

SUMMARY
The transformed state in acute leukemia requires gene regulatory programs involving transcription factors and chromatin modulators. Here, we uncover an IRF8-MEF2D transcriptional circuit as an acute myeloid leukemia (AML)-biased dependency. We discover and characterize the mechanism by which the chromatin “reader” ZMYND8 directly activates IRF8 in parallel with the MYC proto-oncogene through their lineage-specific enhancers. ZMYND8 is essential for AML proliferation in vitro and in vivo and associates with MYC and IRF8 enhancer elements that we define in cell lines and in patient samples. ZMYND8 occupancy at IRF8 and MYC enhancers requires BRD4, a transcription coactivator also necessary for AML proliferation. We show that ZMYND8 binds to the ET domain of BRD4 via its chromatin reader cassette, which in turn is required for proper chromatin occupancy and maintenance of leukemic growth in vivo. Our results rationalize ZMYND8 as a potential therapeutic target for modulating essential transcriptional programs in AML.
In brief
Uncovering transcriptional addictions in cancer can help guide precision therapeutic intervention. Cao et al. used CRISPR screening to reveal how an acute-myeloid-leukemia-essential IRF8-MEF2D transcriptional circuit can be selectively inhibited by perturbing the reader function of an epigenetic regulator, ZMYND8.
Graphical Abstract
INTRODUCTION
The initiation and progression of human cancer is driven by compounding genetic alterations, which ultimately converge on transcriptional and chromatin dysregulation (Bradner et al., 2017; Dawson, 2017). Cancer cells often rely on regulatory proteins, such as transcription factors (TFs) or chromatin regulators (CRs), to control gene expression crucial for sustaining the malignant state. In this manner, cancer cells develop an “addiction” to specific transcriptional programs (Bradner et al., 2017; Dawson and Kouzarides, 2012). A small subset of TFs, referred to as lineage-specific TFs, are expressed in a cell-type-specific manner to coordinate gene expression programs that define a cell state. CRs, including enzyme “writers” and “erasers” that catalyze post-translational modification (PTM) of chromatin, and “readers” that recognize these modifications, are also critical modulators of transcriptional programs. Growing evidence suggests that CRs can similarly function in specialized, cell-type-specific biological pathways. Thus, perturbing TFs or CRs can be a selective method of targeting certain cancer-essential gene expression programs to treat particular malignancies (Bhagwat and Vakoc, 2015; Bradner et al., 2017; Dawson, 2017; Dawson and Kouzarides, 2012).
Acute myeloid leukemia (AML) is an aggressive form of heterogeneous hematopoietic malignancy characterized by aberrant self-renewal and blocked differentiation of myeloid progenitor cells. Cancer genome sequencing projects in AML have revealed frequent genetic alterations in regulators controlling gene transcription, chromatin states, and DNA covalent modifications (Desai et al., 2018; Döhner et al., 2015; Papaemmanuil et al., 2016). While a small fraction of these driver alterations have been pharmaceutically targeted in AML, the majority are not actionable targets for small-molecule inhibitors, underscoring the need to identify cancer dependencies beyond driver genetic lesions. Identification and investigation of cancer-addicted transcription-chromatin regulatory programs, which are not themselves directly associated with genetic alterations, provides another opportunity for therapeutic intervention (Bennett and Licht, 2018; Bhagwat and Vakoc, 2015). For example, targeting transcription coactivator BRD4 with selective chemical inhibitors JQ1 and i-BET promotes anti-leukemia effects via suppression of key oncogenes, such as MYC, BCL2, and CDK6 (Dawson et al., 2011; Mertz et al., 2011; Shi and Vakoc, 2014; Wu and Chiang, 2007; Zuber et al., 2011).
IRF8 is a TF preferentially expressed in hematopoietic cell lineages and critical for normal myeloid cell and B cell development (Holtschke et al., 1996; Lu et al., 2003; Tamura et al., 2000; Wang et al., 2008). IRF8-deficient mice have compromised immune systems and display a chronic-myelogenous-leukemia-like syndrome (Holtschke et al., 1996). Loss of IRF8 facilitates the initiation of acute promyelocytic leukemia by the PML-RARA fusion onco-protein (Gaillard et al., 2018). These examples suggest that IRF8 may play a tumor-suppressor-like role in certain hematopoietic cell types. Myocyte enhancer factor 2D (MEF2D) is a ubiquitously expressed TF. MEF2D-translocated fusion proteins have been implicated in B cell leukemia (Di Giorgio et al., 2018; Gu et al., 2016; Liu et al., 2016), and MEF2D hyperexpression promotes hepatocellular carcinoma cell growth (Ma et al., 2014). Nevertheless, a role for MEF2D in AML has yet to be defined. Zinc finger MYND-type containing 8 (ZMYND8, also known as RACK7 and PRKCBP1) is a CR that contains a reader cassette consisting of a plant homeodomain (PHD), a bromodomain (BD), and a Pro-Trp-Trp-Pro (PWWP) domain in tandem. ZMYND8 uses this reader cassette to recognize PTMs on proteins, with high preference for histones (Li et al., 2016; Savitsky et al., 2016). Previous work has shown that ZMYND8 associates with both repressor and activator complexes to regulate gene expression (Delgado-Benito et al., 2018; Ghosh et al., 2018; Gong et al., 2015; Li et al., 2016). Current studies on the function of ZMYND8 in tumor biology have largely focused on its suppressor role in solid tumors, such as in prostate and breast cancers (Basu et al., 2017b, 2017a; Jin et al., 2019; Li et al., 2016; Shen et al., 2016). To date, the regulatory roles of IRF8, MEF2D, and ZMYND8 have yet to be characterized in the context of hematopoietic malignancies, including in AML.
RESULTS
IRF8 is an AML-biased TF dependency
To identify unexplored transcriptional addiction in AML, we surveyed our previously performed CRISPR-Cas9 negative selection “dropout” screens targeting the DNA-binding domain of 1,427 human TFs (Lu et al., 2018). In order to quantify TF essentiality in cancer cell proliferation, a protein domain essential score (ES) was defined as the average log2 fold change (log2FC) of all Cas9 single guide RNAs (sgRNAs) targeting a given protein domain (Figure 1A). We ranked the screened TFs based on the differential ESs between AML cell lines and non-AML cell lines to identify AML-biased TF dependencies (Figures 1A, S1A, and S1B). While MYB, CEBPA, CBFB, PU.1, ZFP64, and FLI1 have been previously reported to play important roles in AML (Anfossi et al., 1989; Goyama and Mulloy, 2011; Kornblau et al., 2011; Lu et al., 2018; Maiques-Diaz et al., 2018; Ohlsson et al., 2014; Ye et al., 2015; Zhou et al., 2014), the roles of IRF8 and MEF2D have not yet been recognized in this disease. Inspection of IRF8 and MEF2D in a genome-wide CRISPR screening dataset (Project Achilles, DEPMAP) (Meyers et al., 2017) further confirmed their AML-biased essentiality among 721 cancer cell lines representing 26 lineages (Figure S1C).
Figure 1. IRF8 is an AML-biased TF dependency.

(A) Summary of TF-domain-focused CRISPR screens. Genes were ranked by AML-biased ES defined by the difference in a particular domain’s ES in AML versus in non-AML cell lines. CML, chronic myeloid leukemia. Data are from Lu et al. (2018).
(B) Competition-based proliferation assays performed in indicated Cas9+ cell lines. sgRNA+ populations were monitored over time with a GFP co-expression marker. Plotted is the relative sgRNA+ population normalized to the day 3 sgRNA+ population over 21 days. sgNeg, negative control; sgCDK1, positive control.
(C) Design of CRISPR-resistant IRF8 cDNA. Encoded amino acids are labeled in blue at the bottom of the cDNA sequence.
(D) Vectors used for IRF8/dIRF8 cDNA complementation assay. IRF8*, sgIRF8_1-resistant IRF8 cDNA; dIRF8, degradable IRF8 that contains an additional FKBP12G36V domain and a 2×HA tag; Neo, neomycin resistance marker.
(E) Immunoblotting of FKBP12G36V-tagged IRF8, IRF8, or GAPDH (loading control) in whole-cell lysates of MOLM-13 cells transduced with indicated vectors.
(F) Competition-based proliferation assays performed in MOLM-13 Cas9+ cell lines stably expressing empty vector (EV), IRF8*, or dIRF8. sgPCNA, positive control.
(G) Schematic depicting establishment of an inducible IRF8 degradation system in MOLM-13 cells.
(H) Immunoblotting of whole-cell lysate of indicated cells treated with 500 nM dTAG-47 over time.
(I) Competition-based proliferation assay of MOLM-13-dIRF8 versus parental cells treated with either DMSO or 500 nM dTAG-47. Plotted is the relative sgRNA+ population normalized to the day 0 sgRNA+ population.
Data points of line graphs represent the average of three independent biological replicates (n = 3). Error bars represent mean ± SEM. See also Figure S1.
We first validated the requirement of IRF8 in cellular proliferation by performing competition-based proliferation assays in AML cell lines. We confirmed that AML cells transduced with IRF8 sgRNAs were rapidly depleted and outcompeted by parental cells, validating the results of our pooled sgRNA library screen (Figures 1B and S1D). Inspection of the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012) for 39 of the cancer cell lines used in this study revealed heterogeneous expression of IRF8 across cell lines, with high IRF8 expression (IRF8hi, RPKM > 30) in a subset of AML and low IRF8 expression (IRF8low, RPKM < 2) in many solid tumor lines (Figure S1E). Additionally, we found that IRF8 expression was the third highest in AML among a total of 32 cancer types in the The Cancer Genome Atlas (TCGA) dataset, with a broad range of IRF8 expression levels in 173 AML patients (RNA-Seq by Expectation Maximization [RSEM] = 4.82–14.41) (Figure S1F). Moreover, we noted that high IRF8 expression was associated with a subset of AMLs encompassing diverse cytogenetic and driver mutations, including MLL rearrangements and amplifications, CBFB-MYH11 translocations, and RUNX1 mutations (Figures S1G).
To further verify the essential function of IRF8 and the on-target effect of our sgRNAs, we ectopically expressed CRISPR-resistant IRF8 cDNA in leukemia cells (Figures 1C–1F). To enable rapid perturbation of IRF8 in AML, we fused the CRISPR-resistant IRF8 cDNA with an FKBP12G36V domain (dIRF8), which, in the presence of the degradation tag (dTAG) compound, is rapidly degraded (Figures 1D and 1E) (Nabet et al., 2018). We established an endogenous-IRF8 knockout line of MOLM-13 that expresses dIRF8 as a substitute (hereafter referred to MOLM-13-dIRF8 cells) (Figures 1G and 1H). We next mixed a 1:1 ratio of MOLM-13-dIRF8 cells with parental cells and observed that dTAG-induced IRF8 degradation resulted in significant depletion of the MOLM-13-dIRF8 population (Figures 1I and S1H), concurrent with the observed phenotype induced by direct CRISPR-based genetic perturbation. Altogether, these results at both the genetic and protein levels confirmed the essential function of IRF8 in a subset of AML.
IRF8 is enriched at the MEF2D locus and modulatesMEF2D expression
To identify the primary transcriptional targets of IRF8, we performed RNA sequencing (RNA-seq) analysis in MOLM-13-dIRF8 cells after 4 h of dTAG treatment (Figures 1H and 2A). While many known AML genetic dependencies, such as MYB, CEBPA, ZFP64, HOXA9, and MYC, were unchanged, acute depletion of IRF8 led to downregulation of 13 genes with log2FC less than −0.5 (Figures 2A). Among the downregulated genes, MEF2D became our primary focus, because it was nominated as a potential AML-biased TF dependency in both our CRISPR-Cas9 screens (Figure 1A, S1A, and S1C) and the DEPMAP database (Figure 2B). Moreover, we noted that the MEF2D ES was highly correlated with that of IRF8, IRF8hi AML cell lines were most affected by MEF2D depletion, and MEF2D expression was elevated in IRF8hi AML compared to in IRF8low cell lines (Figures 1A, 2C, S1F, and S2B). Analysis of primary human cells revealed that MEF2D expression levels were higher in AML relative to all other cancer types in the TCGA database (Figure S2B).
Figure 2. IRF8 is enriched at the MEF2D locus and modulates MEF2D expression.

(A) RNA-seq analysis of gene expression changes in MOLM-13-dIRF8 cells treated with either DMSO or 500 nM of dTAG-47 for 4 h. Genes are ranked by log2 fold change (n = 3).
(B) ESs of top 12 downregulated genes from DEPMAP dataset with log2 fold change less than —0.5 in IRF8-expressed (TPM > 1) AML cell lines (n = 12). Shown is a box and whisker plot of copy-number-adjusted ESs (CERES). TPM, transcripts per million reads.
(C) Scatterplot of IRF8 and MEF2D essentiality scores in 33 human cancer cell lines extracted from Lu et al. (2018). IRF8hi cell lines are labeled. r, Pearson’s correlation coefficient.
(D) RNA-seq analysis of gene expression changes in indicated cell lines transduced with sgIRF8 or sgNeg for 4 days. sgNeg and two independent sgRNAs targeting IRF8 are used (n = 2 biological replicates per sgRNA). Ranking position from the top most downregulated gene is indicated in parentheses.
(E) GSEA rank plot of RNA-seq data presented in (D). The MEF2D signature is defined as the top 200 downregulated genes upon MEF2D depletion. Normalized enrichment score (NES) and false discovery rate (FDR) q value are shown.
(F) Unbiased GSEA using all signatures from MSigDB v6.1 (Liberzon et al., 2015), together with the MEF2D signature for RNA-seq data presented in (D). Each gene set is represented as a single dot. The MEF2D signature is indicated in red, with numeral rank from the top most-enriched gene set in parentheses.
(G) RNA-seq analysis of gene expression changes in MEF2D-depleted MOLM-13 cells 5 days after transduction of sgNeg or sgMEF2D. Two independent sgRNAs targeting either MEF2D or a negative control locus were used. MEF2D signature genes are indicated with a red box.
(H) Metaplot (top) and density plot (bottom) showing enrichment of IRF8 and H3K27ac surrounding the 23,429 IRF8 ChIP-seq peaks at a ±5-kb interval in MOLM-13 cells. Peaks were ranked by IRF8 ChIP-seq tag counts.
(I) Gene tracks of H3K27ac and IRF8 enrichment at the MEF2D locus in the indicated leukemia cell lines. H3K27ac and IRF8 ChIP-seq tracks in THP-1 cells are extracted from GSE123872.
To investigate the molecular connection between IRF8 and MEF2D in AML, we performed RNA-seq following CRISPR-mediated depletion of either IRF8 or MEF2D. We defined a MEF2D transcriptional signature in AML using the top 200 downregulated genes from MEF2D-deficient cells, many of which are essential genes for AML proliferation (Table S1). We found that IRF8 perturbation caused downregulation of MEF2D mRNA and significant suppression of the MEF2D transcriptional signature (Figures 2D–2G). Of note, IRF8 was among the top downregulated genes in MEF2D knockout cells, which suggests that IRF8 and MEF2D participate in a positive-feedback circuit in AML (Figures 2G). Furthermore, when compared to ~13,000 gene signatures from Molecular Signatures Database (MSigDB) (Liberzon et al., 2015), we found that IRF8 suppression had a strong effect on the MEF2D signature (Figure 2F).
To further corroborate the relationship between IRF8 and MEF2D, as well as identify the genomic occupancy of IRF8, we performed chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) in MOLM-13 cells. Overall, IRF8 enrichment was observed mainly near transcription start sites (TSS), TSS-proximal introns, and intergenic potential enhancer regions and overlapped significantly with the active epigenetic mark H3K27ac (Figures 2H, S2D, and S2E). Motif analysis of IRF8-occupied regions in AML generated a motif sequence similar to motifs obtained from myeloid lineage cells (Figure S2F) (Grajales-Reyes et al., 2015; Langlais et al., 2016). Inspection of the browser track of the IRF8 ChIP-seq revealed the enrichment of IRF8 near the MEF2D TSS, as well as in the promoters of genes whose expression was the most decreased upon rapid IRF8 degradation (Figures 2I and S2G). ChIP-qPCR analysis revealed that rapid degradation of IRF8 by dTAG resulted in significant decrease of IRF8 occupancy near the MEF2D TSS (Figure S2H), further supporting the finding that IRF8 directly regulates MEF2D transcription via proximity to the MEF2D TSS.
CRISPR screens identify ZMYND8 as an AML-biased dependency
While the IRF8-MEF2D transcriptional circuit is an AML-biased vulnerability, pharmacological inhibition of TFs is notably challenging, specifically because of the difficulty in developing effective inhibitors against DNA-binding and transactivation domains (Chen and Koehler, 2020). We therefore sought to identify druggable CRs that function as part of the IRF8-MEF2D pathway. Toward this goal, we performed domain-focused CRISPR screens against 197 CR-associated domains in 19 human cancer cell lines, including IRF8hi and IRF8low leukemias, as well as solid tumor lines, in order to identify AML-biased CR dependencies (Figures 3A and 3B). Upon ranking differential ESs between AML cell lines and non-AML cell lines, we re-identified known AML-biased genetic vulnerabilities: EP300, SETDB1, DOT1L, HBO1, and KDM1A (Figures 3B and S3A) (Bernt et al., 2011; Cuellar et al., 2017; Giotopoulos et al., 2016; Harris et al., 2012; MacPherson et al., 2020; Schenk et al., 2012). Notably, these screens in conjunction with DEPMAP also nominated the BD of ZMYND8 as an AML-biased vulnerability (Figures 3B, S1A, and S3B). Co-essentiality analysis in another independent genome-wide CRISPR screening dataset in AML cell lines (Wang et al., 2017) revealed a positive and reciprocal correlation between ZMYND8 and IRF8, further supporting a potential connection between these two regulators in AML (Figures 3C and S3C).
Figure 3. CRISPR screens identify ZMYND8 as an AML-biased dependency.

(A) Schematic showing the workflow of CRISPR dropout screen.
(B) Summary of CR domain-focused CRISPR screens. Genes were ranked by AML-biased ES. A673 and HS-SY-II screening data were retrieved from Brien et al. (2018).
(C) Correlated essentiality between IRF8 and 3,102 gene ESs from genome-wide CRISPR screens in leukemia cells (Wang et al., 2017). Pan-essential and nonessential genes are excluded. Remaining gene ESs were ranked by Pearson’s correlation coefficient to IRF8 ES.
(D) Competition-based proliferation assays performed in indicated Cas9+ cell lines (n = 3).
(E) Heatmap summarizing the competition-based proliferation assays performed as in Figure 3D (n = 3).
(F) Schematic of in vivo transplantation of MOLM-13 cells infected with sgNeg or sgZMYND8_2.
(G) Flow cytometry analysis of percentage of human CD45+ leukemia cells in BM of recipient mice sacrificed after 9 days post-transplantation (n = 4). Statistical analysis (p value) was performed using an unpaired Student’s t test. BM, bone marrow.
(H) Kaplan-Meier survival curves of recipient mice transplanted with MOLM-13 cells transduced with sgNeg (n = 5) or sgZMYND8_2 (n = 6). The p value was determined by a log-rank Mantel-Cox test.
(I) Colony formation of normal myeloid progenitor cells isolated from constitutively expressing Cas9 mice (n = 3). Statistical analysis was performed using a two-way ANOVA test.
Error bars represent mean ± SEM. See also Figure S3 and Table S2.
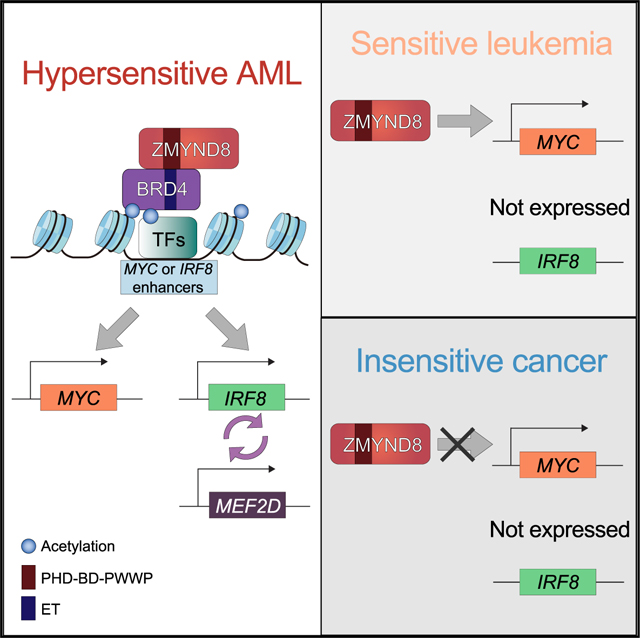
To validate the AML-biased requirements of ZMYND8, we performed competition-based proliferation assays, and consistent with our pooled screening results, we found that perturbation of ZMYND8 caused a distinct pattern of depletion: (1) a subset of leukemia lines were in general the most susceptible to ZMYND8 loss (>3-fold depletion of GFP/sgRNA+, referred to as “hypersensitive”); (2) a subset of leukemia lines were sensitive, but to a lesser extent (1.5- to 3-fold depletion, referred to as “sensitive”); and (3) solid tumor lines did not respond (<1.5-fold change, referred to as “nonsensitive”) (Figures 3D, 3E, and S3D–S3F). Of note, the ZMYND8-hypersensitive leukemia lines all have high IRF8 expression (Figures S1E and S3G). Importantly, ZMYND8 is ubiquitously expressed across cell lines used in our screens (Figure S3H), suggesting that the AML-biased requirement of ZMYND8 cannot be simply attributed to differential gene expression.
We next examined the requirement of ZMYND8 in vivo by injecting sgZMYND8-transduced MOLM-13 cells intravenously into NOD scid gamma (NSG) immunodeficient mice (Figure 3F). We observed a significantly decreased leukemic burden in mice receiving ZMYND8-deficient cells in the bone marrow (Figures 3G and S3I). Accordingly, the mice engrafted with ZMYND8-deficient leukemia had an extended survival (Figure 3H). We noted that leukemia that eventually developed in recipients of sgZMYND8 cells could be explained by positive selection of sgRNA-negative or partially edited sgZMYND8 cells during the experimental time course (Figure S3J–S3L).
To further evaluate the requirement of ZMYND8 in normal hematopoietic cells, we perturbed Zmynd8 in isolated murine normal bone marrow with constitutive Cas9 expression (Figure S3M) and assessed the development of normal myeloid cells in colony-formation assays. In Zmynd8-deficient cells, we found no significant difference in the frequency and absolute number of hematopoietic progenitor colonies (Figure 3I). These results suggest that Zmynd8 is dispensable in normal myeloid development in this short-term assay.
ZMYND8 regulates IRF8 and MYC transcription to sustain AML proliferation
To investigate the primary transcriptional changes of ZMYND8 depletion, we established a dTAG-degradable and CRISPR-resistant ZMYND8 system in MOLM-13 cells (MOLM-13-dZD8) to replace endogenous ZMYND8 (Figures 4A and S4A–S4C). ZMYND8 degradation caused potent cell growth suppression (Figures 4B and S4D). Next, we performed RNA-seq analysis following 4 h of dTAG treatment in MOLM-13-dZD8 cells. Remarkably, the top two downregulated genes upon rapid depletion of dZD8 were IRF8 and MYC, a well-known pan-leukemia required TF, and were the only two of the top 10 downregulated genes required by AML (Figure 4C and data not shown). To validate our RNA-seq results, we performed a time-course analysis of dTAG treatment followed by qRT-PCR in MOLM-13-dZD8 cells and observed a decrease of IRF8 and MYC mRNA levels within 1 h and a 70%–80% decrease in IRF8 and MYC mRNA and protein levels within 24 h (Figures 4D, S4E, and S4F). These data implicate IRF8 and MYC as direct targets of ZMYND8.
Figure 4. ZMYND8 regulates IRF8 and MYC transcription to sustain AML proliferation.

(A) Schematic depicting establishment of an inducible ZMYND8 degradation system in MOLM-13 cells.
(B) Competition-based proliferation assay of MOLM-13-dZD8 versus parental cells.
(C) RNA-seq analysis of gene expression changes in MOLM-13-dZD8 cells treated with either DMSO or 500 nM dTAG-13 for 4 h (n = 2).
(D) Time-course reverse transcriptase quantitative PCR (RT-qPCR) analysis of mRNA expression in MOLM-13-dZD8 cells treated with 500 nM dTAG-47. Relative mRNA levels were normalized to GAPDH levels.
(E) RNA-seq analysis of gene expression changes 5 days after transduction of sgNeg or sgZMYND8. Myeloid-differentiation-associated genes are labeled in blue.
(F) Immunoblotting of ZMYND8, MYC, IRF8, or GAPDH in whole-cell lysates.
(G) GSEA of RNA-seq data presented in Figure 4A. Myc_Targets_Up_Schuhmacher (Schuhmacher et al., 2001) and IRF8_Targets_Up or Myeloid_development_Up (Brown et al., 2006) signatures were used.
(H) Competition-based proliferation assays performed in MOLM-13 cells expressing EV, MYC, IRF8, or MYC+IRF8 and transduced with indicated sgRNAs. EV, empty vector.
Data points in line graphs represent the average of three independent biological replicates (n = 3). Error bars represent mean ± SEM. See also Figure S4 and Table S1.
To evaluate the proportion of IRF8- and MYC-controlled transcriptional programs involved in ZMYND8 addiction in AML, we performed RNA-seq analysis 5 days after CRISPR-mediated ZMYND8 depletion (Figures 4E, 4F, S4G, and S4H). Differential gene expression analysis revealed that loss of ZMYND8 resulted in a decrease in IRF8 expression and an increase in myeloid differentiation associated genes (S100As) selectively in hypersensitive cell lines and a decrease in MYC expression in hypersensitive and sensitive cell lines (Figures 4E and S4H). In contrast, MYC and myeloid differentiation genes were largely unaffected by ZMYND8 depletion in non-leukemia solid tumor cells (Figures S4E, S4F, and S4G). Immunoblotting further supported the notion that AML-biased ZMYND8 dependency was attributed to its regulation of leukemia-specific IRF8 and oncogenic MYC expression (Figures 4F). Gene set enrichment analysis (GSEA) showed that the IRF8-target gene signature, MYC-target signature, and myeloid development signature were significantly altered in majority of the hypersensitive cell lines upon ZMYND8 depletion (Figures 4G and S4I–S4K). Upon ranking the degree of gene signature alteration in ZMYND8-depleted cells, we found that among the most significantly decreased and increased gene signatures across all ZMYND8-deficient hypersensitive cell lines were the IRF8-target and MYC-target signatures, as well as the myeloid differentiation signature, respectively (Figure S4L).
We reasoned that ZMYND8 maintenance of IRF8 and MYC expression provides a plausible explanation for the differential sensitivity to ZMYND8 depletion in different cell lines, where MYC regulation is biased toward AML and IRF8 dependency is specific to IRF8hi leukemia. To further test whether MYC and IRF8 suppression specifically mediates the requirement for ZMYND8 in AML, we ectopically expressed MYC, IRF8, or both in ZMYND8-depleted MOLM-13 cells (Figure S4M). Strikingly, simultaneous overexpression of MYC and IRF8 in combination, but not individually, completely rescued the cell growth inhibition in ZMYND8-deficient leukemia cells (Figure 4H). In contrast, dual cDNA overexpression was not able to rescue the growth arrest in cells that lost either the pan-essential gene PCNA or other AML-biased dependencies (Figures 4H and S4N), suggesting that MYC and/or IRF8 overexpression cannot simply restore all the cytostatic and cytotoxic effects in AML. Collectively, these results provide strong evidence that ZMYND8 directly regulates transcription of IRF8 and MYC in AML.
Genome-wide binding profiles reveal the co-occupancy of ZMYND8 and BRD4 in active enhancer regions
Next, we set out to investigate the molecular activities of ZMYND8 required for IRF8 and MYC regulation in AML. We mapped ZMYND8’s chromatin occupancy by performing cleavage under targets and release using nuclease (CUT&RUN) (Skene et al., 2018) against ZMYND8 and various histone modifications in AML. A meta-analysis revealed that 13,125 ZMYND8-occupied regions in MOLM-13 cells were positively associated with H3K27ac and H3K14ac, another marker of active chromatin (Figure 5A). We observed 8,455 overlapping ZMYND8 peaks between two ZMYND8-hypersensitive AML cell lines, MOLM-13 and THP-1 cells (Figure S5A). Annotation of the ZMYND8-binding sites revealed that a large proportion of sites were in promoter and distal regions likely to be enhancers (Figures 5B and S5B). These results are consistent with prior findings that ZMYND8 can be associated with active genes and enhancer regions (Delgado-Benito et al., 2018; Ghosh et al., 2018; Savitsky et al., 2016; Shen et al., 2016).
Figure 5. Genome-wide binding profiles reveal the co-occupancy of ZMYND8 and BRD4 in active enhancer regions.

(A) Meta-profile (top) and density plot (bottom) of CUT&RUN peaks at 13,125 ZMYND8-occupied regions in MOLM-13 cells. Peaks are ranked by ZMYND8 CUT&RUN tag counts.
(B) Pie chart annotating the distribution of 13,125 ZMYND8 peaks in MOLM-13 cells. TTS, transcription termination site. Other, UTR and non-coding RNA regions.
(C) ZMYND8 CUT&RUN-derived de novo motif analysis in MOLM-13 cells using HOMER. Statistical analysis (p value) was calculated using the binomial test.
(D) Venn diagram displaying CUT&RUN peak overlap between ZMYND8, BRD4, and H3K27ac occupancy in MOLM-13 cells.
(E) Gene tracks of H3K27ac, H3K14ac, BRD4, and ZMYND8 enrichment with 4C-seq analysis at leukemic MYC enhancer locus (ME1-ME5, gray box) in MOLM-13 cells. 4C-seq was performed using MYC promoter as the “viewpoint.”
(F) Top: schematic of dCas9-KRAB-mediated epigenomic silencing. Locations of different sgRNAs targeting H3K27ac-enriched regions +23–86 kb from the IRF8 TSS are shown by red lines. Bottom: gene tracks of H3K27ac, H3K14ac, BRD4, and ZMYND8 enrichment in addition to 4C-seq analysis at the IRF8 locus in MOLM-13 cells. Putative IRF8 enhancer is labeled in a gray box. IE, IRF8 enhancer.
(G) RT-qPCR analysis of mRNA expression of IRF8 in dCas9_KRAB+ MOLM-13 cells transduced with indicated sgRNAs in Figure 5F and harvested after 5 days post-infection. sgIRF8_TSS (purple) targets the IRF8 TSS region. Effective sgRNAs that induce >2-fold downregulation of IRF8 are labeled in green. Relative mRNA levels were normalized to GAPDH levels. sgNeg, negative control; TSS, transcription start site. Plotted are the mean ± SEM (n = 3).
(H) Competition-based proliferation assays performed in dCas9_KRAB+ MOLM-13 cell lines. Cells were transduced with sgNeg (n = 4) or sgIRF8_TSS, sgIR-F8_enh-2, −3, −5, or −9 (n = 2).
(I) Leukemic MYC enhancer (left, ME1-ME5, gray box) or IRF8 enhancer (right, IE, gray box) region in indicated cell lines. H3K27ac ChIP-seq data in THP-1, HEL, and HUH7 cells were extracted from GSE109493, GSE123872, or GSE89212.
Error bars represent mean ± SEM. See also Figure S5.
Using a de novo motif identification tool (Heinz et al., 2010), we deciphered a 12-nt sequence that was most frequently embedded in ZMYND8-occupied regions in leukemia cells (Figures 5C and S5C). Deconvolution of the ZMYND8-derived sequence also revealed enrichment of several hematopoietic-lineage-specific TF-binding motifs, including PU.1, FLI1, CEBPβ, and ERG (Figure S5D), which are known to form enhanceosomes that recruit BRD4 to chromatin (Dawson et al., 2011; Roe et al., 2015; Zuber et al., 2011). Intriguingly, motif analysis in ZMYND8-binding sites in a liver cancer cell line, HUH7, revealed enrichment of a distinct set of TF motifs, including hepatocyte nuclear factors (HNFs) (Figure S5C). These observations implied that the differential chromatin occupancies of ZMYND8 among different cell types might be driven by a diverse set of master TFs. In our above RNA-seq analysis of ZMYND8-perturbed cells (Figure 4E), we noted that the BRD4-dependent gene signature (Rathert et al., 2015) was significantly downregulated in ZMYND8-depleted hypersensitive cell lines (Figure S5E). These two observations thus prompted us to evaluate a potential connection or overlap between BRD4 and ZMYND8 in AML. To this end, we performed BRD4 CUT&RUN and found substantial binding site overlap, ranging from 51.7% to 63.8%, between ZMYND8- and BRD4-occupied sites across diverse AML cell lines (Figures 5A, 5D, and S5F).
To further characterize whether ZMYND8 and BRD4 co-occupy regions essential to AML chromatin regulation, we inspected the known lineage-specific MYC distal enhancer regions, designated as ME1–ME5 (Bahr et al., 2018; Shi et al., 2013). We found that ZMYND8, BRD4, H3K27ac, and H3K14ac histone marks all share similar occupancy status on the MYC ME1–ME5 enhancer clusters in a leukemia-specific manner (Figures 5E), consistent with prior findings demonstrating leukemia-specific BRD4 control of MYC transcription (Bahr et al., 2018; Shi et al., 2013). Using circular chromosome conformation capture followed by next-generation sequencing (4C-seq) (Schwartzman et al., 2016), we set an anchor point at the MYC promoter and confirmed that chromatin loop formation occurs between the ME1–ME5 and the MYC promoter in human AML, in line with our previous observations in murine AML cells (Shi et al., 2013) (Figure 5E).
ZMYND8 regulates IRF8 transcription through a lineage-specific enhancer
We sought to identify the DNA cis-elements underlying the ZMYND8-dependent IRF8 regulation in AML. We surveyed the non-coding regions flanking the IRF8 loci and found potential enhancer regions enriched with H3K27ac marks 23–86 kb downstream of the IRF8 TSS (Figure 5F). We then performed 4C-seq experiments with an anchor set near the IRF8 promoter to nominate any potential cis-elements that were in close physical proximity and therefore would feasibly be able to regulate IRF8 transcription. 4C-seq analysis revealed two distinct peaks, one −83 to −62 kb upstream and the other 76 to 107 kb downstream of the IRF8 TSS, looping to the IRF8 promoter region (Figure 5F). While the upstream 4C peak region appeared to show low enrichment for active histone marks, ZMYND8, or BRD4, the downstream 4C peak region was occupied by all active histone marks, ZMYND8, and BRD4, nominating this region as a potential ZMYND8-dependent cis-regulatory element of IRF8 (Figure 5F).
To functionally test whether the flanking H3K27ac-enriched regions facilitate IRF8 expression, we employed CRISPR interference to perturb the enhancer function in its native environment (Figure 5F) (Fulco et al., 2016; Gilbert et al., 2013). We targeted five potential IRF8 enhancer regions with two or three independent sgRNAs per locus in MOLM-13 cells (Figure 5F). sgRNAs 9–11 nominated a potential enhancer 81–86 kb downstream of the IRF8 TSS, as these sgRNAs most significantly reduced IRF8 mRNA levels to those targeting the TSS site (Figure 5G). Competition assays with individual sgRNA also displayed a similar pattern of proliferation arrest (Figure 5H). Simultaneous transduction of two sgRNAs targeting the lineage-specific MYC and IRF8 enhancers led to an additive growth arrest in MOLM-13 compared with either individual sgRNA (Figures S5G–S5J). Collectively, these data suggest that the ZMYND8-occupied enhancers mediate AML-biased transcriptional regulation of MYC and IRF8 and, moreover, that AML hypersensitivity to ZMYND8 perturbation could be mediated by the additive effect of dual suppression of MYC and IRF8, which is consistent with the observation that individual MYC or IRF8 overexpression can only partially rescue the effect of ZMYND8 deficiency (Figure 4H). Moreover, CUT&RUN signals in the +81- to 86-kb region downstream of the IRF8 TSS revealed an exclusive enrichment of H3K27ac, BRD4, and ZMYND8 in IRF8hi lines, but not in IRF8low lines (Figures 5I, S1E, and S3G). Collectively, the chromatin conformation capture and enhancer perturbation experiments posit the ZMYND8-occupied +81- to 86-kb distal region from the IRF8 TSS as a functionally relevant IRF8 enhancer (defined as IE hereafter).
ZMYND8 occupies active elements in AML through binding the ET domain of BRD4
While ZMYND8 and BRD4 co-occupy similar regions in AML genome-wide (Figures 5A, 5B, and S5B), it remained unclear whether these two regulators were functionally connected on chromatin in AML. To explore their potential relationship on chromatin, we first treated MOLM-13-dZD8 cells for 4 h with either dTAG to deplete ZMYND8 or JQ1 (a selective chemical inhibitor against bromodomain and extra-terminal [BET] family proteins) (Filippakopoulos et al., 2010) to dissociate BRD4 from the chromatin. We then followed with CUT&RUN analysis to determine their resulting genome-wide occupancy. As expected, addition of dTAG led to loss of ZMYND8 chromatin occupancy by ~97.4%, and JQ1 treatment resulted in a decrease of ~70.0% of global BRD4 signals with log2FC less than −1 at the regions with normalized tag count >2 in a DMSO control (Figures 6A and 6B). Interestingly, JQ1 treatment also led to eradication of 93.3% of ZMYND8 from its established binding regions; conversely, BRD4 association at ZMYND8-bound regions was largely preserved following dTAG treatment (Figures 6A and 6B). Immunoblotting indicated that treatment of JQ1 induced rapid MYC reduction, but the ZMYND8 protein level remained unchanged within the first 6 h (Figure 6C). These findings suggest that ZMYND8 occupancy, rather than expression, is altered upon ablation of BRD4 binding at the MYC enhancer. Inspection of the CUT&RUN browser track of MYC and IRF8 enhancer regions revealed the same pattern of ordered BRD4-ZMYND8 binding upon dTAG or JQ1 treatment (Figure 6D), overall suggesting that BRD4’s enhancer-based transcriptional regulation of MYC and IRF8 is mediated by ZMYND8. Analysis of previously reported thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq) data in MOLM-13 and MV4;11 cells treated with JQ1 (Muhar et al., 2018) confirmed both MYC and IRF8 as direct targets of BRD4, consistent with what is known about the BRD4-dependent core transcriptional program (Dawson et al., 2014) (Figure S6A).
Figure 6. ZMYND8 occupies active elements in AML through binding the ET domain of BRD4.

(A) CUT&RUN meta-profile (top) and density plot (bottom) of ZMYND8 and BRD4 enrichment at 8,483 ZMYND8-occupied regions. Cells were treated with DMSO, 500 nM dTAG-47, or 2 μM JQ1 for 4 h.
(B) Violin plot of normalized tag density of ZMYND8 (red) or BRD4 (purple) peaks in Figure 6A. Dots represent median values. p values were calculated by Welch’s two-sided t test.
(C) Immunoblotting of MOLM-13-dZD8 whole-cell lysate treated with DMSO or 2 μM JQ1 over time.
(D) Gene tracks of H3K27ac, ZMYND8, and BRD4 enrichment at the leukemic MYC (left, ME1-ME5, gray box) or IRF8 enhancers (right, IE, gray box) regions in cell populations described in Figure 6A.
(E) Schematic of FLAG-tagged BRD4 variants and truncations used for coIP.
(F) IP-immunoblotting of nuclear lysates prepared from HEK293T cells transfected with indicated vectors for 48 h. Arrowhead represents the expected BRD4-long isoform (BRD4L) band.
(G) IP-MS analysis on nuclear lysates prepared from HEK293T cells transiently expressing the FLAG-ET domain or a streptavidin bead control. ZMYND8 peptide enrichment is labeled in red. Data were extracted from Lambert et al. (2019).
See also Figure S6.
We next sought to determine the specific region or domain of BRD4 that mediates its association with ZMYND8. We transfected a series of FLAG-tagged BRD4 variants and truncations in HEK293T cells (Figure 6E). We confirmed that both long and short isoforms of BRD4 associate with ZMYND8, while the BD1+2 truncation and the ET domain deletion from BRD4-long attenuated any interaction; on the other hand, we found that the ET domain alone was able to successfully precipitate ZMYND8 (Figure 6F). We further explored published immunoprecipitation (IP)-mass spectrometry (MS) of BRD4 (Lambert et al., 2019). IP-MS of either exogenous or endogenous BRD4 showed significant enrichment of ZMYND8 (Figures 6G and S6B), but a BD1+BD2 IP-MS revealed no detectable ZMYND8 peptides (data not shown). Altogether, these data suggest that the BRD4 ET domain is necessary and sufficient to physically associate with ZMYND8 and that ZMYND8 requires BRD4 for proper enhancer occupancy in AML.
The ZMYND8 PHD-BD-PWWP reader cassette is required for association with BRD4 on chromatin and for leukemia growth
We next mapped the ZMYND8 regions responsible for BRD4 interaction. We first truncated ZMYND8’s PHD-BD-PWWP tandem reader cassette, the DUF3544, or the MYND domain (Figure 7A). We found that the ZMYND8 reader cassette truncation, but not the DUF3544 or MYND domain truncations, significantly diminished interactions with BRD4 (Figure 7B). To further dissect the functional importance of the ZMYND8 reader modules in BRD4 binding, we mutagenized specific conserved amino acid residues to alanine while preserving the overall reader structure of the PHD-BD-PWWP cassette (Figure 7A) (Savitsky et al., 2016). While PHD (N107A/D108A), BD (N248A), and PWWP (F308A/W311A) mutant variants were stably expressed in cells, all three showed diminished interaction with BRD4 (Figures 7B), suggesting that each individual reader domain is required to form a stable physical association with BRD4. Similarly, we confirmed that only the full-length (FL) and none of the three reader mutant variants could rescue proliferation in the context of endogenous ZMYND8 depletion (Figures 7C, 7D, and S7A), suggesting that interruption of any one of the reader domains may elicit an anti-AML effect.
Figure 7. ZMYND8 PHD-BD-PWWP reader cassette is required for association with BRD4 on chromatin and leukemia growth.

(A) Schematic of FLAG-tagged constructs used for coIP. FL, full-length. Vertical black bars represent mutagenized amino acid sites.
(B) IP-immunoblotting of nuclear lysates prepared from HEK293T cells. TSA, trichostatin A. Arrowhead represents the expected BRD4 size. (n = 3).
(C) Immunoblotting of nuclear lysates prepared from MOLM-13 cells stably expressing EV, FL, or mutated ZMYND8 cDNA and transduced with sgNeg or sgZMYND8_2. Arrowhead represents the expected ZMYND8 band.
(D) Competition-based proliferation assays performed in MOLM-13 cells stably expressing the indicated cDNA and sgZMYND8_2 (n = 3).
(E) Schematic of in vivo engraftment of MOLM-13 cells expressing FL or BD-mutated (N248A) ZMYND8.
(F) Flow cytometry analysis of human CD45+ leukemia cells in BM of recipient mice (n = 4). Statistical analysis was performed using unpaired Student’s t test. BM, bone marrow.
(G) Kaplan-Meier survival curves of recipient mice transplanted with MOLM-13 cells expressing WT or BD-mutated (N248A) ZMYND8 and transduced with sgNeg or sgZMYND8_2 (n = 6). p value determined by a log-rank Mantel-Cox test.
(H) CUT&RUN meta-profile (top) and density plot (bottom) of ZMYND8 enrichment at 8,454 FL-ZMYND8-occupied regions in MOLM-13 cells. MOLM-13 cells stably expressing FL or mutated ZMYND8 were transduced with sgZMYND8_2 and collected 5 days post-infection.
(I) Violin plot of normalized tag density of ZMYND8 peaks in (H). Black dots represent median values. p values were calculated by Welch’s two-sided t test.
(J and K) Gene tracks of ZMYND8 enrichment at the leukemic MYC (J) or IRF8 (K) enhancer regions in cells described in (H). Enhancer regions are shown in gray boxes.
(L) RT-qPCR analysis of mRNA expression of MYC (top) or IRF8 (bottom) in cells described in (H) (n = 3).
(M and N) Gene track of H3K27ac and ZMYND8 enrichment at MYC (M) and IRF8 (N) regions in two primary AML patient blasts. RNA-seq data are also shown.
(O) Model of how ZMYND8 regulates the IRF8-MEF2D and MYC axis in AML.
Error bars represent mean ± SEM. See also Figure S7.
BD modules share a conserved hydrophobic pocket that is amenable to chemical inhibitor targeting (Fujisawa and Filippakopoulos, 2017; Wimalasena et al., 2020); therefore, we focused on validating the importance of the ZMYND8 BD in vivo for potential therapeutic applications. MOLM-13 cells transduced with the FL or N248A ZMYND8 cDNA along with an ZMYND8 sgRNA targeting the endogenous gene were intravenously injected into NSG immunodeficient mice (Figure 7E). Remarkably, we observed that the BD mutant, displaying similar anti-AML effects to the ZMYND8 knockout, conferred slower disease progression and prolonged mouse survival in comparison with the FL group in vivo (Figures 3F–3H, S3I, 7F, 7G, and S7B).
Moreover, we sought to evaluate whether mutagenesis of the reader domains affects the chromatin-binding capacity of ZMYND8 and therefore suppresses key downstream transcriptional output. To test this possibility, we depleted the endogenous ZMYND8 in MOLM-13 cells expressing the FL, N107A/D108A, N248A, or F308A/W311A variants. Binding profiles for each of the three mutant reader domains revealed a dramatic loss of ZMYND8 occupancy on chromatin, with 80.4%–97.0% reduction in log2FC from −1 to −3.31 (normalized read counts >2 in FL-expressing cells) (Figures 7H and 7I). Examination of CUT&RUN browser tracks confirmed the absence of all three ZMYND8 reader mutants at the IE and ME1-ME5 enhancer regions (Figures 7J and 7K). Furthermore, RT-qPCR analysis showed a significant reduction in IRF8 and MYC mRNA levels in cells expressing reader module mutants, concurrent with corresponding loss of their enhancer occupancies in IE and ME1-ME5 (Figure 7L). Taken together, these results support the notion that ZMYND8 employs its complete PHD-BD-PWWP triple reader cassette to interact with BRD4 and that these factors collaboratively engage with the MYC and IRF8 enhancers as a means to sustain their essential transcriptional programs in AML.
ZMYND8 occupancy, IRF8 expression, and IE accessibility are positively correlated in patient-derived AML cells
To explore the clinical relevance of the leukemia-specific IRF8 and MYC distal enhancers, we analyzed chromatin binding profiles of H3K27ac and ZMYND8 in three primary AML patient samples and normal CD34+ hematopoietic stem and progenitor cells (HSPCs) using CUT&RUN. We observed an overlap in genome-wide occupancies of H3K27ac and ZMYND8 in primary samples (Figures S7D). Motif analysis across all four primary samples revealed a closely related ZMYND8-binding motif also similar to the ones identified in AML cell lines (Figures S7E, 5C, and S5C). In the MYC enhancer cluster, we observed enrichment of both H3K27ac and ZMYND8 at ME1–ME5 regions in all AML patient samples, CD34+ cells from three independent healthy donors, and human GMP and CMP mixed populations (Figures 7M and S7F). These findings were consistent with the evolutionarily conserved functional role of ME1–ME5 in regulating normal and leukemic HSC hierarchies (Bahr et al., 2018). In contrast, the IRF8 IE enhancer region showed heterogeneous ZMYND8 and H3K27ac binding patterns among the primary samples; both marks were enriched in blasts from patients #4943 and #6527, but not in blasts from patient #6610, CD34+ HSPCs, or GMP and CMP cells (Figures 7N and S7G). We noted a positive correlation between the IE active states and IRF8 expression levels (Figures 7N and S7G). These observations were in agreement with the heterogeneous expression pattern of IRF8 observed in the AML cell lines (Figure S1G) and a previous report that the IRF8 gene body was decorated with H3K27ac in only a subset of AML patient samples (McKeown et al., 2017). Additionally, upon inspection of a published assay for transposase-accessible chromatin using sequencing (ATAC-seq) dataset (Corces et al., 2016), we found a positive correlation between either the IE or ME1–ME5 open configuration and the corresponding levels of IRF8 or MYC expression in normal human hematopoiesis (Figures S7H–S7J and S2C). We similarly surveyed MYC and IRF8 enhancer accessibility in combination with their expression during AML evolution in a publicly available dataset of AML primary patient samples (Corces et al., 2016) and found that while MYC expression and ME1–ME5 enhancers remained largely unchanged, IRF8 expression and activity of IE increased during leukemogenesis (Figures S7K–S7L), and IRF8 expression was correlated with IE accessibility in AML blasts (Figures S7M and S7N). These data highlight the clinical relevance of both IRF8 and MYC expression levels and the presence of their respective enhancers in AML patients. Moreover, these findings suggest the possibility that expression of the IRF8 gene and its enhancer activity could serve as a biomarker for predicting therapeutic response to a potential ZMYND8 inhibitor.
DISCUSSION
Here, combining CRISPR-based functional genetic screening, transcriptomic profiling, and chromatin-binding analysis, we uncovered an unexpected IRF8-MEF2D transcriptional regulatory circuit as an AML-biased vulnerability. IRF8 is a TF known to regulate normal myeloid and B cell development (Holtschke et al., 1996; Lu et al., 2003; Tamura et al., 2000; Wang et al., 2008). Recently, MEF2D translocations have been shown to regulate essential precursor B cell receptor signaling in B cell acute lymphoblastic leukemia (Tsuzuki et al., 2020). In AML, we show that IRF8 and MEF2D mutually support transcription, forming a positive-feedback regulatory loop to sustain the oncogenic cell state (Figure 7O). We found that in normal hematopoietic cells, IRF8 and MEF2D were co-upregulated in myeloid progenitors and monocytes, suggesting that leukemia cells might hijack the normal IRF8 and MEF2D transcriptional circuit during oncogenesis.
Previous work has generally documented that ZMYND8 functions as a tumor suppressor in solid tumors. Loss of ZMYND8 was reported to induce expression of metastasis-linked genes to facilitate tumor invasion in prostate cancer (Li et al., 2016), and ZMYND8 was found to modulate enhancer RNA transcription status to prevent enhancer overactivation in breast cancer (Shen et al., 2016). Moreover, ZMYND8 has been implicated in all-trans-retinoic-acid-induced target gene activation and oncogenic repression to suppress breast cancer progression (Basu et al., 2017b, 2017a; Jin et al., 2019). Nevertheless, here, we observed that loss of ZMYND8 results in immediate transcriptional downregulation of IRF8 and MYC in an AML-biased manner. Of note, inspection of the DEPMAP database (Meyers et al., 2017) revealed that ZMYND8 perturbation could also strongly and negatively impact an additional hematopoietic malignancy, multiple myeloma (MM). It has been reported that MM is addicted to both IRF4 (which is homologous to IRF8) and MYC (Holien et al., 2012; Shaffer et al., 2008), while MEF2C is predicted to be an MM-biased dependency (Figure S7O). These observations raise an interesting possibility that ZMYND8 might regulate the IRF-MEF2 and MYC transcriptional programs in a hematopoietic-malignancy-biased manner.
In summary, manipulation of ZMYND8 provides a means to selectively perturb the essential IRF8-MEF2D and MYC transcriptional programs in AML. Through mutagenesis studies, we further identified that all three of the individual and unique reader domains of the PHD/BD/PWWP cassette are required for ZMYND8 to support AML proliferation. Mechanistically, ZMYND8 employs its reader cassette to tether on the ET domain of BRD4 on chromatin. Our findings suggest a non-canonical function of a CR in which it recognizes the modified state of another CR for proper chromatin localization. Given that PHD and PWWP domains can read methylated amino acids and BDs can recognize acetylated lysine residues, it may be possible that ZMYND8’s reader cassette recognizes combinatorial PTMs on the BRD4 ET domain, though those exact PTMs remain unclear and require further investigation. Recent advancements in chemical biology have demonstrated that the discrete reader pocket of the BD and PWWP domains are amenable to chemical perturbation (Clegg et al., 2019; Cochran et al., 2019; Filippakopoulos et al., 2010; de Freitas et al., 2020; Wimalasena et al., 2020). These findings highlight that therapeutic targeting of the reader modules in ZMYND8 can potentially be achieved by selective chemical probes. Overall, targeting the ZMYND8 reader cassette may be a viable and novel potential therapeutic approach to selectively suppress the essential IRF8-MEF2D and MYC transcriptional programs in AML.
Limitations of study
While our in vitro data comparing the effect of loss of ZMYND8 in IRF8hi AML cells and normal bone marrow progenitors suggest that a therapeutic window might exist, much more detailed studies are needed to determine the role of ZMYND8 and the transcriptional circuit we describe in normal hematopoietic differentiation. Ultimately, the definitive assessment of a therapeutic window will require a specific inhibitor and dedicated preclinical studies. Another limitation is that we were unsuccessful in establishing CRISPR-based perturbation of ZMYND8 in primary AML cells. A more in-depth characterization of the effect of ZMYND8 inhibition on primary patient AML cells will also be best performed once a tool compound is available.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Junwei Shi (jushi@upenn.edu)
Material availability
All plasmids will be deposited to Addgene for public requests (Addgene numbers in the key resources table). Transcriptional factor and epigenetic regulator libraries will be available upon requests to the corresponding author.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
|
| |||
| Antibodies | |||
|
| |||
| Rabbit polyclonal anti-ZMYND8 | Sigma-Aldrich | Product #HPAG2G949; Lot #A96431; RRID:AB_1B57223 | |
| Rabbit polyclonal anti-BRD4 | Bethyl | Product #A3G1–9B5A1GG; RRID:AB_262G1B4 | |
| Rabbit monoclonal anti-IRF8 | Abcam | Product #ab2G741B, Lot #GR327157B-1 | |
| Rabbit polyclonal anti-ZMYND8 | Bethyl | Product #A3G2-GB9A; RRID:AB_16G42B2 | |
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat# F1BG4; RRID: AB_262G44 | |
| Mouse monoclonal anti-HA (clone 12CA5) | Laboratory of Gerd Blobel | N/A | |
| Rabbit monoclonal anti-GAPDH | Cell Signaling Technology | Cat# 5G14; Cione# 14C1G; RRID:AB_1G69344B | |
| Rabbit polyclonal anti-FKBP12 antibody | Abcam | Cat# ab24373; RRID:AB_7323B3 | |
| Rabbit monoclonal anti-c-MYC antibody [Y69] | Abcam | Cat# ab32G72; RRID:AB_73165B | |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat# ab4729; RRID:AB_211B29 | |
| APC-R700 Mouse anti-Human CD45 Antibody | BDBiosciences | Cat# 566G41; RRID:AB_2744399 | |
| PerCP anti-mouse CD45 Antibody | Biolegend | Cat# 1G313G; RRID:AB_B93339 | |
| IgG from rabbit serum | Sigma-Aldrich | Cat#: IB14G; RRID:AB_1163661 | |
| Alexa Fluor® 680 Goat anti-mouse IgG (H+L) | Life Technologies | Product #A21G5B; Lot #1692967; RRID:AB_2535724 | |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR | Product #926–3221, Lot #CB121G-G5 | |
| Rabbit polyclonal anti-H3K27me3 | Cell Signaling Tech | Cat#9733S; RRID:AB_2616G29 | |
| Mouse monoclonal anti-H3K14ac, clone 13HH3–1A5 | Millipore | Cat#: MABE351; RRID:AB_26B7B9G | |
| CD34-FITC, 581, ASR | Beckman Coulter | Cat# IM1B7GU; RRID:AB_1G639533 | |
| Human CD38 APCHIT2 | BDBiosciences | Cat# 56G9BG; RRID:AB_1G5B4324 | |
| APC anti-human CD135 (Flt-3/Flk-2) | Biolegend | Cat# 3133GB; RRID:AB_21G4B35 | |
| Rabbit polyclonal anti-FLI1 | Abcam | Cat# ab152B9; RRID:AB_3G1B25 | |
|
| |||
| Chemicais, peptides, and recombinant proteins | |||
|
| |||
| Halt Protease & Phosphatase Inhibitor Cocktail, EDTA- free (100×) | Thermo Fisher Scientific | Ref #78441, Lot #UF284419 | |
| Glycogen | Roche | Ref #10901393001, Lot #11651224 | |
| SuperScript II Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18064014 | |
| AMPure XP | Beckman Coulter | A63880 | |
| Penicillin/ Streptomycin | Thermo Fisher Scientific | 15140122 | |
| Proteinase K | New England Biolabs | P8107S | |
| Puromycin dihydrochloride | Sigma-Aldrich | P8833 | |
| Blasticidin | Invitrogen | R21001 | |
| Geneticin Selective Antibiotic (G418 Sulfate) | Thermo Fisher Scientific | 10131035 | |
| Polyethylenimine, PEI | Polysciences, INC | 23966 | |
| OPTI-MEM | Thermo Fisher Scientific | 31985070 | |
| Hexadimethrine Bromide, Polybrene | Sigma-Aldrich | H9268 | |
| Dynabeads Protein A | Thermo Fisher Scientific | Ref #10002D, Lot #00651865 | |
| TRIzol Reagent | Thermo Fisher Scientific | 15596018 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 polynucleotide kinase | New England Biolabs | M0201L | |
| Agarose, Standard, Low Electroendosmosis (EEO) | Avantor | A426–07 | |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250 | |
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio-Rad | 1610158 | |
| 2XLaemmli Sample Buffer | Bio-Rad | 1610737 | |
| Dimethyl Sulfoxide | Sigma-Aldrich | D2650 | |
| Concanavalin A-coated Magnetic Beads | Bangs Laboratories | BP531 | |
| Digitonin | EMD Millipore | 300410 | |
| Spermidine | Sigma-Aldrich | S2501 | |
| dTAG-13/dTAG-47 | This study | N/A | |
| pA-MN | This study | N/A | |
| Spike-in DNA | Laboratory of Steven Henikoff | N/A | |
| Roche Complete Protease Inhibitor (EDTA-free) tablets | Sigma-Aldrich | 5056489001 | |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210 | |
| Formaldehyde 37% Solution | Avantor | 2106–01 | |
| RNase A | Thermo Fisher Scientific | EN0531 | |
| Phenol/Chloroform/Isoamyl Alcohol | Thermo Fisher Scientific | BP1752I400 | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| Mbol | New England Biolabs | R0147 | |
| Benzonase® Nuclease HC | Millipore | 71205 | |
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | A2220 | |
| 3X FLAG® Peptide | Sigma-Aldrich | F4799 | |
| Trichostatin A | Sigma-Aldrich | T8552 | |
| rmIL3 | Peprotech | 213–13 | |
| rmIL6 | Peprotech | 216–16 | |
| rmSCF | Peprotech | 250–03 | |
| Methylcellulose-based Medium with Recombinant for Mouse | STEMCELL technologies | M3434 | |
|
| |||
| Critical commercial assays | |||
|
| |||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7570 | |
| In-Fusion HD Cloning Kit | Takara Bio | 638909 | |
| 2× Phusion Master Mix | Thermo Scientific | F-548 | |
| Direct-zol RNA Miniprep Plus | Zymo Research | R2072 | |
| QuantSeq 30 mRNA-seq Library Prep Kit for Illumina | Lexogen | 015.96 | |
| Dead Cell Removal Kit | Miltenyi Biotec | 130–090-101 | |
| Agilent High Sensitivity DNA Kit | Agilent | 5067–4626 | |
| QIAquick PCR Purification Kit | QIAGEN | 28104 | |
| Quick-DNA Miniprep Kit | ZYMO Research | D3025 | |
| NucleoSpin Gel and PCR Clean-up Mini Kit | Macherey-Nagel | 740609.250 | |
| Aligent RNA 6000 Nano Kit | Aligent | 5067–1511 | |
| NEBNext® Library Quant Kit for Illumina | NEB | E7630 | |
| NEBNext® Ultra II RNA Library Prep Kit for Illumina® | NEB | E7770 | |
| NEBNext® Poly(A) mRNA Magnetic Isolation Module | NEB | E7490 | |
| TruSeq RNA Sample Prep Kit v.2 | Illumina | RS-122–2001 | |
|
| |||
| Deposited data | |||
|
| |||
| RNA-seq,ChIP-seq, 4C-seq and CUT&RUN data | This study | GSE157636 | |
| RNA-seq data | This study | GSE157249 | |
| ChIP-seq | Tarumoto et al., 2018 | GSE109493 | |
| ChIP-seq | Mohaghegh et al., 2019 | GSE123872 | |
| ChIP-seq | Poppe et al., 2017 | GSE89212 | |
| ChIP-seq and CUT&RUN | Krivtsov et al., 2019 | GSE127508 | |
| RNA-seq | Rathert et al., 2015 | GSE63782 | |
| RNA-seq and ATAC-seq | Corces et al., 2016 | GSE75384 | |
| SLAM-seq | Muharet al., 2018 | GSE100708 | |
| BET JQ1 AP-MS | Lambert et al., 2019 | MSV000081006 | |
| MS of rBRD4 domain | Lambert et al., 2019 | MSV000080986 | |
| MS of endogenouse BET IP-MS in HEK293T cells | Lambert et al., 2019 | MSV000082857 | |
| MS of endogenouse BET IP-MS in K562 cells | Lambert et al., 2019 | MSV000082859 | |
|
| |||
| Experimental models: cell lines | |||
|
| |||
| Human: MOLM-13 | DSMZ | ACC-554 | |
| Human: MV4–11 | ATCC | CRL-9591 | |
| Human: THP1 | ATCC | TIB-202 | |
| Human: NOMO1 | DSMZ | ACC-542 | |
| Human: HEL | ATCC | TIB-180 | |
| Human: OCI-AML3 | DSMZ | ACC-582 | |
| Human: SET-2 | DSMZ | ACC-608 | |
| Human: U937 | ATCC | CRL-1593.2 | |
| Human: K562 | ATCC | CCL-243 | |
| Human:JURKAT | ATCC | TIB-152 | |
| Human: SEM | DSMZ | ACC-546 | |
| Human: REH | ATCC | CRL-8286 | |
| Human: HEK293T | ATCC | CRL-3216 | |
| Human: ML2 | DSMZ | ACC-15 | |
| Human: A549 | ATCC | CCL-185 | |
| Human: HUH7 | JCRB | JCRB0403 | |
| Human: HUH1 | JCRB | JCRB0199 | |
| Human: HepG2 | ATCC | HB-8065 | |
| Human: SK-HEP1 | ATCC | HTB-52 | |
| Human: DMS114 | ATCC | CRL-2066 | |
| Human: NCI-H526 | ATCC | CRL-5811 | |
| Human: NCI-H82 | ATCC | HTB-175 | |
|
| |||
| Experimental models: organisms/strains | |||
|
| |||
| NOD scid gamma | JAX | Stock No: 005557 | |
| Constitutive-Cas9-GFP | JAX | Stock No: 026179 | |
|
| |||
| Oligonucleotides | |||
|
| |||
| sgRNA sequence see Table S3 | This study | N/A | |
| qPCR primers see Table S3 | This study | N/A | |
| 4C-seq primers see Table S3 | This study | N/A | |
|
| |||
| Recombinant DNA | |||
|
| |||
| LentiV_neo_empty | Tarumoto et al., 2018 | Addgene: 108101 | |
| LentiV_neo_ZMYND8 | This study | N/A | |
| LentiV_neo_ZMYND8 (N107A/F108A) | This study | N/A | |
| LentiV_neo_ZMYND8 (N248A) | This study | N/A | |
| LentiV_neo_ZMYND8 (F308A/F311A) | This study | N/A | |
| LentiV_MYC_PGK_blast | This study | N/A | |
| LentiV_neo_IRF8 | This study | N/A | |
| LentiV_Cas9_puro | Tarumoto et al., 2018 | N/A | |
| LRG(Lenti_sgRNA_EFS_GFP) | Tarumoto et al., 2018 | Addgene:65656 | |
| LRG2.1 | Tarumoto et al., 2018 | Addgene:108098 | |
| LRcherry2.1 | Tarumoto et al., 2018 | Addgene:108099 | |
| LentiV_neo_ZMYND8_FKBP12F36V-2×HA | This study | N/A | |
| LentiV_neo_FKBP12F36V-2×HA_IRF8 | This study | N/A | |
| pcDNA3_FLAG_ZMYND8 | This study | N/A | |
| pcDNA3_FLAG_BRD4L | Shen et al., 2015 | N/A | |
| GFP-ZMYND8 | Gong et al., 2015 | Addgene: 65401 | |
| pCRIS-PITChv2-Puro-dTAG vector | Nabet et al., 2018 | Addgene: 91793 | |
| pSL21-mCherry | Chen et al., 2021 | Addgene:164410 | |
|
| |||
| Software and algorithms | |||
|
| |||
| Bowtie2 v2.3.5 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | |
| BEDtools v2.28.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ | |
| Samtools v1.1 | Li et al., 2009 | http://samtools.sourceforge.net | |
| HOMER v4 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ | |
| bcl2fastq Conversion Software, v2.17 | Illumina, Inc. | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html | |
| MACS2 v2.1 | Zhang et al., 2008 | https://github.com/macs3-project/MACS | |
| UCSC Genome Browser | UCSC | http://genome.ucsc.edu/ | |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ | |
| Picard tools v1.96 | Broad Institute | https://github.com/broadinstitute/picard | |
| STAR v2.5.2 | Dobin et al., 2013 | https://github.com/alexdobin/STAR | |
| HTSeq, htseq-count, v0.6.1pl | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ | |
| R Bioconductor DESeq2 package v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| Blacklist | ENCODE | https://sites.google.com/site/anshulkundaje/projects/blacklists | |
| Juicer tools v1.7.6 | Durand et al., 2016 | https://github.com/aidenlab/juicer | |
| MSigDB v6.1 | Liberzon et al., 2015 | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp | |
| Tophat2 | Kim et al., 2013 | http://ccb.jhu.edu/software/tophat/index.shtml | |
| Cufflinks | Trapnell et al., 2013 | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ | |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/index.html | |
| IGVtools, 2.4.10 | Broad Institute | https://software.broadinstitute.org/software/igv/igvtools | |
| FlowJo software, v10.0.7 | FlowJo | N/A | |
| GraphPad Prism 7 | GraphPad Software | N/A | |
Data and code availability
The accession numbers for the RNA-seq, CUT&RUN, ChIP-seq and 4C-seq data in this study are: GSE157249 and GSE157636. Original immunoblot data have been deposited to Mendeley Data: http://data.mendeley.com/v1/datasets/s4w7g7rhr7/draft?a=dbd88495–70d6–41fb-9f6d-5351fd01b557. No original code was developed in association with this study. All other data are available upon requests to the corresponding author.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Around 6–8 week old Female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (Jax 005557) and 12–16 week old male Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh mice (Constitutively expressing Cas9-GFP mice) were purchased from the Jackson Laboratory. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia.
Cell lines
MOLM-13, MV4–11, THP-1, HEL, OCI-AML3, SET-2, U937, K562 and JURKAT were cultured in RPMI-1640 supplemented with 10% Bovine Calf Serum (FCS) and 1% Penicillin/Streptomycin. HEK293T, A549, and HUH7 were cultured in DMEM supplemented with 10% FCS and 1% Penicillin/Streptomycin. NOMO-1, SEM, REH, ML-2, DMS114, NCI-H526 and NCI-H82 were cultured in RPMI-1640 and supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin/Streptomycin. HepG2, HUH1 and SK-HEP1 were cultured in DMEM supplemented with 10% FBS and 1% Penicillin/Streptomycin. Cells were cultured at 37°C with 5% CO2.
AML patient samples
Primary AML specimens were obtained from the Stem Cell and Xenograft Core Facility at The Perelman School of Medicine, University of Pennsylvania, after informed consent in accordance with the Declaration of Helsinki. Protocols used in this study were approved by the University of Pennsylvania’s institutional review board. Samples were frozen in FCS and 10% DMSO in liquid nitrogen until use. Patient clinical follow-up is included in Table S5.
CD34+ HSPCs
CD34+ cells were obtained and purified as previously described (Grevet et al., 2018). Briefly, peripheral blood mononuclear cells were obtained from the University of Pennsylvania Human Immunology Core or from Fred Hutchinson Cancer Research Center, and purified with CD34 MicroBead Kit UltraPure (Miltenyi Biotec). CD34+ cells were cultured in StemSpan SFEM medium (StemCell Technologies) supplemented with 1× CC100 (StemCell Technologies) and 1% Penicillin/Streptomycin.
METHOD DETAILS
sgRNA and plasmid cloning
All Cas9-expressing cancer cell lines were generated through lentiviral delivery of an spCas9 expression vector (Addgene: 108100). For the dCas9-KRAB-based IRF8 enhancer evaluation experiments, control sgRNA were designed to target ~180bp upstream of the IRF8 transcription start site (TSS) and enhancer targeting sgRNAs were designed in the proximity of the H3K27ac-enriched regions +23–86 kb from the IRF8 TSS. All human sgRNAs were cloned by annealing the sense and antisense DNA oligos and ligating them into a BsmbI digested LRG2.1 (Addgene: 108098) or LRcherry2.1 (Addgene:108099) backbone. All murine sgRNAs were cloned into a BbsI digested pSL21-mCherry (Addgene:164410) backbone. All sgRNA sequences are listed in Table S3.
Full length ZMYND8 cDNA was PCR amplified from the GFP-ZMYND8 vector (Addgene: 65401) and cloned into a lentiviral expression vector, LentiV_Neo (Addgene: 108101) using the In-Fusion cloning system (Takara Bio). The full length IRF8 cDNA was cloned directly from a pooled cDNA library in MOLM-13 cells, and into the LentiV_Neo backbone using In-Fusion cloning. The full length MYC cDNA sequence was similarly cloned from the MOLM-13 cDNA library into the modified LentiV_Blast vector (Addgene: 111887) where the P2A is replaced with a PGK promoter. sgRNA-resistant synonymous substitutions N107/F108→AA, N248→A, and F308/F311→AA were introduced into ZMYND8 cDNA through PCR mutagenesis. For the ZMYND8-dTAG/dTAG-IRF8 system, FKBP12F36V-2×HA was PCR amplified from the pCRIS-PITChv2-Puro-dTAG vector (Addgene: 91793) and introduced into sgRNA-resistant ZMYND8_LentiV_Neo or the IRF8_LentiV_Neo vector, respectively. For transient expression experiments using immunoprecipitation, the full length, truncated, or mutated ZMYND8 cDNA was introduced into a pcDNA3 vector (Invitrogen) using the In-Fusion cloning system.
Virus production and transduction
For lentivirus production, HEK293T cells were transfected with the plasmid of interest, along with lentiviral packaging plasmids, pPAX2 and VSVG, and Polyethylenimine at a concentration of 1 mg/mL. Lentivirus produced in 6-well plates comprises 5 μg plasmid, 3.75 μg pPAX2, 2.5 μg VSVG, 40 μL PEI, and 500 μL OPTI-MEM. Lentivirus produced in 10-cm plates comprises 10 μg plasmid, 7.5 μg pPAX2, 5 μg VSVG, 80 μL PEI, and 1 mL OPTI-MEM. Transfected HEK293T were incubated for ~6 hours before media was removed and replenished. Lentivirus was collected at 24 hr, 48 hr, and 72 hr post-transfection and pooled together. For retrovirus production, retrovirus produced in 10-cm plates comprises 10 μg plasmid, 2 μg pCL-Eco, 2 μg VSVG, 40 μL PEI, and 1 mL OPTI-MEM, and virus was collected at 24 and 48 hr post-transfection and pooled together. For viral transduction, filtered virus-embedded supernatant and 4 uL/mL polybrene (2 mg/mL) were applied to indicated cell lines. Cells were spin-infected at room temperature at 650 × g for 25 minutes. Media was replenished at 24 h post infection, administered with appropriate antibiotic selection (1~2 μg/ml puromycin, 20 μg/ml blasticidin or 1 mg/mL G418).
GMP/CMP FACS
CD34+ HSPC cells were further stained with CD34 (1:30), CD38(1:100), CD135(1:100). A FACSAria Fusion Cell Sorter (BioLegend) was used to isolate GMP and CMP mixed populations (CD34+, CD38+, CD135hi).
Domain-focused CRISPR screen
The human CR domain-focused CRISPR sgRNA library was designed based on the conserved domain annotation information retrieved from the NCBI database. Five to six independent sgRNAs were designed to target one or multiple CR domains within individual genes following previously described design principles (Hsu et al., 2013). The final library contained CRISPR sgRNAs against 193 CRs based on a gene list from a previous study (Shi et al., 2015). Pooled sgRNAs were synthesized on an array platform (Twist Bioscience) and cloned into the Bsmb1-digested LRG2.1 vector via the Gibson cloning method (NEB). The representation and identity of the sgRNA library was verified via a deep sequencing analysis (data not shown). The CR domain-focused sgRNA library will be available through Addgene.
spCas9+ cells were generated via lentiviral delivery of the Lenti_Cas9_Puro vector (Addgene: 108110) and selected using puromycin to 100% positivity before sgRNA transduction. The pooled CR library was delivered via lentiviral transduction. To ensure individual cells received only a single copy of sgRNA during transduction, the multiplicity of infection (MOI) was set between 0.3–0.5 (confirmed by measure GFP% on day 3 post-infection). sgRNA-positive cells were cultured for approximately 14 or more doubling times and passaged as needed while maintaining ≥ 1000× representation for each sgRNA. About 1.2 million GFP+ cells were harvested on day 3 post-infection as the initial time point, and again for a final time point 14 or more doubling times after the initial collection. Harvested cell pellets contained both GFP- and GFP+ cells. Cell pellets were washed with PBS and stored at −80°C until genomic DNA extraction. Genomic DNA was extracted using the Quick-DNA Miniprep Kit (ZYMO) according to the manufacturer’s protocol. DNA was eluted with molecular grade PCR water and stored in −20°C for library preparation.
The sequencing library was prepared as previously described (Gier et al., 2020). Briefly, the integrated sgRNA cassette PCR was amplified from the genomic DNA (~300 ng input) with custom stacking barcode incorporation. Each library was amplified with a different barcode to an approximate 100 ng final PCR product. PCR products were gel extracted and purified using the Macherey-Nagel NucleoSpin Gel and PCR Clean-Up mini-kit, eluting with molecular grade PCR water. Eluted DNA was then purified again using the same NucleoSpin kit, eluting with PCR water. lllumina sequencing adaptors were then introduced to the barcode-embedded products with 8 cycles of PCR amplification. Final PCR products were purified using the QIAquick PCR purification kit, eluting with ~30 uL PCR water (QIAGEN). Libraries were analyzed for target product size (~320 bp) and high quality using a Bioanalyzer DNA 1000 kit (Agilent). Library concentration was determined using the Qubit dsDNA HS assay kit (Thermo Fisher). Libraries with different barcodes were pooled to 4 nM using the online Illumina pooling calculator. The 4 nM pooled library was denatured to a 20 pM pool according to the Illumina protocol, and 600 uL of the 20 pM pool was loaded into the cartridge. Libraries were sequenced on either the Miseq or Nextseq 500 platform with 75 bp single- or paired-end reads.
Sequencing reads were de-multiplexed and trimmed to only preserve the sgRNA cassette. Data were further aligned to the reference sgRNA library with no mismatch tolerated as previously described (Shi et al., 2015). All samples were normalized to the same number of total reads. Average log2FC of the sgRNA abundance for each domain or gene (refined as essentiality score, ES) was calculated as previously described (Wang et al., 2017). AML-biased ES was calculated by subtracting average ESs of AML cell lines from that of other cell lines. The chromatin regulator domain screen data from 19 cancer cell lines is provided in Table S2.
Competition-based cell proliferation assay
For individual gene or single enhancer region validation, cell lines stably expressing Cas9 were lentivirally delivered with indicated sgRNAs co-expressed with a GFP reporter. The percentage of GFP+ cells corresponds to the sgRNA representation within the population. GFP measurements in human cell lines were taken on day 3 post-infection and every other day after for 21 days post-infection using a Guava Easycyte HT instrument (Millipore). The fold change in GFP+ population (normalized to day 2 or day 3 post infection) was used for analysis.
For dual CRISPRi experiment, dCas9-KRAB expressing cells were simultaneously transduced with LRG2.1/LRcherry2.1 sgRNA vectors co-expressed with GFP or mCherry fluorescent markers, respectively. The fluorescent signal from the mixed populations containing uninfected, GFP+, mCherry+, and GFP+/mCherry+ cells was measured every 3 days from day 3 to day 15 post-infection using a Guava Easycyte HT instrument (Millipore). The ratios of GFP+, mCherry+ and GFP+/mCherry+ cells were calculated with uninfected cells serving as an internal control.
Colony formation assay
Bone marrow cells were isolated from femurs of 12–16 week old Rosa26-Cas9 knock-in C57BL/6J mice (JAX 026179). Erythroid cells were lysed with ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), and the remaining cells were cultured in IMDM supplemented with 15% FBS, 1% Penicillin/Streptomycin, 10ng/ml rmIL-3, 10 ng/ml rmIL-6, and 20ng/ml rmSCF.
Cells were retrovirally transduced with sgRNA in pSL21-mCherry vectors. On day 2 post-infection, GFP+/mCherry+ cells were sorted by a FACSAria Fusion Cell Sorter, and 25,000 sorted cells were plated into MethoCult GF M3434 media. Following 10 days of incubation, differentiated colonies were counted according to the manufacturer’s instructions and included burst-forming unit-erythroid (BFU-E), CFU-granulocyte, macrophage (CFU-GM), and CFU-granulocyte, erythrocyte, macrophage, megakaryocyte (CFU-GEMM).
Immunoblotting
Cells were lysed using a 25 mm syringe in Laemmli sample buffer (Bio-Rad) containing 5% β-mercaptoethanol. Protein extracts were boiled at 95°C for 7 minutes. Extracts were loaded on either 10% Polyacrylamide gels or 4%–20% precast polyacrylamide Criterion gels (Bio-Rad #5671094), and then transferred to 0.45 μm nitrocellulose membranes before immunoblotting. Membranes were blocked in 5% milk with TBST at room temperature for 30 min. Membranes were incubated with primary antibodies in 5% milk/TBST at 4°C overnight with shaking. Membranes were then washed for 3 cycles of a 5 min PBST wash, and incubated with secondary antibodies in 1× blocking buffer (LI-COR) at room temperature for 45 min. Membranes were again washed in 3 cycles as described before and imaged on the Odyssey® CLx (LI-COR) imager. Quantification was performed on ImageStudioLite.
In vivo transplantation and FACS analysis
For experiments validating the in vivo requirement of ZMYND8 in leukemia cells, MOLM13-Cas9+ cells were lentivirally transduced with LRG2.1-sgRNA-GFP vectors targeting ZMYND8 or a negative control. For experiments evaluating the essential function of the ZMYND8 BD in supporting leukemia cell growth in vivo, MOLM13-Cas9 cells transduced with FL or N248A mutant ZMYND8 cDNA were lentivirally infected with the LRG2.1-sgRNA-GFP vectors targeting ZMYND8. On day 2 post infection before a proliferation suppression phenotype manifests, GFP+ (sgRNA+) populations were FACS sorted and 1 million cells were injected through the tail vein of sublethally irradiated (2.5 Gy) NSG mice. On day 9 post transplantation, 4 mice in each condition were sacrificed and bone marrow was flushed and collected. Red blood cells were lysed with lysis buffer (Roche) on ice for 5 min and remaining cells were stained with human CD45 (1:50) and mouse CD45 (1:500) antibodies. The percentage of GFP- and CD45-positive cells were analyzed on a Cyto-FLEX Flow Cytometer (Beckman). Moribund mice were euthanized, and leukemia burden in bone marrow, spleen, and periphery blood was analyzed, and GFP+ cells collected from bone marrow were sorted with a FACSAria Fusion Cell Sorter for western blotting.
Cell viability assay
To test the effect of dTAG-47 (Huang et al., 2017; Weintraub et al., 2017) treatment on cell growth, 1,000 cells were plated in an opaque-walled 96-well plate and treated with 0.05% dTAG-47 in serial dilution, or with 0.05% DMSO as a normalization control. Cell viability was measured using CellTiter Glo Luminescent Cell Viability Assay kit (Promega) with a BioTek Microplate Reader (Molecular Devices) following the manufacturer’s instructions after a 5-day incubation.
Immunoprecipitation
HEK293T cells were cultured at ~50% confluency in a 10cm plate for transient expression. A mixture of 1 mL OPTI-MEM, 80 μL PEI, and 5 μg of plasmid DNA was added, and the media was replaced and replenished after 6–8 hours. At 48 hr post-transfection, cells were collected and washed one time in 1× PBS. Cells transfected with FL, truncated, or mutated ZMYND8 constructs were treated with 2 μM HDAC inhibitor trichostatin A (TSA, Sigma) for 6 hours prior to cell lysis (Chen et al., 2018). Immunoprecipitation was performed similarly as previously described (Dou et al., 2015; Shen et al., 2015). Briefly, cells were lysed in IP buffer comprising 20 mM Tris (pH 7.5), 137 mM NaCl, 1 mM CaCl2, 1% IGEPAL CA-630, 10% glycerol, 1 mM MgCl2, 1:100 Halt protease, phosphatase inhibitor cocktail (Thermo Scientific), and benzonase (Millipore) at 4°C for 1 hour with rotation. The supernatant was collected, and 2% of the total lysate was saved as input. The supernatant was incubated with 15 μL ANTI-FLAG® M2 Affinity Gel beads (Sigma) at 4°C overnight with rotation. The Affinity gel beads were washed 5 times with the IP buffer, and eluted with 50 μL of 250 μg/mL 3×Flag Peptide (Sigma) at 4°C for 30 min with rotation. The supernatant was collected and boiled with Laemmli sample buffer (Bio-Rad) containing 5% β-mercaptoethanol at 95°C for 5 min.
RNA-seq
For RNA-seq in cell lines transduced with sgRNAs, total RNA was isolated from 1~5 million cells using the Direct-zol RNA Miniprep Plus kit (ZYMO) with DNase I. MOLM-13, MV4;11, OCI-AML3, HEL, K562, and HUH7 cells transduced with sgZMYND8 were harvested on day 5 post-infection, NOMO-1 cell were harvested on day 6, and THP-1 cells were harvested on day 9. MOLM-13 cells transduced with sgMEF2D were harvested on day 5 post-infection. MOLM-13, MV4;11, THP-1, and OCI-AML3 transduced with sgIRF8 were collected on day 4 post-infection.
For targeted degradation, 1 million MOLM13-dIRF8 or MOLM13-dZD8 cells were treated with 500 nM dTAG-47/dTAG-13 (Brunetti et al., 2018; Erb et al., 2017; Nabet et al., 2018) for 4 hours prior to collection, and 0.05% DMSO treated cells were collected as a control. RNA quality was evaluated with the RNA Nano 6000 Bioanalyzer kit (Agilent), and RNA with RIN ≥ 9 was used for further library construction. For cells transduced with sgIRF8, RNA-seq libraries were prepared using TruSeq Sample Prep Kit V2 (Illumina) following the manufacturer’s instruction and starting with 2 μg of total RNA. For all other samples, RNA-seq libraries were prepared using the QuantSeq 3′ mRNA-seq Library Prep Kit (Lexogen) with 2 μg of total RNA according to the manufacturer’s protocol. Library quality was assessed using the High Sensitivity Bioanalyzer kit (Agilent) and libraries with different barcodes were pooled and sequenced on the Nextseq 500 platform with 75 or 150 bp single-end reads.
For RNA-seq in primary patient samples, dead cells were first removed using the Dead Cell Removal Kit (MACS). RNA was isolated from 1–3 million cells with the Direct-zol RNA Miniprep Plus kit (ZYMO). RNA-seq libraries were constructed using NEBNext® Ultra II RNA Library Prep Kit for Illumina. Briefly, 1 μg of total RNA was poly-A selected and fragmented with NEBNext® Poly(A) mRNA Magnetic Isolation Module, followed by first and second strand cDNA synthesis. The libraries were end-repaired, linked with the Illumina adaptors, and PCR amplified with 8 cycles. The libraries were sequenced on the Nextseq 500 platform with 75bp single-end reads or 2×42 paired-end reads.
RT-PCR
RNA was isolated using Direct-zol RNA Miniprep Plus kit (ZYMO) with DNase I treatment according to the manufacturer’s protocol. 2 μg of total RNA was reverse transcribed into cDNA using qScript cDNA SuperMix. RT-PCR was performed with SYBR green PCR master mix on ABI 7900HT standard real-time PCR machine. All results were quantified using ΔCt value normalized to GAPDH. All RT primers are listed in Table S3.
ChIP-seq and ChIP-qPCR
ChIP-seq was performed as previously described (Lu et al., 2018). Briefly, a total of 5×107 leukemia cells were harvested and supplemented with full culture media. Cross-linking was performed with 1% formaldehyde for 20 minutes at room temperature with rotation, and the sample was quenched with glycine for 10 minutes at a final concentration of 0.125 M. The cell pellet was washed with PBS, and lysed with cell lysis buffer (10 mM Tris, pH8.0, 10 mM NaCl, 0.2% NP-40, protease inhibitor) on ice for 10 min. The buffer was removed, and the cell pellet was split into 1×107 cell aliquots in 15 mL falcon tubes and incubated in 1 mL of nuclear lysis buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 1% SDS, protease inhibitor) for at least 10 minutes on ice. Lysed cells were sonicated with a bioruptor for 5 min at 4°C, followed by centrifugation at 16,000 g for 15 minutes at 4°C. Supernatants were combined and diluted with 35 mL IP dilution buffer (20 mM Tris, pH 8.0, 2 mM EDTA, 150 mM NaCI, 1% Triton X-100, 0.01% SDS), 5 mg of IRF8 antibody (Abcam #ab207418), and 40 μL of Protein A magnetic beads (Invitrogen). The mixture was incubated in rotation overnight at 4°C. The beads were subjected to a wash cycle with wash buffers in the following order: one round of IP Wash I buffer (20 mM Tris-HCI pH 8.0, 2mM EDTA, 50 mM NaCI, 1% Triton X-100, 0.1% SDS), two rounds of High salt buffer (20 mM Tris-HCI pH 8.0, 2 mM EDTA, 500 mM NaCI, 1% Triton X-100, 0.1% SDS), one round of IP Wash II buffer (10 mM Tris-HCI pH 8.0, 1 mM EDTA, 0.2 M LiCI, 1% NP-40, 1% Nadeoxycholate), and two rounds of TE pH 8.0. Fragmented chromatin was eluted with 200 mL elution buffer (50 mM Tris 8.0,10 mM EDTA, 1% SDS) with shaking at 600 rpm at 65°C for 15 minutes. The chromatin was reverse-crosslinked with 0.25 M NaCI overnight at 65°C, and non-DNA content was digested with RNase A for 1 hr at 65°C and then with Proteinase K (0.2 mg/mL) for 2 hr at 42°C. DNA was purified with QIAquick PCR purification kit following the manufacturer’s instructions. ChIP-seq library construction was prepared with NEBNext® Ultra II DNA Library Prep Kit for Illumina. Library quality was validated with a high sensitivity Bioanalyzer (Agilent) kit and libraries with different barcodes were pooled and sequenced on the Nextseq 500 platform with 75 bp single-end reads. The IRF8 peak near the MEF2D TSS obtained from IRF8 ChIP-seq was used for qPCR analysis, and input DNA was serially diluted to quantify the relative enrichment of ChIP DNA from DMSO or 500nM dTAG-47 treated cells. Primer sequences and their chromosome coordinates are listed in Table S3.
CUT&RUN
CUT&RUN experiments were performed similarly to a previously described protocol (Skene et al., 2018) with small modifications. For human cell lines and CD34+ HSPC, 1–5×105 cells were used with H3K27ac/H3K14ac/H3K27me3 antibodies, and 2×106 cells were used with ZMYND8, BRD4, and IgG antibodies. For GMP/CMP mixed populations, around 1–5×104 cells were used. Digitonin was diluted at a final concentration of 0.1% (w/v) in the antibody buffer (150 mM NaCl, 20 mM HEPES-KOH, 0.5 mM Spermidine, 2 mM EDTA, protease inhibitor cocktail plus digitonin), and Dig-wash buffer (150 mM NaCl, 20 mM HEPES-KOH, 0.5 mM Spermidine, protease inhibitor cocktail plus digitonin). Live cells from previously frozen primary patient samples or from CD34+ HPSCs were purified with the Dead Cell Removal kit (MACS) before washing. A final concentration of 0.05% of Digitonin was used for subsequent CUT&RUN steps. Cells were washed twice in Wash buffer (150 mM NaCl, 20 mM HEPES-KOH, 0.5 mM Spermidine, 2 mM EDTA, protease inhibitor cocktail) and incubated with 20 μL of Concanavalin A-coated beads for 10 min with rotation at room temperature. Antibody buffer containing indicated antibodies (1 μL H3K27ac, H3K27me3, and H3K14ac; 2 μL ZMYND8, BRD4, Rabbit IgG) was added to the cell-attached beads and incubated at 4°C with rotation overnight. Beads were washed with Dig-wash buffer and incubated with 300 μL Dig-wash buffer containing ~700 ng/ml pA-MN at 4°C for 1 hr. Beads were washed twice with Dig-wash buffer and chilled on ice. Digestion was activated by adding CaCl2, and chromatin was digested for 60 min with a ZMYND8 antibody (Bethyl) or for 30 min with all other antibodies. Equal volume of 2× Stop solution (340 mM NaCl, 4 mM EGTA, 20 mM EDTA, 50 μg/mL RNase A, 0.02% Digitonin, 50 μg/mL Glycogen, 2 pg/mL heterogenous spike-in DNA) was added and beads were incubated at 37°C for 10 min to release the digested chromatin. The supernatant was collected and DNA was extracted through Phenol-Chloroform extraction.
Libraries were constructed with the NEBNext® Ultra II DNA Library Prep Kit for Illumina with small modifications. For H3K27ac, H3K14ac and H3K27me3 libraries, the adaptor was diluted to a ratio of 1:25, and repaired DNA was first selected with 25 μL Ampure Beads (Beckman) and then secondarily selected with 45 μL beads, followed by 9 cycles of PCR amplification for cell lines and HSPCs, and 13–15 cycles for GMP/CMPs. For chromatin regulators, sequencing libraries were constructed as previously described (Liu et al., 2018). Adapters were diluted to a ratio of 1:50 and 10–15 cycles of PCR amplification were used. Library quality was assessed on the High sensitivity Bioanalyzer kit (Agilent) and libraries with different barcodes were pooled and sequenced on the Next-seq 500 platform with 2×42 pair-end reads.
Circular Chromosome Conformation Capture with unique molecular identifier (UMI-4C)
The UMI-4C protocol was performed as previously described (Schwartzman et al., 2016) with small modifications. 5–10 million cells were harvested and cross-linked as described in the above ChIP-seq protocol. Cells were resuspended in lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40 with protease inhibitor) at 4°C for 30 minutes with rotation. Isolated nuclei were washed with lysis buffer, and permeabilized with 200 μL 10% SDS at 62°C for 10 min, which was sequestered by 235 μL 2% Triton X-100 at 37°C for 15 minutes. Chromatin was digested with 15 μL of MboI (R0147) at 37°C for 2 hr, and then an additional 5 μL of MboI was added for 1 more hour. MboI was then heat inactivated at 62°C for 20 minutes. Proximity ligation was performed using the ligation master mix (150μL 10X NEB T4 DNA ligase buffer, 125 μL 10% Triton X-100, 3 μL 50mg/mL BSA, 4000U T4 DNA ligase, 660μL H2O) at room temperature for 4 hr with rotation, and an additional 4000U T4 DNA ligase was applied for 2 hr. The nuclei pellet was centrifuged at 2500 g at 4°C for 5 minutes, resuspended with 200 μL Proteinase K Buffer, and treated with 10 μL proteinase K at 55°C for 45 min, then at 65°C overnight with 900 rpm with shaking.
The next day, 10 μL of RNase A was added and the mixture was incubated at 37°C for 10 minutes. DNA was precipitated with 3M NaAc and cold 100% ethanol, and eluted with 200 μL of Elution Buffer (EB) (10 mM Tris-HCl). 10 μg of DNA was sonicated on the bioruptor for 5 cycles of 30 s on/60 s off, and end repaired with NEB end repair mix (#E6050L) at 20°C for 30 minutes. DNA was size selected with 2.2× Ampure beads and eluted in 76 μL EB. Fragmented DNA was then adenylated with 4 μL of Klenow exo-, 10 μL NEBuffer 2, and 10 μL of 10 mM dATP at 37°C for 30 minutes, followed by CIP (M0525S) treatment at 50°C for 60 minutes. DNA was size selected with 2× Ampure beads, and ligated with Illumina forked indexed adapters using NEB Quick Ligase (#M2200) at room temperature for 15 minutes. Ligated DNA was denatured and selected with 1× Ampure beads. 4C library primers for MYC and IRF8 were designed near the TSS regions 5–15 bp adjacent to the closest MboI cut site (defined as “viewpoint”). Samples underwent two rounds of PCR (20 cycles for the first round, and 15 for the second) and in each round, the 4C primer was paired with the Illumia universal primer. The amplified 4C libraries were purified with 1× Ampure beads. For each viewpoint, two pairs of 4C primers were used and the libraries from the same viewpoint were equally pooled together. 4C libraries were sequenced on the Next-seq 500 platform with 2×42/2×50 paired-end reads, respectively. All 4C-seq primers are listed in Table S3.
RNA-seq, ChIP-seq, CUT&RUN, and 4C-seq analysis
For RNA-Seq analysis, sequencing reads for human cell lines transduced with sgZMYND8, or for MOLM13-dZD8/MOLM13-dIRF8 treated with dTAG were aligned to human genome hg38; primary patient samples were aligned to human genome hg19 using STAR Aligner (Dobin et al., 2013). Raw read counts were generated via HTSeq-count (Anders et al., 2015). RPM-normalized bigwig files were generated using bedGraphToBigWig and were visualized in the UCSC genome browser. Mapped reads were analyzed with DESeq2 (1.14.1) (Love et al., 2014) to identify differentially expressed genes, and raw reads < 5 were filtered out.
In RNA-seq experiments using cells transduced with sgIRF8, raw sequencing reads were mapped to reference human genome hg38 using Tophat2 (Kim et al., 2013). Cuffdiff (Trapnell et al., 2013) was used to analyze differentially expressed genes with structural RNAs masked, and to normalize against total mapped reads. Genes with RPKM > 2 were used for further analysis. Gene set enrichment analysis (GSEA) was performed according to the linked instructions with public signatures in the Molecular Signature Database v6.1 (MSigDB) (Liberzon et al., 2015) and custom gene signatures.
For ChIP-seq and CUT&RUN analysis, reads were aligned to human genome hg19 using Bowtie2 v2.3.5 (Langmead and Salzberg, 2012), following parameters as previously suggested (Skene et al., 2018):–local–very-sensitive-local–no-unal–no-mixed–no-discordant–phred33 -I 10 -X 700 -k1 -N1. Picard tools v1.96 (Broad Institute) was used to remove presumed PCR duplicates using the MarkDuplicates command. Bam files containing uniquely mapped reads were created using Samtools v1.(Li et al., 2009). Fragments of < 40 bp or > 1000 bp were filtered out. Blacklist regions defined by ENCODE, random chromosomes, and mitochondria were removed, and filtered bam files were used for downstream analysis. Read count per million (RPM)-normalized bigwig files were created by bedGraphToBigWig (UCSC).
CUT&RUN and ChIP-Seq signals were called on using MACS v2.1 (Zhang et al., 2008) using the broadPeak setting with p value cutoff of 1e−8 and broad p value cutoff 1e−4 in general. Genes proximal to peaks were annotated against the hg19 genome using annotatePeaks.pl from HOMER v4 (Heinz et al., 2010). ZMYND8 and BRD4 binding motifs were identified using findMotifsGenome.pl from HOMER v4. The venn diagram of comparison of peaks was plotted using Bioconductor package ChIPpeakAnno (Zhu et al., 2010). Heatmaps and metaplots were generated using deepTools plotHeatmap (Ramírez et al., 2016). RPM-normalized bigwig files were created using bedGraphToBigWig (UCSC) and bamCoverage (deepTools), and were used to visualize binding signals. For visualization purposes, the average background was subtracted and scaled bigwig files were used to visualize CUT&RUN signals in MOLM-13, THP-1, HEL, and HUH-7 cells. Tracks were loaded to the UCSC genome browser for visualization.
4C-seq analysis was performed as previously described (Petrovic et al., 2019). Paired-end reads of each replicate were aligned to hg19 and processed as hic file using juicer (Durand et al., 2016). Intrachromosomal contact matrices of chromosome 8 (for MYC) or 16 (for IRF8) were extracted using juicer tools ‘dump’ command at 5 kb resolution without normalization. For each replicate, contacts ± 350 kb around the viewpoint were summed up as total valid contacts. Contacts from the viewpoint were normalized by dividing total valid contacts times 1000. Average normalized contacts of triplicates ± standard deviation were plotted in R using ggplot.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed with either Graphpad Prism 7 or R. For RNA-seq, ChIP-seq, ChIP-qPCR, 4C-seq, RT-PCR, proliferation assay as well as in vivo experiments, statistical parameters including p value, replicates, SEM and FDR are detailed on the figures or annotated in the respective figure legends.
Supplementary Material
Highlights.
IRF8 and MEF2D form a transcriptional circuit to support AML proliferation
A CRISPR screen identifies ZMYND8 as an AML-biased vulnerability
ZMYND8 regulates IRF8 in parallel with MYC via lineage-specific enhancers in AML
ZMYND8 employs its triple reader cassette for enhancer occupancy and cancer growth
ACKNOWLEDGMENTS
We thank Liling Wan and Rahul Kohli for critically reading the manuscript, Tim Somerville and Yali Xu for sharing their experimental conditions for ZMYND8 pull-down, Jelena Petrovic for helpful suggestions on 4C-seq and ChIP-seq protocols, Ryan Corces for sharing processed AML patient ATAC-seq data, and Luke Gilbert for advising the dual CRISPRi experiment. We thank the CHOP flow cytometry core for help with fluorescence-activated cell sorting (FACS). J.S. acknowledges support from the Linda Pechenik Montague Investigator Award and Cold Spring Harbor Laboratory sponsored research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.07.018.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfossi G, Gewirtz AM, and Calabretta B (1989). An oligomer complementary to c-myb-encoded mRNA inhibits proliferation of human myeloid leukemia cell lines. Proc. Natl. Acad. Sci. USA 86, 3379–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr C, von Paleske L, Uslu VV, Remeseiro S, Takayama N, Ng SW, Murison A, Langenfeld K, Petretich M, Scognamiglio R, et al. (2018). A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 553, 515–520. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Leha r, J., Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu M, Sengupta I, Khan MW, Srivastava DK, Chakrabarti P, Roy S, and Das C (2017a). Dual histone reader ZMYND8 inhibits cancer cell invasion by positively regulating epithelial genes. Biochem. J 474, 1919–1934. [DOI] [PubMed] [Google Scholar]
- Basu M, Khan MW, Chakrabarti P, and Das C (2017b). Chromatin reader ZMYND8 is a key target of all trans retinoic acid-mediated inhibition of cancer cell proliferation. Biochim. Biophys. Acta. Gene Regul. Mech 1860, 450–459. [DOI] [PubMed] [Google Scholar]
- Bennett RL, and Licht JD (2018). Targeting Epigenetics in Cancer. Annu. Rev. Pharmacol. Toxicol 58, 187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. (2011). MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat AS, and Vakoc CR (2015). Targeting Transcription Factors in Cancer. Trends Cancer 1, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, Dillon ET, Cagney G, Van Mierlo G, Baltissen MP, et al. (2018). Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 7, e41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Wilkinson CR, Waterman SR, Kok CH, Salerno DG, Diakiw SM, Reynolds B, Scott HS, Tsykin A, Glonek GF, et al. (2006). Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. J. Leukoc. Biol 80, 433–447. [DOI] [PubMed] [Google Scholar]
- Brunetti L, Gundry MC, Sorcini D, Guzman AG, Huang Y-H, Ramabadran R, Gionfriddo I, Mezzasoma F, Milano F, Nabet B, et al. (2018). Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 34, 499–512.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, and Koehler AN (2020). Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med 26, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang B, Bao L, Jin L, Yang M, Peng Y, Kumar A, Wang JE, Wang C, Zou X, et al. (2018). ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J. Clin. Invest 128, 1937–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Arai E, Khan O, Zhang Z, Ngiow SF, He Y, Huang H, Manne S, Cao Z, Baxter AE, et al. (2021). In vivo CD8+ T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg MA, Tomkinson NCO, Prinjha RK, and Humphreys PG (2019). Advancements in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 14, 362–385. [DOI] [PubMed] [Google Scholar]
- Cochran AG, Conery AR, and Sims RJ 3rd (2019). Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov 18, 609–628. [DOI] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar TL, Herzner A-M, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D, et al. (2017). Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol 216, 3535–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA (2017). The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152. [DOI] [PubMed] [Google Scholar]
- Dawson MA, and Kouzarides T (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, Robson SC, Chung CW, Hopf C, Savitski MM, et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, Cannizzaro E, Osaki H, Wiese M, Putwain S, et al. (2014). Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 28, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Freitas RF, Liu Y, Szewczyk MM, Mehta N, Li F, McLeod D, Zepeda-Velázquez C, Dilworth D, Hanley RP, Gibson E, et al. (2020). Discovery of Small-Molecule Antagonists of the PWWP Domain of NSD2. BioRxiv, 2020.11.25.398586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Benito V, Rosen DB, Wang Q, Gazumyan A, Pai JA, Oliveira TY, Sundaravinayagam D, Zhang W, Andreani M, Keller L, et al. (2018). The Chromatin Reader ZMYND8 Regulates Igh Enhancers to Promote Immunoglobulin Class Switch Recombination. Mol. Cell 72, 636–649.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman KV, et al. (2018). Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med 24, 1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio E, Hancock WW, and Brancolini C (2018). MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta BBA - Rev. Cancer 1870, 261–273. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dö hner H, Weisdorf DJ, and Bloomfield CD (2015). Acute Myeloid Leukemia. N. Engl. J. Med 373, 1136–1152. [DOI] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo H-S, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T, and Filippakopoulos P (2017). Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol 18, 246–262. [DOI] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard C, Surianarayanan S, Bentley T, Warr MR, Fitch B, Geng H, Passegué E, de Thé H, and Kogan SC (2018). Identification of IRF8 as a potent tumor suppressor in murine acute promyelocytic leukemia. Blood Adv. 2, 2462–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh K, Tang M, Kumari N, Nandy A, Basu S, Mall DP, Rai K, and Biswas D (2018). Positive regulation of transcription by human ZMYND8 through its association with P-TEFb complex. Cell Rep. 24, 2141–2154.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gier RA, Budinich KA, Evitt NH, Cao Z, Freilich ES, Chen Q, Qi J, Lan Y, Kohli RM, and Shi J (2020). High-performance CRISPR-Cas12a genome editing for combinatorial genetic screening. Nature Communications 11, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giotopoulos G, Chan W-I, Horton SJ, Ruau D, Gallipoli P, Fowler A, Crawley C, Papaemmanuil E, Campbell PJ, Göttgens B, et al. (2016). The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene 35, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Chiu L-Y, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. (2015). Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 29, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S, and Mulloy JC (2011). Molecular pathogenesis of core binding factor leukemia: current knowledge and future prospects. Int. J. Hematol 94, 126–133. [DOI] [PubMed] [Google Scholar]
- Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc W, Kretzer NM, Briseño CG, Durai V, Bagadia P, et al. (2015). Batf3 maintains autoactivation of Irf8 for commitment of a CD8a(+) conventional DC clonogenic progenitor. Nat. Immunol 16, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevet JD, Lan X, Hamagami N, Edwards CR, Sankaranarayanan L, Ji X, Bhardwaj SK, Face CJ, Posocco DF, Abdulmalik O, et al. (2018). Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, McCastlain K, Reshmi SC, Payne-Turner D, Iacobucci I, et al. (2016). Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun 7, 13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. (2012). The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holien T, Våtsveen TK, Hella H, Waage A, and Sundan A (2012). Addiction to c-MYC in multiple myeloma. Blood 120, 2450–2453. [DOI] [PubMed] [Google Scholar]
- Holtschke T, Löhler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch K-P, Gabriele L, Waring JF, et al. (1996). Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87, 307–317. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H-T, Seo H-S, Zhang T, Wang Y, Jiang B, Li Q, Buckley DL, Nabet B, Roberts JM, Paulk J, et al. (2017). MELK is not necessary for the proliferation of basal-like breast cancer cells. eLife 6, e26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Xu X-E, Jiang Y-Z, Liu Y-R, Sun W, Guo Y-J, Ren Y-X, Zuo W-J, Hu X, Huang S-L, et al. (2019). The endogenous retrovirus-derived long noncoding RNA TROJAN promotes triple-negative breast cancer progression via ZMYND8 degradation. Sci. Adv 5, eaat9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblau SM, Qiu YH, Zhang N, Singh N, Faderl S, Ferrajoli A, York H, Qutub AA, Coombes KR, and Watson DK (2011). Abnormal expression of FLI1 protein is an adverse prognostic factor in acute myeloid leukemia. Blood 118, 5604–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, Ross KN, Perner F, Olsen SN, Pritchard T, et al. (2019). A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 36, 660–673.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-P, Picaud S, Fujisawa T, Hou H, Savitsky P, Uusküla-Reimand L, Gupta GD, Abdouni H, Lin Z-Y, Tucholska M, et al. (2019). Interactome Rewiring Following Pharmacological Targeting of BET Bromodomains. Mol. Cell 73, 621–638.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Barreiro LB, and Gros P (2016). The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J. Exp. Med 213, 585–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Li Y, Lv J, Zheng X, Wen H, Shen H, Zhu G, Chen T-Y, Dhar SS, Kan P-Y, et al. (2016). ZMYND8 Reads the Dual Histone Mark H3K4me1-H3K14ac to Antagonize the Expression of Metastasis-Linked Genes. Mol. Cell 63, 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdo H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-F, Wang B-Y, Zhang W-N, Huang J-Y, Li B-S, Zhang M, Jiang L, Li J-F, Wang M-J, Dai Y-J, et al. (2016). Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8, 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Medina KL, Lancki DW, and Singh H (2003). IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 17, 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Klingbeil O, Tarumoto Y, Somerville TDD, Huang Y-H, Wei Y,Wai DC, Low JKK, Milazzo JP, Wu XS, et al. (2018). A transcription factor addiction in leukemia imposed by the MLL promoter sequence. Cancer Cell 34, 970–981.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Liu J, Liu L, Duan G, Wang Q, Xu Y, Xia F, Shan J, Shen J, Yang Z, et al. (2014). Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res. 74, 1452–1462. [DOI] [PubMed] [Google Scholar]
- MacPherson L, Anokye J, Yeung MM, Lam EYN, Chan Y-C, Weng C-F, Yeh P, Knezevic K, Butler MS, Hoegl A, et al. (2020). HBO1 is required for the maintenance of leukaemia stem cells. Nature 577, 266–270. [DOI] [PubMed] [Google Scholar]
- Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, Wiseman DH, Harris WJ, Li Y, Sahoo S, et al. (2018). Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 22, 3641–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown MR, Corces MR, Eaton ML, Fiore C, Lee E, Lopez JT, Chen MW, Smith D, Chan SM, Koenig JL, et al. (2017). Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARα Dependency Targetable by SY-1425, a Potent and Selective RARα Agonist. Cancer Discov. 7, 1136–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, Bergeron L, and Sims RJ 3rd (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H,Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh N, Bray D, Keenan J, Penvose A, Andrilenas KK, Ramlall V, and Siggers T (2019). NextPBM: a platform to study cell-specific transcription factor binding and cooperativity. Nucleic Acids Res. 47, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B, et al. (2018). SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A,Leggett AL, Erb MA, Lawlor MA, Souza A, et al. (2018). The dTAG ystem for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson E, Hasemann MS, Willer A, Lauridsen FKB, Rapin N, Jendholm J, and Porse BT (2014). Initiation of MLL-rearranged AML is dependent on C/EBPα. J. Exp. Med 211, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. (2016). Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med 374, 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic J, Zhou Y, Fasolino M, Goldman N, Schwartz GW, Mumbach MR, Nguyen SC, Rome KS, Sela Y, Zapataro Z, et al. (2019). Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 73, 1174–1190.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe M, Wittig S, Jurida L, Bartkuhn M, Wilhelm J, Müller H, Beuerlein K, Karl N, Bhuju S, Ziebuhr J, et al. (2017). The NF-kB-dependent and -independent transcriptome and chromatin landscapes of human coronavirus 229E-infected cells. PLoS Pathog. 13, e1006286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlŕez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe J-S, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe J-S, Mercan F, Rivera K, Pappin DJ, and Vakoc CR (2015). BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell 58, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky P, Krojer T, Fujisawa T, Lambert J-P, Picaud S, Wang C-Y, Shanle EK, Krajewski K, Friedrichsen H, Kanapin A, et al. (2016). Multivalent Histone and DNA Engagement by a PHD/BRD/PWWP Triple Reader Cassette Recruits ZMYND8 to K14ac-Rich Chromatin. Cell Rep. 17, 2724–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk T, Chen WC, Gö llner S, Howell L, Jin L, Hebestreit K, Klein H-U, Popescu AC, Burnett A, Mills K, et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med 18, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher M, Kohlhuber F, Hö lzel M, Kaiser C, Burtscher H, Jarsch M, Bornkamm GW, Laux G, Polack A, Weidle UH, and Eick D (2001). The transcriptional program of a human B cell line in response to Myc. Nucleic Acids Res. 29, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzman O, Mukamel Z, Oded-Elkayam N, Olivares-Chauvet P, Lubling Y, Landan G, Izraeli S, and Tanay A (2016). UMI-4C for quantitative and targeted chromosomal contact profiling. Nat. Methods 13, 685–691. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. (2008). IRF4 addiction in multiple myeloma. Nature 454, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe J-S, Suzuki Y, Pappin DJ, Joshua-Tor L, and Vakoc CR (2015). NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Mol. Cell 60, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X.u, Z., , et al. (2016). Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell 165, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, and Vakoc CR (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe J-S, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, and Vakoc CR (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol 33, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, and Ozato K (2000). ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity 13, 155–165. [DOI] [PubMed] [Google Scholar]
- Tarumoto Y, Lu B, Somerville TDD, Huang Y-H, Milazzo JP, Wu XS, Klingbeil O, El Demerdash O, Shi J, and Vakoc CR (2018). LKB1, Salt-Inducible Kinases, and MEF2C are linked dependencies in acute myeloid leukemia. Mol. Cell 69, 1017–1027.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki S, Yasuda T, Kojima S, Kawazu M, Akahane K, Inukai T, Imaizumi M, Morishita T, Miyamura K, Ueno T, et al. (2020). Targeting MEF2D-fusion Oncogenic Transcriptional Circuitries in B-cell Precursor Acute Lymphoblastic Leukemia. Blood Cancer Discov. Published online June 10, 2020. 10.1158/2643-3230.BCD-19-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S, Atsumi T, and Morse HC 3rd (2008). IRF8 regulates B-cell lineage specification, commitment, and differentiation. Blood 112, 4028–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day S, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimalasena VK, Wang T, Sigua LH, Durbin AD, and Qi J (2020). Using Chemical Epigenetics to Target Cancer. Mol. Cell 78, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-Y, and Chiang C-M (2007). The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem 282, 13141–13145. [DOI] [PubMed] [Google Scholar]
- Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M, Levantini E, Welner RS, Bach CS, Zhang J, et al. (2015). Hematopoietic Differentiation Is Required for Initiation of Acute Myeloid Leukemia. Cell Stem Cell 17, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE,Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wu J, Li B, Liu D, Yu J, Yan X, Zheng S, Wang J, Zhang L, Zhang L, et al. (2014). PU.1 is essential for MLL leukemia partially via cross-talk with the MEIS/HOX pathway. Leukemia 28, 1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Page s, H., Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the RNA-seq, CUT&RUN, ChIP-seq and 4C-seq data in this study are: GSE157249 and GSE157636. Original immunoblot data have been deposited to Mendeley Data: http://data.mendeley.com/v1/datasets/s4w7g7rhr7/draft?a=dbd88495–70d6–41fb-9f6d-5351fd01b557. No original code was developed in association with this study. All other data are available upon requests to the corresponding author.