

ABSTRACT
Cancer-causing human papillomavirus (HPV) E6 oncoproteins contain a well-characterized phosphoacceptor site within the PDZ (PSD-95/Dlg/ZO-1) binding motif (PBM) at the C terminus of the protein. Previous studies have shown that the threonine or serine residue in the E6 PBM is subject to phosphorylation by several stress-responsive cellular kinases upon the induction of DNA damage in cervical cancer-derived cells. However, there is little information about the regulation of E6 phosphorylation in the absence of DNA damage and whether there may be other pathways by which E6 is phosphorylated. In this study, we demonstrate that loss of E6AP results in a dramatic increase in the levels of phosphorylated E6 (pE6) despite the expected overall reduction in total E6 protein levels. Furthermore, phosphorylation of E6 requires transcriptionally active p53 and occurs in a manner that is dependent upon DNA-dependent protein kinase (DNA PK). These results identify a novel feedback loop, where loss of E6AP results in upregulation of p53, leading to increased levels of E6 phosphorylation, which in turn correlates with increased association with 14-3-3 and inhibition of p53 transcriptional activity.
IMPORTANCE This study demonstrates that the knockdown of E6AP from cervical cancer-derived cells leads to an increase in phosphorylation of the E6 oncoprotein. We show that this phosphorylation of E6 requires p53 transcriptional activity and the enzyme DNA PK. This study therefore defines a feedback loop whereby activation of p53 can induce phosphorylation of E6 and which in turn can inhibit p53 transcriptional activity independently of E6’s ability to target p53 for degradation.
KEYWORDS: HPV, E6, E6AP, PBM, DNA PK, p53
INTRODUCTION
Human papillomaviruses (HPV) are the causative agents of cervical cancer and many other human malignancies. To date, over 200 different HPV types have been identified, of which only a small subset of 12 (also known as high-risk HPV types) are defined by the WHO as being cancer-causing. Among these, HPV-16 and HPV-18 account for approximately 80% of the cervical cancer burden worldwide (1, 2). Over decades, many studies have provided insights into HPV's two oncoproteins, E6 and E7, as key players in HPV-induced cancer progression. E6 and E7 are the only viral genes continually maintained and expressed in HPV-positive cervical cancer lesions (3, 4) and are required for the growth and progression of HPV-positive cervical cancer cells in vitro and in vivo (5, 6). Inhibition of E6/E7 expression induces cellular senescence and apoptosis (7, 8), indicating that cervical cancer cells are “E6/E7 oncogene-addicted” and hence are excellent targets for developing therapeutic interventions to treat HPV-induced malignancies.
E6 and E7 function cooperatively, both being required for the induction of malignancy. They do this by interacting with essential cellular regulators of cell growth, proliferation, and apoptosis control (6, 9, 10). HPV E7 targets pRb for proteasome-mediated degradation and activates several DNA damage response (DDR) pathways (11–13). Consequently, there is an increase in the protein level of the tumor suppressor p53, which is involved in controlling the transition from G1 to S phase and G2 to M phase in the cell cycle (14, 15). Thus, cellular stress generated by HPV E7 leads to p53 activation, which stimulates the activation of cell cycle checkpoint regulators and apoptotic proteins. To counteract this response, high-risk HPV E6 binds directly to p53 and induces its ubiquitination and proteasomal degradation through interaction with the E6-associated protein (E6AP) (10, 16).
The E6 proteins from cancer-causing HPV types, but not from nononcogenic HPV types, also possess a short carboxy-terminal stretch of amino acids, which confers interaction with cellular proteins that contain a PDZ domain (16, 17). Through this PDZ-binding motif (PBM), E6 interacts with various cellular PDZ domain-containing proteins, regulating processes as diverse as cell polarity, cell signaling, cell attachment, and cell proliferation during both the viral life cycle and cancer progression (17–19). According to previous studies, an intact E6 PBM is required for maintaining the viral episome during its life cycle (20, 21). In addition, some studies have demonstrated that, in cases where the E6 PBM was mutated, the inactivation of p53 restored the ability of the cell to maintain the HPV genome episomally, thereby suggesting a possible link between the E6 PBM and p53 function (22, 23). Intriguingly, the E6 PBM is multifunctional, owing to the presence of a phosphoacceptor site embedded within its core. For some time, the phosphorylation of the E6 PBM at the T156 residue was thought to be catalyzed mostly by either protein kinase A (PKA) or AKT activity, depending upon the precise amino acid sequence of the E6 PBM. However, more recently it was shown that DNA damage results in a dramatic increase in the levels of phosphorylated E6, which is mediated by the stress-responsive cellular kinases CHK1 and PKA. (24). In addition, the retention of an intact PBM and phosphoacceptor site within E6 correlates with the ability of E6 to inhibit p53's transcriptional activation of a subset of p53-responsive promoters, thereby linking regulation of the E6 PBM function with inhibition of p53 activity (24).
During the course of our studies to better understand the role of phosphoregulation of E6, we made the surprising observation that loss of E6AP resulted in a dramatic increase in the levels of E6 phosphorylation in HeLa cells. This was found to be dependent upon transcriptionally active p53 and DNA-dependent protein kinase (DNA PK) and suggests a feedback mechanism whereby activation of p53 results in phosphorylation of the E6 PBM, thereby increasing E6’s ability to inhibit p53 transcriptional activity.
RESULTS
E6 is phosphorylated in the absence of E6AP.
Previous studies have shown that HPV E6 oncoproteins are phosphoregulated by several cellular kinases either directly or indirectly (24, 25). E6 is very weakly phosphorylated during the normal cell cycle, but this increases drastically upon the induction of DDR pathways in cervical cancer-derived cell lines (24). Since loss of E6AP induces a potent stress response, involving upregulation of p53, induction of apoptosis, and loss of E6 protein (6, 19), we were interested in investigating how this loss of E6AP might impact signaling to the E6 oncoprotein. To examine this, we decided to knock down the expression of E6AP and E6/E7 in cervical cancer-derived HeLa cells, using small interfering RNAs (siRNAs) targeting E6AP and the E6/E7 oncoproteins. After 72 h, cells were harvested, and the extracts were analyzed by Western blotting, using antibodies raised against HPV-18 E6 to detect both phosphorylated and nonphosphorylated forms. As can be seen in Fig. 1A, knockdown of E6AP and E6/E7 leads to a reduction in the levels of total E6, in agreement with previous studies (19, 26). However, despite an overall reduction in E6 protein levels, we also observed a dramatic increase in the levels of the phosphorylated form of HPV-18 E6 (pE6) upon knockdown of E6AP. However, pE6 remains undetectable in the cells transfected with siE6/E7, similar to the si-luciferase control (Luci). While our antibody does not recognize phosphorylated HPV-16 E6, we nonetheless wanted to ascertain whether similar results could be obtained in a different HPV-18-positive cell line, C4-1. As can be seen from Fig. 1C, loss of E6AP also induces a marked increase in E6 phosphorylation in C4-1 cells, confirming that these effects are not unique to HeLa cells.
FIG 1.

Phosphorylation of HPV-18 E6 upon E6AP knockdown. HeLa (A) and C41 (C) cells were subjected to siRNA-mediated knockdown of E6AP and E6/E7, with luciferase (Luci) as control. After 72 h, cells were harvested and analyzed by Western blotting, using antibodies specific to total E6, pE6, and E6AP; α-tubulin was used as a loading control. (B and D) The histogram shows the statistical significance of changes in pE6 in the absence of E6AP from at least three independent experiments. **, P value < 0.005; ns, nonsignificant; statistically quantified using Student's t test. error bars indicate the standard deviation of the mean.
To ascertain what causes the phosphorylation of E6 upon silencing of E6AP, we performed another knockdown experiment in HeLa cells. This time, we knocked down E6AP together with p53, to see whether E6 is phosphorylated in response to p53 upregulation, since p53 is known to be the major degradation target of the E6/E6AP complex. We also included a double knockdown of siE6AP and siE6/E7 to verify the identity of the pE6 moiety. As can be seen in Fig. 2, loss of E6AP induces a marked increase in the levels of pE6, and this is reduced in the presence of siE6/E7. Most interestingly, this phosphorylation of E6 in the absence of E6AP is dependent upon the presence of p53, since transfection of siRNA p53 abolishes the phosphorylation of E6. Taken together, these results demonstrate that p53 is required for phosphorylation of E6 in the absence of E6AP.
FIG 2.

Knockdown of E6AP induces phosphorylation in response to p53 upregulation. (A) HeLa cells were transfected with siRNAs against E6AP, E6/E7, and p53. After 72 h, cells were harvested and analyzed by Western blotting. Proteins were probed using antibodies specific to pE6, total E6, p53, and E6AP; α-tubulin was used as a loading control. (B) The histogram shows the statistical significance of changes in pE6 levels in the absence of E6AP and in the absence of E6AP + p53 from three independent experiments with a P value of <0.005 (**), statistically quantified using Student's t test; error bars indicate the standard deviation of the mean.
We further validated this in the p53-null H1299 cell line. For this, we first knocked down the E6AP, and after 48 h, cells were transfected with p53, wild-type 18E6, and 18E6 delPBM expression constructs, followed by further incubation for 24 h. Cells were then harvested, and pE6 levels were analyzed using Western blotting. We observed that the levels of pE6 increase markedly in the absence of E6AP and in the presence of the p53 as shown in Fig. 3. Taken together with the data shown in Fig. 2, this suggests that E6, in the absence of E6AP, is phosphorylated in response to the increase in p53 protein levels, and that these effects are independent of the presence or absence of E7.
FIG 3.

Knockdown of E6AP and overexpression of p53 in p53-null H1299 cells lead to increased E6 phosphorylation. (A) H1299 cells were transfected with siE6AP and si-Luci (as control) using Lipofectamine RNAiMax. After 48 h, the same cells were transfected with plasmids expressing Flag-tagged p53, HA-tagged 18E6 (wild type), and HA-tagged 18E6 delPBM, together with a LacZ expression plasmid (transfection efficiency control), followed by further incubation of 24 h. Protein analysis was performed by Western blotting using specific antibodies against p53, p18E6, and HA (for probing 18E6). β-gal was used as a loading control for the overexpressed proteins, while α-tubulin was used as a loading control for E6AP siRNA knockdown. (B) The histogram represents the statistical significance of changes in pE6 in the absence of E6AP and presence of overexpressed p53 protein from at least three independent experiments. **, P value < 0.005; ns, nonsignificant. Statistically quantified using Student's t test; error bars indicate the standard deviation of the mean.
DNA PK phosphorylates E6 in the absence of E6AP.
Having found that E6 is phosphorylated in a p53-dependent manner following knockdown of E6AP, we were interested in identifying the kinase responsible. We had shown previously that various stress-response kinases can strongly phosphorylate E6, including Chk1 and Chk2 via PKA and another, as yet unidentified, kinase activated by cycloheximide treatment (24). Therefore, we initially focused our attention on CHK1 and PKA, repeating the siRNA E6AP transfection in HeLa cells but also including CHK1 and PKA inhibitors. In parallel, as an experimental control, we also induced DNA damage in HeLa cells by treating the cells with H2O2 in the presence of the PKA inhibitor to verify the phosphoregulation of E6 by PKA. The results in Fig. 4A and B show that there are no changes in the levels of pE6 in the presence of either the CHK1 or PKA inhibitors in the absence of E6AP. However, the inhibition of PKA drastically reduces the levels of pE6 in H2O2-treated cells (Fig. 4C) in agreement with the previous study (24). Taken together, these results suggest that neither PKA nor CHK1 kinase is involved in the regulation of pE6 when E6AP is knocked down and p53 is upregulated.
FIG 4.

Neither CHK1 nor PKA phosphorylates E6 in the absence of E6AP. HeLa cells were transfected with siRNAs against E6AP, E6/E7, and luciferase (Luci) as control. After 60 h, CHK1 (100 nM) (A) and PKA (15 μM) (B) inhibitors, and dimethyl sulfoxide (DMSO) as control, were added to the cells, followed by further incubation for 12 h. Cells were then harvested, and samples were analyzed using Western blotting with the antibodies against total E6, pE6, and E6AP; α-tubulin was used as a loading control. (C) HeLa cells were treated with PKA inhibitor (15 μM) for 15 h, followed by treatment with DMSO (as control) or H2O2 (500 μM) for 4 h. Western blots were probed using antibodies specific against total E6, pE6, and α-tubulin (loading control).
In order to search for other potential kinases, we performed an in silico kinase analysis using Netphos 3.1 server software and identified DNA PK as a high-confidence candidate as shown in Fig. 5A. To test this, we performed an in vitro kinase assay using purified DNA PK with purified HPV-18 and HPV-16 E6 glutathione S-transferase (GST) fusion proteins in the presence of radioactively labeled ATP. The results shown in Fig. 5B demonstrate that both HPV-18 and HPV-16 E6 are excellent substrates for phosphorylation by DNA PK in vitro. To confirm that the phosphorylation was on the T156 residue, the assay was repeated using a panel of E6 mutants. The results in Fig. 5C demonstrate that any modification to the E6 PBM reduces phosphorylation by DNA PK and that the phosphoacceptor site is T156.
FIG 5.

DNA PK phosphorylates HPV-18 E6 in vitro. (A) Represents in silico kinase analysis using NET Phos3.0 software for the HPV-18 E6 PBM, specifically the T156 residue. (B) In vitro phosphorylation assay using purified GST fusion proteins for wild-type HPV-16, HPV-18 E6, and HPV-18 E6 mutated within its PBM (C) incubated with purified DNA PK enzyme in the presence of [ϒ-32P] ATP. (Top) Autoradiograms. (Bottom) Coomassie-stained SDS-PAGE gels, indicating the pE6, the GST fusion E6 protein, and GST control.
To ascertain whether DNA PK is involved in E6 phosphorylation in vivo, HeLa cells were transfected with siRNA E6AP, followed by treatment with DNA PK inhibitor for 12 h. The cells were harvested and analyzed by Western blotting. The results in Fig. 6 demonstrate that the levels of pE6 are greatly decreased in the presence of the DNA PK inhibitor, confirming DNA PK as the kinase responsible for phosphorylating E6 after removal of E6AP.
FIG 6.

DNA PK phosphorylates HPV-18 E6 in vivo in the absence of E6AP. (A) HeLa cells were transfected with siRNAs against E6AP, E6/E7, and luciferase as the control. After 60 h, DNA PK inhibitor (10 μM) and DMSO as control were added to the cells, which were incubated for a further 12 h. Cells extracts were then analyzed by Western blotting using antibodies for total E6 and pE6; α-tubulin was used as a loading control. (B) Statistical significance of the pE6 from at least three independent experiments in the presence of DNA PK inhibitor with a P value of <0.005 (**). Statistically quantified using Student's t test; error bars indicate the standard deviation of the mean.
Although all high-risk HPV E6s have a conserved PBM at their carboxy terminus, there is a high degree of variation in their sequences upstream and downstream of the phosphoacceptor site, as shown in Fig. 7A. Having shown that T156, within the HPV-18 E6 PBM, plays a critical role in kinase recognition, we wanted to ascertain whether this is conserved between E6 proteins from different HPV types. To investigate this, we performed a series of in vitro kinase assays using purified DNA PK with different HPV E6 GST fusion proteins in the presence of radiolabeled ATP. As can be seen in Fig. 7B, E6 proteins from HPV-16, HPV-18, HPV-39, and HPV-68 are heavily phosphorylated by DNA PK; those of HPV-33 (potentially due to the presence of alanine at position −1), HPV-51, and HPV-58 are mildly phosphorylated; and E6s from HPV-35 (perhaps because of the presence of glutamic acid downstream of threonine at position −1) and HPV-56 are only weakly phosphorylated. These results suggest that, in agreement with previous studies, the noncanonical residues play critical roles in kinase recognition.
FIG 7.

Different HPV E6 types display different levels of phosphorylation. (A) The carboxy terminus sequence of E6 proteins from different HPV types. (B) The purified GST fusion proteins were incubated with DNA PK enzyme and [γ-32P] ATP. The phosphorylation levels of the E6 proteins were then analyzed by SDS-PAGE and autoradiography. The upper panel shows the autoradiogram of different in vitro phosphorylated 18E6 GST fusion proteins; the lower panel shows the Coomassie blue-stained gel.
Having shown that DNA PK can phosphorylate E6 in vivo following the loss of E6AP, we were interested in determining whether this was specific to loss of E6AP or whether other DNA damage-induced stress responses could trigger DNA PK phosphorylation of E6. To do this, HeLa cells were first treated with the DNA PK inhibitor followed by treatment with teniposide, which we have shown previously can induce a high level of E6 phosphorylation (24). As can be seen from Fig. 8, the DNA PK inhibitor causes a marked decrease in the levels of E6 phosphorylation in response to loss of E6AP; however, DNA PK inhibition has no effect on the levels of pE6 following treatment with teniposide. These results demonstrate that DNA PK activation is a specific response to the loss of E6AP and upregulation of p53, leading to the phosphorylation of E6, and this is not observed in response to other forms of DNA damage-induced stress, such as that induced by teniposide.
FIG 8.

DNA PK does not phosphorylate E6 upon exogenous induction of DNA damage. (A) In the first set, HeLa cells were transfected with siE6AP, siE6/E7, and si-Luci as control. After 48 h, we added DNA PK inhibitor (10 μM) or DMSO as control. A second set of cells was also treated with the DNA PK inhibitor (10 μM) or DMSO as control. Both sets of cells were then incubated for 12 to 15 h and then treated with a DNA-damaging agent, teniposide (at 5 μM), followed by further incubation for 4 h. Cells were then harvested, and analysis was performed by Western blotting using specific antibodies against pE6, total E6, and E6AP, plus α-tubulin as loading control. (B) The histogram represents the statistical significance of changes in pE6 levels upon treating the cells with teniposide in the presence and absence of DNA PK inhibitor. The results from three independent experiments are shown with a P value of <0.005 (***); ns, nonsignificant. Quantification was performed using Student's t test; error bars indicate the standard deviation of the mean.
Transcriptional activity of p53 regulates the phosphorylation of E6 in the absence of E6AP.
Having found that phosphorylation of E6 was dependent upon p53 and DNA PK, we next wanted to determine whether p53 transcriptional activity was required. To examine this, we repeated the knockdown of E6AP in HeLa cells and then treated the cells with α-pifithrin (27, 28) to block p53 transcriptional activity. After a further 48 h, the cells were harvested and pE6 levels ascertained by Western blotting. The results in Fig. 9 show that the levels of pE6 decrease significantly in the presence of α-pifithrin compared with those of control-treated cells. These results demonstrate that loss of E6AP results in a phosphorylation of HPV-18 E6 in a manner that requires both p53 transcriptional activity and DNA PK, although at this stage we do not know whether p53 directly upregulates DNA PK or whether DNA PK is responding to other p53-induced signaling pathways.
FIG 9.

p53 transcriptional activity controls the phosphorylation of E6 upon E6AP knockdown. (A) siRNAs targeting E6AP, E6/E7, and luciferase control were transfected into HeLa cells, followed by incubation for 24 h, after which p53 inhibitor α-pifithrin (30 μM), or DMSO as control, was added to the cells. Cells were then further incubated for 48 h, after which they were harvested and analyzed by Western blotting using antibodies specific to pE6 and total E6; α-tubulin was used as a control. (B) The histogram shows relative statistical significance of changes in pE6 levels in the presence of p53 inhibitor and absence of E6AP from at least three independent experiments with a P value of <0.05 (*); ns, nonsignificant.
The E6/E6AP-mediated degradation of p53 is considered to be a most important mechanism in the initiation and development of cervical cancers. Several studies have shown that disrupting this complex, either by knocking down E6AP or by treating the cells with inhibitors that lead to the accumulation of p53, further induces apoptosis in the cancer cells in a p53-dependent manner (29–33). It was possible that this imbalance between cell proliferation and apoptosis upon the activation of p53 in the absence of E6AP might induce cellular stress, in turn activating the stress-responsive Kinases, further leading to the phosphorylation of E6. To investigate this, we repeated the knockdown experiment in HeLa cells, as described above, and checked for the levels of stress marker pS139γH2AX, which is known to increase upon activation of the DNA damage-signaling pathway and also upon induction of the apoptotic signaling cascade (34). Interestingly, upon analysis, we observed that the levels of pS139γH2AX increase dramatically in the E6AP-knockdown cells compared with those of the Luci control, and decrease significantly upon the knockdown of both E6AP and p53 as shown in Fig. 10A. In order to determine whether these effects were specific for HPV-containing cells, we repeated the assay in U2OS cells, which are HPV negative and contain wild-type p53. As can be seen from Fig. 10B, there were no changes in pS139γH2AX protein levels when either E6AP or E6AP + p53 was knocked down from these cells, indicating that loss of E6AP only has this effect in HPV-positive cells.
FIG 10.

p53 transcriptional activity increases pS139γH2AX levels in the absence of E6AP. siRNAs targeting E6AP and p53 plus si-luciferase (Luci) control were transfected into HeLa (A) and U2OS (B) cells and incubated for 72 h. Cell were then harvested and analyzed by Western blotting. (C) HeLa cells were transfected with siRNA against E6AP and E6/E7 plus Luci as control. After 24 h, cells were treated with p53 inhibitor α-pifithrin (30 μM) for a further 48 h. Cells were then harvested and analyzed by Western blotting. Proteins were probed using specific antibodies, pS139γH2AX, E6AP, pE6, and total E6; α-tubulin was used as a loading control. (D) The histogram represents the statistical significance of the increase in pS139γH2AX in the absence of E6AP and in response to p53 transcriptional activation from three independent experiments. *, P value of <0.05; **, P value of <0.005. Statistically quantified using Student's t test; error bars indicate the standard deviation of the mean.
Next, we wanted to ascertain if it is regulated through the transcriptional activity of p53. To test this, we repeated the siRNA knockdown experiment in the presence and absence of α-pifithrin, an inhibitor of p53 transcriptional activity. Similar to the previous observation, knockdown of E6AP increases the levels of pS139γH2AX, which decreases greatly upon treating the cells with α-pifithrin (Fig. 10C). This further suggests that, in the absence of E6AP, the transcriptional activity of p53 induces stress in the cells that may result in the activation of DNA PK, which eventually phosphorylates E6.
pE6 in the absence of E6AP negatively regulates the transcriptional activation of p53-responsive genes.
Thatte et al. (24), in 2018, showed that phosphorylation of the E6 PBM plays a crucial role in blocking the transcriptional activity of p53-responsive genes. We wanted to determine whether the pool of E6 phosphorylated in the absence of E6AP could functionally block the transcriptional activity of p53. Therefore, we initiated a series of studies to examine whether the phosphorylation of the E6 PBM has any effect on p53 transcriptional transactivation of p21 and mdm2 promoters, using Renilla luciferase as a transfection efficiency control.
To examine this, we first transfected the cells with siE6AP or siScramble (as control) in H1299 p53 null-cells derived from a non-small-cell lung cancer. After 48 h, the cells were transfected with appropriate reporter constructs, together with plasmids expressing p53, wild-type 18E6, and 18E6 delPBM (phospho mutant). After a further 24 h, the cells were harvested, and luciferase activity was measured using the dual-luciferase system (the results are shown in Fig. 11A and B), and the expression profiles of all of the proteins were verified using Western blotting as shown in Fig. 11C. As expected, the transcriptional activity of p53 is inhibited by both wild-type 18E6 and the 18E6 delPBM in the presence of E6AP, since p53 degradation is induced by the E6/E6AP complex. However, in the absence of E6AP, we observed that 18E6 delPBM fails to inhibit the transcriptional activation of p53-responsive genes, while the wild-type E6 retains the ability to inhibit p53 transcriptional activity for both p21 and mdm2. Taken together, these results suggest that the phosphorylation of the E6 PBM plays an essential role in regulating the transcriptional activity of p53 in the absence of E6AP.
FIG 11.

The HPV-18 E6 PBM contributes to inhibition of p53's transcriptional transactivation activity in the absence of E6AP. (A) H1299 cells were transfected with the indicated promoter constructs upstream of a luciferase promoter—p21-Luc (A), Mdm2-Luc (B), together with plasmids expressing p53 and either wild-type HPV-18 E6 or mutant HPV-18 E6 delPBM in the presence or absence of E6AP. The histograms show the results from at least three independent experiments quantified using Student's t test; the error bars indicate standard errors of the mean. Also shown are the P values (*, P < 0.05; ns, nonsignificant) for the changes in relative luciferase activity. (C) Corresponding Western blot for the luciferase analysis confirms the knockdown of E6AP from the cells and expression of pE6, total E6, p53, and β-gal.
DISCUSSION
In this study, we extended earlier studies to investigate the pathways underlying the regulation of E6 phosphorylation in vivo and, in particular, the regulation of phosphorylation at T156 within the E6 PBM. We find that ablation of E6AP expression in HPV-18-positive HeLa and C4-1 cells results in a dramatic increase in the phosphorylation of HPV-18 E6 at this residue. This was somewhat surprising, as previous studies had shown that loss of E6AP results in an overall destabilization of E6 and greatly decreased total levels of E6 protein. Interestingly, the phosphorylated form appears to become the dominant species of E6 under these conditions.
Since one of the major targets of the E6-E6AP complex is p53 (16, 35–37), we proceeded to investigate whether the increase in E6 phosphorylation was linked to p53 activity. We found that siRNA ablation of p53 expression, or inhibition of p53 transcriptional activity, could both inhibit the levels of E6 phosphorylation in the absence of E6AP. This indicates that an intact p53 response is required for inducing E6 phosphorylation at T156, which is particularly intriguing, as this phosphorylation of E6 has been shown previously to contribute to E6's inhibition of p53 transcriptional activity, indicating a feedback loop to enhance E6's inhibition of p53.
Having found that, in the absence of E6AP, phosphorylation of HPV-18 E6 takes place in response to p53 upregulation, we next sought to determine which kinase(s) might be involved in the regulation of E6 phosphorylation. Following from the previous studies (38, 39), we investigated whether CHK1 or PKA is involved in phosphorylating E6 by repeating the knockdown study in the presence of CHK1 and PKA inhibitors. We show that neither CHK1 nor PKA, in this case, is involved in mediating E6 phosphorylation. To identify the kinase involved, we performed an in silico study using the Netphos 3.1 software and identified DNA-dependent protein kinase (DNA PK) as a strong candidate for E6 phosphorylation. Using in vitro kinase assays, we found that DNA PK phosphorylates both HPV-18 and HPV-16 E6 and identified T156 as the phosphoacceptor site in HPV-18 E6. Furthermore, we also analyzed the phosphorylation of different high-risk HPV types with DNA PK kinase. We observed differences in the phosphorylation patterns of different HPV E6s, with E6s from HPV-16, HPV-18, HPV-31, HPV-39, and HPV-68 being highly phosphorylated and E6s from HPV-33, HPV-51, HPV-35, and HPV-58 being phosphorylated to a lesser degree. These results highlight the importance of noncanonical residues of the E6 PBM in susceptibility to phosphorylation and how it varies for different kinases. Moreover, they also provide insight into the differential phosphorylation of different high-risk HPV types, depending on the specific amino acids present in the motif. While phosphospecific antibodies are not available for HPV-16 E6, the efficiency with which it is phosphorylated by DNA PK in vitro strongly suggests that HPV-16 will be similarly regulated in vivo.
In order to ascertain whether DNA PK was required for E6 phosphorylation in vivo, we included DNA PK inhibitor in assays of E6 phosphorylation in the absence of E6AP and found a marked inhibition of E6 phosphorylation. Intriguingly, DNA PK phosphorylation of E6 appeared to be specific to conditions where E6AP is lost and did not occur under other forms of DNA stress response, such as that induced by teniposide.
We next wanted to ascertain whether phosphorylation of E6 actually requires the transcriptional activity of p53 or whether it simply reflects the p53-dependent activation of the DNA damage stress response pathway. We found that the ablation of E6AP from HeLa cells increases the levels of pS139γH2AX but drastically decreases upon silencing both E6AP and p53, which was further validated using an inhibitor of p53 transcriptional activity. This suggests that an intact p53 cellular stress response, initiated by upregulation of pS139γH2AX, was absolutely essential for phosphorylation of E6 in the absence of E6AP. We found that this increase in pS139γH2AX upon knockdown of E6AP in response to p53 activation is specific to HPV-positive cervical cancer cells, since we did not see this effect in HPV-negative U2OS cancer cells harboring wild-type p53.
As noted earlier, this p53-dependent phosphorylation of E6 by DNA PK might indicate the existence of a feedback loop as shown in Fig. 12. Indeed, this would be predicted to result in an increase in pE6 levels, which subsequently inhibit p53's transcriptional activity in a 14-3-3-dependent manner as demonstrated in previous studies (25, 40). While the studies presented here were all performed in HPV-transformed cervical cancer-derived cell lines, we would fully expect a similar pathway of feedback loop regulation to be operable in cells normally infected with HPV, although further studies would be needed to confirm this.
FIG 12.
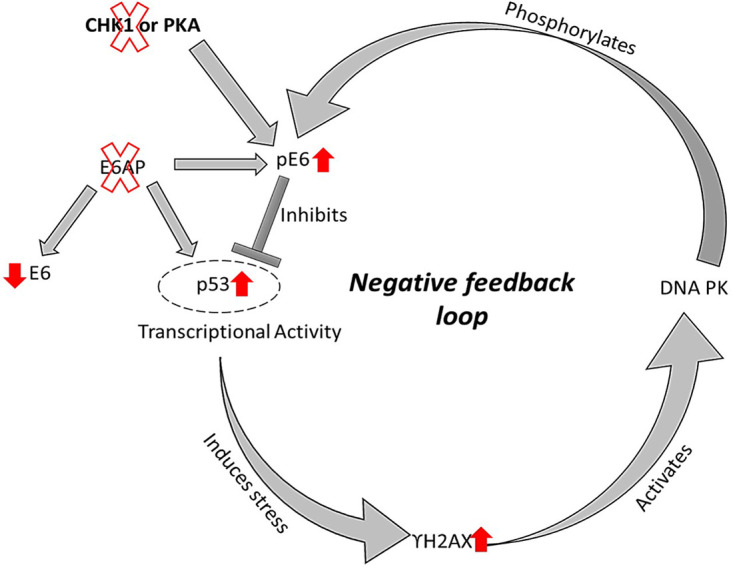
Model summarizing how, in the absence of E6AP, HPV E6 is phosphorylated by DNA PK in cervical cancer-derived cells in response to p53 transcriptional activity.
Using p21 and mdm2 promoter assays, we find that the ability of E6 to block p53 transcriptional activity in the absence of E6AP is absolutely dependent upon an intact PBM, central to which is the T156 phosphorylation site, further supporting previous data showing that phosphorylation of the E6 PBM can inhibit the transcriptional activity of p53 (24). This phosphorylation event will then inhibit E6's interaction with PDZ domain-containing substrates and confer association with 14-3-3 proteins, thereby promoting further inhibition of p53 transcriptional activity; thus, completing a pathway of feedback inhibition.
Current studies aim to understand further the biological relevance of E6 phosphorylation in cervical cancer-derived cell lines. To our surprise, loss of E6AP in HeLa cells induces only very low levels of apoptosis, for which we currently have no explanation. One tempting hypothesis to further explore is whether phospho-E6 can affect the levels of apoptosis in both HPV-18- and HPV-16-positive cancer cells.
In summary, this study provides compelling evidence that the high-risk HPV E6 oncoprotein is phosphoregulated by the kinase DNA PK upon the loss of E6AP in response to p53's transcriptional activation of the DNA damage response pathways. This is the first study to show that the knockdown of E6AP leads to a dramatic increase in the levels of phosphorylated E6 oncoprotein and begins to provide mechanistic insights into how the phosphorylation of E6 oncoprotein controls the transcriptional activity of p53 in a negative feedback loop manner.
MATERIALS AND METHODS
Cell culture.
HeLa, C41, U2OS, and H1299 cells were grown in Dulbecco's modified Eagle's medium (DMEM) (GIBCO; no. 31885-023) supplemented with 10% fetal bovine serum (GIBCO; no. 10270-106), penicillin-streptomycin (100 U mL−1), and glutamine (300 μg mL−1) (GIBCO; no. 10378-016) at 37°C in humidified air incubator containing 10% CO2.
Chemicals, inhibitors, and antibodies.
The inhibitors used for the experiments in this study were as follows: 100 nM UCN01 CHK1 inhibitor (Sigma-Aldrich; no. U6508), 10 μM H-89 PKA inhibitor (Sigma-Aldrich; no. B1427), 30 μM DNA PK inhibitor (Abcam; NU7026), and 30 μM α-pifithrin (Merck; no. p4359). DNA-damaging agent teniposide (Sigma-Aldrich; no. SML0609) was used at a concentration of 5 μM.
Primary antibodies used were rabbit polyclonal HPV-18 E6 phosphospecific antibody (1:500; custom-made by Eurogentec) generated using the H2N-RQERLQRRRET(PO3H2)QV-COOH peptide in rabbits and subjected to affinity purification, mouse monoclonal 18E6 (1:500; Santa Cruz; no. sc365089) and mouse monoclonal p53 (1:2,000; Santa Cruz; no. sc126), mouse monoclonal E6AP (1:500; BD Biosciences; no. 611416), mouse monoclonal α-tubulin (1:10,000; Cell Signaling Technology; no. T5168), rabbit monoclonal pS139γH2AX (1:1,000; Cell Signaling Technology; no. 20E3), mouse monoclonal actinin (1:4,000; Santa Cruz; no. sc17829), mouse monoclonal β-galactosidase (β-Gal) (1:4,000; Promega; no. Z378B), and mouse monoclonal hemagglutinin (HA)-tag peroxidase (1:4,000; Sigma-Aldrich; no. H6533-1VL) followed by horseradish peroxidase (HRP)-conjugated anti-rabbit (1:2,000; Dako; no. P0260) and anti-mouse secondary antibody (1:2,000; Dako; no. P0217).
Plasmid constructs.
The plasmids used were as follows: p21-luciferase, MDM2-luciferase, Renilla luciferase, FLAG-p53, pGWI-18 E6, and pGWI-18 E6 ΔPBM have been described previously (24). The GST, HPV-11 E6 GST, HPV-16 E6 GST, HPV-18 E6 GST, HPV-31 E6 GST, HPV-33 E6 GST, HPV-51 E6 GST, HPV-18 E6 T156E GST, HPV-58 E6 GST, and HPV 18 E6_ S82A_T156E GST fusion proteins have been described previously (25, 40, 41). The 18E6 ET*PBM mutant was generated by using the GeneArt site-directed mutagenesis kit (Invitrogen), and the primer sequences are as follows: 18E6GST ET*_F, CGACGCAGAGAAACATGATATTAAGTATGCATGG; 18E6GST ET*_R, CCATGCATACTTAATATCATGTTTCTCTGCGTCG. GST fusion HPV-35 E6, HPV-39 E6, HPV-56 E6, and HPV-68 E6 proteins were generated by subcloning them from their respective plasmids using BamHI and EcoRI restriction sites (Kind gift from Carina Eklund, Karolinska Institute). The primer sequences used for the same are listed as follows: 35 E6 F, GGTGGATCCATGTTTCAGGACCCAGCTG, and 35 E6_R, GGGGAATTCTTACACCTCGGTTTCTCTACG; 39 E6_F, GTTGGATCCATGGCGCGATTTCACAATC, and 39 E6_R, GGGGAATTCTTATACTTGGGTTTCTCTTCG; 56 E6_F, GGCGGATCCATGGAGCCACAATTCAAC, and 56 E6_R, GGCGAATTCTTATACTGTAGATTCTCTAG; 68 E6_F, GCTGGATCCATGGCGCTATTTCACAAC, and 68 E6_R, GGGGAATTCTTAAACTTGTGTTTCTTGACG.
siRNA transfection.
HeLa cells were seeded at a confluence of 30 to 40% in a 6-well plate and then were transfected with siRNAs against E6AP (Dharmacon; SMARTPool no. L-005137-00-0005), E6/E7 (Dharmacon; no. HR1ZN-000327), and p53 (Dharmacon; no. J-003329-14-0005) at a final concentration of 50 nM using Lipofectamine RNAiMax (Life Sciences Technologies; no. 13778-150). After 72 h incubation, cells were harvested and analyzed by Western blotting.
Western blotting.
Total cell extracts were obtained by lysing the cells directly in a 2× SDS-PAGE sample buffer. Western blotting and processing were then performed as described previously (42) and developed using the ECL detection system (Amersham; no. RPN2106).
In vitro phosphorylation assays.
In vitro phosphorylation of the 18 E6 GST fusion proteins was carried out using a DNA PK protein kinase system (Promega; no. V4106) in the presence of radioactively labeled [γ-32P] ATP. The reaction was carried out according to the manufacturer's protocol. The samples were analyzed by SDS-PAGE and autoradiography.
Dual luciferase assay.
H1299 cells were transfected with appropriate plasmids expressing the luciferase reporters, and cell lysates were collected 24 h posttransfection in lysis buffer. The luciferase assay was performed using the dual-luciferase reporter assay system (Promega; no. E1960) according to the manufacturer's instructions. The firefly luciferase and Renilla luciferase readings were taken using a TD20/20 luminometer by Turner Designs.
Statistical analysis.
All experiments were performed at least three times, and data are shown as mean and standard deviation of the mean. Statistical significance was calculated using the GraphPad Prism software using the unpaired two-tailed Student's t test. A P value below 0.05 was considered statistically significant, and throughout the P values have been defined as follows: *, P < 0.05; **, P < 0.005; ***, P < 0.001. The term “ns” represents a nonsignificant P value above 0.05.
For the quantification of protein levels from Western blots, the films were scanned and the band intensities were measured using ImageJ software. The final relative quantification values are the ratio of the net band to the net loading control bands of tubulin, actinin, and β-gal.
ACKNOWLEDGMENTS
N.S. and A.V. are recipients of ICGEB Arturo Falaschi Fellowships, and A.V. is a registered student with Open University UK.
This work was supported in part by Associazione Italiana per la Ricerca sul Cancro (AIRC) research grant no. IG2019 - ID. 23572 awarded to L.B. The G.D.S. lab is supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC) Special Program 5×1000 (22759), AIRC-IG (22174), Italian University and Research Ministry PRIN-2017HWTP2K_004 and MIUR-ARS01_00876, INTERREG V-A Italia-Austria PCARE (ITAT1050), and Ministero della Salute RF-2019-12368718.
We are very grateful to Miranda Thomas for comments on the manuscript. We also thank Jayashree Thatte for providing the 18E6 ET*PBM mutant plasmid.
Contributor Information
Lawrence Banks, Email: banks@icgeb.org.
Lori Frappier, University of Toronto.
REFERENCES
- 1.Parkin DM, Bray F. 2006. Chapter 2: the burden of HPV-related cancers. Vaccine 24:S11–S25. 10.1016/j.vaccine.2006.05.111. [DOI] [PubMed] [Google Scholar]
- 2.Zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer 2:342–350. 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 3.Durst M, Gissmann L, Ikenberg H, Zur Hausen H. 1983. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci USA 80:3812–3815. 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz E, Freese UK, Gissmann L, Mayer W, Roggenbuck B, Stremlau A, Zur Hausen H. 1985. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 314:111–114. 10.1038/314111a0. [DOI] [PubMed] [Google Scholar]
- 5.Roman A, Munger K. 2013. The papillomavirus E7 proteins. Virology 445:138–168. 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vande Pol SB, Klingelhutz AJ. 2013. Papillomavirus E6 oncoproteins. Virology 445:115–137. 10.1016/j.virol.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamato K, Yamada T, Kizaki M, Ui-Tei K, Natori Y, Fujino M, Nishihara T, Ikeda Y, Nasu Y, Saigo K, Yoshinouchi M. 2008. New highly potent and specific E6 and E7 siRNAs for treatment of HPV16 positive cervical cancer. Cancer Gene Ther 15:140–153. 10.1038/sj.cgt.7701118. [DOI] [PubMed] [Google Scholar]
- 8.Jabbar SF, Abrams L, Glick A, Lambert PF. 2009. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer Res 69:4407–4414. 10.1158/0008-5472.CAN-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. 2001. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 20:7888–7898. 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 10.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 11.Pickering MT, Kowalik TF. 2006. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 25:746–755. 10.1038/sj.onc.1209103. [DOI] [PubMed] [Google Scholar]
- 12.Duensing S, Munger K. 2002. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res 62:7075–7082. [PubMed] [Google Scholar]
- 13.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145:435–446. 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones DL, Thompson DA, Munger K. 1997. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 239:97–107. 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 15.Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer 10:550–560. 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 16.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505. 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 17.Thomas M, Banks L. 2018. Upsetting the balance: when viruses manipulate cell polarity control. J Mol Biol 430:3481–3503. 10.1016/j.jmb.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas M, Narayan N, Pim D, Tomaic V, Massimi P, Nagasaka K, Kranjec C, Gammoh N, Banks L. 2008. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 27:7018–7030. 10.1038/onc.2008.351. [DOI] [PubMed] [Google Scholar]
- 19.Vats A, Trejo-Cerro O, Thomas M, Banks L. 2021. Human papillomavirus E6 and E7: what remains? Tumour Virus Res 11:200213. 10.1016/j.tvr.2021.200213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee C, Laimins LA. 2004. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J Virol 78:12366–12377. 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delury CP, Marsh EK, James CD, Boon SS, Banks L, Knight GL, Roberts S. 2013. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J Virol 87:9463–9472. 10.1128/JVI.01234-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz LD, Rivera Cardona J, Lambert PF. 2013. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog 9:e1003717. 10.1371/journal.ppat.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brimer N, Vande Pol SB. 2014. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal human papillomavirus genome maintenance. J Virol 88:3027–3030. 10.1128/JVI.02360-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thatte J, Massimi P, Thomas M, Boon SS, Banks L. 2018. The human papillomavirus E6 PDZ binding motif links DNA damage response signaling to E6 inhibition of p53 transcriptional activity. J Virol 92:e00465-18. 10.1128/JVI.00465-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boon SS, Tomaic V, Thomas M, Roberts S, Banks L. 2015. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J Virol 89:1579–1586. 10.1128/JVI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tomaic V, Pim D, Banks L. 2009. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 393:7–10. 10.1016/j.virol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Ghiani CA, Kim JY, Liu A, Sandoval J, DeVellis J, Casaccia-Bonnefil P. 2008. Inhibition of p53 transcriptional activity: a potential target for future development of therapeutic strategies for primary demyelination. J Neurosci 28:6118–6127. 10.1523/JNEUROSCI.0184-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leker RR, Aharonowiz M, Greig NH, Ovadia H. 2004. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol 187:478–486. 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 29.Beaudenon S, Huibregtse JM. 2008. HPV E6, E6AP and cervical cancer. BMC Biochem 9(Suppl 1):S4. 10.1186/1471-2091-9-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao CY, Szekely L, Bao W, Selivanova G. 2010. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation. Cancer Res 70:3372–3381. 10.1158/0008-5472.CAN-09-2787. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Kwon JH, Kim EH, Kim ES, Choi KY. 2010. HMGB2 stabilizes p53 by interfering with E6/E6AP-mediated p53 degradation in human papillomavirus-positive HeLa cells. Cancer Lett 292:125–132. 10.1016/j.canlet.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Shai A, Pitot HC, Lambert PF. 2010. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res 70:5064–5073. 10.1158/0008-5472.CAN-09-3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hietanen S, Lain S, Krausz E, Blattner C, Lane DP. 2000. Activation of p53 in cervical carcinoma cells by small molecules. Proc Natl Acad Sci USA 97:8501–8506. 10.1073/pnas.97.15.8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. 2000. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 275:9390–9395. 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 35.Huibregtse JM, Scheffner M, Howley PM. 1993. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol 13:775–784. 10.1128/mcb.13.2.775-784.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 10:4129–4135. 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas M, Pim D, Banks L. 1999. The role of the E6-p53 interaction in the molecular pathogenesis of HPV. Oncogene 18:7690–7700. 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 38.Adhya D, Basu A. 2010. Epigenetic modulation of host: new insights into immune evasion by viruses. J Biosci 35:647–663. 10.1007/s12038-010-0072-9. [DOI] [PubMed] [Google Scholar]
- 39.Basukala O, Sarabia-Vega V, Banks L. 2020. Human papillomavirus oncoproteins and post-translational modifications: generating multifunctional hubs for overriding cellular homeostasis. Biol Chem 401:585–599. 10.1515/hsz-2019-0408. [DOI] [PubMed] [Google Scholar]
- 40.Boon SS, Banks L. 2013. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J Virol 87:1586–1595. 10.1128/JVI.02074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomaic V, Pim D, Thomas M, Massimi P, Myers MP, Banks L. 2011. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J Virol 85:3120–3127. 10.1128/JVI.02004-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vats A, Thatte J, Banks L. 2019. Identification of E6AP-independent degradation targets of HPV E6. J Gen Virol 100:1674–1679. 10.1099/jgv.0.001331. [DOI] [PubMed] [Google Scholar]