

Abstract
Alcohol Use Disorder (AUD) is a multifarious psychiatric condition resulting from complex relationships between genetics, gene expression, neuroadaptations, and environmental influences. Understanding these complex relationships is essential to uncovering the mechanisms involved in the development and progression of AUD, with the ultimate goal of devising effective behavioral and therapeutic interventions. Technical advances in the fields of omics-based research and bioinformatics have yielded insights into gene interactions, biological networks, and cellular responses across humans and animal models. This review highlights several of the newly developed sequencing methodologies and resultant discoveries in neuroscience, as well as the importance of a multi-faceted and integrative approach for determining causal factors in AUD.
Keywords: Alcohol, Bioinformatics, RNA-seq, Gene expression, Drug repurposing
Introduction
Alcohol Use Disorder (AUD) is influenced by both genetic and environmental factors, affecting a series of molecular systems throughout different tissues and cell types. The central nervous system (CNS) is remarkably heterogeneous, consisting of an unknown number of specialized cells that govern behavior. A key challenge in biomedicine is to uncover the identity and function of individual cells, classify cellular populations, and discern their cooperative molecular roles in forming continuously adapting biological networks. The growth and development of high-throughput biological assays, combined with an increasing suite of bioinformatics tools and strategies, are important tools to address these challenges. Furthermore, the rapid proliferation of large-scale datasets is helping to outline the molecular processes that define CNS homeostasis and the pathophysiology of disease.
Since the initial sequencing of the human genome (Lander et al., 2001; Venter et al., 2001), omics-based sciences have received considerable attention, spurring international scientific initiatives and facilitating genome-wide comparisons across diverse human populations and model organisms. Transcriptional regulation of gene expression from DNA is coordinately regulated, dictating the cellular milieu available for shaping physiological processes. Using RNA deep-sequencing (RNA-Seq), the orchestrated arrangement of expressed genes can be systematically collected and quantified from multiple biological samples spanning the entire transcriptome, within a single experiment (Mortazavi, Williams, McCue, Schaeffer, & Wold, 2008; Wang, Gerstein, & Snyder, 2009). RNA transcripts can be assembled de novo or mapped to a reference genome, eliminating the bias for known and protein-coding cDNAs inherent in earlier DNA microarray analyses. Unbiased profiling of the transcriptome provides a comprehensive overview of the affected biological systems across both the coding and non-coding domains. Sequencing studies consistently show that less than 2% of the human genome encodes for proteins; however, a large percentage of the human genome (~80%) is biochemically active (ENCODE Project Consortium, 2012; ENCODE Project Consortium et al., 2007). For example, long non-coding RNAs (lncRNAs), once thought of as transcriptional noise, are gaining recognition as a class of biomolecules with key regulatory functions in transcription, protein localization, subcellular organization, and RNA processing (Wilusz, Sunwoo, & Spector, 2009).
Transcript data can also be incorporated into bioinformatics analyses to discover novel protein functions (Marcotte, 2000). In some instances, transcript levels may actually be a stronger predictor of phenotypic traits than protein levels (Ghazalpour et al., 2011). RNA-Seq and other sequencing-based approaches have seen a precipitous drop in associated laboratory costs, coupled with widespread practices to efficiently 1) interrogate cellular taxonomy, 2) catalog a host of transcripts (mRNAs, non-coding RNAs, and small RNAs), 3) determine the transcriptional structure of genes, novel non-coding RNAs, patterns of alternative splicing, and post-transcriptional modifications, and 4) quantify expression levels in relation to experimental or disease-associated perturbations. A number of scientific initiatives are creating publicly accessible resources, contributing to the widespread use of RNA-Seq for testing hypotheses, and answering biological questions. The Genotype-Tissue Expression (GTEx) project is one of these initiatives (GTEx Consortium, 2013), which can be incorporated into genome-wide association studies (GWAS) to determine the potential functional effects of genetic variants. By profiling thousands of transcriptomes from healthy human donor tissues and cell lines, the GTEx project has established the shared and tissue-specific influences on gene expression due to nearby and distant autosomal genetic loci (Ongen et al., 2017). The genetically emergent expression profiles demonstrate a distinct clustering of brain regions, which are separable from non-brain tissues (GTEx Consortium, 2017). Taking advantage of these publicly available human genotype and tissue-specific gene expression datasets will permit a deeper understanding of the way genetic variants may influence the development of brainbehavior relationships inherent to psychiatric disorders.
Utilizing human postmortem brain tissue
Addiction to alcohol and other substances of abuse is due, at least in part, to persistent changes in gene expression. Establishing associations between human alcoholic subjects and experimental models is essential for studying AUD in a laboratory setting. To accomplish this, high-throughput and integrated systems approaches are needed to characterize the functional state of the brain disease. RNA-Seq of human postmortem hippocampus tissue shows overlapping gene expression signatures related to plasticity between chronic alcohol abuse and cocaine addiction (Zhou, Yuan, Mash, & Goldman, 2011). Molecular networks related to chronic and excessive alcohol drinking behavior exhibit system-wide disruptions in inter-gene connectivities compared with matched non-alcoholic control subjects, suggesting pervasive molecular adaptations in neuronal function. Coherent changes in neuronal gene expression are accompanied by several genes with unknown biological function, including a number of non-coding RNAs. The interrelationship among gene pairs within a gene network is informative for predicting deeply conserved biological processes (Costanzo et al., 2010; Kachroo et al., 2015). Thus, examining gene-gene interactions disrupted by human disease may provide new biological insights for singular genes and rewired systems (see Table 1).
Table 1.
Short list of publicly accessible resources.
| Name | Description | Link |
|---|---|---|
| International Mouse Phenotyping Consortium | A consortium that aims to discover and ascribe biological function to each gene by testing each mutant mouse line | http://www.mousephenotype.org/ |
| The GTex Project | Repository for human genotype, gene expression, and clinical data | https://gtexportal.org/home/ |
| ENCODE | Database for functional elements of the human genome | https://www.encodeproject.org/ |
| The Library of Integrated Network-Based Cellular Signatures (LINCS) | A database of cellular, tissue, and organismal response signatures to various drugs and molecular factors | http://lincsportal.ccs.miami.edu/dcic-portal/ |
| Connectivity Map database (CMap) | Gene expression profiles from thousands of small-molecule compounds and genetic reagents tested in 5 human cell lines | https://www.broadinstitute.org/connectivity-map-cmap |
| CMap and LINCS Unified Environment (CLUE) | Cloud-based infrastructure for integrating LINCS and CMap analyses | https://clue.io/ |
Epigenomic control of gene expression networks, mediated by histone H3 lysine 4 trimethylation (H3K4me3) positioned within promoter regions near active transcription start sites, is characterized in greater detail through separate biological systems for each substance of abuse (Farris, Harris, & Ponomarev, 2015). H3K4me3 and other chromatin markers may overlap single nucleotide polymorphisms (SNPs) that regulate transcriptional properties within gene promoter regions in particular cell types and tissues. Individual and shared-risk variants associated with distinct phenotypic traits are frequently localized within the boundaries of epigenetic marks, influencing the binding of transcription factors to downstream properties among specialized cell types (Brown et al., 2017; Trynka et al., 2013). SNP heritability contributes to common patterns of variation in human gene expression networks across neuropsychiatric disorders (Gandal et al., 2018), with open chromatin regions bearing a larger per-SNP based effect for scoring some polygenic traits (Salvatore et al., 2018). Genetic variants associated with alcohol dependence are enriched within specific gene co-expression networks correlated with lifetime alcohol consumption (Farris, Arasappan, Hunicke-Smith, Harris, & Mayfield, 2015). Combining gene network studies in human alcoholics with epigenetic and single variant analyses will be vital to uncover the shared contributions of genetic and environmental factors capable of driving the molecular adaptations in AUD.
Cross-species conservation for studying AUD
Despite millions of years of evolution, an expansive set of bioinformatics analyses indicates that humans and other species share genomic structure (Gerstein et al., 2014). Greater than 90% of the mouse genome shows evidence for conserved synteny with the human genome, with a nearly identical set of protein-coding homologs (Mouse Genome Sequencing Consortium et al., 2002). Although the expression patterns of human and mouse protein-coding transcripts can differ (Yue et al., 2014), the strong degree of evolutionary conservation supports the utility of mice, and other animal models, for the study of complex traits. Resources such as the International Mouse Phenotyping Consortium extensively characterize genetically engineered mutant mice throughout the entire genome of conserved genes (Dickinson et al., 2016). Over 100 null mouse mutants for individual candidate genes have been studied for alcohol-related behavioral phenotypes, with ~72% of mutants having a known effect in a common model of voluntary alcohol consumption (Crabbe, Phillips, Harris, Arends, & Koob, 2006; Mayfield, Arends, Harris, & Blednov, 2016). The broad array of genes currently implicated is expected, given the polygenic mode of inheritance and the spectrum of molecular systems involved in AUD and other comorbid behaviors. Additionally, similar to findings from human GWAS, animal behavioral phenotypes can vary widely with respect to the null allele carried across mixed genetic backgrounds (Sittig et al., 2016). Complementing single gene-based procedures with systems-oriented approaches strengthens the ability to establish causal determinants of neurobiological substrates in behavior.
Addiction-related alterations in gene expression are time-, tissue-, and substance-dependent (Piechota et al., 2010). Chronic intermittent exposure to ethanol in an animal model of dependence results in temporal changes in gene co-expression networks that may correspond to early epochs of multi-cellular CNS plasticity in the development of AUD (Osterndorff-Kahanek et al., 2015; Smith et al., 2016). Recurring bouts of chronic ethanol exposure, a hallmark of dependence and disease progression, concurrently shift the dynamics between coding and non-coding interaction networks. Akin to protein-coding ensembles, the expression of small non-coding RNA molecules, for example microRNAs (miRNAs), is discretely modulated in rodent models of chronic ethanol exposure (Osterndorff-Kahanek et al., 2018; Tapocik et al., 2013). The transition to excessive ethanol drinking behavior can be altered by directly manipulating the expression of distinct miRNAs (i.e., miR-206 and miR-30a-5p) that converge on the same downstream target, brain-derived neurotrophic factor (Bdnf) (Darcq et al., 2015; Tapocik et al., 2014). Redundancy is a known property of biological networks, imparting a series of checks and balances to ensure integrity of the system as a whole. Paired profiling of mouse prefrontal cortex for protein-coding genes and miRNAs following chronic ethanol consumption demonstrates a reciprocal relationship among multiple biological networks and pathways, analogous to certain features observed in human molecular networks (Nunez et al., 2013). Studying the intricate wiring of biomolecule expression across different model organisms and behavioral paradigms further assists in defining the subsystems involved in the acquisition, maintenance, and recurrence of alcohol dependence (Fig. 1).
Fig. 1.
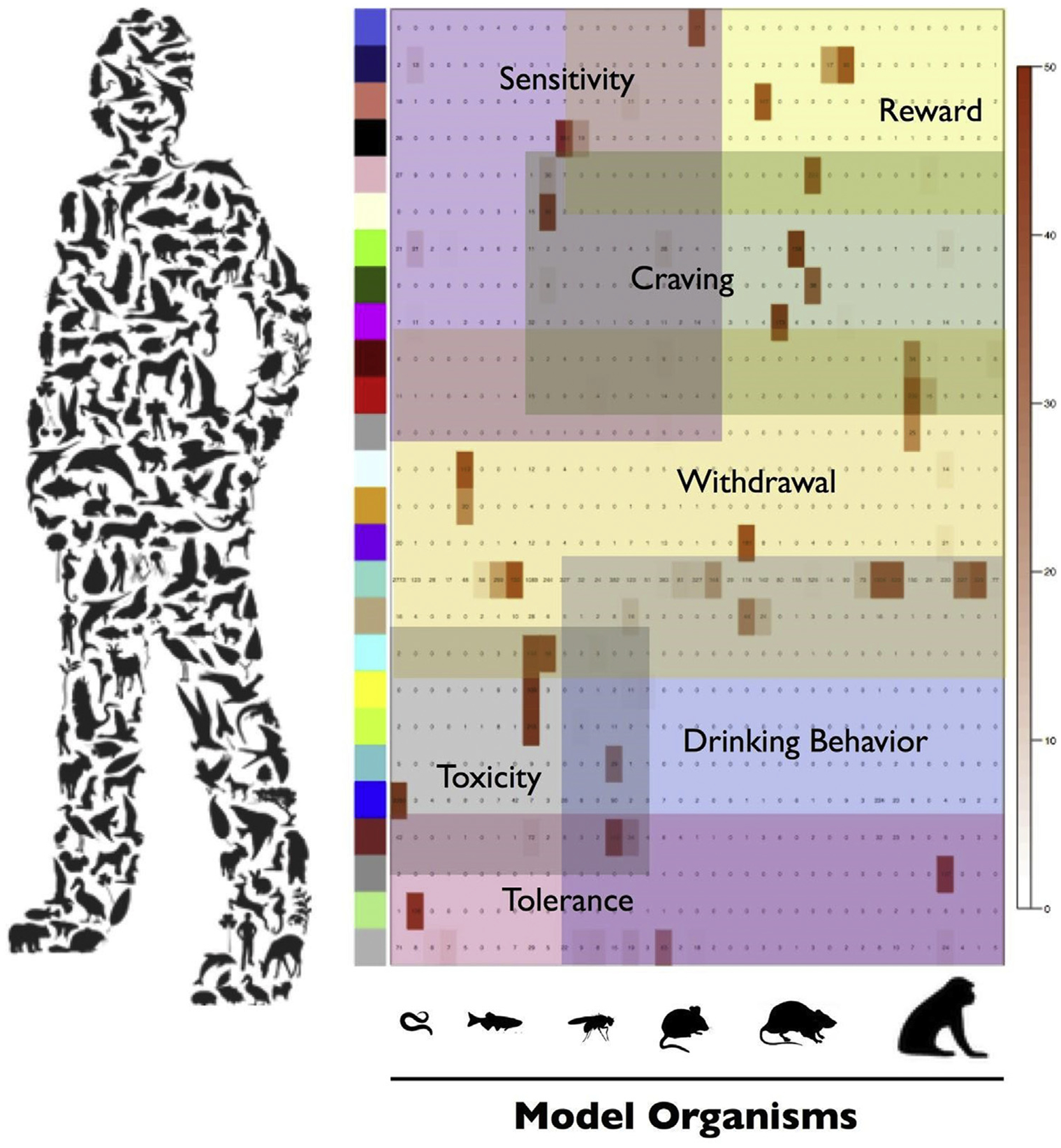
Schematic representation for the human medical diagnosis of alcohol use disorder (AUD) in relation to different animal models used in biomedical research. An AUD diagnosis encapsulates many different phenotypic traits (e.g., ‘craving’ and ‘withdrawal’). Genome-wide studies of model organisms that address latent features of AUD can help decipher coordinated biological activity of interrelated modules found in humans (colored blocks along y-axis).
RNA-seq of cellular populations
Honing the resolution of gene expression networks in CNS cell-types is important for understanding the physiological processes that mediate allostasis. Neurogenomic studies of brain tissue have primarily used whole tissue homogenates, which include neurons, microglia, astrocytes, and oligodendrocytes. Chronic ethanol exposure is linked to activation of the immune system and neuroinflammation (Crews, Lawrimore, Walter, & Coleman, 2017), facilitated by the various cell types in the brain. Behavioral studies also support the role of multiple neuroimmune genes, ascertained from transcriptome profiling of CNS tissue from different species, in the regulation of ethanol consumption (Blednov et al., 2012). Microglia account for a small proportion of the total number of cells in the CNS, but are the primary cells responsible for immune-driven responses. Inhibition of microglial activation by the broad-spectrum antibiotic minocycline decreases ethanol self-administration in mice (Agrawal, Hewetson, George, Syapin, & Bergeson, 2011). RNA-Seq analysis of purified microglial cells from ethanol-consuming mice demonstrates that they possess a unique gene network that is largely undetected in mixed cellular populations (McCarthy, Farris, Blednov, Harris, & Mayfield, 2018). Ethanol consumption induces a constellation of toll-like receptors, Tgf-beta signaling, and Nf-kappa-b signaling genes within microglia. Network connectivity identified the protease factor, sialic acid binding Ig-like lectin H (Siglech), as a putative hub gene with a suspected role in ethanol-induced neuroimmune signaling. These findings complement in vitro studies demonstrating that microglia depletion blunts induction of the pro-inflammatory gene, tumor necrosis factor a (TNF a), and enhances expression of anti-inflammatory genes, interleukin-4 (IL4), and interleukin-10 (IL10) following acute ethanol withdrawal (Walter & Crews, 2017). Further research using microglia-depleted mice is needed to determine the specific in vivo role of microglia during ethanol consumption (Elmore et al., 2014). In addition, in vivo studies are needed to validate the potential roles of Siglech and TGF-β in the ethanol-induced immune responses identified with RNA-Seq.
Pro-inflammatory microglial cells can give rise to reactive macroglial cells known as astrocytes, leading to downstream changes in neuronal morphology and function (Liddelow et al., 2017). Astrocytes participate in a variety of inter- and intra-cellular signaling cascades throughout different regions of the CNS. Excitatory neuronal populations releasing glutamate may evoke the propagation of calcium waves (Cornell-Bell, Finkbeiner, Cooper, & Smith, 1990; Dani, Chernjavsky, & Smith, 1992), prompting a form of bidirectional communication among neighboring cells. Selective stimulation of astrocytes using designer receptors exclusively activated by designer drugs (DREADDS), intended to modify glutamate transmission, attenuates cocaineseeking behavior (Scofield et al., 2015) and ethanol self-administration during periods of abstinence (Bull et al., 2014). Isolation of adult astrocytes from ethanol-consuming mice for RNA-Seq analysis indicates that a distinct set of calcium-related signaling events involving calmodulin binding is altered after voluntary binge-like drinking, which are separate from ethanol-responsive genes found in a heterogeneous cellular suspension (Erickson, Farris, Blednov, Mayfield, & Harris, 2018). Concurrent with the calcium-dependent responses, there was an upregulation of extracellular matrix (ECM) genes in astrocytes after chronic ethanol exposure, consistent with reports of ECM regulation of behavior and synaptic plasticity in the drug-addicted brain (Lasek, 2016). The simultaneous assessment of transcriptome-wide changes in defined cellular populations, such as microglia and astrocytes, underscores the specialized cellular landscapes that are generated by chronic ethanol exposure.
Single-cell sequencing
Creating a complete inventory of the intrinsic machinery for individual cellular phenotypes is a daunting endeavor facing the scientific community. By harnessing many of the advancements in single-cell RNA sequencing (scRNA-Seq), global research efforts such as the Human Cell Atlas are diligently pursuing facets of this goal (Regev et al., 2017). The inherent architecture of the CNS is characterized by a vast assemblage of functionally heterogeneous cell types configured in their local microenvironments (Lovatt et al., 2014). Applying scRNA-Seq to adult and fetal brain samples helps distinguish the signatures of major CNS cell types (Darmanis et al., 2015). Individual nuclei can be obtained from human postmortem brain tissue, permitting analysis of low levels of RNA, and reducing artifacts that may arise from cell dispersion procedures (Krishnaswami et al., 2016). Sequencing 3227 neuronal nuclei from different human postmortem cortical regions uncovered intra-subtype differences between individual neurons (Lake et al., 2016). Neuronal variation may be due to the pronounced somatic mosaicism present within neuronal genomes (Lodato et al., 2015; McConnell et al., 2013), which can gradually accumulate throughout the lifespan of individual cells (Bae et al., 2018; Lodato et al., 2018). Such long-lived postmitotic neuronal populations may be at higher risk for harboring somatic mutations relevant for driving neuropsychiatric disorders (McConnell et al., 2017).
Mouse studies have corroborated the evidence found in humans for somatic mutations in neurons, which differ from several other cell lineages (Hazen et al., 2016). The mechanisms responsible for introducing aberrant single nucleotide variants are unknown; however, depending on the genomic location, such variants may affect alternative splicing, protein function, and transcriptional regulation of gene expression. Comparison of human and mouse dopaminergic neuronal subtypes indicates a strong preservation across species, suggesting the potential for cell replacement therapies (La Manno et al., 2016). Under the control of Cre recombinase, transgenic mice can be used in conjunction with scRNA-Seq to map the molecular terrain of known neuronal cell types, as well as to discover new cell types and cellular markers (Tasic et al., 2016). Importantly, single neuronal expression profiles are strongly associated with their respective morphological and physiological properties (Fuzik et al., 2016; Tasic et al., 2016). Established patterns of gene expression are reproducible large-scale fingerprints for varying activity among neuronal systems (Tyssowski et al., 2018). The scalability of scRNA-Seq and the beginning of the use of tissue-based sequencing may establish a molecular-based atlas of the mammalian brain and identify new, improved cellular markers (Jaitin et al., 2014; Vanlandewijck et al., 2018). Mapping the molecular composition of neuronal ensembles with single-cell sequencing using animal behavioral models of differing disease stages will be essential for recognizing causal factors and relevant molecular approaches for disease intervention.
Medication discovery and repositioning
Leveraging genetics in concert with transcriptome measurements offers a viable approach for treating neuropsychiatric disorders (Breen et al., 2016; So et al., 2017). Transcriptome-based signatures have proven valuable for charting the shared relationships and novel mechanisms of action for many chemical compounds (Lamb et al., 2006). Armed with these experimental datasets, bioinformatics tools can predict and validate large-scale screens that target the druggable genome (Jeong, Moon, Song, & Yoon, 2017; Liu et al., 2017; Sawada, Iwata, Tabei, Yamato, & Yamanishi, 2018). The Library of Integrated Network-Based Cellular Signatures (LINCS) is an example of one scientific resource that provides a set of representative cellular response signatures to chemical and genetic perturbations. The LINCS database includes profiles of gene transcription, cell proliferation, protein binding, and other phenotypes across cell types and pharmacological agents (Subramanian et al., 2017; Vempati et al., 2014). Currently, the dataset contains genome-wide gene expression profiles for more than 40,000 perturbagens and approximately 1200 cells (Koleti et al., 2018). The Connectivity Map database (CMap), a library containing over 1.5 million gene expression profiles from thousands of small-molecule compounds and genetic reagents, tested in multiple cell types, can be queried for perturbations that give a related gene expression response for a given pharmacological agent. To facilitate these analyses, a cloud-based computational infrastructure, known as the CMap and LINCS Unified Environment (CLUE), has been built with user-friendly web applications and software tools to enable researchers to access, analyze, and integrate public data alongside their own experiments. Together, these methods can accelerate the prioritization of novel compounds for further functional and behavioral evaluation, including use of animal models of alcohol drinking behavior (Ferguson et al., 2017). Interrogating the druggability of the genetic and genomic states rooted in disease will guide development of future drug designs and foster drug repositioning to broaden the scope of available treatments.
Conclusions
Individually tailoring specific therapies and empowering prevention is the fundamental goal of precision medicine. Bioinformatics bridges different areas of systems biology, melding aspects of both clinical and basic research. Consolidating information across the fields of genetics, transcriptomics, and neuroscience has already begun to shed light on relevant pathways in disease (Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015), critical developmental circuits (Parikshak et al., 2013; Willsey et al., 2013), and resting-state brain activity (Wang et al., 2015). Human and animal genetics studies, as well as other approaches, have started to embrace open-source resources for understanding the neurobiology of alcohol and substance abuse. For example, a recent GWAS for alcohol consumption demonstrated the potential functional role for significant SNPs in tissue-dependent gene expression (Clarke et al., 2017). Utilizing an assortment of burgeoning databases and repositories obtained from human and animal subjects can answer questions, as well as fuel new, unexplored questions in neuroscience. High-throughput technologies and bioinformatics techniques are constantly evolving, allowing us to delve deeper into the neurobiology of AUD and find improved avenues for treatment. The expansion and implementation of these cross-disciplinary approaches will continue to be beneficial for interpreting the molecular anatomy underlying AUD and other polygenic, comorbid disorders (Salvatore et al., 2018).
Acknowledgments
This work was generously supported by the National Institute of Alcohol Abuse and Alcoholism (NIAAA) grants K99AA024836 and U01AA020926. Additionally, we would like to acknowledge the contributions of Dr. Jody Mayfield for scientific edits and valuable feedback.
References
- Agrawal RG, Hewetson A, George CM, Syapin PJ, & Bergeson SE (2011). Minocycline reduces ethanol drinking. Brain, Behavior, and Immunity, 25(Suppl1), S165–S169. 10.1016/j.bbi.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, et al. (2018). Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science, 359, 550–555. 10.1126/science.aan8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, & Harris RA (2012). Neuroimmune regulation of alcohol consumption: Behavioral validation of genes obtained from genomic studies. Addiction Biology, 17, 108–120. 10.1111/j.1369-1600.2010.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen G, Li Q, Roth BL, O’Donnell P, Didriksen M, Dolmetsch R, et al. (2016). Translating genome-wide association findings into new therapeutics for psychiatry. Nature Neuroscience, 19, 1392–1396. 10.1038/nn.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AA, Viñuela A, Delaneau O, Spector TD, Small KS, & Dermitzakis ET (2017). Predicting causal variants affecting expression by using whole-genome sequencing and RNA-seq from multiple human tissues. Nature Genetics, 49, 1747–1751. 10.1038/ng.3979. [DOI] [PubMed] [Google Scholar]
- Bull C, Freitas KC, Zou S, Poland RS, Syed WA, Urban DJ, et al. (2014). Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology, 39, 2835–2845. 10.1038/npp.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke T-K, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, et al. (2017). Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N = 112 117). Molecular Psychiatry, 22, 1376–1384. 10.1038/mp.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, & Smith SJ (1990). Glutamate induces calcium waves in cultured astrocytes: Long-range glial signaling. Science, 247, 470–473. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. (2010). The genetic landscape of a cell. Science, 327, 425–431. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Harris RA, Arends MA, & Koob GF (2006). Alcohol-related genes: Contributions from studies with genetically engineered mice. Addiction Biology, 11, 195–269. 10.1111/j.1369-1600.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Lawrimore CJ, Walter TJ, & Coleman LG Jr. (2017). The role of neuroimmune signaling in alcoholism. Neuropharmacology, 122, 56–73. 10.1016/j.neuropharm.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JW, Chernjavsky A, & Smith SJ (1992). Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron, 8, 429–440. [DOI] [PubMed] [Google Scholar]
- Darcq E, Warnault V, Phamluong K, Besserer GM, Liu F, & Ron D (2015). MicroRNA-30a-5p in the prefrontal cortex controls the transition from moderate to excessive alcohol consumption. Molecular Psychiatry, 20, 1219–1231. 10.1038/mp.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. (2015). A survey of human brain transcriptome diversity at the single cell level. Proceedings of the National Academy of Sciences of the United States of America, 112, 7285–7290. 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature, 537, 508–514. 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82, 380–397. 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium, Birney E, Stamatoyannapoulos JA, Dutta A, Guigó R, Gingeras TR, et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature, 447, 799–816. 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson EK, Farris SP, Blednov YA, Mayfield RD, & Harris RA (2018). Astrocyte-specific transcriptome responses to chronic ethanol consumption. The Pharmacogenomics Journal. 10.1038/s41397-017-0012-2 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Arasappan D, Hunicke-Smith S, Harris RA, & Mayfield RD (2015). Transcriptome organization for chronic alcohol abuse in human brain. Molecular Psychiatry, 20, 1438–1447. 10.1038/mp.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Harris RA, & Ponomarev I (2015). Epigenetic modulation of brain gene networks for cocaine and alcohol abuse. Frontiers in Neuroscience, 9, 176. 10.3389/fnins.2015.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson LB, Ozburn AR, Ponomarev I, Metten P, Reilly M, Crabbe JC, et al. (2017). Genome-wide expression profiles drive discovery of novel compounds that reduce binge drinking in mice. Neuropsychopharmacology, 43, 1257–1266. 10.1038/npp.2017.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzik J, Zeisel A, Máté Z, Calvigioni D, Yanagawa Y, Szabó G, et al. (2016). Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nature Biotechnology, 34, 175–183. 10.1038/nbt.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, et al. (2018). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science, 359, 693–697. 10.1126/science.aad6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, Brown JB, et al. (2014). Comparative analysis of the transcriptome across distant species. Nature, 512, 445–448. 10.1038/nature13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, et al. (2011). Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genetics, 7, e1001393. 10.1371/journal.pgen.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. (2013). The genotype-tissue expression (GTEx) project. Nature Genetics, 45, 580–585. 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx consortium. (2017). Genetic effects on gene expression across human tissues. Nature, 550, 204–213. 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen JL, Faust GG, Rodriguez AR, Ferguson WC, Shumilina S, Clark RA, et al. (2016). The complete genome sequences, unique mutational spectra, and developmental potency of adult neurons revealed by cloning. Neuron, 89, 1223–1236. 10.1016/j.neuron.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, et al. (2014). Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science, 343, 776–779. 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong E, Moon SU, Song M, & Yoon S (2017). Transcriptome modeling and phenotypic assays for cancer precision medicine. Archives of Pharmacal Research, 40, 906–914. 10.1007/s12272-017-0940-z. [DOI] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, & Marcotte EM (2015). Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science, 348, 921–925. 10.1126/science.aaa0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleti A, Terryn R, Stathias V, Chung C, Cooper DJ, Turner JP, et al. (2018). Data portal for the library of integrated network-based cellular signatures (LINCS) program: Integrated access to diverse large-scale cellular perturbation response data. Nucleic Acids Research, 46, D558–D566. 10.1093/nar/gkx1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, et al. (2016). Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nature Protocols, 11, 499–524. 10.1038/nprot.2016.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, et al. (2016). Molecular diversity of midbrain development in mouse, human, and stem cells. Cell, 167, 566–580.e19. 10.1016/j.cell.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, et al. (2016). Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science, 352, 1586–1590. 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. (2006). The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409, 860–921. 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lasek AW (2016). Effects of ethanol on brain extracellular matrix: Implications for alcohol use disorder. Alcoholism: Clinical and Experimental Research, 40, 2030–2042. 10.1111/acer.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yin X, Zhong J, Guan N, Luo Z, Min L, et al. (2017). Systematic identification and assessment of therapeutic targets for breast cancer based on genome-wide RNA interference transcriptomes. Genes, 8. 10.3390/genes8030086. pii: E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, et al. (2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science, 359, 555–559. 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, et al. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science, 350, 94–98. 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, Ruble BK, Lee J, Dueck H, Kim TK, Fisher S, et al. (2014). Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nature Methods, 11, 190–196. 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM (2000). Computational genetics: Finding protein function by non-homology methods. Current Opinion in Structural Biology, 10, 359–365. [DOI] [PubMed] [Google Scholar]
- Mayfield J, Arends MA, Harris RA, & Blednov YA (2016). Genes and alcohol consumption: Studies with mutant mice. International Review of Neurobiology, 126, 293–355. 10.1016/bs.irn.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy GM, Farris SP, Blednov YA, Harris RA, & Mayfield RD (2018). Microglial-specific transcriptome changes following chronic alcohol consumption. Neuropharmacology, 128, 416–424. 10.1016/j.neuropharm.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, et al. (2013). Mosaic copy number variation in human neurons. Science, 342, 632–637. 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, et al. (2017). Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science, 356. 10.1126/science.aal1641. pii: eaal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, & Wold B (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5, 621–628. 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. (2002). Initial sequencing and comparative analysis of the mouse genome. Nature, 420, 520–562. 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium. (2015). Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature Neuroscience, 18, 199–209. 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez YO, Truitt JM, Gorini G, Ponomareva ON, Blednov YA, Harris RA, et al. (2013). Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genomics, 14, 725. 10.1186/1471-2164-14-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongen H, Brown AA, Delaneau O, Panousis NI, Nica AC, GTEx Consortium, et al. (2017). Estimating the causal tissues for complex traits and diseases. Nature Genetics, 49, 1676–1683. 10.1038/ng.3981. [DOI] [PubMed] [Google Scholar]
- Osterndorff-Kahanek EA, Becker HC, Lopez MF, Farris SP, Tiwari GR, Nunez YO, et al. (2015). Chronic ethanol exposure produces time- and brain region-dependent changes in gene coexpression networks. PLoS One, 10, e0121522. 10.1371/journal.pone.0121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterndorff-Kahanek EA, Tiwari GR, Lopez MF, Becker HC, Harris RA, & Mayfield RD (2018). Long-term ethanol exposure: Temporal pattern of microRNA expression and associated mRNA gene networks in mouse brain. PLoS One, 13, e0190841. 10.1371/journal.pone.0190841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell, 155, 1008–1021. 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, et al. (2010). The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biology, 11, R48. 10.1186/gb-2010-11-5-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. , Human Cell Atlas Meeting Participants. (2017). The Human Cell Atlas. eLife, 6. 10.7554/eLife.27041. pii: e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore JE, Han S, Farris SP, Mignogna KM, Miles MF, & Agrawal A (2018). Beyond genome-wide significance: Integrative approaches to the interpretation and extension of GWAS findings for alcohol use disorder. Addiction Biology. 10.1111/adb.12591 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada R, Iwata M, Tabei Y, Yamato H, & Yamanishi Y (2018). Predicting inhibitory and activatory drug targets by chemically and genetically perturbed transcriptome signatures. Scientific Reports, 8, 156. 10.1038/s41598-017-18315-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, & Kalivas PW (2015). Gq-DREADD selectively initiates glial glutamate release and inhibits cue-induced cocaine seeking. Biological Psychiatry, 78, 441–451. 10.1016/j.biopsych.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittig LJ, Carbonetto P, Engel KA, Krauss KS, Barrios-Camacho CM, & Palmer AA (2016). Genetic background limits generalizability of genotype-phenotype relationships. Neuron, 91, 1253–1259. 10.1016/j.neuron.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Lopez MF, Archer KJ, Wolen AR, Becker HC, & Miles MF (2016). Time-course analysis of brain regional expression network responses to chronic intermittent ethanol and withdrawal: Implications for mechanisms underlying excessive ethanol consumption. PLoS One, 11, e0146257. 10.1371/journal.pone.0146257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So H-C, Chau CK, Chiu W-T, Ho K-S, Lo C-P, Yim SH, et al. (2017). Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nature Neuroscience, 20, 1342–1349. 10.1038/nn.4618. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. (2017). A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell, 171, 1437–1452.e17. 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapocik JD, Barbier E, Flanigan M, Solomon M, Pincus A, Pilling A, et al. (2014). microRNA-206 in rat medial prefrontal cortex regulates BDNF expression and alcohol drinking. Journal of Neuroscience, 34, 4581–4588. 10.1523/JNEUROSCI.0445-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapocik JD, Solomon M, Flanigan M, Meinhardt M, Barbier E, Schank JR, et al. (2013). Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. The Pharmacogenomics Journal, 13, 286–296. 10.1038/tpj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nature Neuroscience, 19, 335–346. 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, et al. (2013). Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genetics, 45, 124–130. 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho JH, Dunn CJ, Poston RG, Carty CE, et al. (2018). Different neuronal activity patterns induce different gene expression programs. Neuron, 98, 530–546.e11. 10.1016/j.neuron.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature, 554, 475–480. 10.1038/nature25739. [DOI] [PubMed] [Google Scholar]
- Vempati UD, Chung C, Mader C, Koleti A, Datar N, Vidović D, et al. (2014). Metadata standard and data exchange specifications to describe, model, and integrate complex and diverse high-throughput screening data from the library of integrated network-based cellular signatures (LINCS). Journal of Biomolecular Screening, 19, 803–816. 10.1177/1087057114522514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. (2001). The sequence of the human genome. Science, 291, 1304–1351. 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Walter TJ, & Crews FT (2017). Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal. Journal of Neuroinflammation, 14, 86. 10.1186/s12974-017-0856-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-Z, Belgard TG, Mao D, Chen L, Berto S, Preuss TM, et al. (2015). Correspondence between resting-state activity and brain gene expression. Neuron, 88, 659–666. 10.1016/j.neuron.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, & Snyder M (2009). RNA-seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics, 10, 57–63. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 155, 997–1007. 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, & Spector DL (2009). Long noncoding RNAs: Functional surprises from the RNA world. Genes & Development, 23, 1494–1504. 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature, 515, 355–364. 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, & Goldman D (2011). Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proceedings of the National Academy of Sciences of the United States of America, 108, 6626–6631. 10.1073/pnas.1018514108. [DOI] [PMC free article] [PubMed] [Google Scholar]