

Abstract
Protein translation is a highly regulated process involving the interaction of numerous genes on every component of the protein translation machinery. Upregulated protein translation is a hallmark of cancer and is implicated in autism spectrum disorder, but the risks of developing each disease do not appear to be correlated with one another. In this study we identified two siblings from the NIH Undiagnosed Diseases Program with loss of function variants in PUS7, a gene previously implicated in the regulation of total protein translation. These patients exhibited a neurodevelopmental phenotype including autism spectrum disorder in the proband. Both patients also had features of Lesch-Nyhan syndrome, including hyperuricemia and self-injurious behavior, but without pathogenic variants in HPRT1. Patient fibroblasts demonstrated upregulation of protein synthesis, including elevated MYC protein, but did not exhibit increased rates of cell proliferation. Interestingly, the dysregulation of protein translation also resulted in mildly decreased levels of HPRT1 protein suggesting an association between dysregulated protein translation and the LNS-like phenotypic findings. These findings strengthen the correlation between neurodevelopmental disease, particularly autism spectrum disorders, and the rate of protein translation.
Keywords: PUS7, Neurodevelopmental Disease, Protein Translation
INTRODUCTION
Many cellular processes are genetically regulated through modifications of DNA and RNA, which in turn affect the abundance, location, and activity of proteins and other effector molecules. While DNA modifications have been extensively studied in recent years, RNA modifications have been more difficult to assess. As methods for sequencing and evaluating RNA have become more robust, it has become apparent that post transcriptional modifications of RNA are critical for fine tuning gene expression. Moreover, disruption of genes involved in this process are now known to be associated with a wide range of human diseases1. RNA pseudouridylation is the most common post transcriptional RNA modification2 and is mediated by enzymes having pseudouridine synthase activity. Depending on the target RNA, dysregulated pseudouridylation can cause diseases ranging from neurodevelopmental disorders to cancer3. Humans possess multiple RNA-independent pseudouridine synthase (PUS) enzymes capable of recognizing target sequences on many RNA species4 without the aid of guide RNA molecules5, however only PUS1, PUS3, and PUS7 have been reported to cause disease in humans.
Pseudouridylation of tRNAs has long been appreciated for stabilizing the three-dimensional structure of tRNA molecules necessary for protein translation. More recently, pseudouridine residues on tRNAs have been associated with a secondary role in the activity of tRNA fragments (tRFs). Functional tRFs help regulate the rate of total protein translation6,7, which is particularly important during differentiation and stress response8. The activity of PUS7 is critical for the activity of tRFs since the majority of its targets are tRNA sequences that are necessary for the function of tRFs9. These tRFs regulate protein translation by controlling the availability of translation initiation factors (eIF4A and eIF4G)10 and are nonfunctional in the absence of PUS7 activity, resulting in increased levels of protein translation9. Dysregulation of protein translation occurs in a number of human diseases11, so dysregulation through disruption of tRF activity is also likely to affect development.
PUS7 deficiency is the most recently described human disease involving PUS enzymes. Patients with PUS7 deficiency (MIM 618342) exhibit decreased pseudouridylation of known PUS7 target sites and pus7 deficient drosophila display behavioral abnormalities12. The previously described cases provide compelling evidence that loss of PUS7 function causes lack of pseudouridine on PUS7 target RNAs, resulting in a neurological phenotype. However, the mechanism by which pathogenic variants in PUS7 lead to disease remains unclear. Previous publications did not evaluate the downstream consequences of improper pseudouridylation of PUS7 targets nor did they link the known role of PUS7 in regulating protein translation with the clinical phenotype.
We report here the first nonconsanguineous siblings who carry loss of function variants in PUS7; we also interrogated the molecular effects of PUS7 deficiency. We confirmed that loss of PUS7 activity in these individuals dysregulated protein translation in their cultured fibroblasts, increased the levels of MYC protein, and decreased levels of HPRT1 protein without impacting the mRNA levels of either gene. Elevated levels of MYC may impair cellular differentiation as has been observed in stem cell lines13 while decreased levels of HPRT1 may contribute to the behavioral phenotype reminiscent of Lesch-Nyhan syndrome (LNS). This study provides the foundation for understanding the molecular mechanism underlying PUS7 deficiency. Furthermore, these results provide insight into the role of pseudouridylation as a regulatory component of global gene expression and into other disease mechanisms involving dysregulation of protein synthesis.
RESULTS
Clinical presentation of PUS7 deficient patients
Two female siblings born to nonconsanguineous parents were referred to the Undiagnosed Diseases Program (UDP)14 at the NIH for a constellation of findings inconsistent with any known disorder at the time. Patients were seen at the UDP at 2 and 4 years of age and presented with slight facial dysmorphisms (Fig 1A) including down slanting palpebral fissures with mild ptosis, upturned nasal tip, down-turned corners of the mouth, micrognathia with a Class 2 malocclusion, and overlapping 1st/2nd and 4th/5th toes. The patients also presented with global developmental delay, hypotonia, postnatal microcephaly, hearing loss, hypoglycemia, hyperuricemia, aggressive/self-injurious behavior, and short stature. Clinical exome sequencing and whole genome sequencing were unable to identify any pathogenic genes of interest. However, reanalysis of the whole genome sequencing data by the UDP revealed that both patients carried two rare, potentially pathogenic variants in PUS7 (NM_019042): c.398+1 G>T (IVS2+1) and c.1160 C>T; p.Thr387Met (T387M). The c.398+1 G>T splice site variant is not present in any genetic database, although c.398+1 G>A has two observations in gnomAD v3.1 (rs1264890888). T387M has two reported observations in gnomAD (rs916775904). Segregation analysis confirmed that the siblings inherited one variant from each non-consanguineous parent, neither of whom presented with any clinical phenotype (Fig 1B). The siblings were born 21 months apart with low birth weight and failure to thrive with difficulty feeding that resulted in the placement of gastrostomy tubes (g-tubes). Hypoglycemia (≥ 40 mg/dL) persisted despite g-tubes and frequent feedings. Both children displayed markedly delayed development. The older sibling walked at 6 years of age while the younger sibling remains non-ambulatory at age 9. Both patients were born with small heads (<1% and 13% centiles respectively) with postnatal growth deceleration (48cm (<−2SD) and 47.8cm (<−4SD) at age 10 and 9 respectively (Fig 1C). Both have hearing loss and are nonverbal. Uric acid levels progressively increased with age in both girls (peaking at 8.7 mg/dL and 6.5 mg/dL (2.3–5.5 mg/dL) at 4 and 6 years respectively, before allopurinol intervention (Sup Fig 1). Both patients have behavioral abnormalities with episodes of self-injurious behavior including face pulling, hair pulling, arm scratching, and finger biting. Due to the clinical overlap with LNS, sequencing of HPRT1 was performed and was normal. These sisters demonstrate significant clinical overlap with recently reported cases of PUS7 deficiency (Sup Table 1) including intellectual disability, speech delay, microcephaly, short stature, and aggressive behavior.
Figure 1. Clinical presentation of PUS7 deficient patients.

A) Both siblings showed mild facial dysmorphology. B) Family pedigree with results of segregation analysis for PUS7 variants. C) Table of significant clinical findings.
Loss of function variants in PUS7 increases total protein translation
To confirm the pathogenicity of c.398+1 G>T, RT-PCR was performed on cDNA generated from RNA extracted from patient fibroblasts. This assay demonstrated that c.398+1 G>T causes the generation of a smaller splice isoform not found in control fibroblasts (Fig 2A). TOPO cloning and Sanger sequencing of RT-PCR products across the exon 2/3 junction demonstrated that c.398+1 G>T disrupted the native donor site and caused utilization of two upstream GT dinucleotides as splice donor sites (Sup Fig 2). These alternatively spliced transcripts introduced either 41 bp or 55 bp deletions into the PUS7 mRNA and both result in frameshift and premature termination (Sup Fig 3). Sanger sequencing of RT-PCR products spanning the site of the paternal missense variant confirmed skewed distribution of PUS7 mRNA such that the maternal allele was significantly reduced compared to the paternal allele (Fig 2B). Thus, we presume that loss of PUS7 expression from the maternal allele is caused by misspliced mRNA that is subsequently degraded by nonsense mediated mRNA decay (NMD). Quantification of total PUS7 mRNA by qRT-PCR demonstrated that the decrease in mRNA from the maternal allele corresponded to a nearly 50% decrease in total PUS7 mRNA relative to control fibroblasts (Fig 2C). Additionally, RNA from the mother’s fibroblasts, which are also heterozygous for c.398+1 G>T, demonstrated a similar decrease in total PUS7 mRNA, confirming that this level of decrease was due to the maternal allele alone.
Figure 2. Loss of function variants in PUS7 increases total protein translation.
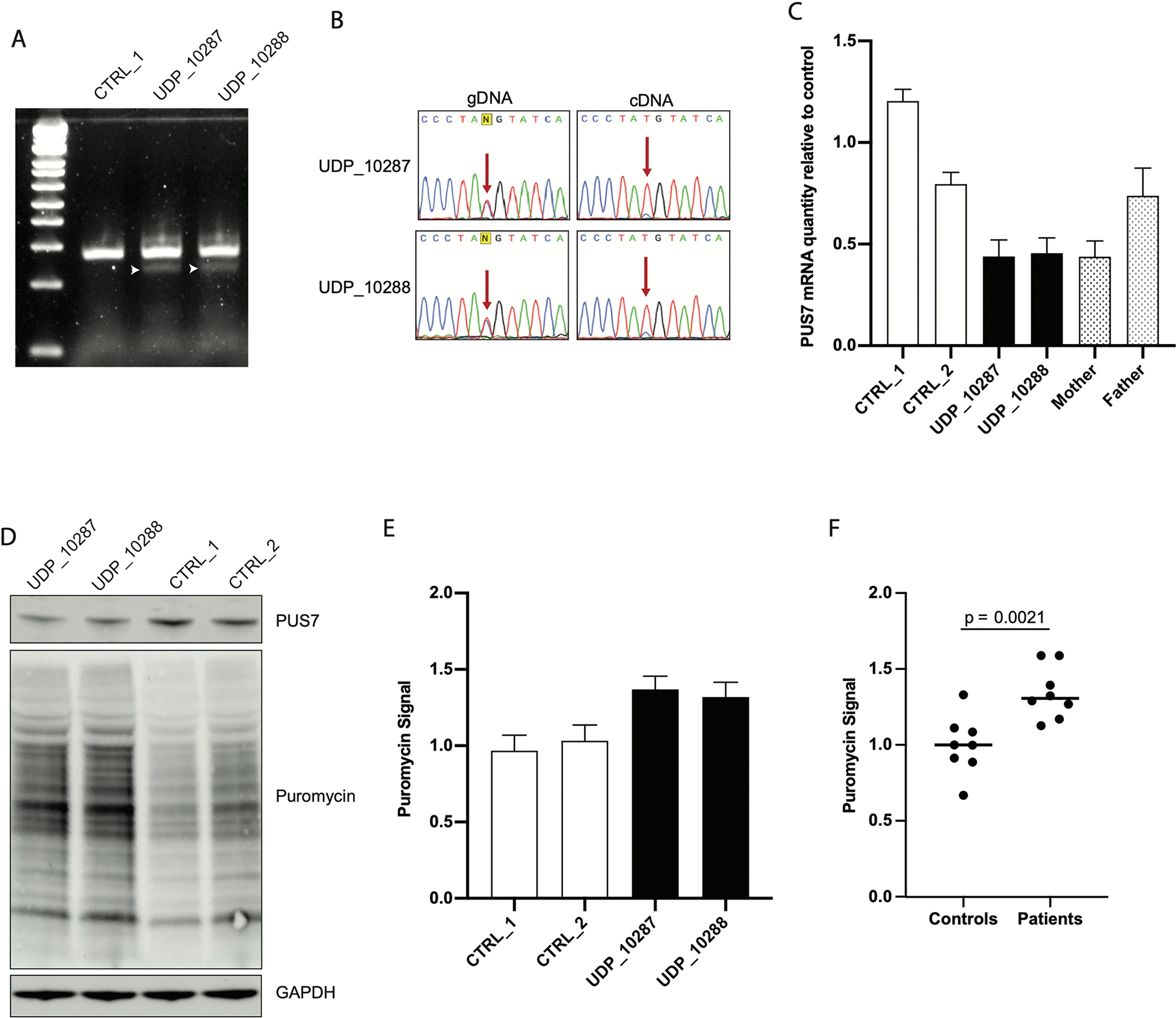
A) RT-PCR of RNA extracted from skin fibroblasts demonstrates the presence of a smaller splice form of PUS7 not detected in control. B) Sanger sequencing of RT-PCR products across the site of T387M shows that the majority of stable PUS7 mRNA is derived from the paternal allele. C) qRT-PCR of total RNA extracted from skin fibroblasts of the two patients, two age matched controls, and both parents demonstrates decreased PUS7 mRNA in individuals carrying the c.398+1 G>T splice variant (N = 9 for each sample); bar plots represent mean ± SEM. D) Western blots of total protein lysates purified from patient fibroblasts demonstrate that patients have decreased PUS7 protein quantity than controls and increased rate of protein translation relative to controls as measured by puromycin incorporation. E) Quantification of SUnSET assays conducted in fibroblasts (N = 4 for each sample); bar plots represent mean ± SEM. F) Combined analysis of SUnSET assay comparing control to patient fibroblast demonstrates statistically significant elevation of puromycin signal in patient cells (p = 2.1×10−3); bars represent mean value.
Decreased PUS7 mRNA resulted in decreased steady state PUS7 protein levels in patient fibroblasts, suggesting that the paternal allele carrying the T387M missense variant has no obvious effect on protein folding or stability. Immunofluorescent staining showed that PUS7 protein localized to the nucleus in patient fibroblasts similar to control fibroblasts (Sup Fig 4). To evaluate PUS7 function, we utilized the non-radioactive SUnSET assay15 to evaluate protein translation. Patient fibroblasts demonstrated elevated protein translation rate and confirmed deficient PUS7 activity (Fig 2D). Quantification of multiple SUnSET blots demonstrated consistent elevation of puromycin signal in patient cells (N = 4 for each cell line) (Fig 2E). Overall, patient fibroblasts demonstrated statistically elevated puromycin signal compared to control fibroblasts (p = 2.1×10−3). Together, these results indicate that our patients carry loss of function variants on both alleles of PUS7 and that loss of function results in dysregulation of protein translation.
Dysregulation of protein translation does not affect cell proliferation because regulation of gene transcription remains active
While dysregulated protein translation can manifest globally, we tested several key proteins to see if expression levels were aberrant (Fig 3A). Elevated protein translation can be associated with increased mTOR activity, which increases phosphorylation of 4EBP116. Unphosphorylated 4EBP1 sequesters eIF4E (RNA cap binding protein), so that it is unable to participate in the translation initiation complex with eIF4A and eIF4G, thereby limiting the rate of protein translation. We observed no difference in the phosphorylation state of 4EBP1, and therefore infer that eIF4E activity is similar between patients and controls (Sup Fig 5), corroborating previous studies that PUS7-based mediation of protein translation acted through regulation of eIF4A and eIF4G and not through eIF4E9. Suspecting that dysregulation of eIF4A and eIF4G was likely driving the observed dysregulation of protein translation, we evaluated the protein levels of genes previously shown to be sensitive to eIF4A (RNA helicase) activity17. We found that MYC protein was elevated in patient cells compared to controls. Because of the phenotypic overlap of our patients with LNS, we also evaluated HPRT1 protein levels and found them to be slightly decreased in patient fibroblasts (Fig 3B). Simultaneous increases in MYC and decreases in HPRT1 demonstrate dysregulation rather than a general increase in protein translation. Increased protein translation and elevation of MYC expression are common markers of tumor cells18. However, we found no significant difference in cell proliferation rates between patient and control fibroblasts (Fig 3C), which in hindsight is not unsurprising given the modest magnitude of difference in MYC protein levels. When we evaluated MYC protein levels at various densities of cell growth, we found that patient fibroblasts were able to downregulate MYC as cells became more confluent (Fig 3D). We also observed that the levels of phosphorylated 4EBP1, another marker of protein translation used as a proxy for mTOR activity16, remained similar between patient and control cells (Sup Fig 5) even with increasing cell density. To confirm that differences in MYC and HPRT1 protein levels were due to dysfunctional protein translation we conducted qRT-PCR on patient and control fibroblasts. We found that MYC mRNA levels were comparable between patient and controls, suggesting that differences in MYC protein levels are driven by dysregulation of protein synthesis and not gene expression. In addition, steady state levels of HPRT1 mRNA measured by qRT-PCR did not correlate with decreased protein levels in the patient cells (Fig 3E). These studies indicate that PUS7 deficiency affects gene expression at the level of translation, not transcription.
Figure 3. Dysregulation of protein translation does not affect cell proliferation because regulation of gene transcription remains active.
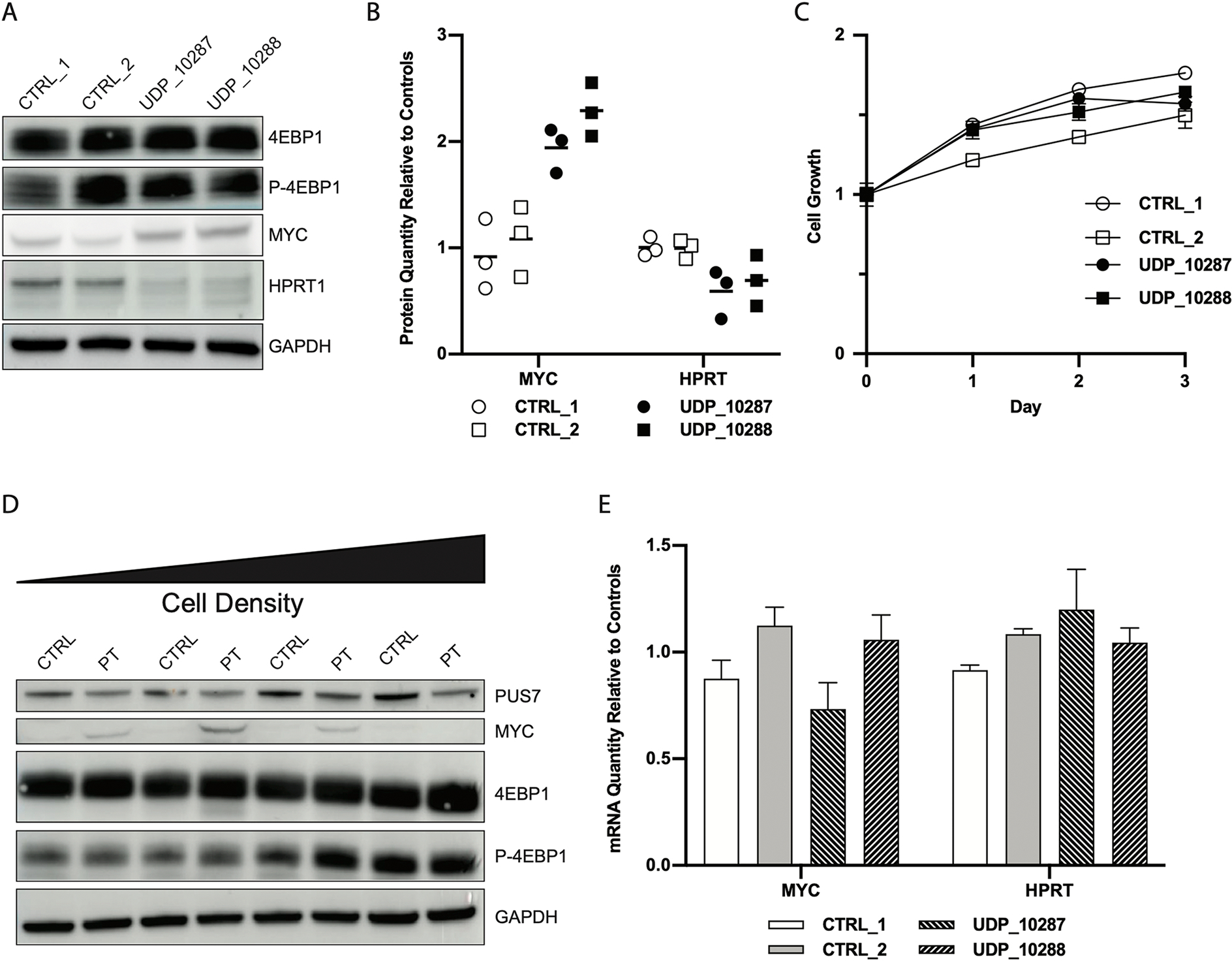
A) Western blot of whole cell lysates from patient and control fibroblasts demonstrates increased expression of MYC and decreased expression of HPRT1 in patient cells relative to controls without alterations to 4EBP1 or phosphorylated 4EBP1. B) Quantification of MYC and HPRT1 protein in control and patient fibroblasts (N = 3 for each sample) demonstrates elevation of MYC (p = 4.9×10−4) and decrease of HPRT1 protein (p = 3.5×10−3); bars represent mean value of each sample. C) Measurements of proliferation of patient and control fibroblasts shows no difference in cell growth (N = 3 for each measurement); error bars represent ± SEM. D) Western blots of whole cell lysates extracted from fibroblasts grown at different densities show that MYC expression is elevated in patient cells but decreases with increasing cell density. Phosphorylated 4EBP1 increases with increasing cell density E) qRT-PCR of demonstrates similar levels of MYC and HPRT1 mRNA between patient and control fibroblasts cells (N = 6); bars represent mean values ± SEM.
PUS7 activity is sufficient to drive molecular findings in HeLa cells
We created an isogenic PUS7 knockout HeLa cell line to confirm that the dysregulation of protein translation in patient cells could be explained by lack of PUS7 activity alone. We transfected HeLa cells constitutively expressing Cas9 with guide RNAs (gRNAs) targeted to PUS7 (Sup Table 2). and isolated a clonal cell line homozygous for a deletion spanning the 3’ end of exon 10 as well as the first 4 bases of intron 10 (Fig 4A). We confirmed that this clone had no detectable PUS7 mRNA by RT-PCR (Fig 4B, Sup Fig 6). Western blot and SUnSET assay demonstrated no detectable PUS7 protein and an increase in protein translation when compared to the parental HeLa cells (Fig 4C and Sup Fig 7). We confirmed that this knockout of PUS7 and subsequent increase in protein translation did not increase cell proliferation (Sup Fig 8). Transient transfection with a vector expressing a reference copy of the PUS7 cDNA sequence (NM_019042) verified the expression and nuclear localization of exogenous PUS7 (Fig 4D). Transient expression of WT-PUS7 restored puromycin signal and MYC protein to levels similar to the parental HeLa cells, demonstrating that PUS7 protein was sufficient to restore regulation of protein translation (Fig 4E and Sup Fig 9). These data demonstrate that loss of PUS7 activity is sufficient to disrupt protein translation in HeLa cells. Total protein translation and MYC protein levels were fully reversed by expression of exogenous PUS7 cDNA.
Figure 4. PUS7 activity is sufficient to drive molecular findings in HeLa cells.
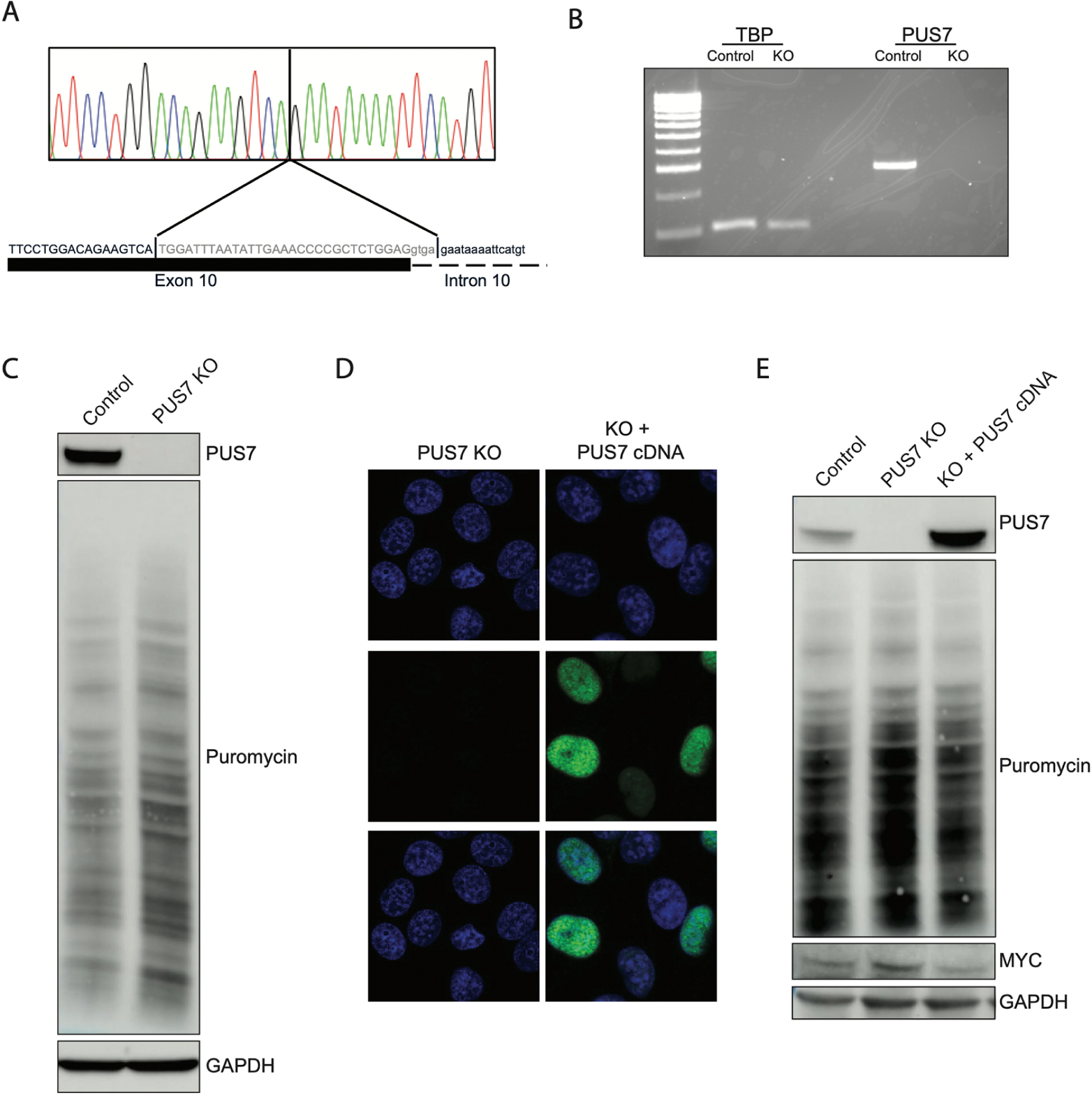
A) Sanger sequencing of genomic DNA extracted from CRISPR induced PUS7 knockout in HeLa cells demonstrates cells are homozygous for a 34 bp deletion at the 3’ end of exon 10 extending into intron 10. B) RT-PCR from total RNA extracted from PUS7 KO cells demonstrating KO cells have no detectable PUS7 mRNA. C) Western blot of whole cell lysates from KO cells demonstrating that cells do not generate detectable stable protein and that protein translation is increased in KO cells. D) Immunocytochemistry of HeLa and KO cells demonstrates lack of PUS7 protein in KO cells. Transient transfection with plasmid expressing PUS7 cDNA demonstrates that expressed protein localizes to the nucleus. Top row: DAPI only, Middle row: anti-PUS7 IHC, Bottom row: merge E) Western blot of whole cell lysates purified from KO cells transiently transfected with PUS7 expression plasmid demonstrates significant overexpression compared to parental HeLa cells. Plasmid expressing PUS7 cDNA is able to decrease puromycin incorporation and decrease MYC protein levels relative to untransfected KO cells.
DISCUSSION
Genes responsible for generating RNA post transcriptional modifications, and consequently regulating gene expression, are associated with a number of Mendelian disorders19. Among them, three disorders of PUS enzymes have been described to date. PUS1 primarily targets mRNA20 and loss of function variants in PUS1 are associated with myopathy, lactic acidosis and sideroblastic anemia (MLASA)21. PUS3 targets tRNAs and loss of function of PUS3 is associated with severe intellectual disability, microcephaly, white matter disease, renal abnormalities, and growth deficiency22,23. PUS7, like PUS3, can modify tRNAs, as well as other noncoding RNAs and mRNAs, and patients carrying loss of function variants in PUS7 manifest with intellectual disability, global developmental delay, aggressive behavior, postnatal microcephaly and growth impairment12,24. More generally, disorders that disrupt post transcriptional modifications of tRNAs are associated with a range of neuropsychiatric diseases25. Recent reports of PUS7 deficiency in humans determined that proper pseudouridylation is essential for normal development of the nervous system by correlating global pseudouridine levels and pseudouridylation status of known PUS7 targets with behavioral findings in a pus7 deficient Drosophila model12. Separate studies in yeast4 and hematopoietic stem cells9 demonstrated that PUS7 plays a role in regulating protein translation, and PUS7 deficiency inhibits cellular differentiation and stress response4. In this study we integrated the results of the previous studies using primary fibroblasts from PUS7-deficient patients to begin to connect molecular mechanisms to clinical phenotypes. In the process, we identified MYC and HPRT1 as genes whose protein expression is perturbed by loss of PUS7 activity.
The patients in this study were referred to the NIH UDP for a constellation of clinical findings including postnatal microcephaly, developmental delay, short stature, speech delay, self-injurious behavior, hearing loss, hypoglycemia, and hyperuricemia. They are compound heterozygous for loss of function variants in PUS7 and the first reported patients born to non-consanguineous parents. We have expanded the phenotype of PUS7 deficiency to include hypoglycemia, hyperuricemia, self-injurious behavior, axonal neuropathy, and ataxia. It is unclear if these additional findings were appreciated due to comprehensive deep phenotyping at the NIH UDP26 or represent a phenotypic expansion from the initial reports of PUS7 deficiency.
Our patients have extremely rare variants in PUS7. We confirmed pathogenicity of the maternally inherited splice variant and inferred pathogenicity of the paternally inherited missense variant through protein translation studies. Because previous reports demonstrated significantly reduced levels of total pseudouridine and pseudouridine at PUS7 target sites in patient cells24 as well as behavioral abnormalities in knockout models12, we focused on dissecting the molecular effects of PUS7 deficiency. PUS7 regulates protein translation through pseudouridylation of tRNA, which allows for the formation of active tRFs and sequestration of the translation initiation factors eIF4A and eIF4G. In the absence of active tRFs, eIF4A and eIF4G constitutively initiate protein translation, resulting in elevated protein translation9. Consistent with this mechanism, patient cells exhibited elevated levels of protein translation.
In addition, we measured expression of MYC protein since its translation has been specifically tied to the activity of eIF4A-dependent protein translation17. We believe that dysregulation of protein translation may also result in decreased levels of certain proteins despite an overall increase in total protein synthesis. Therefore, although the net effect of PUS7 deficiency is an increase in total protein translation, there remained the possibility that some genes might be downregulated; this could be through decreased translational efficiency due to shifts in the translatome or indirectly due to increased MYC activity. We recognized HPRT1 as a second gene of interest because of the clinical similarities of our patients and patients with LNS even though molecular testing for LNS was negative. LNS is a rare X-linked disorder with developmental delay, hyperuricemia, and self-injurious behavior, typically in the form of finger biting27. Our patients presented with clinical symptoms less severe than typically associated with LNS28 patients. Decreased, but not absent, HPRT1 protein translation is a possible explanation for the hyperuricemia and the observed LNS-like behaviors29,30 in the absence of pathogenic variants in HPRT1. The amount of decreased HPRT1 protein observed here is not diagnostic for LNS however could be consistent with the clinical findings, specifically the finger biting and hyperuricemia. Further studies are required to determine the extent of HPRT1 involvement in disease pathology, but we believe it to be an interesting association given the phenotypic overlap with a disease as striking as LNS.
We demonstrate here that loss of function of PUS7 results in dysregulation of protein expression and provide MYC and HPRT1 as two significant examples. We believe dysregulated protein translation is an important component of the molecular mechanism underlying this disease, and is similar to that reported in autism spectrum disorder (ASD)31–34. Interestingly, the proband indeed met criteria for ASD while the younger sibling was too cognitively impaired to formally make an ASD diagnosis.
Although MYC is traditionally considered to be an oncogene35, increased MYC expression due to loss of PUS7 activity did not result in increased cell proliferation, possibly due to other regulatory factors that are not perturbed by loss of PUS7 activity or the modest levels of MYC increase. Pathogenic variants in cancer and overgrowth genes are also found in cases of idiopathic autism without increased incidence of childhood cancer36. This suggests that increased protein translation may contribute to ASD without increasing risk of developing cancer, and that the mechanism underlying PUS7 deficiency may overlap with ASD. Although we are unaware of any studies directly linking HPRT1 expression to protein translation rate, we believe its expression is impacted by loss of PUS7 because we observe decreased protein also without decreased mRNA. Although we focused on MYC and HPRT1 as genes whose expression is dysregulated by PUS7 deficiency, there are likely many more genes whose expression is altered. Since this phenomenon is a result of exaggerated translation without altered transcription, many of the regulatory genetic mechanisms remain intact and are likely preventing more severe disease.
We used CRISPR/Cas9 to generate an independent knockout (KO) cell line from HeLa cells to validate our molecular findings on an isogenic background. Following CRISPR editing, our KO cell lines had no expression of PUS7 mRNA or protein and recapitulated the molecular findings of our fibroblasts, including elevation of MYC protein. Our rescue experiments showed that exogenous PUS7 protein expressed from a plasmid vector could revert the protein translation phenotype, including restoring MYC protein to endogenous levels, when expressed in KO cells. These studies demonstrate that loss of PUS7 activity is sufficient to drive the cellular findings observed in patient cells.
PUS7 deficiency results in a complex neurological phenotype consisting of postnatal microcephaly, developmental delay, intellectual disability, aggressive behavior, mild dysmorphism, and short stature. Our data suggest that PUS7 deficiency dysregulates cap dependent protein translation, a mechanism that is also linked to autism spectrum disorder31,33,34,37. Eukaryotic gene expression is tightly regulated, and disruption of the molecular machinery underlying genetic regulation has been well documented to result in human disease, often with neurological consequences38. Genetic regulation through pseudouridylation of tRNA molecules is still incompletely understood despite pseudouridine being the earliest recognized post transcriptional modification of RNA. In this study, we provide further evidence for the importance of pseudouridylation of tRNA as a regulatory component of global gene expression. We find dysregulated protein translation in patient fibroblasts modestly increases protein expression of MYC and modestly decreases protein expression of HPRT1 without affecting mRNA levels of either gene. Increased activity of MYC may cause blunted differentiation in neurons by reinforcing cells to remain in their undifferentiated state13 while decreased activity of HPRT1 may cause a LNS-like phenotype27. Although we believe that we have identified an aspect of the molecular mechanism underlying this disorder, the complete pathology will be significantly more complex. PUS7 targets a wide range of other RNA species beyond tRNAs, including mRNAs, and those effects have yet to be studied in depth. Exploring the complete scope of genes and pathways whose expression patterns are altered by loss of PUS7 activity will expand our understanding of the basic molecular mechanisms governing gene expression and the complex neurological phenotypes that are associated with disruption of these regulatory mechanisms.
METHODS
Enrollment and consent
The patients were evaluated through the National Institutes of Health Undiagnosed Diseases Program (NIH-UDP) and were enrolled on protocol 15-HG-0150, approved by the National Human Genome Research Institute Institutional Review Board (NHGRI-IRB) with parents providing written informed consent. The parents were consented for skin biopsies under the protocol 76-HG-0238, also approved by the NIH-IRB.
Isolation and culture of primary cells
Primary dermal fibroblast cells from probands and parents were cultured from 3mm punch biopsies taken from the forearm. Unaffected pediatric primary dermal fibroblast cell lines GM05399 (Control 1) and GM01651 (Control 2) (Coriell Institute for Medical Research, Camden, NJ) were used as controls. Fibroblasts were cultured in high glucose DMEM (11965092) with 10% fetal bovine serum (FBS, 10082), 1% nonessential amino acids (11140050,) and 1% pen/strep (1507000063) (all obtained from Gibco/Thermo Fisher Scientific, Gaithersburg, MD) (complete DMEM) at 37°C with 5% CO2.
DNA Extraction of Fibroblasts and HeLa Cells
Cells were pelleted and resuspended in PBS. DNA extraction was performed using the QIAgen DNeasy Blood and Tissue kit (69504, Qiagen Germantown, MD).
PCR and Sanger sequencing
PCR amplification was conducted using Redtaq ReadyMix (R2523 Sigma-Aldrich, St. Louis, MO). PCR cleanup for sequencing was performed using ExoSAP-IT PCR product cleanup reagent (78205, Thermo Fisher Scientific, Waltham, MA). Purified PCR products were sequenced by Sanger sequencing (Eurofins Genomics, Louisville, KY).
Protein purification
Cells were pelleted by centrifugation, washed with 1X PBS, and pelleted a second time. The supernatant was discarded, and the cell pellets were homogenized in RIPA Lysis and Extraction Buffer (89901, Thermo Fisher Scientific, Waltham, MA) supplemented with 1% Halt Protease Inhibitor Cocktail 100X (87786, Invitrogen/Thermo Fisher Scientific, Carlsbad CA). Lysates were cleared by centrifugation.
RNA Extraction
Cells were pelleted by centrifugation and resuspended in Trizol (15596026, Life Technologies/Thermo Fisher Scientific, Grand Island, NY). Suspensions were homogenized using QIA-Shredder spin columns (79656, Qiagen, Germantown, MD). Samples were phase separated with chloroform and proceeded to RNA purification using the QIAgen RNeasy Mini Kit (74104, Qiagen, Germantown, MD).
cDNA synthesis
500 ng of total RNA was converted to cDNA using the iScript cDNA Synthesis kit (1708891, Biorad, Hercules, CA). cDNA generated from fibroblasts was diluted 1:1 and cDNA generated from HeLa cells was diluted 1:10.
qRT-PCR
Diluted cDNA was used for qRT-PCR using SsoAdvance Universal Syber Green Supermix (1725270, Biorad, Hercules, CA) with 100 nM primer (Sup Table 3). TBP was used as the internal experimental control. Quantification was measured using the ABI QuantStudio3 instrument (Thermo Fisher Scientific, Waltham, MA). Data are presented as mean values ± SEM.
Immunocytochemistry
Cells were seeded on coverslips and allowed to grow overnight. Cells were fixed with cold methanol at −20° C for 10 minutes and blocked for 20 minutes with 10% normal goat serum (31873, Life Technologies/Thermo Fisher Grand Island, New York). Coverslips were washed with PBS for 30 minutes and then stained with PUS7 primary antibody (1:500 dilution OTI4C6, Origene Technologies, Rockville, MD) in 2.5% normal goat serum overnight at 4° C with gentle rocking. Cells were washed for 30 minutes with PBS and then stained with secondary anti-mouse antibody conjugated with Alexa-488 fluorophore (1:500 ab150113, abcam, Cambridge, MA) in 2.5% normal goat serum at room temperature for 45 min protected from light. Cells were counter stained with Dapi (ab104139, abcam, Cambridge, MA) and mounted on slides. Images were captured using a Zeiss confocal microscope at 40× magnification.
Western Blots
Protein concentrations were measured using the Pierce BCA assay kit (23225, Life Technologies/Thermo Fisher Grand Island, New York). Samples were denatured with DTT and NuPAGE LDS Sample Buffer (NP0008, Life Technologies/Thermo Fisher, Grand Island, New York) and loaded into a 4–12% Bis-Tris 1.5mm 10 well gel (NW04120, Life Technologies/Thermo Fisher Grand Island, New York) in 5% MOPS (NP0001, Life Technologies/Thermo Fisher Grand Island, New York) or 5% MES (NP0002, Life Technologies/Thermo Fisher Grand Island, New York) for smaller proteins. Gels were transferred onto nitrocellulose membrane using the iBlot 2 Dry Blotting System (Invitrogen/Thermo Fisher Scientific, Carlsbad, CA). Following transfer, the membrane was blocked using 5% milk solution in PBST. Blocking was done for one hour at room temperature or overnight at 4° C. Membranes were incubated in primary antibodies for one hour at room temperature with gentle shaking. Primary antibody was diluted in PBST for PUS7 (1:500 OTI4C6, Origene Technologies, Rockville, MD), GAPDH (1:5,000 EPR1689, abcam, Cambridge, MA), MYC (1:250, D84C12, Cell Signaling Technology, Denvers, MA), HPRT1 (1:5,000 EPR5298, abcam, Cambridge, MA), 4EBP1 (1:5,000 53H11, Cell Signaling Technology, Denvers, MA), phospho-4EBP1 (1:5,000 236B4, Cell Signaling Technology, Denvers, MA). Secondary antibody conjugated with HRP, anti-mouse (ab6728, abcam, Cambridge, MA) or anti-rabbit (ab6721, abcam, Cambridge, MA), was diluted in PBST 1:10,000 in PBST. Blots were visualized by chemiluminescence using Amersham ECL Prime (RPN2236 GE Amersham/Cytiva, Piscataway, NJ) and imaged using an Amersham 680 Blot and Gel Imager.
SUnSET Assay
Cells were treated with culture media containing 10 μg/mL puromycin (ant-pr-1, Invivogen, San Diego, CA) for 30 minutes followed by standard protein purification. 25 μg of whole cell lysates were used for western blot to visualize puromycin incorporation.
Quantification of western blots
Grayscale .tif western blot images were quantified using the ImageQuantTL software from GE. Statistical significance was determined by pooling data from patient and control samples and running a t-test. Lines in figures correspond to mean values.
Cell proliferation measurement
Cell proliferation was measured using the WST-8 reagent (CK04, Dojindo Laboratories, Rockville, MD). Equal numbers of cells were plated into 12 well plates and WST-8 reagent was added to cell lines after growing for 24, 48, 72, or 96 hours. After addition of WST-8 reagent, cells were incubated at 37° C for 2 hours and absorbance was measured at 450 nm. Each cell line was measured in triplicate for each time point.
CRISPR knockout of PUS7 in HeLa cells
HeLa cells expressing Cas9 protein were purchased from GeneCopia (SL503, GeneCopia, Rockville, MD). Guide RNAs targeted to PUS7 were expressed using the pGS-gRNA-Neo vector (Genescript, Piscataway, NJ) and transfected into cells using Lipofectamine 3000 (L3000015, Invitrogen/Thermo Fisher Scientific, Carlsbad, CA). Transfected cells were selected based on neomycin resistance by supplementing culture media with G418 (10131035, Gibco/Thermo Fisher Scientific, Gaithersburg, MD). Neomycin resistant cells were pooled and individual clones were subcloned using cloning cylinders (C1059, Sigma Aldrich Corp, St Louis, MO) and grown independently.
Transient transfection of knockout cells
PUS7 knockout cells were transfected with plasmids expressing PUS7 cDNA using Lipofectamine 3000 (L3000015, Invitrogen/Thermo Fisher Scientific, Carlsbad, CA). RNA and protein were extracted from cells 48 hours post transfection.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients and their family for their participation in this study. This study was supported by the National Human Genome Research Institute (NHGRI) Intramural Research Program (HG200409-01) and the National Institutes of Health (NIH) Undiagnosed Diseases Program, part of the Undiagnosed Diseases Network, which is supported by the Common Fund, Office of the Director, NIH (HG200352).
ABBREVIATIONS
- ASD
Autism Spectrum Disorder
- LNS
Lesch-Nyhan Syndrome
- NHGRI
National Human Genome Research Institute
- NMD
Nonsense Mediated mRNA Decay
- PUS
Pseudouridine Synthase
- tRF
tRNA Fragment
- UDP
Undiagnosed Diseases Program
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Frye M, Harada BT, Behm M, and He C (2018). RNA modifications modulate gene expression during development. Science 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(1998). Modification and editing of RNA (Washington, DC: ASM Press; ). [Google Scholar]
- 3.Torres AG, Batlle E, and Ribas de Pouplana L (2014). Role of tRNA modifications in human diseases. Trends Mol. Med. 20, 306–314. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. (2014). Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell 159, 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behm-Ansmant I, Urban A, Ma X, Yu Y-T, Motorin Y, and Branlant C (2003). The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite–multisubstrate RNA:Ψ-synthase also acting on tRNAs. RNA 9, 1371–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar P, Kuscu C, and Dutta A (2016). Biogenesis and Function of Transfer RNA-Related Fragments (tRFs). Trends Biochem. Sci. 41, 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanov P, Emara MM, Villen J, Gygi SP, and Anderson P (2011). Angiogenin-Induced tRNA Fragments Inhibit Translation Initiation. Mol. Cell 43, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortés-Garrido R, Gkatza N, et al. (2016). Stem cell function and stress response are controlled by protein synthesis. Nature 534, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guzzi N, Cieśla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, Pimková K, Sommarin MNE, Munita R, Lubas M, et al. (2018). Pseudouridylation of tRNA-Derived Fragments Steers Translational Control in Stem Cells. Cell 173, 1204–1216.e26. [DOI] [PubMed] [Google Scholar]
- 10.Sobala A, and Hutvagner G (2013). Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol. 10, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tahmasebi S, Khoutorsky A, Mathews MB, and Sonenberg N (2018). Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 19, 791–807. [DOI] [PubMed] [Google Scholar]
- 12.Brouwer A.P.M. de, Jamra RA, Körtel N, Soyris C, Polla DL, Safra M, Zisso A, Powell CA, Rebelo-Guiomar P, Dinges N, et al. (2018). Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am. J. Hum. Genet. 103, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gahl WA, and Tifft CJ (2011). The NIH Undiagnosed Diseases Program: Lessons Learned. JAMA 305, 1904–1905. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. [DOI] [PubMed] [Google Scholar]
- 16.Fingar DC, Salama S, Tsou C, Harlow E, and Blenis J (2002). Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 16, 1472–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. (2014). RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePinho R, Mitsock L, Hatton K, Ferrier P, Zimmerman K, Legouy E, Tesfaye A, Collum R, Yancopoulos G, Nisen P, et al. (1987). Myc family of cellular oncogenes. J. Cell. Biochem. 33, 257–266. [DOI] [PubMed] [Google Scholar]
- 19.Angelova MT, Dimitrova DG, Dinges N, Lence T, Worpenberg L, Carré C, and Roignant J-Y (2018). The Emerging Field of Epitranscriptomics in Neurodevelopmental and Neuronal Disorders. Front. Bioeng. Biotechnol 6,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Zhu P, Ma S, Song J, Bai J, Sun F, and Yi C (2015). Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, and Zeviani M (2007). Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J. Med. Genet. 44, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Paiva ARB, Lynch DS, Melo US, Lucato LT, Freua F, de Assis BDR, Barcelos I, Listik C, de Castro dos Santos, D., Macedo-Souza, L.I., et al. (2019). PUS3 mutations are associated with intellectual disability, leukoencephalopathy, and nephropathy. Neurol. Genet. 5,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaheen R, Han L, Faqeih E, Ewida N, Alobeid E, Phizicky EM, and Alkuraya FS (2016). A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 135, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaheen R, Tasak M, Maddirevula S, Abdel-Salam GMH, Sayed ISM, Alazami AM, Al-Sheddi T, Alobeid E, Phizicky EM, and Alkuraya FS (2019). PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bednářová A, Hanna M, Durham I, VanCleave T, England A, Chaudhuri A, and Krishnan N (2017). Lost in Translation: Defects in Transfer RNA Modifications and Neurological Disorders. Front. Mol. Neurosci 10,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gahl WA, Mulvihill JJ, Toro C, Markello TC, Wise AL, Ramoni RB, Adams DR, and Tifft CJ (2016). The NIH Undiagnosed Diseases Program and Network: Applications to modern medicine. Mol. Genet. Metab. 117, 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lesch M, and Nyhan WL (1964). A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med. 36, 561–570. [DOI] [PubMed] [Google Scholar]
- 28.Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, and O’Neill JP (2000). The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat. Res. Mutat. Res. 463, 309–326. [DOI] [PubMed] [Google Scholar]
- 29.Ceballos-Picot I, Le Dantec A, Brassier A, Jaïs J-P, Ledroit M, Cahu J, Ea H-K, Daignan-Fornier B, and Pinson B (2015). New biomarkers for early diagnosis of Lesch-Nyhan disease revealed by metabolic analysis on a large cohort of patients. Orphanet J. Rare Dis. 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Page T, Bakay B, and Nyhan WL (1982). Kinetic studies of normal and variant hypoxanthine phosphoribosyltransferases in intact fibroblasts. Anal. Biochem. 122, 144–147. [DOI] [PubMed] [Google Scholar]
- 31.Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. (2013). Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z-X, Kim GH, Tan J-W, Riso AE, Sun Y, Xu EY, Liao G-Y, Xu H, Lee S-H, Do N-Y, et al. (2020). Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat. Commun. 11, 1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, and Klann E (2013). Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogozin IB, Gertz EM, Baranov PV, Poliakov E, and Schaffer AA (2018). Genome-Wide Changes in Protein Translation Efficiency Are Associated with Autism. Genome Biol. Evol. 10, 1902–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayward WS, Neel BG, and Astrin SM (1981). Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature 290, 475–480. [DOI] [PubMed] [Google Scholar]
- 36.Darbro BW, Singh R, Zimmerman MB, Mahajan VB, and Bassuk AG (2016). Autism Linked to Increased Oncogene Mutations but Decreased Cancer Rate. PLoS ONE 11,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y-C, Chang Y-W, and Huang Y-S (2019). Dysregulated Translation in Neurodevelopmental Disorders: An Overview of Autism-Risk Genes Involved in Translation. Dev. Neurobiol. 79, 60–74. [DOI] [PubMed] [Google Scholar]
- 38.Fahrner JA, and Bjornsson HT (2019). Mendelian disorders of the epigenetic machinery: postnatal malleability and therapeutic prospects. Hum. Mol. Genet. 28, R254–R264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.