

Abstract
The vertebrate vasculature displays high organotypic specialization, with the structure and function of blood vessels catering to the specific needs of each tissue. A unique feature of the central nervous system (CNS) vasculature is the blood-brain barrier (BBB). The BBB regulates substance influx and efflux to maintain a homeostatic environment for proper brain function. Here, we review the development and cell biology of the BBB, focusing on the cellular and molecular regulation of barrier formation and its maintenance through adulthood. We summarize unique features of CNS endothelial cells and highlight recent progress and general principles of barrier regulation. Finally, we illustrate why a mechanistic understanding of the development and maintenance of the BBB could provide novel therapeutic opportunities for CNS drug delivery.
Keywords: Blood-brain barrier, Neurovascular unit, Central nervous system, Endothelial cells, Pericytes, Astrocytes
Introduction
The blood brain barrier (BBB) is the interface that separates neural tissue from circulating blood. The BBB comprises of a single layer of endothelial cells that forms the blood vessel wall and maintains a safe and homeostatic milieu for proper neuronal function and synaptic transmission. Impaired barrier function is associated with neurodegenerative diseases, including multiple sclerosis, Alzheimer’s disease and Parkinson’s disease (Sweeney et al 2018, Zhao et al 2015). Conversely, the BBB is a major obstacle for drug delivery into the CNS, impeding the treatment of neurological diseases including neurodegenerative diseases, psychiatric disorders, brain infections, and brain tumors (Banks 2016, Pardridge 2012). Despite the importance of the BBB, our understanding of fundamental mechanisms underlying the formation and maintenance of the BBB remains limited.
Our limited understanding of the BBB can be attributed to its complexity. The BBB is localized to CNS endothelial cells that build the vessel wall and display unique cell biological features. These features distinguish them from peripheral endothelial cells. First, CNS endothelial cells have specialized tight junctions (TJs) to prevent free paracellular passage through the vessel wall. Second, they express designated transporters to regulate the dynamic influx and efflux of specific substrates. Third, they display extremely low rates of transcellular vesicle trafficking, termed transcytosis, to limit transcellular transport through the vessel wall. Lastly, CNS endothelial cells have low expression levels of leukocyte adhesion molecules (LAMs) to limit entry of immune cells into the brain. Recent studies have also implicated the glycocalyx, a negatively charged dense layer of carbohydrates on the luminal side, in preventing immune cell entry, acting as the first line of defense in protecting the brain (Kolarova et al 2014, Kutuzov et al 2018) (Figure 1).
Figure 1 – The Neurovascular Unit.
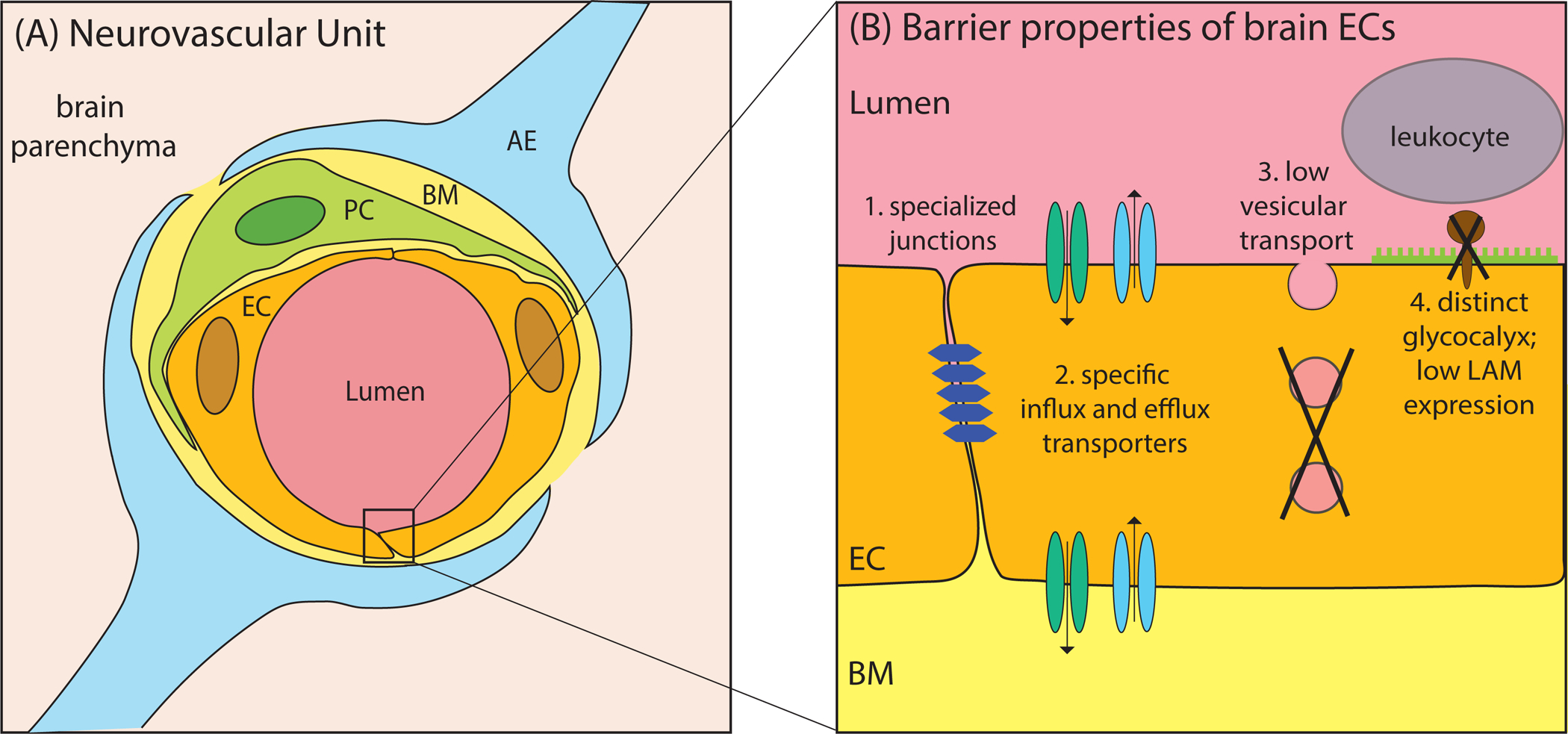
(A) Schematic illustration of a cross-section through the neurovascular unit. Capillary endothelial cells (EC) forming the vessel walls are tightly wrapped by pericytes (PC). These cells are embedded in the basement membrane (BM) and are ensheathed by astrocytic endfeet (AE). (B) Schematic of the CNS endothelium highlighting the four unique properties of the BBB.
However, barrier properties are not intrinsic to endothelial cells. BBB formation and maintenance depend on local perivascular cells and the neuronal environment of the CNS (Armulik et al 2010, Daneman et al 2010, Janzer & Raff 1987, Stewart & Wiley 1981). Blood vessels in the brain are surrounded by pericytes and astrocytes, which serve as the interface between endothelial cells and neurons. Together, these cells form the neurovascular unit (NVU)(Iadecola 2004, Iadecola 2017, Zhao et al 2015) (Figure 1).
In this review, we provide an overview of the cellular and molecular features of the BBB. We first elucidate the specialization of CNS endothelial cells and discuss the cell biology underlying their unique characteristics. We then illustrate the role of other cell types in the NVU that contribute to barrier function. Next, we lay out the development of the BBB, focusing on the signaling pathways and mechanisms that establish the barrier. Finally, we discuss critical questions in the field that require further investigation.
1. Unique barrier properties of CNS endothelial cells
1.1. Specialized tight junctions
Initial tracer injection experiments revealed that brain endothelial cells have specialized TJs that prevent the intercellular passage of tracers from circulation into the brain parenchyma (Reese & Karnovsky 1967). Using ultrastructural electron microscopy analysis, this study revealed that the entry of the injected tracer is sharply halted at TJs between two neighboring endothelial cells, highlighting two aspects: (i) the fact that CNS endothelial cells are the site of the BBB and (ii) the importance of specialized TJs in BBB integrity. Subsequent studies revealed that CNS TJs are specialized to seal contact sites between endothelial cells to prevent paracellular transport of molecules into the CNS, and adherens junctions promote cell-cell adhesion (Brightman & Reese 1969, Lampugnani et al 1992). Earlier studies focusing on junctions in epithelial cells informed much of our understanding about TJs in general and formed the foundation for our understanding of TJs in CNS endothelial cells. Epithelial cells lining peripheral tissues have three kinds of junctions – TJs generally found on the apical side of epithelial cells, desmosomes on the basolateral side and adherens junctions found in between the two (Farquhar & Palade 1963). In contrast, CNS endothelium lacks desmosomes and has a different spatial distribution of junctions than epithelial cells. Specifically, TJs are embedded within adherens junctions and found throughout the cleft between endothelial cells (Schulze & Firth 1993). Further, CNS endothelial cells express unique TJ molecules, indicating differential regulation of CNS barriers.
The four classes of TJ proteins in the CNS are occludin, claudins, tricellulins and junctional adhesion molecules (JAMs). Occludin (Ocln) is enriched in CNS endothelium and was the first transmembrane protein shown to localize exclusively to TJs (Furuse et al 1993). However, Ocln−/− mice develop normal TJs despite having other abnormalities like calcification of the brain (Saitou et al 1998, Saitou et al 2000). A closer examination of Ocln−/− mice revealed mislocalization of tricellulin proteins from tricellular TJs (where three cells meet) to bicellular TJs (between two cells) (Ikenouchi et al 2008). These findings suggest that the role of occludin in TJ formation is complex, and given the structural similarities between tricellulins and occludin (Ikenouchi et al 2005), tricellulins may compensate for occludin deficiency, providing an explanation for the apparently normal TJs in Ocln−/− mice. Consistent with this idea, recent work has demonstrated a role for Lsr, a tricellulin protein enriched at the BBB, in the formation of a functional barrier (Sohet et al 2015).
The surprising finding of functional barriers in Ocln−/− mice fueled the continued search for other TJ proteins, leading to the discovery of claudins (Furuse et al 1998). Of the 27 mammalian claudin genes, claudin-5 (Cldn5) is highly enriched in CNS endothelial cells (Morita et al 1999). Cldn5−/− mice exhibit a size-selective increase in barrier permeability (Nitta et al 2003) implicating claudin-5 as a key component of BBB regulation. These mice display normal vascular development and show no gross abnormalities in peripheral tissues but die shortly after birth, likely due to an impaired BBB (Nitta et al 2003). Finally, JAMs are transmembrane TJ proteins belonging to the immunoglobulin superfamily that regulate leukocyte transmigration across cellular barriers (Martin-Padura et al 1998).
While several TJ proteins are decreased in pathological conditions, our understanding of the basic biology of these proteins in the context of the BBB is still rudimentary. Intracellular trafficking patterns of these proteins and their turn-over rates in the CNS endothelium are unknown. Generating CNS endothelial-specific conditional knockout mouse models for these proteins to determine their contribution in BBB formation as well as its maintenance will provide insight into their specific roles in barrier function.
1.2. Transporters at the BBB
The highly selective nature of the BBB and the high metabolic demand of the brain necessitates other routes of entry for various nutrients to feed and nurture the brain. This is achieved via several influx and efflux transporters expressed on the surface of CNS endothelial cells that drive the active transport of specific solutes and metabolites into the brain. Transport across the BBB generally can be classified into four categories: (i) passive diffusion of lipid soluble molecules such as oxygen, (ii) carrier-mediated transport via solute carrier transporters (SLCs), (iii) selective transport via ATP-binding cassette transporters (ABCs), and (iv) vesicular trafficking by transcytosis (section 1.3). Here we briefly discuss transporters expressed at the BBB. Many of these transporters are ATPases and ATP-binding pumps, indicating a high energy demand to maintain the barrier. Consistent with this, CNS endothelial cells have 4- to 5-fold more mitochondria compared to peripheral endothelial cells (Oldendorf et al 1977).
SLCs are a large class of transporters that facilitate the uptake of nutrients including glucose, amino acids, ions and fatty acids (Campos-Bedolla et al 2014). Owing to the structural complexity and the genetic redundancy of the more than 400 genes in the SLC family, SLCs have been relatively difficult to study. One of the best studied SLCs is SLC2A1 or Glut1 (glucose transporter 1), which transports glucose. The BBB is highly enriched in Glut1 and patients with Glut1 deficiency present with seizures, microcephaly, developmental delays and ataxia (De Vivo et al 1991). While Slc2a1−/− mice die at embryonic stages, mice haploinsufficient for Slc2a1 recapitulate the features seen in patients (Wang et al 2006). Recent work has also shown that lack of Glut1 causes barrier breakdown and prevents clearance of amyloid plaques, thus contributing to the progression of Alzheimer’s disease (Winkler et al 2015).
SLC7A5 or LAT1 (large neutral amino acid transporter 1) is another well-studied transporter in this family. LAT1 transports branched amino acids into the brain, and like Glut1, it is highly enriched in CNS endothelial cells. Recent identification of mutations in LAT1 associated with autism spectrum disorder gave further insight into the role of LAT1. Endothelial specific Slc7A5−/− mice have developmental and neurological abnormalities (Tarlungeanu et al 2016). These mice also display altered amino acid levels in the brain, resulting in reduced mRNA translation with slower protein synthesis. Besides amino acid uptake transporters, the BBB also expresses SLCs that drive the net efflux of amino acids like glutamate and aspartate, which are excitotoxic at high levels. These SLCs are excitatory acidic amino acid transporters (EAATs) that are expressed on the abluminal side of CNS endothelial cells (O’Kane et al 1999) and export glutamate from the brain parenchyma to maintain low levels of glutamate in the brain extracellular fluid. Thus, the roles of these transporters highlight the importance of regulating amino acid concentrations within the brain for neuronal homeostasis.
In contrast to most nutrient transporters, ABC efflux pumps actively clear out metabolic waste from the CNS and hence are usually considered molecular defense systems that protect the brain from exogenous and toxic substances. Among these transporters, P-glycoprotein (P-gp) has been investigated extensively owing to its role in amyloid β (Aβ) efflux from the brain parenchyma. Initial studies observed that P-gp is on the luminal side of BBB capillaries (Beaulieu et al 1997, Cordon-Cardo et al 1989) and promotes the secretion of Aβ (Lam et al 2001), prompting follow-up studies to investigate P-gp function in vivo. Mice lacking P-gp display increased sensitivity to several drugs (Schinkel et al 1996), highlighting the role of P-gp in preventing drugs from entering the brain. Subsequent work has also demonstrated the increased deposition of Aβ in P-gp deficient mice (Cirrito et al 2005). Multiple studies have reported decreased levels of P-gp and other ABC efflux pumps in post-mortem brain tissue samples of Alzheimer’s disease patients (Jeynes & Provias 2011, Wijesuriya et al 2010), implicating the decreased levels of those pumps in promoting disease progression.
Overall, our understanding of the transporters at the BBB is still in early stages. SLCs are thought to make up about 10% of the human genome (Hediger et al 2013), yet the precise roles of most of these proteins is not known. In the case of efflux pumps, it is unclear what triggers their initial loss contributing to disease progression. General questions that remain in this field include: What portion of the nutrients provided by the transporters is consumed by endothelial cells themselves versus other cell types in the NVU? Further, what is the fate of these nutrients once taken up by the endothelial cells and how are they transported to other cell types in the brain? These aspects are completely unknown for most of the transporters at the BBB.
1.3. Low rates of transcytosis
Besides having these designated transporters to deliver specific nutrients to the brain, CNS endothelial cells also have very low rates of vesicle-mediated transcellular trafficking, a process termed transcytosis (Reese & Karnovsky 1967). Recent work demonstrated that these low rates result from an active inhibition of transcytosis in CNS endothelial cells. The mechanism regulating this inhibition at the BBB were largely unknown until recent studies established Mfsd2a as an inhibitor of transcytosis to regulate permeability of the BBB (Andreone et al 2017, Ben-Zvi et al 2014, Chow & Gu 2017). Mfsd2a is a lipid-transporter (Nguyen et al 2014) enriched in CNS endothelial cells and Mfsd2a−/− mice have increased caveolae-mediated vesicular trafficking causing leaky barriers (Andreone et al 2017, Ben-Zvi et al 2014, Chow & Gu 2017). Andreone et al. investigated the specific mechanism by which Mfsd2a inhibits transcytosis: intriguingly, Mfsd2a, by its lipid-transport function, establishes a unique lipid composition in the CNS endothelial cell plasma membrane that inhibits caveolae vesicle formation, thereby suppressing transcytosis (Andreone et al 2017).
Although CNS endothelial cells have limited transcytosis, some macromolecules, such as insulin, low-density lipoprotein (LDL), albumin and iron-bound transferrin, lack specific transporters at the BBB and thus, enter the brain by transcytosis. The passage of these macromolecules demonstrates how the BBB can precisely regulate the transcytotic pathway to allow the transendothelial passage of specific molecules via either receptor-mediated transcytosis or adsorptive transcytosis. In the former, receptors on the luminal cell surface bind to cargo or their ligand. This complex is then endocytosed and trafficked within the endothelial cell. Adsorptive transcytosis, on the other hand, is receptor-independent vesicular trafficking and depends on charged interactions between the ligand and the glycocalyx of endothelial cells. In both cases, after the first step of vesicle formation, the vesicles are trafficked through endosomes and exocytosed into the brain parenchyma. While clathrin- and caveolae-mediated transcytosis are the two major endocytic pathways, other kinds of vesicular structures have also been reported at the ultrastructural level using electron microscopy analyses (Hansen & Nichols 2009). Further, various adaptor proteins play essential roles in each step of these processes. However, it is unclear what determines whether a molecule will be transcytosed and delivered into the brain or sorted into lysosomes for degradation. In the last two decades, researchers have sought to harness receptor-mediated transcytotic pathways to facilitate drug delivery into the brain, particularly the transferrin receptor (TfR) mediated transcytosis pathway (Yu & Watts 2013). Tfr mediated transcytosis has been an attractive system to explore the possibilities of facilitating CNS drug delivery. Antibodies targeting TfR preferentially bind to brain capillaries (Jefferies et al 1984). After binding, the antibody-Tfr complex is shuttled into the brain parenchyma via transcytosis (Friden et al 1991). Bi-specific antibodies targeting TfR and Aβ have been employed to promote the delivery of these antibodies into the CNS and promote the clearance of Aβ plaques (Bien-Ly et al 2014, Niewoehner et al 2014). These studies revealed that both the affinity as well as the valency of antibody binding to the receptor are important in determining the efficient delivery of the antibody into the CNS.
Recent studies have also demonstrated that upregulation of transcytosis is the first step in barrier breakdown. Studies investigating the BBB in models of stroke and brain injury have revealed that few hours post brain insult, transcytotic vesicles in endothelial cells are upregulated causing barrier breakdown (Haley & Lawrence 2017, Knowland et al 2014, Sadeghian et al 2018). Consistent among all of these studies are also the observations that leakage via transcellular pathways precedes breakdown of TJs. While it is possible that these results apply to only specific models such as stroke, it is worth examining what factors cause the early increase in transcytosis at the BBB. Further, identification of these factors will provide a way to manipulate the barrier for CNS drug delivery. With the discovery of BBB regulators like Mfsd2a, future studies focusing on structure-function analysis will provide an entry point for the development of compounds that specifically target proteins to open the barrier for drug delivery. For example, inhibiting Mfsd2a to upregulate transcytosis at the BBB could provide a platform for drug delivery into the brain. Thus, identification of molecules unique to CNS barriers will provide a parts list to effectively harness transcytosis pathways for drug delivery.
1.4. Limited Immune cell trafficking at the BBB
The CNS has low levels of immune cell surveillance when compared to peripheral tissues. Initial studies revealed that extremely low numbers of T-cells enter the brain (Hauser et al 1983, Hickey et al 1991). Further experiments demonstrated that CNS endothelial cells normally express low levels of leukocyte adhesion molecules (LAMs) (Rossler et al 1992), and that LAMs are significantly upregulated in auto-immune diseases such as multiple sclerosis (Cannella et al 1990, Raine et al 1990).
Leukocyte entry into tissues is a highly orchestrated process involving multiple steps and is an early event of pathogenesis in auto-immune diseases. Briefly, leukocytes first survey the vascular wall and signaling molecules such as selectins mediate their initial capture onto the vessel wall. This capture triggers the firm adhesion of the leukocyte to the endothelium via interaction of leukocyte integrins with endothelial membrane proteins such as ICAM-1, ICAM-2 and VCAM-1. Following leukocyte adhesion, diapedesis through the vessel wall occurs, either through paracellular or transcellular route, leading to their entry into the CNS parenchyma. For detailed reviews on immune cell trafficking at the BBB, we direct the readers to other published reviews on this topic (Engelhardt & Wolburg 2004, Ransohoff & Engelhardt 2012).
Leukocyte migration has generally been controversial and it is likely that leukocytes use both paracellular as well as transcellular routes of entry into the CNS (Engelhardt & Wolburg 2004). Recently, Bartholomous et al. used intravital two-photon imaging to follow T-cells in vivo in rat models of experimental autoimmune encephalomyelitis (EAE, a model for multiple sclerosis). Their study revealed a detailed account of the interaction of T-cells with the brain vasculature. T-cells make initial contact with the glycocalyx, crawl along the vascular wall and eventually migrate through the vessel and bind to antigen presenting cells within the brain (Bartholomaus et al 2009). This binding causes the release of cytokines and chemokines which in return recruit more lymphocytes into the CNS. Thus, cytokines and chemokines act in a positive feedback manner to cause leukocyte infiltration into the CNS.
Recent studies have begun to address which cell types in the brain first sense inflammation signals and how these processes impact the BBB. With the initiation of infection, mural cells of blood vessels (pericytes and smooth muscle cells) release the CCL2 chemokine triggering neuroinflammatory responses acting as sensors of the initial infiltration of immune cells (Duan et al 2018). Another recent study exploring the consequence of stress on neuroinflammation and the BBB revealed that in mouse models of social defeat, there is a decrease in claudin-5 expression in some parts of the brain, which in turn promotes the infiltration of cytokines such as interleukin-6 into the CNS parenchyma (Menard et al 2017).
Collectively, these studies highlight the complexity of the neuro-immune axis. We currently know just a fragment of the multi-step, highly coordinated process of how initial leukocyte entry into the CNS causes neuroinflammation and pathology. Further studies are required to delineate the manifold cell types and multiple signaling components that regulate the neuro-immune axis.
2. Barrier regulation by the neurovascular unit
The characteristics described above reflect unique properties of the BBB in endothelial cells. However, early transplantation experiments have shown that these properties are not intrinsic to the CNS endothelium (Stewart & Wiley 1981). Rather, they are induced by the CNS environment. Indeed, while the BBB is localized to CNS endothelial cells, it requires multiple cell types, such as pericytes and astrocytes, working in concert to form a functional barrier. Together, these cells form the neurovascular unit that induces and regulates barrier formation (Figure 1), and which we discuss below.
2.1. Pericytes
Pericytes are mural cells enwrapping capillary blood vessels on their abluminal side. Structurally, pericytes extend processes from their cell body, covering several endothelial cells. In contrast to peripheral tissues, the brain and the retina have the highest pericyte to endothelial cell ratio (Frank et al 1987, Frank et al 1990).They are embedded within the basement membrane of capillary endothelial cells and thus, centrally positioned between endothelial cells, astrocytes and neurons. Although pericytes were first described in the 1870s (Eberth 1871, Rouget 1873), studying them has been difficult due to the lack of specific markers. It is now generally accepted that pericytes are NG2 positive, platelet derived growth factor receptor β (PDGFRβ) positive and alpha smooth muscle actin (α SMA) negative (Armulik et al 2011). Recently, fluorescent Nissl dye NeuroTrace 500/525 has been shown to specifically label pericytes in live mice, allowing in vivo imaging (Damisah et al 2017).
The physical apposition of pericytes and endothelial cells facilitates signaling between them. This signaling plays a role in vessel formation, vessel maturation, pericyte recruitment and pericyte coverage (Gaengel et al 2009). PDGF-B signaling is one such pathway. PDGF-B secreted by endothelial cells recruits pericytes to blood vessels by binding to PDGFRb, a cell surface protein on pericytes (Hellstrom et al 1999, Lindahl et al 1997). While deletion of either gene is perinatally lethal (Leveen et al 1994, Soriano 1994), studies using viable mouse models defective in PDGF-B signaling demonstrated a critical role for pericytes in the formation of the BBB (Armulik et al 2010, Daneman et al 2010). These mice have decreased pericyte coverage and continue to have a dysfunctional barrier through adulthood (Bell et al 2010). These studies also revealed that an intact barrier is highly reliant on the appropriate pericyte coverage of endothelial cells. Consistent with this notion, over-expression of PDGF-B causes an increase in pericyte numbers and results in impaired development of the retinal vasculature, resembling proliferative retinopathy (Edqvist et al 2012, Seo et al 2000).
While the role of pericytes in barrier formation is now well acknowledged, their role in the maintenance of the barrier through adulthood remained unknown until a recent study examined the role of PDGF-B signaling in adult mice (Park et al 2017). Contrary to the role of this pathway in recruiting pericytes during development, endothelial-specific deletion of PDGF-B during adulthood caused neither loss of pericytes from the endothelium, nor the breakdown of the blood-retinal barrier (BRB) or the BBB. This suggests that there likely exist other signaling cues that anchor pericytes onto the endothelium. The authors also induced diphtheria toxin expression in adult pericytes to specifically cause their ablation and found that pericyte depletion had no effect on the adult BRB. However, the authors did note that pericyte-ablated adult retinas were more susceptible to leakage under stress or injury conditions. Pericyte dropout is a hallmark of diabetic retinopathy (Hammes et al 2011) and the general assumption is that it is caused by pericyte loss. However, Park et al suggest that although pericyte loss exacerbates disease progression, it could be a consequence of initial pathogenesis. These findings call for a careful investigation of the role of pericytes in barrier maintenance.
All mouse models used in these studies are mutants causing reduction or ablation of an entire cell type (pericytes) in the NVU. Identifying pericyte genes which do not affect their recruitment to capillaries but play a role in barrier formation will be crucial in dissecting specific mechanisms of how pericytes confer barrier properties to CNS endothelium. One known regulator is the transcription factor Foxf2, which is specifically expressed in CNS pericytes (Reyahi et al 2015). Foxf2−/− mice have decreased PDGFRb signaling, yet they have significantly increased pericyte density, and also fail to form a functional BBB. In stark contrast to the study by Park et al. (Park et al 2017), Foxf2 deletion in adult mice resulted in severe BBB leakage revealing the role of a pericyte-specific gene in BBB maintenance. Additionally, the authors make the intriguing observation that BBB integrity is not affected during the first 3 weeks post Foxf2 deletion but severely compromised after 6 weeks. These findings reveal several compelling ideas: a) the negative correlation between pericyte density and PDGFRb signaling in these mutants indicates that there are other unidentified molecular cues that promote pericyte migration and recruitment b) the mechanism of BBB breakdown in this case is not due to pericyte loss but likely due to a pericyte differentiation defect and c) BBB breakdown weeks after Foxf2 deletion suggests a gradual deterioration of the components regulating barrier integrity, likely due to slow turnover rates of these molecules. In light of these findings, it is important to note that the timing of analysis for a functional barrier after temporal gene/pericyte ablation could impact the conclusion. For example, although Park et al. did not observe a barrier defect at 3 weeks after gene/pericyte ablation, it is possible that the barrier will breakdown several weeks after pericyte ablation.
Pericyte loss is also correlated with BBB breakdown in Alzheimer’s disease (Sagare et al 2013). They play an active role in the clearance of amyloid aggregates via expression of LRP1 (Kanekiyo et al 2012), an Aβ clearance receptor (Deane et al 2004, Shibata et al 2000). CNS pericytes also have a remarkable concentration of acid-phosphatase positive lysosomes (Broadwell & Salcman 1981) which likely indicates robust phagocytic and degradative capabilities. Consistent with this, studies have demonstrated clearance of cellular debris by pericytes (Castejon et al 2005, Mazlo et al 2004). Lastly, several studies have indicated the multipotent differentiation capacity of pericytes (Birbrair et al 2015, Dore-Duffy 2008). However, we still know very little about pericyte differentiation and turnover rates. The last decade in barrier research has uncovered a significant role for pericytes in barrier function. It is becoming increasingly clear that we are only beginning to appreciate the role of pericytes in the CNS and future studies will further evolve our understanding of pericytes in terms of their physiological functions and also their role in pathophysiology.
2.2. Astrocytes
Astrocytes ensheathing capillaries constitute the most abluminal layer of the NVU. They contact the outer basement membrane of the brain vasculature via polarized endfeet that express the water channel aquaporin 4 (Aqp4). Astrocyte endfeet produce extracellular matrix proteins that contribute to the unique basement membrane of brain capillaries (see section 2.3). Given the close contact of astrocyte endfeet with brain capillaries, it is intuitive to assume that they play a role in BBB formation or maintenance. However, it is unlikely that astrocytes contribute to BBB formation in development because they appear postnatally in the brain, well after the barrier has sealed (Yang et al 2013). Rather, it is likely that they are important for barrier maintenance.
An early transplantation study suggests that an astrocytic environment is sufficient to induce barrier properties in newly forming vessels (Janzer & Raff 1987). In this study, astrocytes were transplanted into the eyes of rats. Two weeks after transplantation, newly formed vessels in astrocyte aggregates on the surface of the iris contained a functional barrier. In contrast, transplantation of meningeal cells also led to vascularization of cell aggregates, but these vessels displayed a leaky phenotype (Janzer & Raff 1987). Yet, it is unclear whether the astrocytes themselves induced barrier properties in ECs or whether they recruited additional cells that induced the functional barrier. Along the same line, it is also important to note that blood vessels in the retina develop a functional barrier postnatally (Chow & Gu 2017), when astrocytes are already present there. These growing vessels display a leaky barrier phenotype in the presence of astrocytes (Chow & Gu 2017), indicating that astrocytic signals alone are not sufficient to induce barrier properties.
To date, most evidence for astrocyte contribution to the BBB has been generated in vitro. For example, it has been shown that an astrocyte-derived factor induces endothelial polarization and production of the distinct glycocalyx of CNS endothelium (Yamagata et al 1997). Furthermore, an in vitro study has demonstrated that Sonic hedgehog (Shh) signaling stimulates expression of TJ proteins such as claudin-5 and occludin (Alvarez et al 2011). At the same time, Shh signaling reduces the expression of LAMs and chemokines in endothelial cells which results in the immune quiescent phenotype of brain endothelial cells (Alvarez et al 2011). In vivo, Shh plays a role during embryonic BBB development. In embryonic stages, Shh is highly expressed by neuroprogenitors in the brain, and Shh depletion leads to reduced expression of claudin-5 at embryonic day (E)13.5 (Alvarez et al 2011). Postnatally, endothelial-specific deletion of the Shh receptor Smoothened reduces barrier integrity but does not affect vessel patterning in the brain (Alvarez 2011). While the authors of this study suggest that Shh in the postnatal brain is derived from astrocytes, recent transcriptional profiling studies of different cell types in the CNS show very low mRNA expression of Shh in astrocytes (Zeisel et al 2018, Zhang et al 2014), challenging the proposed mechanism.
Investigating the contribution of astrocytes to barrier maintenance, Yao et al. have shown that laminins specifically secreted by astrocytes maintain BBB integrity (Yao et al 2014). Deletion of astrocytic laminins leads to decreased Aqp4 expression in astrocytes and reduced TJ protein levels in endothelial cells. Mechanistically, the authors suggest that astrocytic laminins inhibit pericyte differentiation via integrin α2 activation which results in a leaky barrier(Yao et al 2014).
Another recent in vivo study investigated brain endothelial cell communication with glial cells during BBB development (Segarra et al 2018). This study shows that neuronal Reelin activates Dab1 in endothelial cells, which causes endothelial cells to deposit laminin α4 to the extracellular matrix. Dab1 conditional knockout mice show barrier leakage and reduced astrocyte endfeet coverage on brain capillaries. The authors conclude that Dab1 activated endothelial cells communicate to glial cells via laminin α4 in order to seal the barrier (Segarra et al 2018). Overall, this model sheds light on the interaction of endothelial and glial cells in BBB development. However, further investigation is needed to rule out that endothelial Dab1 deletion causes cell-autonomous impairment of the BBB.
In summary, there are many indications for a contribution of astrocytes to BBB maintenance, besides the intriguing close proximity of astrocytic endfeet to the vasculature. However, the exact signaling pathways and mechanisms still require a more detailed examination.
2.3. Basement Membrane of the Neurovascular Unit
The basement membrane (BM) is the extracellular matrix (ECM) that provides structural support for the cells of the NVU and is a hub for intercellular communication and signaling pathways between these cells. Structural proteins that make up the ECM of the BM are type IV collagens, fibronectin, laminins and other glycoproteins. Collagen IV and fibronectin are secreted by all three cell types of the NVU and deletion of either of these genes is embryonic lethal (George et al 1993, Poschl et al 2004). Laminins, on the other hand, have several isoforms and the right balance of these isoforms have shown to be important in vessel formation (Thyboll et al 2002), barrier formation (Menezes et al 2014) and regulating leukocyte infiltration into the brain(Wu et al 2009).
The BM has two main families of ECM receptors – dystroglycans and integrins. Together, these receptors enable cell-matrix interactions and link the ECM to the cytoskeleton. The ligands within the ECM bind to these matrix receptors to trigger signaling cascades regulating cell migration, proliferation and cell survival, which in turn regulate barrier function. Integrins are ligand-binding membrane proteins that are heterodimers constituted by α and β subunits. The αv subunit is one of the best studied integrin subunits in the field owing to its role in angiogenensis. Itgav−/− mice die of cerebral hemorrhages (Bader et al 1998) and interestingly, conditional knockouts revealed that deletion of Itgav from neurons and glia, but not from endothelial cells or pericytes recapitulated the phenotype (McCarty et al 2005, McCarty et al 2002). Further, studies have also shown that αv integrins regulate the brain vasculature through activation of TGF-β signaling (reviewed in (Munger & Sheppard 2011, Sheppard 2004). There is also growing evidence for integrin-mediated and ECM-driven regulation of Wnt signaling (Astudillo & Larrain 2014), which in turn modulates barrier properties. For example, deletion of integrin β1 in endothelial cells impairs VE-cadherin signaling which causes disorganization of TJs resulting in a leaky barrier (Yamamoto et al 2015).
Thus, the signaling pathways and cellular interactions occurring within the BM of the NVU are coordinated to regulate barrier function. Not surprisingly, breakdown of the BM is seen in several CNS disorders and diseases (Thomsen et al 2017). This degradation is thought to be due to matrix metalloproteinases (MMPs), which are upregulated in disease conditions. Specifically, increased MMP-2 and MMP-9 have consistently been associated with stroke, ischemia, Alzheimer’s and Parkinson’s disease (Rempe et al 2016). Although targeting MMPs in disease to impede protein degradation is an attractive idea from a therapeutic standpoint, studying MMPs has been difficult owing to lack of suitable reagents to examine their localization or measure their proteolytic activities in vivo. New tools and further research are needed to understand the functional roles of these molecules in BBB regulation.
3. Development of the blood brain barrier
3.1. Barriergenesis
When brain endothelial cells first enter the CNS, they do not inherently display barrier properties. Instead, BBB formation is a gradual process that occurs during embryonic development after exposure to the neural environment. It begins with vascularization of the brain around E11.5. In this process, blood vessels from the perineural vascular plexus invade the brain in a stereotypical manner by sprouting angiogenesis (Daneman et al 2010, Risau 1997). Vascularization is initiated by the release of growth factors from the neural tube. These factors attract vessels to invade via binding to specific receptors on the endothelial cell surface. Shortly after vascular sprouts invade the neural tube, they begin to connect and form a vessel plexus in the brain (Engelhardt 2003).
Numerous signaling pathways such as VEGF, Notch, and ephrin signaling have been shown to play crucial roles in CNS angiogenesis (Engelhardt & Liebner 2014). However, these pathways are not specific to CNS vascularization. Perturbations in these signaling pathways lead to severe vascular defects both in the CNS and in peripheral organs including the most severe form of a barrier breakdown: hemorrhage. Consequently, these phenotypes are likely due to angiogenic defects throughout all tissues as opposed to CNS-specific angiogenic mechanisms.
An elegant model to study barriergenesis specifically in the CNS is the mouse retina. The retina is vascularized radially from the central nerve from postnatal day (P) 0 to P10 (Chow & Gu 2017, Pitulescu et al 2010, Stahl et al 2010). Chow et al. have recently shown by ultrastructural and molecular investigation that invading vessel sprouts already have functional TJs, yet have active transcytosis and therefore do not have a functional barrier. (Chow & Gu 2017). During vascular outgrowth, the immature distal vessels in the retina are leaky while the mature proximal vessels have a sealed barrier. Interestingly, expression of Mfsd2a, a molecule which suppresses transcytosis, correlates with functional barrier formation (Chow & Gu 2017). This model proposes a gradual sealing of the barrier with manifestation of TJs first and inhibition of vesicular transport later (Figure 2).
Figure 2 – Gradual Barrier Formation in the CNS.
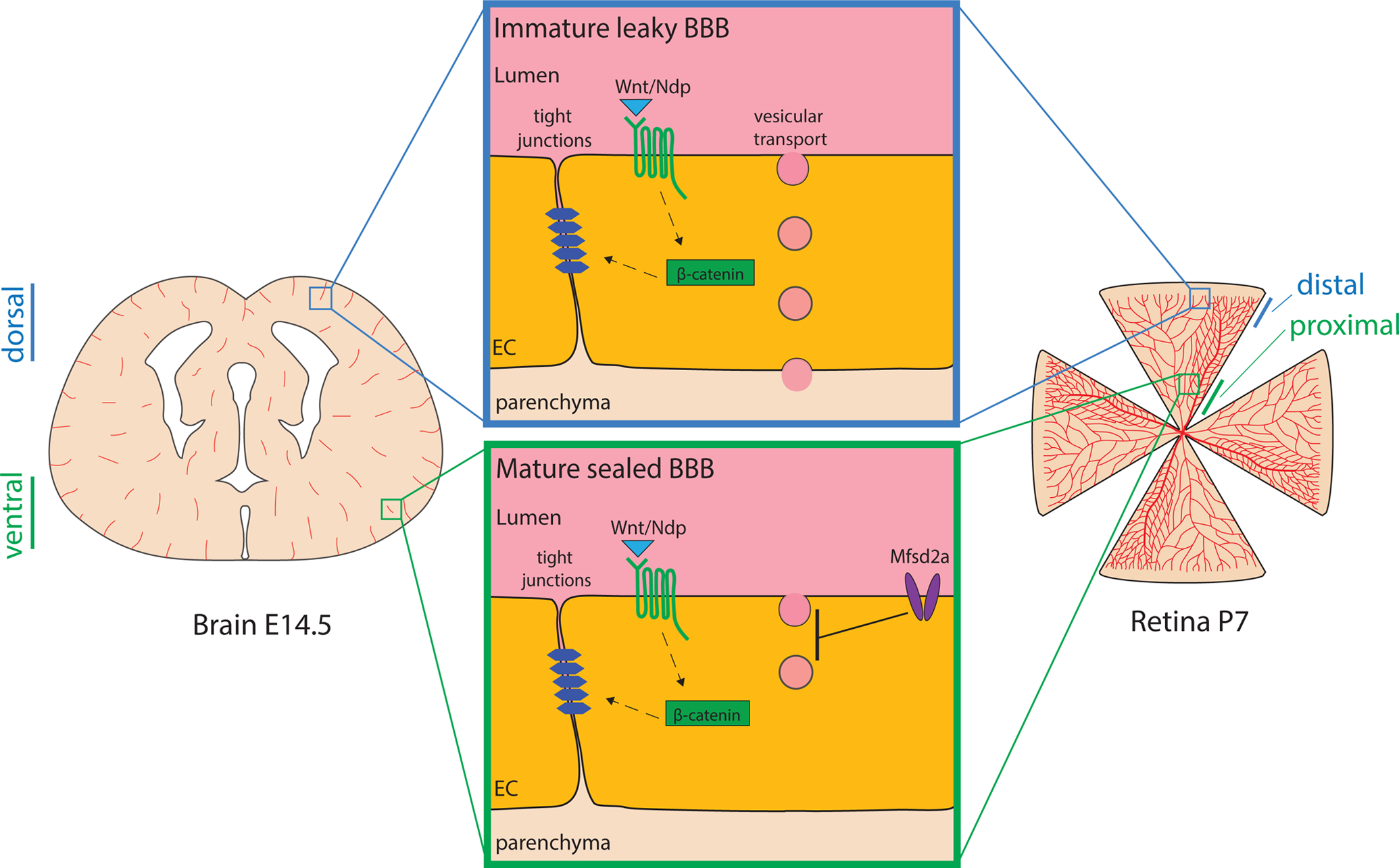
Depiction of a coronal brain section at E14.5 and a flat-mounted retina at P7 highlighting the spatial-temporal gradient in functional barrier formation in the CNS. The blue inset illustrates features of immature, leaky CNS barriers found in dorsal brain regions and the distal retina. Green inset shows features of the mature sealed blood-CNS barriers of ventral embryonic brain and the proximal vessels of the developing retina.
While the brain is much more complex than the retina and the chronology of events that seal the barrier has been debated, it appears that barriergenesis follows a similar gradual process. Initial sprouts invading into the neural tube from the perineural vascular plexus (PNVP) display TJs (Risau 1991, Risau et al 1986) and already show expression of genes associated with barrier function such as Slc2a1, Cldn5 and Abcb1a (Dermietzel et al 1992, Morita et al 1999, Qin & Sato 1995). At the same time, murine brain vessels show transcytotic vesicles before E15.5 and some EM studies have reported fenestrae in the invading vessel sprouts (Stewart & Hayakawa 1987). Moreover, capillaries up to E15.5 express Plvap, a marker associated with transcytosis and endothelial fenestrae (Daneman et al 2010, Hallmann et al 1995, Stan et al 2012). Tracer injections have shown that brain vessels show leakage of tracers only until E15.5 (Ben-Zvi et al 2014, Risau et al 1986). Interestingly, the functional BBB forms in a ventral to dorsal fashion (Ben-Zvi et al 2014, Daneman et al 2010) indicating a spatio-temporal maturation of the barrier (Figure 2).
To summarize, when blood vessels first enter the CNS they immediately acquire functional TJs. This process is mainly controlled by the canonical Wnt signaling pathway, discussed below. In contrast, transcellular transport through endothelial cells via transcytosis is active during earlier stages of development and is then gradually suppressed. Thus, the formation of a functional barrier coincides with the suppression of transcytosis (Figure 2).
Interestingly, while the brain is growing and angiogenesis is still occurring, newly forming vessels immediately display the barrier phenotype. This phenomenon is best demonstrated in the retina. When the intermediate plexus is vascularized between P12 and P17, vessel sprouts invading from the deeper plexus into the intermediate plexus are not leaky although they are still undergoing angiogenesis (Chow & Gu 2017). This means newly formed endothelial cells after initial barrier formation inherit barrier properties from their ‘mother’ cell and they depend on the local microenvironment to maintain these barrier properties (Janzer & Raff 1987, Stewart & Wiley 1981).
3.2. Wnt signaling
Canonical Wnt signaling is, as currently known, the major pathway specifically regulating brain angiogenesis and barriergenesis, but not peripheral angiogenesis (Daneman et al 2009, Liebner et al 2008, Moro et al 2012, Stenman et al 2008). During development, neural progenitor cells secrete Wnt ligands which act on Frizzled receptors on endothelial cells, activating canonical Wnt signaling (Daneman et al 2009, Stenman et al 2008). In this pathway, Wnt ligand binding to their receptors induces β catenin stabilization and downstream gene induction via TCF/LEF transcription factors. It is compelling that the developing brain at E11.5 shows a peculiar expression pattern of Wnt ligands with Wnt7a/b being expressed in the forebrain and ventral neural tube and Wnt1/3/3a/4 being expressed dorsally including the hindbrain (Daneman et al 2009, Stenman et al 2008).
Endothelial-specific knockout mice for canonical Wnt signaling mediator β-catenin (Ctnnb1) show embryonic lethality associated with severe hemorrhaging specifically in the CNS (Daneman et al 2009). Early postnatal EC-specific deletion of Ctnnb1 results in disruption of barrier integrity which correlates with reduced claudin-5 expression (Liebner et al 2008, Zhou et al 2014). Multiple studies have illustrated that the expression of marker genes for a functioning barrier such as Slc2a1, Cldn5 and absence of Plvap are directly regulated by Wnt signaling (Daneman et al 2009, Liebner et al 2008, Wang et al 2018, Zhou et al 2014).
To date, at least two ligand-receptor signaling complexes have been identified that mediate β-catenin stabilization and thereby CNS vascularization and barriergenesis. These pathways show partially redundant functions in activating canonical Wnt signaling. One of the cascades upstream of β-catenin is activated by the ligands Wnt7a and b. These are expressed by astroglia, oligodendrocytes and neurons (Daneman et al 2009, Zhang et al 2014). They act on Frizzled receptors in complex with their co-receptors Lrp5/6, GPR124 and Reck on endothelial cells (Cho et al 2017a, Eubelen et al 2018, Zhou et al 2014). Knockout mice for components of this pathway result in CNS angiogenic defects and leaky CNS barriers. However, these occur in distinct regions of the CNS, namely the cerebral cortex and dorsal spinal cord (Cho et al 2017a, Daneman et al 2009, Stenman et al 2008, Wang et al 2012, Zhou et al 2014). The role of GPR124, the coactivator in this pathway, has been well studied (Anderson et al 2011, Chang et al 2017, Cullen et al 2011, Kuhnert et al 2010, Vanhollebeke et al 2015, Zhou et al 2014). Interestingly, while GPR124 plays a major role in BBB development with knockout mice displaying severe angiogenic defects and tracer leakage in the forebrain, endothelial specific deletion in adult mice does not result in barrier breakdown (Chang et al 2017). This is different from postnatal deletion of Ctnnb1 or Fzd4 (Frizzled 4) both of which result in a compromised BBB (Tran et al 2016, Wang et al 2012). However, in mouse models of ischemic stroke or glioblastoma GPR124 has a protective effect on BBB integrity which is mediated by canonical Wnt signaling (Chang et al 2017). In conclusion, in this pathway Wnt7a/b signals through Frizzled receptors in complex with Lrp5/6, GPR124 and Reck to induce canonical Wnt-signaling.
The second described Wnt pathway in barriergenesis is activated by Norrin (Ndp), a ligand belonging to the TGFb family. Norrin is expressed by astroglia and to some extent by oligodendrocytes in the CNS (Zhang et al 2014). Interestingly, Norrin binds to the Wnt receptor Frizzled4, which is the only member of the Frizzled receptor family that can bind Norrin (Smallwood et al 2007). At the same time, Frizzled4 is still responsive to Wnt ligands and it is presently not known how the two structurally different ligands activate the same downstream response of Frizzled4. Binding of Norrin to Frizzled4, which acts with its coreceptors Lrp5 and Tspan12, leads to downstream β-catenin stabilization (Junge et al 2009, Wang et al 2012, Ye et al 2010). Knockout mice for Ndp, Fzd4 or Tspan12 show severe angiogenic phenotypes and have a dysfunctional barrier in the retina, cerebellum and ventral spinal cord (Junge et al 2009, Wang et al 2012). It has been shown in retina that the Norrin pathway and barrier maturation are modulated by the Wnt inhibitor Apcdd1 (Mazzoni et al 2017). Depleting components of both the Norrin and the Wnt7a/b pathways during development results in severe leakage throughout the brain, including regions such as the brain stem that are not affected by perturbation of either pathway alone (Cho et al 2017a, Wang et al 2018, Zhou et al 2014) (not published yet).
The two different pathways of canonical Wnt activation are regionally active both in mutually exclusive and redundant patterns. The Wnt7a/b pathway is exclusively active in the forebrain and ventral spinal cord, while the norrin pathway is exclusive to the cerebellum, retina and dorsal spinal cord. In the brain stem both pathways are active (Cho et al 2017a, Wang et al 2018, Zhou et al 2014). This local regulation of Wnt signaling might provide avenues to manipulate CNS barriers in a spatially selective manner. Future studies are required to explore this idea further.
Canonical Wnt signaling is the best characterized pathway in BBB formation. The expression of certain transporters and TJ proteins such as Glut1 and claudin-5 also correlates with canonical Wnt activation (Figure 2). However, the mechanisms of how canonical Wnt activation leads to formation of a functional barrier are not well understood. For example, it is not known if Wnt signaling also plays a role in the inhibition of transcytosis, although mutants for Wnt components show increased Plvap expression. Plvap expression commonly correlates with permeable vasculature and it has been associated with transcytosis; however, overexpression of Plvap does not increase brain vessel permeability (Stan et al 2012).
While we are beginning to understand the contribution of canonical Wnt signaling to BBB development, there are other pathways involved that are locally integrated into the formation of a functional barrier. One task for future research will be to understand how these pathways collaborate throughout the CNS and locally in defined brain regions.
Outlook
The advent of new single-cell transcriptomic analyses has allowed researchers to begin to identify the various cell types in the brain and their transcriptional signatures (Saunders et al 2018, Vanlandewijck et al 2018, Zeisel et al 2018). Our next challenges will be to utilize these datasets to identify the specific molecules and signaling pathways that mediate BBB formation and function across cell types. Additionally, identifying endothelial subtype-specific markers might help to understand the spatial distribution of cell subtypes and dissect regional differences in barrier induction. The development of genetic tools to target pericytes, astrocytes or subtypes of endothelial cells will be critical to advance our understanding of barrier function.
As illustrated in this review, the BBB is a highly dynamic structure regulated by multiple cell types. Latest research has found that BBB properties in fruit flies underlie the circadian rhythm (Zhang et al 2018). While the structure of the barrier is very different in flies compared to mammals (O’Brown et al 2018), it will be interesting to see whether the capacity of barrier modulation by circadian signals is conserved across species. As we are beginning to dissect specific roles of different cell types during barrier development, their differential contributions to barrier maintenance through adulthood is unclear. Further, the physiology of the barrier and roles of pericytes and astrocytes in disease progression is unknown. Although barrier breakdown has been observed in several pathologies, it is unclear if this is the initial step of pathogenesis or if this is a consequence of disease progression.
The discovery of novel BBB regulators and a deeper understanding of mechanisms mediating barrier function will likely provide new targets to pharmacologically modulate the barrier in disease conditions. After this initial identification, reliable and robust in vitro systems are needed to develop high-throughput screening of molecules that can change barrier properties. In recent years, a few models have been successful in capturing most, if not all properties of the NVU. These include inducible pluripotent stem cells (Lippmann et al 2012), self-assembling multicellular BBB spheroids (Cho et al 2017b) and microfluidic organ chips (Maoz et al 2018). It remains to be seen if we can use these model systems to screen molecules for drug delivery into the brain and also study basic cell biology of the NVU which can then be investigated in vivo.
Finally, the heterogeneity of the BBB across various brain regions is an unexplored area of research that needs further investigation. The phenotypes of mouse mutants for Wnt- signaling components described above demonstrate that there is molecular heterogeneity in BBB formation across different brain regions. It is presently unknown if the differential development translates into distinct barrier properties during adulthood. Further, there are regions in the brain with permeable vessels serving nearby neurons to sense components in the bloodstream and secrete compounds into circulation. Understanding the molecular and cellular basis of a functional barrier in different parts of the brain will not only identify new players in BBB function but will also uncover unique themes of barrier regulation. Together, these have a huge potential to provide novel ways to manipulate the barrier globally as well as in a region-specific manner. Over the next few decades, we anticipate all of these research areas to reveal new aspects of barrier regulation that will provide a detailed understanding of the BBB in both health and disease.
Acknowledgements
We thank members of the Gu laboratory for helpful comments on the manuscript. We apologize to our colleagues whose research we could not cite or discuss owing to space limitations.
References
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, et al. 2011. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334: 1727–31 [DOI] [PubMed] [Google Scholar]
- Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, et al. 2011. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U S A 108: 2807–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, et al. 2017. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 94: 581–94 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, et al. 2010. Pericytes regulate the blood-brain barrier. Nature 468: 557–61 [DOI] [PubMed] [Google Scholar]
- Armulik A, Mae M, Betsholtz C. 2011. Pericytes and the blood-brain barrier: recent advances and implications for the delivery of CNS therapy. Ther Deliv 2: 419–22 [DOI] [PubMed] [Google Scholar]
- Astudillo P, Larrain J. 2014. Wnt signaling and cell-matrix adhesion. Curr Mol Med 14: 209–20 [DOI] [PubMed] [Google Scholar]
- Bader BL, Rayburn H, Crowley D, Hynes RO. 1998. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95: 507–19 [DOI] [PubMed] [Google Scholar]
- Banks WA. 2016. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 15: 275–92 [DOI] [PubMed] [Google Scholar]
- Bartholomaus I, Kawakami N, Odoardi F, Schlager C, Miljkovic D, et al. 2009. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 462: 94–8 [DOI] [PubMed] [Google Scholar]
- Beaulieu E, Demeule M, Ghitescu L, Beliveau R. 1997. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem J 326 (Pt 2): 539–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, et al. 2010. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68: 409–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, et al. 2014. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509: 507–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, et al. 2014. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 211: 233–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. 2015. Pericytes at the intersection between tissue regeneration and pathology. Clin Sci (Lond) 128: 81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightman MW, Reese TS. 1969. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol 40: 648–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwell RD, Salcman M. 1981. Expanding the definition of the blood-brain barrier to protein. Proc Natl Acad Sci U S A 78: 7820–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. 2014. Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res 45: 610–38 [DOI] [PubMed] [Google Scholar]
- Cannella B, Cross AH, Raine CS. 1990. Upregulation and coexpression of adhesion molecules correlate with relapsing autoimmune demyelination in the central nervous system. J Exp Med 172: 1521–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castejon OJ, Castellano A, Arismendi GJ, Medina Z. 2005. The inflammatory reaction in human traumatic oedematous cerebral cortex. J Submicrosc Cytol Pathol 37: 43–52 [PubMed] [Google Scholar]
- Chang J, Mancuso MR, Maier C, Liang X, Yuki K, et al. 2017. Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat Med 23: 450–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C, Smallwood PM, Nathans J. 2017a. Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 95: 1221–25 [DOI] [PubMed] [Google Scholar]
- Cho CF, Wolfe JM, Fadzen CM, Calligaris D, Hornburg K, et al. 2017b. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat Commun 8: 15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BW, Gu C. 2017. Gradual Suppression of Transcytosis Governs Functional Blood-Retinal Barrier Formation. Neuron 93: 1325–33 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, et al. 2005. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 115: 3285–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, et al. 1989. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A 86: 695–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, et al. 2011. GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci U S A 108: 5759–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damisah EC, Hill RA, Tong L, Murray KN, Grutzendler J. 2017. A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat Neurosci 20: 1023–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. 2009. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 106: 641–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. 2010. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468: 562–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. 1991. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325: 703–9 [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, et al. 2004. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43: 333–44 [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Krause D, Kremer M, Wang C, Stevenson B. 1992. Pattern of glucose transporter (Glut 1) expression in embryonic brains is related to maturation of blood-brain barrier tightness. Dev Dyn 193: 152–63 [DOI] [PubMed] [Google Scholar]
- Dore-Duffy P 2008. Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des 14: 1581–93 [DOI] [PubMed] [Google Scholar]
- Duan L, Zhang XD, Miao WY, Sun YJ, Xiong G, et al. 2018. PDGFRbeta Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 100: 183–200 e8 [DOI] [PubMed] [Google Scholar]
- Eberth CJ. 1871. Handbuch der Lehre von der Geweben des Menschen und der Tiere: Leipzig: Engelmann [Google Scholar]
- Edqvist PH, Niklasson M, Vidal-Sanz M, Hallbook F, Forsberg-Nilsson K. 2012. Platelet-derived growth factor over-expression in retinal progenitors results in abnormal retinal vessel formation. PLoS One 7: e42488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B 2003. Development of the blood-brain barrier. Cell Tissue Res 314: 119–29 [DOI] [PubMed] [Google Scholar]
- Engelhardt B, Liebner S. 2014. Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res 355: 687–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Wolburg H. 2004. Mini-review: Transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol 34: 2955–63 [DOI] [PubMed] [Google Scholar]
- Eubelen M, Bostaille N, Cabochette P, Gauquier A, Tebabi P, et al. 2018. A molecular mechanism for Wnt ligand-specific signaling. Science 361 [DOI] [PubMed] [Google Scholar]
- Farquhar MG, Palade GE. 1963. Junctional complexes in various epithelia. J Cell Biol 17: 375–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RN, Dutta S, Mancini MA. 1987. Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Invest Ophthalmol Vis Sci 28: 1086–91 [PubMed] [Google Scholar]
- Frank RN, Turczyn TJ, Das A. 1990. Pericyte coverage of retinal and cerebral capillaries. Invest Ophthalmol Vis Sci 31: 999–1007 [PubMed] [Google Scholar]
- Friden PM, Walus LR, Musso GF, Taylor MA, Malfroy B, Starzyk RM. 1991. Anti-transferrin receptor antibody and antibody-drug conjugates cross the blood-brain barrier. Proc Natl Acad Sci U S A 88: 4771–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. 1998. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141: 1539–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, et al. 1993. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 123: 1777–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Genove G, Armulik A, Betsholtz C. 2009. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29: 630–8 [DOI] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. 1993. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119: 1079–91 [DOI] [PubMed] [Google Scholar]
- Haley MJ, Lawrence CB. 2017. The blood-brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab 37: 456–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. 1995. Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn 202: 325–32 [DOI] [PubMed] [Google Scholar]
- Hammes HP, Feng Y, Pfister F, Brownlee M. 2011. Diabetic retinopathy: targeting vasoregression. Diabetes 60: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Nichols BJ. 2009. Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci 122: 1713–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles FH, Hoban CJ, Reinherz EL, et al. 1983. Immunohistochemical staining of human brain with monoclonal antibodies that identify lymphocytes, monocytes, and the Ia antigen. J Neuroimmunol 5: 197–205 [DOI] [PubMed] [Google Scholar]
- Hediger MA, Clemencon B, Burrier RE, Bruford EA. 2013. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol Aspects Med 34: 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. 1999. Role of PDGF-B and PDGFRbeta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126: 3047–55 [DOI] [PubMed] [Google Scholar]
- Hickey WF, Hsu BL, Kimura H. 1991. T-lymphocyte entry into the central nervous system. J Neurosci Res 28: 254–60 [DOI] [PubMed] [Google Scholar]
- Iadecola C 2004. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5: 347–60 [DOI] [PubMed] [Google Scholar]
- Iadecola C 2017. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96: 17–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, Tsukita S. 2005. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171: 939–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenouchi J, Sasaki H, Tsukita S, Furuse M, Tsukita S. 2008. Loss of occludin affects tricellular localization of tricellulin. Mol Biol Cell 19: 4687–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzer RC, Raff MC. 1987. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 325: 253–7 [DOI] [PubMed] [Google Scholar]
- Jefferies WA, Brandon MR, Hunt SV, Williams AF, Gatter KC, Mason DY. 1984. Transferrin receptor on endothelium of brain capillaries. Nature 312: 162–3 [DOI] [PubMed] [Google Scholar]
- Jeynes B, Provias J. 2011. An investigation into the role of P-glycoprotein in Alzheimer’s disease lesion pathogenesis. Neurosci Lett 487: 389–93 [DOI] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, et al. 2009. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 139: 299–311 [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Liu CC, Shinohara M, Li J, Bu G. 2012. LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J Neurosci 32: 16458–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, et al. 2014. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 82: 603–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarova H, Ambruzova B, Svihalkova Sindlerova L, Klinke A, Kubala L. 2014. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediators Inflamm 2014: 694312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, et al. 2010. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330: 985–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutuzov N, Flyvbjerg H, Lauritzen M. 2018. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood-brain barrier. Proc Natl Acad Sci U S A 115: E9429–E38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, et al. 2001. beta-Amyloid efflux mediated by p-glycoprotein. J Neurochem 76: 1121–8 [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, et al. 1992. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol 118: 1511–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. 1994. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev 8: 1875–87 [DOI] [PubMed] [Google Scholar]
- Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, et al. 2008. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183: 409–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. 1997. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277: 242–5 [DOI] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, et al. 2012. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol 30: 783–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maoz BM, Herland A, FitzGerald EA, Grevesse T, Vidoudez C, et al. 2018. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat Biotechnol 36: 865–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, et al. 1998. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142: 117–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazlo M, Gasz B, Szigeti A, Zsombok A, Gallyas F. 2004. Debris of “dark” (compacted) neurones are removed from an otherwise undamaged environment mainly by astrocytes via blood vessels. J Neurocytol 33: 557–67 [DOI] [PubMed] [Google Scholar]
- Mazzoni J, Smith JR, Shahriar S, Cutforth T, Ceja B, Agalliu D. 2017. The Wnt Inhibitor Apcdd1 Coordinates Vascular Remodeling and Barrier Maturation of Retinal Blood Vessels. Neuron 96: 1055–69 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, et al. 2005. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132: 165–76 [DOI] [PubMed] [Google Scholar]
- McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, et al. 2002. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol 22: 7667–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, et al. 2017. Social stress induces neurovascular pathology promoting depression. Nat Neurosci 20: 1752–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. 2014. The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood-brain barrier. J Neurosci 34: 15260–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Sasaki H, Furuse M, Tsukita S. 1999. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147: 185–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro E, Ozhan-Kizil G, Mongera A, Beis D, Wierzbicki C, et al. 2012. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev Biol 366: 327–40 [DOI] [PubMed] [Google Scholar]
- Munger JS, Sheppard D. 2011. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 3: a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, et al. 2014. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509: 503–6 [DOI] [PubMed] [Google Scholar]
- Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, et al. 2014. Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81: 49–60 [DOI] [PubMed] [Google Scholar]
- Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, et al. 2003. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol 161: 653–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brown NM, Pfau SJ, Gu C. 2018. Bridging barriers: a comparative look at the blood-brain barrier across organisms. Genes Dev 32: 466–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Kane RL, Martinez-Lopez I, DeJoseph MR, Vina JR, Hawkins RA. 1999. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J Biol Chem 274: 31891–5 [DOI] [PubMed] [Google Scholar]
- Oldendorf WH, Cornford ME, Brown WJ. 1977. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol 1: 409–17 [DOI] [PubMed] [Google Scholar]
- Pardridge WM. 2012. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32: 1959–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DY, Lee J, Kim J, Kim K, Hong S, et al. 2017. Plastic roles of pericytes in the blood-retinal barrier. Nat Commun 8: 15296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Schmidt I, Benedito R, Adams RH. 2010. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat Protoc 5: 1518–34 [DOI] [PubMed] [Google Scholar]
- Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. 2004. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131: 1619–28 [DOI] [PubMed] [Google Scholar]
- Qin Y, Sato TN. 1995. Mouse multidrug resistance 1a/3 gene is the earliest known endothelial cell differentiation marker during blood-brain barrier development. Dev Dyn 202: 172–80 [DOI] [PubMed] [Google Scholar]
- Raine CS, Lee SC, Scheinberg LC, Duijvestin AM, Cross AH. 1990. Adhesion molecules on endothelial cells in the central nervous system: an emerging area in the neuroimmunology of multiple sclerosis. Clin Immunol Immunopathol 57: 173–87 [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Engelhardt B. 2012. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12: 623–35 [DOI] [PubMed] [Google Scholar]
- Reese TS, Karnovsky MJ. 1967. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34: 207–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe RG, Hartz AMS, Bauer B. 2016. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab 36: 1481–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyahi A, Nik AM, Ghiami M, Gritli-Linde A, Ponten F, et al. 2015. Foxf2 Is Required for Brain Pericyte Differentiation and Development and Maintenance of the Blood-Brain Barrier. Dev Cell 34: 19–32 [DOI] [PubMed] [Google Scholar]
- Risau W 1991. Induction of blood-brain barrier endothelial cell differentiation. Ann N Y Acad Sci 633: 405–19 [DOI] [PubMed] [Google Scholar]
- Risau W 1997. Mechanisms of angiogenesis. Nature 386: 671–4 [DOI] [PubMed] [Google Scholar]
- Risau W, Hallmann R, Albrecht U. 1986. Differentiation-dependent expression of proteins in brain endothelium during development of the blood-brain barrier. Dev Biol 117: 537–45 [DOI] [PubMed] [Google Scholar]
- Rossler K, Neuchrist C, Kitz K, Scheiner O, Kraft D, Lassmann H. 1992. Expression of leucocyte adhesion molecules at the human blood-brain barrier (BBB). J Neurosci Res 31: 365–74 [DOI] [PubMed] [Google Scholar]
- Rouget C 1873. Mémoire sur le développement, la structure et les proprietés physiologiques des capillaires sanguins et lymphatiques. Arch Physiol Norm Path 5: 603–63 [Google Scholar]
- Sadeghian H, Lacoste B, Qin T, Toussay X, Rosa R, et al. 2018. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann Neurol 84: 409–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, et al. 2013. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4: 2932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, et al. 1998. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol 141: 397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, et al. 2000. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, et al. 2018. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174: 1015–30 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, Mol CA, van Deemter L. 1996. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 97: 2517–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze C, Firth JA. 1993. Immunohistochemical localization of adherens junction components in blood-brain barrier microvessels of the rat. J Cell Sci 104 (Pt 3): 773–82 [DOI] [PubMed] [Google Scholar]
- Segarra M, Aburto MR, Cop F, Llao-Cid C, Hartl R, et al. 2018. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science 361 [DOI] [PubMed] [Google Scholar]
- Seo MS, Okamoto N, Vinores MA, Vinores SA, Hackett SF, et al. 2000. Photoreceptor-specific expression of platelet-derived growth factor-B results in traction retinal detachment. Am J Pathol 157: 995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D 2004. Roles of alphav integrins in vascular biology and pulmonary pathology. Curr Opin Cell Biol 16: 552–7 [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, et al. 2000. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106: 1489–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Williams J, Xu Q, Leahy DJ, Nathans J. 2007. Mutational analysis of Norrin-Frizzled4 recognition. J Biol Chem 282: 4057–68 [DOI] [PubMed] [Google Scholar]
- Sohet F, Lin C, Munji RN, Lee SY, Ruderisch N, et al. 2015. LSR/angulin-1 is a tricellular tight junction protein involved in blood-brain barrier formation. J Cell Biol 208: 703–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P 1994. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev 8: 1888–96 [DOI] [PubMed] [Google Scholar]
- Stahl A, Connor KM, Sapieha P, Chen J, Dennison RJ, et al. 2010. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci 51: 2813–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan RV, Tse D, Deharvengt SJ, Smits NC, Xu Y, et al. 2012. The diaphragms of fenestrated endothelia: gatekeepers of vascular permeability and blood composition. Dev Cell 23: 1203–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. 2008. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322: 1247–50 [DOI] [PubMed] [Google Scholar]
- Stewart PA, Hayakawa EM. 1987. Interendothelial junctional changes underlie the developmental ‘tightening’ of the blood-brain barrier. Brain Res 429: 271–81 [DOI] [PubMed] [Google Scholar]
- Stewart PA, Wiley MJ. 1981. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail--chick transplantation chimeras. Dev Biol 84: 183–92 [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, Zlokovic BV. 2018. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14: 133–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, et al. 2016. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 167: 1481–94 e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MS, Routhe LJ, Moos T. 2017. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 37: 3300–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyboll J, Kortesmaa J, Cao R, Soininen R, Wang L, et al. 2002. Deletion of the laminin alpha4 chain leads to impaired microvessel maturation. Mol Cell Biol 22: 1194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran KA, Zhang X, Predescu D, Huang X, Machado RF, et al. 2016. Endothelial beta-Catenin Signaling Is Required for Maintaining Adult Blood-Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation 133: 177–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhollebeke B, Stone OA, Bostaille N, Cho C, Zhou Y, et al. 2015. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/beta-catenin pathway during brain angiogenesis. Elife 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, et al. 2018. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554: 475–80 [DOI] [PubMed] [Google Scholar]
- Wang D, Pascual JM, Yang H, Engelstad K, Mao X, et al. 2006. A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet 15: 1169–79 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cho C, Williams J, Zhang C, Junge HJ, Nathans J. 2018. Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood-brain barrier and blood-retina barrier development and maintenance. PNAS [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rattner A, Zhou Y, Williams J, Smallwood PM, Nathans J. 2012. Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151: 1332–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesuriya HC, Bullock JY, Faull RL, Hladky SB, Barrand MA. 2010. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res 1358: 228–38 [DOI] [PubMed] [Google Scholar]
- Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, et al. 2015. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18: 521–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, et al. 2009. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 15: 519–27 [DOI] [PubMed] [Google Scholar]
- Yamagata K, Tagami M, Nara Y, Mitani M, Kubota A, et al. 1997. Astrocyte-conditioned medium induces blood-brain barrier properties in endothelial cells. Clin Exp Pharmacol Physiol 24: 710–3 [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Ehling M, Kato K, Kanai K, van Lessen M, et al. 2015. Integrin beta1 controls VE-cadherin localization and blood vessel stability. Nat Commun 6: 6429. [DOI] [PubMed] [Google Scholar]
- Yang Y, Higashimori H, Morel L. 2013. Developmental maturation of astrocytes and pathogenesis of neurodevelopmental disorders. J Neurodev Disord 5: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Chen ZL, Norris EH, Strickland S. 2014. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun 5: 3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Wang Y, Nathans J. 2010. The Norrin/Frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol Med 16: 417–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Watts RJ. 2013. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10: 459–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, et al. 2018. Molecular Architecture of the Mouse Nervous System. Cell 174: 999–1014 e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yue Z, Arnold DM, Artiushin G, Sehgal A. 2018. A Circadian Clock in the Blood-Brain Barrier Regulates Xenobiotic Efflux. Cell 173: 130–39 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, et al. 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: 11929–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. 2015. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 163: 1064–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, et al. 2014. Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest 124: 3825–46 [DOI] [PMC free article] [PubMed] [Google Scholar]