

Abstract
Respiratory syncytial virus (RSV)-induced immunopathogenesis and disease severity in neonatal mice and human infants have been related to elevated pulmonary IL-33. Thus, targeting IL-33 has been suggested as a potential therapy for respiratory viral infections. Yet, the regulatory mechanisms on IL-33 during early life remain unclear. Here, using a neonatal mouse model of RSV, we demonstrate that IL-1β positively regulates but is not required for RSV-induced expression of pulmonary IL-33 in neonatal mice early after the initial infection. Exogenous IL-1β upregulates RSV-induced IL-33 expression by promoting the proliferation of IL-33+ lung epithelial stem/progenitor cells. These cells are exclusively detected in RSV-infected neonatal rather than adult mice, partially explaining the IL-1β–independent IL-33 expression in RSV-infected adult mice. Furthermore, IL-1β aggravates IL-33–mediated T-helper cell type 2–biased immunopathogenesis upon reinfection. Collectively, our study demonstrates that IL-1β exacerbates IL-33–mediated RSV immunopathogenesis by promoting the proliferation of IL-33+ epithelial stem/progenitor cells in early life.
Keywords: IL-33, IL-1β, respiratory syncytial virus, epithelial stem or progenitor cells, age-dependent RSV immunopathogenesis
Clinical Relevance
By providing mechanistic insights explaining the observation of the positive correlation between respiratory IL-33 and IL-1β in respiratory syncytial virus (RSV)-infected human infants, our current study serves as a reverse translation from bedside to bench in RSV immunopathogenesis research. The knowledge gained from our current research also provides new avenues for future investigation to determine whether IL-1β–mediated proliferation of IL-33+ epithelial stem cells during RSV infection in early life is permanent and, if so, predisposes to the development of certain lung diseases (i.e., asthma).
Respiratory syncytial virus (RSV) is the most common cause of bronchiolitis and pneumonia in infants in the United States and the most common viral cause of pneumonia worldwide (1). Children are most vulnerable to severe RSV bronchiolitis during their infancy. Approximately 80% of the children hospitalized are less than 1 year of age (2). The clinical manifestations of RSV infection in infants range from mild upper respiratory tract illness to severe and potentially life-threatening lower respiratory tract disease (2, 3). Human and animal data have suggested that elevated respiratory IL-33 concentrations are responsible for RSV-induced immunopathogenesis (4, 5). IL-33, a member of the IL-1 cytokine family that includes IL-1β and IL-1α, is highly and constitutively expressed by airway epithelial cells as a full-length precursor (6, 7). Under homeostatic conditions, full-length IL-33 is located in the cell nuclei owing to its nuclear localization sequence (7, 8). As an alarmin, IL-33 is released during cell necrosis (9, 10), or in responses to oxidative and mechanical stress, or via ATP-mediated mechanism during cell activation (11, 12). While IL-33 shares common features with IL-1β, such as a C-terminal IL-1–like cytokine domain and a coreceptor IL-1 receptor accessory protein (6, 13), IL-33 exclusively exerts its effects through IL-1 receptor-like 1 (also known as ST2) (14). While the role of IL-33 in mediating age-variable RSV immunopathogenesis has been well documented (5, 15), little is known regarding the regulation of IL-33 expression during early life, and what is known is contradictory. For example, IL-33 production in adult mice is enhanced in the peritoneal cavity in the absence of IL-1β signaling during the early phase of helminth infection (16). Similarly, exogenous IL-1β has been demonstrated to suppress superconfluence-mediated IL-33 expression by human endothelial vascular cell cultures (17). These studies suggest that IL-1β negatively regulates the expression of IL-33. However, this notion has been challenged by an increasing number of recent human and experimental animal studies. IL-1β has been shown to promote the expression of pulmonary IL-33 and to exacerbate asthma and chronic obstructive pulmonary disease in animals (18). It is also evident that exogenous IL-1β upregulates IL-33 expression of human nasal fibroblast in vitro (19). Intriguingly, a cohort study of 93 RSV-infected infants previously demonstrated a significant positive correlation between IL-33 and IL-1β concentrations in nasal aspirates (5). Prompted by these human and animal data, we sought to determine the role of IL-1β on RSV-induced IL-33 production during early life. To this end, we conducted the current study using the well-established neonatal RSV model that mimics disease observed in human infants (15, 20).
Methods
Full methods are available in the Online Methods section in this article’s data supplement.
Neonatal Mouse Model for RSV Infection
BALB/c mice (Envigo Laboratories) were maintained in a specific-pathogen-free facility in ventilated microisolator cages. The IL-33 citrine mice (cit/wt), generously provided by Dr. Andrew N. J. McKenzie (Medical Research Council, UK), are generated by mating IL33(wt/wt) mice and IL33(cit/cit) mice. Breeders were time-mated, and pups born on the same date were used for experiments. Neonatal mice (5 days old) received 2.5 ng of recombinant IL-1β/g body weight (Biolegend) (21) or 50 μg anakinra (IL-1β receptor antagonist)/g body weight intranasally (16). Three hours later, mice were infected with RSV strain A2 in serum-free media at a dose of 2 × 105 TCID50 (50% tissue culture infectious dose)/g body weight delivered intranasally (hereafter referred to as NR1 + IL-1β or NR1 + Ana, respectively). Control mice received serum-free media at the same schedule and via the same route (NS). Neonatal mice that received vehicle (0.1% BSA in 0.9% NaCl) served as the control groups (NR1 + Veh or NS + Veh, respectively). Mice were reinfected 4 weeks after initial infection (NRR) (Figure 1A). This animal protocol was developed according to the Guide for the Care and Use of Laboratory Animals and was approved by the Institutional Animal Care and Use Committee at Louisiana State University and Pennington Biomedical Research Center, both Association for Accreditation of Laboratory Animal Care accredited.
Figure 1.
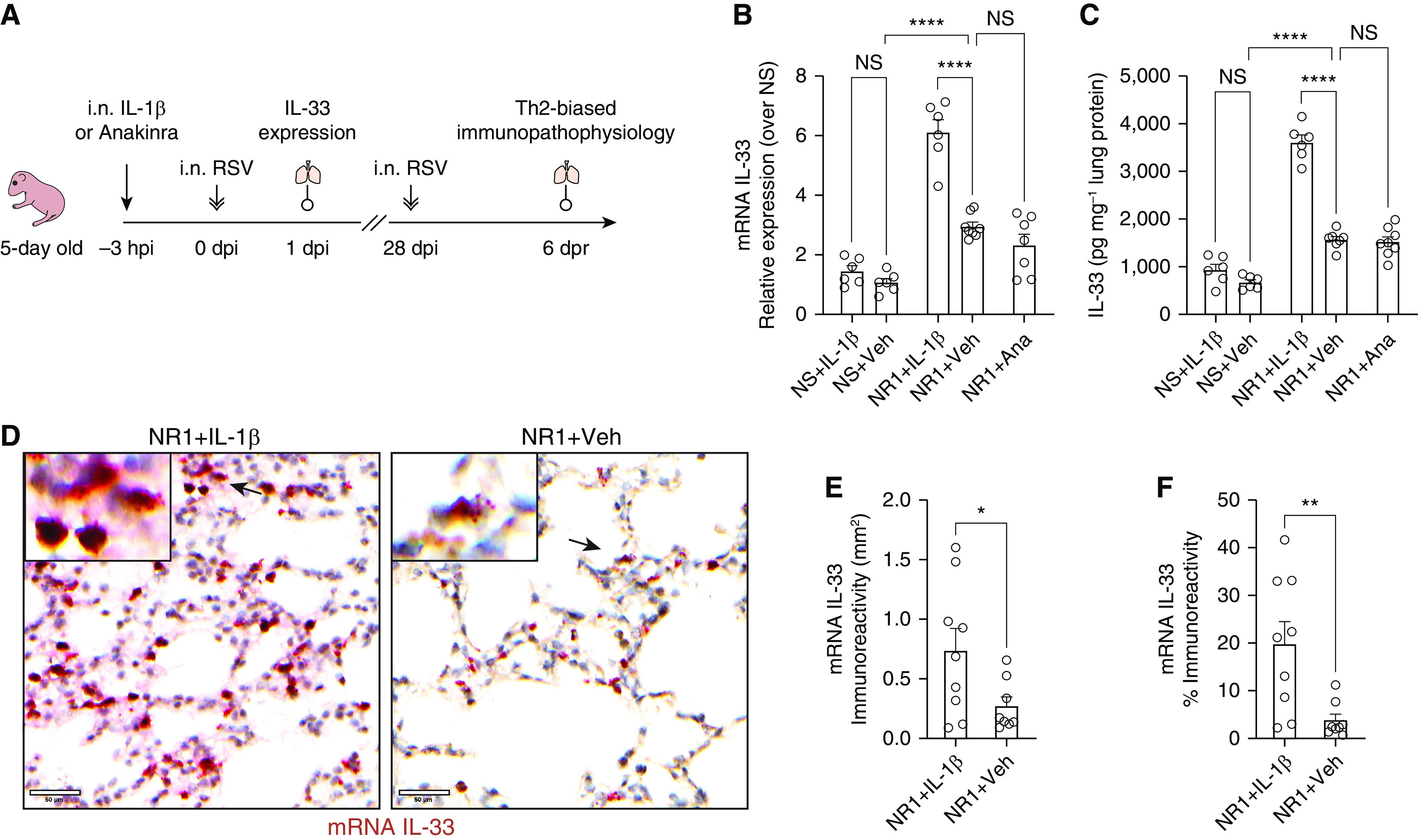
Intranasal treatment with IL-1β before respiratory syncytial virus (RSV) infection significantly boosted the expression of RSV-induced IL-33 at 1 day after infection (dpi) in neonatal mice. (A) Experiment design scheme. Five-day-old pups received either 2.5 ng of recombinant IL-1β/g body weight (Biolegend) or 50 μg anakinra (IL-1β antagonist)/g body weight delivered intranasally (i.n.) 3 hours before i.n. RSV infection. The expression of pulmonary IL-33 was examined at 1 dpi (hereafter referred to as NR1 + IL-1β or NR1 + Ana, respectively). A cohort of mice received vehicle (0.1% BSA in saline) i.n. and served as the control group (NR1 + Veh). We reinfected mice with RSV 4 weeks after initial infection. At 6 days after reinfection (dpr), we profiled pulmonary cell subsets and evaluated the animals’ lung function. (B and C) Bar graphs representing the expression of IL-33 mRNA (B) and protein (C). (D) Representative IL-33 in situ hybridization (ISH) sections of lungs from three independent RSV-infected neonatal mice. IL-33 mRNA was stained as dark red dots indicated by black arrows within the magnified inset (top left). Scale bars, 50 μm. (E and F) Quantification of IL-33 mRNA–positive cells per unit area of lungs. ISH sections were digitized to 20× whole slide images and presented as the staining area (E) and proportions of the whole slide area (F). *P < 0.05, **P < 0.01, and ****P < 0.0001; two-tailed Welch’s t-tests and one-way ANOVA with a Sidak multiple comparisons compared between two or more than two indicated groups, respectively. Ana = anakinra; hpi = hours post-injection; NS = not significant; Veh = vehicle; Th2 = T-helper cell type 2.
mRNA and Protein (IL-33, IL-1β, and Caspase) Expression Degrees
Relative expression degrees of IL-33 mRNA were analyzed using 2−ΔΔCt method. The expression of IL-33, IL-1β, caspase-3, and caspase-7 in whole-lung homogenates were determined by ELISA and Western blot, respectively.
Lung Histopathology
Mouse lungs were retroperfused with PBS and then gravity-inflated with zinc-formalin. Lung sections were stained with periodic acid-Schiff to observe mucus production. Dual in situ hybridization (ISH) and immunohistochemistry (IHC) for Il33 and Caspase-1 were performed on the third stepped tissue section of the lungs from each experimental group.
Lung Function Test
In anesthetized, tracheotomized mice (15), peak airway resistance values in response to increasing doses of inhaled methacholine were measured using the Buxco FinePointe Resistance and Compliance system (DSI, Harvard Bioscience, Inc.) and normalized as deviation from baseline.
Flow Cytometry
Lungs were retroperfused with ice-cold PBS and harvested in ice-cold FACS buffer (5% heat-inactivated FCS in PBS). Lung-derived single-cell suspension was stained with CD16/CD32 Ab, live/dead dyes in PBS for 30 min at 4°C. Subsequently, cells were stained for cell-surface and then intracellular markers with a combination of antibodies (listed in Table E1 in the data supplement). Cells were run on either a three-laser BD Canto-II, or a four-laser BD LSRFortessa X-20, using FACS Diva software (BD Biosciences). Acquired data files were analyzed using FlowJo 10.01 (BD Biosciences).
Statistical Analyses
Raw data were analyzed, and graphs were generated using Prism 9.0.0 (GraphPad). Data are presented as mean ± SEM unless noted otherwise. Normal distribution was examined using the Shapiro-Wilk test. Two-tailed Welch’s t-tests were performed to compare two groups of samples. For three and more groups of samples, either a one-way or a two-way ANOVA with a Sidak multiple comparisons test was conducted. The statistical significance of these tests was represented as P value with ns: non-significant, *P ⩽ 0.05, **P ⩽ 0.01, ***P ⩽ 0.001, and ****P ⩽ 0.0001.
Results and Discussion
IL-1β Positively Regulates but Is Not Required for RSV-induced Expression of Pulmonary IL-33 in Neonatal Mice
We have previously shown that RSV infection induces rapid IL-33 production in neonatal mice, which is not observed in adult mice (15) and which results in an ILC2 response initially upon RSV infection in the neonatal mouse and a T-cytotoxic cell (Tc2) and T-helper cell type 2 (Th2) biased response in both neonatal mice and humans (15, 22). In trying to understand what was inducing IL-33 expression in neonatal mice, we observed differences in IL-1β concentrations. Specially, IL-1β was elevated in adult mice infected with RSV, but not in neonatal mice infected with RSV (Figure E1A). As IL-1β has been shown to negatively regulate the production of IL-33 (16, 17), we administered IL-1β or anakinra (IL-1β receptor antagonist) to neonates just before RSV infection. As RSV infection induced rapid IL-33 expression (i.e., as early as 6 hours after infection) in neonatal mice (15), IL-1β or anakinra was administered intranasally 3 hours before the RSV infection to allow the distribution to the lung before infection (Figure 1A). We expected, based on the existing literature and our adult RSV data, that IL-1β would reduce IL-33 responses upon RSV infection; however, this did not happen in our RSV neonatal model.
In the presence of IL-1β, expression of RSV-induced IL-33 at Day 1 after infection (dpi) was significantly enhanced, as evidenced by the increase in IL-33 mRNA and protein and elevated frequencies of IL-33–producing cells in the lungs following RSV infection (Figures 1B–1F). There was no significant difference between IL-33 concentrations in NS and NS + IL-1β) groups, suggesting that IL-1 β upregulates IL-33 expression only in the setting of RSV infection (Figures 1B and 1C). The different observations in our RSV neonatal model and helminth adult model (16) are more likely attributed to the target organs (pulmonary vs. peritoneal), pathogen (viral vs. parasite infection) and age at initial infection (neonate vs. adult). It has been shown that IL-33 responses to RSV infection (15) are age-dependent as IL-33 production and more robust in neonates than adults.
Interestingly, the blockage of IL-1β signals with anakinra before RSV infection did not alter the expression of pulmonary IL-33 (Figures 1B and 1C). The data suggest that IL-1β enhances but is not required for RSV-induced IL-33 expression in neonatal mice. Similar to our data, a study using house dust mite-induced asthma in mice demonstrated that the absence of IL-1β did not alter the expression of pulmonary IL-33 but significantly diminished the increase in IL-33 concentrations following asthma exacerbation (18). Our current finding was also corroborated by the significantly positive correlation between IL-33 and IL-1β levels in nasal aspirates collected from human infants with severe RSV (5).
IL-1β Has a Long-Lasting Impact Exacerbating IL-33–mediated Th2-biased Immunopathogenesis upon RSV Reinfection
Elevated concentrations of IL-33 have been associated with more severe RSV disease in human infants (4, 5). More severe RSV disease often results in increased airway mucus and dramatic airway obstruction (2, 23–25). Therefore, we anticipated that exogenous IL-1β would enhance IL-33 production and lead to more severe RSV disease. To test this possibility, we continued using our RSV neonatal model, which exhibits a pathophysiological phenotype typically observed in severe human infantile RSV. In our model, RSV-induced lung dysfunction and T-cell responses to RSV infection were limited after the primary infection and peaked at 6 days after reinfection (dpr) or 32 days after primary infection (15, 20). Reinfection with RSV occurs frequently, and neonatal reinfection models have been used by numerous labs (15, 20, 26–29) to determine the role of RSV-specific cell-mediated immunity.
Indeed, administration of IL-1β before RSV infection significantly increased airway mucus production at 6 dpr (Figures 2A and 2B). In addition, the NRR + IL-1β group exhibited significantly greater airway resistance in response to increasing doses of inhaled methacholine than the NRR + Veh group (Figure 2C), indicating airway hyperresponsiveness following RSV infection. As it is evident that IL-1β is a strong inducer of mucin gene in either in vivo (30) or in vitro models (31, 32), we also treated neonatal mice with IL-1β (NSS + IL-1β) and used this group to control for the effect of IL-1β alone on mucus production (Figure 2A) and airway resistance (Figure 2C). We observed no significant difference in mucus production and airway resistance between NSS + IL-1β and NSS + Veh groups, suggesting the limited effect of IL-1β alone on lung function after reinfection. Of note, there are many differences between previous studies (30–32) and our current study. Our IL-1β treatment was a single dose of 2.5 ng/g (∼7 ng/mouse), significantly smaller than that of the previous study (a single dose of 200 ng IL-1β per adult C57BL/6 mouse via tracheostomy). More importantly, although IL-1β–induced expression of mucin genes was assessed at 24 hours after IL-1β treatment in previous studies (30–32), we evaluated mucus production and airway resistance at 32 days after the single dose of IL-1β (6 dpr). The disparities in IL-1β dosing and study designs may reconcile the different observations between previous studies and ours.
Figure 2.

Exogenous IL-1β exacerbates IL-33–mediated RSV immunopathogenesis during early life. (A) Lung sections stained with periodic acid–Schiff (PAS) to observe mucus (bright purple; indicated by black arrowheads). Images were taken at 40×. (B) Quantification of airway mucus in experimental mice. **P < 0.01. (C) Airway resistance in response to increasing doses of inhaled methacholine (MeCh; n = 4–6 per group). The peak airway resistance values in response to increasing doses of inhaled methacholine were measured and normalized as deviation from baseline. *P < 0.05 compared between two indicated groups at the 50 mg/ml dose of methacholine. Data are representative of at least two independent experiments (mean ± SEM). (D and E) Quantification of pulmonary ILC2 cells, including frequencies of pulmonary ILC2 in total pulmonary leukocyte population (D) and the absolute number of ILC2 cells in the lungs (E). *P < 0.05. Data are representative of at least two independent experiments (mean ± SEM). (F) Clusters of T-cytotoxic cell type I (Tc1), (Tc2), Th1, and Th2 from flow cytometry analysis, gated on total CD3+ T cells and visualized by t-distributed stochastic neighbor embedding (tSNE) and assigned specific colors (left panel), the abundance of these clusters in lungs of NRR + IL-1β mice (middle panel), and NRR + Veh mice (right panel). (G) The frequencies of Tc1, Tc2, Th1, and Th2 population in lungs for experimental mice. The intracellular cytokine production of T cells following stimulation with PMA + ionomycin were detected by flow cytometry. The dots on the left panel indicate the positive (dark gray dots) and negative (light gray dots) detection of corresponding cytokines. *P < 0.05 and **P < 0.01, compared between NRR + IL-1β and NRR + Veh group for each respective population (one-way ANOVA with Sidak multiple comparisons test). Data are representative of at least two independent experiments (mean ± SEM). (H and I) Th2/Th1 (H) and Tc2/Tc1 (I) ratio compared between NRR + IL-1β and NRR + Veh group. *P < 0.05 and **P < 0.01. Data are representative of at least two independent experiments (mean ± SEM). ILC2 = type 2 innate lymphoid cells; NRR = mice infected with RSV as neonates and reinfected 4 weeks later; NSS = mice sham infected as neonates and sham reinfected 4 weeks later.
RSV-induced immunopathogenesis has been associated with IL-33–mediated type 2–biased responses (15). IL-33–mediated type 2–biased immune responses are also responsible for other respiratory inflammatory diseases (33–35). Skewed type 2 responses are evidenced by increased accumulation of group 2 innate lymphoid cells (ILC2), Th2 (IL-4+IFN-γ−IL17A−CD4+), and Tc2 (IL-4+IFN-γ−IL17A−CD8+) (5, 15, 22, 33–35). To further investigate whether IL-1β mediated IL-33 exacerbation of type 2 skewed responses, we examined the pulmonary accumulation of ILC2 (Lin−RORγt−CD90.2+ST2+GATA3+CD127+) at 1 dpi and T cell subsets at 6 dpr (Figures E1B and E1C). Our previously published data (15) indicate that IL-33 expression is critical for ILC2 induction. Thus, our data cumulatively suggest that elevated concentrations of IL-1β–enhanced IL-33 expression is responsible for significantly greater frequencies of pulmonary ILC2 in NR + IL-1β group compared with vehicle-treated group (NR + Veh) (Figures 2D and 2E) at 1 dpi. Also, we observed that IL-1β–enhanced IL-33 production led to significantly greater frequencies of pulmonary Tc2 in NRR + IL-1β mice at 6 dpr (Figures 2F and 2G) compared with the NRR + Veh group. In sharp contrast to the Tc2 response observed in NRR + IL-1β mice, the frequencies of Th1 (IL-4−IFN-γ+IL17A−CD4+) and Tc1 (IL-4−IFN-γ+IL17A−CD8+) in lungs of NRR + IL-1β mice were significantly reduced at 6 dpr (Figures 2F and 2G) compared with the NRR + Veh) group. Significant alterations of lung-accumulated Th1, Tc1, and Tc2 frequencies result in the skewed ratio of Th2/Th1 and Tc2/Tc1 in the NRR + IL-1β compared with the NRR + Veh group (Figures 2H and 2I), strongly indicating type 2-biased responses. Although the increase of Th2 frequencies following IL-1β–mediated IL-33 augmentation is not statistically significant (Figure 2G), these collective data regarding ILC2, Th2/Th1, and Tc2/Tc1 ratio strongly suggest that exogenous IL-1β enhanced IL-33 production early after the initial infection (1 dpi) and exacerbated type 2-biased immunopathogenesis after RSV reinfection.
IL-1β Upregulates RSV-induced IL-33 Expression in a Caspase-Independent Manner
Next, we examined potential mechanisms by which exogenous IL-1β upregulates IL-33. In line with previous studies (36–38), we found that exogenous IL-1β significantly enhanced its own production following RSV infection in neonatal mice (Figure E2A). IL-1β can promote its positive feedback loop by upregulating NLRP3 inflammasome, resulting in caspase-1 activation (36). Indeed, using FAM-YVAD-FMK FLICA (BioVerge) (39, 40), which allowed us to assess caspase-1 activation, we found that exogenous IL-1β promotes the activation of caspase-1 in NR mice at 1 dpi (Figure 3A). Paradoxically, active caspase-1, -3, and -7 have been reported to significantly diminish IL-33 bioactivities (41). To assess the possibility that exogenous IL-1β–mediated caspase-1 activation dampens IL-33 bioactivities in our neonatal RSV model, we examined the cellular colocalization of active caspase-1 and IL-33 using IL-33(cit/wt) mice (42) (Figure E2B). We found that the majority of IL-33–producing cells in either the NR + Veh or NR + IL-1β groups are negative for active caspase-1 signal and vice versa (Figures 3B and 3C). Although 1% of pulmonary leukocytes (CD45+CD31−EpCAM−) express active caspase-1, less than 0.05% of the same population produce IL-33 (Figures 3D and 3E). Likewise, more than 14% of distal lung epithelial cells (CD45−CD31−EpCAM+) express IL-33, whereas only 0.05% of the same population harbored active caspase-1 (Figures 3F and 3G). The data demonstrate the distinct cellular origins of IL-33 and active caspase-1 in NR mice regardless of IL-1β pretreatment.
Figure 3.
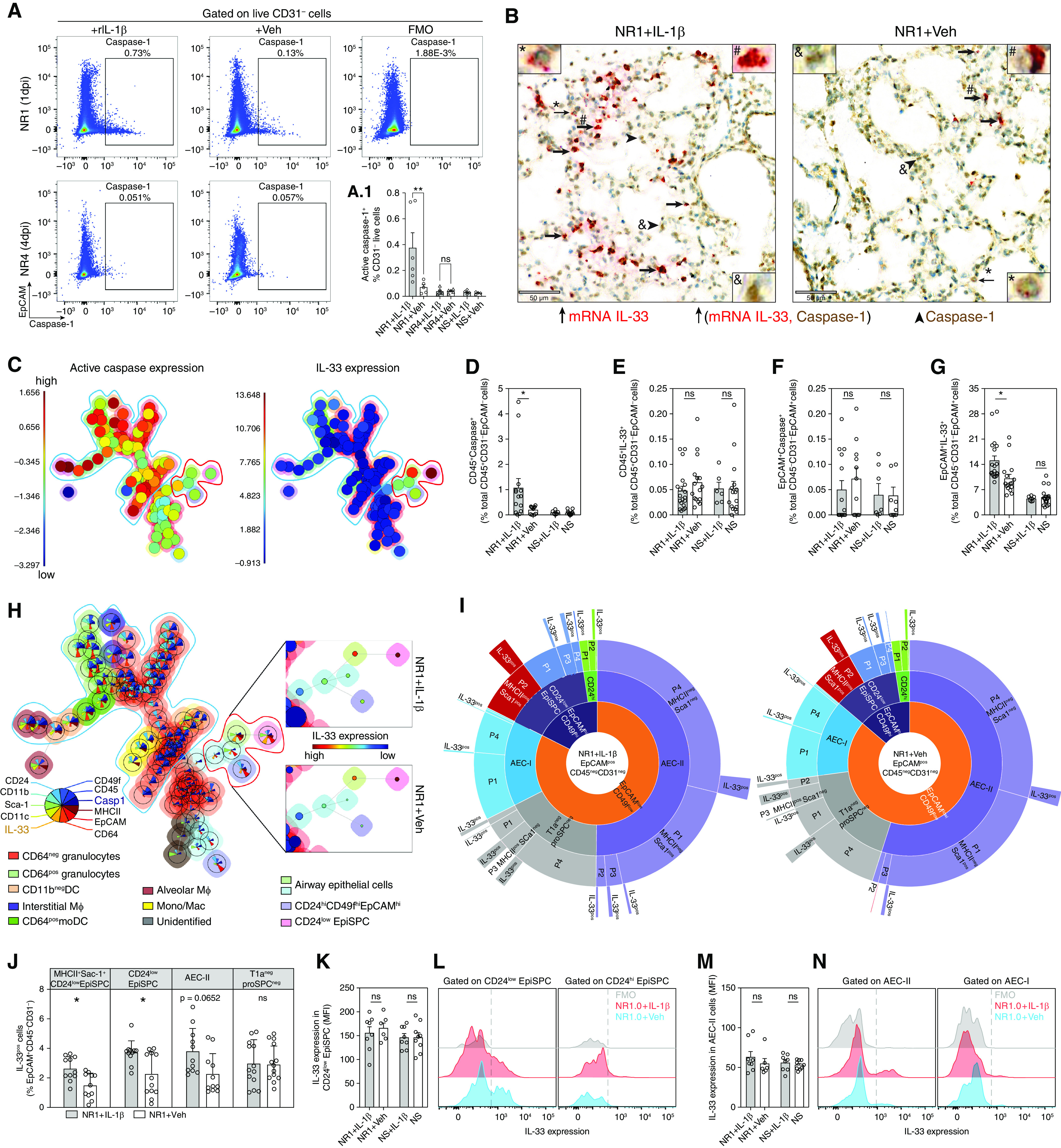
IL-1β enhances IL-33 production by promoting the expansion of IL-33+ epithelial stem/progenitor cells (EpiSPCs). (A) Representative flow cytometric dot plot of total live CD31− cells from lungs of RSV-infected neonatal mice collected at 1 dpi and 4 dpi, gated on epithelial cell adhesion molecule (EpCAM) and active caspase-1. Gates for active caspase-1–positive cells were set using Fluorescence Minus One (FMO) control (top, right panel). (A1) Bar graphs illustrate the frequencies of active caspase-1–positive cells. (B) Representative dual IL-33 mRNA ISH and caspase-1 immunohistochemical sections of lungs from 3 biologically independent RSV-infected neonatal mice. IL-33 mRNA (red) and caspase-1 protein (brown) expressing cells, and collocation of IL-33 mRNA and caspase-1 protein were indicated by thick arrows, arrowheads, and thin arrows with magnified inset (#, &, and *), respectively. Scale bar, 50 μm. (C) FlowSOM visualization of the flow cytometry analysis for the pulmonary cellularity of RSV-infected neonatal mice at 1 dpi, showing the levels of active caspase-1 (left panel) and IL-33 (right panel). (D–G) The frequencies of CD45+ caspase-1+ cells (D), CD45+ IL-33+ cells (E), EpCAM+caspase-1+ (F), and EpCAM+ IL-33+ (G) in lungs of mice with different treatments. Data are representative of n = 12–16, accumulated from at least two independent experiments (mean ± SEM). (H) FlowSOM visualization of the flow cytometry analysis for the pulmonary cellularity of RSV-infected neonatal mice at 1 dpi. The color of the nodes corresponds with the distinct subpopulation. The mean marker values are visualized for each node using star charts. Each part's height indicates the intensity: if the part reaches the border of the circle, the cells have a high expression for that marker (left panel). Expression degrees (median fluorescent intensity) of IL-33 on EpiSPC and airway epithelial cells (AECs) (right top and bottom panels). The color and size of inner cycles represent the intensity of IL-33 expression and the frequencies of that population, respectively. (I) Pie charts illustrate the frequencies of different fractions of EpCAM+CD45−CD31− cells regarding IL-33 expression. (J) Bar graphs illustrate the comparison between NR1 + IL-1β and NR1 + Veh groups regarding the frequencies of IL-33+ cells. Data represent n = 16–19 accumulated from two independent experiments (mean ± SEM). (K–N) Median fluorescent intensity (MFI) levels of IL-33 expression by EpiSPC (K and L) and AEC-II and I (M and N). *P < 0.05 and **P < 0.01; compared between indicated groups using one-way ANOVA with Sidak multiple comparisons test. Data represent two independent experiments (mean ± SEM). DC = dendritic cell.
The majority of active caspase-1 cells are CD64− granulocytes and myeloid subsets, including alveolar and interstitial macrophages, and CD64+ monocyte-derived dendritic cells (DC). In contrast, IL-33 was mainly expressed by CD45−EpCAM+ airway epithelial cells (Figure 3H). The gating strategies and Fluorescence Minus One (FMO) controls are illustrated in Figures E2C, E2D, and E3. In addition, we failed to detect active caspase-3 and -7, which cleave and inactivate IL-33 biological activity (43, 44), and only detected procaspase-3 and -7, regardless of the presence of IL-1β signaling (Figure E4A). Expression degrees of procaspase-3 and -7 also are independent of IL-1β signaling (Figure E4B). Collectively, these data strongly rule out the possibility that IL-1β induces caspase activation and could dampen IL-33 bioactivities in our model.
Exogenous IL-1β Upregulates RSV-induced IL-33 Expression by Promoting the Proliferation of IL-33+ Lung Epithelial Stem/Progenitor Cells
We further fractionated lung epithelial cells into different subsets to investigate the impact of exogenous IL-1β on IL-33 production of CD45−EpCAM+ airway epithelial cells. In line with previous studies (9, 42, 45, 46), our data revealed that IL-33–expressing populations are heterogeneous, including EpCAMint-lowCD49fint-lowPro-SpC+T1a- alveolar type 2 cells (AEC-II), EpCAMint-lowCD49fint-lowPro-SpC−T1a+ (AEC-I) and EpCAMint-lowCD49fint-lowPro-SpC−T1a− airway epithelial cells (T1a−proSPC− cells), EpCAMhiCD49fhiCD24low-int, and EpCAMhiCD49fhiCD24hi cells (Figures 3G and 3H). Among these populations, AEC-II and EpCAMhiCD49fhiCD24low-int are the primary cells expressing IL-33. The gating strategies are illustrated in Figure E4C. Like AEC-II, mouse lung distal EpCAMhiCD49fhiCD24low-int cells have been classified as epithelial stem/progenitor cells (EpiSPC), as evidenced by their high proliferative and renewal capacity (47–50). Subfractionation into MHC-II and stem cells antigen-1 (Sca-1) expression revealed that most IL-33+ AEC-II cells do not express MHC-II or Sca-1 (Figure 3I, P4 population). In sharp contrast, most IL-33+ EpiSPC cells are MHC-II+ and Sca-1+ (Figure 3I, P2 population), suggesting proliferative (51, 52) and antigen-presenting potential (53, 54). The data imply the functional differentiation between these subsets. Intriguingly, we observed that exogenous IL-1β induces a significant expansion of IL-33 expressing cells, including EpiSPC and AEC-II, rather than promoting IL-33 expression per cell (Figures 3J–3N). Indeed, the increase in IL-33–expressing EpiSPC in the NR1 + IL-1β group is almost double that of the NR1 + Veh group (Figure 3J). It is corroborated by the similar increase in mRNA and protein levels of IL-33 (i.e., almost double in NR1 + IL-1β mice) (Figures 1B and 1C).
By examining the cell cycle and the presence of proliferation marker Ki67 (Figures 4A and 4B), we found that most IL-33–expressing cells were in growth phase G1 (Figures 4C and 4D). NR1 + IL-1β pups exhibited significantly greater frequencies of G1 IL-33+EpCAM+ than NR1 + Veh pups did, whereas there was no difference between the two groups regarding the frequencies of G0 IL-33+EpCAM+ (Figures 4E and 4F). As expected, these G1 IL-33+EpCAM+ cells were mainly EpiSPC (33.2%) and AEC-II (62.8%) (Figures 4G and 4H). Consistent with the above data, G1 IL-33+ EpiSPC exhibit high MHC-II and Sca-1 expression degrees, which are significantly limited in the G1 IL-33+AEC-II subset (Figure 4G). The data suggest that IL-1β has mitogenic effects on IL-33+EpiSPC and AEC-II. It remains puzzling why EpCAMint-lowCD49fint-lowAEC-II cells, which express low amounts of Sca-1, therefore, presumably are less proliferative than EpiSPC, appeared to be the predominant subset within G1 IL-33+EpCAM+ population (Figure 4H). Of note, these G1 IL-33+AEC-II cells exhibit high levels of CD24 – a marker for differentiated epithelial cells (48) (Figure 4G). It has been shown that AEC-II cells arise from potent progenitors during early life development and lung injury-induced regeneration (55, 56). Thus, exogenous IL-1β may induce the differentiation of IL-33+ Sca-1+CD24low-int EpiSPC into the more differentiated IL-33+Sca-1−CD24hi AEC-II cells in our model. Although our current study has not been designed to investigate this possibility, the data strongly suggest that IL-1β induces the proliferation of IL-33–expressing EpiSPC and AECII (Figures 4G and 4H). Previous studies corroborate our findings by demonstrating the ability of IL-1β to stimulate the proliferation of different cell types, including glomerular epithelial cells (57), lung fibroblasts (58), gastric epithelial cells (59), epithelial progenitors (60), alveolar epithelial cells (61), and gallbladder cancer cells (62). The role of IL-1β inducing the differentiation of IL-33+CD24lowEpiSPC into IL-33+CD24hi AEC-II in our RSV neonatal model is beyond the scope of the current study. Still, it can be addressed in further lineage-tracing studies using IL-33 and Pro-SpC reporter mice (63).
Figure 4.
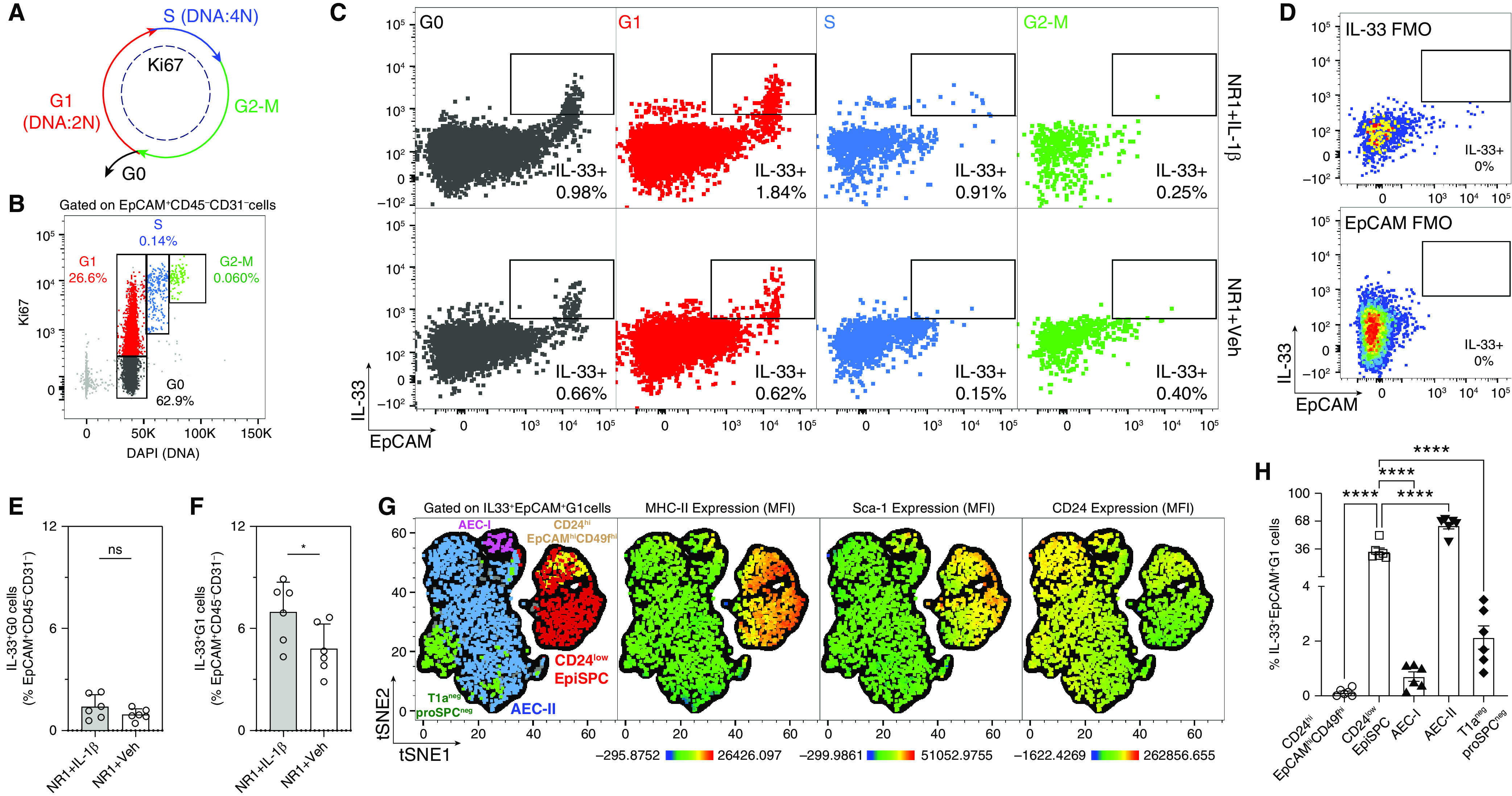
IL-1β induces the proliferation of IL-33+EpiSPC. (A–D) Quantification and characterization of lung epithelial cell proliferation in neonatal mice following RSV infection. (A) Diagram shows the markers of the different phases of the cell cycle; (B and C) representative flow cytometry dot plots showing different cell cycle phases, gated on CD31−CD45−live cells; (D) FMO IL-33 was used to set gatings for IL-33+ cells. (E and F) Frequencies of IL-33+ epithelial cells had undergone resting G0 phase and proliferation G1 phase. *P < 0.05. (G) tSNE clusters of EpiSPC, AEC-II, AEC-I, and T1a−proSPC− epithelial cells from flow cytometry analysis, gated on total IL-33+EpCAM+ cells underwent G1 phase (the first panel from the left). First, the IL-33+EpCAM+ G1 populations in each sample (12 samples: 6 IL-1β and 6 treated mice) were downsampled to the minimum number of IL-33+EpCAM+ G1 events among 12 samples (2,271 events). Second, 2,271 events of the IL-33+EpCAM+G1 population from each sample were concatenated (in total, 12 × 2271 = 27252 events of IL-33+EpCAM+G1 cells). The concatenated population was then subjected to tSNE analysis. The tSNE clusters were evaluated for MHC-II, Sca-1, and CD24 expression. (H) Frequencies of IL-33+EpCAM+ cells underwent G1 phase. ****P < 0.0001 compared between indicated groups using one-way ANOVA with Sidak multiple comparisons test. Data represent two independent experiments (mean ± SEM).
RSV-infected Adult Mice Showed No IL-1β–Dependent Increase of Pulmonary IL-33
It has been shown that RSV-induced IL-33 expression is age dependent (15). RSV infection induces a rapid and significantly greater increase in expression of pulmonary IL-33 in neonatal mice compared with adult mice (15). Confoundingly, compared with infected neonatal mice, RSV-infected adult mice produce significantly higher IL-1β concentrations (Figure E1A), presumably promoting greater production of IL-33 in adult mice. Having demonstrated that IL-1β induces the proliferation of IL-33+EpiSPC, we therefore hypothesized that IL-1β–mediated IL-33 expression is also age variable and associated with the abundance of IL-33+EpiSPC. To investigate the hypothesis, we repeated the same experiments with adult mice. Like neonatal mice, we administered IL-1β intranasally to 6- to 8-week-old mice 3 hours before RSV infection. The expression of pulmonary IL-33 was examined at 1 dpi. Unlike neonatal mice, RSV-infected adult mice showed no IL-1β–dependent increase of pulmonary IL-33 (Figure 5A) and significantly less than that of RSV-infected neonatal mice (Figure 1C). Also, we found that exogenous IL-1β did not alter the abundance of IL-33+EpCAM+, IL-33+CD45+, and IL-33+CD31+ cells (Figures 5B–5D) in RSV-infected adult mice. Collectively, our data demonstrate that IL-1β upregulates RSV-induced IL-33 expression in early life, whereas it failed to alter the IL-33 concentrations in later life. One potential explanation for this difference is that the abundance of EpiSPC capable of expressing IL-33 changes during lung development. The constriction in IL-33+ lung epithelial cells in adulthood has been demonstrated during alveolarization (Postnatal Day 7–14) in mice (64). Accordingly, IL33 has been shown to peak during postnatal alveolarization and gradually decline in adulthood (64). In agreement, our data indicate distinct CD24lowEpiSPC capable of producing IL-33 in neonatal mice (Day 6) but not adult mice (Figures 5E and 5F). We reasoned that despite the production of higher amounts of IL-1β compared with neonatal mice, adult mice have significant constriction in IL33-producing EpiSPC and therefore significantly lower levels of IL-33 in response to RSV. Taken together, we hypothesized that lung IL-33–expressing epithelial stem/progenitor cells differentiate into other subsets and lose their ability to produce IL-33 in adulthood. However, it is also important to remember that there are many immunological variables between these time points, including immaturity of the innate and adaptive immune responses and cellular phenotypes present that play roles in the observed differences in responses to IL-1β between neonatal and adult mice. These notions are beyond the scope of the current study but warrant further investigation.
Figure 5.
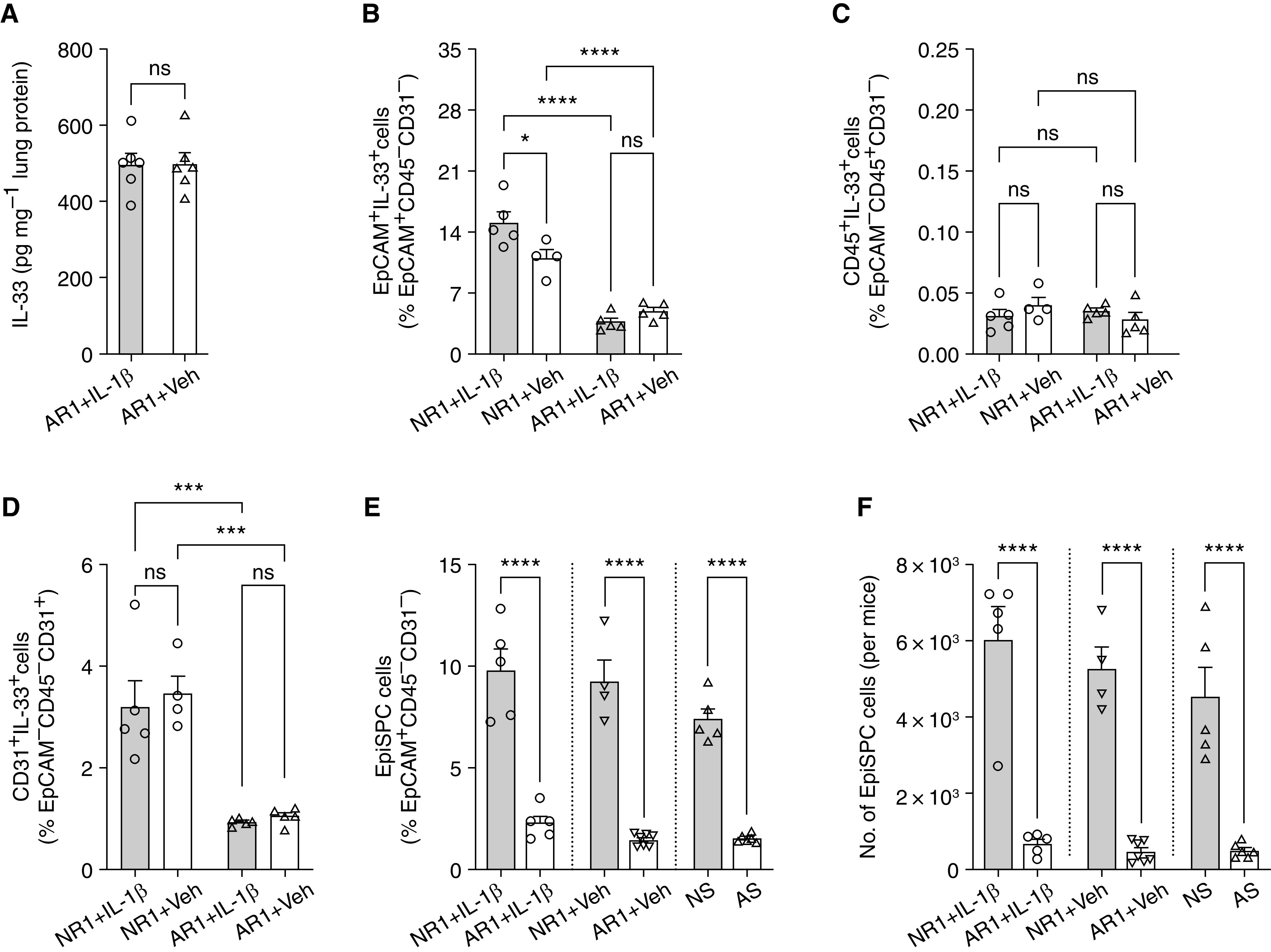
RSV-infected adult mice showed no IL-1β dependent increase of pulmonary IL-33. (A–D) IL-33 expression in RSV-infected adult mice with and without exogenous IL-1β: protein concentrations of IL-33 from whole-lung homogenates (A). The frequencies of IL-33–expressing cell subsets and also EpCAM+ cells (B), CD45+ cells (C), and CD31+ cells (D). *P < 0.05 and ***P < 0.001 compared between indicated groups using one-way ANOVA with Sidak multiple comparisons test. Data represent two independent experiments (mean ± SEM). Circles: neonatal; triangles: adult mice; open bars: vehicle; solid bars: IL-1β–treated groups. (E and F) Frequencies and number of EpiSPC in neonatal and adult mice with different treatments. ****P < 0.0001 compared between indicated groups using one-way ANOVA with Sidak multiple comparisons test. Data represent two independent experiments (mean ± SEM). Cycles: IL-1β; inverted triangles: vehicle; triangles: sham infection groups; solid bars: neonatal; open bars: adult mice. AR1 = adult mice infected with RSV, at 1 dp; AS = adult mice sham infected.
Conclusions
Although the critical role of IL-33 in RSV immunopathologies during early life has been established (5, 15), the regulatory mechanism of IL-33 expression is largely unknown. In recent years, IL-1β and IL-33 became the focus of many studies and are suggested as effective treatment targets (65–67). Therefore, understanding the interaction between IL-1β and IL-33 in mediating RSV-induced immunopathogenesis in early life is essential for successful therapeutics development. Findings from experimental asthma and helminth infection models indicate that IL-33 production occurs dependent on IL-1β signaling in adult mice; however, what is known is contradictory (16, 18). Although we have learned much from these studies, studies evaluating the regulatory role of IL-1β on IL-33 production in adult mice do not provide an accurate reflection of IL-33 production or regulation in neonatal mice and, by extension, human infants with RSV disease.
Our results clearly show that exogenous IL-1β enhances RSV-induced IL-33 expression in early but not later life.
In addition, IL-1β exacerbated IL-33–mediated type 2-biased RSV immunopathogenesis in early life. IL-1β enhances RSV-induced IL-33 expression by promoting the proliferation of IL-33+ EpiSPCs, which are significantly limited in adulthood. Our findings also provide a potential explanation for the age-variable IL-33 expression in lungs, which is largely based on the constriction of IL-33+ epithelial stem/progenitor cells in adulthood, and suggest the involvement of IL-33 in lung development during early life.
There are certain limitations to our study. The possibility that IL-1β upregulates IL-33 expression by promoting the differentiation of IL-33− EpiSPCs into IL-33+ AEC-II cells, which is beyond the scope of our current study, was not assessed. Such future studies are warranted and will strengthen our current findings. It also remains unclear whether IL-1β–mediated expansion of IL-33+ EpiSPCs during RSV infection in early life is permanent and, if so, predisposes neonates/human infants to the development of certain lung diseases (i.e., asthma). These notions are testable and warrant further investigation.
Acknowledgments
Acknowledgment
The authors thank Dr. Andrew McKenzie for producing and supplying them with the IL-33 citrine/citrine mice, and Dr. Clare S. Hardman for sharing the genotyping protocol for IL-33 citrine/citrine mice.
Footnotes
Supported by the National Institutes of Health (NIAID), grant number AI090059, to S.A.C.
Author Contributions: L.D.V., A.T.Q.P., D.R.H., D.T.S., and H.T. performed experiments, analyzed data, provided intellectual input, and contributed to manuscript preparation. L.D.V. and S.A.C conceptualized and directed the study, and drafted the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0313OC on December 3, 2021
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet . 2010;375:1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piedimonte G, Perez MK. Respiratory syncytial virus infection and bronchiolitis. Pediatr Rev . 2014;35:519–530. doi: 10.1542/pir.35-12-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stein RT, Bont LJ, Zar H, Polack FP, Park C, Claxton A, et al. Respiratory syncytial virus hospitalization and mortality: systematic review and meta-analysis. Pediatr Pulmonol . 2017;52:556–569. doi: 10.1002/ppul.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. García-García ML, Calvo C, Moreira A, Cañas JA, Pozo F, Sastre B, et al. Thymic stromal lymphopoietin, IL-33, and periostin in hospitalized infants with viral bronchiolitis. Medicine (Baltimore) . 2017;96:e6787. doi: 10.1097/MD.0000000000006787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vu LD, Siefker D, Jones TL, You D, Taylor R, DeVincenzo J, et al. Elevated levels of type 2 respiratory innate lymphoid cells in human infants with severe respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med . 2019;200:1414–1423. doi: 10.1164/rccm.201812-2366OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X, Hammel M, He Y, Tainer JA, Jeng US, Zhang L, et al. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA . 2013;110:14918–14923. doi: 10.1073/pnas.1308651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Travers J, Rochman M, Miracle CE, Habel JE, Brusilovsky M, Caldwell JM, et al. Chromatin regulates IL-33 release and extracellular cytokine activity. Nat Commun . 2018;9:3244. doi: 10.1038/s41467-018-05485-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cayrol C, Girard J-P. Interleukin-33 (IL-33): a nuclear cytokine from the IL-1 family. Immunol Rev . 2018;281:154–168. doi: 10.1111/imr.12619. [DOI] [PubMed] [Google Scholar]
- 9. Andrews C, McLean MH, Durum SK. Cytokine tuning of intestinal epithelial function. Front Immunol . 2018;9:1270. doi: 10.3389/fimmu.2018.01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Halát G, Haider T, Dedeyan M, Heinz T, Hajdu S, Negrin LL. IL-33 and its increased serum levels as an alarmin for imminent pulmonary complications in polytraumatized patients. World J Emerg Surg . 2019;14:36. doi: 10.1186/s13017-019-0256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uchida M, Anderson EL, Squillace DL, Patil N, Maniak PJ, Iijima K, et al. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy . 2017;72:1521–1531. doi: 10.1111/all.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kouzaki H, Iijima K, Kobayashi T, O’Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol . 2011;186:4375–4387. doi: 10.4049/jimmunol.1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity . 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 14. Günther S, Deredge D, Bowers AL, Luchini A, Bonsor DA, Beadenkopf R, et al. IL-1 family cytokines use distinct molecular mechanisms to signal through their shared co-receptor. Immunity . 2017;47:510–523.e4. doi: 10.1016/j.immuni.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saravia J, You D, Shrestha B, Jaligama S, Siefker D, Lee GI, et al. Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog . 2015;11:e1005217. doi: 10.1371/journal.ppat.1005217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zaiss MM, Maslowski KM, Mosconi I, Guenat N, Marsland BJ, Harris NL. IL-1β suppresses innate IL-25 and IL-33 production and maintains helminth chronicity. PLoS Pathog . 2013;9:e1003531. doi: 10.1371/journal.ppat.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Küchler AM, Pollheimer J, Balogh J, Sponheim J, Manley L, Sorensen DR, et al. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflammatory activation. Am J Pathol . 2008;173:1229–1242. doi: 10.2353/ajpath.2008.080014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mahmutovic Persson I, Menzel M, Ramu S, Cerps S, Akbarshahi H, Uller L. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res . 2018;19:16. doi: 10.1186/s12931-018-0725-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nomura K, Kojima T, Fuchimoto J, Obata K, Keira T, Himi T, et al. Regulation of interleukin-33 and thymic stromal lymphopoietin in human nasal fibroblasts by proinflammatory cytokines. Laryngoscope . 2012;122:1185–1192. doi: 10.1002/lary.23261. [DOI] [PubMed] [Google Scholar]
- 20. You D, Siefker DT, Shrestha B, Saravia J, Cormier SA. Building a better neonatal mouse model to understand infant respiratory syncytial virus disease. Respir Res . 2015;16:91. doi: 10.1186/s12931-015-0244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen K, Eddens T, Trevejo-Nunez G, Way EE, Elsegeiny W, Ricks DM, et al. IL-17 receptor signaling in the lung epithelium is required for mucosal chemokine gradients and pulmonary host defense against K. pneumoniae. Cell Host Microbe . 2016;20:596–605. doi: 10.1016/j.chom.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siefker DT, Vu L, You D, McBride A, Taylor R, Jones TL, et al. Respiratory syncytial virus disease severity is associated with distinct CD8+ T-cell profiles. Am J Respir Crit Care Med . 2020;201:325–334. doi: 10.1164/rccm.201903-0588OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wennergren G, Kristjánsson S. Relationship between respiratory syncytial virus bronchiolitis and future obstructive airway diseases. Eur Respir J . 2001;18:1044–1058. doi: 10.1183/09031936.01.00254101. [DOI] [PubMed] [Google Scholar]
- 24. Jafri HS, Chavez-Bueno S, Mejias A, Gomez AM, Rios AM, Nassi SS, et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis . 2004;189:1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- 25. Pickles RJ, DeVincenzo JP. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J Pathol . 2015;235:266–276. doi: 10.1002/path.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harker JA, Yamaguchi Y, Culley FJ, Tregoning JS, Openshaw PJ. Delayed sequelae of neonatal respiratory syncytial virus infection are dependent on cells of the innate immune system. J Virol . 2014;88:604–611. doi: 10.1128/JVI.02620-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tregoning JS, Yamaguchi Y, Harker J, Wang B, Openshaw PJ. The role of T cells in the enhancement of respiratory syncytial virus infection severity during adult reinfection of neonatally sensitized mice. J Virol . 2008;82:4115–4124. doi: 10.1128/JVI.02313-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Graham BS, Bunton LA, Wright PF, Karzon DT. Reinfection of mice with respiratory syncytial virus. J Med Virol . 1991;34:7–13. doi: 10.1002/jmv.1890340103. [DOI] [PubMed] [Google Scholar]
- 29. Graham BS, Bunton LA, Wright PF, Karzon DT. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J Clin Invest . 1991;88:1026–1033. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song KS, Yoon JH, Kim KS, Ahn DW. c-Ets1 inhibits the interaction of NF-κB and CREB, and downregulates IL-1β-induced MUC5AC overproduction during airway inflammation. Mucosal Immunol . 2012;5:207–215. doi: 10.1038/mi.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim YD, Jeon JY, Woo HJ, Lee JC, Chung JH, Song SY, et al. Interleukin-1beta induces MUC2 gene expression and mucin secretion via activation of PKC-MEK/ERK, and PI3K in human airway epithelial cells. J Korean Med Sci . 2002;17:765–771. doi: 10.3346/jkms.2002.17.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y, Garvin LM, Nickola TJ, Watson AM, Colberg-Poley AM, Rose MC. IL-1β induction of MUC5AC gene expression is mediated by CREB and NF-κB and repressed by dexamethasone. Am J Physiol Lung Cell Mol Physiol . 2014;306:L797–L807. doi: 10.1152/ajplung.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med . 2014;190:1373–1382. doi: 10.1164/rccm.201406-1039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sjöberg LC, Nilsson AZ, Lei Y, Gregory JA, Adner M, Nilsson GP. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep . 2017;7:4219. doi: 10.1038/s41598-017-03674-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Khaitov MR, Gaisina AR, Shilovskiy IP, Smirnov VV, Ramenskaia GV, Nikonova AA, et al. The role of interleukin-33 in pathogenesis of bronchial asthma. New experimental data. Biochemistry (Mosc) . 2018;83:13–25. doi: 10.1134/S0006297918010029. [DOI] [PubMed] [Google Scholar]
- 36. Chen F, Jiang G, Liu H, Li Z, Pei Y, Wang H, et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone Res . 2020;8:10. doi: 10.1038/s41413-020-0087-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jimbo K, Park JS, Yokosuka K, Sato K, Nagata K. Positive feedback loop of interleukin-1beta upregulating production of inflammatory mediators in human intervertebral disc cells in vitro. J Neurosurg Spine . 2005;2:589–595. doi: 10.3171/spi.2005.2.5.0589. [DOI] [PubMed] [Google Scholar]
- 38. Streicher KL, Willmarth NE, Garcia J, Boerner JL, Dewey TG, Ethier SP. Activation of a nuclear factor kappaB/interleukin-1 positive feedback loop by amphiregulin in human breast cancer cells. Mol Cancer Res . 2007;5:847–861. doi: 10.1158/1541-7786.MCR-06-0427. [DOI] [PubMed] [Google Scholar]
- 39. Darzynkiewicz Z, Pozarowski P, Lee BW, Johnson GL. Fluorochrome-labeled inhibitors of caspases: convenient in vitro and in vivo markers of apoptotic cells for cytometric analysis. Methods Mol Biol . 2011;682:103–114. doi: 10.1007/978-1-60327-409-8_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagar A, DeMarco RA, Harton JA. Inflammasome and caspase-1 activity characterization and evaluation: an imaging flow cytometer-based detection and assessment of inflammasome specks and caspase-1 activation. J Immunol . 2019;202:1003–1015. doi: 10.4049/jimmunol.1800973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol . 2015;7:351–365. doi: 10.1093/jmcb/mjv012. [DOI] [PubMed] [Google Scholar]
- 42. Hardman CS, Panova V, McKenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol . 2013;43:488–498. doi: 10.1002/eji.201242863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity . 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 44. Ali S, Nguyen DQ, Falk W, Martin MU. Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem Biophys Res Commun . 2010;391:1512–1516. doi: 10.1016/j.bbrc.2009.12.107. [DOI] [PubMed] [Google Scholar]
- 45. Le Goffic R, Arshad MI, Rauch M, L’Helgoualc’h A, Delmas B, Piquet-Pellorce C, et al. Infection with influenza virus induces IL-33 in murine lungs. Am J Respir Cell Mol Biol . 2011;45:1125–1132. doi: 10.1165/rcmb.2010-0516OC. [DOI] [PubMed] [Google Scholar]
- 46. Heyen L, Müller U, Siegemund S, Schulze B, Protschka M, Alber G, et al. Lung epithelium is the major source of IL-33 and is regulated by IL-33-dependent and IL-33-independent mechanisms in pulmonary cryptococcosis. Pathog Dis . 2016;74:ftw086. doi: 10.1093/femspd/ftw086. [DOI] [PubMed] [Google Scholar]
- 47. Quantius J, Schmoldt C, Vazquez-Armendariz AI, Becker C, El Agha E, Wilhelm J, et al. Influenza virus infects epithelial stem/progenitor cells of the distal lung: impact on Fgfr2b-driven epithelial repair. PLoS Pathog . 2016;12:e1005544. doi: 10.1371/journal.ppat.1005544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci USA . 2010;107:1414–1419. doi: 10.1073/pnas.0909207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest . 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li X, Rossen N, Sinn PL, Hornick AL, Steines BR, Karp PH, et al. Integrin α6β4 identifies human distal lung epithelial progenitor cells with potential as a cell-based therapy for cystic fibrosis lung disease. PLoS One . 2013;8:e83624. doi: 10.1371/journal.pone.0083624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghosh M, Helm KM, Smith RW, Giordanengo MS, Li B, Shen H, et al. A single cell functions as a tissue-specific stem cell and the in vitro niche-forming cell. Am J Respir Cell Mol Biol . 2011;45:459–469. doi: 10.1165/rcmb.2010-0314OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hegab AE, Kubo H, Fujino N, Suzuki T, He M, Kato H, et al. Isolation and characterization of murine multipotent lung stem cells. Stem Cells Dev . 2010;19:523–536. doi: 10.1089/scd.2009.0287. [DOI] [PubMed] [Google Scholar]
- 53. Gereke M, Jung S, Buer J, Bruder D. Alveolar type II epithelial cells present antigen to CD4(+) T cells and induce Foxp3(+) regulatory T cells. Am J Respir Crit Care Med . 2009;179:344–355. doi: 10.1164/rccm.200804-592OC. [DOI] [PubMed] [Google Scholar]
- 54. Wosen JE, Mukhopadhyay D, Macaubas C, Mellins ED. Epithelial MHC class II expression and its role in antigen presentation in the gastrointestinal and respiratory tracts. Front Immunol . 2018;9:2144. doi: 10.3389/fimmu.2018.02144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature . 2014;507:190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yee M, Buczynski BW, O’Reilly MA. Neonatal hyperoxia stimulates the expansion of alveolar epithelial type II cells. Am J Respir Cell Mol Biol . 2014;50:757–766. doi: 10.1165/rcmb.2013-0207OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tateyama F, Yamabe H, Osawa H, Kaizuka M, Shirato K, Okumura K. Interleukin-1β is an autocrine growth factor of rat glomerular epithelial cells in culture. Nephrol Dial Transplant . 2001;16:1149–1155. doi: 10.1093/ndt/16.6.1149. [DOI] [PubMed] [Google Scholar]
- 58. White KE, Ding Q, Moore BB, Peters-Golden M, Ware LB, Matthay MA, et al. Prostaglandin E2 mediates IL-1β-related fibroblast mitogenic effects in acute lung injury through differential utilization of prostanoid receptors. J Immunol . 2008;180:637–646. doi: 10.4049/jimmunol.180.1.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beales ILP. Effect of interlukin-1β on proliferation of gastric epithelial cells in culture. BMC Gastroenterol . 2002;2:7. doi: 10.1186/1471-230X-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jerde TJ, Bushman W. IL-1 induces IGF-dependent epithelial proliferation in prostate development and reactive hyperplasia. Sci Signal . 2009;2:ra49. doi: 10.1126/scisignal.2000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aumiller V, Balsara N, Wilhelm J, Günther A, Königshoff M. WNT/β-catenin signaling induces IL-1β expression by alveolar epithelial cells in pulmonary fibrosis. Am J Respir Cell Mol Biol . 2013;49:96–104. doi: 10.1165/rcmb.2012-0524OC. [DOI] [PubMed] [Google Scholar]
- 62. Guo R, Qin Y, Shi P, Xie J, Chou M, Chen Y. IL-1β promotes proliferation and migration of gallbladder cancer cells via Twist activation. Oncol Lett . 2016;12:4749–4755. doi: 10.3892/ol.2016.5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Salwig I, Spitznagel B, Vazquez-Armendariz AI, Khalooghi K, Guenther S, Herold S, et al. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J . 2019;38:e102099. doi: 10.15252/embj.2019102099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, et al. First-breath-induced type 2 pathways shape the lung immune environment. Cell Rep . 2017;18:1893–1905. doi: 10.1016/j.celrep.2017.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest . 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chan BCL, Lam CWK, Tam L-S, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol . 2019;10:364. doi: 10.3389/fimmu.2019.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Garon EB, Chih-Hsin Yang J, Dubinett SM. The role of interleukin 1β in the pathogenesis of lung cancer. JTO Clin Res Rep . 2020;1:100001. doi: 10.1016/j.jtocrr.2020.100001. [DOI] [PMC free article] [PubMed] [Google Scholar]