

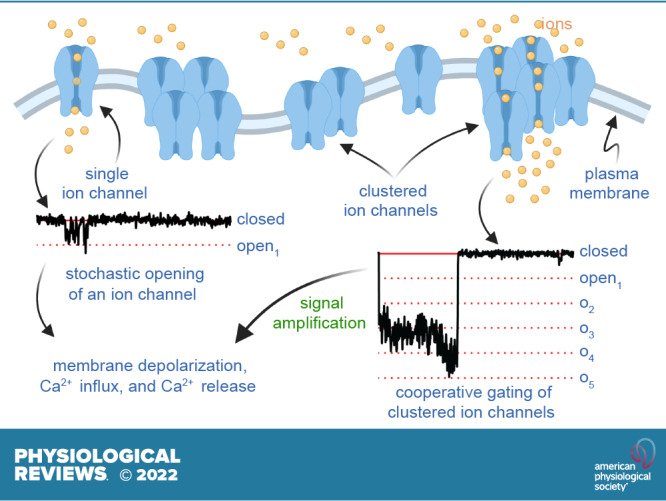
Keywords: calcium signaling, channel clustering, cooperative gating, excitability, stochastic self-assembly
Abstract
Ion channels play a central role in the regulation of nearly every cellular process. Dating back to the classic 1952 Hodgkin–Huxley model of the generation of the action potential, ion channels have always been thought of as independent agents. A myriad of recent experimental findings exploiting advances in electrophysiology, structural biology, and imaging techniques, however, have posed a serious challenge to this long-held axiom, as several classes of ion channels appear to open and close in a coordinated, cooperative manner. Ion channel cooperativity ranges from variable-sized oligomeric cooperative gating in voltage-gated, dihydropyridine-sensitive CaV1.2 and CaV1.3 channels to obligatory dimeric assembly and gating of voltage-gated NaV1.5 channels. Potassium channels, transient receptor potential channels, hyperpolarization cyclic nucleotide-activated channels, ryanodine receptors (RyRs), and inositol trisphosphate receptors (IP3Rs) have also been shown to gate cooperatively. The implications of cooperative gating of these ion channels range from fine-tuning excitation-contraction coupling in muscle cells to regulating cardiac function and vascular tone, to modulation of action potential and conduction velocity in neurons and cardiac cells, and to control of pacemaking activity in the heart. In this review, we discuss the mechanisms leading to cooperative gating of ion channels, their physiological consequences, and how alterations in cooperative gating of ion channels may induce a range of clinically significant pathologies.
CLINICAL HIGHLIGHTS
Ion channels regulate multiple physiological processes including excitability, contraction, neurotransmitter release, and gene expression. Changes in the function of many channels have been linked to pathological conditions such as arrhythmias, epilepsy, and hypertension.
In this review, we discuss a vast and growing body of work that suggests that many ion channels do not gate independently, as originally assumed. Instead, many Ca2+, Na+, and K+ channels seem to open and close cooperatively.
The physiological and pathological implications of cooperative gating are profound. For example, positive cooperativity amplifies Na+ and Ca2+ entry into cardiac muscle. Enhanced cooperativity between clustered dihydropyridine-sensitive Ca2+ channels has been suggested to be critical for sympathetic stimulation of the heart, but when excessive it causes cardiac arrhythmias and hypertension.
Enhanced cooperativity among KNa1.1 channels has been linked to epilepsy.
The development of rational strategies for the targeted modulation of ion channel cooperativity may allow tuning of Ca2+ and electrical signaling in health and disease.
1. INTRODUCTION
Nearly 70 years ago, Hodgkin and Huxley established the foundations of modern biophysics with the advent of their quantitative model for action potential (AP) generation and propagation in neurons (1). This seminal work has been subsequently modified and expanded to form the basis of our understanding of electrical signaling in all excitable cells (2). A central tenet of the Hodgkin–Huxley model was that individual channels gate independently and that the macrocurrents recorded across the cell membrane reflect the ensemble behavior of these quantal conductance elements. Direct testing of the model in general, and the independence principle specifically, became feasible with the advent of the patch-clamp technique (3, 4), which permitted investigators to obtain ultra-low noise current records with high temporal resolution from a small area of membrane (<1 μm2) produced by the putative opening of single ion channels.
Although the Hodgkin–Huxley model accommodates the random simultaneous openings of multiple channels, the probability of a set of channels behaving synchronously is expected to decrease as the number of channels in the set increases. A simple way to explain this is to use the example of flipping a “fair” coin, in which each flip is independent from the other. The probability of observing a tail or a head is 0.5 for every trial, but the probability of observing two tails in a row is 0.52 (i.e., 0.25), that of three tails in a row is 0.53 (i.e., 0.13), and so on. If individual membrane channels behave in an analogous independent fashion, then the current amplitude frequency distributions will be fit with a Poisson function (5). Similarly, the timing between successive channel openings should be uncorrelated. Many ion channels act this way, further fueling the idea that ion channels gate randomly and independently from one another (6).
A limitation of the patch-clamp technique is that it monitors a membrane area of <1 μm2, severely limiting the number of channels that are sampled. Because most ion channels are randomly distributed in the surface membrane, the probability of a patch pipette encompassing a region of membrane containing a group of homogeneous channels is relatively low. Accordingly, recordings of the concerted openings of multiple channels are generally rare but were reported as early as the 1990s for Na+, hyperpolarization-activated cyclic nucleotide-gated (HCN), and Ca2+ channels (7–13).
The development of electron-multiplying charged-coupled device (EMCCD) cameras has facilitated the recording of optical Ca2+ signals from single channels or small clusters of channels with ultrahigh temporal and spatial resolution (14–17). This has afforded direct, detailed observation of rare and low-amplitude Ca2+ signals resulting from the openings of Ca2+-permeable ion channels in the surface membrane and intracellular organelles from relatively large areas of the cell (compared with patch clamp). Indeed, imaging systems such as confocal and evanescent field total internal reflection fluorescence (TIRF) microscopes have been used by multiple groups to detect Ca2+ influx events via small clusters of Ca2+-permeable channels in a wide range of cells (14, 16, 18–26), revealing that cooperative gating of Ca2+ channels is not as rare as previously thought.
At this point, it is important to distinguish the terms “cooperative gating” and “functional coupling” of ion channels. Cooperative gating of ion channels refers to when two or more ion channels from the group open in concert because of physical interactions with one another within a cluster. In this gating modality, gating of one channel is allosterically communicated to other attached channels, increasing their probability of gating. Functional coupling of ion channels refers to interactions between different ion channels in macromolecular complexes displaying cross talk with one another in signaling microdomains. A primary example of functional coupling is the action of Ca2+ flux through one channel group that acts as a second messenger to facilitate gating of a molecularly distinct group of ion channels.
The principal focus of this review is to present the available evidence for cooperative gating of ion channels. We focus on key classes of membrane channels for which there is substantial evidence for this gating modality. We discuss putative mechanisms by which the cooperativity is produced, the physiological implications of cooperative gating in excitable tissues, and possible pathologies that may result from alterations in this behavior. Finally, we identify key gaps in our present knowledge and propose new research avenues for this emerging field of inquiry.
2. STOCHASTIC SELF-ASSEMBLY OF ION CHANNEL CLUSTERS
All models of cooperative gating of ion channels proposed to date are predicated on having the same ion channel aggregated into dense clusters where the centroids of adjacent members are separated by slightly more than the radius of their cross-sectional areas (5, 27, 28). This proximity between the same ion channels within clusters allows physical interactions between adjacent channels to occur, leading to amplification of channel function. Thus, it is pertinent to discuss current views on how clusters of membrane channels are generated.
The complex process by which ion channels are inserted into a membrane begins with the transcription of the genes encoding the pore-forming and accessory subunits into messenger RNA (mRNA). These mRNAs are subsequently translated by ribosomes and transfer RNA (tRNA) molecules associated with the endoplasmic reticulum (ER) to polypeptides. The growing polypeptides of transmembrane channel subunits are progressively translocated and folded into the ER membrane. Once the full protein is synthesized and integrated in the membrane, a vesicle containing this protein forms and fuses with the cis Golgi face (FIGURE 1). As the channel proteins transit through the Golgi stack, they eventually reach the trans Golgi network (TGN). Through this process they undergo posttranslational processing, including N-linked glycosylation. Ion channels leave the Golgi apparatus via vesicles that are transported by molecular motors moving on microtubules that cross over the trans Golgi network and are anchored on the plasma membrane. These vesicles eventually fuse with the surface membrane (29, 30). Once they are delivered to the surface membrane, gating of the new channels can regulate excitability as well as multiple signaling cascades. The ion channels will remain in the plasma membrane until they are removed and either recycled or degraded via endocytic pathways (31, 32).
FIGURE 1.
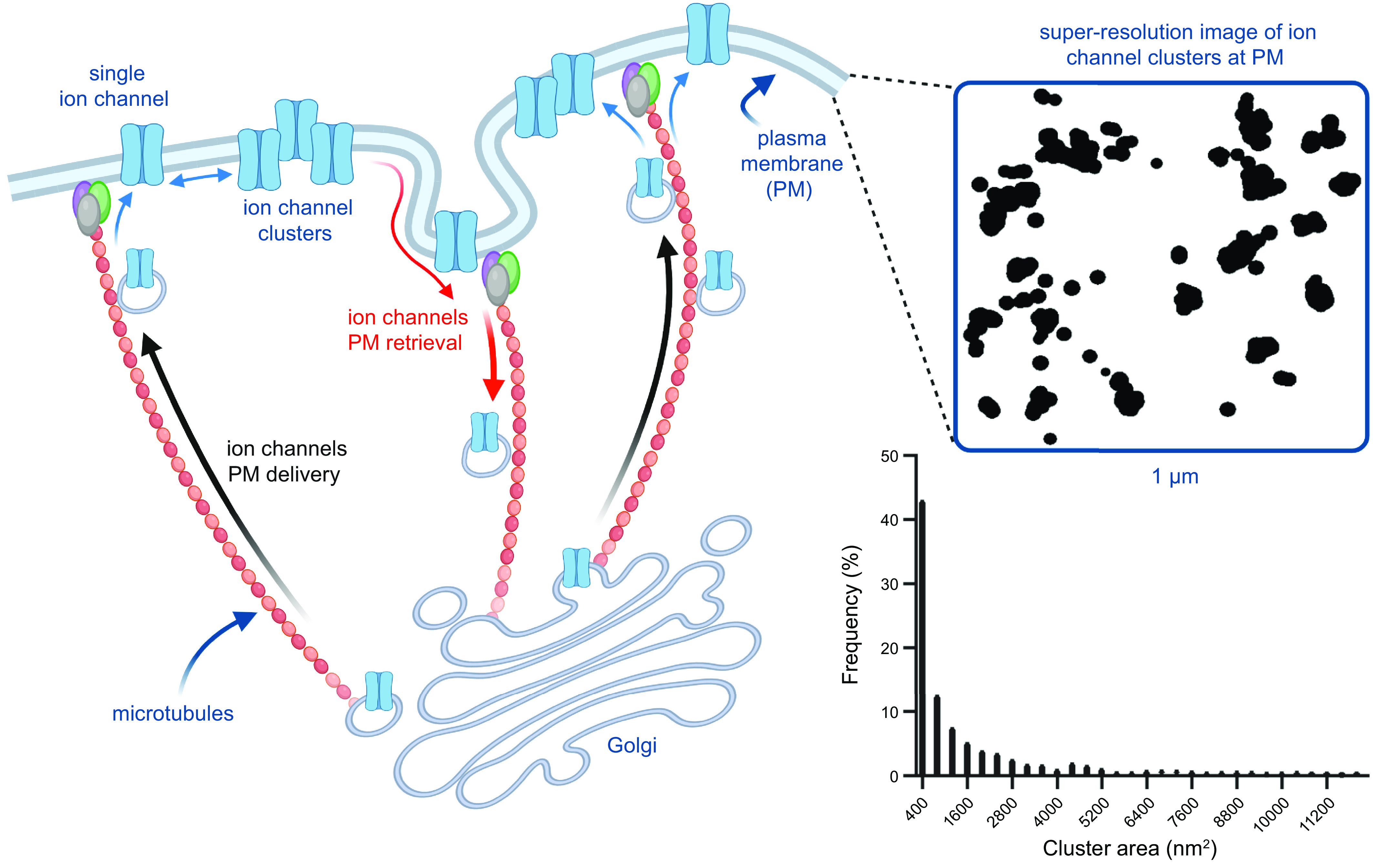
Ion channel proteins translocate to the membrane of the rough endoplasmic reticulum (ER), traffic to the Golgi, and are transported to the surface membrane on microtubules, where they form clusters. Left: cartoon depicting the synthesis, membrane translocation, processing, and trafficking of ion channels to the surface membrane. Top right: superresolution image of ion channel clusters in the surface membrane. Bottom right: histogram of ion channel cluster areas from images like that above. The purple, green, and gray ovals at the tip of the microtubules cartoon represent microtubule-anchoring/binding proteins such as BIN1. Histogram was modified from Sato et al. (5) with permission. Cartoon created with Biorender.com.
There is a large and still growing body of evidence showing that the distributions of ion channels vary along a cell’s membranous surface (33–36). The studies that have examined how ion channels organize in the plasma membrane involved detailed analyses of electron micrographs (35–39), TIRF and confocal images (40), and, more recently, superresolution microscopy (12, 28, 41–46). The majority of ion channels examined to date aggregate into dense clusters (see FIGURE 1). For example, in neurons, N-methyl-d-aspartate (NMDA) channels aggregate in dendritic and somatic membranes (47). Similarly, voltage-gated Na+, Ca2+, and K+ channels that produce action potentials required for neurotransmitter release are specifically expressed in the axon terminals (33, 34, 48, 49). The same is true for striated and smooth muscle, where dihydropyridine-sensitive voltage-gated Ca2+ channels form clusters within the sarcolemma of these cells (36, 38, 42, 45, 46, 50–52).
This clustering of ion channels seems to be critical for multiple physiological processes in both neurons and muscle tissue. Indeed, the clustering of voltage-gated Ca2+ channels is necessary for the amplification of Ca2+ influx that is required to trigger the release neurotransmitter at neuronal terminals and to initiate the process of excitation-contraction (EC) coupling in muscle (11, 15, 24). Furthermore, recent studies indicate that ion channels involved in cooperative signaling cascades cocluster. A notable example is the coclustering of large-conductance Ca2+-activated K+ (BK) channels and CaV1.3 channels in neurons (41, 53–55). In this case, cooperative gating of CaV1.3 channels clusters provides the necessary Ca2+ signal to activate nearby BK channel clusters. We discuss additional examples of this type of organization and signaling modality later in this review (see sects. 4.4 and 8.3).
Recently, Sato et al. (5) and Baddeley et al. (28) implemented a combination of in silico and experimental approaches to investigate the mechanisms regulating channel clustering in the membrane. They found that the distributions of clusters of six types of channels [i.e., CaV1.2, BK, transient receptor potential (TRP) vanilloid (TRPV)4, CaV1.3S, CaV1.3L, and ryanodine receptors (RyRs)] were all described by a single exponential function regardless of their expression in different cell types (FIGURE 1). Inositol 1,4,5-trisphosphate receptors (IP3Rs) have similar distributions in the sarcoplasmic reticulum (SR) and endoplasmic reticulum (56–58). These studies suggest that the presence of CaV1.2, BK, TRPV4, CaV1.3S, RyR, and IP3R clusters in cellular membranes may be the result of a stochastic self-assembly process. Whether active mechanisms of aggregation of ion channels may also contribute to this process is currently unclear. Moreover, although it is possible that not all channels are organized into clusters via stochastic self-assembly processes (59–63), the preponderance of the data suggests that clustering is the default mode of organization of ion channels in the plasma membrane as well as in the endo/sarcoplasmic reticulum of cells. In sects. 3–10, we describe current understanding of clustering and cooperative gating in several classes of ion channels.
3. VOLTAGE-GATED Ca2+ CHANNELS
Voltage-gated Ca2+ (CaV) channels are expressed in excitable cells and nonexcitable cells (reviewed in Ref. 64), where they respond to electrical depolarizations by shifting their voltage sensors upward to open the channel. This movement and/or the resultant Ca2+ influx through the channel pore is an essential trigger for many physiological processes including contraction of skeletal, smooth, and cardiac muscle, hormone secretion, regulation of gene expression, and neurotransmission. The structure and function of CaV channels have been extensively reviewed elsewhere (65–74) and are thus only briefly addressed here.
CaV channels, like the evolutionarily related voltage-gated Na+ (NaV) channels, contain a large, pore-forming α1-subunit that distinguishes the channel within this 10-member channel family (FIGURE 2A) (69). The 10 types of CaV channels are further categorized into three subfamilies, CaV1, CaV2, and CaV3, based on their pharmacological and biophysical profiles as well as their amino acid sequence similarity (75). Channels within each subfamily share >70% homology, whereas across subfamilies the channels are <40% homologous (75). The α1-subunits are 170- to 250-kDa (65, 73) single polypeptides, with intracellularly located NH2 and COOH termini flanking four repeating domains (DI–DIV), each with six transmembrane segments (S1–S6). Voltage sensitivity is conferred by segments S1–S4 (termed the voltage sensing domain; VSD), and S5–S6 and the associated loop from each of the four repeats combine to form the pore domain. Recent cryo-electron microscopy (cryo-EM) studies of purified skeletal muscle CaV1.1 channels have revealed that the four repeat domains of α1S are arranged in a clockwise manner about the central pore (76, 77). This spatial arrangement is thought to be conserved throughout the eukaryotic CaV and NaV channel families (72).
FIGURE 2.
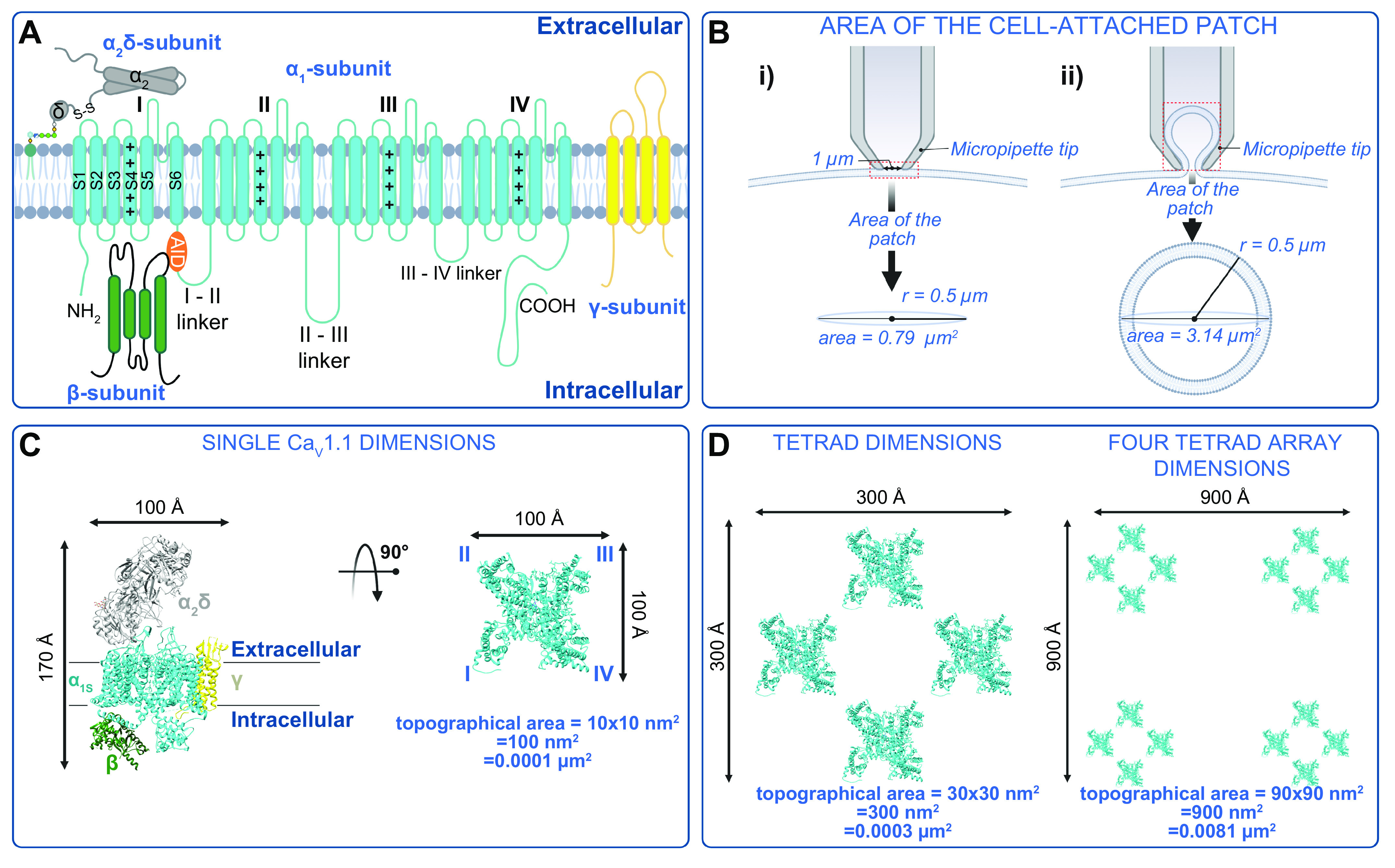
Structure and dimensions of voltage-gated Ca2+ (CaV) channels. A: illustration of the pore-forming CaVα1 and auxiliary CaVβ, CaVα2δ, and CaVγ. AID, alpha-interacting domain. B: 2 illustrations of patch pipettes forming a gigaseal enabling voltage control and single-channel recordings from the underlying patch of membrane. The pipette may enclose either a circle (i) or a 3-dimensional Ω-shaped patch of membrane (ii) that may contain a large number of ion channels. C: dimensions of CaV1.1 (PDB: 5GJV) from Wu et al. (76) (left) and the topological view/footprint of a single α1S pore-forming subunit (right), illustrating that many channels can theoretically be housed in a single patch. D: the dimensions of a single CaV1.1 tetrad (left) and an array of 4 tetrads (right). Figure created with Biorender.com.
Although the α1-subunits dictate the main channel properties, CaV1 and CaV2 channels are multimeric complexes comprised of the α1-subunit along with up to three auxiliary (or accessory) subunits that can modify channel kinetics and play roles in channel trafficking and regulation (FIGURE 2A) (78, 79). These subunits include the cytosolic CaVβ, the CaVα2δ on the extracellular leaflet of the sarcolemma bilayer, and the transmembrane CaVγ. CaV3 channels can be regulated by these accessory subunits (80–82) but do not appear to require coassembly with them (68, 83, 84).
3.1. L-Type Ca2+ Channels
The CaV1 subfamily (CaV1.1–CaV1.4) constitutes the so-called L-type calcium channels, with L signifying that they generate large, long-lasting (slowly inactivating) currents. CaV1 channels are expressed in myocytes, cardiac pacemaker cells, nerves, endocrine cells, the inner ear, and the retina and are characterized by their pharmacological sensitivity to organic Ca2+ channel blockers including dihydropyridines, phenylalkylamines, and benzothiazepines (85). They contain α1-subunits encoded by cacna1s, c, d, and f. Functional channels are expressed in complex with 1:1 stochiometric combinations of various isoforms of CaVβ, CaVα2δ, and sometimes CaVγ (68–70).
Cooperative gating has been observed and reported in a diverse variety of voltage- and ligand-gated ion channels, as we comprehensively review here. It is notable, however, that in recent years major advances in our understanding of this phenomenon have been extracted from studies of voltage-gated calcium channels, and most specifically in three of the L-type calcium channels, namely CaV1.1, CaV1.2, and CaV1.3. We summarize the current thinking in sects. 3.2–3.7.
3.2. CaV1.1 Channel Structure and Function
The skeletal muscle isoform of the L-type Ca2+ channel was the first of the voltage-gated calcium channels to be cloned (86) and coincidentally was also the first to be proposed to exhibit cooperative gating behavior (7). These channels, still sometimes referred as 1,4-dihydropyridine receptors (DHPRs) because of their responsivity to pharmacological agonists and antagonists from this class of drugs, are abundantly expressed in skeletal muscle, where they are physically tethered to juxtapositioned type 1 ryanodine receptors (RyR1s) in the triad junctions. Freeze-fracture electron microscopy studies revealed that CaV1.1s are arranged into tetrads (4 channel groups), with each individual CaV1.1 aligning with a single RyR1 homotetramer (87). The RyR1s are arranged in a checkerboard formation, where every other RyR1 channel is associated with a CaV1.1 tetrad and in which the RyR1s are also reported to exhibit cooperative gating behavior (88) (see sect. 7.2). Because of the physical coupling between the t-tubule-localized CaV1.1 and the sarcoplasmic reticulum (SR)-localized RyR1, Ca2+ influx through CaV1.1 is not mandatory for excitation-contraction (EC) coupling (89). Instead, the action potential-driven movement of the CaV1.1 voltage sensors is allosterically conveyed to the attached RyR1, inducing a conformational change in the RyR1 that results in Ca2+ release from the SR and subsequent activation of the myofilaments. This sarcolemmal Ca2+ influx-independent form of RyR1-mediated Ca2+ entry, where CaV1.1 serves solely as a voltage sensor, has been termed excitation-coupled Ca2+ entry (ECCE) (90). The details of this physical interaction and mechanical gating mechanism are still not fully elucidated despite decades of work to reveal to essential interaction sites. However, recent near-atomic-resolution structural studies of both CaV1.1 (76, 77) and RyR1 (91) have led to the development of a speculative theory on how this may occur (92). Although the details remain unresolved, it is definitively known that there are at least four critical protein components of the triad, including CaV1.1, RyR1, auxiliary subunit of calcium channels CaVβ1a [the one and only CaVβ isoform expressed in skeletal muscle (93)], and adaptor protein STAC3. The absence of any of one of these proteins precludes EC coupling (reviewed in Ref. 94). Pathophysiologies associated with mutations in CaV1.1 include hypokalemic periodic paralysis (HypoPP-1) and malignant hyperthermia (MHS5) (68).
3.3. Cooperativity Among CaV1.1 Channels
Αn early paper in this field from the Schindler group reported cooperativity and clustering of purified CaV1.1 channels when they were isolated from guinea pig skeletal muscle and reconstituted into planar lipid bilayers (7). They observed a time dependence of channel conductance in which larger channel conductance levels emerged over time after bilayer formation: Immediately after planar bilayer formation, channel openings with a 0.9-pS conductance were observed (with 100 mM BaCl2 as the charge carrier in the pipette). This result was suspected to represent the monomeric channel conductance. However, as time passed after membrane formation and more channels were incorporated that presumably had time to cluster and functionally interact, larger conductance values emerged corresponding to oligomeric channel openings of 4, 8, 16, 24, 32, 40, up to 64 channels. Given the propensity of these channels to form tetragonal arrays, it is interesting that each of these oligomeric openings is an even-integer quadruplet of the ∼1-pS single-channel conductance. Independently gating channel openings are predicted to follow a binomial distribution such that, in a multichannel patch, one would expect to observe most frequently a conductance equivalent to a single channel opening, with multiple channel openings occurring with progressively lower frequencies. However, binomial analysis of the conductance amplitude frequency histogram from CaV1.1 channels revealed a positive deviation, indicative of cooperative gating, where the 40-pS conductance was observed to occur more frequently than 4-, 24-, or 32-pS conductance levels. This preference for the 40-pS conductance, equivalent to a simultaneous oligomeric opening of 10 channels, is highly statistically improbable if these channels are independently gating entities.
At first glance, the idea that 64 channels might be captured in a cell-attached patch may seem unlikely. However, some simple calculations confirm that this is not such an unlikely occurrence if one assumes a pipette tip diameter of 1 µm and an area of membrane under the patch that should initially be well approximated by the area of a circle (πr2). However, upon gigaseal as a three-dimensional Ω-shaped section of membrane is pulled into the pipette, the area may be better described by a sphere (4πr2) (FIGURE 2B). Thus, the patch area could be 0.79–3.14 µm2. A key question then is, how many channels or channel tetrads could fit into that space? For single channels, cryo-EM structures of CaV1.1 have revealed their dimensions, confirming their widest aspect as 100 Å (10 nm) (76, 77) (FIGURE 2C). Assuming then that a channel takes up 10 × 10 nm2 = 100 nm2 = 0.0001 µm2 and that they are as densely packed as possible, 7,900–34,400 channels could theoretically occupy the area of a typical patch. Even assuming 1/100th of that density, that still allows 64 channels to easily fit into the area of a typical patch. Knowledge accumulated from EM studies suggests that a CaV1.1 tetrad can fit into a 30 × 30 nm2 = 300 nm2 = 0.0003 µm2 area (95–97) (FIGURE 2D). We also know that tetrads line up with every other RyR1. If we assume a tetragonal array of four tetrads, the dimensions would be 90 × 90 nm2 = 8,100 nm2 = 0.0081 µm2 (FIGURE 2D). To get to 64 channels, that would be 16 tetrads with an assumed array dimension of 120 × 210 nm2 = 25,200 nm2 = 0.0252 µm2, which could still easily fit into the area of a typical patch.
Unfortunately, this paper from the Schindler group (7) is still largely ignored by the skeletal muscle and CaV1.1 field, but it has gained traction in recent years, as evidenced by an increasing number of citations as other groups began to report cooperativity in a diverse variety of ion channels. However, CaV1.1 cooperative gating has not been revisited. We speculate that this may be explained by the fact that, historically, CaV1.1 channels have been difficult to express in heterologous expression systems. This difficulty has recently been obviated by the finding that coexpression of STAC3 permits robust trafficking and recording of these currents in mammalian cell lines including tsA-201 (98). With the emergence of this accessible heterologous expression system that permits the study of CaV1.1 channels in near isolation, the path is clear for a fresh study on CaV1.1 cooperativity. Indeed, many interesting questions as to how the channels interact remain to be answered, including 1) Is the interaction a direct physical coupling between adjacent channels such that the conformational change that occurs when one channel opens is allosterically conveyed to the attached channels, increasing their open probability (Po), or is it an indirect association that requires the presence of another protein that could bridge adjacent channels together? 2) How do adjacent tetrads interact? and 3) Is channel cooperativity affected by receptor signaling cascades? These questions should be addressed in future studies of CaV1.1. Addressing these questions will pave the way to determining whether cooperative gating of native CaV1.1 channels regulates skeletal muscle EC coupling.
3.4. CaV1.2 Channel Structure and Function
CaV1.2 was the second of the L-type channels to be cloned (99), hence the “2” in its name. This chronological nomenclature is maintained through the remainder of the L-type channel subfamily and indeed for each of the three voltage-gated calcium channel subfamilies (75). The α1C-subunit constitutes the pore-forming core of these L-type Ca2+ channels that forms multimers with at least CaVα2δ and CaVβ [with CaVβ2 being the most prevalent isoform in the heart, in particular CaVβ2b (100)] and CaVβ3 in smooth muscle (101) (FIGURE 2A). Associations of CaVα1C with CaVγ are still unclear in primary cells but do appear to occur in transiently transfected HEK293 cells (102). Despite the CaV1.2 channels often being referred to as the cardiac L-type Ca2+ channel, perhaps because they were originally cloned by screening cardiac cDNA libraries for homology with the skeletal muscle isoform, this is somewhat of an exclusionary misnomer given that CaV1.2 channels are expressed in a variety of tissues including vascular and visceral smooth muscle, brain, pancreas, adrenal gland, and, of course, cardiac muscle (65, 68). Within these tissues, CaV1.2 channels are involved in a wide range of fundamental physiological processes including cardiac (103–105) and smooth muscle (15, 74, 104, 106–109) EC coupling, insulin secretion via excitation-secretion coupling (104, 110, 111), and regulation of gene expression via excitation-transcription (ET) coupling (24, 112–115). In the brain, they shape neuronal firing and support neuronal plasticity, learning, and memory formation (116, 117). The critical role of CaV1.2 in many of these processes has been confirmed in mouse models with tissue-specific CaV1.2 knockout (110, 118–120) or in mice with a single T1066Y mutation in their CaVα1C that renders the channels dihydropyridine insensitive (104). This second strategy has been especially helpful in tissues where both CaV1.2 and CaV1.3 are expressed, as there remains no established pharmacological means to unequivocally distinguish between these two channels.
CaVα1C is extensively alternatively spliced, generating several splice variants that create even more structural and functional diversity in these channels (as reviewed in Refs. 121–123). Smooth muscle CaVα1C is predominantly expressed with exon 8, whereas the most highly expressed cardiac CaVα1C has exon 8a instead. Exon 8 confers higher sensitivity to dihydropyridine calcium channel-blocking drugs (123), contributing to their efficacy as antihypertensives. Exon 1 is also spliced to form at least three variants, a long NH2-terminal-encoding 1a (expressed in cardiomyocytes), a short NH2-terminal 1b (expressed in smooth muscle and brain), and an even shorter 1c (expressed in cerebral arteries) (65). The alternative splicing of exon 1 is also thought to alter sensitivity to the nondihydropyridine calcium channel blocker diltiazem. Accordingly, the smooth muscle variant 1b is about twice as sensitive to diltiazem than the cardiac 1a (123).
In cardiac muscle, influx of Ca2+ through CaV1.2 channels is obligatory, as the channels are not mechanically linked to the type 2 cardiac ryanodine receptors (RyR2s) that cluster on the other side of the 12- to 15-nm dyadic cleft (124, 125) on the junctional sarcoplasmic reticulum (jSR). Accordingly, instead of acting exclusively as a voltage sensor, cardiac CaV1.2 channels must open in response to membrane depolarization as the action potential propagates through the myocardium. This allows a small amount of Ca2+ to enter the cell. Influx of Ca2+ through a single CaV1.2 channel is known as a “Ca2+ sparklet” and can be imaged with Ca2+-sensitive fluorescent indicators (14, 126). This Ca2+ entry triggers further “Ca2+-induced Ca2+ release” (CICR) from ryanodine receptors on the SR, producing a “Ca2+ spark” (127–132). Synchronized activation of multiple sparks across the tens of thousands of dyads in a single cardiomyocyte (133), leads to a global elevation in intracellular Ca2+ concentration ([Ca2+]i) that activates the myofilaments and elicits contraction. Cardiac EC coupling has been extensively reviewed elsewhere (see Refs. 103, 134).
Immunogold labeling (38) and freeze-fracture electron microscopy studies (124, 135, 136) have revealed clustering of CaV1.2 channels in heart muscle. This clustering is now frequently examined in superresolution light microscopy studies (42, 50, 51, 137, 138). Channel clustering is a fundamental requirement for cooperative interactions between ion channels. Within clusters, channels are densely packed in close proximity of one another, making functional, physical interactions between adjacent channels plausible. Clustering of CaV1.2 channels in dyadic regions is thought to maximize the probability of triggering release from adjacent RyR2s during EC coupling (11, 42, 139, 140). The relationship between local Ca2+ influx and Ca2+ spark activation has been the subject of intense investigation. The majority of data support the view that, at negative potentials (−50 to −30 mV), where the driving force for Ca2+ influx is high, the opening of a single CaV1.2 channel produces a unitary current (iCa) of 0.5–0.4 pA (126, 141) that is sufficient to activate a spark (142–145). However, in humans, EC coupling takes place during phases 1 and 2 of the AP, when cardiomyocytes are much more depolarized (∼+50 mV). At these potentials, with supraphysiological 20 mM extracellular Ca2+ concentration ([Ca2+]o), a single CaV1.2 channel opening, with an iCa ≈ 150 fA, can activate a spark (126). However, in physiological 2 mM Ca2+, the calculated iCa is only 10 fA (139), ∼15-fold smaller than in 20 mM Ca2+. Is this a sufficient Ca2+ influx to reliably trigger a Ca2+ spark and permit contraction? Work from several laboratories suggests that it is not. Inoue and Bridge (139) reported that Ca2+ influx occurring at the peak of the AP (+50 mV) triggered Ca2+ sparks with a probability (Pspark) of almost unity (0.997) in rabbit ventricular myocytes. Given the low open probability (Po) of CaV1.2 channels at these potentials, they proposed that a cluster of 5–10 channels must be available to activate a spark with a high probability. Sobie and Ramay (140) and Polakova et al. (146) further concluded that because of the low iCa and brevity of the channel openings at these potentials, multiple channels must open simultaneously to reliably trigger a spark. However, the maximum Po of CaV1.2 channels with physiological [Ca2+]o is not unity (108, 139, 141, 147), with some of the higher estimates in unstimulated cells (i.e., cells examined without β-adrenergic agonists) being Po = 0.3. If all CaV1.2 channels gate independently, the probability of 5–10 channels opening simultaneously is much lower than Pspark (i.e., 0.35–0.310). So, how can one reconcile this high probability of a spark with a relatively low probability that multiple CaV1.2 channels open at once? Cooperative gating, as we discuss in sect. 3.5, can cause clusters of cardiac CaV1.2 channels to open simultaneously, driven by the channel in the cluster with the largest Po (11). Thus, cooperativity can explain this discrepancy, and we discuss the evidence for and the mechanisms underlying CaV1.2 channel cooperativity in sect. 3.5.
EC coupling in smooth muscle is different from that of cardiac and skeletal muscle, but one of the common elements is the essential presence and function of CaV1.2 channels (107, 149). Electro-mechanical coupling describes the process through which depolarization-activated Ca2+ influx leads to smooth muscle contraction. CaV1.2 channels are the primary mediators of this response, but Ca2+ influx across the smooth muscle sarcolemma can come from a variety of sources including T-type calcium channels, TRP channels, and Orai (74, 150–152). The incoming Ca2+ binds to calmodulin (CaM) with a stoichiometry of 4:1, and the resultant Ca2+·CaM activates myosin light chain (MLC) kinase (MLCK) that phosphorylates MLC in an ATP-dependent manner. This cascade results in actin-myosin cross-bridge formation and smooth muscle contraction (153). Demonstrating the importance of CaV1.2 channels for development and maintenance of vascular tone and reactivity, L-type calcium channel blockers are effective antihypertensive drugs (154).
Clustering of CaV1.2 channels has also been extensively reported in vascular smooth muscle, where it is also thought to facilitate interactions between adjacent channels (45, 46, 52, 155–157). Furthermore, CaV1.2 clustering has been reported in pancreatic β-cells at sites of secretory granule exocytosis (158), in sensory neurons from the superior cervical ganglia (159), in hippocampal neurons (49, 160–162), and in heterologous cell lines (42, 163–165). The evidence for cooperative interaction of CaV1.2, the mechanisms underlying it, and the physiological implications are discussed in sect. 3.5.
3.5. Cooperativity of CaV1.2 Channels in Health and Disease
The vast majority of the currently published data on CaV1.2 channel cooperativity has been collected in our laboratories, and certainly that is where the idea was first proposed and pursued. We tell the story of it here chronologically as it transpired. The first indication that CaV1.2 channels do not always gate in an independent manner came in 2005 when L. F. Santana’s group began using optical techniques to record CaV1.2 single-channel activity in vascular smooth muscle cells isolated from rat cerebral arteries (FIGURE 3A) (26). Their methods (14) entailed loading cells with a fluorescent Ca2+ indicator dye (in this first case it was fluo-5F) and EGTA via a patch pipette during whole cell voltage clamp, while the cells are held at −70 to −90 mV. TIRF imaging of the cell footprint is then performed at 90–100 Hz with an EMCCD camera. At the chosen membrane potentials, CaV1.2 channels exhibit low Po but high driving force, such that the opening of a channel, when it does occur, generates sufficient Ca2+ influx (with 20 mM external Ca2+) to produce a resolvable signal termed a Ca2+ sparklet when it binds to fluo-5F. The EGTA in the dialyzed solution then buffers the Ca2+ and acts as a sink, preventing the generation of a global Ca2+ transient, which would obscure the detection of single-channel events. This powerful approach, now utilized in multiple laboratories to record sparklets from diverse Ca2+-conducting ion channels (12, 23, 166–170), permits visualization of single-channel activity over the entire coverslip-adhered portion of the sarcolemma, providing a much larger sampling area than one can obtain with a patch pipette recording from a cell-attached patch (17, 20). The change in Ca2+ over time at active regions of interest (ROIs) is then plotted to generate traces that resemble single-channel electrophysiology recordings, albeit with a reduced sampling frequency (FIGURE 3A). With this broader view in which channel activity could be observed and compared across the dorsal sarcolemma, it was obvious that not all CaV1.2 channels behaved in the same way. In ROIs where multiple channels were present, some unusual and unexpected gating behavior was observed in which large, time-correlated multichannel openings occurred frequently. The striking synchrony with which these groups of multiple channels opened and closed suggested that the gating of one channel was influencing the gating of the others. In these regions, the channels also appeared to have a higher Po that generated “persistent” Ca2+ entry, while other sites displayed activity more consistent with the low Po of an independently gating single channel (13, 171). Ca2+ sparklets were eliminated by nifedipine, and the L-type calcium channel agonist BayK 8644 increased the probability of observing a Ca2+ sparklet. The frequency with which openings and/or closings of multiple channels were observed in perfect unison suggested that a subset of CaV1.2 channels were not gating independently but rather cooperatively, or, as we called it at the time, in a “seemingly coupled” manner. We later favored the terminology “cooperative gating” over “coupled gating,” as coupled implies 2 channels, but the simultaneous openings and closings we observed were not limited to 2 channels and often involved larger groups of 6–10 channels (42). Of course, the imaging experiments had a technical limitation in that we could only sample as fast as the camera could read off the data, and even with small imaging areas our technical limit was ∼100 Hz. So, the possibility existed that we were simply sampling too slowly and missing the stepwise notches one might expect to see if a few independently gating channels opened or closed one after the other. Would we still observe these apparently simultaneous multichannel gating events in 10- to 20-kHz single-channel electrophysiology experiments? To address this issue, we made cell-attached patch-clamp electrophysiological recordings and obtained analogous results (FIGURE 3B), and thus the notion that CaV1.2 channels exhibited cooperative gating behavior was born (13). This contention that CaV1.2 channels could gate in a nonindependent manner was, and still is, somewhat controversial, given that it confutes the long-standing Hodgkin–Huxley hypothesis that ion channels gate in a mutually independent manner (1). It has certainly made for some lively discussions at the Biophysical Society Annual Meetings!
FIGURE 3.
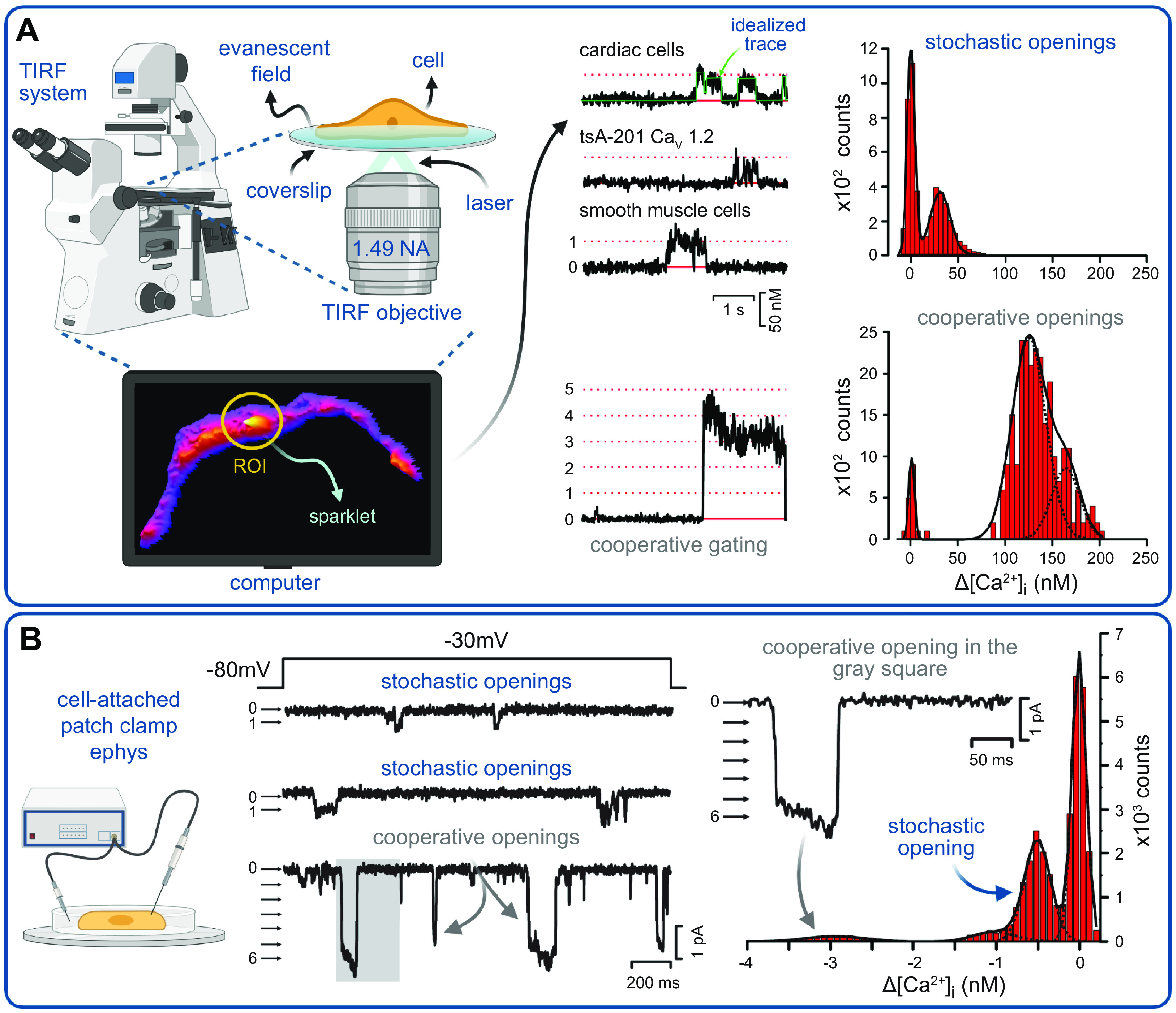
Recording and analysis of CaV1.2 cooperative gating events: total internal reflection fluorescence (TIRF) microscopy configuration (A) and cell-attached patch-clamp electrophysiology (ephys) system (B) for recording Ca2+ sparklets and single-channel currents, respectively. [Ca2+]i, intracellular Ca2+ concentration. Traces and histograms used to detect and analyze CaV1.2 cooperative openings in different cell types are modified from Navedo et al. (13) with permission. Figure created with Biorender.com.
In subsequent studies, the cooperativity hypothesis has been consistently supported and we have delved more deeply into the physiological and pathophysiological mechanisms underlying this noncanonical channel gating behavior. Notably, PKC agonists enhanced the persistent, cooperative gating activity at discrete sites in vascular smooth muscle cells but not all sites, suggesting that some channels were within the range of PKC signaling whereas others were seemingly out of range. Later work (13, 172, 173) revealed that this was because a subset of CaV1.2 channels associate with the A-kinase anchoring protein AKAP5 (previously known as AKAP150 in mice and as AKAP79 in humans), which anchors PKA, PKC, and calcineurin (CaN) (174, 175) next to CaV1.2 channels in vascular smooth muscle, cardiac muscle, and neurons (13, 45, 172, 176–178). In smooth muscle, PKCα-mediated phosphorylation elevates CaV1.2 activity (13, 173, 179, 180). PKCα activity is increased during angiotensin II (ANG II)-induced hypertension, and indeed both acute and chronic ANG II stimulation of smooth muscle cells isolated from mouse mesenteric and cerebral arteries lead to increased Ca2+ influx and increased vascular tone in intact arteries, which is at least partially driven by enhanced cooperative gating behavior of CaV1.2 channels (13, 173, 180). AKAP5 anchoring of PKC is essential for these effects as AKAP5−/− mice are hypotensive, do not develop hypertension in response to chronic ANG II infusion, and do not exhibit enhanced Ca2+ sparklet activity or elevated levels of cooperative gating (13, 173). Furthermore, pharmacological PKC activators enhance cooperative gating behavior, but this response is absent in cells isolated from AKAP5−/− arteries. These studies suggest that cooperative gating of CaV1.2 channels causes ICa amplification during angiotensin-induced hypertension.
In 2010, after many years of observing this unusual gating behavior without a well-defined mechanism to explain it, the hypothesis that CaV1.2 channels can gate cooperatively was finally tested in cardiac and arterial smooth muscle myocytes, and the first mechanistic details began to emerge (13). Cooperativity was visualized in Ca2+ sparklet and single-channel electrophysiology recordings (FIGURE 3). There are several ways to identify positive cooperativity, including binomial and variance analyses, conditional dwell time density analysis, and maximum-likelihood fitting analysis (reviewed in Ref. 181). In binomial analysis, the idea is that if channels gate in an entirely independent manner, then the probability of observing 1 channel opening should be higher than that of observing the simultaneous opening of 2 channels, and in turn there should be a progressively lower probability of observing instances of 3, 4, or 5 channels opening together… and so on. Thus, if one were to plot the all-points amplitude histogram of iCa, independently gating channels should generate a binomial distribution. However, with positive cooperativity, multichannel openings may occur more frequently, and thus the amplitude histogram would deviate from the binomial distribution. This indeed was the case for CaV1.2 channels. However, there appears to be no favored or set number of channels that preferentially undergo cooperative gating events, and often up to 10 channels can be observed to gate coordinately (13, 42). This is in stark contrast to findings in cardiac NaV1.5 channels, where dimers of those channels seem to preferentially gate together in a 14-3-3-dependent manner (10) as discussed in sect. 5. The absence of a single preferential oligomeric state led us to pursue another metric to quantify the degree of cooperativity, namely a coupled Markov chain model. A similar approach had previously been implemented by Chung and Kennedy (182) in their analysis of partially coupled GABA-activated chloride channels. Implementation entails calculating a dimensionless coupling coefficient parameter, kappa (κ), from single-channel activity traces (13). In this model, a κ value of 1 indicates channels that always gate cooperatively, a κ value of 0 indicates channels that always gate independently, and a κ value in the range 1 > κ > 0 indicates a mixture of cooperative and independent openings. This script, written in MATLAB, is freely available as source code accompanying Ref. 42.
In a channel patch or sparklet site, variable numbers of channels open and close coordinately, alongside some independent openings of single channels, and thus the fully cooperative case is seldom observed in an experimental setting with a full sweep of activity. CaV1.2 channel activity in the absence of agonists or other stimulation often shows κ values between ∼0.0 and 0.4, indicating a mixture of channels gating independently and others displaying nonindependent, cooperative gating, even under basal conditions (13, 50, 165). Indeed, in a 2010 study testing the cooperative hypothesis of CaV1.2 channels, the median κ value reported was 0.22 (13). To explore the mechanisms underlying cooperative gating, channels lacking a portion of the COOH-terminal tail between amino acids 1670 and 2171, where AKAP5 was putatively thought to bind to a leucine zipper (LZ) motif (175), were generated, and they failed to display cooperative gating behavior. Furthermore, cooperativity was also absent in AKAP5-null cells (173). Increased fluorescence resonance energy transfer (FRET) between COOH-terminal-tagged CaV1.2-EGFP and CaV1.2-tagRFP correlated with enhanced cooperativity, whereas decreased FRET correlated with more independent gating. These results suggested that physical proximity or interactions of CaV1.2 COOH-terminal tails are a component of the cooperative gating mechanism. Furthermore, PKCα activators were found to enhance cooperative gating, in line with previous findings (13). A subsequent study in tsA-201 cells expressing cardiac CaV1.2 channels found that the dynamic trafficking of these proteins in vesicular structures contributes to cluster formation and cooperative gating behavior (29). Here, disruption of microtubules or actin elements induced a profound reduction in cooperative gating. These results suggest that an intact cytoskeleton facilitates CaV1.2 clustering and cooperative gating. Consistent with this idea, an intact cytoskeleton was recently found to be essential for regulation of CaV1.2 clustering and cooperative gating in response to β-adrenergic stimulation in adult cardiomyocytes (51).
To summarize this first advance in our still-evolving mechanistic understanding of CaV1.2 cooperative interactions, five insights have emerged: 1) physical proximity or interactions between the COOH-terminal tails of adjacent channels is required; 2) cooperativity occurs in the subpopulation of channels that associate with AKAP5; 3) cooperativity requires an intact cytoskeleton; 4) cooperativity is facilitated by channel phosphorylation; and 5) it is regulated by CaM (FIGURE 4).
FIGURE 4.
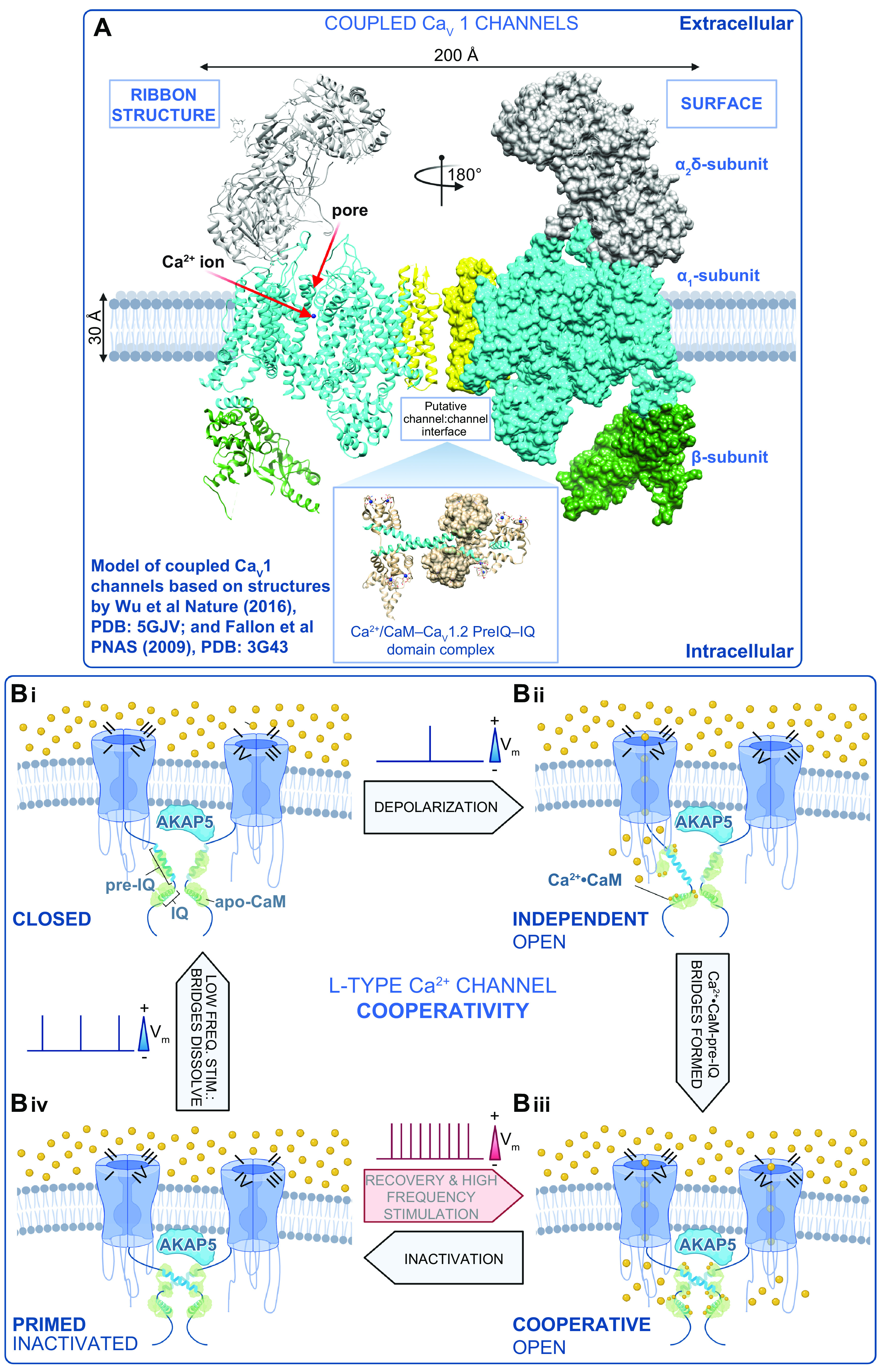
Mechanism of L-type Ca2+ channel cooperative gating. A: model of the interaction between 2 CaV1 channels based on structures by Wu et al. (76) and Fallon et al. (189). B: schematic showing our proposed model of L-type Ca2+ channel cooperative gating based on the analysis of CaV1.2 and CaV1.3 channels (12, 40). Two channels are illustrated for simplicity and are drawn bridged by Ca2+·calmodulin (CaM) in the manner of the 2 published crystal structures of COOH-terminal fragment dimers (189, 190). Although this is speculative at present, we do know that the interactions depend on Ca2+·CaM (12, 42) and intact pre-IQ motifs (12, 42) and occur in AKAP scaffolded microdomains (at least for CaV1.2) (13, 173). For >2 channel multimers to gate coordinately, we postulate that there are at least 2 possibilities: 1) Ca2+·CaM may “daisy-chain” adjacent pre-IQ motifs, utilizing the 2 CaM-binding sites on that motif, or 2) one lobe of Ca2+·CaM may bind to pre-IQ and the other may bind to another of the established CaM binding sites on these channels (for review see Ref. 445). In our scheme, we begin at Bi, where the membrane is at its resting potential and channels are close to one another but not interacting in the closed state. Bii: with depolarization, a subset of the channels stochastically open and Ca2+ flows into the cell, binding to CaM. Biii: Ca2+·CaM facilitates the physical, functional interactions of adjacent channels that depend on binding to the pre-IQ motif. In this functionally cooperative state, the opening of 1 channel is allosterically communicated to the attached channel and they gate coordinately. Biv: CaV channels undergo voltage- and Ca2+-dependent inactivation but remain associated for a time after the Ca2+ signal has decayed, leaving them in a primed state as the cells repolarize. Thus, with high-frequency activation, e.g., in the heart during fight or flight, the channels can immediately transition to the cooperative open state, resulting in an immediate facilitation of Ca2+ influx. During low-frequency stimulation, e.g., during resting heart rate, the physical bridges between adjacent channels in a cluster are dissolved, and the resting confirmation is assumed as the cycle is resumed. Vm, membrane potential. Figure created with Biorender.com.
The contribution of cooperative gating to pathological states was observed in several studies (11, 13, 183) in which CaV1.2 cooperativity was notably enhanced in channels with a gain-of-function mutation [G436R in mice, G406R in humans (71)] associated with the multisystem disorder known as Timothy syndrome (TS) or long QT 8 (LQT8) (184). The enhanced levels of cooperativity of TS channels that incompletely inactivate and thus enhance Ca2+ entry into ventricular myocytes hinted that the cooperativity mechanism may be facilitated by Ca2+ itself. This was indeed shown to be the case in 2015 when electrophysiological recordings made with Ba2+ as the charge carrier in place of Ca2+ failed to display cooperativity and physical channel-channel interactions (42).
With the light-induced fusion of Flavin-binding, Kelch repeat, F box 1 (FKF1) and gigantea (GI) proteins, it was definitively shown that physical interactions between CaV1.2 channel COOH-terminal tails could facilitate cooperative interactions and amplify ICa (11). In this FKF/GI system, which controls flowering during daylight hours in Arabidopsis thaliana, FKF1 undergoes a conformational change upon illumination with blue light that allows it to bind to GI within minutes but dissociate with a half-life (t1/2) of ∼60 h. We tagged CaV1.2 channels with this system to produce CaV1.2-GI and CaV1.2-FKF and expressed them together in tsA-201 cells, finding that ICa was enhanced after blue light (11). Furthermore, channel activity and cooperativity were enhanced after blue light. One of the clear advantages of this system is that it permits recording control currents first and then stimulating with blue light to observe the effects of physical channel-channel interactions in the same cells. Unfortunately, the dissociation of this system is too slow to allow visualization and recording of the unbinding effects, but there are now several, more recently characterized light-activated dimerization systems that dissociate on a more manageable timescale, including the cryptochrome 2 and CIBN system, which reverses with a t1/2 of ∼5.5 min (185, 186). Expression of the GI- and FKF1-fused wild-type (WT) channels in rat ventricular myocytes with biolistic transfection (also known as “gene-gun”) led to larger electric field stimulation (EFS)-evoked Ca2+ transients and enhanced cell shortening after blue light-induced dimerization compared with pre-light. This suggests that more cooperative interactions of CaV1.2 channels can increase inotropy and thereby tune EC coupling.
The finding that physical association between channels could enhance their activity and amplify whole cell Ca2+ influx and inotropy was exciting, but even more so were our findings when we used this system to dimerize WT CaV1.2 with CaV1.2(G436R) TS mutant channels (187). As discussed above, the G436R (or G406R in humans) is a gain-of-function mutation that effects reduced inactivation and increased Po in TS channels (71). When we fused WT channels to TS channels, somewhat unexpectedly, the WT channels exhibited some TS channel properties. This was uncovered by using a dihydropyridine-insensitive WT (T1066Y) channel so that after blue light illumination the individual contribution of WT channels to ICa could be dissected with application of nifedipine (11). We found that WT channels seemed to be strongly influenced by the higher-Po TS channels and actually began to exhibit some deficits in their inactivation in a TS-like manner. Since TS is a heterozygous condition, this finding suggested that having just one high-Po TS channel within a cluster of otherwise WT cooperatively gating channels could make them all behave abnormally and produce an arrhythmogenic substrate. Indeed, biolistic transfection of rat ventricular myocytes with WT-GI and TS-FKF1 channels frequently resulted in arrhythmogenic spontaneous Ca2+ release events and Ca2+ alternans after light-induced channel fusion (187). A later in silico modeling study concluded that aberrant levels of cooperatively gating channels could feasibly prolong action potential (AP) duration and produce Ca2+ alternans (188). TS is a long QT-producing condition that causes AP prolongation and life-threatening arrhythmias. This computational study revealed that CaV1.2 channel cooperativity could play a pivotal role in this pathology.
The observation that channels with the highest Po can exert an influence on the activity of other neighboring channels within a cluster has implications for other modes of channel modulation, for example channel phosphorylation, which is known to produce enhanced Po of CaV1.2 channels in heart, smooth muscle, and neurons. In cardiac muscle, PKA-mediated phosphorylation of CaV1.2 channel complexes has explicitly been linked to enhanced cooperativity of CaV1.2 channels and amplification of Ca2+ influx downstream of β-adrenergic receptor activation (50). In isolated mouse ventricular myocytes, isoproterenol (ISO)-mediated stimulation leads to increased insertion of CaV1.2 from subsarcolemmal reservoirs of endosome-localized channels (51), producing enhanced sarcolemmal expression of CaV1.2, and results in the appearance of large superclusters of CaV1.2 channels in the t-tubule membrane (50, 51). The “superclustering” response depended on PKA activation, as inhibitors of PKA (H-89 and PKAi) prevented their formation (50). Enhanced protein-protein interactions between adjacent CaV1.2 channels were visualized by bimolecular fluorescence complementation. This enhanced physical association facilitates cooperative allosteric interactions between channels, and, accordingly, examination of CaV1.2 single-channel activity in both Ca2+ sparklet and cell-attached patch recordings revealed enhanced cooperative gating of CaV1.2 in response to the β-agonist ISO. Consistent with the idea that cooperative gating is instigated by the highest-Po channel in the cluster, these findings suggest that just a few phosphorylated CaV1.2 channels could have a disproportionately large effect on ICa and add another layer of complexity to β-adrenergic regulation of cardiac CaV1.2.
We have subsequently discovered that Ca2+ and Ca2+·CaM binding to the pre-IQ motif of CaV1.2 channels plays a key role in mediating the physical interaction between adjacent channels within a cluster (42) (FIGURE 4A). Whether Ca2+·CaM acts as a physical bridge between adjacent channels via pre-IQ to pre-IQ interactions is unknown, but a crystallography study supports this model (189). In this structural study, a dimer of CaV1.2 channel COOH-terminal tail segments was found to be bridged by two Ca2+·CaM molecules bound to the pre-IQ motifs. Debate around the validity of this model was sparked by a study suggesting that this dimer had no physiological relevance, noting that channels in Xenopus oocyte cell membrane exclusively expressed as monomers (190). However, Xenopus oocytes are quite distinct from mammalian somatic cells, as they are haploid. Although they have been an extremely useful tool for classical ion channel biophysicists, it is possible that they could lack endogenous expression of a vital element or scaffold that permits channel oligomerization, for example, AKAP5. Furthermore, we have successfully identified oligomeric channel assemblies in mammalian cell lines and in ventricular myocytes with stepwise photobleaching approaches and superresolution microscopy (29, 42, 50), so this may suggest that Xenopus oocytes are not the best platform for studies of mammalian CaV1.2 channel stoichiometry and assembly.
Our current model of CaV1.2 channel interactions has also been informed by FRET experiments that revealed that enhanced associations between COOH-terminal tails were formed dynamically as depolarization-stimulated Ca2+ influx occurred through these voltage-dependent channels (42). With this approach, a subpopulation of CaV1.2 channels were observed to remain closely associated with one another even after the Ca2+ influx that fueled the interactions had subsided because of voltage- and calcium-dependent channel inactivation. Similar results were observed after uncaging Ca2+. We hypothesize that this subpopulation of channels that remain associated with one another even after Ca2+ influx has ceased play a role in facilitation of ICa with successive depolarizing stimuli. This could be important during high-frequency stimulations, for example, during the fight-or-flight response when heart rate is elevated and APs arrive at the ventricular myocardium in more rapid succession than at resting heart rate. We tested this idea with paired-pulse facilitation experiments, and those data were well fit by a model that incorporated the channel recovery from inactivation and the channel “uncoupling” or cluster disassembly, which was informed by the decay kinetics of the FRET signal. Altogether, these results suggest that the degree of facilitation of ICa during high-frequency stimulation is directly proportional to the number of functionally interacting channels.
Recent work has shown that the number of channels in the t-tubule sarcolemma is increased by ∼40% during β-adrenergic receptor stimulation via enhanced sarcolemmal insertion of often intact channel clusters that originate in endosomal channel reservoirs and provide a functional reserve via triggered Rab4a and Rab11a recycling pathways (51). This enhanced channel expression and clustering at the membrane during a fight-or-flight response could cause an even greater degree of facilitation than that provided by the higher-frequency stimulation alone and could efficiently boost inotropy to meet the enhanced hemodynamic and metabolic demands of the body during acutely stressful events. Electrophysiological and imaging studies suggest that this stimulated channel-insertion response is an essential component of β-adrenergic regulation of CaV1.2, as disruption of the cytoskeletal delivery pathways prevented stimulated insertion and superclustering and eliminated the normal ICa augmentation response to ISO and the characteristic leftward shift that is a signature of PKA regulation of cardiac CaV1.2 channels (51).
How this stimulated insertion pathway elides with the recent finding from Steven Marx’s group that β-adrenergic regulation of CaV1.2 channels occurs as a result of disinhibition upon dissociation of phosphorylated Rad from the CaVβ subunit of the channel complex (191) remains to be seen. There have been previous reports that RGK proteins including Rad interfere with CaV channel trafficking and reduce ICa by limiting its expression at the plasma membrane (192, 193). Furthermore, this reduction in channel trafficking is thought to be relieved by Rad phosphorylation and regulated by CaM and 14-3-3 (192). The impact of this on stimulated channel insertion is something that should be disentangled as these two hypotheses are assimilated. Our current understanding of CaV1.2 channel cooperativity is represented in FIGURE 4B.
A recent study has independently corroborated the findings that CaV1.2 channels gate coordinately (194). With the use of scanning patch clamp to record single-channel activity in defined sarcolemmal locations, a strikingly increase in CaV1.2 cooperativity was observed in failing rat ventricular myocyte t-tubule membranes consequent to myocardial infarction (MI). Examination of the all-points histogram reveals that the predominant single-channel openings of control myocytes were almost completely absent from post-MI t-tubule-localized channels, and in their place were doublet and quadruplet channel openings. This overt cooperative gating behavior was extinguished by inhibition of PKA (50). This finding is consonant with our previous report that β-adrenergic receptor stimulation enhanced CaV1.2 channel expression in the t-tubule membranes (51), leading to enhanced channel superclustering and increased cooperative gating and resulting in amplification of ICa (50). Thus, CaV1.2 cooperative gating seems to be a general property of these channels with broad physiological and pathological implications. We foresee that CaV1.2 cooperative gating may be observed in other tissues such as neurons, endocrine cells, and visceral smooth muscle.
Environmental pollutants, such as secondhand smoke, have also been shown to increase CaV1.2 cooperative gating in vascular smooth muscle, and this was associated with elevated myogenic tone of mesenteric arteries (195). Moreover, cooperative channel gating has been implicated in the pathology of hyperglycemia and diabetes. For example, in vascular smooth muscle from humans and mice, high glucose is thought to stimulate a particular pool of PKA that localizes to AKAP5-anchored complexes adjacent to CaV1.2 channels, causing CaVα1C phosphorylation on the COOH-terminal tail at serine residue 1928 (S1928) (45, 46, 52, 155). The consequence of this targeted phosphorylation is enhanced ICa current density and increased myogenic tone. Mice with a nonphosphorylatable S1928A substitution failed to display increased ICa and vasoconstriction responses to high-glucose or high-fat diet metabolic challenges, indicating that this phosphorylation event is required for the enhanced CaV1.2 channel activity and vascular reactivity that likely contributes to the enhanced risk of stroke, hypertension, and heart disease in diabetic patients. So, does cooperative gating of CaV1.2 factor into this enhanced activity? There is some evidence that it does. A study found that hyperglycemia and diabetes produced “persistent” Ca2+ sparklet activity in mouse cerebral artery smooth muscle that could be abolished by PKA inhibitors (172). Close examination of the Ca2+ sparklet recordings from that study shows the hallmarks of cooperative gating behavior including frequent time-correlated openings of multiple CaV1.2 channels and indeed closings. Although the link to cooperativity has not been explicitly stated, it appears to play a role in this pathological response. Future studies should examine this interesting possibility. Moreover, studies should determine whether S1928 phosphorylation plays a role in modulating cooperative gating of CaV1.2 channels in vascular smooth muscle during physiological conditions and in diabetic hyperglycemia. It is tempting to speculate that S1928 phosphorylation may be a key factor underlying cooperative gating of CaV1.2 channels in other cell types such as neurons (196, 197).
3.6. CaV1.3 Channel Structure and Function
The pore-forming subunit of CaV1.3 is CaVα1D encoded by cacna1d (75). CaV1.3 and CaV1.2 are expressed in many of the same tissues, including brain, heart, and endocrine glands (68, 198). CaV1.3 channel inactivation kinetics are slower than CaV1.2, and they display a more left-shifted voltage dependence of activation and begin to open at more hyperpolarized potentials of approximately −60 mV, compared with the approximately −40 mV activation threshold of CaV1.2 (199). However, when recording from cells that have varying proportions of CaV1.2 and 1.3 channels the value falls somewhere in between, and in the absence of a specific pharmacological blocker that can distinguish between these two L-type channels the physiological role of CaV1.3 was unresolved for many years. The generation of a transgenic CaV1.3−/− mouse exposed the functional importance of these channels (reviewed in Ref. 198). Accordingly, the knockout mice displayed two striking phenotypes. First, they were deaf, confirming the critical role of CaV1.3 in the auditory system, where they are expressed on the inner (IHC) and outer hair cells of the cochlea (200). Second, they were bradycardic, a telling consequence of the lack of CaV1.3 in pacemaking sinoatrial node (SAN) and atrioventricular node (AVN) cells, where they play a role in the diastolic depolarization phase of the nodal action potential (200, 201).
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels HCN1, HCN2, and HCN4 that underlie the funny current (If) are also famously critical players in diastolic depolarization, allowing these CaV1.3−/− cells to display automaticity (202) (sect. 9). Thus, CaV1.3 knockout is not lethal because of a total lack of pacemaker activity, but SAN dysfunction is certainly apparent in these animals. Indeed, telemetric recordings from CaV1.3−/− mice, revealed electrocardiograms with high R-R interval variability and sinoatrial bradycardia at low resting heart rates that could be rescued by treatments that increased heart rate, e.g., exercise, or injection of ISO (a β-agonist that mimics sympathetic stimulation) or atropine (a muscarinic receptor blocker that dampens parasympathetic effects) (200). The relatively hyperpolarized threshold of activation of CaV1.3 is thought to contribute to its role in diastolic depolarization in nodal cells, and the more depolarized activation threshold of CaV1.2 explains its inability to compensate for CaV1.3 absence in the SAN and AVN of knockout animals (203).
CaV1.3 channels are also involved in shaping the firing patterns and modulating the resting potentials of neurons in the central nervous system (CNS) (117, 204). In the brain, CaV1.2 channels outnumber CaV1.3 by ∼9:1 and they are often both expressed within the same neuron (205, 206). In the dopaminergic neurons in the substantia nigra, CaV1.3 channels have been implicated in the pathophysiology of Parkinson’s disease (PD), where they orchestrate the rhythmic pacemaking of these cells (207–210).
Splice variants of CaV1.3 include the so-called “long” and “short” isoforms that result from alternative splicing of CaVα1D exon 42 and differ in the length of their COOH-terminal tail, their inactivation kinetics, and their threshold of activation (211). The long CaV1.3L isoform (also known as CaV1.342), has a 695-amino acid COOH-terminal tail containing a COOH-terminal modulatory domain (CTM). Within this domain, interactions between a proximal COOH-terminal regulatory domain (PCRD) and a distal COOH-terminal regulatory domain (PCRD) strongly reduce Ca2+·CaM binding to the IQ motif, resulting in reduced Ca2+-dependent inactivation of these channels (211). The CTM is absent from the short splice variant CaV1.3S (also known as CaV1.342A), which has a 183-amino acid COOH-terminal tail that offers less steric hindrance to Ca2+·CaM-IQ motif interactions. Thus CaV1.3S inactivates more rapidly than CaV1.3L (211). Despite the enhanced inactivation that limits Ca2+ influx, CaV1.3S channels display ∼2.5-fold larger ICa densities than CaV1.3L channels in the absence of any detectable changes in expression levels (211) or unitary current amplitude (12, 212). The current-amplifying effects of positive cooperative gating, exclusively of CaV1.3S and not CaV1.3L, could explain this discrepancy in ICa. Indeed, our group has reported that CaV1.3S channels undergo Ca2+·CaM-dependent cooperative interactions in tsA-201 cells and hippocampal neurons as discussed in detail in sect. 3.7 (12).
3.7. Cooperativity Among CaV1.3 Channels
Almost a decade after our first study demonstrating cooperativity of CaV1.2 channels (26), we investigated whether a similar phenomenon could occur for CaV1.3 channels (12). As noted above, it had already been reported that CaV1.3S channels generated larger ICa density than CaV1.3L channels, which could not be explained by changes in channel expression or unitary current, and so we started there. In whole cell patch-clamp experiments, we noticed that ICa in CaV1.3L-expressing tsA-201 cells was ∼40% smaller than IBa. This was not unexpected given that these channels, like all voltage-gated Ca2+ channels, are more permeable to Ba2+ than Ca2+ (213). However, CaV1.3s only exhibited an ∼15% reduction in current when the extracellular solution was switched from one containing Ba2+ to one containing Ca2+. This finding suggested that Ca2+ might alter the activity of CaV1.3S. We tested this hypothesis in cell-attached patch single-channel recordings and found that NPo (where N represents channel number) in CaV1.3s-expressing cells was ∼1.5-fold higher with Ca2+ as the charge carrier than with Ba2+, whereas no such Ca2+-dependent increase in NPo was observed in CaV1.3L-expressing cells. This was reminiscent of the Ca2+ dependence of CaV1.2 cooperativity we had reported previously, and accordingly we examined CaV1.3 Ca2+ sparklets for the two different isoforms, finding that CaV1.3L channels displayed more overt cooperative gating behavior and had correspondingly higher coupling coefficients than CaV1.3L channels that appeared to largely gate independently. Superresolution microscopy confirmed that both CaV1.3L and CaV1.3S channels cluster in tsA-201 cells, suggesting that although clustering is necessary for cooperative channel interactions, it is not sufficient. CaV1.3 channels were also observed to cluster in cultured rat hippocampal neurons, where stepwise photobleaching of neurons transfected with CaV1.3S-EGFP revealed that clusters of CaV1.3 contained eight channels on average. Consistent with the hypothesis that only the CaV1.3S slice variant undergoes cooperative gating, light-induced dimerization of these channels produced a 1.35-fold amplification in their ICa, whereas similarly evoked dimerization of CaV1.3L channels did not alter ICa. As was the case for CaV1.2 channels, spontaneous physical interactions of CaV1.3S COOH-terminal tails occurred in a manner dependent on Ca2+·CaM binding to the pre-IQ motif. The functional consequences of CaV1.3 channel cooperativity in neurons were assessed by recording action potentials from neurons transfected with CaV1.3S-CIBN and CaV1.3S-CRY2 before and after blue light-induced dimerization. Strikingly, channel fusion was observed to enhance neural excitability and firing rate. It is therefore quite reasonable to posit that cooperative gating of CaV1.3S makes an important contribution to the sustained firing observed in motoneurons and other cells in the CNS (214). A recent publication has shown robust expression of CaV1.3S mRNA in the substantia nigra pars compacta in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease (207), although the physiological role of CaV1.3 cooperativity in cardiac pacemaking remains an important open question.
4. TRANSIENT RECEPTOR POTENTIAL CHANNELS
The transient receptor potential (TRP) channels are a superfamily of ion channels encoded by 28 different genes (215, 216). Sequence homology analyses have revealed that TRP channels can be further classified into six different subfamilies, namely canonical TRP (TRPC), vanilloid TRP (TRPV), melastatin TRP (TRPM), polycystin TRP (TRPP), ankyrin TRP (TRPA), and mucolytic TRP (TRPML) channels (215, 216). Each TRP channel subunit consists of six membrane-spanning helices (S1−S6) (FIGURE 5A) (216). The amino and carboxy terminals of each subunit are located intracellularly and contain several distinctive domains that regulate channel function (217). Several published cryo-EM structures of TRP channels (TRPV1, TRPV2, and TRPA1) at near-atomic resolution support the idea that TRP channels are formed by the assembly of one or more subunits into tetramers around a central pore formed by the S5 and S6 helices (216). The selectivity filter is located in the upper region of the pore and is modulated by endogenous and exogenous ligands (216). Given the ubiquitous expression of a diverse number of TRP channels in a given cell, it is likely that they are formed by homo- and/or heterotetramers.
FIGURE 5.

Model of cooperative gating of transient receptor potential (TRP) channels. A: TRP subunit topology. PIP2, phosphatidylinositol 4,5-bisphosphatase. B: model by which Gq agonists trigger AKAP5-dependent, PKC-mediated cooperative gating of TRPV4 channels. GqPCR, Gq protein-coupled receptor. Figure created with Biorender.com.
TRP channels are nonselective cation-permeable channels with permeability ratios (PCa/PNa) in the range of 0.1–20 (216). Although most TRP channels are permeable to Ca2+, TRPM4 and TRPM5 currents are carried almost exclusively by Na+ ions, with minimal if any contribution made by Ca2+ (215, 216). TRP channels can be activated by numerous intracellular and extracellular stimuli including changes in temperature, different Ca2+ signals, mechanical stress, and chemical cues. TRP channels can act as integrators of different signaling pathways to elicit specific cellular responses (215, 216). Indeed, TRP channels modulate membrane potential, intracellular Ca2+ signaling, generation of action potentials, migration, proliferation, and many other processes. The physiological processes mediated by TRP channel activation include pain and taste sensation, control of vascular tone, immunological responses, and cellular homeostasis (215, 216). Dysfunction of TRP channels leads to several channelopathies, including cardiovascular complications due to gain- and loss-of-function mutations, renal diseases, blindness, atrophy of several tissues, and pain disorders. These pathologies have been extensively discussed and documented in recent comprehensive review papers on TRP channels (215, 216). In sects. 4.1–4.4, we examine examples of studies describing cooperative gating of TRP channels as well as the current understanding of mechanisms underlying this gating modality and its functional implications, topics that have not been previously reviewed.
4.1. TRP Channels Undergo Cooperative Gating
The discovery that TRP channels can engage in cooperative gating was contingent on optical recording techniques, including TIRF and spinning disk confocal imaging (14, 17, 25) that enabled the detection of Ca2+ influx via single or clusters of Ca2+-permeable channels with a high spatial and temporal resolution over a large surface area. The first example of cooperative gating of TRP channels was described for TRPV4 in native mesenteric endothelial cells with an en face preparation from a mouse expressing the GCaMP2 Ca2+ sensor specifically in endothelial cells (166). About the same time, an independent study using pressurized intact cremaster arterioles loaded with the Ca2+ indicator fluo4-AM described the induction of endothelial events produced by the simultaneous opening of two or more TRPV4 channels (218). Subsequently, TRPV4 cooperative gating has been described in cultured human endothelial cells and cultured human airway smooth muscle cells loaded with fluo4-AM (167, 219), native mouse cerebral and mesenteric smooth muscle cells loaded with the fast Ca2+ indicator fluo-5F (43, 44), native mouse and human mesenteric endothelial cells from endothelium-specific GCaMP2 mice or cells loaded with fluo4-AM (220, 221), and native mouse pulmonary endothelial cells from endothelium-specific GCaMP2 mice (222). Intriguingly, TRPV4 activity and the frequency of cooperative gating differ with biological sex and vascular bed (43). TRPV4 activity and cooperative gating is higher in male than female smooth muscle cells. Moreover, TRPV4 activity and cooperative gating are of equal magnitude in cerebral pial and parenchymal smooth muscle cells but completely absent in mesenteric smooth muscle from both sexes. These results highlight important sex-specific and vessel-dependent regulation of TRPV4 biophysical properties that may be general properties of other TRP channels.
Although less extensively studied, there are reports of cooperative gating of TRPA1 channels in mouse cerebral endothelial cells from endothelium-specific GCaMP6f mice or cells loaded with fluo4-AM (168, 170) and TRPV3 in cultured mouse endothelial cells loaded with fluo4-AM (169). General Ca2+ signaling properties of the TRP-mediated events reported in these studies are somewhat different, which is likely due to intrinsic differences in channel activity for different cell types and/or to the use of different acquisition approaches. In TABLE 1, we provide a summary of the unitary current amplitude, number of channels per membrane site, as well as the frequency, amplitude, and spatial spread of the Ca2+ signals produced by different TRP channels in multiple cells. All reports, however, indicate that cooperative gating of TRP channels may result from a two- to four-channel metastructure. These results suggest that cooperative gating appears to be a general property of TRP channels arising from aggregates of a given number of channels.
Table 1.
Properties of Ca2+ signals mediated by TRP channels
| TRP Channel (reference) | Cell Type | Unitary Amplitude | Maximum Number Channels/Site | Open Time/Po | Spatial Spread | Event Frequency |
|---|---|---|---|---|---|---|
| TRPV4 (219) | Human cultured microvascular EC | 0.39 ± 0.01 (ΔF) | ∼5 | 520 ± 40 ms | 6.26 ± 2.01 μm2 | 0.11 ± 0.02 Hz |
| TRPV4 (167) | Human cultured airway SMC | 0.06 (ΔF) | ∼3 | 110–250 ms | 0.7 μm2 | 0.2 ± 0.5 Hz |
| TRPV4 (44) | tsA-201 cells | ∼50 nM | ∼1 | |||
| TRPV4 (44) | Native mouse SMC | ∼48 nM | ∼4 | 12–68 ms | 2–5 sites/cell | |
| TRPV4 (166, 221) | Native mesenteric EC | 0.19 (ΔF) | ∼4 | 37.0 ± 0.7 ms | 11.2 ± 0.4 μm2 | 10–25 sites/field |
| TRPV4 (222) | Native pulmonary EC | 0.29 (ΔF) | ∼4 | 0.03–0.16 (Po) | ∼20 μm2 | 5–30 sites/field |
| TRPV3 (169) | Primary cerebral EC | ∼0.2 (ΔF) | Unknown | 70–170 ms | 0.7 μm2 | 0.05–0.4 Hz |
| TRPA1 (168) | Native cerebral EC | ∼0.25 (ΔF) | ∼3 | 96 ms | 5.02 ± 0.60 μm2 | 1–8 sites/field |
EC, endothelial cell; Po, open probability; SMC, smooth muscle cell; TRP, transient receptor potential; TRPA, ankyrin TRP; TRPV, vanilloid TRP; ▵F, change in fluorescence from the calcium indicator.
4.2. Mechanisms of Cooperative Gating of TRP Channels
TRP channel cooperative gating is predicated on the close proximity of two or more channels and is likely to be supported by an association with a structural protein. Accordingly, there has been a substantial effort to understand the role of the scaffold protein AKAP5 in TRPV4 clustering (5, 43, 44, 221, 223). The focus on AKAP5 stems from work suggesting its association with TRPV4 channels (224) and its role in regulating the extent of cooperative gating of L-type Ca2+ channels (13).
On first principles, the clustering of ion channels must be intimately related to their trafficking properties. The mechanisms underlying the subcellular dynamics and translocation of TRP channels, including TRPV4, have been extensively reviewed (225, 226). However, an emerging concept is that dynamic trafficking of ion channels is essential for sustaining their clustering and cooperative gating (50, 51, 227). By combining experimental and in silico approaches, a model for TRPV4 clustering has been proposed in which channel clustering is mediated by a stochastic self-assembly process (5). This hypothesis is supported by superresolution data showing that TRPV4 cluster size has an exponential distribution indicative of a Poisson process. A similar exponential distribution of cluster sizes has been observed for TRPML1 channels in vascular smooth muscle (228), suggesting that this spatial arrangement is likely to be a general feature of TRP channels. Sato et al. (5) also found that TRPV4 cluster size and density seem to be regulated by a feedback mechanism that controls the size and the number of clusters at the surface membrane. This proposal rests on superresolution data showing that TRPV4 clusters rapidly increased in size and density after transfection in HEK cells but reached a steady state after 24 h. Intriguingly, the accompanying in silico model predicted that TRPV4 channel and cluster dwell times at the membrane would be in the range of minutes (5). This suggests that the insertion and removal of TRPV4 channels and clusters must be dynamic and well controlled to sustain the basal TRPV4 current, as observed for other TRP channels (229–231), and are likely to engender cooperative channel gating. A similar clustering formation mechanism accounts for CaV1.2, CaV1.3, and large-conductance Ca2+-activated K+ (BKCa) channel cluster size in different cells (5). These results suggest a potentially general mechanism for ion channel (including TRP channels) cluster formation, which may influence cooperative gating. This model requires further evaluation.
Existing evidence suggests that elevations in intracellular Ca2+ and AKAP5 contribute to cooperative gating of TRPV4 in mesenteric endothelial and cerebral smooth muscle cells (43, 44, 221, 223). The molecular details underlying the AKAP5-TRPV4 interaction that could sustain cooperative gating are unresolved but may include protein-protein contacts between the carboxy terminals of each protein (224). However, genetic ablation of AKAP5 does not completely abolish the induction of cooperative gating of TRPV4 channels in mesenteric endothelial and cerebral smooth muscle cells (43, 44, 221, 223). Thus, there may be other structural proteins (e.g., α-actinin) that facilitate cooperative gating of TRPV4 channels in the absence of AKAP5, a conjecture that requires experimental confirmation. Furthermore, AKAP5 may be playing a more traditional role for this scaffold in targeting signaling proteins (e.g., PKC, PKA, PP2B) to modulate TRPV4 channel activity and local intracellular Ca2+ concentration that promotes TRPV4 cooperative gating.
Supporting the aforementioned possibility, superresolution imaging in cerebral and mesenteric vascular smooth muscle revealed TRPV4 and AKAP5 clusters of various sizes (43). The association between TRPV4 and AKAP5 clusters is augmented in response to acute ANG II exposure in pial and parenchymal smooth muscle cells (43). This treatment appears to facilitate AKAP5-anchored PKC-dependent stimulation of TRPV4 channel activity and may account for the increased frequency of cooperative gating events in these cells. Conversely, chronic ANG II signaling activation, as during hypertension, promotes an increase in the distance between TRPV4 and AKAP5 clusters that results in a reduction of both channel open probability and the frequency of cooperative events in TRPV4 channels. Intriguingly, the chronic ANG II increase in TRPV4-AKAP5 intermolecular distance in pial and parenchymal smooth muscle is comparable to the basal distance between these proteins in mesenteric cells (43), which is in line with the vessel-specific regulation of TRPV4 channels described above. Thus, the available evidence suggests that TRPV4 activity and cooperative gating are highly dependent on the distance between TRPV4 channel clusters and AKAP5. Accordingly, AKAP5 may compartmentalize TRPV4 and PKC in close proximity to each other to regulate the channel biophysical properties. In agreement with this idea, dialysis of an activated PKC in mesenteric smooth muscle increased TRPV4 current density and likely the frequency of cooperative events (43). These results suggest that regulation of TRPV4 channel function is limited by the availability of AKAP5-anchored PKC near the channel.
On the basis of results described above, we proposed the following model for cooperative gating of TRPV4 channels (FIGURE 5B). In this model, AKAP5, PKC, and TRPV4 clusters form a nanocomplex via a self-assembly, feedback-regulated mechanism. Stimulation of AKAP5-anchored PKC promotes the activity of nearby TRPV4 channels. The resultant increase in Ca2+ influx through a TRPV4 channel primes the other channels within the nanocomplex to undergo as yet undetermined structural rearrangements that facilitate cooperative gating. Considering that PKA increases TRPV4 current density (232), the aforementioned model may have broad implications for regulation of TRPV4 and other TRP channel function and cooperative gating by different G protein-coupled receptors. Because TRPV4 channel activity is modulated by phosphatidylinositol 4,5-bisphosphate (PIP2) (233–236), it would be interesting to determine whether coupled gating of this channel during ANG II/PKC/Ca2+ signaling activation requires PIP2.
4.3. Physiological and Pathological Implications of TRP Cooperative Gating
Cooperative gating of TRP channels amplifies Na+ and Ca2+ influx (15, 23) that can influence membrane potential and intracellular Ca2+-dependent processes. Perhaps not surprisingly, the frequency of cooperative gating of TRP channels is low regardless of the TRP isoform and the cell type (43, 44, 166–170, 219, 221–223). Yet different stimuli can promote cooperative gating of TRP channels. For instance, selective agonists for TRPV4 [e.g., GSK1016790A (43, 44, 167, 219, 221–223)] and TRPA1 [e.g., AITC or 4-HNE (170, 228)] have been shown to increase the frequency of cooperative events. Activation of Gq protein-coupled receptors upstream of PKC with carbachol or ANG II also stimulates TRPV4 cooperative gating (43, 44, 166, 223). The dietary compound carvacrol stimulates TRPV3 cooperative gating (169). Moreover, low pressure was shown to stimulate TRPV4 signals that seem to be produced by the opening of two or more channels, whereas high-pressure conditions prevented/blocked these signals (218).
The increased cooperative gating of TRPV4, TRPV3, and TRPA1 channels can be correlated with endothelium-dependent vasodilation mechanisms in cerebral, mesenteric, cremaster, and pulmonary arteries (166, 168–170, 218, 221, 222). These studies suggest that an increase in cooperative gating of mesenteric and cerebral endothelial TRPV4, TRPV3, and TRPA1 at the myoendothelial projections could amplify local Ca2+ near intermediate-conductance (IK) and small-conductance (SK) Ca2+-activated K+ channels. Activation of IK/SK channels in endothelial cells causes an efflux of K+ ions and hyperpolarization of the plasma membrane of endothelial cells (237, 238). The K+ ions could activate nearby inward-rectifying K+ channels in smooth muscle cells to cause relaxation (149, 239). In addition, endothelial cell hyperpolarization can be transmitted to the adjacent smooth muscle cells via gap junctions at myoendothelial projections to produce relaxation (237, 238). These mechanisms may act in concert to promote vasodilation. In small pulmonary arteries, however, cooperative gating of TRPV4 channels in endothelial cells has been linked with Ca2+-dependent activation of the endothelial nitric oxide synthase (eNOS)/nitric oxide (NO) signaling pathway (222). NO can then diffuse into adjacent smooth muscle cells to activate the guanylyl cyclase/protein kinase G (PKG) pathway, leading to smooth muscle relaxation and vasodilation. Overall, results in mesenteric/cerebral versus pulmonary preparations suggest distinct mechanisms by which cooperative gating of TRP channels may contribute to regulating arterial reactivity.
In smooth muscle, cooperative gating of TRPV4 channels may promote Ca2+ sparks (e.g., Ca2+ release via ryanodine receptors) in the sarcoplasmic reticulum (240). Ca2+ sparks can then trigger spontaneous transient outward currents (STOCs) via activation of large-conductance Ca2+-activated K+ (BKCa) channels, leading to vascular smooth muscle relaxation (149, 241). Consistent with this possibility, it was found that TRPV4 channels oppose ANG II-induced vasoconstriction (44), although the contributions of endothelial versus smooth muscle TRPV4 were not differentiated in this study. Cooperative gating of TRPV4 may also be correlated with activation of the calcineurin/nuclear factor of activated t cell (NFAT) transcription factor in airway smooth muscle as well as proliferation of these cells (167). Thus, cooperative gating of TRP channels may have broad and significant physiological impact on a number of intracellular signaling cascades.
Cooperative gating of TRP channels may play a key role in mediating several pathological conditions. For example, it has been reported that hypoxic conditions stimulate endothelial TRPA1 channel activity, and likely their cooperative gating, to promote vasodilation of cerebral arteries (170). This study also found that hypoxia-induced endothelial TRPA1 activation may be protective against ischemic stroke (170). Cooperative gating of TRPV4 channels in airway smooth muscle may contribute to activation of the calcineurin/NFAT signaling pathway (167). This may lead to airway smooth muscle proliferation that could underlie some of the remodeling observed during asthma. Thus, targeting TRPV4 or preventing TRPV4 cooperative gating might provide novel therapeutic strategies for asthma treatment.
In vascular smooth muscle, TRPV4 activity and cooperative gating are reduced in male cerebral cells in an angiotensin II-induced hypertension mouse model (43). The effects of hypertension on TRPV4 channels properties were correlated with a reduction in the cluster density of TRPV4 channels and AKAP5, as well as an increase in the intermolecular distance between these proteins as determined with superresolution imaging. Intriguingly, hypertensive female vascular smooth muscle from pial and parenchymal branches showed a range of distances between TRPV4 and AKAP5, with all of them exceeding a 200-nm radius (43). This is critical because the overall level of TRPV4 activity, and the likely induction of cooperative channel gating, is determined by the relative distance between the channel and AKAP5, which seems to have a threshold for regulation at ∼200 nm (43). The disruption of vascular smooth muscle TRPV4-AKAP5 signaling during hypertension may prevent optimal activation of Ca2+ sparks and STOCs, resulting in impaired vasodilation (44, 149). Reduced TRPV4 activity and cooperative gating has also been observed in mesenteric endothelial cells from ANG II-induced hypertensive mice (223). As in smooth muscle, the decreased TRPV4 activity and cooperative gating during hypertension was correlated with impaired AKAP5 signaling. Specifically, it was found that AKAP5 disappears from the myoendothelial projections where it facilitates PKC dependent modulation of TRPV4 channels. The absence of AKAP5 at myoendothelial projections during hypertension is likely due to reduced expression of the scaffold rather than its redistribution, as changes in AKAP5-associated signal were not apparent in other endothelial subcellular domains. Disruption of the AKAP5-PKC-TRPV4 axis in endothelial cells during hypertension prevented optimal signaling of TRPV4 Ca2+ to IK/SK channels, leading to impaired transmission of endothelium-dependent hyperpolarization and vasodilation.
A reduction in TRPV4 activity and cooperative gating was also observed in mesenteric endothelial cells from a high-fat diet model of obesity (221). This reduction was correlated with impaired AKAP5-anchored PKC signaling to TRPV4 channels but not with changes in the expression of TRPV4 or AKAP5 or with changes in the association between these proteins. Altered AKAP5-PKC signaling to TRPV4 seems to be mediated by an increase in local peroxynitrite production due to elevated activity of the inducible nitric oxide synthase (iNOS) and the NADPH oxidase 1 (NOX1). It is suspected that increased peroxynitrite oxidizes AKAP5 at cysteine 36 to prevent the interaction of the scaffold with PKC and its subsequent downstream signaling to TRPV4 that will promote cooperative channel gating. These alterations in the AKAP5-PKC-TRPV4 signaling axis by elevated peroxynitrite were found to impair vasodilation and contribute to increased blood pressure in obese mice.
On the basis of the above results, we propose that reduced endothelial and smooth muscle cell TRPV4 channel activity and cooperative gating may be key underlying factors contributing to and promoting vascular dysfunction during hypertension and obesity. Moreover, alterations in TRP channel activity and cooperative gating may have broad implications in modulating many cell and organ functions in health and disease, which remain to be completely elucidated.
4.4. TRP Clustering and Functional Coupling With Other Ion Channels
The coupling of TRP channels with several other ion channels is well documented in the literature (159, 228, 242–250). This cross-coupling between TRP and other ion channels regulates many processes in neuronal, cardiac, and vascular cells in health and disease (215, 251, 252), thus highlighting its significance. Basic understanding of the functional coupling between TRP and other ion channels has been advanced principally through superresolution microscopy that enables the visualization of these proteins at a nanoscale level.
For example, TRPV1 channels were found to cluster in Chinese hamster ovary (CHO) cells overexpressing the TRPV1 subunit and in sensory neurons (159). Using a sophisticated triple labeling approach, the authors found that TRPV1 could form supercomplexes within discrete nanometer regions with clusters of AKAP5 and KCNQ2 channels as well as clusters of AKAP5 and CaV1.2 channels in sensory neurons. These supercomplexes appear to be tethered together by AKAP5, as genetic ablation of this scaffold prevents the close association between TRPV1-KCNQ2 and TRPV1-CaV1.2 complexes in sensory neurons (FIGURE 6, A and B). These results suggest that AKAP5 may serve a dual role as 1) an organizer of signaling complexes to regulate protein function, as traditionally expected (253), and 2) a structural anchor supporting multi-ion channel supercluster formation.
FIGURE 6.
Functional coupling of transient receptor potential (TRP) channels with other ion channels. A: functional coupling between TRPV1 and KCNQ2 modulates neuronal excitability. PIP2, phosphatidylinositol 4,5-bisphosphatase. B: clustering of TRPV1 and CaV1.2 channels regulates TRPV1 desensitization in neurons. C: a tripartite clustering of TRPV1, inositol trisphosphate (IP3) receptor (IP3R), and ANO1 controls membrane depolarization and dorsal root ganglion (DRG) excitability. PLC, phospholipase C. D: mucolytic TRP (TRPML)1s located in immobile lysosomes near ryanodine receptor (RyR) channels control large-conductance (BK) channel activity to regulate vascular smooth muscle contractility. Po, open probability. STOCs, spontaneous transient outward currents. Figure created with Biorender.com.
In a clever series of experiments, it was found that these AKAP5-dependent multi-ion channel superclusters are functionally relevant (159). Activation of TRPV1 channels in sensory neurons led to an AKAP5-dependent inhibition of M currents (159). Although the specific mechanisms for TRPV1-mediated inhibition of M currents were not directly explored in this study, it was suggested that TRPV1 activation could deplete local levels of the phosphoinositide phosphatidylinositol 4,5-bisphosphate (PIP2), likely within the AKAP5-dependent supercluster nanodomain. Because the KCNQ channel open state is highly sensitive to PIP2 (254), the TRPV1-mediated depletion of PIP2 results in suppression of M currents, leading to membrane depolarization and increase in neuronal excitability (FIGURE 6A).
On the other hand, it was also reported that TRPV1 channel activation may lead to activity-dependent desensitization of TRPV1 currents (159). Intriguingly, this was found to require engagement of the CaV1.2 gating machinery and expression of AKAP5. Accordingly, both nifedipine, a CaV1.2 inhibitor that blocks the gating machinery, and genetic ablation of AKAP5 significantly ameliorated activity-dependent TRPV1 desensitization. The mechanisms by which CaV1.2 may promote activity-dependent TRPV1 desensitization are currently unclear but may include local AKAP5-CaV1.2 Ca2+-dependent pathways, AKAP5-mediated physical coupling between both channels leading to conformational changes that alter TRPV1 biophysical properties, or a combination of both (FIGURE 6B).
In independent experiments using triple-labeling superresolution, it was also found that TRPV1 could form a supercomplex with clusters of the plasma membrane Ca2+-activated Cl− channel ANO1 and the endoplasmic reticulum (ER) IP3 receptor (IP3R) in dorsal root ganglion (DRG) neurons (242). Whether AKAP5 contributes to the formation of the TRPV1-ANO1-IP3R supercomplex was not examined, and the mechanisms by which the complex is assembled are unclear at this time. There are data that suggest that TRPV1 activation modulates ANO1 activity via two distinct and likely synergistic pathways (242). First, a local increase in Ca2+ influx via TRPV1 may stimulate ANO1 activity. Second, TRPV1 activation could promote phospholipase C (PLC) activity, leading to activation of IP3Rs. Given the close physical proximity between IP3Rs and ANO1, Ca2+ release via IP3R can maximize ANO1 activity. These concurrent processes are likely to amplify Cl− efflux and membrane depolarization in DRG neurons. The TRPV1-ANO1-IP3R supercomplex may not require physical interaction between channels/receptors but rather a close association in which the local Ca2+ signal generated by TRPV1 and IP3Rs can be sensed by ANO1 (FIGURE 6C).
In vascular smooth muscle, superresolution imaging revealed the presence of TRPML1 clusters in lysosomes that are in close association with ryanodine receptor (RyR) clusters located at the sarcoplasmic reticulum (228). Unexpectedly, TRPML1 channels were found in an immobile lysosomal pool that is thought to be in close proximity to RyRs. It is unclear whether a physical link occurs between TRPML1 and RyR. Nevertheless, this organization places TRPML1 in an ideal position to trigger activation of RyRs to produce Ca2+ sparks [i.e., Ca2+ release via RyR (127, 241)]. The resultant Ca2+ spark can then promote STOCs that affect vascular reactivity and blood pressure (FIGURE 6D).
Taken together, the results described above support the notion that functional, multi-ion channel supercomplexes that include one or several TRP channels are regularly formed in several different cell types. These supercomplexes can play key roles in regulating cellular function in health and disease. This assumption requires further exploration, thus representing a fertile area of research.
5. NaV CHANNELS
The principal physiological function of NaV channels is to regulate sodium ion flux across cell membranes (255). Channel activation results in membrane depolarization that generates the depolarizing phase of the action potential in excitable cells and facilitates the propagation of action potentials along contiguous cellular processes (1). Aberrations in NaV channel function can exert profound effects on membrane excitability and may confer severe dysfunction in nerve and muscle tissues, including loss of sensation, chronic pain, cardiac failure, and paralysis (256).
The biophysical properties of NaV channels have been extensively studied in neurons (e.g., Ref. 257) and heterologous expression systems (e.g., Ref. 258). NaV channels are a ubiquitous class of transmembrane protein complexes composed of an α-subunit that forms the pore of the channel, which is structurally responsible for voltage sensing and gating as well as ion permeation, and one or more auxiliary β-subunits that modulate the voltage dependence, permeability, and kinetics of NaV gating (259–261). In addition, the β-subunits are important regulators in cell adhesion, signal transduction, and channel expression at the plasma membrane (262).
The α-subunits of NaV channels are large proteins of ∼250 kDa composed of four homologous domains (I–IV) consisting of six α-helical transmembrane-spanning segments (S1–S6) (FIGURE 7A) (263). The transmembrane segments are connected via small intracellular and extracellular loops, whereas homologous domains are connected through larger intracellular loops (264). Ten different α-subunit isoforms have been discovered to date that are expressed in different types of cells. Nine of the isoforms generate voltage-gated channels (NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.5, NaV1.6, NaV1.7, NaV1.8, and NaV1.9). The tenth isoform (NaVX) appears to reside within a related protein family, as it does not encode a voltage gate (265).
FIGURE 7.

Voltage-gated Na+ (NaV) channel structure and a model of coupled NaV channels. A: illustration of the pore-forming NaV and auxiliary NaVβ subunits. Location of the 14-3-3 protein is highlighted in the I–II linker. B and C: models of the interaction between 2 NaV channels based on structures by Jiang et al. (446) and Yang et al. (447) and data from Clatot et al. (10) and Allouis et al. (448). Figure created with Biorender.com.
NaV1.1, NaV1.2, NaV1.3, and NaV1.7 isoforms are principally expressed on the membranes of central and peripheral neurons. The NaV 1.4 isoform is expressed predominantly in skeletal muscle cells, whereas the NaV1.6 isoform is expressed in central neurons. NaV1.5 is primarily expressed in the heart and NaV1.8 and NaV1.9 on neurons whose cell bodies reside in the dorsal root ganglia. Tetrodotoxin blocks ion permeation in all of the isoforms, though the binding affinities differ substantially among the isoforms (for review and references, see Ref. 266).
As mentioned above, the α-subunit of NaV channels forms the channel’s pore and is responsible for its voltage sensitivity and selectivity. The S4 transmembrane segments of the α-subunit constitute the voltage sensor in NaV channels, as is the case for other classes of voltage-gated ion channels (263). Depolarization of the membrane leads to a translocation of the positively charged S4 segments toward the outside of the membrane and initiates channel activation (267). The actual movement of the charges within the α-subunit, primarily confined to the intramembrane region of S4 segments, is proposed to produce the gating currents and channel opening (268).
The voltage sensor component of the α-subunit of NaV channels is also associated with the voltage dependence of channel inactivation (269), which plays out on three distinct timescales: fast, slow, and ultraslow inactivation (reviewed in Ref. 270). Inactivation regulates membrane excitability by limiting the pool of “activatable” NaV channels and for fast inactivation is mediated by a group of hydrophobic residues (isoleucine, phenylalanine, and methionine; the IFM motif) localized to the intracellular DIII–DIV linker, forming the “inactivation gate.” In the channel open state, an intracellular binding site for the IFM motif of the inactivation gate is exposed. The subsequent movement and binding of the IFM motif inactivates the channel, as the bound IMF motif blocks the pore of the channel and precludes its reopening until resting state is restored (reviewed in Ref. 255). The mechanisms underlying slow and ultraslow inactivation have not yet been completely described but are thought to involve conformational changes in the pore of the channel (270).
5.1. Persistent Currents Generated by NaV Channels
In addition to generating the fast-inactivating Na+ current (INaT) responsible for the rising phase of the action potential (271), NaV channels are also mediate a “persistent” inward current (INaP) essential for sustaining repetitive firing in neurons (214, 272). The NaV isoforms responsible for INaP in all cell types have not been identified. However, NaV1.6 is a likely contributor in cerebellar Purkinje neurons and in cortical pyramidal neurons (273, 274). In addition to NaV1.6, the other four NaV channel isoforms expressed in central neurons all can produce INaP.
The molecular mechanism leading to INaP is likely to be late channel opening and reopenings during prolonged depolarization, as inferred from the analysis of NaV1.6 channel currents in heterologous HEK-293 cells (275). To date, however, a detailed investigation of the mechanisms underlying INaP in all NaV isoforms has not been performed. Similarly, there is also a lack of mechanistic information on how native NaV channel in neurons generate INaP. It is possible that INaP is facilitated by the cooperative gating of adjacent NaV channels within homonymous channel clusters, but experimental evidence in support of this hypothesis has not been reported.
5.2. Modulation of NaV Channel Activity
The majority of the studies investigating the mechanisms modulating NaV channel activation have focused on the inactivation gate (IG) of the channel. For example, G protein signal transduction pathways activated by monoamines are key regulators of NaV gating. Physical interaction of G protein βγ subunits with the COOH terminal of the channel have been shown to increase INaP (276). Interestingly, Gβγ subunits do not actually alter the voltage dependencies of the rapidly inactivating Na current (INaT). Instead, Gβγ subunits shift the voltage dependence of inactivation of INaP positively with respect to that of INaT. Ma et al. (276) proposed that the Gβγ induces INaP by altering fast inactivation of the channel. The mechanism by which this happens seems to involve the formation of Ca2+-calmodulin (CaM) bridges from the COOH-terminal IQ motif to the DIII–IV linker. This destabilizes the inactivation gate of the channel, shifting inactivation toward more depolarized potentials (258, 277), and thus increasing the number of noninactivating sodium channels.
A recent study involving the cardiac NaV1.5 isoform provides an additional potential mechanism for the induction of INaP (278). In this model, CaM binds to two independent sites located in the intracellular loop that form the inactivation gate in a Ca2+-dependent manner that facilitates recovery from inactivation and also slows down the kinetics of inactivation (278). Future studies should investigate whether a similar mechanism contributes to persistent NaV channel currents by other isoforms in neurons.
5.3. Expression of NaV Channels Within Different Excitable Tissues
The primary locations and clustering properties of the different NaV isoforms have been described in detail in a recent comprehensive review article (279). Our focus here is to identify the cellular compartments of individual cell types where dense aggregations of NaV channels have been observed and the effects these aggregations may have on cell function. NaV1.2 and NaV1.6, for example, are found ubiquitously in neural axons, but NaV1.6 exhibits a particularly dense concentration at two identified sites: 1) the axon initial segment (AIS), where the high density is presumed to be instrumental in action potential initiation (280) and may contribute to the amplification of synaptic inputs (281), and 2) at nodes of Ranvier in myelinated axons, where the high density supports action potential propagation by saltatory conduction (282). The accumulation and stabilization of NaV channels at these sites depend on anchorage to the submembrane cytoskeleton through ankyrin G and on adjacent membrane barriers and, in addition, at nodes of Ranvier, axonal cell adhesion molecules that interact with glial membranes to localize and stabilize the node (279). Other NaV isoforms aggregate in different sites including neuronal dendritic trees, synapses, unmyelinated axons, cardiomyocytes, and other nonneuronal excitable cells (279).
NaV1.4 channels aggregate on the postsynaptic membrane of neuromuscular junctions of skeletal muscle cells, but the scaffolding and anchoring mechanism remain unresolved. Agrin, a neuronal proteoglycan, appears to mediate the aggregation, at least in cultured rat muscle fibers (283). It has also been suggested that these NaV1.4 channels are connected to muscle cell cytoskeletal structures via ankyrin-spectrin and to its extracellular matrix through syntrophin-dystrophin-dystroglycan-agrin (284).
NaV1.5 channels are concentrated in two distinct compartments in cardiomyocytes, the intercalated disk, where NaV1.5 associates with multiple proteins, including ankyrin G and plakophilin 2, as well as the lateral membranes. NaV1.5 engages with the syntrophin-dystrophin complex (285). In the lateral membranes of ventricular myocytes, NaV1.5 channels form clusters of various sizes and densities, the largest of which occur in the crest region of the sarcomere and between which there appears to be little electrical activity (286). With proximity ligation assay (PLA) and stochastic optical reconstruction microscopy (STORM) superresolution microscopy, it has recently been demonstrated that NaV1.6 channels are not only distributed in dense clusters but colocalize with RyR2 and display noticeable alignment with t-tubules (287). Machine learning-based cluster detection and STORM-relative localization analysis (STORM-RLA) demonstrated that ∼60% of NaV1.6 clusters are located within 100 nm of RyR2 (288). These results strongly suggest that NaV1.6 channels are implicated as a component of the Ca2+ cycling nanodomain.
Clusters of NaV1.1 channels associated with ankyrin G and neurofascin have been reported on cells of the inner plexiform layer (IPL) of the retina that are displaced by NaV1.2 channels during development (289). These clusters in the IPL appear to be restricted to AIS-like processes on AII amacrine cells (290) and likely function as spike generators similarly to the conventional AIS on neurons with full axons.
Finally, in the cochlea, NaV1.6 channels are clustered on afferent fibers, whereas NaV1.2 channels are clustered within the membranes of efferent fibers (291). The NaV1.6 channel clusters are concentrated near the axonal terminals of the primary afferent fibers that contact the inner (type I) and outer (type II) hair cells. In addition, NaV1.6 is concentrated in the AIS emanating from the type II ganglion cell soma so that, in effect, both the central and peripheral axon processes of these unmyelinated neurons are richly endowed with NaV1.6 (291). On the efferent cochlea neurons, NaV1.2 clusters are restricted to the axonal endings.
5.4. Cooperative Gating of NaV Channels Within Clusters
Beyond identifying where clusters of NaV channels are found in excitable tissues, we are principally focused in this review on whether NaV channels within dense clusters form functional oligomeric complexes. Evidence for cooperative gating of neuronal NaV isoforms, however, is largely confined to modeling studies that suggest that cooperative gating of NaV channels within dense clusters enhances the capacity of neurons to generate trains of action potentials at high frequencies (292, 293) as well as persistent neural discharge that outlasts the stimulus that triggers it (27).
To our knowledge, the first evidence that this may in fact be the case appeared in a study by Undrovinas and colleagues (9) demonstrating the possible simultaneous opening and closing of two and three channels, presumably NaV1.5, recorded from cell-attached and excised inside-out patches of rat and rabbit cardiomyocytes. The authors offered no mechanism that might account for the synchronized gating but speculated that this behavior might result from a very tight cooperativity in the gating of separate channels. However, the authors were unable to distinguish this possibility from the alternative hypothesis that these recordings represented supraconductance levels that are only coincidentally the same as integral multiples of the unitary conductance.
It has recently been reported that the α-subunits of NaV1.1, NaV1.2, and NaV1.5 channels are expressed in the membrane as dimers that gate in a cooperative fashion (FIGURE 7, B and C) (10). The two α-subunits interact both directly and indirectly via the cytoplasmic protein 14-3-3. Both mutations at the 14-3-3 interaction sites and inhibition by difopein disengage the two NaV α-subunits, which has adduced evidence for dimerization and functional interaction for NaV1.7 channels. As a consequence of these findings, future studies will need to include new analyses and modeling of Na+ currents to acknowledge that the quantal unit is generally a dimer rather than a single channel [i.e., the measured NaV channel conductance is twice the value for an actual single α-subunit pore (294)].
5.5. Pathological Consequences of Altered Clustering of NaV Channels
Alterations in the normal patterns of NaV channel distributions are associated with several pathologies. For example, in chronic demyelinating diseases, large clusters of NaV channels often appear on demyelinated axons (295). Moreover, isoforms other than the normal NaV1.6, such as NaV1.7, are often abnormally expressed along the demyelinated axons (296). Similarly, NaV1.8 channels, which are normally uniformly distributed along the axons of the unmyelinated C fibers innervating nociceptors, display abnormal clustering after injury that appears to be associated with neuropathic pain (297).
In the heart, mutations in SCN5A, the gene encoding cardiac voltage-gated NaV1.5 channel, lead to a wide variety of cardiac arrhythmias, including long QT syndrome type 3 (LQT3), Brugada syndrome, conduction defects, and sick sinus syndrome. It some cases, the pathology may be caused by an alteration in the distribution of these channels within the lateral membranes and t-tubules (298). In other instances, however, it has been proposed that the mutations in SCN5A may lead to reduced INa through a dominant-negative effect exerted by the mutant, nonconducting channels coupling to wild-type channels.
Recent work from the Deschênes laboratory (299) suggests that the heart failure-associated splice variant NaV1.5-G1642X suppresses Na+ currents in heart failure patients by preventing the dimers from gating cooperatively. Similarly, Clatot et al. (300) found that the NaV1.5-L325R mutant, which is associated with Brugada syndrome, dimerizes with wild-type channels and thus leads to a dominant-negative effect, as reflected by a nearly 75% reduction in NaV current density. This was the first evidence that cooperative gating of voltage-gated Na+ channels can also be responsible for the dominant-negative effect leading to arrhythmias.
Finally, Rühlmann et al. (301) discovered that a mutation in the hNaV1.7 channel (A1632E) that causes symptoms of erythromelalgia and paroxysmal extreme pain disorder increases persistent Na+ currents by dimerizing. Preventing dimerization of hNaV1.7-A1632E functionally decoupled the channels and decreased persistent Na+ currents. Functional uncoupling of mutant hNaV1.7/A1632E channel dimers restored their defective allosteric fast inactivation mechanism. Thus, as with CaV1.2 channels, dimerization of mutant NaV1/5/1.7 channels could induce or exacerbate pathological conditions.
6. K+ CHANNELS
Potassium (K+) channels are the most varied and widely distributed ion channels found in the membranes of living cells (2). They are subdivided into four families based on both their structural and functional properties: 1) calcium- or sodium-activated potassium channels (KCa or KNa), 2) inward-rectifying potassium channels (KIr), 3) two-pore domain potassium channels (K2P), and 4) voltage-gated potassium (KV) channels. More than 80 mammalian genes are engaged in encoding their α protein subunits (302–305).
In contrast to the single polypeptide that forms the ionophore of NaV and CaV channels, all K+ channels have a tetrameric structure resulting from the assembly of four identical subunits forming a symmetric (C4) complex organized to form a conducting pore (i.e., a heterotetramer). In some instances, four related, nonidentical protein subunits assemble to form heterotetrameric channels. All K+ channel subunits have a unique pore-loop structure that lines the top of the pore and is responsible for K+ selectivity and permeability (306). The principal physiological functions of K+ channels include setting the resting membrane potential (Vm), repolarization of excitable cells following the rising phase of an action potential, regulation of the cardiac action potential duration, maintenance of vascular tone, and the regulation of many cellular processes including the secretion of hormones (2).
As is the case for other classes of transmembrane channels, K+ channels generally display striking variations in their distributions and relative densities in different cellular compartments and are known to form dense clusters (307, 308). In addition, there have been several reports of alterations in normal gating behavior of K+ channels within clusters that we describe in sects. 6.1–6.7 (181, 309–311).
6.1. KV1 Channels
KV1 channels are concentrated in the axons and synaptic terminals of neurons (312), where they facilitate action potential propagation (e.g., Ref. 313) and neurotransmitter release (e.g., Ref. 314), respectively. KV1 channels are also present in the somatodendritic regions of some CNS neurons (315).
At synaptic terminals, KV1 channels are arranged in distinct clusters that are thought to be formed and maintained through interactions of PSD (postsynaptic density)-95 with a PDZ (postsynaptic density-Dig1-Z0-1) binding motif [X(S/T)XV-COOH] at the channel’s COOH terminus (48, 316). In heterologous cells, the binding induces oligomerization that reduces internalization of expressed KV1.4 channels and causes them to form clusters (317). Similarly, KV1.2 channels aggregate with PSD-95 in presynaptic terminals in the cerebellum (48) and are essential for embedding KV1.4 in rat cortical axons (318). Although PSD-95 is found colocalized with KV1 channels at juxtaparanodal regions along myelinated axons, it is not essential to generate KV1 clusters, as they still appeared at the juxtaparanodal regions of the optic nerve in mice that expressed a truncated, mutant form of PSD-95 (319). KV1.2 clusters are also observed in these mutant mice. At present, it is unclear whether clustering alters KV1 function.
6.2. KV2 Channels
KV2.1 channels are responsible for the delayed-rectifier current in the somatodendritic membranes of neurons (320, 321). They are organized into dense clusters through a phosphorylation-dependent process (322). Furthermore, in hippocampal neurons, it appears that a 28-amino acid sequence on the COOH terminus of the KV2.1 channels provides the requisite signal and possible template for cluster formation (323).
Recent work on the distribution of KV2.1 channels has revealed a more complex pattern of functional segregation: half of the channels appear to be broadly distributed within plasma membrane, whereas the other half are found in dense clusters on the soma, dendrites, and axon initial segment (AIS). The clustering occurs at endoplasmic reticulum (ER)-plasma membrane (PM) junctions created via interaction with VAMP-associated proteins in the expressed in the ER (61, 324). What is most intriguing about KV2.1 channels is that most of these channels are nonconductive in vascular smooth muscle (157) and heterologous expression systems (325). Interestingly, O’Connell, Loftus, and Tamkun (325) proposed that clustered channels are largely nonconducting. This has been referred to as “negative cooperativity” (181, 326). It is unclear, however, whether clustering per se causes negative cooperativity or a parallel signaling event.
6.3. KV7 Channels
KV7 channels are slowly activated delayed-rectifier channels that open at subthreshold levels, contributing to the resting membrane potential and reducing excitability (2). KV7.1 is expressed in multiple cell types such as ventricular myocytes and epithelial cells. KV7.2 is localized to the axon initial segment (AIS) and nodes of Ranvier in both peripheral and central neurons and is often colocalized with KV7.3 (327).
A recent study has shown that KV7.4 channels are arranged in dense clusters in cochlear outer hair cells, within which the channels gate cooperatively (328). The consequences of this cooperative gating are increased gating kinetics and voltage sensitivity. The clustering also appeared to enhance the mechanical sensitivity of the KV7.4 channels, which varied systematically with hair cell position along the basilar membrane. Cooperative channel gating leading to enhanced electromechanical sensitivity could be induced in HEK-293 cells by fusing channels into clusters by optogenetic methods.
6.4. KCa Channels
Large-conductance KCa1.1 channels (also known as BK channels) are activated by Ca2+ and membrane depolarization (2) These channels have been suggested to assemble into macromolecular complexes with the voltage-gated CaV1.2, CaV2.1, and CaV2.2 channels and couple functionally via local Ca2+ signals (329). A recent study suggested that CaV1.3 channels could also activate KCa1.1 (41). Vivas et al. (41) found that when KCa1.1 and CaV1.3 channels were coexpressed in the same cell, these channels were expressed into dense clusters of BK channels surrounded by clusters of CaV1.3 channels, forming a presumed multichannel complex (41, 329).
Similar patterns of coclustering were observed in a heterologous system as well as in both hippocampal and sympathetic neurons in the rat (39). Although this study did not include electrophysiological measurements, the authors suggest that this is a functional consortium, ensuring “tight tracking between local BK channel activation and the gating of CaV1.3 channels at quite negative membrane potentials, facilitating the regulation of neuronal excitability at voltages close to the threshold to fire action potentials.” Future studies should investigate whether, like CaV1.3 channels, CaV1.2, CaV2.1, and CaV2.2 clusters form around or within BK channel clusters in neurons.
6.5. Pathological Consequences of Altered Clustering of KNa1.1 Channels
Mutations of the KNa1.1 (KCNT1) channels are associated with epilepsy (330). In a recent study (331), it was found that nine different mutant KNa1.1 channels linked to epilepsy produced many-fold increase in the current produced by wild-type channels. Although wild-type KNa1.1 channels can gate cooperatively, the authors found that the extent of cooperative gating was much greater for the mutant channels even though their single-channel conductances were reduced (331). These results indicate that changes in the extent to which membrane channels interact in homogeneous clusters may exert as profound an effect on function as changes in the behavior of the individual protein molecules.
6.6. K2P Channels
K2P2.1 and K2P4.1, the thermosensitive and mechanosensitive two-pore potassium channels, have been found to be clustered at the nodes of Ranvier of large-diameter rat trigeminal afferents with density >3,000 times higher than that on the somata (332, 333). These K2P channels are required to generate the repolarization phase of the propagated action potential at the nodes.
6.7. KcsA Channels
KcsA is a prokaryotic K+ channel from the soil bacterium Streptomyces lividans. KcsA is pH activated and has two transmembrane segments that together create a highly selective ion pore (334). Because the amino acid sequence of the selectivity filter of KcsA is highly conserved among both prokaryotic and eukaryotic K+ channels, analysis of the structure of this channel has generated important structural and mechanistic insights on the gating mechanisms of K+ channels in general. Clusters of KcsA channels have been observed (335), and a recent modeling and experimental study (309) suggests that coupled gating within these channel clusters may account for the unusual electrical activity reported for these channels (334, 336).
7. RYANODINE RECEPTORS
7.1. Structure and Function of Ryanodine Receptors
Ryanodine receptors (RyRs) are Ca2+-activated channels expressed in the endo/sarcoplasmic reticulum of many cell types, including neurons and muscle, where they form homotetrameric assemblies (82, 86, 154). The molecular mass of a RyR tetramer is ∼2.2 MDa. The RyR channel has two distinctive domains (337–339). The cytoplasmic domain comprises the largest portion of the channels (∼29 × 29 × 12 nm). Multiple proteins bind to this cytosolic domain (e.g., calmodulin and FKBP12). The second domain is relatively small (∼7 nm) and includes the channel’s transmembrane segments where the pore is located (FIGURE 8A). A detailed review of RyR structure and function has recently been published (340). Here, we focus our discussion on clustering and cooperative gating of RyRs.
FIGURE 8.
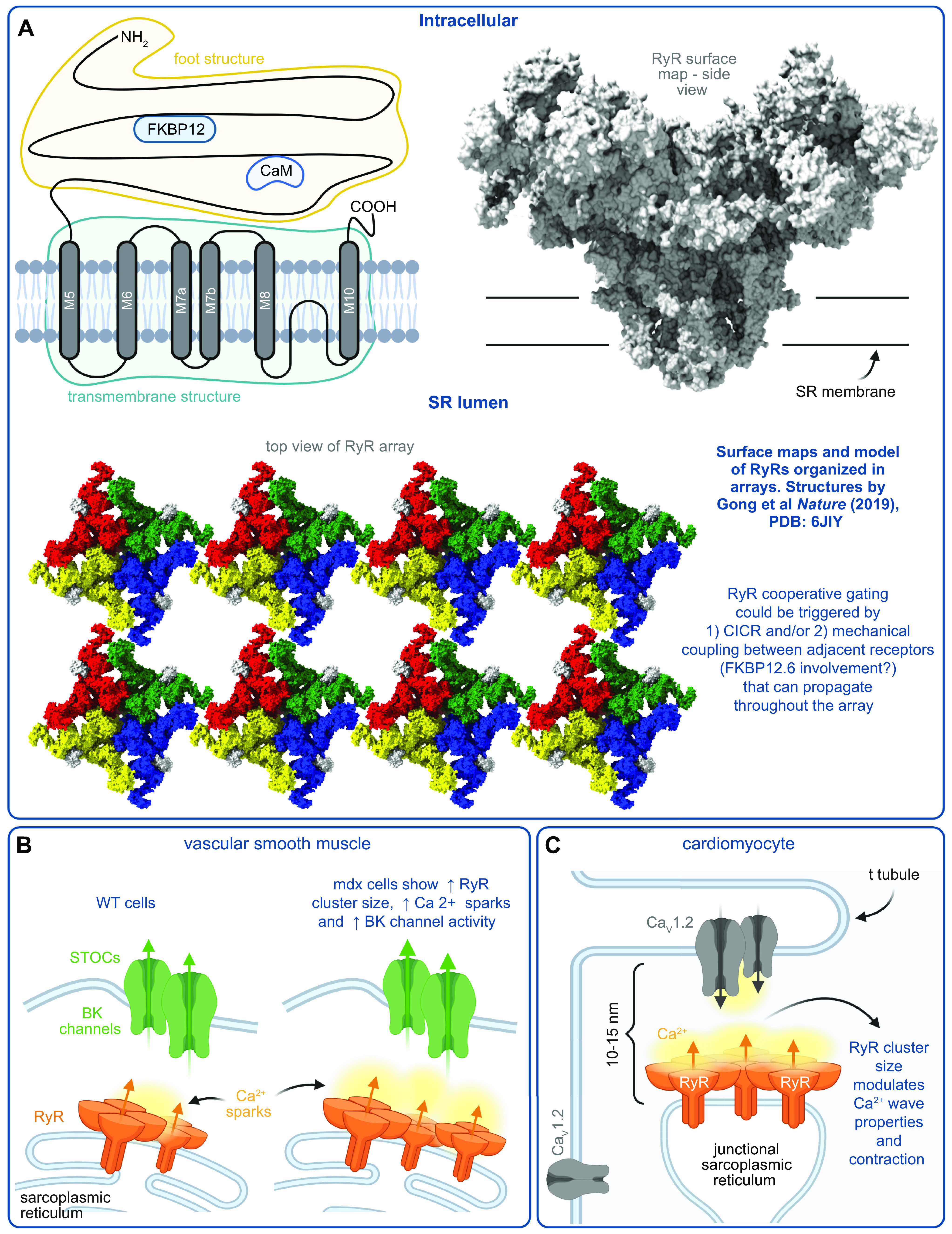
Ryanodine receptor (RyR) structure, organization, and a model of RyR interactions with other ion channels. A: illustration of an RyR subunit and RyR surface map highlighting its 3-dimensional structure. RyRs organize into clusters or arrays that enable cooperative gating. Surface map and structure based on Gong et al. (449) and organization based on Yin et al. (450). CaM, calmodulin; SR, sarcoplasmic reticulum. B: model of RyR organization in wild-type (WT) and mdx cells and its impact on large-conductance (BK) channel activity in vascular smooth muscle. STOCs, spontaneous transient outward currents. C: model of the organization and communication between RyR and CaV1.2 in the t-tubules of cardiac myocytes. Figure created with Biorender.com.
There are three isoforms of RyRs: types 1 (RyR1), 2 (RyR2), and 3 (RyR3). They differ in their tissue and cellular compartment distributions. RyR1s are expressed in skeletal muscle, RyR2s are expressed predominantly in cardiac and smooth muscle, and RyR3s are expressed in neurons as well as smooth muscle (341–343). RyR3 is also expressed at low levels in striated muscle. In general, the shapes and binding sites of modulatory proteins (e.g., calmodulin, FK506-binding proteins) of RyR channels are similar (344, 345).
As intracellular channels, the biophysical properties of RyRs are difficult to study with standard patch-clamp electrophysiological approaches. Accordingly, the most detailed biophysical analyses of RyRs have been done by incorporating them into planar lipid bilayers (346, 347). The three RyR isoforms have many biophysical properties in common, including activation by submicromolar Ca2+ elevations in the cytosolic side of the channel, by ATP, and by caffeine, as well as inhibition by micromolar Mg2+ concentration, ruthenium red, and procaine (341–343). RyRs also display comparable unitary conductances under similar experimental conditions. However, there are some important differences between isoforms, such as the higher sensitivity of RyR2 and RyR3 to cytosolic Ca2+ than RyR1. RyR3s are also more resistant to inactivation by high Ca2+ concentrations (348).
Our understanding of the mechanisms controlling RyR function in intact cells took a major step forward following the development of bright, high-dynamic range Ca2+ indicators such as fluo-3. Cheng et al. (127) used confocal microscopy with high, diffraction-limited resolution to image intracellular Ca2+ in intact rat ventricular myocytes loaded with fluo-3. They discovered that these cells produced localized intracellular Ca2+ elevations, which they called Ca2+ sparks, consequent to the opening of RyR2s (reviewed in Ref. 349). A key feature of Ca2+ sparks was that their amplitude distribution was unimodal, suggesting they were quantal Ca2+ release events. The Ca2+ flux associated with a spark was ∼3 pA, similar to the Ca2+ current produced by the opening of a single RyR in a planar lipid bilayer (350–352). This similarity led them hypothesize that Ca2+ sparks could be produced by the opening of a single RyR or possibly a small cluster of RyRs acting in concert and that Ca2+ sparks are the elementary events underlying excitation-contraction coupling in the heart. Accordingly, a local control model for excitation-contraction coupling was proposed in which the amplitude of whole cell [Ca2+]i transient is graded by the number of Ca2+ sparks activated by Ca2+ influx via L-type CaV1.2 channels (127, 142, 143). Following the publication of the seminal Cheng et al. study (127), Ca2+ sparks have been imaged in multiple cell types, including skeletal muscle (355, 356), smooth muscle (241, 357), and neurons (49, 358).
7.2. Cooperativity Among RyRs
Although the hypothesis that Ca2+ sparks are the elementary events underlying cardiac excitation-contraction coupling was broadly accepted, the issue of whether these events are produced by the opening of a single RyR or a small cluster of RyRs opening simultaneously was the focus of multiple follow-up papers. In an elegant set of experiments, Michael Fill and his team (359) recorded elementary RyR currents in planar lipid bilayers but, unlike in prior studies, under near-physiological conditions (e.g., physiological Mg2+ concentration and Ca2+ gradient, ATP). Their data indicated that the RyR single-channel current was ∼0.25 pA, an order of magnitude smaller than the currents recorded under nonphysiological conditions (∼3 pA). These data strongly suggested that normal Ca2+ sparks were likely produced by a cluster of RyRs opening in synchrony.
The results described above were consistent with classic electron microscopy studies (87, 360, 361) and, more recently, fluorescence superresolution microscopy (28, 49, 133, 362) showing that RyRs organize in clusters/arrays in the endo/sarcoplasmic reticulum of muscle and neurons (FIGURE 8A). In cardiac and smooth muscle, it has been estimated that these clusters contain ∼20–40 ryanodine receptors, respectively (133, 362, 363). Because RyR clusters in neurons subsume about the same area as those observed in muscle cells, the expectation that a similar number of channels is contained in these clusters is quite reasonable. Importantly, the current models of junctional sarcoplasmic reticulum suggest that Ca2+ sparks may be the result of the concerted opening of multiple RyR clusters that collectively form a functional “supercluster” (28, 133).
As Ca2+-activated channels, a plausible mechanism by which a cluster of RyRs could be activated at or near synchrony is Ca2+-induced Ca2+ release. One possibility is that an entire RyR cluster is activated by an adjacent CaV1.2 channel cluster. Similarly, CaV1.2 channels could activate one or a few RyRs in the jSR, which then subsequently activate neighboring RyRs via the Ca2+-induced Ca2+ release mechanism. In this model, Ca2+ is the coupling signal for clustered RyRs. Indeed, Porta et al. (364) found that RyR1 clusters could also undergo cooperative gating in planar lipid bilayers and that the coupling strength was differentially modulated by cytosolic ATP, Mg2+, and Ca2+. Extensive in silico work has been done to simulate how ryanodine-to-ryanodine receptor coupling might operate (365–367).
Marx et al. (88, 368) offered an alternative model of RyR coupling. They showed that multiple RyR1s and RyR2s can be isolated under conditions such that they remained physically coupled to one another in planar lipid bilayers and coordinated their channel openings. The key finding is that the regulatory subunit FK506 binding protein (FKBP12 in skeletal muscle and FKBP12.6 in cardiac muscle) functionally couples multiple RyR1/2 channels. Although the simultaneous activation of RyRs could be mediated entirely through activation of clustered channels by locally released Ca2+, it has been proposed that physical allosteric interactions can also contribute to RyR gating and thus Ca2+ sparks. This FKBP12-based coupling model was extended to suggest that loss of FKBP12/FKBP12.6-decoupled RyRs and increased sarcoplasmic reticulum Ca2+ leak contributes to contraction failure and arrhythmias in the heart (369, 370) and fatigue in skeletal muscle (371). Interestingly, detachment of FKBP12.6 from the RyR2 channel does not lead to the elimination of Ca2+ sparks. Instead, it increases Ca2+ spark frequency and duration (372). Although the bilayer data supporting physical coupling of RyRs are quite compelling, the exact mechanisms by which the opening of RyR channels within clusters are coordinated remain poorly understood.
An important observation made by Baddeley et al. (28), Soeller et al. (133) and Pritchard et al. (363) is that RyRs cluster sizes follow a near-exponential distribution compatible with a stochastic self-assembly process for channel cluster formation similar to that of CaV1.2 and large-conductance Ca2+-activated K+ channels (5). This is significant because it suggests that the two key components of the dyad in cardiac and smooth muscle are formed randomly. Di Maio et al. (373) proposed a model for the formation of Ca2+ release units in striated muscle in which the first step involves the emergence of t-tubules. Stable sarcolemma-sarcoplasmic reticulum junctions are formed by junctophilin-2 (363, 374, 375). This is followed by the recruitment of the junctional SR proteins such as triadin, RyRs, and CaV1.2/1.1 channels but could lead to “silent” dyads if these proteins are not delivered to these sites.
Recent studies on t-tubules considered in conjunction with those on the formation of CaV1.2 and RyR channel clusters suggest an alternative model. t-Tubule formation and random CaV1.2 clustering are possibly early events in the formation of functional Ca2+ release units. Several proteins have been identified to play important roles in t-tubule and dyad formation. These include Caveolin-3 (376–378), dysferlin (379, 380), junctophilin 2 (381), triadin (382), and BIN1 (383–385). BIN1 is a member of the Bin1-Amphiphysin-Rvs (BAR) domain superfamily BIN1, which has been shown to promote t-tubule biogenesis and to serve as anchoring point for microtubules in cardiac muscle. In ventricular myocytes, BIN1 is critical for the trafficking of CaV1.2 channels to the sarcolemma from the Golgi apparatus (383–386). This not only allows for the delivery of CaV1.2 channels to the t-tubules but may also direct the movement of jSR along microtubules anchored to BIN1 toward newly formed CaV1.2 clusters (387). Thus, although RyR-expressing junctional SR membrane may randomly contact the sarcolemma, it only results in stable dyads where CaV1.2 (e.g., cardiac) or BK channel (e.g., smooth muscle) clusters are randomly formed.
A similar process may be at play in ER-plasma membrane junctions in neurons. KV2.1 channels promote the clustering of CaV1.2 channels in the somata of hippocampal neurons. Furthermore, RyR-expressing ER terminals exist at sites where KV2.1 and CaV1.2 channels cocluster (49). It is possible that ER movement is stochastic (388), but, as in muscle, stable ER-plasma membrane junctions only form where KV2.1 and CaV1.2 channels cocluster. The key concept here is that multiple stochastic processes (i.e., CaV1.2 clustering, BK clustering, RyR clustering, and SR/ER mobility) may lead to dyad formation in nerve and muscle.
7.3. Physiological Implications of RyR Cooperative Gating
A central tenet of intracellular Ca2+ signaling is that RyR and IP3R recruitment is hierarchical as follows. Ca2+ sparks are produced by the simultaneous activation of clustered RyRs. Each of the RyRs within these clusters produces an elementary Ca2+ signal, called a “Ca2+ quark” (389, 390). Thus, Ca2+ quarks produce Ca2+ sparks. Whole cell Ca2+ transients are produced by the temporal and spatial summation of multiple independent Ca2+ spark sites (i.e., Ca2+ release units). RyR-mediated Ca2+ waves are produced and propagated by the successive, progressive activation of Ca2+ spark sites along the cell. Thus, current Ca2+ signaling models are based on this Ca2+ quark, Ca2+ spark, whole cell Ca2+ transient/Ca2+ wave hierarchy.
Alterations in the behavior of any of the components of this hierarchical signal cascade could have important implications in Ca2+-dependent cellular events. For example, an increase in the coupling strength between RyRs within a cluster could increase the open probability of these channels and hence increase the amplitude and spatial profile of Ca2+ sparks. This could result in larger Ca2+ transient in cardiac myocytes and hence contraction during the action potential. An increase in Ca2+ spark amplitude could increase the probability of activating nearby Ca2+ release units via Ca2+-induced Ca2+ release (CICR), which could result in a Ca2+ wave. An increase in Ca2+ spark amplitude could increase the probability of CaV1.2 channel coupling, CaV1.2 Ca2+-dependent inactivation, in cardiac myocytes or BK channel activation in smooth muscle. An increase in RyR cluster size would have a similar impact on Ca2+ signaling even in the absence of an increase in coupling strength.
As an example, Pritchard et al. (363) found that RyR2 and BK channels were organized into clusters. The RyR2 clusters that colocalized with BK channel clusters were nearly 1.6-fold larger in smooth muscle cells from the mouse Duchenne muscular dystrophy model (mdx) than in control myocytes (FIGURE 8B). They found that the amplitude and frequency of Ca2+ sparks were greater in smooth muscle cells from mdx mice than those from control mice. This finding was associated with a near loss of myogenic tone in cerebral pial arteries, and parenchymal arterioles from mdx mice failed. However, inhibition of RyR and BK channel activity increased myogenic tone in mdx arteries to control levels. Thus, increased size of RyR2 protein clusters increases Ca2+ spark and BK channel activity and could lead to cerebral microvascular dysfunction.
Similarly, Xie et al. (367) found that the Ca2+ spark frequency and amplitude increased in a nonlinear fashion as the size of RyR clusters increased in ventricular myocytes (FIGURE 8C). The larger RyR clusters are activated at lower Ca2+ levels. However, the larger fluxes associated with larger RyR clusters led to lower SR Ca2+ content. By contrast, the smaller RyR clusters had a higher threshold for activation and a lower associated leak. This increased SR Ca2+ content.
Mathematical modeling suggested that homogeneously large or small RyR clusters decrease the propagation of Ca2+ waves due to low load for large clusters but relatively low excitability for smaller RyR clusters (367). By contrast, their model predicted that expression of a combination of large and small RyR clusters would result in the potentiation of Ca2+ waves, as the enhanced SR Ca2+ load caused by smaller RyR clusters enables Ca2+ wave initiation and propagation from larger RyR clusters. Thus, a heterogeneous distribution of RyR cluster size may increase Ca2+ wave production and propagation and thus afterdepolarizations and triggered arrhythmias.
Kolstad et al. (391) investigated whether the deficit described above could be traced to nanoscale RyR reorganization. They found that failing myocytes had more numerous, smaller RyR clusters compared with those extracted from control rats. The decrease in the size of Ca2+ release units was associated with an increase in optically “silent” SR Ca2+ leak and a decrease in Ca2+ spark numbers during heart failure. Ca2+ sparks in failing cells displayed slow kinetics, as the Ca2+ diffused across dispersed Ca2+ release units. These slow Ca2+ sparks desynchronized the whole cell Ca2+ transient during the action potential. Thus, Kolstad et al. (391) elegantly demonstrated that reorganization of RyRs increases Ca2+ leak and contributes to lower contractility during the development of heart failure.
8. IP3 RECEPTORS
The inositol trisphosphate (IP3)/Ca2+ signaling pathway regulates many cellular processes, including metabolism, secretion, fertilization, proliferation, smooth muscle contraction, memory, and insulin secretion from pancreatic beta cells. Berridge (392), Foskett et al. (393), Narayanan et al. (394), and Woll and Van Petegem (340) have written excellent and extensive articles on inositol 1,4,5-trisphosphate/calcium (IP3/Ca2+) signaling and IP3 receptor (IP3R) structure, function, and pathology in different cell types. Here, we focus our discussion on clustering and cooperative gating of IP3Rs.
8.1. IP3R Structure and Function
Like RyRs, IP3Rs are large tetrameric intracellular proteins. Each IP3R subunit has an apparent molecular mass of ∼300 kDa (395). The cytoplasmic NH2 terminus of each InsP3R molecule accounts for nearly 85% of the total protein mass. The IP3Rs contain six membrane-spanning helices that form the pore of the channel (FIGURE 9A). They also have a relatively short cytoplasmic carboxy terminus. The NH2-terminal region is where the proximal IP3 binding domain is located (396). Three IP3R isoforms have been identified: IP3R types 1–3 (IP3R1, IP3R2, and IP3R3). All three IP3R isoforms cluster and have similar conductances and similar gating kinetics in intact cells (397).
FIGURE 9.

Topology of inositol trisphosphate (IP3) receptors (IP3Rs) and models of IP3R interactions with other ion channels. A: structure of an IP3R subunit. SR, sarcoplasmic reticulum. B: model. IP3 (1) promotes (2) a physical interaction between IP3Rs and canonical transient receptor potential (TRP) (TPRC)3 channels that (3) activates TRPC3 and promotes membrane depolarization, resulting in (4) an increase in the open probability of CaV1.2 channels, an elevation in intracellular Ca2+, and contraction of vascular smooth muscle cells. C: stretch promotes IP3 production and activation of IP3Rs, as well as diacylglycerol (DAG) production leading to activation of TRPC6 channels. The activation of IP3Rs and TRPC6 channels increases local intracellular Ca2+ near melastatin TRP (TRPM)4 channels, which are then activated to cause membrane depolarization and vascular smooth muscle contraction. In this model, it is proposed that IP3Rs, TPRC6, and TRPM4 form a nanocomplex. Figures created with Biorender.com.
Neurotransmitters, vasoactive peptides, and growth factors induce the synthesis of IP3 through the activation of Gq protein-coupled receptors (GqPCRs) or the protein tyrosine kinase-linked receptors that are linked to different phospholipase C (PLC) isoforms (reviewed in Ref. 398). Activation of PLC hydrolyzes the lipid phosphatidylinositol 4,5-bisphosphate, producing diacylglycerol and IP3. IP3 binds and thereby opens IP3Rs to release Ca2+ from the endo/sarcoplasmic reticulum.
An interesting characteristic of IP3Rs is that they are dually activated by IP3 and Ca2+. However, the efficacy of Ca2+ activation of IP3Rs depends on IP3 levels. The cytoplasm has been referred to as an “excitable medium” in which IP3R activation is modulated by IP3 levels and Ca2+-induced Ca2+ release (CICR) (399). IP3Rs, like RyRs, are a critical component of the excitable medium, and their excitability depends on the proximity between IP3Rs as well as local IP3 and Ca2+ levels (399–402).
A key feature of the excitable medium model is that it facilitates the rapid propagation and amplification of local Ca2+ signals via CICR. As proposed by Foskett et al. (393), however, subcellular variations in distance between release sites and excitability of the system may limit the efficacy of local signals to activate nearby IP3Rs via CICR and hence prevent all-or-nothing Ca2+ signaling events. As recently shown by Longden et al. (403), this type of hierarchical IP3R-mediated Ca2+ signaling cascade has important implications in the regulation of the membrane potential of endothelial cells of brain capillaries and hence arteriolar diameter and blood flow during neural activity.
8.2. Cooperativity Among IP3 Receptors
As is the case with RyRs, the spatial organization of IP3Rs and their sensitivity to IP3 and Ca2+ confer a great deal of flexibility in Ca2+ signaling regulation. Local interactions between IP3Rs allow the hierarchical recruitment of IP3Rs so that single IP3Rs respond first, producing “Ca2+ blips,” and then clustered IP3Rs can open together, producing a “Ca2+ puff.” As Ca2+ puffs become more frequent, they give rise to Ca2+ waves (402, 404).
Superresolution imaging suggests that IP3Rs are randomly distributed within the ER, forming clusters of various sizes (56–58). Most IP3R clusters are mobile, however. Low concentrations of IP3 cause IP3Rs to rapidly aggregate reversibly into clusters generally consisting of four closely associated IP3Rs (56). Dickinson et al. (405) proposed that the stochastic opening of any individual IP3R channel within a cluster triggers a Ca2+ puff by activating neighboring IP3Rs via CICR.
When intracellular [Ca2+] is low (i.e., ∼100 nM), most, if not all, clustered IP3Rs are predicted to open independently and with a relatively low open probability (56). Clustered IP3Rs have also shorter open times and appear to be less sensitive to IP3 than single IP3Rs. However, as cytosolic Ca2+ increases, the gating of clustered IP3Rs becomes cooperative. Dynamic regulation of clustering by IP3 retunes IP3R sensitivity to IP3 and Ca2+. This facilitates the recruitment of the elementary IP3R Ca2+ release events.
Consistent with the model outlined above, Wiltgen et al. (404) found that the kinetics of IP3R channel closing differs from that expected for independent gating. They found that upon activation clustered IP3Rs remained open in synchrony for periods much longer than predicted from their individual open lifetimes and that they close simultaneously. Wiltgen et al. (404) proposed that the gating of closely adjacent IP3Rs could not be explained by stochastic attrition mechanisms, Ca2+-inhibition of IP3Rs, and/or local depletion of Ca2+ in the ER lumen. Instead, they postulated that clustered IP3Rs are coupled, likely via allosteric interactions. At present, however, the mechanisms underlying allosteric regulation of clustered IP3Rs are remain unresolved.
8.3. Coupling of IP3 Receptors to Other Channels
IP3Rs, like RyR1s, appear to physically interact with other sarcolemmal channels. Adebiyi et al. (244) found that IP3R1s are positioned in close spatial proximity to sarcolemmal TRPC3 channels. Interestingly, activation of phospholipase C by Endothelin-1 increased IP3 and thus induced direct physical coupling between IP3R1 and sarcolemmal TRPC3 channels in arterial myocytes. This coupling activated TRPC3 channels, which depolarized arterial myocytes and led to the activation of CaV1.2 channels, increased cytosolic Ca2+ levels, and thus vasoconstriction (FIGURE 9B).
In a separate study, Gonzales et al. (247) proposed an indirect path for IP3R-TRPC6/TRPM4 coupling, which is also critical for the development of myogenic tone by arterial smooth muscle in response to increases in intravascular pressure. They demonstrated that an increase in intravascular pressure stretches the sarcolemma of arterial myocytes and activates phospholipase Cγ1. The IP3 produced by the activation of phospholipase Cγ1 makes the junctional SR IP3Rs more sensitive to the Ca2+ entering the cell via TRPC6 channels. This increases IP3R-mediated Ca2+ release, which activates TRPM4 channels. These TRPM4 currents were found to depolarize smooth muscle cells and cause vasoconstriction (FIGURE 9C).
8.4. Physiological Implications of IP3 Receptor Cooperative Gating
The coupling of IP3Rs amplifies and, depending on the coupling strength, grades the amplitude, spatial spread, and frequency of intracellular Ca2+ signals. This could result in stronger contractions in vascular smooth muscle. In neurons and nonexcitable cells, the magnitude of IP3R coupling might alter gene expression and excitability. Notably, a recent study (406) suggested that changes in the nanostructural organization and activity of IP3R1 is linked to cell death in Niemann-Pick type C1 disease. Future studies should investigate whether changes in the clustering of IP3Rs contribute to other pathological conditions via changes in cooperative gating and IP3-mediated Ca2+ signaling in different cell types.
9. HYPERPOLARIZATION-CYCLIC NUCLEOTIDE-ACTIVATED CHANNELS
9.1. HCN Channel Structure and Function
Hyperpolarization-cyclic nucleotide-activated (HCN) channels are tetrameric proteins. Each protomer contains six α-helices (S1–S6) in the transmembrane domain, including a voltage sensor domain composed of S1–S4 and a pore domain composed of S5, a reentrant pore region, and S6. Although HCN channels are members of the voltage-gated potassium channel KV superfamily, they differ from KV channels in several important ways. Most significantly, HCN channels are opened by hyperpolarization rather than by depolarization (407) and HCN channel opening is facilitated by binding of cyclic nucleotides to a COOH-terminal cytosolic cyclic nucleotide binding domain (408, 409).
To date, four HCN isoforms (HCN1–4) have been identified in humans and mice (410). HCN1 is expressed in the hippocampus, neocortex, and cerebellar cortex but also the heart. HCN2 is expressed in midbrain and thalamic neurons. Loss of HCN2 expression nearly eliminates pacemaker currents in thalamocortical neurons and sinoatrial node (SAN) myocytes (411). HCN3 is expressed in ventricular cells (412). HCN4 is the main isoform expressed in the SAN of the heart as well as the thalamic nuclei and basal ganglia (413–416). In addition, several studies have suggested that HCN channels form by heteromultimers: HCN1/HCN2 form heteromeric channels (417), and both HCN1/HCN4 and HCN2/HCN4 heteromultimers have been detected in the SAN and heterologous expression systems (418, 419).
Single-channel recordings of pacemaking currents in native SAN cells revealed single-channel conductance of ∼1 pS for HCN channels, which is very low compared with other voltage-dependent cation channels (77). This is important because it imposes a technical challenge in recording elementary HCN currents with a high signal-to-noise ratio. For this reason, nonstationary fluctuation analysis has been used to estimate the conductance of cloned HCN channels. With this approach, the conductance of HCN2 was estimated to be ∼2.5 pS (160) and ∼0.7 pS for channels underlying HCN currents in neuronal dendrites (178) Thus, the elementary currents produced by most, if not all, HCN channel isoforms are relatively small. This suggests that to alter membrane potential significantly a relatively large number of HCN channels must open simultaneously and/or that the input resistance of the cell membrane be relatively high.
9.2. Cooperativity Among HCN Channels
Dekker and Yellen (8) performed the first rigorous single-channel study of HCN2 channels. Using a nonstationary fluctuation analysis, they detected discrete HCN2 channel openings with amplitudes ranging from 150 to 230 fA. The mean conductance of these openings was ∼1.5 pS at −120 mV in a symmetrical 160 mM K+ bathing solution. A key finding in their study was that multiple HCN2 channels would undergo seemingly correlated openings, suggesting cooperativity between channels.
Dekker and Yellen (8) speculated about the potential mechanisms underlying the coupling of HCN channels. One possibility they raised was that channel coupling resulted from common sensing of a signaling molecule such as cAMP. Experiments performed in excised cell-free patches in both 0 cAMP and saturating 1 mM cAMP, however, ruled out this possibility. Accordingly, Dekker and Yellen (8) proposed that the most likely mechanism for coupling involved direct allosteric communication through direct channel-to-channel interactions mediated by an adaptor or linking protein.
Although similar experiments have not been performed on the HCN1–4 isoforms of the channels, their structural and functional similarities suggest they could also be capable of undergoing cooperative gating. Furthermore, the possibility that HCN1/HCN2 and HCN2/HCN4 could form heteromultimers raises the intriguing possibility that HCN2 may act as coupling agent between HCN1/HCN2 and HCN2/HCN4 heteromultimers (417, 419, 420). Future experiments should determine whether, and under which conditions, native HCN channels undergo cooperative gating in neurons and SAN myocytes.
9.3. Physiological Implications of HCN Cooperative Gating
An important physiological implication of HCN cooperativity is its potential role in modulating the frequency of oscillatory networks in the heart and brain. Here, we focus on the heart’s SAN cells, as they express HCN channels in addition to CaV1.2 and CaV1.3 channels and ryanodine receptors, all of which may undergo cooperative gating. The principal function of the cells in the SAN of the heart is to produce synchronous action potentials (APs) that initiate each heartbeat. This is accomplished despite the findings that the intrinsic excitability of its myocytes and even its vasculature is highly variable (416, 421). Some cells produce APs fairly frequently; others have periods of discharge separated by long periods of inactivity (i.e., bursts). Some cells only produce stochastic spontaneous subthreshold voltage fluctuations, and a relatively small fraction of cells are electrically silent. The combination of diverse electrical modalities within the SAN cell population leads to highly periodic oscillations through a mechanism of entrainment (421, 422) or stochastic resonance (416, 423).
The initiation of the AP in SAN myocytes occurs during diastole and is driven by activation of HCN channel (424, 425) currents (409, 426, 427). Thus, cooperative gating of clustered HCN channels may increase the amplitude of the funny current (If) and hence the rate of diastolic depolarization of spiking pacemaking cells. It is tempting to speculate that the spontaneous subthreshold voltage fluctuations observed by Grainger et al. (416) in SAN myocytes may be produced by the simultaneous opening of coupled HCN channels and to wonder whether or not cooperative gating is more prominent in some SAN cells than others.
Concurrent with HCN activation, Ca2+ sparks (127) activate inward diastolic Na+/Ca2+ exchanger currents (429, 430). These HCN and Na+/Ca2+ exchanger currents depolarize SAN myocytes beyond the activation threshold of voltage-gated CaV3.1 (431), CaV1.3 (201, 432), and, eventually, CaV1.2 (432, 433) channels. This initiates the AP and triggers a transient global increase in [Ca2+]i. Repolarization of the membrane potential during the AP begins by the inactivation of Ca2+ currents and activation of voltage-gated K+ currents. As SAN myocytes approach hyperpolarized potentials, HCN channels are activated, initiating this regenerative process.
In summary, the cooperative activating of HCN, CaV1.2, CaV1.3, and ryanodine receptors influences SA node excitability and hence heart rate. Future experiments should investigate the spatial organization of these channels, whether cooperative gating of these channels varies throughout the node, and how it is influenced by the autonomic nervous system and potentially altered in disease.
10. OTHER CHANNELS REPORTED TO UNDERGO COOPERATIVE GATING
Although we focus on CaV, TRP, NaV, K+, and HCN channels as well as RyRs and IP3Rs in this review, multiple studies have reported nonindependent gating of other channels (see for example Ref. 181). For example, ligand-gated channels including nicotinic acetylcholine receptors (434, 435), NMDA receptors (436), and P2X receptors (437) have also been shown to gate nonindependently. In the case of acetylcholine receptors, the changes in the oligomeric state correlated with receptor activity and corresponding alterations in postsynaptic currents (438). A recent study suggests that clustering of acetylcholine receptors in the postsynaptic membrane is facilitated by their interaction with the scaffold protein rapsyn (439), thus providing a potential structural arrangement that enables coupling of receptors. More recently, there is debate on whether the mechanosensitive Piezo1 channel may undergo cooperative gating (440, 441). Intriguingly, the application of Yoda 1, which is an allosteric modulator of the channel, increases Piezo1 clustering (442). Whether Piezo1 channels undergo cooperative gating and the physiological implications of its clustering remain to be established.
11. SUMMARY, OPEN QUESTIONS, AND FUTURE DIRECTIONS
In this review, we have presented the evidence for cooperative gating of multiple ion channels, discussed the putative mechanisms underlying the phenomenon, and speculated on the physiological consequences of this gating modality. The preponderance of the data suggest that cooperative gating is not a rare event but rather a consequential mode of gating in a wide range of ion channels. Cooperative gating seems to affect every aspect of electrical and Ca2+ signaling in excitable and nonexcitable mammalian cells, i.e., pacemaking, depolarization phase of APs, repolarization, and ER/SR Ca2+ release (see FIGURE 10).
FIGURE 10.

Proposed physiological implications of the cooperative gating of ion channels within membrane clusters. AP, action potential; EC, excitation-contraction; ET, excitation-transcription; βAR, β-adrenergic receptor. Figure created with Biorender.com.
To date, a great deal of evidence for cooperative gating has been adduced from the results of biophysical (i.e., patch-clamp electrophysiology, planar lipid bilayers) and imaging (i.e., FRET, bimolecular complementation assays, optogenetic fusion of channels) experiments. At present, however, detailed structural work on how adjacent channels undergo physical, allosteric interactions that coordinate their openings is scarce. Exceptions are NaV and CaV1 channels, where in both cases the COOH terminal of the channel has been identified as critical for channel-to-channel communication. How exactly the physical contact of two or more CaV1 or NaV channels via their C-tails translates into the increased probability of opening the channels gates remains unclear but may involve S4 transitions, as high-Po channels dominate linked low-Po channels. It is also conceivable that the Ca•CaM linkages between the COOH termini of adjoined channels result in mechanical forces that are transferred to the channel ionophores to induce channel openings.
We know very little about the structural determinants of TRP, HCN, RyR, and IP3R channel coupling. We anticipate that emerging crystallographic and cryo-EM structural data from ion channels combined with structural modeling (e.g., Rosetta and/or molecular dynamics) will provide clues as to the chain of events translating physical contact at specific channel domains to channel gate opening. Importantly, these analyses should lead to the formulation of testable predictions on the mechanisms underlying the coupling of specific channel types in living cells.
Another promising area of investigation is the incorporation of cooperative gating into mathematical models of AP generation, propagation, and Ca2+ signaling that has already begun in the neurosciences (27, 181, 188, 292, 293, 443, 444). It will be extremely interesting to learn how incorporating cooperative gating of HCN, CaV1.3/1.2, and RyRs into mathematical models of cardiac pacemaking, EC coupling, and arrhythmogenesis will inform our understanding of these primary physiological phenomena. In arteries, modeling the impact of cooperative gating of CaV1.2, TRP, IP3R, and RyRs on smooth muscle myogenic tone and propagation of electrical signals through endothelial cells during neurovascular coupling in health and disease may also yield important insights.
To conclude, nearly 70 years after the seminal work by Hodgkin and Huxley established the foundations of modern biophysics with the advent of their quantitative model for action potential generation and propagation in neurons, a growing body of evidence suggests that ion channel gating is not exclusively stochastic and independent. Rather, cooperative gating of ion channels seems to be a general phenomenon that modulates multiple physiological processes including excitability, contraction, arterial diameter, and neuronal coding. Future work on the structural mechanisms together with modeling of the physiological impact of cooperative gating of ion channels in organ function will provide critical insights into how the frequency, duration, and strength of this gating modality is regulated.
GRANTS
This work was supported by grants from the National Institutes of Health to L.F.S. (1R01HL144071, 5R01HL152681, 5R01NS114210, 5R01HL085686, and 1OT2OD026580), R.E.D. (R01AG063796), M.D.B. (1R01NS110953), and M.F.N. (R01HL098200, R01HL121059, R01HL149127, and R01HL161872, and UC-MEXUS CONACYT award).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.E.D., M.F.N., M.D.B., and L.F.S. interpreted results of experiments; prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Jon Sack for critically reading parts of the manuscript.
REFERENCES
- 1.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc., 2001, p. 814. [Google Scholar]
- 3.Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature 260: 799–802, 1976. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 4.Neher E, Sakmann B, Steinbach JH. The extracellular patch clamp: a method for resolving currents through individual open channels in biological membranes. Pflugers Arch 375: 219–228, 1978. doi: 10.1007/BF00584247. [DOI] [PubMed] [Google Scholar]
- 5.Sato D, Hernández-Hernández G, Matsumoto C, Tajada S, Moreno CM, Dixon RE, O’Dwyer S, Navedo MF, Trimmer JS, Clancy CE, Binder MD, Santana LF. A stochastic model of ion channel cluster formation in the plasma membrane. J Gen Physiol 151: 1116–1134, 2019. doi: 10.1085/jgp.201912327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DF, Ermentrout B, Thomas PJ. Stochastic representations of ion channel kinetics and exact stochastic simulation of neuronal dynamics. J Comput Neurosci 38: 67–82, 2015. doi: 10.1007/s10827-014-0528-2. [DOI] [PubMed] [Google Scholar]
- 7.Hymel L, Striessnig J, Glossmann H, Schindler H. Purified skeletal muscle 1,4-dihydropyridine receptor forms phosphorylation-dependent oligomeric calcium channels in planar bilayers. Proc Natl Acad Sci USA 85: 4290–4294, 1988. doi: 10.1073/pnas.85.12.4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dekker JP, Yellen G. Cooperative gating between single HCN pacemaker channels. J Gen Physiol 128: 561–567, 2006. doi: 10.1085/jgp.200609599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ Res 71: 1231–1241, 1992. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- 10.Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschênes I. Voltage-gated sodium channels assemble and gate as dimers. Nat Commun 8: 2077, 2017. doi: 10.1038/s41467-017-02262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci USA 109: 1749–1754, 2012. doi: 10.1073/pnas.1116731109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno CM, Dixon RE, Tajada S, Yuan C, Opitz-Araya X, Binder MD, Santana LF. Ca2+ entry into neurons is facilitated by cooperative gating of clustered CaV1.3 channels. Elife 5: e15744, 2016. doi: 10.7554/eLife.15744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res 106: 748–756, 2010. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nystoriak MA, Nieves-Cintrón M, Navedo MF. Capturing single L-type Ca2+ channel function with optics. Biochim Biophys Acta 1833: 1657–1664, 2013. doi: 10.1016/j.bbamcr.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navedo MF, Santana LF. CaV1.2 sparklets in heart and vascular smooth muscle. J Mol Cell Cardiol 58: 67–76, 2013. doi: 10.1016/j.yjmcc.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santana LF, Navedo MF, Amberg GC, Nieves-Cintrón M, Votaw VS, Ufret-Vincenty CA. Calcium sparklets in arterial smooth muscle. Clin Exp Pharmacol Physiol 35: 1121–1126, 2008. doi: 10.1111/j.1440-1681.2007.04867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demuro A, Parker I. Imaging single-channel calcium microdomains. Cell Calcium 40: 413–422, 2006. doi: 10.1016/j.ceca.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou H, Lifshitz LM, Tuft RA, Fogarty KE, Singer JJ. Visualization of Ca2+ entry through single stretch-activated cation channels. Proc Natl Acad Sci USA 99: 6404–6409, 2002. doi: 10.1073/pnas.092654999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demuro A, Parker I. Imaging the activity and localization of single voltage-gated Ca2+ channels by total internal reflection fluorescence microscopy. Biophys J 86: 3250–3259, 2004. doi: 10.1016/S0006-3495(04)74373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demuro A, Parker I. Optical patch-clamping. Single-channel recording by imaging Ca2+ flux through individual muscle acetylcholine receptor channels. J Gen Physiol 126: 179–192, 2005. doi: 10.1085/jgp.200509331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demuro A, Smith M, Parker I. Single-channel Ca2+ imaging implicates Abeta1-42 amyloid pores in Alzheimer’s disease pathology. J Cell Biol 195: 515–524, 2011. doi: 10.1083/jcb.201104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacquemet G, Baghirov H, Georgiadou M, Sihto H, Peuhu E, Cettour-Janet P, He T, Perälä M, Kronqvist P, Joensuu H, Ivaska J. L-type calcium channels regulate filopodia stability and cancer cell invasion downstream of integrin signalling. Nat Commun 7: 13297, 2016. doi: 10.1038/ncomms13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan MN, Earley S. TRP channel Ca2+ sparklets: fundamental signals underlying endothelium-dependent hyperpolarization. Am J Physiol Cell Physiol 305: C999–C1008, 2013. doi: 10.1152/ajpcell.00273.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navedo MF, Amberg GC. Local regulation of L-type Ca2+ channel sparklets in arterial smooth muscle. Microcirculation 20: 290–298, 2013. doi: 10.1111/micc.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santana LF, Navedo MF. Molecular and biophysical mechanisms of Ca2+ sparklets in smooth muscle. J Mol Cell Cardiol 47: 436–444, 2009. doi: 10.1016/j.yjmcc.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navedo MF, Amberg GC, Votaw VS, Santana LF. Constitutively active L-type Ca2+ channels. Proc Natl Acad Sci USA 102: 11112–11117, 2005. doi: 10.1073/pnas.0500360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfeiffer P, Egorov AV, Lorenz F, Schleimer JH, Draguhn A, Schreiber S. Clusters of cooperative ion channels enable a membrane-potential-based mechanism for short-term memory. Elife 9: e49974, 2020. doi: 10.7554/eLife.49974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci USA 106: 22275–22280, 2009. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh D, Nieves-Cintrón M, Tajada S, Brust-Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF, Navedo MF. Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865: 1341–1355, 2018. doi: 10.1016/j.bbamcr.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman WI, Turcotte DL, Malamud BD. Emergence of patterns in random processes. Phys Rev E Stat Nonlin Soft Matter Phys 86: 026103, 2012. doi: 10.1103/PhysRevE.86.026103. [DOI] [PubMed] [Google Scholar]
- 31.Westhoff M, Dixon RE. Mechanisms and regulation of cardiac CaV1.2 trafficking. Int J Mol Sci 22: 5927, 2021. doi: 10.3390/ijms22115927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balse E, Steele DF, Abriel H, Coulombe A, Fedida D, Hatem SN. Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol Rev 92: 1317–1358, 2012. doi: 10.1152/physrev.00041.2011. [DOI] [PubMed] [Google Scholar]
- 33.Beam KG, Caldwell JH, Campbell DT. Na channels in skeletal muscle concentrated near the neuromuscular junction. Nature 313: 588–590, 1985. doi: 10.1038/313588a0. [DOI] [PubMed] [Google Scholar]
- 34.Catterall WA. Localization of sodium channels in cultured neural cells. J Neurosci 1: 777–783, 1981. doi: 10.1523/JNEUROSCI.01-07-00777.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waxman SG, Ritchie JM. Organization of ion channels in the myelinated nerve fiber. Science 228: 1502–1507, 1985. doi: 10.1126/science.2409596. [DOI] [PubMed] [Google Scholar]
- 36.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol 107: 2587–2600, 1988. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duffin R, Leitch AE, Sheldrake TA, Hallett JM, Meyer C, Fox S, Alessandri AL, Martin MC, Brady HJ, Teixeira MM, Dransfield I, Haslett C, Rossi AG. The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett 583: 2540–2546, 2009. doi: 10.1016/j.febslet.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 38.Gathercole DV, Colling DJ, Skepper JN, Takagishi Y, Levi AJ, Severs NJ. Immunogold-labeled L-type calcium channels are clustered in the surface plasma membrane overlying junctional sarcoplasmic reticulum in guinea-pig myocytes—implications for excitation-contraction coupling in cardiac muscle. J Mol Cell Cardiol 32: 1981–1994, 2000. doi: 10.1006/jmcc.2000.1230. [DOI] [PubMed] [Google Scholar]
- 39.Takagishi Y, Rothery S, Issberner J, Levi A, Severs NJ. Spatial distribution of dihydropyridine receptors in the plasma membrane of guinea pig cardiac myocytes investigated by correlative confocal microscopy and label-fracture electron microscopy. J Electron Microsc (Tokyo) 46: 165–170, 1997. doi: 10.1093/oxfordjournals.jmicro.a023504. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen AO, Arnold W, Shen AC, Yuan SH, Gaver M, Campbell KP. Identification of novel proteins unique to either transverse tubules (TS28) or the sarcolemma (SL50) in rabbit skeletal muscle. J Cell Biol 110: 1173–1185, 1990. doi: 10.1083/jcb.110.4.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vivas O, Moreno CM, Santana LF, Hille B. Proximal clustering between BK and CaV1.3 channels promotes functional coupling and BK channel activation at low voltage. Elife 6: e28029, 2017. doi: 10.7554/eLife.28029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF, Santana LF. Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife 4: e05608, 2015. doi: 10.7554/eLife.05608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, Navedo MF, Santana LF. Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCalpha in arterial myocytes. J Gen Physiol 149: 639–659, 2017. doi: 10.1085/jgp.201611709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J Gen Physiol 143: 559–575, 2014. doi: 10.1085/jgp.201311050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nystoriak MA, Nieves-Cintrón M, Patriarchi T, Buonarati OR, Prada MP, Morotti S, Grandi E, Fernandes JD, Forbush K, Hofmann F, Sasse KC, Scott JD, Ward SM, Hell JW, Navedo MF. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci Signal 10: eaaf9647, 2017. doi: 10.1126/scisignal.aaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, Simó S, Sato D, Sasse KC, Ward SM, Santana LF, Xiang YK, Hell JW, Nieves-Cintrón M, Navedo MF. A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife 8: e42214, 2019. doi: 10.7554/eLife.42214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron 17: 103–113, 1996. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 48.Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 378: 85–88, 1995. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 49.Vierra NC, Kirmiz M, van der List D, Santana LF, Trimmer JS. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife 8: e49953, 2019. doi: 10.7554/eLife.49953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito DW, Hannigan KI, Ghosh D, Xu B, Del Villar SG, Xiang YK, Dickson EJ, Navedo MF, Dixon RE. beta-adrenergic-mediated dynamic augmentation of sarcolemmal CaV1.2 clustering and co-operativity in ventricular myocytes. J Physiol 597: 2139–2162, 2019. doi: 10.1113/JP277283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Del Villar SG, Voelker TL, Westhoff M, Reddy GR, Spooner HC, Navedo MF, Dickson EJ, Dixon RE. beta-Adrenergic control of sarcolemmal CaV1.2 abundance by small GTPase Rab proteins. Proc Natl Acad Sci USA 118: e2017937118, 2021. doi: 10.1073/pnas.2017937118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Syed AU, Reddy GR, Ghosh D, Prada MP, Nystoriak MA, Morotti S, Grandi E, Sirish P, Chiamvimonvat N, Hell JW, Santana LF, Xiang YK, Nieves-Cintrón M, Navedo MF. Adenylyl cyclase 5-generated cAMP controls cerebral vascular reactivity during diabetic hyperglycemia. J Clin Invest 129: 3140–3152, 2019. doi: 10.1172/JCI124705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cox DH. Modeling a Ca2+ channel/BKCa channel complex at the single-complex level. Biophys J 107: 2797–2814, 2014. doi: 10.1016/j.bpj.2014.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sahu G, Asmara H, Zhang FX, Zamponi GW, Turner RW. Activity-dependent facilitation of CaV1.3 calcium channels promotes KCa3.1 activation in hippocampal neurons. J Neurosci 37: 11255–11270, 2017. doi: 10.1523/JNEUROSCI.0967-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plante AE, Whitt JP, Meredith AL. BK channel activation by L-type Ca2+ channels CaV1.2 and CaV1.3 during the subthreshold phase of an action potential. J Neurophysiol 126: 427–439, 2021. doi: 10.1152/jn.00089.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taufiq-Ur-Rahman, Skupin A, Falcke M, Taylor CW. Clustering of InsP3 receptors by InsP3 retunes their regulation by InsP3 and Ca2+. Nature 458: 655–659, 2009. doi: 10.1038/nature07763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thillaiappan NB, Chavda AP, Tovey SC, Prole DL, Taylor CW. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat Commun 8: 1505, 2017. doi: 10.1038/s41467-017-01644-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krishnan V, Ali S, Gonzales AL, Thakore P, Griffin CS, Yamasaki E, Alvarado MG, Johnson MT, Trebak M, Earley S. Peripheral coupling sites formed by stim1 govern the contractility of vascular smooth muscle cells (Preprint). bioRxiv 2005.2025.445565, 2021. doi: 10.1101/2021.05.25.445565. [DOI] [PMC free article] [PubMed]
- 59.Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791–805, 2001. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 60.Nelson AD, Jenkins PM. Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front Cell Neurosci 11: 136, 2017. doi: 10.3389/fncel.2017.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kirmiz M, Vierra NC, Palacio S, Trimmer JS. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum-plasma membrane junctions in mammalian brain neurons. J Neurosci 38: 7562–7584, 2018. doi: 10.1523/JNEUROSCI.0893-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson B, Leek AN, Solé L, Maverick EE, Levine TP, Tamkun MM. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci USA 115: E7331–E7340, 2018. doi: 10.1073/pnas.1805757115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yeramian E, Trautmann A, Claverie P. Acetylcholine receptors are not functionally independent. Biophys J 50: 253–263, 1986. doi: 10.1016/S0006-3495(86)83459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pitt GS, Matsui M, Cao C. Voltage-gated calcium channels in nonexcitable tissues. Annu Rev Physiol 83: 183–203, 2021. doi: 10.1146/annurev-physiol-031620-091043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hofmann F, Flockerzi V, Kahl S, Wegener JW. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol Rev 94: 303–326, 2014. doi: 10.1152/physrev.00016.2013. [DOI] [PubMed] [Google Scholar]
- 66.Pangrsic T, Singer JH, Koschak A. Voltage-gated calcium channels: key players in sensory coding in the retina and the inner ear. Physiol Rev 98: 2063–2096, 2018. doi: 10.1152/physrev.00030.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khosravani H, Zamponi GW. Voltage-gated calcium channels and idiopathic generalized epilepsies. Physiol Rev 86: 941–966, 2006. doi: 10.1152/physrev.00002.2006. [DOI] [PubMed] [Google Scholar]
- 68.Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67: 821–870, 2015. doi: 10.1124/pr.114.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Catterall WA, Lenaeus MJ, Gamal El-Din TM. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu Rev Pharmacol Toxicol 60: 133–154, 2020. doi: 10.1146/annurev-pharmtox-010818-021757. [DOI] [PubMed] [Google Scholar]
- 70.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3: a003947, 2011. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixon RE, Cheng EP, Mercado JL, Santana LF. L-type Ca2+ channel function during Timothy syndrome. Trends Cardiovasc Med 22: 72–76, 2012. doi: 10.1016/j.tcm.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu J, Yan N, Yan Z. Structure-function relationship of the voltage-gated calcium channel Cav1.1 complex. Adv Exp Med Biol 981: 23–39, 2017. doi: 10.1007/978-3-319-55858-5_2. [DOI] [PubMed] [Google Scholar]
- 73.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57: 411–425, 2005. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 74.Ghosh D, Syed AU, Prada MP, Nystoriak MA, Santana LF, Nieves-Cintrón M, Navedo MF. Calcium channels in vascular smooth muscle. Adv Pharmacol 78: 49–87, 2017. doi: 10.1016/bs.apha.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron 25: 533–535, 2000. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 76.Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. Structure of the voltage-gated calcium channel Cav1.1 at 3.6 Å resolution. Nature 537: 191–196, 2016. doi: 10.1038/nature19321. [DOI] [PubMed] [Google Scholar]
- 77.Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M, Yan N. Structure of the voltage-gated calcium channel Cav1.1 complex. Science 350: aad2395, 2015. doi: 10.1126/science.aad2395. [DOI] [PubMed] [Google Scholar]
- 78.Dolphin AC. Voltage-gated calcium channel alpha 2delta subunits: an assessment of proposed novel roles. F1000Res 7: 1830, 2018. doi: 10.12688/f1000research.16104.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol 594: 5369–5390, 2016. doi: 10.1113/JP272262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dubel SJ, Altier C, Chaumont S, Lory P, Bourinet E, Nargeot J. Plasma membrane expression of T-type calcium channel alpha1 subunits is modulated by high voltage-activated auxiliary subunits. J Biol Chem 279: 29263–29269, 2004. doi: 10.1074/jbc.M313450200. [DOI] [PubMed] [Google Scholar]
- 81.Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee JH, Cribbs LL, Volsen SG, Perez-Reyes E. The effect of alpha2-delta and other accessory subunits on expression and properties of the calcium channel alpha1G. J Physiol 519: 35–45, 1999. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hansen JP, Chen RS, Larsen JK, Chu PJ, Janes DM, Weis KE, Best PM. Calcium channel gamma6 subunits are unique modulators of low voltage-activated (Cav3.1) calcium current. J Mol Cell Cardiol 37: 1147–1158, 2004. doi: 10.1016/j.yjmcc.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 83.Lambert RC, Maulet Y, Mouton J, Beattie R, Volsen S, De Waard M, Feltz A. T-type Ca2+ current properties are not modified by Ca2+ channel beta subunit depletion in nodosus ganglion neurons. J Neurosci 17: 6621–6628, 1997. doi: 10.1523/JNEUROSCI.17-17-06621.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strube C. Absence of regulation of the T-type calcium current by Cav1.1, beta1a and gamma1 dihydropyridine receptor subunits in skeletal muscle cells. Pflugers Arch 455: 921–927, 2008. doi: 10.1007/s00424-007-0345-9. [DOI] [PubMed] [Google Scholar]
- 85.Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E; International Union of Pharmacology. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev 55: 579–581, 2003. doi: 10.1124/pr.55.4.8. [DOI] [PubMed] [Google Scholar]
- 86.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature 328: 313–318, 1987. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 87.Franzini-Armstrong C, Nunzi G. Junctional feet and particles in the triads of a fast-twitch muscle fibre. J Muscle Res Cell Motil 4: 233–252, 1983. doi: 10.1007/BF00712033. [DOI] [PubMed] [Google Scholar]
- 88.Marx SO, Ondrias K, Marks AR. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281: 818–821, 1998. doi: 10.1126/science.281.5378.818. [DOI] [PubMed] [Google Scholar]
- 89.Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis (-aminoethyl ether)-N,N'-tetracetic acid. Biochim Biophys Acta 267: 605–608, 1972. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- 90.Bannister RA, Beam KG. CaV1: the atypical prototypical voltage-gated Ca2+ channel. Biochim Biophys Acta 1828: 1587–1597, 2013. doi: 10.1016/j.bbamem.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yan Z, Bai X, Yan C, Wu J, Li Z, Xie T, Peng W, Yin C, Li X, Scheres SH, Shi Y, Yan N. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517: 50–55, 2015. doi: 10.1038/nature14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bai XC, Yan Z, Wu J, Li Z, Yan N. The central domain of RyR1 is the transducer for long-range allosteric gating of channel opening. Cell Res 26: 995–1006, 2016. doi: 10.1038/cr.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Buraei Z, Yang J. Structure and function of the beta subunit of voltage-gated Ca2+ channels. Biochim Biophys Acta 1828: 1530–1540, 2013. doi: 10.1016/j.bbamem.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flucher BE, Campiglio M. STAC proteins: the missing link in skeletal muscle EC coupling and new regulators of calcium channel function. Biochim Biophys Acta Mol Cell Res 1866: 1101–1110, 2019. doi: 10.1016/j.bbamcr.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 95.Franzini-Armstrong C, Kish JW. Alternate disposition of tetrads in peripheral couplings of skeletal muscle. J Muscle Res Cell Motil 16: 319–324, 1995. doi: 10.1007/BF00121140. [DOI] [PubMed] [Google Scholar]
- 96.Paolini C, Protasi F, Franzini-Armstrong C. The relative position of RyR feet and DHPR tetrads in skeletal muscle. J Mol Biol 342: 145–153, 2004. doi: 10.1016/j.jmb.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 97.Perni S, Lavorato M, Beam KG. De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca2+ release. Proc Natl Acad Sci USA 114: 13822–13827, 2017. doi: 10.1073/pnas.1716461115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Polster A, Perni S, Bichraoui H, Beam KG. Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc Natl Acad Sci USA 112: 602–606, 2015. doi: 10.1073/pnas.1423113112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mikami A, Imoto K, Tanabe T, Niidome T, Mori Y, Takeshima H, Narumiya S, Numa S. Primary structure and functional expression of the cardiac dihydropyridine-sensitive calcium channel. Nature 340: 230–233, 1989. doi: 10.1038/340230a0. [DOI] [PubMed] [Google Scholar]
- 100.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marbán E, Yue DT. Novel functional properties of Ca2+ channel beta subunits revealed by their expression in adult rat heart cells. J Physiol 541: 435–452, 2002. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW, Rusch NJ. The beta3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension 61: 137–142, 2013. doi: 10.1161/HYPERTENSIONAHA.112.197863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang L, Katchman A, Morrow JP, Doshi D, Marx SO. Cardiac L-type calcium channel (Cav1.2) associates with gamma subunits. FASEB J 25: 928–936, 2011. doi: 10.1096/fj.10-172353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 104.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renström E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschütz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig J. Isoform-specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430–1439, 2004. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lederer WJ, Cannell MB, Cohen NM, Berlin JR. Excitation-contraction coupling in heart muscle. Mol Cell Biochem 89: 115–119, 1989. doi: 10.1007/BF00220762. [DOI] [PubMed] [Google Scholar]
- 106.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256: 532–535, 1992. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 107.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol Heart Circ Physiol 269: H348–H355, 1995. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 108.Rubart M, Patlak JB, Nelson MT. Ca2+ currents in cerebral artery smooth muscle cells of rat at physiological Ca2+ concentrations. J Gen Physiol 107: 459–472, 1996. doi: 10.1085/jgp.107.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am J Physiol Cell Physiol 259: C3–18, 1990. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- 110.Schulla V, Renström E, Feil R, Feil S, Franklin I, Gjinovci A, Jing XJ, Laux D, Lundquist I, Magnuson MA, Obermüller S, Olofsson CS, Salehi A, Wendt A, Klugbauer N, Wollheim CB, Rorsman P, Hofmann F. Impaired insulin secretion and glucose tolerance in beta cell-selective Cav1.2 Ca2+ channel null mice. EMBO J 22: 3844–3854, 2003. doi: 10.1093/emboj/cdg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nitert MD, Nagorny CL, Wendt A, Eliasson L, Mulder H. CaV1.2 rather than CaV1.3 is coupled to glucose-stimulated insulin secretion in INS-1 832/13 cells. J Mol Endocrinol 41: 1–11, 2008. doi: 10.1677/JME-07-0133. [DOI] [PubMed] [Google Scholar]
- 112.Dolmetsch R. Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE 2003: PE4, 2003. doi: 10.1126/stke.2003.166.pe4. [DOI] [PubMed] [Google Scholar]
- 113.Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep 33: 97–101, 2012. doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW. CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell 149: 1112–1124, 2012. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Santana LF. NFAT-dependent excitation-transcription coupling in heart. Circ Res 103: 681–683, 2008. doi: 10.1161/CIRCRESAHA.108.185090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 25: 9883–9892, 2005. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3: 15–38, 2014. doi: 10.1002/wmts.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wegener JW, Schulla V, Lee TS, Koller A, Feil S, Feil R, Kleppisch T, Klugbauer N, Moosmang S, Welling A, Hofmann F. An essential role of Cav1.2 L-type calcium channel for urinary bladder function. FASEB J 18: 1159–1161, 2004. doi: 10.1096/fj.04-1516fje. [DOI] [PubMed] [Google Scholar]
- 119.Wegener JW, Schulla V, Koller A, Klugbauer N, Feil R, Hofmann F. Control of intestinal motility by the Cav1.2 L-type calcium channel in mice. FASEB J 20: 1260–1262, 2006. doi: 10.1096/fj.05-5292fje. [DOI] [PubMed] [Google Scholar]
- 120.Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, Hofmann F, Klugbauer N. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J 22: 6027–6034, 2003. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch 458: 481–487, 2009. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- 122.Liao P, Yong TF, Liang MC, Yue DT, Soong TW. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc Res 68: 197–203, 2005. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 123.Hu Z, Liang MC, Soong TW. Alternative splicing of L-type CaV1.2 calcium channels: implications in cardiovascular diseases. Genes (Basel) 8: 344, 2017. doi: 10.3390/genes8120344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sun XH, Protasi F, Takahashi M, Takeshima H, Ferguson DG, Franzini-Armstrong C. Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J Cell Biol 129: 659–671, 1995. doi: 10.1083/jcb.129.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jones PP, MacQuaide N, Louch WE. Dyadic plasticity in cardiomyocytes. Front Physiol 9: 1773, 2018. doi: 10.3389/fphys.2018.01773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410: 592–596, 2001. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 127.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262: 740–744, 1993. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 128.Cheng H, Lederer MR, Xiao RP, Gómez AM, Zhou YY, Ziman B, Spurgeon H, Lakatta EG, Lederer WJ. Excitation-contraction coupling in heart: new insights from Ca2+ sparks. Cell Calcium 20: 129–140, 1996. doi: 10.1016/S0143-4160(96)90102-5. [DOI] [PubMed] [Google Scholar]
- 129.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol Cell Physiol 245: C1–C14, 1983. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 130.Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophys J 67: 1942–1956, 1994. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheng H, Cannell MB, Lederer WJ. Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch 428: 415–417, 1994. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- 132.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev 88: 1491–1545, 2008. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 133.Soeller C, Crossman D, Gilbert R, Cannell MB. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc Natl Acad Sci USA 104: 14958–14963, 2007. doi: 10.1073/pnas.0703016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res 121: 181–195, 2017. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Protasi F, Sun XH, Franzini-Armstrong C. Formation and maturation of the calcium release apparatus in developing and adult avian myocardium. Dev Biol 173: 265–278, 1996. doi: 10.1006/dbio.1996.0022. [DOI] [PubMed] [Google Scholar]
- 136.Tijskens P, Meissner G, Franzini-Armstrong C. Location of ryanodine and dihydropyridine receptors in frog myocardium. Biophys J 84: 1079–1092, 2003. doi: 10.1016/S0006-3495(03)74924-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.De La Mata A, Tajada S, O’Dwyer S, Matsumoto C, Dixon RE, Hariharan N, Moreno CM, Santana LF. BIN1 induces the formation of T-tubules and adult-like Ca2+ release units in developing cardiomyocytes. Stem Cells 37: 54–64, 2019. doi: 10.1002/stem.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dixon RE, Vivas O, Hannigan KI, Dickson EJ. Ground state depletion super-resolution imaging in mammalian cells. J Vis Exp 129: 56239, 2017. doi: 10.3791/56239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Inoue M, Bridge JH. Ca2+ sparks in rabbit ventricular myocytes evoked by action potentials: involvement of clusters of L-type Ca2+ channels. Circ Res 92: 532–538, 2003. doi: 10.1161/01.RES.0000064175.70693.EC. [DOI] [PubMed] [Google Scholar]
- 140.Sobie EA, Ramay HR. Excitation-contraction coupling gain in ventricular myocytes: insights from a parsimonious model. J Physiol 587: 1293–1299, 2009. doi: 10.1113/jphysiol.2008.163915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Josephson IR, Guia A, Sobie EA, Lederer WJ, Lakatta EG, Stern MD. Physiologic gating properties of unitary cardiac L-type Ca2+ channels. Biochem Biophys Res Commun 396: 763–766, 2010. doi: 10.1016/j.bbrc.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science 268: 1045–1049, 1995. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 143.López-López JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science 268: 1042–1045, 1995. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 144.Santana LF, Cheng H, Gómez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res 78: 166–171, 1996. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- 145.Collier ML, Thomas AP, Berlin JR. Relationship between L-type Ca2+ current and unitary sarcoplasmic reticulum Ca2+ release events in rat ventricular myocytes. J Physiol 516: 117–128, 1999. doi: 10.1111/j.1469-7793.1999.117aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Poláková E, Zahradníková A Jr, Pavelková J, Zahradník I, Zahradníková A. Local calcium release activation by DHPR calcium channel openings in rat cardiac myocytes. J Physiol 586: 3839–3854, 2008. doi: 10.1113/jphysiol.2007.149989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation by beta-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol 18: 691–710, 1986. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- 149.Tykocki NR, Boerman EM, Jackson WF. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr Physiol 7: 485–581, 2017. doi: 10.1002/cphy.c160011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Amberg GC, Navedo MF. Calcium dynamics in vascular smooth muscle. Microcirculation 20: 281–289, 2013. doi: 10.1111/micc.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sanders KM. Mechanisms of calcium handling in smooth muscles. J Appl Physiol (1985) 91: 1438–1449, 2001. doi: 10.1152/jappl.2001.91.3.1438. [DOI] [PubMed] [Google Scholar]
- 152.Hill-Eubanks DC, Werner ME, Heppner TJ, Nelson MT. Calcium signaling in smooth muscle. Cold Spring Harb Perspect Biol 3: a004549, 2011. doi: 10.1101/cshperspect.a004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83: 1325–1358, 2003. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 154.Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev 68: 476–532, 2016. doi: 10.1124/pr.115.010652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Prada MP, Syed AU, Reddy GR, Martín-Aragón Baudel M, Flores-Tamez VA, Sasse KC, Ward SM, Sirish P, Chiamvimonvat N, Bartels P, Dickson EJ, Hell JW, Scott JD, Santana LF, Xiang YK, Navedo MF, Nieves-Cintrón M. AKAP5 complex facilitates purinergic modulation of vascular L-type Ca2+ channel CaV1.2. Nat Commun 11: 5303, 2020. doi: 10.1038/s41467-020-18947-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sato D, Hernández-Hernández G, Matsumoto C, Tajada S, Moreno CM, Dixon RE, O’Dwyer S, Navedo MF, Trimmer JS, Clancy CE, Binder MD, Santana LF. A stochastic model of ion channel cluster formation in the plasma membrane. J Gen Physiol 151: 1116–1134, 2019. doi: 10.1085/jgp.201912327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.O’Dwyer SC, Palacio S, Matsumoto C, Guarina L, Klug NR, Tajada S, Rosati B, McKinnon D, Trimmer JS, Santana LF. Kv2.1 channels play opposing roles in regulating membrane potential, Ca2+ channel function, and myogenic tone in arterial smooth muscle. Proc Natl Acad Sci USA 117: 3858–3866, 2020. doi: 10.1073/pnas.1917879117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gandasi NR, Yin P, Riz M, Chibalina MV, Cortese G, Lund PE, Matveev V, Rorsman P, Sherman A, Pedersen MG, Barg S. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J Clin Invest 127: 2353–2364, 2017. doi: 10.1172/JCI88491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang J, Carver CM, Choveau FS, Shapiro MS. clustering and functional coupling of diverse ion channels and signaling proteins revealed by super-resolution STORM microscopy in neurons. Neuron 92: 461–478, 2016. doi: 10.1016/j.neuron.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Di Biase V, Tuluc P, Campiglio M, Obermair GJ, Heine M, Flucher BE. Surface traffic of dendritic CaV1.2 calcium channels in hippocampal neurons. J Neurosci 31: 13682–13694, 2011. doi: 10.1523/JNEUROSCI.2300-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Obermair GJ, Szabo Z, Bourinet E, Flucher BE. Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci 19: 2109–2122, 2004. doi: 10.1111/j.0953-816X.2004.03272.x. [DOI] [PubMed] [Google Scholar]
- 162.Wang S, Stanika RI, Wang X, Hagen J, Kennedy MB, Obermair GJ, Colbran RJ, Lee A. Densin-180 controls the trafficking and signaling of L-type voltage-gated Cav1.2 Ca2+ channels at excitatory synapses. J Neurosci 37: 4679–4691, 2017. doi: 10.1523/JNEUROSCI.2583-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Servili E, Trus M, Maayan D, Atlas D. Beta-subunit of the voltage-gated Ca2+ channel Cav1.2 drives signaling to the nucleus via H-Ras. Proc Natl Acad Sci USA 115: E8624–E8633, 2018. doi: 10.1073/pnas.1805380115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sajman J, Trus M, Atlas D, Sherman E. The L-type voltage-gated calcium channel co-localizes with syntaxin 1A in nano-clusters at the plasma membrane. Sci Rep 7: 11350, 2017. doi: 10.1038/s41598-017-10588-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ghosh D, Nieves-Cintrón M, Tajada S, Brust-Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF, Navedo MF. Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865: 1341–1355, 2018. doi: 10.1016/j.bbamcr.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336: 597–601, 2012. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhao L, Sullivan MN, Chase M, Gonzales AL, Earley S. Calcineurin/nuclear factor of activated T cells-coupled vanilloid transient receptor potential channel 4 Ca2+ sparklets stimulate airway smooth muscle cell proliferation. Am J Respir Cell Mol Biol 50: 1064–1075, 2014. doi: 10.1165/rcmb.2013-0416OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sullivan MN, Gonzales AL, Pires PW, Bruhl A, Leo MD, Li W, Oulidi A, Boop FA, Feng Y, Jaggar JH, Welsh DG, Earley S. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci Signal 8: ra2, 2015. doi: 10.1126/scisignal.2005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pires PW, Sullivan MN, Pritchard HA, Robinson JJ, Earley S. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am J Physiol Heart Circ Physiol 309: H2031–H2041, 2015. doi: 10.1152/ajpheart.00140.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Pires PW, Earley S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. Elife 7: e35316, 2018. doi: 10.7554/eLife.35316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Amberg GC, Navedo MF, Nieves-Cintrón M, Molkentin JD, Santana LF. Calcium sparklets regulate local and global calcium in murine arterial smooth muscle. J Physiol 579: 187–201, 2007. doi: 10.1113/jphysiol.2006.124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Navedo MF, Takeda Y, Nieves-Cintrón M, Molkentin JD, Santana LF. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am J Physiol Cell Physiol 298: C211–C220, 2010. doi: 10.1152/ajpcell.00267.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Navedo MF, Nieves-Cintrón M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II induced hypertension. Circ Res 102: e1–e11, 2008. doi: 10.1161/CIRCRESAHA.107.167809. [DOI] [PubMed] [Google Scholar]
- 174.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267: 108–111, 1995. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 175.Oliveria SF, Dell’Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55: 261–275, 2007. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell’Acqua ML. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7: 1577–1588, 2014. doi: 10.1016/j.celrep.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Oliveria SF, Dittmer PJ, Youn DH, Dell’Acqua ML, Sather WA. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J Neurosci 32: 15328–15337, 2012. doi: 10.1523/JNEUROSCI.2302-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry 46: 1635–1646, 2007. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 179.Fish RD, Sperti G, Colucci WS, Clapham DE. Phorbol ester increases the dihydropyridine-sensitive calcium conductance in a vascular smooth muscle cell line. Circ Res 62: 1049–1054, 1988. doi: 10.1161/01.res.62.5.1049. [DOI] [PubMed] [Google Scholar]
- 180.Nieves-Cintrón M, Amberg GC, Navedo MF, Molkentin JD, Santana LF. The control of Ca2+ influx and NFATc3 signaling in arterial smooth muscle during hypertension. Proc Natl Acad Sci USA 105: 15623–15628, 2008. doi: 10.1073/pnas.0808759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Choi KH. Cooperative gating between ion channels. Gen Physiol Biophys 33: 1–12, 2014. doi: 10.4149/gpb_2013076. [DOI] [PubMed] [Google Scholar]
- 182.Chung SH, Kennedy RA. Coupled Markov chain model: characterization of membrane channel currents with multiple conductance sublevels as partially coupled elementary pores. Math Biosci 133: 111–137, 1996. doi: 10.1016/0025-5564(95)00084-4. [DOI] [PubMed] [Google Scholar]
- 183.Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintrón M, Scott JD, Santana LF. Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res 109: 255–261, 2011. doi: 10.1161/CIRCRESAHA.111.248252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119: 19–31, 2004. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 185.Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci USA 109: E2316–E2323, 2012. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Klewer L, Wu YW. Light-Induced dimerization approaches to control cellular processes. Chemistry 25: 12452–12463, 2019. doi: 10.1002/chem.201900562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type CaV1.2 channels. Proc Natl Acad Sci USA 109: 1749–1754, 2012. doi: 10.1073/pnas.1116731109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sato D, Dixon RE, Santana LF, Navedo MF. A model for cooperative gating of L-type Ca2+ channels and its effects on cardiac alternans dynamics. PLoS Comput Biol 14: e1005906, 2018. doi: 10.1371/journal.pcbi.1005906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fallon JL, Baker MR, Xiong L, Loy RE, Yang G, Dirksen RT, Hamilton SL, Quiocho FA. Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+-calmodulins. Proc Natl Acad Sci USA 106: 5135–5140, 2009. doi: 10.1073/pnas.0807487106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim EY, Rumpf CH, Van Petegem F, Arant RJ, Findeisen F, Cooley ES, Isacoff EY, Minor DL Jr.. Multiple C-terminal tail Ca2+/CaMs regulate CaV1.2 function but do not mediate channel dimerization. EMBO J 29: 3924–3938, 2010. doi: 10.1038/emboj.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben-Johny M, Kalocsay M, Marx SO. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577: 695–700, 2020. doi: 10.1038/s41586-020-1947-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Béguin P, Mahalakshmi RN, Nagashima K, Cher DH, Ikeda H, Yamada Y, Seino Y, Hunziker W. Nuclear sequestration of beta-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol 355: 34–46, 2006. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 193.Béguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411: 701–706, 2001. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 194.Medvedev RY, Sanchez-Alonso JL, Mansfield CA, Judina A, Francis AJ, Pagiatakis C, Trayanova N, Glukhov AV, Miragoli M, Faggian G, Gorelik J. Local hyperactivation of L-type Ca2+ channels increases spontaneous Ca2+ release activity and cellular hypertrophy in right ventricular myocytes from heart failure rats. Sci Rep 11: 4840, 2021. doi: 10.1038/s41598-021-84275-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Le T, Martín-Aragón Baudel M, Syed A, Singhrao N, Pan S, Flores-Tamez VA, Burns AE, Man KN, Karey E, Hong J, Hell JW, Pinkerton KE, Chen CY, Nieves-Cintrón M. Secondhand smoke exposure impairs ion channel function and contractility of mesenteric arteries. Function (Oxf) 2: zqab041, 2021. doi: 10.1093/function/zqab041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintrón M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA, Catterall WA, Poomvanicha M, Hofmann F, Navedo MF, Hell JW. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the beta2-adrenergic receptor in neurons. Sci Signal 10: eaaf9659, 2017. doi: 10.1126/scisignal.aaf9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Folci A, Steinberger A, Lee B, Stanika R, Scheruebel S, Campiglio M, Ramprecht C, Pelzmann B, Hell JW, Obermair GJ, Heine M, Di Biase V. Molecular mimicking of C-terminal phosphorylation tunes the surface dynamics of CaV1.2 calcium channels in hippocampal neurons. J Biol Chem 293: 1040–1053, 2018. doi: 10.1074/jbc.M117.799585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2: 233–251, 2008. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- 199.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. Alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276: 22100–22106, 2001. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 200.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102: 89–97, 2000. doi: 10.1016/S0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 201.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA 100: 5543–5548, 2003. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mangoni ME, Nargeot J. Properties of the hyperpolarization-activated current (If) in isolated mouse sino-atrial cells. Cardiovasc Res 52: 51–64, 2001. doi: 10.1016/s0008-6363(01)00370-4. [DOI] [PubMed] [Google Scholar]
- 203.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional roles of Cav1.3 (alpha1D) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res 90: 981–987, 2002. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 204.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans 34: 903–909, 2006. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 205.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123: 949–962, 1993. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75: 407–414, 2009. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 207.Verma A, Ravindranath V. CaV1.3 L-Type calcium channels increase the vulnerability of substantia nigra dopaminergic neurons in MPTP mouse model of Parkinson’s disease. Front Aging Neurosci 11: 382, 2019. doi: 10.3389/fnagi.2019.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27: 645–656, 2007. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29: 11011–11019, 2009. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447: 1081–1086, 2007. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 211.Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A. Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem 283: 20733–20744, 2008. doi: 10.1074/jbc.M802254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, Sartori S, Mangoni ME, Sinnegger-Brauns MJ, Herzig S, Striessnig J, Koschak A. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J Biol Chem 286: 42736–42748, 2011. doi: 10.1074/jbc.M111.269951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sather WA. Selective permeability of voltage-gated calcium channels. In: Voltage-Gated Calcium Channels. Boston, MA: Springer US, 2005, p. 205–218. [Google Scholar]
- 214.Binder MD, Powers RK, Heckman CJ. Nonlinear input-output functions of motoneurons. Physiology (Bethesda) 35: 31–39, 2020. doi: 10.1152/physiol.00026.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 95: 645–690, 2015. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vangeel L, Voets T. Transient receptor potential channels and calcium signaling. Cold Spring Harb Perspect Biol 11: a035048, 2019. doi: 10.1101/cshperspect.a035048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol Biosyst 4: 372–379, 2008. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci USA 109: 18174–18179, 2012. doi: 10.1073/pnas.1211946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sullivan MN, Francis M, Pitts NL, Taylor MS, Earley S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol Pharmacol 82: 464–472, 2012. doi: 10.1124/mol.112.078584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hong K, Cope EL, DeLalio LJ, Marziano C, Isakson BE, Sonkusare SK. TRPV4 (transient receptor potential vanilloid 4) channel-dependent negative feedback mechanism regulates Gq protein-coupled receptor-induced vasoconstriction. Arterioscler Thromb Vasc Biol 38: 542–554, 2018. doi: 10.1161/ATVBAHA.117.310038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ottolini M, Hong K, Cope EL, Daneva Z, DeLalio LJ, Sokolowski JD, Marziano C, Nguyen NY, Altschmied J, Haendeler J, Johnstone SR, Kalani MY, Park MS, Patel RP, Liedtke W, Isakson BE, Sonkusare SK. Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation 141: 1318–1333, 2020. doi: 10.1161/CIRCULATIONAHA.119.043385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, Sonkusare SK. Nitric oxide-dependent feedback loop regulates transient receptor potential vanilloid 4 (TRPV4) channel cooperativity and endothelial function in small pulmonary arteries. J Am Heart Assoc 6: e007157, 2017. doi: 10.1161/JAHA.117.007157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal 7: ra66, 2014. doi: 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang X, Li L, McNaughton PA. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59: 450–461, 2008. doi: 10.1016/j.neuron.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 225.Toro CA, Arias LA, Brauchi S. Sub-cellular distribution and translocation of TRP channels. Curr Pharm Biotechnol 12: 12–23, 2011. doi: 10.2174/138920111793937899. [DOI] [PubMed] [Google Scholar]
- 226.Cayouette S, Boulay G. Intracellular trafficking of TRP channels. Cell Calcium 42: 225–232, 2007. doi: 10.1016/j.ceca.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 227.Ghosh D, Nieves-Cintrón M, Tajada S, Brust-Mascher I, Horne MC, Hell JW, Dixon RE, Santana LF, Navedo MF. Dynamic L-type CaV1.2 channel trafficking facilitates CaV1.2 clustering and cooperative gating. Biochim Biophys Acta Mol Cell Res 1865: 1341–1355, 2018. doi: 10.1016/j.bbamcr.2018.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Thakore P, Pritchard HA, Griffin CS, Yamasaki E, Drumm BT, Lane C, Sanders KM, Feng Earley Y, Earley S. TRPML1 channels initiate calcium sparks in vascular smooth muscle. Sci Signal 13: eaba1015, 2020. doi: 10.1126/scisignal.aba1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ghosh D, Pinto S, Danglot L, Vandewauw I, Segal A, Van Ranst N, Benoit M, Janssens A, Vennekens R, Vanden Berghe P, Galli T, Vriens J, Voets T. VAMP7 regulates constitutive membrane incorporation of the cold-activated channel TRPM8. Nat Commun 7: 10489, 2016. doi: 10.1038/ncomms10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ghosh D, Segal A, Voets T. Distinct modes of perimembrane TRP channel turnover revealed by TIR-FRAP. Sci Rep 4: 7111, 2014. doi: 10.1038/srep07111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bezzerides VJ, Ramsey IS, Kotecha S, Greka A, Clapham DE. Rapid vesicular translocation and insertion of TRP channels. Nat Cell Biol 6: 709–720, 2004. doi: 10.1038/ncb1150. [DOI] [PubMed] [Google Scholar]
- 232.Fan HC, Zhang X, McNaughton PA. Activation of the TRPV4 ion channel is enhanced by phosphorylation. J Biol Chem 284: 27884–27891, 2009. doi: 10.1074/jbc.M109.028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Takahashi N, Hamada-Nakahara S, Itoh Y, Takemura K, Shimada A, Ueda Y, Kitamata M, Matsuoka R, Hanawa-Suetsugu K, Senju Y, Mori MX, Kiyonaka S, Kohda D, Kitao A, Mori Y, Suetsugu S. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P2. Nat Commun 5: 4994, 2014. doi: 10.1038/ncomms5994. [DOI] [PubMed] [Google Scholar]
- 234.Garcia-Elias A, Mrkonjic S, Pardo-Pastor C, Inada H, Hellmich UA, Rubio-Moscardó F, Plata C, Gaudet R, Vicente R, Valverde MA. Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc Natl Acad Sci USA 110: 9553–9558, 2013. doi: 10.1073/pnas.1220231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 7: e38689, 2018. doi: 10.7554/eLife.38689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Goretzki B, Glogowski NA, Diehl E, Duchardt-Ferner E, Hacker C, Gaudet R, Hellmich UA. Structural basis of TRPV4 N terminus interaction with syndapin/PACSIN1-3 and PIP2. Structure 26: 1583–1593.e5, 2018. doi: 10.1016/j.str.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol Sci 16: 23–30, 1995. doi: 10.1016/s0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- 238.Coleman HA, Tare M, Parkington HC. Endothelial potassium channels, endothelium-dependent hyperpolarization and the regulation of vascular tone in health and disease. Clin Exp Pharmacol Physiol 31: 641–649, 2004. doi: 10.1111/j.1440-1681.2004.04053.x. [DOI] [PubMed] [Google Scholar]
- 239.Sancho M, Samson NC, Hald BO, Hashad AM, Marrelli SP, Brett SE, Welsh DG. KIR channels tune electrical communication in cerebral arteries. J Cereb Blood Flow Metab 37: 2171–2184, 2017. doi: 10.1177/0271678X16662041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res 97: 1270–1279, 2005. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- 241.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science 270: 633–637, 1995. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 242.Shah S, Carver CM, Mullen P, Milne S, Lukacs V, Shapiro MS, Gamper N. Local Ca2+ signals couple activation of TRPV1 and ANO1 sensory ion channels. Sci Signal 13: eaaw7963, 2020. doi: 10.1126/scisignal.aaw7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Xi Q, Adebiyi A, Zhao G, Chapman KE, Waters CM, Hassid A, Jaggar JH. IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ Res 102: 1118–1126, 2008. doi: 10.1161/CIRCRESAHA.108.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Adebiyi A, Zhao G, Narayanan D, Thomas-Gatewood CM, Bannister JP, Jaggar JH. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ Res 106: 1603–1612, 2010. doi: 10.1161/CIRCRESAHA.110.216804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Adebiyi A, Thomas-Gatewood CM, Leo MD, Kidd MW, Neeb ZP, Jaggar JH. An elevation in physical coupling of type 1 inositol 1,4,5-trisphosphate (IP3) receptors to transient receptor potential 3 (TRPC3) channels constricts mesenteric arteries in genetic hypertension. Hypertension 60: 1213–1219, 2012. doi: 10.1161/HYPERTENSIONAHA.112.198820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Adebiyi A, Narayanan D, Jaggar JH. Caveolin-1 assembles type 1 inositol 1,4,5-trisphosphate receptors and canonical transient receptor potential 3 channels into a functional signaling complex in arterial smooth muscle cells. J Biol Chem 286: 4341–4348, 2011. doi: 10.1074/jbc.M110.179747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill-Eubanks DC, Nelson MT, Earley S. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal 7: ra49, 2014. doi: 10.1126/scisignal.2004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Hagenacker T, Splettstoesser F, Greffrath W, Treede RD, Büsselberg D. Capsaicin differentially modulates voltage-activated calcium channel currents in dorsal root ganglion neurones of rats. Brain Res 1062: 74–85, 2005. doi: 10.1016/j.brainres.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 249.Wu ZZ, Chen SR, Pan HL. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J Biol Chem 280: 18142–18151, 2005. doi: 10.1074/jbc.M501229200. [DOI] [PubMed] [Google Scholar]
- 250.Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 396: 478–482, 1998. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 251.Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res 108: 265–272, 2011. doi: 10.1161/CIRCRESAHA.110.225888. [DOI] [PubMed] [Google Scholar]
- 252.Vennekens R, Menigoz A, Nilius B. TRPs in the brain. Rev Physiol Biochem Pharmacol 163: 27–64, 2012. doi: 10.1007/112_2012_8. [DOI] [PubMed] [Google Scholar]
- 253.Langeberg LK, Scott JD. Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol 16: 232–244, 2015. doi: 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hille B, Dickson EJ, Kruse M, Vivas O, Suh BC. Phosphoinositides regulate ion channels. Biochim Biophys Acta 1851: 844–856, 2015. doi: 10.1016/j.bbalip.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13–25, 2000. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 256.Matthews E, Balestrini S, Sisodiya SM, Hanna MG. Muscle and brain sodium channelopathies: genetic causes, clinical phenotypes, and management approaches. Lancet Child Adolesc Health 4: 536–547, 2020. doi: 10.1016/S2352-4642(19)30425-0. [DOI] [PubMed] [Google Scholar]
- 257.Moritz AT, Newkirk G, Powers RK, Binder MD. Facilitation of somatic calcium channels can evoke prolonged tail currents in rat hypoglossal motoneurons. J Neurophysiol 98: 1042–1047, 2007. doi: 10.1152/jn.01294.2006. [DOI] [PubMed] [Google Scholar]
- 258.Mantegazza M, Yu FH, Powell AJ, Clare JJ, Catterall WA, Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein betagamma subunits. J Neurosci 25: 3341–3349, 2005. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839–842, 1992. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 260.Hanemaaijer NA, Popovic MA, Wilders X, Grasman S, Pavón Arocas O, Kole MH. Ca2+ entry through NaV channels generates submillisecond axonal Ca2+ signaling. Elife 9: e54566, 2020. doi: 10.7554/eLife.54566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Santana LF, Gómez AM, Lederer WJ. Ca2+ flux through promiscuous cardiac Na+ channels: slip-mode conductance. Science 279: 1027–1033, 1998. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- 262.Davis TH, Chen C, Isom LL. Sodium channel beta1 subunits promote neurite outgrowth in cerebellar granule neurons. J Biol Chem 279: 51424–51432, 2004. doi: 10.1074/jbc.M410830200. [DOI] [PubMed] [Google Scholar]
- 263.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N. and Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 312: 121–127, 1984. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 264.Yu FH, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Biol 4: 207, 2003. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57: 397–409, 2005. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 266.Lee CH, Ruben PC. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels (Austin) 2: 407–412, 2008. doi: 10.4161/chan.2.6.7429. [DOI] [PubMed] [Google Scholar]
- 267.Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature 242: 459–461, 1973. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- 268.Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature 339: 597–603, 1989. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 269.Chanda B, Asamoah OK, Bezanilla F. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J Gen Physiol 123: 217–230, 2004. doi: 10.1085/jgp.200308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Goldin AL. Mechanisms of sodium channel inactivation. Curr Opin Neurobiol 13: 284–290, 2003. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 271.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kuo JJ, Lee RH, Zhang L, Heckman CJ. Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J Physiol 574: 819–834, 2006. doi: 10.1113/jphysiol.2006.107094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17: 4517–4526, 1997. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268–2277, 2001. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chatelier A, Zhao J, Bois P, Chahine M. Biophysical characterisation of the persistent sodium current of the Nav1.6 neuronal sodium channel: a single-channel analysis. Pflugers Arch 460: 77–86, 2010. doi: 10.1007/s00424-010-0801-9. [DOI] [PubMed] [Google Scholar]
- 276.Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein betagamma subunits. Neuron 19: 443–452, 1997. doi: 10.1016/S0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 277.Sarhan MF, Tung CC, Van Petegem F, Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci USA 109: 3558–3563, 2012. doi: 10.1073/pnas.1114748109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Johnson CN, Potet F, Thompson MK, Kroncke BM, Glazer AM, Voehler MW, Knollmann BC, George AL Jr, Chazin WJ. A mechanism of calmodulin modulation of the human cardiac sodium channel. Structure 26: 683–694.e3, 2018. doi: 10.1016/j.str.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Eshed-Eisenbach Y, Peles E. The clustering of voltage-gated sodium channels in various excitable membranes. Dev Neurobiol 81: 427–437, 2021. doi: 10.1002/dneu.22728. [DOI] [PubMed] [Google Scholar]
- 280.Duflocq A, Chareyre F, Giovannini M, Couraud F, Davenne M. Characterization of the axon initial segment (AIS) of motor neurons and identification of a para-AIS and a juxtapara-AIS, organized by protein 4.1B. BMC Biol 9: 66, 2011. doi: 10.1186/1741-7007-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 15: 1065–1076, 1995. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- 282.Hill AS, Nishino A, Nakajo K, Zhang G, Fineman JR, Selzer ME, Okamura Y, Cooper EC. Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates. PLoS Genet 4: e1000317, 2008. doi: 10.1371/journal.pgen.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sharp AA, Caldwell JH. Aggregation of sodium channels induced by a postnatally upregulated isoform of agrin. J Neurosci 16: 6775–6783, 1996. doi: 10.1523/JNEUROSCI.16-21-06775.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kordeli E. The spectrin-based skeleton at the postsynaptic membrane of the neuromuscular junction. Microsc Res Tech 49: 101–107, 2000. doi: 10.1002/(SICI)1097-0029(20000401)49:1<101::AID-JEMT11>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 285.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res 108: 294–304, 2011. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- 286.Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M, Gorelik J. Super-resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res 112: 1112–1120, 2013. doi: 10.1161/CIRCRESAHA.111.300445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Struckman HL, Baine S, Thomas J, Mezache L, Mykytyn K, Györke S, Radwański PB, Veeraraghavan R. Super-resolution imaging using a novel high-fidelity antibody reveals close association of the neuronal sodium channel NaV1.6 with ryanodine receptors in cardiac muscle. Microsc Microanal 26: 157–165, 2020. doi: 10.1017/S1431927619015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Veeraraghavan R, Gourdie RG. Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol Biol Cell 27: 3583–3590, 2016. doi: 10.1091/mbc.e16-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Van Wart A, Boiko T, Trimmer JS, Matthews G. Novel clustering of sodium channel Nav1.1 with ankyrin-G and neurofascin at discrete sites in the inner plexiform layer of the retina. Mol Cell Neurosci 28: 661–673, 2005. doi: 10.1016/j.mcn.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 290.Wu C, Ivanova E, Cui J, Lu Q, Pan ZH. Action potential generation at an axon initial segment-like process in the axonless retinal AII amacrine cell. J Neurosci 31: 14654–14659, 2011. doi: 10.1523/JNEUROSCI.1861-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hossain WA, Antic SD, Yang Y, Rasband MN, Morest DK. Where is the spike generator of the cochlear nerve? Voltage-gated sodium channels in the mouse cochlea. J Neurosci 25: 6857–6868, 2005. doi: 10.1523/JNEUROSCI.0123-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Huang M, Volgushev M, Wolf F. A small fraction of strongly cooperative sodium channels boosts neuronal encoding of high frequencies. PLoS One 7: e37629, 2012. doi: 10.1371/journal.pone.0037629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Naundorf B, Wolf F, Volgushev M. Unique features of action potential initiation in cortical neurons. Nature 440: 1060–1063, 2006. doi: 10.1038/nature04610. [DOI] [PubMed] [Google Scholar]
- 294.Hichri E, Selimi Z, Kucera JP. Modeling the interactions between sodium channels provides insight into the negative dominance of certain channel mutations. Front Physiol 11: 589386, 2020. doi: 10.3389/fphys.2020.589386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Levinson SR, Luo S, Henry MA. The role of sodium channels in chronic pain. Muscle Nerve 46: 155–165, 2012. doi: 10.1002/mus.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Luo S, Perry GM, Levinson SR, Henry MA. Nav1.7 expression is increased in painful human dental pulp. Mol Pain 4: 16, 2008. doi: 10.1186/1744-8069-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Omana-Zapata I, Khabbaz MA, Hunter JC, Clarke DE, Bley KR. Tetrodotoxin inhibits neuropathic ectopic activity in neuromas, dorsal root ganglia and dorsal horn neurons. Pain 72: 41–49, 1997. doi: 10.1016/s0304-3959(97)00012-2. [DOI] [PubMed] [Google Scholar]
- 298.Vermij SH, Rougier JS, Agulló-Pascual E, Rothenberg E, Delmar M, Abriel H. Single-molecule localization of the cardiac voltage-gated sodium channel reveals different modes of reorganization at cardiomyocyte membrane domains. Circ Arrhythm Electrophysiol 13: e008241, 2020. doi: 10.1161/CIRCEP.119.008241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zheng Y, Wan X, Yang D, Ramirez-Navarro A, Liu H, Fu JD, Deschênes I. A heart failure-associated SCN5A splice variant leads to a reduction in sodium current through coupled-gating with the wild-type channel. Front Physiol 12: 661429, 2021. doi: 10.3389/fphys.2021.661429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Clatot J, Zheng Y, Girardeau A, Liu H, Laurita KR, Marionneau C, Deschênes I. Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am J Physiol Heart Circ Physiol 315: H1250–H1257, 2018. doi: 10.1152/ajpheart.00721.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Rühlmann AH, Körner J, Hausmann R, Bebrivenski N, Neuhof C, Detro-Dassen S, Hautvast P, Benasolo CA, Meents J, Machtens JP, Schmalzing G, Lampert A. Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav 1.7 channel mutation. Br J Pharmacol 177: 4481–4496, 2020. doi: 10.1111/bph.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57: 527–540, 2005. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- 303.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 57: 473–508, 2005. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 304.Kubo Y, Adelman JP, Clapham DE, Jan LY, Karschin A, Kurachi Y, Lazdunski M, Nichols CG, Seino S, Vandenberg CA. International Union of Pharmacology. LIV. Nomenclature and molecular relationships of inwardly rectifying potassium channels. Pharmacol Rev 57: 509–526, 2005. doi: 10.1124/pr.57.4.11. [DOI] [PubMed] [Google Scholar]
- 305.Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev 57: 463–472, 2005. doi: 10.1124/pr.57.4.9. [DOI] [PubMed] [Google Scholar]
- 306.Armstrong C. The vision of the pore. Science 280: 56–57, 1998. doi: 10.1126/science.280.5360.56. [DOI] [PubMed] [Google Scholar]
- 307.Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci 7: 548–562, 2006. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- 308.Duménieu M, Oulé M, Kreutz MR, Lopez-Rojas J. The segregated expression of voltage-gated potassium and sodium channels in neuronal membranes: functional implications and regulatory mechanisms. Front Cell Neurosci 11: 115, 2017. doi: 10.3389/fncel.2017.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Molina ML, Giudici AM, Poveda JA, Fernández-Ballester G, Montoya E, Renart ML, Fernández AM, Encinar JA, Riquelme G, Morales A, González-Ros JM. Competing lipid-protein and protein-protein interactions determine clustering and gating patterns in the potassium channel from Streptomyces lividans (KcsA). J Biol Chem 290: 25745–25755, 2015. doi: 10.1074/jbc.M115.669598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Sumino A, Yamamoto D, Iwamoto M, Dewa T, Oiki S. Gating-associated clustering-dispersion dynamics of the KcsA potassium channel in a lipid membrane. J Phys Chem Lett 5: 578–584, 2014. doi: 10.1021/jz402491t. [DOI] [PubMed] [Google Scholar]
- 311.Lewin L, Nsasra E, Golbary E, Hadad U, Orr I, Yifrach O. Molecular and cellular correlates in Kv channel clustering: entropy-based regulation of cluster ion channel density. Sci Rep 10: 11304, 2020. doi: 10.1038/s41598-020-68003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol 66: 477–519, 2004. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- 313.Debanne D, Guérineau NC, Gähwiler BH, Thompson SM. Action-potential propagation gated by an axonal IA-like K+ conductance in hippocampus. Nature 389: 286–289, 1997. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- 314.Lambe EK, Aghajanian GK. The role of Kv1.2-containing potassium channels in serotonin-induced glutamate release from thalamocortical terminals in rat frontal cortex. J Neurosci 21: 9955–9963, 2001. doi: 10.1523/JNEUROSCI.21-24-09955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Wang H, Kunkel DD, Schwartzkroin PA, Tempel BL. Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J Neurosci 14: 4588–4599, 1994. doi: 10.1523/JNEUROSCI.14-08-04588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Tiffany AM, Manganas LN, Kim E, Hsueh YP, Sheng M, Trimmer JS. PSD-95 and SAP97 exhibit distinct mechanisms for regulating K+ channel surface expression and clustering. J Cell Biol 148: 147–158, 2000. doi: 10.1083/jcb.148.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Jugloff DG, Khanna R, Schlichter LC, Jones OT. Internalization of the Kv1.4 potassium channel is suppressed by clustering interactions with PSD-95. J Biol Chem 275: 1357–1364, 2000. doi: 10.1074/jbc.275.2.1357. [DOI] [PubMed] [Google Scholar]
- 318.Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron 23: 149–157, 1999. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- 319.Rasband MN, Park EW, Zhen D, Arbuckle MI, Poliak S, Peles E, Grant SG, Trimmer JS. Clustering of neuronal potassium channels is independent of their interaction with PSD-95. J Cell Biol 159: 663–672, 2002. doi: 10.1083/jcb.200206024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Murakoshi H, Trimmer JS. Identification of the Kv2.1 K+ channel as a major component of the delayed rectifier K+ current in rat hippocampal neurons. J Neurosci 19: 1728–1735, 1999. doi: 10.1523/JNEUROSCI.19-05-01728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2 alpha-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci 22: 10094–10105, 2002. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci 7: 711–718, 2004. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- 323.Lim ST, Antonucci DE, Scannevin RH, Trimmer JS. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25: 385–397, 2000. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- 324.Johnson B, Leek AN, Tamkun MM. Kv2 channels create endoplasmic reticulum/plasma membrane junctions: a brief history of Kv2 channel subcellular localization. Channels (Austin) 13: 88–101, 2019. doi: 10.1080/19336950.2019.1568824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.O’Connell KM, Loftus R, Tamkun MM. Localization-dependent activity of the Kv2.1 delayed-rectifier K+ channel. Proc Natl Acad Sci USA 107: 12351–12356, 2010. doi: 10.1073/pnas.1003028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Zarubin D, Zhuchkova E, Schreiber S. Effects of cooperative ion-channel interactions on the dynamics of excitable membranes. Phys Rev E Stat Nonlin Soft Matter Phys 85: 061904, 2012. doi: 10.1103/PhysRevE.85.061904. [DOI] [PubMed] [Google Scholar]
- 327.Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236–1244, 2004. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Perez-Flores MC, Lee JH, Park S, Zhang XD, Sihn CR, Ledford HA, Wang W, Kim HJ, Timofeyev V, Yarov-Yarovoy V, Chiamvimonvat N, Rabbitt RD, Yamoah EN. Cooperativity of Kv7.4 channels confers ultrafast electromechanical sensitivity and emergent properties in cochlear outer hair cells. Sci Adv 6: eaba1104, 2020. doi: 10.1126/sciadv.aba1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314: 615–620, 2006. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 330.Bausch AE, Dieter R, Nann Y, Hausmann M, Meyerdierks N, Kaczmarek LK, Ruth P, Lukowski R. The sodium-activated potassium channel Slack is required for optimal cognitive flexibility in mice. Learn Mem 22: 323–335, 2015. doi: 10.1101/lm.037820.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9: 1661–1672, 2014. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Kanda H, Ling J, Tonomura S, Noguchi K, Matalon S, Gu JG. TREK-1 and TRAAK are principal K+ channels at the nodes of Ranvier for rapid action potential conduction on mammalian myelinated afferent nerves. Neuron 104: 960–971.e7, 2019. doi: 10.1016/j.neuron.2019.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Brohawn SG, Wang W, Handler A, Campbell EB, Schwarz JR, MacKinnon R. The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. Elife 8: e50403, 2019. doi: 10.7554/eLife.50403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Schrempf H, Schmidt O, Kümmerlen R, Hinnah S, Müller D, Betzler M, Steinkamp T, Wagner R. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J 14: 5170–5178, 1995. doi: 10.1002/j.1460-2075.1995.tb00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hegermann J, Overbeck J, Schrempf H. In vivo monitoring of the potassium channel KcsA in Streptomyces lividans hyphae using immuno-electron microscopy and energy-filtering transmission electron microscopy. Microbiology (Reading) 152: 2831–2841, 2006. doi: 10.1099/mic.0.29002-0. [DOI] [PubMed] [Google Scholar]
- 336.Meuser D, Splitt H, Wagner R, Schrempf H. Exploring the open pore of the potassium channel from Streptomyces lividans. FEBS Lett 462: 447–452, 1999. doi: 10.1016/s0014-5793(99)01579-3. [DOI] [PubMed] [Google Scholar]
- 337.Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S, Wagenknecht T. Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J Cell Biol 127: 411–423, 1994. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Serysheva II, Orlova EV, Chiu W, Sherman MB, Hamilton SL, van Heel M. Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat Struct Biol 2: 18–24, 1995. doi: 10.1038/nsb0195-18. [DOI] [PubMed] [Google Scholar]
- 339.Wagenknecht T, Grassucci R, Frank J, Saito A, Inui M, Fleischer S. Three-dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature 338: 167–170, 1989. doi: 10.1038/338167a0. [DOI] [PubMed] [Google Scholar]
- 340.Woll KA, Van Petegem F. Calcium release channels: structure and function of IP3 receptors and ryanodine receptors. Physiol Rev 102: 209–268, 2022. doi: 10.1152/physrev.00033.2020. [DOI] [PubMed] [Google Scholar]
- 341.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev 82: 893–922, 2002. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 342.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2: a003996, 2010. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Essin K, Gollasch M. Role of ryanodine receptor subtypes in initiation and formation of calcium sparks in arterial smooth muscle: comparison with striated muscle. J Biomed Biotechnol 2009: 135249, 2009. doi: 10.1155/2009/135249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Samsó M, Trujillo R, Gurrola GB, Valdivia HH, Wagenknecht T. Three-dimensional location of the imperatoxin A binding site on the ryanodine receptor. J Cell Biol 146: 493–499, 1999. doi: 10.1083/jcb.146.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem 272: 32463–32471, 1997. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 346.Hymel L, Inui M, Fleischer S, Schindler H. Purified ryanodine receptor of skeletal muscle sarcoplasmic reticulum forms Ca2+-activated oligomeric Ca2+ channels in planar bilayers. Proc Natl Acad Sci USA 85: 441–445, 1988. doi: 10.1073/pnas.85.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature 331: 315–319, 1988. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 348.Chen SR, Li X, Ebisawa K, Zhang L. Functional characterization of the recombinant type 3 Ca2+ release channel (ryanodine receptor) expressed in HEK293 cells. J Biol Chem 272: 24234–24246, 1997. doi: 10.1074/jbc.272.39.24234. [DOI] [PubMed] [Google Scholar]
- 349.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev 88: 1491–1545, 2008. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 350.Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+-release channel: activation by caffeine. Am J Physiol Heart Circ Physiol 256: H328–H333, 1989. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 351.Rousseau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am J Physiol Cell Physiol 253: C364–C368, 1987. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- 352.Tinker A, Lindsay AR, Williams AJ. A model for ionic conduction in the ryanodine receptor channel of sheep cardiac muscle sarcoplasmic reticulum. J Gen Physiol 100: 495–517, 1992. doi: 10.1085/jgp.100.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Klein MG, Cheng H, Santana LF, Jiang YH, Lederer WJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature 379: 455–458, 1996. doi: 10.1038/379455a0. [DOI] [PubMed] [Google Scholar]
- 356.Tsugorka A, Ríos E, Blatter LA. Imaging elementary events of calcium release in skeletal muscle cells. Science 269: 1723–1726, 1995. doi: 10.1126/science.7569901. [DOI] [PubMed] [Google Scholar]
- 357.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol Cell Physiol 278: C235–C256, 2000. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 358.Ouyang K, Zheng H, Qin X, Zhang C, Yang D, Wang X, Wu C, Zhou Z, Cheng H. Ca2+ sparks and secretion in dorsal root ganglion neurons. Proc Natl Acad Sci USA 102: 12259–12264, 2005. doi: 10.1073/pnas.0408494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Mejía-Alvarez R, Kettlun C, Ríos E, Stern M, Fill M. Unitary Ca2+ current through cardiac ryanodine receptor channels under quasi-physiological ionic conditions. J Gen Physiol 113: 177–186, 1999. doi: 10.1085/jgp.113.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Franzini-Armstrong C. Studies of the triad: I. Structure of the junction in frog twitch fibers. J Cell Biol 47: 488–499, 1970. doi: 10.1083/jcb.47.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J 77: 1528–1539, 1999. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Shen X, van den Brink J, Hou Y, Colli D, Le C, Kolstad TR, MacQuaide N, Carlson CR, Kekenes-Huskey PM, Edwards AG, Soeller C, Louch WE. 3D dSTORM imaging reveals novel detail of ryanodine receptor localization in rat cardiac myocytes. J Physiol 597: 399–418, 2019. doi: 10.1113/JP277360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Pritchard HAT, Pires PW, Yamasaki E, Thakore P, Earley S. Nanoscale remodeling of ryanodine receptor cluster size underlies cerebral microvascular dysfunction in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 115: E9745–E9752, 2018. doi: 10.1073/pnas.1804593115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Porta M, Diaz-Sylvester PL, Neumann JT, Escobar AL, Fleischer S, Copello JA. Coupled gating of skeletal muscle ryanodine receptors is modulated by Ca2+, Mg2+, and ATP. Am J Physiol Cell Physiol 303: C682–C697, 2012. doi: 10.1152/ajpcell.00150.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Groff JR, Smith GD. Calcium-dependent inactivation and the dynamics of calcium puffs and sparks. J Theor Biol 253: 483–499, 2008. doi: 10.1016/j.jtbi.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 366.Groff JR, Smith GD. Ryanodine receptor allosteric coupling and the dynamics of calcium sparks. Biophys J 95: 135–154, 2008. doi: 10.1529/biophysj.107.119982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Xie Y, Yang Y, Galice S, Bers DM, Sato D. Size matters: ryanodine receptor cluster size heterogeneity potentiates calcium waves. Biophys J 116: 530–539, 2019. doi: 10.1016/j.bpj.2018.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88: 1151–1158, 2001. doi: 10.1161/hh1101.091268. [DOI] [PubMed] [Google Scholar]
- 369.Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR, Lacampagne A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 107: 1559–1564, 2010. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z, Marks AR. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol 17: 732–747, 2020. doi: 10.1038/s41569-020-0394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Matecki S, Dridi H, Jung B, Saint N, Reiken SR, Scheuermann V, Mrozek S, Santulli G, Umanskaya A, Petrof BJ, Jaber S, Marks AR, Lacampagne A. Leaky ryanodine receptors contribute to diaphragmatic weakness during mechanical ventilation. Proc Natl Acad Sci USA 113: 9069–9074, 2016. doi: 10.1073/pnas.1609707113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Xin HB, Senbonmatsu T, Cheng DS, Wang YX, Copello JA, Ji GJ, Collier ML, Deng KY, Jeyakumar LH, Magnuson MA, Inagami T, Kotlikoff MI, Fleischer S. Oestrogen protects FKBP12.6 null mice from cardiac hypertrophy. Nature 416: 334–338, 2002. doi: 10.1038/416334a. [DOI] [PubMed] [Google Scholar]
- 373.Di Maio A, Karko K, Snopko RM, Mejía-Alvarez R, Franzini-Armstrong C. T-tubule formation in cardiac myocytes: two possible mechanisms? J Muscle Res Cell Motil 28: 231–241, 2007. doi: 10.1007/s10974-007-9121-x. [DOI] [PubMed] [Google Scholar]
- 374.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 154: 1059–1067, 2001. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6: 11–22, 2000. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 376.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem 276: 21425–21433, 2001. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 377.Murphy RM, Mollica JP, Lamb GD. Plasma membrane removal in rat skeletal muscle fibers reveals caveolin-3 hot-spots at the necks of transverse tubules. Exp Cell Res 315: 1015–1028, 2009. doi: 10.1016/j.yexcr.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 378.Zampighi G, Vergara J, Ramón F. On the connection between the transverse tubules and the plasma membrane in frog semitendinosus skeletal muscle. Are caveolae the mouths of the transverse tubule system? J Cell Biol 64: 734–740, 1975. doi: 10.1083/jcb.64.3.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Chase TH, Cox GA, Burzenski L, Foreman O, Shultz LD. Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb-girdle muscular dystrophy 2B. Am J Pathol 175: 2299–2308, 2009. doi: 10.2353/ajpath.2009.080930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Demonbreun AR, Rossi AE, Alvarez MG, Swanson KE, Deveaux HK, Earley JU, Hadhazy M, Vohra R, Walter GA, Pytel P, McNally EM. Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am J Pathol 184: 248–259, 2014. doi: 10.1016/j.ajpath.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Beavers DL, Landstrom AP, Chiang DY, Wehrens XH. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc Res 103: 198–205, 2014. doi: 10.1093/cvr/cvu151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci USA 106: 7636–7641, 2009. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Hong T, Yang H, Zhang SS, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J, Gorelik J, Marbán E, Jan LY, Shaw RM. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med 20: 624–632, 2014. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Muller AJ, Baker JF, DuHadaway JB, Ge K, Farmer G, Donover PS, Meade R, Reid C, Grzanna R, Roach AH, Shah N, Soler AP, Prendergast GC. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol Cell Biol 23: 4295–4306, 2003. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol 8: e1000312, 2010. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Guo J, Tian Q, Barth M, Xian W, Ruppenthal S, Schaefers HJ, Chen Z, Moretti A, Laugwitz KL, Lipp P. Human BIN1 isoforms grow, maintain and regenerate excitation-contraction couplons in adult rat and human stem cell-derived cardiomyocytes. Cardiovasc Res 2021: cvab195, 2021. doi: 10.1093/cvr/cvab195. [DOI] [PubMed] [Google Scholar]
- 387.Drum BM, Yuan C, de la Mata A, Grainger N, Santana LF. Junctional sarcoplasmic reticulum motility in adult mouse ventricular myocytes. Am J Physiol Cell Physiol 318: C598–C604, 2020. doi: 10.1152/ajpcell.00573.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Wu MM, Covington ED, Lewis RS. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol Biol Cell 25: 3672–3685, 2014. doi: 10.1091/mbc.e14-06-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Niggli E. Localized intracellular calcium signaling in muscle: calcium sparks and calcium quarks. Annu Rev Physiol 61: 311–335, 1999. doi: 10.1146/annurev.physiol.61.1.311. [DOI] [PubMed] [Google Scholar]
- 390.Lipp P, Niggli E. Submicroscopic calcium signals as fundamental events of excitation–contraction coupling in guinea-pig cardiac myocytes. J Physiol 492: 31–38, 1996. doi: 10.1113/jphysiol.1996.sp021286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, Norden ES, Cataliotti A, Sjaastad I, Sejersted OM, Edwards AG, Lines GT, Louch WE. Ryanodine receptor dispersion disrupts Ca2+ release in failing cardiac myocytes. Elife 7: e39427, 2018. doi: 10.7554/eLife.39427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev 96: 1261–1296, 2016. doi: 10.1152/physrev.00006.2016. [DOI] [PubMed] [Google Scholar]
- 393.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87: 593–658, 2007. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Narayanan D, Adebiyi A, Jaggar JH. Inositol trisphosphate receptors in smooth muscle cells. Am J Physiol Heart Circ Physiol 302: H2190–H2210, 2012. doi: 10.1152/ajpheart.01146.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature 342: 32–38, 1989. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 396.Bosanac I, Alattia JR, Mal TK, Chan J, Talarico S, Tong FK, Tong KI, Yoshikawa F, Furuichi T, Iwai M, Michikawa T, Mikoshiba K, Ikura M. Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature 420: 696–700, 2002. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 397.Lock JT, Alzayady KJ, Yule DI, Parker I. All three IP3 receptor isoforms generate Ca2+ puffs that display similar characteristics. Sci Signal 11: eaau0344, 2018. doi: 10.1126/scisignal.aau0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Cocco L, Follo MY, Manzoli L, Suh PG. Phosphoinositide-specific phospholipase C in health and disease. J Lipid Res 56: 1853–1860, 2015. doi: 10.1194/jlr.R057984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Lechleiter J, Girard S, Peralta E, Clapham D. Spiral calcium wave propagation and annihilation in Xenopus laevis oocytes. Science 252: 123–126, 1991. doi: 10.1126/science.2011747. [DOI] [PubMed] [Google Scholar]
- 400.Boehning D, Joseph SK, Mak DO, Foskett JK. Single-channel recordings of recombinant inositol trisphosphate receptors in mammalian nuclear envelope. Biophys J 81: 117–124, 2001. doi: 10.1016/s0006-3495(01)75685-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Mak DO, McBride S, Foskett JK. Inositol 1,4,5-trisphosphate [correction of tris-phosphate] activation of inositol trisphosphate [correction of tris-phosphate] receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci USA 95: 15821–15825, 1998. doi: 10.1073/pnas.95.26.15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Smith IF, Parker I. Imaging the quantal substructure of single IP3R channel activity during Ca2+ puffs in intact mammalian cells. Proc Natl Acad Sci USA 106: 6404–6409, 2009. doi: 10.1073/pnas.0810799106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Longden TA, Mughal A, Hennig GW, Harraz OF, Shui B, Lee FK, Lee JC, Reining S, Kotlikoff MI, König GM, Kostenis E, Hill-Eubanks D, Nelson MT. Local IP3 receptor-mediated Ca2+ signals compound to direct blood flow in brain capillaries. Sci Adv 7: eabh0101, 2021. doi: 10.1126/sciadv.abh0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Wiltgen SM, Dickinson GD, Swaminathan D, Parker I. Termination of calcium puffs and coupled closings of inositol trisphosphate receptor channels. Cell Calcium 56: 157–168, 2014. doi: 10.1016/j.ceca.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Dickinson GD, Swaminathan D, Parker I. The probability of triggering calcium puffs is linearly related to the number of inositol trisphosphate receptors in a cluster. Biophys J 102: 1826–1836, 2012. doi: 10.1016/j.bpj.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Tiscione SA, Casas M, Horvath JD, Lam V, Hino K, Ory DS, Santana LF, Simó S, Dixon RE, Dickson EJ. IP3R-driven increases in mitochondrial Ca2+ promote neuronal death in NPC disease. Proc Natl Acad Sci USA 118: e2110629118, 2021. doi: 10.1073/pnas.2110629118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.DiFrancesco D. Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature 324: 470–473, 1986. doi: 10.1038/324470a0. [DOI] [PubMed] [Google Scholar]
- 408.Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature 425: 200–205, 2003. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- 409.DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351: 145–147, 1991. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 410.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89: 847–885, 2009. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- 411.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J 22: 216–224, 2003. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Fenske S, Mader R, Scharr A, Paparizos C, Cao-Ehlker X, Michalakis S, Shaltiel L, Weidinger M, Stieber J, Feil S, Feil R, Hofmann F, Wahl-Schott C, Biel M. HCN3 contributes to the ventricular action potential waveform in the murine heart. Circ Res 109: 1015–1023, 2011. doi: 10.1161/CIRCRESAHA.111.246173. [DOI] [PubMed] [Google Scholar]
- 413.Fenske S, Krause SC, Hassan SI, Becirovic E, Auer F, Bernard R, Kupatt C, Lange P, Ziegler T, Wotjak CT, Zhang H, Hammelmann V, Paparizos C, Biel M, Wahl-Schott CA. Sick sinus syndrome in HCN1-deficient mice. Circulation 128: 2585–2594, 2013. doi: 10.1161/CIRCULATIONAHA.113.003712. [DOI] [PubMed] [Google Scholar]
- 414.Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, Lei M, Escande D, Demolombe S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol 562: 223–234, 2005. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Herrmann S, Layh B, Ludwig A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol 51: 997–1006, 2011. doi: 10.1016/j.yjmcc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 416.Grainger N, Guarina L, Cudmore RH, Santana LF. The organization of the sinoatrial node microvasculature varies regionally to match local myocyte excitability. Function (Oxf) 2: zqab031, 2021. doi: 10.1093/function/zqab031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Ulens C, Tytgat J. Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem 276: 6069–6072, 2001. doi: 10.1074/jbc.C000738200. [DOI] [PubMed] [Google Scholar]
- 418.Much B, Wahl-Schott C, Zong X, Schneider A, Baumann L, Moosmang S, Ludwig A, Biel M. Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem 278: 43781–43786, 2003. doi: 10.1074/jbc.M306958200. [DOI] [PubMed] [Google Scholar]
- 419.Altomare C, Terragni B, Brioschi C, Milanesi R, Pagliuca C, Viscomi C, Moroni A, Baruscotti M, DiFrancesco D. Heteromeric HCN1-HCN4 channels: a comparison with native pacemaker channels from the rabbit sinoatrial node. J Physiol 549: 347–359, 2003. doi: 10.1113/jphysiol.2002.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol 117: 491–504, 2001. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Fenske S, Hennis K, Rötzer RD, Brox VF, Becirovic E, Scharr A, Gruner C, Ziegler T, Mehlfeld V, Brennan J, Efimov IR, Pauža AG, Moser M, Wotjak CT, Kupatt C, Gönner R, Zhang R, Zhang H, Zong X, Biel M, Wahl-Schott C. cAMP-dependent regulation of HCN4 controls the tonic entrainment process in sinoatrial node pacemaker cells. Nat Commun 11: 5555, 2020. doi: 10.1038/s41467-020-19304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Hennis K, Biel M, Wahl-Schott C, Fenske S. Beyond pacemaking: HCN channels in sinoatrial node function. Prog Biophys Mol Biol 166: 51–60, 2021. doi: 10.1016/j.pbiomolbio.2021.03.004. [DOI] [PubMed] [Google Scholar]
- 423.Clancy CE, Santana LF. Evolving discovery of the origin of the heartbeat: a new perspective on sinus rhythm. JACC Clin Electrophysiol 6: 932–934, 2020. doi: 10.1016/j.jacep.2020.07.002. [DOI] [PubMed] [Google Scholar]
- 424.Shi W, Wymore R, Yu H, Wu J, Wymore RT, Pan Z, Robinson RB, Dixon JE, McKinnon D, Cohen IS. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res 85: e1–e6, 1999. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- 425.Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem 268: 1646–1652, 2001. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- 426.Brown H, Difrancesco D. Voltage-clamp investigations of membrane currents underlying pace-maker activity in rabbit sino-atrial node. J Physiol 308: 331–351, 1980. doi: 10.1113/jphysiol.1980.sp013474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Baruscotti M, Bucchi A, Difrancesco D. Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol Ther 107: 59–79, 2005. doi: 10.1016/j.pharmthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 429.Bychkov R, Juhaszova M, Tsutsui K, Coletta C, Stern MD, Maltsev VA, Lakatta EG. Synchronized cardiac impulses emerge from heterogeneous local calcium signals within and among cells of pacemaker tissue. JACC Clin Electrophysiol 6: 907–931, 2020. doi: 10.1016/j.jacep.2020.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res 88: 1254–1258, 2001. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- 431.Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, Le Quang K, Kupfer E, Cohen-Solal A, Vilar J, Shin HS, Escande D, Charpentier F, Nargeot J, Lory P. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ Res 98: 1422–1430, 2006. doi: 10.1161/01.RES.0000225862.14314.49. [DOI] [PubMed] [Google Scholar]
- 432.Christel CJ, Cardona N, Mesirca P, Herrmann S, Hofmann F, Striessnig J, Ludwig A, Mangoni ME, Lee A. Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J Physiol 590: 6327–6342, 2012. doi: 10.1113/jphysiol.2012.239954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Kodama I, Nikmaram MR, Boyett MR, Suzuki R, Honjo H, Owen JM. Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am J Physiol Heart Circ Physiol 272: H2793–H2806, 1997. doi: 10.1152/ajpheart.1997.272.6.H2793. [DOI] [PubMed] [Google Scholar]
- 434.Keleshian AM, Yeo GF, Edeson RO, Madsen BW. Superposition properties of interacting ion channels. Biophys J 67: 634–640, 1994. doi: 10.1016/S0006-3495(94)80523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Keleshian AM, Edeson RO, Liu GJ, Madsen BW. Evidence for cooperativity between nicotinic acetylcholine receptors in patch clamp records. Biophys J 78: 1–12, 2000. doi: 10.1016/S0006-3495(00)76568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Iacobucci GJ, Popescu GK. Spatial coupling tunes NMDA receptor responses via Ca2+ diffusion. J Neurosci 39: 8831–8844, 2019. doi: 10.1523/JNEUROSCI.0901-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Ding S, Sachs F. Evidence for non-independent gating of P2X2 receptors expressed in Xenopus oocytes. BMC Neurosci 3: 17, 2002. doi: 10.1186/1471-2202-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Dunant Y, Garcia-Segura LM, Muller D, Parducz A. Momentary alteration of the postsynaptic membrane during transmission of a single nerve impulse. Proc Natl Acad Sci USA 86: 1717–1720, 1989. doi: 10.1073/pnas.86.5.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Zuber B, Unwin N. Structure and superorganization of acetylcholine receptor-rapsyn complexes. Proc Natl Acad Sci USA 110: 10622–10627, 2013. doi: 10.1073/pnas.1301277110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Lewis AH, Grandl J. Piezo1 ion channels inherently function as independent mechanotransducers. bioRxiv 2006.2003.446975, 2021. doi: 10.1101/2021.06.03.446975. [DOI] [PMC free article] [PubMed]
- 441.Wijerathne T, Ozkan AD, Jiang W, Luo Y, Lacroix JJ. An inter-channel cooperative mechanism mediates PIEZO1’s exquisite mechanosensitivity. bioRxiv 2004.2016.440217, 2021. doi: 10.1101/2021.04.16.440217. [DOI]
- 442.Dumitru AC, Stommen A, Koehler M, Cloos AS, Yang J, Leclercqz A, Tyteca D, Alsteens D. Probing PIEZO1 localization upon activation using high-resolution atomic force and confocal microscopy. Nano Lett 21: 4950–4958, 2021. doi: 10.1021/acs.nanolett.1c00599. [DOI] [PubMed] [Google Scholar]
- 443.Öz P, Huang M, Wolf F. Action potential initiation in a multi-compartmental model with cooperatively gating Na channels in the axon initial segment. J Comput Neurosci 39: 63–75, 2015. doi: 10.1007/s10827-015-0561-9. [DOI] [PubMed] [Google Scholar]
- 444.Parikh J, Kapela A, Tsoukias NM. Stochastic model of endothelial TRPV4 calcium sparklets: effect of bursting and cooperativity on EDH. Biophys J 108: 1566–1576, 2015. doi: 10.1016/j.bpj.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol 143: 679–692, 2014. doi: 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Jiang D, Shi H, Tonggu L, Gamal El-Din TM, Lenaeus MJ, Zhao Y, Yoshioka C, Zheng N, Catterall WA. Structure of the cardiac sodium channel. Cell 180: 122–134.e10, 2020. doi: 10.1016/j.cell.2019.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundström M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci USA 103: 17237–17242, 2006. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Allouis M, Le Bouffant F, Wilders R, Péroz D, Schott JJ, Noireaud J, Le Marec H, Mérot J, Escande D, Baró I. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ Res 98: 1538–1546, 2006. doi: 10.1161/01.RES.0000229244.97497.2c. [DOI] [PubMed] [Google Scholar]
- 449.Gong D, Chi X, Wei J, Zhou G, Huang G, Zhang L, Wang R, Lei J, Chen SR, Yan N. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature 572: 347–351, 2019. doi: 10.1038/s41586-019-1377-y. [DOI] [PubMed] [Google Scholar]
- 450.Yin CC, Blayney LM, Lai FA. Physical coupling between ryanodine receptor-calcium release channels. J Mol Biol 349: 538–546, 2005. doi: 10.1016/j.jmb.2005.04.002. [DOI] [PubMed] [Google Scholar]