

ABSTRACT
Emerging viruses impose global threats to animal and human populations and may bear novel genes with limited homology to known sequences, necessitating the development of novel approaches to infer and test protein functions. This challenge is dramatically evident in tilapia lake virus (TiLV), an emerging “orthomyxo-like” virus that threatens the global tilapia aquaculture and food security of millions of people. The majority of TiLV proteins have no homology to known sequences, impeding functionality assessments. Using a novel bioinformatics approach, we predicted that TiLV’s Protein 4 encodes the nucleoprotein, a factor essential for viral RNA replication. Multiple methodologies revealed the expected properties of orthomyxoviral nucleoproteins. A modified yeast three-hybrid assay detected Protein 4-RNA interactions, which were independent of the RNA sequence, and identified specific positively charged residues involved. Protein 4-RNA interactions were uncovered by R-DeeP and XRNAX methodologies. Immunoelectron microscopy found that multiple Protein 4 copies localized along enriched ribonucleoproteins. TiLV RNA from cells and virions coimmunoprecipitated with Protein 4. Immunofluorescence microscopy detected Protein 4 in the cytoplasm and nuclei, and nuclear Protein 4 increased upon CRM1 inhibition, suggesting CRM1-dependent nuclear export of TiLV RNA. Together, these data reveal TiLV’s nucleoprotein and highlight the ability to infer protein functionality, including novel RNA-binding proteins, in emerging pathogens. These are important in light of the expected discovery of many unknown viruses and the zoonotic potential of such pathogens.
IMPORTANCE Tilapia is an important source of dietary protein, especially in developing countries. Massive losses of tilapia were identified worldwide, risking the food security of millions of people. Tilapia lake virus (TiLV) is an emerging pathogen responsible for these disease outbreaks. TiLV’s genome encodes 10 major proteins, 9 of which show no homology to other known viral or cellular proteins, hindering functionality assessment of these proteins. Here, we describe a novel bioinformatics approach to infer the functionality of TiLV proteins, which predicted Protein 4 as the nucleoprotein, a factor essential for viral RNA replication. We provided experimental support for this prediction by applying multiple molecular, biochemical, and imaging approaches. Overall, we illustrate a strategy for functional analyses in viral discovery. The strategy is important in light of the expected discovery of many unknown viruses and the zoonotic potential of such pathogens.
KEYWORDS: nucleoprotein, tilapia lake virus, RNA-binding protein, emerging virus
INTRODUCTION
In recent decades, more and more emerging viruses have become evident both in the human population and in wildlife, particularly in domestic animals or in wild stock raised by humans in dense conditions. The impact of such emerging viruses on global health and the economy may be devastating and necessitates a rapid response. Tilapia lake virus (TiLV) is an emerging pathogen that threatens the global tilapia aquaculture and the food security of millions of people, in particular in developing countries. Since its discovery in 2014 (1), and the first reports from Israel and Ecuador of the disease it causes (1, 2), TiLV has been detected in 16 countries across four continents (3). Moreover, a more significant global spread of the virus is suspected due to fish export from TiLV-infected hatcheries to over 40 countries (4). TiLV infections may result in extremely high mortality rates in both experimentally infected fish (1, 5, 6) and farmed tilapia (2, 7–9), ranging between 70% to 90%. As tilapia comprise the second most important group of farmed fish worldwide (1, 3, 10), several international agencies, including the Food and Agriculture Organization (FAO) of the United Nations, have issued urgent warnings regarding the global threat that TiLV imposes to food security (3).
The RNA genome of TiLV (1) consists of 10 segments, numbered according to their length: Segment 1 is the longest (1,641 nucleotides [nt]), and Segment 10 is the shortest (465 nt) (10). All 10 segments have conserved, complementary sequences at their 5′ and 3′ termini, and each segment contains one primary open reading frame (ORF). We identified peptides from all 10 predicted proteins by mass spectrometry (10; unpublished data). While Segment 1 ORF shows weak sequence homology to the influenza C virus (ICV) PB1 subunit, remarkably, the other nine ORFs completely lack sequence homology to any other sequence, viral and cellular alike. Nevertheless, several features suggested that TiLV is an “orthomyxo-like” virus (10). These include an enveloped virion; a single-stranded, negative-sense, segmented RNA genome; the presence of similar, complementary sequences at the 5′ and 3′ noncoding termini of all TiLV segments; a short (3 to 5 bases long), uninterrupted uridine stretch present at all of the 5′ ends of TiLV genomic RNA segments; and nuclear and cytoplasmic localization of TiLV mRNA, implying a nuclear site for transcription (1, 10). A later classification has assigned TiLV as a new species (Tilapia tilapinevirus), under the genus Tilapinevirus, family Amnoonviridae and order Articulavirales (11). Recent meta-transcriptomic and data mining analyses have expanded the Amnoonviridae family by identifying transcripts derived from 12 unknown vertebrate viruses, which match multiple genomic segments of TiLV (12, 13).
The nucleoprotein (NP) of negative-sense RNA viruses is essential for their replication. Multiple copies of NP bind the single-strand RNA genome and antigenome to form ribonucleoprotein (RNP) complexes (also called nucleocapsids). In addition to their function in genome encapsidation, NPs interact with multiple viral and cellular factors to enable genome transcription, replication, packaging, and intracellular trafficking (14–22). Several facts hinder the identification of TiLV’s NP: (i) no apparent homology is detected among known viral NPs and any of TiLV ORFs; (ii) there is greater diversity in NP structure for viruses with a segmented RNA genome, compared to viruses with nonsegmented RNA genomes (21); and (iii) NP proteins are characterized by positively charged surfaces, yet, the majority of TiLV proteins have relatively high isoelectric point (pI) values (10). Thus, TiLV is an example of an emerging pathogen for which only minimal information is available regarding the functionality of its proteins. Here, we describe the application of a suite of bioinformatics, genetics, and biochemical tools that identified the protein encoded by Segment 4 of TiLV genome as the NP.
RESULTS
A combined feature analysis predicts Protein 4 or Protein 6 as TiLV’s NP.
Although TiLV is defined as an orthomyxovirus-like agent (10), 9 of its 10 major ORFs show no homology to other known sequences. Even the homology to the ICV PB1 subunit, found in Protein 1 (TiLV proteins are numbered according to the segments from which they are expressed), is low (∼17% amino acid identity [10]). Thus, TiLV is an example of a novel pathogen for which sequence homology cannot be used to deduce the functions of many of its proteins.
Since all negative-stranded RNA viruses encode a single NP, of which multiple copies coassemble with the viral genomic RNA to form nucleocapsids (reviewed in references 21 and 23), we set out to deduce which of TiLV proteins serves as the NP. One common characteristic of orthomyxovirus NPs is a relatively high pI due to the high content of positively charged residues; for example, the 498-residue influenza A virus (IAV) NP (strain A/WSN/33 [24]) has a calculated pI of 9.38 and bears 72 arginines and lysines. However, the calculated pI of most TiLV proteins is high (in fact, eight out of the 10 proteins are basic [10]), making it challenging to identify TiLV NP based on this single criterion.
We reasoned that a given protein’s function is derived from a combination of multiple features and accordingly, proteins with similar characteristics are likely to share similar functions. We harnessed this notion to predict the function of TiLV proteins based on features extracted from their sequences, and compared these features to the characteristics of known proteins of other members of the orthomyxoviridae family. These features included the pI mentioned above, protein size (relative to the total coding size of the virus; see Materials and Methods), and dN/dS, which is the ratio between the rate of nonsynonymous (dN) and synonymous (dS) substitutions (25). The latter reflects the type and level of selection operating on a protein. Our underlying assumption was that viral proteins with similar functions experience similar selection pressures and thus their dN/dS profiles will be similar. For example, positive diversifying selection operates on many viral proteins that directly interact with the adaptive immune system, such as glycoproteins (26), whereas nonstructural proteins tend to be more conserved. Combining these features into a three-dimensional (3D) scatterplot resulted in the clear segregation of the different proteins of the orthomyxoviruses into distinct functional groups (Fig. 1A, colored dots). By superimposing TiLV proteins onto this 3D plot (Fig. 1A, black dots), we assessed the similarity of TiLV proteins to the proteins of the orthomyxoviruses, based on our three features. Reassuringly, we noted that Protein 1, which is known to share homology to the ICV PB1 subunit, was indeed predicted as a polymerase subunit. When focusing on NPs of orthomyxoviruses, our analysis yielded either Protein 4 or Protein 6 as a candidate NP for TiLV (Fig. 1A; see also Fig. 1B for elaborated prediction probabilities); thus, we chose to probe both proteins’ RNA binding activities.
FIG 1.

Combined feature analysis. (A) A 3D scatterplot of IAV, IBV, ICV, IDV, ISAV, Thogotovirus, and TiLV proteins, based on relative ORF length, pI values, and evolutionary rates (dN/dS). Each point corresponds to a single dataset, and the colors represent different protein functions. TiLV ORFs are in black, and the numbers correspond to the protein (segment) number. A total of 431 alignments encompassing 14,025 sequences were used to generate the plot (see Materials and Methods). (B) Quantitative prediction of the linear discriminant analysis. Shown is a heatmap displaying the probability that each of TiLVs’ proteins belongs to one of orthomyxoviruses’ six known protein functions. Colors correspond to the prediction probability.
Protein 4 binds RNA in the yeast three-hybrid system.
To test the above predictions, we evaluated the interactions of Proteins 4 and 6 with RNA in the yeast three-hybrid (Y3H) system that detects protein-RNA interactions (27). To this analysis, we also added Proteins 7 and 8 (predicted as matrix proteins) since these are the two most basic proteins of TiLV (calculated pI values of 9.98 and 9.86, respectively). The Y3H system has successfully been used to analyze interactions of viral protein-RNA pairs (27–30), e.g., the RNA-binding activity of the human immunodeficiency virus (HIV) Gag and nucleocapsid (NC) proteins to HIV RNA encapsidation signal (HIV ψ RNA). In this genetic system, a hybrid RNA molecule bridges two fusion proteins (Fig. 2A). The first fusion protein, named LexA-MS2 coat, contains the LexA DNA-binding domain, fused to the coat protein of the MS2 bacteriophage that specifically interacts with MS2 sequences embedded within the RNA hybrid. The second fusion protein contains an N-terminal Gal4 transcriptional activation domain (Gal4AD) and a C-terminal protein; the latter is tested for binding to a specific sequence inserted into the RNA hybrid. The binding of the two fusion proteins to the same RNA hybrid results in the transcriptional activation of a lacZ reporter located downstream of the LexA DNA binding site. The S. cerevisiae strain L40-coat constitutively expresses the LexA-MS2 coat fusion protein and contains an integrated copy of the LexA-regulated lacZ reporter. To this end, we transformed the yeast strain L40-coat with plasmids expressing the Gal4AD fused to TiLV Protein 4, 6, 7, or 8, and plasmids expressing different RNA hybrids. These RNA hybrids consisted of a shared, primary RNA hybrid sequence linked to different TiLV genomic sequences: Segment 1 middle region (“Segment 1” hybrid) or the 5′ sequence of Segment 7 (“5′ Segment 7” hybrid). The primary RNA hybrid is composed of the 5′ stem-loop structure of the S. cerevisiae RNase P RNA gene (RPR1) leader, a linker (into which, TiLV sequences were inserted), two stem-loop structures that bind the MS2 coat protein, and the RPR1 3′ terminal sequence (27). Qualitative colony-lift filter assay clearly demonstrated that coexpression of Gal4AD-Protein 4 fusion with each of the RNA hybrids resulted in robust activation of the lacZ reporter (depicted by yeast colonies that developed a dark cyan color, Fig. 2B). In contrast, coexpression of Gal4AD-Protein 6, 7, or 8 fusions with the same RNA hybrids did not activate the reporter gene (despite comparable expression levels of all tested TiLV proteins in the yeast, Fig. 2B). To test the RNA-binding specificity of Gal4AD-Protein 4, the assay was repeated with an RNA hybrid containing either the HIV ψ, or only the primary RNA sequence (“Primary RNA”) (27, 28, 30). Here, too, coexpression of these RNA hybrids together with Gal4AD-Protein 4 fusion, but not with Gal4AD-Protein 6, 7, or 8 fusions, resulted in readily detected activation of the lacZ reporter, although this activation was weaker for the Primary RNA hybrid. No such activation occurred when we omitted the plasmid expressing the RNA hybrid from the transformation mix (“No RNA”); excluding the possibility that reporter activation resulted from a direct, RNA-independent interaction between Gal4AD-Protein 4 and LexA-MS2 coat fusion proteins. A quantitative β-galactosidase liquid assay further established these results: we observed a significant activation of the lacZ reporter for Gal4AD-Protein 4 fusion (but not for Gal4AD-Protein 6, 7, or 8 fusions), only if an RNA hybrid was coexpressed with the fusion protein (Fig. 2C).
FIG 2.

Protein 4 binds RNA in the Y3H system. (A) Schematic presentation of the Y3H system. A hybrid RNA molecule bridges the LexA-MS2 coat and the Gal4AD-TiLV chimeric proteins, resulting in transcriptional activation (arrow) of a lacZ reporter (modified from reference 27). (B) Filter assay for β-galactosidase activity. Yeast colonies expressing the indicated RNA-protein pairs were tested for β-galactosidase activity using colony lift colorimetric assay. Upper panels show images of sections of the filters with stained yeast colonies. Lower panels show Western blot analyses of the expression of the HA-tagged, TiLV protein-Gal4AD fusions in yeast, probed with anti-HA and anti-actin (loading control) antibodies. All fusion proteins showed their expected MW (approximately 56, 55, 39, and 37 kDa for Gal4AD fused to Protein 4, 6, 7, and 8, respectively). For Protein 8-Gal4AD, the lower band (asterisk) matches the calculated MW. (C) Quantitative β-galactosidase liquid assay. For each indicated RNA-protein pair, three yeast colonies were randomly picked from a plate of yeast transformants, expanded, and assayed for β-galactosidase activity using the colorimetric liquid assay. The dashed red line indicates the average β-galactosidase activity of no RNA control for all tested proteins. **, P ≤ 0.05; ***, P ≤ 0.005 (Student’s t test, accounting for multiple testing using the Bonferroni correction). (D) Filter assay for β-galactosidase activity in different time and temperature settings. Panels show images of yeast colonies, expressing the HIVΨ RNA and the indicated TiLV proteins, grown at the indicated periods and temperatures and stained as in panel B. HIV NC served as a positive control. (E) Timeline and conditions for testing Protein 4 RNA-binding activity in the Y3H filter and liquid assays.
Notably, while 30 to 32°C is the typical temperature range for the Y3H assay (2 days of yeast growth, followed by β-galactosidase assay) (27–30), the optimal temperature for TiLV replication is 25°C (31). In line with these temperature differences, growing the yeast for two to 3 days at 30°C resulted in relatively strong activation of the lacZ reporter for the positive control (Gal4-HIV NC/HIV ψ pair), but no activation for Gal4-Protein 4, 7, or 8 fusions, coexpressed with HIV ψ RNA (Fig. 2D), presumably due to misfolding or reduced steady-state levels of TiLV protein(s), or both. Performing the assay for 3 days at 25°C resulted in no reporter activation for the TiLV pairs and only weak activation of the positive control, likely because of the suboptimal conditions for yeast growth. Therefore, we adapted the timelines of the filter and the liquid assays to include growth of the yeast at 30°C, followed by incubation at 25°C to allow protein-RNA pairing at the optimal temperature for TiLV replication. This design allowed evident reporter activation for either Gal4-Protein 4 or Gal4-HIV NC fusions, paired with the HIV ψ RNA (Fig. 2D). Accordingly, we applied these timelines (summarized in Fig. 2E) for the experiments described in Fig. 2 and 3.
FIG 3.

Protein 4 mutants with reduced RNA-binding activity in the Y3H system. (A) Random linker insertion mutagenesis. Schematic presentation of Protein 4 ORF (black horizontal line) with randomly inserted linkers (arrows) positions, each disrupted lacZ activation in the Y3H system. A stack of two arrows represents insertions of two different linkers at the same position. An arrow with a pentagon represents two identical insertions and thus, may not represent two independent insertion events. Gray bars denote positions of arginine or lysine residues. Black bars mark the position of positive residues subjected to site-directed mutagenesis. “W” marks a region with multiple in-frame insertions and a deletion. (B) Effects of point mutations in Protein 4 on its RNA-binding activity. Indicated Protein 4 mutants were tested in the Y3H system for lacZ activation by quantitative β-galactosidase liquid assay. Each mutant and its cognate wt clone were tested in parallel for binding to 5′ Segment 7 hybrid RNA. The boxplot presents the average β-galactosidase activity (with error bars; n = 3 yeast colonies per protein-RNA pair). Significance between pairs of wt and mutant was calculated using Student’s t test (***, P ≤ 0.001; NS, not significant). (C) Expression of wt and Protein 4 mutants in the yeast. Protein extracts of yeast expressing wt and the indicated Protein 4 mutants were analyzed by Western blotting with antibodies against Protein 4 and actin.
Mutations in specific positively charged residues hinder Protein 4 binding to RNA in the Y3H system.
Next, we used the Y3H system to screen for mutations in Protein 4 that hamper interaction with RNA. We generated a library of yeast plasmids expressing the Gal4AD-Protein 4 fusion with randomly inserted linkers, encoding in-frame five amino acids additions. Proteins encoded by this library were tested for binding to TiLV RNA (5′ Segment 7 hybrid RNA) in the Y3H system. Of ∼7,000 yeast colonies of transformants, we isolated 45 colonies that did not stain blue by colony-lift filter assay. Sequence analyses of Protein 4 ORF expressing plasmids, extracted from these colonies, revealed 39 ORFs with in-frame linker insertions. Overall, the insertions were distributed along the entire ORF (Fig. 3A), suggesting that multiple sequences in Protein 4 contribute to its RNA-binding activity. For IAV, the structure of NP reveals that positively charged residues, distributed along the NP polypeptide, fold to form the surface of the RNA-binding groove (24); and that mutations in specific basic residues (e.g., R267 and R416) greatly reduce NP RNA-binding activity (32, 33). Analogous organization and function of positively charged residues exist in the NP of a fish orthomyxovirus, the infectious salmon anemia virus (ISAV), and specific mutations in such residues disrupt RNA binding (34). Notably, the distribution of basic residues along Protein 4 polypeptide is also wide (Fig. 3A), and we observed a relatively high number of indel mutations in the region that spans residues K134 and K136 (four different linker insertions, of which one was accompanied by an in-frame deletion, Fig. 3A). Accordingly, we introduced, by site-directed mutagenesis, alanine substitutions in these residues (to generate K134A or K136A mutants). Since three linker insertions flanked residue R158, we also mutated this residue (to generate an R158A mutant). Finally, a cluster of four basic residues (K90, K91, R92, and R94) was mutagenized too to generate the K(90,91)A/R(92,94)A mutant. We introduced these mutations into the pACT2-ORF4 plasmid, and the binding of the parental (wt) or mutant Gal4AD-Protein 4 fusions to 5′ Segment 7 hybrid RNA was tested in the Y3H system, using a quantitative β-galactosidase liquid assay (Fig. 3B). Of note, given that we carried out the site-directed mutagenesis on different clones of the pACT2-ORF4 plasmid (see Materials and Methods), for each mutant, a corresponding control clone with wt sequence was generated and tested in parallel with the mutated cognate plasmid. These analyses demonstrated a significant reduction in β-galactosidase activity for the composite mutant K(90,91)A/R(92,94)A, relative to the wt Gal4AD-Protein 4 fusion (Fig. 3B). Of the single-point mutants, K134A showed a significant and robust reduction in β-galactosidase activity, while K136A or R158A did not (Fig. 3B). Immunoblotting confirmed the expression of all mutant and wt Gal4AD-Protein 4 fusions in the yeast (Fig. 3C).
Altogether, these results demonstrate that TiLV Protein 4 interacts with RNA in the Y3H system in a sequence-independent, temperature-dependent manner and that specific positive residues contribute to Protein 4-RNA interactions.
Multiple copies of Protein 4 are complexed with RNA in infected cells and virions.
To test whether Protein 4 is complexed with RNA in infected cells, we applied the R-DeeP method that screens for “RNA-dependent proteins”—proteins that interact with RNA directly or indirectly (35). This unbiased and enrichment-free method is based on density gradient ultracentrifugation, where complexes of RNA-dependent proteins migrate to different locations in sucrose density gradients, depending on the presence or absence of intact RNA. Accordingly, we infected the tilapia OmB cell line with TiLV, extracted RNA-protein complexes from infected cultures, and separated in sucrose density gradients the extracts that were pretreated or not with RNase A. Then, we determined the migration of Protein 4 in the gradients by immunoblotting fractions of the gradients with αProtein4 antibodies. In the absence of RNase A treatment (and the presence of RNase inhibitor), most of Protein 4 migrated to fractions with relatively high sucrose density (Fig. 4A, –RNase, fractions 10 to 23). In contrast, RNase A treatment shifted the majority of this protein to fractions with lower sucrose densities (Fig. 4A, +RNase, fractions 3 to 11). Such a “left shift” is typical of many proteins enriched in domains linked to RNA binding (35) and suggests that in infected cells, Protein 4 is complexed with RNA.
FIG 4.
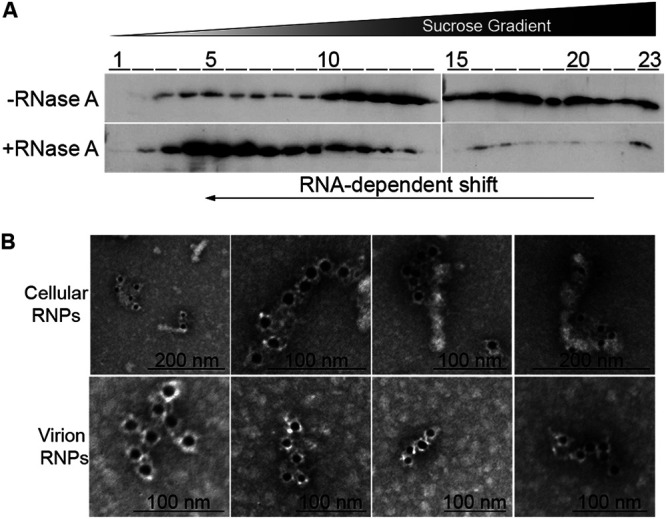
Protein 4 is complexed with RNA. (A) Protein 4 migration in sucrose density gradients. RNA-protein complexes were extracted from TiLV-infected OmB cells, treated or not with RNase A, and separated in sucrose density gradients, by ultracentrifugation, according to the R-DeeP method. Fractions (1–23) of the sucrose gradients were collected from top to bottom (low to high density, respectively). The fractions were analyzed by Western blotting with αProtein4 antibodies. The black arrow at the bottom represents the shift of Protein 4 from dense to light fractions, following RNase A treatment. (B) Negative-staining immuno-EM of RNPs. Fractions of the gradient in panel A (–RNase, fractions 10 to 23), containing cellular RNPs that were extracted by the R-DeeP method and enriched for TiLV RNPs, were pooled, pelleted, and stained by immune-EM with primary αProtein4 antibodies and secondary gold particle-conjugated anti-rabbit antibodies (upper panel). Virion RNPs, extracted as described before (36), were stained as the cellular RNPs (lower panel).
The RNA segments of the IAV genome, multiple copies of the viral NP, and a small number of viral polymerase molecules form rod-like RNP complexes (see, for example, references 15, 36, and 37). Imaging these rod-like structures by negative-staining immunoelectron microscopy (immuno-EM), using primary anti-NP antibodies and secondary gold particles-conjugated antibodies, detected multiple NP molecules along the entire length of the RNP rods (36). To test whether Protein 4 is positioned in a similar pattern with respect to TiLV’s RNPs, obtained by the R-DeeP method, we pooled samples from high-sucrose-density-fractions, enriched for Protein 4-RNA complexes (Fig. 4A, –RNase, fractions 10 to 23). Then, RNPs were pelleted from these pooled samples by ultracentrifugation, stained with primary αProtein4 antibodies and with secondary, gold particle-conjugated, anti-rabbit antibodies, and imaged. The imaging revealed rod-like structures with a clear alignment of the gold particles and these elongated structures (Fig. 4B; cellular RNPs), highly resembling the organization of NP along IAV RNPs (36). The length of the stained rods varied, likely reflecting the different lengths of TiLV genomic RNA segments. RNP filaments of various lengths were reported before for IAV (37). We also observed the same organization of Protein 4 along with rod-like structures by immuno-EM when we isolated RNPs from concentrated TiLV virions, using a method described before for IAV (36) (Fig. 4B; virion RNPs). Altogether, the pattern of Protein 4 staining resembles the one observed for IAV NP and suggests that multiple copies of Protein 4 are part of TiLV RNPs and cover TiLV RNA segments.
Protein 4 directly interacts with RNA in infected cells.
To examine whether Protein 4 directly binds RNA in infected cells, we applied the XRNAX method that purifies cellular cross-linked protein-RNA complexes (38). In this method, proteins and RNAs are extracted from UV-irradiated cells with the classic acid guanidinium thiocyanate-phenol-chloroform extraction mix (39), and a subsequent organic phase separation step separates free RNAs (aqueous phase) and free proteins (organic phase) from cross-linked protein-RNA complexes (enriched in the interphase). To this end, we extracted TiLV-infected OmB cells that were UV-irradiated or not. Only the extract of the UV-irradiated sample formed sponge-like, insoluble interphase, while the sample that was not irradiated formed only fluid-like interphase, as described before (38). We purified the proteins from these interphase fractions and treated them, or not, with DNase or RNase. Next, we analyzed these preparations by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, using αProtein4 antibodies (Fig. 5A). Protein 4 was present in the interphase fraction only when the cells were UV-irradiated, suggesting that it interacts with RNA. In line with this notion, a high molecular weight (MW), Protein 4-reactive band (>250 kDa, marked by a black rectangle in Fig. 5A) was observed only in the UV-cross-linked sample and was sensitive to RNase but not to DNase. Thus, this slow-migrating band is likely composed of high MW complexes of Protein 4 cross-linked to RNA. Of note, in the UV-irradiated samples, we observed an additional Protein 4-reactive band, migrating at Protein 4 calculated MW (∼38 kDa [10], marked by an asterisk in Fig. 5A). This band likely consists of free proteins trapped in the sponge-like interphase, since no additional, silica-based purification steps were included to eliminate such free proteins (38). No reacting bands were observed when we reprobed the membrane with control, rabbit polyclonal antibodies, raised against a TiLV Protein 5-derived peptide (αProtein5 antibodies; see Fig. 5B for immunoblotting specificity of αProtein4 and αProtein5 antibodies and Fig. 5C for reprobing results). Altogether, these results further demonstrate that Protein 4 binds RNA in infected cells. Moreover, the cross-linking by UV suggests a direct binding (40) of Protein 4 to the RNA.
FIG 5.
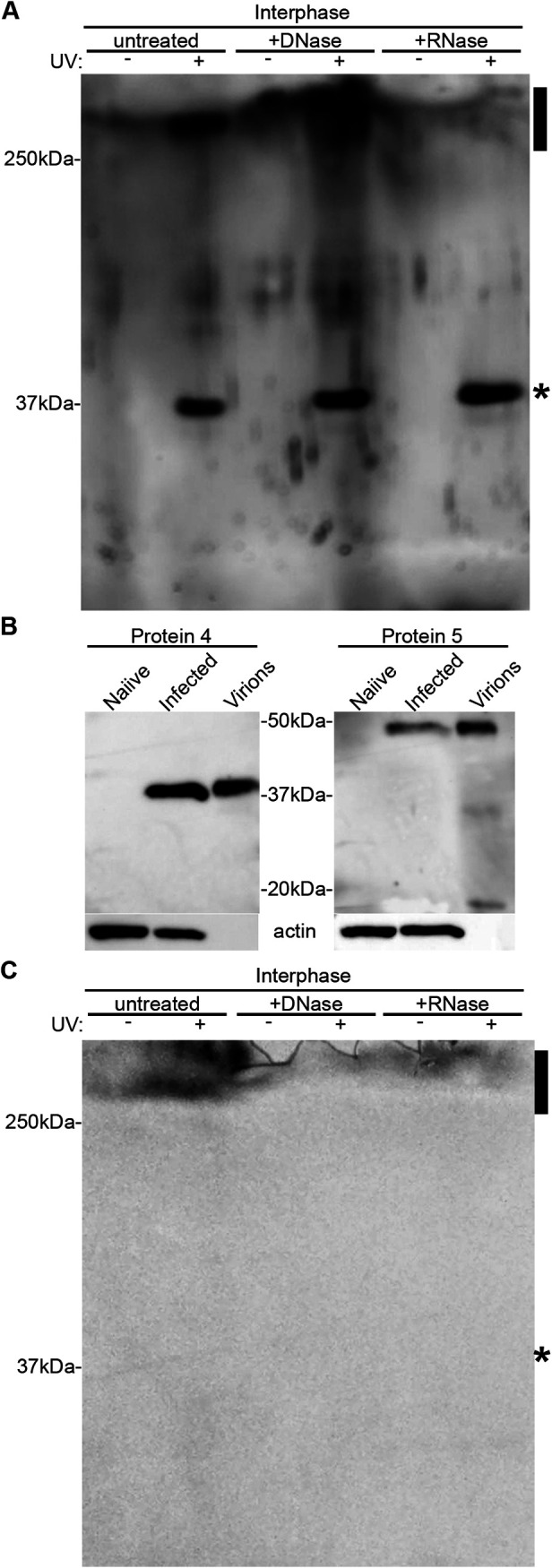
Protein 4 directly interacts with RNA in TiLV-infected cells. (A) TiLV-infected OmB cells were UV-irradiated or not and extracted according to the XRNAX method. Cross-linked protein-RNA complexes were further extracted from a fraction (interphase) enriched with these complexes and were treated, or not, with DNase or RNase. Complexes were analyzed by immunoblotting with αProtein4 antibodies. Reactive bands include free Protein 4 (∼38 kDa; asterisk) and a high-MW, RNase-sensitive complex (>250 kDa; rectangle). (B) TmB cells were infected (MOI = 5) with TiLV (infected), or not (naive), proteins were extracted from the cultured cells at 1 dpi and analyzed by immunoblotting with the indicated antibodies. A pellet of TiLV virions was extracted and analyzed too (Virions). The majority of Protein 5 (calculated MW of ∼38 kDa) appeared as a slower migrating band (∼50 kDa), for both cellular and virions extracts. (C) The blot of Fig. 5A was reprobed with αProtein5 antibodies.
Protein 4 binds TiLV RNA in infected cells and virions.
Next, we tested whether Protein 4 binds TiLV RNA in infected cells. To this end, we infected susceptible E-11 cells (1) with TiLV, and 4 days postinfection (dpi), when first signs of cytopathic effect (CPE) were visible, we lysed the cells with a nondenaturing buffer. Then, we immunoprecipitated Protein 4 from the cleared lysates using αProtein4 antibodies and tested the pellets for the presence of Protein 4 (by immunoblotting) and all 10 segments of TiLV genomic RNA (by qRT-PCR). Controls included immunoprecipitation (IP) of lysates of infected cells, using either αProtein5 antibodies or preimmune rabbit serum (collected before the vaccination with Protein 4), and IP of lysates of uninfected E-11 cells with αProtein4 antibodies. These analyses revealed that the pellet obtained with αProtein4 antibodies contained large amounts of each of the 10 viral RNAs, compared to pellets generated with control antibodies (Fig. 6A). This differential co-IP was in contrast to equal, low levels of actin RNA (CT = ∼26), detected in all pellets (likely reflecting nonspecific binding of this abundant RNA). Accordingly, actin RNA was used as a reference for each pellet, and ΔΔCT values and relative RNA levels (“relative Co-IP”) were calculated (Fig. 6A). Averaging the normalized levels of the 10 RNA segments in each of the pellets revealed a significant co-IP of TiLV RNA with Protein 4, with fold differences of 2 to 3 orders of magnitude, compared to the controls (Fig. 6C). No viral RNAs were detected in the pellet obtained with αProtein4 antibodies from uninfected cells, ensuring the absence of cross-contamination. Immunoblotting revealed an efficient IP of Protein 4 or Protein 5 when the cognate antibodies were used (Fig. 6D). To analyze Protein 4-genomic RNA interactions in TiLV virions, we concentrated virus particles from culture supernatants of TiLV-infected E-11 cells and repeated the above co-IP in nondenaturing conditions with αProtein4, αProtein5, or preimmune antibodies. qRT-PCR (here, virion RNA levels were not normalized to actin RNA) and immunoblotting demonstrated efficient and significant co-IP of Protein 4 with all 10 segments of TiLV RNA genome, compared to controls (Fig. 6B, E, and F). Together, these results suggest that Protein 4 binds the 10 RNA segments of TiLV, both in infected cells and in free virions.
FIG 6.

Co-IP of TiLV RNA with Protein 4. TiLV-infected E-11 cells (A) or virions (B) were lysed in a nondenaturing buffer, and the extracts were immunoprecipitated with αProtein4 or αProtein5 antibodies, or with a preimmune serum. Co-IP of the 10 segments (Seg1-Seg10) of TiLV RNA genome (A and B) and actin mRNA (A) was quantified by qRT-PCR. (A) For each pellet, the ΔΔCT value of each of the viral RNA segments was determined, using actin mRNA as a reference gene (actin CT values were 25.99, 25.84, and 25.97 for preimmune, αProtein5, or αProtein4 pellets, respectively). For each pellet in panel B, the ΔCT value of each of the viral RNA segments was determined. The fold changes of RNA levels in pellets obtained with αProtein4 or αProtein5 antibodies (“relative Co-IP”) in panels A and B were calculated relative to the RNA levels in the preimmune pellet (which was set to 1). (C and E) Boxplots show the average (with error bars) of the co-IP of all 10 viral RNA segments (dots) in the αProtein4 or αProtein5 pellets, relative to the preimmune pellet. ***, P ≤ 0.001 (Student’s t test). (D and F) Immunoblots with αProtein4 or αProtein5 antibodies. Pellets correspond to 4% of the cell or virion extracts, while the input corresponds to 2.5 or 0.8% of the extract analyzed in panels D or F, respectively.
Cytoplasmic and nuclear distribution of Protein 4.
Orthomyxoviruses replicate in the nucleus. The NP escorts the RNA genome to the nucleus and out of the nucleus (41, 42). To monitor the intracellular distribution of Protein 4, we stained TiLV-infected OmB cells at 1 dpi, with primary αProtein4 antibodies and a secondary Alexa Fluor 488-conjugated anti-rabbit antibody; and imaged the cells by fluorescence microscopy. This analysis revealed Protein 4 in both the cytoplasm and the nuclei of infected cells (Fig. 7A and B), while no fluorescent signals were detected in the negative control (Fig. 7A; noninfected), stained cells. Notably, a portion of Protein 4 formed discrete cytoplasmic puncta with variable sizes (Fig. 7A and B). Measuring the average Protein 4 signal in the nucleus and the cytoplasm among infected, untreated cells revealed that 52% of cellular Protein 4 was nuclear (Fig. 7C, UT). This nuclear localization increased to 82% when we treated TiLV-infected cells with leptomycin B (LMB), an inhibitor of nuclear protein export, mediated by CRM1 (43–45) (Fig. 7B and C; LMB). Overall, these results suggest that Protein 4 shuttles between the cytoplasm and the nucleus at one dpi and that it uses CRM1 for nuclear export.
FIG 7.
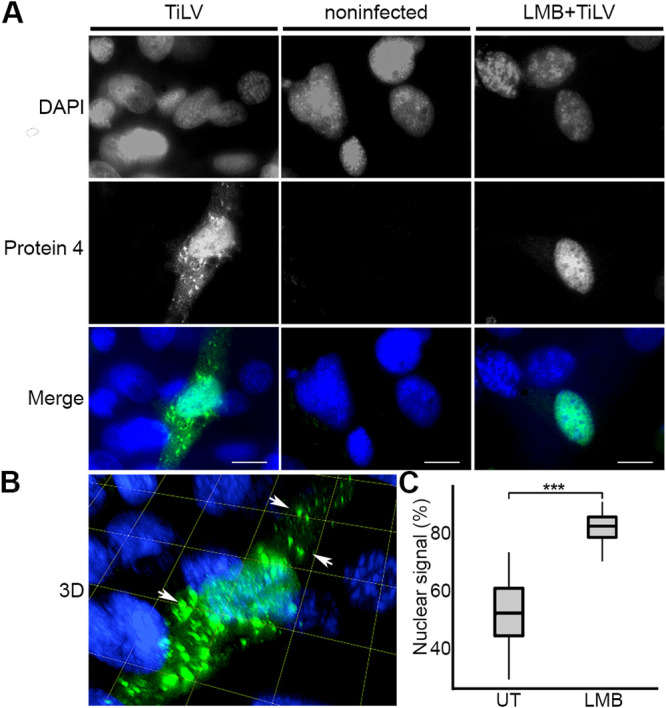
Cytoplasmic and nuclear distribution of Protein 4. (A) OmB cell cultures were infected (MOI = 0.04) with TiLV (TiLV) or not (noninfected) or were treated with 45 nM LMB for 2 h and then infected with TiLV in the presence of LMB (LMB+TiLV). At 24 h postinfection, the cells were stained with αProtein4 antibodies and DAPI and imaged with a confocal microscope. Single optical sections of the cultures are shown, with DAPI and Protein 4 signals in grayscale and merged signals in color. (B) Serial optical sections of the TiLV-infected cell in panel A were reconstituted into a 3D image. Arrows point to Protein 4 puncta. Grid and bars represent 10 μm. (C) Boxplot of the average percentage of the nuclear signal of Protein 4 of 16 TiLV-infected and untreated cells (UT) and 19 infected, LMB-treated cells (LMB), described in panel A. For each cell, the percentage of the nuclear signal was determined by dividing the nuclear signal of Protein 4 by the total signal of this protein in the whole cell. ***, P ≤ 0.001 (Student’s t test).
DISCUSSION
The largest of the 10 major proteins of TiLV has weak homology to the polymerase subunit of ICV and is proposed to subserve that function for TiLV. The functions of the other nine proteins are unknown. The findings presented here are consistent with the identity of Protein 4 as the TiLV NP. These findings include bioinformatic combined feature analysis and experimental characterization of Protein 4 as an RNA-binding protein that binds RNA independently of a specific RNA sequence, interacts with all 10 segments of the TiLV genome, and localizes with RNPs. The migration of Protein 4, extracted from infected cells, in density gradients was typical of RNA-dependent proteins—proteins that are complexed with RNA (35). Indeed, RNase pretreatment of these cellular extracts changed the pattern of Protein 4 migration and shifted it to lesser dense fractions. In addition, Protein 4 from both infected cells and purified virions coimmunoprecipitated in nondenaturing conditions with all 10 segments of the TiLV RNA genome. While these results imply that Protein 4 is complexed with TiLV RNA, they cannot distinguish between direct and indirect interactions. The binding of Protein 4 to RNA in the Y3H system strongly suggests a direct Protein 4-RNA interaction but cannot exclude the presence of an additional cellular factor that mediates this interaction. However, UV-irradiation cross-linked Protein 4 to RNA in infected cells strongly implies a direct interaction (40) between these molecules. Several results suggest that Protein 4 molecules cover TiLV genome in a manner that is independent of the RNA sequence: the localization of multiple copies of Protein 4, along with rod-like assemblies that highly resemble IAV RNPs (36); the interaction of Protein 4 with all 10 genomic RNA segments; and the nonspecific RNA-binding activity of Protein 4 in the Y3H system. Binding single-stranded RNA with no sequence specificity (46) or some specificity to G-rich and U-poor sequences (47) has been demonstrated for IAV NP. The multiple copies of Protein 4 presented in infected cells and virions should assist the development of diagnostics means for TiLV based on antibodies against this protein.
Like IAV NP (48), Protein 4 does not contain canonical consensus sequences of either RGG, KH, arginine-rich, or RRM RNA binding motifs (49). Instead, many positively charged residues likely mediate the interaction with RNA, as is the case for NPs of members of the orthomyxoviridae family, including IAV (24), influenza B virus (IBV) (50), and ISAV (34). These basic residues, scattered throughout the NP polypeptides, fold to make an electropositive, RNA-binding groove. Thus, a similar fold may be essential for Protein 4 RNA binding activity. The linker insertions that disrupted Protein 4 RNA binding activity were scattered throughout ORF4, supporting the notion that multiple regions in Protein 4 contribute to this function. Since many positively charged residues contribute to NP-RNA interaction in the case of IAV, IBV, and ISAV, point mutations in only portion of these basic residues significantly disrupt this interaction (24, 32–34, 50). This is similar to the observation we made for Protein 4 in the Y3H system: specific charged-to-alanine mutations, such as K(90,91)A/R(92,94)A or K134, significantly disrupted the interaction of Protein 4 with RNA, while other point mutations, such as K136A or R158A, did not. Of note, the adaptation, described here, of the Y3H system to study protein-RNA interactions at 25°C should assist the investigation of additional RNA-binding proteins, derived from poikilothermic animals and their pathogens.
During the course of our analyses, we noted an absence of structural homology between IAV/ISAV NPs and Protein 4. The predicted structures of Protein 4 obtained by both AlphaFold (51) or RoseTTAFold (Robetta server [52]) differed substantially one from another and from the solved crystal structures of IAV or ISAV NPs; specifically, we noted very high root mean square deviation (RMSD) values between IAV/ISAV NPs and Protein 4, ranging between 14 and 42 Å (calculated by PyMOL software). The apparent lack of structural homology adds to the lack of sequence homology among TiLV sequences and other known sequences and further demonstrates the uniqueness of TiLV. Moreover, the lack of sequence and structural similarities highlights the importance of the procedures described here that revealed the function for Protein 4.
The fact that an abundant cellular RNA like actin mRNA did not efficiently coimmunoprecipitate with Protein 4 suggests that Protein 4 can preferentially interact with TiLV RNA in infected cells, despite its apparent RNA-binding activity, which is independent of a specific RNA sequence. Such preferential binding may result from coordinated coexpression of NP and viral RNA in a specific cellular compartment(s). Indeed, viruses with negative RNA genome encapsidate their genomes in nucleocapsids concomitantly with viral genome replication (17). In addition, efficient replication of the IAV genome may require nucleolus targeting by NP early in infection (42, 53, 54). The nuclear localization of Protein 4, described here, may provide the cellular compartment needed for TiLV RNP assembly. Currently, we do not know whether specific subnuclear compartments are required for such assembly.
The nuclear and cytoplasmic localization of Protein 4 matches the presence of TiLV RNA in both compartments (10) and supports the notion that Protein 4 functions as NP. Accordingly, we suggest that Protein 4 shuttles TiLV RNA between these two compartments by providing the required nuclear localization signal (NLS) and nuclear export signal (NES). This scenario resembles the cytoplasmic/nuclear distribution of IAV NP (41, 55), mediated by its NLS and NES (reviewed in reference 23). While the balance between NLS and NES activities of IAV NP may be dynamic (23), inhibition of the cellular NES receptor, CRM1/exportin-1, by LMB (43–45), increases the nuclear distribution of IAV NP (56). The nuclear distribution of Protein 4 increased too upon LMB treatment of TiLV-infected cells, further emphasizing the nucleus-cytoplasm shuttling of this protein, its dependency on CRM1 and the overall similarity to influenza virus NP. Currently, we do not know whether Protein 4, like IAV NP, directly binds CRM1 (56), or the identity of specific residues in Protein 4 that function as the NLS or NES. Of note, the matrix (M1) protein of influenza viruses shares some characteristics with the nucleoprotein, including nucleocytoplasmic shuttling as part of the viral RNPs (vRNPs) (57, 58), and thus Protein 4 could have been assigned as TiLV M1 protein. However, we consider this possibility unlikely since M1 does not bind the RNA directly; in contrast, Protein 4 does (demonstrated by the Y3H and XRNAX assays).
Another similarity between TiLV and IAV is that the NPs of the two viruses form discrete puncta. In the case of IAV, after the nuclear-to-cytoplasm export of vRNPs, these complexes form discrete puncta (also named ‘hot spots’) that enlarge upon the progresses of infection, while accumulating different vRNP segments (59–64). Similarly, our immunofluorescence microscopy analyses demonstrate that TiLV protein 4 forms discrete cytoplasmic puncta with various sizes. It will be interesting to determine whether these viral inclusions are also located in close proximity to endoplasmic reticulum (ER) exit sites (63) and if their biogenesis affects (or affected by) the cellular GTPase Rab11 pathway, as was demonstrated for IAV (59, 61–63, 65–68). Overall, the many similarities, described here, that exist between Protein 4 and NPs of influenza viruses further strengthen the idea that TiLV is a member of the orthomyxoviridae family.
In addition to elucidating the probable identity of the NP of a novel virus that is a threat to global food security, our work illustrates a strategy for functional analyses in viral discovery where sequence analysis does not reveal homologies to known viral or cellular proteins. This strategy is important in light of the expected discovery of many unknown viruses (69, 70) and the zoonotic potential (71) of a portion of such pathogens.
MATERIALS AND METHODS
Feature analysis for TiLV and influenza viruses.
Coding sequences of TiLV, influenza D virus (IDV), ISAV and Thogotovirus were downloaded from NCBI (72); coding sequences of IAV, IBV, and ICV were downloaded from the NIAID Influenza Research Database (IRD) (73). Only sequences that maintained a reading frame were further analyzed. The sequences were classified into datasets based on the type of the virus (IAV, IBV, ICV, IDV, ISAV, Thogotovirus, or TiLV) and the type of the ORF (e.g., PB1, M2, or NP). IAV datasets were further classified by the host (e.g., avian or human) and strain (e.g., H1N1 or H5N1). This classification resulted in overall 431 datasets, which were aligned using Mafft alignment (74) with default parameters and, due to computational intensity, the most distant 35 sequences were sampled. Next, we codon-aligned the sequences using PRANK (75) and constructed a maximum-likelihood phylogeny using PhyML (76). Overall, the total number of sequences in our datasets was 14025. Dataset alignments and tree files were uploaded to Zenodo and can be accessed using the following link: https://zenodo.org/record/5811840#.Yc6-TBNBz0o. For each dataset, we calculated three parameters: pI values, the “relative ORF length,” and the dN/dS ratio. Specifically, we determined the average pI for each dataset using an isoelectric point calculator (77). The relative ORF length was calculated by dividing the average length of an ORF (a dataset) by the total coding length of the specific cognate virus (e.g., the average length of PB1 of ICV divided by the total coding length of ICV). For dN/dS calculation, we ran the Selecton software (78) with both the M8 model, which allows for positive selection detection, and the null M8a model. For each dataset, a likelihood ratio test was performed following multiple testing corrections with the false discovery rate (79) to determine which model fits the data better. For each dataset, the dN/dS values were averaged across all positions so that each dataset was represented by one dN/dS value. A 3D scatterplot was generated using ggplot2 (80) and gg3D (https://github.com/AckerDWM/gg3D). Predicting the protein label for each TiLV protein was performed with linear discriminant analysis of the Scikit-learn python module (81).
Y3H plasmids.
TiLV ORFs (of Protein 4, 6, 7, or 8), or portions of TiLV genome (Segment 1 middle region or the 5′ sequence of Segment 7), were amplified from TiLV genomic RNA by RT-PCR (see Table 1 for a list of oligonucleotides used in this work). Amplified ORFs were cloned into BamHI- and EcoRI-digested pACT2 (a yeast expression vector that carries the LEU2 marker; Clontech), using the Gibson Assembly method (82). The resulting plasmids (pACT2-ORF4, 6, 7, or 8) encode fusion proteins with an N-terminal Gal4AD and a C-terminal TiLV protein (Gal4AD-Protein 4, 6 7 or 8, respectively). Plasmids expressing RNA hybrids were generated by homologous recombination in the S. cerevisiae, Y3H strain, L40-coat (27). To this end, pIIIA/MS2-2 (a yeast RNA expression vector that carries the URA3 marker [27]) was linearized with SmaI and SphI and cotransformed into yeast with the above Segment 1- or 7-derived fragments (tailed, by PCR, with sequences homologous to the termini of the linearized pIIIA/MS2-2). The resulting RNA hybrid expressing plasmids were extracted from the yeast with Zymoprep Yeast Plasmid Miniprep I (Zymo Research, catalog no. D2001). All the above plasmids were propagated in E. coli, purified with NucleoBond Xtra Midi Plus (Macherey-Nagel, catalog no. 740412.50), and their inserts’ sequence was verified. RNA hybrids containing either the HIV-1 encapsidation or only the primary sequence of the RNA bridge were described before (27, 28, 30).
TABLE 1.
Oligodeoxynucleotides and their uses
| Oligodeoxynucleotide | Sequence (5′–3′)a | Application |
|---|---|---|
| seg4Fw pACT2homo | ATGGCCATGGAGGCCCCGGGAATGGTGAGAACTACAAAG | Cloning ORF4 in pACT2 |
| Seg4Rev pACT2homo | TCATAGATCTCTCGAGCTCGCTATCTCCCAACAGCCC | Cloning ORF4 in pACT2 |
| seg7Fw pACT2homo | ATGGCCATGGAGGCCCCGGGAATGTCCTACAAGATTGGT | Cloning ORF7 in pACT2 |
| Seg7Rev pACT2homo | TCATAGATCTCTCGAGCTCGTTAGAGTTCAAACGTGATT | Cloning ORF7 in pACT2 |
| seg8Fw pACT2homo | ATGGCCATGGAGGCCCCGGGTATGGCTCAAATCCCAACA | Cloning ORF8 in pACT2 |
| Seg8Rev pACT2homo | TCATAGATCTCTCGAGCTCGTTATTTAAGCATTTCACGG | Cloning ORF8 in pACT2 |
| gen-seg7-Fw-MS2-2-homo | AGAATTCCGGCTAGAACTAGTGGATCCCCCGGGGCAAATCTTTCTCTCATG | Cloning Segment 7 fragment in pIIIA/MS2-2 |
| gen-seg7-Rev-MS2-2-homo | ATCCTCATGTTTTCTAGAGTCGACCTGCAGGCATGCAGCTGTTTCGTTGTCTTA | Cloning Segment 7 fragment in pIIIA/MS2-2 |
| gen-seg1-Fw-MS2-2-homo | AGAATTCCGGCTAGAACTAGTGGATCCCCCGGGCCTGCCATGAGAAAGTAA | Cloning Segment 1 fragment in pIIIA/MS2-2 |
| gen-seg1-Rev-MS2-2-homo | ATCCTCATGTTTTCTAGAGTCGACCTGCAGGCATGCCAAAAGTACCACGCTCAC | Cloning Segment 1 fragment in pIIIA/MS2-2 |
| pACT2 ORF4 FW. pBluescript SK homo (w. EcoRI) | ATCGATAAGCTTGATATCGAATTCCGATGTATAAATGAAAGAAATTG | Cloning ORF4 from pACT2 in pBluescript SK+ |
| pACT2 ORF4 Rev. pBluescript SK homo (w. EcoRI) | GGATCCCCCGGGCTGCAGGAATTCTACCCCACCAAACCCAA | Cloning ORF4 from pACT2 in pBluescript SK+ |
| pACT2 upstream to ORF4 FW | TATCTACGATTCATAGATCT | Insertion detection in pACT2-ORF4 |
| pACT2 downstream to ORF4 REV | TCATATGGCCATGGA | Insertion detection in pACT2-ORF4 |
| A SAMK FW | GGCTCA GCT ATGAAGGTGATAAAAGCCTCAGGG | Site directed mutagenesis- K134A |
| B SAMK Rev | CACCTTCAT AGC TGAGCCCAGTGCAGGCACAC | Site directed mutagenesis- K134A |
| A SKMA FW | GGCTCAAAGATG GCT GTGATAAAAGCCTCAGGG | Site directed mutagenesis- K136A |
| B SKMA Rev | CAC AGC CATCTTTGAGCCCAGTGCAGGCACAC | Site directed mutagenesis- K136A |
| A SAMA Rev | GCCTTCCAGACCTCAATG | Site directed mutagenesis- K134A and K136A |
| B SAMA FW | GGCACCAGAAGCAGGGAT | Site directed mutagenesis- K134A and K136A |
| A LAVCV FW | TTA GCT GTGTGTGTTCGCATTGAGGTCTGGAAGGCTAG | Site directed mutagenesis- R158A |
| A LAVCV Rev | AATCCCCACTCAGATATGG | Site directed mutagenesis- R158A |
| B LAVCV Rev | GCGAACACACAC AGC TAATAGAGAGTTATGGTCCTTC | Site directed mutagenesis- R158A |
| B LAVCV FW | GCCTGCACTGGGCTCAA | Site directed mutagenesis- R158A |
| A ASEAAAEA FW | GAA GCTGCTGCT GAGGCTGAGAACGCTAAGAAATC | Site directed mutagenesis- K(90,91)A/R(92,94)A |
| A ASEAAAEA Rev | GTATTCAGTCCATCTGCT | Site directed mutagenesis- K(90,91)A/R(92,94)A |
| B ASEAAAEA Rev | AGCCTCAGCAGCAGCTTCACTGGCTCTCAGAAC | Site directed mutagenesis- K(90,91)A/R(92,94)A |
| B ASEAAAEA FW | TGTGATATCTGATGCTGCT | Site directed mutagenesis- K(90,91)A/R(92,94)A |
| qSeg1 Fw | TTAGCACCCAGCGGTGG | qPCR of Segment 1 |
| qSeg1 Rev | TGGTCCCTCATAGGCTC | qPCR of Segment 1 |
| qSeg2 Fw | GAGACTTGTCAGCCGTAAGTAAG | qPCR of Segment 2 |
| qSeg2 Rev | GGCAGAACTGAGGTCACAATAA | qPCR of Segment 2 |
| qSeg3 Fw | GTGTACTGTCATCCGCAATCT | qPCR of Segment 3 |
| qSeg3 Rev | GTAAGGACGAGTTCAGGAAAGG | qPCR of Segment 3 |
| qSeg4 Fw | CACTGTTGTCCTCCCTGTTAAA | qPCR of Segment 4 |
| qSeg4 Rev | GTACAAGCTGAACTCCCAAGAA | qPCR of Segment 4 |
| qSeg5 Fw | GTAGCTCTCCAATCACCTCTTC | qPCR of Segment 5 |
| qSeg5 Rev | CGTTCTGCACTGGGTTACA | qPCR of Segment 5 |
| qSeg6 Fw | ACGCTTGACGTGTAGTTTGA | qPCR of Segment 6 |
| qSeg6 Rev | ATGAGTTGGCTTAGGGTGATAAG | qPCR of Segment 6 |
| qSeg7 Fw | GTTGGGTGACTCGTCAATACA | qPCR of Segment 7 |
| qSeg7 Rev | GCCTCGAACGTATGGTTCTT | qPCR of Segment 7 |
| qSeg8 Fw | TGACCCTTGCGGTTTGTTAT | qPCR of Segment 8 |
| qSeg8 Rev | CCGAAAGCGGATAGAGGAAAG | qPCR of Segment 8 |
| qSeg9 Fw | TTCTTGGTGCCTGCCTTT | qPCR of Segment 9 |
| qSeg9 Rev | CAGGAGAGTGCAATGGTGATAG | qPCR of Segment 9 |
| qSeg10 Fw | CCCTTCTTGATCTTCCGACTTC | qPCR of Segment 10 |
| qSeg10 Rev | CAGGATGAGTGTGGCAGATTAT | qPCR of Segment 10 |
| E-11 β-actin-F | CACTGTGCCCATCTACGAG | qPCR of E-11 β-actin |
| E-11 β-actin-R | CCATCTCCTGCTCGAAGTC | qPCR of E-11 β-actin |
TiLV sequences are indicated in italics. Mutated nucleotides are indicated in boldface.
Y3H assay.
Colonies of yeast transformants, selected for uracil (Ura) and leucine (Leu) prototrophy, were analyzed for reporter activation using filter lift assays (27, 28, 30), with the indicated modified incubation times and temperatures. For liquid β-galactosidase assays, yeast transformants (three colonies per condition) were randomly picked from selection plates and grown overnight in a selective medium (5 mL, 25°C). To synchronize the growth of the yeast, the overnight cultures were diluted in a fresh selective medium (OD600 = 0.004) and grown overnight to OD600 of 0.8 to 1 at 25°C. The β-galactosidase activity of the liquid culture was determined with ortho-nitrophenyl-β-galactoside (ONPG) substrate, according to the Clontech Yeast Protocols Handbook (PT3024-1).
Linker insertion mutagenesis.
Using pACT2-ORF4 plasmid as a template, ORF4 and flanking sequences (100 bp long each) were PCR amplified. Using this reaction, we added 40-bp homologous to sequences in pBluescript SK+ plasmid (located upstream and downstream of its EcoRI site) and EcoRI sites to the termini of the amplified fragment. This addition enabled the cloning of the amplified fragment into EcoRI-digested pBluescript SK+ by Gibson assembly reaction. The resulting pBluescript-ORF4 plasmid was subjected to in vitro transposon insertion mutagenesis using MuA transposase (mutation generation system kit; Thermo Scientific, catalog no. F-701). Plasmids with random transposon insertions were transformed into E. coli (JM109 strain) and selected with kanamycin and ampicillin, as the transposon contained the kanamycin resistance gene. Approximately 21,000 kanamycin-resistant colonies were pooled, and their plasmids were extracted. To enrich for ORF4 sequences bearing the transposon insertions, the plasmids (2 μg) were digested with EcoRI and separated in 1% agarose gel, and transposon-containing ORF4 fragments (2.4 kb long) were extracted and cloned into new EcoRI-digested pBluescript SK+ plasmid. The resulting plasmids were transformed and selected as described above. Approximately 3,400 colonies were pooled, and their plasmids were extracted and digested with NotI to remove the majority of the transposon body. The digested plasmids were self-ligated to generate a library of plasmids, encoding ORF4 sequences with randomly inserted, in-frame, 15-bp linker insertions. This library was amplified (electroporated into E. coli, and plasmids were extracted from ∼55,000 ampicillin-resistant colonies) and digested with EcoRI, and ORF4 fragments with random linker insertions were extracted after separation in 1% agarose gel. The sequences that flank ORF4 in these fragments, derived from pACT2 plasmid, allowed the cloning of the fragments into this plasmid by homologous recombination. Accordingly, the fragments were transformed, together with NcoI- and SacI-digested pACT2, into yeast (L40-coat strain) (83), expressing TiLV RNA (5′ Segment 7 hybrid RNA) from plasmid pIIIA/MS2-2. Transformants were grown on selective plates (SD-Leu-Ura), and ∼7,000 colonies were screened for lacZ reporter activation, or lack of activation, using a Y3H, X-Gal colony-lift filter assay. Colonies with no lacZ reporter activation (“white” colonies) were expanded, retested for the lack of reporter activation, and subjected to colony PCR with ORF4-specific primers. Amplified products were sequenced to determine the position of the inserted linkers in ORF4.
Site-directed mutagenesis.
To generate ORF4 sequences with point mutations and to test them in the Y3H assay, we used pACT2-ORF4 plasmids with linker insertions (isolated from the above mutagenesis screen), since these linkers contained a unique NotI site, allowing cloning in the yeast by homologous recombination (see below). Specifically, plasmids pACT2-ORF4 CC38W, CC25W, or CC6W (harboring linker insertions downstream of nucleotide 920, 813 or 731 of Segment 4 genomic RNA, respectively; GenBank accession no. KU751817), were used to generate the ORF4 mutants K(90,91)A/R(92,94)A, K134A, and K136A, or R158A, respectively. Mutation-containing ORF4 fragments were generated by overlapping PCR with primers harboring the indicated mutations (Table 1). The resulting PCR fragments termini contained sequences homologous to ORF4 sequences, found upstream and downstream of the NotI sites in the cognate pACT2-ORF4 CC38W, CC25W, or CC6W plasmids. These plasmids were digested with NotI and cotransformed with their related PCR fragments into yeast L40-coat strain to allow homologous recombination. In the resulting plasmids, the PCR fragment harboring the point mutation(s) replaced the linker-containing sequence. These plasmids were extracted from the yeast, amplified in E. coli, and the sequences of ORF4 mutants were confirmed. Next, the plasmids were tested for loss-of-function (lack of lacZ reporter activation) in the Y3H assay. To verify that this loss of function was the result of the mutations in ORF4 and not of unidentified mutations in the plasmid backbone, ORF4 wild-type (wt) sequences (with no point mutations) were PCR amplified and introduced into NotI-digested, pACT2-ORF4 CC38W, CC25W, or CC6W plasmids to confirm gain of function (activation of the lacZ reporter) of the resulting plasmids in the Y3H assay.
Generation of rabbit polyclonal antibodies.
Polyclonal antibodies were raised in rabbits, according to Tel Aviv University Animal Care guidelines. To generate antibodies against Protein 4 (αProtein4), ORF4 was PCR amplified from the pACT2-ORF4 plasmid and cloned into pET28b(+) bacterial expression vector between NdeI and XhoI restriction sites, thus attaching a 6×His tag to the N terminus of Protein 4. The resulting pET28-ORF4 plasmid was transformed into E. coli BL21(DE3) and grown in LB under kanamycin selection. An overnight starter was diluted 1:50 and grown in 200 mL of LB + kanamycin at 37°C and an optical density at 600 nm (OD600) of 0.5, and protein expression was induced with 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h. Next, the cells were pelleted and resuspended in 4 mL of native suspension buffer (20 mM Tris, 500 mM NaCl [pH 8.0], complete EDTA-free protease inhibitor cocktail [Roche, catalog no. 11836170001], and 0.1% Triton). Lysozyme was added (1 mg/mL final concentration), and the suspension was kept on ice for 20 min with an occasional vortex. Cells were disrupted by sonication on ice, using a microtip-equipped sonicator (Fisher Scientific Series 60 Sonic Dismembrator Model F60, 5 × 10-s pulses with 10-s intervals). The lysate was treated with 10 μg/mL RNase A and 5 μg/mL DNase I (Sigma-Aldrich, catalog no. R4642 and catalog no. D4263, respectively) for 10 min on ice with occasional vortexing and then centrifuged (Sorvall RC 6 plus, SS-34 rotor, 20,200 × g) for 20 min at 4°C. The pellet containing Protein 4 in inclusion bodies was resuspended in 10 mL of Buffer-II (50 mM Na2HPO4, 300 mM NaCl, 8 M urea [pH 8.0]) and stirred at room temperature to solubilize the lysate (∼30 min). The lysate was then centrifuged (20 min at 10,000 × g at room temperature), and the soluble Protein 4 was purified from the clear supernatant by nickel-agarose beads batch purification. Specifically, nickel-agarose slurry (2 mL; Adar-Biotech, catalog no. 1018-25), equilibrated with equilibration buffer (10 mM Tris, 50 mM Na2HPO4, 500 mM NaCl, 8 M urea [pH 8.0]), was mixed with Protein 4 lysate and stirred on a rotary shaker (60 min at 4°C). The beads were washed (on a column) with washing buffer (10 mM Tris, 50 mM Na2HPO4, 500 mM NaCl, 8 M urea, 10 mM imidazole [pH 8.0]) until no protein was detected in the flowthrough, as tested by using Bradford reagent. Protein 4 was eluted with 5 mL of elution buffer (10 mM Tris, 50 mM Na2HPO4, 250 mM NaCl, 8 M urea, 300 mM imidazole [pH 8.0]), collecting 1-mL fractions. Peak fractions were pooled and dialyzed twice at 4°C against dialysis buffer (20 mM Tris, 300 mM NaCl, 0.01% NP-40, 10% glycerol, 1 mM MgCl2 [pH 7.5]; the first dialysis buffer also contained 1 mM dithiothreitol [DTT], while the second did not). Dialyzed protein was stored in aliquots at −20°C. A Bradford assay was used to determine the protein concentration. Two albino New Zealand female rabbits (2 months old) were injected with Protein 4 (100 μg/rabbit) mixed with a 1.5 volume of complete Freund’s adjuvant (Sigma, catalog no. F5881). Immunization was followed by two booster immunizations with the same dose of Protein 4, mixed with incomplete Freund’s adjuvant (Sigma, catalog no. F5506). The rabbits were continuously immunized every 3 to 4 weeks (up to five injections). Rabbit serum samples were collected 1 week after the second boost (and subsequently every 3 weeks). Preimmunized sera were collected before immunization. To generate antibodies against Protein 5 (αProtein5), a peptide derived from the ORF5 sequence was synthesized using a Liberty Blue automated microwave peptide synthesizer (Medicinal Chemistry Laboratory, The Blavatnik Center for Drug Discovery, Tel Aviv University). One rabbit was immunized with KLH-conjugated peptide (100 μg/injection) as described above.
Immunoblotting and antibodies.
Proteins in extracts of yeast, fish cells, and pellets of co-IP or XRNAX experiments were mixed with loading buffer (final concentration of 0.05 M Tris-HCl [pH 6.8], 0.1 M DTT, 2% SDS, 10% glycerol, 0.1% bromophenol blue), heated (100°C, 5 min), separated by SDS-PAGE, and transferred to nitrocellulose membrane (iBlot 2 Transfer Stacks; Invitrogen, catalog no. IB23001). Western blot analyses were performed as previously described (84), with the following modifications: membranes were incubated with primary antibodies overnight at 4°C, and detection of the horseradish peroxidase (HRP), conjugated to the secondary antibodies was performed with Immobilon Forte Western HRP substrate (Millipore, catalog no. WBLUF0500). The primary antibodies included mouse anti-actin (MP Biomedicals, catalog no. 69100; 1:10,000 dilution), mouse anti-HA epitope tag (BioLegend, catalog no. MMS-101R; 1:1,000 dilution), rabbit anti-Protein 4 (1:10,000 dilution), and rabbit anti-Protein 5 (1:1,000 dilution). The secondary antibodies included HRP-conjugated goat anti-mouse antibody (Jackson ImmunoResearch, catalog no. 115-035-003; 1:10,000 dilution) and HRP-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, catalog no. 111-035-003; 1:15,000 dilution).
Cell lines.
The spontaneously immortalized OmB cell line, derived from Mozambique tilapia (Oreochromis mossambicus) brain (85) and the E-11 cell line, derived from the striped snakehead (Ophicephalus striatus) (86), were grown as described previously (31).
Migration of Protein 4 in sucrose density gradients.
TiLV-infected OmB cells (confluent culture in two 10-cm dishes) were trypsinized when the first signs of CPE appeared (4 dpi). Cells were combined, pelleted, and extracted, and the extract was treated, or not, with RNase, according to the R-DeeP method (35), with the following modifications: in the absence of RNase treatment, RNase inhibitor (RNasin; Promega, catalog no. N251B) was added to the extract (100 U/mL); for RNA digestion, only RNase A (Sigma, catalog no. R4642) was used. Treated extracts were loaded on 5 to 50% sucrose density gradients, which were fractionated into 23 fractions (500 μL each) after ultracentrifugation (Beckman Optima XPN-80 ultracentrifugation, SW41 Ti swinging-bucket rotor, 160,000 × g, 18 h, at 4°C). Then, 20-μL portions of each fraction were analyzed by immunoblotting. For immuno-EM, 100-μL portions of the indicated fractions were analyzed.
Negative-staining immuno-EM.
Samples (100 μL each) from fractions of the sucrose density gradient (not treated with RNase and enriched with RNP/Protein 4 complexes), were pooled and mixed with R-DeeP lysis buffer (35) to a final volume of 13 mL. RNPs were pelleted by ultracentrifugation (Beckman Optima XPN-80 ultracentrifugation, SW 41 Ti swinging-bucket rotor, 160,000 × g, 3 h, at 4°C), resuspended in 90 μL of of DNase/RNase-free water, and adsorbed to nickel Formvar/carbon-coated grids (30 μL/grid). RNPs from PEG-concentrated virions (see above) were isolated as described before (36) and adsorbed to the grids. The grids were washed with phosphate-buffered saline (PBS), blocked with bovine serum albumin (BSA) (1%)-containing PBS (30 min), and incubated (40 min at room temperature) with αProtein4 antibodies (diluted 1:500 in blocking solution). Grids were further washed with blocking solution; incubated (40 min at room temperature) with a secondary, 12-nm gold particle-conjugated, goat anti-rabbit antibody (Jackson ImmunoResearch, catalog no. 111-205-144); washed with PBS; fixed with 2.5% glutaraldehyde; and finally washed with PBS and ddH2O. The grids were contrasted with aqueous 2% uranyl acetate and examined with JEM 1400plus transmission electron microscope (Jeol, Japan). Images were captured using SIS Megaview III and the iTEM TEM imaging platform (Olympus).
XRNAX.
Confluent OmB cells, in a 10-cm plate, were infected with TiLV, and when the first signs of CPE appeared (4 dpi), the culture was processed according to the XRNAX method (38) and an online protocol (https://www.xrnax.com/). Briefly, the supernatant was discarded, and cold PBS (6 mL) was added. The cells were irradiated with UV (150 mJ/cm2), trypsinized, and pelleted. The pellet was extracted with acid guanidinium thiocyanate-phenol-chloroform (EZ-RNA kit; Biological Industries, catalog no. 20-400-100), and phases were separated by centrifugation. Aqueous and organic phases (containing free RNA and proteins, respectively) were discarded. The sponge-like, insoluble interphase (enriched for protein-RNA cross-linked complexes) was washed with TE+0.1% SDS buffer (10 mM Tris-Cl, 1 mM EDTA, 0.1% SDS), disintegrated in TE+0.1% SDS and in TE+0.5% SDS buffer (10 mM Tris-Cl, 1 mM EDTA, 0.5% SDS), and isopropanol precipitated according to the XRNAX protocol. The pellet was resuspended in 21 μL of ultrapure water. Equal portions of the cross-linked RNA-Protein suspension were digested with DNase or RNase or left untreated. Specifically, 7 μL of the suspension was mixed with 10 μL of ultrapure water, 2 μL of 10× DNase buffer, and 1 μL of DNase (Baseline-ZERO DNase; Epicentre, catalog no. DB0711K) and then incubated for 1 h at 37°C. Alternatively, 7 μL of the cross-linked RNA-Protein suspension was mixed with 13 μL of ultrapure water and 0.1 μL of RNase A, followed by incubation for 1 h at room temperature. The digested and undigested suspensions were analyzed by immunoblotting with rabbit anti-Protein 4 polyclonal (αProtein4) antibodies.
Co-IP of Protein 4 and TiLV RNA.
TiLV virions (concentrated from the supernatant of a TiLV-infected OmB culture by the PEG method [87]) or TiLV-infected E-11 cells (MOI = 0.8; a confluent culture in a 10-cm plate, trypsinized at 4 dpi and pelleted) were each resuspended in 200 μL of cold nondenaturing lysis buffer (20 mM Tris HCl [pH 8], 137 mM NaCl, 1% Nonidet P-40 [NP-40], 2 mM EDTA, and protease inhibitor cocktail [Merck, catalog no. 11697498001]). The suspensions were incubated on ice (30 min) and centrifuged (20 min, 17,000 × g, 4°C). Then, 60-μL portions of the cleared lysate were transferred to a fresh tube containing 700 μL of lysis buffer and 5 μL of the indicated antibody and rotated (1 h, 4°C). Next, 50 μL of protein A-conjugated magnetic beads (Dynabeads, Invitrogen, catalog no. 10001D) were washed with lysis buffer, rotated with blocking solution (0.1% BSA in PBS, 1 h), washed twice with lysis buffer, resuspended in 100 μL of lysis buffer, and added to each sample. The lysate-beads mixture was rotated (4 h, 4°C), and the beads were pelleted, washed three times in lysis buffer, and resuspended in 40 μL of lysis buffer. Then, 5 μL of the suspension was reverse transcribed (Quantabio, qScript flex cDNA synthesis kit, catalog no. 95049-100). cDNA was diluted (1:10), and 2 μL was subjected to qPCR with a StepOnePlus real-time PCR system (Applied Biosystems), using Fast SYBR green Master Mix (Thermo Fisher Scientific, catalog no. 4385612) and primers specific for each of the 10 segments of TiLV RNA genome or for the cellular actin mRNA. ΔCT or ΔΔCT values were calculated for RNAs immunoprecipitated from virion or cellular preparations, respectively, and the relative expression was calculated (88).
Immunofluorescence.
OmB cells were seeded on collagen-coated, 13-mm glass coverslips (4 mg/mL collagen type I from rat tail; Sigma, catalog no. C3867) in a 24-well plate, treated, or not, with 45 nM LMB (Merck, catalog no. L2913) for 2 h before TiLV addition. At 1 dpi, media with LMB/virus were removed, and cells were washed twice with cold PBS, fixed with 4% paraformaldehyde for 30 min, and washed three times with PBS. The cells were blocked and permeabilized in blocking solution (1% BSA/0.1% Triton X-100 in PBS; 45 min); incubated with αProtein4 antibodies (diluted 1:1,000 in blocking solution; 45 min); washed three times with PBS; and incubated with fluorescently labeled, secondary goat anti-rabbit antibodies (Alexa Fluor 488; Thermo Fisher Scientific, catalog no. A-11034; diluted 1:250 in blocking solution; 45 min). DAPI stain (2.5 μg/mL final concentration) was added to the slides with the secondary antibodies. The cells were washed three times with PBS, and the coverslips were glued to glass slides with fluorescent mounting medium (GBI Labs, catalog no. E18-18). All of these steps were carried out at room temperature. To calculate the nuclear-cytoplasmic distribution of Protein 4, TiLV-infected OmB cells, treated or not with LMB, were stained as described above and imaged with spinning disk confocal (Yokogawa CSU-22 confocal head) microscope (Axiovert 200 M; Carl Zeiss MicroImaging) as described previously (89, 90). Calculations were performed on single confocal midplanes of cells using the DAPI signal to define the nuclei, while the entire cell area was manually defined. Protein 4 signal of selected areas (whole cell, nucleus, and background areas) was measured using the SlideBook program and the Mask function. For each area, the mean pixel intensity of Protein 4 signal and the number of pixels of the area were determined. Total Protein 4 signal of each area was calculated by subtracting the mean pixel intensity of the background from the mean pixel intensity of the selected area and multiplying by the number of pixels per area. The nuclear Protein 4 signal percentage was determined by dividing the Protein 4 signal in the nucleus by the whole-cell Protein 4 signal.
ACKNOWLEDGMENTS
We thank Judith Berman (Tel Aviv University) for helpful discussions, Michal Ucko (Israel Oceanographic and Limnological Research) for E-11 cells, Dietmar Kültz (University of California Davis) for OmB cells, Vered Holdengrebe (Electron Microscopy Core Unit, Faculty of Life Sciences, Tel Aviv University) for negative-staining immuno-EM procedures, and Jeroen Krijgsveld (DKFZ) for advice regarding the XRNAX method.
Contributor Information
Eran Bacharach, Email: eranba@tauex.tau.ac.il.
Anice C. Lowen, Emory University School of Medicine
REFERENCES
- 1.Eyngor M, Zamostiano R, Kembou Tsofack JE, Berkowitz A, Bercovier H, Tinman S, Lev M, Hurvitz A, Galeotti M, Bacharach E, Eldar A. 2014. Identification of a novel RNA virus lethal to tilapia. J Clin Microbiol 52:4137–4146. 10.1128/JCM.00827-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferguson HW, Kabuusu R, Beltran S, Reyes E, Lince JA, del Pozo J. 2014. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): a case report. J Fish Dis 37:583–589. 10.1111/jfd.12142. [DOI] [PubMed] [Google Scholar]
- 3.Surachetpong W, Roy SRK, Nicholson P. 2020. Tilapia lake virus: the story so far. J Fish Dis 43:1115–1132. 10.1111/jfd.13237. [DOI] [PubMed] [Google Scholar]
- 4.Dong HT, Ataguba GA, Khunrae P, Rattanarojpong T, Senapin S. 2017. Evidence of TiLV infection in tilapia hatcheries from 2012 to 2017 reveals probable global spread of the disease. Aquaculture 479:579–583. 10.1016/j.aquaculture.2017.06.035. [DOI] [Google Scholar]
- 5.Waiyamitra P, Piewbang C, Techangamsuwan S, Liew WC, Surachetpong W. 2021. Infection of Tilapia tilapinevirus in Mozambique tilapia (Oreochromis mossambicus), a globally vulnerable fish species. Viruses 13:1104. 10.3390/v13061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tattiyapong P, Dachavichitlead W, Surachetpong W. 2017. Experimental infection of Tilapia Lake Virus (TiLV) in Nile tilapia (Oreochromis niloticus) and red tilapia (Oreochromis spp.). Vet Microbiol 207:170–177. 10.1016/j.vetmic.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 7.Del-Pozo J, Mishra N, Kabuusu R, Cheetham S, Eldar A, Bacharach E, Lipkin WI, Ferguson HW. 2017. Syncytial hepatitis of tilapia (Oreochromis niloticus L.) is associated with orthomyxovirus-like virions in hepatocytes. Vet Pathol 54:164–170. 10.1177/0300985816658100. [DOI] [PubMed] [Google Scholar]
- 8.Surachetpong W, Janetanakit T, Nonthabenjawan N, Tattiyapong P, Sirikanchana K, Amonsin A. 2017. Outbreaks of Tilapia lake virus infection, Thailand, 2015-2016. Emerg Infect Dis 23:1031–1033. 10.3201/eid2306.161278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behera BK, Pradhan PK, Swaminathan TR, Sood N, Paria P, Das A, Verma DK, Kumar R, Yadav MK, Dev AK, Parida PK, Das BK, Lal KK, Jena JK. 2018. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia “Oreochromis niloticus” (Linnaeus 1758) in India. Aquaculture 484:168–174. 10.1016/j.aquaculture.2017.11.025. [DOI] [Google Scholar]
- 10.Bacharach E, Mishra N, Briese T, Zody MC, Kembou Tsofack JE, Zamostiano R, Berkowitz A, Ng J, Nitido A, Corvelo A, Toussaint NC, Abel Nielsen SC, Hornig M, Del Pozo J, Bloom T, Ferguson H, Eldar A, Lipkin WI. 2016. Characterization of a novel orthomyxo-like virus causing mass die-offs of Tilapia. mBio 7:e00431–16. 10.1128/mBio.00431-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfaçon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ. 2017. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch Virol 162:2505–2538. 10.1007/s00705-017-3358-5. [DOI] [PubMed] [Google Scholar]
- 12.Turnbull OMH, Ortiz-Baez AS, Eden J-S, Shi M, Williamson JE, Gaston TF, Zhang Y-Z, Holmes EC, Geoghegan JL. 2020. Meta-transcriptomic identification of divergent Amnoonviridae in fish. Viruses 12:1254. 10.3390/v12111254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz-Baez AS, Eden J-S, Moritz C, Holmes EC. 2020. A divergent articulavirus in an Australian gecko identified using meta-transcriptomics and protein structure comparisons. Viruses 12:613. 10.3390/v12060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turrell L, Lyall JW, Tiley LS, Fodor E, Vreede FT. 2013. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat Commun 4:1591. 10.1038/ncomms2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coloma R, Arranz R, de la Rosa-Trevín JM, Sorzano COS, Munier S, Carlero D, Naffakh N, Ortín J, Martín-Benito J. 2020. Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Nat Microbiol 5:727–734. 10.1038/s41564-020-0675-3. [DOI] [PubMed] [Google Scholar]
- 16.Dadonaite B, Gilbertson B, Knight ML, Trifkovic S, Rockman S, Laederach A, Brown LE, Fodor E, Bauer DLV. 2019. The structure of the influenza A virus genome. Nat Microbiol 4:1781–1789. 10.1038/s41564-019-0513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo M, Terrell JR, Mcmanus SA. 2020. Nucleocapsid structure of negative strand RNA virus. Viruses 12:835. 10.3390/v12080835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreira ÉA, Weber A, Bolte H, Kolesnikova L, Giese S, Lakdawala S, Beer M, Zimmer G, García-Sastre A, Schwemmle M, Juozapaitis M. 2016. A conserved influenza A virus nucleoprotein code controls specific viral genome packaging. Nat Commun 7:12861. 10.1038/ncomms12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine. RNA Biol 8:207–215. 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pflug A, Lukarska M, Resa-Infante P, Reich S, Cusack S. 2017. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res 234:103–117. 10.1016/j.virusres.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Ruigrok RWH, Crépin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Te Velthuis AJW, Grimes JM, Fodor E. 2021. Structural insights into RNA polymerases of negative-sense RNA viruses. Nat Rev Microbiol 19:303–318. 10.1038/s41579-020-00501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Portela A, Digard P. 2002. The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J Gen Virol 83:723–734. 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 24.Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082. 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki Y, Gojobori T. 1999. A method for detecting positive selection at single amino acid sites. Mol Biol Evol 16:1315–1328. 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- 27.SenGupta DJ, Zhang B, Kraemer B, Pochart P, Fields S, Wickens M. 1996. A three-hybrid system to detect RNA-protein interactions in vivo. Proc Natl Acad Sci USA 93:8496–8501. 10.1073/pnas.93.16.8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacharach E, Goff SP. 1998. Binding of the human immunodeficiency virus type 1 Gag protein to the viral RNA encapsidation signal in the yeast three-hybrid system. J Virol 72:6944–6949. 10.1128/JVI.72.8.6944-6949.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans MJ, Bacharach E, Goff SP. 2004. RNA sequences in the Moloney murine leukemia virus genome bound by the Gag precursor protein in the yeast three-hybrid system. J Virol 78:7677–7684. 10.1128/JVI.78.14.7677-7684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mark-Danieli M, Laham N, Kenan-Eichler M, Castiel A, Melamed D, Landau M, Bouvier NM, Evans MJ, Bacharach E. 2005. Single point mutations in the zinc finger motifs of the human immunodeficiency virus type 1 nucleocapsid alter RNA binding specificities of the gag protein and enhance packaging and infectivity. J Virol 79:7756–7767. 10.1128/JVI.79.12.7756-7767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kembou Tsofack JE, Zamostiano R, Watted S, Berkowitz A, Rosenbluth E, Mishra N, Briese T, Lipkin WI, Kabuusu RM, Ferguson H, Del Pozo J, Eldar A, Bacharach E. 2017. Detection of tilapia lake virus in clinical samples by culturing and nested reverse transcription-PCR. J Clin Microbiol 55:759–767. 10.1128/JCM.01808-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elton D, Medcalf L, Bishop K, Harrison D, Digard P. 1999. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J Virol 73:7357–7367. 10.1128/JVI.73.9.7357-7367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng AK-L, Zhang H, Tan K, Li Z, Liu J, Chan PK-S, Li S-M, Chan W-Y, Au SW-N, Joachimiak A, Walz T, Wang J-H, Shaw P-C. 2008. Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J 22:3638–3647. 10.1096/fj.08-112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng W, Olson J, Vakharia V, Tao YJ. 2013. The crystal structure and RNA-binding of an orthomyxovirus nucleoprotein. PLoS Pathog 9:e1003624. 10.1371/journal.ppat.1003624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caudron-Herger M, Rusin SF, Adamo ME, Seiler J, Schmid VK, Barreau E, Kettenbach AN, Diederichs S. 2019. R-DeeP: proteome-wide and quantitative identification of RNA-dependent proteins by density gradient ultracentrifugation. Mol Cell 75:184–199.e10. 10.1016/j.molcel.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugita Y, Sagara H, Noda T, Kawaoka Y. 2013. Configuration of viral ribonucleoprotein complexes within the influenza A virion. J Virol 87:12879–12884. 10.1128/JVI.02096-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallagher JR, Torian U, McCraw DM, Harris AK. 2017. Structural studies of influenza virus RNPs by electron microscopy indicate molecular contortions within NP supra-structures. J Struct Biol 197:294–307. 10.1016/j.jsb.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trendel J, Schwarzl T, Horos R, Prakash A, Bateman A, Hentze MW, Krijgsveld J. 2019. The human RNA-binding proteome and its dynamics during translational arrest. Cell 176:391–403.e19. 10.1016/j.cell.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc 1:581–585. 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 40.Luo M-J, Reed R. 2003. Identification of RNA binding proteins by UV cross-linking. Curr Protoc Mol Biol Chapter 27:Unit 27.2. [DOI] [PubMed] [Google Scholar]
- 41.Neumann G, Castrucci MR, Kawaoka Y. 1997. Nuclear import and export of influenza virus nucleoprotein. J Virol 71:9690–9700. 10.1128/JVI.71.12.9690-9700.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Josset L, Frobert E, Rosa-Calatrava M. 2008. Influenza A replication and host nuclear compartments: many changes and many questions. J Clin Virol 43:381–390. 10.1016/j.jcv.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Kudo N, Wolff B, Sekimoto T, Schreiner EP, Yoneda Y, Yanagida M, Horinouchi S, Yoshida M. 1998. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp Cell Res 242:540–547. 10.1006/excr.1998.4136. [DOI] [PubMed] [Google Scholar]
- 44.Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060. 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 45.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. 1997. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390:308–311. 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 46.Baudin F, Bach C, Cusack S, Ruigrok RW. 1994. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J 13:3158–3165. 10.1002/j.1460-2075.1994.tb06614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee N, Le Sage V, Nanni AV, Snyder DJ, Cooper VS, Lakdawala SS. 2017. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res 45:8968–8977. 10.1093/nar/gkx584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albo C, Valencia A, Portela A. 1995. Identification of an RNA binding region within the N-terminal third of the influenza A virus nucleoprotein. J Virol 69:3799–3806. 10.1128/JVI.69.6.3799-3806.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burd CG, Dreyfuss G. 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265:615–621. 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- 50.Ng AK-L, Lam MK-H, Zhang H, Liu J, Au SW-N, Chan PK-S, Wang J, Shaw P-C. 2012. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J Virol 86:6758–6767. 10.1128/JVI.00073-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim DE, Chivian D, Baker D. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davey J, Colman A, Dimmock NJ. 1985. Location of influenza virus M, NP, and NS1 proteins in microinjected cells. J Gen Virol 66:2319–2334. 10.1099/0022-1317-66-11-2319. [DOI] [PubMed] [Google Scholar]
- 54.Ozawa M, Fujii K, Muramoto Y, Yamada S, Yamayoshi S, Takada A, Goto H, Horimoto T, Kawaoka Y. 2007. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J Virol 81:30–41. 10.1128/JVI.01434-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whittaker G, Bui M, Helenius A. 1996. Nuclear trafficking of influenza virus ribonucleoproteins in heterokaryons. J Virol 70:2743–2756. 10.1128/JVI.70.5.2743-2756.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elton D, Simpson-Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. 2001. Interaction of the influenza virus nucleoprotein with the cellular CRM1-mediated nuclear export pathway. J Virol 75:408–419. 10.1128/JVI.75.1.408-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Yu M, Zheng W, Liu W. 2015. Nucleocytoplasmic shuttling of influenza A virus proteins. Viruses 7:2668–2682. 10.3390/v7052668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sakaguchi A, Hirayama E, Hiraki A, Ishida Y, Kim J. 2003. Nuclear export of influenza viral ribonucleoprotein is temperature-dependently inhibited by dissociation of viral matrix protein. Virology 306:244–253. 10.1016/s0042-6822(02)00013-2. [DOI] [PubMed] [Google Scholar]
- 59.Amorim MJ, Bruce EA, Read EKC, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J Virol 85:4143–4156. 10.1128/JVI.02606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chou Y-y, Heaton NS, Gao Q, Palese P, Singer RH, Singer R, Lionnet T. 2013. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog 9:e1003358. 10.1371/journal.ppat.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eisfeld AJ, Kawakami E, Watanabe T, Neumann G, Kawaoka Y. 2011. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J Virol 85:6117–6126. 10.1128/JVI.00378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vale-Costa S, Alenquer M, Sousa AL, Kellen B, Ramalho J, Tranfield EM, Amorim MJ. 2016. Influenza A virus ribonucleoproteins modulate host recycling by competing with Rab11 effectors. J Cell Sci 129:1697–1710. 10.1242/jcs.188409. [DOI] [PubMed] [Google Scholar]
- 63.Alenquer M, Vale-Costa S, Etibor TA, Ferreira F, Sousa AL, Amorim MJ. 2019. Influenza A virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat Commun 10:1629. 10.1038/s41467-019-09549-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lakdawala SS, Wu Y, Wawrzusin P, Kabat J, Broadbent AJ, Lamirande EW, Fodor E, Altan-Bonnet N, Shroff H, Subbarao K. 2014. Influenza a virus assembly intermediates fuse in the cytoplasm. PLoS Pathog 10:e1003971. 10.1371/journal.ppat.1003971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Castro Martin IF, Fournier G, Sachse M, Pizarro-Cerda J, Risco C, Naffakh N. 2017. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat Commun 8:1396. 10.1038/s41467-017-01557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Momose F, Sekimoto T, Ohkura T, Jo S, Kawaguchi A, Nagata K, Morikawa Y. 2011. Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS One 6:e21123. 10.1371/journal.pone.0021123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avilov SV, Moisy D, Munier S, Schraidt O, Naffakh N, Cusack S. 2012. Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J Virol 86:1433–1448. 10.1128/JVI.05820-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kawaguchi A, Hirohama M, Harada Y, Osari S, Nagata K. 2015. Influenza virus induces cholesterol-enriched endocytic recycling compartments for budozone formation via cell cycle-independent centrosome maturation. PLoS Pathog 11:e1005284. 10.1371/journal.ppat.1005284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, Ojeda-Flores R, Arrigo NC, Islam A, Ali Khan S, Hosseini P, Bogich TL, Olival KJ, Sanchez-Leon MD, Karesh WB, Goldstein T, Luby SP, Morse SS, Mazet JAK, Daszak P, Lipkin WI. 2013. A strategy to estimate unknown viral diversity in mammals. mBio 4:e00598-13. 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delwart E. 2013. A roadmap to the human virome. PLoS Pathog 9:e1003146. 10.1371/journal.ppat.1003146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, Daszak P. 2017. Host and viral traits predict zoonotic spillover from mammals. Nature 546:646–650. 10.1038/nature22975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.NCBI Resource Coordinators. 2016. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44:D7–D19. 10.1093/nar/gkv1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, Aevermann BD, Anderson TK, Burke DF, Dauphin G, Gu Z, He S, Kumar S, Larsen CN, Lee AJ, Li X, Macken C, Mahaffey C, Pickett BE, Reardon B, Smith T, Stewart L, Suloway C, Sun G, Tong L, Vincent AL, Walters B, Zaremba S, Zhao H, Zhou L, Zmasek C, Klem EB, Scheuermann RH. 2017. Influenza Research Database: an integrated bioinformatics resource for influenza virus research. Nucleic Acids Res 10.1093/nar/gkw857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katoh K, Rozewicki J, Yamada KD. 2017. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166. 10.1093/bib/bbx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Löytynoja A. 2014. Phylogeny-aware alignment with PRANK. Methods Mol Biol 1079:155–170. 10.1007/978-1-62703-646-7_10. [DOI] [PubMed] [Google Scholar]
- 76.Guindon S, Delsuc F, Dufayard J-F, Gascuel O. 2009. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137. 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 77.Kozlowski LP. 2016. IPC: isoelectric point calculator. Biol Direct 11:55. 10.1186/s13062-016-0159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stern A, Doron-Faigenboim A, Erez E, Martz E, Bacharach E, Pupko T. 2007. Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res 35:W506–W511. 10.1093/nar/gkm382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodological) 57:289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 80.Villanueva RAM, Chen ZJ. 2019. ggplot2: elegant graphics for data analysis, 2nd ed. Measurement Interdiscip Res Perspect 17:160–167. 10.1080/15366367.2019.1565254. [DOI] [Google Scholar]
- 81.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. 2011. Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830. [Google Scholar]
- 82.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 84.Melamed D, Mark-Danieli M, Kenan-Eichler M, Kraus O, Castiel A, Laham N, Pupko T, Glaser F, Ben-Tal N, Bacharach E. 2004. The conserved carboxy terminus of the capsid domain of human immunodeficiency virus type 1 Gag protein is important for virion assembly and release. J Virol 78:9675–9688. 10.1128/JVI.78.18.9675-9688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gardell AM, Qin Q, Rice RH, Li J, Kültz D. 2014. Derivation and osmotolerance characterization of three immortalized tilapia (Oreochromis mossambicus) cell lines. PLoS One 9:e95919. 10.1371/journal.pone.0095919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iwamoto T, Nakai T, Mori K, Arimoto M, Furusawa I. 2000. Cloning of the fish cell line SSN-1 for piscine nodaviruses. Dis Aquat Organ 43:81–89. 10.3354/dao043081. [DOI] [PubMed] [Google Scholar]
- 87.Deboosere N, Horm SV, Pinon A, Gachet J, Coldefy C, Buchy P, Vialette M. 2011. Development and validation of a concentration method for the detection of influenza a viruses from large volumes of surface water. Appl Environ Microbiol 77:3802–3808. 10.1128/AEM.02484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔC(T) method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 89.Elis E, Ehrlich M, Bacharach E. 2015. Dynamics and restriction of murine leukemia virus cores in mitotic and interphase cells. Retrovirology 12:95. 10.1186/s12977-015-0220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prizan-Ravid A, Elis E, Laham-Karam N, Selig S, Ehrlich M, Bacharach E. 2010. The Gag cleavage product, p12, is a functional constituent of the murine leukemia virus pre-integration complex. PLoS Pathog 6:e1001183. 10.1371/journal.ppat.1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]