

Abstract
Syntrophus aciditrophicus is a model syntrophic bacterium that degrades fatty and aromatic acids into acetate, CO2, formate, and H2 that are utilized by methanogens and other hydrogen-consuming microbes. S. aciditrophicus benzoate degradation proceeds by a multistep pathway with many intermediate reactive acyl-coenzyme A species (RACS) that can potentially Nε-acylate lysine residues. Herein, we describe the identification and characterization of acyl-lysine modifications that correspond to RACS in the benzoate degradation pathway. The amounts of modified peptides are sufficient to analyze the post-translational modifications without antibody enrichment, enabling a range of acylations located, presumably, on the most extensively acylated proteins throughout the proteome to be studied. Seven types of acyl modifications were identified, six of which correspond directly to RACS that are intermediates in the benzoate degradation pathway including 3-hydroxypimeloylation, a modification first identified in this system. Indeed, benzoate-degrading enzymes are heavily represented among the acylated proteins. A total of 125 sites were identified in 60 proteins. Functional deacylase enzymes are present in the proteome, indicating a potential regulatory system/mechanism by which S. aciditrophicus modulates acylation. Uniquely, Nε-acyl-lysine RACS are highly abundant in these syntrophic bacteria, raising the compelling possibility that post-translational modifications modulate benzoate degradation in this and potentially other, syntrophic bacteria. Our results outline candidates for further study of how acylations impact syntrophic consortia.
Keywords: lysine acylation, syntrophs, sirtuins, Syntrophus aciditrophicus
Abbreviations: 2D-PAGE, two-dimensional polyacrylamide gel electrophoresis; ABC, ammonium bicarbonate; ATP, adenosine triphosphate; BCL, benzoate-CoA ligase; BLAST, Basic Local Alignment Search Tool; CoA, Coenzyme A; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; MALDI, matrix-assisted laser desorption/ionization; PPI, protein–protein interaction; PTM, post-translational modification; RACS, reactive acyl-coenzyme A species
Graphical Abstract
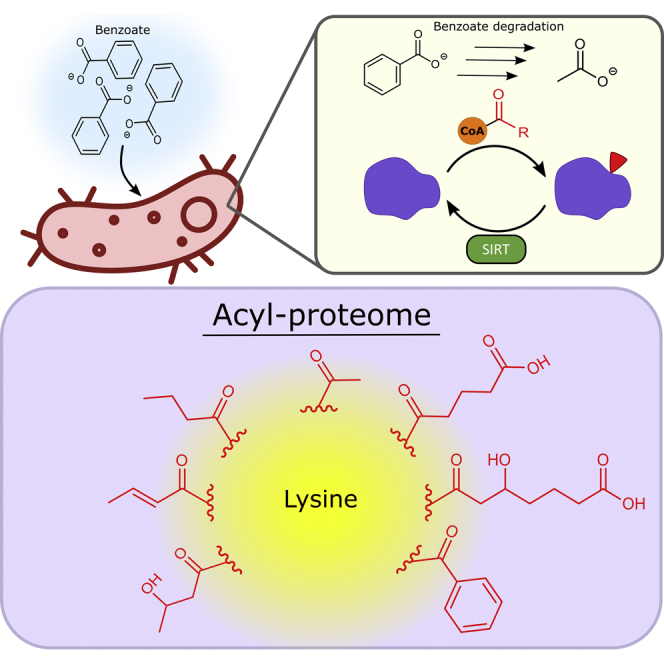
Highlights
-
•
Abundant lysine modifications in microbes enable unbiased global acylation profiling.
-
•
Seven types of acyl modifications are found; six from benzoate degradation intermediates.
-
•
Benzoate-degrading enzymes are prominent among the 60 acylated proteins.
-
•
Abundant acylation/active deacylases suggest PTMs modulate syntrophic metabolism.
In Brief
Syntrophus aciditrophicus is a syntrophic bacterium degrading fatty and aromatic acids into acetate, CO2, formate, and H2, consumed by methanogenic archaea. The syntroph’s acyl-lysine modifications were analyzed with a workflow avoiding antibody enrichment, enabling unbiased global acylation profiling. Seven acyl modification types were identified, six corresponding to reactive acyl-CoA species intermediates in benzoate degradation. Benzoate-degrading enzymes were also prominent among the 60 acylated proteins. The abundant acylations and active deacylases suggest that post-translational modifications directly regulate syntrophic benzoate degradation.
Syntrophic bacteria serve critical roles in bioremediation and carbon cycling from anaerobic environments (1, 2, 3, 4, 5, 6). They degrade a broad range of aliphatic and aromatic acids to hydrogen, formate, CO2, and acetate by cooperation with hydrogen- and/or formate-consuming microbes such as archaea which generate methane (2, 3). Here, the presence of methanogens and/or other hydrogen- and/or formate-consuming partners is required to maintain low hydrogen and formate concentrations, such that syntrophic substrate degradation can occur spontaneously (4). This obligate partnership between members of the microbial community exists because anaerobic degradation is thermodynamically unfavorable when hydrogen or formate levels are high (3, 5). Direct electron transfer between syntrophic microorganisms and their partner microorganisms is also possible (7). Importantly, degrading syntrophic substrates requires multiple enzymatically catalyzed reactions to be performed on many acyl-coenzyme A (CoA) intermediates. Even when hydrogen and formate levels are low, the essential acyl-CoA oxidations of syntrophic metabolism are weakly exergonic with free-energy changes close to thermodynamic equilibrium (4). The energetic challenges of living at the edge of thermodynamic feasibility make syntrophic microbes interesting models for exploring energy conservation and metabolic regulation (6).
Syntrophus aciditrophicus is an anaerobic, gram-negative bacterium that degrades fatty, aromatic, and alicyclic (cyclohexane-1-carboxylate) acids to acetate, CO2, formate, and hydrogen when grown in coculture with hydrogen- and/or formate-consuming partner microorganisms. S. aciditrophicus can grow in pure culture with crotonate or benzoate (8, 9, 10) and can use benzoate as an electron acceptor to form cyclohexane-1-carboxylate (11). Previous work elucidated the pathways for crotonate, benzoate, and cyclohexane-1-carboxylate metabolism (12, 13, 14) and found that S. aciditrophicus uses many of the same enzymes to degrade and to synthesize benzoate and cyclohexane carboxylate (10, 12).
Due to challenges in isolating, manipulating, and cultivating syntrophic microorganisms, questions still remain regarding the regulatory mechanisms involved in the important anaerobic degradations. Oxidizing benzoate and other substrates generates reduced cofactors (nicotinamide adenine dinucleotide reduced and flavin adenine dinucleotide reduced) that, if unregulated, could cause stress and cellular dysfunction (15). Intermediates of substrate metabolism can also stress cells. Various acid substrates in S. aciditrophicus are activated through their respective CoA derivative, which serves as a scaffold through subsequent conversions, until acetate, CO2, hydrogen, and formate are released (6, 16). The intermediates of these pathways are a series of reactive acyl-CoA species (RACS), with potential to modify the nucleophilic side chain of lysine (17). Given the multiple stressors that S. aciditrophicus cells experience in carbon metabolism and that its enzymes catalyze in either direction, depending on the substrate and environmental conditions (10, 12), it is suspected that previously undescribed pathway regulation is involved. The reversibility of syntrophic metabolism and its correspondingly low energy yields makes us consider that post-translational modifications (PTMs), specifically acylations associated with RACS intermediates, may play a role modulating degradations and syntheses in the cell.
Identifying and characterizing PTMs is most often done using mass spectrometry. At the systems level, proteomics can capture PTMs across all proteins, but a system presenting a wide range of acylations brings analytical complications, including potential sequence misidentifications due to isomeric and/or isobaric combinations (18, 19). Ambiguities must be addressed to confidently ascribe sequences, modifications, and modified residues in data. Increases in theoretical search space and scoring thresholds needed to satisfy false-discovery rate cutoffs for database searching are also considerations for proteomes possessing a broad variety of acylations (20).
Here, we take a systems-level approach to identify post-translational acylations associated with syntrophic benzoate metabolism by S. aciditrophicus. Without using PTM-specific enrichment procedures, we identify a wide range of acylations by targeting acyl-CoAs that are known to be involved in fatty and aromatic acid degradation in this and other microbes (16, 21). Using marker ions diagnostic of lysine acylation increases confidence in the PTMs identified (22). Acylations previously undocumented across the bacterial domain are identified. An understanding of how the modifications are regulated is currently lacking, although bacterial sirtuins are known to deacylate promiscuously (23, 24). To this end, we identified one sirtuin homolog (from the two present in the genome) that displays deacylase activity.
Experimental Procedures
Media, Cultivation, and Cell Harvest
Pure cultures of S. aciditrophicus strain SB (DSM 26646) were grown in 500-ml Schott bottles with 250 ml of a basal medium (25) with 20 mM crotonate or 10 mM crotonate plus 2 mM benzoate. S. aciditrophicus was grown in coculture with Methanospirillum hungatei JF1 (ATCC 27890) in 2-L Schott bottles with 1 L of Tanner’s mineral medium with 14 mM benzoate. Tanner’s mineral medium was used for large culture volumes to avoid chemical precipitants that form in large volumes of the basal medium. Tanner’s mineral medium (26) contained (per liter) 10 ml of Tanner’s minerals, 5 ml of Tanner’s metals, 10 ml of Tanner’s vitamins, 1 ml of 0.1% resazurin, 3.75 g of NaHCO3, and 20 ml of cysteine sulfide (2.5%) solution (27). Both media were adjusted with NaOH and HCl to pH 7.1 to 7.3. The anaerobic procedures of Balch and Wolfe (28) were used to prepare media and solutions and to inoculate and sample cultures. The headspace was pressurized with N2/CO2 (80%:20% v/v) to 70 kPa, and cultures were incubated at 37 °C without shaking.
Cultures were grown to mid-log phase and harvested by centrifugation (8000g for 20 min at 4 °C) under strict anaerobic conditions. The cell pellet was washed twice by resuspending in 50 mM anoxic potassium phosphate buffer (pH 7.5) and centrifuging as described previously. The final cell pellet was resuspended in anoxic potassium phosphate buffer, transferred to cryovials, and stored in −80 °C (29). Culture manipulations were performed in an anaerobic Coy chamber, and all centrifuge steps were done with sealed, anoxic, centrifuge tubes (28).
Two-Dimensional Polyacrylamide Gel Electrophoresis of S. aciditrophicus
Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) was performed, and protein-containing spots identified as described in the Supplemental Materials section of the study by James et al. (12).
Preparing Tryptic Peptides for Non–Gel-Based Proteomics
S. aciditrophicus SB coculture with M. hungatei JF1 on benzoate substrate was described previously (12, 29). Frozen cell pellets were resuspended in lysis buffer containing 4.0% (v/v) ammonium lauryl sulfate, 0.1% sodium deoxycholate (w/v), 5 mM tris(2-carboxyethyl)phosphine, and 100 mM ammonium bicarbonate (ABC). Proteins were digested via the enhanced filter-aided sample preparation method, similar to that described by Erde et al. (30, 31). Briefly, lysate was buffer-exchanged into 8 M urea, 0.1% (w/v) sodium deoxycholate, and 0.1% (w/v) n-octyl glucoside using a 10-kDa Microcon ultrafiltration unit (Millipore). Proteins were alkylated with 17 mM iodoacetamide for an hour at room temperature, after which they were exchanged into 0.1% (w/v) sodium deoxycholate/0.1% (w/v) n-octyl glucoside in 100 mM ABC. Trypsin (1:100 w/w) was added, and the solution was incubated overnight at 37 °C. Ethyl acetate extraction was used to remove detergents, as previously described (30, 31).
The resulting peptides were separated off-line via hydrophilic interaction liquid chromatography. Fifty micrograms of peptides (quantified with Pierce Quantitative Fluorometric Peptide Assay) were deposited onto a BioPureSPN MACRO PolyHYDROXYETHYL A column (The Nest Group) in 90% acetonitrile and 150 mM ammonium formate, pH 3. Peptides were eluted in six fractions using ammonium formate buffers sequentially decreasing in organic content: 80% acetonitrile, 78% acetonitrile, 74% acetonitrile, 71% acetonitrile, 40% acetonitrile, and 35.5% acetonitrile. Fractions one and six were combined. No antibody affinity fractionations were performed to enrich acylated peptides.
Liquid Chromatography and Tandem Mass Spectrometry
Peptides were mass-measured and fragmented using high-performance liquid chromatography tandem mass spectrometry. Using an EASY nLC1000 liquid chromatography system (Thermo Scientific), peptides (200 ng) were loaded onto an Acclaim PepMap100 C18 trap column (Thermo Scientific, Product #16-494-6, 75 μm × 2 cm, 100 Å) and separated on an Acclaim PepMap RSLC C18 analytical column (Thermo Scientific, Product #03-251-873, 75 μm × 25 cm, 100 Å). Buffer A (0.1% formic acid) and buffer B (0.1% formic acid in acetonitrile) were mixed and delivered at 300 nl min−1 in a gradient of 3 to 20% B for 62 min, 20 to 30% B for 31 min, 30 to 50% B for 5 min, and 50 to 80% B for 2 min.
A Q Exactive (Thermo Fisher Scientific) quadrupole-orbitrap mass spectrometer was operated using data-dependent acquisition mode. MS scans (m/z 300–1800) were acquired at 70,000 resolution, with an automatic gain control target set to 1E6 and a maximum fill time of 100 ms. The ten most abundant precursor ions were dissociated sequentially using higher energy collisional dissociation at a normalized collisional energy of 27 (unless otherwise indicated), and MS/MS spectra were acquired at 17,500 resolution with an automatic gain control target of 1E5 at a maximum fill time of 80 ms. The mass spectrometry data from 2D gels were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD025631. Shotgun datasets submitted through MassIVE are identified as PXD025603.
Database Searching
Q Exactive ∗.RAW files were analyzed using ProteomeDiscoverer (version 1.4) employing Matrix Science’s Mascot search algorithm (32). A database concatenating UniProt S. aciditrophicus and M. hungatei protein sequences (as of July 8, 2019) to the sequences of common contaminants was used for searches. The total number of entries in this database was 6479. Parameters used for the search were enzyme name, trypsin; maximum missed cleavage sites, two; precursor mass tolerance, 10 ppm; fragment mass tolerance, 0.02 Da; and variable methionine oxidation and cysteine carbamidomethylation. Searches also considered variable acyl-lysine modifications, all of which are shown in Table 1. These modifications were selected from RACS identified in the aromatic acid–degrading pathways of a variety of bacteria. Spectra matched to acylated peptides with Mascot ion scores ≥20 were subjected to manual examination, the criteria of which prioritized the presence of immonium ions and product ions spanning the modification as well as from regions that do not contain the modification. A score of 20 is associated with an expectation value of ≥0.1, chosen to broadly collect spectra of potentially modified peptides.
Table 1.
Acylations considered
| Acyl modification | Chemical formula | Monoisotopic mass shift | Average mass shift | Immonium ion | Cyclized immonium ion |
|---|---|---|---|---|---|
| Acetyl | C2H2O | 42.01056 | 42.03677 | 143.1179 | 126.0913 |
| Crotonyl | C4H4O | 68.02621 | 68.07413 | 169.1336 | 152.1070 |
| Acetoacetyl | C4H4O2 | 84.02113 | 84.07353 | 185.1285 | 168.1019 |
| Succinyl | C4H4O3 | 100.016 | 100.0729 | 201.1234 | 184.0968 |
| Butyryl | C4H6O | 70.04186 | 70.09001 | 171.1492 | 154.1226 |
| 3-Hydroxybutyryl | C4H6O2 | 86.03678 | 86.08942 | 187.1441 | 170.1176 |
| Glutaconyl | C5H4O3 | 112.016 | 112.0837 | 213.1234 | 196.0968 |
| Glutaryl | C5H6O3 | 114.0317 | 114.0996 | 215.1391 | 198.1125 |
| Benzoyl | C7H4O | 104.0262 | 104.1063 | 205.1336 | 188.1070 |
| Cyclohexa-1,5-diene-1-carboxyl | C7H6O | 106.0419 | 106.1222 | 207.1493 | 190.1227 |
| 6-Oxocyclohex-1-ene-1-carboxyl | C7H6O2 | 122.0368 | 122.1216 | 223.1442 | 206.1176 |
| Cyclohex-1-ene-1-carboxyl | C7H8O | 108.0575 | 108.1381 | 209.1649 | 192.1383 |
| 6-Hydroxycyclohex-1-ene-1-carboxyl | C7H8O2 | 124.0524 | 124.1375 | 225.1598 | 208.1332 |
| 2-Oxocyclohexane-carboxyl | C7H8O2 | 124.0524 | 124.1375 | 225.1598 | 208.1332 |
| 2-Heptenedioyl | C7H8O3 | 140.0473 | 140.1369 | 241.1547 | 224.1281 |
| 3-Oxopimelyl | C7H8O4 | 156.0423 | 156.1363 | 257.1497 | 240.1231 |
| Cyclohexane-1-carboxyl | C7H10O | 110.0732 | 110.154 | 211.1806 | 194.1540 |
| 2-Hydroxycyclohexane-carboxyl | C7H10O2 | 126.0681 | 126.1534 | 227.1755 | 210.1489 |
| Pimelyl | C7H10O3 | 142.063 | 142.1528 | 243.1704 | 226.1438 |
| 3-Hydroxypimelyl | C7H10O4 | 158.0579 | 158.1522 | 259.1653 | 242.1387 |
Bioinformatic Analyses
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to annotate relevant pathways associated with the acylated proteins identified. UniProt accessions were converted to KEGG identifiers and mapped to the KEGG pathway database using KEGG Mapper (33). To identify Gene Ontology (GO)/KEGG pathways frequently represented by acylated proteins, functional enrichment was performed by the StringApp (version 1.6.0) in Cytoscape (version 3.8.2), and p-values were corrected for multiple testing within each category using the Benjamini–Hochberg procedure (34).
To identify sequence motifs enriched in acylated peptides, the motif-x algorithm (35) integrated into the MoMo Modification Motifs program (version 5.3.0) on the MEME suite platform (36) was used to examine sequences of acyl-21-mers (ten amino acids upstream and downstream of the corresponding acylation site) (37) from the Uniprot S. aciditrophicus database. The other parameters were set to default values: p-value threshold: 0.000001, the minimum number of occurrences for residue/position pair: 10.
All acylated proteins identified were searched against the STRING database (version 11.0) to reveal potential protein–protein interactions (PPIs) (38). Only protein interactions found among the acylated proteins were selected, removing any external candidate interactions. Only interactions with at least medium confidence (>0.4) were included. The interaction network was visualized in Cytoscape (version 3.8.2).
Cloning, Expression, and Purification of Syn_00042 and Syn_01020
Two S. aciditrophicus sirtuin candidate genes annotated as sir2 family proteins were identified in the genome by homology search. The genes were obtained via polymerase chain reaction (PCR) of genomic DNA, cloned into plasmid pMAPLE21, and expressed in Escherichia coli strain BL21 as previously described by Arbing et al. (39). The recombinant proteins were purified by Ni-affinity chromatography followed by size exclusion (Superdex 75) and Q-Sepharose anion exchange. Purity was greater than 95% as visualized by SDS-PAGE. The apparent protein sizes were consistent with the predicted gene/protein sizes.
Sirtuin Assay (anhydride)
Acyl-insulins were prepared from the corresponding acyl-anhydrides; for example, glutaric anhydride was used for glutaryl-lysine, using methods adapted from Baeza et al. (40). About 25 μmol of anhydride was added to 100 μl of a 1-mg/ml solution of human insulin (Alfa Aesar, J67626) in 100 mM ABC. After incubating at 4 °C for 20 min, the solution was readjusted to pH ∼8 using an ammonium hydroxide solution. Anhydride addition, incubation, and pH adjustment were repeated twice more. Ostensible O-acylation was reversed by adding 50% w/v of hydroxylamine hydrochloride in H2O (adjusted with ammonium hydroxide to pH 7–8). Following overnight, room-temperature incubation, the modified products were buffer-exchanged into 100 mM ABC using 3-kD molecular weight cut-off Amicon spin filters (Millipore).
For the matrix-assisted laser desorption/ionization (MALDI)–based activity assay, an approximately 27 μM solution of acyl-modified insulin was mixed with 0.24 μM recombinantly expressed SYN_00042 and excess oxidized nicotinamide adenine dinucleotide (NAD+) in 100 mM ABC. The solution was incubated at 37 °C for 2 h. Sample aliquots were mixed 1:1 with saturated sinapinic acid in 50% acetonitrile/0.1% trifluoroacetic acid and spotted onto a MALDI sample stage. MALDI mass spectra were obtained with an Applied BioSystems Voyager-DE STR time-of-flight mass spectrometer.
Time-series measurements of glutaryl-insulin masses used a Synapt G2-Si quadrupole time-of-flight mass spectrometer (Waters) with electrospray ionization. The assay was performed at 37 °C for the period of time indicated and quenched with 50% acetonitrile. The glutaryl-insulin was diluted to a final concentration of 10 μM for mass analysis with 20 μM ubiquitin added as internal standard. The solution was delivered by direct infusion.
Experimental Design and Statistical Rationale
S. aciditrophicus cells were grown in pure culture on crotonate as a sole carbon source or with crotonate/benzoate to analyze via 2D-PAGE. Cells were cocultured with M. hungatei and grown on benzoate for mass spectrometry proteomic analysis. Three samples from each culture condition were grown, and for each biological sample, three technical replicates were analyzed. As the priority was to maximize the identification of potentially low stoichiometry PTMs, offline hydrophilic interaction liquid chromatography was used to increase the depth of the identifiable proteome. Resulting data were searched for the acyl modifications shown in Table 1. To identify PTMs, mass spectra matched to acylated peptides with Mascot ion scores ≥20 were subjected to manual examination. Modifications were considered present if the tandem mass spectra included acyl-lysine–associated immonium ions (22).
Results
2D-Gel Analyses Suggest Proteins Are Modified Heterogeneously
S. aciditrophicus cell lysates from axenic cultivation on benzoate supplemented with crotonate (9) were harvested and subjected to 2D-PAGE (Fig. 1) (41, 42). Spots were excised, digested with trypsin in-gel, and identified using tandem mass spectrometry (12). Frequently, spots were composed of multiple proteins, limiting the value of densitometry for evaluating abundance. In some spots, MS identified seven or more proteins with two or more tryptic peptides (or a single tryptic peptide identified from an MS/MS spectrum matched to that in a different spot, known to contain that protein, displaying 2+ tryptic peptides) (supplemental Table S1). Abundant proteins are listed in Table 2. Within the darkest spots, some MS heterogeneity suggestive of small variations in sequence was also observed, potentially reflecting high error rates in transcription and/or translation. Little is known about this latter effect.
Fig. 1.

Proteomic patterns of S. aciditrophicus as detected by 2D-PAGE. Two-dimensional gel analysis of the S. aciditrophicus proteome for cells grown on benzoate supplemented with crotonate. The x-axis is separated by isoelectric focusing (IEF), whereas the y-axis is separated by SDS-PAGE. Selected proteins present in the spots are listed in Table 2. 2D-PAGE, two-dimensional polyacrylamide gel electrophoresis.
Table 2.
Proteins identified in two-dimensional gel spots
| Spot number | Locus tag | Protein function |
|---|---|---|
| 1 | SYN_03116 | Hypothetical exported protein |
| 2 | SYN_00544 | ATP synthase beta chain |
| 3 | SYN_02966 | Phosphoenolpyruvate synthase |
| 4 | SYN_01983 | Chaperone protein |
| 5 | SYN_03223 | 60-kDa chaperonin |
| 6 | SYN_00198 | Porin |
| 7 | SYN_01709 | Ketol-acid reductoisomerase |
| 8 | SYN_00480 | Acyl-CoA dehydrogenase |
| 9 | SYN_01909 | 60-kDa chaperonin 3 (GroEL3) |
| 10 | SYN_00546 | ATP synthase subunit alpha 1 |
| 11 | SYN_00983 | Elongation factor Tu |
| 12 | SYN_02896 | Benzoate-CoA ligase (Bcl2) |
| 13 | SYN_01681 | Acetyl-CoA acetyltransferase |
| 14 | SYN_02586 | Cyclohexane-1-carbonyl-CoA dehydrogenase |
| 15 | SYN_01653 | Enoyl-CoA hydratase |
| 16 | SYN_02635 | Acetyl-CoA synthetase (Acs1) |
| 17 | SYN_03128 | Cyclohexane-1-carboxylate-CoA ligase (12) |
| 18 | SYN_01654 | 6-Oxocyclohex-1-ene-1-carbonyl-CoA hydrolase |
| 19a | SYN_02898, SYN_02896 | 4-Hydroxybenzoate-CoA ligase/benzoate-CoA ligase (Bcl2) |
| 20a | SYN_01681 | Acetyl-CoA acetyltransferase |
| 21a | SYN_02642 | Acetyl-CoA acetyltransferase |
| 22a | SYN_01310 | 3-Hydroxyacyl-CoA dehydrogenase |
| 23a | SYN_01780, SYN_02636 | Polyribonucleotide nucleotidyltransferase, electron transfer flavoprotein beta-subunit |
Gel spots numbered in Figure 1 were excised, and their identities determined by mass spectrometry on a QStar-XL quadrupole time-of-flight mass spectrometer.
Denotes proteins identified that were acetylated.
These analyses revealed many instances in which the same protein was identified from multiple spots at the same molecular size but different isoelectric points. This pattern, often seen with glycoproteins (43), suggests heterogeneous PTMs, but we found no evidence of S. aciditrophicus glycoproteins. Spot trains might strike some as reminiscent of carbamylation, but the artifacts were easily ruled out. Trains do not accompany every spot, and MS/MS analyses found protein carbamylation to be insignificant. Some of these proteins were identified as housekeeping proteins (Table 2), while others have roles in the catabolism of benzoate and other aromatic and fatty acids. Given the high abundance of RACS in these pathways, acyl-lysine modifications were considered possible. Acylation will neutralize the positive charge on lysine, or, in the cases of acidic RACS, such as glutaryl-CoA, will switch lysine to a negatively charged site. Both types of acylation reduce protein isoelectric points, shifting migration toward the anode (the left in standard 2D gel images); hence, a distribution of many and differently charged acylations would give a pattern similar to that seen in Figure 1.
Digests for spot 20 (Fig. 1) established that the acetylated and nonacetylated peptides YGTKacPEDLALIR and YGTKPEDLALIR of the acetyl-CoA acetyltransferase SYN01681 were both present. That both acetylated and nonacetylated lysines were present could suggest that migration-compensating acylations or modifications reside at other sites on some of the SYN01681 protein in this spot. From spot 21, peptides TAVGAFGGSLKacGVR and MoxAKacLAPVFK of the acetyl-CoA acetyltransferase SYN02642 were observed bearing acetyl lysines. Nonacylated TAVGAFGGSLK and LAPVFK were also observed in the spot, but at lower signal levels. Two acetylated sites were also observed from SYN01310 in spot 22, from peptides ATEEFSKacQLGK and LMoxAGSIKacK of 3-hydroxyacyl-CoA dehydrogenase. A corresponding unmodified peptide, GYYTSDETFKATEEFSK, was also observed in this spot, although neither unacetylated LMAGSIK nor LMoxAGSIK was found. From the spot 23 proteins SYN_01788 and SYN_02636, acetylated peptides TAAALEALEKacR and YASLPGIMKacAK were detected, respectively. Unacetylated TAAALEALEK was also detected from the digested spot, but not the unacetylated SYN_02636 peptide. Most interestingly, peptide TATGKacIQR, found in spot 19, could be attributed to SYN02896 and/or SYN02898. These benzoate-CoA ligases are 77% identical and are both present in this spot, as ascertained from their unique peptides. Despite strong signals from the acylated peptide, no unacetylated peptides spanning the TATGK segment were recovered. Supplemental Spectra S1 present annotated tandem mass spectra from spots 20 to 24.
Shotgun Proteomics Identifies Many Acyl Modifications in the S. aciditrophicus Proteome
Three biological replicates of protein lysates from S. aciditrophicus/M. hungatei cocultures utilizing benzoate as the carbon source were analyzed in three technical replicates each by liquid chromatography tandem mass spectrometry data-dependent acquisition (supplemental Tables S2 and S3). Analyses for one biological replicate incorporated stepped collision energies to increase the diagnostic immonium ion abundances from acyl-lysines (22). Speculating that peptides acylated by reactive intermediates from benzoate degradation might be observable, we sought evidence by including acylation mass shifts in database searches. Table 1 illustrates the acylation shifts that were considered known intermediates for S. aciditrophicus, for related organisms, and common acylations. A total of 125 sites were identified in 60 different proteins, and each site was modified by at least one of the acyl groups (supplemental Tables S4 and S5; supplemental Spectra S2). Acylated peptides comprise 3.2% of the unique peptides identified (supplemental Table S3). The acetylated and 3-hydroxypimelylated species predominated in both numbers of proteins and numbers of modified sites. All of the PTMs found in each biological replicate are listed in Table 3, along with information about the supporting immonium ions. Acylations corresponding to seven of the 20 searched intermediates were found (Table 3). Modifications were detected in biological replicates reproducibly (supplemental Fig. S1 and supplemental Table S4). Peptides with missed cleavages enabled elution time comparisons between modified peptides and their unmodified counterparts. Twenty-eight of these peptide pairs were identified; in 26 pairs, the acylated peptide eluted later by an average of 9.37 min (supplemental Table S6).
Table 3.
Summary of acyl-lysine modifications identified in S. aciditrophicus
| Modification | Proteins identified | Sites identified | Cyclized immonium ion m/z | Peptides with cyclized immonium ion (%) |
|---|---|---|---|---|
| Benzoylation | 1 | 2 | 188.107 | 100% |
| 3-Hydroxypimelylation | 16 | 20 | 224.129, 242.139 | 75%, 55% |
| Glutarylation | 4 | 6 | 198.112 | 100% |
| Crotonylation | 2 | 2 | 152.107 | 100% |
| 3-Hydroxybutyrylation | 4 | 6 | 170.118 | 83% |
| Acetylation | 48 | 104 | 126.091 | 94% |
| Butyrylation | 3 | 3 | 154.123 | 33% |
For each type of modification, the number of unique proteins bearing the modification and the total number of sites observed to be modified are presented. For the modified sequences, the percentages of MS/MS spectra that contained the diagnostic ions are stated.
PTMs can be misidentified in complex proteomic datasets due to chimeric precursors, isobaric peptides, spectral complexity, and insufficient sequence-related ions (18). The large search space traversed in seeking multiple, variable modifications from shotgun datasets challenges confident assignments. To meet these challenges, we relied upon the presence of immonium ions specific for each putative acyl modification. Immonium and immonium-like ions are strong indicators of acyl-lysine (22). Proportions of putative acyl-modified spectra displaying diagnostic immonium ions are presented in Table 3. We also examined sequences upstream and downstream of each acylated residue to determine if there were sequence preferences for these modifications, and indeed, we found that nearby glycines favored lysine acetylation (supplemental Fig. S3).
Pathway Analysis of Modified Proteins
A functional annotation analysis classified proteins by KEGG pathways and GO. The GO functional classification grouped acylated proteins into three categories: biological process, molecular function, and cellular component. Most acylated proteins mapped onto KEGG pathways had roles in metabolic processes, as expected. Benzoate degradation, oxidative phosphorylation, and carbon metabolism were highly enriched (Fig. 2A). GO enrichment analyses of all acylated proteins revealed biological processes primarily related to the synthesis of metabolites, as well as energy production (Fig. 2B). Processes related to nucleotide metabolism were also highly enriched, in line with recent reports that nucleotide-binding regions increase acylation of proximate lysines (44). Altogether, the findings suggest that these acylated proteins are generally involved in carbon metabolism, degrading aromatic and fatty acids.
Fig. 2.

Functional enrichment of acylated proteins.A, Gene Ontology biological processes that are enriched. B, enriched KEGG pathway processes. C, a functional protein association network generated using the STRING database. Proteins involved in oxidative phosphorylation are colored in blue. Proteins involved in degrading benzoate are red. KEGG, Kyoto Encyclopedia of Genes and Genomes.
Acyl-lysine modifications have been shown to impact PPIs and affect the formation of enzyme complexes, altering their role in cellular physiology. To determine connections between the acylated proteins and to elucidate their functional PPI networks, we generated an interaction network based on the STRING database, visualized through Cytoscape (Fig. 2C). Within the network, there are two highly interconnected clusters, benzoate degradation and oxidative phosphorylation (Fig. 2C). These strong physiological interactions among all modified proteins led us to further investigate the role of protein acylation in the degradation of aromatic compounds.
Benzoate Degradation Enzymes Are Heavily Acylated
Proteins across many pathways are modified by different acyl groups at varying levels, summarized by the heat map in Figure 3A. Acetylation is found in the widest range of functional pathways, with the highest number of acetyl-lysines found in proteins related to the biosynthesis of secondary metabolites. Other acyl modifications are enriched in benzoate degradation pathway proteins, corresponding to the reactive acyl-CoA intermediates from the pathway (16). These findings imply that high local concentrations of intermediates may modify proteins spontaneously.
Fig. 3.

Mapping sites of protein lysine acylation based on COG functional pathway analysis.A, a heatmap indicating the pathways (KEGG Ontology) involved for the acyl-modified protein identified. The color indicates the number of proteins in each category that have the specified acylation. B, modified sites identified in the “upper” and “lower” benzoate pathway. Bar plots illustrate the range of modifications found on associated enzymes. One square indicates one site of modification, and each color represents a different acylation. RACS modification types are indicated in the upper right corner boxed color code. COG, clusters of orthologous groups;KEGG, Kyoto Encyclopedia of Genes and Genomes; RACS, reactive acyl-coenzyme A species.
The largest number and widest variety of acyl modifications were found in benzoate degradation (Fig. 3B). Ten different proteins in the pathway were observed bearing several different acyl modifications. Acylated proteins included benzoate-CoA ligase, the initial step in the pathway. Several of the other acylated proteins are members of gene clusters involved in benzoic acid metabolism (bam genes) that are found in numerous anaerobic delta-proteobacteria (45, 46). The gene products of bamR (SYN_01653), bamQ (SYN_01655), and bamA (SYN_01654), which successively reduce cyclohexa-1,5-diene-1-carboxyl-CoA to 3-hydroxypimelyl-CoA, display modifications. All three gene products are acetylated, those of bamQ and bamA are glutarylated and 3-hydroxypimelylated, and that from bamA is also 3-hydroxybutyrylated and benzoylated. Interestingly, all five modifications for the bamA product occur at the same site, lysine 261 (supplemental Fig. S3 and supplemental Spectra S2). Peptides containing these lysine residues were also observed unmodified.
S. aciditrophicus uses acetyl-CoA synthetase (acs1, SYN_02635) to make adenosine triphosphate (ATP) from acetyl-CoA. Other bacteria, in contrast, use phosphate acetyltransferase and acetate kinase, enzymes that are absent in the S. aciditrophicus genome. Using acs1 to produce ATP is also remarkable because acetyl-CoA synthetases were previously thought to function only in activating acetate to acetyl-CoA, not in the reverse direction (ATP-forming direction) (29). The rates at which ATP is produced when acetyl-CoA is the limiting substrate differ between purified and recombinantly expressed Asc1 (Vmax of 7.5 and 1.2, respectively and Km of 0.41 and 1.34 mM, respectively) (29), suggesting that there are factors associated with the in vivo protein that are not recapitulated when the enzyme is recombinantly expressed. PTMs may be one such factor. Indeed, six different sites on this protein were seen in acylated forms, all of which included acetylation, except for one site that was only butyrylated.
Proteins involved in energy conservation were also heavily acetylated. ATP synthase, Atp1, is a large complex that contains many components. Acetylation sites were identified from several subunits of Atp1, namely on the alpha (SYN_00546), beta (SYN_00544), and gamma (SYN_00545) chains (supplemental Table S5). Two homologs of ATP synthase B (the beta subunit) were also acetylated (SYN_00548 and SYN_00549).
Benzoate-CoA Ligase Reveals a Highly Modified Residue in a Conserved Region
Five of the seven acyl modifications identified in S. aciditrophicus proteins appeared in the sequence TATGKIQR, present in paralogs Bcl1 (SYN_02898) and Bcl2 (SYN_02896), which differ in their activity toward different fatty and aromatic acids. The modified lysine is critical to benzoate-CoA ligase (BCL) function (47, 48), catalyzing the first step of benzoate degradation. Immonium ions present in the spectra shown in Figure 4A confirm that the unique mass shifts do, indeed, result from acylation and are not induced by misidentifications that may result from additive side reactions that occur during sample processing; for example, formylation from exposure to high concentrations of formic acid or 12-Da mass shifts from exposure to formaldehyde (49, 50). This modification site was further investigated to determine what functional role, if any, the PTMs play. Basic Local Alignment Search Tool (BLAST) sequence alignments against closely related BCLs (47) indicated that the lysine falls in a highly conserved region (Fig. 4B). Previous Burkholderia xenovorans structural studies of benzoate-bound BCL (Bxe_A1419), a homolog with 46% sequence identity to Bcl1 and 44% to Bcl2, indicated that this conserved lysine is located near the benzoate carboxyl (Fig. 4C) and is hypothesized to coordinate to benzoate during enzymatic activity (47). Acylation both neutralizes the ε-amino side chain and adds a bulky group disrupting coordination to the carboxylate. The Rhodopseudomonas palustris BCL (BadA), with 43% sequence identity to Bcl1 and 42% to Bcl2, has also been found acetylated at that residue (K512). Studies in vitro established that acetylation abolishes enzymatic activity (48), suggesting possible feedback inhibition of the enzyme and, in turn, the pathway.
Fig. 4.

Modified benzoate-CoA ligase (BCL).A, HCD spectra of acylated TATGKIQR from SYN_02896 and SYN_02898. The benzoylated spectrum (top) was collected with a stepped NCE method of 27 V/40 V. Other spectra were collected at 27 NCE. Immonium ions of modifications are denoted in red. B, sequence alignment of known BCLs across microbial systems. The line above the sequence (TATGKIQR) denotes the peptide identified in (A). The asterisk (∗) denotes the corresponding modified lysine residue identified in S. aciditrophicus. C, crystal structure of a closely related BCL from Burkholderia xenovorans, determined by Bains and Boulanger (47), indicates that the acyl-lysine is proximal to a benzoate bound to the active site. BCL, benzoate-CoA ligase; HCD, higher energy collisional dissociation; NCE, normalized collision energy.
S. aciditrophicus Sirtuins Deacylate Promiscuously
Given the ubiquity of acyl modifications in S. aciditrophicus, an ability to control and/or regulate acyl modifications would seem to be advantageous and critical. Sirtuin proteins have been shown to possess generalized deacylase activity, often promiscuous with respect to both sequence context and acyl modification (23, 51, 52). Bacterial systems that contain sirtuin homologs are typically denoted as cobB or as SIR2 family (24, 53). To determine if a deacylase capability is present, the S. aciditrophicus genome was searched with BLAST to determine if E. coli cobB homologs were present and SYN_00042 and SYN_01020 were found. A sequence alignment between these proteins displays their similarities (supplemental Fig. S4). SYN_00042 and SYN_01020 were recombinantly expressed to assay activity by mass spectrometry (Experimental Procedures).
Insulin, a dual-chain protein with one lysine and two free NH2-termini, was synthetically acylated using acyl anhydrides. Insulin was selected as it only has a single lysine residue and has been well characterized as a standard protein used in intact mass spectrometry work. Acylated insulin was incubated with recombinantly produced SYN_00042 and its cofactor, NAD+. MALDI mass spectra of glutarylated insulin showed a 114-Da decrease in mass after incubation with SYN_00042 (Fig. 5A and supplemental Table S7). The recombinant protein displayed deacylase activity against butyryl and succinyl acylations, as well (supplemental Fig. S5 and supplemental Table S7, A–G), but did not appear to act on shorter acetyl or propionyl chains. A quantitative assay of SYN_00042 gene product deacylase activity on glutaryl-insulin, performed by electrospray ionization MS, clearly shows time-dependent deglutarylase activity (Fig. 5B and supplemental Table S7, H–M). SYN_01020 was expressed in an attempt to assess its function, but did not display reproducible activity.
Fig. 5.

S. aciditrophicus sirtuin SYN_00042 shows in vitro deacylase activity.A, MALDI-MS of insulin and modified insulin deacylated by sirtuin (Syn_00042). The peak corresponding to “Glutarylated Nε - Lysine” reflects the mass shift from three glutaryl modifications (+342 Da), two at the N-termini and one at the single lysine. B, ESI-MS of the 4+ charge states 0, 2, 15 and 60 min after sirtuin addition. Purple peak clusters (left) correspond to insulin modified at the two N-termini; red clusters identify insulin modified at all three sites. Ammonium adducts (+17 Da) are also present. ESI, electrospray ionization; MALDI, matrix-assisted laser desorption/ionization; MS, mass spectrometry.
Discussion
PTMs can modulate protein function in response to cellular or environmental changes and may occur spontaneously due to cellular conditions or simply over time (17, 54, 55, 56). Acylation is one class of modification that can be spontaneously induced in the presence of acyl phosphate or RACS or can be enzyme-mediated by a lysine acyltransferase (KAT) (57). Regardless of its origin, acylation can affect enzymatic activity profoundly. In vitro work has shown that modifying lysine side chains near catalytic regions of bacterial enzymes can directly alter function (58, 59, 60). Work in other systems has shown more subtle but equally significant acylation effects: acetylation can disrupt enzyme complexes, thereby altering activity (61, 62, 63, 64). Acetyl-lysine has been identified as a ubiquitous modification in bacteria (65). Most acetylated proteins in our system were involved in the synthesis of secondary metabolites (Fig. 3A), which is sensible given that acetyl-CoA is an intermediate metabolite in many of those pathways. Benzoyl- (51), glutaryl- (66), hydroxybutyryl- (67, 68), crotonyl- (69, 70), and butyryl-lysine (71, 72) have shown function in some systems under certain conditions, although the studies have been less extensive than those for acetyl-lysine, and few, if any, bacterial studies have reported their occurrence. A 3-hydroxypimelylation has previously been reported in S. aciditrophicus by our laboratory (22). Detecting these modifications across biological replicates without pre-enrichment suggests that they are uniquely prevalent and abundant in this system. While butyryl-CoA is not expected to be an intermediate in the degradation pathway, S. aciditrophicus has genes for butyrate dehydrogenase activity (3, 8) that could synthesize butyryl-CoA from excess crotonyl-CoA or reduced cofactors (8, 12).
To begin to understand how these modifications affect global cellular function requires a comprehensive and unbiased catalog of acylations. Recording a biological system’s acyl modifications comprehensively presents many challenges. Low stoichiometries of acyl modifications in many model systems have required enrichment to identify and characterize acylated peptides (66, 73, 74, 75, 76, 77, 78, 79, 80, 81). Pan-specific antibodies are valuable for enriching specifically modified peptides but have some limitations. Antibody cross-reactivity may lessen the modification specificity of enriched peptides, a problem for Western blotting, but not mass spectrometry. Sequence-dependent binding efficiencies can bias results and challenge quantification strategies. Our syntrophic system provides a unique opportunity to investigate the array of acylations without enrichment, bypassing technical challenges in other systems. The experimental approach described herein using RACS as a predictor for lysine acylations complements antibody enrichment and utilizes diagnostic marker ions to validate putative modifications proposed from an enlarged search space.
Many of the S. aciditrophicus proteins decorated with acyl modifications function to degrade benzoate, a process that could result in carbon or reductive stress, if unregulated. Modulating enzymatic activity through protein acylation would be a fitting and elegant mechanism for global metabolic regulation, given the limited energy available to these cells and the high concentrations of acyl-CoA intermediates, a feature also evidenced by the unusual ability of this species to use acetyl-CoA to form ATP (29). The benzoate degradation pathway is a likely candidate for this level of regulation as its constituents frequently interact with RACS. While we have identified a wide range of acyl modifications in this system, the biological role that these modifications play has yet to be explored. We can, however, provide some insight into the functional effect of certain acylation sites by drawing on similar modifications in other systems. BCL acylation, specifically acetylation, has been identified as a means of negative feedback inhibition, stopping benzoate-CoA ligase activity in R. palustris by directly inhibiting enzymatic catalysis (48). The large number of acyl modifications we report at this site may indicate that when its respective acyl-intermediate builds, the PTM can act as a brake to slow or stop degradation, thereby acting as a negative feedback inhibitor to mitigate carbon and reductive stress. BCL also consumes ATP, creating a direct link between the energetic state of the cell and aromatic degradation. Additionally, the multiple sites of acetylation found on ATP synthase subunits further hint at a link between bioenergetics and acylation as it, too, demonstrates a relationship between the reactive metabolites and oxidative phosphorylation.
Beyond energetics, the cellular reductive state may also be regulated by the acylation of enzymes. Benzoate degradation generates nicotinamide adenine dinucleotide reduced and flavin adenine dinucleotide reduced, but excess reducing capacity stresses cells. A buildup of intermediates that subsequently modifies and slows the rates of enzymatic catalysis would safeguard against damaging reductive stress (15, 82, 83). This link is further enforced by the presence of sirtuins, whose activity is non–energy-conserving, and relies upon the oxidized cofactor NAD+ for activity (84), innately linking the extent of protein acylation to cellular redox state. Interestingly, the sirtuin assayed in this system appears to have a bias for longer chain acylations, particularly glutarylation, a trait that is shared with Sirt4 and Sirt5 in mammalian systems (66, 85, 86). Although S. aciditrophicus sirtuins were identified by homology to CobB, their activities deviate notably from those of the E. coli enzyme, as illustrated by their action on only longer acyl groups, not acetyl. Previous studies indicated that sirtuin specificity across species including E. coli CobB, mammalian Sirt5, and Archaeoglobus fulgidus Sir2Af2 demonstrates that while also having deacetylase activity, their selectivity toward long-chain deacylation is dependent on a YxxR motif present in the binding pocket (87, 88). However, in the S. aciditrophicus sirtuins, no such motif exists (supplemental Fig. S4), suggesting a different substrate selection mechanism.
The PTMs identified derive from acyl-CoA intermediates at steps which require the loss of 2 [H] (release of two reducing equivalents) (16). Hydrogen buildup is a limiting factor in the syntrophic metabolism, as is evidenced by the need for a hydrogen-scavenging partner (4). It is therefore expected that some metabolites would accumulate under high hydrogen concentrations, providing another link between the metabolic conditions of the cell and the observed acyl modifications. Interestingly, the lower benzoate pathway degrading 3-hydroxypimelyl-CoA (Fig. 2C) is conserved across many anaerobes that degrade benzoate and other aromatic compounds (89, 90). The acyl modifications we identified on these proteins suggest that the modifications are likely present in other bacteria sharing the same or similar metabolic pathways, especially under cellular conditions that induce an accumulation of intermediates. S. aciditrophicus has demonstrated a wide array of acyl modifications within its proteome. Further investigation into the function of these acylations presents an opportunity to understand how these essential environmental microbes regulate their metabolism. Other syntrophs also contain critical pathways with RACS intermediates (91, 92, 93), and a wider investigation of other syntrophs may further reveal the role RACS play in bacterial metabolism.
Data Availability
All data were uploaded to ProteomeXchange (proteomexchange.org) either through PRIDE (2D-gel dataset; PXD025631) or through MassIVE (Shotgun dataset; PXD025603).
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We are grateful for the 2D-PAGE and mass spectrometry analyses performed by Dr Yanan Yang. We would like to thank Dr Mark Arbing and Annie Shin of the UCLA-DOE Institute Protein Expression Technology Center (supported by grant 447147-EL-21341 from the US Department of Energy) for expression and purification of Syntrophus aciditrophicus SYN_00042 and SYN_01020.
Author contributions
M. J. M., R. P. G., J. A. L., and R. R. O. L. conceptualization; J. M. M., H. H. N., M. J. M., R. P. G., J. A. L., and R. R. O. L. methodology; J. M. M., J. Y. F., H. H. N., N. Q. W., H. M., K. L. J., and R. R. O. L. investigation; J. M. M., J. Y. F., H. H. N., and R. R. O. L. data curation; J. M. M., J. Y. F., N. Q. W., H. M., K. L. J., M. J. M., R. P. G., and R. R. O. L. formal analysis; J. M. M., J. Y. F., M. J. M., R. P. G., J. A. L., and R. R. O. L. writing–original draft; J. M. M., J. Y. F., N. Q. W., H. M., K. L. J., M. J. M., R. P. G., J. A. L., and R. R. O. L. writing–review and editing.
Funding and additional information
Funding from the Department of Energy Office of Science (BER) contract DE-FC-02-02ER63421 (to J. A. L. and R. P. G; UCLA/DOE Institute for Genomics and Proteomics), NIH Ruth L. Kirschstein National Research Service 18 Award (to J. Y. F.; GM007185), NSF Award 1911781 to R. P. G and M. J. M, and NSF Graduate Research Fellowship (to J. Y. F.; DGE-1650604) are acknowledged. Also acknowledged are the National Institute of Health awards GM104610 and GM085402 to R. O. L. and J. A. L. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Present address for Kimberly James: Department of Microbiology and Cell Science, University of Florida, Florida 32611, USA.
Present address for Housna Mouttaki: Novartis, Sandoz GmbH, Biochemiestrasse 10, Kundl 6250, Austria.
Supplemental Data
References
- 1.Stams A.J.M., Sousa D.Z., Kleerebezem R., Plugge C.M. Role of syntrophic microbial communities in high-rate methanogenic bioreactors. Water Sci. Technol. 2012;66:352–362. doi: 10.2166/wst.2012.192. [DOI] [PubMed] [Google Scholar]
- 2.McInerney M.J., Sieber J.R., Gunsalus R.P. Syntrophy in anaerobic global carbon cycles. Curr. Opin. Biotechnol. 2009;20:623–632. doi: 10.1016/j.copbio.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McInerney M.J., Rohlin L., Mouttaki H., Kim U., Krupp R.S., Rios-Hernandez L., Sieber J., Struchtemeyer C.G., Bhattacharyya A., Campbell J.W., Gunsalus R.P. The genome of Syntrophus aciditrophicus: Life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7600–7605. doi: 10.1073/pnas.0610456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schink B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997;61:262–280. doi: 10.1128/mmbr.61.2.262-280.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stams A.J.M., Plugge C.M. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 2009;7:568–577. doi: 10.1038/nrmicro2166. [DOI] [PubMed] [Google Scholar]
- 6.Jackson B.E., McInerney M.J. Anaerobic microbial metabolism can proceed close to thermodynamic limits. Nature. 2002;415:454–456. doi: 10.1038/415454a. [DOI] [PubMed] [Google Scholar]
- 7.Walker D.J.F., Nevin K.P., Holmes D.E., Rotaru A.E., Ward J.E., Woodard T.L., Zhu J., Ueki T., Nonnenmann S.S., McInerney M.J., Lovley D.R. Syntrophus conductive pili demonstrate that common hydrogen-donating syntrophs can have a direct electron transfer option. ISME J. 2020;14:837–846. doi: 10.1038/s41396-019-0575-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jackson B.E., Bhupathiraju V.K., Tanner R.S., Woese C.R., McInerney M.J. Syntrophus aciditrophicus sp. nov., a new anaerobic bacterium that degrades fatty acids and benzoate in syntrophic association with hydrogen-using microorganisms. Arch. Microbiol. 1999;171:107–114. doi: 10.1007/s002030050685. [DOI] [PubMed] [Google Scholar]
- 9.Elshahed M.S., McInerney M.J. Benzoate fermentation by the anaerobic bacterium Syntrophus aciditrophicus in the absence of hydrogen-using microorganisms. Appl. Environ. Microbiol. 2001;67:5520–5525. doi: 10.1128/AEM.67.12.5520-5525.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouttaki H., Nanny M.A., McInerney M.J. Cyclohexane carboxylate and benzoate formation from crotonate in Syntrophus aciditrophicus. Appl. Environ. Microbiol. 2007;73:930–938. doi: 10.1128/AEM.02227-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mouttaki H., Nanny M.A., McInerney M.J. Use of benzoate as an electron acceptor by Syntrophus aciditrophicus grown in pure culture with crotonate. Environ. Microbiol. 2008;10:3265–3274. doi: 10.1111/j.1462-2920.2008.01716.x. [DOI] [PubMed] [Google Scholar]
- 12.James K.L., Kung J.W., Crable B.R., Mouttaki H., Sieber J.R., Nguyen H.H., Yang Y., Xie Y., Erde J., Wofford N.Q., Karr E.A., Loo J.A., Ogorzalek Loo R.R., Gunsalus R.P., McInerney M.J. Syntrophus aciditrophicus uses the same enzymes in a reversible manner to degrade and synthesize aromatic and alicyclic acids. Environ. Microbiol. 2019;21:1833–1846. doi: 10.1111/1462-2920.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters F., Shinoda Y., McInerney M.J., Boll M. Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: Evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J. Bacteriol. 2007;189:1055–1060. doi: 10.1128/JB.01467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuntze K., Shinoda Y., Moutakki H., McInerney M.J., Vogt C., Richnow H.H., Boll M. 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: Characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol. 2008;10:1547–1556. doi: 10.1111/j.1462-2920.2008.01570.x. [DOI] [PubMed] [Google Scholar]
- 15.Mavi P.S., Singh S., Kumar A. Reductive stress: New insights in physiology and drug tolerance of Mycobacterium. Antioxid. Redox Signal. 2020;32:1348–1366. doi: 10.1089/ars.2019.7867. [DOI] [PubMed] [Google Scholar]
- 16.Elshahed M.S., Bhupathiraju V.K., Wofford N.Q., Nanny M.A., McInerney M.J. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by Syntrophus aciditrophicus strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 2001;67:1728–1738. doi: 10.1128/AEM.67.4.1728-1738.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trub A.G., Hirschey M.D. Reactive acyl-CoA species modify proteins and induce carbon stress. Trends Biochem. Sci. 2018;43:369–379. doi: 10.1016/j.tibs.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim M.S., Zhong J., Pandey A. Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics. 2016;16:700–714. doi: 10.1002/pmic.201500355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S., Tan M., Dai L., Kwon O.K., Yang J.S., Zhao Y., Chen Y. MS/MS of synthetic peptide is not sufficient to confirm new types of protein modifications. J. Proteome Res. 2013;12:1007–1013. doi: 10.1021/pr300667e. [DOI] [PubMed] [Google Scholar]
- 20.Fu Y. Bayesian false discovery rates for post-translational modification proteomics. Stat. Interf. 2012;5:47–59. [Google Scholar]
- 21.Harwood C.S., Burchhardt G., Herrmann H., Fuchs G. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol. Rev. 1998;22:439–458. [Google Scholar]
- 22.Muroski J.M., Fu J.Y., Nguyen H.H., Ogorzalek Loo R.R., Loo J.A. Leveraging immonium ions for targeting acyl-lysine modifications in proteomic datasets. Proteomics. 2021;21:2000111. doi: 10.1002/pmic.202000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bheda P., Jing H., Wolberger C., Lin H. The substrate specificity of sirtuins. Annu. Rev. Biochem. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. [DOI] [PubMed] [Google Scholar]
- 24.Zhao K., Chai X., Marmorstein R. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J. Mol. Biol. 2004;337:731–741. doi: 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 25.Sieber J.R., Le H.M., Mcinerney M.J. The importance of hydrogen and formate transfer for syntrophic fatty, aromatic and alicyclic metabolism. Environ. Microbiol. 2014;16:177–188. doi: 10.1111/1462-2920.12269. [DOI] [PubMed] [Google Scholar]
- 26.Tanner R.S. In: Manual of Environmental Microbiology. 3rd Ed. Hurst C., Crawford R., Garland J., Lipson D., Mills A., Stetzenbach L., editors. American Society of Microbiology; Washington, DC: 2007. Cultivation of bacteria and fungi; pp. 69–78. [Google Scholar]
- 27.McInerney M.J., Bryant M.P., Pfennig N. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Arch. Microbiol. 1979;122:129–135. doi: 10.1007/s002030050685. [DOI] [PubMed] [Google Scholar]
- 28.Balch W.E., Wolfe R.S. New approach to the cultivation of methanogenic bacteria: 2 Mercaptoethanesulfonic acid (HS CoM) dependent growth of methanobacterium ruminantium in a pressurized atmosphere. Appl. Environ. Microbiol. 1976;32:781–791. doi: 10.1128/aem.32.6.781-791.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James K.L., Ríos-Hernández L.A., Wofford N.Q., Mouttaki H., Sieber J.R., Sheik C.S., Nguyen H.H., Yang Y., Xie Y., Erde J., Rohlin L., Karr E.A., Loo J.A., Loo R.R.O., Hurst G.B., et al. Pyrophosphate-dependent ATP formation from acetyl coenzyme a in Syntrophus aciditrophicus, a new twist on ATP formation. mBio. 2016;7 doi: 10.1128/mBio.01208-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erde J., Loo R.R.O., Loo J.A. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 2014;13:1885–1895. doi: 10.1021/pr4010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erde J., Loo R.R.O., Loo J.A. Improving proteome coverage and sample recovery with enhanced FASP (eFASP) for quantitative proteomic experiments. Methods Mol. Biol. 2017;1550:11–18. doi: 10.1007/978-1-4939-6747-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perkins D.N., Pappin D.J.C., Creasy D.M., Cottrell J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 33.Kanehisa M., Sato Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020;29:28–35. doi: 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Doncheva N.T., Morris J.H., Gorodkin J., Jensen L.J. Cytoscape StringApp: Network analysis and visualization of proteomics data. J. Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz D., Gygi S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 36.Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng A., Grant C.E., Noble W.S., Bailey T.L. MoMo: Discovery of statistically significant post-translational modification motifs. Bioinformatics. 2019;35:2774–2782. doi: 10.1093/bioinformatics/bty1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., Jensen L.J., Von Mering C. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arbing M.A., Chan S., Harris L., Kuo E., Zhou T.T., Ahn C.J., Nguyen L., He Q., Lu J., Menchavez P.T., Shin A., Holton T., Sawaya M.R., Cascio D., Eisenberg D. Heterologous expression of mycobacterial Esx complexes in Escherichia coli for structural studies is facilitated by the use of maltose binding protein fusions. PLoS One. 2013;8 doi: 10.1371/journal.pone.0081753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baeza J., Smallegan M.J., Denu J.M. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 2015;10:122–128. doi: 10.1021/cb500848p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Farrell P.H. High resolution two dimensional electrophoresis of proteins. J. Biol. Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 42.Ogorzalek Loo R.R., Cavalcoli J.D., VanBogelen R.A., Mitchell C., Loo J.A., Moldover B., Andrews P.C. Virtual 2-D gel electrophoresis: Visualization and analysis of the E. coli proteome by mass spectrometry. Anal. Chem. 2001;73:4063–4070. doi: 10.1021/ac0101858. [DOI] [PubMed] [Google Scholar]
- 43.Kleinert P., Kuster T., Arnold D., Jaeken J., Heizmann C.W., Troxler H. Effect of glycosylation on the protein pattern in 2-D-gel electrophoresis. Proteomics. 2007;7:15–22. doi: 10.1002/pmic.200600297. [DOI] [PubMed] [Google Scholar]
- 44.James A.M., Smith A.C., Ding S., Houghton J.W., Robinson A.J., Antrobus R., Fearnley I.M., Murphy M.P. Nucleotide-binding sites can enhance N-acylation of nearby protein lysine residues. Sci. Rep. 2020;10:20254. doi: 10.1038/s41598-020-77261-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wischgoll S., Heintz D., Peters F., Erxleben A., Sarnighausen E., Reski R., Van Dorsselaer A., Boll M. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 2005;58:1238–1252. doi: 10.1111/j.1365-2958.2005.04909.x. [DOI] [PubMed] [Google Scholar]
- 46.Carmona M., Zamarro M.T., Blázquez B., Durante-Rodríguez G., Juárez J.F., Valderrama J.A., Barragán M.J.L., García J.L., Díaz E. Anaerobic catabolism of aromatic compounds: A genetic and genomic view. Microbiol. Mol. Biol. Rev. 2009;73:71–133. doi: 10.1128/MMBR.00021-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bains J., Boulanger M.J. Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400: Defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol. 2007;373:965–977. doi: 10.1016/j.jmb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Crosby H.A., Heiniger E.K., Harwood C.S., Escalante-Semerena J.C. Reversible Nε-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris. Mol. Microbiol. 2010;76:874–888. doi: 10.1111/j.1365-2958.2010.07127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du Y., Wang F., May K., Xu W., Liu H. Determination of deamidation artifacts introduced by sample preparation using 18O-labeling and tandem mass spectrometry analysis. Anal. Chem. 2012;84:6355–6360. doi: 10.1021/ac3013362. [DOI] [PubMed] [Google Scholar]
- 50.Tang L., Wu Z., Wang J., Zhang X. Formaldehyde derivatization, an unexpected side reaction during filter-aided sample preparation. Anal. Chem. 2020;92:12120–12125. doi: 10.1021/acs.analchem.0c01981. [DOI] [PubMed] [Google Scholar]
- 51.Huang H., Zhang D., Wang Y., Perez-Neut M., Han Z., Zheng Y.G., Hao Q., Zhao Y. Lysine benzoylation is a histone mark regulated by SIRT2. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-05567-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathias R.A., Greco T.M., Oberstein A., Budayeva H.G., Chakrabarti R., Rowland E.A., Kang Y., Shenk T., Cristea I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell. 2014;159:1615–1625. doi: 10.1016/j.cell.2014.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tucker A.C., Escalante-Semerena J.C. Biologically active isoforms of CobB sirtuin deacetylase in Salmonella enterica and Erwinia amylovora. J. Bacteriol. 2010;192:6200–6208. doi: 10.1128/JB.00874-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindner H., Helliger W. Age-dependent deamidation of asparagine residues in proteins. Exp. Gerontol. 2001;36:1551–1563. doi: 10.1016/s0531-5565(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 55.Aswad D.W., Paranandi M.V., Schurter B.T. Isoaspartate in peptides and proteins: Formation, significance, and analysis. J. Pharm. Biomed. Anal. 2000;21:1129–1136. doi: 10.1016/s0731-7085(99)00230-7. [DOI] [PubMed] [Google Scholar]
- 56.Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christensen D.G., Baumgartner J.T., Xie X., Jew K.M., Basisty N., Schilling B., Kuhn M.L., Wolfe A.J. Mechanisms, detection, and relevance of protein acetylation in prokaryotes. mBio. 2019;10 doi: 10.1128/mBio.02708-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Christensen D.G., Xie X., Basisty N., Byrnes J., McSweeney S., Schilling B., Wolfe A.J. Post-translational protein acetylation: An elegant mechanism for bacteria to dynamically regulate metabolic functions. Front. Microbiol. 2019;10:1604. doi: 10.3389/fmicb.2019.01604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garrity J., Gardner J.G., Hawse W., Wolberger C., Escalante-Semerena J.C. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 60.Gardner J.G., Grundy F.J., Henkin T.M., Escalante-Semerena J.C. Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD+ involvement in Bacillus subtilis. J. Bacteriol. 2006;188:5460–5468. doi: 10.1128/JB.00215-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitata R.B., Dimayacyac-Esleta B.R.T., Choong W.-K., Tsai C.-F., Lin T.-D., Tsou C.-C., Weng S.-H., Chen Y.-J., Yang P.-C., Arco S.D., Nesvizhskii A.I., Sung T.-Y., Chen Y.-J. Mining missing membrane proteins by high-pH reverse-phase StageTip fractionation and multiple reaction monitoring mass spectrometry. J. Proteome Res. 2015;14:3658–3669. doi: 10.1021/acs.jproteome.5b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu Y., Zou X., Dean A.E., Brien J.O., Gao Y., Tran E.L., Park S.H., Liu G., Kieffer M.B., Jiang H., Stauffer M.E., Hart R., Quan S., Satchell K.J.F., Horikoshi N., et al. Lysine 68 acetylation directs MnSOD as a tetrameric detoxification complex versus a monomeric tumor promoter. Nat. Commun. 2019;10:2399. doi: 10.1038/s41467-019-10352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang X., Yuan Z., Zhang Y., Yong S., Salas-Burgos A., Koomen J., Olashaw N., Parsons J.T., Yang X.J., Dent S.R., Yao T.P., Lane W.S., Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.VanDrisse C.M., Escalante-Semerena J.C. Protein acetylation in bacteria. Annu. Rev. Microbiol. 2019;73:111–132. doi: 10.1146/annurev-micro-020518-115526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan M., Peng C., Anderson K.A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G.R., Green M.F., Madsen A.S., Schmiesing J., et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu K., Li F., Sun Q., Lin N., Han H., You K., Tian F., Mao Z., Li T., Tong T., Geng M., Zhao Y., Gu W., Zhao W. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019;10:243. doi: 10.1038/s41419-019-1463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., Qi S., Li J., Colak G., Chen Y., Xia C., Peng C., Ruan H., Kirkey M., Wang D., et al. Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun C.F., Xu W.F., Zhao Q.W., Luo S., Chen X.A., Li Y.Q., Mao X.M. Crotonylation of key metabolic enzymes regulates carbon catabolite repression in Streptomyces roseosporus. Commun. Biol. 2020;3:192. doi: 10.1038/s42003-020-0924-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sabari B.R., Tang Z., Huang H., Yong-Gonzalez V., Molina H., Kong H.E., Dai L., Shimada M., Cross J.R., Zhao Y., Roeder R.G., Allis C.D. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C., Falck J.R., Peng J., Gu W., Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu J.Y., Xu Z., Liu X.X., Tan M., Ye B.C. Protein acetylation and butyrylation regulate the phenotype and metabolic shifts of the endospore-forming Clostridium acetobutylicum. Mol. Cell. Proteomics. 2018;17:1156–1169. doi: 10.1074/mcp.RA117.000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weinert B.T., Satpathy S., Hansen B.K., Lyon D., Jensen L.J., Choudhary C. Accurate quantification of site-specific acetylation stoichiometry reveals the impact of Sirtuin deacetylase CobB on the E. coli acetylome. Mol. Cell. Proteomics. 2017;16:759–769. doi: 10.1074/mcp.M117.067587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hansen B.K., Gupta R., Baldus L., Lyon D., Narita T., Lammers M., Choudhary C., Weinert B.T. Analysis of human acetylation stoichiometry defines mechanistic constraints on protein regulation. Nat. Commun. 2019;10:1055. doi: 10.1038/s41467-019-09024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weinert B.T., Iesmantavicius V., Moustafa T., Schölz C., Wagner S.A., Magnes C., Zechner R., Choudhary C. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol. Syst. Biol. 2014;10:716. doi: 10.1002/msb.134766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sadhukhan S., Liu X., Ryu D., Nelson O.D., Stupinski J.A., Li Z., Chen W., Zhang S., Weiss R.S., Locasale J.W., Auwerx J., Lin H. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U. S. A. 2016;113:4320–4325. doi: 10.1073/pnas.1519858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishida Y., Rardin M.J., Carrico C., He W., Sahu A.K., Gut P., Najjar R., Fitch M., Hellerstein M., Gibson B.W., Verdin E. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell. 2015;59:321–332. doi: 10.1016/j.molcel.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schilling B., Christensen D., Davis R., Sahu A.K., Hu L.I., Walker-Peddakotla A., Sorensen D.J., Zemaitaitis B., Gibson B.W., Wolfe A.J. Protein acetylation dynamics in response to carbon overflow in Escherichia coli. Mol. Microbiol. 2015;98:847–863. doi: 10.1111/mmi.13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basisty N., Meyer J.G., Wei L., Gibson B.W., Schilling B. Simultaneous quantification of the acetylome and succinylome by ‘one-pot’ affinity enrichment. Proteomics. 2018;18:1800123. doi: 10.1002/pmic.201800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cao J., Wang T., Wang Q., Zheng X., Huang L. Functional insights into protein acetylation in the hyperthermophilic archaeon Sulfolobus islandicus. Mol. Cell. Proteomics. 2019;18:1572–1587. doi: 10.1074/mcp.RA119.001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M., Ding J., Czyz D., Hu R., Ye Z., He M., et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rajasekaran N.S., Connell P., Christians E.S., Yan L.J., Taylor R.P., Orosz A., Zhang X.Q., Stevenson T.J., Peshock R.M., Leopold J.A., Barry W.H., Loscalzo J., Odelberg S.J., Benjamin I.J. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trotter E.W., Grant C.M. Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 2002;46:869–878. doi: 10.1046/j.1365-2958.2002.03216.x. [DOI] [PubMed] [Google Scholar]
- 84.Sauve A.A., Wolberger C., Schramm V.L., Boeke J.D. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 85.Anderson K.A., Huynh F.K., Fisher-Wellman K., Stuart J.D., Peterson B.S., Douros J.D., Wagner G.R., Thompson J.W., Madsen A.S., Green M.F., Sivley R.M., Ilkayeva O.R., Stevens R.D., Backos D.S., Capra J.A., et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017;25:838–855.e15. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pannek M., Simic Z., Fuszard M., Meleshin M., Rotili D., Mai A., Schutkowski M., Steegborn C. Crystal structures of the mitochondrial deacylase Sirtuin 4 reveal isoform-specific acyl recognition and regulation features. Nat. Commun. 2017;8:1513. doi: 10.1038/s41467-017-01701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Colak G., Xie Z., Zhu A.Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y.H., Lin H., Zhao Y., Tan M. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics. 2013;12:3509–3520. doi: 10.1074/mcp.M113.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ringel A.E., Roman C., Wolberger C. Alternate deacylating specificities of the archaeal sirtuins Sir2Af1 and Sir2Af2. Protein Sci. 2014;23:1686–1697. doi: 10.1002/pro.2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gallus C., Schink B. Anaerobic degradation of pimelate by newly isolated denitrifying bacteria. Microbiology. 1994;140:409–416. doi: 10.1099/13500872-140-2-409. [DOI] [PubMed] [Google Scholar]
- 90.Harrison F.H., Harwood C.S. The pimFABCDE operon from Rhodopseudomonas palustris mediates dicarboxylic acid degradation and participates in anaerobic benzoate degradation. Microbiology. 2005;151:727–736. doi: 10.1099/mic.0.27731-0. [DOI] [PubMed] [Google Scholar]
- 91.Schöcke L., Schink B. Energetics and biochemistry of fermentative benzoate degradation by Syntrophus gentianae. Arch. Microbiol. 1999;171:331–337. [Google Scholar]
- 92.Müller N., Worm P., Schink B., Stams A.J.M., Plugge C.M. Syntrophic butyrate and propionate oxidation processes: From genomes to reaction mechanisms. Environ. Microbiol. Rep. 2010;2:489–499. doi: 10.1111/j.1758-2229.2010.00147.x. [DOI] [PubMed] [Google Scholar]
- 93.Boll M., Geiger R., Junghare M., Schink B. Microbial degradation of phthalates: Biochemistry and environmental implications. Environ. Microbiol. Rep. 2020;12:3–15. doi: 10.1111/1758-2229.12787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data were uploaded to ProteomeXchange (proteomexchange.org) either through PRIDE (2D-gel dataset; PXD025631) or through MassIVE (Shotgun dataset; PXD025603).