

SUMMARY
Predicting phenotype from genotype represents the epitome of biological questions. Comparative genomics of appropriate model organisms holds the promise of making it possible. However, the high heterozygosity of many Eukaryotes currently prohibits assembling their genomes. Here, we report the 376 Mb genome sequence of Papilio glaucus (Pgl), the first sequenced genome from the Papilionidae family. We obtained the genome from a wild-caught specimen using a cost-effective strategy that overcomes the high (2%) heterozygosity problem. Comparative analyses suggest the molecular bases of various phenotypic traits, including terpene production in the Papilionidae-specific organ, osmeterium. Comparison of Pgl and Papilio canadensis transcriptomes reveals mutation hotspots (4% genes) associated with their divergence: four key circadian clock proteins are enriched in inter-species mutations and likely responsible for the difference in pupal diapause. Finally, the Pgl genome confirms Papilio appalachiensis as a hybrid of Pgl and Pca, but suggests it inherited 3/4 of its genes from Pca.
In Brief
The swallowtail butterfly has attracted broad attention from people. Now, Cong et al. present the genome of the Eastern Tiger Swallowtail obtained with a protocol that works for highly heterozygous genomes. Comparative analyses provide insights to caterpillar chemical defense, winter sleep of pupae, speciation mechanisms, and hybrid species.
Graphical Abstract
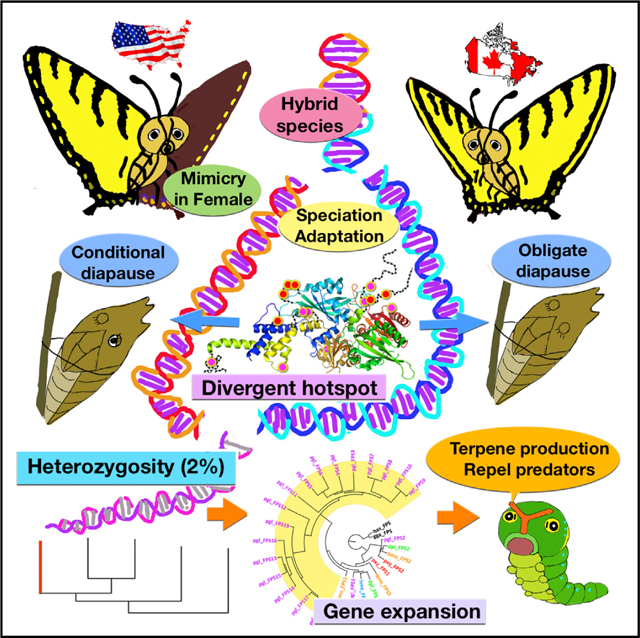
INTRODUCTION
An organism in all of its complexity of morphological and behavioral traits develops through interaction between its genetic makeup and the environment. Unraveling and predicting these traits from the genotype chart the future of biological research. The success of such prediction depends on the ability to routinely sequence and analyze the genomes of thousands of individuals from selected model organisms. In this quest, butterflies and moths, which have relatively small genomes but complex life cycles and diverse wing patterns, are emerging as powerful models. A new paradigm that gene exchange between species is pivotal in the evolution of adaptation (Heliconius Genome, 2012), and anticipation of using comparative genomics to uncover molecular mechanisms responsible for complex traits are fueling excitement in the field (Kunte et al., 2014; You et al., 2013; Zhan et al., 2011).
A showy North American butterfly, the Eastern Tiger Swallowtail, Papilio glaucus (Pgl), is honored as the state insect in five states in the US (Figures 1A and 1B). Pgl has remarkable morphological and behavioral features at all stages of its development. Like other swallowtails, the Pgl caterpillar possesses a fleshy, fork-shaped osmeterium. Upon threat, this organ everts to emit malodorous, predator-repelling terpenes (Eisner and Meinwald, 1965). The caterpillar mimics a snake, with this tongue-like osmeterium and two eyespots on the thorax. The Pgl chrysalis undergoes conditional diapauses (Hagen et al., 1991). Female adults of Pgl are dimorphic between a yellow form and a melanic form to mimic the unpalatable Pipevine Swallowtail, Battus philenor (Brower, 1958).
Figure 1. The Pgl Genome Is Highly Heterozygous.

(A and B) (A) Dorsal and (B) ventral aspects of the sequenced Pgl specimen that was preserved after tissue sampling.
(C) Histogram of 17-mer frequency in all the sequence reads before (blue curve) and after (red curve) error correction, which corrected the sequencing errors but not the heterozygosity problem. The peak on the left (about 30) represents frequency distribution of 17-mers from heterozygous regions, and the peak on the right (about 60) is for homozygous regions. The relative height of the two peaks is an indicator for heterozygosity level. The black lines indicate the height of the two peaks (heterozygous peak and homozygous peak) in a similar graph for Pxy (estimated from Figure S3 in You et al., 2013).
(D) Percentage of SNPs in 1,000-bp overlapping windows.
Pgl and its sister species, Papilio canadensis (Pca), diverged 0.6 million years ago (Kunte et al., 2011), and yet developed substantial differences in thermal preference, caterpillar food plants, body size, and female mimicry (Hagen et al., 1991). Unlike Pgl, Pca undergoes obligate pupal diapause. However, Pgl and Pca hybridize in a narrow zone where they meet. A hybrid species from the Appalachian Mountains, Papilio appalachiensis (Pap), was described recently (Pavulaan and Wright, 2002). These three species offer a model system to study evolution, hybridization, and speciation, and these studies will benefit from the decoding of a Papilio genome.
RESULTS AND DISCUSSION
A Cost-Effective Protocol for De Novo Sequencing and Assembly of Highly Heterozygous Genomes
Despite rapid development of next generation sequencing techniques, assembling highly heterozygous genomes remains a challenge. Many insects have large, widespread, and morphologically variable populations with high heterozygosity (Allendorf, 1986). Extensive inbreeding was used to overcome this problem in the Heliconius melpomene (Hme)2 and Bombyx mori (Bmo) (International Silkworm Genome Consortium, 2008; Xia et al., 2004) genome projects, while the highly hetergozygous Plutella xylostella (Pxy) genome was cloned into over 100,000 fosmids and required 114 Illumina lanes to sequence (You et al., 2013). These laborious and expensive procedures impede acquisition of numerous eukaryotic genomes for data-driven discoveries.
The Pgl genome is comparable to Pxy in terms of size and heterozygosity, and the latter is reflected by the histogram of K-mer (K bp segment) frequencies in the sequence reads (Figure 1C) (Kajitani et al., 2014). K-mers from heterozygous regions of maternal and paternal chromosomes will be different, while those from homozygous regions will have identical sequences. Therefore, the distribution of K-mer frequency in the sequencing data will form two peaks: heterozygous and homozygous, and the relative height of the two peaks reflects the heterozygosity level in the sequenced genome.
Our protocol allowed us to obtain the Pgl genome with quality comparable to other Lepidoptera using DNA from a single wild-caught specimen and sequence data from a single Illumina lane. Briefly, we extracted DNA from a piece of Pgl adult thoracic muscle. Paired-end libraries (250 and 500 bp) and three mate-pair libraries (2, 6, and 15 kb) made with a modified Cre-lox-based protocol (Van Nieuwerburgh et al., 2012) were sequenced at both ends for 150 bp (Table S1A). After the removal of low quality sequences and error correction (Table S1B), we used Platanus (Kajitani et al., 2014) software designed for highly heterozygous genomes (Table S1C) to assemble the reads. The primary assembly contained a number of shorter scaffolds with significantly lower coverage (Figures S1C and S1D), representing highly heterozygous regions that were not merged to equivalent segments in the homologous chromosomes. Using in-house scripts, these scaffolds were merged to obtain the final assembly (details in Experimental Procedures and Supplemental Information).
Genome Quality Assessment and Gene Annotation
We assembled a 376 Mb genome of Pgl and compared its quality and content with published Lepidoptera genomes (Table 1; Tables S2A and S2B). The scaffold N50 of Pgl is 230 kb, comparable to other butterfly genomes, but shorter than the Pxy genome. However, despite a larger N50, the Pxy genome assembly is incomplete as measured by the presence of CEGMA (Core Eukaryotic Genes Mapping Approach) genes (Parra et al., 2007), Cytoplasmic Ribosomal Proteins and independently assembled transcripts, while the Pgl genome rivals the best in terms of completeness. The residue coverage of CEGMA genes (Table S2C) by single Pgl scaffolds is the same as the current Bmo assembly with an N50 of 3.7 Mb, indicating that the quality of the Pgl draft is sufficient for protein annotation and comparative analysis.
Table 1.
Quality and Composition of Lepidoptera Genomes
| Feature | Pgl | Dpl a | Hme | Bmo | Pxy |
|---|---|---|---|---|---|
|
| |||||
| Genome size (Mb) | 376 | 249 | 274 | 480 | 394 |
| Heterozygosity (%) | 1.80 | 0.55 | na | na | ~2b |
| Scaffold N50 (kb) | 230 | 207 | 277 | 27 (3,700c) | 734 |
| CEGMA (%) | 99.3 | 99.3 | 98.0 | 99.3 | 98.0 |
| CEGMA coverage by single scaffold (%) | 85.6 | 85.9 | 85.6 | 85.6 | 81.7 |
| Cytoplasmic ribosomal proteins (%) | 100 | 100 | 95.7 | 98.9 | 94.6 |
| De novo assembled transcripts (%) | 98 | 96 | na | 98 | 83 |
| Repeat content (%) | 22.2 | 16.3 | 24.9 | 44.1 | 34.0 |
| Number of proteins (k) | 15.7 | 15.1 | 12.8 | 14.3 | 18.1 |
The Pgl genome is highly heterozygous with an overall SNP rate of 2%. The distribution of SNPs in the genome is prominently nonrandom (Figure 1D; Figure S1E), with a SNP rate of 0.8% in the coding regions (Table S3A). 505 protein-coding genes have significantly more (false discovery rate <0.1) SNPs than the average (Table S3B). Enriched GO terms show that many of these genes encode enzymes (Table S3C; Figures S1F and S1G). They are also significantly enriched in proteins involved in the detection of stimuli (chemosensory ionotropic receptors), which may be related to the wide range of food plants they feed on. Repeats constitute 22% of the Pgl genome (Table S1D), which is similar to other butterflies, but less than in moth genomes. We predicted 15,695 protein-coding genes in the Pgl genome (Table S1E) and annotated the function for 11,975 of them (Table S1F).
Comparative Analysis of Lepidoptera Genomes Reveals Genetic Bases for Morphological Traits
We compared the Pgl protein set with other Lepidoptera. Phylogenetic trees built both from an alignment of the 3,858 universal single-copy orthologs (Figure 2A; Figure S2A) and from synteny (Figure S2B; Table S4A) indicate that Pgl groups with other butterflies. Except Pxy, the other four species share over 85% of genes in micro-syntenic blocks. All the Pgl Hox genes that are expected to be linked are on the same scaffold (Figure 2B; Figures S2C and S2D; Table S4B), indicating the good quality of Pgl assembly.
Figure 2. Comparative Analysis of Lepidoptera Genomes and Protein Families that Underwent Gene Expansion in Pgl.

(A) Evolutionary tree based on the concatenated alignment of universal single-copy orthologs.
(B) Arrangement of Hox genes on scaffolds of all published Lepidoptera genomes. Orthologs are shown as boxes of the same color; a box surrounded by dashed lines represents a gene that is missing in the genome assembly; double boxes in the same position indicate gene duplications and “//” marks the boundaries between different scaffolds.
(C) Phylogenetic tree of Opsins.
(D) Phylogenetic tree of eclosion hormones.
(E) A typical reaction of isoprenoid biosynthesis enzymes.
(F) Phylogenetic tree of isoprenoid biosynthesis enzymes. Abbreviations of the species and protein names are used as labels in the phylogenetic trees. See also Figures S2 and S3 and Table S4.
The Pgl genome reveals expansions in several protein families (Table S4C). Previous studies identified six opsins from Pgl (Briscoe, 2000), whereas the genome assembly suggests nine (Table S4D). Pgl has more green-light-sensitive opsins, which may indicate a more advanced color perception. The identified opsins cluster into four groups (Figure 2C): in addition to the previously reported UV, blue- and green-light-sensitive opsins, we discovered another group of putative UV-sensitive opsins similar to the Drosophila Rh7 (Brody and Cravchik, 2000). Other notable gene expansions include the eclosion hormone (Figure 2D; Table S4E) that triggers the emergence of adults (Truman, 2005), and circadian clock-controlled proteins that are involved in timing of the eclosion (Myers et al., 2003). Different eclosion hormone copies may vary in temporal-spatial distribution and impart complex regulation of eclosion, allowing Pgl to diapause conditionally in response to external stimuli.
The largest expansion involves farnesyl pyrophosphate synthase (FPPS) homologs belonging to a family of isoprenoid biosynthesis enzymes that synthesize steroids and terpenes (Figure 2E) (Dhar et al., 2013). The 24 Pgl FPPS genes cluster at several genomic loci (Table S4F). The FPPS proteins are predicted to adopt an isprenoid synthase fold (Figures S3A and S3B) with fully preserved catalytic sites in 19 of them (Zhang et al., 2013b). The amino acids lining the FPPS substrate-binding sites are less conserved, implying diverse substrate specificity (Figure S3C). RNA sequencing (RNA-seq) data indicate that this gene expansion occurs in other Papilionidae species (Table S4G). The Papilio-specific FPPS enzymes form a clade in the evolutionary tree (Figure 2F), and we hypothesized that they could function in a pathway to synthesize predator-repelling terpenes secreted by the osmeterium, a Papilionidae-specific organ among butterflies.
Speciation between Papilio glaucus and Papilio canadensis
We have built an isolation-with-migration model (Hey and Nielsen, 2007) (Table S5A) using all transcripts shared by the Pgl and Pca to simulate their evolutionary history (Figure S4A). The model predicts that they diverged approximately half a million years ago and underwent dramatic increases in the effective population size, resulting in high DNA variability. The model also suggests gene flow between Pgl and Pca, consistent with their successful mating in the lab and the discovery of a hybrid species, Pap.
At the whole-transcriptome level, Pgl and Pca are easily distinguishable. Combining all 8,230 transcripts shared among the three Pgl (including the one for whole-genome sequencing) and two Pca specimens, the average variation rate and dN/dS ratio within species are significantly lower (p < 10−4) than the inter-species values (Table S5B; Figures S4B and S4C). However, due to the closeness of intra- and interspecies variation rates, these two species are indistinguishable by most individual genes. Only 351 (4.3%, Table S5C) transcripts display higher inter-species variation in both protein and DNA. As the number of samples being analyzed increases, the number of such genes will become even smaller. Therefore, only a small fraction of genes, which we term “divergence hotspots,” drive the speciation and adaptation to different environments. The divergence hotspots are mostly conserved within species (Figure 3A). Overlap between divergence hotspots and positively selected loci in Pgl and Pca (Table S5D) is small (11.1%), because 97.5% of these loci reflect adaptive evolution within either Pgl or Pca (Figure S4D).
Figure 3. Divergence Hotspots and Associated GO Terms.

(A) Venn diagram showing divergence hotspots and highly variable proteins within species.
(B) Enriched GO terms (biological processes) associated with divergence hotspots. GO terms are grouped in space by similarity in meaning and colored by the level of significance as shown by the label at the upper left corner. Annotations are shown for the most significantly enriched terms (false discovery rate <0.2).
The divergence hotspots show significant (p < 10−2) enrichment in 57 GO terms (Figure 3B; Figure S4E; Table S5E). The GO terms suggest that Pgl and Pca differ in defense against xenobiotics (e.g., GO: 0009410), insecticides (e.g., GO: 0017143), and bacteria (e.g., GO: 009617), which is consistent with the fact that they are exposed to different food plants, plant defense compounds, insecticides, and bacteria.
The GO term, “eclosion rhythm” is among the most significantly enriched. This GO term is associated with four divergence hotspots that are the central players in the circadian clock system: CLOCK, CYCLE, PEROID, and TIMELESS (Zhu et al., 2008) (Figures 4A and 4B; Figures S5A–S5D). These proteins regulate the timing for adults to hatch from pupae (opposite to diapause) and the temperature preference rhythm in Drosophila (Blanchardon et al., 2001; Kaneko et al., 2012). Mapping amino acid differences between species to 3D structure templates shows that these mutations concentrate on one side of the CLOCK/CYCLE complex (Huang et al., 2012), forming clusters on the surface (Figure 4C). A similar distribution of mutation sites is observed in PERIOD (Figure S5E). The surface clustering of mutations suggest that they likely modify interactions between circadian clock proteins and other regulators. Differences in modulation of this timing system could determine the different phenotypes: obligate diapause versus conditional diapause.
Figure 4. Circadian Clock System Could Explain Differences in Diapause between Pgl and Pca.

(A) Domain diagram of CLOCK, CYCLE, PERIOD, and TIMELESS. Mutations within species are marked by green flags, and positions that are conserved within but differ between species are marked by red flags.
(B) Circadian clock system. CRY, cryptochrome proteins.
(C) Map of inter-species mutations on the spatial structure template (PDB id: 4F3L) of CLOCK/CYCLE complex. The mutations are marked by red (CLOCK) and pink (CYCLE) dots, and the approximate position of disordered loops is shown as black beads on threads.
See also Figure S5.
Novel Nuclear DNA Barcodes for Insect Identification
The widely used mitochondrial DNA barcode encoding part of cytochrome c oxidase subunit 1 (COI) is routinely used for insect identification and cryptic species discovery. However, maternally inherited mitochondrial DNA can be transferred between species via cellular symbionts (Whitworth et al., 2007), hybridization, and backcrossing and therefore may have a history different from the whole organism. Consequently, tests with COI barcodes need to be supplemented with work based on nuclear barcodes. The commonly used nuclear markers for insects include 18s rRNA, wingless, EF1a genes and non-coding ITS1 and ITS2. However, these nuclear barcodes fail to distinguish closely related species such as Pgl and Pca, which are clearly separated by COI barcodes. In a quest for nuclear barcodes, we searched for long (>150 bp) exons that: (1) are present in most genomes as confidently identifiable and alignable single-copy orthologs; (2) differ between many pairs of closely related insect species, but are less variable within species (Figure S6A).
Out of 22,731 long exons shared by Pgl and Pca specimens, only 236 can confidently (p < 0.05) distinguish the two species, and only 41 have a higher discriminating power than the COI barcode (Table S6A) in either binomial tests or inter-species divergence level. We used 56 insect genomes forming 460 close species pairs (Table S6B) to further reduce the candidate list. Finally, 11 nuclear barcodes were selected (Table 2; Table S6C). In addition to their ability to distinguish sister species, most of them represent the phylogeny of insects better than the COI barcode (Figures S6B–S6E).
Table 2.
Nuclear Barcode Candidates and the COI Barcode
| Gene ID and Exon Number | Length (bp) | Test Pgl versus Pca | Validation in Others | FlyBase Ortholog | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| p value | V1 (%) | V2 (%) | A1 (%) | A2 (%) | V3 (%) | |||
|
| ||||||||
| pgl3382.3_e1 | 1,005 | 0 | 2.33 | 0.1 | 88.9 | 66.7 | 0.25 | CG12084 |
| pgl6760.2_e9 | 232 | 2.2E-3 | 2.4 | 0.43 | 66.7 | 62.5 | 0.72 | CG32346 |
| pgl170951.1_e1 | 1,482 | 6.4E-14 | 2.18 | 0.34 | 88.9 | 66.7 | 0.72 | CG12529 |
| pgl667.7_e7 | 1,131 | 5.9E-8 | 1.97 | 0.44 | 88.9 | 88.9 | 0.90 | CG32498 |
| pgl6877.1_e1 | 2,372 | 0 | 1.36 | 0.21 | 87.5 | 100 | 0.40 | CG16940 |
| pgl2859.6_e2 | 1,653 | 9.3E-9 | 1.28 | 0.24 | 88.9 | 100 | 0.70 | CG6294 |
| pgl5965.3_e17 | 1,604 | 4.4E-9 | 1.18 | 0.19 | 77.8 | 88.9 | 0.31 | CG7020 |
| pgl170945.2_e6 | 1,224 | 2.9E-7 | 1.13 | 0.16 | 88.9 | 88.9 | 0.35 | CG32707 |
| pgl6110.7_e1 | 2,616 | 0 | 0.98 | 0.11 | 100 | 100 | 0.46 | CG8434 |
| pgl6877.3_e2 | 1,347 | 3.3E-9 | 0.91 | 0.07 | 100 | 100 | 0.24 | CG3889 |
| pgl12124.2_e10 | 2,336 | 9.4E-11 | 0.9 | 0.13 | 62.5 | 66.7 | 0.60 | CG3696 |
| COI | 654 | 3.1E-6 | 2.14 | 0.46 | 75 | 28.6 | 0.62 | CG34067 |
V1, minimum inter-species variation rate; V2, maximum intra-species variation rate; V3, minimum variation rate between other closely related species pairs; A1, agreement between the topology of the evolutionary tree for Drosophila species built on this barcode and that built on whole-genome data; A2, similar to A1 but for the Anopheles species. See also Figure S6 and Table S6.
Reference Genome Provides New Insights into Hybrid Species Papilio appalachiensis
Using the Pgl genome as a reference, we compared the transcripts of Pap and its parental species (Figure 5A; Table S7A), Pgl and Pca. Based on the 7,410 shared transcripts, Pap is more similar to Pca than to Pgl. The two Pap specimens show the lowest variability, which agrees with it being a distinct species with a smaller effective population size, rather than a result of presently continuing hybridizations of Pgl and Pca. However, the situation may change as more specimens from different localities are sequenced, and more sequences from additional species are needed to fully understand speciation in these Papilio species.
Figure 5. Reference Genome Supports Pap as a Hybrid Species.

(A) Variation within and between species over all common transcripts shared between Pgl, Pca, and Pap.
(B) Venn diagram of statistically supported Pca-originated, Pgl-originated proteins in Pap, and proteins that are significantly different between Pca and Pgl.
(C) Percentage of Pgl-like genes in the neighborhood of confident (p < 0.05) Pgl-originated genes is significantly (p < 0.01) higher than that in the neighborhood of randomly selected samples of genes. Pgl-like genes are defined as those with average sequence identity to Pgl genes higher than the average sequence identity to Pca genes by at least 0.2%. See also Figure S7 and Table S7.
High intra-species variation hinders attribution of a Pap gene to its parental species by marginally higher sequence identity alone. We assign the origin of a Pap gene to a particular species if its sequence is similar to those from this species but different (p < 0.05) from the other species. Thus, we detected 207 Pca-originated and 70 Pgl-originated transcripts that are shared by both Pap specimens (Tables S7B and S7C). Stringent tests with Bonferroni correction show similar hybrid composition of Pap genes. Despite the small number of confidently assigned genes, they represent a majority (86%) of genes that are significantly different between Pgl and Pca (Figure 5B). Only eight Pap genes are significantly (p < 0.05) different from both parental species (Table S7D). As shown in Figure 5B, Pgl-originated genes in one Pap specimen are never Pca-originated in another Pap specimen, suggesting that the Pap population may not be a mixture of hybrids where the same loci could contain both Pgl- and Pca-originated alleles.
We used the Pgl assembly to analyze the genomic distribution of statistically supported Pgl-originated genes. These genes are significantly (p < 10−7) more likely to cluster in the genome compared to randomly selected gene sets of the same size. In addition, neighborhoods of these Pgl-originated genes are enriched in genes with higher similarity to Pgl (Figure 5C; Figure S7A). This trend of clustering is even more prominent for Pca-originated genes (Figures S7B and S7C), suggesting that the linkage between some loci was kept at the time when Pap was originating by hybridization.
The results support a hybrid origin of Pap with about 72% genes inherited from Pca, explaining the higher morphological and behavioral similarity between Pap and Pca. For instance, Pap with obligate diapause has all four divergence hotspots involved in the circadian clock system inherited from Pca, offering additional evidence for the proposed functional role of these genes. In contrast, the Pgl 6-phosphogluconate dehydrogenase (6PGD), which was shown to be closely linked to the melanic female-enabling gene on the Z chromosome (Hagen and Scriber, 1989), is inherited from Pgl. This link could be relevant to the observed Pgl-like black females in Pap from West Virginia, where the two Pap specimens were collected (Zhang et al., 2013a). Two putative transcriptional factors in the neighborhood of 6PGD (Table S7E) are candidate regulators of melanic female trait.
EXPERIMENTAL PROCEDURES
See the Supplemental Information for detailed protocols. In-house scripts and intermediate results are available at http://prodata.swmed.edu/LepDB/. Academic users can use them freely.
Library Preparation and Sequencing
We dissected approximately 80 mg of muscle from the thorax of a freshly caught and frozen male Papilio glaucus (USA: Texas, Denton County, Lake Ray Roberts State Park, Greenbelt Corridor along Elm Fork of the Trinity River, 33.2536, −97.0434, 4-VIII-2013) and preserved the remaining nearly intact specimen. The specimen will be deposited in the National Museum of Natural History, Smithsonian Institution, Washington, DC, USA (USNM). We extracted genomic DNA from this piece of tissue with the ChargeSwitch gDNA mini tissue kit and obtained approximately 20 μg DNA. 250- and 500-bp paired-end libraries were prepared using enzymes from NEBNext Modules and following the Illumina Tru-Seq DNA sample preparation guide. 2-, 6-, and 15-kb mate-pair libraries were prepared using a protocol that was modified from a previously published Cre-Lox-based method (Van Nieuwerburgh et al., 2012). For the 250-, 500-, 2-, 6-, and 15-kb libraries, approximately 1, 1, 1.7, 3, and 7.2 μg of DNA were used, respectively. After quantifying the DNA concentration with the KAPA Library Quantification Kit, we mixed 250-, 500-, 2-, 6-, and 15-kb libraries to get the final library with relative molar concentration 40:20:8:4:3. The final library was sent to the genomics core facility at UT Southwestern Medical Center to sequence 150 bp at both ends with a rapid run on Illumina HiSeq1500.
Genome Assembly and Quality Assessment
We removed the sequence reads that did not pass the purity filter and classified the rest according to their TruSeq adaptor indices. Mate-pair libraries were processed by the Delox script (Van Nieuwerburgh et al., 2012) to remove the loxP sequences and separate the true mate-pair reads from paired-end reads. All reads were processed by mirabait (Chevreux et al., 1999) to remove contamination from the TruSeq adapters, fastq_quality_trimmer to remove low quality portions at both ends, JELLYFISH (Marçais and Kingsford, 2011) to obtain k-mer frequencies in all the libraries, and QUAKE (Kelley et al., 2010) to correct sequencing errors. The data processing resulted in nine libraries that were supplied to Platanus (Kajitani et al., 2014) for genome assembly: 250- and 500-bp paired-end libraries, three paired-end and three mate-pair li- braries from 2-, 6-, and 15-kb libraries, and a single-end library containing all reads whose pairs were removed in the process.
The size of the genome assembled by Platanus was larger than the expected size based on the 15-mer coverage by the reads used for assembly. We mapped these reads to this initial assembly with Bowtie2 (Langmead and Salzberg, 2012) and calculated the coverage of each scaffold with the help of SAMtools (Li et al., 2009). Many short scaffolds in the assembly showed coverage that was about half of the expected value; they likely came from highly heterozygous regions that were not merged to the equivalent segments in the homologous chromosomes. We merged them into other scaffolds if they could be fully aligned to another significantly less covered region (coverage >90% and uncovered region <500 bp) in a longer scaffold with high sequence identity (>95%). Similar problems occurred in the initial Heliconius Melpomene genome assembly, and a similar strategy was used to improve it (Heliconius Genome, 2012).
We obtained the most recent versions of all published Lepidoptera genomes, including Bombyx mori, Danaus plexippus, Heliconius melpomene, and Plutella xylostella (Duan et al., 2010; Heliconius Genome, 2012; International Silkworm Genome Consortium, 2008; Tang et al., 2014; You et al., 2013; Zhan et al., 2011; Zhan and Reppert, 2013). Their quality was evaluated and compared with the Pgl genome. Using the criteria applied in the Monarch butterfly genome paper (Zhan et al., 2011), we estimated the completeness of these genomes based on their coverage of independently obtained transcripts, CEGMA (Parra et al., 2007) genes, and the Cytoplasmic Ribosomal Proteins.
Transcriptome Assembly
We used the reads from the previously published RNA-seq libraries of two Pgl, two Pca, two Pap, and two Papilio polytes specimens (Zhang et al., 2013a). After removal of contamination from TruSeq adapters and the low-quality portion of the reads, we applied three methods to assemble the transcriptomes: (1) de novo assembly by Trinity (Haas et al., 2013), (2) reference-based assembly by TopHat (Kim et al., 2013) (v.2.0.10) and Cufflinks (Roberts et al., 2011) (v.2.2.1), and (3) reference-guided assembly by Trinity. The results from all three methods were then integrated by PASA (Haas et al., 2008). For the more distantly related Papilio polytes, only de novo assembly was performed.
Identification of Repeats and Gene Annotation
Two approaches were used to identify repeats in the Pgl genome: the RepeatModeler (Smit and Hubley, 2010) pipeline and in-house scripts that extracted regions with coverage four times more than expected. These repeats were submitted to the CENSOR (Jurka et al., 1996) server to assign them to the repeat classification hierarchy. The species-specific repeat library and repeats classified in RepBase (Jurka et al., 2005) (v.18.12) were used to mask repeats in the genome by RepeatMasker (Smit et al., 2010).
From the transcriptome of each Pgl specimen, we obtained two sets of transcript-based annotations from two pipelines: TopHat followed by Cufflinks and Trinity followed by PASA. In addition, we obtained five sets of homology-based annotations by aligning protein sets from all four published Lepidoptera and Drosophila melanogaster (Misra et al., 2002) to the Pgl genome with exonerate (Slater and Birney, 2005). Proteins from the entire UniRef90 (Suzek et al., 2007) database were used to generate another set of gene predictions by genblastG (She et al., 2011). Integrating the evidence from transcripts and homologs, we manually curated and selected 1,313 confident gene models to train de novo gene predictors: AUGUSTUS (Stanke et al., 2006), SNAP (Korf, 2004), and GlimmerHMM (Majoros et al., 2004). These trained predictors, the self-trained Genemark (Besemer and Borodovsky, 2005) and a consensus-based pipeline, Maker (Cantarel et al., 2008) were used to generate another five sets of gene models. Homology and transcript evidence was supplied to AUGUSTUS, SNAP, and Maker to boost their performance. In total, we generated 15 sets of gene predictions and integrated them with EvidenceModeller (Haas et al., 2008) to generate the final gene models.
We predicted the function of Pgl proteins by transferring annotations and GO terms from the closest BLAST hit (E-value <10−5) in the Swissprot (UniProt Consortium, 2014) database and FlyBase (St Pierre et al., 2014), respectively. Finally, we performed InterproScan (Jones et al., 2014) to identify conserved protein domains and functional motifs, to predict coiled coils, transmembrane helices, and signal peptides, to detect homologous 3D structures, to assign Pgl proteins to protein families, and to map them to metabolic pathways.
Detection of SNPs
We mapped all the sequence reads used to assemble the Pgl genome to the Pgl scaffolds with Bowtie2, and, using both SAMtools followed by BCFtools (Li et al., 2009) and the Genome Analysis Toolkit (DePristo et al., 2011), we identified SNPs and indels that happened to only one of the homologous chromosomes. To analyze the distribution of SNPs, we divided the genome into exons, introns, repeats, and intergenic regions. The percentage of SNPs in 1,000-bp windows in the genome and the distances between neighboring SNPs were used to reflect this distribution, and a random distribution of SNPs was simulated in silico as a control. We identified proteins with significantly more SNPs with binomial tests (p = average percentage of SNPs in all exons, m = number of SNPs in the exons of a protein, N = length of a protein) followed by false discovery rate (FDR) tests (Storey and Tibshirani, 2003). We considered proteins with Q-values from FDR tests smaller than 0.1 to be significantly enriched in SNPs. Enriched GO terms associated with these SNP-enriched proteins were identified by means of another binomial test (p = probability of this GO term being associated with any Pgl protein, m = number of SNP-enriched proteins associated with this GO term, N = number of SNP-enriched proteins). The significantly enriched GO terms (p < 0.01) were submitted to the REVIGO (Supek et al., 2011) web server.
Comparison of Lepidoptera Genomes
We used OrthoMCL (Li et al., 2003) to identify the orthologous groups from the Lepidoptera species. We aligned the protein sequences of universal single-copy orthologs with MAFFT (Katoh and Standley, 2013) and built an evolutionary tree from the concatenated alignment with PhyML 3.0 (Guindon et al., 2010) (JTT model [Jones et al., 1992]). We calculated the percentage of genes in micro-syntenic blocks and identified gene inversion events. The frequency of gene inversions was used to build a synteny-based tree using BioNJ (Gascuel, 1997). In a case study, we identified the homeodomains from Hox genes in all published Lepidoptera and in the unpublished Manduca sexta and Helicoverpa punctigera genomes (BCM-HGSC), using homeodomains from Drosophila in the HomeoDB (Zhong and Holland, 2011) as reference. Orthologous groups among these homeodomains were detected using a phylogenetic tree built by PhyML (JTT model) on the MAFFT alignment.
If two OrthoMCL-defined orthologous groups overlapped in the Drosophila proteins that they mapped to, we merged them into one family. The total number and total length of proteins in a family were used to identify expanded gene families in Pgl. The most significantly expanded gene families with well-defined functions were further investigated to include all relevant proteins using reciprocal BLAST results and function annotations. Proteins encoded by the genome but missed in the protein sets were predicted with the help of genblastG. Protein sequences from each family were aligned with MAFFT or PROMALS3D (Pei et al., 2008). Evolutionary trees were built with PhyML (JTT model) and visualized in FigTree.
Speciation between Papilio glaucus and Papilio canadensis
We used PASA to map the transcripts from two Pgl and two Pca specimens to the genome and considered transcripts to be orthologous if they mapped to the same loci. We used MAFFT and BLAST to align the orthologous transcripts and extracted the positions that were consistently aligned by both methods and shared among all specimens to obtain the final alignments. We discarded any alignments that were shorter than 50 bp. From each alignment, we calculated the pairwise identities on both the DNA and protein levels and identified transcripts, namely, “divergence hotspots,” that were more similar between any pairs from the same species than from different species. We identified the enriched GO terms associated with these proteins using both binomial tests as described above and the DAVID Bioinformatics Resources (Huang et al., 2009). Significantly enriched GO terms (p < 0.01) were visualized in REVIGO. Important divergence hotspots were submitted to MESSA (Cong and Grishin, 2012) to perform secondary structure and disordered region prediction, domain identification, and 3D structure prediction. In addition, we used PAML (Yang, 2007) to identify proteins with positively selected sites, with a model that allowed the dN/dS ratio (ω) to vary among sites. To study the evolutionary history of Pgl and Pca, we removed protein-coding genes with signs of recombination detected by PhiPack (Bruen et al., 2006), and randomly divided the rest into 20 data sets and applied IMa2 (Hey and Nielsen, 2007) and IMfig (Hey, 2010) to build and visualize the isolation with migration models.
Selection of Nuclear DNA Barcodes for Insect Identification
We selected nuclear DNA barcodes from exons that were longer than 150 bp and shared among all Pgl and Pca specimens. We performed binomial tests (p = maximal percentage of intra-species mutations, m = minimal number of inter-species mutations, N = length of the exon) to select exons that could confidently distinguish Pgl and Pca. Exons that showed lower p values in this test or higher inter-species variation than the COI barcode were considered to be nuclear barcode candidates. We further removed exons without unique and confident (identity >40%, coverage >80%) orthologs in other Lepidoptera genomes and those that could not distinguish other closely related insect species. For the final barcode candidates, we tested their ability to reflect the phylogeny by comparing the evolutionary trees (PhyML based on the MAFFT alignment) built on them to those based on whole-genome data for Drosophila species in FlyBase and Anopheles species in Vectorbase (Neafsey et al., 2013), using TOPD/FMTS (Puigbòet al., 2007) to quantify the similarity in the topology of the trees.
Studies of Papilio appalachiensis, the Hybrid Species
We detected and aligned orthologs among all Pgl, Pca, and Pap specimens using the procedure described above. We assigned a Pap transcript to Pgl if it was significantly (p < 0.05) different from all orthologs in Pca in a binomial test (p = maximum intra-species variation rate for Pgl and Pca, m = minimum mutation number compared to orthologs in Pca, n = length of the transcript) and not different (p > 0.05) from the Pgl transcripts in another test (p = maximum intra-species variation rate for Pgl and Pca, m = maximum mutation number compared to orthologs in Pgl, n = length of the transcript). Pca-origi- nated genes were assigned similarly. The numbers of statistically supported Pgl- and Pca-originated transcripts were used to estimate the fraction of Pap genes that were inherited from Pca.
We further tested if the Pgl-like genes tend to cluster in the genome by comparing their distribution to random samples. In the first test, we counted the number of Pgl-originated genes (p < 0.05) that have other Pgl-originated genes (p < 0.05) in the same scaffold. In the second test, we identified genes that showed higher sequence identities to orthologs from Pgl than to those from Pca by a certain cutoff (0.2%, 0.4%, and 0.6%) and defined them as Pgl-like genes. We calculated the percentage of Pgl-like genes [Pgl-like genes/(Pgl-like genes + Pca-like genes)] in the neighborhood of the confident Pgl-originated (p < 0.05) genes and random samples of the same size. Similar tests were performed for Pca-like genes as well.
Supplementary Material
Highlights.
Genome sequence of Eastern Tiger Swallowtail, the first for the Papilionidae family
A cost-effective protocol to sequence and assemble highly heterozygous genomes
Molecular basis for terpene synthesis in the Papilionidae- specific organ, osmeterium
Mutations in circadian clock proteins that dictate different timing of pupal diapause
ACKNOWLEDGMENTS
We acknowledge Texas Parks and Wildlife Department (Natural Resources Program Director David H. Riskind) for the permit #08–02Rev that makes research based on material collected in Texas State Parks possible. We thank Jeremy Semeiks for suggestions on sequence data processing, Johann Deisenhofer, Lisa N. Kinch, R. Dustin Schaeffer, and Raquel Bromberg for critical suggestions and proofreading of the manuscript. Q.C. is a Howard Hughes Medical Institute International Student Research fellow. This work was supported in part by the NIH (GM094575 to N.V.G.) and the Welch Foundation (I-1505 to N.V.G.).
Footnotes
ACCESSION NUMBERS
The genome sequence has been deposited at DDBJ/EMBL/GenBank under the accession number JWHW00000000. The version described in this paper is version JWHW01000000. The sequencing experiments have been deposited in the NCBI SRA database under accession numbers SRX806850, SRX806851, SRX806914, SRX806951, SRX807040, SRX807049, and SRX807050. The sequence reads to assemble the genome have been deposited at the same database under accession numbers: SRR1707280, SRR1707287, SRR1707316, SRR1707405, SRR1707569, SRR1707572, and SRR1707573.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.01.026.
REFERENCES
- Allendorf FW (1986). Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol. 5, 181–190. [Google Scholar]
- Besemer J, and Borodovsky M. (2005). GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33, W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchardon E, Grima B, Klarsfeld A, Chélot E, Hardin PE, Préat T, and Rouyer F. (2001). Defining the role of Drosophila lateral neurons in the control of circadian rhythms in motor activity and eclosion by targeted genetic ablation and PERIOD protein overexpression. Eur. J. Neurosci. 13, 871–888. [DOI] [PubMed] [Google Scholar]
- Briscoe AD (2000). Six opsins from the butterfly Papilio glaucus: molecular phylogenetic evidence for paralogous origins of red-sensitive visual pigments in insects. J. Mol. Evol. 51, 110–121. [DOI] [PubMed] [Google Scholar]
- Brody T, and Cravchik A. (2000). Drosophila melanogaster G protein- coupled receptors. J. Cell Biol. 150, F83–F88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower JVZ (1958). Experimental studies of mimicry in some North American butterflies: Part II. Battus philenor and Papilio troilus, P. polyxenes and P. glaucus. Evolution 12, 123–136. [Google Scholar]
- Bruen T, Phillipe H, and Bryant D. (2006). A quick and robust statistical test to detect the presence of recombination. Genetics 172, 2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, and Yandell M. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevreux B, Wetter T, and Suhai S. (1999). Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Computer Science and Biology: Proceedings of the German Conference on Bioinformatics 99, 45–56. [Google Scholar]
- Cong Q, and Grishin NV (2012). MESSA: MEta-Server for protein sequence analysis. BMC Biol. 10, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A frame-work for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar MK, Koul A, and Kaul S. (2013). Farnesyl pyrophosphate synthase: a key enzyme in isoprenoid biosynthetic pathway and potential molecular target for drug development. New Biotechnol. 30, 114–123. [DOI] [PubMed] [Google Scholar]
- Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, and Xia Q. (2010). SilkDB v2.0: a platform for silkworm (Bombyx mori ) genome biology. Nucleic Acids Res. 38, D453–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner T, and Meinwald YC (1965). Defensive Secretion of a Caterpillar (Papilio). Science 150, 1733–1735. [DOI] [PubMed] [Google Scholar]
- Gascuel O. (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, and Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, and Wortman JR (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen RH, and Scriber JM (1989). Sex-linked diapause, color, and allozyme loci in Papilio glaucus: linkage analysis and significance in a hybrid zone. J. Hered. 80, 179–185. [Google Scholar]
- Hagen RH, Lederhouse RC, Bossart JL, and Scriber JM (1991). Papilio canadensis and P. glaucus are distinct species. J. Lepid. Soc. 45, 245–258. [Google Scholar]
- Heliconius Genome C; Heliconius Genome Consortium (2012). Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J. (2010). The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 27, 921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, and Nielsen R. (2007). Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 104, 2785–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang N, Chelliah Y, Shan Y, Taylor CA, Yoo SH, Partch C, Green CB, Zhang H, and Takahashi JS (2012). Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 337, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Silkworm Genome Consortium (2008). The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem. Mol. Biol. 38, 1036–1045. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, and Thornton JM (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. [DOI] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Klonowski P, Dagman V, and Pelton P. (1996). CENSOR—a pro- gram for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 20, 119–121. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, and Walichiewicz J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Head LM, Ling J, Tang X, Liu Y, Hardin PE, Emery P, and Hamada FN (2012). Circadian rhythm of temperature preference and its neural control in Drosophila. Curr. Biol. 22, 1851–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, and Salzberg SL (2010). Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 11, R116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte K, Shea C, Aardema ML, Scriber JM, Juenger TE, Gilbert LE, and Kronforst MR (2011). Sex chromosome mosaicism and hybrid speciation among tiger swallowtail butterflies. PLoS Genet. 7, e1002274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, and Kronforst MR (2014). doublesex is a mimicry supergene. Nature 507, 229–232. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr., and Roos DS (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, and Salzberg SL (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Marçais G, and Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al. (2002). Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome biology 3, RESEARCH0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EM, Yu J, and Sehgal A. (2003). Circadian control of eclosion: interaction between a central and peripheral clock in Drosophila melanogaster. Current biology: CB 13, 526–533. [DOI] [PubMed] [Google Scholar]
- Neafsey DE, Christophides GK, Collins FH, Emrich SJ, Fontaine MC, Gelbart W, Hahn MW, Howell PI, Kafatos FC, Lawson D, et al. (2013). The evolution of the Anopheles 16 genomes project. G3 3, 1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, and Korf I. (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Pavulaan H, and Wright DM (2002). Pterourus appalachiensis (Papilionidae: Papilioninae), a new swallowtail butterfly from the Appalachian region of the United States. Taxonomic Report of the International Lepidoptera Survery 3, 1–10. [Google Scholar]
- Pei J, Kim BH, and Grishin NV (2008). PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbò P, Garcia-Vallvé S, and McInerney JO (2007). TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics 23, 1556–1558. [DOI] [PubMed] [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, and Pachter L. (2011). Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 27, 2325–2329. [DOI] [PubMed] [Google Scholar]
- She R, Chu JS, Uyar B, Wang J, Wang K, and Chen N. (2011). gen-BlastG: using BLAST searches to build homologous gene models. Bioinformatics 27, 2141–2143. [DOI] [PubMed] [Google Scholar]
- Slater GS, and Birney E. (2005). Automated generation of heuristics for bio- logical sequence comparison. BMC Bioinformatics 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, and Hubley R. (2010). RepeatModeler Open-1.0 (Seattle: The Institute for Systems Biology; ). [Google Scholar]
- Smit AFA, Hubley R, and Green P. (2010). RepeatMasker Open-3.0 (Seattle: The Institute for Systems Biology; ). [Google Scholar]
- St Pierre SE, Ponting L, Stefancsik R, and McQuilton P; FlyBase Consortium (2014). FlyBase 102—advanced approaches to interrogating FlyBase. Nucleic Acids Res. 42, D780–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schöffmann O, Morgenstern B, and Waack S. (2006). Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, and Šmuc T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Huang H, McGarvey P, Mazumder R, and Wu CH (2007). UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23, 1282–1288. [DOI] [PubMed] [Google Scholar]
- Tang W, Yu L, He W, Yang G, Ke F, Baxter SW, You S, Douglas CJ, and You M. (2014). DBM-DB: the diamondback moth genome database. Database 2014, bat087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman JW (2005). Hormonal control of insect ecdysis: endocrine cascades for coordinating behavior with physiology. Vitam. Horm. 73, 1–30. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium (2014). Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nieuwerburgh F, Thompson RC, Ledesma J, Deforce D, Gaasterland T, Ordoukhanian P, and Head SR (2012). Illumina mate-paired DNA sequencing-library preparation using Cre-Lox recombination. Nucleic Acids Res. 40, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth TL, Dawson RD, Magalon H, and Baudry E. (2007). DNA barcoding cannot reliably identify species of the blowfly genus Protocalliphora (Diptera: Calliphoridae). Proc. Biol. Sci. 274, 1731–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Zhou Z, Lu C, Cheng D, Dai F, Li B, Zhao P, Zha X, Cheng T, Chai C, et al. ; Biology Analysis Group (2004). A draft sequence for the genome of the domesticated silkworm (Bombyx mori). Science 306, 1937–1940. [DOI] [PubMed] [Google Scholar]
- Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- You M, Yue Z, He W, Yang X, Yang G, Xie M, Zhan D, Baxter SW, Vasseur L, Gurr GM, et al. (2013). A heterozygous moth genome provides insights into herbivory and detoxification. Nat. Genet. 45, 220–225. [DOI] [PubMed] [Google Scholar]
- Zhan S, and Reppert SM (2013). MonarchBase: the monarch butterfly genome database. Nucleic Acids Res. 41, D758–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan S, Merlin C, Boore JL, and Reppert SM (2011). The monarch butterfly genome yields insights into long-distance migration. Cell 147, 1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Kunte K, and Kronforst MR (2013a). Genome-wide characterization of adaptation and speciation in tiger swallowtail butterflies using de novo transcriptome assemblies. Genome Biol. Evol. 5, 1233–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhu W, Liu YL, Wang H, Wang K, Li K, No JH, Ayong L, Gulati A, Pang R, et al. (2013b). Chemo-immunotherapeutic anti-malarials targeting isoprenoid biosynthesis. ACS medicinal chemistry letters 4, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong YF, and Holland PW (2011). HomeoDB2: functional expansion of a comparative homeobox gene database for evolutionary developmental biology. Evol. Dev. 13, 567–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Sauman I, Yuan Q, Casselman A, Emery-Le M, Emery P, and Reppert SM (2008). Cryptochromes define a novel circadian clock mechanism in monarch butterflies that may underlie sun compass navigation. PLoS Biol. 6, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.