

Summary
Homeostasis of gut microbiota is crucial in maintaining human health. Alterations, or “dysbiosis,” are increasingly implicated in human diseases, such as cancer, inflammatory bowel diseases, and, more recently, neurological disorders. In ischemic stroke patients, gut microbial profiles are markedly different compared to healthy controls, whereas manipulation of microbiota in animal models of stroke modulates outcome, further implicating microbiota in stroke pathobiology. Despite this, evidence for the involvement of specific microbes or microbial products and microbial signatures have yet to be identified, likely owing to differences in methodology, data analysis, and confounding variables between different studies. Here, we provide a set of guidelines to enable researchers to conduct high-quality, reproducible, and transparent microbiota studies, focusing on 16S rRNA sequencing in the emerging subfield of the stroke-microbiota. In doing so, we aim to facilitate novel and reproducible associations between the microbiota and brain diseases, including stroke, and translation into clinical practice.
Subject areas: Neuroscience, Clinical neuroscience, Microbiome
Graphical abstract
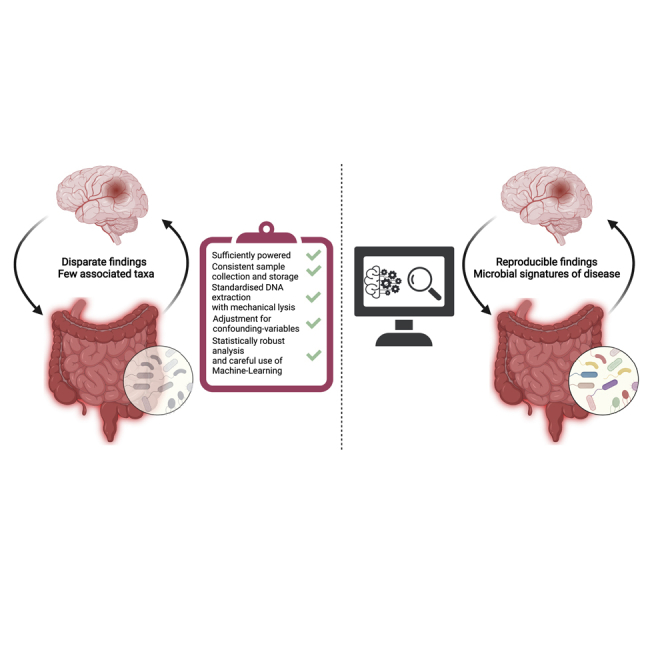
Highlights
-
•
Guidelines for reproducible stroke-microbiota research in patients and animal models
-
•
Current best practices for 16S rRNA profiling and analysis
-
•
Easy-to-use, freely available bioinformatics pipeline for gut microbiota analysis
Neuroscience; Clinical neuroscience; Microbiome
Introduction
Besides select sterile sites, the human body is colonized by complex communities of microbes, termed the microbiota that encompasses bacteria, fungi, and viruses. These microbial communities have co-evolved with us playing a crucial role in health and disease (Claesson et al., 2017). Characterization of the microbiota has been the subject of intensive research in the past decade, generating large volumes of data and with the advent of next-generation sequencing and bioinformatic methods to analyze such data sets; these communities are being characterized in-ever greater detail. Microbiota data, however, are highly complex and high-dimensional, and affected by a multitude of confounding factors; thus, careful consideration of sample collection, study design, and bioinformatic analysis is necessary to ensure robust and reproducible results (Ghosh et al., 2020; Vujkovic-Cvijin et al., 2020).
Analysis of high-throughput microbial data often requires specialist training but progress has been made on lowering the barrier of entry, particularly for biologists who may lack the statistical or bioinformatic expertise necessary to conduct these analyses (Bolyen et al., 2019; Lagkouvardos et al., 2016; Schloss et al., 2009). Although commendable, this, in combination with the surge in attention surrounding the microbiota, has led to an increase in underpowered, associative, and often overinterpreted studies that limit translational impact (Schloss, 2018). Fecal microbiota transplantation (FMT) and probiotics are the only microbiota-based therapies currently used in clinical practice. FMT is solely used to treat Clostridioides difficile infection, whereas despite widespread use of probiotics, evidence regarding their efficacy as a treatment is currently lacking (Gupta et al., 2016). Clinically recommended uses are currently limited to formulations containing Lactobacillus and Bifidobacterium to prevent necrotizing enterocolitis in preterm infants (Su et al., 2020). Regardless, the recent adoption and development of orthogonal methods to characterize microbiota, such as gnotobiotic mice and metabolomics have allowed elucidation of the mechanisms underlying microbiota-disease associations (Chu et al., 2019). Fully embracing these methods and ensuring microbiota studies are well designed will ensure further development and establishment of emerging subfields of microbiota research, especially in fields addressing complex gut-brain interactions, such as the involvement of the microbiota in ischemic stroke (Delgado Jiménez and Benakis, 2021).
Recently, the interaction between the gut microbiota and CNS – the gut brain axis – has been implicated in the etiology of neurological diseases, particularly those with a neuroinflammatory basis such as cerebral infarction (Benakis et al., 2016; Cryan et al., 2020; Houlden et al., 2016; Singh et al., 2016; Xu et al., 2021). Indeed, patients with severe stroke display significantly altered microbiota compared to healthy age-matched controls. Xia et al. identified dysbiotic communities in stroke patients, with the level of dysbiosis predicting stroke severity (Xia et al., 2019). In murine studies, manipulation of microbiota with antibiotics before experimental stroke improved the outcome. Further, the microbiota affects the development of brain lesions by regulating polarization of gut T cells that traffic to the brain post-stroke (Benakis et al., 2016, 2020). Despite the implication of the microbiota in stroke severity and outcome, identification of both taxonomic signatures and functional mechanisms of disease remains to be seen. Uncovering a stroke-associated microbiota signature holds the potential to utilize specific gut bacteria as therapeutic interventions, impacting host pathobiology and stroke outcome (Figure 1). High-level associations such as an increase in members of Enterobacteriaceae and a decrease in butyrate-producing bacteria have been shown by several groups but insights at lower taxonomic resolution are lacking (Haak et al., 2021; Xu et al., 2021). Two recent studies, however, have linked specific microbes or their metabolites to stroke severity and outcome. Post-stroke expansion of Escherichia coli and immune stimulation via lipopolysaccharide was linked to systemic inflammation and impaired outcome (Xu et al., 2021). High levels of the metabolite trimethylamine-N-oxide (TMAO) – a gut microbial metabolite known to be associated with cardiovascular disease – increased infarct volume and led to a poorer functional outcome (Zhu et al., 2021). Together, these studies provide significant evidence for an important role of the microbiota in stroke pathogenesis and treatment, yet a distinct microbial signature has not been identified, hampered by disparate findings between existing studies, likely owing to differences in methodologies, low sample sizes, and unacknowledged confounding variables (Sadler et al., 2017; Singh et al., 2016; Stanley et al., 2018).
Figure 1.

The gut microbiota in ischemic stroke
A summary of current knowledge on the involvement of the gut microbiota in stroke and outstanding questions which microbiota signatures identified by 16S rRNA sequencing can answer (created with BioRender.com)
In general, existing stroke studies have been conducted to a high methodological standard. Nevertheless, more carefully designed and conducted investigations into the role of the microbiota in stroke will enable reproducibility between different studies and further generation of mechanistic insights. This review aims to provide a framework for conducting well-designed microbiota studies in general, but with a focus on aspects specific to stroke research. Furthermore, we aim to facilitate cross-study comparison and reproducibility of stroke-microbiota studies, supporting translation into clinical practice. Importantly, this work presents a publicly available toolbox and reproducible bioinformatic pipeline to analyze 16S rRNA gene sequencing data applicable to all microbiota studies.
Pitfalls in stroke-microbiota studies
Each step in the process of conducting a microbiota study can introduce bias, impacting downstream results. The sheer variety in sample collection methods, DNA extraction, and sequencing itself can be overwhelming for those new to the field. By discussing each step in-detail, outlining where problems can occur and how, and suggesting best practices while also providing a detailed step-by-step microbiota analysis from stroke fecal samples, we aim to provide a set of guidelines and our own recommendations (Box 1) for stroke researchers who wish to enter the field, enabling them to conduct well-designed and executed studies. Importantly, we believe our guidelines and bioinformatic pipeline will be of interest beyond the field of microbiota stroke research.
Box 1. Recommendations for sample collection and processing.
| Design | Recommendations | Further reading |
|---|---|---|
| Sample size | Consult a statistician or utilize tools such as powmic or Micropower to estimate sample size before beginning study | (Chen, 2020; Debelius et al., 2016; Kelly et al., 2015) |
| Sample collection method | Stool samples: fresh sample | (Claesson et al., 2017; Liang et al., 2020) |
| Tissue samples: whole biopsies rather than mucosal scrapes are preferable | ||
| Sample storage | Store samples immediately at −80°C or if study design requires RT storage, store in 95% ethanol | (Marotz et al., 2021; Pollock et al., 2018) |
| DNA extraction method | Use a mechanical lysis method and try to ensure samples are processed with the same kit | (Gerasimidis et al., 2016; Nearing et al., 2021) |
| Controls | Prudent use of negative and positive controls. We recommend at least one extraction control per batch and additional water controls during library preparation and sequencing | (Bedarf et al., 2021; de Goffaude et al., 2018) |
| Sequencer | Illumina MiSeq/HiSeq™ machines are appropriate for most 16S studies | (Caporaso et al., 2012; Pollock et al., 2018) |
| Hypervariable region | V1-V2/V3, V4, and V3-V4 are all commonly used and suitable for animal or human studies | (Abellan-Schneyder et al., 2021) |
Study design
Sample size and experimental design is one of the most important steps in conducting a microbiome study but often overlooked (Debelius et al., 2016). For human microbiota analysis, it is crucial to define the type of study (i.e., cross-sectional, or longitudinal) and the question of interest as this will impact processing and analysis downstream. A cross-sectional study of mucosal biopsies will require different extraction methods and controls than a longitudinal follow-up study of stool samples. Thus, the experimental design needs to consider the following points: (1) Will this be a longitudinal or cross-sectional study? (2) How will samples be preserved and within what time frame will they be processed? (3) Which readouts are planned, 16S, Shotgun metagenomics, Metabolomics or multiple? If using 16S which hypervariable region will be used? (4) Is the study well powered and controlled? (5) Are potential confounding variables well documented and can they be accounted for? By ensuring all these questions are sufficiently answered, robustness and reproducibility of results can be significantly improved.
Many confounding variables can impact microbiome composition, potentially obscuring genuine patterns or leading to false conclusions. Factors such as lifestyle (Vujkovic-Cvijin et al., 2020), geography (He et al., 2018), diet (Asnicar et al., 2021a), and medication (Forslund et al., 2021) all impact microbiota composition. In stroke patients, stool transit time and frequency, sex, age, and smoking may also confound results, either alone or in an additive or combinatorial manner. Importantly, many of these factors are either impacted by or are risk factors for stroke themselves (Asnicar et al., 2021b; Ghosh et al., 2020; Hankey, 2020; Lim et al., 2012; Phan et al., 2019; Vujkovic-Cvijin et al., 2020). Several other factors associated with stroke, such as severity, reduced food intake post-admission, infection, underlying co-morbidities, and post-stroke medication and treatment, could also introduce significant variability in microbiota composition (Chamorro et al., 2007; FOOD Trial Collaboration, 2003; Vujkovic-Cvijin et al., 2020). Antibiotics certainly impact gut microbiota; however, current, or past usage (e.g., in the last two months) is included in the exclusion criteria of most if not all existing stroke-microbiota studies. Despite this, recent work has suggested that antibiotics may have a longer lasting impact on microbial composition, which should be taken into account (Vujkovic-Cvijin et al., 2020). At this stage, it is important to systematically collect metadata from patients to ensure that confounding variables can be controlled for (Michel et al., 2010). An overview of considerations during this stage and our recommendations are listed in Table 1. This is particularly important for human studies but also in animal models as well. It is well established that co-housing of mice homogenizes microbiota composition as mice are coprophagic, hence experimental findings must be replicated across multiple cages to ensure treatment/condition associated effects are reproducible (Robertson et al., 2019). Stroke specific factors also apply to animal models; mice subjected to severe stroke (transient occlusion of the middle cerebral artery) (Jackman et al., 2011) have a reduced food intake in the acute response (Lourbopoulos et al., 2016) in comparison to their sham counterparts, in part owing to sickness behavior (Roth et al., 2020), which likely impacts microbiota. To our knowledge, no stroke-microbiota study to date has accounted for this, however, controlling food intake, for example, by fasting sham mice, can mitigate this effect, improving the reliability of results. Stress induced by surgery and anesthesia also alter microbial composition, however using sham mice as a control likely reduces the impact of this (Singh et al., 2016). Nevertheless, it can be useful to include a non-surgical control in addition. Stroke severity itself (as quantified by infarct volume) also differentially impacts microbiota. Microbial composition of animals with small infarct volumes, induced by permanent middle cerebral artery occlusion (pMCAO) resemble sham mice (Singh et al., 2016). Infarct size also depends on the gender and age of experimental animals; thus, mice must be age and gender matched (Manwani et al., 2013). Examples of these considerations and our recommendations to mitigate them are compiled in Table 2.
Table 1.
Experimental design considerations for human studies
| Considerations | Recommendations |
|---|---|
| Co-morbidities | Record any significant existing or prior medical conditions |
| Age | Ensure control groups are age matched |
| Sex | Stroke severity and outcome is often worse in women and may need to be adjusted for (Phan et al., 2019) |
| Geography | Ensure individuals reside in the same region/country or stratify by location (He et al., 2018) |
| Food intake | Patients with severe stroke may have reduced food intake, potentially confounding results (FOOD Trial Collaboration, 2003) |
| Stool consistency | Record stool consistency if possible (Lim et al., 2012; Vujkovic-Cvijin et al., 2020) |
| Infection | Post-stroke infections are common and should be recorded (Chamorro et al., 2007) |
| Medication | Existing medication may confound results, patients with a history of antibiotic use in the past six months should be excluded (Forslund et al., 2021; Vujkovic-Cvijin et al., 2020) |
| Treatment | Treatment given – i.e., Thrombolysis should be recorded and tested for an effect on the microbiota. Surgery likely additionally impacts microbiota and should be noted |
| Alcohol use/smoking | History of alcohol use and smoking are independent stroke risk factors but may also impact microbiota composition (Hankey, 2020; Vujkovic-Cvijin et al., 2020) |
Table 2.
Experimental design considerations for animal studies
| Considerations | Recommendations |
|---|---|
| Microbiota standardization | Ensure external animals are acclimatized to facility for at least 1 week before experiment (Montonye et al., 2018) |
| Mouse congenital background and source | Comparison of microbiota composition between mice from same genetic background or same commercial vendors/facilities (Robertson et al., 2019) |
| Food | Standardization and sterilization (autoclave) of mouse diets |
| Litter and cage effect | Co-housing of animals of different treatment/conditions ensures microbiota shifts are not due to cage effect (Robertson et al., 2019) |
| Randomization of experimental groups | |
| Replication of findings across multiple cages of different litters | |
| Age and sex of mice | Use animals of similar age and the same sex, as both factors can impact stroke outcome (Manwani et al., 2013) |
| Timing of sample collection | Ensure samples are collected at approximately the same time of day to limit variation due to circadian rhythm (Liang et al., 2015) |
| Experimental model | Avoid direct comparisons of different experimental stroke models as some models impact microbiota composition more than others (Singh et al., 2016) |
| Food intake | Monitor weight loss and food intake after stroke/sham surgery by fasting of sham mice or weighing food |
| Include weight loss as a covariate in analyses if significantly different between groups | |
| Anesthesia | Record and standardize the dose, duration, and type of anesthesia given during surgical procedures |
Sample size
Estimating sample size is another often overlooked step, yet the validity and reproducibility of findings is highly dependent on this. Compounding this problem, very few software packages or tools currently exist to calculate sample size specifically for microbiota studies. Several R packages have been developed to solve this problem. Micropower, allows users to estimate the sample size required to detect differences in beta-diversity using PERMANOVA (Kelly et al., 2015). The recently published Powmic also enables power analysis for differential abundance testing (Chen, 2020). Despite this, only a limited number of microbiome studies have utilized these tools (Debelius et al., 2016). In stroke, the severity appears to correlate with the magnitude of alterations in microbiota composition, thus when dealing with high variation in severity, this must be taken into consideration when deciding which samples to sequence (Singh et al., 2016; Xia et al., 2019; Yin et al., 2015). The microbiota also shows high variability for several weeks post-stroke (Xu et al., 2021). For longitudinal studies, this necessitates a larger number of total samples or utilizing multiple baseline samples to assess inherent variability. Well-planned and powered experiments and thoughtfully collected metadata underpin high quality studies as all downstream steps can be affected by choices made at this stage.
Sample type, collection, and storage
Owing to the relatively low cost and apparent ease of analysis, 16S rRNA sequencing is commonly used to profile microbial communities, providing a high-level, but low-resolution, overview of taxonomic composition. More in-depth methods such as shotgun metagenomics meta-transcriptomics and meta-proteomics provide information on both microbial composition and function but can be significantly more costly and require specialist resources to process and analyze data (Knight et al., 2018). Increasingly utilized in microbiome research, the chemical composition of the microbiome can also be measured via untargeted metabolomics (Bauermeister et al., 2021). Not all sample types are amenable to each method of profiling microbial communities; however, the choice of readout impacts the sample type that can be collected and dictates study design. In the stroke-microbiota field, there is currently a lack of diversity in sample types, particularly in humans, likely owing to logistical and ethical constraints. Results derived from stool samples are potentially limited in their translational impact as stool may not necessarily represent microbes involved in disease pathology (Claesson et al., 2017). Furthermore, stool samples provide a snapshot of the entire gastrointestinal tract but likely do not represent each compartment equally, reflecting distal gut sites better than proximal gut, which should be taken into consideration with respect to the scientific questions being asked. Here we focus on taxonomic profiling via 16S rRNA sequencing that, along with metabolomics and meta-proteomics, has the greatest breadth in terms of suitable sample types.
The method used for sample collection and subsequent storage can also introduce technical variation (Liang et al., 2020). Stool collection is the most used sample type, particularly in clinical studies and is likely to be the most appropriate for human stroke-microbiota studies, owing to the difficulties of obtaining other relevant sample types. Multiple methods exist to sample stool; a specimen can be obtained directly or via rectal swab, yet results are conflicting on the impact of each on microbial composition. Rectal swabs, when processed quickly, reflect stool microbiota composition relatively well; however, Fair et al. have recently shown that rectal swabs may not accurately represent the microbiota of critically ill patients (Bokulich et al., 2019; Fair et al., 2019). As such, in stroke-microbiota studies, stool samples are preferred over rectal swabs, where possible.
In animal studies, there is often greater flexibility over sample type and collection; thus, the method chosen needs to be aligned with the scientific question. For example, profiling mucosal associated bacteria is more useful when studying the involvement of the microbiota in modulating intestinal immune response, post-stroke mucosal tissue impairment, and bacterial translocation (Houlden et al., 2016; Stanley et al., 2016). Several mucosal associated taxa, such as Segmented filamentous bacteria (SFB) in mice, induce intestinal T-cells under homeostatic conditions, as well as in stroke and are not reliably detected in stool (Goto et al., 2014; Sadler et al., 2017). Tissue and other ntestinal samples, such as cecal content, probably approximate local communities better than stool; however, obtaining these sample types requires sacrifice, restricting their use to cross-sectional studies. Regardless of the subject or sampling method, it is crucial to ensure samples are collected in the same manner and that comparisons between different collection methods are avoided.
Different sample storage conditions can additionally impact microbial abundances and composition (Chen et al., 2019; Liang et al., 2020; Marotz et al., 2021). In principle, extraction from fresh samples is best, however this is almost never feasible in practice. Ideally, samples should therefore be frozen at −80°C before extraction. For some study designs, this may present difficulties, particularly where samples are collected by patients themselves. For room temperature preservation, OMNIGENE Gut (Choo et al., 2015), 95% ethanol (Marotz et al., 2021), and FTA cards (Vogtmann et al., 2016) all suitably maintain composition during storage. As the most widely available and cost-effective method, we recommend storing samples in 95% ethanol at a ratio of 2:1 for stroke-microbiota studies where immediate storage at −80°C is not practical. As with sample collection, it is important to keep storage methods consistent between samples.
DNA extraction
Choice of DNA extraction kit can be another source of unwanted technical variation (Gerasimidis et al., 2016; Mackenzie et al., 2015). Various commercial kits are available, utilizing several methods of lysis – chemical, enzymatic, or mechanical – each significantly differing in resulting microbial composition. Gram positive and endospore forming bacteria are more resistant to lysis, particularly to chemical and enzymatic methods. Moreover, abundances of certain taxa also vary with the DNA extraction kit used. Mackenzie et al. compared five different commonly used extraction methods, showing that the abundance of Firmicutes and Bacteroidetes varied according to the method used (Mackenzie et al., 2015). Nevertheless, between-sample variation was greater than technical variation attributed to kit. Gerasimidis et al. reported similar findings and suggest that among the methods tested there was no one “best” method for all purposes (Gerasimidis et al., 2016). However, we recommend the use of a kit employing a mechanical-based lysis method. The inclusion of this step is associated with the increased quantity of microbial DNA, higher bacterial diversity, and increased recovery of Gram-positive taxa (Pollock et al., 2018). Ultimately, any approach needs to be optimized for sample type, have a high DNA yield, and not be biased toward any particular taxon.
Low-biomass samples such as biopsies present additional challenges owing to the presence of contamination in extraction reagents, recently termed the “kitome” (de Goffau et al., 2018; Nearing et al., 2021). Salter et al. elegantly highlighted this problem using a mock community dilution series, spiked with an unusual Salmonella strain (Salter et al., 2014). With increasing dilution, true biological reads were drowned out by contamination. Multiple studies have verified these findings, since finding that the kitome varies according to both batch and kit (Glassing et al., 2016; Weiss et al., 2014). For any study, we recommend a prudent use of negative extraction controls and to process all samples in one batch. Mock community controls can also be useful, particularly when optimizing methods. Negative and positive controls are essential for low biomass studies as contaminating bacterial DNA can mask true biological signal or lead to false conclusions (Salter et al., 2014). Bacterial DNA has been identified in many sterile sites previously thought to be sterile, including the brain tissue. However, evidence for a “brain microbiome” is scant and additional work has been unable to separate contamination from true signal (Bedarf et al., 2021).
Library preparation and sequencing
Library preparation and sequencing are generally outsourced to a core facility or commercial sequencing provider, but the choice of hypervariable region(s) and primer pair – in the case of 16S sequencing – and sequencing platform is left to the researcher. As we are focused on amplicon sequencing of the bacterial component of the microbiome, other technologies are outside the scope of this review. Readers are referred to the following in-depth articles on metagenomics (Quince et al., 2017), meta-transcriptomics (Zhang et al., 2021), and metabolomics (Bauermeister et al., 2021). The 16S rRNA gene comprises nine hypervariable regions, V1–V9. The region and primer pair used will have a significant impact on both phylogenetic resolution and the taxa that are detected (Abellan-Schneyder et al., 2021; Chen et al., 2019). No existing primer pair are universal, and some pairs may miss biologically relevant taxa, for example, the commonly used 27f primer, amplifying the V1 region can miss some Bifidobacteria (Chen et al., 2019). Similarly, there is no gold standard hypervariable region, although V1–V2/V3, V3–4, and V4 are among the most commonly used regions in human and mouse microbiota studies that may aid comparison with existing published data (Abellan-Schneyder et al., 2021). Larger regions also increase the identification of certain taxa. The number of PCR cycles used to generate 16S amplicons will also affect microbial composition. A high number of cycles increases the possibility of bias, chimera formation and the impact of contamination (Sze and Schloss, 2019). However, this is generally only an issue for low biomass samples such as tissue biopsies, as the low input may require a higher number of cycles to produce sufficient input for sequencing, and pooling of PCR products can be used to mitigate this issue (Kennedy et al., 2014). The choice of primer pair and hypervariable region is mostly down to personal preference; however, for certain environments, some primer pairs are preferred. V1–V2 is recommended for oral microbiota, for example, as it produces the most faithful representation of oral communities (Wade and Prosdocimi, 2020). In any case, researchers should be aware of the pros and cons of each pair and choose accordingly.
The majority of 16S studies are sequenced on using Illumina MiSeq machines, owing to their high throughput, accuracy, and length of reads (up to 2 x 300 bp) (Caporaso et al., 2012). The chosen platform depends on the scientific question being asked but generally, MiSeq will be the most appropriate. Illumina HiSeq machines are also appropriate for both 16S and Shotgun metagenomics studies, offering higher accuracy at the expense of increased cost and shorter reads (Caporaso et al., 2012; Quince et al., 2017). However, recent newer Nextseq and Novaseq Illumina machines are being increasingly utilized in metagenomics, owing to reduced costs and similar output (Quince et al., 2017). In Illumina machines, reads can be sequenced in either single- or paired-end mode. We recommend opting for paired end as this can increase the coverage and amplicon length. Longer-read technologies such as Nanopore and PacBio sequencing are becoming more commonplace in recent years but traditionally suffered from high error rates and an underdeveloped bioinformatics ecosystem, encumbering analysis. However, this has improved vastly, with both Nanopore and PacBio sequencing achieving accuracies of >99.9% on full-length 16S sequences as of 2021 (Karst et al., 2021). Improvements to bioinformatics tools have also facilitated pre-processing and analysis but only a handful of microbiota analysis packages accept these kinds of data (Callahan et al., 2019). In the coming years, full-length 16S sequencing will likely be a viable alternative to short amplicon sequencing, and further enable the identification of taxonomic signatures in stroke and other diseases.
Bioinformatic tools for microbiota analysis
In this section, we outline methods and tools used for raw data processing and statistical analysis of microbiota data. As the application of these tools can require significant background knowledge, even those with well-designed microbiota studies can run into problems with their analyses. Here we provide an explanation of techniques and tools used and suggest various software packages and pipelines with extensive tutorials and documentation to enable researchers unfamiliar with the field to make informed choices. As microbiota analysis contains a substantial amount of field-specific jargon, we provide a glossary below explaining commonly used terms (Box 2).
Box 2. Glossary.
16S: The 16S rRNA small ribosomal subunit. Containing a combination of conserved and hypervariable regions it can be used as a molecular barcode to profile bacteria and archaea.
Alpha diversity: Species diversity within samples. Measures include Richness, Chao1, Shannon, and Simpson diversity.
ASV: Amplicon sequence variant. Error-corrected sequence representing the true biological sequence.
AUC (AUCROC): Area under the receiver operator curve. A measure used to assess the ability of a machine learning models to distinguish between classes.
Beta diversity: Between sample diversity. Metrics include Bray-Curtis and UniFrac.
Bray-Curtis: A count-based dissimilarity metric (beta diversity), based on the fraction of overabundant counts.
Chao1: An alpha diversity metric, which estimates species richness based on abundance of individuals belonging to a given class. Accounts for abundance and evenness.
Compositional data: Data composed of strictly positive numbers with a fixed sum (i.e., 1), conveying only relative information.
CLR transform: Centered-log ratio transform. A data transformation computed by calculating the log-ratio of each taxon relative to the geometric mean.
Cross-validation: A method of estimating the generalizability of a machine learning model by resampling limited data.
Differential abundance: Identification of bacteria that differ in abundance between groups.
Evenness: An alpha diversity metric, assessing how similar abundance distributions are between samples.
Faith’|'s phylogenetic diversity: A phylogenetic measure of alpha diversity, calculated as the sum of branch lengths in the phylogenetic tree.
False discovery rate (FDR): The proportion of findings that are falsely identified as significant.
NMDS: Non-metric multidimensional scaling. An ordination method that attempts to represent the dissimilarity between samples, as closely as possible in a low-dimensional space.
OTU: Operational taxonomic unit. A cluster of similar 16S sequences (usually 97%) approximating a bacterial/archaeal genera or species.
PCoA (MDS): Principal coordinate analysis (also known as metric multidimensional scaling). An ordination method that attempts to preserve distance between samples in a low dimensional Euclidean space.
PERMANOVA: Non-parametric multivariate ANOVA. Used to assess whether beta diversity metrics differ between groups.
Machine learning: Use of data and algorithms to learn from data without explicit instruction. Machine learning models can be used to identify patterns in data and predict future trends.
Metagenomics: Measurement of the entire gene content present in a sample (DNA).
Metatranscriptomics: Measurement of actively transcribed genes in a sample (RNA).
Metabolomics: Measurement of the small molecule component (<2,000 Da) of a sample.
Shannon diversity: A measure of alpha diversity, accounting for both the number of observed species and species evenness.
Simpson diversity: An alpha diversity metric, measuring the relative abundance of species comprising sample richness.
Training/Test sets: Data splits used to train and test machine learning classifiers. The training set allows the model to learn patterns in the data while the test set’s purpose is to estimate model performance on unseen data.
Rarefaction curve: A plot of species richness against read depth used to assess whether sampling depth was adequate. A plateau indicates that no further diversity is likely to be detected with increased depth.
Richness: An alpha diversity metric measuring the total number of observed species.
ROC: A visual aid to assess machine learning classifier performance where the true positive rate is plotted against the false positive rate.
UniFrac: A phylogenetic beta-diversity metric measuring the fraction of unique branches in a phylogenetic tree. The extensions weighted and generalized UniFrac also take relative abundance into account.
Raw data and pre-processing
Raw 16S rRNA sequencing data will generally be received in a fastq format, which requires substantial quality control and pre-processing before any inferences regarding microbiota composition can be made. A multitude of pipelines and software packages are available, with varying levels of skill and technical knowledge required for use. Although pipelines with a graphical user interface (GUI) may seem like an attractive option, we maintain that it is better to invest the time to learn how to use more advanced and flexible tools, as this necessitates a greater understanding of the data. Widely used, well-maintained tools with extensive documentation such as QIIME2 (Bolyen et al., 2019), mothur (Schloss et al., 2009), and DADA2 (Callahan et al., 2016) are among some of the best options to pre-process and/or analyze microbiota data. A summary of all tools listed in this section and their use can be found in Box 3.
Box 3. Bioinformatic tools.
| Use | Tools | References |
|---|---|---|
| Quality Control | FastQC, MultiQC | (Andrews, 2010; Ewels et al., 2016) |
| Primer/adapter trimming | Cutadapt, Trimmomatic | (Bolger et al., 2014; Martin, 2011) |
| Amplicon denoising | DADA2, Deblur, UNOISE2 | (Amir et al., 2017; Callahan et al., 2019; Edgar, 2016) |
| Taxonomy databases | RDP, SILVA, Greengenes | (Cole et al., 2013; DeSantis et al., 2006; Quast et al., 2012) |
| Phylogeny | FastTree | (Price et al., 2010) |
| Analysis suites | QIIME2, mothur, phyloseq | (Bolyen et al., 2019; McMurdie and Holmes, 2013; Schloss et al., 2009) |
| Pipelines | IMNGS, nf-core/ampliseq | (Lagkouvardos et al., 2016; Straub et al., 2020) |
| Differential abundance | ALDEx2, ANCOM/ANCOM-BC, DESeq2, gneiss, LEfSe, MaAsLin2, selbal | (Fernandes et al., 2014; Lin and Peddada, 2020b; Love et al., 2014; Mallick et al., 2021; Mandal et al., 2015; Morton et al., 2017; Rivera-Pinto et al., 2018; Segata et al., 2011) |
| Machine learning | mikropml, SIAMCAT | (Topçuoğlu et al., 2021; Wirbel et al., 2021) |
| Data repositories | ENA/SRA | (Leinonen et al., 2011) |
| Tools for reproducible research | GitHub/GitLab, Rmarkdown, Jupyter notebooks | (Allaire et al., 2021; Kluyver et al., 2016) |
A crucial starting point in any analysis of sequencing data is the assessment of data quality (Figure 2, step 1). Tools such as FastQC and the extension package MultiQC enable an overview of sequence length, quality, and adapter content (Andrews, 2010; Ewels et al., 2016). Low quality-reads, bases, and adapter sequences can then be removed using tools such as Cutadapt or Trimmomatic (Figure 2, step 2) (Bolger et al., 2014; Martin, 2011). Complete analysis suites such as QIIME2 and mothur wrap these programs or include their own scripts to perform these tasks. Low-quality or improperly filtered data can lead to various problems with downstream analyses, potentially inflating the diversity or hampering taxonomic classification.
Figure 2.

Data analysis pipeline
Overview of the data-analysis pipelines provided with this study, displaying each step in the analysis pipeline and the software used. Some examples of the kinds of figures which can generated with our pipeline are highlighted in the last step. Two versions of the same pipeline are provided, one written in R (left) and one in python via QIIME2 (right), which wraps the individual analysis steps in one software package (created with BioRender.com)
Cleaned data can then be denoised, dereplicated, merged, if sequenced using paired reads, and then used to infer Amplicon sequence variants (ASVs) or alternatively clustered into 97% operational taxonomic units (OTUs) (Figure 2, steps 2 and 3). It is generally recommended to use ASVs as they increase the resolution and are comparable between experiments. DADA2 (Callahan et al., 2016), Deblur (Amir et al., 2017), and UNOISE2 (Edgar, 2016) are among the most widely used tools for ASV inference and all provide extensive documentation. OTUs are still a viable option for community-level analyses such as alpha and beta diversity and have been demonstrated to generate similar conclusions as ASVs from the same samples (Glassman and Martiny, 2018). Recent work utilizing mock communities recommends applying abundance filtering at 0.25% relative abundance to remove spurious ASVs or OTUs, as low abundant sequencing artifacts can impact downstream analyses, and we recommend including this step in any analysis (Reitmeier et al., 2021). After merging, using paired-end reads (Figure 2, step 4), ASVs or OTUs are assigned taxonomy, usually by a machine learning model trained on representative sequences (Wang et al., 2007). Most approaches use some variation of naive bayes by default, and, recently, Ziemski et al. demonstrated alternative models do not significantly improve on this (Figure 2, step 5). In this case, other methods should only be used with clear justification (Ziemski et al., 2021).
The choice of reference database, however, has a significant impact on the resulting taxonomic assignments. The Ribosomal database project (RDP) (Cole et al., 2013), SILVA (Quast et al., 2012), and Greengenes (DeSantis et al., 2006) are among the most comprehensive and most widely used reference databases. Using mock communities, Abellan-Schneyder et al. demonstrated considerable discordance in taxonomic assignment between databases, showing that RDP and SILVA performed consistently better than Greengenes (Abellan-Schneyder et al., 2021). Although there is no gold-standard database, we recommend using RDP or SILVA, as these are both larger and more recently updated than Greengenes. To generate a phylogenetic tree, ASV or OTU sequences are first aligned, and a tool like FastTree can be used to construct the phylogeny (Figure 2, step 5) (Price et al., 2010).
Each choice made during raw data processing will inevitably impact the data and it is critical that these choices are justified and well documented to enhance thee validity and reproducibility of findings. Using analysis pipelines in the form of scripts can help with this by providing a record of each step in the process. QIIME2 and containerized pipelines, such as the nextflow-based nf-core/ampliseq, extend this concept further by including provenance tracking and enabling fully reproducible analysis across various computing infrastructures in the case of nf-core (Bolyen et al., 2019; Straub et al., 2020). We recommend using such pipelines as they maintain the best balance between the ease of use and reproducibility.
Statistical analysis and interpretation
After ASV inference and taxonomic assignment, a matrix containing samples and the abundance of each unique taxon is generated. Microbiota data are generally high-dimensional, often containing thousands of ASVs and sparse, violating the assumptions of many common statistical tests. Additionally, each sample has differing read counts that must be managed before proceeding with any analysis. It is important to note that the number of reads is an additional important step in assessing the quality of the data. Defining a minimum threshold is difficult as different environments will vary in the number of reads recovered; however, work analyzing complex soil microbiomes has demonstrated even 2,000 reads per sample to be sufficient to capture the diversity of most samples (Caporaso et al., 2010). Plotting a rarefaction curve can also aid in assessing sampling effort.
To ensure measured differences are not an artifact of different read depth between samples, the data must be subjected to normalization before proceeding with downstream analyses.
Normalization is a contentious topic in the microbiota field, with no universally agreed-upon method. Rarefaction, which is randomly subsampling each sample to an even read depth, is very commonly used and yet has received criticism, as it discards valid data and reduces statistical power (McMurdie and Holmes, 2014). Scaling methods such as cumulative sum scaling or converting data into relative abundances avoid the problem of data loss but may lead to increased compositional bias and a higher false discovery rate (FDR). Methods such as centered log-ratio (clr) transform mitigate compositional effects but are not compatible with many alpha and beta diversity metrics. As with many other steps in microbiome data processing and analysis, there is no gold standard, and readers are referred to the following publications that discuss the pros and cons of each in detail (Lin and Peddada, 2020a; McMurdie and Holmes, 2014; Weiss et al., 2017). The choice of normalization method should depend on the desired downstream analyses, with more advanced analyses potentially requiring a mixture of normalization strategies.
Assessing microbial community structure
After normalization, variations in the community-level structure can be assessed using alpha and beta diversity metrics. Alpha diversity measures diversity within samples. Commonly used metrics such as richness, Shannon index and Faith’s phylogenetic diversity (Faith’s PD) measure different aspects and can be used complementary to each other (Finotello et al., 2016). Richness is a simple measure of the number of observed species, whereas the Shannon index takes both species richness and evenness into account. Faith’s PD integrates phylogenetic information to estimate species diversity. Other metrics such as Chao1 and Simpson index are also often used; however, they are analogous to richness and Shannon, respectively, and are redundant if used in combination (Finotello et al., 2016). Beta diversity, on the other hand, measures the pairwise dissimilarity between samples and is used to assess how similar or different two given microbial communities are. The choice of metric can significantly influence the results obtained; thus, the metric used should be justified by the biological question. Bray-Curtis, a counts-based metric, measures the compositional dissimilarity between sites/groups while phylogenetic methods, such as UniFrac, take the genetic relationship between species into account (Lozupone and Knight, 2005). Weighted and Generalized UniFrac are extensions of this method, which also account for abundance into, differing in terms of their sensitivity to lowly and highly abundant lineages (Chen et al., 2012; Lozupone et al., 2007). Differences in beta diversity can be visualized using ordination methods like principal coordinates analysis (PCoA; also known as multidimensional scaling) or mon-metric multidimensional scaling (NMDS). Both NMDS and PCoA are suitable for all commonly used metrics; however, PCoA can generally only detect linear trends, whereas NMDS additionally detects non-linear patterns. Additionally, using PCoA with non-Euclidean distance metrics such as Bray-Curtis can produce negative eigenvalues, which cannot be represented meaningfully, potentially hindering interpretation. In this case, additional corrections must be applied before plotting. Researchers should be aware of the basics of each beta diversity metric and choose an appropriate ordination method accordingly.
Differential abundance
Beyond community-level changes, a common goal is to identify taxa that are associated with disease or differentially abundant between sample groupings. A recent meta-analysis of human microbiota studies found that among the 224 publications included 34 different methods of differential abundance (DA) were used, with LEfSe being the most common (Bardenhorst et al., 2021; Segata et al., 2011). This magnitude of different options can seem overwhelming, often leading to researchers choosing sub-optimal methods. Methods developed for RNAseq data such as DESeq2 have also been widely applied to microbiota data, which have been shown to perform poorly (Lin and Peddada, 2020a; Love et al., 2014), in part owing to not accounting for data compositionality. Although there is still some debate over whether microbiota data are truly compositional, compositionally aware methods are becoming more widely used, leading to the development of DA tools accounting for this (Gloor et al., 2017; Lovell et al., 2020). Compositional data refer to data that sum to a fixed constant e.g., 1 or 100, as with relative abundances. As these data are proportional, an increase in the abundance of one species necessitates a decrease in others, which can be misinterpreted by non-compositional methods. Novel methods such as ANCOM – including its recent extension ANCOM-BC – and ALDEx2, account for the compositional nature of microbiota data through the use of log-ratios, which makes the data scale invariant (Fernandes et al., 2014; Lin and Peddada, 2020b; Mandal et al., 2015). Other approaches such as selbal and gneiss utilize a concept known as “Balances,” whereby the log-ratios of groups of taxa rather than individuals are associated with a response variable, enabling compositionally-aware identification of microbial signatures (Morton et al., 2017; Rivera-Pinto et al., 2018). We recommend using compositional methods as they are generally more conservative, reducing the potential number of false positives. Regardless of the DA method used, controlling the FDR is a crucial step to ensure that the number of false positives is limited. Kleine Bardenhorst et al. found almost half included studies did not correct for multiple-testing (Bardenhorst et al., 2021). Owing to the high dimensionality of microbiota data, significant results can be expected just by chance; therefore, it is essential to use an appropriate method to control type I error. Most DA packages include this by default but researchers should always be aware of the problem of multiple-testing and ensure the FDR is adequately controlled for. The impact of multiple testing can also be mitigated by including an additional abundance and/or prevalence filtering step prior to DA testing, removing taxa below a given threshold. For example, in our laboratory, we commonly use a prevalence filter, retaining taxa present in more than a third of samples; however, we recommend defining thresholds operationally as, in some cases or environments, low abundant taxa may be relevant to outcome.
Machine learning for the prediction of disease-associated microbiota
Machine learning (ML) is a promising method to identify potential microbial biomarkers associated with disease but is perhaps the most abused analysis technique in microbiota research. Many studies report extremely high classification accuracies, using metrics such as Area under the receiver operator curve (AUC), but very few report low performance. The complexity and high variability of microbiota data would imply that this is likely to arise from other factors rather than the true biological signal (Quinn, 2021). Many microbiota studies have relatively low sample sizes or imbalanced groups, which, when combined with the large number of features (i.e., ASVs or OTUs), present significant difficulties when training ML models, leading to overfitting (Teschendorff, 2019). The minimum sample size to use depends on the complexity of both the given data set and the model used. As a rule of thumb, we do not recommend training models on data sets with a sample size <40 (Wirbel et al., 2021). Compounding this issue, small sample sizes can often lead to researchers omitting test sets, artificially inflating performance and leading to unverified results. Another common mistake is test set leakage, where information from the test set is included in the training set, which can similarly inflate model accuracy. Estimates of accuracy can be obtained by using cross-validation, where the data set is repeatedly split into training and test sets and the accuracy averaged across each fold. However, in many cases, this is used incorrectly, also often owing to test set leakage, or not at all (Quinn, 2021). Finally, supervised feature selection – that is, preselecting features that are differentially abundant or show high feature importance – is often performed before training an ML model, for example, by filtering data sets to only include differentially abundant taxa, again leading to overly optimistic model accuracy (Wirbel et al., 2021). Feature selection should be nested within the cross-validation process to ensure reliable estimates of model accuracy.
Clinical stroke-microbiota studies using of ML have generally demonstrated acceptable use of cross-validation and test and training sets, however many have performed supervised feature selection (Li et al., 2019; Xu et al., 2019; Yin et al., 2015). Together, these issues hamper identification of accurate microbial signatures of disease. The correct use of ML models and larger sample sizes will allow the identification of robust bacterial signatures of stroke, as already demonstrated for many other diseases (Asnicar et al., 2021a; Ghosh et al., 2020). To ensure the correct use of ML, researchers can make use of R packages like mikropml and SIAMCAT that automate most of the steps where researchers unfamiliar with ML can make mistakes (Topçuoğlu et al., 2021; Wirbel et al., 2021).
Longitudinal analysis of microbiota data
Longitudinal sampling designs enable additional insights, enabling concurrent analysis of both inter and intra-individual variability, however in stroke, temporal analyses have been limited thus far, yet are likely crucial in identifying microbes involved in modulating outcome. Such designs present additional analysis challenges, owing to the inherent autocorrelation of temporal data, which violates the assumptions of many common statistical tests. Care must therefore be taken to ensure the correct tests are used, particularly for DA or ML analyses.
Microbiome-specific methods or packages for time-series analysis are currently scarce; however, recent versions of QIIME2 ship with a plugin for longitudinal analysis: q2-longitudinal that utilizes various methods to measure the temporal stability and identify DA features (Bokulich et al., 2018). Most of the existing DA methods mentioned above are either not suitable at all for temporal data, or not suitable without modifying default parameters. ALDEx2, ANCOM-BC, and MaAsLin2, however, all implement models that can be used to perform longitudinal DA (Fernandes et al., 2014; Lin and Peddada, 2020b; Mallick et al., 2021). For a more detailed review and tutorial on analysis of time-series data, we refer the readers to Coenen et al. (2020).
Controlling for confounding variables
Confounding variables can lead to false identification of differentially abundant taxa or exaggerate AUC, in the case of ML. Unfortunately, confounders are plentiful in microbiome research, which can make it difficult to discern true signal from noise (Vujkovic-Cvijin et al., 2020). As discussed above, it is important to collect as much detailed and relevant metadata as possible as this can be extremely helpful at this step in the analysis. We recommend always plotting data before beginning any further analyses. For example, when conducting alpha or beta diversity analyses, it can be informative to generate PCoA/NMDS plots colored by additional variables than those of direct relevance to the scientific question of the study, to assess the potential impact of confounding variables. However, this will only highlight confounders thatcontribute significantly to variation in the data. In some cases, careful grouping of samples, or, dependent on sample size, exploring subsets of groups can mitigate the impact of confounding variables. We recommend using packages where confounders can be included as interactions, such as gneiss, ANCOM-BC, or MaAsLin2 (Lin and Peddada, 2020b; Mallick et al., 2021; Morton et al., 2017).
Fostering reproducibility and transparency
To ensure the reproducibility and validation of the findings, raw data and associated metadata need to be openly accessible before publication via public repositories such as the Sequence read archive/European Nucleotide archive (SRA/ENA) (Leinonen et al., 2011). At the very least, every software package and the specific version used need to be listed in the methods of any resulting manuscript. In the case of automated pipelines, this information should be sufficient to reproduce the analysis. However, for bespoke analyses, pipeline scripts used to process the raw data should be shared in a publicly available repository such as GitHub or GitLab. Documenting each step in the analysis process is also important and further fosters reproducibility and transparency. Various tools are available to aid in this process and we strongly recommend making full use of these. Writing code in Rmarkdown or Jupyter Notebooks/Jupyter Lab allows the inclusion of text in markdown format between cells of code, facilitating thorough documentation of the analyses performed (Allaire et al., 2021; Kluyver et al., 2016). Using a version control software, such as Git or Mercurial, is also encouraged and enables both researchers and collaborators to better track analyses. Similarly, a analysis code should also be uploaded to a public repository after publication. Ensuring clear and detailed reporting will boost the stroke-microbiota field, facilitating novel insights into the interaction between gut bacteria and stroke. Moreover, consistent and rigorous standards will enable meta-analyses allowing identification of microbial signatures of stroke and stroke outcome.
Best practices – example pipeline
Theory and recommendations are necessary to help guide stroke-microbiota researchers toward making the correct decisions regarding their data; however, practical advice and tutorials for some steps are often lacking. To this end, we have designed and conducted our own stroke-microbiota study in mice, investigating differentially abundant taxa, so that other researchers can use this as a template for their own studies.
Here, we designed a simple experiment to identify enriched taxa in mice subjected to experimental stroke. Mice were randomly split into two groups, and either sham surgery or transient middle cerebral artery occlusion (tMCAO) was performed (Llovera et al., 2021). Stool samples were collected 3 days after surgery and frozen at −80°C before extraction and subsequent sequencing of the V4 region of the 16S rRNA gene.
To enable a wider number of researchers to utilize our pipeline, we have provided two versions of our analysis pipeline, one for those familiar with R and another for those more familiar with python. Each step in the analysis pipeline is summarized above (Figure 2).
Results summary
To confirm the presence of ischemic stroke, we performed cresyl violet staining of brains from mice subjected to tMCAO. All mice developed moderately large lesions in the ipsilateral hemisphere (mean 40.6 ± 12.6 mm3; Figure 3A). Previous work comparing the microbiota of stroke and sham mice has identified broad changes in microbial community structure but a limited loss of sample diversity. This suggests that modulation of stroke and stroke outcome by the microbiota is mediated by changes in microbial composition, rather than a loss of taxa. Thus, to confirm the absence of changes in overall diversity and evenness, we calculated richness, Shannon effective, and Faith’s PD assessing the observed number of species, diversity, and evenness and phylogenetic diversity. No significant differences were observed between sham and stroke mice using any alpha diversity metric (Figure 3B), which is in line with previous findings from murine studies (Houlden et al., 2016; Singh et al., 2016).
Figure 3.

Disruption of community structure and composition post-stroke
(A) Representative cresyl violet stained sections, 3 days after tMCAO. Scale bar: 4 mm
(B) Alpha diversity measurements between stroke and sham mice showing richness (left), Shannon effective (center), and Faith’s phylogenetic diversity (right). Individual points dots represent a single mouse. Error bars represent the median +/− 1.5 multiplied by the IQR. Outliers are highlighted by an empty square
(C) Non-metric multidimensional scaling plot of Generalized UniFrac distance colored by group (Adonis PERMANOVA R2 = 0.12, p-value = 0.018)
(D) Family-level relative abundance in sham and stroke mice. Low abundant families were grouped with each other
(E) Significant differentially abundant taxa between stroke and sham mice, identified by ANCOM-BC (corrected p-value <0.05). Log2 Fold-change between conditions is shown on the x axis
We next sought to assess differences in global community structure between sham and stroke mice. Unsupervised analysis based on Generalized UniFrac distance revealed stroke samples clearly separated from controls. Consistent with prior studies showing a differential impact of stroke severity on gut microbiota, mice with larger infarcts tended to be further from sham mice than those with less severe strokes (Figure 3C). To visualize taxonomic changes driving this difference, we plotted the taxonomic composition of each sample by experimental group. It is important to note that the conclusions made from these types of visualizations are generally limited; however, they can provide a useful overview of potentially interesting changes in relative abundance. Shifts in relative abundance at the family level in stroke appeared relatively minor, implying microbial shifts occur at lower taxonomic ranks (Figure 3D). Finally, we sought to investigate changes in microbial abundance in detail. Using ANCOM-BC, a compositional DA method, we identified five ASVs with a corrected p-value < 0.05, in stroke mice (Figure 3E). These were classified as Blautia, unknown Lachnospiraceae, Parabacteroides, unknown Rikenellaceae and Alistipes. We additionally identified 17 ASVs decreased in stroke mice, several of which are known butyrate producers, previously identified as decreased in stroke patients (Haak et al., 2021).
Concluding remarks
Here we have provided a blueprint for conducting stroke-microbiota studies along with an example data set recapitulating and extending current findings. Adherence to these guidelines will enable further insights on the involvement of the microbiota in stroke, facilitating targeted manipulation, with the potential to improve stroke outcomes. Beyond 16S rRNA sequencing, we encourage further adoption of other technologies such as shotgun metagenomics, meta-transcriptomics, and metabolomics/meta-proteomics coupled with multi-omic integration in stroke-microbiota research. Multi-omic integration of these different data types aids in functional understanding of microbial factors influencing a disease. Additionally, reductionist approaches such as gnotobiotic mice can help identify individual microbes that can alter a disease or disease course. Employing these technologies in the stroke-microbiota field will further mechanistic insights into the interplay between stroke and the microbiota.
We hope that this manuscript will lead to an improvement in the consistency, transparency, and reproducibility of methods and believe this is required before consistent microbial signatures can be identified, laying the groundwork for microbiota-based treatments to become a reality. This, combined with the adoption of methods to delineate functional mechanisms, will enable the translation of stroke-microbiota research into clinical practice.
Limitations of the study
The limitations of this study include the focus on 16S rRNA sequencing at the expense of other, more detailed, profiling techniques such as shotgun metagenomics. In addition, our data were collected at a single timepoint, which may miss important microbial shifts associated with stroke that a longitudinal sampling design would uncover.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| DNeasy PowerLyzer PowerSoil Kit | QIAGEN | Cat. No. / ID: 12855-50 |
| Deposited data | ||
| Raw sequencing data | This publication | ENA: PRJEB48735 |
| Processed data and metadata | This publication | https://github.com/adamsorbie/Stroke_Microbiota_reproducibility/tree/main/data |
| Software and algorithms | ||
| Powmic | (Chen, 2020) | https://github.com/lichen-lab/powmic |
| FastQC/MultiQC | (Andrews, 2010; Ewels et al., 2016) | https://github.com/s-andrews/FastQC/https://multiqc.info/ |
| Cutadapt | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| DADA2 | (Callahan et al., 2016) | https://benjjneb.github.io/dada2/index.html |
| QIIME2 | (Bolyen et al., 2019) | https://qiime2.org/ |
| Phyloseq | (McMurdie and Holmes, 2013) | https://joey711.github.io/phyloseq/ |
| R | The R foundation | https://www.r-project.org/ |
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| BioRender | BioRender | https://biorender.com/ |
| Other | ||
| Chow | Ssniff | V1574-300 |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Corinne Benakis (Corinne.Benakis@med.uni-muenchen.de).
Material availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
All experimental protocols were approved by the responsible governmental department (Regierung von Oberbayern, Munich, Germany). C57BL/6 male mice were purchased from Charles-River Laboratories (Sulzfeld, Germany), and acclimatized for 1 week. Mice were fed a standard chow diet (Ssniff, Soest, Germany) fed ad libitum and housed under SPF conditions (12 h light-dark cycles at 22 ± 2°C). Mice were co-housed until surgery and subsequently placed in separate cages for 72h to allow food intake and weight loss to be monitored.
Middle cerebral artery occlusion
Transient middle cerebral artery occlusion (tMCAO) was induced in 8-12 week old mice as previously described with monitoring cerebral blood flow by transcranial laser Doppler flowmetry (Jackman et al., 2011; Llovera et al., 2021). Briefly, mice were anesthetized with 1.5–2% isoflurane, and rectal temperature maintained at 37 ± 0.5°C for the duration of surgery. A silicon-coated filament (#602112PK5Re, Doccol) was inserted into the left external carotid artery and advanced until obstructing the MCA together with ligation of the common carotid artery for 45 min. Regional cerebral blood flow (CBF, bregma coordinates: 2-mm posterior, 5-mm lateral) was recorded by transcranial laser Doppler flowmetry (PF 5010 LDPM, Periflux System 5000, Perimed) at induction of ischemia, before filament removal and at reperfusion. When MCA occlusion was confirmed (a residual CBF <20%) animals were placed in a recovery chamber at 37°C for 45 min (until filament removal). To allow reperfusion, mice were anesthetized after occlusion and the filament was removed. Mice were included if the residual CBF is <20% before reperfusion and CBF recovery >80% within 10 min of reperfusion. Following tMCAO, mice were placed in temperature-controlled recovery cages for 2 h to prevent post-surgery hypothermia. Sham surgery was performed as described above including ligation of the external and common arteries, and introduction of the filament (in/out). At 72h after surgery, mice were euthanized, brains were collected and immediately frozen at –80°C for cresyl violet staining.
Method details
Infarct volume quantification
Brains tissues were sectioned (30-μm thick at 600-μm intervals) on a cryotome for cresyl violet staining as previously described (Benakis et al., 2020). Quantification of the infarct volume (corrected for swelling and tissue loss) was performed blinded using ImageJ.
Power analysis
Sample size estimation to detect differentially abundant taxa was carried out using powmic (Chen, 2020). Parameters were estimated by fitting a subset of a previously published dataset (Xu et al., 2021), selected to closely mirror our own study design, to a zero-inflated negative binomial model. These parameters were subsequently used to generate synthetic datasets with 1000 ASVs. Estimation of the false discovery rate (FDR) and the true and false-positive rates was then performed using the powmic function with default parameters, testing a range of sample sizes (six, eight, ten and twelve). We used a true-positive rate threshold of 0.8 to determine sufficient power.
DNA extraction and 16S sequencing
DNA was extracted from fecal samples using a QIAGEN DNeasy Powersoil Kit and quantified using a NanoDrop ND-1000 Spectrophotometer (Thermo Fisher, United States). Sequencing was performed by the Beijing Genomics Institute (BGI) according to standard procedures. Briefly, the V4 region of the 16S rRNA gene was amplified using the 515F- GTGCCAGCMGCCGCGGTAA and 806R-GGACTACHVGGGTWTCTAAT primer pair and sequenced on an Illumina HiSeq™ 2500 machine using 2x250 bp paired end reads.
Quality of raw reads were checked with MultiQC (Ewels et al., 2016) before trimming with Cutadapt (Martin, 2011). Subsequently, ASVs were inferred using DADA2, and filtered using a threshold of 0.25% relative abundance to remove spurious sequences (Reitmeier et al., 2021). The remaining ASVs were assigned taxonomy using the SILVA database version 138 (Callahan et al., 2016; Quast et al., 2012) and a phylogenetic tree inferred using FastTree2 with the GTR+CAT model (Price et al., 2010). Steps were identical in the python-based pipeline, except that QIIME2 was used, which wraps the aforementioned tools.
Quantification and statistical analysis
Alpha (richness and Faith’s PD) and beta diversity (Generalized UniFrac) were calculated using R or QIIME2 (Bolyen et al., 2019). Statistical analysis of alpha and beta diversity was performed using Wilcoxon-tests and PERMANOVA respectively. Differential abundance analysis was performed using ANCOM-BC (Lin and Peddada, 2020a) using the Benjamini Hochberg method to control the FDR.
Acknowledgments
The authors thank Dr. Melanie Schirmer (Technical University of Munich) for her important inputs and proofreading and Dr. Monica Weiler for technical assistance. Figures were created using BioRender (Toronto). Funding Acquisition: This study was supported by the European Commission H2020MSCA-IF 2016 753893 MetaBiota (C.B.).
Author contributions
A.S.: conceptualization, data curation, formal analysis, investigation, methodology, supervision, visualization, writing original draft; R.D.: investigation, methodology; C.B.: funding acquisition, supervision, project administration, conceptualization, manuscript review, and editing.
Declaration of interests
The authors declare no competing interests.
Published: April 15, 2022
Data and code availability
-
•
Raw sequencing data were deposited in the European Nucleotide Archive under project number: PRJEB48735 and are publicly available as of the date of publication.
-
•
Scripts used for data processing and analysis are publicly available on GitHub and are publicly available at the following url: https://github.com/adamsorbie/Stroke_Microbiota_reproducibility.
-
•
Any additional information required, is available from the lead contact upon request.
References
- Abellan-Schneyder I., Matchado M.S., Reitmeier S., Sommer A., Sewald Z., Baumbach J., List M., Neuhaus K. Primer, pipelines, parameters: issues in 16S rRNA gene sequencing. Msphere. 2021;6:e01202–e01220. doi: 10.1128/msphere.01202-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaire J., Xie Y., McPherson J., Luraschi J., Ushey K., Atkins A., Wickham H., Cheng J., Chang W., Iannone R. 2021. Rmarkdown: Dynamic Documents for R. R Package. version 2.11. [Google Scholar]
- Amir A., McDonald D., Navas-Molina J.A., Kopylova E., Morton J.T., Xu Z.Z., Kightley E.P., Thompson L.R., Hyde E.R., Gonzalez A., et al. Deblur rapidly resolves single-nucleotide community sequence patterns. Msystems. 2017;2 doi: 10.1128/msystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FASTQC: A Quality Control Tool for High Throughput Sequence Data. [Google Scholar]
- Asnicar F., Berry S.E., Valdes A.M., Nguyen L.H., Piccinno G., Drew D.A., Leeming E., Gibson R., Roy C.L., Khatib H.A., et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat. Med. 2021;27:321–332. doi: 10.1038/s41591-020-01183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asnicar F., Leeming E.R., Dimidi E., Mazidi M., Franks P.W., Khatib H.A., Valdes A.M., Davies R., Bakker E., Francis L., et al. Blue poo: impact of gut transit time on the gut microbiome using a novel marker. Gut. 2021;70:1665–1674. doi: 10.1136/gutjnl-2020-323877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardenhorst S.K., Berger T., Klawonn F., Vital M., Karch A., Rübsamen N. Data analysis strategies for microbiome studies in human populations-a systematic review of current practice. Msystems. 2021;6:20. doi: 10.1128/msystems.01154-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauermeister A., Mannochio-Russo H., Costa-Lotufo L.V., Jarmusch A.K., Dorrestein P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 2021:1–18. doi: 10.1038/s41579-021-00621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedarf J.R., Beraza N., Khazneh H., Özkurt E., Baker D., Borger V., Wüllner U., Hildebrand F. Much ado about nothing? Off-target amplification can lead to false-positive bacterial brain microbiome detection in healthy and Parkinson’s disease individuals. Microbiome. 2021;9:75. doi: 10.1186/s40168-021-01012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C., Brea D., Caballero S., Faraco G., Moore J., Murphy M., Sita G., Racchumi G., Ling L., Pamer E.G., et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 2016;22:516–523. doi: 10.1038/nm.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C., Poon C., Lane D., Brea D., Sita G., Moore J., Murphy M., Racchumi G., Iadecola C., Anrather J. Distinct commensal bacterial signature in the gut is associated with acute and long-term protection from ischemic stroke. Stroke. 2020;51:1844–1854. doi: 10.1161/strokeaha.120.029262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich N.A., Dillon M.R., Zhang Y., Rideout J.R., Bolyen E., Li H., Albert P.S., Caporaso J.G. q2-longitudinal: longitudinal and paired-sample analyses of microbiome data. Msystems. 2018;3 doi: 10.1128/msystems.00219-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich N.A., Maldonado J., Kang D.-W., Krajmalnik-Brown R., Caporaso J.G. Rapidly processed stool swabs approximate stool microbiota profiles. Msphere. 2019;4:e00208–e00219. doi: 10.1128/msphere.00208-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., McMurdie P.J., Rosen M.J., Han A.W., Johnson A.J.A., Holmes S.P. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B.J., Wong J., Heiner C., Oh S., Theriot C.M., Gulati A.S., McGill S.K., Dougherty M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019;47:e103. doi: 10.1093/nar/gkz569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Huntley J., Fierer N., Owens S.M., Betley J., Fraser L., Bauer M., et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Lauber C.L., Walters W.A., Berg-Lyons D., Lozupone C.A., Turnbaugh P.J., Fierer N., Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. P Natl. Acad. Sci. U S A. 2010;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A., Urra X., Planas A.M. Infection after acute ischemic stroke: a manifestation of brain-induced immunodepression. Stroke. 2007;38:1097–1103. doi: 10.1161/01.str.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- Chen J., Bittinger K., Charlson E.S., Hoffmann C., Lewis J., Wu G.D., Collman R.G., Bushman F.D., Li H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics. 2012;28:2106–2113. doi: 10.1093/bioinformatics/bts342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. powmic: an R package for power assessment in microbiome case–control studies. Bioinformatics. 2020;36:3563–3565. doi: 10.1093/bioinformatics/btaa197. [DOI] [PubMed] [Google Scholar]
- Chen Z., Hui P.C., Hui M., Yeoh Y.K., Wong P.Y., Chan M.C.W., Wong M.C.S., Ng S.C., Chan F.K.L., Chan P.K.S. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. Msystems. 2019;4 doi: 10.1128/msystems.00271-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo J.M., Leong L.E., Rogers G.B. Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 2015;5:16350. doi: 10.1038/srep16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C., Murdock M.H., Jing D., Won T.H., Chung H., Kressel A.M., Tsaava T., Addorisio M.E., Putzel G.G., Zhou L., et al. The microbiota regulate neuronal function and fear extinction learning. Nature. 2019;574:543–548. doi: 10.1038/s41586-019-1644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson M.J., Clooney A.G., O’Toole P.W. A clinician’s guide to microbiome analysis. Nat. Rev. Gastroenterol. Hepatol. 2017;14:585–595. doi: 10.1038/nrgastro.2017.97. [DOI] [PubMed] [Google Scholar]
- Coenen A.R., Hu S.K., Luo E., Muratore D., Weitz J.S. A primer for microbiome time-series analysis. Front. Genet. 2020;11:310. doi: 10.3389/fgene.2020.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J.R., Wang Q., Fish J.A., Chai B., McGarrell D.M., Sun Y., Brown C.T., Porras-Alfaro A., Kuske C.R., Tiedje J.M. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2013;42:D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J.F., O’Riordan K.J., Sandhu K., Peterson V., Dinan T.G. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19:179–194. doi: 10.1016/s1474-4422(19)30356-4. [DOI] [PubMed] [Google Scholar]
- Debelius J., Song S.J., Vazquez-Baeza Y., Xu Z.Z., Gonzalez A., Knight R. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 2016;17:217. doi: 10.1186/s13059-016-1086-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado Jiménez R., Benakis C. The gut ecosystem: a critical player in stroke. Neuromol Med. 2021;23:236–241. doi: 10.1007/s12017-020-08633-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., Andersen G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microb. 2006;72:5069–5072. doi: 10.1128/aem.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. Biorxiv. 2016 doi: 10.1101/081257. Preprint at. [DOI] [Google Scholar]
- Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair K., Dunlap D.G., Fitch A., Bogdanovich T., Methé B., Morris A., McVerry B.J., Kitsios G.D. Rectal swabs from critically ill patients provide discordant representations of the gut microbiome compared to stool samples. Msphere. 2019;4 doi: 10.1128/msphere.00358-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A.D., Reid J.N., Macklaim J.M., McMurrough T.A., Edgell D.R., Gloor G.B. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome. 2014;2:15. doi: 10.1186/2049-2618-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotello F., Mastrorilli E., Camillo B.D. Measuring the diversity of the human microbiota with targeted next-generation sequencing. Brief Bioinform. 2016:Bbw119. doi: 10.1093/bib/bbw119. [DOI] [PubMed] [Google Scholar]
- FOOD Trial Collaboration Poor nutritional status on admission predicts poor outcomes after stroke: observational data from the food trial. Stroke. 2003;34:1450–1456. doi: 10.1161/01.str.0000074037.49197.8c. [DOI] [PubMed] [Google Scholar]
- Forslund S.K., Chakaroun R., Zimmermann-Kogadeeva M., Markó L., Aron-Wisnewsky J., Nielsen T., Moitinho-Silva L., Schmidt T.S.B., Falony G., Vieira-Silva S., et al. Combinatorial, additive and dose-dependent drug-microbiome associations. Nature. 2021;600:500–505. doi: 10.1038/s41586-021-04177-9. [DOI] [PubMed] [Google Scholar]
- Gerasimidis K., Bertz M., Quince C., Brunner K., Bruce A., Combet E., Calus S., Loman N., Ijaz U.Z. The effect of DNA extraction methodology on gut microbiota research applications. Bmc Res. Notes. 2016;9:365. doi: 10.1186/s13104-016-2171-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T.S., Das M., Jeffery I.B., O’Toole P.W. Adjusting for age improves identification of gut microbiome alterations in multiple diseases. Elife. 2020;9 doi: 10.7554/elife.50240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassing A., Dowd S.E., Galandiuk S., Davis B., Chiodini R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016;8:24. doi: 10.1186/s13099-016-0103-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassman S.I., Martiny J.B.H. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. Msphere. 2018;3 doi: 10.1128/msphere.00148-18. e00148–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Goffau M.C., Lager S., Salter S.J., Wagner J., Kronbichler A., Charnock-Jones D.S., Peacock S.J., Smith G.C.S., Parkhill J. Recognizing the reagent microbiome. Nat. Microbiol. 2018;3:851–853. doi: 10.1038/s41564-018-0202-y. [DOI] [PubMed] [Google Scholar]
- Goto Y., Panea C., Nakato G., Cebula A., Lee C., Diez M.G., Laufer T.M., Ignatowicz L., Ivanov I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Allen-Vercoe E., Petrof E.O. Fecal microbiota transplantation: in perspective. Ther. Adv. Gastroenter. 2016;9:229–239. doi: 10.1177/1756283x15607414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak B.W., Westendorp W.F., van Engelen T.S.R., Brands X., Brouwer M.C., Vermeij J.-D., Hugenholtz F., Verhoeven A., Derks R.J., Giera M., et al. Disruptions of anaerobic gut bacteria are associated with stroke and post-stroke infection: a prospective case–control study. Transl. Stroke Res. 2021;12:581–592. doi: 10.1007/s12975-020-00863-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankey G.J. Population impact of potentially modifiable risk factors for stroke. Stroke. 2020;51:719–728. doi: 10.1161/strokeaha.119.024154. [DOI] [PubMed] [Google Scholar]
- He Y., Wu W., Zheng H.-M., Li P., McDonald D., Sheng H.-F., Chen M.-X., Chen Z.-H., Ji G.-Y., Zheng Z.-D.-X., et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018;24:1532–1535. doi: 10.1038/s41591-018-0164-x. [DOI] [PubMed] [Google Scholar]
- Houlden A., Goldrick M., Brough D., Vizi E.S., Lénárt N., Martinecz B., Roberts I.S., Denes A. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun. 2016;57:10–20. doi: 10.1016/j.bbi.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman K., Kunz A., Iadecola C. Neurodegeneration, methods and protocols. Methods Mol. Biol. 2011;793:195–209. doi: 10.1007/978-1-61779-328-8_13. [DOI] [PubMed] [Google Scholar]
- Karst S.M., Ziels R.M., Kirkegaard R.H., Sørensen E.A., McDonald D., Zhu Q., Knight R., Albertsen M. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods. 2021;18:165–169. doi: 10.1038/s41592-020-01041-y. [DOI] [PubMed] [Google Scholar]
- Kelly B.J., Gross R., Bittinger K., Sherrill-Mix S., Lewis J.D., Collman R.G., Bushman F.D., Li H. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinform Oxf. Engl. 2015;31:2461–2468. doi: 10.1093/bioinformatics/btv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy K., Hall M.W., Lynch M.D.J., Moreno-Hagelsieb G., Neufeld J.D. Evaluating bias of illumina-based bacterial 16S rRNA gene profiles. Appl. Environ. Microb. 2014;80:5717–5722. doi: 10.1128/aem.01451-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluyver T., Ragan-Kelley B., Pérez F., Granger B., Bussonnier M., Frederic J., Kelley K., Hamrick J., Grout J., Corlay S., et al. Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOS Press: 2016. Jupyter Notebooks—A Publishing Format for Reproducible computational Workflows; pp. 87–90. [Google Scholar]
- Knight R., Vrbanac A., Taylor B.C., Aksenov A., Callewaert C., Debelius J., Gonzalez A., Kosciolek T., McCall L.-I., McDonald D., et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018;16:410–422. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- Lagkouvardos I., Joseph D., Kapfhammer M., Giritli S., Horn M., Haller D., Clavel T. IMNGS: a comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep-UK. 2016;6:33721. doi: 10.1038/srep33721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R., Akhtar R., Birney E., Bower L., Cerdeno-Tárraga A., Cheng Y., Cleland I., Faruque N., Goodgame N., Gibson R., et al. The European nucleotide archive. Nucleic Acids Res. 2011;39:D28–D31. doi: 10.1093/nar/gkq967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Wang X., Sun C., Wu X., Lu M., Si Y., Ye X., Wang T., Yu X., Zhao X., et al. Change of intestinal microbiota in cerebral ischemic stroke patients. Bmc Microbiol. 2019;19:191. doi: 10.1186/s12866-019-1552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Bushman F.D., FitzGerald G.A. Rhythmicity of the intestinal microbiota is regulated by gender and the host circadian clock. Proc. Natl. Acad Sci. 2015;112:10479–10484. doi: 10.1073/pnas.1501305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y., Dong T., Chen M., He L., Wang T., Liu X., Chang H., Mao J.-H., Hang B., Snijders A.M., et al. Systematic analysis of impact of sampling regions and storage methods on fecal gut microbiome and metabolome profiles. Msphere. 2020;5 doi: 10.1128/msphere.00763-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y.H., Kim D.H., Lee M.Y., Joo M.C. Bowel dysfunction and colon transit time in brain-injured patients. Ann. Rehabil. Med. 2012;36:371. doi: 10.5535/arm.2012.36.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Peddada S.D. Analysis of microbial compositions: a review of normalization and differential abundance analysis. Npj Biofilms Microbiomes. 2020;6:60. doi: 10.1038/s41522-020-00160-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Peddada S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020;11:3514. doi: 10.1038/s41467-020-17041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovera G., Simats A., Liesz A. Modeling stroke in mice: transient middle cerebral artery occlusion via the external carotid artery. J. Vis. Exp. 2021 doi: 10.3791/62573. [DOI] [PubMed] [Google Scholar]
- Lourbopoulos A., Mamrak U., Roth S., Balbi M., Shrouder J., Liesz A., Hellal F., Plesnila N. Inadequate food and water intake determine mortality following stroke in mice. J. Cereb. Blood Flow Metab. 2016;37:2084–2097. doi: 10.1177/0271678x16660986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell D.R., Chua X.-Y., McGrath A. Counts: an outstanding challenge for log-ratio analysis of compositional data in the molecular biosciences. Nar Genom. Bioinform. 2020;2 doi: 10.1093/nargab/lqaa040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 2005;71:8228–8235. doi: 10.1128/aem.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C.A., Hamady M., Kelley S.T., Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microb. 2007;73:1576–1585. doi: 10.1128/aem.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie B.W., Waite D.W., Taylor M.W. Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front. Microbiol. 2015;6:130. doi: 10.3389/fmicb.2015.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick H., Rahnavard A., McIver L.J., Ma S., Zhang Y., Nguyen L.H., Tickle T.L., Weingart G., Ren B., Schwager E.H., et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021;17 doi: 10.1371/journal.pcbi.1009442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S., Treuren W.V., White R.A., Eggesbø M., Knight R., Peddada S.D. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health D. 2015;26:27663. doi: 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwani B., Liu F., Scranton V., Hammond M.D., Sansing L.H., McCullough L.D. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp. Neurol. 2013;249:120–131. doi: 10.1016/j.expneurol.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz C., Cavagnero K.J., Song S.J., McDonald D., Wandro S., Humphrey G., Bryant M., Ackermann G., Diaz E., Knight R. Evaluation of the effect of storage methods on fecal, saliva, and skin microbiome composition. Msystems. 2021;6 doi: 10.1128/msystems.01329-20. e01329–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- McMurdie P.J., Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014;10 doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie P.J., Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel P., Odier C., Rutgers M., Reichhart M., Maeder P., Meuli R., Wintermark M., Maghraoui A., Faouzi M., Croquelois A., et al. The acute STroke registry and analysis of Lausanne (ASTRAL): design and baseline analysis of an ischemic stroke registry including acute multimodal imaging. Stroke. 2010;41:2491–2498. doi: 10.1161/strokeaha.110.596189. [DOI] [PubMed] [Google Scholar]
- Montonye D.R., Ericsson A.C., Busi S.B., Lutz C., Wardwell K., Franklin C.L. Acclimation and institutionalization of the mouse microbiota following transportation. Front. Microbiol. 2018;9:1085. doi: 10.3389/fmicb.2018.01085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton J.T., Sanders J., Quinn R.A., McDonald D., Gonzalez A., Vázquez-Baeza Y., Navas-Molina J.A., Song S.J., Metcalf J.L., Hyde E.R., et al. Balance trees reveal microbial niche differentiation. Msystems. 2017;2 doi: 10.1128/msystems.00162-16. e00162–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nearing J.T., Comeau A.M., Langille M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome. 2021;9:113. doi: 10.1186/s40168-021-01059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan H.T., Reeves M.J., Blizzard C.L., Thrift A.G., Cadilhac D.A., Sturm J., Otahal P., Rothwell P., Bejot Y., Cabral N.L., et al. Sex differences in severity of stroke in the INSTRUCT study: a meta-analysis of individual participant data. J. Am. Heart Assoc. 2019;8 doi: 10.1161/jaha.118.010235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J., Glendinning L., Wisedchanwet T., Watson M. The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microb. 2018;84 doi: 10.1128/aem.02627-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. Plos One. 2010;5 doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017;35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- Quinn T.P. Stool studies don’t pass the sniff test: a systematic review of human gut microbiome research suggests widespread misuse of machine learning. Arxiv. 2021 doi: 10.48550/arXiv.2107.03611. Preprint at. [DOI] [Google Scholar]
- Reitmeier S., Hitch T.C.A., Treichel N., Fikas N., Hausmann B., Ramer-Tait A.E., Neuhaus K., Berry D., Haller D., Lagkouvardos I., et al. Handling of spurious sequences affects the outcome of high-throughput 16S rRNA gene amplicon profiling. Isme Commun. 2021;1:31. doi: 10.1038/s43705-021-00033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Pinto J., Egozcue J.J., Pawlowsky-Glahn V., Paredes R., Noguera-Julian M., Calle M.L. Balances: a new perspective for microbiome analysis. Msystems. 2018;3 doi: 10.1128/msystems.00053-18. e00053–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson S.J., Lemire P., Maughan H., Goethel A., Turpin W., Bedrani L., Guttman D.S., Croitoru K., Girardin S.E., Philpott D.J. Comparison of Co-housing and Littermate methods for microbiota standardization in mouse models. Cell Rep. 2019;27:1910–1919.e2. doi: 10.1016/j.celrep.2019.04.023. [DOI] [PubMed] [Google Scholar]
- Roth S., Yang J., Cramer J., Malik R., Liesz A. Detection of cytokine-induced sickness behavior after ischemic stroke by an optimized behavioral assessment battery. Brain Behav. Immun. 2020;91:668–672. doi: 10.1016/j.bbi.2020.11.016. [DOI] [PubMed] [Google Scholar]
- Sadler R., Singh V., Benakis C., Garzetti D., Brea D., Stecher B., Anrather J., Liesz A. Microbiota differences between commercial breeders impacts the post-stroke immune response. Brain Behav. Immun. 2017;66:23–30. doi: 10.1016/j.bbi.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Salter S.J., Cox M.J., Turek E.M., Calus S.T., Cookson W.O., Moffatt M.F., Turner P., Parkhill J., Loman N.J., Walker A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P.D. Identifying and overcoming threats to reproducibility, replicability, robustness, and generalizability in microbiome research. Mbio. 2018;9 doi: 10.1128/mbio.00525-18. e00525–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss P.D., Westcott S.L., Ryabin T., Hall J.R., Hartmann M., Hollister E.B., Lesniewski R.A., Oakley B.B., Parks D.H., Robinson C.J., et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009;75:7537–7541. doi: 10.1128/aem.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V., Roth S., Llovera G., Sadler R., Garzetti D., Stecher B., Dichgans M., Liesz A. Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci. 2016;36:7428–7440. doi: 10.1523/jneurosci.1114-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley D., Mason L.J., Mackin K.E., Srikhanta Y.N., Lyras D., Prakash M.D., Nurgali K., Venegas A., Hill M.D., Moore R.J., et al. Translocation and dissemination of commensal bacteria in post-stroke infection. Nat. Med. 2016;22:1277–1284. doi: 10.1038/nm.4194. [DOI] [PubMed] [Google Scholar]
- Stanley D., Moore R.J., Wong C.H.Y. An insight into intestinal mucosal microbiota disruption after stroke. Sci. Rep. 2018;8:568. doi: 10.1038/s41598-017-18904-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub D., Blackwell N., Langarica-Fuentes A., Peltzer A., Nahnsen S., Kleindienst S. Interpretations of environmental microbial community studies are biased by the selected 16S rRNA (gene) amplicon sequencing pipeline. Front. Microbiol. 2020;11:550420. doi: 10.3389/fmicb.2020.550420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su G.L., Ko C.W., Bercik P., Falck-Ytter Y., Sultan S., Weizman A.V., Morgan R.L. AGA clinical practice guidelines on the role of probiotics in the management of gastrointestinal disorders. Gastroenterology. 2020;159:697–705. doi: 10.1053/j.gastro.2020.05.059. [DOI] [PubMed] [Google Scholar]
- Sze M.A., Schloss P.D. The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. Msphere. 2019;4 doi: 10.1128/msphere.00163-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff A.E. Avoiding common pitfalls in machine learning omic data science. Nat. Mater. 2019;18:422–427. doi: 10.1038/s41563-018-0241-z. [DOI] [PubMed] [Google Scholar]
- Topçuoğlu B.D., Lapp Z., Sovacool K.L., Snitkin E., Wiens J., Schloss P.D. Mikropml: user-friendly R package for supervised machine learning pipelines. J. Open Source Softw. 2021;6:3073. doi: 10.21105/joss.03073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtmann E., Chen J., Amir A., Shi J., Abnet C.C., Nelson H., Knight R., Chia N., Sinha R. Comparison of collection methods for fecal samples in microbiome studies. Am. J. Epidemiol. 2016;185:115–123. doi: 10.1093/aje/kww177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujkovic-Cvijin I., Sklar J., Jiang L., Natarajan L., Knight R., Belkaid Y. Host variables confound gut microbiota studies of human disease. Nature. 2020;587:448–454. doi: 10.1038/s41586-020-2881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade W.G., Prosdocimi E.M. Profiling of oral bacterial communities. J. Dent. Res. 2020;99:621–629. doi: 10.1177/0022034520914594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Garrity G.M., Tiedje J.M., Cole J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007;73:5261–5267. doi: 10.1128/aem.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S., Amir A., Hyde E.R., Metcalf J.L., Song S.J., Knight R. Tracking down the sources of experimental contamination in microbiome studies. Genome Biol. 2014;15:564. doi: 10.1186/s13059-014-0564-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A., Lozupone C., Zaneveld J.R., Vázquez-Baeza Y., Birmingham A., et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbel J., Zych K., Essex M., Karcher N., Kartal E., Salazar G., Bork P., Sunagawa S., Zeller G. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021;22:93. doi: 10.1186/s13059-021-02306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia G.-H., You C., Gao X.-X., Zeng X.-L., Zhu J.-J., Xu K.-Y., Tan C.-H., Xu R.-T., Wu Q.-H., Zhou H.-W., et al. Stroke dysbiosis index (SDI) in gut microbiome are associated with brain injury and prognosis of stroke. Front. Neurol. 2019;10:397. doi: 10.3389/fneur.2019.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Gao X., Xia G., Chen M., Zeng N., Wang S., You C., Tian X., Di H., Tang W., et al. Rapid gut dysbiosis induced by stroke exacerbates brain infarction in turn. Gut. 2021;70:1486–1494. doi: 10.1136/gutjnl-2020-323263. [DOI] [PubMed] [Google Scholar]
- Xu R., Tan C., Zhu J., Zeng X., Gao X., Wu Q., Chen Q., Wang H., Zhou H., He Y., et al. Dysbiosis of the intestinal microbiota in neurocritically ill patients and the risk for death. Crit. Care. 2019;23:195. doi: 10.1186/s13054-019-2488-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Liao S., He Y., Wang S., Xia G., Liu F., Zhu J., You C., Chen Q., Zhou L., et al. Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J. Am. Heart Assoc. 2015;4 doi: 10.1161/jaha.115.002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Thompson K.N., Branck T., Yan Y., Nguyen L.H., Franzosa E.A., Huttenhower C. Metatranscriptomics for the human microbiome and microbial community functional profiling. Annu. Rev. Biomed. Data Sci. 2021;4:279–311. doi: 10.1146/annurev-biodatasci-031121-103035. [DOI] [PubMed] [Google Scholar]
- Zhu W., Romano K.A., Li L., Buffa J.A., Sangwan N., Prakash P., Tittle A.N., Li X.S., Fu X., Androjna C., et al. Gut microbes impact stroke severity via the trimethylamine N-oxide pathway. Cell Host Microbe. 2021;29:1199–1208.e5. doi: 10.1016/j.chom.2021.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemski M., Wisanwanichthan T., Bokulich N.A., Kaehler B.D. Beating naive bayes at taxonomic classification of 16S rRNA gene sequences. Front. Microbiol. 2021;12:644487. doi: 10.3389/fmicb.2021.644487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
Raw sequencing data were deposited in the European Nucleotide Archive under project number: PRJEB48735 and are publicly available as of the date of publication.
-
•
Scripts used for data processing and analysis are publicly available on GitHub and are publicly available at the following url: https://github.com/adamsorbie/Stroke_Microbiota_reproducibility.
-
•
Any additional information required, is available from the lead contact upon request.