

Summary
Background
Polycythaemia vera is a myeloproliferative neoplasm characterised by excessive proliferation of erythroid, myeloid, and megakaryocytic components in the bone marrow due to mutations in the Janus kinase 2 (JAK2) gene. Ruxolitinib, a JAK 1 and JAK 2 inhibitor, showed superiority over best available therapy in a phase 2 study in patients with polycythaemia vera who were resistant to or intolerant of hydroxyurea. We aimed to compare the long-term safety and efficacy of ruxolitinib with best available therapy in patients with polycythaemia vera who were resistant to or intolerant of hydroxyurea.
Methods
We report the 5-year results for a randomised, open-label, phase 3 study (RESPONSE) that enrolled patients at 109 sites across North America, South America, Europe, and the Asia-Pacific region. Patients (18 years or older) with polycythaemia vera who were resistant to or intolerant of hydroxyurea were randomly assigned 1:1 to receive either ruxolitinib or best available therapy. Patients randomly assigned to the ruxolitinib group received the drug orally at a starting dose of 10 mg twice a day. Single-agent best available therapy comprised hydroxyurea, interferon or pegylated interferon, pipobroman, anagrelide, approved immunomodulators, or observation without pharmacological treatment. The primary endpoint, composite response (patients who achieved both haematocrit control without phlebotomy and 35% or more reduction from baseline in spleen volume) at 32 weeeks was previously reported. Patients receiving best available therapy could cross over to ruxolitinib after week 32. We assessed the durability of primary composite response, complete haematological remission, overall clinicohaematological response, overall survival, patient-reported outcomes, and safety after 5-years of follow-up. This study is registered with ClinicalTrials.gov, NCT01243944.
Findings
We enrolled patients between Oct 27, 2010, and Feb 13, 2013, and the study concluded on Feb 9, 2018. Of 342 individuals screened for eligibility, 222 patients were randomly assigned to receive ruxolitinib (n=110, 50%) or best available therapy (n=112, 50%). The median time since polycythaemia vera diagnosis was 8·2 years (IQR 3·9–12·3) in the ruxolitinib group and 9·3 years (4·9–13·8) in the best available therapy group. 98 (88%) of 112 patients initially randomly assigned to best available therapy crossed over to receive ruxolitinib and no patient remained on best available therapy after 80 weeks of study. Among 25 primary responders in the ruxolitinib group, six had progressed at the time of final analysis. At 5 years, the probability of maintaining primary composite response was 74% (95% CI 51–88). The probability of maintaining complete haematological remission was 55% (95% CI 32–73) and the probability of maintaining overall clinicohaematological responses was 67% (54–77). In the intention-to-treat analysis not accounting for crossover, the probability of survival at 5 years was 91·9% (84·4–95·9) with ruxolitinib therapy and 91·0% (82·8–95·4) with best available therapy. Anaemia was the most common adverse event in patients receiving ruxolitinib (rates per 100 patient-years of exposure were 8·9 for ruxolitinib and 8·8 for the crossover population), though most anaemia events were mild to moderate in severity (grade 1 or 2 anaemia rates per 100 patient-years of exposure were 8·0 for ruxolitinib and 8·2 for the crossover population). Non-haematological adverse events were generally lower with long-term ruxolitinib treatment than with best available therapy. Thromboembolic events were lower in the ruxolitinib group than the best available therapy group. There were two on-treatment deaths in the ruxolitinib group. One of these deaths was due to gastric adenocarcinoma, which was assessed by the investigator as related to ruxolitinib treatment.
Interpretation
We showed that ruxolitinib is a safe and effective long-term treatment option for patients with polycythaemia vera who are resistant to or intolerant of hydroxyurea. Taken together, ruxolitinib treatment offers the first widely approved therapeutic alternative for this post-hydroxyurea patient population.
Introduction
Polycythaemia vera is a clonal myeloproliferative neoplasm that arises because of mutations in the Janus kinase 2 (JAK2) gene, and is primarily characterised by an elevation in the red blood cell mass.1 A rise in white blood cell and platelet counts is seen in approximately 40% of patients with polycythaemia vera.2 Splenomegaly is a common feature of advanced disease.3 Patients with polycythaemia vera have a substantial symptom burden,3 increased risk of thromboembolic events,4 and shortened survival.5 Hence, the main goals of therapy are to ease the symptom burden, reduce the risk of thromboembolic events, and minimise the transformation to myelofibrosis or acute myeloid leukaemia.1 In patients at high risk, either hydroxyurea or interferon alfa is the recommended therapy.6,7 Approximately 25% of patients at high risk given the first-line therapy (hydroxyurea or interferon) become resistant to or intolerant of treatment.8–10 In addition, many patients have persisting polycythaemia vera associated symptoms despite being given standard therapies.11
Ruxolitinib, a JAK1 and JAK2 inhibitor, has shown efficacy in treating patients with polycythaemia vera who were resistant to or intolerant of hydroxyurea in the large, randomised, phase 3 RESPONSE study (Randomised Study of Efficacy and Safety in Polycythemia Vera with JAK Inhibitor INCB018424 versus Best Supportive Care).12 In the primary analysis of RESPONSE, ruxolitinib was superior to best available therapy in providing haematocrit control along with a 35% or more reduction in spleen volume from baseline at week 32 (primary endpoint 22·7% vs 0·9% of patients in the ruxolitinib vs best available therapy group; p<0·001) in patients with polycythaemia vera who were inadequately controlled with hydroxyurea.12 These results supported the United States Food and Drug Administration and the European Medicines Agency approvals of ruxolitinib for the treatment of polycythaemia vera in patients who are resistant to or intolerant of hydroxyurea.12–14
The long-term follow-up in RESPONSE showed that ruxolitinib provided durable haematocrit control, spleen volume reduction, complete haematological remission, and clinicohaematological response in these patients with an acceptable safety profile.15,16 The analyses from RESPONSE-2 (a phase 3 study in patients with polycythaemia vera who were resistant to or intolerant of hydroxyurea and had a non-palpable spleen) further confirmed the benefits of ruxolitinib in patients with polycythaemia vera who were inadequately controlled with hydroxyurea.17,18 The European LeukemiaNet and National Comprehensive Cancer Network guidelines now include ruxolitinib as the recommended treatment for patients who are resistant to or intolerant of hydroxyurea.7,19
Here, we present the long-term efficacy and safety results from a planned analysis after all patients completed 256 weeks (approximately 5 years) of treatment or had discontinued from the RESPONSE study.
Methods
Study design and participants
We did an international, multicentre, randomised, open-label, phase 3 study (RESPONSE) comparing the safety and efficacy of ruxolitinib with best available therapy in patients with polycythaemia vera (appendix p 7). Patients were enrolled at 109 sites across North America, South America, Europe, and the Asia-Pacific region (appendix p 15–18). The methods of this study have been published previously.12 The study population comprised patients (18 years and older) with polycythaemia vera who were resistant to or intolerant of hydroxyurea as per modified European LeukemiaNet criteria; have required phlebotomy at least two times in the 24 weeks before screening and at least one time in the 16 weeks before screening, with the most distant and the most recent phlebotomy at least 4 weeks apart, or the most recent phlebotomy within the 16 weeks before screening and a haematocrit more than 45% at screening.12 Patients were excluded if they had received prior JAK-inhibitor therapy, PEG-IFN-α-2a within 5 weeks of screening, or ₃₂P therapy, and patients who were pregnant, lactating, or had inadequate liver or renal function. An inadequate response to hydroxyurea was defined as a dose of 2 g or more per day or a maximum tolerated dose of less than 2 g per day resulting in at least one of the following: need for phlebotomy to maintain haematocrit at less than 45%; platelet count more than 400 × 109 cells per L and white blood cell count more than 10 × 109 cells per L; and failure to reduce splenomegaly extending more than 10 cm below the costal margin by more than 50%. The unacceptable side-effects from hydroxyurea were defined as at least one of the following: absolute neutrophil count less than 1·0 × 109 cells per L; platelet count less than 100 × 109 cells per L, or haemoglobin less than 100 g/L at the lowest dose of hydroxyurea required for a response; presence of leg ulcers or other unacceptable hydroxyurea-related non-haematological toxicities. The resistance to and intolerance of hydroxyurea were planned as the stratification factors during randomisation and no formal analysis was planned to compare the two subgroups.
The study was approved by the central ethics committee or institutional review board at each participating institution and was done in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Randomisation and masking
Patients were stratified by hydroxyurea resistance or hydroxyurea intolerance as categorised at the screening visit, and randomly assigned in a 1:1 ratio, to receive ruxolitinib or single-agent best available therapy at the physician’s discretion. The treating physician made a clinical judgment while deciding the single agent therapy for the patient as best available therapy. The trial was open label and neither investigators nor participants were masked to study treatment.
Procedures
Patients randomly assigned to the ruxolitinib group received the drug orally at a starting dose of 10 mg twice a day. Dose adjustments for safety and efficacy reasons were allowed in patients receiving ruxolitinib. A standardised dosing regimen was used to determine dose adjustments for safety and efficacy so that each participant was titrated to their most appropriate dose. Single-agent best available therapy comprised hydroxyurea, interferon or pegylated interferon, pipobroman, anagrelide, approved immunomodulators, or observation without pharmacological treatment. All randomly assigned patients received a low dose of aspirin (75–150 mg per day) unless medically contraindicated.
Patients assigned to best available therapy were permitted to cross over to ruxolitinib from week 32 if they did not meet the primary endpoint or after week 32 in cases of phlebotomy eligibility or splenomegaly progression, or both. At week 80, patients receiving best available therapy who did not cross over discontinued the study. The data from the pre-planned analyses (week 80 and week 208) have been published previously.15,16 Adverse events were assessed according to the National Cancer Institute Common Toxicity Criteria for Adverse Events version 3.0. The safety results were summarised for the patients randomly assigned to ruxolitinib and separately for all patients after crossover from best available therapy to ruxolitinib. For patients randomly assigned to best available therapy, safety results were summarised for the duration of randomised treatment until crossover.
Outcomes
The composite primary endpoint was the proportion of patients achieving both (1) haematocrit control without phlebotomy (defined as no phlebotomy eligibility between weeks 8 and 32 with ≤1 phlebotomy eligibility from randomisation to week 8; phlebotomy eligibility was defined as haematocrit >45% and ≥3 percentage points higher than baseline or >48%, whichever was lower) and (2) 35% or more reduction from baseline in spleen volume (as measured by MRI or CT scan) at week 32. The proportion of patients who reached complete haematological remission (defined as haematocrit control, platelet count ≤400 × 109 cells per L, and white blood cell count ≤10 × 109 cells per L) was a key secondary endpoint. Overall clinicohaematological response was the additional secondary endpoint and was defined by spleen volume reduction of 35% or more by imaging (MRI or CT), platelet count ≤400 × 109 cells per L, and white blood cell count ≤10 × 109 cells per L, or absence of phlebotomy eligibility, or both.
Since all patients randomly assigned to best available therapy either crossed over to ruxolitinib or discontinued the treatment by week 80, this analysis evaluated the durability of efficacy in the ruxolitinib group only, including durability of the primary response, primary response components (haematocrit control and spleen volume reduction), complete haematological remission, and overall clinicohaematological response. Overall survival, an exploratory endpoint, was defined as the time from the date of randomisation to the date of death from any cause. The longer-term efficacy assessments also included the changes in JAK2 Val617Phe allele burden from baseline and patient-reported outcomes (European Organization for the Research and Treatment of Cancer quality of life questionnaire functional scores and Pruritus Symptom Impact Scale).
Statistical analysis
The methods of the statistical analysis have been published previously.12 Briefly, the primary and secondary endpoints were analysed based on the principle of intention-to-treat. For efficacy analyses, all patients that were randomly assigned to receive ruxolitinib were combined as one group regardless of their titrated dose, and all patients that were randomly assigned to receive best available therapy were combined as one group regardless of their initial or subsequent therapy. Safety was assessed in all patients who received at least one dose of assigned treatment. Assessments of change and percentage change from baseline included all patients with baseline measurements; changes in individual symptom scores included only patients with baseline values greater than 0. Patients with missing assessments were not included in the analyses. Duration of primary response was defined as the time from the first occurrence when both components of the primary endpoint were met to the date of the first documented disease progression. Kaplan-Meier estimates of duration of primary response along with 95% CIs were presented for the responders in the ruxolitinib group only as prespecified in the protocol. Kaplan-Meier estimates of duration of complete haematological remission and overall clinicohaematological responses along with 95% CIs were also presented. For the overall survival analysis, hazard ratios (HRs) and 95% CIs were calculated from stratified Cox proportional hazards using the Wald test. The Kaplan-Meier estimates of median overall survival along with 95% CIs were calculated by treatment. Since this study was not designed to address specific biomarker-related hypotheses, the analysis of these data was viewed as exploratory and hypotheses generating.
Role of the funding source
The study was designed by academic investigators and representatives of the funder. All authors had full access to the data for interpretation and analysis, were involved in development and approval of the report, and had the final responsibility for the decision to submit for publication. All authors vouch for the accuracy and completeness of the reported data, and attest that the study conformed to the protocol and statistical analysis plan.
Results
We enrolled patients between Oct 27, 2010 (first patient first visit), and Feb 13, 2013, and the study concluded on Feb 9, 2018 (last patient last visit). Of 342 individuals screened for eligibility, 222 patients were randomly assigned to receive either ruxolitinib (n=110, 50%) or best available therapy (n=112, 50%). The baseline characteristics of patients and the primary results of the study have been reported previously12 and were mostly balanced between the treatment groups (appendix p 2). At baseline, patients randomly assigned to ruxolitinib were reported to have longer previous exposure to hydroxyurea compared with best available therapy (median 162·9 weeks, IQR 52·9–382·0 vs 145·6 weeks, 42·9–365·4) and higher frequency of previous non-melanoma skin cancer or precancerous skin conditions (11% vs 6%). The median time since polycythaemia vera diagnosis was 8·2 years (IQR 3·9–12·3) in the ruxolitinib group and 9·3 years (4·9–13·8) in the best available therapy group.
The median dose intensity was 22·5 mg per day (IQR 18·7–28·7) in patients who were randomly assigned to ruxolitinib, in which 68 (62%) of 110 patients required a dose reduction or interruption and 88 (80%) of 110 patients required a dose increase at some point of time during the study. After the primary analysis at week 32, 98 (88%) of 112 patients initially randomly assigned to best available therapy crossed over to receive ruxolitinib and no patient remained on best available therapy after 80 weeks of study. In the crossover population, the median dose intensity of ruxolitinib was 19·8 mg per day (IQR 15·4–27·8). 66 (67%) of 98 crossover patients required reduction or interruption and 68 (69%) required increase in dose at some point of time during the study.
The median time to crossover from best available therapy to ruxolitinib was 34·7 weeks (95% CI 33·9–35·3). At study completion, 72 (66%) of 110 patients in the ruxolitinib group and 64 (65%) of 98 patients who crossed over from the best available therapy group to receive ruxolitinib completed 5 years of on-study treatment (figure 1; appendix p 3). The main reasons for premature discontinuation before completion of 5 years in the ruxolitinib group (median exposure 255 weeks, IQR 158–256) and crossover group (220 weeks, 135–223) were adverse events (16 [15%] of 110 and 16 [16%] of 98; regardless of study drug relationship), disease progression (12 [11%] of 110 and 9 [9%] of 98), and patient decision (6 [6%] of 110 and 6 [6%] of 98). The lack of efficacy (100 [89%] of 112) primarily led to the treatment discontinuations in the best available therapy group (median exposure 34 weeks, IQR 32–36).
Figure 1: Trial profile.
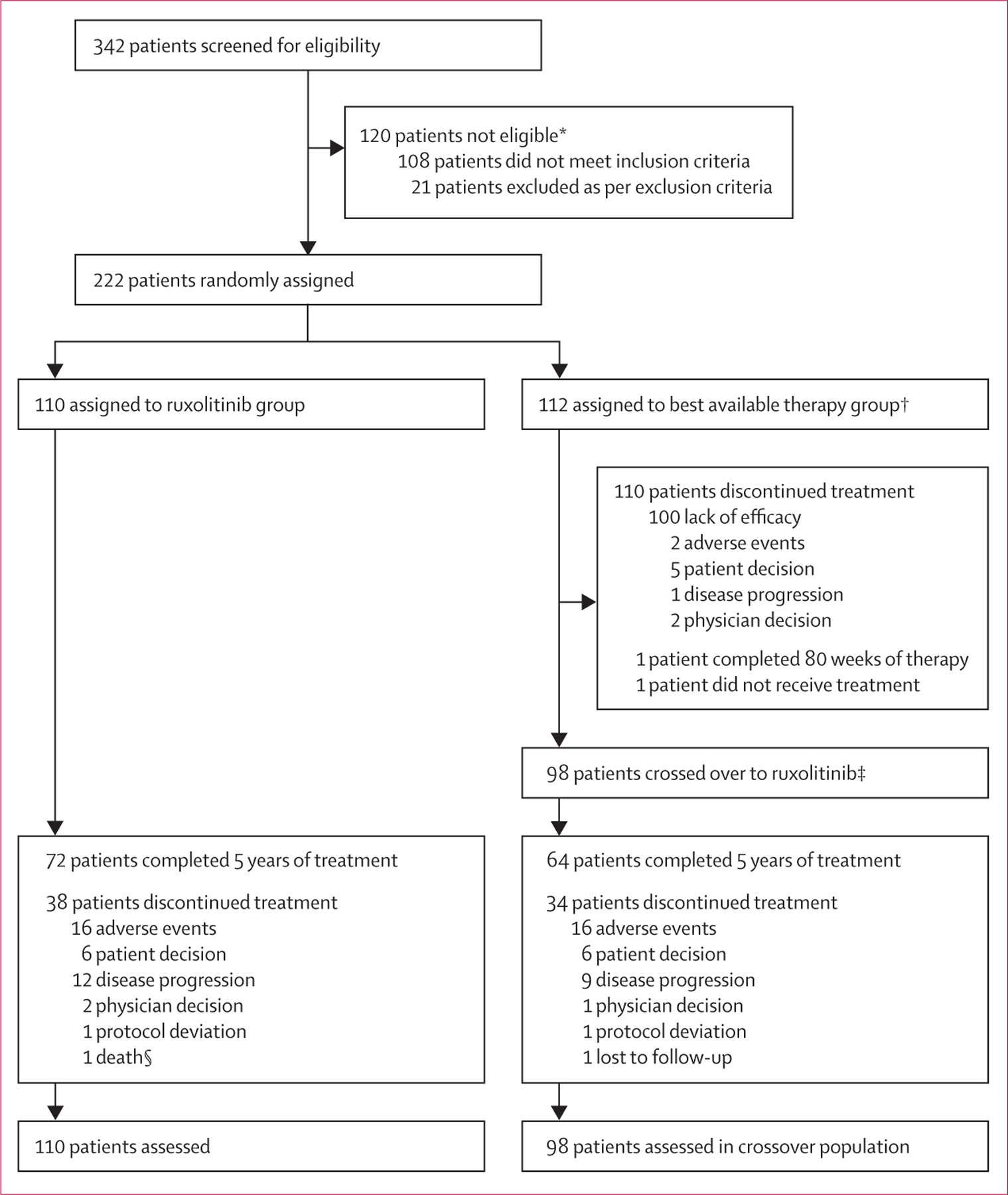
*Reasons for ineligibility are described in detail in the appendix (p 1); patients might have more than one reason for ineligibility. †One patient was randomly assigned to best available therapy but did not receive study treatment. Initial best available therapy comprised hydroxyurea (n=66), interferon or pegylated interferon (n=13), anagrelide (n=8), immunomodulators (n=5), pipobroman (n=2), and observation (n=17). ‡98 of 112 patients were eligible for cross over to ruxolitinib. §One patient in the ruxolitinib group, determined by the investigator to have discontinued the study treatment because of adverse events, died afterwards.
As reported previously,12 the primary endpoint was reached in 25 (23%) patients randomly assigned to ruxolitinib and one patient (1%) receiving best available therapy at week 32 (p<0·0001). In the primary analysis, 66 (60%) patients randomly assigned to ruxolitinib showed haematocrit control compared with 21 (19%) patients randomly assigned to best available therapy, whereas 44 (40%) patients given ruxolitinib showed a spleen response compared with one (1%) patient given best available therapy. At the time of data cutoff, six (24%) of 25 primary responders had progressed (progression criteria were phlebotomy eligibility, progression of splenomegaly, or both). Duration of maintaining primary response at 224 weeks (starting from week 32) was 74% (95% CI 51–88). The median duration of primary response was not reached at the time of study completion (figure 2A).
Figure 2: Response in patients treated with ruxolitinib.
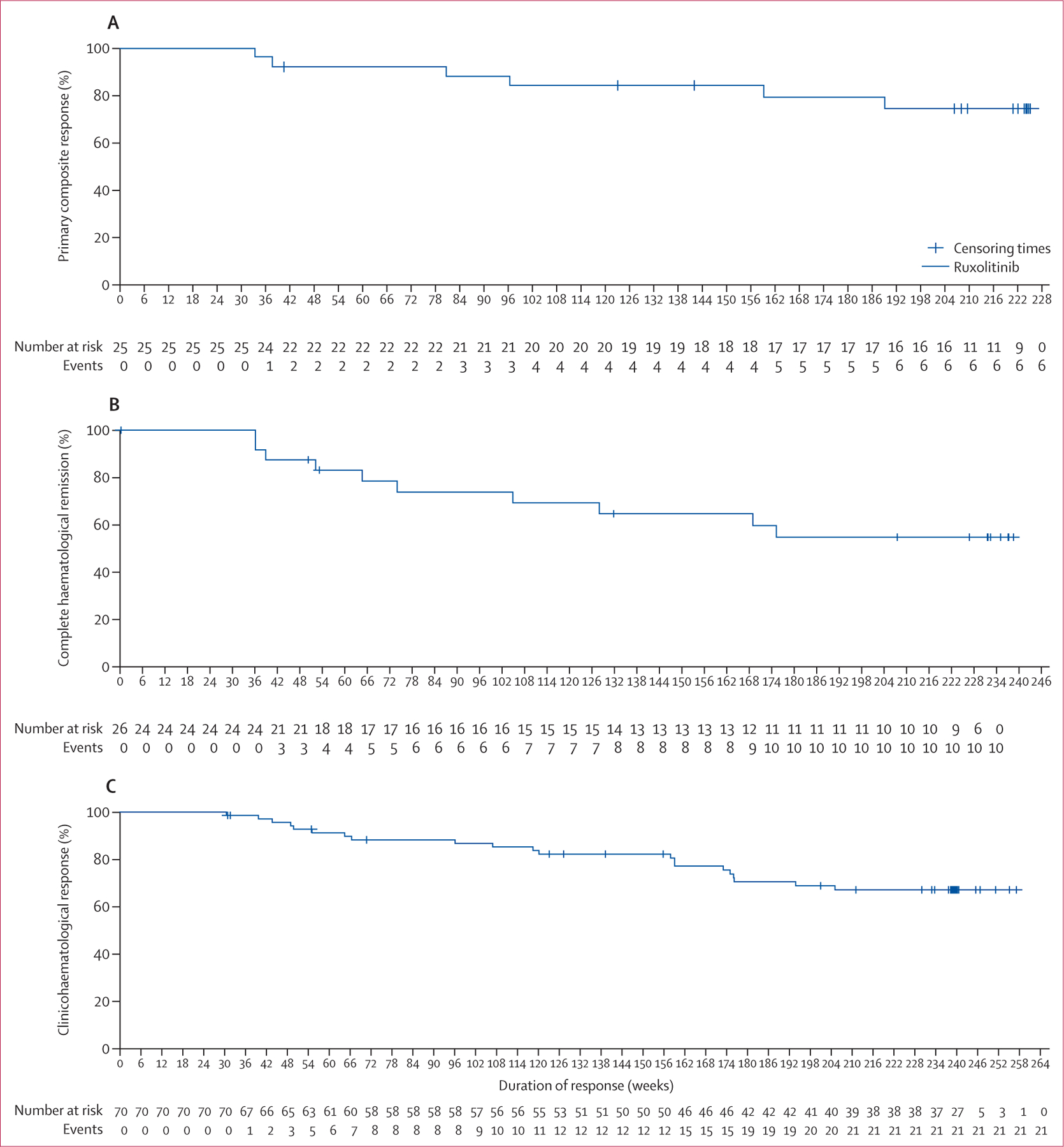
(A) Primary response (patients who achieved both haematocrit control without phlebotomy and 35% or more reduction from baseline in spleen volume). There were 25 responders, six events, and 19 censored. (B) Complete haematological remission. There were 26 responders, ten events, and 16 censored. (C) Overall clinicohaematological response. There were 70 responders, 21 events, and 49 censored. Crosses indicate censored patients.
Duration of complete haematological remission (haematocrit control, platelet count ≤400 × 109 cells per L, and white blood cell count ≤10 × 109 cells per L) at 224 weeks (starting from week 32) was 55% (95% CI 32–73). Of 26 (24%) patients who had complete haematological remission at week 32, ten (38%) progressed by week 256 (figure 2B). Of the 66 (60%) patients who had haematocrit control at week 32, 16 (24%) had progressed by week 256, and duration of haematocrit control at 224 weeks (starting from week 32) was 73% (95% CI 60–83; appendix p 8). In the ruxolitinib group, 78 (83%) of 94 patients (94 patients were evaluable after week 80 up until the week 256 visit) required no phlebotomies and only six (6%) of 94 patients needed three or more after week 80 up until the week 256 visit. Similarly, 69 (87%) of 79 patients (79 patients were evaluable after week 80 up until the week 256 visit) who crossed over to ruxolitinib from best available therapy remained phlebotomy free, with only six (8%) of 79 patients needing three or more phlebotomies at week 224 of crossover (appendix p 9). 63 (64%) of 98 patients who crossed over to ruxolitinib had haematocrit control after 32 weeks. Overall, there were fewer phlebotomies required in patients who were either randomly assigned to ruxolitinib or crossed over to ruxolitinib as compared with best available therapy. Of the 87 patients with a white blood cell count more than 10 × 109 cells per L at baseline, 36 (41%) had a white blood cell count of 10 × 109 cells per L less than at week 256. 25 (46%) of 54 patients with platelet counts greater than 400 × 109 cells per L at baseline reduced platelet counts to less than 400 × 109 cells per L by week 256.
Among the 70 (64%) patients who had an overall clinicohaematological response at week 32, 21 (30%) had progressed by week 256. The probability of maintaining clinicohaematological response at 224 weeks (starting from week 32) was 67% (95% CI 54–77), and the median duration of clinicohaematological response was not reached (figure 2C). The probability of maintaining at least a 35% reduction in the spleen volume at week 224 (starting from week 32) was 72% (34–91; appendix p 10). In the ruxolitinib group (98 [89%]) and crossover population (84 [86%]), most patients showed a decrease in spleen volume at some point of time during the study, whereas only 55 (49%) of the patients given best available therapy showed a reduction in spleen volume (appendix p 11).
Overall, there were ten (9%) deaths in the ruxolitinib group and nine (8%) deaths in the best available therapy group during the study or in the survival follow-up phase (appendix p 4). In the intention-to-treat analysis, not accounting for crossover, the Kaplan-Meier estimates for overall survival at 5 years were 91·9% (95% CI 84·4–95·9) in the ruxolitinib group and 91·0% (82·8–95·4) in the best available therapy group (HR 0·95, 95% CI 0·38–2·41, figure 3). The survival follow-up was only applicable to those patients who completed or discontinued study treatment before week 256 or continued until the time when the individual week 256 visit from randomisation would have been reached. The majority of the patients were discontinued from the study at or before their individual week 256 visit and were censored, therefore there is a substantial drop in the number of patients at risk after week 256. The estimates beyond week 256 were highly variable, and do not suggest any significant differences between the ruxolitinib and best available therapy groups. There were 110 patients, ten events, and 100 censored for ruxolitinib and 112 patients, nine events, and 103 censored for best available therapy. For week 256, there were 77 patients at risk and eight events for ruxolitinib and 71 patients at risk and eight events for best available therapy. There were 98 patients on best available therapy who crossed over to ruxolitinib.
Figure 3: Overall survival by intention-to-treat analysis.
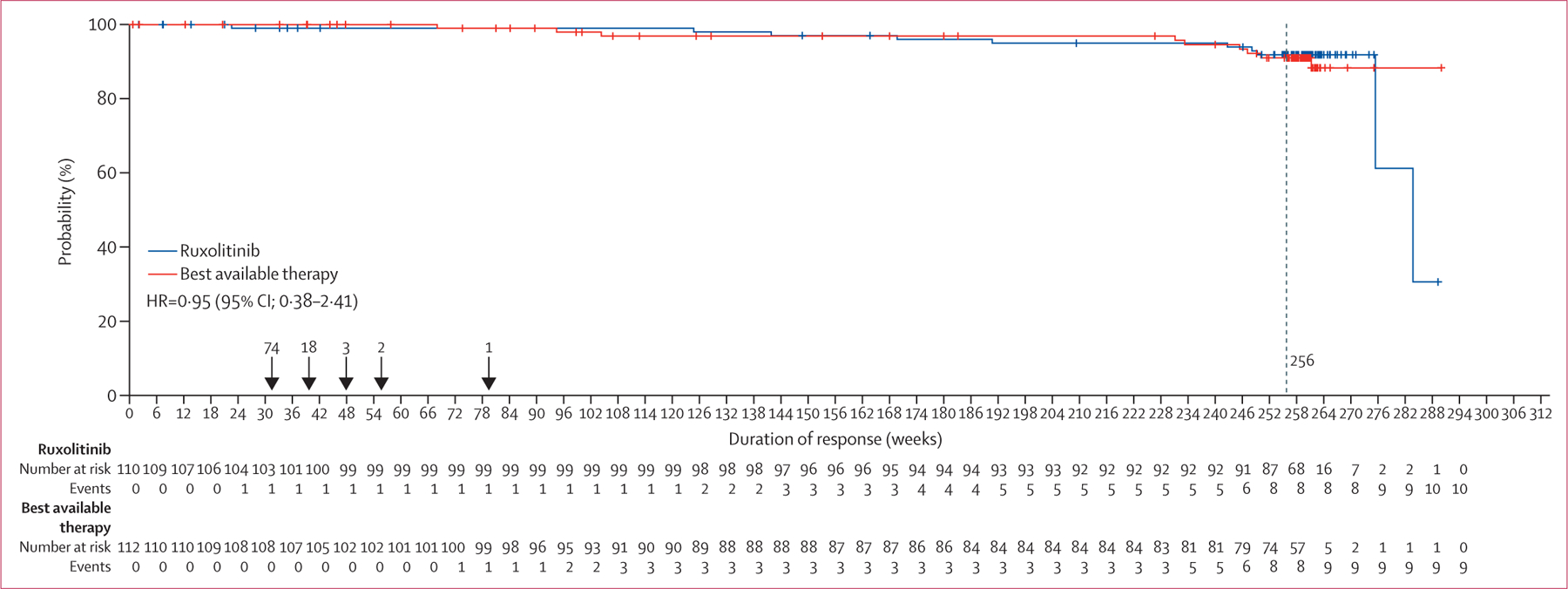
HR=hazard ratio. Crosses indicate censored patients.
At baseline, 104 (95%) patients were JAK2 Val617Phe positive with a mean allele burden of 76%. Over the course of treatment, the mean JAK2 Val617Phe allele burden decreased consistently in patients who were given ruxolitinib (appendix p 12). At the time of study completion (week 256), the mean percentage change from baseline in allele burden (negative value indicates improvement) was −38% (SD 38·64, n=66) in the ruxolitinib group. The patients who crossed over from best available therapy to ruxolitinib showed a reduction of −23% (SD 40·5, n=64). In the best available therapy group at week 32, the mean percentage change from baseline in the JAK2 Val617Phe allele burden was 1·18 (SD 25·33, n=80). Likewise, the improvements in the measures of quality of life seen at week 32 were maintained by the end of the study in patients originally randomly assigned to ruxolitinib (appendix pp 13–14). As assessed by the Pruritus Symptom Impact Scale, 42 (40%) patients given ruxolitinib maintained improvement (ie, very much improved and much improved responses) of pruritus up until week 256 (appendix p 13). The improvements in scores on the European Organization for the Research and Treatment of Cancer quality of life questionnaire global health status–quality of life scale were also sustained in some patients by week 256 (appendix p 14).
Because patients given best available therapy crossed over to ruxolitinib (median crossover time 34·7 weeks, 95% CI 33·9–35·3), it is important to consider the safety findings in the context of difference in exposure duration between the ruxolitinib and best available therapy groups. There was no relevant increase in the exposure-adjusted rates of adverse events with longer exposure compared with previous reports, and there were no new or unexpected adverse events. Being consistent with the previous reports and given the mechanism of action of ruxolitinib, anaemia was the most common adverse event in patients receiving ruxolitinib (including those who received ruxolitinib after crossover, table 1). Of note, most anaemia events were mild to moderate in severity, and four patients in both the ruxolitinib group and crossover population showed grade 3 or 4 new or worsening of haemoglobin from baseline. The exposure-adjusted rates for thrombocytopenia were higher in the best available therapy group (16·3 per 100 patient-years) than the ruxolitinib group (4·4 per 100 patient-years) or the crossover patients (1·2 per 100 patient-years).
Table 1:
Exposure-adjusted rates (per 100 patient-years) of common adverse events
| Ruxolitinib rate (n=110)* |
Best available therapy rate (n=111)† |
Crossover rate (n=98)‡ |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| All grades | Grade 1–2 | Grade 3–4 | All grades | Grade 1–2 | Grade 3–4 | All grades | Grade 1–2 | Grade 3–4 | |
| Haematological adverse events | |||||||||
| Anaemia | 8·9 | 8·0 | 0·9 | 5·4 | 5·4 | 0·0 | 8·8 | 8·2 | 0·6 |
| Thrombocytopenia | 4·4 | 3·2 | 1·2 | 16·3 | 13·6 | 2·7 | 1·2 | 0·9 | 0·3 |
| Non-haematological adverse events | |||||||||
| Pruritus | 7·0 | 6·5 | 0·5 | 32·6 | 27·2 | 5·4 | 6·1 | 6·1 | 0·0 |
| Diarrhoea | 7·0 | 6·8 | 0·2 | 12·2 | 10·8 | 1·4 | 3·6 | 3·6 | 0·0 |
| Increased weight | 6·1 | 5·4 | 0·7 | 1·4 | 1·4 | 0·0 | 4·2 | 3·6 | 0·6 |
| Headache | 5·8 | 5·3 | 0·5 | 28·5 | 27·1 | 1·4 | 5·2 | 5·2 | 0·0 |
| Arthralgia | 5·6 | 5·4 | 0·2 | 10·9 | 9·5 | 1·4 | 3·3 | 3·0 | 0·3 |
| Fatigue | 5·1 | 4·9 | 0·2 | 23·1 | 19·0 | 4·1 | 3·9 | 3·9 | 0·0 |
| Muscle spasms | 5·1 | 4·9 | 0·2 | 9·5 | 9·5 | 0·0 | 3·3 | 3·3 | 0·0 |
| Pyrexia | 4·0 | 3·8 | 0·2 | 6·8 | 6·8 | 0·0 | 3·3 | 3·0 | 0·3 |
| Dizziness | 4·0 | 4·0 | 0·0 | 15·0 | 15·0 | 0·0 | 6·1 | 6·1 | 0·0 |
| Back pain | 4·0 | 3·8 | 0·2 | 6·8 | 6·8 | 0·0 | 5·5 | 5·2 | 0·3 |
| Hypertension | 4·0 | 3·5 | 0·5 | 5·4 | 4·0 | 1·4 | 4·5 | 3·6 | 0·9 |
| Abdominal pain | 3·7 | 3·2 | 0·5 | 17·7 | 17·7 | 0·0 | 3·0 | 2·7 | 0·3 |
| Nausea | 3·5 | 3·3 | 0·2 | 5·4 | 5·4 | 0·0 | 2·1 | 2·1 | 0·0 |
| Night sweats | 3·0 | 3·0 | 0·0 | 12·2 | 12·2 | 0·0 | 1·8 | 1·8 | 0·0 |
| Pain in extremity | 2·3 | 2·1 | 0·2 | 5·4 | 5·4 | 0·0 | 3·3 | 3·3 | 0·0 |
| Decreased appetite | 2·1 | 1·9 | 0·2 | 8·2 | 8·2 | 0·0 | 1·5 | 1·5 | 0·0 |
| Musculoskeletal pain | 1·9 | 1·7 | 0·2 | 5·4 | 5·4 | 0·0 | 1·8 | 1·8 | 0·0 |
| Myalgia | 1·6 | 1·6 | 0·0 | 10·9 | 10·9 | 0·0 | 1·2 | 1·2 | 0·0 |
| Paraesthesia | 1·6 | 1·6 | 0·0 | 9·5 | 9·5 | 0·0 | 2·4 | 2·1 | 0·3 |
| Vertigo | 1·6 | 1·6 | 0·0 | 5·4 | 5·4 | 0·0 | 1·2 | 1·2 | 0·0 |
| Abdominal distension | 1·4 | 1·2 | 0·2 | 5·4 | 5·4 | 0·0 | 0·3 | 0·3 | 0·0 |
| Vomiting | 1·4 | 1·4 | 0·0 | 5·4 | 5·4 | 0·0 | 2·4 | 2·1 | 0·3 |
| Peripheral neuropathy | 1·4 | 1·4 | 0·0 | 6·8 | 5·4 | 1·4 | 0·6 | 0·6 | 0·0 |
| Bone pain | 0·9 | 0·9 | 0·0 | 8·2 | 6·8 | 1·4 | 1·2 | 0·9 | 0·3 |
| Hyperuricaemia | 0·7 | 0·5 | 0·2 | 6·8 | 4·1 | 2·7 | 0·9 | 0·9 | 0·0 |
| Gout | 0·2 | 0·2 | 0·0 | 6·8 | 4·1 | 2·7 | 0·3 | 0·3 | 0·0 |
| All infections | 18·9 | 15·4 | 3·5 | 59·8 | 55·7 | 4·1 | 19·1 | 13·0 | 6·1 |
| Herpes zoster infection | 4·7 | 4·2 | 0·5 | 0·0 | 0·0 | 0·0 | 3·9 | 3·3 | 0·6 |
| Nasopharyngitis | 4·4 | 4·4 | 0·0 | 12·2 | 12·2 | 0·0 | 4·2 | 4·2 | 0·0 |
| Bronchitis | 3·3 | 3·3 | 0·0 | 6·8 | 6·8 | 0·0 | 3·9 | 3·6 | 0·3 |
| Upper respiratory tract infection | 2·3 | 2·3 | 0·0 | 6·8 | 6·8 | 0·0 | 2·4 | 2·4 | 0·0 |
| Cellulitis | 0·2 | 0·0 | 0·2 | 5·4 | 4·0 | 1·4 | 0·6 | 0·0 | 0·6 |
Data are exposure adjusted rates. Adverse events occurring at a rate of ≥5 per 100 patient-years of exposure in any group, regardless of relationship to study drug. Adjusted rates were calculated as the number of patients with events per 100 patient-year of exposure.
Exposure=428 4 patient-years.
Exposure=73 6 patient-years.
Exposure=329 9 patient-years.
The rates of non-haematological adverse events were generally lower with the longer-term ruxolitinib treatment than those in the best available therapy group (table 1). The most common non-haematological adverse events (exposure-adjusted rate ≥5 per 100 patient-years) in the ruxolitinib group and crossover population, respectively, were pruritus (7·0 and 6·1), diarrhoea (7·0 and 3·6), increased weight (6·1 and 4·2), headache (5·8 and 5·2), arthralgia (5·6 and 3·3), fatigue (5·1 and 3·9), and muscle spasms (5·1 and 3·3). The rates of infections were generally lower in patients given ruxolitinib (per 100 patient-years of exposure 18·9 in the ruxolitinib group and 19·1 in crossover population) than those in the best available therapy group (59·8 per 100 patient-years), except herpes zoster infection, which was more common in the patients given ruxolitinib (table 1). The overall rates of serious adverse events per 100 patient-years of exposure were 10·3 in the ruxolitinib group versus 13·6 in the best available therapy group versus 13·0 in the crossover population (table 2). The most frequent adverse events leading to dose adjustment or interruption of ruxolitinib occurring in 3% or more of patients were anaemia, thrombocytopenia, and pruritus. The exposure-adjusted rates (per 100 patient-years) of thromboembolic events were lower in patients given ruxolitinib (1·2) and the crossover population (2·7) than patients given best available therapy (8·2). Thromboembolic events are presented in table 3.
Table 2:
Exposure-adjusted rates (per 100 patient-years) of serious adverse events
| Ruxolitinib rate (n=110)* |
Best available therapy rate (n=111)† |
Crossover rate (n=98)‡ |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| All grades | Grade 1 or 2 | Grade 3 or 4 | All grades | Grade 1 or 2 | Grade 3 or 4 | All grades | Grade 1 or 2 | Grade 3 or 4 | |
| Pneumonia | 1·2 | 0·0 | 1·2 | 1·4 | 0·0 | 1·4 | 1·8 | 0·0 | 1·8 |
| Squamous cell carcinoma | 0·9 | 0·0 | 0·9 | 0·0 | 0·0 | 0·0 | 0·3 | 0·0 | 0·3 |
| Atrial fibrillation | 0·7 | 0·0 | 0·7 | 1·4 | 0·0 | 1·4 | 0·3 | 0·3 | 0·0 |
| Basal cell carcinoma | 0·7 | 0·2 | 0·5 | 0·0 | 0·0 | 0·0 | 0·3 | 0·0 | 0·3 |
| Rectal haemorrhage | 0·5 | 0·0 | 0·5 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 |
| Chest pain | 0·5 | 0·3 | 0·2 | 0·0 | 0·0 | 0·0 | 0·3 | 0·0 | 0·3 |
| Metastatic squamous cell carcinoma | 0·5 | 0·0 | 0·5 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 |
| Squamous cell carcinoma of skin | 0·5 | 0·0 | 0·5 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 |
| Dehydration | 0·5 | 0·3 | 0·2 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 |
| Cellulitis | 0·2 | 0·0 | 0·2 | 1·4 | 0·0 | 1·4 | 0·6 | 0·0 | 0·6 |
| Herpes zoster | 0·2 | 0·0 | 0·2 | 0·0 | 0·0 | 0·0 | 0·6 | 0·3 | 0·3 |
| Urinary tract infection | 0·2 | 0·0 | 0·2 | 0·0 | 0·0 | 0·0 | 0·6 | 0·0 | 0·6 |
| Diverticulitis | 0·2 | 0·0 | 0·2 | 1·4 | 0·0 | 1·4 | 0·3 | 0·0 | 0·3 |
| Malignant melanoma | 0·2 | 0·0 | 0·2 | 1·4 | 0·0 | 1·4 | 0·0 | 0·0 | 0·0 |
| Prostate cancer | 0·2 | 0·0 | 0·2 | 0·0 | 0·0 | 0·0 | 0·6 | 0·0 | 0·6 |
| Subdural hematoma | 0·0 | 0·0 | 0·0 | 1·4 | 1·4 | 0·0 | 0·0 | 0·0 | 0·0 |
| Gout | 0·0 | 0·0 | 0·0 | 1·4 | 0·0 | 1·4 | 0·0 | 0·0 | 0·0 |
| Pulmonary embolism | 0·0 | 0·0 | 0·0 | 1·4 | 0·0 | 1·4 | 0·0 | 0·0 | 0·0 |
| Deep vein thrombosis | 0·0 | 0·0 | 0·0 | 1·4 | 0·0 | 1·4 | 0·0 | 0·0 | 0·0 |
| Bladder disorder | 0·0 | 0·0 | 0·0 | 1·4 | 1·4 | 0·0 | 0·0 | 0·0 | 0·0 |
| Abdominal Pain | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·6 | 0·3 | 0·3 |
| Dyspnoea | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·6 | 0·6 | 0·0 |
| Epistaxis | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·6 | 0·0 | 0·6 |
| Acute myocardial infarction | 0·0 | 0·0 | 0·0 | 1·4 | 0·0 | 1·4 | 0·0 | 0·0 | 0·0 |
| Transient ischaemic attack | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·6 | 0·0 | 0·6 |
| Varicella zoster virus infection | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·0 | 0·6 | 0·0 | 0·6 |
| Gastroenteritis | 0·0 | 0·0 | 0·0 | 1·4 | 1·4 | 0·0 | 0·0 | 0·0 | 0·0 |
Data are exposure adjusted rates. Adverse events occurring at a rate of ≥0 5 per 100 patient-years of exposure in any group, regardless of relationship to study drug. Adjusted rates were calculated as the number of patients with events per 100 patient-years of exposure.
Exposure=428 4 patient-years.
Exposure=73 6 patient-years.
Exposure=329 9 patient-years.
Table 3:
Exposure-adjusted rates (per 100 patient-year) of thromboembolic events
| Ruxolitinib rate (n=110)* |
Best available therapy rate (n=111)† |
Crossover rate (n=98)‡ |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| All grades | Grade 1 or 2 | Grade 3 or 4 | All grades | Grade 1 or 2 | Grade 3 or 4 | All grades | Grade 1 or 2 | Grade 3 or 4 | |
| All thromboembolic events | 5 (1·2) | 2 (0·5) | 3 (0·7) | 6 (8·2) | 4 (5·5) | 2 (2·7) | 9 (2·7) | 4 (1·2) | 5 (1·5) |
| Cerebral infarction | 1 (0·2) | 0 | 1 (0·2) | 0 | 0 | 0 | 0 | 0 | 0 |
| Ischaemic stroke | 1 (0·2) | 1 (0·2) | 0 | 0 | 0 | 0 | 1 (0·3) | 0 | 1 (0·3) |
| Transient ischaemic attack | 0 | 0 | 0 | 0 | 0 | 0 | 2 (0·6) | 0 | 2 (0·6) |
| Portal vein thrombosis | 1 (0·2) | 0 | 1 (0·2) | 0 | 0 | 0 | 0 | 0 | 0 |
| Pulmonary embolism | 1 (0·2) | 0 | 1 (0·2) | 1 (1·4) | 0 | 1 (1·4) | 0 | 0 | 0 |
| Retinal vascular thrombosis | 1 (0·2) | 1 (0·2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Myocardial infarction | 0 | 0 | 0 | 0 | 0 | 0 | 2 (0·6) | 1 (0·3) | 1 (0·3) |
| Acute myocardial infarction | 0 | 0 | 0 | 1 (1·4) | 0 | 1 (1·4) | 0 | 0 | 0 |
| Deep vein thrombosis | 0 | 0 | 0 | 2 (2·7) | 1 (1·3) | 1 (1·4) | 1 (0·3) | 1 (0·3) | 0 |
| Thrombophlebitis | 0 | 0 | 0 | 1 (1·4) | 1 (1·4) | 0 | 1 (0·3) | 1 (0·3) | 0 |
| Thrombosis | 0 | 0 | 0 | 1 (1·4) | 1 (1·4) | 0 | 1 (0·3) | 1 (0·3) | 0 |
| Bone infarction | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0·3) | 1 (0·3) | 0 |
| Coronary artery occlusion | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0·3) | 1 (0·3) | 0 |
| Disseminated intravascular coagulation | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0·3) | 0 | 1 (0·3) |
| Splenic infarction | 0 | 0 | 0 | 1 (1·4) | 1 (1·4) | 0 | 0 | 0 | 0 |
Data are n (rate). Adjusted rates were calculated as the number of patients with events per 100 patient-year of exposure. Events occurring at a rate of 0・2 per 100 patient-years of exposure in any group. MedDRA version 19.1 was used to code the events.
Exposure=428 4 patient-years.
Exposure=73 6 patient-years.
Exposure=329 9 patient-years.
The rates of secondary malignancies per 100 patient-years of exposure were 7·0 in those originally randomly assigned to ruxolitinib, 4·1 with best available therapy, and 4·5 in the crossover population (appendix p 5). The rates of non-melanoma skin cancer were 5·1 in those originally randomly assigned to ruxolitinib, 2·7 with best available therapy, and 2·7 in the crossover population. Among the patients with a history of non-melanoma skin cancer, rates of non-melanoma skin cancer (per 100 patient-years of exposure) were 18·6 in ruxolitinib group, 28·5 in best available therapy group, and 13·4 in crossover population. Among the patients without a history of non-melanoma skin cancer, rates of non-melanoma skin cancer (per 100 patient-years of exposure) were 3·6 in ruxolitinib group, 1·4 in the best available therapy group, and 2·0 in crossover population (appendix p 6). The rates of transformation to myelofibrosis and acute myeloid leukaemia (per 100 patient-years) were 2·1 and 0·2 in the ruxolitinib group, 1·8 and 0·6 in the crossover population, and 1·4 and 0·0 in the best available therapy group, respectively. One patient in the crossover population was diagnosed with lymphoplasmacytoid lymphoma or immunocytoma (grade 2) 35 days after the last dose of ruxolitinib. The event was reported as a serious adverse event and was assessed to be not related to the study treatment.
There were two on-treatment deaths in the ruxolitinib group. One of these deaths was due to gastric adenocarcinoma, which was assessed by the investigator as related to ruxolitinib treatment. The second death was due to a malignant neoplasm. During the treatment, the CT of the patient’s thorax revealed the presence of a bronchopulmonary malignant tumour confirming the diagnosis of malignant neoplasm. 9 days after the last dose of the ruxolitinib, the patient died because of multiple comorbidities, with malignant neoplasm being the contributory factor. The investigator did not suspect an association between the event (malignant neoplasm) and ruxolitinib treatment. In the crossover population, four patients had fatal adverse events leading to four on-treatment deaths (2 patients had pneumonia, 1 had a CNS haemorrhage, and 1 had hypovolaemic shock). None of these deaths were considered to be related to ruxolitinib treatment. No patients died while on best available therapy treatment.
Discussion
The results from this final analysis of the RESPONSE study add to the evidence for the efficacy and safety of ruxolitinib in patients with polycythaemia vera who are inadequately controlled with hydroxyurea either because of resistance or intolerance. The primary analysis of this study showed the superiority of ruxolitinib compared with best available therapy12 (including interferon)20 in terms of achieving haematocrit control, spleen response, complete haematological remission, and overall clinicohaematological response. These 5-year findings showed that the primary response, complete haematological remission, and overall clinicohaematological response were maintained with long-term ruxolitinib therapy. In addition, modest reductions in JAK2 Val617Phe allele burden and improvements in quality of life parameters were observed with longer-term ruxolitinib use, indicating greater overall benefits with the long-term treatment. The obvious limitation here is the quality of life data being collected at week 256 or at the end of treatment visit after week 32. It is worth noting that the patient population included in this study are patients with polycythaemia vera who had significant splenomegaly at baseline and therefore had advanced disease (some might in fact be developing myelofibrosis).
The benefits of ruxolitinib treatment were not limited to patients who were initially randomly assigned to ruxolitinib, but were also observed in the crossover patients. Many of the crossover patients did not require phlebotomy after 32 weeks of crossover. This finding is in agreement with the previously published subanalysis from RESPONSE by Verstovsek and colleagues.21 These findings showed that non-responders in the ruxolitinib group had a greater median duration of time to subsequent phlebotomy eligibility (52 weeks) than non-responders in the best available therapy group (21 weeks). Hence, it is plausible that patients from the crossover population might not have reached haematocrit control while on best available therapy, but showed clinical improvement after crossing over to ruxolitinib. Similar to the improvement in phlebotomy requirement, a reduction in allele burden was also observed in these patients after crossing over to ruxolitinib.
A previous study showed that patients resistant to hydroxyurea (but not intolerant) have a 5·6-times increased risk of death.9 A subsequent analysis by the same research group showed that patients fulfilling the unified definition of resistance or intolerance criteria did not have worse survival compared with those patients who were not resistant or intolerant.8 The additional analysis of subgroups from the above-mentioned study8 further revealed that patients who were hydroxyurea-resistant or hydroxyurea-intolerant who develop cytopenia had significantly lower survival rates (63%) compared with those who responded to hydroxyurea. Althoug cross-trial comparisons might be done with caution, the observed survival at 5 years with ruxolitinib treatment in the RESPONSE study appears to be higher than the survival previously reported in the hydroxyurea-resistant or hydroxyurea-intolerant population. In our study, it was not possible to make any direct comparisons with best available therapy, as most patients given best available therapy crossed over to ruxolitinib after week 32. Due to extensive crossover of patients from best available therapy, the observed HR from this analysis represents a conservative estimate of ruxolitinib benefit and warrants further exploration.
Nearly 65% of patients given ruxolitinib completed this long-term treatment period as per protocol, with only 15% of patients discontinuing the study drug because of an adverse event. The long-term safety and tolerability of ruxolitinib was consistent with the previous findings.12,16 As expected from the mechanism of action of ruxolitinib,1 the most common haematological adverse events were anaemia and thrombocytopenia but these rarely led to the treatment discontinuation. With longer-term follow-up, the rates of non-haematological adverse events and infection (except herpes zoster) were lower in the ruxolitinib group than the best available therapy group. Most of the herpes zoster infections were grade 1 or 2 and were resolved. As the evidence is increasing over time for better control of infections using active screening techniques, prophylaxis, or treatment modalities at symptom onset and patient education, these measures could have possibly contributed to controlled infection rates observed over time in the RESPONSE study. Although the RESPONSE study was not designed to evaluate the reduction in thrombotic events, the lower rate of thromboembolic events, durable haematocrit control, and complete haematological remission with ruxolitinib treatment could potentially benefit in minimising the risk of a thromboembolic event.
The rates of non-melanoma skin cancer were higher in the patients originally randomly assigned to ruxolitinib and who did not have previous history of non-melanoma skin cancer at baseline. Based on the observation made from a small number of patients in the ruxolitinib group, who had longer prior exposure to hydroxyurea at baseline, it is plausible that previous hydroxyurea treatment could be an underlying contributory factor for an increased rate of non-melanoma skin cancer in the ruxolitinib group.22 Overall, no new long-term safety signals were detected in this analysis.
Taken together, the long-term efficacy and safety data from the completed RESPONSE study support that ruxolitinib is a safe and effective therapeutic option for patients with polycythaemia vera who are resistant to or intolerant of hydroxyurea treatment.
Supplementary Material
Research in context.
Evidence before this study
We searched PubMed for articles published between July 1, 2003, and July 1, 2018 with no language restrictions. We searched “polycythemia vera” AND “hydroxyurea” AND “resistance”, “polycythemia vera” AND “second-line”, and “polycythemia vera” AND “phase 3”. We included all completed and ongoing studies. The therapeutic goals for patients with polycythaemia vera are to prevent vascular events, to improve symptom burden, and to delay disease progression. Hydroxyurea has been a long-standing cytoreductive agent in the first-line setting for patients at high risk. In 2011, the European LeukemiaNet also recommended interferon as the first-line option in patients at high risk with polycythaemia vera who are in need of cytoreductive therapy. However, 25% of patients at high risk given the first-line therapy (hydroxyurea or interferon) become resistant to or intolerant of treatment. For patients who do not tolerate or are resistant to hydroxyurea or interferon, therapeutic options remain limited. The available cytoreductive agents have rarely been compared in a randomised study, and their use is supported by little prospective evidence. More than 95% of patients with polycythaemia vera possess the JAK2 Val617Phe mutation; hence, ruxolitinib, a JAK 1 and JAK2 inhibitor, was investigated in patients with polycythaemia vera who were refractory to or intolerant of hydroxyurea. A phase 2 study (INCB 18424–256) in patients with polycythaemia vera who were refractory or intolerant to hydroxyurea showed that treatment with ruxolitinib was effective and well tolerated, and can result in normalisation of haematocrit, white blood cell count, and platelet count while reducing the need for phlebotomy. In this study, splenomegaly was improved with ruxolitinib treatment. On the basis of the emerging efficacy and safety evidence from this study, the registration phase 3 RESPONSE study was designed to compare the long-term efficacy and safety of ruxolitinib to best available therapy in patients with polycythaemia vera who were refractory to or intolerant of hydroxyurea.
Added value of this study
In the primary analysis of RESPONSE, ruxolitinib was superior to best available therapy in patients with polycythaemia vera who were inadequately controlled with hydroxyurea. These results supported the United States Food and Drug Administration and the European Medicines Agency approvals of ruxolitinib for the treatment of polycythaemia vera in patients who are resistant to or intolerant of hydroxyurea. The European LeukemiaNet and National Comprehensive Cancer Network guidelines now include ruxolitinib as the recommended treatment for patients who are resistant to or intolerant of hydroxyurea. The results from this final analysis after 5 years of follow-up show that the primary response, complete haematological remission, and clinicohaematological response were durable in these patients with long-term ruxolitinib therapy. In RESPONSE, the exposure-adjusted rates (per 100 patient-years) of thromboembolic events were lower in patients given ruxolitinib compared with patients given best available therapy. Additionally, sustained reductions in JAK2 Val617Phe allele burden and improvements in quality of life parameters were observed with longer-term ruxolitinib use.
Implications of all the available evidence
Ruxolitinib is a safe and effective long-term treatment option for patients with polycythaemia vera who are resistant to or intolerant of hydroxyurea treatment. Our results indicated a greater benefit with long-term treatment. The long-term safety and tolerability of ruxolitinib were consistent with the previous reports, and no new safety signals were reported.
Acknowledgments
The study is supported by Novartis Pharmaceuticals Corporation. We thank all the investigators of the study, and also thank Ambrin Fatima, of Novartis Healthcare Pvt Ltd, for providing medical editorial and writing support with this manuscript.
Funding
Novartis Pharmaceuticals Corporation.
Footnotes
Declaration of interests
JJK reports membership on an entity’s board of directors or advisory committees and research funding for Novartis, AOP Orphan, and Celgene. MH reports honoraria and research funding from Otsuka Pharmaceutical, Kyowa-Kirin, Chugai, Astellas, Takeda, MSD, Sumitomo Dainippon, Pfizer, and Daiichisankyo: research funding from Taiho, Teijin, Eisai, Japan Blood Products Organization, and Nihon Pharmaceutical; honoraria from BMS, Jansenn, Celgene, Mochida, Ono, Sanofi, Nippon Shinyaku, Mundi Pharma, and Alexion. FP reports research funding and speakers bureau from Novartis and personal fees from Celgene. TM reports consultancy for AbbVie, BMS, Janssen-Cilag, Novartis, Pfizer, and Takeda. CNH reports consultancy, honoraria, research funding, and speakers bureau from Novartis; consultancy, honoraria, and speakers bureau from Celgene; consultancy and honoraria from Roche and CTI BioPharma; personal fees from AOP, Jannsen, Sierra Oncology, Promedior. RM reports consultancy for Novartis, Sierra Oncology, and LaJolla; and research funding from Incyte, Abbvie, Genentech, and Celgene. CBM reports honoraria and speakers bureau from Novartis; and consultancy, honoraria, research funding, and speakers bureau from Incyte. FP reports consultancy and speakers bureau for Novartis, Celgene, Roche, and Janssen. MG reports membership on an entity’s board of directors or advisory committees and speakers bureau for Novartis. KK reports honoraria for Novartis. ER reports a speakers bureau for Novartis. NF, TD, and MW report employment and equity ownership for Novartis. AMV reports speakers bureau, and membership on an entity’s board of directors or advisory committees for Novartis and CTI BioPharma; and membership on an entity’s board of directors or advisory committees for Celgene and Incyte. SV reports consultancy for Novartis, Constellation, Pragmatist, Sierra, Incyte, and Celegene; and honoraria and research funding for Incyte, Roche, NS Pharma, Celgene, Gilead, Promedior, CTI Biopharma, Genentech, and Blueprint Medicines. CB, PZ, BM, VR, SD, and IWB declare no competing interests.
Data sharing
This trial data availability is according to the criteria and process described on the ClinicalStudyDataRequest website. Novartis is committed to sharing with qualified external researchers, access to patient-level data and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymised to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations.
Contributor Information
Jean-Jacques Kiladjian, Hôpital Saint-Louis, Assitance Publique - Hôpitaux de Paris, Université de Paris, Inserm, Paris, France.
Pierre Zachee, Ziekenhuis Netwerk Antwerpen, Stuivenberg, Antwerp, Belgium.
Masayuki Hino, Department of Clinical Hematology, Osaka City University Graduate School of Medicine, Osaka, Japan.
Fabrizio Pane, University of Naples Federico II, Naples, Italy.
Tamas Masszi, 3rd Department of Internal Medicine, Semmelweis University, Budapest, Hungary.
Claire N Harrison, Guy’s and St Thomas’ NHS Foundation Trust, London, UK.
Ruben Mesa, UT Health San Antonio Cancer Center, San Antonio, TX, USA.
Carole B Miller, Saint Agnes Cancer Institute, Baltimore, MD, USA.
Francesco Passamonti, Ospedale di Circolo e Fondazione Macchi, Varese, Italy.
Simon Durrant, Royal Brisbane and Women’s Hospital, Brisbane, Australia.
Martin Griesshammer, Johannes Wesling Clinic, University of Bochum, Minden, Germany.
Keita Kirito, Department of Hematology and Oncology, University of Yamanashi, Japan.
Carlos Besses, Haematology Department, Hospital del Mar-IMIM, Universidad Autónoma de Barcelona, Barcelona, Spain.
Beatriz Moiraghi, Hospital Jose Maria Ramos Mejia, Buenos Aires, Argentina.
Elisa Rumi, Department of Haematology Oncology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy.
Vittorio Rosti, Center for the Study of Myelofibrosis, IRCCS Policlinico San Matteo Foundation, Pavia, Italy.
Igor Wolfgang Blau, Medical Department, Division of Hematology, Oncology, and Tumor Immunology, Charité Universitätsmedizin Berlin, Germany.
Nathalie Francillard, Novartis Pharma SAS, Rueil Malmaison, France.
Tuochuan Dong, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA.
Monika Wroclawska, Novartis Pharma AG, Basel, Switzerland.
Alessandro M Vannucchi, University of Florence, Florence, Italy.
Srdan Verstovsek, The University of Texas MD Anderson Cancer Center, Houston, TX, USA.
References
- 1.Spivak JL. Myeloproliferative neoplasms. N Engl J Med 2017; 376: 2168–81. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013; 27: 1874–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geyer H, Scherber R, Kosiorek H, et al. Symptomatic profiles of patients with polycythemia vera: implications of inadequately controlled disease. J Clin Oncol 2016; 34: 151–59. [DOI] [PubMed] [Google Scholar]
- 4.Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005; 23: 2224–32. [DOI] [PubMed] [Google Scholar]
- 5.Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med 2004; 117: 755–61. [DOI] [PubMed] [Google Scholar]
- 6.Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol 2011; 29: 761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbui T, Tefferi A, Vannucchi AM, et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia 2018; 32: 1057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez-Larrán A, Kerguelen A, Hernández-Boluda JC, et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br J Haematol 2016; 172: 786–93. [DOI] [PubMed] [Google Scholar]
- 9.Alvarez-Larrán A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 2012; 119: 1363–69. [DOI] [PubMed] [Google Scholar]
- 10.Reiter A, Harrison C. How we identify and manage patients with inadequately controlled polycythemia vera. Curr Hematol Malig Rep 2016; 11: 356–67. [DOI] [PubMed] [Google Scholar]
- 11.Scherber RM, Geyer HL, Dueck AC, et al. The potential role of hematocrit control on symptom burden among polycythemia vera patients: insights from the CYTO-PV and MPN-SAF patient cohorts. Leuk Lymphoma 2017; 58: 1481–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 2015; 372: 1670–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Incyte. Jakafi (ruxolitinib): package insert. 2019. https://www.jakafi.com/pdf/prescribing-information.pdf (accessed Jan 9, 2020).
- 14.Novartis. Jakavi (ruxolitinib): SmPC. 2017. https://www.ema.europa.eu/en/documents/product-information/jakavi-epar-product-information_en.pdf (accessed Jan 9, 2020).
- 15.Kiladjian JJ, Verstovsek S, Griesshammer M, et al. Results from the 208-week (4-year) follow-up of RESPONSE trial, a phase 3 study comparing ruxolitinib (rux) with best available therapy (BAT) for the treatment of polycythemia vera (PV). Blood 2017; 130 (suppl): 322 (abstr). [Google Scholar]
- 16.Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 2016; 101: 821–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griesshammer M, Saydam G, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80-week follow-up from the RESPONSE-2 trial. Ann Hematol 2018; 97: 1591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Passamonti F, Griesshammer M, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol 2017; 18: 88–99. [DOI] [PubMed] [Google Scholar]
- 19.Mesa R, Jamieson C, Bhatia R, et al. Myeloproliferative Neoplasms, Version 2. 2018 Featured updates to the NCCN guidelines. J Natl Compr Canc Netw 2017; 15: 1193–207. [DOI] [PubMed] [Google Scholar]
- 20.Kiladjian JJ, Guglielmelli P, Griesshammer M, et al. Efficacy and safety of ruxolitinib after and vs interferon use in the RESPONSE studies. Ann Hematol 2018; 97: 617–27. [DOI] [PubMed] [Google Scholar]
- 21.Verstovsek S, Mesa R, Jones MM, et al. Ruxolitinib efficacy by hematocrit control in patients with polycythemia vera: an analysis of the RESPONSE trial. Blood 2014; 124 (suppl): 3201 (abstr).25287708 [Google Scholar]
- 22.Antonioli E, Guglielmelli P, Pieri L, et al. Hydroxyurea-related toxicity in 3,411 patients with Ph’-negative MPN. Am J Hematol 2012; 87: 552–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.