cells, 800 ng PE2-expressing plasmid, 200 ng pegRNA-expressing plasmid, and 83 ng nicking sgRNA-expressing plasmid were nucleofected in a final volume of 20 μL in a 16-well nucleovette strip (Lonza). HeLa cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 2 × 105 cells per sample (program CN-114), according to the manufacturer’s protocol. U2OS cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 2 × 105 cells per sample (program DN-100), according to the manufacturer’s protocol. K562 cells were nucleofected using the SE Cell Line 4D-Nucleofector X Kit (Lonza) with 2 × 105 cells per sample (program FF-120), according to the manufacturer’s protocol.
Patient-derived fibroblasts were electroporated with mRNA-encoding PE2 and synthetic pegRNA and nicking sgRNA as described above for HEK293T cells using an SE cell line kit and 100,000 cells which were centrifuged at 100 x g for 10 min. Additionally, 40 μL of recovered cells were added to a 48 well plate instead of 25. In all cases, cells were cultured 3 days following transfection, after which the media was removed, and cells were washed with PBS (pH 7.4 at 23 °C) and subsequently lysed by the addition of 50 μL for 96-well plates or 150 μL for 48-well plates of freshly prepared lysis buffer (10 mM Tris-HCl, pH 8 at 23 °C; 0.05% SDS; 25 μg mL−1 Proteinase K (Qiagen)), and incubating at 37 °C for 1 hour or more, after which Proteinase K was inactivated over 30 minutes at 80 °C. The resulting gDNA was stored at −20 °C until used.
Lentivirus preparation and transduction.
Lentiviral transfer plasmids were designed to contain a pegRNA or epegRNA under expression from a human U6 promoter and a PuroR–T2A–BFP marker under expression from the EF1α core promoter. To package lentivirus, HEK293T cells were seeded on 6-well plates (Corning) at 7 × 105 cells per well in DMEM supplemented with 10% FBS. At 60% confluency 16 hr after seeding, cells were transfected with 12 μL Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol and 1.33 μg lentiviral transfer plasmid, 0.67 μg pMD2.G (Addgene #12259), and 1 μg psPAX2 (Addgene #12260). 6 hr after transfection, media was exchanged with DMEM supplemented with 10% FBS. 48 hr after transfection, viral supernatant was centrifuged at 3000 g for 15 min to remove cellular debris, filtered through a 0.45 μm PVDF filter (Corning), and stored at −80 °C.
To transduce cells with pegRNAs or epegRNAs, 2 × 106 HEK293T cells were infected with 20 μL lentivirus in DMEM supplemented with 10% FBS and 8 μg/mL polybrene (Sigma-Aldrich), and centrifuged at 1,000 × g for 2 hr at 33 °C. 24 hr following transduction, cells were passaged into DMEM supplemented with 10% FBS and 2 μg/mL puromycin (Thermo Fisher Scientific) to begin selection. BFP fluorescence was monitored using a CytoFLEX S Flow Cytometer (Beckman Couolter) to ensure a multiplicity of infection of 0.2. After 4 days of puromycin selection, transduced HEK293T cells were seeded on 96-well plates (Corning) at 1.6 × 104 cells per well in DMEM supplemented with 10% FBS. 20 hr after seeding, cells were transfected at 60–80% confluency with 200 ng pCMV–PE2 plasmid and 0.5 μL Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. To extract genomic DNA 120 hr following transfection, cells were washed with PBS (pH 7.4 at 23 °C) and lysed in 10 mM Tris-HCl, pH 8.0 at 23 °C; 0.05% SDS; 800 units/μL proteinase K (New England BioLabs) at 37 °C for 1.5 h, followed by enzyme inactivation at 80 °C for 30 min.
High-throughput DNA sequencing of genomic DNA samples.
Genomic sites of interest were amplified from genomic DNA samples and sequenced on an Illumina MiSeq as previously described14. Cas9 off-target sites for HEK3, EMX1, and FANCF were previously identified via Guide-Seq28. Primers used for mammalian cell genomic DNA amplification are listed in Supplementary Table 3 and amplicons are listed in Supplementary Table 4. Sequencing reads were demultiplexed using MiSeq Reporter (Illumina). Alignment of amplicon sequences to a reference sequence was performed using CRISPResso2 (ref. 54). For all prime editing yield quantifications, editing efficiency was calculated as the percentage of reads with the desired editing without indels out of the total number of reads with an average phred score of at least thirty. For quantification of point mutation editing, CRISPResso2 was run in standard mode with “discard_indel_reads” on. Editing yield was calculated as the percentage of non-discarded reads containing the edit divided by total reads. For insertion or deletion edits, CRISPResso2 was run in HDR mode using the desired allele as the expected allele, and with “discard_indel_reads” on. Editing yield was calculated as the percentage of HDR aligned reads divided by total reads. For all experiments, indel frequency was calculated as the number of discarded reads divided by the total number of reads. For experiments involving PE2, reads were analyzed for indels within 10 nucleotides up- and downstream of the pegRNA nick site, inclusive. For experiments involving PE3, reads were analyzed for indels between 10 nucleotides upstream of the pegRNA nick site and downstream from the sgRNA nick site, inclusive. Off-target editing was quantified as described previously14.
RTqPCR of total RNA.
10,000 HEK293T cells per well were seeded in 96-well plates. 16–24 hours post-seeding, cells were transfected at approximately 60% confluency with 0.5 μL of Lipofectamine 2000, 200 ng of PE2 plasmid and 40 ng of either pegRNA or epegRNA plasmid according to the manufacturer’s protocols. After three days, total RNA was isolated using the AllPrep DNA/RNA/miRNA universal kit (Qiagen). The Power SYBR Green Cells-to-CT kit (Thermo Fisher Scientific) was used to generate cDNA using random hexamers and to perform qPCR with forward and reverse primers that amplify the pegRNA scaffold according to the manufacturer’s protocols. Primer sequences are available in Supplemental Table 5.
In vitro exonuclease susceptibility assays.
pegRNAs or epegRNAs containing either mpknot or evopreQ1 were prepared using the HiScribe T7 Quick High Yield RNA synthesis kit (New England BioLabs) from PCR-amplified templates containing a T7 promoter sequence per the manufacturer’s protocols followed by purification via the Monarch RNA Cleanup kit (New England BioLabs). Nuclear extracts were prepared from 3 million HEK293T cells grown to 70–80% confluency per the manufacturer’s protocols using the EpiQuik Nuclear Extraction kit (EpiGentek). Assays were carried out in 10 μL reactions containing 20 mM Tris-HCl (pH 7.5 at 23 °C), 5 mM MgCl2, 50 mM NaCl, 2 mM DTT, 1 mM NTP and 0.8 U/μL RNaseOUT Recombinant Ribonuclease Inhibitor (40 U/μL; ThermoFisher Scientific) to inhibit endonuclease activity. 3 μL of fresh nuclear lysate was used to degrade 0.5 μg of RNA substrate per reaction. Followed by the incubation of reaction mixtures at 37 °C for 20 min, degradation products were resolved on 2.0% agarose gels stained with SYBR Gold. The extent of degradation was determined using ImageJ software (NIH). To determine whether Cas9 could protect the sgRNA scaffold from degradation by exonucleases, 1 nM of pegRNA or epegRNA was incubated in the presence or absence of 100 nM nCas9 at room temperature for 10 min to enable the binding of nCas9-H840A to pegRNA. Degradation assays were carried out in 10-μl reaction mixtures containing 20 mM Tris-HCl (pH 7.5 at 23 °C), 5 mM MgCl2, 50 mM NaCl, 2 mM DTT, 1 mM dNTP, and and 0.8 U/μL RNaseOUT Recombinant Ribonuclease Inhibitor. 3 μL of fresh nuclear lysate was used to degrade Cas9 nickase-bound pegRNA or epegRNA. Followed by the incubation of reaction mixtures at 37 °C for 10 min, 1 μL of Protease K solution (Qiagen) was added to the reaction mixture and incubated at room temperature for 10 min to inactivate the nucleases. Total remaining RNA was isolated using the Monarch RNA Cleanup Kit (New England Biolabs) for analysis by RT-qPCR.
Detection of pegRNAs and epegRNAs in cellular lysates via northern blot.
HEK293T cells were transfected with plasmids encoding PE2 and pegRNA or epegRNA as described above. Cells were lysed after 3 days and total RNA isolated using the AllPrep DNA/RNA/miRNA universal kit (Qiagen) following the manufacturer’s instructions. The amount of PE2 mRNA was determined using RT-qPCR and this value was used to normalize lysates to the same concentration of PE2 mRNA. Lysates were separated by PAGE using a 10% denaturing PAGE gel (Criterion, Biorad). An ssRNA ladder was 3’ labeled with digoxigenin-ddUTP using terminal transferase (DIG Oligonucleotide 3’-End Labeling Kit, Roche) and used as a marker. Other markers used were in vitro transcribed pegRNA and epegRNA templating at +1 FLAG tag insertion at HEK3 and HEK293T cellular lysates containing HEK3-targeted sgRNA.Transfer and crosslinking of RNAs to the northern blot membrane largely followed previously described procedures for detection of small RNAs55. RNAs were transferred to a positively charged nylon membrane (Roche) using a Trans-Blot SD semi-dry gel at 20V for 1 hr (1–3 mA/cm2). RNAs were then crosslinked to the membrane by soaking in an aqueous solution of 0.162 M EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, Sigma Aldrich) and 0.17 M 1-methylimidazole (Sigma Aldrich, pH 8.0 at 23 °C) at 60 °C for 1 hr. Blots were briefly rinsed several times in DEPC-treated water and then pre-hybridized in Ultrahyb hybridization buffer (northern Max kit, Thermo Fisher) at 68 °C for 2 hours. An RNA probe complementary to 64 nt of the sgRNA scaffold (Supplementary Table 5) was generated and body-labeled with digoxigenin-UTP via in vitro transcription with T7 and using the DIG northern starter kit (Roche). 5 pmol of labeled probe was added to 0.5 mL of Ultrahyb buffer and incubated for 5 min at 70 °C before being added to the pre-hybridized blot at 68 °C. Hybridization was allowed to proceed overnight. Blots were then washed twice in Low Stringency wash solution (northernMax kit, equivalent to 2x SSC and 0.1% SDS) for 5 min at room temperature and twice in High Stringency wash solution (northernMax kit, equivalent to 0.1x SSC and 0.1% SDS) for 15 min at 68 °C. Blots were then rinsed in washing buffer (0.1 M maleic acid, 0.15 M NaCl, 0.3% (v/v) Tween 20, pH 7.5 at 23 °C) for 5 min at room temperature and then incubated in blocking buffer (DIG northern starter kit) for 30 min at room temperature. Blots were then incubated in blocking buffer supplemented with anti-digoxigenin-AP antibody (DIG northern Starter Kit) for 30 min at room temperature and then washed with washing buffer for 15 min at room temperature twice. Blots were then equilibrated in detection buffer (0.1 M Tris-HCl, 0.1 M NaCl, pH 9.5 23 °C) for 5 min before being removed to a development folder. CDP-Star was then added dropwise to the blot, and the blot was covered and incubated at room temperature for 5 min to overnight before being imaged with a Biorad ChemiDoc MP. Levels of pegRNA and epegRNA were determined by densitometry using ImageJ.
epegRNA binding assays.
Microscale Thermophoresis (MST) analysis was conducted using a Monolith NT.Automated (Nanotemper) with premium-coated capillaries to determine Cas9 binding affinities for pegRNAs and epegRNAs or for dsDNA when complexed with pegRNAs or epegRNAs. Binding reactions were conducted at 25 °C in 20 μL of HBS-P buffer (10 mM HEPES pH 7.4 at 23 °C, 150 mM NaCl, 0.05% v/v Surfactant P20) containing 1 mM MgCl2. Mg2+ concentration was chosen to mimic the estimated free Mg2+ concentration in human cells56. For RNA binding experiments, RNAs were in vitro transcribed as described previously and 3′-labeled with pCp-Cy5 (Jena Bioscience) using T4 RNA ligase 1 (New England BioLabs) per the manufacturer’s protocols, modified to include 10 μM pCp-Cy5 and followed by purification with the Monarch RNA Cleanup kit (New England BioLabs). 1 nM Cy5-labeled RNA was denatured for 3 minutes at 72 °C, rested on ice for 1 minute, and then incubated with SpCas9-H840A (Integrated DNA Technologies) for 30 minutes at 25 °C prior to MST analysis. For dsDNA binding experiments, the dsDNA substrate was assembled by slow annealing of a Cy5-labeled reverse oligo and an unlabeled forward oligo corresponding to the HEK3 genomic locus (Supplementary Table 5). Cas9 RNP was formed by incubating dSpCas9 (Integrated DNA Technologies) for 30 minutes at 25 °C in HBS-P buffer with 1 mM MgCl2 and 50-fold molar excess of unlabeled pegRNA or epegRNA to maintain saturated RNA binding conditions. Cas9 RNP was then incubated with 1 nM Cy5-labeled dsDNA substrate for 30 minutes at 25 °C prior to MST analysis. Cy5 exhibits substantial temperature related intensity change (TRIC), which allows for a sufficient amplification of signal to detect protein/pegRNA or dsDNA/RNP interactions. Generally, the laser excitation energy was set to 20 % and the IR laser power was set to high for all readings. All measurements were performed in triplicate, with the dilution series for each replicate made separately, using serial dilutions of either dSpCas9 or SpCas9-H840A from 100 nM to 0 nM. Data were analyzed in Prism 9 by performing logistic regression on the log([protein]) and signal change, with the 0 nM concentration datapoint set to 0.1 nM for regression purposes.
Cas9-based transcriptional activation.
10,000 HEK293T cells per well were seeded in 96-well black-wall plates (Corning). 16–24 hours post-seeding, cells were transfected at approximately 60% confluency with .5 μL of Lipofectamine 2000 according to the manufacturer’s protocols and 100 ng of dCas9–VPR plasmid, 30 ng of GFP reporter plasmid, 15 ng of iRFP plasmid, and 20 ng of sgRNA, pegRNA, or epegRNA plasmid. After three days, cells were measured for GFP and iRFP fluorescence using an Infinite M1000 Pro microplate reader (Tecan). GFP fluorescence was normalized to iRFP fluorescence after subtracting background fluorescence signal from untreated cells.
Terminal deoxynucleotidyl transferase assay.
10,000 HEK293T cells per well were seeded in 96-well plates. 16–24 hours post-seeding, cells were transfected at approximately 60% confluency with 0.5 μL of Lipofectamine 2000 according to the manufacturer’s protocols and 200 ng of PE2 plasmid and 40 ng of either pegRNA or epegRNA plasmid. After 24 hr, genomic DNA was isolated from the cells using the Agencourt DNAdvance kit (Beckman Coulter) according to the manufacturer’s instructions. 3′ termini were tailed with guanosine using terminal deoxynucleotidyl transferase (New England Biolabs) according to the manufacturer’s instructions. The samples were then purified again using the Agencourt DNAdvance kit prior to PCR amplification for high-throughput DNA sequencing using a locus-specific forward primer and an oligo-C (C18) reverse primer. Primer sequences are available in Supplemental Table 5. Prime editing intermediates were quantified using a custom python script available in Supplementary Note 3.
Linker design via pegLIT.
To design epegRNA linker sequences, we wrote a custom algorithm, pegRNA Linker Identification Tool, or pegLIT, that searches for linker sequences of a specified length that minimize base pairing with the remainder of the pegRNA. To reduce computing time, this procedure uses simulated annealing to maximize subscores57, each of which corresponds to a subsequence of the pegRNA: spacer, PBS, template, or scaffold. Accordingly, different linker sequences may be generated for the same epegRNA if the algorithm is run multiple times. During optimization, the higher-scoring linker in any pair of linkers was determined by comparing their discretized subscores in order of the following subsequence priority: spacer, PBS, template, and then scaffold. Each subscore is calculated, using base pair probabilities calculated by ViennaRNA 2.0 (ref. 23) under standard parameters (37 °C, 1 M NaCl, 0.05 M MgCl2), as the complement of the mean probability that a nucleotide in the linker forms a base pair with any nucleotide in the pegRNA subsequence under consideration, where the mean is taken over all bases in the linker. Linker sequences with AC content < 50% and those that would result in a pegRNA containing four of the same nucleotide consecutively are removed from consideration39,58. Optionally, the algorithm performs hierarchical agglomerative clustering on the 100 highest-scoring linkers and outputs one linker per cluster in order to promote sequence diversity in the final output. Our Python implementation of pegLIT is publicly accessible at liugroup.us and the code can be found in Supplementary Note 2 or at github.com/sshen8/peglit.
Data availability
High-throughput sequencing data have been deposited to the NCBI Sequence Read Archive database at PRJNA707486. Plasmids encoding select pegRNA expression vectors and golden-gate cloning vectors have been deposited to Addgene for distribution.
Code availability
A Python implementation of pegLIT is publicly accessible at peglit.liugroup.us and the code can be found in Supplementary Note 2 or at github.com/sshen8/peglit.
Supplementary Material
Acknowledgments
This work was supported by U.S. NIH U01Al142756, RM1HG009490, R01EB031172, and R35GM118062, the Howard Hughes Medical Institute, and the Loulou Foundation. J.W.N. and A.V.A. were supported by Jane Coffin Childs postdoctoral fellowships, P.B.R., S.P.S, K.A.E., and P.J.C. were supported by NSF graduate fellowships, G.A.N. was supported by a Helen Hay Whitney postdoctoral fellowship. We thank Dr. Anahita Vieira for assistance editing this manuscript, Mary O’Reilly, Elena Berg and the Broad Institute Pattern team for help with figure design, Sean McGreary and Kehui Xiang for helpful discussions on northern blot procedures, and Max Shen for helpful discussions on pegLIT coding.
Footnotes
Competing Interests
The authors are co-inventors on patents filed by the Broad Institute on prime editing. D.R.L. is a consultant and co-founder of Prime Medicine, Beam Therapeutics, and Pairwise Plants, companies that use genome editing. A.V.A. is currently an employee of Prime Medicine.
Supplementary Information Supplementary Discussion, Supplementary Figures 1–16, Supplementary Tables 1–6, Supplementary Notes 1–3
References
- 1.Komor AC, Badran AH & Liu DR CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anzalone AV, Koblan LW & Liu DR Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Cullot G et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun 10, 1136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosicki M, Tomberg K & Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boroviak K, Fu B, Yang F, Doe B & Bradley A Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci Rep 7, 12867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enache OM et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet 52, 662–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24, 927–930 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24, 939–946 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Leibowitz ML et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet 10.1038/s41588-021-00838-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgio G & Teboul L Anticipating and Identifying Collateral Damage in Genome Editing. Trends Genet 36, 905–914 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox DB, Platt RJ & Zhang F Therapeutic genome editing: prospects and challenges. Nat Med 21, 121–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houseley J & Tollervey D The many pathways of RNA degradation. Cell 136, 763–776 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Hendel A et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat Biotechnol 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geisberg JV, Moqtaderi Z, Fan X, Ozsolak F & Struhl K Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell 156, 812–824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X & Bartel DP Widespread Influence of 3′-End Structures on Mammalian mRNA Processing and Stability. Cell 169, 905–917 e911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown JA et al. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat Struct Mol Biol 21, 633–640 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth A et al. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat Struct Mol Biol 14, 308–317 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Anzalone AV, Lin AJ, Zairis S, Rabadan R & Cornish VW Reprogramming eukaryotic translation with ligand-responsive synthetic RNA switches. Nat Methods 13, 453–458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houck-Loomis B et al. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature 480, 561–564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorenz R et al. ViennaRNA package 2.0. Algorithms Mol Biol 6, 26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schene IF et al. Prime editing for functional repair in patient-derived disease models. Nat Commun 11, 5352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim DY, Moon SB, Ko JH, Kim YS & Kim D Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res 48, 10576–10589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao P et al. Prime editing in mice reveals the essentiality of a single base in driving tissue specific gene expression. Genome Biol 22, 83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin Q, et al. High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat Biotechnol (2021). 10.1038/s41587-021-00868-w. [DOI] [PubMed] [Google Scholar]
- 28.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid M & Jensen TH The exosome: a multipurpose RNA-decay machine. Trends Biochem Sci 33, 501–510 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Ibrahim H, Wilusz J & Wilusz CJ RNA recognition by 3′-to-5′ exonucleases: the substrate perspective. Biochim biophys acta 1779, 256–265 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green L, Kim CH, Bustamante C & Tinoco I Jr. Characterization of the mechanical unfolding of RNA pseudoknots. J Mol Biol 375, 511–528 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chavez A et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12, 326–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nahar S et al. A G-quadruplex motif at the 3′ end of sgRNAs improves CRISPR-Cas9 based genome editing efficiency. Chem Commun 54, 2377–2380 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Steckelberg AL et al. A folded viral noncoding RNA blocks host cell exoribonucleases through a conformationally dynamic RNA structure. Proc Natl Acad Sci USA 115, 6404–6409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cate JH et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science 273, 1678–1685 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Fedor MJ & Westhof E Ribozymes: the first 20 years. Mol Cell 10, 703–704 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Chen B et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jost M et al. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat Biotechnol 38, 355–364 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basila M, Kelley ML & Smith AVB Minimal 2′-O-methyl phosphorothioate linkage modification pattern of synthetic guide RNAs for increased stability and efficient CRISPR-Cas9 gene editing avoiding cellular toxicity. PLoS One 12, e0188593 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mead S et al. A novel protective prion protein variant that colocalizes with Kuru exposure. N Engl J Med 361, 2056–2065 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Asante EA et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522, 478–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HK et al. Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol (2020). [DOI] [PubMed] [Google Scholar]
- 44.Jonsson T et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Abifadel M et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 34, 154–156 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Bustami J et al. Cholesteryl ester transfer protein (CETP) I405V polymorphism and cardiovascular disease in eastern European Caucasians - a cross-sectional study. BMC Geriatr 16, 144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flannick J et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet 46, 357–363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakuntabhai A et al. A variant in the CD209 promoter is associated with severity of dengue disease. Nat Genet 37, 507–513 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olson HE et al. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr Neurol 97, 18–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al-Saaidi R et al. The LMNA mutation p.Arg321Ter associated with dilated cardiomyopathy leads to reduced expression and a skewed ratio of lamin A and lamin C proteins. Exp Cell Res 319, 3010–3019 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Ip JPK, Mellios N & Sur M Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 19, 368–382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christodoulou J, Grimm A, Maher T & Bennetts B RettBASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat 21, 466–472 (2003). [DOI] [PubMed] [Google Scholar]
Method References
- 53.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol 38, 892–900 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Clement K et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang W & Bartel DP The menu of features that define primary microRNAs and enable de novo design of microRNA genes. Mol Cell 60, 131–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romani AMP Cellular magnesium homeostasis. Arch Biochem Biophys 512, 1–23 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bertsimas D & Tsitsiklis J Simulated Annealing. Stat Sci 8, 10–15 (1993). [Google Scholar]
- 58.Fedor MJ & Westhof E Ribozymes: the first 20 years. Mol Cell 10, 703–704 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data have been deposited to the NCBI Sequence Read Archive database at PRJNA707486. Plasmids encoding select pegRNA expression vectors and golden-gate cloning vectors have been deposited to Addgene for distribution.