

Abstract
Cardiometabolic diseases are the leading cause of death worldwide. Despite a known genetic component, our understanding of these diseases remains incomplete. Here we analyzed the contribution of rare variants to 57 diseases and 26 cardiometabolic traits, using data from 200,337 UK Biobank participants with whole-exome sequencing. We identified 57 gene-based associations, with broad replication of novel signals in Geisinger MyCode. There was a striking risk associated with mutations in known Mendelian disease genes, including MYBPC3, LDLR, GCK, PKD1 and TTN. Many genes showed independent convergence of rare and common variant evidence, including an association between GIGYF1 and type 2 diabetes. We identified several large-effect associations for height, and 18 unique genes associated with blood lipid or glucose levels. Finally, we found that between 1.0 and 2.4% of participants carried rare potentially pathogenic variants for cardiometabolic disorders. These findings may facilitate studies aimed at therapeutics and screening of these common disorders.
Over the past decade, genome-wide association studies (GWAS) have provided critical insights into the genetic architecture of cardiometabolic traits and diseases, through the identification of thousands of associated genomic loci1-7. Typically, these studies focused on common variants, which individually often confer small effect sizes and do not always directly implicate causal genes. Familial linkage and targeted sequencing studies have identified numerous Mendelian causes of cardiovascular disease8-11, although such studies have been limited by small sample size. A handful of larger case-control studies have had some success in discovery of genes harboring large-effect variants for adult-onset disease, for example, for myocardial infarction12 and diabetes13.
Analysis of large-scale population-based sequencing data offers multiple advantages over conventional common variant association studies. First, sequencing provides the opportunity to identify rare and ultra-rare genetic variants, which often would not have been genotyped, or typed inaccurately, by array genotyping and imputation14. Second, exome sequencing—which is focused on the protein-coding regions of the genome—may directly implicate genes in phenotype variability through burden testing of multiple rare protein-coding variants15. Third, analysis of rare coding variation can help establish the directionality of impaired gene function through the analysis of loss-of-function alleles, a feature that can be informative both for understanding disease mechanisms and for potential therapeutic targeting. In cardiovascular research, a paradigmatic example of this approach is PCSK9, which progressed from gene discovery to an available therapeutic in just over a decade16-18. Finally, sequencing at scale enables assessment of penetrance, risk, and carrier frequencies for rare deleterious genetic variation14,19.
Here we present an analysis of the second release of whole-exome sequencing data from the UK Biobank, a population-based study, consisting of sequencing data on approximately 200,000 individuals20. Through exome-wide gene-based analyses, we evaluate the contributions of rare (minor allele frequency (MAF) < 0.1%) damaging variants to 83 traits, including anthropometric traits, cardiometabolic diseases, metabolic blood biomarkers, electrocardiographic traits and cardiac magnetic resonance imaging traits. We replicate novel signals in the Geisinger MyCode cohort (also known as the DiscovEHR study), comprising 166,000 participants with exome sequencing data. Finally, we describe the frequency of mutations in genes underlying cardiovascular diseases and monogenic diabetes.
Results
Exome-wide rare variant analysis of 83 traits identifies 57 significant associations.
After sample level quality control procedures, we identified 200,337 UK Biobank participants with a mean age of 57 years at enrollment and 68 years at last follow-up, of which 55% were female and 87% were of white-British European ancestry (Supplementary Fig. 1). Table 1 provides the baseline characteristics and case number for each representative disease phenotype. A total of 12,756,075 distinct autosomal genetic variants were available from the exome sequencing data after quality control, of which 12,553,131 had a MAF < 0.1%.
Table 1 ∣.
Baseline characteristics of participants in the UK Biobank with exome sequencing data
| Participants, n | 200,337 |
| Female, n (%) | 110,359 (55.1) |
| European ancestry, n (%) | 174,879 (87.3) |
| Age at baseline, mean (s.d.) | 56.97 (8.10) |
| Age at last follow-up, mean (s.d.) | 67.82 (8.07) |
| Height in cm, mean (s.d.) | 168.47 (9.27) |
| Body mass index, median (IQR) | 26.70 (5.72) |
| Cardiovascular and metabolic diseases | |
| Atrial fibrillation, n (%) | 12,277 (6.1) |
| Supraventricular arrhythmia, n (%) | 2,075 (1.0) |
| Ventricular arrhythmia, n (%) | 2,072 (1.0) |
| Mitral valve disease, n (%) | 3,898 (1.9) |
| Hypertension, n (%) | 74,347 (37.1) |
| Heart failure, n (%) | 5,344 (2.7) |
| Dilated cardiomyopathy, n (%) | 377 (0.2) |
| Hypertrophic cardiomyopathy, n (%) | 220 (0.1) |
| Myocardial Infarction, n (%) | 6,238 (3.1) |
| Hypercholesterolemia, n (%) | 42,799 (21.4) |
| Diabetes type 2, n (%) | 14,607 (7.3) |
| Hypothyroidism, n (%) | 14,097 (7.0) |
| Malignancy | |
| Breast cancer, n (%) | 8,112 (7.4) |
| Colorectal cancer, n (%) | 2,733 (1.4) |
| Other medical conditions | |
| Chronic kidney disease, n (%) | 6,415 (3.2) |
| Cataracts, n (%) | 21,762 (10.9) |
Values are presented as number (percentage) unless otherwise specified. s.d., standard deviation; IQR, interquartile range.
We performed exome-wide gene-based tests across 57 medical conditions (Supplementary Tables 1 and 2) and 26 quantitative cardiometabolic traits (Supplementary Table 3), testing rare loss-of-function (LOF) and missense variants. Exome-wide gene-based analyses showed good calibration of P-values across all performed tests (Supplementary Figs. 2-4 and Supplementary Note), although three traits (height, weight and QTc) showed moderate inflation of test statistics (1.1 ≤ λ < 1.25; Supplementary Fig. 4). In an attempt to identify the cause of the inflation, we analyzed rare synonymous variants. Exome-wide analysis of rare synonymous variation produced good calibration of test statistics (Supplementary Fig. 5), indicating that a large proportion of the observed inflation was due to polygenicity rather than bias. We then performed a number of additional sensitivity analyses. When we restricted our analyses to individuals of homogeneous white-British European ancestry, all identified gene-phenotype associations still showed strong evidence of association with comparable effect size estimates (Supplementary Tables 4 and 5 Supplementary Figs. 6 and 7). Furthermore, when restricting to LOF variants only, we generally observed comparable effect size estimates across the significant associations (Supplementary Figs. 8 and 9). One notable exception was the association between GCK and type 2 diabetes, for which effect sizes were attenuated by the inclusion of predicted-deleterious missense variants.
In total, we identified 57 significant associations across 83 traits at an overall FDR Q < 0.01, which was equivalent to P < 5.44 x 10−7. All tests reaching Q < 0.05 are displayed in Supplementary Tables 4 and 5. Of the 57 significant associations, 42 were known, while 15 represented novel associations (Tables 2 and 3). Given the many parallel analyses of the same dataset, we define ‘novel’ to indicate rare variant associations that were not described prior to the release of the UK Biobank exomes. Across the disease associations where rare variants were associated with increased disease risk, the median OR was large at 5.6 (Q1-Q3: 2.4-13.0). The median absolute beta for significant quantitative associations was 0.63 s.d. (Q1-Q3: 0.43-0.85).
Table 2 ∣.
Gene associations with cardiometabolic and other diseases at FDR Q-value < 0.01
| Trait | Gene | Carriers | Cases among carriers (%) |
Cases among noncarriers (%) |
OR [95% CI] | P-value | Ref |
|---|---|---|---|---|---|---|---|
| Known associations | |||||||
| Hypertrophic cardiomyopathy | MYBPC3 | 93 | 9 (9.68) | 211 (0.11) | 120.38 [55.94-231.86] | 2.39 x 10−16 | 65,66 |
| Heart failure | TTN | 1,858 | 121 (6.51) | 5,223 (2.63) | 2.64 [2.15-3.20] | 2.14 x 10−18 | 67,68 |
| Dilated cardiomyopathy | TTN | 1,741 | 38 (2.11) | 339 (0.17) | 12.20 [8.44-17.09] | 4.14 x 10−27 | 68 |
| Atrial fibrillation | TTN | 1,858 | 211 (11.36) | 12,066 (6.08) | 2.06 [1.75-2.40] | 8.23 x 10−18 | 19,57,69 |
| Ventricular arrhythmia | TTN | 1,858 | 47 (2.53) | 2,025 (1.02) | 2.45 [1.77-3.30] | 4.18 x 10−8 | 28 |
| Diabetes type 2 | GCK | 64 | 31 (48.44) | 14,576 (7.28) | 13.98 [8.33-23.42] | 1.80 x 10−19 | 70 |
| Chronic kidney disease | PKD1 | 51 | 24 (47.06) | 6,391 (3.19) | 40.33 [21.27-76.24] | 3.54 x 10−25 | 71-73 |
| Hypercholesterolemia | LDLR | 104 | 74 (71.15) | 42,725 (21.34) | 13.11 [8.28-21.26] | 3.53 x 10−31 | 74-76 |
| APOB | 247 | 6 (2.43) | 42,793 (21.39) | 0.08 [0.03-0.18] | 4.27 x 10−13 | 77 | |
| PCSK9 | 258 | 20 (7.75) | 42,779 (21.38) | 0.26 [0.15-0.42] | 2.78 x 10−8 | 78 | |
| Hypothyroidism | TSHR | 304 | 48 (15.79) | 14,049 (7.02) | 2.53 [1.79-3.49] | 2.34 x 10−8 | 79 |
| TG | 785 | 97 (12.36) | 14,000 (7.02) | 1.83 [1.45-2.28] | 3.18 x 10−7 | 80,81 | |
| Novel associations * | |||||||
| Diabetes type 2 | GIGYF1 | 55 | 16 (29.09) | 14,591 (7.29) | 5.61 [2.90-10.32] | 3.04 x 10−7 | |
| CCAR2 | 26 | 11 (42.31) | 14,596 (7.29) | 12.79 [5.63-28.44] | 5.43 x 10−8 | ||
| Supraventricular tachycardia | TTN | 1,858 | 46 (2.48) | 2,029 (1.02) | 2.40 [1.73-3.23] | 7.88 x 10−8 | |
| Mitral valve disease | TTN | 1,858 | 81 (4.36) | 3,817 (1.92) | 2.31 [1.80-2.91] | 4.74 x 10−11 | |
P-values were computed using the saddle point approximation and were obtained from logistic mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs) and a sparse kinship matrix. P-values shown are unadjusted for multiple testing. Odds ratios (OR) and 95% confidence intervals (95% CI) were obtained from Firth’s regression models adjusting for sex, age, sequencing batch and associated PCs among unrelated individuals.
Novel indicates that rare variant associations were not reported prior to the release of UK Biobank exomes. Ref, references.
Table 3 ∣.
Gene associations for quantitative cardiometabolic traits at FDR Q-value < 0.01
| Trait | Gene | Carriers | Effect in s.d. [95% CI] | P-value | Ref |
|---|---|---|---|---|---|
| Known associations | |||||
| Height, cm | ADAMTS17 | 173 | −0.34 [−0.45, −0.24] | 6.14 x 10−11 | 82,83 |
| ACAN | 42 | −1.17 [−1.38, −0.96] | 6.86 x 10−28 | 84 | |
| NPR2 | 114 | −0.62 [−0.75, −0.49] | 1.26 x 10−21 | 85-87 | |
| IGF1R | 51 | −0.49 [−0.68, −0.30] | 4.56 x 10−7 | 88 | |
| ADAMTS10 | 24 | −0.76 [−1.03, −0.48] | 9.35 x 10−8 | 89,90 | |
| Glucose | GCK | 56 | 1.22 [0.96, 1.48] | 1.91 x 10−20 | 91 |
| HDL | ABCA1 | 236 | −0.92 [−1.03, −0.80] | 5.90 x 10−55 | 92 |
| APOA5 | 156 | −0.57 [−0.71, −0.43] | 4.30 x 10−15 | 12 | |
| ANGPTL3 | 310 | −0.35 [−0.45, −0.25] | 1.47 x 10−11 | 93 | |
| PLIN1 | 315 | 0.40 [0.30, 0.49] | 8.01 x 10−15 | 37 | |
| LCAT | 27 | −1.05 [−1.39, −0.71] | 1.34 x 10−9 | 94 | |
| LPL | 78 | −0.83 [−1.03, −0.63] | 3.68 x 10−16 | 92 | |
| LIPC | 320 | 0.51 [0.41, 0.60] | 1.23 x 10−23 | 95 | |
| APOB | 201 | 0.37 [0.25, 0.50] | 5.28 x 10−9 | 96 | |
| CETP | 58 | 0.89 [0.66, 1.12] | 6.80 x 10−14 | 97 | |
| LDL | PCSK9 | 245 | −0.94 [−1.06, −0.81] | 9.19 x 10−49 | 16 |
| ANGPTL3 | 363 | −0.42 [−0.52, −0.32] | 7.98 x 10−16 | 93 | |
| APOB | 237 | −2.24 [−2.37, −2.11] | 2.94 x 10−262 | 98 | |
| LDLR | 98 | 0.66 [0.47, 0.86] | 4.06 x 10−11 | 99 | |
| Triglycerides | APOA5 | 180 | 0.91 [0.77, 1.05] | 2.31 x 10−36 | 12,100 |
| ANGPTL3 | 363 | −0.67 [−0.77, −0.57] | 2.03 x 10−39 | 93 | |
| ANGPTL4 | 173 | −0.45 [−0.59, −0.30] | 1.32 x 10−9 | 35 | |
| APOB | 237 | −1.45 [−1.57, −1.32] | 3.21 x 10−117 | 77 | |
| LPL | 86 | 0.65 [0.45, 0.85] | 4.68 x 10−10 | 100 | |
| APOC3 | 22 | −1.17 [−1.58, −0.77] | 1.42 x 10−8 | 101 | |
| PDE3B | 224 | −0.33 [−0.46, −0.20] | 3.63 x 10−7 | 4 | |
| Lipoprotein (a) | LPA | 307 | −0.35 [−0.46, −0.24] | 4.30 x 10−10 | 102 |
| LVEF | TTN | 179 | −0.44 [−0.57, −0.30] | 5.01 x 10−10 | 56,64 |
| Pulse rate | TTN | 1,707 | 0.20 [0.15, 0.25] | 8.82 x 10−17 | 19 |
| Novel associations * | |||||
| Height, cm | SCUBE3 | 71 | −0.57 [−0.73, −0.41] | 5.02 x 10−12 | |
| PIEZO1 | 574 | −0.20 [−0.25, −0.14] | 1.39 x 10−11 | ||
| IRS1 | 47 | −0.58 [−0.78, −0.38] | 7.81 x 10−9 | ||
| ANGPTL2 | 119 | −0.44 [−0.57, −0.32] | 4.88 x 10−12 | ||
| PAPPA | 36 | −0.66 [−0.89, −0.43] | 1.28 x 10−8 | ||
| ZFAT | 53 | 0.58 [0.40, 0.77] | 1.04 x 10−9 | ||
| DTL | 72 | −0.67 [−0.83, −0.51] | 3.42 x 10−16 | ||
| Igf-1 | JAK2 | 70 | −0.77 [−1.00, −0.55] | 1.64 x 10−11 | |
| Glucose | GIGYF1 | 51 | 0.79 [0.52, 1.06] | 9.45 x 10−9 | |
| HDL | NR1H3 | 352 | 0.39 [0.30, 0.49] | 3.27 x 10−16 | |
| LDL | GIGYF1 | 52 | −0.79 [−1.06, −0.52] | 9.02 x 10−9 | |
P-values, effect sizes and 95% confidence intervals (95% CI) were obtained from score tests from linear mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs), MRI serial number (for MRI traits) and a sparse kinship matrix. P-values shown are unadjusted for multiple testing.
Novel indicates that rare variant associations were not reported prior to the release of UK Biobank exomes. s.d., standard deviation; Ref, reference; LVEF, left ventricular ejection fraction; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Igf-1, insulin-like growth factor 1.
Identified associations represent burdens of multiple rare variants and are independent of common variation.
To assess the stability of our results to changes in the analytic approach, we performed a leave-one-variant-out (LOVO) analyses for each of the significant associations. For example, when analyzing the relation between TSHR and hypothyroidism, the highest LOVO P-value attained for the significant binary associations was P = 7.9 x 10−4 after removing p.Trp546Ter (Supplementary Tables 4 and 6). Similarly, the highest LOVO P-value for the significant quantitative associations was P = 5.7 x 10−4 for LCAT/high-density lipoprotein after removing p.Tyr107Ter (Supplementary Tables 5 and 7). Importantly, the novel associations that we identified remained robust in a LOVO analysis (Supplementary Fig. 10). Thus, the genes significantly associated with diseases or traits were identified due to a burden of multiple contributing rare variants, although in certain cases—such as the associations of ANGPTL2 with height and NR1H3 with high-density-lipoprotein—single variants were important. The complete LOVO results for all variants, as well as variant frequencies and annotations, are presented in Supplementary Tables 6 and 7.
We then aimed to evaluate whether the identified rare variant signals were independent of nearby common variants. We first performed common variant analyses for common (MAF > 0.5%) imputed variants within ± 500 kb of the gene; we then performed the rare variant association tests with the index common variants as additional covariates (Methods). Overall, we found that the effect size estimates, and P-values, were not significantly changed after conditioning on nearby common variants (Supplementary Tables 8 and 9). Interestingly, the association of LPA with lipoprotein(a) became more significant (P = 4.3 x 10−10, β = −0.35; Pconditional = 7.7 x 10−23, βconditional = −0.45), as did the suggestive associations KCNQ1 with QTc, BSN with BMI, and BSN with weight (Pconditional < 5.44 x 10−7; Supplementary Table 9).
Independent replication in the Geisinger MyCode cohort.
To replicate gene-based associations that we identified in the UK Biobank, we utilized data from the Geisinger MyCode cohort from the Geisinger health system21 (also known as the DiscovEHR cohort). MyCode is a healthcare-based cohort with high quality whole-exome sequencing data for 166,661 adults (aged 57 ± 18 years, 61% female) of primarily European ancestry (95%) linked to longitudinal electronic health records. We put forward the 15 associations that were unreported prior to the release of UK Biobank exomes. Of 14 testable associations, 13 (93%) replicated in MyCode at P < 0.05 with consistent direction of effect (Table 4).
Table 4 ∣.
Replication of novel associations in the Geisinger MyCode cohort
| Binary traits | ||||
|---|---|---|---|---|
| Trait | Gene | Carriers | OR [95% CI] | P-value |
| Diabetes type 2 | GIGYF1 | 152 | 3.18 [2.22, 4.54] | 1.98 x 10−9 |
| CCAR2 | 24 | 0.75 [0.24, 2.33] | 0.62 | |
| Supraventricular tachycardia | TTN | 3,059 | 1.35 [1.08, 1.70] | 0.01 |
| Mitral valve disease | TTN | 3,059 | 1.45 [1.18, 1.78] | 8.26 x 10−4 |
| Quantitative traits | ||||
| Trait | Gene | Carriers | Effect in s.d. [95% CI] | P-value |
| Height, cm | SCUBE3 | 153 | −0.67 [−0.95, −0.39] | 3.90 x 10−6 |
| PIEZO1 | 577 | −0.35 [−0.50, −0.21] | 1.97 x 10−6 | |
| IRS1 | 36 | −0.67 [−1.24, −0.11] | 0.02 | |
| ANGPTL2 | 78 | −1.02 [−1.41, −0.64] | 2.45 x 10−7 | |
| PAPPA | 69 | −0.47 [−0.89, −0.05] | 0.03 | |
| ZFAT | 42 | 0.79 [0.24, 1.34] | 5.04 x 10−3 | |
| DTL | 45 | −0.90 [−1.41, −0.39] | 5.69 x 10−4 | |
| Glucose | GIGYF1 | 102 | 0.55 [0.37, 0.73] | 1.37 x 10−9 |
| HDL | NR1H3 | 122 | 0.38 [0.22, 0.54] | 2.97 x 10−6 |
| PLIN1 * | 97 | 0.36 [0.18, 0.54] | 9.29 x 10−5 | |
| LDL | GIGYF1 | 105 | −0.29 [−0.47, −0.11] | 1.76 x 10−3 |
P-values, odds ratios (OR) and 95% confidence intervals (95% CI) were obtained from whole-genome ridge-regression models implemented in REGENIE, further adjusting for sex, age and associated principal components (PCs). P-values shown are unadjusted for multiple testing. Novel indicates that rare variant associations were not reported prior to the release of UK Biobank exomes. Association test between Igf-1 and JAK2 was not performed due to low cumulative minor allele counts (cMAC); cMAC among individuals with Igf-1 was 2.
The association between PLIN1 and HDL was added to replication because the direction of effect (increased HDL) was different to the direction reported in family-based studies of partial lipodystrophy.
Rare variants confer substantial risk and penetrance for cardiometabolic and other disorders.
From the exome-wide analyses of 57 curated medical conditions in the UK Biobank, we observed a number of well-described gene-disease associations (Fig. 1 and Table 2). For cardiac diseases, these include the association between dilated and hypertrophic cardiomyopathy with the genes TTN and MYBPC3, respectively. For metabolic disorders involving cholesterol transport, glucose regulation, and thyroid disorders, associations were noted with LDLR, APOB, PCSK9, GCK, TSHR and TG. Interestingly, biallelic mutations in TSHR and TG are known to cause penetrant congenital thyroid disease (MIM 275200 and 274700), while our findings indicate that heterozygote carriers may also be at 2 to 3-fold increased odds of hypothyroidism. Accordingly, large-effect missense variants in both genes have been found to affect thyroid stimulating hormone levels22, and common variants near both genes are associated with thyroid disease, including autoimmune thyroiditis23.
Figure 1 ∣. Rare genetic variation for 57 cardiometabolic and other medical disorders in the UK Biobank.

Multiple-trait Manhattan plot representing the results from exome-wide gene-based tests for each phenotype. Phenotypes are labelled on the x-axis and the −log10 of the P-value for each test on the y-axis. Variants included in the gene-based test are restricted to loss-of-function and predicted-deleterious missense variants. P-values were computed using the saddle point approximation and were obtained from logistic mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs) and a sparse kinship matrix. P-values shown are two-sided and unadjusted for multiple testing. The red line indicates the significance threshold at a Benjamini-Hochberg false-discovery-rate (FDR) of 1% across all tests across all binary and quantitative traits, while the blue line represents the suggestive threshold at FDR 5%. An arrow pointing upwards indicates that rare variants were associated with increased risk of disease, while arrows pointing downward indicate decreased risk.
In many cases, the phenotypic penetrance of deleterious variants and the increased risk of disease was striking. For example, 71% of LDLR mutation carriers had hypercholesterolemia (OR 13.1, 95%CI 8.3-21.3), 45% of MIP variant carriers had cataracts (OR 7.6, 95%CI 3.4-16.7) and 47% of PKD1 variant carriers had chronic kidney disease (OR 40.3, 95%CI 21.3-76.2) (Supplementary Table 10 and Supplementary Figs. 11 and 12). Indeed, PKD1 mutations, while known for penetrant autosomal dominant polycystic kidney disease, have recently been suggested to exhibit incomplete penetrance24 (Supplementary Note). GCK mutation carriers had over 14-fold increased odds (95%CI 8.3-23.4) of type 2 diabetes, with over 48% of carriers having a diagnosis. GCK variants are known to cause maturity-onset diabetes of the young25 and have also previously been found enriched in type 2 diabetes cases26,27 (Supplementary Note). TTN mutations were associated with a several-fold increased risk of multiple cardiovascular disorders, including an over 11-fold increased odds of dilated cardiomyopathy (MIM 604145), and a more than doubling in the risk of heart failure, atrial fibrillation19, and ventricular arrhythmia28. TTN variants also had novel associations with the risk of supraventricular tachycardia (OR 2.5, P = 2.9 x 10−9) and mitral valve disease (OR 2.3, P = 2.6 x 10−11), findings which were replicated in MyCode (OR 1.4, P = 0.01 and OR 1.5, P = 8.3 x 10−4, respectively). While these specific associations have not been reported before, they may be related to diagnoses of atrial fibrillation or dilated cardiomyopathy. Of note, all known and novel TTN associations became stronger (markedly higher ORs and lower P-values) after restricting to exons highly expressed in cardiac left ventricular tissue (Supplementary Note).
Rare variants in GIGYF1 are associated with diabetes risk.
We further identified two gene associations for diabetes, which were not reported prior to the release of UK Biobank exomes. Rare mutations in GIGYF1 were significantly associated with an increased risk of type 2 diabetes (55 carriers, OR 5.6, P = 3.0 x 10−7), and also associated significantly with higher blood glucose levels (β = 0.8 s.d., P = 9.5 x 10−9) and lower low-density lipoprotein levels (β = −0.8 s.d., P = 9.02 x 10−9) (Tables 2 and 3 and Fig. 3). GIGYF1 further showed suggestive evidence of association with lower insulin-like growth factor-1 levels (Fig. 3). Common variants near GIGYF1 also associated with diabetes risk and glucose levels7 (Supplementary Table 11). Using GTEx, we found that the top common variants in this locus were strong expression-QTLs for GIGYF1 in multiple relevant tissues (adipose tissue, pancreas, thyroid, skeletal muscle), with the expression-lowering alleles showing consistency with the observed LOF association (Supplementary Note and Supplementary Table 12). The protein product of GIGYF1 regulates insulin-like-growth factor signaling29 and interacts with Grb10, a protein that has been implicated in insulin signaling, glucose tolerance and insulin resistance30. GIGYF1 LOFs have previously been linked to autism31, although none of the carriers in the present study had ICD code diagnoses relevant to autism or developmental delay. In MyCode, the association between rare variants in GIGYF1 and type 2 diabetes was replicated (OR 3.2, P = 2.0 x 10−9), as was the association with glucose (β = 0.5, P = 1.4 x 10−9). We further leveraged summary data from a previous exome sequencing study of diabetes13, and found additional evidence of independent replication for GIGYF1 LOF variants (9 carriers, OR 8.6, P = 7.8 x 10−3). Across all three studies, GIGYF1 variants were robustly associated with type 2 diabetes (OR 3.8, P = 4.1 x 10−16; Extended Data Fig. 1).
Figure 3 ∣. Pleiotropy of rare variants in metabolic genes.

This heatmap shows association results for genes associated at false-discovery rate (FDR) Q-value < 0.05 with any metabolic trait in our primary analysis, across a range of relevant metabolic traits. P-values were obtained from score tests in linear mixed effects models (quantitative traits) or saddle point approximation in logistic mixed effects models (binary traits), adjusting for sex, age, sequencing batch, associated principal components (PCs) and a sparse kinship matrix. P-values shown are two-sided and unadjusted for multiple testing, while Q-values represent false-discovery rate (FDR) adjusted two-sided P-values by Benjamini-Hochberg method. Effect sizes for quantitative traits (β) were obtained from the same linear model, while odds ratios (OR) for binary traits were obtained from Firth’s regression models adjusting for sex, age, sequencing batch and associated PCs among unrelated samples. A small dot indicates nominal significance (P < 0.05), a large dot indicates P < 0.00014 (0.05/ 350 tests), while a black square indicates FDR Q-value < 0.01 in the primary discovery phase. Red indicates β > 0 (quantitative traits) or OR > 1 (binary traits), while blue indicates β < 0 (quantitative traits) or OR < 1 (binary traits). LDL, low-density lipoprotein; HDL, high-density lipoprotein; TG, triglycerides; Lp(a), lipoprotein (a); Igf-1, insulin-like growth factor 1; Hyperchol, hypercholesterolemia; CAD, coronary artery disease; PVD, peripheral vascular disease; Isch. Stroke, ischemic stroke; T2D, type 2 diabetes.
Mutations in CCAR2, also known as KIAA1967 and DBC1, were also associated with diabetes risk in the UK Biobank (26 carriers, OR 12.8, P = 5.4 x 10−8). Common variants near CCAR2 associated with diabetes risk as well7 (Supplementary Table 11), and the top common variant in this locus was found to be a significant expression-QTL for CCAR2 in multiple relevant tissues (Supplementary Table 12 and Supplementary Note). Previous studies have suggested that CCAR2 regulates a glucose metabolic gene network, and that the gene is downregulated in cells from diabetic patients32. Furthermore, Ccar2 knockout mice develop a metabolic phenotype, including obesity, elevated glucose and insulin resistance33. However, we were not able to replicate the association between CCAR2 rare variants and diabetes in MyCode (P = 0.62). Therefore, the role for CCAR2 remains uncertain, and further studies are necessary to dissect the contribution of CCAR2 mutations to human diabetes.
Rare variants confer large effect sizes for quantitative cardiometabolic and anthropometric traits.
In our primary analyses, we identified 18 unique genes that were significantly associated with blood lipid or glucose levels (Table 3 and Fig. 2). Rare variants conferred large effect sizes, ranging from 0.3 to 2.2 s.d., and in many cases showed pleiotropy across multiple metabolic traits (Fig. 3 and Supplementary Table 13). As expected, APOB (β = −2.2 s.d. for low-density lipoprotein and β = −1.4 s.d. for triglycerides), APOC3 (β = −1.2 s.d. for triglycerides) and GCK (β = 1.2 s.d. for glucose) were among genes conferring the largest effect sizes.
Figure 2 ∣. Rare genetic variation for 26 quantitative cardiometabolic traits in the UK Biobank.

Multiple-trait Manhattan plot representing the results from exome-wide gene-based tests for each phenotype. Phenotypes are labelled on the x-axis and the −log10 of the P-value for each test on the y-axis. Variants included in the gene-based test are restricted to loss-of-function and predicted-deleterious missense variants. P-values were obtained from score tests from linear mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs), MRI serial number (for MRI traits) and a sparse kinship matrix. P-values shown are two-sided and unadjusted for multiple testing. The red line indicates the significance threshold at a Benjamini-Hochberg false-discovery rate (FDR) of 1% across all tests across all binary and quantitative traits, while the blue line represents the suggestive threshold at FDR 5%. For height, suggestively associated genes are not annotated with gene names for clarity. An arrow pointing upwards indicates that rare variants were associated with higher value for a given quantitative trait, while arrows pointing downward indicate lower value. BMI, body mass index; HDL, high-density lipoprotein; LDL, low-density lipoprotein; Igf-1, insulin-like growth factor 1; RR, RR interval; Pdur, P-wave duration; PQ, PQ interval; QRS, QRS-complex duration; QTc, Bazett-corrected QT interval; LVEDV, left ventricular end-diastolic volume; LVEDVi, body-surface-area indexed left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume; LVESVi, body-surface-area indexed left ventricular end-systolic volume; SV, stroke volume; SVi, body-surface-area indexed stroke volume; LVEF, left ventricular ejection fraction.
Our analysis revealed several genes that have been proposed as potential therapeutic targets or for which lipid-lowering therapeutics are in development, such as ANGPTL334, ANGPTL435 and PCSK916-18. Other notable findings included PDE3B, in which damaging variants were associated with lower triglyceride levels (β = −0.3 s.d., P = 3.6 x 10−7), consistent with previous reports4. PDE3B variants have also been associated with improved body fat distribution, making it an interesting therapeutic target36. Of note, our findings are independent of previous UK Biobank analyses for this gene, as the functional4,36 stop-gain variant p.Arg783Ter was not included in the current analysis based on MAF filters. Indeed, conditioning on p.Arg783Ter, and nearby common variants, did not strongly affect the results (β = −0.3 s.d., P = 4.3 x 10−6) (Supplementary Table 9).
We further found that rare mutations in PLIN1 were associated with elevated high-density lipoprotein levels (β = 0.4 s.d., P = 8.01 x 10−15), an association that was confirmed in MyCode (β = 0.4 s.d., P = 9.29 x 10−5). Furthermore, PLIN1 variants associated suggestively with decreased triglyceride levels (β = −0.2 s.d., P =1.6 x 10−5) and nominally with lower risk of hypercholesterolemia (OR 0.69, P = 9.0 x 10−3) and lower risk of coronary artery disease (OR 0.56, P = 0.03) (Fig. 3 and Supplementary Table 13). PLIN1 frameshift variants have paradoxically been linked to a phenotype of partial lipodystrophy with low levels of high-density lipoprotein and high triglycerides37. Our findings, however, are consistent with the notion that PLIN1 haploinsufficiency does not cause partial lipodystrophy38 and indicate that PLIN1 inhibition39 might represent a potential target for lipid therapeutics.
Among the many associations between rare variants and cardiometabolic traits, lipid associations for GIGYF1 (low-density lipoprotein; β = −0.8 s.d., P = 9.02 x 10−9) and NR1H3 (high-density lipoprotein; β = 0.4 s.d., P = 3.27 x 10−16) were also novel, and replicated in MyCode (GIGYF1: β = −0.3 s.d., P = 1.76 x 10−3; NR1H3: β = 0.4 s.d., P = 2.97 x 10−6, respectively). Common variants near both genes show concordant associations with blood lipid levels40,41 (Supplementary Table 11). Furthermore, top common variants in both loci are significant expression- or splice-QTLs for these genes in relevant tissues (Supplementary Table 12 and Supplementary Note). NR1H3 encodes the liver X receptor alpha, a regulator of cholesterol homeostasis in the liver42. For GIGYF1, our findings indicate that damaging variants may be beneficial for blood lipids (e.g. lower low-density lipoprotein), yet harmful for glucose homeostasis (e.g. higher glucose and increased risk of diabetes). Interestingly, an inverse association between cholesterol and diabetes risk has been observed previously for common variants43,44, and for lipid medications such as statins45,46. A multivariate GWAS analyzing diabetes and concurrent lower low-density lipoprotein in the UK Biobank previously identified a locus overlapping GIGYF147.
We further found 7 novel rare variant associations for height (Table 3 and Fig. 2), all of which replicated at P < 0.05 in MyCode (Table 4). While common and low-frequency variant analyses have already implicated DTL, ZFAT, PIEZO1, SCUBE3, ANGPTL2, IRS1 and PAPPA in standing height48-50 (Supplementary Table 11 and Supplementary Note), our results indicate that rare variation in these genes may confer substantial effects, with absolute effect sizes ranging from 0.2 to 1.0 s.d. (Tables 3 and 4). Interestingly, IRS1 and ANGPTL2 represent genes of possible interest to cardiometabolic health. Irs1 knockout mice exhibit both impaired growth and insulin resistance51, while ANGPTL2 is involved in many metabolic processes, including cardiac energy metabolism and heart failure52.
Over 1% of individuals carry putatively pathogenic rare variants for cardiometabolic disease.
Given the high prevalence and morbidity of cardiometabolic disease in the general population, we then sought to quantify carrier frequencies and disease associations for putatively pathogenic variation in the UK Biobank. Among 71 cardiovascular disease associated genes included on typical sequencing panels for arrhythmias, cardiomyopathies and hypercholesterolemia, we identified 55 genes that were reported for autosomal dominant Mendelian inheritance (Supplementary Table 14). Similarly, we found 13 genes reported for dominant forms of adult-onset diabetes. For each gene, we collapsed carrier status for rare LOF, pathogenic and likely pathogenic variants (Methods) and performed association tests with relevant diseases. Genes associated with a relevant disease at Q < 0.01 (P < 3.0 x 10−4) included TTN, MYBPC3, MYH7, LDLR, DSP, SCN5A, PKP2, GCK and HNF1A (Fig. 4 and Extended Data Fig. 2). The median OR for significant associations was 4.5 (Q1-Q3: 3.9-18.8) and became OR 8.2 (Q1-Q3: 4.1-22.9) after excluding TTN associations.
Figure 4 ∣. Putatively pathogenic variants in Mendelian cardiovascular disease genes in the UK Biobank.

The top of the figure is a bar chart showing carrier frequencies for rare LOF, pathogenic and likely pathogenic variants in genes reported for dominant inheritance of arrhythmia, cardiomyopathy or hypercholesterolemia. The absolute number of carriers in the UK Biobank is shown above each bar. Bar charts are stacked to visualize carriers of LOFs reported in ClinVar as likely pathogenic or pathogenic (P/LP), carriers of LOFs not reported in ClinVar as P/LP, and carriers of other P/LP variants (missense, inframe indels, noncanonical or low-confidence LOFs, etc). The bottom of the figure is a pruned heatmap showing association results between these variants and cardiovascular outcomes that reach nominal significance (P < 0.05). P-values were computed using the saddle point approximation and were obtained from logistic mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs) and a sparse kinship matrix. P-values shown are two-sided and unadjusted for multiple testing. Odds ratios (OR) were obtained from Firth’s regression models adjusting for sex, age, sequencing batch and associated PCs among unrelated samples. A small dot represents nominal significance (P < 0.05), while a large dot represents P < 0.005. A square represents significant at an FDR Q-value of < 0.01. Blue indicates OR < 1, while red indicates OR > 1. For clarity, tests with P > 0.05 or an OR between 0.7 and 1.43 have been made white. HF, heart failure; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; AF, atrial fibrillation; SVT, supraventricular tachycardia; AV, atrioventricular; ICD, implantable cardioverter-defibrillator; CHD, congenital heart disease; PVD, peripheral vascular disease; Isch., Ischemic; CAD, coronary artery disease; MI, myocardial infarction.
As expected, variants in TTN were most common, as 0.42% of the samples (840 carriers) carried a truncating variant located in one of the cardiac exons (Fig. 4 and Supplementary Table 15). MYBPC3 variants, which associated with hypertrophic cardiomyopathy (OR 88.9, P = 2.2 x 10−26) and several related phenotypes (Supplementary Table 16), were carried by 0.12% (244 carriers). Putatively pathogenic variants in LDLR were observed in 0.12% of individuals (236 carriers) and showed associations with coronary artery disease (OR 3.7, P = 6.8 x 10−8), and myocardial infarction (OR 4.0, P = 1.5 x 10−6). PKP2 variants, carried by 0.12% of individuals (235 carriers), showed an association with ventricular arrhythmias (OR 4.4, P = 2.21 x 10−4). SCN5A (0.10%) and DSP (0.06%) showed associations with conduction defects and dilated cardiomyopathy, respectively. GCK (0.02%) and HNF1A (0.01%) both were associated with type 2 diabetes (Extended Data Fig. 2).
The penetrance of putatively pathogenic cardiovascular disease variants was generally modest (<15%), especially when compared to previous estimates from family-member based analyses, although GCK and HNF1A variants conferred high absolute risks of diabetes (64 and 44% penetrance, respectively) (Supplementary Note, Supplementary Tables 10 and 15, and Supplementary Fig. 13). The yield of relevant LOFs and known pathogenic variants among disease cases was low for common diseases such as diabetes and atrial fibrillation (generally <5%), while dilated and hypertrophic cardiomyopathy both had rare variant yields of greater than 10% (Supplementary Note, Supplementary Fig. 14 and Supplementary Table 17).
Overall, 2.4% of samples (n = 4,855) carried a putatively pathogenic variant in any of the 68 panel genes included in our analysis (Fig. 4, Extended Data Fig. 2 and Supplementary Table 15). This statistic includes a number of genes with limited or disputed evidence of pathogenicity (e.g KCNE2), as well as LOFs for a number of genes where truncation is not an established mechanism of dominant disease (e.g. MYL2, MYL3, MYL4). As such, this number represents an upper-bound estimate. We then restricted our analysis to genes which associated significantly with a relevant phenotype at Q < 0.01. When restricting only to associated genes, we arrive at a lower-bound estimate of 2,098 carriers, or 1.0% of samples, as carriers of putatively pathogenic variants for cardiometabolic disease.
Discussion
The availability of exome sequencing data in nearly 200,000 individuals from the UK Biobank has provided an unparalleled opportunity to explore the genetic basis of common diseases using many distinct analytic approaches53-55. Through exome-wide gene-based analysis of very rare genetic variants, we replicate many known Mendelian gene-trait associations for cardiometabolic disorders in the UK Biobank. We also identify several large-effect associations that were not previously reported prior to the release of the UK Biobank exome data, and which were broadly replicated in the independent Geisinger MyCode cohort. We further quantify the frequency of rare pathogenic variation and show that between 1.0% and 2.4% of individuals carry potentially high-impact putatively pathogenic rare variants for cardiovascular diseases and diabetes.
Our findings permit a number of conclusions. First, our findings show the value of large-scale population sequencing for identifying key contributors to cardiometabolic disease, as well as the relative odds of disease associated with Mendelian mutations. For example, through exome-wide analyses, we identified large effect associations for rare MYBPC3 variants with hypertrophic cardiomyopathy (OR 120, MIM 115197), LDLR mutations with hypercholesterolemia (OR 13, MIM 143890), PKD1 mutations with chronic kidney disease (OR 40, MIM 173900) and GCK with diabetes (OR 14, MIM 125853). In a targeted analysis of panel genes, we further found markedly increased disease risk (OR > 5) for multiple genes, including TTN, MYH7, DSP, SCN5A and HNF1A. Our data also allowed estimation of rare variant penetrance, strengths and limitations of which are discussed in detail in the Supplementary Note. These results highlight the potential of large-scale population-based sequencing for assessment of risk and pathogenicity associated with genes and variants.
Second, we identified associations for large-effect coding variants with cardiometabolic traits, which were not reported prior to the release of large-scale exome data. Rare variants in GIGYF1 were associated with marked increased risk of type 2 diabetes in discovery and replication datasets, with ORs ranging from 3.2 to 8.6. While GIGYF1 is among the hundreds of loci identified for diabetes through GWAS7, previous human genetic studies have not explicitly prioritized this gene prior to release of the UK Biobank exomes. In contrast, our findings directly implicate GIGYF1, a known regulator of insulin-like growth factor signalling29, in the pathogenesis of human diabetes. We also highlight novel large-effect associations for standing height, which showed similar convergence between evidence from rare and common genetic variants. We further identified several rare variant associations for blood lipids that have not been previously described through population-based association testing, including GIGYF1, NR1H3 and PLIN1, as well as genes which have been put forward as potential therapeutic targets or for which therapeutics are under development, such as PDE3B, ANGPTL3 and ANGPTL4. Taken together, these results show the added value of exome sequencing for identifying genes with important roles in disease pathogenesis and therapeutic targeting.
Third, we quantify carrier frequencies of rare pathogenic variants for cardiometabolic diseases and show that a meaningful proportion of individuals carry genetic variants underlying cardiovascular disease and diabetes. LOF, pathogenic or likely pathogenic variants in cardiomyopathy, arrhythmia, hypercholesterolemia or diabetes genes were carried by 2.4% of individuals. Even when restricting to genes that show evidence of association with relevant outcomes, we identify 1.0% of UK Biobank participants as carriers of disease-causing variation. Consistent with previous reports, TTN LOF variants were relatively common, accounting for nearly half of this group or 0.42% of individuals19,56,57. However, another ~0.5-2% of individuals may carry deleterious variation in other cardiometabolic disease genes. A previous analysis in a smaller subset of the UK Biobank already showed that ~2% of individuals carry clinically-actionable variants in 59 important Mendelian disease genes14. Our results were focused on a larger list of cardiometabolic disease genes, while incorporating population-based associations. These studies show the potential for large-scale sequencing to identify a meaningful proportion of individuals at high risk of disease morbidity and mortality.
Fourth, our results have several analytical implications for rare variant analysis in large biobanks. We found that our rare variant associations were not negatively affected by adjustment for nearby common variation, likely due to our strict variant filter at MAF < 0.1% in both the UK Biobank and gnomAD58, as employed previously15. In addition, effect sizes for LOF and missense variants were comparable to the effect sizes for LOF variants alone, which is likely a reflection of both variant frequency filters and strict inclusion filters based on 30 in silico missense prediction tools. Finally, we note that our gene-based implementation of the saddle point approximation controlled well for test statistic inflation, even for extremely imbalanced phenotypes; we employed Firth’s regression to yield accurate OR estimates for rare variants. Indeed, recent analytical developments have shown the value of these methods for genetic analyses in large biobanks59,60. We have made our code for gene-based burden testing using the saddle point approximation, based on GENESIS code61, available through the repository https://github.com/seanjosephjurgens/UKBB_200KWES_CVD.
In spite of the large sample size of over 200,000 sequenced individuals, we note that rare variant discovery power was still limited. For example, we identified only 3 associations for type 2 diabetes and 1 for atrial fibrillation at test-wide significance, despite having over 12,000 cases for both phenotypes. This may reflect a modest contribution of rare variants to phenotypic variability, or of a distributed contribution over many genes. In support of the latter, a previous exome sequencing study of type 2 diabetes13 estimated that over 75,000 sequenced cases, or over 600,000 samples from population-based biobanks, would be necessary to identify known diabetes drug targets at 80% statistical power. Therefore, UK Biobank analyses utilizing data from all 500,000 samples may prove particularly fruitful for complex diseases in the future.
Our study has several other potential limitations. First, participants in the UK Biobank are largely middle-aged individuals of European ancestry. As such, our findings may not be broadly applicable to all age strata and ancestries. Second, disease status in the UK Biobank relies on self-reports, ICD codes, operation codes, and death registry codes. As a consequence, some misclassification for disease phenotypes is possible. However, previous efforts using the same phenotypic definitions in GWAS for a number of analyzed diseases replicated well-described genetic loci for common variants2,62,63. Furthermore, many of the exome-wide significant rare variant associations presented here are well-described Mendelian gene-trait associations. Third, there is potential for ascertainment bias among participants in the UK Biobank, making it unlikely that the study perfectly reflects the overall middle-aged UK population. The ascertainment of UK Biobank participants would be anticipated to attenuate rather than inflate effect sizes and penetrance estimates, as discussed in detail in the Supplementary Note. Fourth, we acknowledge that alternate methods for defining diseases or traits are feasible such as analyzing all ICD or Phecodes. However, we used a set of curated disease definitions that builds from prior work and has in many cases been validated and replicated5,19,64.
In conclusion, large-scale sequencing has enabled the dissection of the rare genetic contributors to cardiometabolic traits and diseases. We confirm many Mendelian gene-disease associations in an unselected, population-based cohort. Furthermore, we also identified and replicated novel large-effect associations for several traits, including diabetes, blood lipids and standing height. Finally, we found that a considerable portion of individuals carry putatively pathogenic variants in cardiomyopathy, arrhythmia, hypercholesterolemia and diabetes genes. In the future, our findings may facilitate studies aimed at therapeutics and screening of cardiovascular and metabolic disorders.
Methods
Study population and phenotypes.
The UK Biobank is a large population-based prospective cohort study from the United Kingdom with deep phenotypic and genetic data on approximately 500,000 individuals aged 40-69 at enrollment103. Curated disease phenotypes were defined using reports from medical history interviews, in- and outpatient ICD-9 and -10 codes, operation codes, and death registry records (Supplementary Table 1). For diseases and medical conditions, case status was determined at last follow-up. The breakdown of prevalent and incident cases, as well as the mean age at disease onset, are presented in Supplementary Table 2. Age at disease onset was defined as the earliest of either (i) age when electronic health records or death registry records first reported billing codes, or (ii) age at second or third visit if defined during a UK Biobank visit. Phenotypes defined at the first (baseline) UK Biobank visit were considered missing for age at onset. The UK Biobank further provides access to a wide range of other phenotypic data, including anthropometric measurements, electrocardiographic intervals, metabolic biomarkers, and cardiac magnetic resonance imaging data. The UK Biobank resource was approved by the UK Biobank Research Ethics Committee and all participants provided written informed consent to participate. Use of UK Biobank data was performed under application number 17488 and was approved by the local Massachusetts General Hospital Institutional Review Board.
Sequencing and quality control.
Whole-exome sequencing was performed on over 200,000 participants from the UK Biobank20, for which the methods have been described for the earlier release of data from approximately 50,000 individuals14. The revised version of the IDT xGen Exome Research Panel v1.0 was used to capture exomes with over 20X coverage at 95% of sites. Because the 200K dataset released by the UK Biobank had been subject to limited quality-control and filtering, we applied an extensive genotype, variant and sample level pipeline to produce a high-quality dataset for analysis, for which the methods are described in detail in the Supplementary Note. Briefly, we set low-quality genotypes to missing, after which we removed variants based on call rate (<90%), Hardy-Weinberg equilibrium test (P < 1 x 10−15), presence in low-complexity regions, and minor allele count (≥1). Sample-level quality-control consisted of removal of samples that had withdrawn their consent, were duplicates, had a mismatch between exome sequencing and genotyping array data, had a mismatch between genetically inferred and self-reported sex, had low call rates or were outliers (outside 8 standard deviations from the mean) for a number of additional metrics (Supplementary Note). Of the 200,642 individuals with exome sequencing who passed the internal quality-control, we excluded an additional 305 samples based on our filters, leaving 200,337 individuals. We also defined an unrelated subset of this cohort (Supplementary Note), which included 185,990 individuals.
Variant annotation.
The protein consequences of variants were annotated using dbNSFP104 (version 4.1a) and the Loss-of-Function Transcript Effect Estimator58 (LOFTEE) plug-in implemented in the Variant Effect Predictor105 (VEP; version 95) (https://github.com/konradjk/loftee). VEP was used to ascertain the most severe consequence of a given variant for each gene transcript. LOFTEE was implemented to identify high-confidence loss-of-function variants (LOF), which include frameshift indels, stopgain variants and splice site disrupting variants. We also removed any LOFs flagged by LOFTEE as dubious, such as LOFs affecting poorly conserved exons and splice variants affecting NAGNAG sites or non-canonical splice regions. Missense variants were annotated using 30 in silico prediction tools included in the dbNSFP database. We collapsed information from the 30 tools into a single value, representing the percentage of tools which predicted a given missense variant was deleterious (Supplementary Note). A missense variant was considered damaging if at least 90% of in silico prediction tools predicted it to be deleterious.
Rare variant burden analyses.
To identify genes and rare genetic variation relevant to cardiometabolic diseases and traits, we performed association tests between a curated binary or quantitative phenotype and rare variants using gene-based collapsing tests. Variants were considered rare if they had minor allele frequency (MAF) <0.1% in the UK Biobank exome sequencing dataset, and <0.1% in each major continental population in gnomAD58 (version 2 exomes). In our primary analysis, we collapsed carriers of LOF variants and predicted-damaging missense variants into a single variable by sample, for each gene. For a given binary phenotype, genes with ≥20 rare variant carriers were analyzed using a logistic mixed-effects model implemented in GENESIS61 (version 2.18.0), adjusting for age, sex, sequencing batch (first 50K vs remaining 150K) and significantly associated (P < 0.05) ancestral principal components (Supplementary Note). For analyses of MRI data, we additionally adjusted for MRI serial number. Missing genetic data were imputed to zero. We accounted for relatedness by including a sparse kinship matrix as a random effect (Supplementary Note), and P-values were computed using the saddle point approximation to account for case-control imbalance106. Odds ratios (OR) and confidence intervals for binary traits were estimated using Firth’s bias-reduced logistic regression107 in the unrelated subset of the cohort. Quantitative traits were inverse-rank normalized and analyzed using a linear mixed-effects model in GENESIS, implementing the same fixed and random effects and using score tests. For two traits, high-density lipoprotein and lipoprotein(a), the mixed-effects nullmodel failed to converge. We therefore ran these traits using standard linear regression in the unrelated subset of the cohort.
We conducted exome-wide association tests for a curated set of 57 binary disease phenotypes and 26 quantitative traits. The binary disease phenotypes have an emphasis on cardiac diseases, vascular disease, metabolic disorders, and also include a range of additional conditions (Supplementary Table 2). We further analyzed 26 quantitative traits, including anthropometric measurements, metabolic blood markers, electrocardiographic intervals and magnetic resonance imaging traits. Anthropometric (including height, weight, body mass index, systolic blood pressure and diastolic blood pressure), metabolic biomarker data (high-density lipoprotein, low-density lipoprotein, triglycerides, glucose, insulin-like-growth-factor 1) and pulse rate measurements were available in a range of 150,000 and 200,000 individuals. Approximately 22,700 individuals had 12-lead resting electrocardiographic data available, including the RR interval, P-wave duration, QRS complex duration and Bazett-corrected QT interval. Previously derived magnetic resonance imaging measurements for left ventricle5 and thoracic aorta108 were available for approximately 21,000 and 20,000 individuals, respectively. The exact breakdown of samples for each trait is presented in Supplementary Table 3. All performed tests were two-sided unless otherwise specified. To determine statistical significance, we applied a Benjamini-Hochberg false discovery rate (FDR) to compute Q-values from two-sided P-values across all performed tests (all genes for all traits combined). Tests with Q < 0.01 were considered significant. Associations with 0.01 ≤ Q < 0.05 were considered suggestive.
Sensitivity analyses for variant annotation and ancestry.
For all identified significant associations, we ran a number of sensitivity analyses. First, we restricted analyses to LOF variants only to evaluate the consistency of effect sizes for the combined set of LOFs and damaging missense variants as compared to LOFs only. Second, we restricted analyses to individuals of white-British European ancestry only, as determined by previous principal component analysis63, to evaluate whether results were affected strongly by our multi-ancestry approach.
Leave-one-variant-out analysis.
To assess the robustness of our gene-based results to changes in the variant mask, we performed leave-one-variant-out (LOVO) analyses. For all significant associations, we iteratively reran the association test for each variant included in the original mask, after removing that variant from the mask. We defined the maximum LOVO P-value as the highest P-value attained for a given gene-phenotype association by this procedure. The variant that was removed to attain the maximum LOVO P-value was considered the most important variant for the gene-phenotype association.
Conditional analyses adjusting for nearby common variation.
To show that the identified rare variant signals were independent of nearby common variants, we reran the significant gene-phenotype associations while conditioning on common variants in the region. For a given gene-phenotype association, we first ran common variant (MAF > 0.5%) association analyses in the genomic region 500 kb downstream and upstream of the identified gene, using UK Biobank imputed data (Supplementary Note). We then clumped and thresholded the results to identify independent index common variants within the region using the --clump function in PLINK109, utilizing cutoffs of P < 1 x 10−5 and r2< 0.01. Gene-based rare variant association analyses were then rerun within individuals who had both exome sequencing and imputed data available, adding each of the clumped common variants to the model as fixed-effect covariates.
Replication of novel rare variant signals.
We sought to replicate novel rare variant associations within the Geisinger MyCode cohort. The MyCode Community Health Initiative is a research study of the Geisinger health system in central and northeastern Pennsylvania21,110. Started in 2007, the study is open to any Geisinger patient—through opt-in informed consent—including both primary and specialty care clinics, and has enrolled over 280,000 participants to date. Through the DiscovEHR collaboration with Regeneron Genetics Center, whole-exome sequencing from collected blood samples has been completed for approximately 175,000 participants to date, and linked with health information from the Geisinger electronic health record (1996–present). This study leveraged exome data for over 166,661 adult (≥18) individuals uniformly sequenced using an IDT exome capture platform and who passed subsequent central quality control procedures. The Geisinger Institutional Review Board approved the MyCode project and the present analysis.
Given the many parallel analyses of our discovery dataset, we define ‘novel’ to indicate rare variant associations not described prior to the release of the UK Biobank exome data. Using this definition, we identified 15 novel associations, of which 14 were testable in MyCode (those with adequate number of samples available). Gene-based collapsing tests were performed in this cohort, including rare LOF and predicted-deleterious missense variants, as described for the UK Biobank discovery analysis (except for the use of LOFTEE). We tested the 14 novel gene-phenotype associations using REGENIE59, implementing a logistic whole-genome ridge regression model (which accounts for the relatedness among study samples) and further including age, sex, and associated PCs (1-4) as additional fixed-effects. The null model was fit using genome-wide genotype data (MAF > 0.05) from the same patients acquired on the Illumina GSA v2 chip. We additionally replicated the association between PLIN1 and high-density lipoprotein, because the direction of effect for PLIN1 LOF variants was different to the reported effect from small studies focused on partial lipodystrophy37. Therefore, in sum, we attempted replication for 15 rare variant associations in the MyCode cohort.
We further utilized the Type 2 Diabetes Knowledge Portal (T2DKP)111 to seek direct replication of novel rare variant associations for type 2 diabetes. We focused on the dataset described in a previous large scale whole exome sequencing analysis of type 2 diabetes (n = 20,791 cases and 24,440 controls)13, using the portal to look up results for rare LOF variants.
Pathogenic variation for cardiometabolic genes in the population.
Given the high prevalence of cardiometabolic disease, we then sought to quantify carrier frequencies for putatively pathogenic variation in cardiovascular genes in the population. We analyzed genes included on typical sequencing panels for Mendelian cardiovascular disease, namely the Invitae Arrhythmia and Cardiomyopathy panel and the Invitae Hypercholesterolemia panel (accessed on 10 November 2020). We then restricted to genes reported for autosomal dominant modes of inheritance in the Online Mendelian Inheritance in Man (OMIM) database (accessed on 10 November 2020). Using ClinVar, we identified carriers of rare (MAF < 0.1% in the exome sequencing dataset and MAF < 0.1% in the continental gnomAD populations) pathogenic or likely pathogenic variants in each genes in the UK Biobank (Supplementary Note). We collapsed carrier status for LOFs (affecting canonical gene transcripts), pathogenic and likely pathogenic variants for each gene, with a few exceptions: for TTN, we restricted to LOF variants located in exons highly expressed in cardiac tissue112 (Supplementary Note); for RYR2, PCSK9, APOB, MYH7 and TTR, analyses were restricted to pathogenic and likely pathogenic variants only given well-characterized gain-of-function or non-truncating mechanisms causing dominant cardiovascular disease113-116. After collapsing as described, individuals harboring these variants were considered carriers of putatively pathogenic rare variants. Next, we calculated the percentage of the study sample that carried putatively pathogenic variants in each gene.
We also analyzed monogenic diabetes genes in a similar manner. We used genes included on the Invitae Monogenic Diabetes panel that were reported in the OMIM database to be associated with autosomal dominant forms of type 2 diabetes, insulin-dependent diabetes, or Maturity-Onset Diabetes of the Young. Again, we collapsed carrier status for LOF, pathogenic and likely pathogenic variants for each included gene, with a few exceptions. For ABCC8 and KCNJ11, we restricted to pathogenic and likely pathogenic variants only, based on known gain-of-function mechanisms causing dominant diabetes117,118.
We then employed the same logistic mixed-model approach described above to identify associations between putatively pathogenic variants in genes and a range of relevant diseases and outcomes (20 diseases for the cardiovascular analysis and 3 diseases for the diabetes analysis). Significance was determined using a separate FDR correction, taking into account all tests performed for the cardiovascular and diabetes analysis combined. Associations at Q < 0.01 were considered significant. Firth’s regression was used to estimate ORs and CIs in the unrelated subset of the sample.
Data Availability
Summary results for the main analyses have been made available through the Cardiovascular Disease Knowledge Portal (https://cvd.hugeamp.org/downloads.html; direct download using https://personal.broadinstitute.org/ryank/Ellinor_ukbb_200k_exome.zip). Access to individual-level UK Biobank data, both phenotypic and genetic, is available to bona fide researchers through application on the UK Biobank website (https://www.ukbiobank.ac.uk). The exome sequencing data can be found in the UK Biobank showcase portal https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=170. Additional information about registration for access to the data is available at http://www.ukbiobank.ac.uk/register-apply/. Use of UK Biobank data was performed under application number 17488.
Summary statistics from previous GWAS which were utilized in this study are publicly available through the Type 2 Diabetes Knowledge Portal (https://t2d.hugeamp.org); MAGMA results referenced in this manuscript were downloaded on 7 December 2020, while index single variant results were downloaded on 7 June 2021.
Other datasets utilized in this manuscript include: the dbNSFP database version 4.1a (https://sites.google.com/site/jpopgen/dbNSFP); gnomAD exomes version 2.1 (https://gnomad.broadinstitute.org/downloads); the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) downloaded in November 2020; the Invitae Arrhythmia and Cardiomyopathy panel (https://www.invitae.com/en/physician/tests/02101/) and the Invitae Hypercholesterolemia panel (https://www.invitae.com/en/physician/tests/02401/) accessed on 10 November 2020; the Invitae Monogenic Diabetes panel (https://www.invitae.com/pt/physician/tests/55001/) accessed in January 2021; the Online Mendelian Inheritance in Man (OMIM) database (omim.org) accessed on 10 November 2020; Ensembl release 95 (https://gnomad.broadinstitute.org/downloads); and the GTEx dataset version 8 (https://gtexportal.org/home/).
Code Availability
The code used for gene-based analyses is an adaptation of the R package GENESIS version 2.18 (https://rdrr.io/bioc/GENESIS/man/GENESIS-package.html), and has been made available through the following GitHub repository: https://github.com/seanjosephjurgens/UKBB_200KWES_CVD. Quality-control of individual level data was performed using Hail version 0.2 (https://hail.is), PLINK version 2.0.a (https://www.cog-genomics.org/plink/2.0/), and KING version 2.2.5 (https://www.kingrelatedness.com/Download.shtml). Variant annotation was performed using VEP version 95 (https://github.com/Ensembl/ensembl-vep) with the LOFTEE plug-in (https://github.com/konradjk/loftee). All analyses that were run in R were run in R version 4.0 (https://www.r-project.org).
Extended Data
Extended Data Fig. 1. Meta-analysis results for GIGYF1 rare variants and type 2 diabetes across three cohorts.
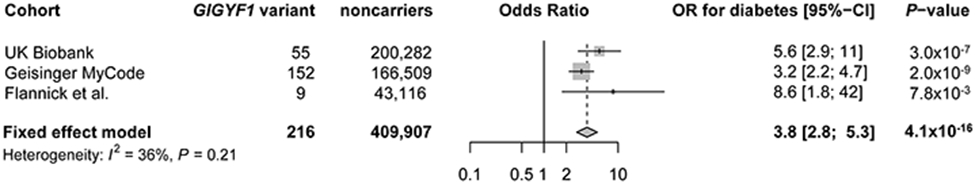
Data are presented in a forest plot, with study specific odds ratios (OR) with 95% confidence intervals (95% CI), and a meta-analysis OR shown with a diamond where the edges of the diamond show the meta-analysis 95% CI. Meta-analysis results are obtained from an inverse-variance weighted fixed-effects meta-analysis approach. Study-specific and meta-analysis P-values are two-sided and unadjusted for multiple testing. To evaluate heterogeneity between studies, an I2 index for heterogeneity and a P-value from Cochran’s Q test are provided, which show limited evidence of heterogeneity.
Extended Data Fig. 2. Carrier frequencies of putatively pathogenic variants in monogenic diabetes genes.

The top of the graph is a bar chart showing carrier frequencies for loss-of-function (LOF) variants and pathogenic/likely pathogenic (P/LP) variants for genes in which variants are known to cause dominant type 2 diabetes or maturity-onset diabetes of the young (MODY). For ABCC8 and KCNJ11, analyses were restricted to previously reported P/LP variants only. The bottom of the graph is a pruned heatmap showing associations between such variants with diabetes and chronic kidney disease, where blue indicates lower risk of disease and red indicates increased risk of the disease. P-values were computed using saddle point approximation and were obtained from logistic mixed effects models, adjusting for sex, age, sequencing batch, associated principal components (PCs), a sparse kinship matrix. P-values shown are two-sided and unadjusted for multiple testing. Odds ratios (OR) were obtained from Firth’s regression models adjusting for sex, age, sequencing batch and associated PCs among unrelated samples. For clarity, associations with P > 0.05 and 0.7 < OR < 1.43 have been made white. Only GCK (45 carriers) and HNF1A (29 carriers) showed robust associations with diabetes. Of note, PDX1 carriers are driven by a single likely pathogenic missense variant, p.Cys18Arg (n = 112 carriers). Our results therefore indicate that this allele specifically does not represent a highly penetrant pathogenic variant, but do not necessarily translate to the 13 carriers of LOF variants.
Supplementary Material
Acknowledgements
We gratefully thank all UK Biobank and MyCode participants, as this study would not have been possible without their contributions. This work was supported by funding from the Fondation Leducq (14CVD01), by grants from the National Institutes of Health (1RO1HL092577, K24HL105780) and by a grant from the American Heart Association (18SFRN34110082) to P.T.E. This work was further supported by a grant from the National Institutes of Health (1R01HL139731) and by a grant from the American Heart Association (18SFRN34250007) to S.A.L. This work was also supported by an American Heart Association Strategically Focused Research Networks (SFRN) postdoctoral fellowship (18SFRN34110082) to L.-C.W. and A.W.H. This work was supported by the John S. LaDue Memorial Fellowship for Cardiovascular Research, a Sarnoff Scholar award from the Sarnoff Cardiovascular Research Foundation, and by a National Institutes of Health grant (K08HL159346) to J.P.P. This work was further supported by a grant from the National Institutes of Health (1K08HL153937) and a grant from the American Heart Association (862032) to K.G.A. This work was supported by a National Institutes of Health grant (T32HL007604) to V.N. This work was also supported by student scholarships from the Dutch Heart Foundation (Nederlandse Hartstichting) and the Amsterdams Universiteitsfonds to S.J.J. This work was supported by the BioData Ecosystem fellowship to S.H.C.
Consortium Author Information
Regeneron Genetics Center
Authors shown in alphabetical order by surname
Goncalo Abecasis6, Xiaodong Bai6, Suganthi Balasubramanian6, Aris Baras6, Christina Beechert6, Boris Boutkov6, Michael Cantor6, Giovanni Coppola6, Tanima De6, Andrew Deubler6, Aris Economides6, Gisu Eom6, Manuel A. R. Ferreira6, Caitlin Forsythe6, Erin D. Fuller6, Zhenhua Gu6, Lukas Habegger6, Alicia Hawes6, Marcus B. Jones6, Katia Karalis6, Shareef Khalid6, Olga Krasheninina6, Rouel Lanche6, Michael Lattari6, Dadong Li6, Alexander Lopez6, Luca A. Lotta6, Kia Manoochehri6, Adam J. Mansfield6, Evan K. Maxwell6, Jason Mighty6, Lyndon J. Mitnaul6, Mona Nafde6, Jonas Nielsen6, Sean O’Keeffe6, Max Orelus6, John D. Overton6, Maria Sotiropoulos Padilla6, Razvan Panea6, Tommy Polanco6, Manasi Pradhan6, Ayesha Rasool6, Jeffrey G. Reid6, William Salerno6, Thomas D. Schleicher6, Alan Shuldiner6, Katherine Siminovitch6, Jeffrey C. Staples6, Ricardo H. Ulloa6, Niek Verweij6, Louis Widom6 and Sarah E. Wolf6
Footnotes
Competing Interests Statement
P.T.E. has received sponsored research support from Bayer AG and IBM Research. P.T.E. has also served on advisory boards or consulted for Bayer AG, MyoKardia and Novartis. S.A.L. receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer AG, Boehringer Ingelheim, Fitbit, and IBM, and has consulted for Bristol Myers Squibb / Pfizer, Bayer AG, and Blackstone Life Sciences. L.-C.W. is supported by a grant from IBM to the Broad Institute. The remaining authors have no relevant competing interests to disclose.
References
- 1.Locke AE et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roselli C et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet 50, 1225–1233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah S et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun 11, 163 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klarin D et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet 50, 1514–1523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pirruccello JP et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun 11, 2254 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ntalla I et al. Multi-ancestry GWAS of the electrocardiographic PR interval identifies 202 loci underlying cardiac conduction. Nat. Commun 11, 2542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vujkovic M et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet 52, 680–691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrier L et al. Mapping of a novel gene for familial hypertrophic cardiomyopathy to chromosome 11. Nat. Genet 4, 311–313 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Ahlberg G et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat. Commun 9, 4316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keating M et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 252, 704–706 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Gerull B et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet 36, 1162–1164 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Do R et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 518, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flannick J et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Hout CV et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cirulli ET et al. Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat. Commun 11, 542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen JC, Boerwinkle E, Mosley TH Jr. & Hobbs HH Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med 354, 1264–1272 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Lambert G, Sjouke B, Choque B, Kastelein JJ & Hovingh GK The PCSK9 decade. J. Lipid Res 53, 2515–2524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y & Liu ZP PCSK9 inhibitors: novel therapeutic strategies for lowering LDL cholesterol. Mini Rev. Med. Chem 19, 165–176 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Choi SH et al. Monogenic and polygenic contributions to atrial fibrillation risk: results from a national biobank. Circ. Res 126, 200–209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szustakowski JD et al. Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat. Genet 53, 942–948 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Carey DJ et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet. Med 18, 906–913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W et al. GWAS of thyroid stimulating hormone highlights pleiotropic effects and inverse association with thyroid cancer. Nat. Commun 11, 3981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwangbo Y & Park YJ Genome-wide association studies of autoimmune thyroid diseases, thyroid function, and thyroid cancer. Endocrinol. Metab. (Seoul) 33, 175–184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallawaarachchi AC, Furlong TJ, Shine J, Harris PC & Cowley MJ Population data improves variant interpretation in autosomal dominant polycystic kidney disease. Genet. Med 21, 1425–1434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakera AJ et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 38, 1383–1392 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Bansal V et al. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med. 15, 213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnefond A et al. Pathogenic variants in actionable MODY genes are associated with type 2 diabetes. Nat. Metab 2, 1126–1134 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Corden B et al. Association of Titin-truncating genetic variants with life-threatening cardiac arrhythmias in patients with dilated cardiomyopathy and implanted defibrillators. JAMA Netw. Open 2, e196520 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giovannone B et al. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J. Biol. Chem 278, 31564–31573 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Plasschaert RN & Bartolomei MS Tissue-specific regulation and function of Grb10 during growth and neuronal commitment. Proc. Natl. Acad. Sci. USA 112, 6841–6847 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satterstrom FK et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basu S et al. DBC1, p300, HDAC3, and Siah1 coordinately regulate ELL stability and function for expression of its target genes. Proc. Natl. Acad. Sci. USA 117, 6509–6520 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiang L et al. Hepatic SirT1-dependent gain of function of stearoyl-CoA desaturase-1 conveys dysmetabolic and tumor progression functions. Cell Rep. 11, 1797–1808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lang W & Frishman WH Angiopoietin-like 3 protein inhibition: a new frontier in lipid-lowering treatment. Cardiol. Rev 27, 211–217 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Dewey FE et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med 374, 1123–1133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emdin CA et al. Analysis of predicted loss-of-function variants in UK Biobank identifies variants protective for disease. Nat. Commun 9, 1613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gandotra S et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med 364, 740–748 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laver TW et al. PLIN1 haploinsufficiency is not associated with lipodystrophy. J. Clin. Endocrinol. Metab 103, 3225–3230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noureldein MH In silico discovery of a perilipin 1 inhibitor to be used as a new treatment for obesity. Eur. Rev. Med. Pharmacol. Sci 18, 457–460 (2014). [PubMed] [Google Scholar]
- 40.Richardson TG et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 17, e1003062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabatti C et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet 41, 35–46 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu R, Ou Z, Ruan X & Gong J Role of liver X receptors in cholesterol efflux and inflammatory signaling. Mol. Med. Rep 5, 895–900 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lotta LA et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: a meta-analysis. JAMA 316, 1383–1391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu DJ et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet 49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmadizar F et al. Associations of statin use with glycaemic traits and incident type 2 diabetes. Br. J. Clin. Pharmacol 85, 993–1002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sattar N et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet 375, 735–742 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Klimentidis YC et al. Phenotypic and genetic characterization of lower LDL cholesterol and increased type 2 diabetes risk in the UK Biobank. Diabetes 69, 2194–2205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lango Allen H et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kichaev G et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet 104, 65–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marouli E et al. Rare and low-frequency coding variants alter human adult height. Nature 542, 186–190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamemoto H et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372, 182–186 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Tian Z et al. ANGPTL2 activity in cardiac pathologies accelerates heart failure by perturbing cardiac function and energy metabolism. Nat. Commun 7, 13016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 597, 527–532 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y et al. GIGYF1 loss of function is associated with clonal mosaicism and adverse metabolic health. Nat. Commun 12, 4178 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schiabor Barrett KM et al. Positive predictive value highlights four novel candidates for actionable genetic screening from analysis of 220,000 clinicogenomic records. Genet. Med 23, 2300–2308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schafer S et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet 49, 46–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haggerty CM et al. Genomics-first evaluation of heart disease associated with Titin-truncating variants. Circulation 140, 42–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mbatchou J et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet 53, 1097–1103 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Zhao Z et al. UK Biobank whole-exome sequence binary phenome analysis with robust region-based rare-variant test. Am. J. Hum. Genet 106, 3–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gogarten SM et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weng LC et al. Heritability of atrial fibrillation. Circ. Cardiovasc. Genet 10, e001838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aragam KG et al. Phenotypic refinement of heart failure in a national biobank facilitates genetic discovery. Circulation doi: 10.1161/CIRCULATIONAHA.118.035774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pirruccello JP et al. Titin truncating variants in adults without known congestive heart failure. J. Am. Coll. Cardiol 75, 1239–1241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanjore RR, Rangaraju A, Kerkar PG, Calambur N & Nallari P MYBPC3 gene variations in hypertrophic cardiomyopathy patients in India. Can. J. Cardiol 24, 127–130 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Richard P et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Gerull B et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet 30, 201–204 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Herman DS et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med 366, 619–628 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi SH et al. Association between Titin loss-of-function variants and early-onset atrial fibrillation. JAMA 320, 2354–2364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vionnet N et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature 356, 721–722 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Reeders ST et al. Regional localization of the autosomal dominant polycystic kidney disease locus. Genomics 3, 150–155 (1988). [DOI] [PubMed] [Google Scholar]
- 72.Breuning MH et al. Improved early diagnosis of adult polycystic kidney disease with flanking DNA markers. Lancet 2, 1359–1361 (1987). [DOI] [PubMed] [Google Scholar]
- 73.Reeders ST et al. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 317, 542–524 (1985). [DOI] [PubMed] [Google Scholar]
- 74.Hopkins PN et al. A novel LDLR mutation, H190Y, in a Utah kindred with familial hypercholesterolemia. J. Hum. Genet 44, 364–367 (1999). [DOI] [PubMed] [Google Scholar]
- 75.Callis M et al. Mutation analysis in familial hypercholesterolemia patients of different ancestries: identification of three novel LDLR gene mutations. Mol. Cell. Probes 12, 149–152 (1998). [DOI] [PubMed] [Google Scholar]
- 76.Jensen HK, Jensen LG, Hansen PS, Faergeman O & Gregersen N An Iranian-Armenian LDLR frameshift mutation causing familial hypercholesterolemia. Clin. Genet 49, 88–90 (1996). [DOI] [PubMed] [Google Scholar]
- 77.Peloso GM et al. Rare protein-truncating variants in APOB, lower low-density lipoprotein cholesterol, and protection against coronary heart disease. Circ. Genom. Precis. Med 12, e002376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patni N, Ahmad Z & Wilson DP Genetics and Dyslipidemia. in Endotext (eds. Feingold KR et al.) (South Dartmouth (MA), 2000). [Google Scholar]
- 79.Narumi S et al. TSHR mutations as a cause of congenital hypothyroidism in Japan: a population-based genetic epidemiology study. J. Clin. Endocrinol. Metab 94, 1317–1323 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Heo S, Jang JH & Yu J Congenital hypothyroidism due to thyroglobulin deficiency: a case report with a novel mutation in TG gene. Ann. Pediatr. Endocrinol. Metab 24, 199–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watanabe Y et al. A novel mutation in the TG gene (G2322S) causing congenital hypothyroidism in a Sudanese family: a case report. BMC Med. Genet 19, 69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shah MH, Bhat V, Shetty JS & Kumar A Whole exome sequencing identifies a novel splice-site mutation in ADAMTS17 in an Indian family with Weill-Marchesani syndrome. Mol. Vis 20, 790–796 (2014). [PMC free article] [PubMed] [Google Scholar]
- 83.Khan AO, Aldahmesh MA, Al-Ghadeer H, Mohamed JY & Alkuraya FS Familial spherophakia with short stature caused by a novel homozygous ADAMTS17 mutation. Ophthalmic. Genet 33, 235–239 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Crippa M et al. A balanced reciprocal translocation t(10;15)(q22.3;q26.1) interrupting ACAN gene in a family with proportionate short stature. J. Endocrinol. Invest 41, 929–936 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Hwang IT et al. Role of NPR2 mutation in idiopathic short stature: Identification of two novel mutations. Mol. Genet. Genomic Med 8, e1146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang SR et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum. Mutat 36, 474–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Olney RC et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J. Clin. Endocrinol. Metab 91, 1229–1232 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Klammt J, Kiess W & Pfäffle R IGF1R mutations as cause of SGA. Best Pract. Res. Clin. Endocrinol. Metab 25, 191–206 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Steinkellner H et al. Identification and molecular characterisation of a homozygous missense mutation in the ADAMTS10 gene in a patient with Weill-Marchesani syndrome. Eur. J. Hum. Genet 23, 1186–1191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morales J et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet 85, 558–568 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gloyn AL Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum. Mutat 22, 353–362 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Reddy MV et al. Exome sequencing identifies 2 rare variants for low high-density lipoprotein cholesterol in an extended family. Circ. Cardiovasc. Genet 5, 538–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Musunuru K et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med 363, 2220–2227 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cohen JC et al. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305, 869–872 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Guerra R, Wang J, Grundy SM & Cohen JC A hepatic lipase (LIPC) allele associated with high plasma concentrations of high density lipoprotein cholesterol. Proc. Natl. Acad. Sci. USA 94, 4532–4537 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Whitfield AJ, Barrett PH, van Bockxmeer FM & Burnett JR Lipid disorders and mutations in the APOB gene. Clin. Chem 50, 1725–1732 (2004). [DOI] [PubMed] [Google Scholar]
- 97.Inazu A et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N. Engl. J. Med 323, 1234–1238 (1990). [DOI] [PubMed] [Google Scholar]
- 98.Myant NB Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis 104, 1–18 (1993). [DOI] [PubMed] [Google Scholar]
- 99.Hobbs HH, Brown MS & Goldstein JL Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat 1, 445–66 (1992). [DOI] [PubMed] [Google Scholar]
- 100.Dron JS & Hegele RA Genetics of hypertriglyceridemia. Front. Endocrinol. (Lausanne) 11, 455 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crosby J et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med 371, 22–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berg K Lp(a) lipoprotein: an overview. Chem. Phys. Lipids 67-68, 9–16 (1994). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 103.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu X, Wu C, Li C & Boerwinkle E dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat 37, 235–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou W et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet 50, 1335–1341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heinze G A comparative investigation of methods for logistic regression with separated or nearly separated data. Statistics in Medicine 25, 4216–4226 (2006). [DOI] [PubMed] [Google Scholar]
- 108.Pirruccello JP et al. Deep learning enables genetic analysis of the human thoracic aorta. Nat Genet. doi: 10.1038/s41588-021-00962-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dewey FE et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354, aaf6814 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Type 2 Diabetes Knowledge Portal. Accessed in December 2020 and June 2021; http://www.type2diabetesgenetics.org
- 112.Roberts AM et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med 7, 270ra6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Priori SG & Chen SR Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res 108, 871–883 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sharifi M, Futema M, Nair D & Humphries SE Genetic architecture of familial hypercholesterolaemia. Curr. Cardiol. Rep 19, 44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Walsh R et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med 19, 192–203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ton VK, Mukherjee M & Judge DP Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin. Med. Insights Cardiol 8, 39–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ellard S et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am. J. Hum. Genet 81, 375–382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ashcroft FM ATP-sensitive potassium channelopathies: focus on insulin secretion. J. Clin. Invest 115, 2047–2058 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary results for the main analyses have been made available through the Cardiovascular Disease Knowledge Portal (https://cvd.hugeamp.org/downloads.html; direct download using https://personal.broadinstitute.org/ryank/Ellinor_ukbb_200k_exome.zip). Access to individual-level UK Biobank data, both phenotypic and genetic, is available to bona fide researchers through application on the UK Biobank website (https://www.ukbiobank.ac.uk). The exome sequencing data can be found in the UK Biobank showcase portal https://biobank.ndph.ox.ac.uk/showcase/label.cgi?id=170. Additional information about registration for access to the data is available at http://www.ukbiobank.ac.uk/register-apply/. Use of UK Biobank data was performed under application number 17488.
Summary statistics from previous GWAS which were utilized in this study are publicly available through the Type 2 Diabetes Knowledge Portal (https://t2d.hugeamp.org); MAGMA results referenced in this manuscript were downloaded on 7 December 2020, while index single variant results were downloaded on 7 June 2021.
Other datasets utilized in this manuscript include: the dbNSFP database version 4.1a (https://sites.google.com/site/jpopgen/dbNSFP); gnomAD exomes version 2.1 (https://gnomad.broadinstitute.org/downloads); the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) downloaded in November 2020; the Invitae Arrhythmia and Cardiomyopathy panel (https://www.invitae.com/en/physician/tests/02101/) and the Invitae Hypercholesterolemia panel (https://www.invitae.com/en/physician/tests/02401/) accessed on 10 November 2020; the Invitae Monogenic Diabetes panel (https://www.invitae.com/pt/physician/tests/55001/) accessed in January 2021; the Online Mendelian Inheritance in Man (OMIM) database (omim.org) accessed on 10 November 2020; Ensembl release 95 (https://gnomad.broadinstitute.org/downloads); and the GTEx dataset version 8 (https://gtexportal.org/home/).