

ABSTRACT
We previously reported that hepatitis C virus (HCV) infection activates the reactive oxygen species (ROS)/c-Jun N-terminal kinase (JNK) signaling pathway. However, the roles of ROS/JNK activation in the HCV life cycle remain unclear. We sought to identify a novel role of the ROS/JNK signaling pathway in the HCV life cycle. Immunoblot analysis revealed that HCV-induced ROS/JNK activation promoted phosphorylation of Itch, a HECT-type E3 ubiquitin ligase, leading to activation of Itch. The small interfering RNA (siRNA) knockdown of Itch significantly reduced the extracellular HCV infectivity titers, HCV RNA, and HCV core protein without affecting intracellular HCV infectivity titers, HCV RNA, and HCV proteins, suggesting that Itch is involved in the release of HCV particles. HCV-mediated JNK/Itch activation specifically promoted polyubiquitylation of an AAA-type ATPase, VPS4A, but not VPS4B, required to form multivesicular bodies. Site-directed mutagenesis revealed that two lysine residues (K23 and K121) on VPS4A were important for VPS4A polyubiquitylation. The siRNA knockdown of VPS4A, but not VPS4B, significantly reduced extracellular HCV infectivity titers. Coimmunoprecipitation analysis revealed that HCV infection specifically enhanced the interaction between CHMP1B, a subunit of endosomal sorting complexes required for transport (ESCRT)-III complex, and VPS4A, but not VPS4B, whereas VPS4A K23R/K121R greatly reduced the interaction with CHMP1B. HCV infection significantly increased ATPase activity of VPS4A, but not VPS4A K23R/K121R or VPS4B, suggesting that HCV-mediated polyubiquitylation of VPS4A contributes to activation of VPS4A. Taken together, we propose that the HCV-induced ROS/JNK/Itch signaling pathway promotes VPS4A polyubiquitylation, leading to enhanced VPS4A-CHMP1B interaction and promotion of VPS4A ATPase activity, thereby promoting the release of HCV particles.
IMPORTANCE The ROS/JNK signaling pathway contributes to liver diseases, including steatosis, metabolic disorders, and hepatocellular carcinoma. We previously reported that HCV activates the ROS/JNK signaling pathway, leading to the enhancement of hepatic gluconeogenesis and apoptosis induction. This study further demonstrates that the HCV-induced ROS/JNK signaling pathway activates the E3 ubiquitin ligase Itch to promote release of HCV particles via polyubiquitylation of VPS4A. We provide evidence suggesting that HCV infection promotes the ROS/JNK/Itch signaling pathway and ESCRT/VPS4A machinery to release infectious HCV particles. Our results may lead to a better understanding of the mechanistic details of HCV particle release.
KEYWORDS: hepatitis C virus, ROS/JNK, Itch, ESCRT, VPS4A
INTRODUCTION
The World Health Organization estimates over 70 million people worldwide are chronically infected with hepatitis C virus (HCV), emphasizing that HCV still represents a major public health problem (1). HCV infection often causes chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (HCC). Despite the availability of highly effective direct-acting antivirals (DAAs) for HCV treatment, emerging DAA resistance and limited access to DAAs therapy in developing countries still impede global HCV elimination (2, 3).
HCV is a positive-sense single-stranded RNA virus that belongs to the genus Hepacivirus of the family Flaviviridae. The HCV genome consists of 9.6-kb RNA encoding a single polyprotein of about 3,010 amino acids (aa), which is processed by viral proteases and cellular signalases to produce three structural proteins (Core, E1, and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (4).
The precise mechanisms by which infectious HCV particles are released from infected hepatocytes have not been fully understood. Extensive research has shown that the Golgi is important for HCV release (5, 6). HCV particles are assembled in the endoplasmic reticulum (ER), transported to the Golgi compartment in COPII vesicles, and finally released through the Golgi secretory pathway (7). Notably, recent evidence indicates that late endosomes/multivesicular bodies (MVBs), which are associated with the release of exosomes, play a major role in HCV release (6, 8, 9). MVB formation is regulated by members of endosomal sorting complexes required for transport (ESCRT) machinery that mediate membrane abscission processes and intraluminal budding on endosomal membranes. The ESCRT machinery consists of five protein complexes, termed ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III, and VPS4. The AAA ATPase VPS4 (two isoforms, VPS4A and VPS4B) disassembles the ESCRT-III complex from the MVB membrane, thereby driving membrane fission and recycling of ESCRT-III subunits (10, 11). HCV hijacks the ESCRT machinery, especially the VPS4 ATPase, to direct assembled viral particles into MVBs for subsequent release (12, 13).
We previously reported that HCV infection induces reactive oxygen species (ROS) production and activates the c-Jun N-terminal kinase (JNK) signaling pathway, leading to enhancement of hepatic gluconeogenesis (14) and induction of apoptosis (15, 16). In addition, a growing body of evidence suggests that the JNK pathway contributes to the development of HCC (17–20). However, the roles of the ROS/JNK signaling pathway in the HCV life cycle are still unclear.
JNK regulates various physiological processes, including cell death and survival, differentiation, proliferation, and carcinogenesis, by phosphorylating targets, including c-Jun (21), p53 (22), and c-Myc (23). Itch is a member of the neural precursor cell-expressed developmentally downregulated protein 4 (NEDD4) family of HECT-type E3 ligases with an N-terminal lipid-interacting C2 domain, four WW domains for protein-protein interaction, and a C-terminal catalytic HECT domain (24). Under physiological conditions, Itch WW domains restrict the interdomain mobility of the HECT domain, locking Itch in a closed inactive conformation (25). JNK phosphorylates Itch at Ser199, Thr222, and Ser232, leading to a conformational change that weakens the interaction between the WW and HECT domains, thereby augmenting the catalytic activity of Itch (25, 26).
In this study, we demonstrate that the HCV-induced ROS/JNK signaling pathway activates the E3 ubiquitin ligase Itch to promote release of HCV particles via polyubiquitylation of VPS4A. Thus, we propose that HCV infection activates the ROS/JNK/Itch/VPS4A signaling pathway to facilitate the release of infectious HCV particles.
RESULTS
HCV infection promotes phosphorylation of HECT-type E3 ubiquitin ligase Itch via ROS/JNK signaling pathway.
To identify a novel role for the ROS/JNK signaling pathway in the HCV life cycle, we first examined whether HCV infection induced phosphorylation and activation of Itch in Huh-7.5 cells. Immunoblot showed that the amount of phosphorylated Itch at Thr222 markedly increased in HCV J6/JFH1-infected Huh-7.5 cells at 3 days postinfection (dpi), 6 dpi, and 8 dpi compared to those in the mock-infected control cells (Fig. 1A, 1st panel, lanes 2, 4, and 6). The total expression levels of Itch were unchanged (Fig. 1A, 2nd panel, lanes 1 to 6). We found that c-Jun, a key substrate for JNK, was phosphorylated and activated in HCV-infected cells compared to the mock-infected control cells (Fig. 1A, 3rd panel, lanes 2, 4, and 6). The total expression levels of c-Jun in HCV-infected cells were also higher than those in the mock-infected control cells (Fig. 1A, 4th panel, lanes 2, 4, and 6). These results suggest that HCV infection activates the ROS/JNK signaling pathway and promotes phosphorylation of Itch at Thr-222.
FIG 1.
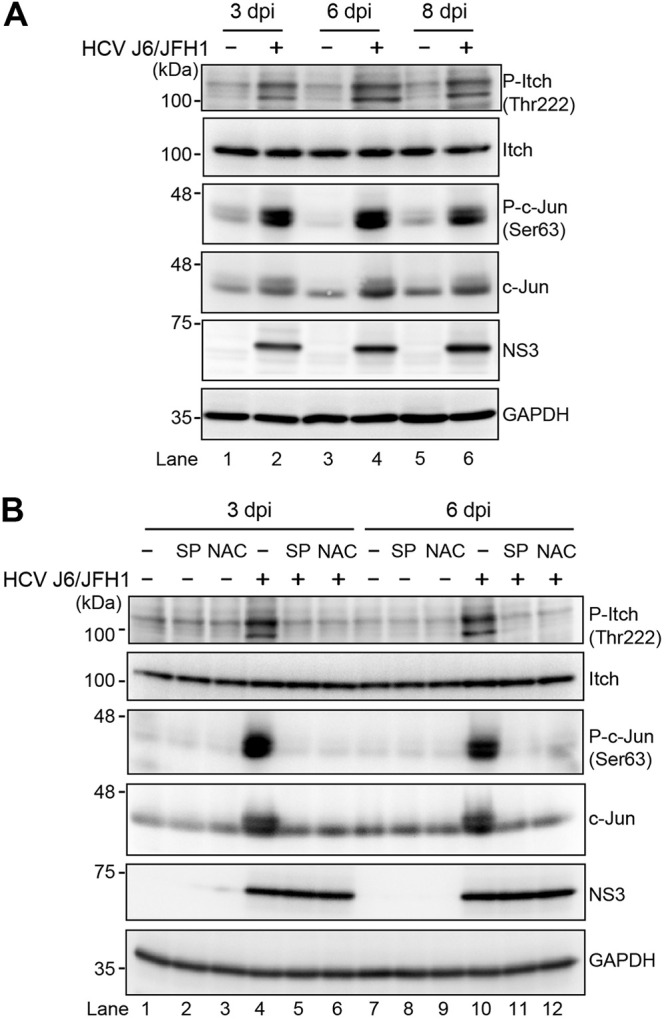
HCV infection promotes phosphorylation of HECT-type E3 ubiquitin ligase Itch via the ROS/JNK signaling pathway. (A) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3, 6, and 8 days postinfection (dpi), the cells were harvested, and the cell lysates were analyzed by immunoblotting with the indicated antibodies. The level of GAPDH served as a loading control. (B) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 dpi and 6 dpi, the cells were harvested with or without pretreatment with JNK inhibitor SP600125 (SP; 30 μM for 30 h) or antioxidant N-acetyl cysteine (NAC; 5 mM for 8 h). The cell lysates were analyzed by immunoblotting with the indicated antibodies. The level of GAPDH served as a loading control.
To determine whether ROS/JNK signaling pathway induces Itch activation, we treated HCV-infected cells with the specific JNK inhibitor SP600125 or the antioxidant N-acetyl cysteine (NAC). We performed immunoblot analysis to determine the phosphorylation status of Itch in HCV-infected cells at 3 dpi and 6 dpi. Treatment with SP600125 or NAC clearly inhibited HCV-induced phosphorylation of c-Jun and the levels of c-Jun (Fig. 1B, 3rd and 4th panels, lanes 4 to 6 and lanes 10 to 12). In parallel, the HCV-induced Itch phosphorylation at Thr222 was markedly reduced after treatment with SP600125 or NAC (Fig. 1B, 1st panel, lanes 4 to 6 and lanes 10 to 12). These results suggest that HCV infection promotes phosphorylation and activation of Itch via the ROS/JNK signaling pathway.
Knockdown of Itch does not affect HCV replication but decreases the release of HCV particles.
To determine the effect of Itch on the HCV life cycle, Huh-7.5 cells were infected with HCV J6/JFH1 in the presence or absence of Itch small interfering RNA (siRNA). We examined the amount of intracellular viral proteins, HCV infectivity titers, and HCV RNA, as well as extracellular HCV infectivity titers, HCV RNA, and HCV core protein. Endogenous Itch expression was markedly reduced by Itch siRNA in HCV-infected cells (Fig. 2A, 1st panel, lanes 2, 4, 6, and 8). Knockdown of Itch did not show any effect on the levels of intracellular HCV core protein and NS3 protein (Fig. 2A, 2nd and 3rd panels, lanes 1 to 8), amount of intracellular HCV infectivity titers (Fig. 2B), and HCV RNA (Fig. 2C). These results suggest that Itch does not affect HCV replication. We further examined the effect of Itch on HCV replication in HCV full-genome replicon (FGR) Con1 (RCYM1) cells. Itch siRNA efficiently knocked down endogenous Itch expression level in HCV FGR-harboring cells (Fig. 2D, 1st panel, lane 2). However, knockdown of Itch did not show any effect on the amount of NS3 protein (Fig. 2D, 2nd panel, lanes 1 and 2), and intracellular HCV RNA (Fig. 2E). These results indicate that Itch is not involved in HCV replication.
FIG 2.

Knockdown of Itch does not affect HCV replication. (A) Huh-7.5 cells were transfected with 40 nM Itch siRNA or control siRNA. At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI of 2. The cells were harvested at 1, 3, 5, and 7 dpi. The cell lysates were analyzed by immunoblotting with the indicated antibodies. The level of GAPDH served as a loading control. (B) Cell pellets were washed and disrupted by freeze-thaw, and then the intracellular virus infectivity was measured by a focus-forming assay. (C) Total cellular RNA was extracted, and HCV RNA was quantitated by RT-qPCR. Amounts of intracellular HCV RNA were normalized to amounts of GAPDH mRNA. (D) Huh-7 cells stably harboring an HCV-1b RNA full-genome replicon (FGR) derived from Con1 (RCYM1) were transfected with 40 nM Itch siRNA or control siRNA. At 48 h posttransfection, the cells were harvested. The cell lysates were analyzed by immunoblotting with the indicated antibodies. The level of GAPDH served as a loading control. (E) Total cellular RNA was extracted, and HCV RNA was quantitated by RT-qPCR. Amounts of intracellular HCV RNA were normalized to amounts of GAPDH mRNA.
Interestingly, knockdown of Itch markedly decreased the amount of extracellular HCV infectivity titers (Fig. 3A), HCV RNA (Fig. 3B), and HCV core protein (Fig. 3C). Notably, knockdown of Itch did not show any significant effect on the amount of extracellular apolipoprotein E (ApoE) (Fig. 3D). These results suggest that the decreased HCV infectivity titers by Itch knockdown are not due to the downregulation of ApoE, a cellular factor for enhancement of extracellular HCV infectivity (27). To determine whether the decreased extracellular HCV infectivity titers resulted from depletion of Itch, we transfected a plasmid encoding FLAG-tagged siRNA-resistant Itch (FLAG-Itch-R) into HCV-infected cells in the presence or absence of Itch siRNA (Fig. 3E). Immunoblot analysis showed the expression levels of endogenous Itch and FLAG-Itch-R (Fig. 3E, 1st panel, lanes 1 to 4). Overexpression of the FLAG-Itch-R recovered the extracellular HCV infectivity titers (Fig. 3F) and extracellular HCV RNA levels (Fig. 3G). These results suggest that Itch is involved in the release of infectious HCV particles, but not HCV replication.
FIG 3.

Knockdown of Itch decreases the release of HCV particles. (A) Huh-7.5 cells were transfected with 40 nM Itch siRNA or control siRNA. At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI of 2. The culture supernatants were collected at 1, 3, 5, and 7 dpi. The extracellular virus infectivity was measured by a focus-forming assay. (B) Extracellular HCV RNA was extracted and quantitated by RT-qPCR. The value for the control siRNA-transfected cells at 1 dpi was arbitrarily expressed as 1.0. (C) The amount of extracellular HCV core protein was measured by core ELISA. The value for the control siRNA-transfected cells at 1 dpi was arbitrarily expressed as 1.0. Data represent means ± SEM of data from three independent experiments; **, P < 0.01 compared with the controls. (D) The amount of extracellular ApoE protein was measured by the human ApoE ELISA kit. (E) Huh-7.5 cells were transfected with 40 nM Itch siRNA or control siRNA. At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were transfected with Itch siRNA-resistant plasmid pCAG-FLAG-Itch-R. At 72 h after plasmid transfection, the cells were harvested, and cell lysates were analyzed by immunoblotting with anti-Itch MAb, anti-NS3 MAb, and anti-GAPDH MAb. The level of GAPDH served as a loading control. (F) Culture supernatants were collected, and the extracellular virus infectivity was measured by a focus-forming assay. (G) The extracellular HCV RNA was extracted and quantitated by RT-qPCR (G). Data represent means ± SEM of data from three independent experiments.
Inhibition of the ROS/JNK signaling pathway decreases the release of infectious HCV particles.
To determine the role of the ROS/JNK signaling pathway in the release of infectious HCV particles, we examined the effects of the antioxidant NAC and the JNK inhibitor SP600125 on the release of infectious HCV particles at 3 dpi and 6 dpi. Inhibition of the ROS/JNK signaling pathway by the antioxidant NAC or the JNK inhibitor SP600125 significantly decreased extracellular HCV infectivity titers (Fig. 4A), but not intracellular HCV infectivity titers (Fig. 4B) and HCV RNA (Fig. 4C). To test whether combined treatment of HCV-infected cells with JNK inhibitor SP600125 and Itch siRNA leads to further reduced release of HCV particles, we treated HCV-infected cells with JNK inhibitor SP600125 in the presence or absence of Itch siRNA at 3 dpi and 6 dpi, respectively. Endogenous Itch expression was reduced by Itch siRNA in HCV-infected cells (Fig. 4D, top right panel, lanes 2, 4, 6, and 8). Combined treatment with SP600125 and Itch siRNA caused a slight decrease in extracellular HCV infectivity titers compared to SP600125 alone treatment or knockdown of Itch alone (Fig. 4D, left panel). These results suggest that ROS/JNK signaling pathway promotes the release of infectious HCV particles, but not HCV replication.
FIG 4.

Inhibition of the ROS/JNK signaling pathway decreases the release of infectious HCV particles. Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 and 6 dpi, the cells and the culture supernatants were harvested with or without pretreatment with JNK inhibitor SP600125 (SP; 30 μM for 30 h) or antioxidant NAC (5 mM for 8 h). The extracellular virus infectivity (A) and intracellular virus infectivity (B) were measured by a focus-forming assay. (C) Intracellular HCV RNA was quantitated by RT-qPCR. Amounts of intracellular HCV RNA were normalized to amounts of GAPDH mRNA. The value for the untreated control cells was arbitrarily expressed as 1.0. (D) Huh-7.5 cells were transfected with 40 nM Itch siRNA or control siRNA. At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 and 6 dpi, the cells and the culture supernatants were harvested with or without pretreatment with JNK inhibitor SP600125 (SP; 30 μM for 30 h). (D, Left) Extracellular virus infectivity was measured by a focus-forming assay. (D, Right) Cell lysates were analyzed by immunoblotting with anti-Itch MAb and anti-GAPDH MAb. The level of GAPDH served as a loading control. Data represent means ± SEM of data from three independent experiments, *, P < 0.05 compared with the untreated control.
HCV infection specifically promotes VPS4A polyubiquitylation via JNK/Itch signaling pathway.
To determine whether HCV-induced activated Itch promotes the polyubiquitylation of AAA ATPase VPS4A or VPS4B, we performed cell-based ubiquitylation assays. We infected Huh-7.5 cells with HCV J6/JFH1 and then transfected cells with pcDNA-VPS4A-FLAG or pcDNA-VPS4B-FLAG together with a plasmid pRK5-HA-ubiquitin (Ub) encoding HA-tagged Ub. HCV-infected cells or mock-infected control cells at 5 days after virus infection were treated with the specific JNK inhibitor SP600125. Cell lysates were immunoprecipitated with anti-FLAG beads and immunoblotted with anti-HA polyclonal antibody to detect ubiquitylated VAP4A-FLAG and VPS4B-FLAG proteins, respectively (Fig. 5A). HCV infection markedly increased VPS4A polyubiquitylation compared to the mock-infected control cells (Fig. 5A, 1st panel, lanes 3 and 9). Moreover, treatment of the HCV-infected cells with the JNK inhibitor SP600125 greatly decreased HCV-induced VPS4A polyubiquitylation (Fig. 5A, 1st panel, lanes 11 and 9), suggesting that the JNK/Itch signaling pathway is involved in VPS4A polyubiquitylation. HCV infection did not show any effect on the VPS4B polyubiquitylation (Fig. 5A, 1st panel, lanes 4 and 10). Treatment with JNK inhibitor SP600125 inhibited HCV-induced increased Itch phosphorylation (Fig. 5A, 5th panel, lanes 11 and 12), phosphorylation of c-Jun, and the levels of c-Jun (Fig. 5A, 7th and 8th panels, lanes 11 and 12). These results suggest that HCV infection specifically promotes VPS4A polyubiquitylation via the JNK/Itch signaling pathway.
FIG 5.

HCV infection specifically promotes VPS4A polyubiquitylation via the JNK/Itch signaling pathway. (A) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were transfected with pcDNA-VPS4A-FLAG or pcDNA-VPS4B-FLAG together with pRK5-HA-Ub as indicated. At 4 days postinfection, HCV-infected cells and uninfected control cells were treated with JNK inhibitor SP600125 (SP; 30 μM). The cells were harvested at 30 h after treatment with SP600125, and cell lysates were immunoprecipitated with anti-FLAG beads, followed by immunoblotting with anti-HA PAb (1st panel) or anti-FLAG PAb (2nd panel). Input samples were immunoblotted with anti-HA PAb (3rd panel), anti-FLAG PAb (4th panel), anti-phospho-Itch (T222) PAb (5th panel), anti-Itch MAb (6th panel), anti-phospho-c-Jun (S63) RMAb (7th panel), anti-c-Jun RMAb (8th panel), anti-NS3 MAb (9th panel), and anti-GAPDH MAb (10th panel), respectively. The level of GAPDH served as a loading control. HC, immunoglobulin heavy chain. (B) Huh-7.5 cells were transfected with 40 nM Itch siRNA or control siRNA. At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI at 2. At 3 h postinfection, cells were cotransfected with pcDNA-VPS4A-FLAG and pRK5-HA-Ub as indicated. At 4 days after plasmid transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-FLAG beads, followed by immunoblotting with anti-HA PAb (1st panel) or anti-FLAG PAb (2nd panel). Input samples were immunoblotted with anti-HA PAb (3rd panel), anti-FLAG PAb (4th panel), anti-Itch MAb (5th panel), anti-NS3 MAb (6th panel), and anti-GAPDH MAb (7th panel), respectively. The level of GAPDH served as a loading control.
To further validate the effect of Itch on VPS4A polyubiquitylation, we transfected HCV-infected Huh7.5 cells or mock-infected control cells with pcDNA-VPS4A-FLAG together with pRK5-HA-Ub in the presence or absence of Itch siRNA. Knockdown of Itch resulted in a decrease in HCV-induced VPS4A polyubiquitylation (Fig. 5B, 1st panel, lanes 2 and 4), suggesting that Itch is involved in VPS4A polyubiquitylation in HCV-infected cells.
Knockdown of VPS4A decreases the release of infectious HCV particles.
To determine a role of VPS4A in the release of infectious HCV particles, Huh7.5 cells were infected with HCV J6/JFH1 in the presence of VPS4A siRNA and VPS4B siRNA, respectively. Endogenous expression of VPS4A and VPS4B in HCV-infected cells was greatly reduced by siRNAs (Fig. 6A, 1st panel, lane 2; 2nd panel, lane 3). Notably, knockdown of VPS4A, but not VPS4B, significantly decreased the extracellular HCV infectivity titers (Fig. 6B). We confirmed that neither VPS4A nor VPS4B knockdown affected the intracellular HCV NS3 protein levels (Fig. 6A, 3rd panel, lanes 1 to 3), intracellular HCV infectivity titers (Fig. 6C), and intracellular HCV RNA levels (Fig. 6D). These results suggest that VPS4A, but not VPS4B, is involved in the release of infectious HCV particles.
FIG 6.
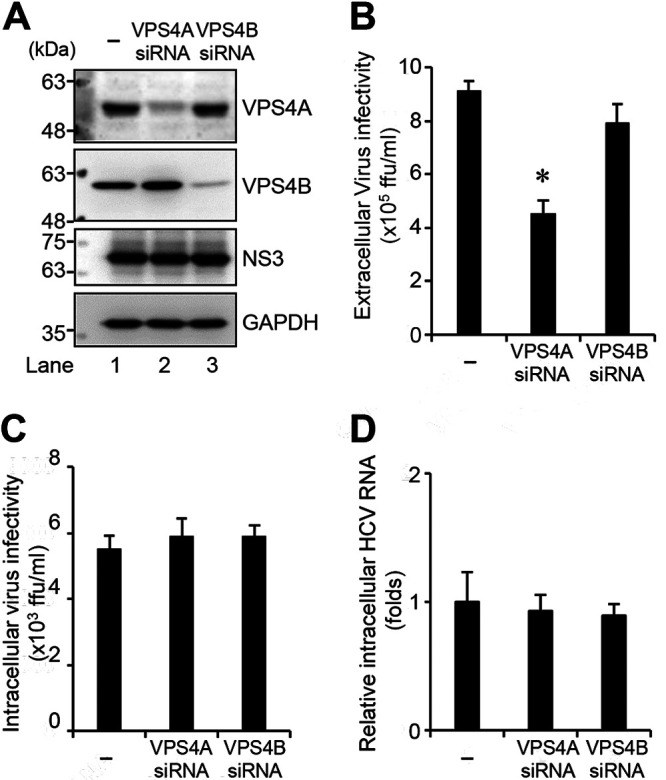
Knockdown of VPS4A decreases the release of infectious HCV particles. (A) Huh-7.5 cells were transfected with either VPS4A siRNA (80 nM) or VPS4B siRNA (40 nM). At 24 h posttransfection, cells were infected with HCV J6/JFH1 at an MOI of 2. The cell lysate and the culture supernatants were harvested at 6 dpi. The cell lysates were analyzed by immunoblotting with the indicated antibodies. The level of GAPDH served as a loading control. (B and C) Extracellular virus infectivity (B) and intracellular virus infectivity (C) were measured by a focus-forming assay. (D) Intracellular HCV RNA was quantitated by RT-qPCR. Amounts of intracellular HCV RNA were normalized to amounts of GAPDH mRNA. The value for the control cells was arbitrarily expressed as 1.0. Data represent means ± SEM of data from three independent experiments, *, P < 0.05, compared with the control.
HCV induces polyubiquitylation of VPS4A via the K6-, K11-, K27-, and K29-linkage polyubiquitin chains.
To determine the types of polyubiquitin chains on VPS4A, HCV-infected cells or mock-infected control cells were transfected with pcDNA-VPS4A-FLAG together with a series of ubiquitin K-only constructs in which only the indicated lysine was encoded, while the other six lysine residues were mutated to arginine residues. K0, in which all lysine residues are mutated to arginine residues, represents no lysine available. Cell lysates were immunoprecipitated with anti-FLAG beads and immunoblotted with anti-HA polyclonal antibody to detect ubiquitylated VPS4A-FLAG. Interestingly, polyubiquitylation of VPS4A-FLAG was detected using wild-type (WT) and K6-, K11-, K27-, and K29-Ub upon HCV infection (Fig. 7A, 1st panel, lanes 6, 8, and 10; Fig. 7B, 1st panel, lanes 2 and 4). No polyubiquitylation of VPS4A-FLAG was detected using other ubiquitin plasmids, such as K0-, K33-, K48-, and K63-Ub. These results suggest that VPS4A is polyubiquitylated via the K6-, K11-, K27-, and K29-linkage polyubiquitin chains in HCV-infected cells.
FIG 7.

HCV induces polyubiquitylation of VPS4A via the K6-, K11-, K27-, and K29-linkage polyubiquitin chains. (A and B) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were cotransfected with pcDNA-VPS4A-FLAG and the indicated HA-tagged Ub plasmids. At 4 days transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-FLAG beads, followed by immunoblotting with anti-HA PAb (1st panel) or anti-FLAG PAb (2nd panel). Input samples were immunoblotted with anti-HA PAb (3rd panel), anti-FLAG PAb (4th panel), anti-NS3 MAb (5th panel), or anti-GAPDH MAb (6th panel), respectively. The level of GAPDH served as a loading control. HC, immunoglobulin heavy chain. (C) HCV-infected cells and mock-infected control cells were transfected with pcDNA-VPS4A-FLAG. The cells were treated with 50 μg/mL cycloheximide (CHX) at 48 h after transfection. The cell lysates were harvested at 0, 8, 16, and 24 h after treatment with CHX, followed by immunoblotting with anti-FLAG PAb (top), anti-p53 MAb (middle), and anti-GAPDH MAb (bottom). The level of GAPDH served as a loading control. (D) Specific signals were quantitated by densitometry, and the percentage of remaining VPS4A-FLAG, p53, and GAPDH at each time was compared with that at the starting point, respectively. Closed triangles, HCV-infected cells; closed circles, mock-infected control cells. Red lines, blue lines, and black lines indicate VPS4A-FLAG, p53, and GAPDH, respectively. H, HCV; M, mock.
To determine whether HCV-induced VPS4A polyubiquitylation affects the stability of VPS4A, we performed kinetic analysis using the protein synthesis inhibitor cycloheximide (CHX). VPS4A-FLAG protein was expressed in HCV-infected cells or mock-infected control cells. The CHX half-life experiment demonstrated no significant difference in the half-life of VPS4A-FLAG protein in HCV-infected cells and mock-infected control cells (Fig. 7C, 1st panel, and Fig. 7D, red lines). On the other hand, the half-life of endogenous mutant p53 protein was shortened in HCV-infected cells compared to those in the mock-infected control cells (Fig. 7C, 2nd panel, and Fig. 7D, blue lines). This result is consistent with a previous report (28) that p53 is rapidly degraded in HCV-infected Huh-7.5 cells. On the one hand, the half-life of endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein did not show any significant change upon HCV infection (Fig. 7C, 3rd panel, and Fig. 7D, black lines). These results suggest that HCV-mediated VPS4A polyubiquitylation does not affect the stability of VPS4A.
Lys23 and Lys121 of VPS4A are ubiquitylation acceptor sites for HCV-mediated VPS4A polyubiquitylation.
To determine the ubiquitylation acceptor sites for HCV-mediated VPS4A polyubiquitylation, we aligned the amino acid sequences of VPS4A and VPS4B. VPS4A and VPS4B have 40 Lys residues, and 37 Lys residues are common between these two isoforms (Fig. 8A, highlighted in blue). In addition, we found that Lys23, Lys82, and Lys121 are specific Lys residues for VPS4A (Fig. 8A, highlighted in pink). Therefore, we hypothesized that these three Lys residues are candidates for ubiquitylation acceptor sites.
FIG 8.

Lys23 and Lys121 of VPS4A are ubiquitylation acceptor sites for HCV-mediated VPS4A polyubiquitylation. (A) Amino acid sequence alignment of VPS4A (GenBank accession number NM_013245.3) and VPS4B (GenBank accession number NM_004869.4). Common Lys residues are highlighted in blue, specific Lys residues for VPS4A are highlighted in pink, and specific Lys residues for VPS4B are highlighted in yellow. Numbers indicate the position of amino acid residues. (B and C) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were transfected with expression plasmids for each FLAG-tagged VPS4A single-point mutant (B) or double-point mutant (C) together with HA-tagged Ub plasmid. At 4 days transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-FLAG beads, followed by immunoblotting with anti-HA PAb (1st panel) or anti-FLAG PAb (2nd panel). Input samples were immunoblotted with anti-HA PAb (3rd panel), anti-FLAG PAb (4th panel), anti-NS3 MAb (5th panel), and anti-GAPDH MAb (6th panel), respectively. The level of GAPDH served as a loading control. HC, immunoglobulin heavy chain.
We constructed three VPS4A mutants, containing a point mutation of Lys to Arg at Lys23, Lys82, and Lys121, respectively. The cell-based ubiquitylation assays, coupled with immunoblotting, revealed that these three single-point mutants were polyubiquitylated (Fig. 8B, 1st panel, lanes 6, 8, and 10). We further constructed three double-point mutants, VPS4A K23R/K82R, VPS4A K23R/K121R, and VPS4A K82R/K121R, respectively. Notably, the double-point mutant VPS4A K23R/K121R, but not VPS4A K23R/K82R and VPS4A K82R/K121R, markedly decreased HCV-induced VPS4A polyubiquitylation (Fig. 8C, 1st panel, lanes 6, 8, and 10). These results suggest that Lys23 and Lys121 of VPS4A are ubiquitylation acceptor sites for HCV-mediated VPS4A polyubiquitylation.
HCV infection enhances the interaction between VPS4A and CHMP1B via VPS4A polyubiquitylation.
To address the mechanism underlying VPS4A polyubiquitylation-enhanced release of infectious HCV particles, we performed coimmunoprecipitation analysis using plasmids for nine human ESCRT-III proteins (CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4B, CHMP5, CHMP6, and CHMP7). HCV-infected cells or mock-infected control cells were expressed with VPS4A-Myc-His6 together with FLAG-tagged ESCRT-III proteins, as indicated in Fig. 9A and B. At 4 days postinfection, cells were harvested, and cell lysates were immunoprecipitated with anti-Myc beads. Immunoprecipitation analysis revealed that FLAG-CHMP1B was coimmunoprecipitated with VPS4A-Myc-His6 in the mock-infected control cells (Fig. 9A, 1st panel, lane 5). Furthermore, the coprecipitation of FLAG-CHMP1B with VPS4A-Myc-His6 using anti-Myc beads increased by 1.8-fold in HCV-infected cells, suggesting that HCV infection enhances the interaction between VPS4A and CHMP1B (Fig. 9A, 1st panel, lane 6). No other FLAG-tagged ESCRT-III proteins were coprecipitated with VPS4A-Myc-His6 in either control cells or HCV-infected cells (Fig. 9A, 1st panel, lanes 3, 4, and 7 to 10; Fig. 9B, 1st panel, lanes 1 to 8). These results suggest that VPS4A specifically interacts with CHMP1B upon HCV infection.
FIG 9.

HCV infection enhances the interaction between VPS4A and CHMP1B via VPS4A polyubiquitylation. (A and B) Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were cotransfected with pEF1A-VPS4A-myc-His6 and the indicated 3×FLAG-tagged CHMP plasmids. At 4 days transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-Myc beads, followed by immunoblotting with anti-FLAG PAb (1st panel) or anti-VPS4A PAb (2nd panel). The relative levels of coimmunoprecipitated FLAG-tagged CHMP1B protein were quantitated by densitometry and indicated below in the respective lanes. Input samples were immunoblotted with anti-FLAG PAb (3rd panel), anti-Myc MAb (4th panel), anti-NS3 MAb (5th panel), and anti-GAPDH MAb (6th panel), respectively. The level of GAPDH served as a loading control. HC, immunoglobulin heavy chain; LC, immunoglobulin light chain. Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, cells were cotransfected with pCMV-3×FLAG-CHMP7 (C) or pCMV-3×FLAG-CHMP1B (D) and pEF1A-VPS4A-Myc-His6, or pEF1A-VPS4A (K23R/K121R)-Myc-His6, or pEF1A-VPS4B-Myc-His6 as indicated. At 4 days transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-Myc beads, followed by immunoblotting with anti-FLAG PAb (1st panel) or anti-Myc MAb (2nd panel). The relative levels of coimmunoprecipitated FLAG-tagged CHMP1B protein were quantitated by densitometry and indicated below in the respective lanes. Input samples were immunoblotted with anti-FLAG PAb (3rd panel), anti-Myc MAb (4th panel), anti-NS3 MAb (5th panel), anti-GAPDH MAb (6th panel), respectively. The level of GAPDH served as a loading control. HC, immunoglobulin heavy chain.
We noticed that a very faint FLAG-CHMP7 band was detected after coimmunoprecipitation with VPS4A-Myc-His6 in the mock-infected control cells and in HCV-infected cells (Fig. 9B, 1st panel, lanes 9 and 10). To determine whether the FLAG-CHMP7 band detected after coimmunoprecipitation with anti-Myc-beads was specific or not, we performed coimmunoprecipitation with anti-Myc-beads in the presence or absence of VPS4A-Myc-His6 (Fig. 9C). Immunoprecipitation analysis revealed that FLAG-CHMP7 band was detected even in the absence of VPS4A-Myc-His6 (Fig. 9C, 1st panel, lanes 1 and 2). We thus concluded that FLAG-CHMP7 nonspecifically bound to anti-Myc beads and was not coimmunoprecipitated with VPS4A-Myc-His6.
To further determine whether polyubiquitylation of VPS4A is important for the interaction between VPS4A and CHMP1B, we transfected HCV-infected cells with pEF1A-VPS4A-Myc-His6, pEF1A-VPS4A (K23R/K121R)-Myc-His6, or pEF1A-VPS4B-Myc-His6 together with pCMV-3×FLAG-CHMP1B. Immunoprecipitation analysis using anti-Myc beads revealed that less FLAG-CHMP1B was coimmunoprecipitated with VPS4A (K23R/K121R)-Myc-His6 than wild-type VPS4A-Myc-His6 upon HCV infection (Fig. 9D, 1st panel, lanes 4 and 6). FLAG-CHMP1B was hardly coprecipitated with VPS4B-Myc-His6 in HCV-infected cells (Fig. 9D, 1st panel, lane 8). These results suggest that HCV infection enhances the interaction between VPS4A and CHMP1B via VPS4A polyubiquitylation.
HCV infection enhances the ATPase activity of VPS4A via VPS4A polyubiquitylation.
VPS4A ATP hydrolysis is required for disassembly of the ESCRT-III complex from the MVB membrane. To determine the effect of HCV infection on the ATPase activity of VPS4A, HCV-infected cells or mock-infected control cells were transfected with pEF1A-VPS4A-Myc-His6, pEF1A-VPS4A (K23R/K121R)-Myc-His6, or pEF1A-VPS4B-Myc-His6, respectively. At 4 days postinfection, the cells were lysed, and VPS4A-Myc-His6, VPS4A K23R/K121R-Myc-His6, and VPS4B-Myc-His6 proteins were purified using anti-Myc beads, respectively. After elution with Myc peptide (Fig. 10A, 2nd panel), these Myc-tagged proteins were precipitated with 3.2 M ammonium sulfate followed by resuspension in ATPase assay buffer (Fig. 10A, 1st panel). The ATPase assay showed that ATPase activity of VPS4A was significantly increased in HCV-infected cells compared with the control cells (Fig. 10B). On the other hand, HCV infection did not affect ATPase activity of VPS4A K23R/K121R or VPS4B (Fig. 10B). These results suggest that HCV infection enhances ATPase activity of VPS4A via VPS4A polyubiquitylation.
FIG 10.

HCV infection enhances the ATPase activity of VPS4A via VPS4A polyubiquitylation. Huh-7.5 cells were infected with HCV J6/JFH1 at an MOI of 2. At 3 h postinfection, HCV-infected cells or mock-infected control cells were transfected with pEF1A-VPS4A-Myc-His6, pEF1A-VPS4A (K23R/K121R)-Myc-His6, or pEF1A-VPS4B-Myc-His6 as indicated. At 4 days after transfection, cells were harvested, and cell lysates were immunoprecipitated with anti-Myc beads, followed by elution with Myc peptide. The eluate was precipitated with 3.2 M ammonium sulfate, followed by resuspending in ATPase assay buffer. (A) The purified proteins were subjected to immunoblotting with anti-Myc MAb (1st panel, after precipitation with ammonium sulfate; 2nd panel, after elution with Myc peptide). Input samples were immunoblotted with anti-Myc MAb (3rd panel), anti-NS3 MAb (4th panel), and anti-GAPDH MAb (5th panel), respectively. The level of GAPDH served as a loading control. Arrows indicate immunoglobulin heavy chain. (B) ATPase activities of VPS4A-Myc-His6, VPS4A K23R/K121R-Myc-His6, or VPS4B-Myc-His6 expressed in HCV-infected cells or mock-infected controls cells were measured by ATPase assay. The value of mock-infected controls cells was used as background and was arbitrarily defined as 0. Background-subtracted ATPase activities were shown. Data represent means ± SEM of data from three independent experiments, *, P < 0.05.
Taken together, we propose a model in which the HCV-induced ROS/JNK signaling pathway activates the E3 ubiquitin ligase Itch to promote VPS4A polyubiquitylation, leading to enhanced VPS4A-CHMP1B interaction and VPS4A ATPase activity, thereby promoting the release of HCV particles (Fig. 11).
FIG 11.
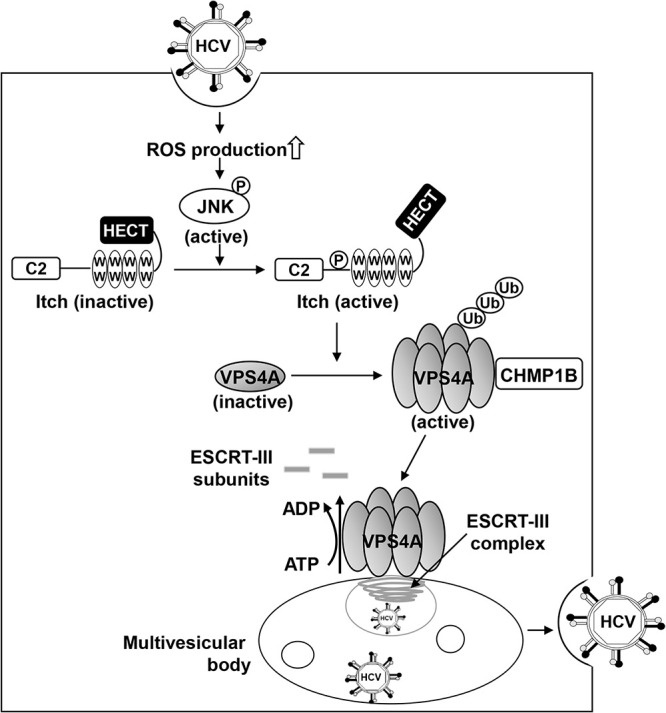
A proposed mechanism of the ROS/JNK/Itch signaling pathway-mediated HCV particle release. HCV infection causes an increase in ROS production and JNK activation, which phosphorylates Itch, a HECT-type E3 ligase with a C2 domain, four WW domains, and a HECT domain, leading to a conformational change and activation of Itch. Activated Itch promotes polyubiquitylation of VPS4A, leading to an increased interaction between VPS4A and CHMP1B, which may be involved in the promotion of VPS4A ATPase activity and formation of VPS4A hexamer. Activated VPS4A dissociates ESCRT-III complex from endosomal membranes, resulting in membrane scission and formation of the multivesicular body. Subsequently, ESCRT pathway-mediated release of HCV particles is enhanced. P, phosphorylation; Ub, ubiquitylation.
DISCUSSION
In this study, we demonstrated that HECT-type E3 ubiquitin ligase Itch was activated by the HCV infection-induced ROS/JNK signaling pathway (Fig. 1). Importantly, Itch is involved in the release of infectious HCV particles (Fig. 3). To explore the molecular mechanism of Itch-mediated HCV particle release, we investigated the effect of Itch on the polyubiquitylation of VPS4. Cell-based ubiquitylation assays showed that the HCV-induced JNK/Itch signaling pathway specifically promoted polyubiquitylation of VPS4A, but not VPS4B (Fig. 5). In addition, VPS4A, but not VPS4B, was involved in releasing HCV particles (Fig. 6). Next, we explored the possible involvement of the VPS4A polyubiquitylation in the activation of VPS4A ATPase activity. Immunoprecipitation analysis revealed that HCV infection specifically enhanced the interaction between VPS4A and CHMP1B via VPS4A polyubiquitylation (Fig. 9). Moreover, HCV infection significantly enhanced ATPase activity of VPS4A, but not VPS4B (Fig. 10). Collectively, our results suggest that the ROS/JNK/Itch signaling pathway enhances the release of HCV particles via VPS4A polyubiquitylation to enhance its interaction with CHMP1B and ATPase activity (Fig. 11). To our knowledge, this is the first study to clarify the role of Itch in the release of infectious HCV particles.
Several studies demonstrated possible links between Itch and virus budding and release. For example, Itch induces influenza A virus release from endosomes through ubiquitylation of viral M1 protein (29). Itch also facilitates Ebola virus budding (30) and nuclear egress of Epstein-Barr virus (31) through interaction with viral proteins. In this study, we further expanded the role of Itch in the release of HCV particles.
The ESCRT machinery is required for the biogenesis of MVBs, a special subset of late endosomes, generated by the inward budding of numerous intraluminal vesicles. It is well-known that many enveloped RNA viruses, including human immunodeficiency virus type 1 (HIV-1), utilize ESCRT machinery to acquire their membrane envelopes and affect assembly and release following membrane scission (32–34). In addition, ESCRT machinery is also hijacked by nonenveloped RNA viruses, such as hepatitis A virus (35), bluetongue virus (BTV) (36), and enveloped DNA viruses, including hepatitis B virus (37), for facilitation of intracellular budding or release. Importantly, the release of infectious HCV particles depends on components of the ESCRT machinery, such as HRS (ESCRT-0) (8), TSG101 (ESCRT-I) (12), ESCRT-III (12, 13), Alix (a protein that bridges ESCRT-I and ESCRT-III) (9, 12), and VPS4 (12, 13), while the assembly of intracellular infectious virus particles may be independent of the late ESCRT pathway that mediates membrane fission (12, 13). Moreover, HCV particles have been observed to traffic to recycling endosomes (38, 39).
Consistent with previous studies, we found that VPS4, either VPS4A or VPS4B, did not show any significant effect on HCV replication as well as intracellular HCV assembly. Importantly, our data showed that VPS4A, but not VPS4B, was involved in infectious HCV particle release (Fig. 6), suggesting that VPS4A specifically plays a role in the trafficking of HCV particles into a secretory compartment MVB. VPS4A ATP hydrolysis is required for disassembly of the ESCRT-III complex from the MVB membrane (40). Although the mechanistic details of VPS4A activation need further investigation, there is an increasing body of evidence that interaction between VPS4A and ESCRT-III proteins, including CHMP1B, induces VPS4A ATPase activity through relief of VPS4A autoinhibition (41, 42). It is noteworthy that HCV infection greatly enhanced the interaction between VPS4A and CHMP1B (Fig. 9) and promoted VPS4A ATPase activity (Fig. 10). We reason that HCV infection may specifically promote VPS4A ATP hydrolysis activity via association with CHMP1B. In addition, we demonstrated evidence suggesting that VPS4A polyubiquitylation is involved in the enhancement of interaction between VPS4A and CHMP1B and the promotion of VPS4A ATPase activity. In this context, VPS4A has been reported to interact directly with HCV NS5A protein (9). Interestingly, a part of the NS5A-containing low-density HCV particles most likely exit the cells via fusion of MVB to the plasma membrane (43). These findings further highlight the importance of VPS4A in the release of HCV particles.
We noted that although knockdown of Itch markedly decreased the release of HCV particles, still, substantial amounts of infectious HCV particles were left (Fig. 3) since it is known that only a part of HCV particles might be released by the MVB and a part by the conventional secretory pathway, namely, Golgi secretory pathway (5, 6). HCV particles are transported from the ER to the Golgi in COPII vesicles to embark on the Golgi secretory route (7). The roles of unconventional secretion pathways in HCV particle release also have been investigated. HCV might directly remodel the Golgi and initiate an autophagy-related release pathway (44, 45). These studies suggest that there might be more than one pathway that could be used by assembled HCV particles for release. However, it is challenging to clearly delineate the exact route of HCV particle release.
Based on our findings, we propose that HCV-induced ROS/JNK/Itch signaling pathway promotes VPS4A polyubiquitylation, leading to enhanced VPS4A-CHMP1B interaction and promotion of VPS4A ATPase activity, thereby upregulating the release of HCV particles. Our results may lead to a better understanding of the mechanistic details of HCV particle release and allow the development of a novel antiviral therapeutic strategy.
MATERIALS AND METHODS
Cell culture and virus.
A human hepatoma cell line, Huh-7.5 (46), was kindly provided by C.M. Rice (The Rockefeller University, NY). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (high glucose) with l-glutamine (Fujifilm Wako Pure Chemical Industries, Osaka, Japan) and supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Biowest, Nuaillé, France), 100 units/mL penicillin, 100 μg/mL streptomycin (Gibco, Grand Island, NY), and 0.1 mM nonessential amino acids (Gibco). Cells were transfected with plasmid DNA using FuGene 6 transfection reagents (Promega, Madison, WI).
The pFL-J6/JFH1 plasmid, which encodes the entire viral genome of a chimeric strain of HCV-2a, JFH1 (47), was kindly provided by C.M. Rice. The HCV genome RNA was synthesized in vitro using pFL-J6/JFH1 as a template and was transfected into Huh-7.5 cells by electroporation (15, 48, 49). The virus produced in the culture supernatant was used for infection experiments (15, 49). Virus infection was performed at a multiplicity of infection (MOI) of 2 in the infection experiments. Huh-7 cells stably harboring an HCV-1b FGR derived from Con1 (RCYM1) (50) were also used. The FGR cells express all of the HCV proteins ranging from the core protein to NS5B.
Expression plasmids.
The expression plasmid pMD18-human Itch was purchased from Sino Biological Inc. (Wayne, PA). The cDNA fragment of Itch was amplified by PCR using pMD18-human Itch as a template. The primer sequences were as follows: sense primer, 5′-TCGAGCTCAGCGGCCGCCATGTCTGACAGTGGATCACA-3′, and antisense primer, 5′-TCGGAATTCGCGGCCCTTGTCCAAATCCTT-3′. The amplified PCR product was purified and inserted into the NotI site of pCAG-FLAG using the In-Fusion HD cloning kit (Clontech, Mountain View, CA). To generate an Itch siRNA-resistant expression plasmid, pCAG-FLAG-Itch-R, that contains two silent mutations in the Itch siRNA target sequence, nucleotide substitutions were introduced to wild-type FLAG-Itch expression plasmid by using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The primer sequences were as follows: sense primer (FLAG-Itch-R), 5′-CTGCCACCATACAAGAGTTACGAGCAACTGAAGGAAAAG-3′, antisense primer (FLAG-Itch-R), and 5′-CTTTTCCTTCAGTTGCTCGTAACTCTTGTATGGTGGCAG-3′. The expression plasmids pcDNA-VPS4A-FLAG and pcDNA-VPS4B-FLAG have been described previously (51). The single-point mutants of VPS4A K23R, K82R, and K121R were constructed by overlap extension PCR using pcDNA-VPS4A-FLAG as a template. The specific primers used for that PCR were as follows: sense primer (K23R), 5′-CAGAGGAGGACAAAGCCAGGAACTACGAGGAGGCGCTGC-3′; antisense primer (K23R), 5′-GCAGCGCCTCCTCGTAGTTCCTGGCTTTGTCCTCCTCTG-3′; sense primer (K82R), 5′-AGCAAAGAGAAACACGGCAGGAAGCCAGTCAAAGAGAACC-3′; antisense primer (K82R), 5′-GGTTCTCTTTGACTGGCTTCCTGCCGTGTTTCTCTTTGCT-3′; sense primer (K121R), 5′-GGGTGCCGTCGTGATGGAGAGGCCCAACATACGGTGGAA-3′; and antisense primer (K121R), 5′-TTCCACCGTATGTTGGGCCTCTCCATCACGACGGCACCC-3′. The double-point mutants of VPS4A K23R/K82R, K23R/K121R, and K82R/K121R were constructed by overlap extension PCR using pcDNA-VPS4A (K23R)-FLAG or pcDNA-VPS4A (K82R)-FLAG as the templates. The specific primers used for that PCR were the same as mentioned above. The cDNA fragments of VPS4A, VPS4A K23R/K121R, and VPS4B were amplified by PCR using pcDNA-VPS4A-FLAG, pcDNA-VPS4A (K23R/K121R)-FLAG, and pcDNA-VPS4B-FLAG as the templates, respectively. The amplified PCR products were inserted into the BamHI and EcoRI sites of pEF1A-Myc-His6 (Invitrogen, Carlsbad, CA) using the In-Fusion HD cloning kit (Clontech). The expression plasmids for 3×FLAG-tagged CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4B, CHMP5, CHMP6, and CHMP7 have been described previously (52–55). N-terminal HA-tagged ubiquitin (Ub) expression plasmids, pRK5-HA-Ub-WT, pRK5-HA-Ub-K0, pRK5-HA-Ub-K6, pRK5-HA-Ub-K11, pRK5-HA-Ub-K27, pRK5-HA-Ub-K29, pRK5-HA-Ub-K33, pRK5-HA-Ub-K48, and pRK5-HA-Ub-K63, were purchased from Addgene (Watertown, MA).
Antibodies.
The mouse monoclonal antibodies (MAbs) used in this study were anti-NS3 MAb (catalog no. MAB8691; Millipore, Billerica, MA), anti-Itch MAb (catalog no. 611198; BD Biosciences, San Jose, CA), anti-c-Myc MAb (catalog no. 9E10; Santa Cruz Biotechnology, Santa Cruz, CA), anti-VPS4B MAb (catalog no. sc-377162; Santa Cruz Biotechnology), anti-HCV core MAb (clone 2H9) (48), anti-p53 (Ab-6) (catalog no. OP43; Millipore), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) MAb (catalog no. 14-25524; Fujifilm Wako Pure Chemical Industries). The rabbit monoclonal antibodies (RMAbs) used in this study were anti-c-Jun RMAb (catalog no. 9165; Cell Signaling Technology, Beverly, MA) and anti-phospho-c-Jun (Ser63) RMAb (catalog no. 9261; Cell Signaling Technology). The rabbit polyclonal antibodies (PAbs) used in this study were anti-HA PAb (catalog no. H-6908; Sigma, St. Louis, MO), anti-FLAG PAb (catalog no. 2368; Cell Signaling Technology), anti-phospho-Itch-(pT222) PAb (catalog no. AB10050; Millipore), and anti-VPS4A PAb (catalog no. ABS1646; Millipore). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Cell Signaling Technology) and HRP-conjugated anti-rabbit IgG (Cell Signaling Technology) were used as secondary antibodies.
Immunoblot analysis.
Immunoblot analysis was performed as described previously (15, 56). The cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (Millipore). The membranes were incubated with a primary antibody, followed by incubation with an HRP-conjugated secondary antibody. The positive bands were visualized using enhanced chemiluminescence Western blotting detection reagents (ECL; GE Healthcare, Buckinghamshire, UK). The intensity of bands was quantified using NIH ImageJ software (Java 1.8.0_112).
Cell-based ubiquitylation assay.
Cell-based ubiquitylation assays were performed as described previously (57, 58). Cultured cells were lysed with a buffer containing 120 mM NaCl, 50 mM HEPES (pH 7.2), 1 mM EDTA (pH 8.0), 1% NP-40, 0.5% sodium deoxycholic acid, 10 mM N-ethylmaleimide (Sigma), and protease inhibitor cocktail (Roche, Mannheim, Germany) for 30 min on ice. The lysates were centrifuged at 20,400 × g for 20 min at 4°C. To dissociate proteins, 1% SDS was added to the lysates, which were then heated at 100°C for 10 min and diluted 10-fold with the lysis buffer. FLAG-tagged VPS4A, VPS4A mutants, and VPS4B were immunoprecipitated with anti-FLAG M2 affinity gel (Sigma) at 4°C overnight, respectively. Immunoprecipitates were analyzed by immunoblotting using anti-HA PAb to detect ubiquitylated FLAG-tagged VPS4A, VPS4A mutants, and VPS4B, respectively.
Immunoprecipitation.
Cultured cells were lysed with a buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM EDTA (pH 8.0), 0.1% SDS, 1% sodium deoxycholic acid, 1% Triton X-100, and protease inhibitor cocktail (Roche) for 30 min on ice. The lysates were centrifuged at 20,400 × g for 20 min at 4°C, and the supernatant was immunoprecipitated with anti-Myc tag beads (MBL, Nagoya, Japan) at 4°C overnight. After being washed with the lysis buffer five times, the immunoprecipitates were analyzed by immunoblotting.
siRNA transfection.
HCV-infected Huh-7.5 cells or mock-infected control cells were transfected with 40 nM Itch siRNA (catalog no. SI00141085; Qiagen, Valencia, CA), 80 nM VPS4A siRNA (catalog no. M-013092-00-0005; Dharmacon, Lafayette, CO), or 40 nM VPS4B siRNA (catalog no. SI03144456; Qiagen), using Lipofectamine RNAiMAX transfection reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Allstars negative-control siRNA (Qiagen) was used as a control. All siRNAs were transfected into cells once.
Titration of extracellular and intracellular HCV infectivity.
The extracellular virus was isolated from cell culture supernatants, collected, and centrifuged at 3,000 × g for 10 min at 4°C to remove cell debris. The intracellular virus was isolated by freeze-thaw of cell lysates (59). Briefly, HCV-infected cells were washed with phosphate-buffered saline (PBS), harvested by trypsin treatment, and centrifuged at 1,000 × g for 5 min at 4°C. To release intracellular virus particles, the cells pellet was resuspended in complete DMEM and subjected to five cycles of freeze and thaw using liquid nitrogen and a 37°C water bath. Cell debris was removed by centrifugation at 20,400 × g for 20 min at 4°C.
Samples were then used to inoculate Huh-7.5 cells in triplicate. The inoculum was incubated with cells for 3 h at 37°C and then supplemented with fresh complete DMEM. At 24 h postinoculation, the cells were fixed in cold 100% methanol for 20 min at −20°C. The fixed cells were subjected to indirect immunofluorescence analysis to detect HCV core using anti-HCV core MAb (clone 2H9). The secondary antibody used was Alexa Fluor 488-conjugated goat anti-mouse IgG (catalog no. A11001; Molecular Probes, Eugene, OR). The viral titer is expressed as focus-forming units per milliliter of the sample (FFU/mL), determined by the average number of HCV core-positive foci detected.
Quantification of extracellular and intracellular HCV RNA.
To quantitate extracellular HCV RNA, culture supernatants were collected from HCV-infected cells, and viral RNA was isolated using a QIAamp viral RNA minikit (Qiagen). The total cellular RNA was isolated using a ReliaPrep RNA cell miniprep system (Promega) according to the manufacturer’s instructions. The cDNA was generated using a GoScript reverse transcription system (Promega). Real-time quantitative PCR (RT-qPCR) was performed using TB Green Premix Ex Taq II (Tli RNaseH Plus) (TaKaRa Bio, Shiga, Japan) with SYBR green chemistry on the StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) as reported previously (56). The primer sequences were as follows: HCV, 5′-AGACGTATTGAGGTCCATGC-3′ and 5′-CCGCAGCGACGGTGCTGATAG-3′. As an internal control, human GAPDH gene expression levels were measured using the primers 5′-GCCATCAATGACCCCTTCATT-3′ and 5′-TCTCGCTCCTGGAAGATGG-3′.
Quantification of extracellular HCV core protein and ApoE protein.
The HCV core protein and ApoE protein in the culture supernatants were quantified by the enzyme immunoassay using the Ortho HCV core antigen enzyme-linked immunosorbent assay (ELISA) (Ortho-Clinical Diagnostics, Raritan, NJ) and human ApoE ELISA kit (Cell Biolabs, San Diego, CA), respectively.
CHX half-life experiment.
To examine the half-life of VPS4A protein, HCV-infected cells and mock-infected control cells were transfected with pcDNA-VPS4A-FLAG. At 72 h posttransfection, cells were treated with 50 μg/mL cycloheximide (CHX). The cells at zero time points were harvested immediately after treatment with CHX. Cells from subsequent time points were incubated in a medium containing CHX at 37°C for 8, 16, and 24 h, as indicated.
ATPase activity assay.
HCV-infected cells or mock-infected control cells were transfected with pEF1A-VPS4A-Myc-His6, pEF1A-VPS4A (K23R/K121R)-Myc-His6, or pEF1A-VPS4B-Myc-His6 and harvested at 4 days postinfection. The harvested cells were lysed with a lysis buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 1 mM EDTA (pH 8.0), 0.1% SDS, 1% sodium deoxycholic acid, 1% Triton X-100, and protease inhibitor cocktail (Roche) for 30 min on ice. The lysate was centrifuged at 20,400 × g for 20 min at 4°C, and the supernatant was immunoprecipitated with anti-Myc tag beads (MBL) at 4°C overnight. After the beads were washed, the Myc-tagged proteins were eluted with the Myc peptide. The eluate was precipitated with 3.2 M ammonium sulfate for 20 min on ice. After centrifugation at 10,000 × g for 10 min at 4°C, the precipitates were resuspended in ATPase assay buffer. ATPase activities of Myc-tagged VPS4A, VPS4A K23R/K121R, and VPS4B proteins were measured by using the ATPase assay kit (catalog no. ab234055; Abcam, Cambridge, UK), according to the manufacturer’s instructions. Briefly, purified Myc-tagged VPS4A, VPS4A K23R/K121R, and VPS4B were incubated with ATPase substrate at 25°C for 30 min. After adding ATPase assay developer, ATPase activities of all samples were estimated by measuring optical density at 650 nm (OD650) on a SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA). The value of mock-infected controls cells was used as background and was arbitrarily defined as 0. ATPase activities of Myc-tagged VPS4A, VPS4A K23R/K121R, and VPS4B were calculated as background subtracted.
Statistical analysis.
Results were expressed as means ± standard errors of the means (SEM). Statistical significance was evaluated by analysis of variance (ANOVA) and was defined as a P value of <0.05.
ACKNOWLEDGMENTS
We thank Y. Kozaki for the secretarial work.
This work was supported by the Program Basic and Clinical Research on Hepatitis from the Japan Agency for Medical Research and Development (AMED) under grant nos. JP21fk0210053, JP21fk0210090, and JP21fk0310104 and by a grant from the KAKENHI under grant number 20K07514. A.A. is supported by the Program for Nurture of Next Generation Leaders Guiding Medical Innovation in Asia of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan. This study was also supported by a grant from Hyogo Science and Technology Association.
L.D. and I.S. conceived and designed the experiments. L.D. carried out most of the experiments. Y.L., A.A., C.M., and T.A. assisted the constructions and the data analysis. M.M., T.W., H.S., and M.M. contributed to the materials. L.D. and I.S. wrote the manuscript. All the authors contributed to the manuscript and approved the submitted version.
Contributor Information
Ikuo Shoji, Email: ishoji@med.kobe-u.ac.jp.
J.-H. James Ou, University of Southern California
REFERENCES
- 1.World Health Organization. 2017. Global hepatitis report. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Vermehren J, Park JS, Jacobson IM, Zeuzem S. 2018. Challenges and perspectives of direct antivirals for the treatment of hepatitis C virus infection. J Hepatol 69:1178–1187. 10.1016/j.jhep.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Heffernan A, Cooke GS, Nayagam S, Thursz M, Hallett TB. 2019. Scaling up prevention and treatment towards the elimination of hepatitis C: a global mathematical model. Lancet 393:1319–1329. 10.1016/S0140-6736(18)32277-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mankouri J, Walter C, Stewart H, Bentham M, Park WS, Heo WD, Fukuda M, Griffin S, Harris M. 2016. Release of infectious hepatitis C virus from Huh7 cells occurs via a trans-Golgi network-to-endosome pathway independent of very-low-density lipoprotein secretion. J Virol 90:7159–7170. 10.1128/JVI.00826-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunz M, Ritter M, Schindler M. 2021. HCV egress - unconventional secretion of assembled viral particles. Trends Microbiol. 10.1016/j.tim.2021.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Syed GH, Khan M, Yang S, Siddiqui A. 2017. Hepatitis C virus lipoviroparticles assemble in the endoplasmic reticulum (ER) and bud off from the ER to the Golgi compartment in COPII vesicles. J Virol 91:e00499-17. 10.1128/JVI.00499-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamai K, Shiina M, Tanaka N, Nakano T, Yamamoto A, Kondo Y, Kakazu E, Inoue J, Fukushima K, Sano K, Ueno Y, Shimosegawa T, Sugamura K. 2012. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 422:377–385. 10.1016/j.virol.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Barouch-Bentov R, Neveu G, Xiao F, Beer M, Bekerman E, Schor S, Campbell J, Boonyaratanakornkit J, Lindenbach B, Lu A, Jacob Y, Einav S. 2016. Hepatitis C virus proteins interact with the endosomal sorting complex required for transport (ESCRT) machinery via ubiquitination to facilitate viral envelopment. mBio 7:e01456-16. 10.1128/mBio.01456-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurley JH, Hanson PI. 2010. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat Rev Mol Cell Biol 11:556–566. 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. 2017. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci 42:42–56. 10.1016/j.tibs.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Ariumi Y, Kuroki M, Maki M, Ikeda M, Dansako H, Wakita T, Kato N. 2011. The ESCRT system is required for hepatitis C virus production. PLoS One 6:e14517. 10.1371/journal.pone.0014517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corless L, Crump CM, Griffin SD, Harris M. 2010. Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J Gen Virol 91:362–372. 10.1099/vir.0.017285-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng L, Shoji I, Ogawa W, Kaneda S, Soga T, Jiang DP, Ide YH, Hotta H. 2011. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. J Virol 85:8556–8568. 10.1128/JVI.00146-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng L, Adachi T, Kitayama K, Bungyoku Y, Kitazawa S, Ishido S, Shoji I, Hotta H. 2008. Hepatitis C virus infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway. J Virol 82:10375–10385. 10.1128/JVI.00395-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng L, Chen M, Tanaka M, Ku Y, Itoh T, Shoji I, Hotta H. 2015. HCV upregulates Bim through the ROS/JNK signalling pathway, leading to Bax-mediated apoptosis. J Gen Virol 96:2670–2683. 10.1099/jgv.0.000221. [DOI] [PubMed] [Google Scholar]
- 17.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. 2008. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest 118:3943–3953. 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakurai T, Maeda S, Chang L, Karin M. 2006. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci USA 103:10544–10551. 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seki E, Brenner DA, Karin M. 2012. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 143:307–320. 10.1053/j.gastro.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das M, Garlick DS, Greiner DL, Davis RJ. 2011. The role of JNK in the development of hepatocellular carcinoma. Genes Dev 25:634–645. 10.1101/gad.1989311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morton S, Davis RJ, McLaren A, Cohen P. 2003. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J 22:3876–3886. 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchs SY, Adler V, Pincus MR, Ronai Z. 1998. MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci USA 95:10541–10546. 10.1073/pnas.95.18.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. 1999. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem 274:32580–32587. 10.1074/jbc.274.46.32580. [DOI] [PubMed] [Google Scholar]
- 24.Bernassola F, Karin M, Ciechanover A, Melino G. 2008. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14:10–21. 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Zhu K, Shan Z, Chen X, Cai Y, Cui L, Yao W, Wang Z, Shi P, Tian C, Lou J, Xie Y, Wen W. 2017. Allosteric auto-inhibition and activation of the Nedd4 family E3 ligase Itch. EMBO Rep 18:1618–1630. 10.15252/embr.201744454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher E, Gao M, Liu YC, Karin M. 2006. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci USA 103:1717–1722. 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. 2010. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol 84:10999–11009. 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aydin Y, Chatterjee A, Chandra PK, Chava S, Chen W, Tandon A, Dash A, Chedid M, Moehlen MW, Regenstein F, Balart LA, Cohen A, Lu H, Wu T, Dash S. 2017. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol Commun 1:256–269. 10.1002/hep4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. 2013. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci USA 110:17516–17521. 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han Z, Sagum CA, Bedford MT, Sidhu SS, Sudol M, Harty RN. 2016. ITCH E3 ubiquitin ligase interacts with Ebola virus VP40 to regulate budding. J Virol 90:9163–9171. 10.1128/JVI.01078-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee CP, Liu GT, Kung HN, Liu PT, Liao YT, Chow LP, Chang LS, Chang YH, Chang CW, Shu WC, Angers A, Farina A, Lin SF, Tsai CH, Bouamr F, Chen MR. 2016. The ubiquitin ligase Itch and ubiquitination regulate BFRF1-mediated nuclear envelope modification for Epstein-Barr virus maturation. J Virol 90:8994–9007. 10.1128/JVI.01235-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welsch S, Muller B, Krausslich HG. 2007. More than one door - budding of enveloped viruses through cellular membranes. FEBS Lett 581:2089–2097. 10.1016/j.febslet.2007.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 35.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walker C, Lanford RE, Lemon SM. 2013. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496:367–371. 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wirblich C, Bhattacharya B, Roy P. 2006. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J Virol 80:460–473. 10.1128/JVI.80.1.460-473.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. 2007. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc Natl Acad Sci USA 104:10205–10210. 10.1073/pnas.0704000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog 8:e1002466. 10.1371/journal.ppat.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai CK, Jeng KS, Machida K, Lai MM. 2010. Hepatitis C virus egress and release depend on endosomal trafficking of core protein. J Virol 84:11590–11598. 10.1128/JVI.00587-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Babst M, Wendland B, Estepa EJ, Emr SD. 1998. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J 17:2982–2993. 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merrill SA, Hanson PI. 2010. Activation of human VPS4A by ESCRT-III proteins reveals ability of substrates to relieve enzyme autoinhibition. J Biol Chem 285:35428–35438. 10.1074/jbc.M110.126318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norgan AP, Davies BA, Azmi IF, Schroeder AS, Payne JA, Lynch GM, Xu Z, Katzmann DJ. 2013. Relief of autoinhibition enhances Vta1 activation of Vps4 via the Vps4 stimulatory element. J Biol Chem 288:26147–26156. 10.1074/jbc.M113.494112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai CK, Saxena V, Tseng CH, Jeng KS, Kohara M, Lai MM. 2014. Nonstructural protein 5A is incorporated into hepatitis C virus low-density particle through interaction with core protein and microtubules during intracellular transport. PLoS One 9:e99022. 10.1371/journal.pone.0099022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen MD, Johnsen IB, Stiberg KA, Sherstova T, Wakita T, Richard GM, Kandasamy RK, Meurs EF, Anthonsen MW. 2017. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc Natl Acad Sci USA 114:E3462–E3471. 10.1073/pnas.1616683114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shrivastava S, Devhare P, Sujijantarat N, Steele R, Kwon YC, Ray R, Ray RB. 2016. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J Virol 90:1387–1396. 10.1128/JVI.02383-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buonocore L, Blight KJ, Rice CM, Rose JK. 2002. Characterization of vesicular stomatitis virus recombinants that express and incorporate high levels of hepatitis C virus glycoproteins. J Virol 76:6865–6872. 10.1128/jvi.76.14.6865-6872.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 48.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bungyoku Y, Shoji I, Makine T, Adachi T, Hayashida K, Nagano-Fujii M, Ide YH, Deng L, Hotta H. 2009. Efficient production of infectious hepatitis C virus with adaptive mutations in cultured hepatoma cells. J Gen Virol 90:1681–1691. 10.1099/vir.0.010983-0. [DOI] [PubMed] [Google Scholar]
- 50.Murakami K, Ishii K, Ishihara Y, Yoshizaki S, Tanaka K, Gotoh Y, Aizaki H, Kohara M, Yoshioka H, Mori Y, Manabe N, Shoji I, Sata T, Bartenschlager R, Matsuura Y, Miyamura T, Suzuki T. 2006. Production of infectious hepatitis C virus particles in three-dimensional cultures of the cell line carrying the genome-length dicistronic viral RNA of genotype 1b. Virology 351:381–392. 10.1016/j.virol.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 51.Kamada K, Shoji I, Deng L, Aoki C, Ratnoglik SL, Wakita T, Hotta H. 2012. Generation of a recombinant reporter hepatitis C virus useful for the analyses of virus entry, intra-cellular replication and virion production. Microbes Infect 14:69–78. 10.1016/j.micinf.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Yorikawa C, Takaya E, Osako Y, Tanaka R, Terasawa Y, Hamakubo T, Mochizuki Y, Iwanari H, Kodama T, Maeda T, Hitomi K, Shibata H, Maki M. 2008. Human calpain 7/PalBH associates with a subset of ESCRT-III-related proteins in its N-terminal region and partly localizes to endocytic membrane compartments. J Biochem 143:731–745. 10.1093/jb/mvn030. [DOI] [PubMed] [Google Scholar]
- 53.Katoh K, Shibata H, Suzuki H, Nara A, Ishidoh K, Kominami E, Yoshimori T, Maki M. 2003. The ALG-2-interacting protein Alix associates with CHMP4b, a human homologue of yeast Snf7 that is involved in multivesicular body sorting. J Biol Chem 278:39104–39113. 10.1074/jbc.M301604200. [DOI] [PubMed] [Google Scholar]
- 54.Okumura M, Takahashi T, Shibata H, Maki M. 2013. Mammalian ESCRT-III-related protein IST1 has a distinctive met-pro repeat sequence that is essential for interaction with ALG-2 in the presence of Ca2+. Biosci Biotechnol Biochem 77:1049–1054. 10.1271/bbb.130022. [DOI] [PubMed] [Google Scholar]
- 55.Horii M, Shibata H, Kobayashi R, Katoh K, Yorikawa C, Yasuda J, Maki M. 2006. CHMP7, a novel ESCRT-III-related protein, associates with CHMP4b and functions in the endosomal sorting pathway. Biochem J 400:23–32. 10.1042/BJ20060897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng L, Gan X, Ito M, Chen M, Aly HH, Matsui C, Abe T, Watashi K, Wakita T, Suzuki T, Okamoto T, Matsuura Y, Mizokami M, Shoji I, Hotta H. 2019. Peroxiredoxin 1, a novel HBx-interacting protein, interacts with exosome component 5 and negatively regulates hepatitis B virus (HBV) propagation through degradation of HBV RNA. J Virol 93:e02203-18. 10.1128/JVI.02203-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirakura M, Murakami K, Ichimura T, Suzuki R, Shimoji T, Fukuda K, Abe K, Sato S, Fukasawa M, Yamakawa Y, Nishijima M, Moriishi K, Matsuura Y, Wakita T, Suzuki T, Howley PM, Miyamura T, Shoji I. 2007. E6AP ubiquitin ligase mediates ubiquitylation and degradation of hepatitis C virus core protein. J Virol 81:1174–1185. 10.1128/JVI.01684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wertz IE, O'Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, Dixit VM. 2004. Human de-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science 303:1371–1374. 10.1126/science.1093549. [DOI] [PubMed] [Google Scholar]
- 59.Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. 2010. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog 6:e1001087. 10.1371/journal.ppat.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]