

ABSTRACT
Mechanisms of protein homeostasis are crucial for overseeing the clearance of misfolded and toxic proteins over the lifetime of an organism, thereby ensuring the health of neurons and other cells of the central nervous system. The highly conserved pathway of autophagy is particularly necessary for preventing and counteracting pathogenic insults that may lead to neurodegeneration. In line with this, mutations in genes that encode essential autophagy factors result in impaired autophagy and lead to neurodegenerative conditions such as amyotrophic lateral sclerosis (ALS). However, the mechanistic details underlying the neuroprotective role of autophagy, neuronal resistance to autophagy induction, and the neuron-specific effects of autophagy-impairing mutations remain incompletely defined. Further, the manner and extent to which non-cell autonomous effects of autophagy dysfunction contribute to ALS pathogenesis are not fully understood. Here, we review the current understanding of the interplay between autophagy and ALS pathogenesis by providing an overview of critical steps in the autophagy pathway, with special focus on pivotal factors impaired by ALS-causing mutations, their physiologic effects on autophagy in disease models, and the cell type-specific mechanisms regulating autophagy in non-neuronal cells which, when impaired, can contribute to neurodegeneration. This review thereby provides a framework not only to guide further investigations of neuronal autophagy but also to refine therapeutic strategies for ALS and related neurodegenerative diseases.
Abbreviations: ALS: amyotrophic lateral sclerosis; Atg: autophagy-related; CHMP2B: charged multivesicular body protein 2B; DPR: dipeptide repeat; FTD: frontotemporal dementia; iPSC: induced pluripotent stem cell; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; PINK1: PTEN induced kinase 1; RNP: ribonuclear protein; sALS: sporadic ALS; SPHK1: sphingosine kinase 1; TARDBP/TDP-43: TAR DNA binding protein; TBK1: TANK-binding kinase 1; TFEB: transcription factor EB; ULK: unc-51 like autophagy activating kinase; UPR: unfolded protein response; UPS: ubiquitin-proteasome system; VCP: valosin containing protein.
KEYWORDS: Amyotrophic lateral sclerosis, C9orf72, CHMP2B, macroautophagy, mitophagy, myelinophagy, neuronal autophagy, optineurin, SQSTM1/p62, TBK1
Introduction
The diverse cellular processes involved in protein homeostasis are critical for maintaining neuronal health and preventing neurodegenerative disease. Genetic, biochemical and pathophysiological evidence in model systems have repeatedly shown that dysfunction of these homeostatic mechanisms leads to the accumulation of misfolded proteins and neurodegeneration in multiple contexts. Of particular interest among proteostasis pathways that have been characterized to date is the process known as autophagy, the term for which was first coined by Christian de Duve in 1963 [1,2] from the Greek αὐτός (the reflexive pronoun, for the “self”) and φαγεῖν (to eat). This self-consumptive pathway maintains cellular and organismal integrity in the setting of various developmental events or stressors by degrading cellular components and organelles for reuse or redistribution. The ability of autophagy to address disparate needs of the cell arising during stress and development is achieved through specialization among the major autophagy subtypes – designated macroautophagy, microautophagy, and chaperone-mediated autophagy – together with additional organelle- or compartment-specific mechanisms (e.g., mitophagy, granulophagy, pexophagy, reticulophagy, aggrephagy, nucleophagy, and xenophagy, among others). Moreover, autophagy is an essential pathway, as illustrated by embryonic lethality in animals missing key components of the autophagic machinery [3–9], and the high degree of evolutionary conservation across eukaryotes [10].
The individual stages of autophagy, each with its own set of regulatory factors, provide numerous potential points of dysfunction that can confer vulnerability to disease. Furthermore, mounting evidence shows that autophagy is regulated by distinct mechanisms in different cell types [11–26], and these cell type-specific mechanisms underlie the differential vulnerability of separate tissues to autophagy mutations, resulting in heterogenous manifestations of disease. Accordingly, to fully understand the implications of autophagy regulation and autophagy disruption in amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases, we will review the current understanding of mammalian autophagy, with a particular focus on pivotal regulatory features that differ between specific cell types. The clinicopathological features of ALS are beyond the scope of this review and are described elsewhere [27–30]. Here, we will first provide an overview of the autophagy process and describe essential components of each stage of the pathway. Second, we will focus on specific pathway components for which there are ALS-related mutations and describe the physiologic sequelae of these mutations. We will also delineate the extent to which autophagy is affected by common ALS-related genes, as demonstrated by cell and animal models of ALS. Third, we will explore autophagy machinery in the nervous system on a cell type-specific basis to highlight tissue-specific phenotypes caused by autophagy disruption, how these relate to ALS and neurodegeneration, and identify potential targets for selective modulation of autophagy in each cell type. Finally, we will conclude by integrating the concepts examined in this review to provide frameworks for investigating remaining critical questions surrounding autophagy and neuronal proteostasis, thereby informing strategies for developing effective autophagy-based therapies in neurodegenerative disease.
Overview of autophagy
Macroautophagy, often referred to simply as autophagy, is an evolutionarily conserved eukaryotic pathway for bulk and selective degradation of proteins and organelles [31]. It is characterized by a multi-step process in which cytoplasm and its contents are sequestered within double-membraned autophagosomes (Figure 1). The first step in autophagosome formation involves induction of a nascent autophagosome, or phagophore, which is regulated by a multimeric structure known as the phagophore assembly site (PAS) [32–35]. This complex is composed of three ULK (unc-51 like autophagy activating kinases; ULK1, ULK2, and ULK3), ATG13 (autophagy related 13), ATG101, and RB1CC1/FIP200 [36]. The initiation step begins with the dissociation of MTOR (mechanistic target of rapamycin kinase) from the PAS, cued by nutrient starvation or treatment with rapamycin or its various derivatives [36,37]. Next, nucleation of the nascent phagophore is orchestrated by a class III phosphatidylinositol 3-kinase (PtdIns3K) complex containing BECN1, PIK3C3/VPS34, PIK3R4/p150, NRBF2 and either ATG14 or UVRAG [38,39]. Expansion then occurs through maturation of the extending phagophore membrane guided by the ATG12–ATG5-ATG16L1 complex, catalyzing insertion of MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) into the phagophore membrane [40,41]. Before membrane insertion, the unbound and immature LC3 precursor protein (LC3-I) undergoes proteolytic processing by ATG4, and conjugation to phosphatidylethanolamine through ATG7 and ATG3. The resulting mature, lipidated isoform (LC3-II) is incorporated into the phagophore membrane [41].
Figure 1.
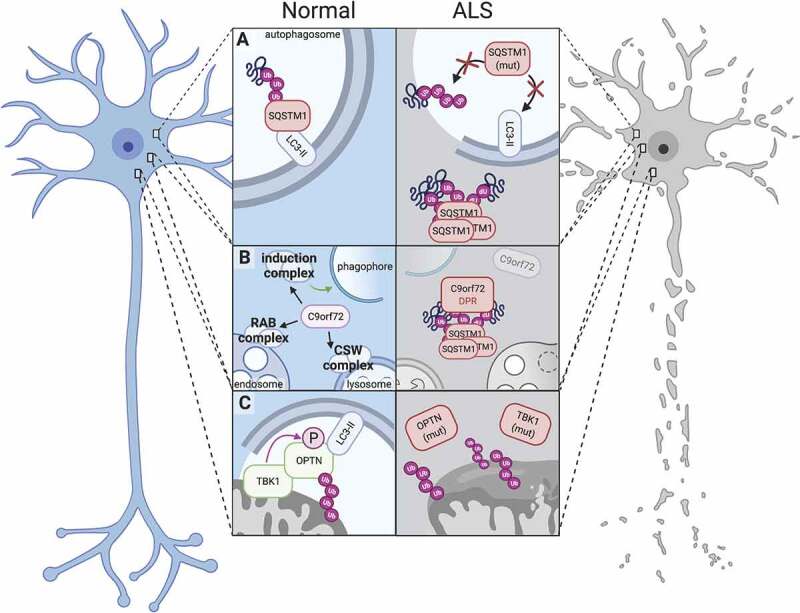
Dysfunction of autophagy-related proteins impairs proteostasis and leads to neurotoxicity in ALS. (A) Under normal conditions, SQSTM1 serves as a receptor protein in selective autophagy and binds both LC3-II and polyubiquitinated proteins, thereby targeting ubiquitinated substrates to phagophores (left); Mutations in SQSTM1 abrogate SQSTM1’s binding activities (right top) or result in the aggregation of SQSTM1 into ubiquitin-positive inclusions (right bottom). (B) The C9orf72 protein participates in several autophagy-related complexes, including the autophagy induction complex (ULK1-RAB1A) that promotes autophagosome biogenesis, the RAB7-RAB11 complex (RAB complex) that regulates endosome maturation, and the C9orf72-SMCR8-WDR41 (CSW) complex that regulates lysosomal dynamics and autophagic flux (left). Disease-associated C9orf72 mutations reduce C9orf72 protein levels (right), while dipeptide repeat proteins generated from the C9orf72 expansion localize to SQSTM1- and ubiquitin-positive inclusions (right). (C) In normal mitophagy, TBK1 binds and phosphorylates OPTN, enhancing its affinity for polyubiquitinated mitochondria and LC3-II (left). TBK1 and OPTN mutations perturb these functions and compromise efficient mitophagy, leading to failed mitochondrial clearance and dysfunctional mitochondria.
In the sequestration phase, the extending ends of the phagophore fuse with one another, forming a double membrane-bound vesicle called an autophagosome that surrounds a segment of cytoplasm, together with its macromolecular and organellular constituents [42]. Molecular motors transport autophagosomes along microtubules to coordinate fusion with lysosomes and form autolysosomes, within which the vesicular constituents are exposed to hydrolytic proteases for degradation [43,44]. Fusion of the autophagosome and lysosome is mediated by the homotypic fusion and protein sorting (HOPS) complex, whose subunits include VPS11, VPS16, VPS18, VPS33A, VPS39, and VPS41 [45,46]. Dismantled cellular components in the autolysosome lumen are actively exported for reconstitution into new macromolecules or for energy metabolism [42]. Global regulation of the myriad effectors and phases of autophagy is overseen by TFEB (transcription factor EB), a basic helix-loop-helix leucine zipper that directs expression of autophagy and lysosomal genes comprising the coordinated lysosomal expression and regulation (CLEAR) network by binding to a conserved promoter motif [47,48]. Further details regarding TFEB activity and regulation are reviewed elsewhere [49–52].
Although autophagic cargo can be trapped passively within the autophagosome in nonselective autophagy, direct substrate recruitment to phagophores in selective autophagy is mediated by a series of receptor proteins (e.g., SQSTM1/p62 [sequestosome 1], NBR1, AMBRA1) that contain both an LC3-interacting region (LIR) and separate motifs for binding polyubiquitinated proteins [41]. These receptor proteins therefore serve as molecular scaffolds for targeting ubiquitinated proteins to phagophores for degradation via autophagy rather than the proteasome.
Additional autophagic specialization is achieved through chaperone-mediated autophagy (CMA), microautophagy, targeted degradation of mitochondria in mitophagy, and degradation of ribonucleoprotein particles (RNPs) in granulophagy. Client proteins that harbor an internal or masked amino acid motif (lysine-phenylalanine-glutamate-arginine-glutamine, or KFERQ) are recognized by the molecular chaperone HSPA8/HSC70 upon partial protein unfolding, enabling CMA. Once bound by HSPA8, the protein is targeted to the lysosome via interaction between HSPA8 and the lysosomal protein LAMP2A [53]. The substrate protein is fully unfolded by HSPA8, LAMP2, and additional co-chaperones as it translocates into the lysosomal lumen for degradation [54]. In microautophagy, lysosomes directly capture cargo from the cytoplasm by membrane invagination to form intraluminal “autophagic tubes,” which expand and pinch off to form microvesicles within the lysosome that are ultimately degraded by lysosomal hydrolases [55].
Physiologic and chemical insults to mitochondria – via reactive oxygen species, disruption of the inner membrane potential in disease models, or treatment with protonophores such as carbonyl cyanide 3-chlorophenylhydrazone (CCCP) – triggers mitophagy. Upon mitochondrial damage, PINK1 (PTEN induced kinase 1) is recruited to the outer mitochondrial membrane [56], where it phosphorylates and activates the E3 ubiquitin ligase PRKN/parkin, which in turn ubiquitinates mitochondrial surface proteins [24]. These polyubiquitin chains are also phosphorylated by PINK1, and the resultant phospho-ubiquitin moieties not only bind the receptor proteins OPTN (optineurin), CALCOCO2/NDP52, and SQSTM1, but also activate TBK1 (TANK-binding kinase 1) [57]. OPTN, CALCOCO2, and SQSTM1 contain ubiquitin-binding domains and LIR motifs, thereby providing a means for delivering ubiquitinated mitochondria to autophagosomes [58]. Additionally, TBK1 amplifies the signaling cascade for mitophagy by phosphorylating OPTN and SQSTM1 (which enhances binding to ubiquitin and LC3) and promoting TBK1 autophosphorylation in a positive-feedback manner [58,59]. Interestingly, degradation of neuronal mitochondria can occur in a non-cell autonomous fashion, as demonstrated by the astrocytic engulfment and degradation of mitochondria excreted from retinal ganglion cells [60].
Essential processes such as translation may stall during specific phases of development or when cells are exposed to certain stressors (heat shock, ER perturbation, proteasomal dysfunction, or hyperosmolar exposure). To prevent potentially toxic errors in translation and facilitate mRNA degradation, cells upregulate granulophagy, a distinct subtype of autophagy that acts on ribonuclear protein (RNP) granules [61]. Several different RNPs are granulophagy substrates, including cytoplasmic stress granules and processing bodies, which dynamically assemble to sequester non-translating mRNA-ribonuclear protein complexes (mRNPs), translation initiation factors, translational repressors, and mRNA decay machinery until the stress resolves or is removed [62]. In granulophagy, RNP granules that contain RNA-binding proteins and their associated mRNAs are targeted for degradation via VCP/p97 (valosin containing protein), a chaperone-like, type II AAA+ ATPase that binds and delivers large ribosomal subunits to autophagosomes [63–66]. Granulophagy can also serve a compensatory role in the setting of dysfunctional macroautophagy [67].
Key autophagy factors and disease-causing mutations in ALS
The long-term health of neurons is inextricably reliant on intact proteostasis. This notion is underscored by the fact that nearly every neurodegenerative disorder is marked by the accumulation of misfolded proteins within or adjacent to affected neurons and glia, in spite of heterogeneous clinical and pathological characteristics among these diseases [68,69]. The abnormal and age-dependent, progressive formation of protein inclusions observed in disease-affected regions of the nervous system implies that proteostasis mechanisms become overwhelmed or dysfunctional with time [68–73]. Molecular and histopathologic analyses of patient tissues demonstrate mislocalized or accumulated autophagy machinery, pathogenic mutations underlying familial neurodegenerative diseases disable known autophagy-related factors, and genetically ablating autophagy effectors produce neurodegeneration in cell and animal model systems [74–81], supporting a role for autophagy dysfunction in the development of age-related neurodegenerative diseases. Accordingly, precisely defining the functions performed by key autophagy effectors in each phase of autophagy, and the dysfunction conferred by genetic mutations in each factor in relation to disease phenotypes, is essential for the development of effective, autophagy-based therapies for neurodegenerative diseases. Furthermore, the degree to which autophagy is differentially regulated in a cell type-specific manner, and how each of the myriad autophagy effectors functions in distinct cell types, remains insufficiently explored, potentially contributing to the disappointing results of indiscriminate autophagy modulation in disease models to date. Ultimately, these are crucial considerations when deciding on discrete targets, maximizing clinically meaningful benefits, and minimizing off-target toxicity. Below, we consider the physiological and pathophysiological relevance of autophagy-related factors in the development of two neurodegenerative disorders, ALS and frontotemporal dementia (FTD), since these disorders often share a common background, and arise due to mutations in genes encoding fundamental components of the autophagy pathway. We will also discuss the impact of autophagy on ALS-FTD in a cell type-specific manner, with particular attention paid to autophagy effectors and their function in discrete cell types within the central nervous system (CNS).
SQSTM1/p62
Selective autophagy relies on autophagy receptor proteins that recognize ubiquitinated cargo and target them to the autophagy machinery [82]. The LC3-interacting region (LIR) enables autophagy receptors to interact with the Atg8 family of proteins within the nascent autophagosome. The ubiquitin-binding protein SQSTM1 harbors a well-characterized LIR [83,84]. Besides its LIR domain, SQSTM1 contains an N‐terminal Phox1 and Bem1p (PB1) domain, a zinc finger (ZZ), a TRAF6 (TNF receptor associated factor 6)‐binding (TB) motif, a KEAP1‐interacting region (KIR), and a ubiquitin‐associated (UBA) domain [85,86]. Homodimerization of the UBA domain maintains SQSTM1 in an inactive state by preventing its interaction with ubiquitin [87,88]. Upon cargo protein ubiquitination, the UBA domain of SQSTM1 is phosphorylated by ULK1 on serine 407, liberating the protein from dimeric inactivation [89]. Subsequently, TBK1, CSNK2 (casein kinase 2), or ULK1 phosphorylate the UBA domain on serine 403, increasing the affinity of SQSTM1 for ubiquitin chains [59,90]. The interactions between SQSTM1, ubiquitinated cargo proteins, and membrane-bound LC3-II are reinforced by homopolymerization of the SQSTM1 PB1 domain [91]. The resulting complex undergoes liquid-liquid phase separation (LLPS), facilitating assembly of the phagophore [92,93]. Incorporation of SQSTM1 into the autophagosome leads to its degradation upon fusion with lysosomes, along with its ubiquitinated targets. As such, SQSTM1 levels inversely correlate with autophagy efficiency, and are often used to estimate autophagy flux [84,94,95].
In addition to targeting ubiquitinated proteins, SQSTM1 also mediates the degradation of ubiquitinated bacteria and dysfunctional mitochondria [96]. As discussed above, the selective degradation of mitochondria by autophagy, or mitophagy, is regulated by the serine/threonine protein kinase PINK1 and the ubiquitin E3 ligase PRKN [97,98]. During PRKN-dependent mitophagy, SQSTM1 is recruited by TBK1 phosphorylation on serine 403 and is essential for the clearance of polyubiquitinated mitochondria [59,99].
SQSTM1 is also involved in several key physiological signaling pathways, including MTOR-dependent signaling, regulation of the ubiquitin-proteasome system (UPS), inflammation, the response to oxidative stress, and apoptosis [86,100–102]. When amino acids are abundant, SQSTM1 is phosphorylated and forms a signaling hub on the lysosomal membrane through its interaction with RPTOR, which in turn recruits and activates MTORC1 on the surface of the lysosome [103–105]. Additionally, SQSTM1 directly associates with the proteasome through its PB1 domain [106–108] and is itself a proteasome substrate [109]. Under conditions of proteasomal impairment, or when the UPS is overwhelmed, SQSTM1 delivers ubiquitinated substrates to the lysosome for autophagic degradation, thereby providing a crucial link between these two cellular degradation pathways.
SQSTM1 is also significantly upregulated during oxidative stress. The transcription factor NFE2L2/NRF2 (nuclear factor erythroid-derived 2-like 2) is an essential component of the oxidative stress response and is normally degraded by the UPS via its interactions with KEAP1 (kelch-like ECH-associated protein 1) [110]. Under oxidative stress, KEAP1 releases NRF2 to translocate to the nucleus and activate the transcription of genes involved in the antioxidant response, including SQSTM1 [110–112]. SQSTM1 interacts with KEAP1 and displaces NRF2, promoting its own transcription as well as other genes involved in the antioxidant response [110]. This positive feedback loop significantly enhances the cellular capacity for selective autophagy, contributing to the clearance of damaged organelles and proteins, and eventually recovery from stress [111,113].
SQSTM1/p62 in disease
SQSTM1 is a major component of neuronal and glial cytoplasmic inclusions that characterize many neurological disorders such as Alzheimer disease and Parkinson disease [114,115]. Furthermore, SQSTM1-positive inclusions are found in the majority of patients with ALS and FTD, and often overlap with inclusions that stain for ubiquitin and TARDBP/TDP-43 (TAR DNA binding protein), an essential RNA-binding protein integrally connected with ALS-FTD [82,116–118] (Figure 1). In the most commonly inherited form of ALS and FTD due to mutations in the C9orf72 gene (described below), SQSTM1 accumulates together with ubiquitin in neurons and glia [119–121] as well as muscle fibers [122]. In fact, individuals with disease-associated C9orf72 mutations display a significantly greater burden of SQSTM1-positive glial inclusions than those with sporadic ALS [82].
Consistent with the pivotal links between SQSTM1 and several distinct signaling pathways, SQSTM1 mutations result in multiple overlapping disease phenotypes. SQSTM1 mutations are not only a rare cause of ALS-FTD, accounting for ~1-3.5% of cases with or without family history [123–125], but also Paget’s disease of bone (PDB), a chronic and progressive skeletal disorder characterized by disorganized bone turnover [126,127], and inclusion body myositis (IBM) [128]. Rather than being mutually exclusive, each of these conditions may co-exist with one another, resulting in a spectrum of overlapping clinical signs collectively referred to as “multisystem proteinopathy” [129]. In contrast to PDB-associated mutations that affect mainly the UBA domain [127], SQSTM1 mutations identified in ALS-FTD affect several different regions. ALS-FTD-associated SQSTM1 mutations frequently affect the UBA and LIR domains involved in autophagy and include both missense and nonsense mutations [123–125]. SQSTM1 mutations affecting the UBA domain impair ubiquitin recognition [130], whereas mutations within the LIR domain impact LC3 recognition and substrate delivery to the autophagosomes [84,131]. Additionally, mutations detected in the SQSTM1 promoter region are associated with reduced SQSTM1 protein levels in familial ALS-FTD [132,133].
Evidence from animal models strengthens the association between SQSTM1, autophagy impairment, and multisystem proteinopathy. sqstm1-knockout mice display reduced numbers of osteoclasts as well as cognitive impairment and anxiety, associated with hyperphosphorylated MAPT/tau and neurofibrillary tangles [134–136]. Knockdown of the SQSTM1 ortholog in zebrafish leads to an ALS-like phenotype consisting of locomotor and motor neuron axonal abnormalities. Overexpression of wild-type human SQSTM1 or application of the autophagy activator rapamycin rescues these phenotypes [137]. On the other hand, overexpression of SQSTM1 variants carrying disease-associated mutations in the UBA domain exhibit a PDB-like skeletal disorder [138,139] as well as impaired spatial learning and long-term memory [140]. Additional knock-in models involving the introduction of disease-associated mutations into endogenously expressed SQSTM1 are needed to clarify its function in select cell types, and how pathogenic SQSTM1 mutations result in neuronal dysfunction as well as disorders of bone and muscle.
C9orf72
In 2011, the seminal discovery of disease-associated expansion mutations within the C9orf72 gene uncovered the most common genetic cause of inherited ALS and FTD, accounting for 23–47% of familial cases and 4–21% of sporadic disease. Unaffected individuals carry 2–30 repeated stretches of GGGGCC within the first intron of the gene, but patients with C9orf72-associated ALS-FTD harbor several hundred to thousands of repeats [141,142].
The C9orf72 protein belongs to the DENN (differentially expressed in normal and neoplastic cells) family [143,144] and binds the GTPases RAB7 and RAB11, which are involved in endosome maturation and recycling [145]. C9orf72 also forms a complex with SMCR8, a guanine nucleotide exchange factor, and WDR41, a WD40 protein essential for targeting the C9orf72-SMCR8-WDR41 complex to lysosomes [146–150]. Here, this complex interacts with RB1CC1-ULK1-ATG13-ATG101, which together are involved in autophagosome formation [149]. Separately, the C9orf72-SMCR8-WDR41 complex acts as a guanine nucleotide exchange factor for RAB8A and RAB39B, regulating vesicle trafficking and autophagic flux [148]. Additionally, through its interactions with ULK1 and RAB1A, a GTP-binding protein involved in vesicular protein trafficking and MTOR regulation [151,152], C9orf72 promotes autophagy initiation [153]. Together, these findings suggest that C9orf72 assists in the formation of nascent autophagosomes, consistent with the finding of impaired autophagy in murine cortical neurons [148] and human iPSC-derived neurons [154] upon C9orf72 knockdown.
C9orf72 in disease
The C9orf72 hexanucleotide repeat expansion exhibits substantial pleiotropy at the clinical and molecular levels. For instance, C9orf72 expansion mutations are associated not only with ALS/FTLD, but also with symptoms reminiscent of Parkinson disease, Alzheimer disease, Huntington disease, and corticobasal syndrome, reinforcing the imperfect connection between clinical and molecular phenotypes and emphasizing the need for molecular diagnostics [155–164]. ALS-FTD due to C9orf72 mutations displays TARDBP pathology, consisting of granular neuronal cytoplasmic inclusions, diffuse neuronal cytoplasmic staining, and glial cytoplasmic inclusions rich in TARDBP [120]. A unique hallmark of C9orf72-related disease is the presence of cytoplasmic star-like inclusions positive for SQSMT1 and ubiquitin, but not TARDBP, in cerebellar granule neurons and other cell types (Figure 1). Nevertheless, the impact of these inclusions is uncertain, as cerebellar degeneration and clinical phenotypes attributable to cerebellar dysfunction are lacking in C9orf72-related disease.
Although the C9orf72 mutation occurs within a non-coding region of the gene, the unique secondary structure of the expansion triggers ribosome stalling and the initiation of translation through a process known as repeat-associated non-AUG (RAN) translation. As there is no true start codon, the GGGGCC expansion is translated in all three reading frames into repeating elements of two peptides: GP, GA and GR (sense), and PG, PR and PA (antisense) [165–168]. Antibodies specific for each dipeptide repeat (DPR) also stain SQSTM1-positive inclusions, including the star-like inclusions unique to C9orf72-related disease, suggesting that DPRs are targeted for clearance by the UPS and/or autophagy [119,169–171] (Figure 1). Furthermore, the loss of C9orf72 function and consequent impairment in autophagy may enhance DPR accumulation in C9orf72 mutation carriers, acting synergistically to accentuate neurodegeneration [154,172,173]. On the other hand, DPR inclusions rarely co-label with TARDBP pathological inclusions and are mainly found in areas of the CNS that are not pathologically affected [171,174–177], arguing against a direct role for DPRs in neurodegeneration.
C9orf72 mutations are associated with reduced basal autophagy and enhanced sensitivity to autophagy inhibitors in human iPSC-derived neurons [153,178]. Paradoxically, however, c9orf72 knockout in mouse (which models the loss of function attributed to the hexanucleotide repeat expansion) enhances autophagy by de-repressing MTOR and, in turn, increasing TFEB nuclear translocation [179,180]. In addition, conditioned medium from astrocytes differentiated from patient-derived iPSC lines (two of which harbor C9orf72 expansion mutations) applied to HEK293T cells reduces LC3-II while increasing SQSTM1 levels [181], suggesting non-cell autonomous inhibition of autophagy by astrocytes in ALS. Overall, further studies are needed to determine if disease-associated C9orf72 expansions affect autophagy through additional means independent of loss-of-function mechanisms and whether the C9orf72 protein may have cell type-, species-specific, and non-cell autonomous effects that explain these discrepancies.
Deletion of the zebrafish and C. elegans homologs of C9orf72 results in impaired locomotion and motor axon degeneration [182,183], implying that the C9orf72 protein is essential for motor neuron function and survival in these systems. In contrast, deletion of the mouse C9orf72 ortholog leads to splenomegaly, lymphadenopathy, dysregulation of macrophages and microglia, and excess production of inflammatory cytokines [184–187]. While c9orf72 knockout animals exhibit a shortened lifespan [184,185,187], no specific neuronal defects have been identified in these mice. On the other hand, loss of endogenous murine C9orf72 results in glial activation, impaired autophagy and toxicity when combined with the C9orf72 expansion mutation [173]. Further, neurons derived from iPSCs carrying the C9orf72 repeat expansion recapitulate key features of cellular pathology associated with ALS, including hyperexcitability [154,188,189] and decreased viability upon exposure to excitotoxic agents such as glutamate [154,190,191]. These results are consistent with those in animal models, suggesting that the endogenously encoded C9orf72 repeat expansion mutation leads to toxicity by both gain and loss of function mechanisms.
TBK1
TBK1/NAK/T2K (TANK binding kinase 1) is a cytoplasmically-localized homodimeric multidomain protein with an ubiquitin-like domain, a N-terminal serine/threonine kinase domain, and two coiled-coil domains [192]. TBK1 is expressed in all tissues, but at much higher levels in neurons of the hippocampus, cortex and lateral ventricle. Moderate levels of the protein have been detected in the glial cells of the cortex and cerebellum [193]. Through its phosphorylation of the autophagy receptors OPTN, SQSTM1, and CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) [58,59,194], TBK1 regulates several aspects of autophagy, including macroautophagy, xenophagy and mitophagy [59,90,148,194–196]. TBK1-dependent effects on autophagy and autophagy receptor proteins are spatially and functionally regulated by interactions with the ULK1 initiation complex [148,150,153,197]. TBK1 also phosphorylates SMCR8, which forms a complex with C9orf72 and WDR41, thereby enhancing autophagy initiation [148]. In neurons, TBK1 knockdown enhances the accumulation of SQSTM1, which can be rescued by phosphomimetic SMCR8 mutations [148], suggesting that SMCR8 phosphorylation is crucial for TBK1’s effects on autophagy.
In addition to its role in modulating autophagy, TBK1 regulates innate immunity by phosphorylating IRF3 (interferon regulatory factor 3), triggering nuclear localization of IRF3 and the subsequent production of IFNA2/IFNα (interferon alpha 2) and IFNB/IFNβ. These events, in turn, stimulate the expression of interferon-inducible genes, including ISG15, whose protein product interacts with SQSTM1 and HDAC6 [198,199]. Whether TBK1 acts on autophagy or innate immunity may partially depend on its associations with specific adaptor proteins such as NAP1L1 (nucleosome assembly protein 1), TANK and TBKBP1/SINTBAD that bind in a mutually exclusive manner [58,200,201].
TBK1 in disease
TBK1 mutations were initially described in conditions with prominent neuroinflammatory responses, including two forms of glaucoma [192,202,203] and herpes simplex encephalitis [204]. Whole exome sequencing studies revealed that mutations in TBK1 are also responsible for ~1% of patients with familial or otherwise sporadic ALS [205,206]. Supporting the link between ALS and FTD, TBK1 mutations have also been identified in FTD patients, and in fact occur more frequently in patients with combined ALS-FTD than in those with ALS alone [207–209]. Neuropathological analysis of CNS tissue from ALS patients with TBK1 mutations shows SQSTM1- and TARDBP-positive inclusions in motor neurons and glia [206,210,211], along with reduced TBK1 mRNA and protein, suggesting haploinsufficiency mechanisms [205,206]. Consistent with this, most ALS-associated TBK1 mutations affect the kinase and the coiled-coiled domains, both of which are vital for TBK1 function [206].
In vitro, silencing of TBK1 or ALS-related TBK1 variants reduces the recruitment of OPTN and LC3B to damaged mitochondria [212,213], thereby impairing mitophagy and resulting in the accumulation of defective mitochondria (Figure 1). Tbk1 haploinsufficiency impairs motor neuron autophagy and accelerates clinical phenotypes as well as the accumulation of mutant SOD1 in transgenic ALS mouse models [214,215]. Unexpectedly, however, TBK1 loss of function also prolongs the disease course by reducing neuroinflammation, suggesting that the dual functions of TBK1 in autophagy and inflammation independently and inversely impact ALS pathogenesis. In keeping with this, Tbk1 deletion disrupts the migration of T-cells [216] that may exert anti-inflammatory effects in ALS models [192,217].
Other ALS-related mutations and their relation to autophagy
UBQLN2
UBQLN2 (ubiquilin 2) is an adaptor protein that binds polyubiquitinated substrates via a C-terminal ubiquitin-associated (UBA) domain, and directs them to the UPS for degradation through an N-terminal ubiquitin-like (UBL) domain [218]. UBQLN2 can also act as an autophagy receptor by directly interacting with LC3 in autophagosomes [219,220]. Rare UBQLN2 mutations are associated with familial forms of ALS and ALS-FTD [221–223], and UBQLN2 colocalizes with SQSTM1 and ubiquitin in neuronal aggregates characteristic of both ALS and ALS-FTD [221,224]. Overexpression of ALS-linked UBQLN2 mutants interrupts the interaction between UBQLN2 and ATG9-ATG16L1 [225], while also triggering intracellular inclusions [226,227] and motor defects in vitro and in vivo [226]. In rats, overexpression of ALS mutant UBQLN2 leads to motor neuron loss and accumulation of autophagy substrates such as SQSTM1 and LC3-II [228]. Consistent with a central role for ubiquilins in autophagy, UBQLN2 modulates MTORC1 activity, lysosomal acidification, and, in turn, autophagosome maturation [229–231].
FUS
FUS (Fused in Sarcoma) is a DNA/RNA-binding protein that participates in DNA damage, RNA transport, and RNA splicing. As with TARDBP, mutations in the gene encoding FUS cause familial ALS with FUS-positive pathology, but neuronal inclusions in sporadic disease show little FUS staining. The majority of disease-associated FUS mutations affect the C-terminal nuclear localization signal, resulting in cytosolic mislocalization and aggregation of mutant FUS [232–234]. In cultured neurons, mutant FUS impairs stress granules clearance; this effect is exacerbated by genetic loss of ATG5, enhancing stress granule accumulation and, ultimately, leading to cell death [235]. Conversely, rapamycin-mediated activation of autophagy ameliorates these features, suggesting that FUS-positive inclusions are autophagy substrates [235]. Supporting this, autophagy activation via PI3K, AKT or MTOR inhibition reverses stress granule accumulation caused by ALS-linked mutant FUS [236], and autophagy induction extends survival in a Drosophila model of FUS-ALS [236]. Intriguingly, FUS may exert a direct influence on autophagy initiation and autophagosome elongation by maintaining expression of factors including RB1CC1 and ATG16L1, which are diminished after CRISPR-mediated knockout of Fus but restored with re-introduction of wild-type FUS in N2A cells [237].
VCP
VCP/p97/Cdc48 (valosin containing protein) is involved in a wide range of essential cellular processes, including DNA replication and repair, cell cycle regulation, and protein clearance [238,239]. Mutations in VCP most commonly lead to the combined disorder of inclusion body myopathy, Paget disease of the bone, and FTD (IBMPFD) [240], but are also responsible for ~1-2% of familial ALS cases [241–243]. Thus, as with SQSTM1, OPTN, HNRNPA2B1, MATR3, and TIA1, VCP mutations result in pleiotropic phenotypes affecting muscle, nerve, bone and brain. Histopathological analyses consistently illustrate TARDBP, ubiquitin and SQSTM1-positive inclusions in VCP-related diseases [244–247]. Moreover, muscle from IBMPFD patients carrying VCP mutations [246] and transgenic mice expressing VCP mutants [244,248,249] display accumulations of SQSTM1- and LC3-positive autophagosomes and abnormal mitochondria [250,251]. Together, these observations suggest a conserved pattern of autophagy dysfunction in association with VCP mutations, implying a crucial role for VCP in autophagy and the maintenance of protein homeostasis.
Supporting this, several studies have highlighted key functions for VCP in autophagosome maturation. In IBMPFD murine muscle lysates, VCP mutations disrupt autophagosome maturation and fusion with lysosomes, but not the formation of early autophagosome precursors [246,249]. Similarly, enlarged and acidified autophagic vacuoles accumulate in cells expressing disease-associated VCP mutants, suggesting a late-stage block in the fusion of lysosomes with autophagosomes [249]. Motor neurons and astrocytes derived from iPSCs carrying pathogenic VCP mutations demonstrate cytoplasmic TARDBP accumulation, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, and oxidative stress, ultimately leading to cell death [252]. Neurons may not be the only cell type affected by VCP mutations in these conditions – mutant VCP iPSC-derived astrocytes were unable to sufficiently support motor neuron survival, indicating the presence of non-cell autonomous mechanisms of neurodegeneration in VCP-related diseases [252].
OPTN
OPTN has emerged as a key player in the regulation of various cellular mechanisms, including vesicular trafficking, maintenance of the Golgi, NFKB/NF-κB signaling and immune response [253]. OPTN is an autophagy receptor that binds ubiquitinated cargo (via its UBAN domain) and LC3 (via its LIR domain) [194,254,255], although it can also act independently of target ubiquitination [254]. Upon mitochondrial damage, OPTN translocates to the surface of mitochondria, an interaction that is stabilized via TBK1-mediated phosphorylation, facilitating the engulfment of mitochondria into phagophores [256]. OPTN also directly interacts with SQSTM1 to form an autophagy receptor complex that accelerates autophagy flux [257]. Genetic analyses have uncovered >20 OPTN mutations in ALS patient cohorts, accounting for ~3% of familial disease and ~1% of sporadic disease [258]. Interestingly, the majority of pathogenic OPTN mutations are located in its UBAN domain and may impair ubiquitin binding [259], suggesting that defects in the targeting of ubiquitinated substrates could be important for ALS pathogenesis. In addition, ALS-associated OPTN mutations enhance microglial NFKB1 expression [260], which may accentuate neuronal loss secondary to neuroinflammation [261].
CHMP2B
CHMP2B (charged multivesicular body protein 2B) is a subunit of the endosomal sorting complex required for transport-III (ESCRT-III) [262]. ESCRT-III is one of the four multiprotein complexes required for multivesicular body formation, and is responsible for the membrane deformation that enables autophagosome initiation and endolysosomal trafficking [263]. CHMP2B mutations were initially associated with rare cases of chromosome 3-linked familial FTD [264,265], but CHMP2B mutations were subsequently identified in ALS patients with a lower motor neuron predominant syndrome [266,267]. Although TARDBP is typically absent from neuronal inclusions found in CHMP2B-associated disease, SQSTM1-positive oligodendroglial inclusions are found in the motor cortex of ALS patients with CHMP2B mutations [267]. Motor neurons from CHMP2B mutant ALS patients show increased calcium concentrations, reduced ATP availability, downregulation of the MAPK/p38 signaling pathway and impaired autophagy initiation [266]. Furthermore, overexpression of CHMP2B mutants leads to the formation of large cytoplasmic vacuoles and increased LC3-II protein levels [266], suggesting that disease-associated CHMP2B mutations disrupt autophagy. In agreement with these observations, CHMP2B downregulation or expression of disease-associated CHMP2B mutants in Drosophila and mammalian cell lines results in aberrant endosomal structures, impaired autophagosome maturation, and cytosolic TARDBP aggregates [264,265,268,269].
Transgenic mice expressing mutant CHMP2B develop progressive neurological defects, axonal pathology, early mortality and neuronal loss reminiscent of FTD [270,271]. These mice also develop lysosomal storage pathology characterized by large neuronal aggregates derived from the endosomal system [272], and early microglial proliferation leading to a pro-inflammatory phenotype at later stages [270]. In contrast, few phenotypes were noted in chmp2b knockout animals [271], suggesting predominantly gain of function toxicity in association with pathogenic CHMP2B mutations.
Autophagy in the context of ALS
Disrupted autophagy and proteostasis are not only apparent in experimental models of familial ALS, with defined genetic mutations impairing autophagy effectors or imparting proteotoxic stress, but also likely play a key role in the pathogenesis of sporadic ALS (sALS). In humans with sALS, pathologic inclusions found in motor neurons are surrounded by LC3 and SQSTM1 [273], suggesting failed or stalled clearance via autophagy. SQSTM1-positive inclusions are also ubiquitin-positive, consistent with a disruption in the UPS [118]. Furthermore, biochemical and histopathologic examination of frontotemporal cortex and spinal cord in sALS patients reveals that ubiquitin-positive inclusions contain TARDBP [274,275], which is itself involved in the regulation of autophagy [276]. Markers of oxidative stress, ER stress, and the UPR are upregulated in spinal cords and anterior horn cells from human patients with sALS [277–280]. Accumulations of dysmorphic and enlarged mitochondria, many with deficiencies of critical oxidative enzymes, in anterior horn cells, intramuscular nerves, and skeletal muscle are characteristic of sALS [281–285]. Such mitochondrial abnormalities, together with observations of OPTN-positive inclusions and de novo OPTN mutations found in sALS [286–289], are suggestive of attenuated mitophagy. Collectively, these findings indicate autophagy dysfunction compounded by failures in additional proteostatic mechanisms, which may together lead to the degeneration of susceptible neuronal populations in sALS.
In familial ALS, apart from the ALS-associated mutations that directly affect autophagy-related genes, genetic mutations in TARDBP and SOD1 deserve particular mention not only because they are responsible for some of the more common familial forms of ALS but also because of their intrinsic connections with autophagy. Mutations in TARDBP, which encodes the transactive response DNA binding protein 43 kDa (TDP-43), are the third most common cause of familial autosomal dominant ALS and account for rare cases of sporadic ALS [290–293]. Mutations in the SOD1 gene, which encode the free radical/reactive oxygen species scavenger superoxide dismutase 1 [294,295], represent the second most common cause of familial ALS in Europeans and the most common cause in those of Asian descent [296]. Disease models involving mutations in either of these genes have been indispensable for uncovering fundamental mechanisms of ALS pathogenesis. Models of TARDBP proteinopathy range from transient transfections in primary rat cortical cultures [297], to motor neurons [298] and astrocytes [299] differentiated from patient-derived induced pluripotent stem cells (iPSCs) or murine embryonic stem cells [300], to transgenic or virally transduced rodents overexpressing wild-type [301–316], truncated [317,318], or mutant TARDBP harboring the A315T [301,308,319], Q331K [310], M337V [301,310,311,320–322], G348C [308] mutations, knock-in mutant TARDBPN390D [300], or tardbp knockouts [323–328] (these models are reviewed in further detail elsewhere [329–334]). SOD1 model systems include human and murine motor neuron cultures [335,336], and transgenic mice expressing SOD1 with the G93A [214,215,335–338], G86R [335,339], or H46R/H48Q [340] mutations (also reviewed elsewhere [341–344]).
Several studies employing these models point to cell-autonomous toxicity in both neurons and astrocytes in TARDBP- and SOD1-related ALS, in addition to non-cell autonomous mechanisms, implying that therapies should target both cell types for optimal effect. iPSC-derived motor neurons harboring the ALS-associated TARDBPM337V mutation demonstrate an accumulation of insoluble mutant TARDBP at native expression levels [298], which in previous models has not only been closely linked to neurodegeneration [297], but also increases the risk of death in iPSC-derived astrocytes [299]. Furthermore, glial TARDBP inclusions predominantly reside in oligodendrocytes [345,346], and expression of TARDBPM337V in astrocytes exerts cell autonomous toxicity [299]. Supporting the notion of non-cell autonomous contributions to disease, transplantation of SOD1G93A astrocytes into the spinal cords of wild-type mice induced motor neuron loss [347]. In addition, transplantation of wild-type astrocytes into SOD1G93A rat spinal cords was neuroprotective [348], and limited overexpression of NRF2 in astrocytes mitigated neurodegeneration in SOD1G93A and SOD1H46R/H48Q mice [340].
These models also revealed key abnormalities in autophagy that occur in the context of ALS pathogenesis and suggest that autophagy modulation can dramatically influence ALS-related neurodegeneration. For example, loss of nuclear TARDBP leads to reduced ATG7 expression [349], and embryonic stem cell-derived motor neurons expressing TARDBPN390D show increased levels of the autophagy suppressor, BCL2, and reduced autophagic activity [300]. TARDBP downregulation promotes TFEB nuclear localization and TFEB target gene expression, but it also reduces DCTN1 (dynactin subunit 1) expression, thereby blocking autolysosome formation and impairing autophagic flux [350]. Transgenic expression of a C-terminal TARDBP fragment commonly associated with ALS and FTD leads to decreases in several autophagy effectors, including ATG3, ATG7, LC3, SQSTM1, and BECN1 [351]. Together, these findings link autophagy dysfunction to TARDBP pathology in ALS and suggest that correcting autophagy dysregulation or promoting the autophagic degradation of TARDBP may be of therapeutic benefit in ALS.
To this end, inducing autophagy with rapamycin reduces TARDBP accumulation and mislocalization in vitro [352] and in vivo [353], whereas inhibiting autophagy with 3-methyladenine impairs TARDBP degradation [352]. Autophagic degradation of TARDBP is enhanced by downregulation of HSP90AA1 and CDC37, suggesting that these chaperones are key suppressors of autophagy and may accentuate TARDBP-related toxicity in ALS [354]. HSPB8 overexpression also enhances autophagic clearance of TARDBP via elevations in TFEB, SQSTM1, and LC3 [355,356]. Importantly, autophagy induction rescues relevant outcomes in models that do not involve protein overexpression, but rather native mutant forms of TARDBP associated with familial disease. For example, inducing autophagy using novel compounds belonging to the benzoxazine class of molecules, including 10-NCP, enhance TARDBPM337V degradation and extend survival in rodent and human neurons [357]. Inhibition of MTOR likewise reduces TARDBP aggregation and toxicity in multiple cell and animal models of ALS [353,358,359]. Similarly, the phosphodiesterase inhibitor ibudilast induces autophagy by promoting TFEB activity, which enhances clearance of TARDBP aggregates and reduces cytotoxicity [360].
Although stimulating autophagy improves outcomes in TARDBP ALS models, autophagy induction has complex effects in ALS due to SOD1 mutations. Inducing autophagy with lithium [361] rescues SOD1G93A-related pathology and cell death, and overexpression of TFEB enhances LC3 expression and prevents cytotoxicity in SOD1G93A-expressing NSC-34 cells [362]. Furthermore, antagonizing the unfolded protein response (UPR) through deletion of XBP1 [335] triggers autophagy and the clearance of SOD1 inclusions, in the process delaying disease onset and improving survival. Although these studies suggest that therapies capable of stimulating autophagy hold great promise for SOD1-related ALS, many caveats still remain. For instance, neurodegeneration may itself stimulate autophagy, and it is unclear whether this response is beneficial or maladaptive [78]. In a SOD1G93A transgenic mouse model of ALS, motor neurons of symptomatic animals show increased levels of LC3-II and autophagic vacuoles compared to controls or pre-symptomatic mice. Although elevated LC3-II in this context could represent compromised autophagy flux, reduced levels of active/phosphorylated markers of the MTOR pathway [337] suggest autophagy induction. In addition, rapamycin facilitates motor neuron degeneration and rapid disease progression in SOD1G93A ALS mice [338], and excess mitophagy accelerates neurotoxicity and metabolic compromise in ALS models [363]. It is possible that overactive autophagy in SOD1-related ALS may generate toxic fragments of SOD1 or accelerate the atrophy of denervated muscle [71,364]. Regardless, the nature of autophagy induction in these biologic settings, whether representing a coping response to antecedent proteotoxicity or an active component of pathogenesis at disease onset (or both), remains incompletely defined.
Interestingly, mutant SOD1 may act in combination with autophagy dysfunction to promote motor neuron loss in ALS. Ubiquitous expression of ALS-related TBK1 mutations, heterozygous TBK1 deletion, or motor neuron-specific TBK1 deletion result in loss of function phenotypes but are insufficient to produce ALS-related neurodegeneration in mice [214,215]. However, in the setting of the SOD1G93A mutation, loss of TBK1 or pathogenic TBK1 mutations accentuate several pathologic features of ALS (disease onset, muscle denervation) while ameliorating others (inflammation, early death) [214]. Similarly, conditional deletion of Atg7 in mouse motor neurons accentuates early ALS pathology but mitigates glial inflammation and early death [336]. These findings not only suggest that impaired autophagy sensitizes the nervous system to key pathogenic events occurring early in ALS pathogenesis, but also highlight the importance of neuroinflammation in disease progression [215].
The interplay between autophagy and TARDBP or SOD1 mutations is complex and influences ALS pathogenesis within and between different cell types of the nervous system. Identifying whether and how specific autophagy factors are differentially affected by TARDBP and SOD1 mutations, and to what extent these alterations are salient for neurons, glia, and/or muscle, is critical for refining therapeutic strategies and targeting cell type-specific regulators. In so doing, future ALS therapies may be able to effectively induce autophagy in the relevant cell types without adversely affecting others, maximizing on-target benefits while reducing off-target side effects that would be unavoidable with indiscriminate autophagy inducers.
Cell type-specific regulation of autophagy
Genetic disruption of key autophagy effectors is sufficient to cause ALS, highlighting the integral relationship between autophagy and neuronal survival. Moreover, individual ALS mutations affect all phases of autophagy, from early (C9orf72), intermediate (SQSTM1, CHMP2B), and late (VCP) stages of the pathway. Since neuronal death and dysfunction occur regardless of the point at which autophagy is impaired by disease-causing mutations, enhancing flux through the pathway as a whole is indispensable for preventing neurodegeneration. Notably, with global expression of germline mutations associated with ALS, neural and non-neural tissues manifest the detrimental effects of compromised autophagy [11], but to varying extents [365,366]: SQSTM1 mutations affect muscle, bone, and liver [125,367,368]; C9orf72 deficiency leads to lymphocytic inflammation, lymphadenopathy, and splenomegaly [149]; CHMP2B mutations induce oligodendroglial pathology [267]; OPTN mutations produce microglial activation and glaucoma [288]; and VCP mutations cause extensive cardiac and skeletal muscle pathology [248]. Since a given autophagy-related mutation is identical in all tissues, the heterogeneous manifestations of autophagy dysfunction between tissues may relate to differences in autophagy regulation specific to each cell type. Below we describe cell type-specific mechanisms regulating autophagy, emphasizing the distinct phenotypes observed in each tissue upon autophagy dysfunction, and outlining therapeutic strategies for selectively modulating autophagy within predetermined cell types. A summary of key cell type-specific regulators is included in Figure 2 and Table 1.
Figure 2.
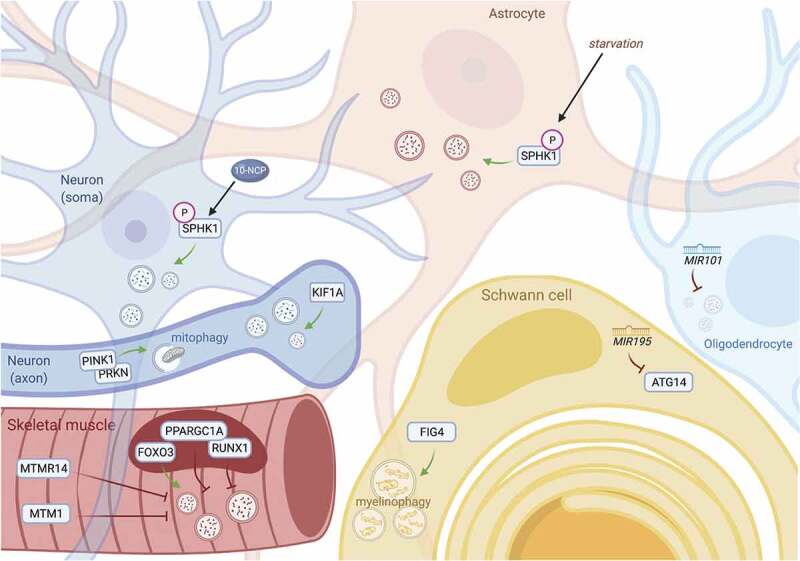
Distinct factors regulate autophagy among different cell types of the nervous system. In each of the cells which comprise the central and peripheral nervous systems, autophagy is differentially regulated by cell type-specific effectors. In neurons (top left), modulation of SPHK1 by phenoxazine compounds such as 10-NCP, but not starvation, potently induces autophagy. In contrast, nutrient deprivation is sufficient to promote SPHK1 signaling and autophagy induction in astrocytes (top center). In the axonal compartment of neurons, KIF1A and PINK1-PRKN are critical for facilitating local autophagic activity (left, middle). MicroRNAs such as MIR101 and MIR195 suppress oligodendrocytic and Schwann cell autophagy, respectively (right). Schwann cells also clear myelin debris through myelinophagy, a unique form of selective autophagy that is dependent on FIG4 (bottom). In muscle, specific transcription factors can exert activating (FOXO3) or suppressive (PPARGC1A, RUNX1) effects on autophagy, and phosphatases such as MTM1 and MTMR14 inhibit autophagy by recycling phosphoinositides needed for autophagy induction (bottom left).
Table 1.
Role of starvation-induced MTOR inhibition in various cell types of the nervous system
| cell type | Starvation | MTOR inhibition | common factors | cell type-specific factors | |
|---|---|---|---|---|---|
| Neurons | Insensitive or mild activation [385] |
Insensitive [370] | AMPK TFEB ULK1 PIK3C3/VPS34 BECN1 ATG5 ATG7 LC3 ATG12 ATG14 SQSTM1 VCP LAMP2 OPTN |
PINK1, PRKN (axonal mitophagy) [24] KIF1A (synaptic autophagy) [22,25] HAP1, MAPK8IP1 (axonal transport) [23,26] SPHK1 (benzoxazine treatment) [11] |
|
| Glia | Astrocytes | Activates [404] | Activates [404] | SPHK1 (starvation induction) [11] | |
| Oligodendrocytes | Activates [450] | Activates [530] | MIR101 [12] | ||
| Schwann cells | Activates [465] | Activates [467,468] | FIG4 [13] MIR195 [14,15] | ||
| Skeletal muscle | Activates [475] | Activates [482] | RUNX1 [474] MTM1 [17] MTMR14 [20] PPARGC1A [19] AKT, FOXO3 [16,18] |
||
Neurons
Neurons exhibit specialized topology that includes not only dendrites capable of extensive arborization, but also axons that extend at times over a meter in length [369]. As a result, autophagy must operate within the contexts of these localized compartments since the autophagy-lysosomal system of the soma is metabolically and temporally incapable of meeting all dendritic, axonal and synaptic demands [370]. Another critical factor that uniquely impacts neuronal autophagy is the requirement of neurons to maintain protein homeostasis for the entire lifespan of an organism; this arises from the extended post-mitotic state of neurons, and the lack of their renewal and replacement [371]. Neuronal autophagy also exhibits physiologic characteristics that are fundamentally distinct from those in other cellular types, as detailed further below.
Autophagic activity varies significantly within each subcellular compartment of the neuron. Autophagy initiation typically begins outside of the soma, in the distal axon tip [370,372,373]. Newly-formed autophagosomes in neuronal processes undergo retrograde transport to the soma and proximal segments of neurites to fuse with somatodendritic lysosomes for cargo degradation, with maturation and acidification of the autophagosomal lumen developing en passant [370,372,374]. Axonal autophagy is dependent on specialized enzymatic apparatuses, including the PINK1-PRKN complex for localized mitophagy [24], KIF1A/UNC-104 for autophagosome formation at the synapse [22,25], and the HTT-HAP1 complex and MAPK8IP1/JIP1 for retrograde transport of autophagosomes [23,26]. Autophagy is also necessary for maintaining the structural integrity of the axon [375], as Purkinje-specific deletion of Atg7 (encoding an essential E1-like enzyme required for activating ATG12, ATG8, and LC3-I) results in Purkinje cell-autonomous axonal dystrophy and degeneration in mice [376]. In addition, autophagy helps eliminate synapses during development [377,378], but can also accentuate axon degeneration during excitotoxic stress [375].
Baseline autophagy in neurons constitutively operates at a highly processive level, such that maturing autophagosomes are rapidly incorporated into autolysosomes, which represent the majority of the autophagic vesicle population after autophagy induction or flux inhibition [379]. Basal autophagy in neurons is essential for maintaining long-term neuronal health, since disruption of baseline autophagic activity in vivo causes neurodegeneration and the accumulation of inclusions containing polyubiquitinated proteins [380,381]. With increasing age, basal autophagy and the capacity for efficient autophagosome maturation progressively deteriorate in neurons, leading to accumulation of malformed and immature autophagic vesicles, implying reduced proteostatic reserve over time [382].
Neuronal autophagy is distinct in several additional physiologic respects. For example, despite serving as a potent autophagy inducer in most cell types [383], starvation is an ineffective means of stimulating autophagy in neurons, even in the face of starvation-related downregulation of neuronal MTOR signaling [370]. Similarly, rapamycin and related MTOR inhibitors, as well as AKT inhibitors, are ineffective inducers of neuronal autophagy [338,384]. Alternative methods have proven more successful in neurons, however, including selective MTORC1 inhibition via everolimus [385–387], or modulation of MTOR- and AKT-independent pathways with 10-NCP, an N10-substituted derivative of the heterocyclic dye compound and antipsychotic analog phenoxazine [384]. These observations imply the presence of unknown, cell type-specific pathways regulating autophagy in neurons [388].
Glia
In addition to neurons, the central and peripheral nervous systems (CNS and PNS) are populated with glial cells in approximately equal proportions to neurons [389] and are comprised of several subtypes: astrocytes, oligodendrocytes and Schwann cells (which myelinate neurons in the CNS and PNS, respectively), ependymal cells, microglia, enteric glia, and satellite cells [390,391]. For the purposes of this review, we will examine the current state of knowledge regarding glial autophagy in astrocytes, microglia, oligodendrocytes and Schwann cells.
Astrocytes. Astrocytes are the most abundant subtype of glial cells and provide structural and metabolic support for neurons through several mechanisms, including neurotransmitter clearance, free radical scavenging, modulating neuronal excitability, maintaining ion homeostasis, regulating synaptic maintenance and plasticity, paracrine and autocrine signaling, blood-brain barrier formation, modulating inflammation and injury with reactive astrogliosis, among other functions [391]. Astrocytes are enriched in intermediate filaments (including glial fibrillary acidic protein, GFAP) and exhibit morphologic heterogeneity ranging from intricately branching processes of protoplasmic astrocytes in gray matter to longer but less elaborate processes of fibrous astrocytes in white matter [392–396]. Not only do astrocytes and neurons harbor extensive morphologic and physiologic differences, but the regulatory pathways coordinating autophagy and the physiologic consequences of autophagy activation in each cell type may also differ considerably, depending on the pharmacologic or pathologic context:
Adaptive proteostasis: Similar to neurons [397], autophagy is activated in astrocytes to compensate for inhibition of the UPS [398]. Such compensatory stimulation may underlie the activation of astrocyte autophagy observed in Alexander disease, a pediatric leukodystrophy caused by GFAP mutations which lead to accumulation of Rosenthal fibers and, in turn, proteasomal impairment [399].
Mitochondrial homeostasis: Mitochondrial compromise and ATP synthase inhibition with oligomycin A stimulate autophagy to a greater extent in astrocytes than in neurons, although both cell types activate autophagy to similar extents in the face of ischemia [400]. In reactive gliosis, a process unique to astrocytes, intact mitochondrial health is essential and is maintained through autophagy, such that astrocyte-specific deletion of Atg7 perturbs the balance of mitochondrial dynamics to favor fission over fusion and thereby reduces oxidative capacity [401]. Unlike neurons, astrocytic loss of Atg7 or Atg5 is insufficient to cause cell death or the accumulation of misfolded and ubiquitinated proteins [380,381], implying greater proteostatic reserve in astrocytes compared to neurons.
Differential signaling cascades: In contrast to neurons, MTOR inhibition and starvation robustly activate autophagy in astrocytes [402], whereas MTOR-independent activation of autophagy with benzoxazines (e.g., 10-NCP and fluphenazine) takes place in neurons but not astrocytes [11]. These differences in autophagy may be due to enhanced phosphorylation of astrocytic SPHK1/SK1 (sphingosine kinase 1) with starvation, whereas benzoxazines activate neuronal (but not astrocytic) SPHK1 to induce autophagy [11]. Notably, SPHK1 signaling may promote autophagy in SH-SY5Y cells and fibroblasts, but its role in mature neurons is incompletely elucidated [403–406]. Notably, although benzoxazines induce differential responses in SPHK1 phosphorylation and autophagy activation, these compounds are sufficient to reduce TARDBPM337V-associated toxicity in iPSC-derived astrocytes [357], presumably through autophagy upregulation.
Non-cell autonomous effects on neuronal autophagy and survival: Astrocytes regulate neuronal autophagy and exert non-cell autonomous, neuroprotective effects. In the context of neurodegeneration, astrocytes take up aggregate-prone MAPT/tau, amyloid beta (Aβ) and SNCA (synuclein alpha), and degrade these proteins through autophagy [407–412]. Furthermore, not only is autophagy responsible for degrading HTT in astrocytes, but it also promotes glutamate transporter expression in these cells [413], thereby providing greater capacity for astrocytes to buffer and mitigate glutamate-mediated excitotoxicity in disease. In transgenic mice overexpressing mutant SOD1 associated with ALS, astrocytes display more SOD1-positive inclusions than do neurons, and at an earlier stage [414]. Selective clearance of SOD1-positive inclusions from astrocytes is sufficient to slow disease progression [414], while conditioned media from SOD1-expressing astrocytes is toxic to cultured human motor neurons in vitro [181,415],, implying that astrocytes are both necessary and sufficient for neurodegeneration in this ALS model. Notably, reactive astrocytes from SOD1 transgenic mice secrete TGFB1, which stimulates MTOR signaling and impairs autophagy in co-cultured motor neurons [415], suggesting that astrocytes can exacerbate neuronal toxicity by compromising neuronal proteostasis in a non-cell autonomous fashion.
Further characterization of astrocyte-specific pathways will provide critical information for honing the precision of therapies for ALS by targeting neuron-specific autophagy in particular, potentiating the beneficial cell autonomous and non-cell autonomous effects of astrocyte autophagy, or both.
Microglia. Microglia are macrophage-like cells that comprise 10–15% of the CNS, and mediate diverse functions ranging from innate immunity and phagocytosis to synaptic pruning and the regulation of synaptic activity [416,417]. Activation of microglia through genetic, infectious, inflammatory, or physical stimuli, leads to stereotypical changes in their morphology, accompanied by proliferation, migration, and the release of proinflammatory cytokines and chemokines [417–419]. In neurodegenerative diseases, microglial activation may accentuate neuron loss by promoting and propagating regional and local neuroinflammation [420,421]. Consistent with this, ALS patients exhibit elevated macrophage markers in their CSF compared to unaffected controls [422], presumably due to increased CNS microglia or their activation, as suggested by positron emission tomography (PET) imaging using radiotracers specific for activated microglia [423,424]. Moreover, loss of function mutations in TREM2 (triggering receptor expressed on myeloid cells 2), which affect microglia phagocytosis and inflammation, are risk factors for ALS [425–427], confirming the pivotal role played by microglia in ALS pathogenesis.
Phagocytosis: Autophagy and phagocytosis share key morphological and mechanistic features [428]. For instance, both pathways rely on vesicular structures to deliver contents to lysosomes for degradation. Microglia actively clear apoptotic cells, myelin and synaptic debris, axonal fragments and protein deposits from the CNS through phagocytosis [428–430]. In line with these connections between phagocytosis and autophagy, microglial phagocytosis of Aβ deposits in Alzheimer disease models [430,431] requires the autophagy-related factors BECN1, ATG7 and LC3 [432,433].
Neuroinflammation: The activation of CNS microglia and astrocytes, and the subsequent production of pro-inflammatory cytokines and interleukins (neuroinflammation), is intricately connected to ALS pathogenesis. In pre-symptomatic mutant SOD1 transgenic mice, activated microglia initially display neuroprotective properties that slow disease progression; however, with time the continued and unabridged neuroinflammation only enhances neurodegeneration and accelerates disease progression [434–436]. Transplanted wild-type microglia effectively extend the survival of mutant SOD1 transgenic mice [437], confirming the importance of microglial activation in ALS pathogenesis. Supporting this, ALS patients carrying C9orf72 mutations display microglial activation that correlates with disease progression [438]. The C9orf72 protein – itself closely tied to autophagy, as discussed above – is highly expressed in myeloid cells such as microglia. C9orf72-deficient mice display lysosomal accumulation and a hyperactive immune response characterized by enhanced production of IL1B and IL6 [184,185,187,439], a phenotype recapitulated by myeloid-specific C9orf72 knockout, indicating the significance of C9orf72 in maintaining physiological immune responses [440]. Emphasizing the link between autophagy and inflammation, impaired autophagy in C9orf72-deficient myeloid cells leads to the accumulation of the pro-inflammatory STING (stimulator of interferon genes) protein [440]. Likewise, ALS-associated mutations in TBK1, OPTN, SQSTM1, and VCP all affect autophagy as well as microglial function and innate immunity [192,205,206,441–443]. Collectively, these findings reinforce the convergence between inflammation and autophagy in microglia, and their importance for ALS pathogenesis.
Oligodendrocytes. Oligodendrocytes comprise 45–75% of the glial population, depending on the region of cerebrum or brainstem sampled, and are essential for CNS axon myelination [389]. Although much of oligodendrocyte autophagy remains unknown, this process is important for several aspects of development and disease:
Developmental CNS myelination: Autophagic activity increases during progenitor cell differentiation into mature oligodendrocytes [444]. During oligodendroglial precursor (OPC) differentiation, autophagy becomes most active in distal OPC processes, and autophagosomes are trafficked proximally to the soma in a manner similar to neurons [444]. Conditional oligodendrocyte-specific deletion of Atg5 in mice leads to incomplete differentiation, reduced myelination, abnormal myelin compaction, oligodendrocytic death, and shortened life span [444]. In addition, oligodendrocyte autophagy, OPC differentiation, and myelination during development are all regulated by MTOR [445–447]. Consistent with these findings, autophagy activation enhances oligodendrocyte survival and promotes myelination in a rat model of demyelinating disease [448].
Non-cell autonomous trophic support: Loss of autophagy with oligodendrocyte-specific deletion of Atg5 impairs motor recovery after spinal cord contusion in mice [449], but rapamycin fails to enhance recovery [449] despite effective stimulation of autophagy in these cells [449–451]. Paradoxically, suppression of autophagy with NTF3 (neurotrophin 3) disinhibits oligodendrocyte proliferation and promotes motor recovery in a separate model of spinal cord contusion [452]. These observations suggest that oligodendrocyte autophagy may play a necessary but insufficient role in post-injury repair of the spinal cord [453], but excessive autophagy in oligodendrocytes may lead to neuronal toxicity in a non-cell autonomous manner.
Impaired proteostasis in neurodegeneration: In multiple system atrophy (MSA), the pathologic hallmark consists of cytoplasmic SNCA inclusions in oligodendrocytes [12], and the accumulation of these inclusions is exacerbated by autophagy inhibition, either pharmacologically with chloroquine or genetically by MIR101 overexpression [12,450,454]. Separately, oligodendrocytes in ALS patients demonstrate inclusions containing TARDBP and FUS [455–457] and lower levels of SLC16A1/MCT1 (solute carrier family 16 member 1), an essential lactate transporter in the CNS whose expression is regulated by autophagy [458]. These findings constitute indirect evidence for impaired oligodendrocyte autophagy in ALS; however, direct assessment of oligodendrocyte autophagy in ALS models has not been fully explored.
Current knowledge regarding oligodendrocyte-specific mechanisms of regulating autophagy is limited. Additional studies are needed to establish the specific autophagy factors that distinguish autophagy in oligodendrocytes from that in neurons.
Schwann cells. Details regarding Schwann cell-specific autophagy are also limited and are largely devoted to examining Schwann cells in development and after traumatic injury to peripheral nerves:
Developmental PNS myelination: During development, autophagy is required for refining and culling of Schwann cell-mediated myelination, and conditional Atg7 deletion in mouse Schwann cells leads to cytoplasmic swelling, ribosomal accumulation, dysmorphic ER, and dysmyelination [459]. Furthermore, conditional deletion of Pik3c3 in murine Schwann cells not only results in hypomyelination, but also in non-cell autonomous disruption of neuronal autophagic flux and dysregulated neuronal trafficking of ERBB2 or ERBB3 receptor tyrosine kinases [460]. Additional non-cell autonomous effects are observed with Schwann cell-specific PTEN deletion or EGFR overexpression, which attenuate peripheral nerve autophagy and lead to disorganization of the nascent neuromuscular junction [461].
Peripheral nerve trauma: After nerve injury, Schwann cells oversee the capture of myelin debris in autophagosomes in a process termed “myelinophagy” [459,462,463], and this function is impaired by Schwann cell-specific Atg7 deletion [459,464]. Remyelination after peripheral nerve injury is hindered by injury-induced upregulation of the microRNA Mir195a, which inhibits Atg14 and, in turn, suppresses Schwann cell autophagy [14,15]. Conversely, stimulating Schwann cell autophagy with rapamycin promotes myelination and improves peripheral nerve regeneration [465,466].
Inherited demyelinating neuropathies: Abnormal Schwann cell autophagy is also observed in dysmyelinating forms of inherited polyneuropathies. For instance, in Charcot-Marie-Tooth (CMT) type 1A caused by duplication of PMP22 (peripheral myelin protein 22), Schwann cell autophagy is aberrantly elevated and associated with higher levels of JUN/c-Jun, which may be part of a compensatory, neuroprotective response to the pathogenic effects of the PMP22 duplication [467–469]. In CMT4J, autosomal recessive mutations in the gene encoding FIG4, a phosphatase required for recycling phosphoinositides involved in vesicle trafficking, lead to incompetent myelinophagy [13]. Autosomal dominant FIG4 mutations are associated with rare forms of familial ALS [470], suggesting that inefficient myelinophagy may result in the loss of motor neurons and muscle atrophy.
Together, these studies indicate that targeting Schwann cell autophagy in development or following traumatic injury has strong therapeutic potential not only for enhancing remyelination, but also for promoting axonal regrowth through non-cell autonomous mechanisms. Even so, treatments for specific peripheral myelination disorders must be individualized based on the specific pathologic context and the perceived need to enhance deficient autophagy, or conversely slow overactive autophagy.
Skeletal muscle
Skeletal muscle comprises another integral component of the peripheral nervous system. Similar to neurons, muscle fibers must contend with life-long metabolic needs due to their post-mitotic state, but unlike neurons these cells must adapt to mechanical stressors arising from weight-bearing activity and locomotion [471]. Structural and oxidative maintenance, adaptive hypertrophy, senescent sarcopenia, and pathogenic atrophy from neuromuscular disease all appear to require regulatory changes in autophagy, and the mechanisms underlying these different physiologic states and their impact on proteostasis will be the focus of this section.
- Muscle atrophy: There is conflicting evidence as to whether autophagy contributes to or mitigates muscle atrophy in ALS. On the one hand, skeletal muscle denervation causes neurogenic atrophy and may activate autophagy; such a protective response (mounted by the transcription factor RUNX1) limits muscle wasting after denervation by suppressing autophagy. Conversely, genetic ablation of RUNX1 disinhibits autophagy and worsens atrophy [472]. Starvation-related atrophy is also associated with upregulated autophagy, which in turn is controlled by the transcription factor FOXO3 and is independent of MTOR activity [473]. On the other hand, neurogenic atrophy can be partially mitigated by adrenergic stimulation, which activates autophagy through Atg7 and counteracts denervation-induced inhibition of autophagic flux [474]. Autophagy appears to be required for muscle regeneration, since autophagy inhibition with 3-methyladenine blunts recovery in mice after myotoxic injury [475]. Similarly, ulk1 knockout impairs muscle regeneration in zebrafish [476], and conditional Atg7 deletion in mouse skeletal muscle leads to atrophy, weakness, and accumulation of unhealthy mitochondria and sarcoplasmic reticulum [477]. Analogously, conditional Atg5 deletion in mouse skeletal muscle leads to accumulation of ubiquitin-positive inclusions and ultrastructural disorganization [478]. The discrepancies suggested by these paradoxical findings may indicate that autophagy is required for muscle maintenance and homeostasis, but there is a limit or threshold beyond which autophagy stimulation may exacerbate fiber atrophy.
- Neuromuscular disease: In ALS, skeletal muscle-specific expression of SOD1G93A is associated with atrophy and elevated autophagy [479], but these effects are mitigated by siRNA-mediated MAP1LC3B knockdown and the subsequent reduction of autophagy flux [479]. In addition, inherited myopathies demonstrate muscle pathology that includes markers of elevated autophagy. For instance, centronuclear myopathy is caused by mutations in the genes encoding MTM1 (myotubularin1) or MTMR14/Jumpy (myotubularin related protein 14) [17,20]. These loss-of-function mutations prevent these enzymes from metabolizing phosphoinositides required for autophagy, thereby disinhibiting autophagy flux and muscle atrophy [17,20]. In contrast, enhancing autophagy in mouse models of Duchenne muscular dystrophy (DMD) pharmacologically with rapamycin or 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, an AMP analog which stimulates AMPK) improves muscle strength and contractility [480,481]. Moreover, PPARGC1A/PGC1A (PPARG coactivator 1A) overexpression drives non-MTOR-dependent autophagy by increasing TFEB nuclear localization in dystrophin-deficient muscle fibers [19]; however, the extent to which this manipulation rescues DMD-related pathology, atrophy, or weakness was not assessed. These discrepant results may be due to effects of the genetic background unique to each disease, resulting in excessive autophagy stimulation in centronuclear myopathy, but insufficient autophagy in DMD. In either case, deviation from normal proteostasis results in muscle pathology.
- Autophagy and exercise: The positive effect of exercise on autophagy was first noted in 1984, with the observation of autophagosome accumulation after exercise in rat skeletal muscle [482]. Similar effects have been seen in humans and mice after strenuous exercise, interpreted as a response to exercise-related muscle breakdown [482,483]. Exercise triggers autophagy by activating ULK1 and dissociating BCL2 from BECN1, thereby disinhibiting the initiation complex [484,485]. In muscle biopsies collected from human volunteers after endurance exercise, measurements of autophagy markers revealed downregulated AKT and MTOR signaling, elevated ATG5, ATG12, and LC3-II [486]. Similarly, vastus lateralis biopsies taken from human runners after a 200-kilometer race showed increased transcription of autophagy factors ATG4B, ATG12, GABARAPL1, LC3B, CSTL (cathepsin L), BNIP3, and BNIP3L [487], which may be an adaptive response to muscle injury related to long-distance running. Exercise also potentiates autophagy activation due to starvation and the resulting reduction in plasma insulin, which in turn downregulates AKT and FOXO3 signaling [16,18]. Autophagy induction is not only seen with endurance exercise, but also acute bouts of exercise, and this short-term increase in autophagy appears to require PPARGC1A [488].
- Autophagy and aging: Aging is associated with a decline in muscle mass (termed sarcopenia) and mitochondrial function, both of which are associated with dysregulated autophagy [489]. However, there remains considerable controversy as to whether these events arise due to impaired or overactive autophagy with age. On the one hand, skeletal muscle autophagy declines with aging in Drosophila and can be rescued by FOXO overexpression or constitutive activation of EIF4EBP1 [490]. Mouse muscle also shows age-related decline in autophagy machinery (LC3, BECN1, and LAMP2), and these deficiencies are partially mitigated by 8 weeks of treadmill exercise [491]. Age-related declines in MFN2 (mitofusin 2), which is associated with macroautophagy and mitophagy [492], together with mitochondrial fragmentation and mitochondrial fission machinery in muscle, indicate inefficient mitophagic flux in aging muscle [493]. Supporting this, skeletal muscle from aged rats exhibits lower LC3 expression, suggesting reduced levels of basal autophagy and an overall decreased capacity for autophagy [494].
On the other hand, sarcopenia involves autophagy of muscle structural components, and attenuation of autophagy with PPARGC1A overexpression mitigates sarcopenia [495]. In aged rats, muscle tissues display elevated basal levels of proteolysis, higher expression of autophagy proteins and the accumulation of unfolded protein response elements. These changes are accompanied by reduced adaptive reserve in response to denervation, with impaired autophagy induction and lower levels of mitophagy compared to young rats [496]. Aged rats also show elevated basal mitophagy, higher levels of mitophagy proteins, higher TFEB expression and lysosomal markers than young mice; these changes were reduced with chronic and forced contractile activity induced by implanted stimulators [497]. Taken together, the mixed evidence describing age-related changes in muscle autophagy emphasizes the need for further investigations of the regulatory machinery overseeing autophagy in muscle. This issue is particularly prominent given the potential for nonselective autophagy-inducing therapies to be used for highly prevalent age-related neurodegenerative conditions, and their unknown effects upon sarcopenia in susceptible populations.
Concluding remarks: remaining questions & therapeutic implications
Although autophagy affords much potential for disease-modifying benefit in neurodegenerative disease, additional challenges arise when administering autophagy-modulating drugs on a clinical basis. For instance, cell and animal models may not faithfully reproduce autophagy regulation in healthy subjects or humans with disease. Additionally, treatment with autophagy modulating drugs runs the risk of incurring intolerable side effects and toxicities, especially when given on a long-term basis that may be required to prevent or delay late-onset neurodegenerative diseases.
Autophagy induction using MTOR inhibitors is currently being tested in ALS – a phase II trial of rapamycin (sirolimus) for ALS patients began in 2018 [498]. Results from this trial have yet to be announced or published, but the use of rapamycin in ALS is complicated not only by its potential for insufficient induction of neuronal autophagy per se, but also its capacity for modulating multiple MTOR-dependent downstream pathways (Figure 3). Rapamycin has been extensively utilized for transplant rejection prophylaxis and graft-versus-host disease, as well as seizure control and reduction of tumors in tuberous sclerosis complex patients [499–501]. As a result, the drawbacks of chronic rapamcyin therapy – including cytopenias, nephrotoxicity, metabolic syndrome, dermatitis, and pneumonitis [502,503] – are well known. Clinicians may therefore be required to implement regular monitoring of serum drug levels, blood pressure, skin exams, complete blood counts, liver and renal function, cholesterol, and triglycerides, as is commonly performed in those taking rapamycin for FDA-approved indications [504].
Figure 3.
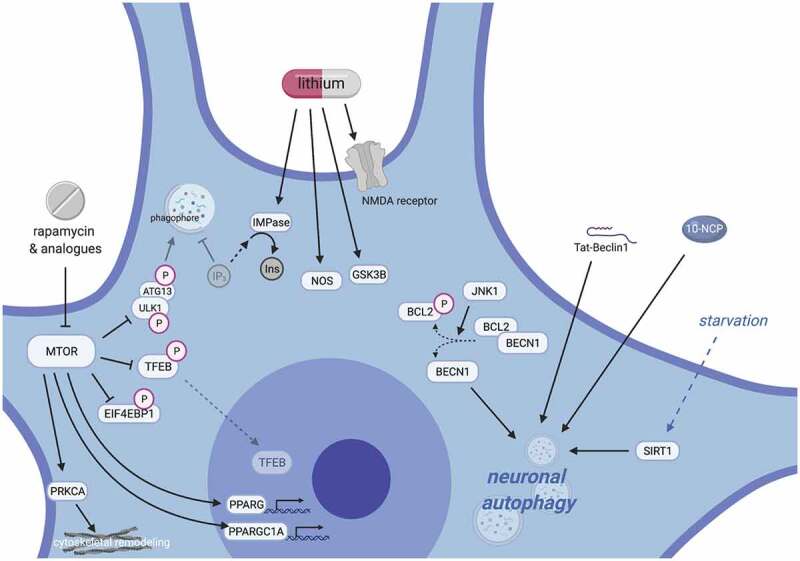
Mechanisms of neuronal autophagy and their impact for therapeutic design in ALS. Pharmacodynamic considerations limit the use of currently available drugs for modulating autophagy. Inhibitors of MTOR inhibitors (rapalogs) only weakly activate autophagy in neurons, and their wide-ranging effects target myriad cellular pathways, including growth signaling, translation, stress responses, transcriptional regulation, and cytoskeletal remodeling (left). Such pleiotropy leads to well-documented and multi-systemic toxicities, especially with long-term use; however, newer rapalogs may enable more specific targeting and selective autophagy modulation. Similarly, lithium has numerous multi-target effects, including depletion of IP3 through IMPase, activating nitric oxide synthase, inhibiting GSK3B, stimulating NMDA receptors and enhancing glutamatergic tone, among many others (middle). This results in a narrow therapeutic index for lithium, potentially explaining its apparent lack of neuroprotective effects in human trials for neurodegenerative disease to date. To reduce off-target effects, recent efforts have focused on MTOR-independent strategies for stimulating autophagy, including modulation of SIRT1, phenoxazine compounds such as 10-NCP, or synthetic peptides such as Tat-Beclin 1 (right).
Additional methods for bypassing MTOR to modulate autophagy exist, however, and should be explored in the context of ALS. Despite early enthusiasm over lithium in patients and mouse models [505,506], five subsequent trials showed no clinical benefit in survival or neuromuscular outcomes for ALS patients [507–511]. These negative trials, together with lithium’s narrow therapeutic index, severely limit the therapeutic utility of lithium for ALS (Figure 3). Modulation of amino acids represents an alternative strategy for regulating autophagy in an MTOR-independent fashion: amino acid starvation induces autophagy, whereas amino acid provision results in RPS6KB/p70S6K phosphorylation and autophagy suppression [512]. Neither event is affected by rapamycin, suggesting that MTOR is dispensable for this pathway [512]. 10-NCP and related compounds are also capable of inducing neuronal autophagy through an MTOR- and AKT-independent signaling cascade [357,384]. The array of MTOR-independent methods for stimulating autophagy also includes direct stimulation of AKT and BECN1, activating MAPK8/JNK1 to dissociate BCL2 from (and thereby disinhibit) BECN1, applying the synthetic peptide Tat-Beclin 1, SIRT1-associated starvation, and disrupting nonsense-mediated RNA decay [513–517] (Figure 3). Notably, with the exception of 10-NCP, the above strategies have been tested almost exclusively in non-neuronal model systems. Therefore, the applicability of these biologic phenomena to neurons, and whether manipulating MTOR-independent effectors and pathways are sufficient to modulate neuronal autophagy, must be investigated in relevant neuronal model systems to inform their suitability for therapy development.
Given the limitations demonstrated by therapeutic strategies empirically trialed to date, the rational design of autophagy inducers for neurodegenerative disease will require several key advances. First, cell type-specific regulatory mechanisms controlling autophagy flux must be elucidated in neurons, astrocytes, microglia and muscle. Second, the means to accurately measure autophagy induction in each cell type must be established. Traditional measures relying upon the accumulation of pathway intermediates such as LC3-II cannot discriminate between true induction and late-stage inhibition of autophagosome maturation and should therefore be utilized cautiously in estimating the ability of compounds to stimulate autophagy. Third, these approaches must be adapted for high-throughput screening, enabling the unbiased identification of effective autophagy inducers in multiple cell types [518–520]. Fourth, the effectiveness of these compounds must be tested rigorously in model systems that do not depend upon the overexpression of disease-associated proteins, since this approach may inappropriately emphasize strategies that reduce the non-physiological accumulation of exogenous proteins. In this light, the use of alternative model systems, such as iPSCs and mature cells differentiated from iPSCs, provides a unique advantage, enabling investigations of heretofore untestable aspects of neuronal biology and cell type-specific autophagy regulation.
Funding Statement
This work was supported by the National Institute of Neurological Disorders and Stroke [2R25NS089450-06].
References
- [1].De Duve C, Wattiaux R.. Functions of lysosomes. Annu Rev Physiol. 1966;28(1):435–492. [DOI] [PubMed] [Google Scholar]
- [2].Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4(6):740–743. [DOI] [PubMed] [Google Scholar]
- [3].Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–1125. DOI: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- [4].Gan B, Peng X, Nagy T, et al. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol. 2006;175(1):121–133. DOI: 10.1083/jcb.200604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. DOI: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- [6].Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. DOI: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. DOI: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- [8].Tsukamoto S, Kuma A, Murakami M, et al. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321(5885):117–120. DOI: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- [9].Yue Z, Jin S, Yang C, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–15082. DOI: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hughes T, Rusten TE. Origin and evolution of self-consumption: autophagy. Adv Exp Med Biol. 2007;607:111–118. [DOI] [PubMed] [Google Scholar]
- [11].Moruno-Manchon JF, Uzor NE, Ambati CR, et al. Sphingosine kinase 1-associated autophagy differs between neurons and astrocytes. Cell Death Dis. 2018;9(5):521. DOI: 10.1038/s41419-018-0599-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Valera E, Spencer B, Mott J, et al. MicroRNA-101 modulates autophagy and oligodendroglial alpha-synuclein accumulation in multiple system atrophy. Front Mol Neurosci. 2017;10:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vaccari I, Carbone A, Previtali SC, et al. Loss of Fig4 in both Schwann cells and motor neurons contributes to CMT4J neuropathy. Hum Mol Genet. 2015;24(2):383–396. DOI: 10.1093/hmg/ddu451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bremer J, O’Connor T, Tiberi C, et al. Ablation of Dicer from murine Schwann cells increases their proliferation while blocking myelination. PLoS One. 2010;5(8):e12450. DOI: 10.1371/journal.pone.0012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi G, Shi J, Liu K, et al. Increased miR-195 aggravates neuropathic pain by inhibiting autophagy following peripheral nerve injury. Glia. 2013;61(4):504–512. DOI: 10.1002/glia.22451. [DOI] [PubMed] [Google Scholar]
- [16].Jamart C, Naslain D, Gilson H, et al. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab. 2013;305(8):E964–74. DOI: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- [17].Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13(2):175–182. DOI: 10.1038/ng0696-175. [DOI] [PubMed] [Google Scholar]
- [18].Pagano AF, Py G, Bernardi H, et al. Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med Sci Sports Exerc. 2014;46(7):1314–1325. DOI: 10.1249/MSS.0000000000000237. [DOI] [PubMed] [Google Scholar]
- [19].Spaulding HR, Ludwig AK, Hollinger K, et al. PGC-1alpha overexpression increases transcription factor EB nuclear localization and lysosome abundance in dystrophin-deficient skeletal muscle. Physiol Rep. 2020;8(4):e14383. DOI: 10.14814/phy2.14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vergne I, Roberts E, Elmaoued RA, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. Embo J. 2009;28(15):2244–2258. DOI: 10.1038/emboj.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang H, Dai J, Hou S, et al. Treatment of hepatocellular carcinoma with adenoviral vector-mediated Flt3 ligand gene therapy. Cancer Gene Ther. 2005;12(9):769–777. DOI: 10.1038/sj.cgt.7700843. [DOI] [PubMed] [Google Scholar]
- [22].Ashrafi G, Schlehe JS, LaVoie MJ, et al. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206(5):655–670. DOI: 10.1083/jcb.201401070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fu MM, Holzbaur EL. MAPK8IP1/JIP1 regulates the trafficking of autophagosomes in neurons. Autophagy. 2014;10(11):2079–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rub C, Wilkening A, Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017;367(1):111–123. [DOI] [PubMed] [Google Scholar]
- [25].Stavoe AK, Hill SE, Hall DH, et al. KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell. 2016;38(2):171–185. DOI: 10.1016/j.devcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34(4):1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hirano A. Neuropathology of ALS: an overview. Neurology. 1996;47(4 Suppl 2):S63–6. [DOI] [PubMed] [Google Scholar]
- [28].Laferriere F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. 2015;145:w14054. [DOI] [PubMed] [Google Scholar]
- [29].Peters OM, Ghasemi M, Brown RH Jr. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125(5):1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131(8):1975–1978. DOI: 10.1016/j.clinph.2020.04.005. [DOI] [PubMed] [Google Scholar]
- [31].Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124(2):161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ganley IG, Lam Du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. DOI: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. DOI: 10.1091/mbc.e08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. DOI: 10.1091/mbc.e08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584(7):1302–1312. [DOI] [PubMed] [Google Scholar]
- [39].Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–5372. DOI: 10.1091/mbc.e08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(9):859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80(1):125–156. [DOI] [PubMed] [Google Scholar]
- [42].Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [44].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774. [DOI] [PubMed] [Google Scholar]
- [45].Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. DOI: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol. 2000;151(3):551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [48].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. DOI: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lapierre LR, Kumsta C, Sandri M, et al. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy. 2015;11(6):867–880. DOI: 10.1080/15548627.2015.1034410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Medina DL, Ballabio A. Lysosomal calcium regulates autophagy. Autophagy. 2015;11(6):970–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. Embo J. 2018;37(11):11. DOI: 10.15252/embj.201798804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016;283(13):2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69(7):1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kondapalli C, Kazlauskaite A, Zhang N, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2(5):120080. DOI: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dengjel J, Abeliovich H. Roles of mitophagy in cellular physiology and development. Cell Tissue Res. 2017;367(1):95–109. [DOI] [PubMed] [Google Scholar]
- [58].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015;60(1):7–20. DOI: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Matsumoto G, Shimogori T, Hattori N, et al. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet. 2015;24(15):4429–4442. DOI: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- [60].Davis CH, Kim KY, Bushong EA, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111(26):9633–9638. DOI: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4(9):a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Erickson SL, Lykke-Andersen J. Cytoplasmic mRNP granules at a glance. J Cell Sci. 2011;124(3):293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Buchan JR, Kolaitis RM, Taylor JP, et al. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153(7):1461–1474. DOI: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. [DOI] [PubMed] [Google Scholar]
- [65].Mijaljica D, Prescott M, Devenish RJV. ATPase engagement in autophagic processes. Autophagy. 2011;7(6):666–668. [DOI] [PubMed] [Google Scholar]
- [66].Ossareh-Nazari B, Bonizec M, Cohen M, et al. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep. 2010;11(7):548–554. DOI: 10.1038/embor.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kaushik S, Massey AC, Mizushima N, et al. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19(5):2179–2192. DOI: 10.1091/mbc.e07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991–1995. [DOI] [PubMed] [Google Scholar]
- [69].Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998;21(12):516–520. [DOI] [PubMed] [Google Scholar]
- [70].Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47(3):e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gadad BS, Britton GB, Rao KS. Targeting oligomers in neurodegenerative disorders: lessons from α-synuclein, tau, and amyloid-β peptide. J Alzheimers Dis. 2011;24(Suppl 2):223–232. [DOI] [PubMed] [Google Scholar]
- [72].Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(S7):S10–7. [DOI] [PubMed] [Google Scholar]
- [73].Yerbury JJ, Ooi L, Dillin A, et al. Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem. 2016;137(4):489–505. DOI: 10.1111/jnc.13575. [DOI] [PubMed] [Google Scholar]
- [74].Corti O, Blomgren K, Poletti A, et al. Autophagy in neurodegeneration: new insights underpinning therapy for neurological diseases. J Neurochem. 2020;154(4):354–371. DOI: 10.1111/jnc.15002. [DOI] [PubMed] [Google Scholar]
- [75].Finkbeiner S. The autophagy lysosomal pathway and neurodegeneration. Cold Spring Harb Perspect Biol. 2020;12(3):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Frake RA, Ricketts T, Menzies FM, et al. Autophagy and neurodegeneration. J Clin Invest. 2015;125(1):65–74. DOI: 10.1172/JCI73944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694–2701. DOI: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- [78].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Martini-Stoica H, Xu Y, Ballabio A, et al. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39(4):221–234. DOI: 10.1016/j.tins.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Menzies FM, Fleming A, Caricasole A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5):1015–1034. [DOI] [PubMed] [Google Scholar]
- [81].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16(6):345–357. [DOI] [PubMed] [Google Scholar]
- [82].Lorente Pons A, Higginbottom A, Cooper-Knock J, et al. Oligodendrocyte pathology exceeds axonal pathology in white matter in human amyotrophic lateral sclerosis. J Pathol. 2020;251(3):262–271. DOI: 10.1002/path.5455. [DOI] [PubMed] [Google Scholar]
- [83].Lippai M, Lőw P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int. 2014;2014:832704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. DOI: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- [85].Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–624. [DOI] [PubMed] [Google Scholar]
- [86].Sánchez-Martín P, Komatsu M. p62/SQSTM1 - steering the cell through health and disease. J Cell Sci. 2018;131(21):21. [DOI] [PubMed] [Google Scholar]
- [87].Long J, Garner TP, Pandya MJ, et al. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-kappaB signalling. J Mol Biol. 2010;396(1):178–194. DOI: 10.1016/j.jmb.2009.11.032. [DOI] [PubMed] [Google Scholar]
- [88].Isogai S, Morimoto D, Arita K, et al. Crystal structure of the ubiquitin-associated (UBA) domain of p62 and its interaction with ubiquitin. J Biol Chem. 2011;286(36):31864–31874. DOI: 10.1074/jbc.M111.259630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987. DOI: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37(2):223–234. DOI: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wurzer B, Zaffagnini G, Fracchiolla D, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sun D, Wu R, Zheng J, et al. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018;28(4):405–415. DOI: 10.1038/s41422-018-0017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Zaffagnini G, Savova A, Danieli A, et al. p62 filaments capture and present ubiquitinated cargos for autophagy. Embo J. 2018;37(5):5. DOI: 10.15252/embj.201798308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. [DOI] [PubMed] [Google Scholar]
- [96].Zheng YT, Shahnazari S, Brech A, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183(9):5909–5916. DOI: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- [97].Deng H, Dodson MW, Huang H, et al. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105(38):14503–14508. DOI: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yang Y, Gehrke S, Imai Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103(28):10793–10798. DOI: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. DOI: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- [100].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sánchez-Martín P, Saito T, Komatsu M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. Febs J. 2019;286(1):8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–146. DOI: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Linares JF, Duran A, Reina-Campos M, et al. Amino acid activation of mTORC1 by a PB1-domain-driven kinase complex cascade. Cell Rep. 2015;12(8):1339–1352. DOI: 10.1016/j.celrep.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Linares JF, Duran A, Yajima T, et al. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51(3):283–296. DOI: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Myeku N, Figueiredo-Pereira ME. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J Biol Chem. 2011;286(25):22426–22440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10(3):431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Seibenhener ML, Babu JR, Geetha T, et al. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24(18):8055–8068. DOI: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lee J, Kim HR, Quinley C, et al. Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem. 2012;287(6):4033–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bellezza I, Giambanco I, Minelli A, et al. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):721–733. DOI: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- [111].Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. DOI: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Jo C, Kim S, Cho SJ, et al. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett. 2014;588(17):3081–3088. DOI: 10.1016/j.febslet.2014.06.036. [DOI] [PubMed] [Google Scholar]
- [113].Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. DOI: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- [114].Kuusisto E, Suuronen T, Salminen A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun. 2001;280(1):223–228. [DOI] [PubMed] [Google Scholar]
- [115].Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160(1):255–263. DOI: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Arai T, Nonaka T, Hasegawa M, et al. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett. 2003;342(1–2):41–44. DOI: 10.1016/S0304-3940(03)00216-7. [DOI] [PubMed] [Google Scholar]
- [117].Hiji M, Takahashi T, Fukuba H, et al. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008;116(2):183–191. DOI: 10.1007/s00401-008-0402-2. [DOI] [PubMed] [Google Scholar]
- [118].Mizuno Y, Amari M, Takatama M, et al. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci. 2006;249(1):13–18. DOI: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- [119].Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122(6):691–702. DOI: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- [120].Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol. 2014;127(3):347–357. [DOI] [PubMed] [Google Scholar]
- [121].Troakes C, Maekawa S, Wijesekera L, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2012;32(5):505–514. DOI: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- [122].Türk M, Haaker G, Winter L, et al. C9ORF72-ALS: P62- and ubiquitin-aggregation pathology in skeletal muscle. Muscle Nerve. 2014;50(3):454–455. DOI: 10.1002/mus.24283. [DOI] [PubMed] [Google Scholar]
- [123].Le Ber I, Camuzat A, Guerreiro R, et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013;70(11):1403–1410. DOI: 10.1001/jamaneurol.2013.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446. DOI: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- [125].Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–522. [DOI] [PubMed] [Google Scholar]
- [126].Laurin N, Brown JP, Morissette J, et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70(6):1582–1588. DOI: 10.1086/340731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Rea SL, Majcher V, Searle MS, et al. SQSTM1 mutations–bridging Paget disease of bone and ALS/FTLD. Exp Cell Res. 2014;325(1):27–37. DOI: 10.1016/j.yexcr.2014.01.020. [DOI] [PubMed] [Google Scholar]
- [128].Gang Q, Bettencourt C, Machado PM, et al. Rare variants in SQSTM1 and VCP genes and risk of sporadic inclusion body myositis. Neurobiol Aging. 2016;47:218.e1–218.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467–473. DOI: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Rea SL, Walsh JP, Layfield R, et al. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of Paget’s disease of bone. Endocr Rev. 2013;34(4):501–524. [DOI] [PubMed] [Google Scholar]
- [131].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857. DOI: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- [132].Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis. 2009;35(2):302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Rubino E, Rainero I, Chiò A, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–1562. DOI: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Babu A, Wang Q, Muralidharan R, et al. Chitosan coated polylactic acid nanoparticle-mediated combinatorial delivery of cisplatin and siRNA/Plasmid DNA chemosensitizes cisplatin-resistant human ovarian cancer cells. Mol Pharm. 2014;11(8):2720–2733. DOI: 10.1021/mp500259e. [DOI] [PubMed] [Google Scholar]
- [135].Durán A, Serrano M, Leitges M, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6(2):303–309. DOI: 10.1016/S1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- [136].Rodriguez A, Durán A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3(3):211–222. DOI: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- [137].Lattante S, De Calbiac H, Le Ber I, et al. Sqstm1 knock-down causes a locomotor phenotype ameliorated by rapamycin in a zebrafish model of ALS/FTLD. Hum Mol Genet. 2015;24(6):1682–1690. DOI: 10.1093/hmg/ddu580. [DOI] [PubMed] [Google Scholar]
- [138].Daroszewska A, Van ‘T Hof RJ, Rojas JA, et al. A point mutation in the ubiquitin-associated domain of SQSMT1 is sufficient to cause a Paget’s disease-like disorder in mice. Hum Mol Genet. 2011;20(14):2734–2744. DOI: 10.1093/hmg/ddr172. [DOI] [PubMed] [Google Scholar]
- [139].Kurihara N, Hiruma Y, Zhou H, et al. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J Clin Invest. 2007;117(1):133–142. DOI: 10.1172/JCI28267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Seibenhener ML, Zhao T, Du Y, et al. Behavioral effects of SQSTM1/p62 overexpression in mice: support for a mitochondrial role in depression and anxiety. Behav Brain Res. 2013;248:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. DOI: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. DOI: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Levine TP, Daniels RD, Gatta AT, et al. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503. DOI: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Zhang D, Iyer LM, He F, et al. Discovery of Novel DENN Proteins: implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease. Front Genet. 2012;3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Farg MA, Sundaramoorthy V, Sultana JM, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–3595. DOI: 10.1093/hmg/ddu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Almeida S, Gao FB. Lost & found: C9ORF72 and the autophagy pathway in ALS/FTD. Embo J. 2016;35(12):1251–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Amick J, Tharkeshwar AK, Amaya C, et al. WDR41 supports lysosomal response to changes in amino acid availability. Mol Biol Cell. 2018;29(18):2213–2227. DOI: 10.1091/mbc.E17-12-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sellier C, Campanari ML, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35(12):1276–1297. DOI: 10.15252/embj.201593350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016;4(1):51. DOI: 10.1186/s40478-016-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2(9):e1601167. DOI: 10.1126/sciadv.1601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Goud B, M M. Small GTP-binding proteins and their role in transport. Curr Opin Cell Biol. 1991;3(4):626–633. [DOI] [PubMed] [Google Scholar]
- [152].Thomas JD, Zhang YJ, Wei YH, et al. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell. 2014;26(5):754–769. DOI: 10.1016/j.ccell.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Webster CP, Smith EF, Bauer CS, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. Embo J. 2016;35(15):1656–1676. DOI: 10.15252/embj.201694401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Shi Y, Lin S, Staats KA, et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018;24(3):313–325. DOI: 10.1038/nm.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Anor CJ, Xi Z, Zhang M, et al. Mutation analysis of C9orf72 in patients with corticobasal syndrome. Neurobiol Aging. 2015;36(10):2905.e1–5. DOI: 10.1016/j.neurobiolaging.2015.06.008. [DOI] [PubMed] [Google Scholar]
- [156].Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet. 2013;92(3):345–353. DOI: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Cacace R, Van Cauwenberghe C, Bettens K, et al. C9orf72 G4C2 repeat expansions in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2013;34(6):1712.e1–7. DOI: 10.1016/j.neurobiolaging.2012.12.019. [DOI] [PubMed] [Google Scholar]
- [158].Harms M, Benitez BA, Cairns N, et al. C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 2013;70(6):736–741. DOI: 10.1001/2013.jamaneurol.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Kostić VS, Dobričić V, Stanković I, et al. C9orf72 expansion as a possible genetic cause of Huntington disease phenocopy syndrome. J Neurol. 2014;261(10):1917–1921. DOI: 10.1007/s00415-014-7430-8. [DOI] [PubMed] [Google Scholar]
- [160].Lesage S, Le Ber I, Condroyer C, et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain. 2013;136(2):385–391. DOI: 10.1093/brain/aws357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Lindquist SG, Duno M, Batbayli M, et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin Genet. 2013;83(3):279–283. DOI: 10.1111/j.1399-0004.2012.01903.x. [DOI] [PubMed] [Google Scholar]
- [162].Luigetti M, Quaranta D, Conte A, et al. Frontotemporal dementia, Parkinsonism and lower motor neuron involvement in a patient with C9ORF72 expansion. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(1):66–69. DOI: 10.3109/17482968.2012.692383. [DOI] [PubMed] [Google Scholar]
- [163].O’Dowd S, Curtin D, Waite AJ, et al. C9ORF72 expansion in amyotrophic lateral sclerosis/frontotemporal dementia also causes parkinsonism. Mov Disord. 2012;27(8):1072–1074. DOI: 10.1002/mds.25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292–299. DOI: 10.1212/WNL.0000000000000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. DOI: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Kwon I, Xiang S, Kato M, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345(6201):1139–1145. DOI: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Mori K, Arzberger T, Grässer FA, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–893. DOI: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- [168].Wen X, Tan W, Westergard T, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–1225. DOI: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Mackenzie IR, Arzberger T, Kremmer E, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126(6):859–879. DOI: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- [170].Mahoney CJ, Downey LE, Ridgway GR, et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. 2012;4(5):41. DOI: 10.1186/alzrt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Mann DM, Rollinson S, Robinson A, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1(1):68. DOI: 10.1186/2051-5960-1-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Boivin M, Pfister V, Gaucherot A, et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. Embo J. 2020;39(4):e100574. DOI: 10.15252/embj.2018100574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Zhu Q, Jiang J, Gendron TF, et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020;23(5):615–624. DOI: 10.1038/s41593-020-0619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Davidson Y, Robinson AC, Liu X, et al. Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP-43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol. 2016;42(3):242–254. DOI: 10.1111/nan.12292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Gendron TF, Van Blitterswijk M, Bieniek KF, et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 2015;130(4):559–573. DOI: 10.1007/s00401-015-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Gomez-Deza J, Lee YB, Troakes C, et al. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun. 2015;3(1):38. DOI: 10.1186/s40478-015-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Mackenzie IR, Frick P, Grässer FA, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130(6):845–861. DOI: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- [178].Almeida S, Gascon E, Tran H, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–399. DOI: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Ji YJ, Ugolino J, Brady NR, et al. Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy. 2017;13(7):1254–1255. DOI: 10.1080/15548627.2017.1299312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet. 2016;12(11):e1006443. DOI: 10.1371/journal.pgen.1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Madill M, McDonagh K, Ma J, et al. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol Brain. 2017;10(1):22. DOI: 10.1186/s13041-017-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Ciura S, Lattante S, Le Ber I, et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol. 2013;74(2):180–187. DOI: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- [183].Therrien M, Rouleau GA, Dion PA, et al. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8(12):e83450. DOI: 10.1371/journal.pone.0083450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Atanasio A, Decman V, White D, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6(1):23204. DOI: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Burberry A, Suzuki N, Wang JY, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8(347):347ra93. DOI: 10.1126/scitranslmed.aaf6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].O’Rourke JG, Bogdanik L, Muhammad A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901. DOI: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Sudria-Lopez E, Koppers M, De Wit M, et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016;132(1):145–147. DOI: 10.1007/s00401-016-1581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Devlin AC, Burr K, Borooah S, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015;6(1):5999. DOI: 10.1038/ncomms6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7(1):1–11. DOI: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. DOI: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Selvaraj BT, Livesey MR, Zhao C, et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat Commun. 2018;9(1):347. DOI: 10.1038/s41467-017-02729-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. [DOI] [PubMed] [Google Scholar]
- [194].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113(15):4039–4044. DOI: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34(5):680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. DOI: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Jung J, Nayak A, Schaeffer V, et al. Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. Elife. 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Nakashima H, Nguyen T, Goins WF, et al. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem. 2015;290(3):1485–1495. DOI: 10.1074/jbc.M114.593871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Xu D, Zhang T, Xiao J, et al. Modification of BECN1 by ISG15 plays a crucial role in autophagy regulation by type I IFN/interferon. Autophagy. 2015;11(4):617–628. DOI: 10.1080/15548627.2015.1023982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Goncalves A, Bürckstümmer T, Dixit E, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6(9):e23971. DOI: 10.1371/journal.pone.0023971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Kim JY, Beg AA, Haura EB, et al. IKKϵ and TBK1, as novel therapeutic targets in the treatment of non-small cell lung cancer. Expert Opin Ther Targets. 2013;17(10):1109–1112. [DOI] [PubMed] [Google Scholar]
- [202].Awadalla MS, Fingert JH, Roos BE, et al. Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am J Ophthalmol. 2015;159(1):124–30.e1. DOI: 10.1016/j.ajo.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Ritch R, Darbro B, Menon G, et al. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol. 2014;132(5):544–548. DOI: 10.1001/jamaophthalmol.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Herman M, Ciancanelli M, Ou YH, et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med. 2012;209(9):1567–1582. DOI: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–1441. DOI: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. DOI: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- [207].Gijselinck I, Van Mossevelde S, Van Der Zee J, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–2125. DOI: 10.1212/WNL.0000000000002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Le Ber I, De Septenville A, Millecamps S, et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging. 2015;36(11):3116.e5–3116.e8. DOI: 10.1016/j.neurobiolaging.2015.08.009. [DOI] [PubMed] [Google Scholar]
- [209].Pottier C, Bieniek KF, Finch N, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77–92. DOI: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Gijselinck I, Van Mossevelde S, Van Der Zee J, et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry. 2016;21(8):1112–1124. DOI: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Van Mossevelde S, Van Der Zee J, Gijselinck I, et al. Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain. 2016;139(2):452–467. DOI: 10.1093/brain/awv358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. DOI: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Moore AS, Holzbaur EL. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy. 2016;12(10):1956–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Brenner D, Sieverding K, Bruno C, et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med. 2019;216(2):267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Gerbino V, Kaunga E, Ye J, et al. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron. 2020;106(5):789–805.e5. [DOI] [PubMed] [Google Scholar]
- [216].Yu J, Zhou X, Chang M, et al. Regulation of T-cell activation and migration by the kinase TBK1 during neuroinflammation. Nat Commun. 2015;6(1):6074. DOI: 10.1038/ncomms7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Komine O, Yamanaka K. Neuroinflammation in motor neuron disease. Nagoya J Med Sci. 2015;77(4):537–549. [PMC free article] [PubMed] [Google Scholar]
- [218].Walters KJ, Goh AM, Wang Q, et al. Ubiquitin family proteins and their relationship to the proteasome: a structural perspective. Biochim Biophys Acta. 2004;1695(1–3):73–87. DOI: 10.1016/j.bbamcr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- [219].N’Diaye EN, Kajihara KK, Hsieh I, et al. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10(2):173–179. DOI: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Rothenberg C, Srinivasan D, Mah L, et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010;19(16):3219–3232. DOI: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. DOI: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Millecamps S, Corcia P, Cazeneuve C, et al. Mutations in UBQLN2 are rare in French amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(4):839.e1–3. DOI: 10.1016/j.neurobiolaging.2011.11.010. [DOI] [PubMed] [Google Scholar]
- [223].Daoud H, Suhail H, Szuto A, et al. UBQLN2 mutations are rare in French and French-Canadian amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33(9):2230.e1–2230.e5. DOI: 10.1016/j.neurobiolaging.2012.03.015. [DOI] [PubMed] [Google Scholar]
- [224].Mizusawa H, Nakamura H, Wakayama I, et al. Skein-like inclusions in the anterior horn cells in motor neuron disease. J Neurol Sci. 1991;105(1):14–21. DOI: 10.1016/0022-510X(91)90112-K. [DOI] [PubMed] [Google Scholar]
- [225].Osaka M, Ito D, Suzuki N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem Biophys Res Commun. 2016;472(2):324–331. [DOI] [PubMed] [Google Scholar]
- [226].Ceballos-Diaz C, Rosario AM, Park HJ, et al. Viral expression of ALS-linked ubiquilin-2 mutants causes inclusion pathology and behavioral deficits in mice. Mol Neurodegener. 2015;10(1):25. DOI: 10.1186/s13024-015-0026-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Picher-Martel V, Dutta K, Phaneuf D, et al. Ubiquilin-2 drives NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol Brain. 2015;8(1):71. DOI: 10.1186/s13041-015-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Wu Q, Liu M, Huang C, et al. Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015;129(3):417–428. DOI: 10.1007/s00401-014-1367-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Şentürk M, Lin G, Zuo Z, et al. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol. 2019;21(3):384–396. DOI: 10.1038/s41556-019-0281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Wu JJ, Cai A, Greenslade JE, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci U S A. 2020;117(26):15230–15241. DOI: 10.1073/pnas.1917371117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Yang W, Hamilton JL, Kopil C, et al. Current and projected future economic burden of Parkinson’s disease in the U. S NPJ Parkinsons Dis. 2020;6(1):15. DOI: 10.1038/s41531-020-0117-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Dormann D, Madl T, Valori CF, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. Embo J. 2012;31(22):4258–4275. DOI: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Huey ED, Ferrari R, Moreno JH, et al. FUS and TDP43 genetic variability in FTD and CBS. Neurobiol Aging. 2012;33(5):1016.e9–17. DOI: 10.1016/j.neurobiolaging.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Ryu HH, Jun MH, Min KJ, et al. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging. 2014;35(12):2822–2831. DOI: 10.1016/j.neurobiolaging.2014.07.026. [DOI] [PubMed] [Google Scholar]
- [236].Marrone L, Poser I, Casci I, et al. Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell Reports. 2018;10(2):375–389. DOI: 10.1016/j.stemcr.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Arenas A, Kuang L, Zhang J, et al. FUS regulates autophagy by mediating the transcription of genes critical to the autophagosome formation. J Neurochem. 2020;157(3):752–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Halawani D, Latterich M. p97: the cell’s molecular purgatory?. Mol Cell. 2006;22(6):713–717. [DOI] [PubMed] [Google Scholar]
- [239].Kakizuka A. Roles of VCP in human neurodegenerative disorders. Biochem Soc Trans. 2008;36(1):105–108. [DOI] [PubMed] [Google Scholar]
- [240].Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381. DOI: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- [241].DeJesus-Hernandez M, Desaro P, Johnston A, et al. Novel p.Ile151Val mutation in VCP in a patient of African American descent with sporadic ALS. Neurology. 2011;77(11):1102–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Koppers M, Groen EJ, Van Vught PW, et al. Screening for rare variants in the coding region of ALS-associated genes at 9p21.2 and 19p13.3. Neurobiol Aging. 2013;34(5):1518.e5–7. DOI: 10.1016/j.neurobiolaging.2012.09.018. [DOI] [PubMed] [Google Scholar]
- [243].Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. DOI: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Custer SK, Neumann M, Lu H, et al. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010;19(9):1741–1755. DOI: 10.1093/hmg/ddq050. [DOI] [PubMed] [Google Scholar]
- [245].Hübbers CU, Clemen CS, Kesper K, et al. Pathological consequences of VCP mutations on human striated muscle. Brain. 2007;130(2):381–393. DOI: 10.1093/brain/awl238. [DOI] [PubMed] [Google Scholar]
- [246].Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009;187(6):875–888. DOI: 10.1083/jcb.200908115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Schröder R, Watts GD, Mehta SG, et al. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol. 2005;57(3):457–461. DOI: 10.1002/ana.20407. [DOI] [PubMed] [Google Scholar]
- [248].Nalbandian A, Llewellyn KJ, Kitazawa M, et al. The homozygote VCP(R155H/R155H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One. 2012;7(9):e46308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Tresse E, Salomons FA, Vesa J, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy. 2010;6(2):217–227. DOI: 10.4161/auto.6.2.11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Nalbandian A, Nguyen C, Katheria V, et al. Exercise training reverses skeletal muscle atrophy in an experimental model of VCP disease. PLoS One. 2013;8(10):e76187. DOI: 10.1371/journal.pone.0076187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Yin HZ, Nalbandian A, Hsu CI, et al. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012;3(8):e374. DOI: 10.1038/cddis.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Hall CE, Yao Z, Choi M, et al. Progressive motor neuron pathology and the role of astrocytes in a human stem cell model of VCP-related ALS. Cell Rep. 2017;19(9):1739–1749. DOI: 10.1016/j.celrep.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Kachaner D, Génin P, Laplantine E, et al. Toward an integrative view of Optineurin functions. Cell Cycle. 2012;11(15):2808–2818. DOI: 10.4161/cc.20946. [DOI] [PubMed] [Google Scholar]
- [254].Korac J, Schaeffer V, Kovacevic I, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(2):580–592. DOI: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. [DOI] [PubMed] [Google Scholar]
- [256].Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Liu Z, Chen P, Gao H, et al. Ubiquitylation of autophagy receptor optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014;26(1):106–120. DOI: 10.1016/j.ccr.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226. DOI: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- [259].Evans CS, Holzbaur EL. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife. 2020;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Sako W, Ito H, Yoshida M, et al. Nuclear factor κ B expression in patients with sporadic amyotrophic lateral sclerosis and hereditary amyotrophic lateral sclerosis with optineurin mutations. Clin Neuropathol. 2012;31(6):418–423. DOI: 10.5414/NP300493. [DOI] [PubMed] [Google Scholar]
- [261].Akizuki M, Yamashita H, Uemura K, et al. Optineurin suppression causes neuronal cell death via NF-κB pathway. J Neurochem. 2013;126(6):699–704. DOI: 10.1111/jnc.12326. [DOI] [PubMed] [Google Scholar]
- [262].Ghazi-Tabatabai S, Obita T, Pobbati AV, et al. Evolution and assembly of ESCRTs. Biochem Soc Trans. 2009;37(1):151–155. DOI: 10.1042/BST0370151. [DOI] [PubMed] [Google Scholar]
- [263].Odorizzi G. Membrane manipulations by the ESCRT machinery. F1000Res. 2015;4(F1000 Faculty Rev):516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. DOI: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- [265].Van Der Zee J, Urwin H, Engelborghs S, et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet. 2008;17(2):313–322. DOI: 10.1093/hmg/ddm309. [DOI] [PubMed] [Google Scholar]
- [266].Cox LE, Ferraiuolo L, Goodall EF, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One. 2010;5(3):e9872. DOI: 10.1371/journal.pone.0009872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67(6):1074–1077. DOI: 10.1212/01.wnl.0000231510.89311.8b. [DOI] [PubMed] [Google Scholar]
- [268].Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179(3):485–500. DOI: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Lee JA, Beigneux A, Ahmad ST, et al. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17(18):1561–1567. DOI: 10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- [270].Clayton EL, Mancuso R, Nielsen TT, et al. Early microgliosis precedes neuronal loss and behavioural impairment in mice with a frontotemporal dementia-causing CHMP2B mutation. Hum Mol Genet. 2017;26(5):873–887. DOI: 10.1093/hmg/ddx003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Ghazi-Noori S, Froud KE, Mizielinska S, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain. 2012;135(3):819–832. DOI: 10.1093/brain/aws006. [DOI] [PubMed] [Google Scholar]
- [272].Clayton EL, Mizielinska S, Edgar JR, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015;130(4):511–523. DOI: 10.1007/s00401-015-1475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2011;70(5):349–359. [DOI] [PubMed] [Google Scholar]
- [274].Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–611. DOI: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- [275].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. DOI: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- [276].Ying Z, Xia Q, Hao Z, et al. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy. 2016;12(4):707–708. DOI: 10.1080/15548627.2016.1151596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Ito Y, Yamada M, Tanaka H, et al. Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol Dis. 2009;36(3):470–476. DOI: 10.1016/j.nbd.2009.08.013. [DOI] [PubMed] [Google Scholar]
- [278].Atkin JD, Farg MA, Walker AK, et al. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30(3):400–407. DOI: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- [279].Sasaki S. Endoplasmic reticulum stress in motor neurons of the spinal cord in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2010;69(4):346–355. [DOI] [PubMed] [Google Scholar]
- [280].Ilieva EV, Ayala V, Jové M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130(Pt 12):3111–3123. DOI: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- [281].Atsumi T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1981;55(3):193–198. [DOI] [PubMed] [Google Scholar]
- [282].Chung MJ, Suh YL. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol. 2002;26(1):3–7. [DOI] [PubMed] [Google Scholar]
- [283].Napoli L, Crugnola V, Lamperti C, et al. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(12):1612–1613. DOI: 10.1001/archneur.68.12.1612. [DOI] [PubMed] [Google Scholar]
- [284].Sasaki S, Iwata M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1996;204(1–2):53–56. [DOI] [PubMed] [Google Scholar]
- [285].Wiedemann FR, Winkler K, Kuznetsov AV, et al. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J Neurol Sci. 1998;156(1):65–72. DOI: 10.1016/S0022-510X(98)00008-2. [DOI] [PubMed] [Google Scholar]
- [286].Deng HX, Bigio EH, Zhai H, et al. Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch Neurol. 2011;68(8):1057–1061. DOI: 10.1001/archneurol.2011.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Li C, Ji Y, Tang L, et al. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16(7–8):485–489. DOI: 10.3109/21678421.2015.1089909. [DOI] [PubMed] [Google Scholar]
- [288].Toth RP, Atkin JD. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front Immunol. 2018;9:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Yang L, Cheng Y, Jia X, et al. Four novel optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in Mainland China. Neurobiol Aging. 2021;97:149.e1–149.e8. [DOI] [PubMed] [Google Scholar]
- [290].Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. DOI: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- [291].Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. DOI: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].Van Deerlin VM, Leverenz JB, Bekris LM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7(5):409–416. DOI: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Yokoseki A, Shiga A, Tan CF, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63(4):538–542. DOI: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- [294].Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. DOI: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- [295].Sea K, Sohn SH, Durazo A, et al. Insights into the role of the unusual disulfide bond in copper-zinc superoxide dismutase. J Biol Chem. 2015;290(4):2405–2418. DOI: 10.1074/jbc.M114.588798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Zou ZY, Zhou ZR, Che CH, et al. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–549. DOI: 10.1136/jnnp-2016-315018. [DOI] [PubMed] [Google Scholar]
- [297].Barmada SJ, Skibinski G, Korb E, et al. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci. 2010;30(2):639–649. DOI: 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Bilican B, Serio A, Barmada SJ, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 2012;109(15):5803–5808. DOI: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Serio A, Bilican B, Barmada SJ, et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2013;110(12):4697–4702. DOI: 10.1073/pnas.1300398110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Huang SL, Wu LS, Lee M, et al. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol Commun. 2020;8(1):3. DOI: 10.1186/s40478-020-0881-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Stallings NR, Puttaparthi K, Luther CM, et al. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol Dis. 2010;40(2):404–414. DOI: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- [302].Xu YF, Gendron TF, Zhang YJ, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30(32):10851–10859. DOI: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–3863. DOI: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Shan X, Chiang PM, Price DL, et al. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A. 2010;107(37):16325–16330. DOI: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Tsai KJ, Yang CH, Fang YH, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207(8):1661–1673. DOI: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Igaz LM, Kwong LK, Lee EB, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121(2):726–738. DOI: 10.1172/JCI44867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Tian T, Huang C, Tong J, et al. TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci. 2011;7(2):234–243. DOI: 10.7150/ijbs.7.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Swarup V, Phaneuf D, Bareil C, et al. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(9):2610–2626. DOI: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- [309].Cannon A, Yang B, Knight J, et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol. 2012;123(6):807–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Arnold ES, Ling SC, Huelga SC, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110(8):E736–45. DOI: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Zhou H, Huang C, Chen H, et al. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6(3):e1000887. DOI: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Uchida A, Sasaguri H, Kimura N, et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain. 2012;135(3):833–846. DOI: 10.1093/brain/awr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Dayton RD, Wang DB, Cain CD, et al. Frontotemporal lobar degeneration-related proteins induce only subtle memory-related deficits when bilaterally overexpressed in the dorsal hippocampus. Exp Neurol. 2012;233(2):807–814. DOI: 10.1016/j.expneurol.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Herman AM, Khandelwal PJ, Rebeck GW, et al. Wild type TDP-43 induces neuro-inflammation and alters APP metabolism in lentiviral gene transfer models. Exp Neurol. 2012;235(1):297–305. DOI: 10.1016/j.expneurol.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Tatom JB, Wang DB, Dayton RD, et al. Mimicking aspects of frontotemporal lobar degeneration and Lou Gehrig’s disease in rats via TDP-43 overexpression. Mol Ther. 2009;17(4):607–613. DOI: 10.1038/mt.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Wang DB, Dayton RD, Henning PP, et al. Expansive gene transfer in the rat CNS rapidly produces amyotrophic lateral sclerosis relevant sequelae when TDP-43 is overexpressed. Mol Ther. 2010;18(12):2064–2074. DOI: 10.1038/mt.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Dayton RD, Gitcho MA, Orchard EA, et al. Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol Ther. 2013;21(7):1324–1334. DOI: 10.1038/mt.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Caccamo A, Majumder S, Oddo S. Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am J Pathol. 2012;180(1):293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [319].Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106(44):18809–18814. DOI: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Xu YF, Zhang YJ, Lin WL, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6(1):73. DOI: 10.1186/1750-1326-6-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Janssens J, Wils H, Kleinberger G, et al. Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol Neurobiol. 2013;48(1):22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Huang C, Tong J, Bi F, et al. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–118. DOI: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Chiang PM, Ling J, Jeong YH, et al. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107(37):16320–16324. DOI: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Iguchi Y, Katsuno M, Niwa J, et al. Loss of TDP-43 causes age-dependent progressive motor neuron degeneration. Brain. 2013;136(5):1371–1382. DOI: 10.1093/brain/awt029. [DOI] [PubMed] [Google Scholar]
- [325].Kraemer BC, Schuck T, Wheeler JM, et al. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010;119(4):409–419. DOI: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Sephton CF, Good SK, Atkin S, et al. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285(9):6826–6834. DOI: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Wu LS, Cheng WC, Hou SC, et al. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48(1):56–62. DOI: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- [328].Wu LS, Cheng WC, Shen CK. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem. 2012;287(33):27335–27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [329].Wegorzewska I, Baloh RHTDP. 43-based animal models of neurodegeneration: new insights into ALS pathology and pathophysiology. Neurodegener Dis. 2011;8(4):262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans. 2013;41(6):1536–1540. [DOI] [PubMed] [Google Scholar]
- [331].Casci I, Pandey UB. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 2015;1607:47–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Chen H, Kankel MW, Su SC, et al. Exploring the genetics and non-cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018;25(4):648–662. DOI: 10.1038/s41418-018-0060-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Liu YC, Chiang PM, Tsai KJ. Disease animal models of TDP-43 proteinopathy and their pre-clinical applications. Int J Mol Sci. 2013;14(10):20079–20111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Tan RH, Ke YD, Ittner LM, et al. ALS/FTLD: experimental models and reality. Acta Neuropathol. 2017;133(2):177–196. DOI: 10.1007/s00401-016-1666-6. [DOI] [PubMed] [Google Scholar]
- [335].Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23(19):2294–2306. DOI: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Rudnick ND, Griffey CJ, Guarnieri P, et al. Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc Natl Acad Sci U S A. 2017;114(39):E8294–e8303. DOI: 10.1073/pnas.1704294114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Morimoto N, Nagai M, Ohta Y, et al. Increased autophagy in transgenic mice with a G93A mutant SOD1 gene. Brain Res. 2007;1167:112–117. [DOI] [PubMed] [Google Scholar]
- [338].Zhang X, Li L, Chen S, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7(4):412–425. DOI: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- [339].Gong YH, Parsadanian AS, Andreeva A, et al. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20(2):660–665. DOI: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Vargas MR, Johnson DA, Sirkis DW, et al. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28(50):13574–13581. DOI: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Julien JP, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762(11–12):1013–1024. [DOI] [PubMed] [Google Scholar]
- [342].Morrice JR, Gregory-Evans CY, Shaw CA. Animal models of amyotrophic lateral sclerosis: a comparison of model validity. Neural Regen Res. 2018;13(12):2050–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Philips T, Rothstein JD. Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol. 2015;69(1):5.67.1–5.67.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Van Damme P, Robberecht W, Van Den Bosch L. and Van Den Bosch L. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech. 2017;10(5):537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [345].Nishihira Y, Tan CF, Onodera O, et al. Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008;116(2):169–182. DOI: 10.1007/s00401-008-0385-z. [DOI] [PubMed] [Google Scholar]
- [346].Zhang H, Tan CF, Mori F, et al. TDP-43-immunoreactive neuronal and glial inclusions in the neostriatum in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008;115(1):115–122. DOI: 10.1007/s00401-007-0285-7. [DOI] [PubMed] [Google Scholar]
- [347].Papadeas ST, Kraig SE, O’Banion C, et al. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc Natl Acad Sci U S A. 2011;108(43):17803–17808. DOI: 10.1073/pnas.1103141108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [348].Lepore AC, Rauck B, Dejea C, et al. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11(11):1294–1301. DOI: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [349].Bose JK, Huang CC, Shen CK. Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem. 2011;286(52):44441–44448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Xia Q, Wang H, Hao Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. Embo J. 2016;35(2):121–142. DOI: 10.15252/embj.201591998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Caccamo A, Shaw DM, Guarino F, et al. Reduced protein turnover mediates functional deficits in transgenic mice expressing the 25 kDa C-terminal fragment of TDP-43. Hum Mol Genet. 2015;24(16):4625–4635. DOI: 10.1093/hmg/ddv193. [DOI] [PubMed] [Google Scholar]
- [352].Caccamo A, Majumder S, Deng JJ, et al. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J Biol Chem. 2009;284(40):27416–27424. DOI: 10.1074/jbc.M109.031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109(37):15024–15029. DOI: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Jinwal UK, Abisambra JF, Zhang J, et al. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J Biol Chem. 2012;287(29):24814–24820. DOI: 10.1074/jbc.M112.367268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [355].Crippa V, Carra S, Rusmini P, et al. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy. 2010;6(7):958–960. DOI: 10.4161/auto.6.7.13042. [DOI] [PubMed] [Google Scholar]
- [356].Crippa V, D’Agostino VG, Cristofani R, et al. Transcriptional induction of the heat shock protein B8 mediates the clearance of misfolded proteins responsible for motor neuron diseases. Sci Rep. 2016;6(1):22827. DOI: 10.1038/srep22827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [357].Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677–685. DOI: 10.1038/nchembio.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Urushitani M, Sato T, Bamba H, et al. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J Neurosci Res. 2010;88(4):784–797. DOI: 10.1002/jnr.22243. [DOI] [PubMed] [Google Scholar]
- [359].Wang X, Fan H, Ying Z, et al. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett. 2010;469(1):112–116. DOI: 10.1016/j.neulet.2009.11.055. [DOI] [PubMed] [Google Scholar]
- [360].Chen Y, Wang H, Ying Z, et al. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: the new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem Biophys Res Commun. 2020;526(1):231–238. DOI: 10.1016/j.bbrc.2020.03.051. [DOI] [PubMed] [Google Scholar]
- [361].Natale G, Lenzi P, Lazzeri G, et al. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front Cell Neurosci. 2015;9:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [362].Chen Y, Liu H, Guan Y, et al. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am J Transl Res. 2015;7(9):1574–1587. [PMC free article] [PubMed] [Google Scholar]
- [363].Guo X, Sun X, Hu D, et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat Commun. 2016;7(1):12646. DOI: 10.1038/ncomms12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [364].Forloni G, Artuso V, La Vitola P, et al. Oligomeropathies and pathogenesis of Alzheimer and Parkinson’s diseases. Mov Disord. 2016;31(6):771–781. DOI: 10.1002/mds.26624. [DOI] [PubMed] [Google Scholar]
- [365].Corcia P, Couratier P, Blasco H, et al. Genetics of amyotrophic lateral sclerosis. Rev Neurol (Paris). 2017;173(5):254–262. DOI: 10.1016/j.neurol.2017.03.030. [DOI] [PubMed] [Google Scholar]
- [366].Redler RL, Dokholyan NV. The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog Mol Biol Transl Sci. 2012;107:215–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [367].Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85(8):665–674. DOI: 10.1212/WNL.0000000000001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [368].Manley S, Williams JA, Ding WX. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med (Maywood). 2013;238(5):525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [369].Lodish HF, Berk A, Zipursky SL, et al. Molecular Cell Biology. New York: W.H. Freeman; 2000. 1084. [Google Scholar]
- [370].Maday S, Holzbaur EL. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci. 2016;36(22):5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Horgusluoglu E, Nudelman K, Nho K, et al. Adult neurogenesis and neurodegenerative diseases: a systems biology perspective. Am J Med Genet B Neuropsychiatr Genet. 2017;174(1):93–112. DOI: 10.1002/ajmg.b.32429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [372].Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196(4):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [373].Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell. 2014;30(1):71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Maday S, Holzbaur EL. Autophagosome assembly and cargo capture in the distal axon. Autophagy. 2012;8(5):858–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [375].Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26(31):8057–8068. DOI: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104(36):14489–14494. DOI: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187(1):71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Tang G, Gudsnuk K, Kuo SH, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. 2014;83(5):1131–1143. DOI: 10.1016/j.neuron.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. DOI: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [380].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. DOI: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- [381].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. DOI: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [382].Stavoe AK, Gopal PP, Gubas A, et al. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. Elife. 2019;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [383].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–1111. DOI: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [384].Tsvetkov AS, Miller J, Arrasate M, et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107(39):16982–16987. DOI: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [385].Roscic A, Baldo B, Crochemore C, et al. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119(2):398–407. DOI: 10.1111/j.1471-4159.2011.07435.x. [DOI] [PubMed] [Google Scholar]
- [386].Fox JH, Connor T, Chopra V, et al. The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington’s disease. Mol Neurodegener. 2010;5(1):26. DOI: 10.1186/1750-1326-5-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [387].Arriola Apelo SI, Neuman JC, Baar EL, et al. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell. 2016;15(1):28–38. DOI: 10.1111/acel.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [388].Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Von Bartheld CS, Bahney J, Herculano-Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524(18):3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [390].Jessen KR. Glial cells. Int J Biochem Cell Biol. 2004;36(10):1861–1867. [DOI] [PubMed] [Google Scholar]
- [391].Von Bernhardi R, Eugenin-von Bernhardi J, Flores B, et al. Glial Cells and Integrity of the Nervous System. Adv Exp Med Biol. 2016;949:1–24. [DOI] [PubMed] [Google Scholar]
- [392].Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [393].Yong VW, Yong FP, Olivier A, et al. Morphologic heterogeneity of human adult astrocytes in culture: correlation with HLA-DR expression. J Neurosci Res. 1990;27(4):678–688. DOI: 10.1002/jnr.490270428. [DOI] [PubMed] [Google Scholar]
- [394].Miller RH, Zhang H, Fok-Seang J. Glial cell heterogeneity in the mammalian spinal cord. Perspect Dev Neurobiol. 1994;2(3):225–231. [PubMed] [Google Scholar]
- [395].Miller RH, Raff MC. Fibrous and protoplasmic astrocytes are biochemically and developmentally distinct. J Neurosci. 1984;4(2):585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [396].Vaughn JE, Pease DC. Electron microscopy of classically stained astrocytes. J Comp Neurol. 1967;131(2):143–154. [DOI] [PubMed] [Google Scholar]
- [397].Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. 2008;1782(12):691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [398].Janen SB, Chaachouay H, Richter-Landsberg C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia. 2010;58(14):1766–1774. [DOI] [PubMed] [Google Scholar]
- [399].Tang G, Yue Z, Talloczy Z, et al. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17(11):1540–1555. DOI: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [400].Pamenter ME, Perkins GA, McGinness AK, et al. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 2012;7(12):e51469. DOI: 10.1371/journal.pone.0051469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [401].Motori E, Puyal J, Toni N, et al. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013;18(6):844–859. DOI: 10.1016/j.cmet.2013.11.005. [DOI] [PubMed] [Google Scholar]
- [402].Kulkarni A, Dong A, Kulkarni VV, et al. Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy. 2019;16(9):1651–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [403].Lavieu G, Scarlatti F, Sala G, et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281(13):8518–8527. DOI: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- [404].Lima S, Milstien S, Spiegel S. Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking. J Biol Chem. 2017;292(8):3074–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [405].Moruno Manchon JF, Uzor NE, Finkbeiner S, et al. SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy. 2016;12(8):1418–1424. DOI: 10.1080/15548627.2016.1183082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [406].Young MM, Takahashi Y, Fox TE, et al. Sphingosine Kinase 1 Cooperates with Autophagy to Maintain Endocytic Membrane Trafficking. Cell Rep. 2016;17(6):1532–1545. DOI: 10.1016/j.celrep.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [407].Koistinaho M, Lin S, Wu X, et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10(7):719–726. DOI: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- [408].Nielsen HM, Veerhuis R, Holmqvist B, et al. Binding and uptake of A beta1-42 by primary human astrocytes in vitro. Glia. 2009;57(9):978–988. DOI: 10.1002/glia.20822. [DOI] [PubMed] [Google Scholar]
- [409].Wyss-Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–457. DOI: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- [410].Lee HJ, Suk JE, Patrick C, et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285(12):9262–9272. DOI: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [411].Braidy N, Gai WP, Xu YH, et al. Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl Neurodegener. 2013;2(1):20. DOI: 10.1186/2047-9158-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [412].Lindstrom V, Gustafsson G, Sanders LH, et al. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci. 2017;82:143–156. [DOI] [PubMed] [Google Scholar]
- [413].Chen LL, Wu JC, Wang LH, et al. Rapamycin prevents the mutant huntingtin-suppressed GLT-1 expression in cultured astrocytes. Acta Pharmacol Sin. 2012;33(3):385–392. DOI: 10.1038/aps.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [414].Bruijn LI, Becher MW, Lee MK, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18(2):327–338. DOI: 10.1016/S0896-6273(00)80272-X. [DOI] [PubMed] [Google Scholar]
- [415].Tripathi P, Rodriguez-Muela N, Klim JR, et al. Reactive astrocytes promote ALS-like degeneration and intracellular protein aggregation in human motor neurons by disrupting autophagy through TGF-beta1. Stem Cell Reports. 2017;9(2):667–680. DOI: 10.1016/j.stemcr.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [416].Hanamsagar R, Bilbo SD. Environment matters: microglia function and dysfunction in a changing world. Curr Opin Neurobiol. 2017;47:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [417].Schafer DP, Stevens B. Microglia Function in Central Nervous System Development and Plasticity. Cold Spring Harb Perspect Biol. 2015;7(10):a020545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [418].Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. DOI: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- [419].Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10(3):253–263. [DOI] [PubMed] [Google Scholar]
- [420].Gleichman AJ, Carmichael ST. Glia in neurodegeneration: drivers of disease or along for the ride?. Neurobiol Dis. 2020;142:104957. [DOI] [PubMed] [Google Scholar]
- [421].Haukedal H, Freude K. Implications of Microglia in amyotrophic lateral sclerosis and frontotemporal dementia. J Mol Biol. 2019;431(9):1818–1829. [DOI] [PubMed] [Google Scholar]
- [422].Thompson AG, Gray E, Thézénas ML, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol. 2018;83(2):258–268. DOI: 10.1002/ana.25143. [DOI] [PubMed] [Google Scholar]
- [423].Corcia P, Tauber C, Vercoullie J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7(12):e52941. DOI: 10.1371/journal.pone.0052941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [424].Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15(3):601–609. DOI: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- [425].Cady J, Koval ED, Benitez BA, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(4):449–453. DOI: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [426].Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3(6):445–453. [DOI] [PubMed] [Google Scholar]
- [427].Kleinberger G, Yamanishi Y, Suárez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra86. DOI: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- [428].Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and Microglia: novel Partners in Neurodegeneration and Aging. Int J Mol Sci. 2017;18(3):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [429].Etchegaray JI, Elguero EJ, Tran JA, et al. Defective phagocytic corpse processing results in neurodegeneration and can be rescued by TORC1 activation. J Neurosci. 2016;36(11):3170–3183. DOI: 10.1523/JNEUROSCI.1912-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [430].Sierra A, Abiega O, Shahraz A, et al. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [431].Solé-Domènech S, Cruz DL, Capetillo-Zarate E, et al. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res Rev. 2016;32:89–103. DOI: 10.1016/j.arr.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [432].Cho MH, Cho K, Kang HJ, et al. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10(10):1761–1775. DOI: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [433].Lucin KM, O’Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79(5):873–886. DOI: 10.1016/j.neuron.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [434].Haidet-phillips AM, Hester ME, Miranda CJ, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824–828. DOI: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [435].Henkel JS, Beers DR, Zhao W, et al. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4(4):389–398. DOI: 10.1007/s11481-009-9171-5. [DOI] [PubMed] [Google Scholar]
- [436].Yamanaka K, Chun SJ, Boillee S, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–253. DOI: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [437].Beers DR, Henkel JS, Xiao Q, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103(43):16021–16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [438].Brettschneider J, Toledo JB, Van Deerlin VM, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7(6):e39216. DOI: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [439].O’Rourke JG, Bogdanik L, Yáñez A, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–1329. DOI: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [440].McCauley ME, O’Rourke JG, Yáñez A, et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020;585(7823):96–101. DOI: 10.1038/s41586-020-2625-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [441].Asai T, Tomita Y, Nakatsuka S, et al. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J Cancer Res. 2002;93(3):296–304. DOI: 10.1111/j.1349-7006.2002.tb02172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [442].Duran A, Linares JF, Galvez AS, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–354. DOI: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- [443].Zhu G, Wu CJ, Zhao Y, et al. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007;17(16):1438–1443. DOI: 10.1016/j.cub.2007.07.041. [DOI] [PubMed] [Google Scholar]
- [444].Bankston AN, Forston MD, Howard RM, et al. Autophagy is essential for oligodendrocyte differentiation, survival, and proper myelination. Glia. 2019;67(9):1745–1759. DOI: 10.1002/glia.23646. [DOI] [PubMed] [Google Scholar]
- [445].Dello Russo C, Lisi L, Feinstein DL, et al. mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia. 2013;61(3):301–311. DOI: 10.1002/glia.22433. [DOI] [PubMed] [Google Scholar]
- [446].Figlia G, Gerber D, Suter U. Myelination and mTOR. Glia. 2018;66(4):693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [447].Tyler WA, Gangoli N, Gokina P, et al. Activation of the mammalian target of rapamycin (mTOR) is essential for oligodendrocyte differentiation. J Neurosci. 2009;29(19):6367–6378. DOI: 10.1523/JNEUROSCI.0234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [448].Smith CM, Mayer JA, Duncan ID. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J Neurosci. 2013;33(18):8088–8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [449].Saraswat Ohri S, Bankston AN, Mullins SA, et al. Blocking Autophagy in Oligodendrocytes Limits Functional Recovery after Spinal Cord Injury. J Neurosci. 2018;38(26):5900–5912. DOI: 10.1523/JNEUROSCI.0679-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [450].Pukaß K, Goldbaum O, Richter-Landsberg C. Mitochondrial impairment and oxidative stress compromise autophagosomal degradation of α-synuclein in oligodendroglial cells. J Neurochem. 2015;135(1):194–205. [DOI] [PubMed] [Google Scholar]
- [451].Riedel M, Goldbaum O, Schwarz L, et al. 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PLoS One. 2010;5(1):e8753. DOI: 10.1371/journal.pone.0008753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [452].Cong Y, Wang C, Wang J, et al. NT-3 promotes oligodendrocyte proliferation and nerve function recovery after spinal cord injury by inhibiting autophagy pathway. J Surg Res. 2020;247:128–135. [DOI] [PubMed] [Google Scholar]
- [453].Liu S, Sarkar C, Dinizo M, et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6(1):e1582. DOI: 10.1038/cddis.2014.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [454].Kaji S, Maki T, Kinoshita H, et al. Pathological endogenous alpha-synuclein accumulation in oligodendrocyte precursor cells potentially induces inclusions in multiple system atrophy. Stem Cell Reports. 2018;10(2):356–365. DOI: 10.1016/j.stemcr.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [455].Mackenzie IR, Ansorge O, Strong M, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122(1):87–98. DOI: 10.1007/s00401-011-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [456].Mackenzie IR, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–434. DOI: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- [457].Seilhean D, Cazeneuve C, Thuries V, et al. Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol. 2009;118(4):561–573. DOI: 10.1007/s00401-009-0545-9. [DOI] [PubMed] [Google Scholar]
- [458].Lee Y, Morrison BM, Li Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487(7408):443–448. DOI: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [459].Jang SY, Shin YK, Park SY, et al. Autophagy is involved in the reduction of myelinating Schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS One. 2015;10(1):e0116624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [460].Logan AM, Mammel AE, Robinson DC, et al. Schwann cell-specific deletion of the endosomal PI 3-kinase Vps34 leads to delayed radial sorting of axons, arrested myelination, and abnormal ErbB2-ErbB3 tyrosine kinase signaling. Glia. 2017;65(9):1452–1470. DOI: 10.1002/glia.23173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [461].Zhang SJ, Li XX, Yu Y, et al. Schwann cell-specific PTEN and EGFR dysfunctions affect neuromuscular junction development by impairing Agrin signaling and autophagy. Biochem Biophys Res Commun. 2019;515(1):50–56. DOI: 10.1016/j.bbrc.2019.05.014. [DOI] [PubMed] [Google Scholar]
- [462].Brosius Lutz A, Chung WS, Sloan SA, et al. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci U S A. 2017;114(38):E8072–e8080. DOI: 10.1073/pnas.1710566114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [463].Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol. 2015;210(1):153–168. DOI: 10.1083/jcb.201503019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [464].Jang SY, Yoon BA, Shin YK, et al. Schwann cell dedifferentiation-associated demyelination leads to exocytotic myelin clearance in inflammatory segmental demyelination. Glia. 2017;65(11):1848–1862. DOI: 10.1002/glia.23200. [DOI] [PubMed] [Google Scholar]
- [465].Huang HC, Chen L, Zhang HX, et al. Autophagy promotes peripheral nerve regeneration and motor recovery following sciatic nerve crush injury in rats. J Mol Neurosci. 2016;58(4):416–423. DOI: 10.1007/s12031-015-0672-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [466].Marinelli S, Nazio F, Tinari A, et al. Schwann cell autophagy counteracts the onset and chronification of neuropathic pain. Pain. 2014;155(1):93–107. DOI: 10.1016/j.pain.2013.09.013. [DOI] [PubMed] [Google Scholar]
- [467].Hantke J, Carty L, Wagstaff LJ, et al. c-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain. 2014;137(11):2922–2937. DOI: 10.1093/brain/awu257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [468].Hutton EJ, Carty L, Laura M, et al. c-Jun expression in human neuropathies: a pilot study. J Peripher Nerv Syst. 2011;16(4):295–303. DOI: 10.1111/j.1529-8027.2011.00360.x. [DOI] [PubMed] [Google Scholar]
- [469].Lee S, Bazick H, Chittoor-Vinod V, et al. Elevated peripheral myelin protein 22, reduced mitotic potential, and proteasome impairment in dermal fibroblasts from charcot-marie-tooth disease type 1A patients. Am J Pathol. 2018;188(3):728–738. DOI: 10.1016/j.ajpath.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [470].Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84(1):85–88. DOI: 10.1016/j.ajhg.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [471].Fernandez AM, LeRoith D. Skeletal muscle. Adv Exp Med Biol. 2005;567:117–147. [DOI] [PubMed] [Google Scholar]
- [472].Wang X, Blagden C, Fan J, et al. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev. 2005;19(14):1715–1722. DOI: 10.1101/gad.1318305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [473].Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–471. DOI: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- [474].Campos JC, Baehr LM, Ferreira ND, et al. beta2 -adrenoceptor activation improves skeletal muscle autophagy in neurogenic myopathy. Faseb J. 2020;34(4):5628–5641. DOI: 10.1096/fj.201902305R. [DOI] [PubMed] [Google Scholar]
- [475].Nichenko AS, Southern WM, Atuan M, et al. Mitochondrial maintenance via autophagy contributes to functional skeletal muscle regeneration and remodeling. Am J Physiol Cell Physiol. 2016;311(2):C190–200. DOI: 10.1152/ajpcell.00066.2016. [DOI] [PubMed] [Google Scholar]
- [476].Saera-Vila A, Kish PE, Louie KW, et al. Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy. 2016;12(10):1864–1875. DOI: 10.1080/15548627.2016.1207015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [477].Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10(6):507–515. DOI: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- [478].Raben N, Hill V, Shea L, et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17(24):3897–3908. DOI: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [479].Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8(5):425–436. DOI: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- [480].Bibee KP, Cheng YJ, Ching JK, et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. Faseb J. 2014;28(5):2047–2061. DOI: 10.1096/fj.13-237388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [481].Pauly M, Daussin F, Burelle Y, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol. 2012;181(2):583–592. DOI: 10.1016/j.ajpath.2012.04.004. [DOI] [PubMed] [Google Scholar]
- [482].Salminen A, Vihko V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch B Cell Pathol Incl Mol Pathol. 1984;45(1):97–106. [DOI] [PubMed] [Google Scholar]
- [483].Fry CS, Drummond MJ, Glynn EL, et al. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J Gerontol A Biol Sci Med Sci. 2013;68(5):599–607. DOI: 10.1093/gerona/gls209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [484].Moller AB, Vendelbo MH, Christensen B, et al. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J Appl Physiol. 1985;118(8):971–979. DOI: 10.1152/japplphysiol.01116.2014. [DOI] [PubMed] [Google Scholar]
- [485].He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–515. DOI: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [486].Jamart C, Francaux M, Millet GY, et al. Modulation of autophagy and ubiquitin-proteasome pathways during ultra-endurance running. J Appl Physiol (1985). 2012;112(9):1529–1537. DOI: 10.1152/japplphysiol.00952.2011 [DOI] [PubMed] [Google Scholar]
- [487].Jamart C, Benoit N, Raymackers JM, et al. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur J Appl Physiol. 2012;112(8):3173–3177. DOI: 10.1007/s00421-011-2287-3. [DOI] [PubMed] [Google Scholar]
- [488].Vainshtein A, Tryon LD, Pauly M, et al. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308(9):C710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [489].Fan J, Kou X, Jia S, et al. Autophagy as a Potential Target for Sarcopenia. J Cell Physiol. 2016;231(7):1450–1459. DOI: 10.1002/jcp.25260. [DOI] [PubMed] [Google Scholar]
- [490].Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143(5):813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [491].Kim YA, Kim YS, Oh SL, et al. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem. 2013;69(4):697–705. DOI: 10.1007/s13105-013-0246-7. [DOI] [PubMed] [Google Scholar]
- [492].Sebastian D, Sorianello E, Segales J, et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. Embo J. 2016;35(15):1677–1693. DOI: 10.15252/embj.201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [493].Halling JF, Ringholm S, Olesen J, et al. Exercise training protects against aging-induced mitochondrial fragmentation in mouse skeletal muscle in a PGC-1alpha dependent manner. Exp Gerontol. 2017;96:1–6. [DOI] [PubMed] [Google Scholar]
- [494].Russ DW, Boyd IM, McCoy KM, et al. Muscle-specificity of age-related changes in markers of autophagy and sphingolipid metabolism. Biogerontology. 2015;16(6):747–759. DOI: 10.1007/s10522-015-9598-4. [DOI] [PubMed] [Google Scholar]
- [495].Wenz T, Rossi SG, Rotundo RL, et al. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci U S A. 2009;106(48):20405–20410. DOI: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [496].O’Leary MF, Vainshtein A, Iqbal S, et al. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304(5):C422–30. DOI: 10.1152/ajpcell.00240.2012. [DOI] [PubMed] [Google Scholar]
- [497].Carter HN, Kim Y, Erlich AT, et al. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J Physiol. 2018;596(16):3567–3584. DOI: 10.1113/JP275998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [498].Mandrioli J, D’Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis: protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore). 2018;97(24):e11119. DOI: 10.1097/MD.0000000000011119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [499].Li M, Zhou Y, Chen C, et al. Efficacy and safety of mTOR inhibitors (rapamycin and its analogues) for tuberous sclerosis complex: a meta-analysis. Orphanet J Rare Dis. 2019;14(1):39. DOI: 10.1186/s13023-019-1012-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [500].Lutz M, Mielke S. New perspectives on the use of mTOR inhibitors in allogeneic haematopoietic stem cell transplantation and graft-versus-host disease. Br J Clin Pharmacol. 2016;82(5):1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [501].Nguyen LS, Vautier M, Allenbach Y, et al. Sirolimus and mTOR Inhibitors: a Review of Side Effects and Specific Management in Solid Organ Transplantation. Drug Saf. 2019;42(7):813–825. DOI: 10.1007/s40264-019-00810-9. [DOI] [PubMed] [Google Scholar]
- [502].Sofroniadou S, Goldsmith D. Mammalian target of rapamycin (mTOR) inhibitors: potential uses and a review of haematological adverse effects. Drug Saf. 2011;34(2):97–115. [DOI] [PubMed] [Google Scholar]
- [503].Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12(2):177–186. [DOI] [PubMed] [Google Scholar]
- [504].Kahan BD, Napoli KL, Kelly PA, et al. Therapeutic drug monitoring of sirolimus: correlations with efficacy and toxicity. Clin Transplant. 2000;14(2):97–109. DOI: 10.1034/j.1399-0012.2000.140201.x. [DOI] [PubMed] [Google Scholar]
- [505].Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105(6):2052–2057. DOI: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [506].Fornai F, Longone P, Ferrucci M, et al. Autophagy and amyotrophic lateral sclerosis: the multiple roles of lithium. Autophagy. 2008;4(4):527–530. DOI: 10.4161/auto.5923. [DOI] [PubMed] [Google Scholar]
- [507].Aggarwal SP, Zinman L, Simpson E, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(5):481–488. DOI: 10.1016/S1474-4422(10)70068-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [508].Chio A, Borghero G, Calvo A, et al. Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose-finding trial. Neurology. 2010;75(7):619–625. DOI: 10.1212/WNL.0b013e3181ed9e7c. [DOI] [PubMed] [Google Scholar]
- [509].Miller RG, Moore DH, Forshew DA, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis: examining a more efficient trial design. Neurology. 2011;77(10):973–979. DOI: 10.1212/WNL.0b013e31822dc7a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [510].Morrison KE, Dhariwal S, Hornabrook R, et al. Lithium in patients with amyotrophic lateral sclerosis (LiCALS): a phase 3 multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2013;12(4):339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [511].Verstraete E, Veldink JH, Huisman MH, et al. Lithium lacks effect on survival in amyotrophic lateral sclerosis: a phase IIb randomised sequential trial. J Neurol Neurosurg Psychiatry. 2012;83(5):557–564. DOI: 10.1136/jnnp-2011-302021. [DOI] [PubMed] [Google Scholar]
- [512].Kanazawa T, Taneike I, Akaishi R, et al. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 2004;279(9):8452–8459. DOI: 10.1074/jbc.M306337200. [DOI] [PubMed] [Google Scholar]
- [513].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [514].Wang RC, Wei Y, An Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959. DOI: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [515].Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. DOI: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [516].Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. DOI: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [517].Wengrod J, Martin L, Wang D, et al. Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol. 2013;33(11):2128–2135. DOI: 10.1128/MCB.00174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [518].Daub A, Sharma P, Finkbeiner S. High-content screening of primary neurons: ready for prime time. Curr Opin Neurobiol. 2009;19(5):537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [519].Engle SJ, Vincent F. Small molecule screening in human induced pluripotent stem cell-derived terminal cell types. J Biol Chem. 2014;289(8):4562–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [520].Sarkar S, Perlstein EO, Imarisio S, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol. 2007;3(6):331–338. DOI: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]