

SUMMARY
Human pluripotent stem cells (hPSCs) can self-renew indefinitely or induced to differentiate. We previously showed that exogenous glutamine (Gln) withdrawal biased hPSC differentiation toward ectoderm and away from mesoderm. We uncovered that while all three germ lineages are capable of de novo Gln synthesis, only ectoderm generates sufficient Gln to sustain cell viability and differentiation, clarifying lineage fate restrictions under Gln withdrawal. Furthermore, Gln acts as a signaling molecule for ectoderm that supersedes lineage-specifying cytokine induction. In contrast, Gln in mesoderm and endoderm is the preferred precursor of α-ketoglutarate without a direct signaling role. Our work raises a question about whether the nutrient environment functions directly in cell differentiation during development. Interestingly, transcriptome analysis of a gastrulation stage human embryo shows unique Gln enzyme-encoding gene expression patterns may also distinguish germ lineages in vivo. Together, our study suggests that intracellular Gln may help coordinate differentiation of the three germ layers.
Keywords: pluripotent stem cell, cell fate, glutamine, nutrient, development, auxotroph, prototroph
Graphical Abstract
eTOC Blurb
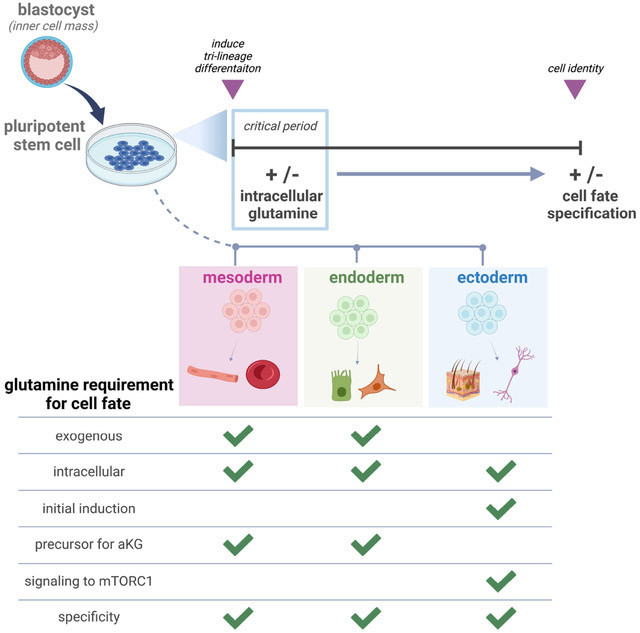
Lu et al. report that cell type specific Gln requirements distinguish the three embryonic germ lineages upon pluripotency exit. They further uncover Gln acts as a signaling molecule to activate mTORC1 and enable downstream ectoderm, but not mesoderm nor endoderm, differentiation.
INTRODUCTION
Human pluripotent stem cells (hPSCs) self-renew indefinitely or can be induced to differentiate into the three embryonic germ lineages, mesoderm, definitive endoderm (endoderm), and ectoderm (Takahashi et al., 2007; Thomson et al., 1998). Multiple studies show that specific nutrients can supply specific metabolic pathways to generate enzyme cofactors or substrates that enable or amplify PSC self-renewal or differentiation signals (Carey et al., 2015; Chantranupong et al., 2015; Moussaieff et al., 2015; Reid et al., 2017; Shiraki et al., 2014; TeSlaa et al., 2016; Vozza et al., 2014; Wellen et al., 2010). We recently reported that mesoderm, endoderm, and ectoderm all consume Gln supplied in cell culture media, yet exogenous Gln deprivation suppressed mesoderm and endoderm lineages and promoted the ectoderm lineage upon spontaneous non-directed differentiation without affecting cell growth or proliferation. This result indicated that the lack of environmental Gln skewed cell fate specification (Lu et al., 2019), with a mechanism for Gln control of stem cell plasticity and lineage differentiation potential unknown.
Multiple case studies have shown the importance of Gln availability in fetal development. For example, inborn glutamine synthetase (GS) enzyme deficiency resulted in severe abnormalities in ectoderm-derived brain development and infant death in two unrelated newborns (Haberle et al., 2005; Haberle et al., 2006). Although the role of Gln as a metabolic precursor and anaplerotic fuel is well known, whether Gln itself has a direct role in stem cell fate determination remains unknown. A recent study reported non-metabolized glucose controlled lineage specification of totipotent stem cells in cleavage stage mouse embryos. Glucose as a nutrient, independent of glycolysis, initiated signaling and transcription to induce trophectoderm, which formed the extraembryonic structures of the placenta but not the embryonic inner cell mass (Chi et al., 2020). Accordingly, our finding that Gln withdrawal induced one cell lineage over others led to a more general question of whether nutrients, without conversion to their metabolic derivatives, can regulate embryonic cell fates.
Here, we report that Gln functions as a signaling molecule that enables the acquisition of ectoderm fate. The requirement for a Gln signal in ectoderm induction supersedes differentiation cues from lineage-inducing cytokines present in the media environment. In contrast, Gln has no direct signaling role and is mainly converted to α-KG to drive mesoderm and endoderm specification. Gln also cannot be substituted by another nutrient, carbon source, or amino acid in tri-lineage differentiation, reinforcing its specificity in germ layer development. Additionally, we uncover that gene expression signatures of Gln utilization are differentially regulated in each germ lineage in vivo. Transcriptome signature analysis of a rare gastrulation stage human embryo resembles enzyme-encoding gene expression patterns found in our in vitro studies, generating the hypothesis that distinct Gln utilization may characterize germ lineages in vivo. Thus, our study suggests that nutrient signaling, particularly Gln signaling, may coordinate and facilitate germ layer fate determination.
RESULTS
Germ lineages have different Gln dependencies
We investigated Gln processing in hPSCs and the three embryonic germ lineages. hPSCs were directed to differentiate into mesoderm, endoderm, and ectoderm cells using nutrient-equivalent base media and lineage-inducing cytokines (Fig. 1A). We previously confirmed that this differentiation method generates homogenous, lineage-specific cell populations by RNA-Seq and transcription factor biomarker expression analyses (Lu et al., 2019), and also cross-referenced transcript profiles with an independent study to verify the reproducibility of this chemically defined differentiation system (Cliff et al., 2017).
Figure 1. The germ lineages have distinct dependencies on exogenous Gln and catabolism.

(A) hPSC differentiation into three embryonic germ lineages mesoderm, endoderm, and ectoderm in nutrient-equivalent culture media.
(B) Schematic of Glc and Gln entry into the TCA cycle. Gln catabolism and anabolism pathways highlighted in grey box. Dichloroacetate (DCA) increases pyruvate flux into the TCA cycle.
(C) Heatmap of differentially expressed glutaminolysis enzyme transcripts. Values represent the Z-score of variance stabilizing transformation (VST) normalized RNA-Seq counts across each gene listed (n=6). Genes shown are significantly altered between pairwise combinations of hPSC, mesoderm, endoderm, and mesoderm (p < 0.05).
(D) qRT-PCR analysis of GLS1 and GLS2 isoenzymes in mesoderm, endoderm, and ectoderm throughout differentiation.
(E) Cell viability of D5 mesoderm, D3 endoderm, and D5 ectoderm cells grown in Gln-free (−Gln) or Gln-supplemented with added 1 μM CB-839 glutaminase inhibitor (+GLS1i) culture media relative to Gln-supplemented (Ctr).
(F-G) Cell viability and differentiation quantifications of H9-derived (F) endoderm (SOX17+) and (G) mesoderm (CD34+, SNAI2/SLUG+) cells in Gln-free and metabolic rescue conditions.
(H) Metabolite set variation analysis of ectoderm cells in Gln-free (−Gln) or added CB-389 (+GLS1i) conditions. Scores represent pathway enrichment difference between the given treatment to baseline vehicle (Ctr) (n = 3). Pathways displayed are significantly enriched either with −Gln or +GLSi treatment relative to Ctr (p < 0.05).
Data represent mean ± SD of n ≥ 3 biological replicates. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. The p values were determined by (C) Wald test with Benjamini-Hochberg FDR correction, (D) two-way ANOVA, (E-G) one-way ANOVA with correction for multiple comparisons, or (H) moderated t-statistics determined using empirical Bayes linear modelling.
To begin, we examined how each lineage handles Gln from the culture media. Two genes, GLS (herein GLS1) and GLS2, encode the first enzyme in Gln metabolism, glutaminase (GLS) and its isoenzymes, GLS1 (low Km) and GLS2 (high Km) (Fig. 1B). GLS amidohydrolase activity converts Gln into ammonia and glutamate (Glu), which can be processed into α-KG for tricarboxylic acid (TCA) cycle anaplerosis or other uses (Katt et al., 2017). We used RNA-Seq to identify differential expression of glutaminolysis-related and Gln metabolism genes between hPSCs and lineage-induced progeny cells (Fig. 1C). hPSC differentiation repressed GLS2 expression in all lineages, whereas induction of GLS1 occurred in mesoderm and endoderm compared to slight repression in ectoderm during differentiation (Figs. 1C, 1D). This lineage-specific isoenzyme switching suggested that increased GLS1 expression in mesoderm and endoderm could favor exogenous Gln processing, whereas ectoderm may depend less upon Gln from the external environment, consistent with our prior results (Lu et al., 2019).
To study whether ectoderm depends on external Gln, hPSCs were induced to differentiate in Gln-supplemented culture media (Ctr), Gln-free media (−Gln, except for trace/negligible amounts in Matrigel), or Gln-supplemented culture media with added GLS1 inhibitor, CB-839 (+GLS1i). Mesoderm perished in Gln-free and glutaminolysis-inhibited conditions (Figs. 1E, S1A, S1B) and endoderm had a similar but less drastic response to Gln perturbations (Figs. 1E, S1A). In contrast, ectoderm viability was unaffected by Gln-free and glutaminolysis-inhibited conditions (Figs. 1E, S1A, S1B). Our prior 13C-isotopologue tracing studies showed that Gln supplied the TCA cycle, with significant Gln-derived carbons and negligible glucose-derived carbons incorporated into α-KG in all three germ lineages (Lu et al., 2019). To clarify whether mesoderm and endoderm cell death is from a dependence on Gln fueling the TCA cycle, the culture media was supplemented with cell permeable dimethyl α-KG (dm-α-KG) or pyruvate. Addition of dm-α-KG or pyruvate, but not dichloroacetate (DCA; Fig. 1B) alone, to Gln-free media fully rescued endoderm viability (Figs. 1F, S1C). To quantify differentiation efficacy, differentiated cells were gated by lineage-defining transcription factor positive versus negative populations, which was set by hPSC expression levels. Only dm-α-KG addition to Gln-free conditions resulted in recovery of endoderm differentiation, as measured by the population of SOX17+ cells, whereas added pyruvate restored viability but not endoderm differentiation (Fig. 1F). Similarly, only dm-α-KG, but not pyruvate, supplementation in Gln-free media completely restored both viability and biomarker validated mesoderm differentiation, as measured by populations of CD34+ and SNAI2/SLUG+ cells (Figs. 1G, S1C). These data indicate that the Gln carbon backbone, retained through glutaminolysis conversion to Glu and then to α-KG, is necessary for mesoderm and endoderm viability and differentiation. This substrate preference indicates that endoderm and mesoderm, but not ectoderm, favor α-KG derivation from Gln specifically since glucose/pyruvate-derived carbons cannot substitute for Gln-derived carbons, at least as an alternative TCA cycle fuel or for other metabolic processes.
To study the observed exogenous Gln independence of ectoderm viability, we examined steady-state metabolite patterns in Gln-free and glutaminolysis-inhibited conditions using ultra-high performance liquid chromatography-mass spectrometry (UHPLC-MS) and quantified a panel of 153 central carbon metabolites. Metabolite set variation analysis (MSVA) showed that TCA cycle, amino acid, and pyruvate metabolic pathway activities were elevated in Gln-free compared to Gln-supplemented media (Figs. 1H, S1D), possibly through re-routing of glucose-derived carbons to maintain TCA metabolite levels. In contrast, glutaminolysis inhibition did not show significant metabolic pathway changes compared to no treatment (Figs. 1H, S1E), except for increased purine and pyrimidine metabolism. This suggested that Gln blocked from GLS1 enzymatic conversion to Glu is shunted toward enhanced nucleotide biosynthesis, possibly supporting proliferation (Figs. 1H, S1E). Notably, the Gln-free and glutaminolysis-inhibited metabolomes are distinct; a Gln-free environment results in decreased nucleotide biosynthesis and increased TCA cycle metabolites compared to GLS1 inhibition (Figs. 1H, S1F). Together, these findings indicate that exogenous Gln withdrawal results in widespread metabolic rewiring and that Gln oxidation in the TCA cycle is not required for ectoderm viability.
Sufficient Gln synthesis distinguishes ectoderm from mesoderm and endoderm
Compensatory metabolic adaptations and sustained ectoderm viability in Gln-free media strongly suggested that ectoderm obtains Gln by de novo synthesis from GS conversion of Glu and ammonia to Gln (Fig. 1B) (Bott et al., 2015; Fu et al., 2019; Issaq et al., 2019; Kung et al., 2011; Stadtman, 2004; Tardito et al., 2015). Consistent with this assessment, GS enzyme is highly expressed in Gln-free, but not in Gln-supplemented, media during ectoderm differentiation (Figs. 2A, S2A). Equivalent lineage-specifying PAX6, and suppression of pluripotent OCT4 and NANOG, transcription factor expression also indicated that downstream ectoderm differentiation was unaffected by Gln-free conditions (Figs. 2A, S2A). Prior to widespread cell death, mesoderm and endoderm cells also upregulate GS in response to exogenous Gln withdrawal, an unexpected finding because of the dependence of mesoderm and endoderm cells on exogenous Gln for survival (Fig. S2B). However, heavy isotopologue tracing showed low levels of 13C5-Gln derived from 13C5-glutamic acid at steady state in mesoderm and endoderm cells in Gln-free media (Figs. 2B, S2C). Additionally, there is minimal derivation of 13C5-α-KG from 13C5-glutamic acid in mesoderm and endoderm cells, indicating that even though these lineage cell types are capable of de novo Gln synthesis, insufficient levels of Gln and/or α-KG are generated, resulting in cell death upon exogenous Gln withdrawal (Figs. 1F-G, 2B, S2C). Combined, the data suggests that ectoderm uniquely responds to exogenous Gln withdrawal by synthesizing sufficient endogenous Gln, in contrast to an insufficient Gln synthesis adaptation that occurs during mesoderm and endoderm differentiation.
Figure 2. Sufficient Gln synthesis distinguish ectoderm from other germ lineages.

(A) Immunoblot of GS, PAX6, and OCT4 expression during H9-derived ectoderm differentiation in Gln-free conditions.
(B) Difference in 13C5-Gln and 13C5-α-KG amounts derived from 13C5-glutamic acid in Gln-free (−Gln) relative to supplemented (Ctr) conditions.
(C-D) (Left) Complete Gln-starvation (−G+M) is attained by culturing cells in Gln-free media and inhibiting de novo Gln synthesis with 1 mM MSO. (Right) Cell viability of (C) mesoderm, endoderm and (D) differentiation of ectoderm cells grown in Gln-free (−Gln) conditions with 50 μM DON or 1 mM MSO relative to Gln-supplemented (Ctr) media.
(E) Metabolite abundance quantified by UHPLC-MS in differentiated ectoderm cells treated in Gln-supplemented (Ctr) or 1h Gln-starvation (−G+M) conditions.
(F) (Top) Schematic of Gln-starvation (−G+M) supplemented with individual cell-permeable Gln-derived metabolites. (Bottom) Cell viability of H9-derived D5 ectoderm cells differentiated in Gln-starvation and supplemented with GlcN6P, dm-α-KG, nucleosides, or GSH.
(G) Cell viability and percentage of ectoderm (PAX6+MAP2B+ double positive (dp), PAX6+SOX1+ (n=2)) and pluripotent (OCT4+) biomarkers in H9-derived D5 ectoderm cells grown in Gln-starvation with added GlcN6P or both GlcN6P and nucleosides.
Data represent mean ± SD of n ≥ 3 biological replicates unless indicated otherwise. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. The p values were determined by (B-D, F, G) one-way ANOVA, or (E) unpaired two-tailed Student’s t test with correction for multiple comparisons.
Requirement of intracellular Gln for all germ lineages
We next asked whether ectoderm required Gln at all. Cells were differentiated in the presence of 6-Diazo-5-oxo-L-norleucine (DON), a Gln mimetic that alkylates Gln utilizing enzymes, resulting in >95% death of mesoderm, endoderm, and ectoderm cells (Figs. 2C, 2D). Pharmacological inhibition of GS with MSO in Gln-free media also compromised mesoderm, endoderm, and ectoderm cell viability (Figs. 2C, 2D). This hinted that Gln, and/or the by-products of Gln metabolism, is still required for ectoderm, similar to mesoderm and endoderm. The small percentage (< 2%) of residual viable cells at the end of differentiation grown with either Gln mimetic (Ctr+DON) or Gln-starvation (−G+M) showed significantly decreased populations of respective lineage-specifying transcription factors SNAI2/SLUG (mesoderm), SOX17 (endoderm), and PAX6 (ectoderm) (Figs. 2D, S2D). This indicated that intracellular Gln supports germ lineage cell survival and differentiation potential. Mesoderm and endoderm require exogenous Gln as metabolic fuel for glutaminolysis-derived α-KG (Figs. 1F-1G), whereas ectoderm synthesizes Gln for role(s) other than glutaminolysis and its reaction products (Figs. 1E, 2A-B, S2A, S2C).
We performed steady-state metabolite profiling by UHPLC-MS following acute Gln-starvation (−G+M) to identify altered Gln-derived metabolites in ectoderm at the initiation of the differentiation process. Aside from Gln, the largest decrease in metabolites occurred for α-KG, hexosamine biosynthesis pathway (HBP) intermediates, purine nucleosides, and glutathione redox status (GSH/GSSG) (Figs. 2E). Metabolite rescue was attempted by supplementing Gln-starved ectoderm with cell-permeable forms of these Gln-derived metabolites (Fig. 2F) (Qie et al., 2019). Verification of uptake and conversion to related metabolites was confirmed for all supplementations, with the exception of GSH, which did not significantly alter intracellular GSH and GSSG levels (Fig. S2E). The addition of HBP metabolite glucosamine-6-phosphate (GlcN6P) yielded the largest rescue of ectoderm cell viability with a single agent at ~57% (Figs. 2F, S2F), and the combination of GlcN6P and nucleosides resulted in the highest ectoderm cell survival at ~69% compared to control conditions (Figs. 2G, S2F), although full rescue was not achieved with any metabolite or binary combination (Figs. 2F-2G, S2F). Despite partial viability rescue with GlcN6P supplementation, pluripotency is not completely repressed by D5 of differentiation in live-gated Gln-starved (−G+M) ectoderm cells under this metabolite rescue condition, as measured by a residual OCT4+ cell population (Figs. 2G). Furthermore, supplementation with combined GlcN6P and nucleosides showed significantly decreased populations of PAX6+MAP2B+ and PAX6+SOX1+ ectoderm cells compared to Gln-supplemented (Ctr) and Gln-free (−Gln) conditions by D5 of differentiation (Figs. 2G, S2F). Together, the data show that replenishment of HBP and nucleosides partially restored Gln-starved ectoderm cell viability, but did not restore differentiation, suggesting that Gln has a role in ectoderm generation in addition to a role as a precursor for metabolites.
Initial Gln absence impairs ectoderm differentiation
Impaired ectoderm differentiation could be a consequence of reduced cell viability due to chronic Gln-starvation. Alternatively, Gln absence could directly impact the ectoderm specification program(s). To examine these possibilities, we measured differentiation status while cell viability and cell cycle progression were progressively altered as Gln-starvation time increased (Figs. 3A, S3A). We quantified ectoderm differentiation status after 24h of Gln-starvation (−G+M), a time point with equivalent viable ectoderm cells relative to control conditions (Fig. 3A). Flow cytometry of live-gated cells showed decreased ectoderm (PAX6+MAP2B+, PAX6+SOX1+) cells, and qRT-PCR showed decreased expression of MAP2B, PAX6, and OTX2 transcripts in Gln-starvation compared to Gln-supplemented and Gln-free conditions (Figs. 3A, S3B). This differentiation impairment increased with starvation time, and pluripotency biomarkers (OCT4+NANOG+, OCT4+TRA1-81+) were suppressed with differentiation regardless of Gln availability (Figs. 3A). Combined, these findings showed that reduced ectoderm differentiation and cell cycle drop out occurred within 24h of Gln-starvation.
Figure 3. Gln is required at the initiation of ectoderm, but not mesoderm and endoderm.

(A) Cell viability and percentage of ectoderm (PAX6+MAP2B+, PAX6+SOX1+, OCT4− SOX2+) and pluripotent (OCT4+NANOG+, OCT4+TRA1-81+) biomarkers in H9-derived ectoderm cells grown continuously in Gln-starvation for D1 (24h), D2 (48h), and D3 (72h).
(B) (Top) Schematic of pulsed Gln-starvation experiments. Ectoderm cells were Gln-starved for the first 24h of differentiation then switched to Gln-supplemented media (Gln-repletion, −G+M→Ctr) until D5. (Bottom) Cell viability of H9-derived D5 ectoderm cells after initial 24h Gln-starvation.
(C) Percentage of ectoderm (PAX6+MAP2B+, PAX6+SOX1+) and pluripotent (OCT4+NANOG+, OCT4+TRA1-81+) biomarkers in H9-derived D5 ectoderm cells after initial 24h Gln-starvation (−G+M→Ctr).
(D) Representative immunofluorescence images of ectoderm and pluripotent biomarkers in H9-derived D5 ectoderm cells grown in Gln-supplemented (Ctr), Gln-free (−Gln), or Gln-repletion after initial 24h Gln-starvation (−G+M→Ctr). (Top) PAX6 (magenta, ectoderm;) MAP2B (green, ectoderm); NANOG (cyan, pluripotent). (Bottom) OCT4 (magenta, pluripotent); SOX2 (green, ectoderm/pluripotent); NANOG (cyan, pluripotent). Scale bar indicates 50 μm.
(E) H9-derived ectoderm expressing shRNA targeting GLUL (shRNA GLUL #1; shRNA GLUL #2) or a non-targeting control (NTC). Cell viability and percentage of PAX6+MAP2B+ D5 ectoderm cells in Gln-supplemented (Ctr), Gln-free (−Gln), or Gln-repletion after initial 14h Gln-free treatment (Gln→Ctr).
(F) Percentage of proliferating H9-derived ectoderm cells (EdU+ staining) immediately after 14h Gln-free or Gln-starvation treatment.
(G) Nascent protein synthesis in H9-derived ectoderm cells immediately after 14h Gln-free, Gln-starvation, or blasticidin treatment relative to Ctr.
(H) ATP levels, quantified by UHPLC-MS, in ectoderm cells after Gln-starvation.
(I) Cell viability and percentage of H9-derived D5 mesoderm (CD34+, SNAI2/SLUG+) and D3 endoderm (SOX17+) cells after initial 14h Gln-free (−Gln→Ctr) or Gln-starvation (−G+M→Ctr) treatment.
(J) Representative immunofluorescence images of pluripotent and lineage-specific biomarkers in H9-derived (Top) D5 mesoderm and (Bottom) D3 endoderm cells grown in Gln-supplemented (Ctr), Gln-free (−Gln), or Gln-repletion follow initial 14h Gln-starvation (−G+M→Ctr). (Top) OCT4 (magenta, pluripotent); SNAI2/SLUG (green, mesoderm). (Bottom) FOXA2 (magenta, endoderm); SOX17 (green, endoderm); NANOG (cyan, pluripotent). Scale bar indicates 50 μm.
(K) Timing of Gln requirement for downstream lineage cell identity.
Data represent mean ± SD of n ≥ 3 biological replicates. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. The p values were determined by (A-C, F-G, I) one-way ANOVA, (E) two-way ANOVA with correction for multiple comparisons or (H) unpaired two-tailed Student’s t test.
To assess a temporal requirement for Gln in implementing ectoderm fate, we replenished Gln after pulsed Gln-starvation while retaining inducing cytokines throughout this differentiation condition (Gln-repletion) (Fig. 3B). After 24h of initial Gln-starvation, Gln-repletion (−G+M-→Ctr) completely rescued the viability and cell cycle of ectoderm cells by D5 (Figs. 3B, S3C). However, ectoderm differentiation was not rescued at D5 following Gln-repletion, as indicated by significantly decreased ectoderm specifying biomarkers (PAX6, MAP2B, SOX1, SOX2) quantified by flow cytometry and immunofluorescence imaging (Figs. 3C, 3D, S3D). Pluripotency exit occurred regardless of initial Gln availability during initial ectoderm differentiation as indicated by suppressed pluripotency biomarkers (OCT4, NANOG, TRA-81, SOX2) relative to hPSC progenitors, although Gln-repletion resulted in slight residual pluripotency biomarker expression compared to Gln-supplemented and Gln-free conditions (Figs. 3C, 3D, S3E). Additionally, impaired ectoderm differentiation does not compensate with alternative lineage-specific biomarker (SOX17, endoderm; CD34, mesoderm) expression, confirming the specific failure for an ectoderm fate (Fig. S3E). To exclude off-target effects of MSO treatment in Gln-starved conditions, H9 hPSCs containing short hairpin RNA (shRNA) knockdown of GLUL, encoding for GS, were differentiated for 5 days in Gln-free media, and equally led to ectoderm cell death (Figs. 3E, S3F, S3G). We found that a shorter, initial Gln-starvation period of 14h followed by Gln-repletion (−Gln-→Ctr) was sufficient to impair ectoderm differentiation, as quantified by a decreased population of PAX6+MAP2B+ cells (Fig. 3E). Additionally, supplementation with Gln-derived metabolites during the initial 14h Gln-starvation period (−G+M+GlcN−6P+nuc →Ctr) equally resulted in impaired ectoderm differentiation (Fig. S3H). Impaired ectoderm differentiation was independent of cell proliferation, nascent protein synthesis, and ATP levels, which were all unaffected immediately after 14h of Gln-starvation and at later time points after Gln-repletion (Figs. 3F-H, S3I-S3J). These results suggest that Gln is required within the first 14h of directed ectoderm differentiation to activate the ectoderm program, a fate that could not be rescued by Gln restoration following this window of time.
To study the ectoderm specificity of this temporal Gln requirement in the larger context of germ layer differentiation, mesoderm and endoderm were grown in initial 14h Gln-free (−Gln) and Gln-starvation (−G+M) media conditions followed by Gln-repletion (→Ctr). Strikingly, initial Gln absence did not affect subsequent mesoderm nor endoderm specification (Figs. 3I-3J). This demonstrates that a temporal Gln requirement is specific to ectoderm differentiation and highlights distinct signatures of Gln dependency for each respective germ lineage cell fate (Fig. 3K).
Ectoderm differentiation requires Gln-dependent mTORC1 signaling
Because addition of Gln-derived metabolites and delayed Gln add-back failed to restore ectoderm fate specification, we considered an alternative possibility that Gln could signal to activate a nutrient-sensing pathway, similar to glucose signaling in trophectoderm (Chi et al., 2020). Recent work showed that Gln is a substrate that activates the mammalian target of rapamycin complex 1 (mTORC1). mTORC1 activation by Gln occurs through Rag GTPase-dependent activation by Gln exchange for exogenous leucine and arginine, or by Rag GTPase-independent activation directly from a Gln interaction with Arf1 GTPase (Jewell et al., 2015; Meng et al., 2018; Saxton and Sabatini, 2017). We observed that phosphorylation of the canonical mTORC1 substrate, ribosomal protein S6 kinase 1 (S6K1-Thr389), in 24h Gln-starved ectoderm is significantly reduced, indicating mTORC1 inhibition (Figs. 4A, S4A). Pulsed addition of mTORC1 inhibitor rapamycin during initial 14h of tri-lineage differentiation replicated initial Gln-starvation (−G+M→Ctr), with impaired ectoderm and unaffected mesoderm and endoderm differentiation (Figs. 3C-3E, 3I-3K, S3D-S3E, 4B). This result suggested that initial mTORC1 activation, like intracellular Gln presence, is critical for and specific to ectoderm differentiation.
Figure 4. Ectoderm differentiation requires initial Gln-dependent mTORC1 signaling.

(A) Representative immunofluorescence images of mTORC activation in H9-derived ectoderm cells grown for 24h in Gln-supplemented (Ctr), Gln-free (−Gln), Gln-starvation (−G+M), or 200 nM rapamycin treatment (Ctr+Rapa). DAPI (blue, nucleus); ACTIN (green, cytoskeleton); pS6K1-Thr389 (yellow, mTORC1 activation). Scale bar indicates 50 μm.
(B) Percentage of H9-derived D5 mesoderm (CD34+SNAI2/SLUG+), D3 endoderm (SOX17+FOXA2+), and D5 ectoderm (PAX6+MAP2B+, PAX6+SOX1+) cells after initial 14h 200 nM rapamycin treatment.
(C) Representative immunofluorescence images of mTORC activation in H9-derived WT-Raptor or Raptor-Rheb15 ectoderm cells grown for 12h in Gln-supplemented (Ctr) or Gln-starvation (−G+M). DAPI (blue, nucleus); ACTIN (green, cytoskeleton); pS6K1-Thr389 (yellow, mTORC1 activation); FLAG tag (magenta; transduced cell line reporter). Scale bar indicates 50 μm.
(D) Percentage of H9-derived D5 WT-Raptor or Raptor-Rheb15 ectoderm (PAX6+MAP2B+FLAG+, PAX6+SOX1+FLAG+) cells after initial 14h Gln-starvation (−G+M→Ctr).
(E) Representative immunofluorescence images of ectoderm biomarkers in H9-derived D5 (Top) WT-Raptor or (Bottom) Raptor-Rheb15 ectoderm cells grown in Gln-supplemented (Ctr) or Gln-repletion follow initial 14h Gln-starvation (−G+M→Ctr). (Left) MAP2B (green, ectoderm;) PAX6 (magenta, ectoderm); FLAG (grey, transduced cell line reporter). (Bottom) SOX2 (green, ectoderm/pluripotent); OTX2 (magenta, ectoderm), FLAG (grey, transduced cell line reporter). Scale bar indicates 50 μm.
(F) Percentage of ectoderm (PAX6+MAP2B+FLAG+) and pluripotent (SOX2− OCT4+FLAG+) biomarkers in H9-derived D5 WT-Raptor or Raptor-Rheb15 ectoderm cells after initial 14h Gln-starvation with increasing concentrations of exogenous Gln supplementation.
(G) Percentage of H9-derived D5 ectoderm (PAX6+MAP2B+, PAX6+NESTIN+) cells after initial (−G+M→Ctr), intermediate (Ctr→−G+M→Ctr), or late (Ctr→−G+M) 14h Gln-starvation.
(H) Arginine (Arg) or Leucine (Leu) were removed from culture media for 14h then switched to Arg/Leu-supplemented conditions (AA repletion) until D5. Percentage of (Left) H9 or (Right) UCLA1-derived D5 WT-Raptor or Raptor-Rheb15 ectoderm (PAX6+MAP2B+FLAG+) cells after initial 14h Gln-starvation, Arg deprivation, or Leu deprivation.
(I) Cell viability and percentage of H9-derived WT-Raptor or Raptor-Rheb15 D5 mesoderm (CD34+SNAI2/SLUG+FLAG+) and D3 endoderm (SOX17+FOXA2+FLAG+) cells.
(J) Timing of Gln requirement for downstream lineage cell identity. Gln is required as a metabolite precursor in mesoderm and endoderm cells and as a mTORC1 signaling activator in ectoderm cells. Related to Figure 3K.
Data represent mean ± SD of n ≥ 3 biological replicates. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. The p values were determined by (B, I) unpaired two-tailed Student’s t test, (D, F, H) two-way ANOVA, or (G) one-way ANOVA with correction for multiple comparisons.
To investigate whether Gln-dependent mTORC1 activation is essential for ectoderm differentiation, we transduced two separate hPSC lines, H9 and UCLA1, with a constitutively active mTORC1 expression vector, Raptor-Rheb15, and a control, wild-type (WT) Raptor {Sancak et al., 2010). Construct expression was verified by puromycin selection and DYKDDDDK (FLAG) tag detection (Fig. S4B), and hPSCs were directed toward ectoderm differentiation with an initial 14h Gln-starvation. As anticipated, mTORC1 activation was sustained in Raptor-Rheb15 but not in WT-Raptor transduced hPSCs differentiated into ectoderm with Gln-starvation (Fig. 4C, S4C). After an initial 14h Gln-starvation, Gln-repletion (−G+M→Ctr) completely rescued the viability and cell cycle of ectoderm cells at D5 (Figs. S4D-S4E). Gln-repletion in WT-Raptor ectoderm cells resulted in significantly reduced lineage-specifying biomarkers (PAX6, MAP2B, SOX1, SOX2, OTX2) at D5, whereas Raptor-Rheb15 ectoderm cells grown under the same experimental conditions showed minimal, non-significant changes in ectoderm specification compared to Gln-supplemented and Gln-free conditions at D5 (Figs. 4D-4E, S4F). These results indicated that, in addition to nascent protein synthesis being unaffected within an acute 14h Gln-starvation period (Figs. 3G, S3J), the lack of Gln incorporation into polypeptides was not the cause of impaired ectoderm differentiation because successful ectoderm specification occurred in Raptor-Rheb15 hPSCs following transient Gln absence.
To evaluate the concentration of Gln that impacts differentiation, we performed Gln add-back titrations. Reports suggest that the primary source of Gln in the embryo and fetus is derived from placental conversion of Glu to Gln, with maternal and fetal Gln concentrations ranging from 0.8-1.0 mM (Cruzat et al., 2018; Holm et al., 2017; McIntyre et al., 2020; Neu, 2001). This concentration range is consistent with the amount of Gln required to restore differentiation in transient 14h Gln-starved ectoderm cells at D5, with full recovery of PAX6+MAP2B+ and SOX2+OCT4− cells starting at 0.8 mM Gln (Figs. 4F, S4G-S4H).
We tested whether only the first hours of ectoderm specification depend on intracellular Gln by performing Gln-starvation studies in the intermediate and late stages of differentiation. Results indicate that only early transient (−G+M→Ctr), but not intermediate (Ctr→−G+M→Ctr) nor late stage (Ctr→−G+M), Gln absence impaired ectoderm differentiation (Figs. 4G, S4I). This suggests that Gln-dependent mTORC1 activation is an early requirement for downstream functional ectoderm specification.
We next asked whether the initial absence of other mTORC1 sensed amino acids impacted ectoderm differentiation. Since leucine (Leu) and/or arginine (Arg) signaling to activate mTORC1 is well-characterized (Wolfson and Sabatini, 2017), H9 and UCLA1 hPSCs were differentiated in Leu- or Arg-free media for the first 14h, followed by repletion until D5. We observed that from these mTORC1 sensed amino acids, only the initial absence of Gln, and not Leu nor Arg, impaired initial mTORC1 activation and downstream ectoderm differentiation, indicating specificity for the Gln requirement (Figs. 4H, S4J-S4K).
Finally, we confirmed that constitutively active mTORC1 is not compatible with mesoderm nor endoderm directed-differentiation, as indicated by significantly decreased levels of cell viability in Raptor-Rheb15 compared to WT-Raptor transduced cells. At the differentiation endpoint, the remaining ~20% live Raptor-Rheb15 mesoderm cells did not exhibit significantly decreased lineage nor pluripotency biomarker expression, whereas the ~60% live endoderm cells showed decreased lineage and pluripotency biomarker expression (Figs. 4I, S4L). This result supports a working model in which early mTORC1 activity is critical for ectoderm differentiation but is detrimental to mesoderm and endoderm differentiation (Figs. 4A, S4A, S4M), highlighting distinct requirements for germ lineage cell fates. The data showed that a Gln signal activates mTORC1 within the initial hours of differentiation induction to produce ectoderm (Fig. 4J). Gln itself, but not the by-products of Gln metabolism, were required for ectoderm differentiation.
Transcriptome signatures suggest unique, lineage-specific Gln utilization in vivo
Our experimental model of Gln withdrawal and add-back is a useful tool to identify nutrient specificities in vitro, similar to gain and loss of function genetic studies, and provided predictions for potential differences in Gln dependency for each germ layer in vivo. Although hPSC differentiation is a reproducible method for studying human gastrulation (Taniguchi et al., 2019), cross-referencing hPSC results to available in vivo data would add further support for lineage-specific Gln utilization. To generate hypotheses for potential translation of these hPSC study results, we compared hPSC findings with human embryo development at approximately the same post-implantation stage (Mascetti and Pedersen, 2016; Taniguchi et al., 2019). To do this, we queried the single cell RNA-Seq transcriptome of a gastrulating human embryo ~16-19 days post fertilization (Tyser et al., 2020). Since hPSC derived mesoderm and endoderm depend on exogenous Gln, and ectoderm can synthesize sufficient Gln de novo, we used these distinguishing features as a proxy for Gln utilization patterns. We calculated the ratio of Gln degradation [averaged (GLS1 + GLS2) transcripts] to endogenous Gln synthesis [GLUL transcripts] to correlate the tendencies for Gln auxotrophy versus prototrophy in each lineage. In both in vitro (Fig. 5A) and queried in vivo (Fig. 5B) transcriptome datasets, we found that Gln degradation transcripts were much lower than Gln synthesis transcripts in ectoderm, but not in mesoderm nor endoderm related lineages, as quantified by the [averaged (GLS1 + GLS2)]/GLUL ratio. A comparison of our in vitro hPSC-derived germ lineage model versus the in vivo human gastrula sample showed similar clustering of Gln degradation to synthesis ratios in corresponding lineages. Thus, relative lineage-specific Gln consumption and synthesis transcript patterns identified in the in vitro model and in vivo sample may be maintained in the human embryo (Fig. 5C), and presents an enticing hypothesis for future studies.
Figure 5. Distinct transcript patterns of Gln utilization characterize germ lineages in vivo.

(A) Ectoderm but not endoderm and mesoderm cells exhibit increased Gln synthesis (GLUL transcripts) relative to Gln consumption (GLS1 + GLS2 transcripts) in vitro. The average GLS1 and GLS2 expression was divided by GLUL expression for each lineage and values were normalized to hPSC control.
(B) Ectoderm cells exhibit increased Gln synthesis (GLUL transcripts) relative to Gln consumption (GLS + GLS2 transcripts) in vivo. Single cell RNA-sequencing data from Tyser et al., 2020 were used to calculate the same ratio as in (A). The values were normalized to the average Epiblast ratio. Lineages were taken from Tyser et al., 2020.
(C) Ectoderm cells exhibit similar trends in Gln synthesis and Gln consumption in both in vitro and in vivo contexts. Results from (A) and (B) are shown with a reference line drawn at y = x.
(D) Working model of germ lineage cell fate dependent on Gln.
Data represent mean ± SD. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. The p values were determined by (A-B) unpaired two-tailed Student’s t test.
DISCUSSION
Our current understanding of how metabolism permits cell state transitions between pluripotency and differentiation is fragmented and limited. A particular knowledge gap is that previous studies focused largely on the metabolic roles of Gln and its by-products in maintaining pluripotency (Carey et al., 2015; Marsboom et al., 2016; Tohyama et al., 2016; Vardhana et al., 2019) or regulating multipotent cell lineages (Johnson et al., 2018; Oburoglu et al., 2014; Yu et al., 2019). Here, we report that Gln itself is a required signaling molecule in ectoderm differentiation. We demonstrate that Gln synthesis and consumption is differentially regulated between lineages in vitro, and possibly in vivo, and that Gln has an irreversible temporal requirement in ectoderm, but not mesoderm nor endoderm, lineage differentiation (Fig. 5D).
In vivo evidence for the developmental significance of Gln and mTORC1 has recently been reported, in which dysregulated Gln-Glu cycling and mTORC1 signaling contributed to placental dysfunction and fetal growth restriction (McIntyre et al., 2020). Additionally, prior studies showed that mTORC1 inhibition favors PSC differentiation to mesoderm and endoderm lineages, yet a mechanism has never been reported (Jung et al., 2016; Nazareth et al., 2016; Zhou et al., 2009). Our results show that a Gln activating signal for mTORC1 enables ectoderm, whereas this nutrient-signaling activation is detrimental for mesoderm and endoderm fates, as an attempt to direct differentiation of constitutively active mTORC1 Raptor-Rheb15 hPSCs into mesoderm and endoderm lineages led to widespread cell death. Gln is essential for mTORC1 activation within the first hours of ectoderm induction, because a transient absence of Gln in this initial period, but not in later time periods, caused slight residual pluripotency biomarker retention and diminished ectoderm-specifying biomarker expression. In contrast to this strict ectoderm time requirement for Gln, mesoderm and endoderm cells achieve full differentiation even with an initial Gln absence (Figs. 4J, 5D, S5A-S5E, S6A).
Gln is a non-essential amino acid not previously implicated in germ lineage differentiation, but here we reveal it is conditionally essential and depends upon cell fate context. A divergence in Gln essentiality depending on germ lineage was unexpected since all three lineages initiate from a common pluripotent progenitor cell. We speculate on the possibility of a Gln-specific symbiotic relationship amongst the three germ lineages during early in vivo development. Specifically, ectoderm may generate de novo Gln to support nearby mesoderm and endoderm cells dependent on exogenous Gln, which could influence tissue patterning. This hypothesis is supported by our previous media footprint profiling data showing high secretion of Gln in ectoderm and high consumption of Gln in mesoderm (Lu et al., 2019). Patterns of Gln metabolism during blastocyst development may be one of multiple input signals for embryos experiencing nutrient gradients and discontinuities. This tempting possibility is also suggested, but not yet proven, by human embryo transcriptome data showing the Gln synthesis to consumption expression signature is higher in ectoderm compared to mesoderm and endoderm lineages, which is at least consistent with our in vitro model system (Figs. 5A-C). Thus, our data begin building a framework for how temporal and/or spatial fluctuations in Gln could potentially impact normal development.
In conclusion, our findings uncover a new testable model in which a nutrient acts as a signaling molecule that could differentially coordinate the development of the three embryonic germ lineages. Our study provides several testable hypotheses for nutrient-specific signaling cues as contributors to organizational structure and fidelity in early mammalian development. These include targeting key Gln gatekeeping enzymes in in vivo development models, and future investigations of additional nutrient specificities implicated in cell fate decisions. These findings could also provide more logical manipulations of the metabolite environment and pathway fluxes in vitro to produce higher quality hPSC derivatives for disease modeling, tissue engineering, and eventual therapeutic usage.
LIMITATIONS
Here, we report that specific Gln requirements define the three embryonic germ lineages upon in vitro pluripotency exit. We further uncover Gln acts as a signaling molecule to activate mTORC1 and enable downstream ectoderm differentiation. However, two main limitations temper our study conclusions. First, to the best of our knowledge, transient Gln removal does not occur during in vivo development. Thus, this experimental perturbation does not reflect endogenous regulation, but was a useful tool to identify nutrient specificities of different cell types. Second, we present correlative evidence linking GLS1, GLS2, GLUL RNA expressions to potential functional in vivo trilineage Gln dependency. This is a study limitation since transcript levels do not necessarily equate to protein abundance/activity. We note that this comparison of relative gene expression made between embryo tissue and hPSC-derived cells may inspire future work regarding in vivo tissue-specific regulation of Gln metabolism.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents will be addressed by the corresponding author, Michael A. Teitell (MTeitell@mednet.ucla.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw RNA-Seq data for nutrient balanced hPSC and tri-lineage differentiation to ectoderm, mesoderm, and endoderm were deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE127270. All raw and processed UHPLC-MS metabolomics data from this study are in Excel spreadsheet format as Supplemental Table 1. All data were analyzed with standard programs and packages, as detailed above. All R Jupyter notebooks and supplemental files used to process RNA-Seq and UHPLC-MS data will be uploaded upon manuscript press to Github at https://github.com/fahsan/GlnFateGatekeeper. All raw images, immunoblots, and values for quantification are available from the corresponding author and lead contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Human embryonic stem cell (hESCs) lines H9 (WA09 – Female, RRID:CVCL_9773) and UCLA1 (Female, RRID:CVCL_9951) were grown in six-well tissue culture-treated polystyrene microplates (Falcon, 08-772-1B). Polystyrene plates were coated with feeder-free Matrigel basement membrane matrix (Corning, CB-40234A) diluted at 1:60 in DMEM/F-12 w/o glutamine (Gibco, 21331020), and incubated for 30 m at 37°C. hESCs were cultured in mTeSR Plus (Stemcell Technologies, 05825), with media changes every 48h, to 80% confluency at 5% CO2 in a 37°C incubator. For passaging, hESCs were washed with DPBS 1x w/o calcium & magnesium (Corning, 21031CV) and removed from culture plates using Gentle Cell Dissociation Reagent (Stemcell Technologies, 07174) at 37°C for 5 m. Cells were passaged onto Matrigel-coated plates in mTeSR Plus as described above. Cells were tested for mycoplasma (Lonza, LT07-418) every two weeks.
Tri-lineage directed differentiation
Twelve-well tissue culture-treated polystyrene microplates (Falcon, 08-772-29) were coated in Matrigel basement membrane matrix. hESCs at 75%-85% confluency were removed from culture plates and dissociated into single cells using Gentle Cell Dissociation Reagent at 37°C for 10 min. Gentle Cell Dissociation Reagent was diluted in an equal volume of DMEM/F-12 (Gibco, 11320033). hESCs were scraped and centrifuged at 450 x g for 5 m, and cell number was quantified with a hemocytometer. Cells were reseeded at 125,000 cells/cm2 onto Matrigel-coated plates in mTeSR Plus with 10 μM ROCK inhibitor (Y-27632; Stemcell Technologies, 72304).
Ectoderm differentiation
To generate ectoderm, seeded cells were changed to a chemically-defined nutrient-balanced differentiation media 48 h after seeding. Cells were differentiated over a five-day period, cultured in DMEM/F12 supplemented with 450 μM 1-Thioglycerol (Millipore Sigma, M6145), 1 mg/mL BSA (Gibco, 15260037), 0.11 μM 2-mercaptoethanol (Gibco, 21985023), 1% Glutamax (Gibco, 35050061), 1% N-2 supplement (Gibco, 17502048), 2% B-27 supplement (Gibco, 17504044), 10 μM SB43154 (Stemgent, 04-0010), and 0.2 μM Dorsomorphin (Stemgent, 04-0024). Media was sterilized through a 0.22 μm filter (Denville, 1210N26), and changed every 24 h until D5 differentiation.
Mesoderm differentiation
To generate mesoderm, seeded cells were changed to a chemically-defined nutrient-balanced differentiation media 48 h after seeding. Cells were differentiated over a five-day period, cultured in DMEM/F12 supplemented with 450 μM 1-Thioglycerol, 1 mg/mL BSA, 0.11 μM 2-mercaptoethanol, 1% Glutamax, 0.7 μg/mL insulin (Millipore Sigma, 11376497001), 15 μg/mL transferrin (Millipore Sigma, 10652202001), 1 mL/100 mL chemically-defined lipid concentrate (Gibco, 11905031), 100 ng/mL VEGF-165 (Stemcell Technologies, 78073.1), 100 ng/mL BMP4 (Peprotech, 120-05ET), and 20 ng/mL FGF2 (Peprotech, 78003). During the first 24 h of differentiation, media was supplemented with 100 ng/mL Activin A (Stemcell Technologies, 78001). Media was sterilized through a 0.22 μm filter (Denville, 1210N26), and changed every 24 h until D5 differentiation.
Endoderm differentiation
To generate endoderm, seeded cells were changed to a chemically-defined nutrient-balanced differentiation media 48 h after seeding. Cells were differentiated over a three-day period, cultured in DMEM/F12 supplemented with 450 μM 1-Thioglycerol, 1 mg/mL BSA, 0.11 μM 2-mercaptoethanol, 1% Glutamax, 0.7 μg/mL insulin (Millipore Sigma, 11376497001), 15 μg/mL transferrin (Millipore Sigma, 10652202001), 1 mL/100 mL chemically-defined lipid concentrate (Gibco, 11905031). During the first 24 h of differentiation, media was supplemented with 100 ng/mL Activin A, 2 μM CHIR99021 (Stemcell Technologies, 72052), and 50 nM PI-103 (Fisher Scientific, 29-301). After 24 h of differentiation, media was supplemented with 100 ng/mL Activin A and 250 nM LDN 1931189 (Stemgent, 04-0074). Media was sterilized through a 0.22 μm filter (Denville, 1210N26), changed every 24 h until D3 differentiation.
METHOD DETAILS
Glutamine perturbations
For all glutamine-free conditions, differentiation media (as described above) was made with DMEM/F-12 w/o glutamine and in the absence of 1% Glutamax (note: trace/negligible amounts of glutamine may be present in Matrigel). To achieve glutamine-starvation conditions, glutamine-free media was supplemented with 1 mM L-methionine sulfoximine (Millipore Sigma, 91016). Metabolite rescue experiments were performed under glutamine-starvation media in the presence of 8 mM dimethyl 2-oxoglutarate (Millipore Sigma, 349631), 60 μL/mL Embryomax nucleosides (Millipore Sigma, ES-008), 10 mM D-glucosamine 6-phosphate (Millipore Sigma, G5509), 2 mM D-(+)-glucosamine hydrochloride (Sigma-Aldrich, G1514), or 1 mM L-glutathione reduced (Millipore Sigma, G6529). Gln utilization was blocked by addition of 50 μM 6-diazo-5-oxo-L-norleucine (Millipore Sigma, D2141) or 1 μM CB-839 (Selleckchem, S7655) to either Gln-supplemented or Gln-free media. Gln, Arg, or Leu starvation/deprivation timed pulse experiments were performed by culturing cells in respective deprivation conditions for an initial 14 h or 24 h, washed with PBS, and then grown in Gln, Arg, or Leu replete conditions until D5 of differentiation.
Intracellular flow cytometry
Cells were dissociated using Gentle Cell Dissociation Reagent for 10 m, diluted in sterile dPBS, and pelleted at 450 x g for 5 m. Supernatant was aspirated off, and cells were re-suspended in 250 μL of Fixation/Permeabilization solution (BD Biosciences, 554714) and incubated at 4°C for 30 m. Following fixation, cells were washed with 1x BD Perm/Wash Buffer (BD Biosciences, 554714) and pelleted at 450 x g for 5 m. Cells were re-suspended in 100 uL of 1x BD Perm/Wash Buffer. Conjugated antibodies were incubated with fixed cells for 45 m at 4°C in the dark. Samples were processed on a LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (FlowJo, Inc.). For cell viability quantifications, values were generated from EF780 Fixable Viability Dye negative (EF780−) cell populations or from Annexin A5 and 7AAD double negative (%AV- 7AAD- dn) cell populations. For cell cycle quantifications, values were determined based on 7AAD-stained DNA content and histone H3 (phospho-S28). Experiments were performed at least three independent times. The following conjugated antibodies were used: Alexa Fluor 647 Rat anti-histone H3 (pS28) (BD Biosciences, 558217), APC anti-DYKDDDDK tag (Biolegend, 637308), PE Mouse anti-SNAI2/Slug (BD Biosciences, 564615), Alexa Fluor 488 Mouse anti-human Sox17 (BD Biosciences, 562205), PerCPCy5.5 Mouse anti-human PAX6 (BD Biosciences, 562388), human Pax6 PE-conjugated antibody (R&D Systems, IC8150P-100), Alexa Fluor 488 mouse anti-MAP2B (BD Biosciences, 560399), PE Mouse anti-human Sox1 (BD Biosciences, 561592), Alexa Fluor 488 mouse anti-Oct3/4 (BD Biosciences, 560253), V450 mouse anti-Sox2 (BD Biosciences, 561610), PerCP-Cy5.5 mouse IgG2a, κ isotype control (BD Biosciences, 558020), Alexa Fluor 700 mouse IgG1, κ isotype control (BD Biosciences, 557882), Alexa Fluor 488 mouse IgG1 κ isotype control (BD Biosciences, 557782), PE mouse IgG1 k isotype control (BD Biosciences, 556680), V450 mouse IgG1, and κ isotype control (BD Biosciences, 560373). For flow cytometry quantification of phosphorylated mTORC1 substrates, the following antibodies were used: rabbit anti-phospho-p70 S6 kinase (Thr389) (108D2) (Cell Signaling, 9234) and Alexa Fluor 568 goat anti-rabbit IgG (H+L) (Invitrogen, A11036, 1/1000).
Immunofluorescence
Cells were differentiated in 35 mm μ-Dish on No. 1.5 ibitreat polymer coverslips (ibidi GmbH, 81156, 80416) coated with Matrigel (Corning, CB-40234A). Cells were fixed in 4% methanol-free formaldehyde (Thermo Scientific, 28906) for 10-15 min at RT. Cells were washed with PBS, permeabilized with 0.3% Triton X-100 in PBS for 10 min at RT, washed again with PBS, and incubated in blocking buffer (5% normal donkey serum in PBS) for 1hr at RT. Cells were incubated with primary antibodies diluted in blocking buffer overnight at 4C, washed with PBS, then incubated with secondary antibodies diluted in blocking buffer for 1hr at RT. Cells were counterstained and mounted with ibidi Mounting Medium with DAPI (ibidi GmbH, 50011, nD 1.42–1.44). The following antibodies were used: rabbit anti-OCT-4 (Cell Signaling, 610517, 1/200), mouse anti-OCT-4 (Cell Signaling, 2750, 1/200), goat anti-human NANOG (R&D Systems, AF1997, 5 μg/mL), mouse anti-SOX2 (Cell Signaling, 4900, 1/400), rabbit anti-PAX6 (Cell Signaling, 60433, 1/200), mouse anti-MAP2 (Invitrogen, MA5-12826, 1/500), rabbit anti-OTX2 (Abcam, ab114138, 4 μg/mL), goat anti-Brachyury/TBXT (R&D Systems, AF2085, 15 μg/mL), rabbit anti-SLUG/SNAI2 (Cell Signaling, 9585, 1/400), rabbit anti-SLUG/SNAI2 (Cell Signaling, 9585, 1/400), mouse anti-SOX17 (Invitrogen, MA5-24891, 1/100), rabbit anti-FoxA2/HNF3β (Cell Signaling, 8186, 1/400), mouse anti-DYKDDDDK Tag (Cell Signaling, 8146, 1/200), rabbit anti-phospho-p70 S6 kinase (Thr389) (Cell Signaling, 9234). Actin counterstain was done following ActinRed™ 555 ReadyProbes® Reagent Protocol (Rhodamine phalloidin) (Invitrogen, R37112). All secondary antibodies were from sourced from donkey host of the Alexa Flour series (Life technologies, A-31573, A-21202, A-21432, A-11055, A-31570).
Confocal microscopy image acquisition and processing
Imaging was performed on Leica TCS SP8 DLS (Digital LightSheet) confocal microscope, equipped with a Galvo X resonant scanner and sCMOS camera controlled by the Leica Application Suite (LAS) X software, using a HC PL APO CS2 63x/1.40 OIL lens and a TD 488/552/638 main beamsplitter. Pinhole size was set to 1AU with an 8,000 Hz scan speed. 2048 x 2048 8- or 16-bit images at 1.25 zoom were acquired using HyD (410nm - 527nm, 655nm - 685nm) and PMT (567nm - 607nm) detectors. All samples with the same immunolabeling and/or compared to each other were acquired with the same settings. DAPI was excited with Diode 405 nm laser line and emission collected with HyD detector (455±45nm). Alexa Flour 488 was excited with OPSL 488 nm laser line and emission collected with HyD detector (518±13nm) or PMT (518±21nm). Alexa Flour 555 was excited with OPSL 552 nm laser line and emission collected with HyD detector (584±23nm) or PMT (569±10nm). Alexa Flour 647 was excited with Diode 638 nm laser line and emission collected with HyD detector (670±15nm).
All compared image sets had identical processing in shift phase (arithmetic mean), denoising (median filtering with radius of 2-6 pixels), and histogram normalization (brightness and contrast) using the Leica Application Suite (LAS) X software according to field standards (Aaron and Chew, 2021; Schmied and Jambor, 2020). All experiment interpretations and data analysis were done on raw image files. False color LUTs and image merges were done on the Leica Application Suite (LAS) X software.
qRT-PCR
Cells were grown to between 70% and 80% confluence and harvested as described above. RNA was isolated using the RNeasy Mini Kit (Qiagen, 74104) and RNase-free DNase (Qiagen, 79254) following the manufacturer’s protocols. RNA was quantified using a Nanodrop spectrophotometer (Thermo Scientific). cDNA was synthesized with 2 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad, 1708841). qRT-PCR was performed on a LightCycler480 (Roche) using SYBR green (Roche, 04887352001). Experiments were performed at least three independent times. Primers were used as follows: RPLPO, forward, 5’-CAGATTGGCTACCCAACTGTT-3’, reverse, 5’-GGAAGGTGTAATCCGTCTCCAC-3’; OCT4, forward, 5’-CAAAGCAGAAACCCTCGTGC-3’, reverse, 5’-TCTCACTCGGTTCTCGATACTG-3’; NANOG, forward, 5’-CCCCAGCCTTTACTCTTCCTA-3’, reverse, 5’-CCAGGTTGAATTGTTCCAGGTC-3’; SOX2, forward, 5'-TACAGCATGTCCTACTCGCAG-3’, reverse, 5’-GAGGAAGAGGTAACCACAGGG-3’; MAP2, forward, 5’-CTCAGGACCGCTAACAGAGG-3’, reverse, 5’-CATTGGCGCTTCGGACAA-3’; OTX2, forward, 5’-CAAAGTGAGACCTGCCAAAAAGA-3’, reverse, 5’-TGGACAAGGGATCTGACAGTG-3’; PAX6, forward, 5’-TGGGCAGGTATTACGAGACTG-3’, reverse, 5’-ACTCCCGCTTATACTGGGCTA-3’; GLS1, forward, 5’-TGGTGGCCTCAGGTGAAAAT-3’, reverse, 5’-CCAAGCTAGGTAACAGACCCTGTTT-3’; GLS2, forward, 5’-AACGAATCCCTATCCACAAGTTCA-3’, reverse, 5’- GCAGTCCAGTGGCCTTTAGTG-3’; GLUL, forward, 5’- AAGAGTTGCCTGAGTGGAATTTC-3’, reverse, 5’-AGCTTGTTAGGGTCCTTACGG-3’.
Immunoblot
Cells were grown to between 70% and 80% confluence and harvested as described above. Cells were lysed in SDS buffer (40 mM Tris-HCl pH 6.8, 3% glycerol, 1% SDS), 1x protease inhibitor (Sigma, P8340) and 1x phosphatase inhibitor (Sigma, P5726 and Sigma, P0044). Protein concentrations were quantified using a BCA assay (Pierce). 50 μg aliquots were supplemented with 5% 2-mercaptoethanol and loaded onto gels at 90 V for 20 m followed by 120 V for 75 m. Protein was transferred to a nitrocellulose membrane at 250 mA for 2 h. Membranes were blocked with Odyssey Blocking Buffer TBS (Li-Cor, 927-50010) for 30 m and incubated in primary antibodies overnight at 4°C. Following incubation, membranes were washed in TBST for 20 m. IRDye-conjugated secondary antibodies IRDye 680RD donkey anti-rabbit IgG (H + L) (LiCor, 926-68703, 1/10,000) and IRDye® 800CW goat anti-mouse IgG (H + L) (LiCor, 925-32210, 1/10,000) were added to membranes, incubated for 1 h at RT, and washed in TBST for 20 m. All images were captured with an Odyssey Fc (Licor). Experiments were performed at least three independent times. The following antibodies were used: rabbit anti-pyruvate dehydrogenase E1-alpha subunit (phospho S293) (ab177461, 1/500), rabbit anti-pyruvate dehydrogenase E1-alpha subunit (ab168379, 1/500), mouse anti-glutamine synthetase (BD Biosciences, 610517, 1/500), rabbit anti-PAX6 (Cell Signaling, 60433, 1/500), mouse anti-β-tubulin (Millipore Sigma, T4026, 1/10,000), rabbit anti-phospho-p70 S6 kinase (Thr389) (108D2) (Cell Signaling, 9234, 1/500), rabbit anti-p70 S6 kinase (49D7) (Cell Signaling, 2708, 1/500).
Click chemistry reactions
EdU labeling:
Samples were cultured for 4 h with fresh media supplemented with 5 μM EdU (Invitrogen, A10044), harvested and fixed in 100 μL 4% phosphate-buffered paraformaldehyde for 15 m at RT, permeabilized in 100 μL of saponin-based permeabilization wash for 15 m at RT, and proceeded to Click-iT reaction labeling.
L-Azidohomoalanine (AHA) labeling:
Samples were pulsed for 15 m with DMEM/F-12 w/o L-methionine (US Biological Life Sciences, D9807-05A) differentiation media for optimized AHA labeling. Then, samples were cultured in DMEM/F-12 w/o L-methionine differentiation media with 1 mM AHA (Sigma-Aldrich, 900892; labeling condition) or 0.1 mM L-methionine (Sigma-Aldrich, M5308; control condition) for 1 h. As a negative control, nascent protein synthesis was blocked by addition of 1 μM Blasticidin S HCL (Gibco, A1113903) to AHA-supplemented media. As with EdU labeling, samples were harvested and fixed in 100 μL 4% phosphate-buffered paraformaldehyde for 15 m at RT, permeabilized in 100 μL of saponin-based permeabilization wash for 15 m at RT, and proceeded to Click-iT reaction labeling.
Click chemistry reaction
For EdU labeling, Alexa Fluor 488 azide (Invitrogen, A10266) was used. For AHA labeling, Alexa Fluor 488 Alkyne (Invitrogen, A10267) was used. Freshly prepared Click-iT reagent mix was made with saponin-based wash reagent, 100 mM copper(II) sulfate (Sigma-Aldrich, C1297), Click-iT buffer additive (Invitrogen, C10269), and 1 μM Alexa Fluor 488 azide or Alexa Fluor 488 Alkyne. Following permeabilization, samples were incubated with 500 μL reagent for 30 m at RT in the dark, then washed twice with saponin-based wash reagent. Samples were then stored in saponin-based wash reagent at 4°C in the dark for flow cytometry and/or further intracellular antibody labeling.
shRNA knockdown
pLKO.1 vectors containing shRNA targeting GLUL RNA transcripts were purchased from Sigma-Aldrich (Sigma, SHCLNG-NM_002065 TRCN0000343991 and SHCLNG-NM_002065, TRCN0000343992). pLK0.5 vectors containing shRNA control targeting non-mammalian turboGFP were also purchased from Addgene (Sigma, SHC 202). Vectors were packaged along with pCMV-dR8.2 dvpr (Addgene, 8455) and pCMV-VSV-G (Addgene, 8454) into HEK293FT cells. HEK293FT cells were seeded into 100 mm dishes and grown to 65% - 80% confluence in DMEM with L-Glutamine and Sodium Pyruvate (Corning, MT10013CV) supplemented with 10% Fetal Bovine Serum (Omega Scientific, FB-11), 1% Glutamax, and 1% non-essential amino acids (Gibco, 1140). HEK293FT cells were transfected with 829 μL of .020 μg/μl plasmid solution (8 μg shRNA plasmids, 8 μg pCMV-dR8.9, 2 μg pCMV-VSVG) in OptiMEM (Gibco, 11058021) and Fugene HD (Promega, E2312). Media was collected 48 h and 60 h after transfection and concentrated using Lenti-X Concentrator (Clontech, 631231). hESC lines grown until 80% confluency were transduced with 5 – 10 x 105 lentiviral particles and 6 μg/mL polybrene for 8 h on D1. Transduction was repeated on D2 and D3, doubling viral concentration each successive day. Transduced cell lines were selected with 1 μg/mL puromycin on D5 – 8.
cDNA transfection
pLJM1 vectors FLAG Raptor Rheb 15, FLAG Raptor, and Empty were purchased from Addgene (26634, 26633, 91980). Vectors were a gift from the laboratories of David Sabatini and Joshua Mendell. Vectors were packaged along with pCMV-dR8.2 dvpr (Addgene, 8455) and pCMV-VSV-G (Addgene, 8454) into HEK293FT cells. HEK293FT cells were seeded into 100 mm dishes and grown to 65% - 80% confluence in DMEM with L-Glutamine and Sodium Pyruvate (Corning, MT10013CV) supplemented with 10% Fetal Bovine Serum (Omega Scientific, FB-11), 1% Glutamax, and 1% non-essential amino acids (Gibco, 1140). HEK293FT cells were transfected with 829 μL of .020 μg/μl plasmid solution (8 μg shRNA plasmids, 8 μg pCMV-dR8.9, 2 μg pCMV-VSVG) in OptiMEM (Gibco, 11058021) and Fugene HD (Promega, E2312). Media was collected 48 h and 60 h after transfection and concentrated using Lenti-X Concentrator (Clontech, 631231). hESC lines grown until 80% confluency were transduced with 5 – 10 x 105 lentiviral particles and 6 μg/mL polybrene for 8h on D1. Transduction was repeated on D2 and D3, doubling viral concentration each successive day. Transduced cell lines were selected with 1 μg/mL puromycin on D5 – 8.
Media preparation for metabolite extraction and UHPLC-MS processing
For Gln-free conditions, ectoderm differentiation media (as described above) was made with DMEM/F-12 w/o glutamine and in the absence of 1% Glutamax. For glutaminolysis-inhibited conditions, ectoderm differentiation media (as described above) was made with DMEM/F-12 supplemented with 1 μM CB-839. To achieve Gln-starvation conditions, Gln depleted media was additionally supplemented with 1 mM L-methionine sulfoximine (Millipore Sigma, 91016). For Gln-replete conditions, ectoderm differentiation media was made with DMEM/F-12 w/o glutamine and .01% Glutamax. For Glu conversion to Gln isotopologue tracing, cells were labeled with 0.5mM 13C5-Glu for 1 h under defined Gln perturbation conditions.
Metabolite extraction and UHPLC-MS processing
Ultra-high performance liquid chromatography and mass spectrometry was performed as previously described (Lu et al., 2019). To extract intracellular metabolites, cells were washed with warm PBS for less than 10 s, rinsed with ice-cold 150 mM ammonium acetate (pH 7.3), and quenched with 1 mL/well of cold 80% MeOH in water. Wells were scraped and contents transferred to Eppendorf tubes and vortexed for 10 s. 5 nmol D/L-norvaline was added to each cell suspension as an internal control, and tubes were centrifuged at 16,000 x g for 15 m at 4°C. The supernatant was transferred into glass vials and dried under a vacuum. Dried metabolites were re-suspended in 50% ACN:water and one-tenth was loaded onto a Luna 3um NH2 100A (150 × 2.0 mm) column (Phenomenex). The chromatographic separation was performed on an Ultimate 3000RSLC or a Vanquish Flex (Thermo Scientific) with mobile phases A (5 mM NH4AcO pH 9.9) and B (ACN) and a flow rate of 200 μl/min. A linear gradient from 15% A to 95% A over 18 m was followed by 9 m isocratic flow at 95% A and re-equilibration to 15% A. Metabolites were detection with a Thermo Scientific Q Exactive mass spectrometer run with polarity switching (+3.5 kV/− 3.5 kV) in full scan mode with an m/z range of 65-975. TraceFinder 4.1 (Thermo Scientific) was used to quantify the targeted metabolites by area under the curve using expected retention time and accurate mass measurements (< 5 ppm). Experiments were performed with three biological replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Metabolomics data analysis
Metabolomics analysis was performed as previously described (Waters et al., 2019). Data analysis was performed using the statistical language R v3.6.3 and Bioconductor v3.6.1 packages(Huber et al., 2015; R Core Team, 2017). Metabolite abundance was normalized per μg of protein content per metabolite extraction, and metabolites not detected were set to zero.
Pathway-level relative amounts metabolite set variation analysis (MSVA) was performed using R Bioconductor package GSVA v1.26.0 (Hanzelmann et al., 2013). Metabolite normalized relative abundances were standardized using a log2(normalized amounts + 1) transformation, and metabolites per sample were converted to a pathways per sample matrix using function gsva() with parameters “method = gsva, rnaseq = FALSE, abs.ranking = FALSE, min.sz = 5, max.sz = 500”. GSVA pathway enrichment scores were then extracted and significance testing between conditions was calculated using R Bioconductor package limma v3.34.9, fitting a linear model to each metabolite and assessing differences in normalized abundance using an empirical Bayes moderated F-statistic with an adjusted P value threshold of 0.05, using the Benjamini-Hochberg false discovery rate of 0.05 (Benjamini and Hochberg, 1995; Ritchie et al., 2015). Pathway metabolite sets were constructed using the KEGG Compound Database and derived from the existing Metabolite Pathway Enrichment Analysis (MPEA) toolbox (Kanehisa et al., 2012; Kankainen et al., 2011).
RNA-Seq analysis
RNA-Seq data analysis was performed as previously described (Lu et al., 2019). TPM values were extracted from GEO:GSE127270 for genes that were determined to be differentially expressed via statistical testing using R Bioconductor package DESeq2 v1.18.1 (Love et al., 2014). Differentially expressed genes were identified using Wald significance testing, with an adjusted P value threshold below 0.05 for one or more comparisons. A variance stabilized transform matrix (VST) was generated using DESeq2, and subset for the differentially expressed genes (DEGs) identified above. VST values were row scaled by subtracting the average mean per gene row between samples, and then averaged per condition. Heatmaps were generated using R Bioconductor package pheatmap v1.0.8 (Kolde, 2015).
Comparison of Gln synthesis to Gln consumption
RNA-Seq data from a previous publication (Lu et al., 2019) and single cell RNA-Seq data from Tyser et al. 2020 (Tyser et al., 2020) were used to query the ratio of Gln consumption transcripts [average(GLS1 + GLS2)] to Gln synthesis transcripts [GLUL]. For the single cell RNA-Seq data, the ratio was calculated for each individual cell and then normalized to the average Epiblast value. For bulk RNA-Seq, each replicate was normalized to the average hPSC value.
Statistical testing
Statistical testing for all data except for RNA-Seq and UHPLC-MS metabolomics was performed using Prism 7 and 8 (Graphpad). Details for statistical testing of RNA-Seq and UHPLC-MS metabolomics data are included in the section above. Unless otherwise noted, each experiment is n=3, where n represents the number of independent biological replicates. Statistical details can be found in each figure legend. Parametric data were analyzed using Student’s t tests or one-/two-way ANOVA with multiple comparisons correction. Metabolite pathway enrichment values were quantified using empirical Bayes linear modelling with multiple hypothesis correction. P values ≤ 0.05 were considered significant for all data assayed. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Supplementary Material
Supplemental Table 1. Spreadsheet of normalized transcript abundance values from RNA-Sequencing data and UHPLC-MS metabolomics relative abundance, pathway enrichment values, and isotopologue tracing amounts. Related to Figures 1, 2, 3 and Supplemental Figure 2.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Glutamine Synthetase; 1/500 | BD Biosciences | Cat#610517; RRID:AB_2313837 |
| Rabbit anti-PAX6; 1/500 | Cell Signaling Technology | Cat#60433; RRID:AB_2797599 |
| Rabbit anti-Pyruvate Dehydrogenase E1-alpha subunit (phospho S293); 1/500 | Abcam | Cat#ab177461; RRID:AB_2756339 |
| Rabbit anti-Phospho-p70 S6 Kinase (Thr389) (108D2); 1/500 | Cell Signaling Technology | Cat#9234; RRID:AB_2269803 |
| Rabbit anti-p70 S6 Kinase (49D7); 1/500 | Cell Signaling Technology | Cat#2708; RRID:AB_390722 |
| Rabbit anti-OCT-4; 1/200 | Cell Signaling Technology | Cat#2750; RRID:AB_823583 |
| Mouse anti-OCT-4 (D7O5Z); 1/200 | Cell Signaling Technology | Cat#75463; RRID:AB_2799870 |
| Goat anti-human NANOG; 5 μg/mL | R&D Systems | Cat#AF1997; RRID:AB_355097 |
| Mouse anti-SOX2; 1/400 | Cell Signaling Technology | Cat#4900; RRID:AB_10560516 |
| Mouse anti-MAP2; 1/500 | Invitrogen | Cat#MA5-12826; RRID:AB_10976831 |
| Rabbit anti-OTX2; 4 μg/mL | Abcam | Cat#ab114138 |
| Goat anti-Brachyury/TBXT; 15 μg/mL | R&D Systems | Cat# AF2085; RRID:AB_2200235 |
| Rabbit anti-SLUG/SNAI2; 1/400 | Cell Signaling Technology | Cat#9585; RRID:AB_2239535 |
| Mouse anti-SOX17; 1/100 | Invitrogen | Cat#MA5-24891; RRID:AB_2725395 |
| Rabbit anti-FoxA2/HNF3β (D56D6); 1/400 | Cell Signaling Technology | Cat#8186; RRID:AB_10891055 |
| Mouse anti-β-tubulin; 1/1000 | Millipore Sigma | Cat#T4026; RRID:AB_477577 |
| Alexa Fluor 647 Rat Anti-Histone H3 (pS28) | BD Biosciences | Cat#558217; RRID:AB_397065 |
| APC anti-DYKDDDDK Tag Antibody | Biolegend | Cat#637308; RRID:AB_2561497 |
| PE Mouse Anti-SNAI2/Slug | BD Biosciences | Cat#564615; RRID:AB_2738866 |
| Alexa Fluor 488 Mouse Anti-Human Sox17 | BD Biosciences | Cat#562205; RRID:AB_10893402 |
| PerCPCy5.5 Mouse anti-Human PAX6 | BD Biosciences | Cat#562388; RRID:AB_11153319 |
| Alexa Fluor 488 Mouse anti-MAP2B | BD Biosciences | Cat#560399; RRID:AB_1645358 |
| PE Mouse anti-Human Sox1 | BD Biosciences | Cat#561592; RRID:AB_10714631 |
| Alexa Fluor 488 Mouse anti-Oct3/4 | BD Biosciences | Cat#560253; RRID:AB_1645304 |
| V450 Mouse Anti-Sox2 | BD Biosciences | Cat#561610; RRID:AB_10712763 |
| PerCP-Cy5.5 Mouse IgG2a, κ Isotype Control | BD Biosciences | Cat#558020; RRID:AB_396989 |
| Alexa Fluor 700 Mouse IgG1, κ Isotype Control | BD Biosciences | Cat#557882; RRID:AB_396920 |
| Alexa Fluor 488 Mouse IgG1 κ Isotype Control | BD Biosciences | Cat#557782; RRID:AB_396870 |
| PE Mouse IgG1 k Isotype Control | BD Biosciences | Cat#554680 RRID:AB_395506 |
| V450 Mouse IgG1, κ Isotype Control | BD Biosciences | Cat#560373; RRID:AB_1645606 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fixable Viability Dye APC-Cy7 eFluor780 | Thermofisher | Cat#65-0865-14 |
| 7-AAD Staining Solution | BD Pharmingen | Cat#559925 |
| ActinRed™ 555 ReadyProbes® Reagent Protocol (Rhodamine phalloidin) | Invitrogen | Cat#R37112 |
| Gentle Cell Dissociation Reagent | Stemcell Technologies | Cat#07174 |
| mTeSR1 Plus | Stemcell Technologies | Cat#05825 |
| DMEM/F12 | Gibco | Cat#11320033 |
| ROCK Inhibitor; Y-27632 | Stemcell Technologies | Cat#72304 |
| Monothioglycerol | Millipore Sigma | Cat#M6145 |
| BSA Fraction V, 7.5% | Gibco | Cat#15260037 |
| 2-mercaptoethanol | Thermofisher | Cat#21985-023 |
| GlutaMAX Supplement | Thermofisher | Cat#35050-061 |
| N2 Supplement | Gibco | Cat#17502048 |
| B27 Supplement | Gibco | Cat#17504044 |
| SB43154 | Stemgent | Cat#04-0010 |
| Dorsomorphin | Stemgent | Cat#04-0024 |
| Human Recombinant Insulin | Millipore Sigma | Cat#11376497001 |
| Transferrin from human serum | Millipore Sigma | Cat#10652202001 |
| Chemically Defined Lipid Concentrate (CDLC) | Gibco | Cat#11905031 |
| Human Recombinant VEGF-165 | Stemcell Technologies | Cat#78073.1 |
| Human Recombinant BMP-4 | PeproTech | Cat#120-05ET |
| Human Recombinant FGF2 | Stemcell Technologies | Cat#78003 |
| Human Recombinant Activin A | Stemcell Technologies | Cat#78001.1 |
| CHIR99021 (GSK3i) | Stemcell Technologies | Cat#72052 |
| PI-103 Hydrochloride | Tocris/Fisher Scientific | Cat#29-301 |
| Stemolecule LDN-1931189 | Stemgent | Cat#04-0074 |
| 10% Fetal Bovine Serum | Omega Scientific | Cat#FB-11 |
| MEM Non-Essential Amino Acids | Gibco | Cat#11140-050 |
| Matrigel | Corning/Fisher Scientific | Cat#CB-4023A |
| RNAse-Free DNase Set | Qiagen | Cat#79254 |
| DMEM/F12, no glutamine | Gibco | Cat#21331020 |
| L-GLUTAMINE (AMIDE-15N, 98%) | Cambridge Isotope Laboratories | Cat#NLM-577 |
| DPBS 1x w/o calcium & magnesium | Corning | Cat#21031CV |
| L-methionine sulfoximine | Millipore Sigma | Cat#91016 |
| Dimethyl 2-oxoglutarate | Millipore Sigma | Cat#ES-008 |
| Glucosamine 6-phosphate | Millipore Sigma | Cat#G5509 |
| L-Glutathione reduced | Millipore Sigma | Cat#G6529 |
| Ammonium Chloride | Millipore Sigma | Cat#A9434 |
| 6-Diazo-5-oxo-L-norleucine | Millipore Sigma | Cat#D2141 |
| CB-839 | Selleckchem | Cat#S7655 |
| 1x BD Perm/Wash Buffer | BD Biosciences | Cat#554714 |
| SYBR Green | Roche | Cat#04887352001 |
| 1x protease inhibitor | Sigma | Cat#P8340 |
| 1x phosphatase inhibitor | Sigma | Cat#P5726 |
| 1x phosphatase inhibitor | Sigma | Cat#P0044 |
| Odyssey Blocking Buffer TBS | Li-Cor | Cat#927-50010 |
| DMEM with L-Glutamine and Sodium Pyruvate | Corning | Cat#MT10013CV |
| OptiMEM | Gibco | Cat#11058021 |
| Fugene | Promega | Cat#E2312 |
| Non Essential Amino Acids | Gibco | Cat#1140 |
| Lenti-X Concentrator | Clontech | Cat#631231 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#555028 |
| iSCRIPT cDNA Synthesis Kit | Bio-Rad | Cat#1708841 |
| MycoAlert Detection Kit | Lonza | Cat#LT08-418 |
| BCA Protein Assay Kit | Pierce/Thermofisher | Cat#23225 |
| Deposited Data | ||
| RNA-Sequencing Data for Nutrient Balanced hPSC and Trilineage Differentiation to EN/EC/ME | (Lu et al., 2019) | GEO: GSE127270 |
| UHPLC-MS quantification of intracellular abundance of metabolites. | This Manuscript | Supplemental Table S1 |
| Experimental Models: Cell Lines | ||
| H9 (WA09); Female | UCLA BSCRC hESC Core Bank | RRID:CVCL_9773 |
| UCLA-1; Female | UCLA BSCRC hESC Core Bank | RRID:CVCL_9951 |
| HEK293FT | Life Technologies | Cat#31985 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Primers for RT-qPCR, see Method Details | IDT | N/A |
| Recombinant DNA | ||
| pLKO-shGLUL-1 | Sigma-Aldrich | SHCLNG-NM 002065 TRCN0000343991 |
| pLKO-shGLUL-2 | Sigma-Aldrich | SHCLNG-NM 002065, TRCN0000343992 |
| shRNA NTC (non-mammalian turboGFP) | N/A | Addgene: SH202 |
| pCMV-VSV-G | Laboratory of Robert Weinberg(Stewart et al., 2003) | Addgene: 8454 |
| pCMV-dR8.2 dvpr | Laboratory of Robert Weinberg(Stewart et al., 2003) | Addgene: 8455 |
| pLJM1 FLAG Raptor Rheb 15 | Laboratory of David Sabatini(Sancak et al., 2010) | Addgene: 26634 |
| pLJM1 FLAG Raptor | Laboratory of David Sabatini(Sancak et al., 2010) | Addgene: 26633 |
| pLJM1 Empty | Laboratory of Joshua Mendell(Golden et al., 2017) | Addgene: 91980 |
| Software and Algorithms | ||
| Seahorse Wave Desktop Software | Agilent Technologies | https://www.agilent.com/en/products/cell-analysis/cell-analysis-software/data-analysis/wave-desktop-2-6 |
| Leica Application Suite (LAS) X Software | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Salmon v0.9.1 | (Patro et al., 2017) | https://combine-lab.github.io/salmon/ |
| Tximport v1.6.0 | (Soneson et al., 2015) | http://bioconductor.org/packages/release/bioc/html/tximport.html |
| DESeq2 v1.18.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| MitoMiner v4.0 | (Calvo et al., 2016) | http://mitominer.mrc-mbu.cam.ac.uk/release-4.0/begin.do |
| Pheatmap v1.0.8 | (Kolde, 2015) | https://github.com/raivokolde/pheatmap |
| Gplots v3.0.1 | (Gregory Warnes, 2016) | https://cran.r-project.org/web/packages/gplots/index.html |
| GSVA v1.26.0 | (Hanzelmann et al., 2013) | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| Prism 7 and 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| TraceFinder v3.3 | Thermofisher | https://www.thermofisher.com/order/catalog/product/OPTON-30491 |
| Limma v3.34.9 | (Ritchie et al., 2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R v3.4.4 | The R Project for Statistical Computing | https://www.r-project.org |
| Bioconductor v3.6 | (Huber et al., 2015) | https://www.bioconductor.org |
| Other | ||
HIGHLIGHTS.
All three lineages make de novo Gln, but only ectoderm can survive in Gln-free media.
Intracellular Gln is more crucial than cytokines for enabling ectoderm specification.
Exogenous Gln is the preferred precursor for α-KG in mesoderm and endoderm.
Transcriptome of a human embryo shows unique Gln enzyme-encoding expression patterns.
ACKNOWLEDGEMENTS
V.L. is supported by F31HD097960 and UCLA Graduate Division. V.L and I.J.R. are supported by UCLA Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research. M.A.T. is supported by the Air Force Office of Scientific Research (FA9550-15-1-0406), by the Department of Defense (W81XWH2110139) and by the National Institutes of Health (R01GM073981, R21CA227480, R01GM127985, and P30CA016042). Confocal laser scanning microscopy was performed at the Advanced Light Microscopy/Spectroscopy Laboratory and the Leica Microsystems Center of Excellence at the California NanoSystems Institute at UCLA with funding support from NIH Shared Instrumentation Grant S10OD025017 and NSF Major Research Instrumentation grant CHE-0722519. We thank Jinghua Tang (BSCRC, UCLA) for hPSC lines, Tom Graeber and Johanna ten Hoeve (Metabolomics Center, UCLA) for metabolomics processing, and Justin Golovato and Stephen Benz (NantOmics, LLC.) for RNA-Seq, library preparation, and pre-processing. We thank Kathrin Plath, William Tu, Caius Radu, Heather Christofk, David Nathanson, William Lowry, Tara TeSlaa, Woosuk Kim and Jason S. Hong for helpful discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aaron J, and Chew TL (2021). A guide to accurate reporting in digital image processing - can anyone reproduce your quantitative analysis? J Cell Sci 134. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57, 289–300. [Google Scholar]
- Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al. (2015). Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab 22, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, and Thompson CB (2015). Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, and Sabatini DM (2015). Nutrient-sensing mechanisms across evolution. Cell 161, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi F, Sharpley MS, Nagaraj R, Roy SS, and Banerjee U (2020). Glycolysis-Independent Glucose Metabolism Distinguishes TE from ICM Fate during Mammalian Embryogenesis. Dev Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, Prestegaard JH, and Dalton S (2017). MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 21, 502–516 e509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, and Newsholme P (2018). Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B, Zhou Q, He J, Qiu Y, Wen M, et al. (2019). Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep 28, 1136–1143 e1134. [DOI] [PubMed] [Google Scholar]
- Golden RJ, Chen B, Li T, Braun J, Manjunath H, Chen X, Wu J, Schmid V, Chang TC, Kopp F, et al. (2017). An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 542, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory Warnes BB, Lodewijk Bonebakker, Robert Gentleman, Wolfgang Huber, Andy Liaw, Thomas Lumley, Martin Maechler, Arni Magnusson, Steffen Moeller, Marc Schwartz, Bill Venables (2016). gplots: Various R Programming Tools for Plotting Data (CRAN: R-Project). [Google Scholar]
- Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, Gelot A, Suc AL, Hohne W, Schliess F, et al. (2005). Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353, 1926–1933. [DOI] [PubMed] [Google Scholar]
- Haberle J, Gorg B, Toutain A, Rutsch F, Benoist JF, Gelot A, Suc AL, Koch HG, Schliess F, and Haussinger D (2006). Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis 29, 352–358. [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm MB, Bastani NE, Holme AM, Zucknick M, Jansson T, Refsum H, Morkrid L, Blomhoff R, Henriksen T, and Michelsen TM (2017). Uptake and release of amino acids in the fetal-placental unit in human pregnancies. PLoS One 12, e0185760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaq SH, Mendoza A, Fox SD, and Helman LJ (2019). Glutamine synthetase is necessary for sarcoma adaptation to glutamine deprivation and tumor growth. Oncogenesis 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, Tagliabracci VS, and Guan KL (2015). Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JH, Kang KW, Kim J, Hong SC, Park Y, and Kim BS (2016). CXCR2 Inhibition in Human Pluripotent Stem Cells Induces Predominant Differentiation to Mesoderm and Endoderm Through Repression of mTOR, beta-Catenin, and hTERT Activities. Stem Cells Dev 25, 1006–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, and Tanabe M (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankainen M, Gopalacharyulu P, Holm L, and Oresic M (2011). MPEA--metabolite pathway enrichment analysis. Bioinformatics 27, 1878–1879. [DOI] [PubMed] [Google Scholar]
- Katt WP, Lukey MJ, and Cerione RA (2017). A tale of two glutaminases: homologous enzymes with distinct roles in tumorigenesis. Future Med Chem 9, 223–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2015). Package ‘pheatmap’ - Pretty Heatmaps (CRAN: R-Project). [Google Scholar]
- Kung HN, Marks JR, and Chi JT (2011). Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet 7, e1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu V, Dahan P, Ahsan FM, Patananan AN, Roy IJ, Torres A Jr., Nguyen RMT, Huang D, Braas D, and Teitell MA (2019). Mitochondrial metabolism and glutamine are essential for mesoderm differentiation of human pluripotent stem cells. Cell Res 29, 596–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsboom G, Zhang GF, Pohl-Avila N, Zhang Y, Yuan Y, Kang H, Hao B, Brunengraber H, Malik AB, and Rehman J (2016). Glutamine Metabolism Regulates the Pluripotency Transcription Factor OCT4. Cell Rep 16, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascetti VL, and Pedersen RA (2016). Human-Mouse Chimerism Validates Human Stem Cell Pluripotency. Cell Stem Cell 18, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre KR, Vincent KMM, Hayward CE, Li X, Sibley CP, Desforges M, Greenwood SL, and Dilworth MR (2020). Human placental uptake of glutamine and glutamate is reduced in fetal growth restriction. Sci Rep 10, 16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng D, Frank AR, and Jewell JL (2018). mTOR signaling in stem and progenitor cells. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, et al. (2015). Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab 21, 392–402. [DOI] [PubMed] [Google Scholar]
- Nazareth EJP, Rahman N, Yin T, and Zandstra PW (2016). A Multi-Lineage Screen Reveals mTORC1 Inhibition Enhances Human Pluripotent Stem Cell Mesendoderm and Blood Progenitor Production. Stem Cell Reports 6, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu J (2001). Glutamine in the fetus and critically ill low birth weight neonate: metabolism and mechanism of action. J Nutr 131, 2585S–2589S; discussion 2590S. [DOI] [PubMed] [Google Scholar]
- Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, Mamede J, Cretenet G, Mongellaz C, An X, et al. (2014). Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell 15, 169–184. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qie S, He D, and Sang N (2019). Overview of Glutamine Dependency and Metabolic Rescue Protocols. Methods Mol Biol 1928, 427–439. [DOI] [PubMed] [Google Scholar]
- R Core Team (2017). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Reid MA, Dai Z, and Locasale JW (2017). The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, and Sabatini DM (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmied C, and Jambor HK (2020). Effective image visualization for publications - a workflow using open access tools and concepts. F1000Res 9, 1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, and Kume S (2014). Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 19, 780–794. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtman ER (2004). Regulation of Glutamine Synthetase Activity. EcoSal Plus 1. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Heemskerk I, and Gumucio DL (2019). Opening the black box: Stem cell-based modeling of human post-implantation development. J Cell Biol 218, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO, Weinstock A, Wagner A, et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol 17, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Chaikovsky AC, Lipchina I, Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D, and Teitell MA (2016). alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab 24, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, and Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Tohyama S, Fujita J, Hishiki T, Matsuura T, Hattori F, Ohno R, Kanazawa H, Seki T, Nakajima K, Kishino Y, et al. (2016). Glutamine Oxidation Is Indispensable for Survival of Human Pluripotent Stem Cells. Cell Metab 23, 663–674. [DOI] [PubMed] [Google Scholar]
- Tyser RCV, Mahammadov E, Nakanoh S, Vallier L, Scialdone A, and Srinivas S (2020). A spatially resolved single cell atlas of human gastrulation. bioRxiv. [Google Scholar]
- Vardhana SA, Arnold PK, Rosen BP, Chen Y, Carey BW, Huangfu D, Fontaine CC, Thompson CB, and Finley LWS (2019). Glutamine independence is a selectable feature of pluripotent stem cells. Nat Metab 1, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, et al. (2014). UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A 111, 960–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LR, Ahsan FM, Ten Hoeve J, Hong JS, Kim DNH, Minasyan A, Braas D, Graeber TG, Zangle TA, and Teitell MA (2019). Ampk regulates IgD expression but not energy stress with B cell activation. Sci Rep 9, 8176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, and Thompson CB (2010). The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24, 2784–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, and Sabatini DM (2017). The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab 26, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, Zhang H, Knudsen E, Zhang GF, Hilton MJ, et al. (2019). Glutamine Metabolism Regulates Proliferation and Lineage Allocation in Skeletal Stem Cells. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Su P, Wang L, Chen J, Zimmermann M, Genbacev O, Afonja O, Horne MC, Tanaka T, Duan E, et al. (2009). mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc Natl Acad Sci U S A 106, 7840–7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Spreadsheet of normalized transcript abundance values from RNA-Sequencing data and UHPLC-MS metabolomics relative abundance, pathway enrichment values, and isotopologue tracing amounts. Related to Figures 1, 2, 3 and Supplemental Figure 2.
Data Availability Statement
Raw RNA-Seq data for nutrient balanced hPSC and tri-lineage differentiation to ectoderm, mesoderm, and endoderm were deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE127270. All raw and processed UHPLC-MS metabolomics data from this study are in Excel spreadsheet format as Supplemental Table 1. All data were analyzed with standard programs and packages, as detailed above. All R Jupyter notebooks and supplemental files used to process RNA-Seq and UHPLC-MS data will be uploaded upon manuscript press to Github at https://github.com/fahsan/GlnFateGatekeeper. All raw images, immunoblots, and values for quantification are available from the corresponding author and lead contact on request.