

Abstract
Atopic dermatitis (AD) is the most common chronic skin inflammatory disease, with a profound impact on patients’ quality of life. AD varies considerably in clinical course, age of onset and degree to which it is accompanied by allergic and non-allergic comorbidities. Skin barrier impairment in both lesional and nonlesional skin is now recognized as a critical and often early feature of AD. This may be explained by a number of abnormalities identified within both the stratum corneum and stratum granulosum layers of the epidermis. The goal of this review is to provide an overview of key barrier defects in AD, starting with a historical perspective. We will also highlight some of the commonly used methods to characterize and quantify skin barrier function. There is ample opportunity for further investigative work which we call out throughout this review. These include: quantifying the relative impact of individual epidermal abnormalities and putting this in a more holistic view with physiological measures of barrier function, as well as determining whether these barrier-specific endotypes predict clinical phenotypes (e.g. age of onset, natural history, comorbidities, response to therapies, etc). Mechanistic studies with new (and in development) AD therapies that specifically target immune pathways, Staphylococcus aureus abundance and/or skin barrier will help us understand the dynamic crosstalk between these compartments and their relative importance in AD.
Keywords: Atopic dermatitis, Skin barrier, Stratum corneum, Tight junctions, Type 2 inflammation
Introduction
Epidermal barrier dysfunction is increasingly recognized as a key determinant of Atopic Dermatitis (AD, also referred as eczema) pathogenesis. Since the earliest reports of this disease, physicians have noted features that would suggest that the skin barrier was impaired; such as xerotic skin, the need for and benefit from emollients and the reduced skin irritancy threshold. Starting in the late 1970s, with the development of methodology to quantify skin barrier features, evidence has accumulated that AD subjects have skin barrier abnormalities. However, the underlying mechanisms for these abnormalities remain only partially understood. Many studies have implicated cutaneous inflammation as a major contributor to epidermal barrier dysfunction. Unfortunately, modeling the dynamic and highly complex human epidermal differentiation process ex-vivo or in-vitro is still not ideal.
This review will provide an overview of the skin barrier defects in AD, starting from a historical perspective and covering what is known about the AD clinical phenotype(s) and endotypes associated with barrier impairment. A number of noninvasive clinical devices or techniques are available to measure different biophysical properties such as Trans Epidermal Water Loss (TEWL), capacitance (or skin hydration), skin pH. We will discuss several studies that use these measures to quantify AD skin barrier dysfunction. We will highlight research opportunities that would help define the contribution of skin barrier defects to AD pathogenesis and clinical phenotypes of AD. The new generation of more targeted and highly effective AD therapies provides an unprecedented research opportunity to evaluate the mechanisms responsible for AD skin barrier defects.
Historical perspective on AD skin barrier defects
Reports of AD like features (i.e. itchy, exudative, eczematous dermatitis) date back over 2000 years, with possibly the first report by the Roman historian, Suetonius (69–140 A.D.). Since then, numerous descriptions of skin lesions akin to AD can be traced over the centuries and among various races and ethnicities.1 Besnier coined the term “prurigo diasthésique” in 1892, as a clinically distinct entity to be differentiated from other allergic (asthma and hay-fever) and skin conditions (urticaria and ichthyosis).2,3 However, it was not until the early 1900s that the term “atopic dermatitis” was more commonly used.
In early 1900s, studies were undertaken to determine whether there was a relationship between food allergy (FA) or asthma and sensitization to food proteins. Blackfan observed that asthma patients who were food sensitized often had a history of eczema during infancy. This observation led him to evaluate the relationship between eczema and food sensitization.4 Key observations from his studies were that young patients with significant eczema were much more likely to have positive reactions to multiple foods, and that eliminating these from the diet improved the eczema.4 Subsequently, a number of other studies also demonstrated a high frequency of positive reaction to various food proteins in patients with eczema.5,6 One study made a puzzling observation, namely, that 42 of the 46 infants with a positive skin reaction to egg white had never eaten egg.6 This observation has now been confirmed for other common food allergens and suggest that sensitization may occur through “leaky” and inflamed AD skin and not through the gastro-intestinal tract as was originally assumed.7–10
Rasch referred to AD as Besnier’s prurigo in 1914,2,3 and made the observation that even the non-eczematous skin was xerotic. Histologic analysis of AD lesions documented changes in both the epidermis (acanthosis) and dermis (inflammatory infiltrates characterized by mononuclear cells and eosinophils) which were also observed in nonlesional (NL) skin.11–15
It was not until 1978, when TEWL measurements were first performed on both lesional (L) and NL skin of AD patients that the scientific community began to accept that AD patients had a skin barrier defect and that this observed throughout the skin integument.16 This accelerated interest in skin barrier research and lead to seminal observations such as NL TEWL values correlating with AD severity.11,17 Investigators also evaluated the changes observed in TEWL values in response to graded mechanical disruption of the stratum corneum (SC); originally using sandpaper18 and later using a tape stripping approach.19,20 This skin barrier assessment, is now referred to as SC integrity assay (Fig. 1).19,20 Based on these findings and others, ultrastructural and biochemical characterizations of the SC in AD patients became a focus for many laboratories worldwide. These studies revealed three features that may explain these barrier observations in AD patients: 1) increased number of intact lamellar bodies (LBs) in the uppermost cells of the stratum granulosum (SG), 2) reduced LBs fusing to the apical cell membrane, suggesting that the exocytosis of LBs is disturbed,21,22 and 3) alterations in the SC lipid composition (e.g. reduced ceramide 1) and ultrastructural conformation (Fig. 2).23–25
Fig. 1.
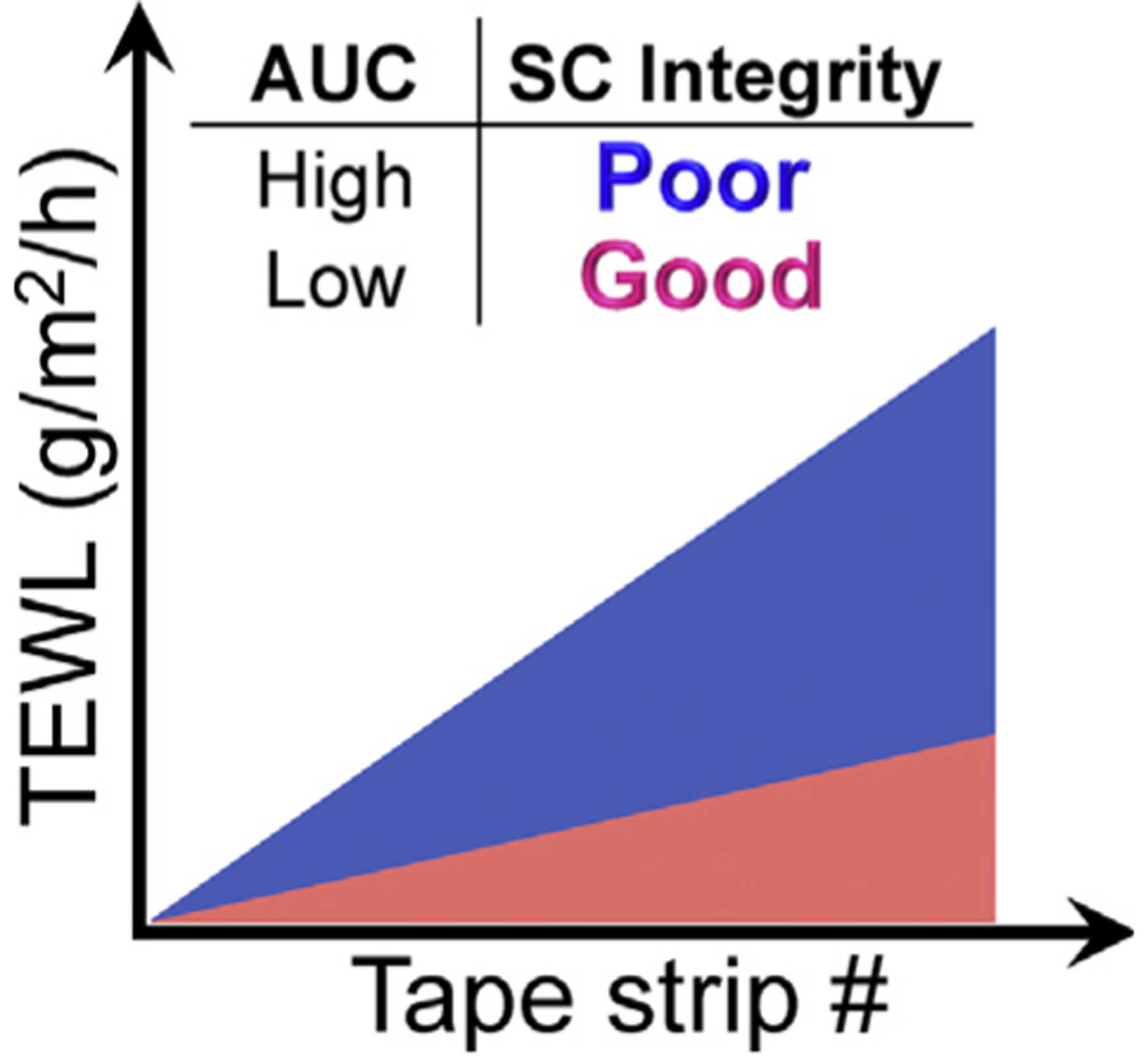
The SC integrity assay. This illustrates the area under the curve (AUC) which integrates TEWL changes after sequential tape strippings. A larger AUC with a steeper slope (Blue) reflects poor (or reduced) SC integrity comparing to smaller AUC (pink).
Fig. 2.

The key components of the SC and SG that are important for skin barrier function are shown in this figure. Using the f-TKD model, we speculate how inflammation (even the NL AD skin) contributes to a disrupted barrier. In healthy skin, keratinocytes become more flattened as they differentiate and assume a f-TKD shape in the SG where TJ are assembled. LBs fuse with the cell membrane, which are indicated by circles with black stripes, and are seen at the SC-SG interface. They release their contents at about the same time f-TKD corneocytes emerge. AD skin inflammation disrupts the differentiation process: 1) affecting keratinocytes flattening and ultimately the f-TKD shape which should be seen at the SG layers, 2) TJ assembly is disrupted, 3) which in turn affects the SC-SG interface where LB contents are released, leading to reduced LB exocytosis, 4) cornification of f-TKD SG1 cells is incomplete, leading to alterations in the SC. All of these features contribute to defective skin barrier observed in AD skin. f-TKD, flattened tetrakaidecahedron; KC, keratinocyte; LB, lamellar body; SC, stratum corneum; SG, stratum granulosum; TJ, tight junction.
The debate on which epidermal structures were responsible for skin barrier function continued with observations that paracellular diffusion of tracers (such as ions, molecules or dextrans) across the epidermis was occluded at the level of the SG or at the interface between the SG and SC.26–29 In 1971, Hashimoto used lanthanum to identify the ultrastructural site where the tracer stopped and he found it did not move beyond the SG layer.26 It is now recognized that this is where tight junctions (TJ) form, and where LB contents are deposited.27 TJ mediate cell–cell adhesion and in so doing provide a barrier to water, solute and macromolecular diffusion and also establish a polarity to the skin epithelium. In 2002, Furuse et al. developed a mouse deficient in claudin-1 (CLDN1), the most highly expressed epidermal TJ transmembrane protein (Cldn1−/− mouse).30 Cldn1−/− mice died within 1 day of birth, which was due to excessive water loss through the skin. These mice had a profound skin barrier abnormality as demonstrated by markedly increased TEWL and permeability to biotin (MW = ~600 Da). This observation solidified the importance of TJ function and CLDN1 in a healthy skin barrier. Subsequent Cldn1−/− mouse studies demonstrated that this TJ defect lead to SC barrier defects.31 If TJ were a critical structure preventing insensible water loss through the skin, there had to be a mechanism to explain how this function was maintained during the orderly process of epidermal maturation. To address this conundrum, Yokouchi et al. proposed a flattened Kelvin’s tetrakaidecahedron (f-TKD) model, by which TJ-bearing SG keratinocytes could maintain skin barrier homeostasis in healthy skin.32 A key feature of this model is that the shape of TJ-bearing SG cells resembles a flattened Kelvin’s tetrakaidecahedron that was originally described as the shape observed for corneocytes,33–35 and this shape is maintained as SG cells differentiate into corneocytes. This results in highly orderly stacked columns of corneocytes and suggests that SG may influence SC cells. Schematic representation in Figure 2 summarizing SC and SG defects in AD as it compared to healthy skin.
In the late 1980s and early 1990s, a number of studies demonstrated high expression of IgE, IL-4 and IL-13 in the skin of AD patients.36–39 Now several decades later, the importance of type 2 immunity in AD pathogenesis has been firmly established with the use of biologics that target one or more Th2 cytokines (e.g. dupilumab, tralokinumab, lebrikizumab and nemolizumab).40–47 Whether and how these cytokines affect the complex and inter-dependent epidermal structures that result in a healthy barrier is still being studied.
What is known about epidermal abnormality in AD?
The epidermis is composed of basilar keratinocytes that are highly proliferative and differentiate as they move toward the skin surface. This differentiation is characterized by the orderly expression of specific proteins, intercellular junction complexes, lipids and proteases/antiproteases. This terminates at the SC layer, where enucleated corneocytes are organized in vertical stacks embedded in a well-organized extracellular matrix composed of lipids.48 The membranes around the corneocytes are formed by highly cross-linked insoluble protein structures and are referred to as the cornified envelope. The lipid/protein ratio in the SC as well as the SC thickness are thought to play a role in skin barrier function (i.e. TEWL).49 Other AD abnormalities include epidermal hyper-proliferation (resulting in acanthosis), increased skin pH, enhanced protease activity, and increased sodium concentration. This last observation, coupled with the observation that high sodium chloride promotes Th2 responses (e.g. enhanced IL-4 and IL-13 production and suppressed IFN-γ) by regulating transcription factor activation, makes it worthy of further study. Sodium was detected in frozen skin biopsies using sophisticated high-resolution neutron measurements. The mechanism(s) responsible for this increased sodium chloride is currently unknown.50 Several review articles have summarized AD skin barrier defects,51–53 and therefore, we will discuss just the major AD skin barrier features.
Stratum corneum defects in AD
The epidermal differentiation complex (EDC) is a chromosomal region located at 1q21.3 that harbors over fifty genes encoding proteins expressed in late keratinocyte differentiation.54 This includes genes for cornified envelop proteins such as loricrin, involucrin and small proline-rich proteins (SPRRs), as well as genes encoding calcium-binding proteins of the S100A family, late cornified envelope (LCE) and multifunctional proteins such as filaggrin (FLG) and trichohyalin.55
In 2005, Sugiura et al. identified reduced expression of cornified envelope genes (i.e., FLG, LORICRIN, LCE) and upregulation of S100A7 and A8 in AD skin from DNA microarray analysis.56 Other studies have validated and extended this observation, demonstrating dysregulation of a number of EDC genes in both L and NL skin of AD subjects. One, if not the most, striking finding has been the strong association between the loss-of-function FLG mutations and risk of AD; initially reported in a North European cohort and later reproduced in other (but not all) populations.57–59 Mutations in FLG confer a significantly increased risk not only of developing AD but publications have also suggested this is true for other allergic disorders such as asthma, allergic rhinitis and FA.60,61 The association with respiratory diseases (allergic rhinitis and asthma) is controversial as several studies have shown that when you exclude patients with history or active AD from these airway disease cohorts the FLG association was lost.62,63 Importantly, genome-wide association study suggests that in addition to FLG, other EDC gene variants are risk factors for AD (e.g. FLG2, SPRR3, LELP1).64–68
In addition to SC proteins, a number of studies have identified lipid abnormalities in AD skin. Under homeostatic conditions, the SC lipids are enriched for cholesterol (about 25% by weight), free fatty acids (FFAs; about 25% by weight) and ceramides (about 50% by weight) with smaller amounts of cholesterol sulfate and phospholids. These lipids are organized into a 3-dimensional structure of stacked lipid layers or lamellae precisely arranged around protein elements. Both the synthesis and organization of epidermal lipids are critical for the optimal skin barrier function.69–71 AD subjects have abnormalities in both lipid composition and architectural organization.72
Recently, Berdyshev et al. demonstrated an increased proportion of short-chain CER [NS], non-hydroxy fatty acid sphingosine ceramides, that consists fatty acid with carbon chain ranging from 14 to 24 (N-14:0 to N-24:0), sphingomyelins, and lysophosphati-dylcholines that consists fatty acid with carbon chain ranging from 14 to 22 (14:0 to 22:0 LPC), and reduced proportion of long-chain species in AD skin when compared with healthy controls. In vitro, treatment of human keratinocytes with IL-4 and IL-13 induced similar changes, further supporting the role of these cytokines in AD pathogenesis.73
Much of the AD lipid research has focused on the ceramides abnormalities49 and led to development of numerous ceramide-containing moisturizers, with the assumption they would improve dry skin and barrier function.74 There is a real need for randomized double blind placebo control studies of these products to evaluate their effect on skin barrier function and AD clinical improvement.
The filaggrin dilemma: the strongest genetic association whose contribution to AD pathogenesis is still unclear
Over the past 15 years, a number of publications have demonstrated reduced, and in some cases complete absence, of FLG expression in Ichthyosis Vulgaris (IV) and AD subjects. We know that the majority of AD subjects have reduced FLG expression on either a genetic61 or acquired basis (e.g. in response to inflammation including Th2 cytokines).75
Within corneocytes, FLG aggregates with keratin filaments providing a scaffold for the assembly of other structural proteins (i.e. involucrin, loricrin, and SPRPs) which are then cross-linked by transglutaminases to form the cornified envelop.76 AD subjects with FLG loss-of-function mutations have abnormal corneocyte morphology and reduced natural moisturizing factors (NMFs; histidine, pyrrolidone-5-carboxylic acid and urocanic acid) that contribute to SC hydration.77 What remained to be fully understood is the biological consequence of reduced FLG expression in AD pathogenesis and perhaps in other atopic conditions. Several murine models have been employed to study FLG function(s). The first attempt was with the flaky tail (ft) mice.78,79 Homozygous flaky tail ft/ft mice showed AD like phenotype with dry and flaky skin, skin barrier defects (increased TEWL), high serum IgE and enhanced epicutaneous sensitization to protein antigen.80 Initially only the Flg mutation (Flg5303delA, resulting in hypomorphic FLG expression) was identified in this spontaneously occurring mouse model,81 but further studies uncovered an additional mutation, namely Tmem 79/Matt (linked to Flg mutation on murine chromosome 3). Two independent groups showed that the Tmem79 mutation alone was associated with matted fur and AD-like dermatitis with high IgE.82,83 The targeted inactivation of Flg in C57BL/6 mice resulted in the expected IV-like phenotype but failed to reproduce the AD-like phenotype.84 To further clarify the role of Flg deficiency in skin barrier function and inflammation, recently Muhandes et al. generated Flg−/− deficient mice on a pure BALB/c background (BALB/c Flg−/−) and were able to demonstrate that even this pro-allergic mouse strain did not spontaneously develop dermatitis or skin barrier impairment following Flg knockout.85 BALB/c Flg−/− presented with transitory ichthyosis (dry and scaly skin) early in life, but it resolved in older animals. Increase TEWL and reduction of keratohyalin granules were observed in older mice (16–18 weeks). BALB/c Flg−/− show no signs of atopy, with normal IgE at baseline and even after skin colonization with Staphylococcus aureus (S. aureus) or after OVA-epicutaneous sensitization. However, these mice had reduced microbial diversity (Shannon diversity index) associated with reductions in the Lachnospiraceae or Muribaculaceae families. Interestingly, keratinocytes isolated from BALB/c Flg−/− showed alterations in gene expression with enrichment of type-2 immune gene. Altogether these data suggest that FLG knockout alone is not sufficient to cause an AD-like phenotype in mice. The authors proposed the intriguing hypothesis that to cause atopy in the context of FLG-deficiency additional atopypromoting gene variants are required.85
These observations are consistent with what is observed in humans who carry a FLG mutation, with some developing early onset AD or IV (or both) but many having no obvious skin phenotype. A research opportunity is to explore the gene–gene and gene-environment effects of FLG mutations on risk for AD and IV and impact on disease phenotype and endotypes.
Tight junction abnormalities in AD
TJs are an intercellular junctional complex found in all epithelial and endothelial cells and composed of transmembrane proteins (e.g. claudins, occludin, junctional adhesion molecules) linked to cytoplasmic proteins (e.g. ZO-1).86 In all epithelial surfaces (e.g. gut, airway, blood, urogenital) TJs control paracellular flux (i.e. passage of molecules through intercellular space between adjunct cells) and epithelial permeability (i.e. selective permeable barrier that limits penetration of harmful molecules). TJs also have a fence function, which is required to generate cell-membrane polarity.87 The TJ fence function in keratinocytes is still controversial, as the fence function could be relevant at either the level of a single cell or the entire epidermis.88 In addition to barrier, TJs play a role in proliferation, migration and differentiation of keratinocytes.88,89 Disruption of epithelial TJs has been observed in most of the allergic disorders: asthma, chronic rhinosinusitis, eosinophilic esophagitis and AD.90,91
About a decade after Furuse et al. generated Cldn1−/−mice and demonstrated that they had a profound skin barrier defect,30 a new human CLDN1-deficient disease characterized by ichthyosis and AD-like features was discovered (Neonatal Ichthyosis and Sclerosing Cholangitis syndrome or NISCH).92 In 2011, our group identified electrophysiologic barrier defects (i.e. increased permeability and reduced transepithelial resistance) in the NL skin of AD subjects which was seen in association with reduced expression of CLDN1 and CLDN23.93 Subsequently, studies demonstrated a dose dependent role of CLDN1 in skin barrier function, assessed by TEER and permeability assays in murine keratinocytes in which the claudin-1 expression was altered94 which was also confirmed in AD subjects.95 We also noted that CLDN1 inversely correlated serum biomarkers of type 2 immunity.93 This suggests a dynamic crosstalk between epithelial barrier and type 2 inflammation. The role epithelium plays in tissue inflammation has been more extensively evaluated for intestinal epithelium, and the effects of a number of environmental factors have been studied.96 How breakdown of the TJ barrier functions affect antigen penetration and immune responses is an active area of investigation, with clear implications for AD and even epicutaneous administration of vaccine and drug delivery strategies.97
Genetic and acquired mechanisms have been reported to cause TJ defects in AD subjects. Polymorphisms of CLDN1 associate with AD both in Black/African American (strongest association) and White/European American subjects in a US population.93 One of the SNPs (rs893051) associated with AD severity in the population of African descent in the USA cohort, was later associated with childhood onset AD in an Ethiopian population.98
The expression of CLDN1 and other TJ components are dynamically regulated during differentiation of keratinocytes, and are also influenced by the inflammatory milieu in AD skin. In vitro studies have shown that several cytokines relevant to AD modulate CLDN1 expression and TJ function (with IL-33, IFN-γ and IL-4 reducing and IL-17A enhancing).99–101 We also observed that TLR2 activation enhanced TJ function, suggesting positive feedback of innate immune pathways on skin barrier function which may be particularly beneficial to respond to pathogens like S. aureus. We and others have hypothesized that this protective mechanism may be defective in AD subjects who express defective TLR2 responses.102–105 Enhanced endogenous protease activity (e.g. Trypsin-like and Chymotrypsin-like serine proteases)106 or exogenous (e.g. protease active allergens such as house dust mite or cockroach or protease produced by Bacteria)107 in the skin has also been discussed as possible pathway to disrupt TJ in AD, via a direct enzymatic mechanism or following activation of the protease activated receptor 2.106,108–112
Whether there is a dynamic interaction between TJ function (or expression of CLDN family members) and FLG expression (or mutations) is an important but not entirely unanswered question. Most studies have not observed an effect of FLG-deficiency on TJ function and/or composition. This was true in the Flg-deficient mouse model113 and in human subjects with AD.114 Interestingly, reduced immunoreactivity for two TJ proteins (occludin and ZO-1) was observed in IV subjects.114 However, reduced CLDN1 expression affects SC barrier function, including processing of pro-FLG.31 An important challenge will be to uncover the mechanism(s) responsible for TJ impairment in AD, and investigating the crosstalk between TJ and SC barriers as well between TJ and the immune system. Ultimately, the goal is to develop targeted treatments that improve skin barrier function and are safe and improve disease severity.
Nonlesional AD skin is not unaffected
Clinically unaffected skin is in fact “abnormal” in AD as compared to non-AD subjects. This has been shown at the functional (i.e. TEWL and skin irritation)115 and molecular level.116,117 Of note, in addition to skin barrier abnormalities, NL skin also is characterized by inflammation representing both innate and adaptive pathways (mostly skewed toward Th2, Th17/Th22).118,119
Several studies have reported a strong correlation between degree of NL skin barrier disruption (e.g. increased TEWL) and disease severity both in adult and pediatric cohorts.115,120 Understandably “correlations” do not address causality or confer directionality and therefore skin barrier defects may simply be a consequence of diseases severity or in contrast it could be a key driver of AD severity. Studies have started to investigate the clinical phenotype of subjects with greater barrier disruption. Recently has emerged that in AD patients NL barrier dysfunction is associated with other allergic comorbidities. In a large AD pediatric cohort (Mechanisms of Progression from Atopic Dermatitis to Asthma in Children; n = 400), greater barrier impairment (e.g. increased TEWL and decreased FLG expression), and more severe disease (SCORAD) was observed in AD subjects who were poly-sensitized (specifically to peanut, egg, cat or dog) than children with no AD or with AD and other allergen sensitizations.121 Another study found that pediatric AD patients with FA have greater skin barrier abnormalities (increased TEWL, reduced FLG breakdown products and reduced CER [EOS], esterified ω-hydroxy fatty acid sphingosine ceramides/CER [NS], non-hydroxy fatty acid sphingosine ceramides, ratio) as compared to AD without FA or non-AD controls.122
Another important feature of NL AD skin barrier disruption is the association with microbial dysbiosis and more specifically, S. aureus colonization.123,124 Skin infections are commonly associated with AD flares and treatment of these is important for the management of the disease.125 As part of the Atopic Dermatitis Research Network, we investigated the phenotype and endotype of AD subjects colonized with S. aureus in a multicenter, cross-sectional study.126 We found that AD patients who were S. aureus colonized had significantly greater skin barrier impairment in NL skin and greater systemic immune activation, type 2 immunity and allergen sensitization than those who were not colonized.
A number of SC alterations, commonly seen in AD subjects, have been shown to promote S. aureus adhesion to the skin. For example, S. aureus isolated from AD skin binds to corneocytes more avidly when NMF levels are reduced which was thought to be due to the unmasking of adhesion sites (N-terminal region of corneodesmosin).127,128 Disturbances in skin lipids (i.e. lower levels of CER [AH]C38, α-hydroxy fatty acid 6-hydroxy-sphingosine ceramides, with 38 total carbons and CER [AP]C40, α-hydroxy fatty acid phytosphingosine ceramides, with 40 total carbons and two unsaturated FFAs, FFA16:1 and FFA18:1) strongly associated with S. aureus colonized AD vs uncolonized AD subjects.129 Using a multi-omics approach Altunbulakli et al. observed an inverse relationship between Staphylococcus species and expression of TJ components (CLDN4, CLDN5, TJP1, and TJP2); with CLDN4 and TJP1 negatively correlated with S. aureus and positively with Staphylococcus epidermidis.130 Further studies are needed to better understand the role these TJ proteins play in chronic S. aureus skin colonization. It is important to acknowledge that this interaction between the host (epidermis) and microbes is not unidirectional, but in fact bidirectional with the skin microbiome and particularly S. aureus, impacting epidermal inflammation and skin barrier composition and function.131
Methods to assess skin barrier in vivo
Non-invasive clinical devices are available to measure different biophysical properties such as TEWL, capacitance, and skin surface pH for a skin barrier assessment.
Transepidermal water loss measures the amount of water evaporating from the skin surface. This instrument has been widely utilized to assess skin barrier in AD subjects. Numerous studies have demonstrated that even NL AD skin has higher TEWL values than healthy individuals.115,122,126 As mentioned earlier, TEWL values are also higher in AD subjects who are colonized with S. aureus (based on routine cultures) vs those that are not colonized126 and also higher in AD subjects who have FA vs those without.122 While FLG mutations are highly predictive of those at risk for AD,61 the relevance of reduced FLG expression to barrier abnormalities such as increased TEWL is hotly debated with most observations in humans and refined mouse models showing no relationship.132–135 Interestingly, subjects with asthma and/or allergic rhinitis do not have TEWL abnormalities suggesting that skin inflammation may responsible for the abnormal TEWL values in AD subjects.136,137 This could also suggest that in subjects with those atopic airway diseases (without concomitant AD) their allergen sensitization likely occurs via the respiratory mucosa rather than the skin.
The SC Integrity Assay assesses the cohesiveness of corneocytes. Tape-stripping technique was initially used to demonstrate that TEWL increases as you remove SC layers.19,20 The SC integrity assay combines repeated TEWL measurements following sequential tape stripping.138 An abnormal SC integrity assay is characterized by a steeper slope of TEWL measurements following the sequential tape strips and is often represented as the area under the curve (AUC). Therefore, a higher AUC value indicates a poor (or reduced) SC integrity comparing to a lower AUC value as illustrated in Figure 1. Subjects with higher AUC presumably have more SC layers removed with each tape strip or have a blunted barrier repair response. The SC integrity assay can be abnormal even when “basal” TEWL measurements are normal. For example, basal TEWL values in NL skin were not significantly different between AD subjects with and without FA, while the SC integrity assay showed that those subjects with FA had a significantly higher TEWL AUC than those without FA.122 This tape-stripping technique has recently been employed as a much less invasive way of sampling the skin with the goal of characterizing genomic, transcriptomic, lipidomic, proteomic and/or even metabolomic profiles of both L and NL skin in AD subjects. This approach is particularly appealing in children where skin biopsies can be more challenging.139–144
SC hydration and pH are relatively simple and rapid assessments often used to fully characterize skin barrier function. Instruments that measure SC hydration emit high frequency (3.5 MHz), to measure either skin conductance or capacitance.145 SC hydration is reduced in AD skin and inversely correlates with FLG degradation products (NMF).133 The optimal skin pH is slightly acidic (4–6), while AD skin is more neutral and sometimes even basic in its pH.146 Acidification is important for direct and indirect anti-bacterial actions, optimal activity of serine proteases and ceramide levels.
New non-invasive methods used to evaluate the epidermis in vivo
Confocal Raman spectroscopy uses the vibrational signatures of chemical groups such as lipids, amino acids, NMF and water in the SC of the human skin to quantify them.147,148 This technology was uses to show reduced levels of NMF in AD children who carry one of the FLG null mutation(s), making this a potential assay to identify carriers of FLG mutations.134,149 Electrical impedance spectroscopy (EIS), is a new non-invasive method currently used for the detection of melanoma and uses alternating currents of various frequencies to measure overall resistance within a tissue.150 EIS has recently been used to evaluate skin barrier in AD subjects.151 The EIS score was significantly reduced in NL and L AD skin compared to healthy controls and inversely correlated with TEWL, disease severity (SCORAD) and serum CCL13.
In summary, we have a plethora of tools to measure a wide range of physiological features of the human epidermis but exactly what abnormal readings mean mechanistically for each of these is still not entirely clear. Which of these would most accurately reflect a skin surface that is more permissive and responsive to environmental allergens? Additionally, some of these assays require equipment that is both expensive and operator-dependent making their widespread use problematic. Nevertheless, we can appreciate that each of these assays have demonstrated how distinctly different even NL AD skin is than healthy controls and we can hope that what they reflect biologically will be better understood in the years to come.
How barrier defects lead to AD
The “outside-in” barrier hypothesis postulates that a leaky skin barrier allows penetration of toxins, allergens and microbes which then elicits an immune response. This response likely arises from allergen uptake by immature DCs, made possible by the extension of DC dendrites to the skin surface.152 When this happens to an individual predisposed to AD (genetically, epigenetically or environmentally), the ensuing response will lead to type 2 inflammation. A number of studies have demonstrated that barrier defects promote type 2 immunity, at least in part, because of the epidermal production of alarmins (IL-33, TSLP and IL-25).153–156 Consistent with this hypothesis, epicutaneous allergen sensitization of mice requires mechanical disruption of the skin with either tape-stripping, acetone or occlusion or a combination of these.157 Tape-stripping activates Langerhans cells and induces movement of Langerhans cells’ dendrites through TJ to the skin surface.152 Disruption of TJs also promotes the extension of sensory nerve endings to the skin surface where they are more readily activated by both host and environmental factors that induce itch.158 The contrasting hypothesis, often referred to as the “inside-out” theory, suggests that the epidermal defects including barrier dysfunction are the consequence of tissue inflammation. In other words, more dermal inflammation - greater barrier defects which may result in a feed forward loop of enhanced inflammatory responses to microbes, irritants and allergens. Further support for this theory comes from the observation that anti-inflammatory treatments from topical (corticosteroids and calcineurin antagonists) have been shown to improve barrier function159–163; while data on the effect of systemic treatment on skin barrier functions are lacking at this time as studies have mainly looked at molecular changes after intervention.163 Additional support for this hypothesis comes from the evidence that elevated levels of the type 2 alarmin, TSLP from skin tape strips taken from infants predict risk for AD.164
We might also learn something from the study of rare monogenetic disorders caused by mutations in skin barrier or immune genes (Table 1). It is interesting to note that a number of these syndromes have some similarities with AD subjects and therefore they provide clues about the role of a single mutated protein in AD features. This also suggests that AD features can emerge from a combinatorial effect of either epidermal barrier and/or immune gene abnormalities, providing further support for both AD hypotheses (inside-out and outside-in).
Table 1.
Genodermatoses that have AD features.
| Disease OMIM | Gene/Protein | Skin Phenotype† | tIgE | Severe AD |
|---|---|---|---|---|
| Barrier related gene defects | ||||
| Ichthyosis Prematurity Syndrome | SLC27A4/FATP4 | - Greasy, thick vernix caseosa-like scale (at birth) - Thick caseous, desquamating skin (neonate) - Erythrodermic, swollen skin (neonate) - Non-scaly generalized ichthyosis - Follicular hyperkeratosis - Hyperpigmentation - Pruritis |
High | YES |
| Netherton Syndrome | SPINK5/LEKTI | - Generalized erythroderma - Ichthyosis linearis circumflexa - Congenital lamellar ichthyosis - Urticaria |
High | YES |
| Peeling Skin Type B | CDSN/Corneodesmosin | - Superficial generalized lifelong skin peeling - Erythema - Pruritus - Vesicular lesions |
High | YES |
| Severe dermatitis, multiple allergies and metabolic wasting (SAM) syndrome | DSG1/Desmoglein 1 | - Erythroderma, generalized congenital - Erosions - Scaling - Hyperkeratotic yellowish papules and plaques arranged linearly on palms and fingers - Hyperkeratosis of weight-bearing areas of soles - Recurrent skin infections |
High | YES |
| NON-Barrier related gene defects | ||||
| Autoinflammation, immune dysregulation, and eosinophilia (AIIDE) | JAK1/JAK1 | - Atopic dermatitis - Pruritis |
Normal | YES |
| Hyper-IgE recurrent infection syndrome-1 (HIES1), autosomal dominant | STAT3/STAT3 | - Eczema, severe - Recurrent skin abscesses |
High | YES |
| Hyper-IgE recurrent infection syndrome-2 (HIES2), autosomal recessive | DOCK8/DOCK8 | - Eczema, severe - Atopic dermatitis - Skin abscesses, recurrent |
High | YES |
| Hyper-IgE recurrent infection syndrome-3 (HIES3), autosomal recessive | ZNF341/ZNF341 | - Atopic dermatitis - Eczema - Excoriated lesions - Skin infections - Atrophic hypopigmented scars - Pruritis - S. aureus infections - Candidal infections |
Elevated | YES |
| Hyper-IgE recurrent infection syndrome-4 (HIES4), autosomal recessive | IL6ST/IL6ST | - Eczema - Recurrent skin infections - Atopic dermatitis |
Elevated | YES |
| IMD11B | CARD11/CARD11 | - Atopic dermatitis, severe - Molluscum infection - Abscesses |
High | YES |
| IMD23 | PGM3/PGM3 | - Dermatitis - Atopy - Cutaneous vasculitis - Erythema multiforme major - Recurrent Infection |
Elevated | YES |
| IPEX syndrome | FOXP3/Foxp3 | - Eczema - Atopy |
High | YES |
| Loeys-Dietz syndrome 1 and 2 |
TGFBR1/TGFBR1 TGFBR2/TGFBR2 |
- Velvety texture - Translucent skin |
Normal or Elevated | YES |
| Omenn syndrome |
RAG1/RAG1 RAG2/RAG2 DCLRE1C/DCLRE1C |
- Generalized erythroderma - Pachydermia |
High | YES |
| Pentasomy X | Pentasomy X | Normal or Elevated | YES (mild) | |
| STAT5B deficiency | STAT5B/STAT5B | - Severe eczema - Pyoderma - Severe viral infection (VZV) - Thin, fine hair |
High | YES |
| Wiskott-Aldrich syndrome | WAS/WASp | - Eczema - Petechiae - Purpura |
High | YES |
CARD11, Caspase Recruitment Domain Family Member 11; CDSN, Corneodesmosin; DOCK8, Dedicator Of Cytokinesis 8; DCLRE1C, DNA Cross-Link Repair 1C; DSG1, Desmoglein 1; FATP4, Fatty Acid Transport Protein 4; FOXP3, Forkhead Box P3; IL6ST, Interleukin 6 Cytokine Family Signal Transducer; IMD, Immunodeficiency; IPEX, Immune dysregulation, polyendocrinopathy, enteropathy, X-linked; LEKTI, Lymphoepithelial Kazal-Type-Related Inhibitor; PGM3, Phosphoglucomutase 3; RAG1, Recombination Activating 1; RAG2, Recombination Activating 2; SLC27A4, Solute Carrier Family 27 Member 4; SPINK5, Serine Peptidase Inhibitor Kazal Type 5; STAT3, Signal Transducer And Activator Of Transcription 3; STAT5B, Signal Transducer And Activator Of Transcription 5B; TGFBR1, Transforming Growth Factor Beta Receptor 1; TGFBR2, Transforming Growth Factor Beta Receptor 2; WAS, Wiskott-Aldrich Syndrome Protein Actin Nucleation Promoting Factor; WASp, Wiskott–Aldrich Syndrome protein; ZNF341, Zinc Finger Protein 341.
As reported in OMIM.
In summary, we accept the overwhelming evidence that AD subjects skin barrier is impaired. But a more holistic view of this barrier abnormality, taking into account the combinatorial effects of the components that make up a healthy epidermal barrier will be enlightening. In other words, how do these abnormalities individually and collectively contribute to AD pathogenesis? To achieve these lofty goals, we will need more robust and sophisticated tools to better characterize and quantify this barrier abnormality in human subjects (and not mice whose epidermis does not faithfully model human skin). Combining these barrier assays with a multiomic (i.e. transcriptomic, lipidomics, metabolomics, metagenomics) approach will help us understand the drivers of this seminal AD feature. This will enable us to ask a number of key questions. What is the relative import of SC constituents (corneocyte compaction, lipid composition and conformation, endogeneous proteases/antiproteases ratio) vs intercellular junctional complexes (TJ, corneodesmosomes and adherens junctions) in AD phenotypes (such as severity) or endotypes? Do you need more than one epidermal barrier defect to develop AD? Is there a barrier defect threshold needed to become allergen sensitized and is that different for allergen elicitation?
Until the mechanism(s) of skin barrier defects are fully elucidated it will be difficult to answer these questions. A greater understanding of AD pathogenesis is informing the development of more targeted treatments. In Figure 3, we propose a simplified model with therapies directed at the immune system, the skin barrier or both to obtain an optimal clinical improvement. Considering the dynamic cross talk between barrier and immune system it is reasonable to speculate that BOTH need to be addressed to reach an optimal clinical improvement and stop the feed-forward loop we think contributes to the chronic relapsing/remitting course seen in our AD patients. Although better itch management as well as normalization of the microbial dysbiosis may also be critical to achieve the best clinical outcome, since they were not discussed in this review, we have omitted them from this overview.
Fig. 3.

The optimal clinical improvement will likely only be achieved with treatment strategies that target immune system, skin barrier or both. Here we propose a simplified model with therapies targeting the immune system (IL4/IL-13 blockade) or the skin barrier (emollients) or both (JAK inhibitors and AhR agonist). Therapies directed at itch or microbial dysbiosis are not included since they were not discussed in this review.
With the development of highly targeted treatments, which are primarily directed at inflammatory mediators, we have an opportunity to clarify the relative import of these mediators on skin barrier physiology and composition. One such therapy is dupilumab (Dupixent, Regeneron Inc.), a fully humanized monoclonal antibody targeting IL-4Rα, a shared subunit for both IL-4 and IL-13 receptors. Pivotal trials have demonstrated significant clinical improvement and a favorable safety profile in moderate to severe AD subjects, and confirms the central role of IL-4 and IL-13 in this disease.40,41 Mechanistic studies showed that blocking IL-4/IL-13 signaling suppressed systemic type-2 inflammation,165 increased microbial diversity, reduced S. aureus abundance166 and normalized AD-associated epidermal transcriptomic abnormalities.167 Specifically, dupilumab increased the expression of FLG, LEKTI (proteases inhibitor) and HBD-3 (antimicrobial peptide) within 6–8 weeks of treatment.168 In a small study, 12 weeks of Dupilumab treatment significantly increased SC hydration (L and NL) and this barrier improvement correlated with EASI improvement.169 These studies suggest that Th2 inflammation is responsible for a number of epidermal abnormalities observed in AD subjects. Future studies will need to investigate how changes seen at early time points may predict later improvement in barrier and disease severity.
Both topical and oral Janus kinase (JAK) inhibitors that target the JAK/STAT signaling cascade common to many inflammatory mediator and growth factor receptors, have shown efficacy in clinical trials for patients with moderate-to-severe AD.170 The JAK inhibitors (depending on their JAK specificity) target multiple cytokines pathways including Th2 (IL-4, IL-13, IL-31 and TSLP), Th22 (IL-22), and Th1 (IFN-γ, IL-12, and IL-23) and therefore are much less selective than biologics.
Several studies have suggested that JAK/STAT signaling may be important for skin barrier. In the NC/Nga AD mouse model, the topical application of a JAK inhibitor (JTE-052) reduced TEWL, increased NMF and resulted in clinical improvements.171 Ruxolitinib, a selective JAK1/2 inhibitor, was able to reverse TJ disruption and CLDN1 downregulation induced by IFN-γ in primary human keratinocytes.100 In a small AD clinical trial, ASN002 (an oral dual JAK/SYK inhibitor) demonstrated clinical improvement and a qualitative reversal of some epidermal barrier abnormalities (e.g. hyperplasia, reduced FLG and increased Keratin 16) as early as 15 days into treatment.172
Tapinarof, is a topical aryl hydrocarbon receptor (AhR) agonist is currently in trials for the treatment of psoriasis and AD. In a phase 2b AD trial, clinical improvement (EASI, IGA and pruritus score) was seen after 12 weeks of treatment.173 AhR is a ligand-activated transcription factor, highly expressed in keratinocytes. Activation of AhR promotes differentiation and skin barrier repair. Tapinarof treatment of human keratinocytes enhances the expression of FLG and loricrin.174
Emollients and moisturizers are a cornerstone of AD management and are recommended to try to reverse the xerosis and potentially improve the skin barrier. However, there are few studies that directly address their mechanism of action, even though their clinical benefit in AD is not questioned.175 Research is ongoing to investigate whether the early use of emollients in high-risk infants will prevent AD development or FA. At this time results are controversial possibly because critical differences in the study design, control groups, emollients used (and their frequency of applications), duration, and outcomes. In a seminal randomized controlled trial in the United States and United Kingdom 124 neonates at high risk for AD were enrolled. Caregivers in the treatment arm were instructed to apply emollient therapy (full body) daily starting within 3 weeks of birth for 6 months; whereas, in the control arm they were asked to use no emollients. Remarkably, a significant protective effect was found with daily emollient use on cumulative AD incidence (relative risk, 0.50; 95% CI, 0.28–0.9).176 In contrast, larger studies (Barrier Enhancement for Eczema Prevention [BEEP] and PEBBLES) showed no benefit from daily emollient use during the first year of life in high-risk children, but the control groups were not forbidden to use moisturizers.177,178 The Enquiring About Tolerance (EAT) study recently demonstrated that greater moisturizing frequency was associated with greater food allergen sensitization.179 This has been argued to arise by introduction of allergens to the skin surface with parental moisturizing. Further investigations into the impact of emollient use on AD and FA prevention are ongoing.178,180 Should these studies confirm the initial evidence of a protective effect of emollients in AD prevention, they will have profound impact in public health by reducing the overall burden of these common allergic diseases.
Conclusion
A cardinal AD feature is global alteration in epidermal biology, which among other things leads to a leaky barrier and inflammation that promotes a type 2 immune response. This is at least in part, responsible for AD subjects poly-sensitization, widespread xerosis, S. aureus colonization and pruritus. The epidermal components responsible for the barrier function of the skin are numerous and highly integrated. Sorting out the hierarchy and relevance of each of these as they relate to barrier function and ultimately AD severity (and phenotypes) is the goal. The AD treatment pipeline is impressive and expanding daily. It includes therapies that are intended to target one or several of the key features of this disease with the majority focusing on inflammation or pruritus and only more recently targeting skin dysbiosis or epidermal abnormalities. We believe the future is very bright, not just because we can more effectively treat AD patients, but because these more targeted therapies will lead to a much more refined and nuanced understanding of AD pathogenesis. This of course will only happen if we design mechanistic studies of the most highly effective AD therapies. These studies should be longitudinal intervention trials that capture mechanistic readouts coupled with multi-omics at early timepoints to model how they impact objective measures of disease severity.
Funding
NIAID funding for LAB (U01AI152011 and U19AI117673).
Footnotes
Conflict of interest
LAB is a Consultant for Galderma, Janssen, Leo Pharma, Lilly, Regeneron, Sanofi/Genzyme, Sanofi-Aventis, and an Investigator for Abbvie, Astra-Zeneca, Dermtech, Kiniksa, Leo Pharma, Pfizer, Regeneron and Sanofi. ADB is an Investigator for Dermira, Kiniksa, Novartis and Pfizer. TY has no conflict of interest.
References
- 1.Kramer ON, Strom MA, Ladizinski B, Lio PA. The history of atopic dermatitis. Clin Dermatol 2017;35:344–8. [DOI] [PubMed] [Google Scholar]
- 2.Barber HW, Oriel GH. A clinical and biochemical study of allergy. Lancet 1928;212:1064–70. [Google Scholar]
- 3.Rackemann FM. Eczema. N Engl J Med 1945;232:649–54. [Google Scholar]
- 4.Blackfan KD. Cutaneous reaction from proteins in eczema. Am J Dis Child 1916;11:441–54. [Google Scholar]
- 5.Ramirez MA. Protein sensitization in eczema: report of seventy-eight cases. Arch Derm Syphilol 1920;2:365–7. [Google Scholar]
- 6.Hill LW, Sulzberger MB. Evolution of atopic dermatitis. Arch Derm Syphilol 1935;32:451–63. [Google Scholar]
- 7.Alvaro M, Garcia-Paba MB, Giner MT, Piquer M, Dominguez O, Lozano J, et al. Tolerance to egg proteins in egg-sensitized infants without previous consumption. Allergy 2014;69:1350–6. [DOI] [PubMed] [Google Scholar]
- 8.Brough HA, Simpson A, Makinson K, Hankinson J, Brown S, Douiri A, et al. Peanut allergy: effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J Allergy Clin Immunol 2014;134:867–75. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brough HA, Liu AH, Sicherer S, Makinson K, Douiri A, Brown SJ, et al. Atopic dermatitis increases the effect of exposure to peanut antigen in dust on peanut sensitization and likely peanut allergy. J Allergy Clin Immunol 2015;135:164–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brough HA, Kull I, Richards K, Hallner E, Soderhall C, Douiri A, et al. Environmental peanut exposure increases the risk of peanut sensitization in high-risk children. Clin Exp Allergy 2018;48:586–93. [DOI] [PubMed] [Google Scholar]
- 11.Finlay AY, Nicholls S, King CS, Marks R. The ‘dry’ non-eczematous skin associated with atopic eczema. Br J Dermatol 1980;103:249–56. [DOI] [PubMed] [Google Scholar]
- 12.Leung DY, Bhan AK, Schneeberger EE, Geha RS. Characterization of the mononuclear cell infiltrate in atopic dermatitis using monoclonal antibodies. J Allergy Clin Immunol 1983;71:47–56. [DOI] [PubMed] [Google Scholar]
- 13.van Neste D, Douka M, Rahier J, Staquet MJ. Epidermal changes in atopic dermatitis. Acta Derm Venereol Suppl (Stockh) 1985;114:67–71. [DOI] [PubMed] [Google Scholar]
- 14.Uehara M Clinical and histological features of dry skin in atopic dermatitis. Acta Derm Venereol Suppl (Stockh) 1985;114:82–6. [DOI] [PubMed] [Google Scholar]
- 15.Leiferman KM, Ackerman SJ, Sampson HA, Haugen HS, Venencie PY, Gleich GJ. Dermal deposition of eosinophil-granule major basic protein in atopic dermatitis. Comparison with onchocerciasis. N Engl J Med 1985;313: 282–5. [DOI] [PubMed] [Google Scholar]
- 16.Abe T, Ohkido M, Yamamoto K. Studies on skin surface barrier functions:–skin surface lipids and transepidermal water loss in atopic skin during childhood. J Dermatol 1978;5:223–9. [DOI] [PubMed] [Google Scholar]
- 17.Agner T Susceptibility of atopic dermatitis patients to irritant dermatitis caused by sodium lauryl sulphate. Acta Derm Venereol 1991;71:296–300. [PubMed] [Google Scholar]
- 18.Winsor T, Burch GE. Differential roles of layers of human epigastric skin on diffusion rate of water. Arch Intern Med 1944;74:428–36. [Google Scholar]
- 19.Blank IH. Further observations on factors which influence the water content of the stratum corneum. J Invest Dermatol 1953;21:259–71. [DOI] [PubMed] [Google Scholar]
- 20.Monash S, Blank H. Location and reformation of the epithelial barrier to water vapor. AMA Arch Derm 1958;78:710–4. [DOI] [PubMed] [Google Scholar]
- 21.Fartasch M, Diepgen TL. The barrier function in atopic dry skin. Disturbance of membrane-coating granule exocytosis and formation of epidermal lipids? Acta Derm Venereol Suppl (Stockh) 1992;176:26–31. [PubMed] [Google Scholar]
- 22.Fartasch M, Bassukas ID, Diepgen TL. Disturbed extruding mechanism of lamellar bodies in dry non-eczematous skin of atopics. Br J Dermatol 1992;127:221–7. [DOI] [PubMed] [Google Scholar]
- 23.Melnik B, Hollmann J, Plewig G. Decreased stratum corneum ceramides in atopic individuals–a pathobiochemical factor in xerosis? Br J Dermatol 1988;119:547–9. [DOI] [PubMed] [Google Scholar]
- 24.Imokawa G, Abe A, Jin K, Higaki Y, Kawashima M, Hidano A. Decreased level of ceramides in stratum corneum of atopic dermatitis: an etiologic factor in atopic dry skin? J Invest Dermatol 1991;96:523–6. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto A, Serizawa S, Ito M, Sato Y. Stratum corneum lipid abnormalities in atopic dermatitis. Arch Dermatol Res 1991;283:219–23. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto K Intercellular spaces of the human epidermis as demonstrated with lanthanum. J Invest Dermatol 1971;57:17–31. [DOI] [PubMed] [Google Scholar]
- 27.Elias PM, Friend DS. The permeability barrier in mammalian epidermis. J Cell Biol 1975;65:180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirschner N, Houdek P, Fromm M, Moll I, Brandner JM. Tight junctions form a barrier in human epidermis. Eur J Cell Biol 2010;89:839–42. [DOI] [PubMed] [Google Scholar]
- 29.Kirschner N, Rosenthal R, Furuse M, Moll I, Fromm M, Brandner JM. Contribution of tight junction proteins to ion, macromolecule, and water barrier in keratinocytes. J Invest Dermatol 2013;133:1161–9. [DOI] [PubMed] [Google Scholar]
- 30.Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 2002;156:1099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugawara T, Iwamoto N, Akashi M, Kojima T, Hisatsune J, Sugai M, et al. Tight junction dysfunction in the stratum granulosum leads to aberrant stratum corneum barrier function in claudin-1-deficient mice. J Dermatol Sci 2013;70: 12–8. [DOI] [PubMed] [Google Scholar]
- 32.Yokouchi M, Atsugi T, Logtestijn MV, Tanaka RJ, Kajimura M, Suematsu M, et al. Epidermal cell turnover across tight junctions based on Kelvin’s tetrakaidecahedron cell shape. Elife 2016;5:e19593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menton DN. A liquid film model of tetrakaidecahedral packing to account for the establishment of epidermal cell columns. J Invest Dermatol 1976;66: 283–91. [DOI] [PubMed] [Google Scholar]
- 34.Menton DN. A minimum-surface mechanism to account for the organization of cells into columns in the mammalian epidermis. Am J Anat 1976;145:1–22. [DOI] [PubMed] [Google Scholar]
- 35.Allen TD, Potten CS. Significance of cell shape in tissue architecture. Nature 1976;264:545–7. [DOI] [PubMed] [Google Scholar]
- 36.Ogawa H, Yoshiike T. A speculative view of atopic dermatitis: barrier dysfunction in pathogenesis. J Dermatol Sci 1993;5:197–204. [DOI] [PubMed] [Google Scholar]
- 37.Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest 1994;94: 870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reinhold U, Kukel S, Goeden B, Neumann U, Kreysel HW. Functional characterization of skin-infiltrating lymphocytes in atopic dermatitis. Clin Exp Immunol 1991;86:444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruynzeel-Koomen C, van Wichen DF, Toonstra J, Berrens L, Bruynzeel PL. The presence of IgE molecules on epidermal Langerhans cells in patients with atopic dermatitis. Arch Dermatol Res 1986;278:199–205. [DOI] [PubMed] [Google Scholar]
- 40.Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 2014;371:130–9. [DOI] [PubMed] [Google Scholar]
- 41.Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med 2016;375:2335–48. [DOI] [PubMed] [Google Scholar]
- 42.Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol 2019;143:135–41. [DOI] [PubMed] [Google Scholar]
- 43.Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol 2021;184:437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taieb A, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol 2018;78:863–71. e11. [DOI] [PubMed] [Google Scholar]
- 45.Guttman-Yassky E, Blauvelt A, Eichenfield LF, Paller AS, Armstrong AW, Drew J, et al. Efficacy and safety of lebrikizumab, a high-affinity Interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis: a phase 2b randomized clinical trial. JAMA Dermatol 2020;156:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruzicka T, Hanifin JM, Furue M, Pulka G, Mlynarczyk I, Wollenberg A, et al. Anti-Interleukin-31 receptor A antibody for atopic dermatitis. N Engl J Med 2017;376:826–35. [DOI] [PubMed] [Google Scholar]
- 47.Kabashima K, Matsumura T, Komazaki H, Kawashima M. The Nemolizumab JP01, JP02 Study Group. Nemolizumab plus topical agents in patients with atopic dermatitis and moderate-to-severe pruritus provide improvement in pruritus and signs of atopic dermatitis for up to 68 weeks: results from two phase III, long-term studies. Br J Dermatol 2021. 10.1111/bjd.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Smeden J, Janssens M, Gooris GS, Bouwstra JA. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim Biophys Acta 2014;1841:295–313. [DOI] [PubMed] [Google Scholar]
- 49.Janssens M, van Smeden J, Puppels GJ, Lavrijsen AP, Caspers PJ, Bouwstra JA. Lipid to protein ratio plays an important role in the skin barrier function in patients with atopic eczema. Br J Dermatol 2014;170:1248–55. [DOI] [PubMed] [Google Scholar]
- 50.Matthias J, Maul J, Noster R, Meinl H, Chao YY, Gerstenberg H, et al. Sodium chloride is an ionic checkpoint for human TH2 cells and shapes the atopic skin microenvironment. Sci Transl Med 2019;11:eaau0683. [DOI] [PubMed] [Google Scholar]
- 51.Luger T, Amagai M, Dreno B, Dagnelie MA, Liao W, Kabashima K, et al. Atopic dermatitis: role of the skin barrier, environment, microbiome, and therapeutic agents. J Dermatol Sci 2021;102:142–57. [DOI] [PubMed] [Google Scholar]
- 52.Leung DYM, Berdyshev E, Goleva E. Cutaneous barrier dysfunction in allergic diseases. J Allergy Clin Immunol 2020;145:1485–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nomura T, Kabashima K. Advances in atopic dermatitis in 2019–2020: endotypes from skin barrier, ethnicity, properties of antigen, cytokine profiles, microbiome, and engagement of immune cells. J Allergy Clin Immunol 2021;148(6):1451–562. [DOI] [PubMed] [Google Scholar]
- 54.Toulza E, Mattiuzzo NR, Galliano MF, Jonca N, Dossat C, Jacob D, et al. Large-scale identification of human genes implicated in epidermal barrier function. Genome Biol 2007;8:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kypriotou M, Huber M, Hohl D. The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol 2012;21:643–9. [DOI] [PubMed] [Google Scholar]
- 56.Sugiura H, Ebise H, Tazawa T, Tanaka K, Sugiura Y, Uehara M, et al. Large-scale DNA microarray analysis of atopic skin lesions shows overexpression of an epidermal differentiation gene cluster in the alternative pathway and lack of protective gene expression in the cornified envelope. Br J Dermatol 2005;152: 146–9. [DOI] [PubMed] [Google Scholar]
- 57.Akiyama M FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol 2010;162:472–7. [DOI] [PubMed] [Google Scholar]
- 58.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006;38: 441–6. [DOI] [PubMed] [Google Scholar]
- 59.Meng L, Wang L, Tang H, Tang X, Jiang X, Zhao J, et al. Filaggrin gene mutation c.3321delA is associated with various clinical features of atopic dermatitis in the Chinese Han population. PLoS One 2014;9:e98235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Osawa R, Akiyama M, Shimizu H. Filaggrin gene defects and the risk of developing allergic disorders. Allergol Int 2011;60:1–9. [DOI] [PubMed] [Google Scholar]
- 61.Irvine AD, McLean WH, Leung DY. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011;365:1315–27. [DOI] [PubMed] [Google Scholar]
- 62.Weidinger S, O’Sullivan M, Illig T, Baurecht H, Depner M, Rodriguez E, et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol 2008;121:1203–9. e1. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H, Guo Y, Wang W, Shi M, Chen X, Yao Z. Mutations in the filaggrin gene in Han Chinese patients with atopic dermatitis. Allergy 2011;66:420–7. [DOI] [PubMed] [Google Scholar]
- 64.Guttman-Yassky E, Suarez-Farinas M, Chiricozzi A, Nograles KE, Shemer A, Fuentes-Duculan J, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol 2009;124:1235–44. e58. [DOI] [PubMed] [Google Scholar]
- 65.Marenholz I, Rivera VA, Esparza-Gordillo J, Bauerfeind A, Lee-Kirsch MA, Ciechanowicz A, et al. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J Invest Dermatol 2011;131:1644–9. [DOI] [PubMed] [Google Scholar]
- 66.Trzeciak M, Wesserling M, Bandurski T, Glen J, Nowicki R, Pawelczyk T. Association of a single nucleotide polymorphism in a late cornified envelope-like proline-rich 1 gene (LELP1) with atopic dermatitis. Acta Derm Venereol 2016;96:459–63. [DOI] [PubMed] [Google Scholar]
- 67.Brown SJ, Elias MS, Bradley M. Genetics in atopic dermatitis: historical perspective and future prospects. Acta Derm Venereol 2020;100:adv00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Margolis DJ, Gupta J, Apter AJ, Ganguly T, Hoffstad O, Papadopoulos M, et al. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol 2014;133:784–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robson KJ, Stewart ME, Michelsen S, Lazo ND, Downing DT. 6-Hydroxy-4-sphingenine in human epidermal ceramides. J Lipid Res 1994;35:2060–8. [PubMed] [Google Scholar]
- 70.Uchida Y, Holleran WM. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci 2008;51:77–87. [DOI] [PubMed] [Google Scholar]
- 71.Di Nardo A, Wertz P, Giannetti A, Seidenari S. Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm Venereol 1998;78:27–30. [DOI] [PubMed] [Google Scholar]
- 72.van Smeden J, Bouwstra JA. Stratum corneum lipids: their role for the skin barrier function in healthy subjects and atopic dermatitis patients. Curr Probl Dermatol 2016;49:8–26. [DOI] [PubMed] [Google Scholar]
- 73.Berdyshev E, Goleva E, Bronova I, Dyjack N, Rios C, Jung J, et al. Lipid abnormalities in atopic skin are driven by type 2 cytokines. JCI Insight 2018;3: e98006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujii M The pathogenic and therapeutic implications of ceramide abnormalities in atopic dermatitis. Cells 2021;10:2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 2009;124:R7–12. [DOI] [PubMed] [Google Scholar]
- 76.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005;6:328–40. [DOI] [PubMed] [Google Scholar]
- 77.Riethmuller C, McAleer MA, Koppes SA, Abdayem R, Franz J, Haftek M, et al. Filaggrin breakdown products determine corneocyte conformation in patients with atopic dermatitis. J Allergy Clin Immunol 2015;136:1573–80. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Presland RB, Boggess D, Lewis SP, Hull C, Fleckman P, Sundberg JP. Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris. J Invest Dermatol 2000;115:1072–81. [DOI] [PubMed] [Google Scholar]
- 79.Sundberg JP. The flaky tail (ft) mutation. In: Sundberg JP, editor. Handbook of Mouse Mutations with Skin and Hair Abnormalities: Animal Models and Biomedical Tools. 1st ed. Ann Arbor, Michigan, USA: CRC Press; 1994. p. 269–73. [Google Scholar]
- 80.Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol 2009;124: 485–93. 93 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet 2009;41:602–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sasaki T, Shiohama A, Kubo A, Kawasaki H, Ishida-Yamamoto A, Yamada T, et al. A homozygous nonsense mutation in the gene for Tmem79, a component for the lamellar granule secretory system, produces spontaneous eczema in an experimental model of atopic dermatitis. J Allergy Clin Immunol 2013;132:1111–20. e4. [DOI] [PubMed] [Google Scholar]
- 83.Saunders SP, Goh CS, Brown SJ, Palmer CN, Porter RM, Cole C, et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J Allergy Clin Immunol 2013;132:1121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kawasaki H, Nagao K, Kubo A, Hata T, Shimizu A, Mizuno H, et al. Altered stratum corneum barrier and enhanced percutaneous immune responses in filaggrin-null mice. J Allergy Clin Immunol 2012;129:1538–46. e6. [DOI] [PubMed] [Google Scholar]
- 85.Muhandes L, Chapsa M, Pippel M, Behrendt R, Ge Y, Dahl A, et al. Low threshold for cutaneous allergen sensitization but no spontaneous dermatitis or atopy in FLG-deficient mice. J Invest Dermatol 2021;141:2611–9. e2. [DOI] [PubMed] [Google Scholar]
- 86.Panwar S, Sharma S, Tripathi P. Role of barrier integrity and dysfunctions in maintaining the healthy gut and their health outcomes. Front Physiol 2021;12: 715611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Otani T, Nguyen TP, Tokuda S, Sugihara K, Sugawara T, Furuse K, et al. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J Cell Biol 2019;218:3372–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kirschner N, Brandner JM. Barriers and more: functions of tight junction proteins in the skin. Ann N Y Acad Sci 2012;1257:158–66. [DOI] [PubMed] [Google Scholar]
- 89.Volksdorf T, Heilmann J, Eming SA, Schawjinski K, Zorn-Kruppa M, Ueck C, et al. Tight junction proteins claudin-1 and occludin are important for cutaneous wound healing. Am J Pathol 2017;187:1301–12. [DOI] [PubMed] [Google Scholar]
- 90.Sugita K, Kabashima K. Tight junctions in the development of asthma, chronic rhinosinusitis, atopic dermatitis, eosinophilic esophagitis, and inflammatory bowel diseases. J Leukoc Biol 2020;107:749–62. [DOI] [PubMed] [Google Scholar]
- 91.Hellings PW, Steelant B. Epithelial barriers in allergy and asthma. J Allergy Clin Immunol 2020;145:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirchmeier P, Sayar E, Hotz A, Hausser I, Islek A, Yilmaz A, et al. Novel mutation in the CLDN1 gene in a Turkish family with neonatal ichthyosis sclerosing cholangitis (NISCH) syndrome. Br J Dermatol 2014;170:976–8. [DOI] [PubMed] [Google Scholar]
- 93.De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2011;127:773–86. e1–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tokumasu R, Tamura A, Tsukita S. Time- and dose-dependent claudin contribution to biological functions: lessons from claudin-1 in skin. Tissue Barriers 2017;5:e1336194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bergmann S, von Buenau B, Vidal YSS, Haftek M, Wladykowski E, Houdek P, et al. Claudin-1 decrease impacts epidermal barrier function in atopic dermatitis lesions dose-dependently. Sci Rep 2020;10:2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen L, Turner JR. Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am J Physiol Gastrointest Liver Physiol 2006;290:G577–82. [DOI] [PubMed] [Google Scholar]
- 97.Brewer MG, Anderson EA, Pandya RP, De Benedetto A, Yoshida T, Hilimire TA, et al. Peptides derived from the tight junction protein CLDN1 disrupt the skin barrier and promote responsiveness to an epicutaneous vaccine. J Invest Dermatol 2020;140:361–9. e3. [DOI] [PubMed] [Google Scholar]
- 98.Asad S, Winge MC, Wahlgren CF, Bilcha KD, Nordenskjold M, Taylan F, et al. The tight junction gene Claudin-1 is associated with atopic dermatitis among Ethiopians. J Eur Acad Dermatol Venereol 2016;30:1939–41. [DOI] [PubMed] [Google Scholar]
- 99.Ryu WI, Lee H, Bae HC, Jeon J, Ryu HJ, Kim J, et al. IL-33 down-regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J Dermatol Sci 2018;90:313–22. [DOI] [PubMed] [Google Scholar]
- 100.Mizutani Y, Takagi N, Nagata H, Inoue S. Interferon-gamma downregulates tight junction function, which is rescued by interleukin-17A. Exp Dermatol 2021;30:1754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brewer MG, Yoshida T, Kuo FI, Fridy S, Beck LA, De Benedetto A. Antagonistic effects of IL-4 on IL-17A-mediated enhancement of epidermal tight junction function. Int J Mol Sci 2019;20:4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair. J Invest Dermatol 2013;133:988–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yuki T, Yoshida H, Akazawa Y, Komiya A, Sugiyama Y, Inoue S. Activation of TLR2 enhances tight junction barrier in epidermal keratinocytes. J Immunol 2011;187:3230–7. [DOI] [PubMed] [Google Scholar]
- 104.Niebuhr M, Heratizadeh A, Wichmann K, Satzger I, Werfel T. Intrinsic alterations of pro-inflammatory mediators in unstimulated and TLR-2 stimulated keratinocytes from atopic dermatitis patients. Exp Dermatol 2011;20: 468–72. [DOI] [PubMed] [Google Scholar]
- 105.Panzer R, Blobel C, Folster-Holst R, Proksch E. TLR2 and TLR4 expression in atopic dermatitis, contact dermatitis and psoriasis. Exp Dermatol 2014;23: 364–6. [DOI] [PubMed] [Google Scholar]
- 106.Nomura H, Suganuma M, Takeichi T, Kono M, Isokane Y, Sunagawa K, et al. Multifaceted analyses of epidermal serine protease activity in patients with atopic dermatitis. Int J Mol Sci 2020;21:913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cau L, Williams MR, Butcher AM, Nakatsuji T, Kavanaugh JS, Cheng JY, et al. Staphylococcus epidermidis protease EcpA can be a deleterious component of the skin microbiome in atopic dermatitis. J Allergy Clin Immunol 2021;147: 955–66. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nadeau P, Henehan M, De Benedetto A. Activation of protease-activated receptor 2 leads to impairment of keratinocyte tight junction integrity. J Allergy Clin Immunol 2018;142:281–4. e7. [DOI] [PubMed] [Google Scholar]
- 109.Henehan M, De Benedetto A. Update on protease-activated receptor 2 in cutaneous barrier, differentiation, tumorigenesis and pigmentation, and its role in related dermatologic diseases. Exp Dermatol 2019;28:877–85. [DOI] [PubMed] [Google Scholar]
- 110.Abu Khweek A, Kim E, Joldrichsen MR, Amer AO, Boyaka PN. Insights into mucosal innate immune responses in house dust mite-mediated allergic asthma. Front Immunol 2020;11:534501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hovnanian A Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res 2013;351:289–300. [DOI] [PubMed] [Google Scholar]
- 112.Smith L, Gatault S, Casals-Diaz L, Kelly PA, Camerer E, Metais C, et al. House dust mite-treated PAR2 over-expressor mouse: a novel model of atopic dermatitis. Exp Dermatol 2019;28:1298–308. [DOI] [PubMed] [Google Scholar]
- 113.Yokouchi M, Kubo A, Kawasaki H, Yoshida K, Ishii K, Furuse M, et al. Epidermal tight junction barrier function is altered by skin inflammation, but not by filaggrin-deficient stratum corneum. J Dermatol Sci 2015;77:28–36. [DOI] [PubMed] [Google Scholar]
- 114.Gruber R, Elias PM, Crumrine D, Lin TK, Brandner JM, Hachem JP, et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am J Pathol 2011;178:2252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Seidenari S, Giusti G. Objective assessment of the skin of children affected by atopic dermatitis: a study of pH, capacitance and TEWL in eczematous and clinically uninvolved skin. Acta Derm Venereol 1995;75:429–33. [DOI] [PubMed] [Google Scholar]
- 116.Suarez-Farinas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles K, Cardinale I, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol 2011;127:954–64. e1–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.He H, Del Duca E, Diaz A, Kim HJ, Gay-Mimbrera J, Zhang N, et al. Mild atopic dermatitis lacks systemic inflammation and shows reduced nonlesional skin abnormalities. J Allergy Clin Immunol 2021;147:1369–80. [DOI] [PubMed] [Google Scholar]
- 118.Pavel AB, Zhou L, Diaz A, Ungar B, Dan J, He H, et al. The proteomic skin profile of moderate-to-severe atopic dermatitis patients shows an inflammatory signature. J Am Acad Dermatol 2020;82:690–9. [DOI] [PubMed] [Google Scholar]
- 119.Brunner PM, Emerson RO, Tipton C, Garcet S, Khattri S, Coats I, et al. Nonlesional atopic dermatitis skin shares similar T-cell clones with lesional tissues. Allergy 2017;72:2017–25. [DOI] [PubMed] [Google Scholar]
- 120.Addor FA, Takaoka R, Rivitti EA, Aoki V. Atopic dermatitis: correlation between non-damaged skin barrier function and disease activity. Int J Dermatol 2012;51:672–6. [DOI] [PubMed] [Google Scholar]
- 121.Sherenian MG, Kothari A, Biagini JM, Kroner JW, Baatyrbek Kyzy A, Johannson E, et al. Sensitization to peanut, egg or pets is associated with skin barrier dysfunction in children with atopic dermatitis. Clin Exp Allergy 2021;51:666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leung DYM, Calatroni A, Zaramela LS, LeBeau PK, Dyjack N, Brar K, et al. The nonlesional skin surface distinguishes atopic dermatitis with food allergy as a unique endotype. Sci Transl Med 2019;11:eaav2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol 2019;143:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Blicharz L, Rudnicka L, Czuwara J, Waskiel-Burnat A, Goldust M, Olszewska M, et al. The influence of microbiome dysbiosis and bacterial biofilms on epidermal barrier function in atopic dermatitis-An Update. Int J Mol Sci 2021;22:8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Alexander H, Paller AS, Traidl-Hoffmann C, Beck LA, De Benedetto A, Dhar S, et al. The role of bacterial skin infections in atopic dermatitis: expert statement and review from the International Eczema Council Skin Infection Group. Br J Dermatol 2020;182:1331–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simpson EL, Villarreal M, Jepson B, Rafaels N, David G, Hanifin J, et al. Patients with atopic dermatitis colonized with Staphylococcus aureus have a distinct phenotype and endotype. J Invest Dermatol 2018;138:2224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Feuillie C, Vitry P, McAleer MA, Kezic S, Irvine AD, Geoghegan JA, et al. Adhesion of Staphylococcus aureus to corneocytes from atopic dermatitis patients is controlled by natural moisturizing factor levels. mBio 2018;9: e01184–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Towell AM, Feuillie C, Vitry P, Da Costa TM, Mathelie-Guinlet M, Kezic S, et al. Staphylococcus aureus binds to the N-terminal region of corneodesmosin to adhere to the stratum corneum in atopic dermatitis. Proc Natl Acad Sci U S A 2021;118: e2014444118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li S, Villarreal M, Stewart S, Choi J, Ganguli-Indra G, Babineau DC, et al. Altered composition of epidermal lipids correlates with Staphylococcus aureus colonization status in atopic dermatitis. Br J Dermatol 2017;177:e125–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Altunbulakli C, Reiger M, Neumann AU, Garzorz-Stark N, Fleming M, Huelpuesch C, et al. Relations between epidermal barrier dysregulation and Staphylococcus species-dominated microbiome dysbiosis in patients with atopic dermatitis. J Allergy Clin Immunol 2018;142:1643–7. e12. [DOI] [PubMed] [Google Scholar]
- 131.Darlenski R, Kozyrskyj AL, Fluhr JW, Caraballo L. Association between barrier impairment and skin microbiota in atopic dermatitis from a global perspective: unmet needs and open questions. J Allergy Clin Immunol 2021;148(6): 1387–93. [DOI] [PubMed] [Google Scholar]
- 132.Flohr C, England K, Radulovic S, McLean WH, Campbel LE, Barker J, et al. Filaggrin loss-of-function mutations are associated with early-onset eczema, eczema severity and transepidermal water loss at 3 months of age. Br J Dermatol 2010;163:1333–6. [DOI] [PubMed] [Google Scholar]
- 133.Nemoto-Hasebe I, Akiyama M, Nomura T, Sandilands A, McLean WH, Shimizu H. Clinical severity correlates with impaired barrier in filaggrin-related eczema. J Invest Dermatol 2009;129:682–9. [DOI] [PubMed] [Google Scholar]
- 134.O’Regan GM, Kemperman PM, Sandilands A, Chen H, Campbell LE, Kroboth K, et al. Raman profiles of the stratum corneum define 3 filaggrin genotype-determined atopic dermatitis endophenotypes. J Allergy Clin Immunol 2010;126:574–80. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hubiche T, Ged C, Benard A, Leaute-Labreze C, McElreavey K, de Verneuil H, et al. Analysis of SPINK 5, KLK 7 and FLG genotypes in a French atopic dermatitis cohort. Acta Derm Venereol 2007;87:499–505. [DOI] [PubMed] [Google Scholar]
- 136.Seidenari S, Belletti B, Schiavi ME. Skin reactivity to sodium lauryl sulfate in patients with respiratory atopy. J Am Acad Dermatol 1996;35:47–52. [DOI] [PubMed] [Google Scholar]
- 137.Gupta J, Grube E, Ericksen MB, Stevenson MD, Lucky AW, Sheth AP, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol 2008;121:725–30. e2. [DOI] [PubMed] [Google Scholar]
- 138.Danby SG, Al-Enezi T, Sultan A, Chittock J, Kennedy K, Cork MJ. The effect of aqueous cream BP on the skin barrier in volunteers with a previous history of atopic dermatitis. Br J Dermatol 2011;165:329–34. [DOI] [PubMed] [Google Scholar]
- 139.Lyubchenko T, Collins HK, Goleva E, Leung DYM. Skin tape sampling technique identifies proinflammatory cytokines in atopic dermatitis skin. Ann Allergy Asthma Immunol 2021;126:46–53. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pavel AB, Renert-Yuval Y, Wu J, Del Duca E, Diaz A, Lefferdink R, et al. Tape strips from early-onset pediatric atopic dermatitis highlight disease abnormalities in nonlesional skin. Allergy 2021;76:314–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Berdyshev E, Goleva E, Bronova I, Bronoff AS, Hoffman BC, Ramirez-Gama MA, et al. Unique skin abnormality in patients with peanut allergy but no atopic dermatitis. J Allergy Clin Immunol 2021;147:361–7. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.He H, Olesen CM, Pavel AB, Clausen ML, Wu J, Estrada Y, et al. Tape-strip proteomic profiling of atopic dermatitis on dupilumab identifies minimally invasive biomarkers. Front Immunol 2020;11:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guttman-Yassky E, Diaz A, Pavel AB, Fernandes M, Lefferdink R, Erickson T, et al. Use of tape strips to detect immune and barrier abnormalities in the skin of children with early-onset atopic dermatitis. JAMA Dermatol 2019;155:1358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hulshof L, Hack DP, Hasnoe QCJ, Dontje B, Jakasa I, Riethmuller C, et al. A minimally invasive tool to study immune response and skin barrier in children with atopic dermatitis. Br J Dermatol 2019;180:621–30. [DOI] [PubMed] [Google Scholar]
- 145.Tagami H Electrical measurement of the hydration state of the skin surface in vivo. Br J Dermatol 2014;171(Suppl 3):29–33. [DOI] [PubMed] [Google Scholar]
- 146.Panther DJ, Jacob SE. The importance of acidification in atopic eczema: an underexplored avenue for treatment. J Clin Med 2015;4:970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Caspers PJ, Lucassen GW, Carter EA, Bruining HA, Puppels GJ. In vivo confocal Raman microspectroscopy of the skin: noninvasive determination of molecular concentration profiles. J Invest Dermatol 2001;116:434–42. [DOI] [PubMed] [Google Scholar]
- 148.Egawa M Raman microscopy for skin evaluation. Analyst 2021;146:1142–50. [DOI] [PubMed] [Google Scholar]
- 149.Ni Chaoimh C, Nico C, Puppels GJ, Caspers PJ, Wong X, Common JE, et al. In vivo Raman spectroscopy discriminates between FLG loss-of-function carriers vs wild-type in day 1–4 neonates. Ann Allergy Asthma Immunol 2020;124:500–4. [DOI] [PubMed] [Google Scholar]
- 150.Braun RP, Mangana J, Goldinger S, French L, Dummer R, Marghoob AA. Electrical impedance spectroscopy in skin cancer diagnosis. Dermatol Clin 2017;35:489–93. [DOI] [PubMed] [Google Scholar]
- 151.Rinaldi AO, Korsfeldt A, Ward S, Burla D, Dreher A, Gautschi M, et al. Electrical impedance spectroscopy for the characterization of skin barrier in atopic dermatitis. Allergy 2021;76:3066–79. [DOI] [PubMed] [Google Scholar]
- 152.Kubo A, Nagao K, Yokouchi M, Sasaki H, Amagai M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med 2009;206:2937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Elias PM, Steinhoff M. Outside-to-inside” (and now back to “outside”) pathogenic mechanisms in atopic dermatitis. J Invest Dermatol 2008;128:1067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.De Benedetto A, Kubo A, Beck LA. Skin barrier disruption: a requirement for allergen sensitization? J Invest Dermatol 2012;132:949–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Akdis CA, Arkwright PD, Bruggen MC, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy 2020;75:1582–605. [DOI] [PubMed] [Google Scholar]
- 156.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 2013;14:536–42. [DOI] [PubMed] [Google Scholar]
- 157.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest 1998;101:1614–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Takahashi S, Ishida A, Kubo A, Kawasaki H, Ochiai S, Nakayama M, et al. Homeostatic pruning and activity of epidermal nerves are dysregulated in barrier-impaired skin during chronic itch development. Sci Rep 2019;9: 8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jensen JM, Weppner M, Dahnhardt-Pfeiffer S, Neumann C, Brautigam M, Schwarz T, et al. Effects of pimecrolimus compared with triamcinolone acetonide cream on skin barrier structure in atopic dermatitis: a randomized, double-blind, right-left arm trial. Acta Derm Venereol 2013;93:515–9. [DOI] [PubMed] [Google Scholar]
- 160.Aschoff R, Schwanebeck U, Brautigam M, Meurer M. Skin physiological parameters confirm the therapeutic efficacy of pimecrolimus cream 1% in patients with mild-to-moderate atopic dermatitis. Exp Dermatol 2009;18: 24–9. [DOI] [PubMed] [Google Scholar]
- 161.Jensen JM, Pfeiffer S, Witt M, Brautigam M, Neumann C, Weichenthal M, et al. Different effects of pimecrolimus and betamethasone on the skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol 2009;123:1124–33. [DOI] [PubMed] [Google Scholar]
- 162.Bissonnette R, Pavel AB, Diaz A, Werth JL, Zang C, Vranic I, et al. Crisaborole and atopic dermatitis skin biomarkers: an intrapatient randomized trial. J Allergy Clin Immunol 2019;144:1274–89. [DOI] [PubMed] [Google Scholar]
- 163.Guttman-Yassky E, Ungar B, Malik K, Dickstein D, Suprun M, Estrada YD, et al. Molecular signatures order the potency of topically applied anti-inflammatory drugs in patients with atopic dermatitis. J Allergy Clin Immunol 2017;140:1032–42. e13. [DOI] [PubMed] [Google Scholar]
- 164.Kim J, Kim BE, Lee J, Han Y, Jun HY, Kim H, et al. Epidermal thymic stromal lymphopoietin predicts the development of atopic dermatitis during infancy. J Allergy Clin Immunol 2016;137:1282–5. e4. [DOI] [PubMed] [Google Scholar]
- 165.Hamilton JD, Suarez-Farinas M, Dhingra N, Cardinale I, Li X, Kostic A, et al. Dupilumab improves the molecular signature in skin of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol 2014;134: 1293–300. [DOI] [PubMed] [Google Scholar]
- 166.Callewaert C, Nakatsuji T, Knight R, Kosciolek T, Vrbanac A, Kotol P, et al. IL-4Ralpha blockade by dupilumab decreases Staphylococcus aureus colonization and increases microbial diversity in atopic dermatitis. J Invest Dermatol 2020;140:191–202. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Guttman-Yassky E, Bissonnette R, Ungar B, Suarez-Farinas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol 2019;143:155–72. [DOI] [PubMed] [Google Scholar]
- 168.Rohner MH, Thormann K, Cazzaniga S, Yousefi S, Simon HU, Schlapbach C, et al. Dupilumab reduces inflammation and restores the skin barrier in patients with atopic dermatitis. Allergy 2021;76:1268–70. [DOI] [PubMed] [Google Scholar]
- 169.Lee SJ, Kim SE, Shin KO, Park K, Lee SE. Dupilumab therapy improves stratum corneum hydration and skin dysbiosis in patients with atopic dermatitis. Allergy Asthma Immunol Res 2021;13:762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chovatiya R, Paller AS. JAK inhibitors in the treatment of atopic dermatitis. J Allergy Clin Immunol 2021;148:927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol 2015;136:667–77. e7. [DOI] [PubMed] [Google Scholar]
- 172.Pavel AB, Song T, Kim HJ, Del Duca E, Krueger JG, Dubin C, et al. Oral Janus kinase/SYK inhibition (ASN002) suppresses inflammation and improves epidermal barrier markers in patients with atopic dermatitis. J Allergy Clin Immunol 2019;144:1011–24. [DOI] [PubMed] [Google Scholar]
- 173.Paller AS, Stein Gold L, Soung J, Tallman AM, Rubenstein DS, Gooderham M. Efficacy and patient-reported outcomes from a phase 2b, randomized clinical trial of tapinarof cream for the treatment of adolescents and adults with atopic dermatitis. J Am Acad Dermatol 2021;84:632–8. [DOI] [PubMed] [Google Scholar]
- 174.Furue M, Hashimoto-Hachiya A, Tsuji G. Aryl hydrocarbon receptor in atopic dermatitis and psoriasis. Int J Mol Sci 2019;20:5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.van Zuuren EJ, Fedorowicz Z, Christensen R, Lavrijsen A, Arents BWM. Emollients and moisturisers for eczema. Cochrane Database Syst Rev 2017;2: CD012119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Simpson EL, Chalmers JR, Hanifin JM, Thomas KS, Cork MJ, McLean WH, et al. Emollient enhancement of the skin barrier from birth offers effective atopic dermatitis prevention. J Allergy Clin Immunol 2014;134:818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chalmers JR, Haines RH, Bradshaw LE, Montgomery AA, Thomas KS, Brown SJ, et al. Daily emollient during infancy for prevention of eczema: the BEEP randomised controlled trial. Lancet 2020;395:962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lowe AJ, Su JC, Allen KJ, Abramson MJ, Cranswick N, Robertson CF, et al. A randomized trial of a barrier lipid replacement strategy for the prevention of atopic dermatitis and allergic sensitization: the PEBBLES pilot study. Br J Dermatol 2018;178:e19–21. [DOI] [PubMed] [Google Scholar]
- 179.Perkin MR, Logan K, Marrs T, Radulovic S, Craven J, Boyle RJ, et al. Association of frequent moisturizer use in early infancy with the development of food allergy. J Allergy Clin Immunol 2021;147:967–76. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Skjerven HO, Rehbinder EM, Vettukattil R, LeBlanc M, Granum B, Haugen G, et al. Skin emollient and early complementary feeding to prevent infant atopic dermatitis (PreventADALL): a factorial, multicentre, cluster-randomised trial. Lancet 2020;395:951–61. [DOI] [PubMed] [Google Scholar]