

Abstract
Live imaging of single RNA from birth to death brought important advances in our understanding of the spatiotemporal regulation of gene expression. These studies have provided a comprehensive understanding of RNA metabolism by describing the process step by step. Most of these studies used for live imaging a genetically encoded RNA-tagging system fused to fluorescent proteins. One of the best characterized RNA-tagging systems is derived from the bacteriophage MS2 and it allows single RNA imaging in real-time and live cells. This system has been successfully used to track the different steps of mRNA processing in many living organisms. The recent development of optimized MS2 and MCP variants now allows the labeling of endogenous RNAs and their imaging without modifying their behavior. In this chapter, we discuss the improvements in detecting single mRNAs with different variants of MCP and fluorescent proteins that we tested in yeast and mammalian cells. Moreover, we describe protocols using MS2-MCP systems improved for real-time imaging of single mRNAs and transcription dynamics in S. cerevisiae and mammalian cells, respectively.
Keywords: MS2-MCP system, mRNA labeling, Single molecule, Single cell, S. cerevisiae, Mammalian cells, Gene expression, mRNA localization, Transcription
1. Introduction
Cells are the basic unit of life. Within a single cell, networks of molecules control how the environment is sensed through signaling and how cells adapt via metabolic changes and modulation of gene expression. Quantitative methods are required to model these fundamental processes, which often involve only tens or less molecules [1–3]. To detect variations in gene expression, one approach is to measure mRNA levels. Even though these are not always a proxy for protein expression, i.e., when a delay exists between mRNA accumulation and protein production [4], mRNA measurements provide information about the rate of transcription, accumulation, and decay, revealing modes of gene expression regulation. Bulk mRNA measurements, i.e., northern blots, quantitative PCR, and RNA sequencing, are informative to measure multiple mRNA species from a single RNA preparation and to perform relative comparisons of mRNA levels in different conditions. However, these approaches, which average millions of cells, have fundamental limitations when it comes to precisely measuring RNAs at the level of single cells, or within cellular compartments, i.e., nucleus vs. cytoplasm, cellular protrusions such as the bud of S. cerevisiae, the leading edge of fibroblast, or neuronal dendrites and axons.
To achieve quantitative subcellular mRNA measurements, several approaches based on fluorescence microscopy have been developed over the past decades. In fixed cells, a standard method to visualize and count individual mRNAs is single-molecule fluorescent in situ hybridization (smFISH) [5, 6]. Briefly, this approach allows detecting single endogenous mRNAs by hybridizing tens of fluorescently labeled DNA oligos onto the target molecule. By using sensitive digital cameras and wide-field microscopy, it is possible to detect single mRNAs as diffraction-limited spots, allowing their subcellular localization and quantification in thousands of cells. This method can be applied to single isolated cells (i.e., [7–13]) as well as to tissues [14–16]. Several modified smFISH protocols exist, which use DNA probes of different lengths and complexities (i.e., 20 mer, 50 mer, branched DNA, RNA scope [8, 10, 17]) or fluorescence amplification systems to detect weak signals (i.e., hybridization chain reaction, HCR [18]). An important difference between these techniques is whether the probes are fluorescently labeled or not, in which case they need to be detected with a secondary fluorescent oligo. A simple, reliable, and affordable protocol that uses indirect labeling has recently been published [19]. Furthermore, smFISH can be multiplexed to simultaneously visualize different mRNA species within single cells (up to 10,000; [15, 20–24]), or it can also be combined to protein detection by immunofluorescence (see refs. 25, 26 and this issue). These approaches revealed asymmetric RNA distribution within single cells, as well as significant cell-to-cell variability existing in tissue or even isogenic populations [27–32]. Furthermore, by using smFISH, other aspects of gene expression have been characterized, such as the “bursty” nature of transcription [8, 9, 33, 34], the mechanisms controlling mRNA export form the nucleus to the cytoplasm [35, 36], as well as the control of mRNA degradation [37, 38]. For more in-depth reviews see refs. 1, 2, 39; see also Chapter 1 by Bleckmann et al. and Chapter 4 by Tutucci and Singer.
However, fixed cells provide limited information about highly dynamic and rare events controlling mRNA metabolism. To follow mRNAs in living cells, several labeling strategies have been developed over the past decades. The best characterized system is a genetically encoded reporter based on the multimerization of RNA stem-loops derived from the bacteriophage MS2 [40–42]. To visualize single mRNAs in living cells, 24 MS2 stem-loops are used to tag an RNA of interest, which is then detected by co-expression of a specific RNA-binding protein, the MS2 coat protein (MCP), fused to fluorescent proteins (FP, i.e., eGFP, mCherry, tdTomato, HALO, photoactivatable proteins) [1, 39]. Several MS2 array variants are available, and the recommendation for using a particular one depends on the model organism, the mRNA, and the step of the mRNA life cycle under investigation [1, 43]. High-affinity MS2-MCP variants have been successfully used to measure the dynamics of mRNA transcription and splicing [44–46], export [47, 48], localization [49, 50], and translation in mammalian cells [51–55]. They are also recommended if the experimental setup involves FRAP. However, several groups reported that the high-affinity MS2-MCP variants are not optimal to visualize mRNAs in rapidly dividing organisms such as S. cerevisiae [56–59]. For this reason, we recently generated an improved MS2 array (MS2-binding sites V6, MBSV6) with decreased affinity for MCP. This allowed measuring the half-life of rapidly decaying mRNAs while preserving single-molecule mRNA detection in living cells [43, 58]. Low-affinity MS2-MCP variants have also been used to tag mRNAs in mammalian cells, specifically to generate arrays containing up to 128 MS2 stem-loops in a single transcript, with the aim of monitoring transcription with high temporal resolution for long periods of time and with minimal photo-bleaching [60].
To visualize more than one mRNA species at the time, in single living cells, several orthogonal systems are available. Another system was generated by multimerizing RNA stem-loops derived from the bacteriophage PP7, detected by the cognate protein PP7 coat protein (PCP) fused to fluorescent proteins [61]. This reporter has been used to study transcription dynamics [62], mRNA export [63], and translation [64, 65]. It has also been used to create a homozygous mice where the immediate early gene Arc was endogenously tagged with 24 PP7 loops, allowing to visualize its response to synaptic activity [66]. Alternative genetically encoded RNA labeling strategies use arrays generated from other RNA sequences, such as the BglG stem-loop [67], the λ BoxB RNA [68], and the U1A loop [69, 70]. Other RNA labeling methods are reviewed elsewhere [1, 43, 54].
The optimization of single mRNA visualization in living cells relies also on the expansion of the FP and fluorophore palette [71, 72]. In a recent publication, the brightness, photostability, pH resistance, and monomeric properties of more than 40 FP have been systematically quantified [73]. These measurements are valuable to design mRNA imaging reporters with precise photochromatic properties. Furthermore, the development of fluorescent dyes partly bypasses some of the common problems encountered using fluorescent proteins, i.e., wide excitation and emission spectrum, sensitivity to photobleaching, tendency to multimerization since these molecules generally have small size, high brightness and photostability, and narrow spectrum.
In the following paragraphs we report our tests aimed at optimizing the visualization of single mRNAs in living eukaryotic cells. We tested several MCP variants in the model organism S. cerevisiae as well as multiple green FP fused to MCP both in S. cerevisiae and mammalian cell lines. These comparisons revealed that for efficient mRNA detection, MCP variants with high affinity for the RNA reporter remain the best option. In addition, we found that the green FP Envy shows improved brightness compared to other GFP variants, both in yeast and mammalian cells.
1.1. mRNA Detection Using Different MCP Variants
We previously demonstrated that it is possible to efficiently detect single mRNAs in S. cerevisiae by using the latest MS2 variant, MBSV6, in combination with the expression of MCP-NLS-2xyeGFP [43, 58] (Fig. 1a–d). To improve the long-term detection of single mRNAs in living cells (i.e., brightness and photostability) we generated other MCP constructs that we compared to MCP-NLS-2xyeGFP for their brightness and propensity to form aggregates. Even though we find that the MCP-NLS-2xyeGFP performs better than other constructs tested thus far, here we report the advantages and disadvantages of other tested reporters.
Fig. 1.
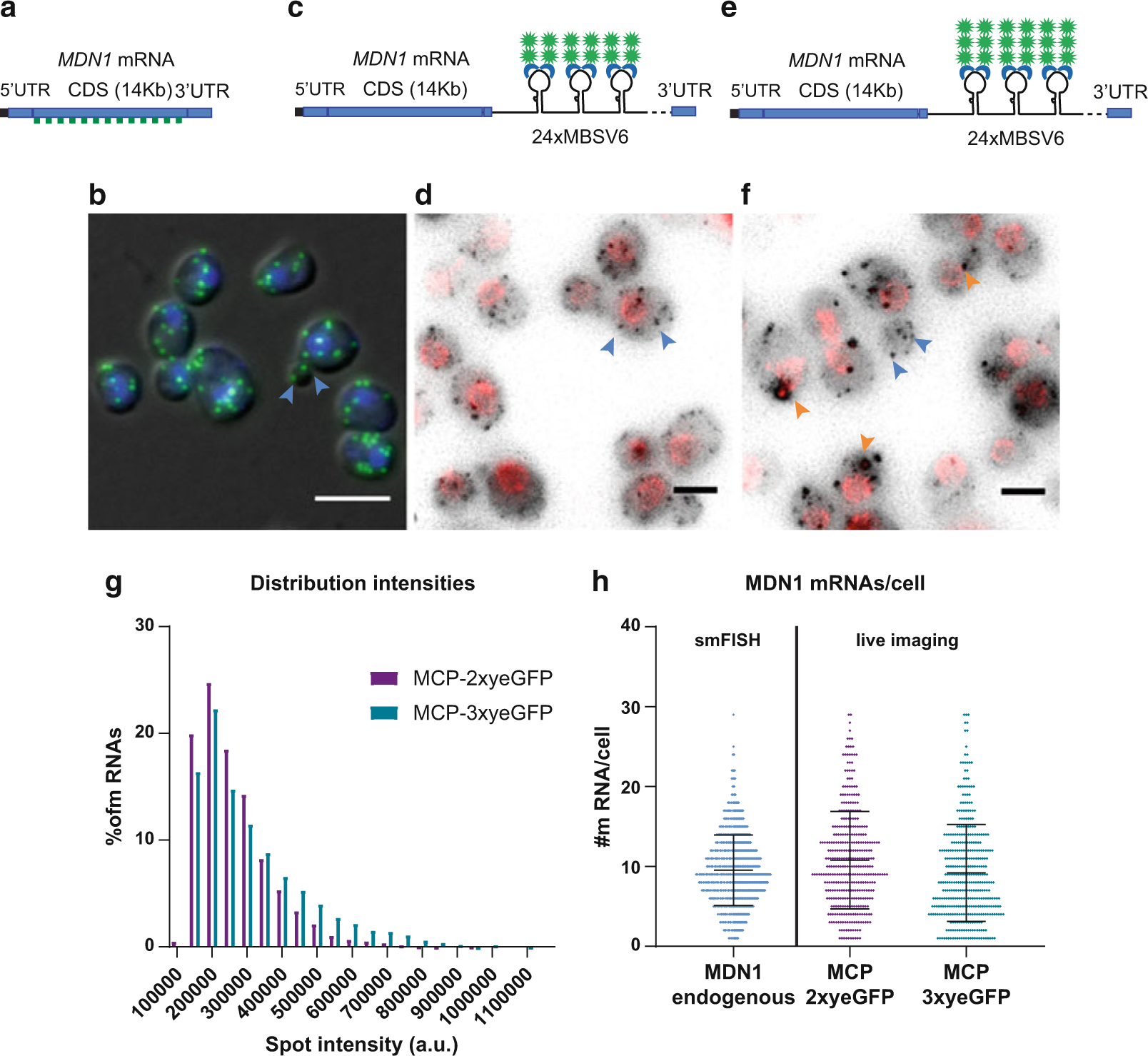
MDN1 mRNA detection using the MS2-MCP system in S. cerevisiae. (a) Schematic representation of the MDN1 locus. smFISH probes labeled with Q760 anneal all along the CDS. Probe list is provided in [58]. (b) Overlap of the DAPI signal in the nucleus (blue), smFISH for the MDN1 CDS (green) with the differential interference contrast (DIC) image. Single mRNAs are indicated with blue arrowheads. Scale bar 5 μm. (c) Schematic representation of MDN1 locus tagged at the 3’UTR with 24×MBSV6. mRNAs are detected in living cells by co-expression of the plasmid MCP-NLS-2xyeGFP. (d) MDN1 mRNAs are shown in gray, and the nuclear pore protein Nup49 is shown in red. Max projection of a 15 Z-stack. Rolling average background subtraction (rolling ball radius = 50) was performed for the two channels. Single mRNAs are indicated by blue arrowheads. Scale bar 3 μm. (e) Schematic representation of MDN1 locus tagged at the 3’UTR with 24×MBSV6. mRNAs are detected by co-expression of the plasmid MCP-NLS-3xyeGFP. (f) MDN1 mRNAs are shown in gray, and the nuclear pore protein Nup49 is shown in red. Max projection of a 15 Z-stack. Rolling average background subtraction (rolling ball radius = 50) was performed for the two channels. Selected single mRNAs are indicated by blue arrowheads. MCP-containing granules are indicated in orange arrowheads. Scale bar 3 μm. (g) Distribution of MDN1 mRNA intensities measured by live imaging in expressing cells. Purple bars, MDN1 24×MBSV6 co-expressing MCP-NLS-2xyeGFP. Mean ± S.D. 263,373 ± 109,890 a.u. number of spots = 4186. Green bars, MDN1 24×MBSV6 co-expressing MCP-NLS-3xyeGFP. Mean ± S.D. 3,100,950 ± 155,349 a.u. number of spots = 3414. (h) Comparison of MDN1 mRNA/cell counted in fixed cells vs. live imaging. mRNAs per cell counted by smFISH in wild-type cells vs. MDN1 mRNAs counted in cells expressing MDN1 24×MBSV6 co-expressing MCP-NLS-2xyeGFP (purple, n = 368) or MDN1 24×MBSV6 co-expressing MCP-NLS-3xyeGFP (green, n = 390)
To improve the brightness of single mRNAs, we generated an MCP variant fused to 3xyeGFP. This plasmid was transformed in the yeast strain expressing MDN1 tagged with 24×MBSV6 and Nup49-tdTomato (Fig. 1e, f). We compared the brightness of MDN1 mRNAs detected with either MCP-2xyeGFP or MCP-3xyeGFP (Fig. 1g). As expected, mRNAs labeled with MCP-3xyeGFP shows a 25% increase in brightness compared to mRNAs labeled with MCP-2xyeGFP (310,950 ± 155,349 a.u. and 263,373 ± 109,890 a.u., respectively). In addition, the number of MDN1 mRNAs per cell counted with the MCP-3xyeGFP reporter is similar to the mRNAs counted with MCP-2xyeGFP or by smFISH (mean ± S.D. 9.2 ± 6.1 mRNAs/cell, Fig. 1h). However, we found that the MCP-3xyeGFP reporter has the tendency to induce cytoplasmic aggregates, likely due to the propensity of GFP to multimerize (Fig. 1f, orange arrowheads). These aggregates are similar to the ones that we previously described [58] and that can lead to artifactual conclusions about mRNA localization in yeast. It may still be possible to use the MCP-3xyeGFP reporter for mRNAs that are less abundant than MDN1, but we recommend always comparing the live imaging results to smFISH to avoid false conclusions.
1.2. Comparison of FP Variants in Yeast
Many FP variants now exist, and we tested a few promising ones to see whether this would improve the signal: sfGFP, mNG2, eGFP, muGFP, and Envy. sfGFP is the superfolder GFP and muGFP is one of its ultrastable monomeric variants [74]. mNG is a bright and stable variant derived from a Branchiostoma lanceolatum fluorescent protein [75], and Envy is an FP that performs particularly well in S. cerevisiae [76]. The cDNA coding for these fluorescent proteins was cloned into pET296, replacing the yeGFP tag, such that they are expressed as monomeric variants lacking NLS (MCP-1xFP). The plasmids were then transformed into yeast cells expressing the DOA1 gene tagged with 24×MBSV6 as described in [58]. Cells were then grown, fixed, and observed under the microscope (Fig. 2a–c). We found that mRNAs were undetectable with MCP-1xeGFP, MCP-1xsfGFP, and MCP-1xmuGFP, while they were nicely visible with the MCP-1xEnvy and MCP-1xmNG2 (with best results for MCP-1xEnvy).
Fig. 2.
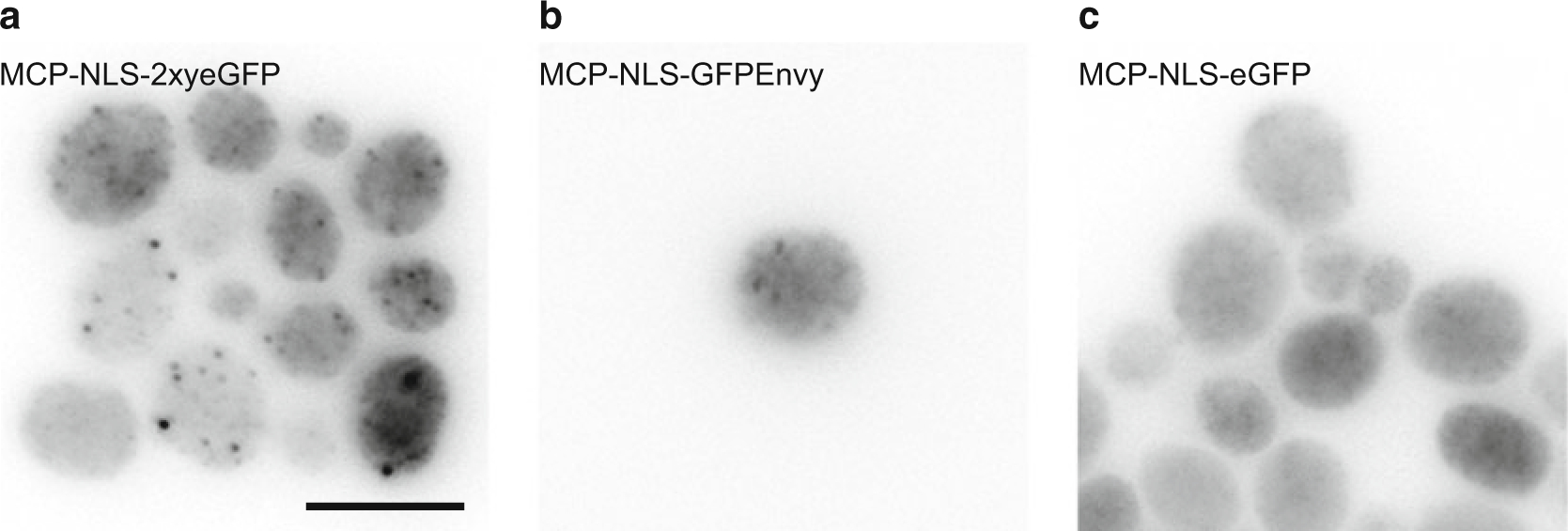
Comparison of MCP-FP variants in yeast. Images are from yeast cells expressing the DOA1 gene tagged with 24×MBSV6 transformed with (a) MCP-NLS-2xyeGFP plasmid, (b) MCP-NLS-GFPEnvy plasmid, (c) MCP-NLS-eGFP plasmid. Panels are maximum intensity projections of Z-stacks. Scale bar 5 μm
1.3. Comparison of Low-and High-Affinity MCP Variants in Yeast
The original MS2 system was designed with high-affinity variants of both the MBS and the MCP [40]. Indeed, this MCP variant carried the V29I mutation that enhances binding stability by 5–10-fold [77]. Since a too high affinity of the MCP-MBS interaction was found to cause artifacts in yeast [58], we tested the effect of reverting the V29I mutation. A plasmid expressing MCP-I29V-2xyeGFP was generated and transformed in yeast cells expressing the DOA1 gene tagged with MBV6. No signals corresponding to single mRNAs could be detected, suggesting that the affinity of the MS2V6/MCP-I29V is probably too low for an efficient mRNA detection. Overall, MBSV6/MCP-V29I appears to have the best affinity compromise to allow for an artifact-free mRNA detection in yeast.
1.4. Comparison of FP Variants in Mammalian Cells
MCP-FP variants displayed distinct performance in yeast, and we thus tested how these variants [74, 76, 78–81] performed in mammalian cells (Fig. 3 and Table 1). Transient transfections of the different NLS-MCP-FP variant constructs were evaluated in a HeLa H9 cell line without RNA reporter gene (Fig. 3, top panels). A diffuse fluorescent signal was seen in the nucleus, indicating a good solubility of all NLS-MCP-FP variant constructs. When the various NLS-MCP-FP were expressed in a derivative of this cell line expressing the HIV-1 MS2×64 RNA reporter (Figs. 3 and 4, middle panels), the NLS-MCP-GFPEnvy showed the highest contrast (see signal quantifications in Fig. 5), followed by the NLS-MCP-eGFP and NLS-MCP-sfGFP. Single RNA molecules were only barely detectable with the NLS-MCP-muGFP and NLS-MCP-mNG construct, while single RNA molecules were observed by smFISH (Fig. 3, bottom panels).
Fig. 3.
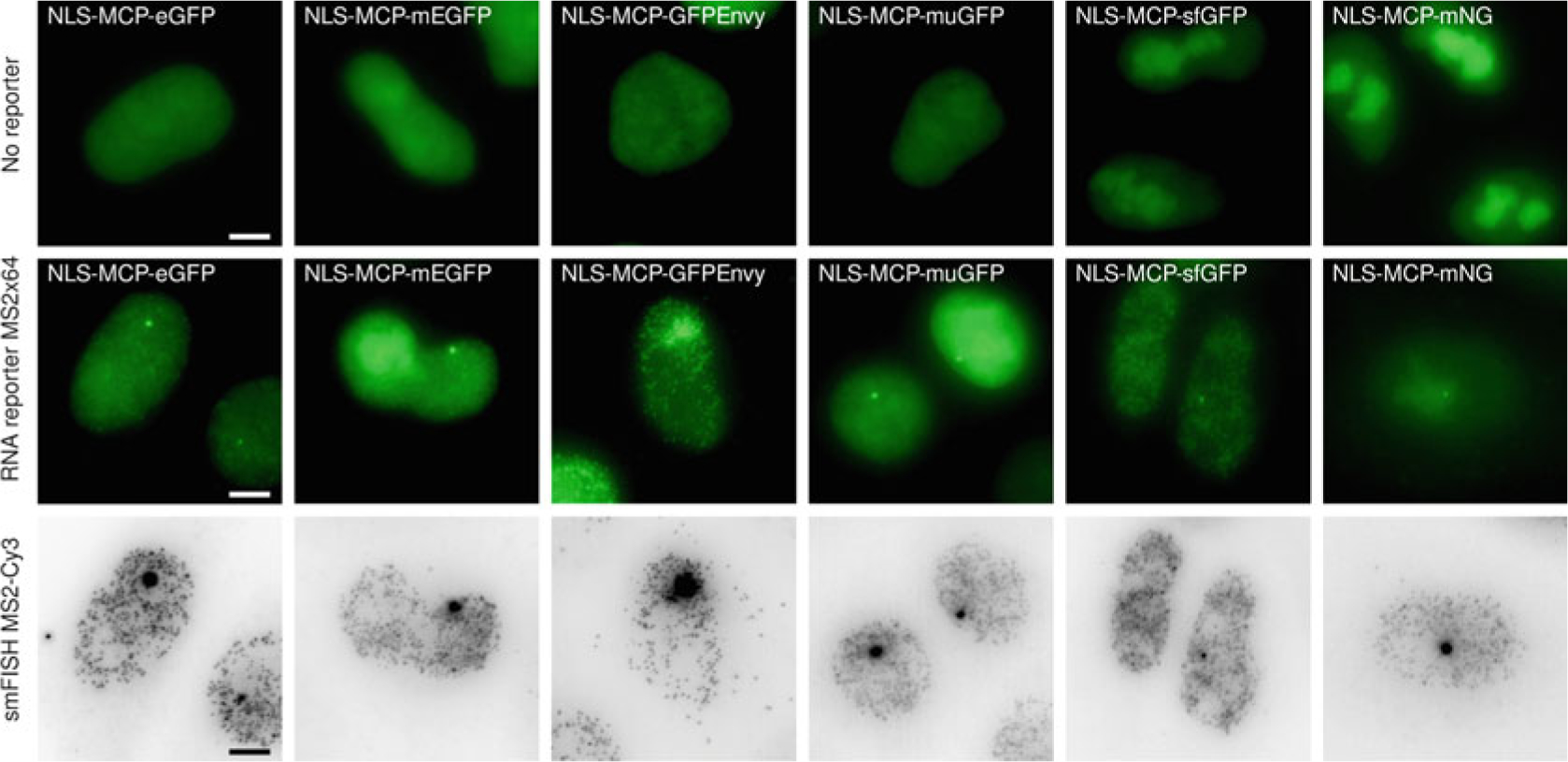
Comparison of MCP-FP variants in mammalian cells. Images are maximum intensity projections from HeLa Flp-in cells transfected with the indicated NLS-MCP-FP variant constructs. Top panels: Images are from cells expressing the indicated NLS-MCP-FP variants without RNA reporter (parental HeLa H9 AAVS1-Tat cell line). Middle panels: Images are from cells expressing the indicated NLS-MCP-FP variant in HeLa H9 AAVS1-Tat cell line expressing the HIV-1 MS2×64 reporter. Bottom panels: Images are smFISH signals obtained with probes against the MS2 tag. Scale bar 5 μm. Contrast adjustment is identical for all images of the top and middle panels
Table 1.
List of the FP variants
| NLS-MCP-FP variants | FP plasmid from | References for the FP |
|---|---|---|
|
| ||
| eGFP | Gift from Lionnet T./Singer RH | Cormack et al. (1996) [78] |
| mEGFP | Addgene #54610 | Zacharias et al. (2002) [79] |
| sfGFP | Addgene #60907 | Pedelacq et al. (2006) [80] |
| GFPenvy | Addgene #60782 | Slubowski et al. (2015) [76] |
| muGFP | Gift from Scott DJ | Scott etal. (2018) [74] |
| mNG | pUC57-mNeonGreen | Shaner et al. (2013) [81] |
Fig. 4.
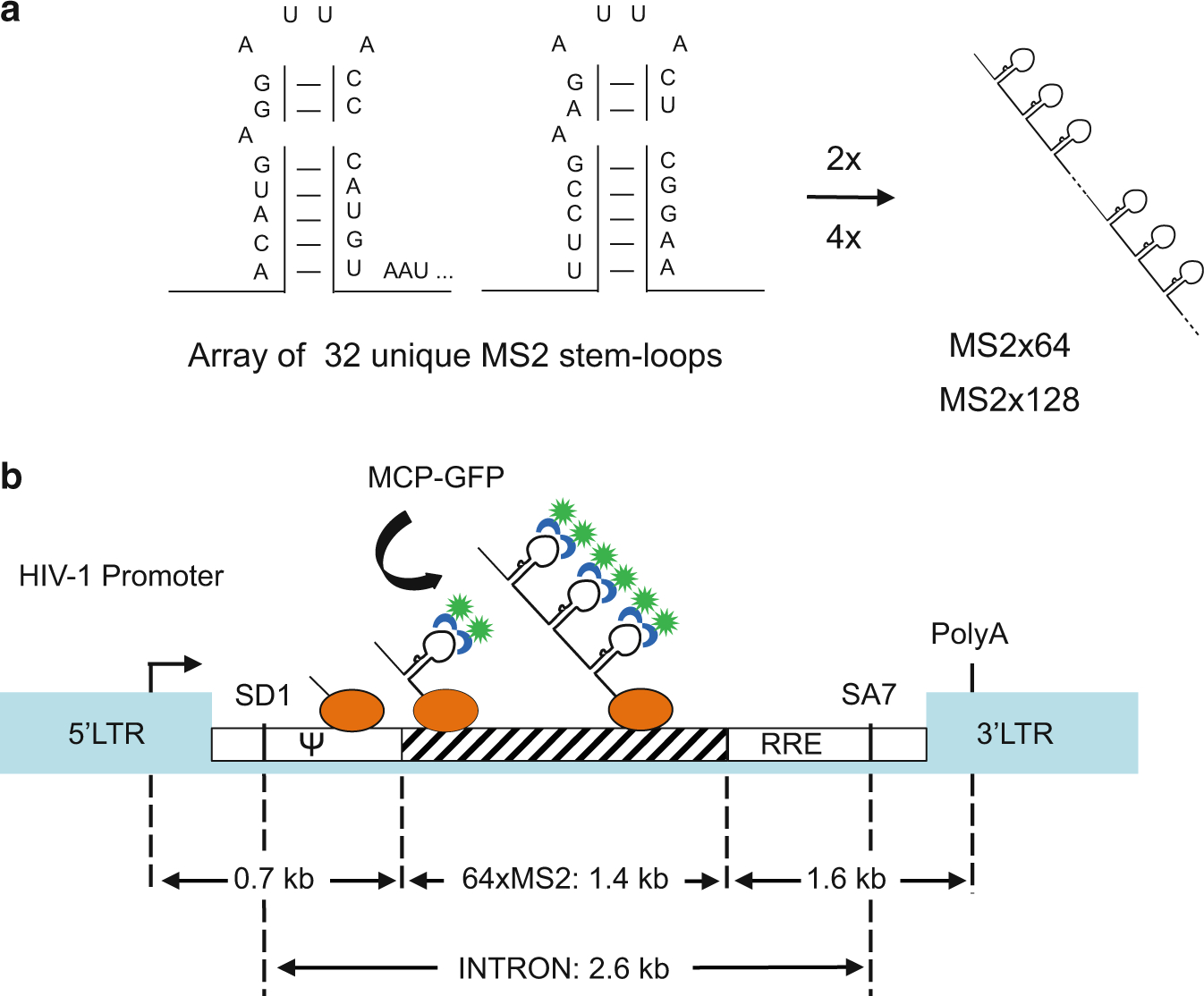
Schematic representation of pIntro-MS2×64 RNA reporter construct. (a) Generation of the MS2×64 RNA tag. The MS2×32 stem-loop sequence with low-affinity binding to MCP is multimerized to generate repeats of 64 and 128 stem-loops. (b) Schematic of the pIntro-MS2×64 reporter construct. The striped box represents the MS2 repeat; the green spot represents the GFP fused to MCP (blue); the orange oval represents RNAPII with the nascent RNAs. LTR The HIV-1 long terminal repeat; SD1 The major HIV-1 splice donor; SA7 The last splice acceptor; Ψ Packaging sequence; RRE Rev-responsive element
Fig. 5.
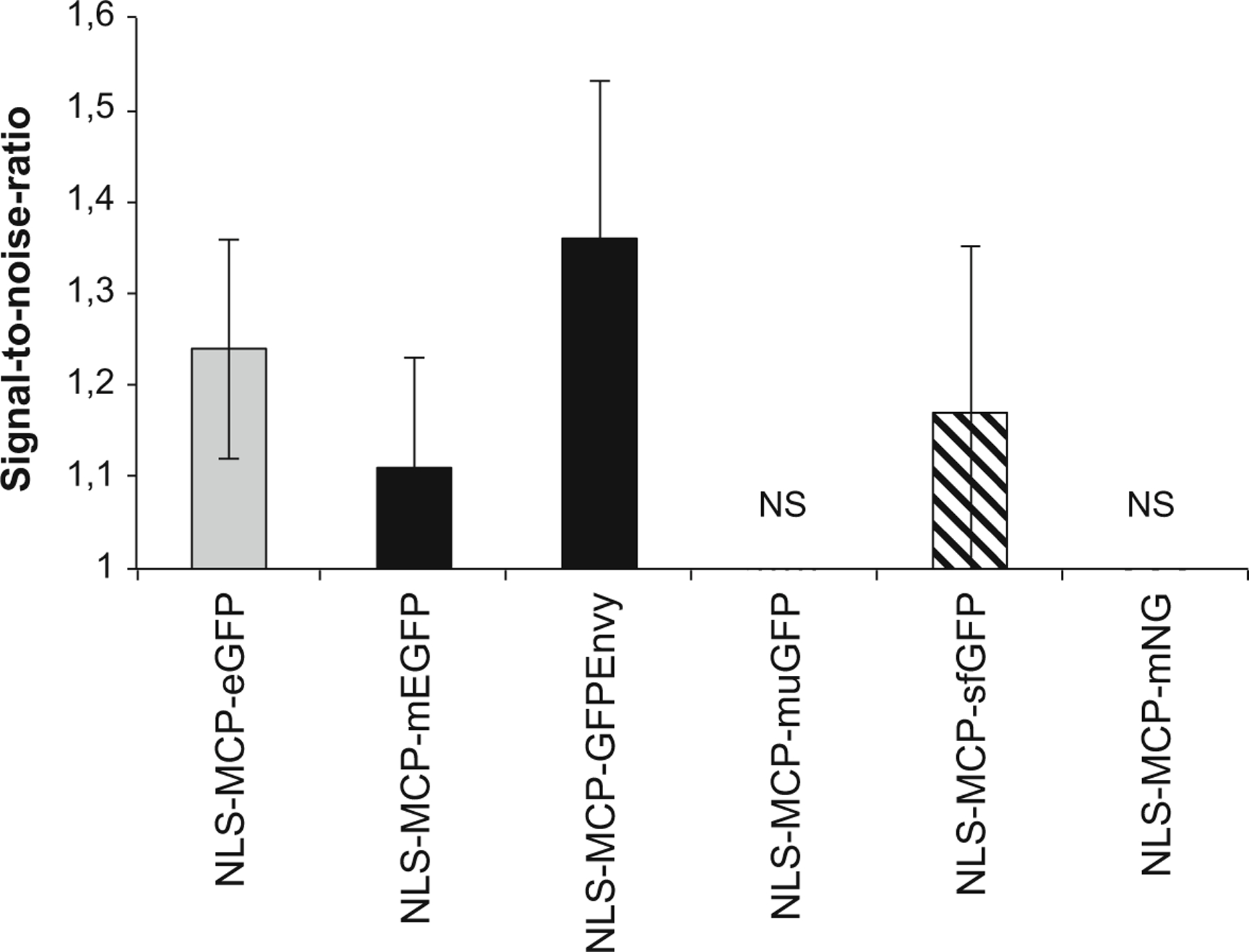
Signal-to-noise ratio (SNR) of single RNA molecule detection with MCP-FP variants. The bar plots show the mean values of the SNR of single RNA molecules (error bar: standard deviation). NS: nonsignificant detection of single RNAs occurred with the NLS-MCP-muGFP and NLS-MCP-mNG constructs. To measure the intensity of single RNA molecules, a straight line with a width of 2 pixels was used, and the maximal value of the intensity plot was recorded. This value was divided over the mean gray value measured in the nucleus of the cell following the application of a Gaussian blur of 3 pixels (maximum intensity projected images). The background value (outside the cell nucleus) was subtracted from each of these values (total of 133 single molecules quantified per construct)
In light of these recent developments, we describe here protocols to visualize and quantify mRNAs labeled with the low-affinity MS2 systems that we recently developed both for S. cerevisiae and mammalian cells. By describing the visualization of mRNA in these model organisms, we highlight the general rules and recommendations that can improve live imaging of single mRNAs. While elsewhere we described in depth the steps required to endogenously tag an mRNA with the MS2 reporter in yeast [43, 58] or in mammalian cells [49], here we focused on the protocols used to perform live imaging and to quantify the number and the brightness of single mRNAs and transcription sites.
For S. cerevisiae we outline a protocol for the visualization of mRNAs labeled with the low-affinity 24×MBSV6 (see Subheadings 2.1 and 3.1). The constitutive and well-characterized mRNA MDN1 is used as a model gene. MDN1 was endogenously tagged with 24×MBSV6 as previously described in [58]. Here, we detail how to transform yeast cells with the constructs expressing MCP-FP (Subheading 3.1.1), grow cells for live imaging (Subheading 3.1.2), prepare the coated dishes (Subheading 3.1.3), perform live imaging (Subheading 3.1.4), and count single mRNAs in living cells (Subheading 3.1.5).
For mammalian cells, we outline a protocol for the visualization of mRNAs labeled with the low-affinity MS2×64 reporter, which is particularly useful to analyze transcription dynamics (see Subheadings 2.2 and 3.2). To this end, we used a stable HeLa H9 AAVS1-Tat cell line expressing the HIV-1 MS2×64 reporter gene [60]. Here, we describe how to produce lentiviruses encoding the MCP-FP (Subheading 3.2.1), how to generate a stable cell line expressing the MCP-FP construct (Subheading 3.2.2), how to select a cell line with optimal MCP-FP expression (Subheading 3.2.3), how to prepare the cells (Subheading 3.2.4), and how to perform live image acquisition (Subheading 3.2.5).
2. Materials
2.1. Materials for Visualizing MBSV6-Labeled mRNAs in Yeast
Yeast cells: All strains used in this protocol are derived from the S. cerevisiae background BY4741 MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0.
YPD medium: 50 g/L of the YPD mix (i.e., Clonetech). Sterilize by autoclaving.
LEU medium (dropout media lacking leucine): 6.7 g/L Yeast nitrogen base (YNB) with ammonium sulfate, dropout mix lacking leucine, 20 g/L glucose. Sterilize by autoclaving.
LEU plates (dropout agar plates): 6.7 g/L Yeast nitrogen base (YNB) with ammonium sulfate, dropout mix lacking leucine, 20 g/L glucose, 20 g/L of bacteriological agar. Sterilize by autoclaving.
100% Glycerol stock: Sterilize by autoclaving. Store at room temperature, protected from light.
Plasmids: pET264-pUC 24×MS2V6 Loxp KANr Loxp (Addgene ID:104393), pET251-pUC 12×MS2V6 Loxp KANr Loxp (Addgene ID:104392); pET296-YcpLac111 CYC1p-MCP-NLS-2xyeGFP (yeast-optimized eGFP) (Addgene ID:104394); pET511-YcpLac111 CYC1p-MCP-NLS-3xyeGFP; pET518-YcpLac11-CYC1p-MCP-1x-eGFP; pET519-YcpLac11-CYC1p-MCP-1xEnvy; pET521-YcpLac11-CYC1p-MCP-1xmuGFP; pET522-YcpLac11-CYC1p-MCP-1xmNG; pET523-YcpLac11-CYC1p-MCP-1xsfGFP (constructs available upon request).
Lithium-TE: 100 mM LiAc, 10 mM Tris–HCl, pH 7.5, 1 mM EDTA. Sterilize by autoclaving.
Lithium-TE-PEG: 100 mM LiAc, 10 mM Tris, pH 7.5, 1 mM EDTA, 50% PEG 3350–4000. Sterilize by autoclaving.
Salmon sperm DNA (ssDNA): 10 mg/mL Lyophilized, sheared, organically extracted, and denatured ssDNA is resuspended in double-distilled water (DDW). Store 100 μL aliquots at −20 °C.
Centrifuges (table top): Up to 20,000 × g for samples 1.5 mL.
Heat blocks at 42 °C and 95 °C.
Temperature-controlled shaker for yeast cultures.
Temperature-controlled, Delta-T dishes (i.e., Bioptech Cat# 04200417C).
Concanavalin A (ConA) stock: 10 mg/mL in sterile DDW (10× stock). Store 500 μL aliquots at −20 °C.
ConA-coated plate activation solution: 50 mM CaCl2, 50 mM MnCl2 in DDW. Filter sterilize, store at room temperature.
Fluorescent wide-field microscope of choice for live-cell image acquisition (see Note 1).
Image analysis software: FISH-quant [82], free software developed in the MATLAB programming language (MathWorks). Download the FISH-quant package (http://code.google.com/p/fish-quant/) together with the MCRInstaller, which allows one to run a MATLAB algorithm without separately installing MATLAB onto the computer.
Image analysis software: Fiji (Java software for image-processing analysis; freely available at https://fiji.sc/).
Image analysis software: CellProfiler [83], for cell outline generation (freely available at https://cellprofiler.org/).
Image analysis software: For image deconvolution use a software such as the Huygens Software Suite (https://svi.nl/HomePage).
2.2. Materials for Visualizing MS2-Labeled RNAs in Mammalian Cells, with a Focus on Analyzing Transcription Dynamics
Transcription reporter constructs containing MS2 stem-loop repeats: pIntro-MS2×64 and pIntro-MS2×128 [60]. These plasmids are available upon request (see Fig. 4).
Lentiviral plasmid for MCP fused to a fluorescent protein: pHAGE-Ubc-NLS-MCP-GFP (available upon request) or pHAGE-Ubc-NLS-tdMCP-GFP (Addgene #40649).
Packaging plasmids for lentivirus production (available upon request): pHDM-tat1b (helper plasmid for lentiviral vector, HIV tat driven by CMV promoter); pRC-CMV-rev1b (helper plasmid for lentiviral vector, rev1b driven by CMV promoter); pHDM-Hgpm2 (helper plasmid for lentiviral vector, has codon-optimized HIV gag-pol driven by CMV promoter); pHDM-G (helper plasmid for lentiviral vectors, VSV-G driven by CMV promoter).
XL1-Blue competent cells (see Note 2).
HeLa Flp-in H9 cell line [60], allowing the generation of isogenic stable cell lines by genomic integration in a single integrated Flp recombination target (FRT) site from pFRT/Lac Zeo, under zeocin selection (100 μg/mL) (see Notes 3 and 4).
DMEM + GlutaMAX supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (P/S; 10 U/mL), in a humidified CO2 incubator at 37 °C for propagation conditions.
HEK-293T cell line for lentiviral production.
1× Phosphate-buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4 (without Ca2+ and Mg2+).
Trypsin (0.25%)/EDTA (1 mM).
Transfection reagent: JetPRIME® (Polyplus transfection).
Syringes for filtering (5 mL).
Sterile falcon tube (15 mL).
0.45 μm Cellulose acetate or polyethersulfone filters.
Lenti-X™ Concentrator (Clontech).
Polybrene.
10% Bleach.
Live-cell imaging medium (showing lower background fluorescence) supplemented with 10% FBS.
32% (w/v) Paraformaldehyde (PFA): Store at room temperature, protected from light.
Vectashield mounting medium with DAPI.
Glass microscopy slides.
Noncoated 22 × 22 mm coverslips.
25 mm diameter non-coated coverslips (0.17 mm thick).
Epifluorescent microscope of choice for acquisition of still pictures (see Note 5).
Fluorescent microscope of choice for live-cell image acquisition (see Note 6).
smFISH analysis software (FISH-quant [82], see above) Fiji image-processing software (freely available at https://fiji.sc/).
3. Methods
3.1. Visualizing MBSV6-Labeled mRNAs in Yeast
3.1.1. Yeast Transformation
Grow yeast expressing the mRNA of interest tagged with 24×MBSV6 in 5 mL of YPD at 26 °C until an OD600 of 0.6–0.8 is reached. Do not use cells grown over OD600 >1. The transformation efficiency will be significantly reduced. The method to tag the mRNA with the MS2 system and to verify that the expression of the mRNA is not affected is described in more details in [43, 58] (see Note 7).
Centrifuge the cells for 3 min at 7000 × g. Discard the supernatant and resuspend the cells in 5 mL of lithium-TE.
Centrifuge for 3 min at 1000 × g and resuspend in 150 μL of lithium-TE.
Put 450 μL of lithium-TE-PEG in an Eppendorf tube.
Add 5 μL of 10 mg/mL ssDNA denatured at 95 C for 10 min.
Add 500 ng–1 μg of plasmid expressing MCP-GFP (i.e., YcpLac111 MCP-NLS-2xyeGFP) for each transformation.
Add 150 μL of cells to the tube and mix by gentle vortexing (speed 5 out of 10).
Incubate at room temperature for 30 min.
Heat shock the cells at 42 C for 15 min.
Centrifuge the cells for 3 min at 7000 × g.
Discard the supernatant, resuspend the pellet in 100 μL of DDW, and plate the entire transformation on selective LEU plates.
Incubate at 26 C for 3–4 days. The transformed cells can be used to start cultures for live imaging (see Note 8).
3.1.2. Growing Yeast Cells for Live Imaging
Grow a low-density culture of the yeast strain expressing the endogenously tagged mRNA and MCP-GFP in selective LEU medium overnight at 26 °C. Apply constant shaking at 210 rpm. It is important to keep the cells growing in exponential phase (OD600 <1) at all times.
In the morning, dilute the cells with fresh medium to an OD600 ~0.1 and allow to grow until OD600 0.2–0.3. At this concentration, the autofluorescence of the cells is minimal and the expression of the MCP is homogenous (see Note 9).
3.1.3. Coating of Delta-T Dishes and Plating of Cells
Incubate the Delta-T dishes with 400 μL of ConA at a final concentration 1 mg/mL for 10 min at room temperature.
Aspirate the excess and let the dish air-dry completely.
Activate the ConA coating, by incubating the dish with 400 μL of ConA activating solution, for 10 min at room temperature (see Note 10).
Aspirate the excess and let the dish air-dry completely.
Wash the dish twice with sterile DDW and let air-dry completely.
Plate 500 μL of cells at OD600 0.2–0.3 (see Note 11).
Place the dish on the microscope stage and let the cells attach for at least 15–30 min. Using the Delta-T temperature control system, allow the temperature to stabilize at 26 °C. It is important to wait until cells attach and restart the cell cycle.
3.1.4. Live Imaging Acquisition
The live imaging conditions need to be adapted based on the expression of the mRNA under investigation. We recommend testing the mRNA of interest first by smFISH. Here, we outline the imaging conditions for the constitutive MDN1 mRNA. We previously characterized the expression of this mRNA by smFISH [8, 58] and this protocol is detailed in Chapter 4 of this book as well as in [43, 58]. Figure 1a, b shows an example of smFISH for the MDN1 mRNA in wild-type cells, for side-by-side comparison with the live imaging results shown in Fig. 1c–f. For live imaging, the MDN1 mRNA was tagged at the 3’ UTR with 24×MBSV6 (Fig. 1c) [58]. To distinguish the nucleus from the cytoplasm, the nuclear pore protein Nup49 is endogenously fused to the red fluorescent protein tdTomato (YET443 MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0 NUP49::NUP49-tdTomato KAN- CRE recombined MDN1:: MDN1 3UTR 24MS2V6 KAN- CRE recombined; Ycp-Lac111 CYC1p MCP-NLS-2xyeGFP).
For short-term imaging and fast acquisition to follow mRNAs with high temporal resolution, stream one single Z-plane, 50 ms exposure. Detection of single mRNAs tagged with MCP-GFP is achieved by using 10% of 100 mW 491 laser (~1–2 mW/cm2 measured at the objective). To detect Nup49-tdTomato, use 1% of 50 mW 561 laser (~0.5 mW/cm2 measured at the objective). Under these conditions the mRNAs in the cytoplasm and the transcription sites in the nucleus can be visualized for 2–3 min before significant photo-bleaching occurs (see Supplementary Video 1).
For long-term imaging, i.e., over the course of a complete cell cycle, and to cover the whole cell width, take 15 Z-stacks every 0.5 μm every 2 min (~90 Z-stacks total). An exposure of 50 ms for each Z-plane was used under our conditions. To visualize MBSV6-MCP-NLS-2xyeGFP-labeled mRNAs use a 491 nm wavelength laser. For visualization of single mRNA molecules, set the laser to 10% power (~1–2 mW/cm2 measured at the objective). To detect Nup49-tdTomato, use 1% of 50 mW 561 laser (~0.5 mW/cm2 measured at the objective). Acquire Z-planes at different stage positions and use them to detect the number, the position, and the brightness of mRNAs in living cells (Fig. 1d and see Note 12).
3.1.5. Imaging Analysis
To improve the signal-to-noise ratio, restore the images using a deconvolution software such as the Huygens software package. Automatically compute the theoretical point spread function based on your microscope settings. Restore the images using the classic maximum likelihood estimation algorithm (i.e., number of iterations = 99; signal/noise ratio = 15).
To measure the number, position, and brightness of the mRNAs in single cells, use the freely available software FISH-quant running on Matlab [82]. Deconvolved images can be analyzed with FISH-quant without further filtering. Cell outlines can be created using FISH-quant or using the freely available software CellProfiler [83]. Recent versions of FISH-quant have a built-in plug-in converting CellProfiler outlines for FISH-quant analysis. 3D gaussian fitting of the single spots generates a text file containing the x,y position, brightness, and number of spots identified in each cell. In our experiments, the counting of MDN1 mRNA molecules per cell detected with MCP-2xyeGFP revealed a mean ± S.D. = 10.8 ± 6.1 mRNAs/cell. Counting the MDN1 mRNAs per cell by smFISH gave similar results, i.e., a mean ± S.D. = 9.5 ± 4.4 mRNAs/cell (Fig. 1h). These results show that live imaging faithfully reports on MDN1 mRNA expression.
3.2. Visualizing MS2-Labeled RNAs in Mammalian Cells, with a Focus on Analyzing Transcription Dynamics
3.2.1. Lentiviral Production of MCP-GFP
Seed HEK-293T cells at 4 × 106 cells in 100 mm tissue culture plate and incubate cells for 24 h.
Change HEK-293T cells into 5 mL of fresh medium without antibiotics.
Transfect HEK-293T cells with JetPRIME (Polyplus transfection; according to the supplier’s recommendations) by preparing a mix of 10 μg DNA total with 8 μg of pHAGE-Ubc-NLS-MCP-GFP, 0.4 μg of pHDM-Hgpm2, 0.4 μg of pHDM-tat1b, 0.4 μg of pRC-CMV-rev1b, 0.8 μg of pHDM-G, and 500 μL jetPRIME buffer.
Mix by vortexing, add 20 μL of JetPRIME, vortex for 5 s, spin down briefly, and incubate for 10 min at room temperature.
Add the transfection mix dropwise onto the HEK-293T cells. Gently rock the plate back and forth and incubate at 37 °C for 24 h (see Note 13).
Harvest medium containing lentivirus, filter it through a 0.45 μm filter into a sterile falcon tube, and keep it at 4 °C.
Add 5 mL of fresh medium without antibiotics to the packaging cells and incubate at 37 °C for 24 h for a new round of lentivirus production.
Repeat steps 6 and 7 once. After the harvest of virus, discard the HEK-293T cells with 10% bleach.
Pool the viral harvests of this and the previous days.
To concentrate lentiviral stocks, add Lenti-X Concentrator (Clontech) to viral harvest and incubate at 4 °C for 30 min to overnight.
Centrifuge the mixture at low speed (1500 × g for 45 min at 4 °C) and discard the supernatant.
Resuspend the pellet in DMEM and aliquot for titration and single-use aliquots. Store at −80 °C (see Note 14).
3.2.2. Generating Stable Cell Lines Expressing MCP-GFP in MS2×64 RNA Reporter Cell Line
Seed MS2×64 RNA reporter cells (see Note 4) to a low density (5000 cells per well) in a 12-well plate.
Thaw a lentiviral single-use aliquot at 37 °C, prepare a range of virus dilutions (e.g., 1:10; 1:50; 1:100) in DMEM (the volume of 300 μL is enough to cover a 12 well-plate dish) without serum and 6 μg/mL of polybrene (see Note 15), and mix well.
Remove the medium from cells, rinse once with DMEM, remove and add the virus dilution, and incubate at 37 °C. Tilt the vessel back and forth to mix the virus every 20 min. After 2 h, add 1 mL of fresh medium with serum and allow cells to recover overnight.
Change medium the next day and treat all culture supernatant as hazardous waste for several days afterward (5 days). Expand and passage cells as normal for a week.
3.2.3. Screening Polyclonal Population Expressing MCP-GFP
Grow a fraction of the cells on 22 × 22 mm coverslips in a 6-well plate dish.
Wash briefly in 1× PBS and fix the cells with PFA 4% in PBS for 20 min at room temperature.
Wash briefly in 1× PBS, add 20 μL of Vectashield mounting medium on glass microscopy slides, and mount the coverslips upside down.
Using an epifluorescence microscope, select the pool of cells based on GFP expression (see Note 16).
3.2.4. Growing Cells for Live Imaging
Split the cells to 50% confluence 1 day before imaging on a 25 mm diameter coverslips or a glass-bottomed tissue culture plates (based on the microscope used).
Set up the incubator chamber of the microscope at 37 °C and 5% CO2 1 h before starting your experiment to avoid thermal fluctuation leading to optical instability and cellular stress.
Set up the coverslips on the microscopic chamber and 30 min before starting imaging replace the media with live-cell imaging medium supplemented with 10% FBS and P/S.
3.2.5. Live Image Acquisition
Determine experimental parameters for live-cell imaging. To minimize photobleaching, the light intensity and the exposure time need to be set to the lowest values allowing visualization of single pre-mRNA molecules in the nucleus. These parameters are dependent on the microscope used (see Note 6) and the light source and should be strictly determined when starting imaging (see Note 17). This should be done at the beginning of each experiment.
For each cell, determine the correct focal plane, as well as the boundaries of the Z-stacks (see Note 18). For transcriptional studies, two types of movies can be recorded: short movies (fast acquisition) where one Z-stack is recorded every 3 s for 30 min and long movies where one Z-stack is recorded every 3 min for >8 h (see Note 19).
Analyze the time-lapse movies of transcription sites with dedicated software tools that are available upon request (MS2-quant, RampFinder, RampFitter, ON-quant, see ref. 60) (see Note 20).
Supplementary Material
Acknowledgments
This work was supported by NIH Grant GM57071 to R.H.S., and by an ANRS Grant to E.B. E.T. was supported by Swiss National Science Foundation Fellowships P2GEP3_155692 and P300PA_164717. X.P. was supported by a fellowship from the Labex EpiGenMed Montpellier/Université de Montpellier, and E.B. had a travel grant from the Philippe Foundation.
Footnotes
Yeast microscopy experiments were performed on a home-built microscope built around an IX71 stand (Olympus). For excitation, a 491 nm laser (CalypsoTM, Cobolt) and a 561 nm laser (JiveTM, Cobolt) were combined and controlled by an acoustic-optic tunable filter (AOTF, AOTFnC-400.650-TN, AA Opto-electronic) before being coupled into a single-mode optical fiber (Qioptiq). The output of the fiber was collimated and delivered through the back port of the microscope and reflected into an Olympus 150× 1.45 N.A. oil immersion objective lens with a dichroic mirror (zt405/488/561rpc, 2 mm substrate, Chroma). The tube lens (180 mm focal length) was removed from the microscope and placed outside of the right port. A triple-band notch emission filter (zet405/488/561 nm) was used to filter the scattered laser light. A dichroic mirror (T560LPXR, 3 mm substrate, Chroma) was used to split the fluorescence onto two precisely aligned EMCCDs (Andor iXon, Model DU-897 U-CS0, pixel size 16 μm) mounted on alignment stages (x, y, z, θ-, and φ-angle). Emission filters FF03–525/50–25 and FF01–607/70–25 (Semrock) were placed in front of green and red channel cameras, respectively. The two cameras were triggered for exposure with a TTL pulse generated on a DAQ board (Measurement Computing). The microscope was equipped with a piezo stage (ASI) for fast z-stack and a Delta-T incubation system (Bioptech) for live-cell imaging. The microscope (AOTF, DAQ, stage, and cameras) was automated with the software MetaMorph (Molecular Devices).
XL1-blue competent cells are bacteria of choice for transformation of plasmid containing MS2 repeats. Note that the bacteria can be grown at 30 °C if the plasmid is unstable.
Other Flp-in mammalian cell lines can be used.
A stable Flag-Tat-expressing cell line was created by CRISPR genome editing using an AAVS1 repair vector [60] in HeLa Flp-in H9 cell line (available upon request). Individual clones were picked and analyzed by immunofluorescence with an anti-Flag antibody. One clone was further characterized and used for the following experiments. Isogenic stable cell lines expressing the HIV-1 MS2×64 reporter gene were created using the Flp-in system in a HeLa H9 AAVS1-Tat cell line [60]. Flp-in integrants were selected on hygromycin (150 μg/mL). The MS2 tag is located in the intron of the reporter and thus labels only the pre-mRNA. Note that splicing of this reporter occurs post-transcriptionally and is not disturbed by the MS2 repeat [60]. We found that this setup was the most appropriate to visualize transcription because it allows the use of large tags without compromising the mRNA fate. Individual clones were picked and analyzed by in situ hybridization. One clone was further characterized and used for the following experiments.
A ZEISS Axioimager Z1 wide-field microscope equipped witha Plan Apochromat 63x objective, N.A. 1.4 oil-immersion objective (ZEISS), was used with a ZEISS VSG HBO 100/001.26E illuminating System, and Zyla 4.2 sCMOS Camera (2048 × 2048 pixels; 6.5 μm pixel size, from Andor). We acquire data using 21 optical sections with a z-step size of 0.3 μm. MetaMorph (Molecular Devices) software is used for instrument control as well as image acquisition.
An inverted OMX Deltavision microscope in time-lapse mode with temperature-controlled chamber with CO2, together with a ×100, N.A. 1.4 objective and EMCCD cameras Evolve 512 × 512, was used for live-cell imaging. Spinning disk confocal or HiLo microscopes equipped with 60× or 100× objectives with a N.A. >1.3 are also suitable.
To maximize the brightness of the tagged mRNAs, we recommend tagging the mRNA of interest with 24×MBSV6. However, as we discussed in previous publications [43, 58], for mRNAs that are strongly expressed and that have a short half-life (i.e., GAL1 mRNA), tagging with 24 stem-loops can be suboptimal. This is because when many mRNAs have to be rapidly degraded, the presence of 24 MS2 loops, even if they are low-affinity variant, can cause a delay in the degradation of the MS2 array. In this case the mRNAs can be tagged with 12×MBSV6. We recommend always testing whether the insertion of the MS2 loops affects the stability, the localization, and the expression of the mRNA of interest by comparing the tagged mRNA (with or without the expression of MCP) to the untagged mRNA by smFISH [43, 58].
Transformed cells can be frozen at this stage. Glycerol stocks can be prepared by mixing 1 mL of exponentially growing culture with 1 mL of 60% glycerol in YPD. Mix thoroughly and freeze in cryo-tubes at −80 °C. We did not notice a reduction in live imaging quality if the cells are thawed instead of using fresh transformations.
Background strains that are ADE2+, thus not producing the red pigment accumulating in mutant cells, show reduced cellular background during live imaging.
For short-term imaging, not requiring strict temperature control, other types of glass-bottom dishes can be used, i.e., MatTek.
High-quality and reproducible imaging is achieved only if the-cells are imaged while growing exponentially.
The parameters for imaging single mRNAs in living cells (laser power, exposure time) can be modified to increase the fluorescence intensity of single mRNAs. Choose parameters that will keep the fluorescence intensity signal in the dynamic range of the camera while minimizing photo-bleaching of the sample.
At this point active lentivirus is being produced. Strict adherence to biosafety class II is necessary. All materials in contact with virus-containing liquid must be bleached prior to disposal.
Viruses may be stored at 4 °C for short periods (hours to days). For long-term storage, aliquots should be frozen at −80 °C.
Polybrene increases the efficiency of retrovirus-mediated gene transfer; the conditions should be optimized for each cell type.
Expression levels of the MCP-FP transgene should show a strong spot for the transcription site, smaller dots throughout the nucleus (single-molecule mRNA), and a low nuclear background of free unbound MCP-FP. If needed, the best pool can be re-sorted by FACS to obtain a more homogenous population. Positive cells can be FACS-sorted upon a single-cell clonal dilution to isolate pure clones. Select the most suitable sub-cloned cells that display the optimal signal-to-noise ratio for the live-cell imaging acquisition.
We recommend a stable integration of the MCP-GFP with viral infection followed by sorting a pool of low-expressing MCP-FP cells. This gives much better results than a crude transient transfection. Optimization of the MCP-GFP expression further improves the signal-to-noise ratio of single-molecule detection and enhances the quality of the data recorded.
We recommend stacks of 11 planes with a Z-spacing of 0.6 μm. This size allows accurate quantification of single mRNA molecules when using 100×, N.A. 1.4 objectives.
These short and long movies are important if one wants to capture the entire promoter dynamics (see ref. 60), and they are required since at high temporal resolution, bleaching limits the acquisitions to about 30 min.
After being processed, each movie should be checked to ensure that no drift or loss of focus happened during the acquisition (especially for long-term movies).
Electronic supplementary material: The online version of this chapter (https://doi.org/10.1007/978-1-0716-0712-1_7) contains supplementary material, which is available to authorized users.
References
- 1.Tutucci E, Livingston NM, Singer RH, Wu B (2018) Imaging mRNA in vivo, from birth to death. Annu Rev Biophys 47:85–106. 10.1146/annurev-biophys-070317-033037 [DOI] [PubMed] [Google Scholar]
- 2.Vera M, Biswas J, Senecal A, Singer RH, Park HY (2016) Single-cell and single-molecule analysis of gene expression regulation. Annu Rev Genet 50:267–291. 10.1146/annurev-genet-120215-034854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raj A, van Oudenaarden A (2008) Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135(2):216–226. 10.1016/j.cell.2008.09.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Beyer A, Aebersold R (2016) On the dependency of cellular protein levels on mRNA abundance. Cell 165(3):535–550. 10.1016/j.cell.2016.03.014 [DOI] [PubMed] [Google Scholar]
- 5.Femino AM, Fay FS, Fogarty K, Singer RH (1998) Visualization of single RNA transcripts in situ. Science 280(5363):585–590 [DOI] [PubMed] [Google Scholar]
- 6.Femino AM, Fogarty K, Lifshitz LM, Carrington W, Singer RH (2003) Visualization of single molecules of mRNA in situ. Methods Enzymol 361:245–304 [DOI] [PubMed] [Google Scholar]
- 7.Gandhi SJ, Zenklusen D, Lionnet T, Singer RH (2011) Transcription of functionally related constitutive genes is not coordinated. Nat Struct Mol Biol 18(1):27–34. 10.1038/nsmb.1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zenklusen D, Larson DR, Singer RH (2008) Single-RNA counting reveals alternative modes of gene expression in yeast. Nat Struct Mol Biol 15(12):1263–1271. 10.1038/nsmb.1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senecal A, Munsky B, Proux F, Ly N, Braye FE, Zimmer C, Mueller F, Darzacq X (2014) Transcription factors modulate c-Fos transcriptional bursts. Cell Rep 8(1):75–83. 10.1016/j.celrep.2014.05.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5(10):877–879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L, Titlow J, Ennis D, Smith C, Mitchell J, Young FL, Waddell S, Ish-Horowicz D, Davis I (2017) Single molecule fluorescence in situ hybridisation for quantitating post-transcriptional regulation in Drosophila brains. Methods 126:166–176. 10.1016/j.ymeth.2017.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buxbaum AR, Wu B, Singer RH (2014) Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science 343:419–422. 10.1126/science.1242939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duncan S, Olsson TSG, Hartley M, Dean C, Rosa S (2016) A method for detecting single mRNA molecules in Arabidopsis thaliana. Plant Methods 12:13. 10.1186/s13007-016-0114-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moor AE, Golan M, Massasa EE, Lemze D, Weizman T, Shenhav R, Baydatch S, Mizrahi O, Winkler R, Golani O, Stern-Ginossar N, Itzkovitz S (2017) Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357:1299–1303. 10.1126/science.aan2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffitt JR, Hao J, Bambah-Mukku D, Lu T, Dulac C, Zhuang X (2016) High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc Natl Acad Sci U S A 113(50):14456–14461. 10.1073/pnas.1617699113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long X, Colonell J, Wong AM, Singer RH, Lionnet T (2017) Quantitative mRNA imaging throughout the entire Drosophila brain. Nat Methods 14(7):703–706. 10.1038/nmeth.4309 [DOI] [PubMed] [Google Scholar]
- 17.Battich N, Stoeger T, Pelkmans L (2013) Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat Methods 10(11):1127–1133. 10.1038/nmeth.2657 [DOI] [PubMed] [Google Scholar]
- 18.Choi HM, Chang JY, Trinh le A, Padilla JE, Fraser SE, Pierce NA (2010) Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat Biotechnol 28(11):1208–1212. 10.1038/nbt.1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsanov N, Samacoits A, Chouaib R, Traboulsi AM, Gostan T, Weber C, Zimmer C, Zibara K, Walter T, Peter M, Bertrand E, Mueller F (2016) smiFISH and FISH-quant - a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res 44(22):e165. 10.1093/nar/gkw784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, Zhuang X (2016) High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc Natl Acad Sci U S A 113(39):11046–11051. 10.1073/pnas.1612826113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L (2014) Single-cell in situ RNA profiling by sequential hybridization. Nat Methods 11(4):360–361. 10.1038/nmeth.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah S, Lubeck E, Zhou W, Cai L (2017) seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron 94(4):752–758. 10.1016/j.neuron.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 23.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC, Cai L (2019) Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568:235–239. 10.1038/s41586-019-1049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah S, Takei Y, Zhou W, Lubeck E, Yun J, Eng CL, Koulena N, Cronin C, Karp C, Liaw EJ, Amin M, Cai L (2018) Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174(2):363–376. 10.1016/j.cell.2018.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayer LV, Batish M, Formel SK, Bratu DP (2015) Single-molecule RNA in situ hybridization (smFISH) and immunofluorescence (IF) in the Drosophila egg chamber. Methods Mol Biol 1328:125–136. 10.1007/978-1-4939-2851-4_9 [DOI] [PubMed] [Google Scholar]
- 26.Eliscovich C, Shenoy SM, Singer RH (2017) Imaging mRNA and protein interactions within neurons. Proc Natl Acad Sci U S A 114(10):E1875–E1884. 10.1073/pnas.1621440114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buxbaum AR, Haimovich G, Singer RH (2015) In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol 16(2):95–109. 10.1038/nrm3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weil TT, Parton RM, Davis I (2010) Making the message clear: visualizing mRNA localization. Trends Cell Biol 20(7):380–390. 10.1016/j.tcb.2010.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin KC, Ephrussi A (2009) mRNA localization: gene expression in the spatial dimension. Cell 136(4):719–730. 10.1016/j.cell.2009.01.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecuyer E, Yoshida H, Parthasarathy N, Alm C, Babak T, Cerovina T, Hughes TR, Tomancak P, Krause HM (2007) Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131(1):174–187. 10.1016/j.cell.2007.08.003 [DOI] [PubMed] [Google Scholar]
- 31.Samacoits A, Chouaib R, Safieddine A, Traboulsi AM, Ouyang W, Zimmer C, Peter M, Bertrand E, Walter T, Mueller F (2018) A computational framework to study sub-cellular RNA localization. Nat Commun 9(1):4584. 10.1038/s41467-018-06868-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das S, Singer RH, Yoon YJ (2019) The travels of mRNAs in neurons: do they know where they are going? Curr Opin Neurobiol 57:110–116. 10.1016/j.conb.2019.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahar Halpern K, Tanami S, Landen S, Chapal M, Szlak L, Hutzler A, Nizhberg A, Itzkovitz S (2015) Bursty gene expression in the intact mammalian liver. Mol Cell 58(1):147–156. 10.1016/j.molcel.2015.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S (2006) Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4(10):e309. 10.1371/journal.pbio.0040309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul B, Montpetit B (2016) Altered RNA processing and export lead to retention of mRNAs near transcription sites and nuclear pore complexes or within the nucleolus. Mol Biol Cell 27(17):2742–2756. 10.1091/mbc.E16-04-0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bahar Halpern K, Caspi I, Lemze D, Levy M, Landen S, Elinav E, Ulitsky I, Itzkovitz S (2015) Nuclear retention of mRNA in mammalian tissues. Cell Rep 13(12):2653–2662. 10.1016/j.celrep.2015.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trcek T, Larson DR, Moldon A, Query CC, Singer RH (2011) Single-molecule mRNA decay measurements reveal promoter-regulated mRNA stability in yeast. Cell 147(7):1484–1497. 10.1016/j.cell.2011.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trcek T, Sato H, Singer RH, Maquat LE (2013) Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev 27(5):541–551. 10.1101/gad.209635.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pichon X, Lagha M, Mueller F, Bertrand E (2018) A growing toolbox to image gene expression in single cells: sensitive approaches for demanding challenges. Mol Cell 71(3):468–480. 10.1016/j.molcel.2018.07.022 [DOI] [PubMed] [Google Scholar]
- 40.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM (1998) Localization of ASH1 mRNA particles in living yeast. Mol Cell 2(4):437–445 [DOI] [PubMed] [Google Scholar]
- 41.Peabody DS (1993) The RNA binding site of bacteriophage MS2 coat protein. EMBO J 12(2):595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fusco D, Bertrand E, Singer RH (2004) Imaging of single mRNAs in the cytoplasm of living cells. Prog Mol Subcell Biol 35:135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tutucci E, Vera M, Singer RH (2018) Single-mRNA detection in living S. cerevisiae using a re-engineered MS2 system. Nat Protoc 13(10):2268–2296. 10.1038/s41596-018-0037-2 [DOI] [PubMed] [Google Scholar]
- 44.Brody Y, Neufeld N, Bieberstein N, Causse SZ, Bohnlein EM, Neugebauer KM, Darzacq X, Shav-Tal Y (2011) The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol 9(1):e1000573. 10.1371/journal.pbio.1000573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH (2007) In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14(9):796–806. 10.1038/nsmb1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt U, Basyuk E, Robert MC, Yoshida M, Villemin JP, Auboeuf D, Aitken S, Bertrand E (2011) Real-time imaging of cotranscriptional splicing reveals a kinetic model that reduces noise: implications for alternative splicing regulation. J. Cell Biol 193(5):819–829. 10.1083/jcb.201009012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grunwald D, Singer RH (2010) In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport. Nature 467(7315):604–607. 10.1038/nature09438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith C, Lari A, Derrer CP, Ouwehand A, Rossouw A, Huisman M, Dange T, Hopman M, Joseph A, Zenklusen D, Weis K, Grunwald D, Montpetit B (2015) In vivo single-particle imaging of nuclear mRNA export in budding yeast demonstrates an essential role for Mex67p. J Cell Biol 211(6):1121–1130. 10.1083/jcb.201503135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, Wells AL, Chao JA, Park HY, de Turris V, Lopez-Jones M, Singer RH (2011) A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods 8(2):165–170. 10.1038/nmeth.1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park HY, Lim H, Yoon YJ, Follenzi A, Nwokafor C, Lopez-Jones M, Meng X, Singer RH (2014) Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science 343(6169):422–424. 10.1126/science.1239200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halstead JM, Lionnet T, Wilbertz JH, Wippich F, Ephrussi A, Singer RH, Chao JA (2015) Translation. An RNA biosensor for imaging the first round of translation from single cells to living animals. Science 347(6228):1367–1671. 10.1126/science.aaa3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katz ZB, English BP, Lionnet T, Yoon YJ, Monnier N, Ovryn B, Bathe M, Singer RH (2016) Mapping translation ‘hot-spots’ in live cells by tracking single molecules of mRNA and ribosomes. elife 5:e10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, Lavis LD, Grimm JB, Viswanathan S, Looger LL, Lionnet T, Stasevich TJ (2016) Real-time quantification of single RNA translation dynamics in living cells. Science 352(6292):1425–1429. 10.1126/science.aaf0899 [DOI] [PubMed] [Google Scholar]
- 54.Pichon X, Bastide A, Safieddine A, Chouaib R, Samacoits A, Basyuk E, Peter M, Mueller F, Bertrand E (2016) Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J Cell Biol 214(6):769–781. 10.1083/jcb.201605024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu B, Eliscovich C, Yoon YJ, Singer RH (2016) Translation dynamics of single mRNAs in live cells and neurons. Science 352(6292):1430–1435. 10.1126/science.aaf1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia JF, Parker R (2015) MS2 coat proteins bound to yeast mRNAs block 5’ to 3’ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system. RNA 21(8):1393–1395. 10.1261/rna.051797.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haimovich G, Zabezhinsky D, Haas B, Slobodin B, Purushothaman P, Fan L, Levin JZ, Nusbaum C, Gerst JE (2016) Use of the MS2 aptamer and coat protein for RNA localization in yeast: A response to “MS2 coat proteins bound to yeast mRNAs block 5’ to 3’ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system”. RNA 22(5):660–666. 10.1261/rna.055095.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tutucci E, Vera M, Biswas J, Garcia J, Parker R, Singer RH (2018) An improved MS2 system for accurate reporting of the mRNA life cycle. Nat Methods 15(1):81–89. 10.1038/nmeth.4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinrich S, Sidler CL, Azzalin CM, Weis K (2017) Stem-loop RNA labeling can affect nuclear and cytoplasmic mRNA processing. RNA 23(2):134–141. 10.1261/rna.057786.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tantale K, Mueller F, Kozulic-Pirher A, Lesne A, Victor JM, Robert MC, Capozi S, Chouaib R, Backer V, Mateos-Langerak J, Darzacq X, Zimmer C, Basyuk E, Bertrand E (2016) A single-molecule view of transcription reveals convoys of RNA polymerases and multiscale bursting. Nat Commun 7:12248. 10.1038/ncomms12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chao JA, Patskovsky Y, Almo SC, Singer RH (2008) Structural basis for the coevolution of a viral RNA-protein complex. Nat Struct Mol Biol 15(1):103–105. 10.1038/nsmb1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH (2011) Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332(6028):475–478. 10.1126/science.1202142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saroufim MA, Bensidoun P, Raymond P, Rahman S, Krause MR, Oeffinger M, Zenklusen D (2015) The nuclear basket mediates perinuclear mRNA scanning in budding yeast. J Cell Biol 211(6):1131–1140. 10.1083/jcb.201503070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang C, Han B, Zhou R, Zhuang X (2016) Real-time imaging of translation on single mrna transcripts in live cells. Cell 165(4):990–1001. 10.1016/j.cell.2016.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan X, Hoek TA, Vale RD, Tanenbaum ME (2016) Dynamics of translation of single mRNA molecules In vivo. Cell 165(4):976–989. 10.1016/j.cell.2016.04.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das S, Moon HC, Singer RH, Park HY (2018) A transgenic mouse for imaging activitydependent dynamics of endogenous Arc mRNA in live neurons. Sci Adv 4(6):eaar3448. 10.1126/sciadv.aar3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen J, Nikolaitchik O, Singh J, Wright A, Bencsics CE, Coffin JM, Ni N, Lockett S, Pathak VK, Hu WS (2009) High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc Natl Acad Sci U S A 106(32):13535–13540. 10.1073/pnas.0906822106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lange S, Katayama Y, Schmid M, Burkacky O, Brauchle C, Lamb DC, Jansen RP (2008) Simultaneous transport of different localized mRNA species revealed by live-cell imaging. Traffic 9(8):1256–1267. 10.1111/j.1600-0854.2008.00763.x [DOI] [PubMed] [Google Scholar]
- 69.Brodsky AS, Silver PA (2002) Identifying proteins that affect mRNA localization in living cells. Methods 26(2):151–155. 10.1016/S1046-2023(02)00017-8 [DOI] [PubMed] [Google Scholar]
- 70.Takizawa PA, Vale RD (2000) The myosin motor, Myo4p, binds Ash1 mRNA via the adapter protein, She3p. Proc Natl Acad Sci U S A 97(10):5273–5278. 10.1073/pnas.080585897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu X, Zhang J, Tsien RY (2017) The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem Sci 42(2):111–129. 10.1016/j.tibs.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Specht EA, Braselmann E, Palmer AE (2017) A critical and comparative review of fluorescent tools for live-cell imaging. Annu Rev Physiol 79:93–117. 10.1146/annurev-physiol-022516-034055 [DOI] [PubMed] [Google Scholar]
- 73.Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, Kremers GJ, Davidson MW, Ustione A, Piston DW (2016) Quantitative assessment of fluorescent proteins. Nat Methods 13(7):557–562. 10.1038/nmeth.3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scott DJ, Gunn NJ, Yong KJ, Wimmer VC, Veldhuis NA, Challis LM, Haidar M, Petrou S, Bathgate RAD, Griffin MDW (2018) A novel ultra-stable, monomeric green fluorescent protein for direct volumetric imaging of whole organs using CLARITY. Sci Rep 8(1):667. 10.1038/s41598-017-18045-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng S, Sekine S, Pessino V, Li H, Leonetti MD, Huang B (2017) Improved split fluorescent proteins for endogenous protein labeling. Nat Commun 8(1):370. 10.1038/s41467-017-00494-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Slubowski CJ, Funk AD, Roesner JM, Paulissen SM, Huang LS (2015) Plasmids for C-terminal tagging in Saccharomyces cerevisiae that contain improved GFP proteins, Envy and Ivy. Yeast 32(4):379–387. 10.1002/yea.3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim F, Peabody DS (1994) Mutations that increase the affinity of a translational repressor for RNA. Nucleic Acids Res 22(18):3748–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cormack BP, Valdivia RH, Falkow S (1996) FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33–38 [DOI] [PubMed] [Google Scholar]
- 79.Zacharias DA, Violin JD, Newton AC, Tsien RY (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296(5569):913–916. 10.1126/science.1068539 [DOI] [PubMed] [Google Scholar]
- 80.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS (2006) Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24(1):79–88. 10.1038/nbt1172 [DOI] [PubMed] [Google Scholar]
- 81.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10(5):407–409. 10.1038/nmeth.2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mueller F, Senecal A, Tantale K, Marie-Nelly H, Ly N, Collin O, Basyuk E, Bertrand E, Darzacq X, Zimmer C (2013) FISH-quant: automatic counting of transcripts in 3D FISH images. Nat Methods 10(4):277–278. 10.1038/nmeth.2406 [DOI] [PubMed] [Google Scholar]
- 83.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7(10):R100. 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.