

Abstract
Podocyte damage is strongly associated with the progression of diabetic nephropathy. Mitotic catastrophe plays an essential role in accelerating podocyte loss and detachment from the glomerular basement membrane. In the current study, we observed that the long non-coding RNA (lncRNA) MIAT was noticeably upregulated in the plasma and kidney tissues of patients with diabetic nephropathy, and this upregulation was accompanied by higher albumin/creatinine ratios and serum creatinine levels. By generating CRISPR-Cas9 Miat-knockout (KO) mice in vivo and employing vectors in vitro, we found that the depletion of Miat expression significantly restored slit-diaphragm integrity, attenuated foot process effacement, prevented dedifferentiation, and suppressed mitotic catastrophe in podocytes during hyperglycemia. The mechanistic investigation revealed that Miat increased Sox4 expression and subsequently regulated p53 ubiquitination and acetylation, thereby inhibiting the downstream factors CyclinB/cdc2 by enhancing p21cip1/waf1 activity, and that Miat interacted with Sox4 by sponging miR-130b-3p. Additionally, the inhibition of miR-130b-3p with an antagomir in vivo effectively enhanced glomerular podocyte injury and mitotic dysfunction, eventually exacerbating proteinuria. Based on these findings, MIAT may represent a therapeutic target for diabetic nephropathy.
Keywords: MT, diabetic nephropathy, MIAT, podocyte, mitotic catastrophe, Sox4, p53, miR-130b-3p, ubiquitination, acetylation
Graphical abstract
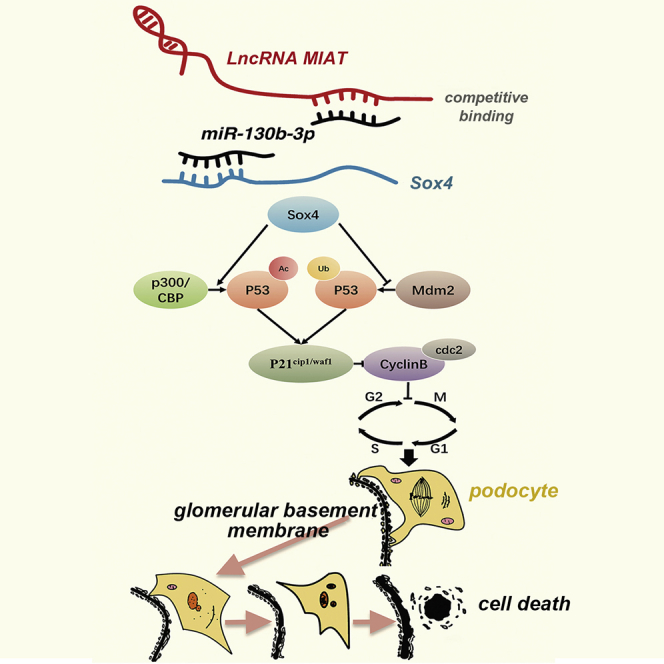
Wang and colleagues showed that the lncRNA MIAT was expressed at high levels in individuals with diabetic nephropathy (DN) and participated in the progression of DN. Miat depletion significantly ameliorated proteinuria by preventing podocyte injury and mitigating podocyte mitotic catastrophe, suggesting that MIAT may represent a therapeutic target for DN.
Introduction
Diabetic nephropathy (DN) is becoming the leading cause of end-stage renal disease (ESRD) in developed countries.1,2 Proteinuria is directly related to podocyte damage and results in podocyte foot process effacement (FPE).3, 4, 5 Mitotic catastrophe is a mechanism of delayed mitosis-related cell death that occurs because of the premature or inappropriate entry of cells into mitosis and the loss of cell-cycle checkpoints; mitotic catastrophe is triggered by chemical or physical stresses that lead to abnormal chromosome segregation. The morphological features of mitotic catastrophe include multiple centrosomes, misaligned chromosomes, aberrant mitotic spindles, micronuclei, or irregularly shaped nuclei.6,7 To date, many renal pathologists have identified occasional mitotic, aneuploid, or binucleated podocytes in individuals with focal segmental glomerular sclerosis (FSGS), minimal-change disease (MCD), and immunoglobulin A (IgA) nephropathy.8
The long non-coding RNA (lncRNA) MIAT, a key gene that is responsible for myocardial infarction,9 is an intergenic lncRNA that is highly conserved across species, including mice and humans.10 MIAT is involved in regulating pathological angiogenesis in individuals with diabetes mellitus (DM) and in exacerbating retinal vessel impairment in mice with DM.11 Moreover, MIAT is closely associated with cell-cycle arrest, a characteristic of mitotic catastrophe, by regulating the percentage of cells in the G0/G1 phase.12,13 All these data emphasized the importance of MIAT as a critical regulator of mitotic progression. However, the precise mechanism by which MIAT participates in podocyte injury and mitotic dysregulation in DN has not been elucidated.
p53 is involved in several fundamental cellular signaling pathways,14,15 among which the most meaningful pathways are apoptosis regulation and cell-cycle arrest.16,17 Sox4, which is upstream of p53,18 is a member of the SRY-related high-mobility group (HMG)-box (SOX) transcription factor family and participates in many cellular processes.19, 20, 21 Notably, good evidence supports a role for a Sox4-induced increase in plasma glucose levels in blocking insulin secretion into the extracellular space via STXBP6 in subjects with type 2 diabetes.22 Hence, additional efforts to elucidate the interaction between Sox4 and p53 during mitotic progression in DN are needed. Based on this information, we hypothesize that MIAT may have notable biological functions in mitotic dysregulation in podocytes by regulating Sox4/p53 during DN. In this study, we characterized the pattern of MIAT expression and investigated its role in podocyte injury and mitotic dysregulation in DN.
Results
MIAT is expressed at high levels in individuals with DN and participates in DN progression
RNA fluorescence in situ hybridization (RNA-FISH) showed that Miat was simultaneously located in the cytoplasm and nucleus (Figure 1A); in contrast, the reference transcript (18S mRNA) was located in the cytoplasm. The fluorescence intensity of Miat increased after high-glucose (HG) (30 mmol/L) stimulation compared with low-glucose (LG) (5.5 mmol/L) stimulation, indicating that Miat was expressed at high levels in podocytes exposed to HG conditions. Similar results were observed using real-time PCR (Figure 1B). Next, we detected the expression of Miat and its relative position to podocin in paraffin-embedded mouse renal tissues. The results revealed that Miat was distributed in the nucleus and cytoplasm and that its expression increased in the podocytes of wild-type, streptozotocin-treated (WT-STZ) mice compared with those of WT, control-treated (WT-CTL) mice at 12 weeks after the onset of diabetes (Figure 1C). Additionally, Miat expression was significantly increased in primary podocytes from WT-STZ mice at 12 weeks compared with those from WT-CTL mice, which further confirmed increased levels of Miat following STZ stimulation (Figure 1D). As shown in Figure 1E, no significant difference in Miat levels was observed between the control and STZ mice at 4 weeks, while a clear increase in Miat expression occurred after 6 weeks as diabetes progressed. This discrepancy between the two groups of mice was more apparent at 12 weeks. As MIAT is highly conserved between mice and humans, we detected its expression in clinical subjects. Compared with the plasma from healthy controls, the plasma from patients with DN exhibited noticeably upregulated MIAT expression (Figure 1F) with microalbuminuria (30–300 mg/g) and macroalbuminuria (>300 mg/g) rather than normal urinary albumin (<30 mg/g), according to the albumin/creatinine ratio (ACR) levels (Figure 1G). These results suggested that MIAT may exert its effects as microalbuminuria progresses to macroalbuminuria in patients with DN. The robust increase in MIAT levels in the plasma from patients with DN was accompanied by an elevated ACR (R = 0.538, p < 0.01) and increased serum creatinine (Scr) levels (R = 0.272, p = 0.034) (Figures 1H and 1I). Moreover, we observed significantly increased MIAT expression in the kidney tissues of patients with DN compared with those of healthy subjects (Figure 1J). The subjects shown in Figure 1J, whose renal biopsies or normal kidney tissues were collected, are part of a subgroup of the subjects shown in Figure 1F. Important information and the clinical characteristics of the subjects are listed in Table S1.
Figure 1.

MIAT is expressed at high levels in individuals with DN and participates in DN progression
(A) RNA-FISH revealed the location and expression of Miat under LG or HG conditions by confocal laser scanning microscope (n = 4). Scale bar, 25 μm. (B) Real-time PCR showed the mRNA levels of Miat in podocytes after LG or HG stimulation for 48 h (n = 7). (C) RNA-FISH illustrated the distribution of Miat and its relative position to podocin in paraffin-embedded mouse sections by confocal laser scanning microscope (n = 3). The podocytes were labeled with arrows. Scale bar, 25 μm. (D) The expression of Miat in primary podocytes from WT-CTL and WT-STZ mice at 12 weeks after the onset of diabetes (n = 3). (E) Expression of Miat in the kidney tissues from WT-CTL mice at 4 (n = 7), 6 (n = 7), 8 (n = 7), and 12 weeks (n = 8) and WT-STZ mice at 4 (n = 8), 6 (n = 8), 8 (n = 9), and 12 weeks (n = 8) after the onset of diabetes. (F) Levels of MIAT in the plasma from clinical patients with DN (n = 61) and their healthy counterparts (n = 22). (G) MIAT expression in the plasma from control subjects (n = 22) and patients with DN presenting normal urinary albumin levels (n = 20), microalbuminuria (n = 21), and macroalbuminuria (n = 20). (H) The correlation between MIAT expression and ACR (R = 0.538, p < 0.01). (I) The correlation between MIAT expression and Scr levels (R = 0.272, p = 0.034). (J) The expression of MIAT in the kidney tissues of clinical patients with DN (n = 20) and healthy subjects (n = 11). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Miat expression is closely related to podocyte injury
We observed that podocalyxin was present in the urinary sediment from patients with DN (Figure 2A, left panel), and urinary synaptopodin mRNA levels were increased in patients with DN (Figure 2A, right panel), suggesting that podocytes may be injured during DN.23,24 The clinical characteristics of the clinical subjects included in the urine sediment study are presented in Table S2. Immunofluorescence staining showed that de novo Desmin expression was induced in the cytoplasm of podocytes by HG, and a decreased fluorescence intensity was observed after Miat knockdown (Figure 2B). Synaptopodin was expressed at high levels in podocytes treated with LG, and this expression was altered by Miat knockdown (Figure 2C). HG stimulation decreased the staining of podocin and ZO-1, and this effect was reversed by Miat knockdown (Figures 2D and 2E). Western-blot analysis revealed reductions in podocin, synaptopodin, and ZO-1 levels under HG conditions, and these effects were accompanied by increased expression of Desmin (Figure 2F), indicating that podocytes were injured under HG conditions; however, Miat shRNA mitigated podocyte injury. Transwell assays indicated higher podocyte motility in HG-treated podocytes than in LG-treated podocytes, but Miat depletion decreased the migratory ability of podocytes (Figure 2G). Furthermore, fluorescein isothiocyanate (FITC)-phalloidin staining showed that the organization of F-actin in podocytes changed from bundles along the cell axis under LG conditions to dense cortical staining under HG conditions for 48 h, and transfection of the Miat shRNA partially reversed this rearrangement of F-actin (Figure 2H). Moreover, as shown in Figure 2I, the levels of injury-associated proteins were altered in podocytes infected with a Miat-overexpressing lentivirus (oeMiat), and this effect was accompanied by increased migration (Figure 2J) and F-actin rearrangement in podocytes (Figure 2K). The efficiencies of the oeMiat lentivirus and Miat short hairpin RNA (shRNA) lentivirus were confirmed using real-time PCR (Figures S1A and S1B).
Figure 2.

Effects of Miat on podocyte injury
(A) Real-time PCR detected the levels of podocalyxin (left panel) and synaptopodin (right panel) in the urinary sediment from patients with DN (n = 25) and healthy individuals (n = 17). (B–E) Immunofluorescence staining showed the intensities of Desmin (B), synaptopodin (C), podocin (D), and ZO-1 (E) (n = 4). Scale bar, 50 μm. (F) Western blotting demonstrated the expression of Desmin, synaptopodin, podocin, and ZO-1 under different conditions (n = 3). (G) The motility of podocytes was determined using Transwell assays (n = 5). Scale bars, 100 μm. (H) FITC-phalloidin staining showed the organization of F-actin in podocytes (n = 3). Scale bar, 50 μm. (I) Podocyte injury-associated proteins were detected using western blotting (n = 3). (J) The motility of podocytes under different conditions was detected using Transwell assays (n = 5). Scale bars, 100 μm. (K) The organization of F-actin was determined using FITC-phalloidin staining (n = 3). Scale bar, 50 μm. Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Miat depletion ameliorates podocyte injury in vivo
With the aid of the CRISPR-Cas9 Miat knockout technique and diabetes model establishment, we further verified the role of Miat in vivo; genotype identification of the Miat-knockout (Miat-KO) mice is illustrated in Figure S1C. No clear differences in hair growth and appearance were observed between the WT and the Miat-KO mice (Figure S1D). Table S3 shows no differences in the systolic blood pressure (SBP) and weight levels and no differences in the trends of spontaneous proteinuria or diabetes between these two strains. As shown using transmission electron microscope (TEM), the podocyte foot processes (FPs) were extensively effaced, retracted, and fused in the WT-STZ mouse glomeruli at 12 weeks, and these effects were accompanied by a thickened glomerular basement membrane (GBM), but the changes were substantially ameliorated in the KO-STZ mice (Figure 3A). Periodic acid-Schiff (PAS) staining showed an expansion of endothelial cells in the WT-STZ mice at 6 weeks, and these cells were present prior to apparent mesangial proliferation, matrix augmentation, and capillary stenosis in the glomerular area at 12 weeks; however, Miat depletion prevented this phenomenon (Figure 3B). Consistent with the results of PAS staining, Masson’s trichrome staining showed marked collagen fibrosis and inflammatory cell invasion in the DN model mice at 12 weeks compared with the control mice (Figure 3C), while the KO-STZ mice exhibited reduced STZ-induced glomerular injury at 6, 8, and 12 weeks. Additionally, the typical and representative morphology of podocytes in clinical patients was detected using PAS staining and TEM (Figures 3D and 3E). Consistently, the fluorescence intensities of podocin, synaptopodin, and ZO-1 were decreased, along with increased staining for Desmin and a decreased level of WT-1, in the WT-STZ groups compared with the control groups at 12 weeks. Miat deficiency reversed these trends in the expression of these proteins (Figures 3F–3J and S1E). The protein levels of these injury markers in primary podocytes were confirmed with immunofluorescence staining, whereas KO of Miat clearly attenuated these effects, as described above (Figure 3K). In addition, mouse proteinuria, which occurred at approximately 8 weeks, was attenuated in the KO-STZ mice compared with the WT-STZ mice, and the pronounced changes in podocytes at approximately 12 weeks were also attenuated (Table S3).
Figure 3.

Miat depletion ameliorates podocyte injury in vivo
(A) Morphological changes in the FPs and GBM were observed under TEM. The GBM thickness and podocyte effacement were qualified (n = 3). Scale bar, 1 μm. (B) PAS staining of tissues from WT and KO mice. The relative mesangial area and glomerular volume were qualified (n = 4). Scale bar, 50 μm. (C) Masson’s trichrome staining of tissues from WT and KO mice (n = 4). Scale bar, 50 μm. (D) PAS staining of tissues from clinical patients with different levels of progression. Scale bar, 100 μm. (E) TEM image of tissues from clinical patients with different levels of progression. Scale bar, 2 μm. (F–I) Immunofluorescence staining for Desmin (F), podocin (G), synaptopodin (H), and ZO-1 (I) in WT and KO mice at 12 weeks (n = 3). Scale bar, 50 μm. (J) The expression of WT-1 in glomeruli of WT and KO mice at 12 weeks (n = 3). Scale bar, 50 μm. (K) Western blotting exemplified the expression of Desmin, podocin, synaptopodin, and ZO-1 in primary podocytes of WT and KO mice at 12 weeks (n = 3). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Silencing of Miat mitigates podocyte mitotic catastrophe via the Sox4 pathway in vitro
As shown in Figure 4A, cultured podocytes exhibited a higher apoptosis rate under HG conditions, and the rate was decreased when the cells were treated with the Miat shRNA lentivirus. The flow-cytometry analysis showed that when podocytes were grown under LG conditions, 8.98% of the population was in the G2/M phase, and approximately 24.24% of the podocytes were arrested at the G2/M phase when grown in the presence of HG. However, this HG-induced G2/M phase arrest was prevented by pretreatment with the Miat shRNA lentivirus (Figure 4B). Immunostaining of mitotic processes confirmed that immature podocytes (33°C) proliferated by forming normal mitotic spindles and exhibited an appropriate chromosomal arrangement in the metaphase plate. Mature podocytes cultured at 37°C did not proliferate under LG conditions, but HG triggered the formation of abnormal mitotic spindles and aberrant nuclear shapes, including multiple centrosomes (multipolar spindles), micronuclei, and spindle collapse in interphase cells. However, the podocytes treated with the Miat shRNA remained H3-Ser10-negative and lacked mitotic spindles, indicating a non-mitotic state (Figure 4C). The percentage of anomalous mitotic cells was quantified and is shown in Figure 4D. P21cip1/waf1, the first transcriptional target of p53 to be identified,25,26 not only mediates the G1/S checkpoint transition but also plays a crucial role in the G2 checkpoint via the CyclinB/cdc2 complex.27,28 HG simultaneously increased the expression of Sox4, p53, and p21cip1/waf1 and decreased the expression of CyclinB/cdc2, whereas Miat shRNA transfection partially alleviated these changes (Figures 4E–4J).
Figure 4.

Effects of Miat on podocyte mitotic catastrophe are mediated by Sox4 in vitro
(A) Flow cytometry showed the apoptosis rate of podocytes (n = 4). (B) Flow cytometry illustrated the cell-cycle progression of podocytes (n = 3). (C) Immunostaining of mitosis with antibodies against α-tubulin (green; to indicate mitotic spindles) and histone-3 phosphorylated at serine 10 (H3-Ser10) (red; to indicate metaphase) (n = 3). Scale bar, 50 μm. (D) The percentages of podocytes with normal or abnormal mitotic processes were monitored and analyzed (n = 3). Scale bar, 10 μm. (E) Western blotting depicted the expression of Sox4, p53, p21cip1/waf1, CyclinB, and cdc2 (n = 3). (F–J) Levels of the Sox4 (n = 3), p53 (n = 5), p21cip1/waf1 (n = 4), CyclinB (n = 4), and cdc2 (n = 3) mRNAs. Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Podocyte injury and mitotic catastrophe require Sox4 in vitro
oeMiat led to increased expression of Sox4, p53, and p21cip1/waf1 and decreased expression of CyclinB/cdc2, which were reversed by transfection of the Sox4 shRNA (Figure 5A). The efficiency of the Sox4 shRNA lentivirus was confirmed by real-time PCR and western blotting (Figures S1F and S1G). Overexpression of Miat resulted in a clear accumulation of cells in G2/M phase, while transfection of the Sox4 shRNA reduced the population of podocytes arrested in G2/M phase (Figure 5B), suggesting that G2-phase arrest occurs together with reduced CyclinB/cdc2 activity and that Sox4 may be partially required to inhibit the G2/M transition triggered by the increase in Miat expression. The phenotype of mitotic catastrophe was determined by immunostaining, as described above. Podocytes cultured at 33°C proliferated normally and formed correct mitotic spindles and chromosomal alignment but did not undergo mitosis at 37°C. However, aberrant mitotic spindles, one of the features of mitotic catastrophe, were observed in Miat-overexpressing podocytes, and this effect was blocked by the Sox4 shRNA (Figure 5C). The percentages of podocytes undergoing normal or abnormal mitotic processes are shown in Figure 5D. Concurrently, the dense cortical rearrangement of F-actin induced by Miat overexpression was mitigated by Sox4 shRNA (Figure 5E). The expression of Desmin was decreased in the podocytes pretreated with the Sox4 shRNA, while the podocin, synaptopodin, ZO-1, and p-cadherin levels were obviously increased following Sox4 depletion (Figures 5F–5I). Additionally, inhibition of Sox4 attenuated the HG-induced increase in podocyte mobility (Figure 5J). The flow-cytometry analysis revealed that podocytes exhibited a lower apoptosis rate and reduced G2/M-phase arrest from 28.49% in the Sox4 scramble group to 12.95% in the Sox4 shRNA group (Figures 5K and 5L). Based on these results, Sox4 at least partially mediates HG-induced podocyte injury and G2/M-phase arrest. Additionally, immunostaining showed that spindle collapse, a characteristic of mitotic catastrophe, was induced by HG in podocytes compared with immature podocytes cultured at 33°C, and this abnormal mitotic process was abrogated by the transfection of the Sox4 shRNA (Figures 5M and 5N). Taken together, these results indicate that podocyte injury and mitotic catastrophe require Sox4 in vitro.
Figure 5.

Podocyte injury and mitotic catastrophe require Sox4 in vitro
(A) Western-blot analysis showed the expression of Sox4, p53, p21cip1/waf1, CyclinB, and cdc2 (n = 3). (B) Flow cytometry analysis illustrated the cell-cycle progression of podocytes (n = 3). (C) Immunostaining of mitosis with antibodies against α-tubulin (green) and H3-Ser10 (red) (n = 3). Scale bar, 10 μm. (D)The percentages of podocytes with normal or abnormal mitotic processes were monitored and analyzed (n = 3). (E) Morphological changes in the podocyte cytoskeleton (n = 3). Scale bar, 50 μm. (F) The expression of Desmin, podocin, synaptopodin, and ZO-1 was measured using western blotting (n = 3). (G–I) Immunofluorescence staining was performed to determine the intensities of podocin (G), Desmin (H), and p-cadherin (I) under HG conditions (n = 4). Scale bar, 50 μm. (J) Cell migration was detected using Transwell assays (n = 5). Scale bar, 100 μm. (K) Flow cytometry revealed the apoptosis rate of podocytes (n = 5). (L) Flow cytometry clarified the proportion of podocytes in G2/M phase (n = 4). (M) Immunostaining illustrated morphological changes associated with normal or abnormal mitosis using antibodies against α-tubulin (green) and H3-Ser10 (red) (n = 3). Scale bar, 10 μm. (N) The percentages of podocytes with normal or abnormal mitotic processes were monitored and analyzed (n = 3). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Sox4 expression is critical for p53 stabilization and function
We further investigated whether Sox4 mediates G2/M-phase progression via p53 stabilization and function. As shown in Figure 6A, Sox4 suppression led to a decrease in p53 and p21cip1/waf1 expression at the protein level without an obvious alteration in p53 mRNA levels (Figure 6B). Simultaneously, the half-life of p53 was substantially shortened following Sox4 depletion, as determined by cycloheximide (CHX) at the indicated time points (Figure 6C). All of the aforementioned data indicate that Sox4 potentially modulates p53 at the posttranslational level. Furthermore, podocytes were transfected with pG13-Luc (containing a generic p53-responsive element), MG15-Luc (containing a mutated p53 binding sequence), Sox4 negative control (Sox4-NC), and Sox4 overexpressing (Sox4-OE) vectors, as indicated. As shown in Figure 6D, Sox4 overexpression evidently amplified the endogenous transcriptional activity of p53, whereas Sox4 overexpression had no effect on the transcriptional activity of MG15-Luc. Next, we verified the relationship between endogenously expressed Sox4 and p53 in podocytes via immunoprecipitation (IP) with an anti-Sox4 antibody (Figure 6E). We explored whether Sox4 regulates the ubiquitination of p53 in the presence of MG-132, a specific proteasome inhibitor, and found that the amount of ubiquitinated p53 was significantly increased due to Sox4 repression, and the Sox4 shRNA substantially reduced the effect of HG on p53 ubiquitination (Figure 6F). Therefore, Sox4 inhibits the ubiquitination of p53. As Mdm2 is a vital protein in mediating podocyte mitotic catastrophe and modulating p53,29, 30, 31 we then analyzed whether Sox4 stabilized p53 by inhibiting in Mdm2-mediated p53 ubiquitination. As shown in Figure 6G, the p53 protein level was reduced when Mdm2 was cotransfected into podocytes, while its expression was increased with increasing amounts of the Myc-Sox4 plasmid, indicating that the interaction between Sox4 and p53 is pivotal for the inhibitory effect of Sox4 on Mdm2-mediated p53 degradation. Next, using MG-132, we observed that the coprecipitation of p53 with Mdm2 was effectively abrogated when Sox4 was overexpressed (Figure 6H), indicating that Sox4 inhibits the interaction between p53 and Mdm2. In addition, the silencing of Sox4 led to decreased levels of p53 acetylation following HG treatment (Figure 6I), suggesting that Sox4 stabilized p53 at least in part by increasing p53 acetylation at the Lys-373 and -382 residues. The acetylation of p53 is principally mediated by the acetyltransferases CBP and p300.32 We then observed that the expression of Sox4 strengthened the interaction of p53 with CBP or p300, indicating that Sox4 may function as a cofactor of p53 by recruiting CBP/p300 and promoting the formation of the p53-CBP/p300 complex to mediate the acetylation of p53 (Figure 6J).
Figure 6.

Sox4 is critical for p53 stabilization and function
(A–D) Western blotting (A) and real-time PCR illustrated the expression of Sox4 (B), p53 (C), and p21cip1/waf1 (D) under HG conditions (n = 3). (E) Lysates from scramble shRNA or Sox4 shRNA-transfected podocytes treated with 20 mg/mL cycloheximide (CHX) at the indicated time points (0, 20, 40, and 60 min; left panel), and quantification of the relative p53 levels were quantified (right panel; n = 3). (F) Luciferase reporter assay revealed the interaction between Sox4 and p53 transcriptional activity in podocytes (n = 3). (G) Sox4 and p53 were endogenously expressed in podocytes, as shown by immunoprecipitation (IP) with an anti-Sox4 antibody (n = 3). (H) Podocytes were transfected with His-ubiquitin with or without Sox4 shRNA for 24 h and were incubated in the presence or absence of HG for another 48 h, followed by treatment with 20 μmmol/L MG-132 for 8 h. Cell lysates were immunoprecipitated and analyzed using immunoblotting with an anti-p53 antibody (n = 3). IB, immunoblotting. (I) Podocytes were transfected with FLAG-p53, GFP, HA-Mdm2, and Myc-Sox4 vectors (1, 2, and 4 μg) for 24 h, and the expression of p53 in podocyte lysates was analyzed using IB (n = 3). (J) Coprecipitation of p53 with Mdm2 was observed after Sox4 overexpression (2 and 4 μg) in podocytes in the presence of MG-132 for 8 h. Cell lysates were IP with an anti-HA antibody and were subjected to IB (n = 3). (K) Podocytes transfected with the control or Sox4 shRNA were treated with or without HG, and the levels of p53 acetylation at the Lys-373 and -382 residues were analyzed using IB with an anti-acetyl-p53 (Lys-373 or -382) antibody (n = 3). (L) Podocytes were transfected with FLAG-p53, Myc-Sox4, HA-CBP, and HA-p300 plasmids for 24 h. Cell lysates were IP with an anti-HA antibody and IB with an anti-FLAG antibody, and the whole-cell lysates (WCLs) were analyzed using IB (n = 3). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Miat promotes Sox4 expression by targeting miR-130b-3p in vitro
Cytoplasmic lncRNAs usually serve as competing endogenous RNAs (ceRNAs), functioning as sponges of miRNAs to modulate target gene expression. Here, we used TargetScan, miRanda, PicTar, and starBase to further elucidate the miRNA target recognition sequences in Miat and in the 3′ UTR of Sox4. One such candidate was miR-130b-3p. First, we observed markedly reduced expression of hsa-miR-130b-3p in the plasma from patients with DN compared with that from healthy controls, which was opposite to the finding for MIAT (Figure 7A). The clinical information of the subjects is illustrated in Table S1. We constructed a Miat-mutant (MUT) vector containing a triple mutation in the miR-130b-3p binding site, and the luciferase reporter assay revealed that miR-130b-3p overexpression decreased the luciferase activity of the Miat-WT vector but not that of the Miat-MUT vector in HEK293 cells (Figure 7B). As expected, RNA-binding protein IP (RIP) assays with an antibody against Ago2, a core constituent of the RNA-induced silencing complex (RISC), revealed that Miat and miR-130b-3p were notably enriched in Ago2-IP in contrast to control IgG-IP in podocytes (Figure 7C). We also observed that the Miat-WT plasmid significantly inhibited miR-130b-3p expression compared with the Miat-MUT plasmid when the podocytes were cotransfected with miR-130b-3p mimic (Figure 7D), while the change trend of Sox4 mRNA was opposite to that of miR-130b-3p (Figure S1H). Next, a dual-luciferase reporter assay illustrated that the miR-130b-3p mimic suppressed the expression of Sox4, but the inhibitory effect of miR-130b-3p was abrogated by transfecting the Sox4 3′ UTR-MUT plasmid into HEK293 cells (Figure 7E). Moreover, the western blotting and real-time PCR results showed decreased Sox4 levels in cultured podocytes pretreated with miR-130b-3p (Figures 7F and 7G). Finally, as shown in Figure 7H, Miat-WT significantly increased Sox4 expression, while point mutations in Miat completely blocked this change. Moreover, inhibition of miR-130b-3p reversed the suppression of Sox4 expression induced by the Miat shRNA (Figure 7I). Conversely, miR-130b-3p overexpression decreased the Sox4 protein levels compared with those observed in the Miat lentivirus group (Figure 7J).
Figure 7.

Miat promotes Sox4 expression by targeting miR-130b-3p in vitro
(A) The levels of miR-130b-3p in the plasma from healthy controls (n = 22, −0.8066 ± 0.4477) and clinical patients with DN (n = 61, −1.124 ± 0.4351). (B) A luciferase reporter assay was performed to evaluate the interaction between miR-130b-3p and Miat in HEK293 cells (n = 3). (C) RIP revealed the interaction of Miat and miR-130b-3p in podocytes (n = 3). (D) Real-time PCR revealed the expression of miR-130b-3p in podocytes in the presence of miR-130b-3p mimic and Miat-WT plasmid or Miat-MUT plasmid (n = 3). (E) Dual-luciferase reporter assay showed the interaction between miR-130b-3p and the Sox4 3′ UTR in HEK293 cells (n = 3). (F) Western blotting manifested the expression of Sox4 in podocytes after pretreatment with the miR-130b-3p mimic (n = 3). (G) Real-time PCR revealed Sox4 mRNA expression in podocytes (n = 5). (H) Western blotting was used to assess Sox4 expression in different groups of podocytes (n = 3). EV, empty vector. (I) The expression of Sox4 in podocytes was revealed by western blotting (n = 3). (J) The expression of Sox4 was measured in podocytes using western blotting (n = 3). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Miat enhances podocyte injury and G2/M-phase arrest by binding to miR-130b-3p in vivo
Because proteinuria with pronounced alterations in podocytes already occurred before 12 weeks, as described above, we intravenously injected a miR-130b-3p antagomir (10 nmol/g) into Miat-KO-STZ mice twice a week for 12 weeks, and the efficiency of the miR-130b-3p antagomir was confirmed in primary podocytes using real-time PCR (Figure S1I). Miat and Sox4 levels in mouse kidneys were detected (Figures S1J and S1K). Using TEM, we observed that the GBM thickness and morphological lesions of podocyte FPs were exacerbated by the miR-130b-3p antagomir, in sharp contrast to those from the Miat-KO mice (Figure 8A). Similarly, PAS staining showed that the miR-130b-3p antagomir exacerbated mesangial proliferation, matrix accumulation, and capillary stenosis in the glomerular area (Figure 8B); in addition, Masson’s trichrome staining showed that collagen fibrosis and inflammatory cell invasion were aggravated by miR-130b-3p inhibition (Figure 8C). Furthermore, miR-130b-3p depletion reduced the fluorescence intensities of the podocyte injury marker podocin, synaptopodin, and ZO-1, decreased the average level of WT-1, and increased Desmin staining (Figures 8D–8H and S1L). Additionally, the results of the western-blot analysis of primary podocytes were consistent with those of immunofluorescence staining (Figure 8I). As shown in Figure 8J, the expression of Sox4, p53, and p21cip1/waf1 in primary podocytes was increased by the miR-130b-3p antagomir, and the CyclinB/cdc2 complex was noticeably impaired. Importantly, flow cytometry showed increased percentages of primary podocytes in the G2/M phase, from 6.67% in the WT-CTL mice to 20.26% in the WT-STZ mice; in contrast, the percentages of primary podocytes in the G2/M phase decreased to 10.99% after Miat KO, but miR-130b-3p deficiency increased this percentage (Figure 8K). Overall, mouse proteinuria, as indicated by urinary total protein (UTP) levels, was decreased in Miat-KO mice, but the loss of miR-130b-3p function exerted the opposite effects at 12 weeks (Table S4).
Figure 8.

Miat enhances podocyte injury and G2/M-phase arrest by binding to miR-130b-3p in vivo
(A) Morphological changes in the FPs and GBM of the mice at 12 weeks were observed under TEM. The GBM thickness and podocyte effacement were qualified (n = 3). Scale bar, 1 μm. (B) PAS staining of mouse tissues at 12 weeks. The relative mesangial area and glomerular volume were qualified (n = 4). Scale bar, 50 μm. (C) Masson’s trichrome staining of mouse tissues at 12 weeks (n = 4). Scale bar, 50 μm. (D–G) Immunofluorescence staining for the podocyte-specific markers Desmin (D), podocin (E), synaptopodin (F), and ZO-1 (G) in mice at 12 weeks (n = 3). Scale bar, 50 μm. (H) Immunofluorescence staining showed the expression of WT-1 in the mouse glomeruli at 12 weeks (n = 3). Scale bar, 50 μm. (I) Levels of injury markers were confirmed in primary mouse podocytes at 12 weeks using western blotting (n = 3). (J) Western-blot analysis showed the expression of Sox4, p53, p21cip1/waf1, CyclinB, and cdc2 in primary mouse podocytes at 12 weeks (n = 3). (K) Cell-cycle progression of primary mouse podocytes at 12 weeks was assessed using flow cytometry (n = 4). Error bars represent ±SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Discussion
In the current study, we observed that MIAT contributed to podocyte injury and mitotic catastrophe while Miat depletion significantly ameliorated podocyte injury and aberrant mitosis by restoring the SD integrity, attenuating FPE, preventing dedifferentiation, and suppressing G2/M-phase arrest. We further explored the interaction among MIAT, miR-130b-3p, and Sox4, which consequently leads to podocyte mitotic catastrophe associated with the progression of DN.
DN often begins with proteinuria, an early sign of renal injury that constitutes a risk factor for the further progressive destruction of the kidney.33,34 The onset of proteinuria in patients with DN is caused by damage to podocytes, which are terminally differentiated epithelial cells that reside in the glomeruli.35,36 Once podocytes are injured, with FPE and slit-diaphragm (SD) disruption, kidney function deteriorates, subsequently leading to progressive glomerular damage, which is characterized by increased podocyte apoptosis.37,38 Consistent with the aforementioned findings, our study showed that podocytes were impaired due to hyperglycemia and the ratios of apoptotic podocytes were markedly increased, with a simultaneous retraction of the FPs and mutation of the SD-associated differentiation proteins. Importantly, HG induced the rearrangement of F-actin, which is recognized as an indicator of SD instability and dynamic restructuring of the FP configuration, while the depletion of Miat reversed this hypermotility in vitro. Our experiments showed that the reduced expression of Miat in vivo mitigated podocyte injury and albuminuria by interfering with the plasticity of the podocyte cytoskeleton and maintaining podocyte integrity by stabilizing SD-associated differentiation markers.
Anomalous mitotic entry before the completion of DNA replication may result in mitotic catastrophe and is correlated with multipolar spindle and giant, multinuclear cells.39 In addition, mitotic catastrophe is a non-lethal process, but it includes a crossroad that might drive cells into necrosis or apoptosis,40 and the inhibition of apoptosis may mitigate asymmetrical division and the aneuploid cell generation.41 Our study indicated that the increased apoptotic rate of HG-treated cells was an outcome of both podocyte injury and mitotic catastrophe. Many published data suggest that prolonged activation of the CyclinB/cdc2 complex is a molecular feature of mitotic catastrophe,42,43 and while our study provided evidence that podocytes underwent aberrant mitosis following HG treatment, these effects were accompanied by elevated p53/p21cip1/waf1 levels but decreased CyclinB/cdc2 levels. To our knowledge, a few possible factors may account for the decreased CyclinB/cdc2 levels. On the one hand, DNA damage occurs in podocytes in the context of hyperglycemia,44 and incomplete DNA replication or DNA strand breaks stimulate the G2 DNA-damage checkpoint to block CyclinB/cdc2 activation and entry into mitosis through a p53/p21cip1/waf1-dependent pathway.28 On the other hand, when the arrest was prolonged, CyclinB/cdc2 would not escape from a slow but incessant proteosomal degradation, and podocytes exited mitosis without cell division, becoming multinucleated.45 Moreover, consistent with our study, the inactivation of CyclinB/cdc2 is associated with increased apoptosis to some extent.46
Intriguingly, Mulay et al. concluded that Mdm2 mediates mitotic catastrophe by driving podocytes into the cell cycle and leading to podocyte aneuploidy,30 which appears to be consistent with the findings from our study. We provided evidence that Mdm2 ubiquitinated p53 to accelerate its degradation. Consistent with a previous report,47 we further showed that Sox4 strengthened the formation of the p53-CBP/p300 complex to enhance p53 acetylation. Notably, mitosis is a delicate process that must be orchestrated by a highly coordinated series of events, and a high degree of variability seems to occur in the molecular cascades that are activated during mitotic catastrophe; among them, p53 signaling shows a superior sensitivity.48,49 In our study, the use of Miat shRNA and Sox4 shRNA did not completely reverse mitotic arrest in podocytes. As such, we speculated that p53, which is downstream of MIAT and Sox4, may be involved in other signaling pathways to regulate mitotic progression in the presence of HG. Studies exploring whether other abnormalities in the expression of p53-related proteins exist during DN will be interesting.
In recent years, accumulating evidence has suggested that aberrant levels of MIAT may exert various biological effects on multiple diseases;11,50, 51, 52 among them, the ceRNA regulatory system is a vital mechanism of MIAT in disease recurrence and progression. For example, elevated MIAT levels mediate microvascular dysfunction by sponging miR-150-5p in diabetic retinopathy11 and are associated with decreased miR-24 levels in cardiac fibrosis,52 while our research elucidated that MIAT might sponge miR-130b-3p to exert its biological effects. In addition to the ceRNA mechanism involving miR-130b-3p, MIAT may participate in DN through another mechanism owing to the fact that it was partially retained in the nuclei of podocytes.53 Notably, MIAT has been shown to be involved in other kidney diseases; for instance, MIAT drives renal fibrosis by affecting myofibroblast formation in chronic kidney disease,54 and high MIAT expression is related to a poor prognosis of patients with kidney renal clear cell carcinoma.55 However, our study highlighted the role of MIAT in podocyte injury and mitotic catastrophe during DN.
Furthermore, our results prompted us to inquire as to what factors contribute to elevated MIAT expression in the setting of DN. Based on previous studies, many mechanisms are responsible for the abnormal expression of lncRNAs, such as (1) miRNAs,56 (2) functional proteins, i.e., the p65 subunit of nuclear factor κB (NF-κB),57 and (3) genetic changes.58 Strikingly, the lncRNA MIAT was reported to be a target gene of ALKBH1-modulated m6A in atherosclerosis;59 therefore, many factors may facilitate the binding of transcription factors to the MIAT promoter and increase its transcription under hyperglycemic conditions; the concrete mechanisms will be elucidated and proposed in the future. In summary, the comprehensive regulatory network of MIAT, including both the upstream and downstream networks, should receive more attention in the future.
Several limitations to our study must be acknowledged. (1) Our study was conducted at a single center with a limited number of patients, and future large-scale and independent cohorts are needed to validate our findings. (2) We did not explore whether and how the increased expression of MIAT under hyperglycemic conditions was induced by miRNAs, functional proteins, or epigenetic changes. (3) Last, other cells including proximal tubular epithelial cells, glomerular endothelial cells, and mesangial cells,60, 61, 62 which have not been included in our studies but may also play a critical role in contributing to DN progression.
Collectively, all of the approaches described above provide plausible insights into the mechanism by which MIAT specifically contributes to the podocyte injury and mitotic catastrophe involved in the progression of DN, suggesting that approaches targeting MIAT may be a potentially effective therapeutic strategy for patients with DN.
Materials and methods
Clinical specimens
The plasma study included 61 patients with DN and 22 healthy control individuals; from these subjects, 11 freshly frozen normal kidney samples (>2 cm adjacent to urological neoplasms) and 20 DN renal biopsy samples were collected. The urine sediment study included 25 patients with DN and 17 healthy subjects. All participants were consecutively recruited from Shandong Provincial Hospital between December 2018 and September 2020. Signed informed consent was obtained from all patients, and the study was approved by the Institutional Ethical Review Boards (approval ID: NSFC: no. 2017-083).
CRISPR-Cas9 Miat-KO mouse
The mouse Miat gene is located on chromosome 5, and seven exons have been identified. One kilobase upstream of exons 1 to 3 was selected as the target site, and the gRNA target sequences were as follows:
gRNA1 (matches forward strand of the gene): CCCTGACAGGGAAGTATCCGAGG;
gRNA2 (matches reverse strand of the gene): CAGCGATCCCCGACGCCTGGAGG.
The gRNA targeting vector was constructed and confirmed by sequencing. Then, the Cas9 mRNA and gRNA generated by in vitro transcription were coinjected into fertilized mouse eggs via a microinjection technique to generate targeted KO offspring. The F0 founder animals, identified by PCR followed by sequence analysis, were bred with WT mice at 8 weeks to test germline transmission and generate F1 animals. The heterozygous F1 mice were intercrossed to generate homozygous F2 mice. The pups were genotyped by PCR followed by sequence analysis with the following primers:
Forward primer: 5′-AGATGCAGAGTGACAAACTACCC-3′;
Reverse primer 1: 5′-CTCAGGCATTTCTTTTGCTTTTGGG-3′;
Reverse primer 2: 5′-GACAACAGAATGGGCTCTCTCAC-3′.
The PCR products were analyzed by agarose gel electrophoresis.
Animal model and treatment
All experiments were carried out following the Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of Shandong University (approval ID: NSFC: no. 2018-051). At 8 weeks of age, all mice (155 male C57BL/6 mice and CRISPR-Cas9 Miat-KO mice) were given ad libitum access to food and water, housed under standard conditions, and randomly assigned to receive STZ (Sigma-Aldrich, St Louis, MO, USA) dissolved in citrate buffer (50 mg/kg) or citrate buffer alone by intraperitoneal injection for 5 consecutive days. The glucose, SBP, and urinary protein levels of the mice were detected as previously described,63 and we euthanized the animals at 4 (WT-CTL, n = 7; WT-STZ, n = 8; KO-CTL, n = 7; KO-STZ, n = 8); 6 (WT-CTL, n = 7; WT-STZ, n = 8; KO-CTL, n = 8; KO-STZ, n = 7); 8 (WT-CTL, n = 7; WT-STZ, n = 9; KO-CTL, n = 8; KO-STZ, n = 9); and 12 weeks (WT-CTL, n = 8; WT-STZ, n = 8; KO-CTL, n = 7; KO-STZ, n = 8) after diabetes onset (blood glucose level ≥16.7 mmol/L on 2 consecutive days). In addition, miR-130b-3p antagomir (10 nmol/g) or miR-NC was intravenously injected into mice by tail vein twice a week for 12 weeks, and the mice were divided into five groups: WT-CTL (n = 6), WT-STZ (n = 7), KO-STZ (n = 7), KO-STZ + miR-130b-3p antagomir (n = 6), and KO-STZ + NC (n = 5).
Cell culture
As previously described,63 conditionally immortalized mouse podocytes (provided by Professor Peter Mundel, Massachusetts General Hospital, Boston, MA, USA) were cultured on type I collagen-coated plates in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin (Life Technologies, Carlsbad, CA, USA) under permissive conditions at 33°C with 10 U/mL mouse recombinant γ-interferon (Pepro Tech, East Windsor, NJ, USA). Podocyte differentiation was induced by culturing the cells in type I collagen-coated plates under non-permissive conditions at 37°C for at least 14 days without γ-interferon. Before being exposed to other pharmacological treatments, the differentiated cells were cultured in serum-free RPMI 1640 medium for 24 h. To explore the effect of HG on podocytes, cells were exposed to LG medium (5.5 mM glucose) or HG medium (30 mM glucose) for 48 h. HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS at 37°C under humidified conditions with 5% CO2. Cell lines were authenticated by short tandem repeat profiling and were tested for mycoplasma contamination regularly using the Mycoplasma Detection Kit (Solarbio, Beijing, China). For primary podocytes, the separation of glomeruli from mouse renal cortices was performed stepwise with filters with 250, 100, and 70 μm pores before incubation in type I collagen-coated plates in RPMI 1640 medium. The isolated glomeruli were removed 7 days later, and the cells were filtered through a filter with 40 μm pores after trypsinization.
RNA-FISH
RNA-FISH was performed with a RiboTM Fluorescence In Situ Hybridization Kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions. In brief, mature podocytes were cultured at appropriate densities on slides on the bottoms of the wells of 24-well plates. Then, 4% paraformaldehyde was added and incubated for 5 min to fix the different podocytes when the cell confluence reached 60%–70%, and then the cells were incubated with 0.5% Triton X-100 for 5 min. Then, 200 μL prehybridization solution was added to the cells and incubated at 37°C for 30 min in the dark. The probe hybridization solution was prepared by adding the Miat FISH Probe or Internal Reference FISH Probe to the hybridization solution, which was preheated at 37°C. The prehybridization solution was discarded, and 100 μL probe hybridization solution was added and incubated overnight at 37°C to hybridize the podocytes. The cells were carefully washed 3 times with hybridization wash solutions I, II, and III at 42°C in the dark. Images were observed and captured by confocal laser scanning microscope (Leica, Wetzlar, Germany) after the nuclei were stained for 10 min with a 4,6-diamidino-2-phenylindole (DAPI) working solution.
Real-time PCR
Kidney tissues were quickly ground in liquid nitrogen, and total RNA was extracted using the TRIpure Reagent (Takara, Dalian, China) according to the manufacturer’s instructions. Plasma RNA was extracted with a BIOG cfRNA Easy Kit (BIOG, Changzhou, China). Urine from clinical subjects was centrifuged at 3,200 × g for 20 min, and the sediment was washed with PBS before centrifugation at 12,000 × g for 5 min. Then, the total RNA was extracted and qualified by the measurement of A260/A280 ratio, which indicated a high purity of the extracted RNA ranged from 1.8 to 2.0. RNA samples were reverse transcribed and amplified as previously described.63 β-actin and/or U6 were used as internal controls, and the relative expression of different genes was calculated following the 2–ΔΔCT method or following a log transformation after the 2–ΔCT method. The primer sequences are listed in Table S5.
Immunofluorescence
The protocol was previously described in detail.63 The following antibodies were used: ZO-1 (Invitrogen, Carlsbad, CA, USA), Desmin, podocin, WT-1, α-tublin, H3-Ser10, goat anti-rabbit IgG H&L (Alexa Fluor 594 or 488), goat anti-mouse IgG H&L (Alexa Fluor 488; Abcam, Cambridge, MA, USA), synaptopodin, and p-cadherin (Proteintech, Rosemont, IL, USA). The level of WT-1 was measured in 5 glomeruli per mouse, and ImageJ 10.2 software was used to measure the intensity of immunostaining in the glomeruli.
Western blotting and IP
For western blotting, the detailed protocol was previously described.63 For IP, podocytes were lysed in IP lysis buffer before being briefly sonicated, and the supernatants were incubated with antibodies and protein A/G-Sepharose beads after centrifugation. Finally, the immunocomplexes were washed and immunoblotted with antibodies. The following antibodies were used: ZO-1 (Invitrogen, Calrsbad, CA, USA), Desmin, podocin, Sox4, p21cip1/waf1 (Abcam, Cambridge, MA, USA), β-actin, synaptopodin, cdc2, CyclinB, p53, GFP-tag, hemagglutinin (HA)-tag, Myc-tag, FLAG tag, and HRP-conjugated goat anti-rabbit/mouse IgG (Proteintech, Rosemont, IL, USA).
Transwell assays
Cell migration was assessed with Transwell chambers (8 μm pores, Corning, Tewksbury, MA, USA). Podocytes exposed to different treatments were suspended in 200 μL RPMI 1640 (serum-free), seeded in the upper chambers, and incubated at 37°C for 48 h, and the lower chambers were filled with 600 μL RPMI 1640 (supplemented with 10% FBS). Then, the non-migratory cells on the upper surface of the filters were removed with cotton swabs; the migrated podocytes on the lower side of the filters were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and stained with hematoxylin. The number of migrated cells was counted in three random fields under a phase-contrast microscope (Leica Microsystems, Mannheim, Germany).
Histology and immunohistochemistry
PAS and Masson’s trichrome staining (Solarbio, Beijing, China) were performed according to the detailed protocol. The relative mesangial areas (%) were calculated as follows: relative mesangial areas (%) = mesangial area/glomerular area × 100%, where the mesangial area was defined as the PAS-positive and nucleus-free area in the mesangium.64 The relative mesangial areas (%) were measured in 10 glomeruli per mouse. The glomerular cross-sectional area (GA) was used to determine the glomerular tuft volume (GV) value with the formula: , where β is the shape coefficient (1.38), k is the size distribution coefficient (1.1), and GA was calculated as the average area of 10 cortical glomeruli per mouse with ImageJ 10.2 software.64
TEM
Fresh renal tissue samples less than 1 cubic millimeter in size were fixed in 2.5% glutaraldehyde (buffered pH 7.4) overnight at 4°C, postfixed in 1% osmium tetroxide at 4°C, dehydrated with gradient ethanol, and embedded in Epon 812. Ultrathin specimens were cut, stained with uranium acetate and lead citrate, and examined under a TEM (Hitachi, Japan). Three glomeruli where the GBM was displayed in the best cross-section were selected in each mouse, and the FP widths of glomeruli were calculated as follows: , where ΣGBM length is the total GBM length measured in one glomerulus, Σsilts is the total number of slits counted, and π/4 is the correction factor to correct the assumed random orientation by which the FP were sectioned.65 The mean thickness of the GBM was measured in 5 parts of the GBM per glomerulus in 3 glomeruli per mouse with ImageJ 10.2 software.
Flow cytometry
For cell-cycle progression, 70% cold ethanol was added to fix the cells, then the cells were mixed with propidium iodide (PI) staining solution in the presence of RNase A (10 μg/mL). After incubating the podocytes at 37°C, a FACScan flow cytometer (Becton Dickinson [BD], San Jose, CA, USA) was used to determine cell-cycle progression. For apoptosis analysis, the cells were processed according to the PE Annexin V Apoptosis Detection Kit I (BD, San Jose, CA, USA) and analyzed by a FACScan flow cytometer.
RIP
The RIP assay was performed using the Magna RIPTMRNA-Binding Protein Immunoprecipitation Kit (Millipore, Burlington, MA, USA) according to the detailed protocol.
Luciferase reporter assays
The Mus-pmirGLO-Miat-WT (Miat-WT) plasmid, pmirGLO-Miat-MUT (Miat-MUT) plasmid, pmirGLO-Sox4-WT/pmirGLO-Sox4-MUT (Sox4-3′-UTR-WT/MUT) plasmids, miR-130b-3p mimic (miR-130b-3p), miR-130b-3p NC (miR-NC), Sox4-NC plasmid, Sox4-OE plasmid, pG13-Luc plasmid, and MG15-Luc plasmid were provided by RiboBio (Guangzhou, China). The Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI, USA) was utilized in HEK293 cells to detect the luciferase and Renilla signals.
Vectors constructions and transfection
The Miat shRNA lentivirus, oeMiat, Sox4 shRNA lentivirus, His-ubiquitin, FLAG-p53, GFP, HA-Mdm2, and Myc-Sox4 plasmids, the miR-130b-3p inhibitors, and the NCs were constructed by Cyagen Biosciences (Guangzhou, China). The miR-130b-3p antagomir and controls were designed by RiboBio (Guangzhou, China). The HA-CBP and HA-p300 plasmids were designed by Genomeditech (Shanghai, China). The miR-130b-3p mimic was constructed as described above. Podocyte transfection with the plasmids or miRNA was carried out with the aid of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) or HiPerFect Transfection Reagent (Qiagen, Hilden, Germany).
Statistical analysis
All data were analyzed by SPSS 25.0 software and GraphPad Prism 6.0. Data with a Gaussian distribution are shown as the mean ± SD, and the median and interquartile range are reported for data with a skewed distribution. Statistical analyses were performed by Student’s t test, Mann-Whitney U test, Kruskal-Wallis test, and one-way ANOVA and least significant difference (LSD) t test, and the correlations among the MIAT expression, ACR, and Scr levels were analyzed with Pearson’s correlation analysis. p < 0.05 was considered to indicate statistical significance.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (grants 81770723, 81873615, and 82070744), the Academic Promotion Programme of Shandong First Medical University (no. 2019QL022), and the Taishan Scholars Program (nos. ts201712090 and tsqn201812138). All authors approved the final version of the manuscript.
Author contributions
Z.W.: methodology, investigation, writing – original draft; Y.C.: validation, formal analysis, methodology; Y.L.: formal analysis, validation; B.L.: investigation, validation; J.Z.: methodology, formal analysis; X.L.: methodology; J.L.: validation; Q.Y.: methodology, investigation; Z.L.: conceptualization, writing – review & editing, funding acquisition; R.W.: conceptualization, supervision, funding acquisition.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.03.001.
Contributor Information
Zhimei Lv, Email: lyuzhimei@126.com.
Rong Wang, Email: wangrong_sd@126.com.
Supplemental information
References
- 1.Singh D.K., Winocour P., Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat. Rev. Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- 2.Hussain F., Castledine C., van Schalkwyk D., Sinha M.D., Lewis M., Inward C. UK Renal Registry 12th Annual Report (December 2009): chapter 15: clinical, haematological and biochemical parameters in patients receiving renal replacement therapy in paediatric centres in the UK in 2008: national and centre-specific analyses. Nephron Clin. Pract. 2010;115:c289–c308. doi: 10.1159/000301238. [DOI] [PubMed] [Google Scholar]
- 3.Faul C., Asanuma K., Yanagida-Asanuma E., Kim K., Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Suleiman H.Y., Roth R., Jain S., Heuser J.E., Shaw A.S., Miner J.H. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight. 2017;2:e94137. doi: 10.1172/jci.insight.94137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viberti G.C., Hill R.D., Jarrett R.J., Argyropoulos A., Mahmud U., Keen H. Microalbuminuria as a predictor of clinical nephropathy in insulin-dependent diabetes mellitus. Lancet. 1982;1:1430–1432. doi: 10.1016/s0140-6736(82)92450-3. [DOI] [PubMed] [Google Scholar]
- 6.Castedo M., Perfettini J.L., Roumier T., Andreau K., Medema R., Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 7.Vitale I., Galluzzi L., Castedo M., Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 8.Liapis H., Romagnani P., Anders H.-J. New insights into the pathology of podocyte loss. Am. J. Pathol. 2013;183:1364–1374. doi: 10.1016/j.ajpath.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii N., Ozaki K., Sato H., Mizuno H., Susumu S., Takahashi A., Miyamoto Y., Ikegawa S., Kamatani N., Hori M., et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 10.Rapicavoli N.A., Poth E.M., Blackshaw S. The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev. Biol. 2010;10:49. doi: 10.1186/1471-213X-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan B., Yao J., Liu J.-Y., Li X.-M., Wang X.-Q., Li Y.-J., Tao Z.-F., Song Y.-C., Chen Q., Jiang Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ. Res. 2015;116:1143–1156. doi: 10.1161/CIRCRESAHA.116.305510. [DOI] [PubMed] [Google Scholar]
- 12.Zhang C., Xie L., Fu Y., Yang J., Cui Y. lncRNA MIAT promotes esophageal squamous cell carcinoma progression by regulating miR-1301-3p/INCENP axis and interacting with SOX2. J. Cell. Physiol. 2020;235:7933–7944. doi: 10.1002/jcp.29448. [DOI] [PubMed] [Google Scholar]
- 13.Zhong X., Ma X., Zhang L., Li Y., Li Y., He R. MIAT promotes proliferation and hinders apoptosis by modulating miR-181b/STAT3 axis in ox-LDL-induced atherosclerosis cell models. Biomed. Pharmacother. 2018;97:1078–1085. doi: 10.1016/j.biopha.2017.11.052. [DOI] [PubMed] [Google Scholar]
- 14.Vousden K.H., Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 15.Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 16.Krause K., Wasner M., Reinhard W., Haugwitz U., Dohna C.L.Z., Mossner J., Engeland K. The tumour suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res. 2000;28:4410–4418. doi: 10.1093/nar/28.22.4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer M., Quaas M., Steiner L., Engeland K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016;44:164–174. doi: 10.1093/nar/gkv927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chetty C., Dontula R., Gujrati M., Dinh D.H., Lakka S.S. Blockade of SOX4 mediated DNA repair by SPARC enhances radioresponse in medulloblastoma. Cancer Lett. 2012;323:188–198. doi: 10.1016/j.canlet.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ha Thi H.T., Kim H.Y., Kim Y.M., Hong S. MicroRNA-130a modulates a radiosensitivity of rectal cancer by targeting SOX4. Neoplasia. 2019;21:882–892. doi: 10.1016/j.neo.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vervoort S.J., Lourenco A.R., Tufegdzic Vidakovic A., Mocholi E., Sandoval J.L., Rueda O.M., Frederiks C., Pals C., Peeters J.G.C., Caldas C., et al. SOX4 can redirect TGF-beta-mediated SMAD3-transcriptional output in a context-dependent manner to promote tumorigenesis. Nucleic Acids Res. 2018;46:9578–9590. doi: 10.1093/nar/gky755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong H., Hu J., Wang L., Qi M., Lu N., Tan X., Yang M., Bai X., Zhan X., Han B. SOX4 is activated by C-MYC in prostate cancer. Med. Oncol. 2019;36:92. doi: 10.1007/s12032-019-1317-6. [DOI] [PubMed] [Google Scholar]
- 22.Collins S.C., Do H.W., Hastoy B., Hugill A., Adam J., Chibalina M.V., Galvanovskis J., Godazgar M., Lee S., Goldsworthy M., et al. Increased expression of the diabetes gene SOX4 reduces insulin secretion by impaired fusion pore expansion. Diabetes. 2016;65:1952–1961. doi: 10.2337/db15-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger D., Thibodeau J.F., Holterman C.E., Burns K.D., Touyz R.M., Kennedy C.R. Urinary podocyte microparticles identify prealbuminuric diabetic glomerular injury. J. Am. Soc. Nephrol. 2014;25:1401–1407. doi: 10.1681/ASN.2013070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lytvyn Y., Xiao F., Kennedy C.R., Perkins B.A., Reich H.N., Scholey J.W., Cherney D.Z., Burger D. Assessment of urinary microparticles in normotensive patients with type 1 diabetes. Diabetologia. 2017;60:581–584. doi: 10.1007/s00125-016-4190-2. [DOI] [PubMed] [Google Scholar]
- 25.Bertoli C., Skotheim J.M., de Bruin R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013;14:518–528. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgakilas A.G., Martin O.A., Bonner W.M. p21: a two-faced genome guardian. Trends Mol. Med. 2017;23:310–319. doi: 10.1016/j.molmed.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Niculescu A.B., Chen X.B., Smeets M., Hengst L., Prives C., Reed S.I. Effects of p21(Cip1/Waf1) at both the G(1)/S and the G(2)/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smits V.A.J., Klompmaker R., Vallenius T., Rijksen G., Makela T.P., Medema R.H. p21 Inhibits Thr(161) phosphorylation of Cdc2 to enforce the G(2) DNA damage checkpoint. J. Biol. Chem. 2000;275:30638–30643. doi: 10.1074/jbc.M005437200. [DOI] [PubMed] [Google Scholar]
- 29.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 30.Mulay S.R., Thomasova D., Ryu M., Kulkarni O.P., Migliorini A., Bruns H., Gröbmayr R., Lazzeri E., Lasagni L., Liapis H., et al. Podocyte loss involves MDM2-driven mitotic catastrophe. J. Pathol. 2013;230:322–335. doi: 10.1002/path.4193. [DOI] [PubMed] [Google Scholar]
- 31.Tian H., Tackmann N.R., Jin A., Zheng J., Zhang Y. Inactivation of the MDM2 RING domain enhances p53 transcriptional activity in mice. J. Biol. Chem. 2017;292:21614–21622. doi: 10.1074/jbc.RA117.000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caramori M.L., Fioretto P., Mauer M. Enhancing the predictive value of urinary albumin for diabetic nephropathy. J. Am. Soc. Nephrol. 2006;17:339–352. doi: 10.1681/ASN.2005101075. [DOI] [PubMed] [Google Scholar]
- 34.Lavoz C., Sanchez Matus Y., Orejudo M., Carpio J.D., Droguet A., Egido J., Mezzano S., Ruiz-Ortega M. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019;95:1418–1432. doi: 10.1016/j.kint.2018.12.031. [DOI] [PubMed] [Google Scholar]
- 35.Zhou L., Chen X., Lu M., Wu Q., Yuan Q., Hu C., Miao J., Zhang Y., Li H., Hou F.F., et al. Wnt/beta-catenin links oxidative stress to podocyte injury and proteinuria. Kidney Int. 2019;95:830–845. doi: 10.1016/j.kint.2018.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rennke H.G. How does glomerular epithelial-cell injury contribute to progressive glomerular damage. Kidney Int. 1994;45:S58–S63. [PubMed] [Google Scholar]
- 37.Wharram B.L., Goyal M., Wiggins J.E., Sanden S.K., Hussain S., Filipiak W.E., Saunders T.L., Dysko R.C., Kohno K., Holzman L.B., et al. Podocyte. depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 38.Nagata M. Podocyte injury and its consequences. Kidney Int. 2016;89:1221–1230. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Rainey M.D., Bennett D., O'Dea R., Zanchetta M.E., Voisin M., Seoighe C., Santocanale C. ATR restrains DNA synthesis and mitotic catastrophe in response to CDC7 inhibition. Cell Rep. 2020;32:108096. doi: 10.1016/j.celrep.2020.108096. [DOI] [PubMed] [Google Scholar]
- 40.Denisenko T.V., Sorokina I.V., Gogvadze V., Zhivotovsky B. Mitotic catastrophe and cancer drug resistance: a link that must to be broken. Drug Resist. Updat. 2016;24:1–12. doi: 10.1016/j.drup.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Castedo M., Perfettini J.L., Roumier T., Valent A., Raslova H., Yakushijin K., Horne D., Feunteun J., Lenoir G., Medema R., et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- 42.Yoshikawa R., Kusunoki M., Yanagi H., Noda M., Furuyama J., Yamamura T., Hashimoto-Tamaoki T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: a novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001;61:1029–1037. [PubMed] [Google Scholar]
- 43.Nabha S.M., Mohammad R.M., Dandashi M.H., Coupaye-Gerard B., Aboukameel A., Pettit G.R., Al-Katib A.M. Combretastatin-A4 prodrug induces mitotic catastrophe in chronic lymphocytic leukemia cell line independent of caspase activation and poly(ADP-ribose) polymerase cleavage. Clin. Cancer Res. 2002;8:2735–2741. [PubMed] [Google Scholar]
- 44.Lorenzi M., Montisano D.F., Toledo S., Barrieux A. High glucose induces DNA damage in cultured human endothelial cells. J. Clin. Invest. 1986;77:322–325. doi: 10.1172/JCI112295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brito D.A., Rieder C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X., Chen H., Zhang Z., Xu D., Duan J., Li X., Yang L., Hua R., Cheng J., Li Q. Isorhamnetin promotes estrogen biosynthesis and proliferation in porcine granulosa cells via the PI3K/Akt signaling pathway. J. Agric. Food Chem. 2021;69:6535–6542. doi: 10.1021/acs.jafc.1c01543. [DOI] [PubMed] [Google Scholar]
- 47.Pan X., Zhao J., Zhang W.N., Li H.Y., Mu R., Zhou T., Zhang H.Y., Gong W.L., Yu M., Man J.H., et al. Induction of SOX4 by DNA damage is critical for p53 stabilization and function. Proc. Natl. Acad. Sci. U S A. 2009;106:3788–3793. doi: 10.1073/pnas.0810147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Portugal J., Mansilla S., Bataller M. Mechanisms of drug-induced mitotic catastrophe in cancer cells. Curr. Pharm. Des. 2010;16:69–78. doi: 10.2174/138161210789941801. [DOI] [PubMed] [Google Scholar]
- 49.Liu L., Charville G.W., Cheung T.H., Yoo B., Santos P.J., Schroeder M., Rando T.A. Impaired Notch signaling leads to a decrease in p53 activity and mitotic catastrophe in aged muscle stem cells. Cell Stem Cell. 2018;23:544–556.e4. doi: 10.1016/j.stem.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu M., Li N., Luo P., Jing W., Wen X., Liang C., Tu J. Peripheral blood leukocyte expression of lncRNA MIAT and its diagnostic and prognostic value in ischemic stroke. J. Stroke Cerebrovasc. Dis. 2018;27:326–337. doi: 10.1016/j.jstrokecerebrovasdis.2017.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H.-Y., Zheng F.-S., Yang W., Lu J.-B. The long non-coding RNA MIAT regulates zinc finger E-box binding homeobox 1 expression by sponging miR-150 and promoteing cell invasion in non-small-cell lung cancer. Gene. 2017;633:61–65. doi: 10.1016/j.gene.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Qu X., Du Y., Shu Y., Gao M., Sun F., Luo S., Yang T., Zhan L., Yuan Y., Chu W., et al. MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci. Rep. 2017;7:42657. doi: 10.1038/srep42657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mercer T.R., Mattick J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 54.Bijkerk R., Au Y.W., Stam W., Duijs J., Koudijs A., Lievers E., Rabelink T.J., van Zonneveld A.J. Long non-coding RNAs rian and Miat mediate myofibroblast formation in kidney fibrosis. Front. Pharmacol. 2019;10:215. doi: 10.3389/fphar.2019.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang D., Zeng S., Hu X. Identification of a three-long noncoding RNA prognostic model involved competitive endogenous RNA in kidney renal clear cell carcinoma. Cancer Cell Int. 2020;20:319. doi: 10.1186/s12935-020-01423-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia L.F., Wei S.B., Gan Y.H., Guo Y., Gong K., Mitchelson K., Cheng J., Yu G.Y. Expression, regulation and roles of miR-26a and MEG3 in tongue squamous cell carcinoma. Int. J. Cancer. 2014;135:2282–2293. doi: 10.1002/ijc.28667. [DOI] [PubMed] [Google Scholar]
- 57.Majumder S., Hadden M.J., Thieme K., Batchu S.N., Niveditha D., Chowdhury S., Yerra V.G., Advani S.L., Bowskill B.B., Liu Y., et al. Dysregulated expression but redundant function of the long non-coding RNA HOTAIR in diabetic kidney disease. Diabetologia. 2019;62:2129–2142. doi: 10.1007/s00125-019-4967-1. [DOI] [PubMed] [Google Scholar]
- 58.Fan Z., Chen X., Liu L., Zhu C., Xu J., Yin X., Sheng Y., Zhu Z., Wen L., Zuo X., et al. Association of the polymorphism rs13259960 in SLEAR with predisposition to systemic lupus Erythematosus. Arthritis Rheumatol. 2020;72:985–996. doi: 10.1002/art.41200. [DOI] [PubMed] [Google Scholar]
- 59.Wu L., Pei Y., Zhu Y., Jiang M., Wang C., Cui W., Zhang D. Association of N(6)-methyladenine DNA with plaque progression in atherosclerosis via myocardial infarction-associated transcripts. Cell Death Dis. 2019;10:909. doi: 10.1038/s41419-019-2152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao L., Zhu X., Yang S., Liu F., Zhou Z., Zhan M., Xie P., Zhang D., Li J., Song P., et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes. 2014;63:1366–1380. doi: 10.2337/db13-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lenoir O., Jasiek M., Henique C., Guyonnet L., Hartleben B., Bork T., Chipont A., Flosseau K., Bensaada I., Schmitt A., et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy. 2015;11:1130–1145. doi: 10.1080/15548627.2015.1049799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsai Y.C., Kuo M.C., Hung W.W., Wu L.Y., Wu P.H., Chang W.A., Kuo P.L., Hsu Y.L. High glucose induces mesangial cell apoptosis through miR-15b-5p and promotes diabetic nephropathy by extracellular vesicle delivery. Mol. Ther. 2020;28:963–974. doi: 10.1016/j.ymthe.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lv Z., Hu M., Fan M., Li X., Lin J., Zhen J., Wang Z., Jin H., Wang R. Podocyte-specific Rac1 deficiency ameliorates podocyte damage and proteinuria in STZ-induced diabetic nephropathy in mice. Cell Death Dis. 2018;9:342. doi: 10.1038/s41419-018-0353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Awazu M., Omori S., Ishikura K., Hida M., Fujita H. The lack of cyclin kinase inhibitor p27(Kip1) ameliorates progression of diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:699–708. doi: 10.1097/01.asn.0000051726.41601.c0. [DOI] [PubMed] [Google Scholar]
- 65.Mallipattu S.K., Liu R., Zhong Y., Chen E.Y., D'Agati V., Kaufman L., Ma'ayan A., Klotman P.E., Chuang P.Y., He J.C. Expression of HIV transgene aggravates kidney injury in diabetic mice. Kidney Int. 2013;83:626–634. doi: 10.1038/ki.2012.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.