

Abstract
Mitochondria are essential for the activity, function and viability of eukaryotic cells and mitochondrial dysfunction is involved in the pathogenesis of acute kidney injury (AKI) and chronic kidney disease, as well as in abnormal kidney repair after AKI. Multiple quality control mechanisms, including antioxidant defence, protein quality control, mitochondrial DNA repair, mitochondrial dynamics, mitophagy and mitochondrial biogenesis, have evolved to preserve mitochondrial homeostasis under physiological and pathological conditions. Loss of these mechanisms may induce mitochondrial damage and dysfunction, leading to cell death, tissue injury and, potentially, organ failure. Accumulating evidence suggests a role of disturbances in mitochondrial quality control in the pathogenesis of AKI, incomplete or maladaptive kidney repair and chronic kidney disease. Moreover, specific interventions that target mitochondrial quality control mechanisms to preserve and restore mitochondrial function have emerged as promising therapeutic strategies to prevent and treat kidney injury and accelerate kidney repair. However, clinical translation of these findings is challenging owing to potential adverse effects, unclear mechanisms of action and a lack of knowledge of the specific roles and regulation of mitochondrial quality control mechanisms in kidney resident and circulating cell types during injury and repair of the kidney.
Mitochondria are intracellular organelles that have key roles in the production of ATP, the regulation of various catabolic and anabolic processes and in the maintenance of cellular calcium and redox homeostasis. They also act as central hubs that coordinate signalling cascades that regulate cell survival and cell death pathways. Thus, maintaining mitochondrial integrity and function is critical for cellular homeostasis.
Mitochondria are a major intracellular source and a primary target of reactive oxygen species (ROS), which makes them extremely vulnerable to damage during physiological adaptations and stressful conditions. Multiple quality control mechanisms have evolved within mitochondria to counteract stress and preserve organelle integrity and function, including antioxidant defence, protein quality control, mitochondrial DNA (mtDNA) repair, mitochondrial dynamics (fusion and fission), mitophagy and mitochondrial biogenesis. These quality control mechanisms act at the molecular or organelle levels and work coordinately to maintain a healthy mitochondrial population. Loss of mitochondrial quality control may induce mitochondrial damage and dysfunction, leading to cell death, tissue injury and possible organ failure1,2.
The kidney has an essential role in maintaining body homeostasis, mainly through the removal of waste metabolic products via glomerular filtration and the subsequent reabsorption of the glomerular filtrate, including water, ions and nutrients, in the proximal tubules and thick ascending limbs of the loop of Henle. This active reabsorption of large quantities of solutes results in high energy demand that can only be met by mitochondrial oxidative metabolism. Active transport is greatest in the proximal tubules, which have very limited capacity for glycolysis; thus, aerobic respiration is the primary mechanism of ATP production3,4. The proximal tubules are rich in mitochondria and maintenance of mitochondrial homeostasis and quality control is vital for normal kidney function.
Acute kidney injury (AKI) is characterized by an abrupt loss of kidney function, which is often caused by ischaemia–reperfusion (IR), sepsis or nephrotoxins. AKI is associated with high mortality but may also contribute substantially to the development and progression of chronic kidney disease (CKD)5. Evidence from clinical and experimental studies suggests that incomplete or maladaptive repair after AKI leads to tubulointerstitial fibrosis and ultimately to CKD6,7. Moreover, mitochondrial damage or dysfunction may have a role in the pathogenesis of kidney injury owing to AKI or CKD and in abnormal kidney repair after injury1,8,9. In this Review, we summarize current knowledge of the role of mitochondrial quality control mechanisms in kidney injury and repair and highlight their potential as therapeutic targets.
Mitochondria in tissue injury and repair
Mitochondria regulate a variety of cellular processes that are closely associated with tissue injury and repair, such as cell death, cell proliferation and differentiation, metabolic adaptation and inflammation (FIG. 1).
Fig. 1 |. Mitochondrial functions and the effects of mitochondrial damage.
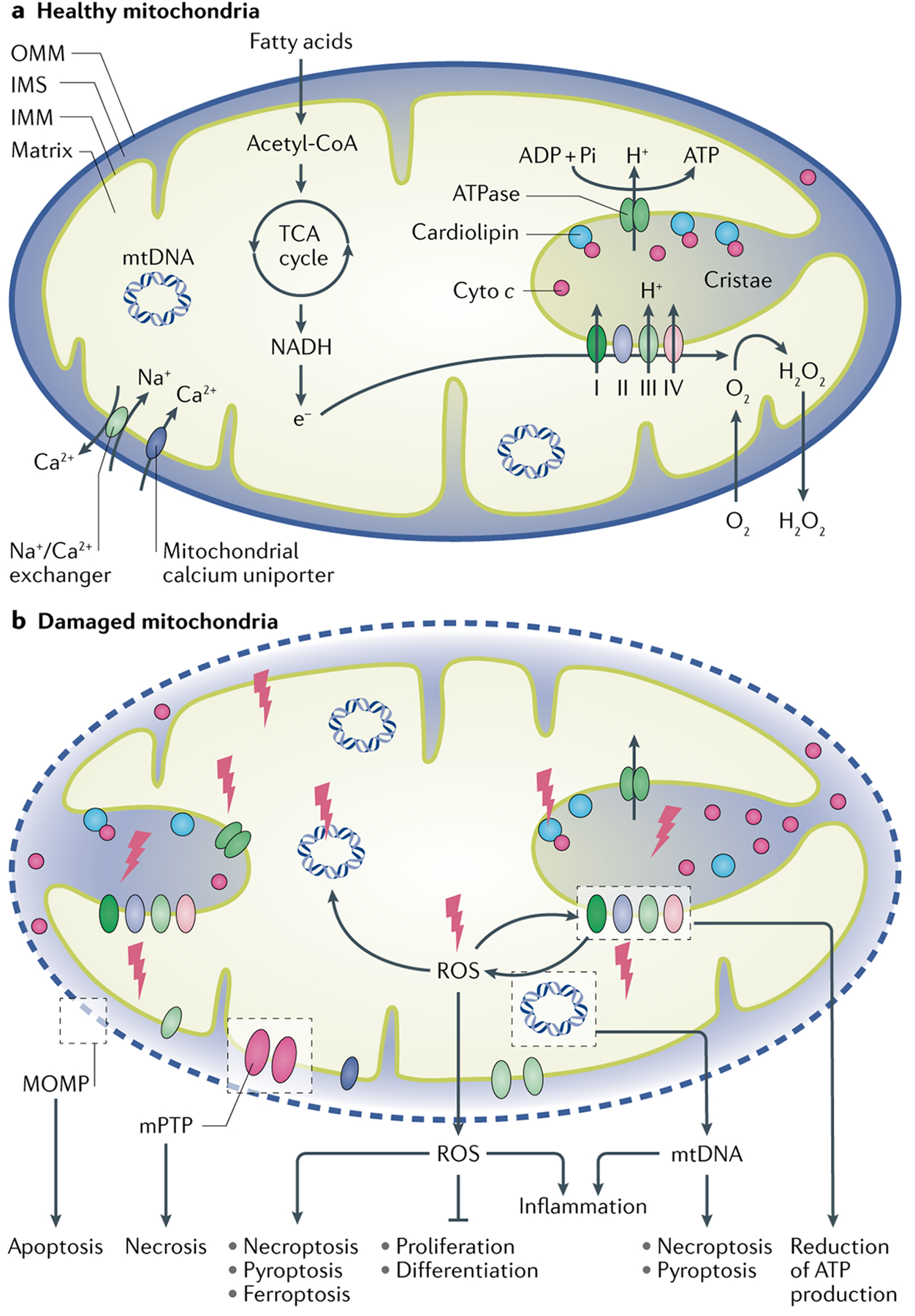
a | Mitochondria have a key role in the generation of energy in the form of ATP. The nicotinamide adenine dinucleotides (NADHs) that are formed by fatty acid oxidation and the tricarboxylic acid (TCA) cycle in the matrix of the mitochondria pass their electrons to O2 via the electron transport chain comprising complexes I–IV, resulting in the generation of a proton gradient across the inner mitochondrial membrane (IMM) for ATP production. Cytochrome c (cyto c) exists in its free form in the intermembrane space (IMS) or is anchored to the IMM through interaction with cardiolipin, where it acts as an electron carrier between respiratory complexes III and IV. Mitochondria are a major source of reactive oxygen species (ROS). Electrons that leak from the electron transport chain react with O2 to form a superoxide anion, which is transformed into H2O2 by the enzymatic antioxidant superoxidase. Emission of mitochondrial H2O2 to the cytosol is essential for maintaining redox homeostasis and may also have a role in signalling pathways. Mitochondria also have important roles in maintaining cellular calcium homeostasis. b | In damaged mitochondria, ROS induce cardiolipin peroxidation, which converts cyto c from an electron carrier into a peroxidase that further oxidizes cardiolipin. This process contributes to the development of mitochondrial outer membrane permeabilization (MOMP) and the subsequent release of pro-apoptotic factors such as cyto c from the IMS into the cytosol, resulting in caspase activation and apoptosis. Mitochondrial permeability transition at the IMM drives necrosis. An increase in mitochondrial ROS production by damaged mitochondria may also induce other forms of cell death, including necroptosis, pyroptosis and ferroptosis, as well as inflammation. Release of mitochondrial ROS may impair cell proliferation and/or differentiation through the regulation of various signalling pathways. Mitochondrial DNA (mtDNA) released from damaged mitochondria is a potential activator of necroptosis and ferroptosis and can also induce inflammation. Mitochondrial damage reduces ATP production and can result in the energetic failure of cells. mPTP, mitochondrial permeability transition pore; OMM, outer mitochondrial membrane.
Cell death.
Mitochondria regulate various forms of cell death, including apoptosis and necrosis. Apoptosis is a regulated cell death process that can occur via intrinsic or extrinsic pathways. Intrinsic apoptosis is initiated by mitochondrial outer membrane permeabilization (MOMP), which releases pro-apoptotic factors such as cytochrome c and second mitochondria-derived activator of caspase (SMAC; also known as Diablo homologue, mitochondrial) from the intermembrane space (IMS) into the cytosol, where they activate caspase 9 and subsequent executioner caspases10. Under physiological conditions, cytochrome c either exists in its free form in the IMS or is anchored in the inner mitochondrial membrane (IMM) through binding to cardiolipin and acts as an electron carrier between respiratory complexes III and IV in the electron transport chain (ETC)11,12. Upon mitochondrial damage, ROS accumulation results in conversion of cytochrome c into a peroxidase that oxidizes cardiolipin, ultimately leading to the release of cytochrome c from the IMS into the cytosol, caspase activation and apoptosis11,12.
Electron transport chain.
(ETC). A series of four protein complexes (complex I–IV) embedded in the inner mitochondrial membrane that transfer electrons from electron donors to electron acceptors via redox reactions. This process drives the transfer of protons across the inner mitochondrial membrane to produce ATP.
MOMP can result from the pore-forming activity of pro-apoptotic members of the BCL-2 protein family, such as the apoptosis regulators BAX (also known as BCL-2-like protein 4) and BAK1 (also known as BCL-2 homologous antagonist/killer) or from high amplitude swelling of the matrix owing to increased permeability of the IMM to small solutes, a phenomenon termed mitochondrial permeability transition13. Extrinsic apoptosis occurs in response to perturbations of the extracellular microenvironment that are detected and relayed by plasma membrane receptors (also known as death receptors), resulting in activation of caspase 8 and downstream executioner caspases10. The intrinsic pathway of apoptosis may be linked to the extrinsic pathway through caspase 8-mediated cleavage of BH3-interacting domain death agonist (BID) and consequent translocation of the truncated BID to mitochondria, which induces MOMP, providing an amplification loop for apoptosis14.
Necrosis was traditionally classified as a passive, unregulated cell death process that is characterized morphologically by cell swelling and plasma membrane rupture. However, various forms of regulated necrosis have now been identified. One such form, necroptosis, is critically dependent on receptor-interacting serine-threonine kinase 1/3 (RIPK1/3)-mediated activation of mixed line-age kinase domain-like protein (MLKL)10. Mitochondrial ROS (mtROS) and mtDNA have been shown to induce necroptosis. In tumour necrosis factor (TNF)-induced necroptosis, mtROS-driven autophosphorylation of RIPK1 is essential for RIPK3 recruitment into the necrosome to induce necroptosis15. In addition, TNF-induced release of mtDNA into the cytosol can activate DNA sensors to enhance RIPK3–MLKL-dependent necroptosis16. Mitochondrial permeability transition is typically associated with necrosis17.
Mitochondria also regulate pyroptosis, which depends on the formation of plasma membrane pores by members of the gasdermin protein family and often occurs as a consequence of inflammatory caspase activation10,18. In addition, mitochondria may have a role in ferroptosis, a regulated form of necrosis that is initiated by iron-dependent lipid peroxidation and controlled by phospholipid hydroperoxide glutathione peroxidase (GPX4)10,19.
Animal studies have demonstrated an involvement of apoptosis, pyroptosis and ferroptosis in AKI20–23. However, the role of mitochondria in various forms of regulated necrosis during kidney injury and repair remains unclear.
Cell proliferation and differentiation.
To repair tissue injury, cells must enter and progress through the cell cycle to proliferate and then differentiate. Dynamic changes in mitochondrial morphology have a role in the regulation of cell cycle events. Mitochondria have been shown to form a hyperfused network during the gap 1 (G1) phase to DNA replicating phase (S) transition24. In normal rat kidney epithelial cells, reducing mitochondrial membrane potential using an uncoupling agent (carbonyl cyanide-4-(trifluoromethoxy) phenylhydra-zone) specifically blocked progression from the late G1 to the S phase24. Inhibition of dynamin-1-like protein (DRP1), the primary regulator of mitochondrial fission, for 48 h, prolonged formation of hyperfused mitochondria and disrupted mitotic chromosome alignment and S phase entry, resulting in cell cycle arrest in the human colon carcinoma cell line HCT116 (REF.24). In addition, mitochondrial clearance by autophagy accompanied by a bioenergetic shift from oxidative phosphorylation (OXPHOS) to anaerobic metabolism seemed to be essential for cell dedifferentiation and proliferation25. A reduction in mitochondrial number, together with an increase in glycolysis, was observed in kidney tubules that regenerated after ischaemia–reperfusion injury (IRI)4. This reduction was reversed during normal repair of tubules but persisted and became progressively more severe in tubule cells that failed to redifferentiate, suggesting a role of mitochondrial regression in kidney repair after AKI.
Hyperfused mitochondria.
A network of elongated and highly connected mitochondria, which can result from increased fusion and/or reduced fission, and represent an adaptive response against stress.
Oxidative phosphorylation.
(OXPHOS). A metabolic process in which the energy transferred by electrons from electron donors to electron acceptors through the electron transport chain via redox reactions drives the transport of protons across the inner mitochondrial membrane to generate a potential energy gradient. ATP synthase uses this energy to transform ADP into ATP in a phosphorylation reaction.
Mitochondrial regulation of cell proliferation and differentiation is mainly dependent on the control of energy metabolism and mtROS production. The hyperfused network of mitochondria that forms at the G1–S transition of the cell cycle is electrically continuous, with greater ATP output than mitochondria at any other cell cycle stage24. This high ATP output may meet the high energy demand for DNA replication. At the G2–M phase transition, mitochondria undergo fragmentation, possibly owing to a reduction in energy demand or a compensational increase in glycolysis.
Mitochondria are a major intracellular source of ROS in both physiological and pathological conditions. Depending on their concentrations and pulse duration, ROS exert different effects on cell proliferation and differentiation. Low levels of ROS may activate various cell proliferation signalling pathways, possibly through redox modulation of cysteine residues in transcription factors, such as nuclear factor-κB (NF-κB), hypoxia-inducible factor 1 (HIF1) and protein kinases (for example, RACα serine/threonine-protein kinase (ATK)). Moderate levels of ROS may activate cellular tumour antigen p53 to induce cell cycle arrest, whereas high levels of ROS cause oxidative stress and ultimately cell death26,27.
Inflammation.
Inflammation is a complex biological response that is essential for repairing tissue after injury. However, chronic inflammation is associated with a range of diseases, including CKD and diabetes mellitus28. Mitochondria act as a central hub of pro-inflammatory signalling. mtROS are activators of inflammation that promote pro-inflammatory gene expression, activating NF-κB signalling and the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome29,30. Damage-associated molecular patterns derived from mitochondria, including formyl peptides and mtDNA, can bind to Toll-like receptors or nucleotide-binding oligomerization domain-containing protein (NOD)-like receptors, leading to inflammation31. Mitochondria also have roles in controlling the development, activation, differentiation and survival of diverse immune cell types, including T lymphocytes32.
Several studies have demonstrated an important role of mitochondria in the regulation of inflammation during kidney injury. In mouse models of cisplatin-induced AKI and in cultured tubular cells exposed to cisplatin, mtDNA from damaged mitochondria in kidney tubular cells leaked into the cytosol, probably through BAX-activated pores in the outer mitochondrial membrane (OMM), resulting in activation of the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes protein (STING) cytosolic DNA sensing pathway and thereby triggering kidney inflammation and AKI progression33. Similarly, in mice with defective mitochondria in kidney tubule cells owing to tubule-specific knockout of transcription factor A, mitochondrial (Tfam), mtDNA was released into the cytosol and activated the cGAS–STING pathway, resulting in kidney inflammation and fibrosis34.
Mitochondria in kidney injury and repair
AKI is characterized by sublethal and lethal injury of the kidney tubules. After injury, surviving tubular cells undergo dedifferentiation followed by proliferation, migration and differentiation into fully mature tubular cells to repair injured kidney tubules. Complete kidney repair after mild injury leads to full functional recovery, whereas severe or recurrent episodic AKI is usually associated with incomplete or maladaptive repair, which leads to nephron loss, tubulointerstitial fibrosis and, ultimately, progression to CKD.
Kidney injury and repair are complex and multi-factorial, involving an interplay between microvascular, tubular and inflammatory factors, as well as various signalling pathways35,36. Accumulating evidence suggests that mitochondrial dysfunction contributes critically to the pathogenesis of AKI and incomplete kidney repair after AKI (TABLES 1,2). First, mitochondrial pathology occurs before detectable kidney dysfunction and persists in kidney tubules that do not recover after AKI. For example, in a mouse model of glycerol-induced AKI, disruption of mitochondrial respiration and abnormalities in mitochondrial ultrastructure were detected in kidneys 3 h after glycerol injection before evidence of kidney injury was present37. Disruption of mitochondrial homeostasis persisted until 144 h after glycerol injection or IR-induced AKI in mice38. In addition, a 2020 study demonstrated that chronic impairment of mitochondrial bioenergetics and β-oxidation in kidneys promoted experimental AKI to CKD transition following folic acid treatment39. Second, mitochondrial protection before the onset of kidney injury (for example, via inhibition of mitochondrial fragmentation through genetic or pharmacological approaches) protects against AKI40,41. Third, mitochondrial protection after AKI mitigates the transition to CKD. For example, after ischaemic AKI, inhibition of mitochondrial fragmentation in proximal tubular cells or treatment with mitochondria-targeting antioxidants were shown to attenuate kidney fibrosis in rodent models41,42.
Table 1 |.
The effects of genetic modulation of mitochondrial quality control mechanisms on AKI and kidney repair
| Genetic modulation | Effect on mitochondrial quality control | Mouse AKI model | Effect on AKI and repair | Ref. |
|---|---|---|---|---|
| Drp1 deletion in PTECs | Suppressed mitochondrial fission in PTECs | IRI | Attenuated AKI | 41 |
| Inducible Drp1 deletion in PTECs after IR | Suppressed mitochondrial fission in PTECs | IRI | Attenuated post-AKI kidney fibrosis | 41 |
| Global Pink1 deletion | Loss of PINK1-parkin pathway of mitophagy | IRI | Aggravated AKI | 141 |
| Cisplatin | Aggravated AKI | 144 | ||
| Contrast | Aggravated AKI | 143 | ||
| Global Park2 deletion | Loss of PINK1-parkin pathway of mitophagy | IRI | Aggravated AKI | 141 |
| Cisplatin | Aggravated AKI | 144 | ||
| Contrast | Aggravated AKI | 143 | ||
| CLP | Aggravated AKI | 147 | ||
| Pink1 and Park2 deletion | Loss of PINK1-parkin pathway of mitophagy | IRI | Aggravated AKI | 141 |
| Pgc1a deletion in PTECs | Suppressed mitochondrial biogenesis in PTECs | LPS | Aggravated AKI and decreased kidney recovery | 79 |
| Global Pgc1a deletion | Suppressed mitochondrial biogenesis | LPS | Aggravated AKI and decreased kidney recovery | 79 |
| IRI | Aggravated AKI | 160 | ||
| Inducible Pgc1a overexpression in PTECs | Enhanced mitochondrial biogenesis in PTECs | IRI | Attenuated AKI | 160 |
| Global Oma1 deletion | Reduced OPA1 proteolysis and mitochondrial fragmentation | IRI | Attenuated AKI | 112 |
AKI, acute kidney injury; CLP, caecal ligation and puncture; IRI, ischaemia–reperfusion injury; LPS, lipopolysaccharide; OPA1, dynamin-like 120 kDa protein, mitochondrial; PINK1, serine/threonine-protein kinase PINK1, mitochondrial; PTEC, proximal tubule epithelial cells.
Table 2 |.
The effects of pharmacological modulation of mitochondrial quality control on AKI and repair
| Agent | Mechanism | AKI model | Administration | Effect on AKI and repair | Refs |
|---|---|---|---|---|---|
| MitoQ | Mitochondria-targeted antioxidant | IRI (mouse) | Tail vein injection 15 min before ischaemia | Attenuated AKI | 178 |
| Cisplatin (mouse) | IP administration 1 h before cisplatin administration | Attenuated AKI | 180 | ||
| Mito-CP | Mitochondria-targeted antioxidant | Cisplatin (mouse) | IP administration 1 h before cisplatin administration | Attenuated AKI | 180 |
| GC4419 | Mitochondrial-specific SOD mimetic | Cisplatin (mouse) | IP injection, once daily starting 5 days before cisplatin administration | Attenuated AKI and accelerated kidney repair | 50 |
| SkQR1 | Mitochondria-targeted antioxidant | IRI (mouse) | IP injection 3 h before ischaemia and at 1, 13, 25 and 37 h | Attenuated AKI | 179 |
| Gentamicin (mouse) | IP injection daily 3 h before gentamycin injection for 6 days | Attenuated AKI | 179 | ||
| SS-31 | Specifically binds to cardiolipin, protects cristae curvature, stabilizes mitochondrial structure, facilitates electron transport and minimizes ROS production | IRI (rat) | SC injection 30 min before ischaemia, at the onset of reperfusion and 2 h after reperfusion | Attenuated AKI | 181 |
| SC injection 30 min before ischaemia and at the onset of reperfusion | Attenuated AKI | 55,182 | |||
| Starting at the onset of reperfusion for 4 weeks using a subcutaneously implanted osmotic pump | Reduced post-AKI kidney fibrosis | 55 | |||
| Starting 1 month after ischaemia for 6 weeks using a subcutaneously implanted osmotic pump | Attenuated post-AKI kidney fibrosis | 42 | |||
| Mitochonic acid 5 | Binds to mitofilin in the IMM, facilitates ATP production | IRI (mouse) | Oral gavage 3 h before ischaemia | Attenuated AKI | 218 |
| Cisplatin (mouse) | Oral gavage starting at cisplatin injection, daily for 3 days | Attenuated AKI | 218 | ||
| mdivi-1 | Inhibits DRP1 activity and suppresses mitochondrial fission | IRI (mouse) | IP injection 1 h prior to ischaemia | Attenuated AKI | 40 |
| SRT1720 | Activates SIRT1 and induces mitochondrial biogenesis | IRI (rat) | IP injection starting at 24 h after reperfusion, daily for 2 or 5 days | Attenuated AKI | 205 |
| LY344864 | 5-HT1F agonist, induces mitochondrial biogenesis | IRI (mouse) | IP injection starting at 24 h after reperfusion, daily for 5 days | Attenuated AKI and accelerated kidney repair | 217 |
| Formoterol | β2 adrenergic receptor agonist, induces mitochondrial biogenesis | IRI (mouse) | IP administration starting at 24 h after surgery, daily for 5 days | Accelerated kidney repair | 161 |
| Niacinamide | NAD+ precursor, improves mitochondrial oxidative metabolism | IRI (mouse) | IP administration once daily for 4 days with the final dose an hour prior to surgery | Attenuated AKI | 160 |
| Single IP dose 18 h after the onset of reperfusion | Attenuated AKI | 160 | |||
| Cisplatin (mouse) | IP administration 24 h before and at the time of cisplatin administration | Attenuated AKI | 160 | ||
| TES-1025 | ACMSD inhibitor, boosts in vivo synthesis of NAD+ | Cisplatin (mouse) | Supplementation of chow diet starting 10 days before cisplatin administration | Attenuated AKI | 224 |
| IRI (mouse) | Supplementation of chow diet starting 10 days before surgery | Attenuated AKI | 224 |
5-HT1F, 5-hydroxytryptamine receptor 1F; AKI, acute kidney injury; ACMSD, 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase; DRP1, dynamin-1-like protein; IMM, inner mitochondrial membrane; IP, intraperitoneal; IRI, ischaemia-reperfusion injury; NAD, nicotinamide adenine dinucleotide; ROS, reactive oxygen species; SC, subcutaneous; SOD, superoxide dismutase.
Mitochondrial dysfunction also contributes to the development of CKD, regardless of aetiology. Mitochondrial ultrastructural changes and functional impairment are common features in the kidneys of animal models and patients with CKD. Moreover, genetic and pharmacological interventions that improve mitochondrial functions have been shown to attenuate kidney dysfunction in animal models1,9.
Mitochondrial antioxidant defence
The ETC, particularly on complexes I and III, is the major site of ROS generation within mitochondria43. Electrons that leak from the ETC react with oxygen (O2) to form superoxide anions, which can be dismutated to hydrogen peroxide (H2O2) by superoxidase dismutases (SODs). H2O2 can be further reduced to water by antioxidant catalase, glutathione peroxidases (GPXs) and peroxiredoxin.
Under physiological conditions, the mitochondrial antioxidant defence system keeps ROS levels low within the organelles and enables emission of low levels of mitochondrial H2O2 into the cytosol, where they act as cell survival signalling molecules44–46. Under conditions of stress when mtROS production overwhelms the capacity of mitochondrial antioxidant defence or the antioxidant defence system is impaired, ROS accumulate. Although O2•− is fairly unreactive, it can react rapidly with nitric oxide to form the potent oxidant and nitrating agent peroxynitrite and other reactive nitrogen species that have high reactivity with various biomolecules. H2O2 can damage enzymes by oxidizing their thiol groups47 and can produce highly reactive hydroxyl radicals in the presence of Fe2+ cations via the Fenton reaction48. Increased mitochondrial H2O2 emission to the cytosol as a result of mtROS accumulation expands oxidative damage outside of the mitochondria. As a result, a balance of ROS production and scavenging within mitochondria is essential for maintaining mitochondrial function and cell viability.
Fenton reaction.
A catalytic process that converts hydrogen peroxide (H2O2) into highly reactive hydroxyl free radicals in the presence of ferrous iron (Fe(II)).
Role in AKI and kidney repair.
Experimental studies have demonstrated an inadequate ROS-scavenging capacity within mitochondria in kidney tubules during AKI and subsequent kidney repair, as demonstrated by increases in mtROS in tubular cells and the beneficial effects of supplementation of exogenous mitochondria-targeted antioxidants during these processes. In a mouse model of kidney fibrosis induced by IR, a persistent increase in ROS and oxidative stress, accompanied by a sustained reduction in the activities of SOD1, SOD2 and catalase, was shown in kidney tissue 16 days after ischaemia49. Supplementation of a cell-permeable SOD mimetic from 48 h to 14 days after IR dramatically reduced kidney fibrosis in this model. Treatment of mice with cisplatin resulted in a persistent increase in mtROS in the kidney tubules for up to 1 month, whereas treatment with a mitochondrial-specific SOD mimetic (GC4419) ameliorated AKI induced by a single dose of cisplatin and kidney fibrosis induced by repeated cisplatin doses50. Together, these findings suggest that the mitochondrial antioxidant defence system is impaired or overwhelmed during kidney injury and repair and that this inadequacy contributes to pathological processes.
Peroxisomes are intracellular organelles that contain antioxidant enzymes (particularly catalase) and a fatty acid β-oxidation machinery in which electrons from various metabolites reduce O2 to H2O2. Studies have demonstrated that peroxisomes have a role in the regulation of redox homeostasis in kidney tubular cells and peroxisome damage might contribute to AKI51. Reductions in peroxisome number and activity, as well as downregulation of catalase, were shown in the kidneys of mice with AKI induced by cisplatin or IRI52. Proximal tubule-specific overexpression of NAD-dependent protein deacetylase sirtuin 1 (SIRT1) restored peroxisome number and function in kidney tubular cells, which led to upregulation of catalase expression and mitigation of ROS and kidney injury in these models. Notably, overexpression of SIRT1 failed to restore mitochondrial number but preserved mitochondrial proteins as a secondary effect of ROS reduction via catalase, suggesting that peroxisome damage-induced ROS contributes to mitochondrial damage and AKI. The potential relationship between peroxisomes and mitochondria in the pathogenesis of kidney diseases awaits further investigation.
Excessive mtROS cause oxidative damage to mitochondrial components, which further increases mtROS production, forming a vicious circle, with an increased tendency towards cell death. Cardiolipin is a unique phospholipid that exclusively anchors in the IMM and has critical roles in the regulation of mitochondrial cristae formation and organization of the respiratory chain complex. Interaction with cardiolipin anchors cytochrome c in the IMM and thus prevents its release into the cytosol11,53. ROS induce cardiolipin peroxidation, which converts cytochrome c from an electron carrier into a peroxidase that enhances cardiolipin peroxidation, ultimately leading to mitochondrial dysfunction and cell death11,53. Experimental studies suggest that ROS-mediated peroxidation of cardiolipin contributes critically to the development of AKI and incomplete kidney repair after AKI. Administration of SS-31, a cell-permeable compound that selectively binds to cardiolipin and prevents its peroxidation11,53, has been shown to protect against kidney injury in various models of AKI42,54. In addition, treatment with SS-31 after kidney injury protected against AKI to CKD transition in rat IRI models42,55.
Mitochondrial cristae.
Folds of the inner mitochondrial membrane that increase the surface area in which oxidative phosphorylation can occur and thus enhance the capacity of the mitochondrion to synthesize ATP.
In contrast to the acute destructive effects of high levels of ROS, a moderate increase in mtROS might regulate signalling pathways that are involved in kidney injury and incomplete kidney repair. mtROS have been shown to activate hypoxia-inducible factor 1α (HIF1α) in response to hypoxia56, the NLRP3-inflammasome pathway, which induces inflammation, cytokine production and innate immune responses57, and the transforming growth factor-β (TGFβ) pathway, which has pro-fibrotic effects in disease conditions58. Increased levels of ROS can also active p53, which has a critical role in AKI pathogenesis and incomplete kidney repair after AKI59. However, the precise role of mtROS in the regulation of these signalling pathways during AKI and subsequent kidney repair awaits in-depth investigation.
Role in CKD.
Experimental evidence suggests that the mitochondrial antioxidant defence system is impaired or overwhelmed in the kidney during CKD. An increase in mtROS production is a common feature of CKD60,61 and augmentation of mitochondrial antioxidant defence capacity via supplementation of mitochondria-targeted antioxidants, such as MitoQ, MitoTEMPO or SS-31, has been shown to relieve mitochondrial dysfunction and attenuate kidney injury in animal models of diabetic kidney disease (DKD) and unilateral ureteral obstruction (UUO)-induced CKD62–66. Together, these findings suggest an inadequate ROS-scavenging capacity within the kidney mitochondria during CKD. Although mitochondrial superoxide anion production has been reported to be reduced in kidney tissues from mice with streptozotocin-induced diabetes and in Akita diabetic mice67, excessive mtROS production is generally thought to be associated with mitochondrial dysfunction, which ultimately causes cellular damage and progression of kidney disease.
Mitochondrial protein quality control
Mitochondrial function is highly dependent on protein homeostasis within the organelles. However, mitochondrial protein homeostasis is challenging in both physiological and pathological conditions because of the complex mitochondrial proteome, which consists of proteins encoded by nuclear and mitochondrial genomes, and the continuous exposure of mitochondrial proteins to mtROS. Coordinated gene expression between nuclear and mitochondrial genomes, efficient importing of nuclear-encoded mitochondrial proteins from the cytosol into the mitochondria, accurate protein sorting to distinct mitochondrial subcompartments and protein maturation by folding, as well as correct assembly of relevant proteins into functional complexes, are essential for mitochondrial protein homeostasis during mitochondrial biogenesis and remodelling. Efficient refolding or removal of damaged proteins to prevent deleterious protein aggregation within the organelles is also critically important for normal mitochondrial function. The mitochondrial protein quality control system consists of chaperones that catalyse protein folding and ATP-dependent proteases that remove unwanted and unrepaired proteins68 (FIG. 2). When the capacity of this quality control system is overwhelmed by excessive amounts of unfolded or misfolded proteins, the mitochondrial unfolded protein response (UPRmt) is induced. In the UPRmt, signals released from mitochondria trigger transcription of nuclear genes that encode mitochondrial chaperones to expand the mitochondrial protein folding capacity and thereby prevent deleterious protein aggregation within the organelles69. Impaired capacity of mitochondrial protein quality control systems may compromise mitochondrial function and jeopardize cell viability; such impairment has been associated with mitochondria-related and age-related degenerative diseases such as Parkinson disease and Alzheimer disease70.
Fig. 2 |. Mitochondrial quality control.
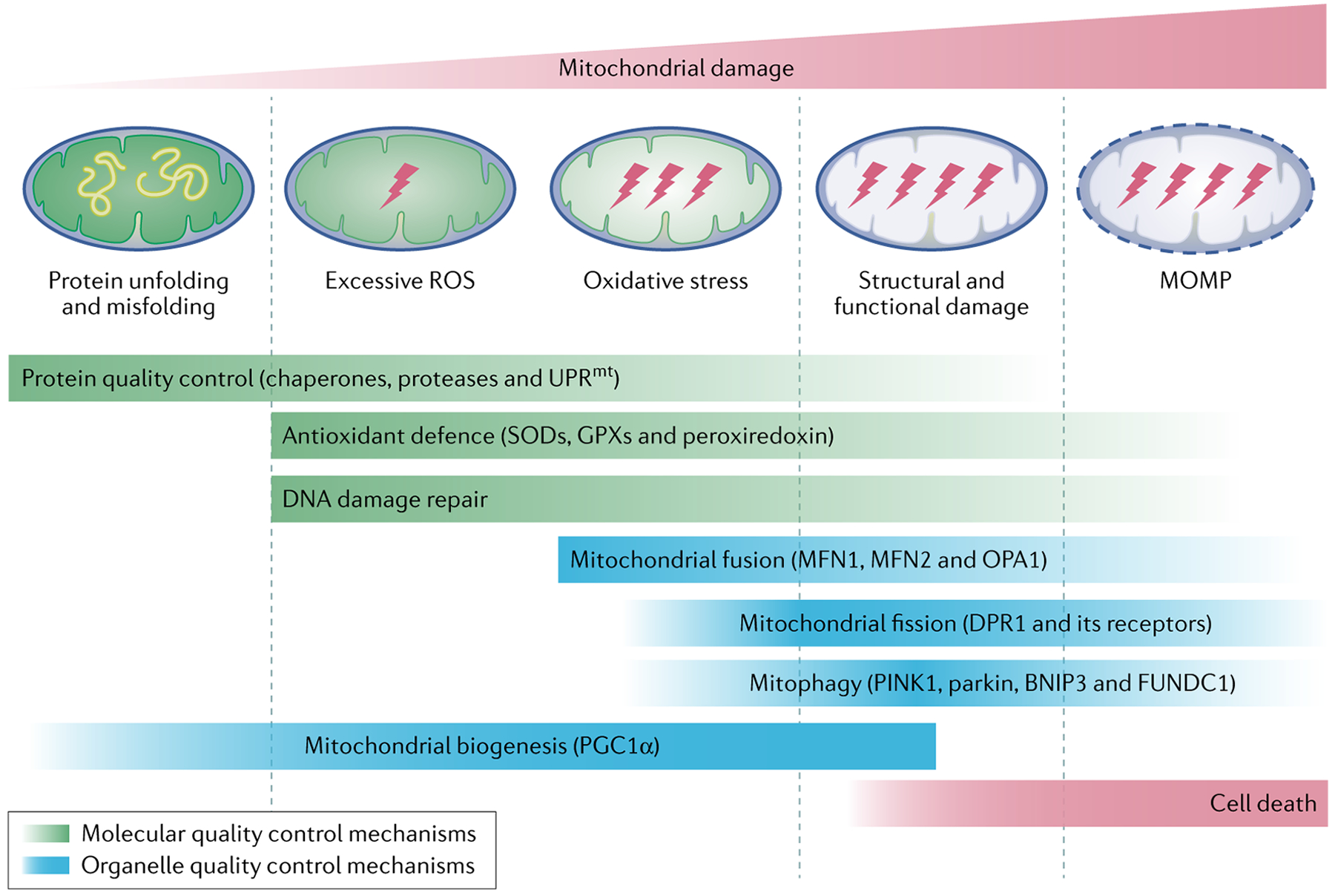
The mitochondrial quality control system consists of molecular and organelle quality control mechanisms. Protein quality control is maintained by chaperones that catalyse protein folding and ATP-dependent proteases that remove unwanted and damaged proteins. In settings where the capacity of protein quality control is overwhelmed, the mitochondrial unfolded protein response (UPRmt) is induced. In this response, signals released from mitochondria trigger the transcription of nuclear genes that encode mitochondrial chaperones to enhance the protein-folding capacity. The mitochondrial antioxidant defence system consisting of superoxidase dismutases (SODs), glutathione peroxidases (GPXs) and peroxiredoxin limits reactive oxygen species (ROS) levels within the organelles and the DNA damage repair machinery repairs damaged mitochondrial DNA. When these molecular quality control mechanisms fail to restore mitochondrial homeostasis, organelle quality control mechanisms are activated. Mitochondrial fusion mediated by mitofusin 1 (MFN1), MFN2 and dynamin-like 120 kDa protein, mitochondrial (OPA1) mitigates organelle stress by enabling the contents of damaged mitochondria to be combined with those of healthy mitochondria for complementation. Fission, which is regulated by cytosolic dynamin-1-like protein (DRP1) and its receptors, segregates damaged parts of the mitochondrial network, which are then removed by mitophagy. Mitophagy is mediated by the serine/threonine-protein kinase PINK1, mitochondrial (PINK1)–parkin pathway and mitophagy receptors, including BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL-2-interacting protein 3-like and FUN14 domain-containing 1 (FUNDC1). Mitochondrial biogenesis depends on specific transcription factors, including peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α). When mitochondrial damage exceeds the capacity of mitochondria quality control or when mitochondrial quality control is defective, cell death ensues. MOMP, mitochondrial outer membrane permeabilization.
Role in AKI and kidney repair.
Disturbances in mitochondrial protein homeostasis usually arise from either mutations that alter mitochondrial protein sequences or accumulation of mtROS that alter protein structure by direct oxidative modification. Recessive mutations in TRAP1, which encodes heat shock protein 75 kDa, mitochondrial (TRAP1; also known as HSP75), a mitochondrial chaperone that is highly expressed in the proximal tubules and thick medullary ascending limbs of the loop of Henle, have been linked to congenital abnormalities of the kidney and urinary tract71, suggesting a critical role of TRAP1 in kidney tubular pathophysiology. Upregulation of TRAP1 and heat shock 70 kDa protein 9 (HSPA9) was detected in kidney tissue from animals with AKI47,72,73, suggesting a possible occurrence of UPRmt in this disorder.
Notably, the endoplasmic reticulum unfolded protein response (UPRER) has been implicated in the pathogenesis of AKI and incomplete kidney repair after AKI74–76. Although the UPRmt and UPRER operate within distinct organelles and involve organelle-specific chaperones and proteases, both pathways are rapidly activated and crosstalk with each other in response to extrinsic stimuli, such as hypoxia, oxidative stress and metabolic disorders77,78, which are present in kidney tubules during AKI. The induction of UPRER during AKI and subsequent kidney repair might therefore suggest that activation of the UPRmt also occurs under these conditions. Moreover, mitochondrial biogenesis is critically important for kidney recovery after AKI79. Given the essential role of mitochondrial protein quality control in polypeptide sorting, folding and subsequent assembly into functional complexes during mitochondrial biogenesis, it is conceivable that efficient pro-protein folding activity within mitochondria is essential for kidney recovery. However, the functional association between the mitochondrial protein quality control system, AKI and kidney repair remains largely unknown.
Role in CKD.
In mice, UUO induced loss of TRAP1 expression in kidney tissues, which correlated with the progression of fibrotic injury80. Another study showed that increased expression of TRAP1 in kidney tubules attenuated mitochondrial dysfunction and kidney fibrosis in UUO mice81. These findings suggest that impairment of mitochondrial protein quality control facilitates CKD progression. However, the precise role and regulation of mitochondrial quality control in the development and progression of CKD require further investigation.
Mitochondrial DNA repair
In comparison with nuclear DNA, mtDNA is particularly vulnerable to oxidative stress because of its lack of protective histone proteins and close proximity to the site of ROS production. As mtDNA encodes 13 proteins of the ETC, mtDNA damage may impair OXPHOS. Moreover, once the fraction of damaged mtDNA in an individual cell exceeds a certain threshold, cell death ensues82. Efficient repair or removal of damaged mtDNA is therefore essential for preserving normal mitochondrial function and cell survival. In mammalian cells, mitochondria possess most of the DNA repair pathways that are available in the nucleus, including base excision repair, mismatch repair, homologous recombination and non-homologous end joining83. These mechanisms are responsible for repairing distinct mtDNA lesions and involve both nuclear gene- and mitochondrial gene-encoded enzymes. In the case of severe, irreparable damage, mtDNA can be degraded through a mitophagy- and autophagy-independent manner84. The molecular mechanism that underlies autophagy-independent mtDNA degradation has not been characterized. Defects in mtDNA repair impair mitochondrial function, increase vulnerability to cell death and have been implicated in human diseases such as cancer82.
Role in AKI and kidney repair.
Nuclear DNA damage and the subsequent DNA damage response have been demonstrated to have important roles in AKI and kidney repair85,86. mtDNA damage also occurs in the kidneys during AKI and kidney fibrosis. Increased levels of 8-hydroxy-2-deoxy-guanosine, a marker of oxidative DNA damage, in kidney mtDNA were identified shortly after reperfusion in rat models of kidney IRI87,88 and in mice with Staphylococcus aureus sepsis-induced kidney injury89. In addition, mtDNA deletion was found in kidney tubular cells during adefovir-induced nephrotoxicity90 and intraperitoneal administration of cisplatin was shown to decrease kidney mtDNA content in mice91. As mtDNA deletions are most likely to be the result of the repair of double-strand breaks by recombination-based processes92, mtDNA deletion during AKI and kidney fibrosis suggests activation of mtDNA repair. Consistent with this hypothesis, mitochondrial levels of the base excision repair proteins N-glycosylase/DNA lyase and uracil DNA glycosylase were increased in kidney tissues following septic AKI89. In rats with kidney IRI, levels of mtDNA oxidation (shown by 8-hydroxy-2-deoxyguanosine staining) decreased during the recovery of kidney function, suggesting efficient mtDNA damage repair88. Although further investigation is needed, together these findings suggest that mtDNA damage and activation of mtDNA repair processes occur during kidney injury and repair.
Role in CKD.
mtDNA abnormalities can cause tubular atrophy and tubulointerstitial fibrosis93,94. Accumulation of oxidative mtDNA lesions and loss of mtDNA copy number have been reported in the kidneys of animal models with various aetiologies of CKD, including DKD95,96. Moreover, lower mtDNA copy number in peripheral blood was associated with a higher incidence of CKD in a population-based study of US adults96. These findings suggest that reducing mtDNA damage and/or enhancing mtDNA repair might be beneficial for the prevention and treatment of CKD. In addition, expression of Y box-binding protein 1, which has a key role in mtDNA damage repair, was dramatically increased in the kidney tissues of patients with DKD or CKD and UUO mice compared with normal human and mouse kidney tissues, respectively80, suggesting activation of mtDNA repair machinery in these settings. However, the role and regulation of mtDNA repair in CKD remains largely unknown.
Mitochondrial dynamics
Mitochondria are highly dynamic organelles that constantly undergo fusion and fission (FIG. 3). These processes are an important adaptive response to metabolic or environmental stresses and also a critical defence mechanism to preserve a mitochondrial population that is healthy overall, especially when quality control mechanisms fail to repair or remove damaged mitochondrial components. Mitochondrial fusion facilitates the exchange of metabolites and substrates between mitochondria to ensure optimal function of the mitochondrial network and is required for the complementation of oxidatively damaged mitochondrial components to mitigate organelle stress97,98. Mitochondrial fission is required for mitotic segregation of mitochondria into daughter cells and separation of damaged or dysfunctional parts of mitochondria for autophagic degradation40,99. Mitochondrial fission may also facilitate apoptosis in response to severe cellular stress.
Fig. 3 |. Mitochondrial fusion and fission.
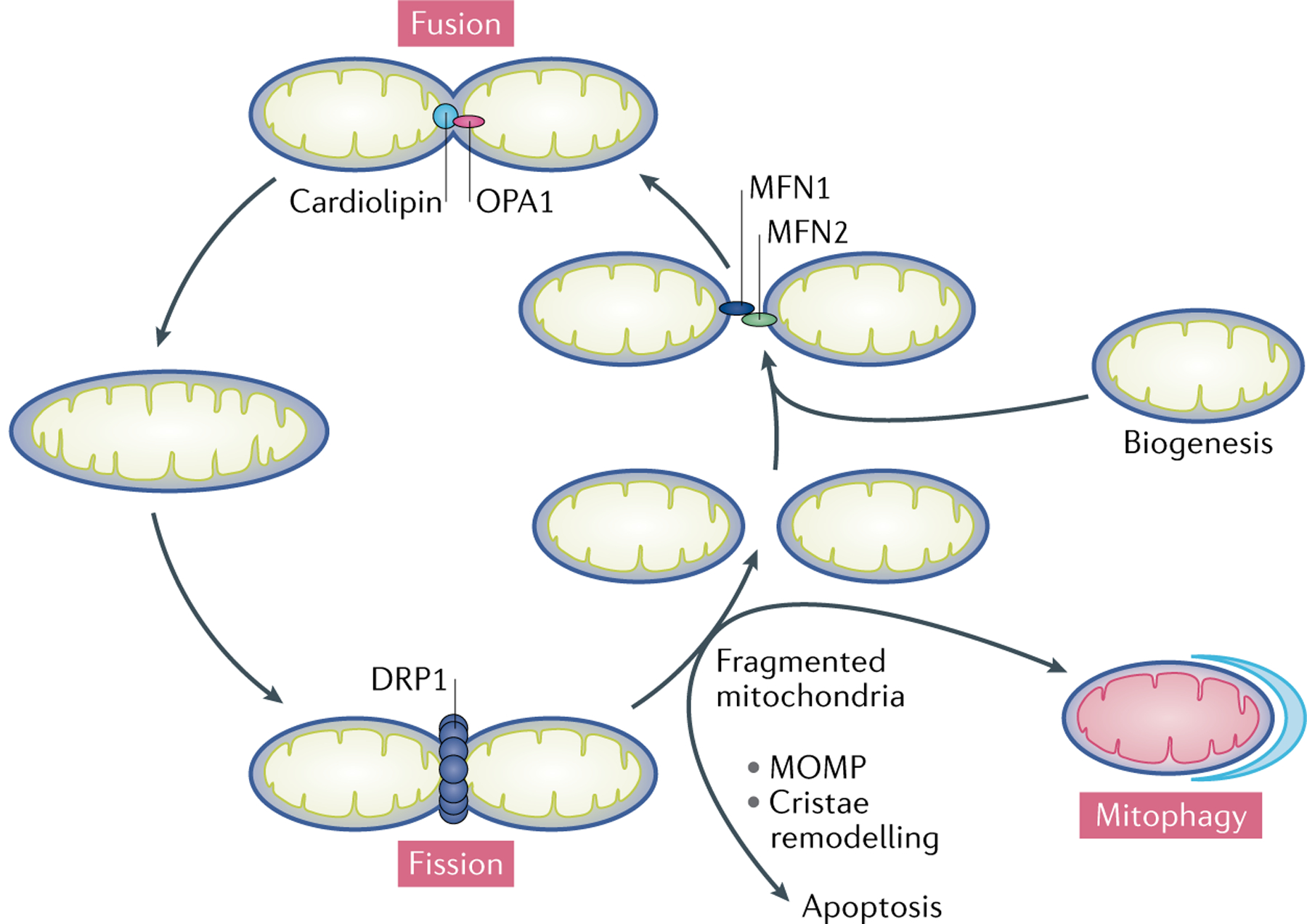
During fusion, mitofusin 1 (MFN1) and MFN2 expressed on two adjacent mitochondria interact to tether the organelles. GTP hydrolysis-induced conformational changes in the MFNs drive the docking and contact of the outer mitochondrial membranes (OMMs). The MFNs then oligomerize to fuse the OMMs. Following OMM fusion, inner mitochondrial membrane fusion is mediated by dynamin-like 120 kDa protein, mitochondrial (OPA1), which interacts with cardiolipin. Mitochondrial fusion facilitates the exchange of metabolites and substrates between mitochondria to ensure optimal functioning of the mitochondrial network and is also required for the complementation of damaged mitochondrial components to mitigate mitochondrial stress. During fission, dynamin-1-like protein 1 (DRP1) is recruited from the cytosol to the mitochondria, where it oligomerizes to form a ring-like structure around the OMM that utilizes the energy from GTP hydrolysis to constrict the organelle. Mitochondrial fission is required to separate damaged or dysfunctional components of mitochondria for selective autophagic degradation via mitophagy. Mitochondrial fragmentation, as a result of excessive mitochondrial fission over fusion, leads to mitochondrial outer membrane permeabilization (MOMP) and/or cristae remodelling, ultimately resulting in cell death.
Mitochondrial fusion and fission are governed by members of a family of conserved GTPases. Fission is a multistep process in which DRP1 has an essential role. During fission, DRP1 is recruited from the cytosol to the OMM at mitochondria–endoplasmic reticulum contact sites by its receptors mitochondrial fission factor (MFF), mitochondrial dynamics protein MID49 and/or MID51. DRP1 then oligomerizes to form a ring-like structure around the mitochondria that utilizes the energy from GTP hydrolysis to constrict the organelle100,101. Dynamin 2 is then recruited to the mitochondrial constriction neck to sever the OMM102. DRP1 may also have severing ability sufficient for mitochondrial fission103. Our studies suggest an involvement of endophilin B1 (also known as BIF1) in IMM fission. We found that endophilin B1 translocates to mitochondria during cell stress and binds prohibitin 2 on the IMM, resulting in release of the metalloendopeptidase OMA1 and subsequent proteolysis of the IMM fusion protein dynamin-like 120 kDa protein, mitochondrial (OPA1)104. Another study suggested that IMM fission is regulated by calcium influx105.
Mitochondrial fusion involves fusion of the OMM, which is mediated by mitofusin 1 (MFN1) and MFN2, and fusion of the IMM, which is mediated by OPA1. During fusion, MFNs in the OMM of two adjacent mitochondria interact to tether the organelles. GTP hydrolysis-induced conformational changes in the MFNs drive docking of the OMMs and increase the contact surface area. Subsequently, MFNs oligomerize to ensure OMM fusion. Following OMM fusion, interactions between OPA1 and cardiolipin tether the IMMs for fusion106. Disruption of the balance between fusion and fission alters mitochondrial morphology and impairs mitochondrial function and cell viability; such disruption has been linked to neurodegenerative diseases and cancer107.
Role in AKI and kidney repair.
Mitochondrial fragmentation resulting from excessive fission and/or suppression of fusion has been implicated as a key event in mitochondrial damage and kidney tubule injury during AKI40,108. In rodent models of AKI induced by kidney IR or cisplatin toxicity, mitochondrial fragmentation occurred prior to tubular cell apoptosis and inhibition of fission attenuated tubular cell death and kidney injury40. Consistent with this finding, proximal tubule-specific deletion of Drp1 protected mice against kidney IR-induced tubular cell death, inflammation and kidney injury, and accelerated kidney recovery41.
Upstream regulation of mitochondrial fragmentation following AKI has also been investigated in kidney tubular cells. In mice, cisplatin-induced AKI was associated with downregulation of SIRT3, which could be prevented by the antioxidant acetyl-l-carnitine or the AMP-activated protein kinase (AMPK) agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR)109. AICAR and acetyl-l-carnitine attenuated cisplatin-induced kidney injury and mitochondrial fragmentation in wild-type mice but not in Sirt3-knockout mice, suggesting that downregulation of SIRT3 contributes critically to mitochondrial fragmentation in tubular cells during kidney injury. In cultured kidney tubular cells, overexpression of SIRT3 prevented cisplatin-induced recruitment of DRP1 to the OMM109.
Mitochondrial uncoupling protein 2 (UCP2) might also be a negative regulator of mitochondrial fragmentation. Following kidney IR, Ucp2-deficient mice exhibited more severe mitochondrial fragmentation and kidney injury than did wild-type mice, whereas mice with increased kidney expression of UCP2 showed reduced mitochondrial fragmentation and less severe kidney injury110. Overexpression of UCP2 reduced the recruitment of DRP1 to the mitochondria of kidney tubular cells following kidney IR. Protein numb homologue (NUMB) might also negatively regulate DRP1. In mice, proximal tubule-specific knockout of Numb increased mitochondrial fragmentation in kidney tubule cells under normal conditions111. Moreover, following cisplatin treatment, Numb-knockout mice showed more severe mitochondrial fragmentation than wild-type mice. NUMB deletion induced Rho-associated protein kinase 1 (ROCK1)-mediated phosphorylation of DRP1 and thereby increased DRP1 translocation to the mitochondria and mitochondrial fragmentation111. Mitochondrial fission process protein 1 (also known as MTP18) has also been shown to mediate mitochondrial fission in models of ischaemic AKI108.
Arrest of mitochondrial fusion also potentiates mitochondrial fragmentation and tubular cell death in AKI. We showed activation of OMA1 and proteolysis of OPA1 in mice following ischaemic AKI112. Following kidney IR, OPA1 proteolysis, mitochondrial fragmentation and kidney injury were attenuated in Oma1-knockout mice compared with wild-type mice. We also found that in mice, kidney IRI induced translocation of endophilin B1 to the mitochondria and OPA1 proteolysis, leading to mitochondrial fragmentation and kidney tubular cell apoptosis104. In addition, in vitro studies showed that Mfn2 deficiency enhanced ATP-depletion-induced cell injury and death113. However, proximal tubule-specific Mfn2-knockout mice showed less severe kidney injury and greater survival than wild-type mice following kidney IR114. Mfn2 deficiency stimulated mitogen-activated protein kinase (MAPK) signalling pathway-dependent tubular cell proliferation after AKI in these mice, which might accelerate kidney repair and thereby outweigh the adverse effect of the inhibition of mitochondrial fusion, leading to kidney protection114. These findings suggest that therapeutic modulation of proteins with roles in mitochondrial dynamics to inhibit mitochondrial fission might also inhibit cell proliferation. Collectively, the data available indicate that mitochondrial fragmentation as a result of fission activation and fusion arrest contributes critically to tubular injury in AKI.
Disruption of mitochondrial dynamics also has a role in maladaptive kidney repair after AKI. Following kidney IRI, an increase in DRP1, along with a reduction in MFN2 in kidney tissue, was detected in rodent models38. Notably, these changes persisted, even after recovery of kidney function, suggesting a continuous shift of mitochondrial dynamics to fission after AKI. Moreover, induction of proximal tubule-specific Drp1 deletion after unilateral kidney IR resulted in a dramatic reduction in progression to kidney fibrosis in mice, suggesting that Drp1 deficiency and associated preservation of mitochondrial dynamics improved kidney repair after AKI41. Collectively, these findings suggest that mitochondrial fragmentation potentiates AKI and maladaptive kidney repair after AKI.
Mitochondrial fragmentation may trigger cell death by several related mechanisms. First, mitochondrial fragmentation may induce MOMP. In rat kidney proximal tubular cells, mitochondrial fragmentation facilitated BAX insertion and oligomerization in mitochondria, resulting in MOMP and apoptosis115. Fragmentation may also trigger remodelling of mitochondrial cristae and release of cytochrome c, leading to apoptosis116. OPA1 oligomers have an important role in maintaining cristae junctions and excessive OPA1 proteolysis by OMA1 induces opening of these junctions and release of cytochrome c117. DRP1-dependent mitochondrial fission has also been implicated in cristae remodelling during apoptosis118. DRP1 activation and excessive OPA1 proteolysis have been shown to contribute to kidney tubular cell death in AKI108,112, suggesting that cristae remodelling might contribute to AKI-associated cell death.
Cristae junctions.
Narrow, neck-like structures that connect the cristae membranes to the inner mitochondrial membrane. Cristae junctions act as a diffusion barrier that maintains the asymmetric protein composition between the inner mitochondrial membrane and cristae membranes and limits the diffusion of molecules, such as cytochrome c, from the intracristae space into the intermembrane space.
In addition to acute effects resulting in cell death, the sustained upregulation of fission-related proteins after AKI, together with the beneficial effect of kidney tubule-specific Drp1 ablation after AKI38,41, suggest that mitochondrial fragmentation has chronic adverse effects in the kidney. As mentioned above, mitochondrial fragmentation in kidney tubular cells might reduce energy metabolism and increase ROS formation, which could promote tissue damage, inflammation and maladaptive kidney repair. The precise mechanisms that underlie the deleterious effects of mitochondrial fragmentation in kidney repair await in-depth investigation.
Role in CKD.
Enhanced mitochondrial fragmentation in kidney tubular cells and podocytes has been reported in experimental models of DKD and in kidney biopsy samples from patients with DKD119,120. Moreover, Drp1 knockout in podocytes blocked mitochondrial fragmentation, improved mitochondrial fitness and protected against DKD progression in mice121. DRP1 was phosphorylated at serine 600 in DKD mice and mutation of this serine to alanine reduced mitochondrial fission and ameliorated DKD, highlighting an essential role of DRP1 phosphorylation at serine 600 in mitochondrial fragmentation and DKD progression122. Consistent with these findings, pharmacological inhibition of DRP1 protected against DKD progression in mice121,123.
Mitochondrial fragmentation was also detected in the kidney tubules in experimental models of UUO or TGFβ-induced CKD124,125 and in the interstitial fibroblasts of fibrotic kidneys from patients with CKD and UUO mice126. In the UUO mouse kidney, increased phosphorylation of DRP1 at serine 616 stimulated mitochondrial fission, which promoted fibroblast activation and proliferation, suggesting a role in kidney fibrosis126. These findings suggest that mitochondrial fragmentation owing to a loss of mitochondrial dynamics has a critical role in the progression of CKD.
Mitophagy
Mitophagy is a mechanism of selective degradation of excessive and defective mitochondria via the autophagy pathway. In addition to eliminating unwanted mitochondria during development and adjusting mitochondrial number to changes in metabolic demand127,128, mitophagy acts as a critical component of mitochondrial quality control mechanisms that identifies and tags severely damaged mitochondria for prompt elimination. Defects in mitophagy have been implicated in a variety of human disorders129.
Mitophagy requires efficient coordination of mitochondrial recognition with subsequent engulfment of targeted mitochondria within autophagosomes (FIG. 4). Two major mechanisms for labelling mitochondria and delivering them to autophagosomes have been defined, one of which is regulated by the serine/threonine-protein kinase PINK1, mitochondrial (PINK1)–E3 ubiquitin-protein ligase parkin pathway and the other by mitophagy receptors including BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L), FUN14 domain-containing 1 (FUNDC1) and E3 ubiquitin-protein ligase SMURF1 (SMURF1)129,130.
Fig. 4 |. Molecular mechanisms of mitophagy.
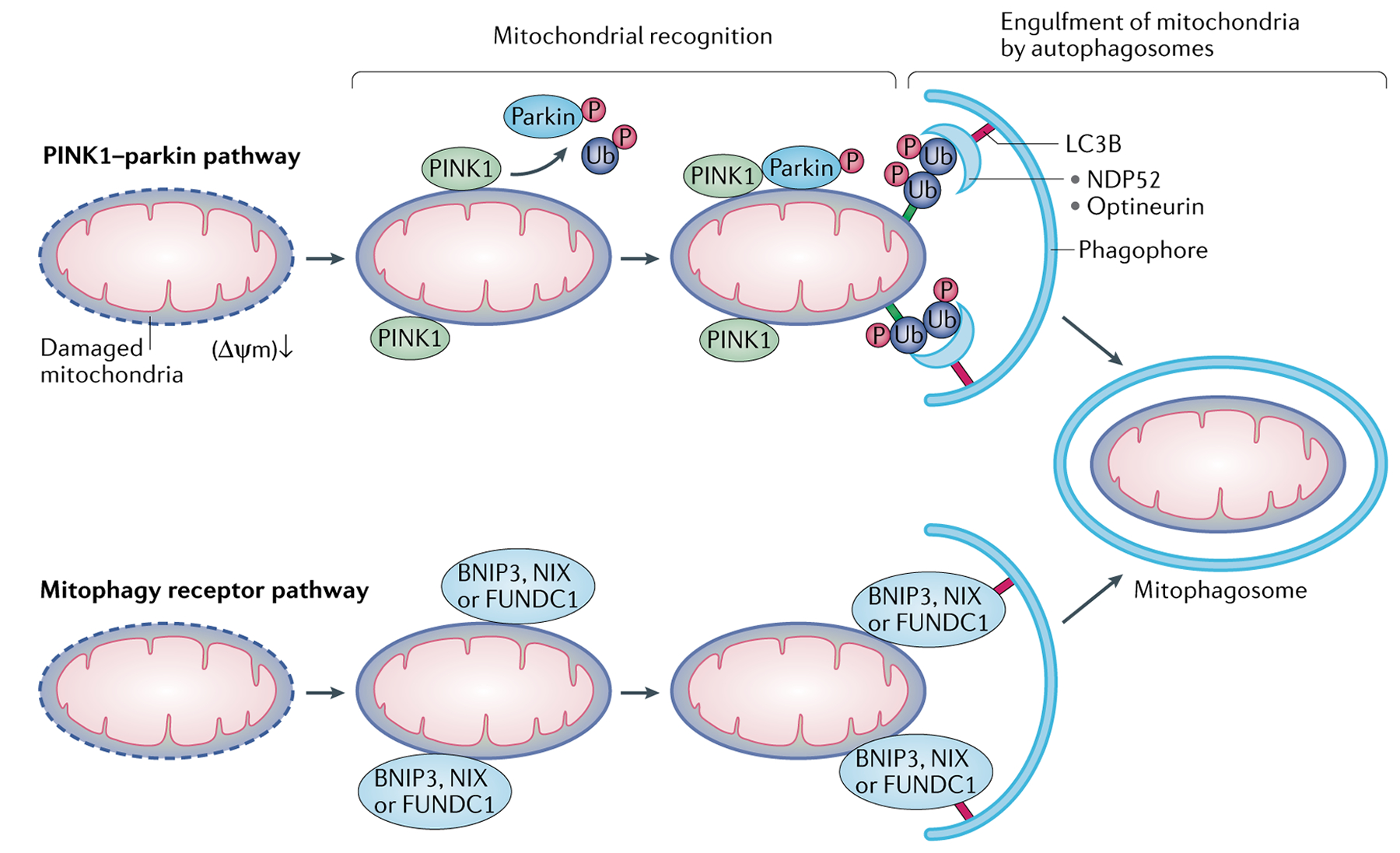
Mitophagy requires efficient mitochondrial recognition and sequestration of target mitochondria within autophagosomes. There are two major mechanisms for mitochondrial priming in mitophagy. In the serine/threonine-protein kinase PINK1, mitochondrial (PINK1)–parkin pathway, mitochondrial damage or depolarization leads to impairment of PINK1 import into mitochondria, resulting in PINK1 accumulation on the outer mitochondrial membrane (OMM). PINK1 then recruits parkin from the cytosol and activates its E3 ligase activity via phosphorylation. Upon activation, parkin catalyses the formation of poly-ubiquitin chains on OMM proteins, which are then recognized by adaptor proteins, such as calcium-binding and coiled-coil domain-containing protein 2 (NDP52) and optineurin on autophagic phagophores, resulting in formation of the mitophagosome. In the mitophagy receptor pathway, BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3-like and FUN14 domain-containing 1 (FUNDC1) in the OMM directly bridge mitochondria to autophagosomes via their interactions with MAP1A/MAP1B LC3B (LC3B).
PINK1 is a mitochondrial protein kinase that is constitutively imported into mitochondria, where it is cleaved by protease presenilins-associated rhomboid-like protein, mitochondrial (PARL). Parkin is a cytosolic ubiquitin E3 ligase. Upon mitochondrial damage or depolarization, PINK1 import into mitochondria is prevented and thus it accumulates in the OMM. PINK1 recruits parkin from the cytosol to the damaged mitochondria and activates its E3 ligase activity via phosphorylation of parkin and ubiquitin131,132. Activated parkin builds poly-ubiquitin chains on the OMM proteins, which in turn recruit receptor proteins, such as calcium-binding and coiled-coil domain-containing protein 2 (NDP52) and optineurin. These receptors simultaneously bind to poly-ubiquitin chains in mitochondria and microtubule-associated proteins 1A/1B light chain 3B (LC3B) on autophagosome membranes, resulting in engulfment of targeted mitochondria within autophagosomes, subsequent autophagosome–lysosome fusion and degradation of the polyubiquitinated mitochondria by lysosomal hydrolases133,134. The PINK1–parkin pathway is also involved in a mitophagy-independent quality control pathway in which damaged mitochondrial cargo is enclosed into mitochondria-derived vesicles for lysosomal degradation135–137. However, the involvement of this quality control mechanism in the kidney has not been determined. Mitophagy receptors are also recruited to the OMM of damaged mitochondria, where they can bridge the mitochondria to LC3B on autophagosome membranes via their N-terminal LC3-interacting regions, ultimately targeting mitochondria for autophagosome engulfment and degradation by lysosomal hydrolases. Experimental evidence suggests that crosstalk between the PINK1–parkin pathway and mitophagy receptors has a role in regulating mitophagy138,139.
Role in AKI and kidney repair.
Accumulating evidence suggests an important role of mitophagy in the pathogenesis of AKI. In mouse models that underwent bilateral kidney ischaemia for 30 min followed by reperfusion for up to 48 h, we showed an increase in autophagy flux accompanied with increases in autophagosomes with engulfed mitochondria and degradation of mitochondrial proteins in proximal tubular cells140,141. By contrast, another study demonstrated that both autophagy and mitophagy were suppressed in kidney tubules in a mouse model of AKI that was induced by 30 min of bilateral kidney ischaemia followed by 24 h of reperfusion142. The cause of the discrepancy remains unclear, but the differing durations of reperfusion might have a role.
Induction of mitophagy in kidney tubular cells was also detected in models of nephrotoxic AKI induced by cisplatin or contrast medium143,144. An increase in mitophagy in kidney tubular cells in the early stages of septic AKI followed by mitophagy impairment in the later phases of AKI was reported in a mouse model of septic AKI induced by caecal ligation and puncture (CLP)145. Peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) was identified as a positive regulator of mitophagy that induces transcription factor EB (TFEB)-mediated lysosome biogenesis in kidney tubular cells following cisplatin-induced AKI in mice146. Further studies demonstrated that kidney IR-, cisplatin- and contrast medium-induced mitophagy in kidney proximal tubules was partially abrogated in mice with knockout of Pink1 or Park2 (which encodes parkin)141,143,144. Bnip3 knockout also reduced tubular cell mitophagy in a mouse model of ischaemic AKI140. These findings suggest that both the PINK1–parkin pathway and mitophagy receptors are involved in the regulation of mitophagy in kidney tubules. Pink1, Park2 or Bnip3 deficiency aggravated IR-induced kidney injury and enhanced mitochondrial damage, ROS production and apoptosis in tubular cells, as well as tubulointerstitial inflammation140,141. Pink1 or Park2 deficiency also worsened cisplatin-, contrast medium- or sepsis-induced kidney injury143,144,147. Collectively, these data suggest a critical role of mitophagy in mitochondrial quality control mechanisms that maintain tubular cell viability and function in AKI.
Mitochondrial loss, together with increased autophagosome formation, has been reported in regenerating proximal tubule cells after kidney IRI4. These abnormalities were resolved in normal repaired tubules but persisted and became progressively more severe in tubule cells that failed to differentiate, suggesting a role of mitophagy in kidney repair after AKI. The precise role of mitophagy and its regulation in kidney repair after AKI awaits future investigation.
Mitophagy is generally believed to be a defence mechanism under pathological conditions. It is conceivable that during the initial stage of kidney injury, mitophagy is induced to ensure quality control by removing damaged mitochondria, thereby preventing ROS accumulation and release of pro-apoptotic factors and ultimately reducing tubular cell injury and death. Mitophagy induction may also prevent the release of mtDNA from damaged mitochondria, thus mitigating the immune activation and inflammation that have been implicated in AKI pathogenesis. However, as the kidney injury progresses, the number of damaged mitochondria might exceed the capacity of mitophagy, or mitophagy might become impaired, leading to increased cell death as a mechanism to minimize tissue damage. Activation of mitophagy might also induce the metabolic switch from OXPHOS towards aerobic glycolysis that occurs during cell proliferation, differentiation and dedifferentiation148,149, thereby contributing to tissue repair.
Role in CKD.
Accumulating evidence suggests an impairment of mitophagy in DKD. Reductions in PINK1 and parkin expression, as well as reduced autophagic vesicle formation, were reported in high-glucose-treated proximal tubular cells and in the kidney tubules of diabetic mice65,120. Expression of optineurin was also reduced in kidney tubular cells following high-glucose treatment and in the kidney tubules of biopsy samples from patients with DKD150,151. Moreover, overexpression of optineurin enhanced mitophagy in high-glucose-treated kidney tubular cells, leading to attenuation of cellular senescence, mtROS accumulation and NLRP3 inflammasome activation150,151. These observations support a beneficial role of mitophagy in kidney tubular cells in DKD.
Evidence also suggests an impairment of mitophagy in podocytes in DKD. PINK1 expression was reduced after high glucose exposure in cultured podocytes and in the podocytes of mice with streptozotocin-induced DKD152,153. Notably, overexpression of forkhead box protein O1 (FOXO1) in podocytes restored PINK1 expression and activated the PINK1–parkin pathway of mitophagy, leading to attenuation of mitochondrial dysfunction and podocyte injury in streptozotocin-induced DKD152. Progranulin was also downregulated in the kidney tissues of mice and patients with DKD and was identified as an upstream positive regulator of mitophagy in podocytes via activation of the SIRT1–PGC1α–FOXO1 signalling pathway154. Despite these interesting findings, the precise role and regulation of mitophagy in DKD awaits further investigation.
Mitophagy has also been implicated in the development of non-diabetic CKD. Increased mitophagosome formation, together with increased levels of mitochondrial PINK1 and parkin, as well as autophagy, was shown in the kidney tubules of UUO mice and in hypoxia-exposed proximal tubular cells, suggesting activation of mitophagy in these settings155. By contrast, another study reported that parkin levels and autophagy were reduced in kidney tissue from UUO mice and in the mitochondrial fraction of TGFβ1-treated macrophages156. These contradictory findings suggest context- and cell-type-specific alterations of mitophagy in CKD. However, both studies showed that loss of PINK1 or parkin aggravated kidney injury in UUO mice, supporting a protective role of mitophagy in CKD.
Impairment of mitophagy in kidney tubular cells in UUO mice enhanced the accumulation of damaged mitochondria and mtROS, which induced TGFβ1 expression and exacerbated kidney injury155. In addition to accumulation of abnormal mitochondria and mtROS, failure of mitophagy in macrophages in UUO mice increased the expression of rapamycin-insensitive companion of mTOR, which promoted the differentiation of macrophages towards the profibrotic M2 phenotype for extracellular matrix production and thus progression of kidney fibrosis156. These findings suggest cell-type-specific roles of mitophagy in CKD. Further research using kidney cell-type-specific, mitophagy-deficient animals is needed to improve our understanding of the role of mitophagy in CKD.
Mitochondrial biogenesis
Mitochondrial biogenesis is the generation of new mitochondrial mass and replication of mtDNA through the proliferation of existing mitochondria157. The master regulator of mitochondrial biogenesis, PGC1α, directly regulates an array of transcription factors to modulate expression of nuclear genes that are required for this process, including nuclear respiratory factor 1 (NRF1), nuclear factor erythroid 2-related factor 2 (NRF2), peroxisome proliferator-activated receptor-α (PPARα), steroid hormone receptor ERR1 and transcriptional repressor protein YY1 (REF.158) (FIG. 5).
Fig. 5 |. Regulation of mitochondrial biogenesis during AKI and repair.
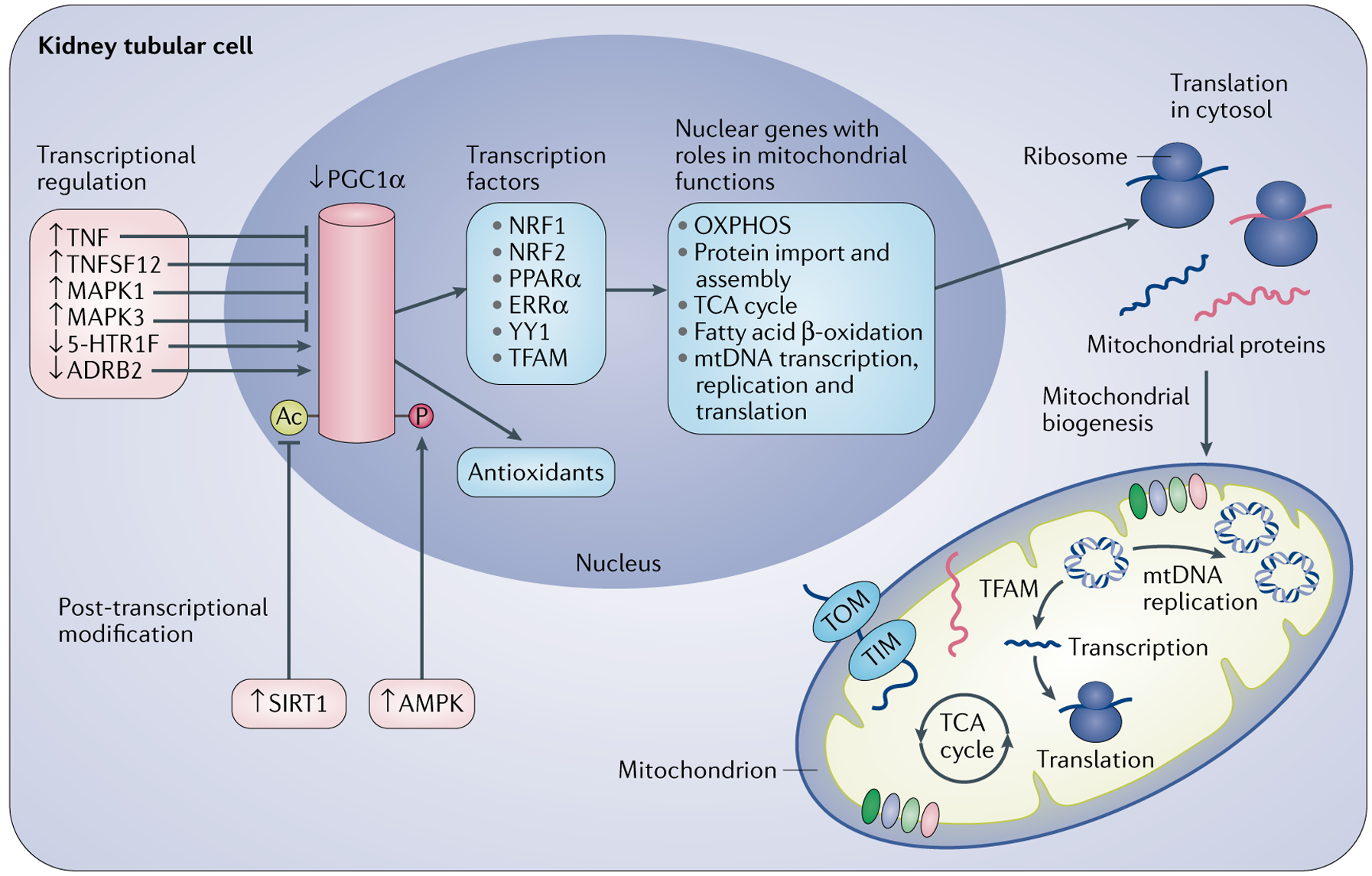
Peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) is the master regulator of mitochondrial biogenesis. PGC1α activates the expression of transcription factors that transactivate nuclear genes for fatty acid β-oxidation, the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), mitochondrial DNA (mtDNA) transcription, replication and translation, and mitochondrial protein import and assembly. Transcription factor A, mitochondrial (TFAM) specifically regulates mitochondrial genome replication. The nuclear gene-encoded proteins are transported into mitochondria through translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM). Acute kidney injury (AKI) and repair are associated with the upregulation of tumour necrosis factor (TNF) and tumour necrosis factor ligand superfamily member 12 (TNFSF12), activation of mitogen-activated protein kinase 1 (MAPK1) and MAPK3, and downregulation of 5-hydroxytryptamine receptor 1F (5-HT1F), which suppress PGC1A transcription. β2 adrenergic receptor (ADRB2) positively regulates PGC1A transcription. Sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK) activate PGC1α through deacetylation and phosphorylation, respectively. PGC1α also regulates the expression of antioxidant proteins, such as superoxide dismutase and glutathione peroxidase. ERRα, oestrogen-related receptor-α; NRF1, nuclear respiratory factor 1; NRF2, nuclear factor erythroid 2-related factor 2; PPARα, peroxisome proliferator-activated receptor-α.
Role in AKI and kidney repair.
Accumulating evidence supports a beneficial role of mitochondrial biogenesis in kidney injury and repair after AKI. PGC1α is highly expressed in the proximal tubules159 where mitochondria are abundant. In mouse models of septic AKI induced by lipopolysaccharide or CLP, the levels of PGC1α and downstream OXPHOS genes in the kidney were suppressed proportionally to the degree of kidney injury and were restored to normal levels during kidney recovery79, suggesting a negative correlation between PGC1α expression in kidney tubules and AKI severity. Reduced kidney expression of PGC1α was also observed in animal models of kidney IRI or cisplatin-induced AKI compared with controls159,160 and in kidney biopsy samples from patients with AKI compared with normal human kidney tissue sections160. Further studies demonstrated that global or tubule-specific Pgc1a deficiency delayed kidney recovery following lipopolysaccharide-induced AKI in mice79. Global PGC1A deficiency also reduced kidney recovery following IRI, whereas transgenic expression of PGC1α in tubular cells facilitated kidney repair and recovery after IRI160. Similarly, pharmacological activation of PGC1α accelerated kidney function recovery after IRI in mice161. PGC1A deficiency worsened mitochondrial damage, whereas transgenic expression of PGC1A improved mitochondrial function and increased mitochondrial mass in experimental models of AKI162, suggesting a key role of PGC1α in maintaining mitochondrial function, at least in part via regulation of mitochondrial biogenesis163. Mitochondria that have been newly generated by biogenesis may replace damaged and degraded mitochondria that have undergone mitophagy during AKI and thus contribute to the repopulation of kidney tubular cells with adequate numbers of mitochondria to meet the increased metabolic and energy demands of tubular recovery after acute injury.
A plethora of stimuli, including ATP depletion, ROS, hypoxia, nitric oxide, cyclic guanosine monophosphate and nutrient deprivation are potential inducers of PGC1α expression164. Although ATP depletion, ROS accumulation and hypoxia occur in the kidney during injury, PGC1α expression is suppressed during AKI and restored during kidney recovery79. This finding suggests that an evolving balance of suppressive and inductive factors might determine the temporal profile of PGC1α expression during AKI and kidney repair. Emerging evidence suggests that the pro-inflammatory cytokine tumour necrosis factor (TNF)79,165 and tumour necrosis factor ligand superfamily member 12 (TNFSF12; also known as TWEAK) suppress PGC1A expression during AKI166. Several studies have also shown that kidney IR-induced activation of mitogen-activated protein kinase 3 (MAPK3; also known as ERK1) and MAPK1 (also known as ERK2) contribute to the downregulation of PGC1α pathways and ultimately kidney injury in mouse models167,168. By contrast, 5-hydroxytryptamine receptor 1F (5-HT1F; encoded by Htr1f) and β2 adrenergic receptor (encoded by Adrb2) are positive regulators of Pgc1a expression in mouse kidney cells169,170. Kidney proximal tubule-specific Htr1f or Adrb2 knockout reduced mitochondrial numbers in these cells under physiological conditions and to a greater extent in a mouse model of kidney IRI, and also increased the severity of kidney injury in this model169,170. In addition to transcriptional regulation of PGC1A expression, post-translational modifications, including phosphorylation, ubiquitylation, methylation, acetylation and GlcNAcylation, have important roles in regulating PGC1α activity171.
Role in CKD.
Kidney tissue from patients with DKD showed a reduction in PGC1α expression, compared with kidney tissue from patients with minimal change disease172. Downregulation of podocyte PGC1α was also shown in patients and mouse models of DKD173. The long non-coding RNA Tug1 might be an important positive regulator of Pgc1a expression in podocytes in DKD. Interaction between Tug1 and PGC1α has been shown to promote binding of PGC1α to its gene promoter and enhance Pgc1a expression174. Moreover, podocyte-specific Tug1 overexpression reduced DKD pathology and progression and increased levels of PGC1α in a mouse model174. Pharmacological approaches to restoring PGC1α expression also attenuated diabetic kidney injury in mice67. However, podocyte-specific induction of Pgc1a overexpression in mice resulted in alteration of mitochondrial properties, including the formation of giant mitochondria and increased expression of ETC and mitochondrial fusion genes, and increased podocyte proliferation and dedifferentiation, which led to albuminuria and glomerulosclerosis173. These findings suggest the existence of a critical window of PGC1α activity in podocytes for glomerular health.
Downregulation of Pgc1a and related genes involved in fatty acid oxidation was also reported in kidney tissue of animals and patients with CKD175,176. Transgenic expression of Pgc1a in kidney tubular cells improved fatty acid oxidation, ATP production and kidney pathology in mouse models of CKD induced by folic acid toxicity or by proximal tubule-specific overexpression of the intracellular fragment of neurogenic locus notch homologue protein 1 (NOTCH1)176. Further work showed that activation of a TGFβ–SMAD3-dependent pathway repressed Pgc1a transcription in kidney tubules in these models176.
Kidney distal tubule-specific deletion of Stk11, which encodes serine/threonine-protein kinase STK11 (also known as liver kinase B1), also resulted in a significant decrease in Pgc1a expression in mice, suggesting that STK11 is a positive regulator of Pgc1a177. Notably, mice with distal tubule-specific Stk11 deletion developed CKD and expression of STK11 was reduced in kidney biopsy samples from patients with CKD, suggesting that STK11 may preserve PGC1A expression to prevent CKD177. In addition, the transcription factor HES1 was shown to negatively regulate Pgc1a transcription through direct binding to the Pgc1a promoter in kidney tubular cells in a mouse model of CKD induced by NOTCH1 overexpression in kidney tubules175. These findings suggest that downregulation of PGC1α facilitates the pathogenesis and progression of CKD. As PGC1α has multiple functions, the specific role of PGC1α-induced mitochondrial biogenesis in kidney protection requires further investigation.
Therapeutic outlook
Given the critical role of mitochondrial dysfunction in kidney injury and abnormal kidney repair, specific interventions that target mitochondrial quality control mechanisms to preserve and restore mitochondrial function have emerged as promising therapeutic strategies to prevent and treat kidney injury, as well as accelerate kidney repair. A variety of compounds that target mitochondrial quality control mechanisms have been shown to protect against kidney injury and/or to accelerate kidney repair in AKI and CKD (TABLE 2; FIG. 6).
Fig. 6 |. Targeting mitochondrial quality control mechanisms to protect against kidney injury and accelerate kidney repair in AKI and CKD.
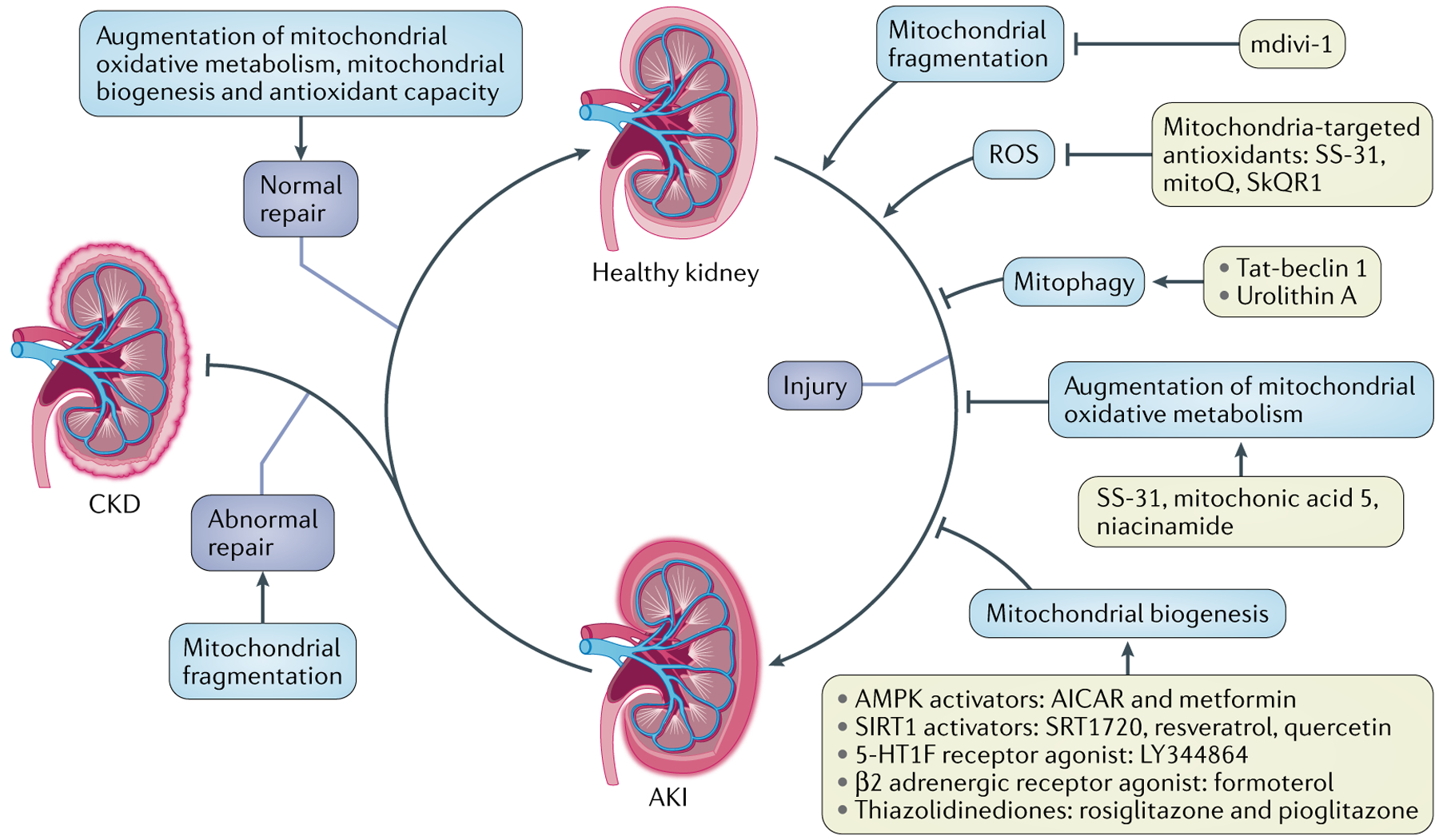
Potential kidney-protective strategies include inhibition of mitochondrial fragmentation using mdivi-1, augmentation of mitochondrial antioxidant capacity using mitochondrial-target antioxidants (for example, SS-31, mitoQ and SkQR1), enhancement of mitophagy using urolithin A or TAT-beclin 1 peptide, enhancement of mitochondrial biogenesis using AMP-activated protein kinase (AMPK) activators (5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) or metformin), sirtuin 1 (SIRT1) activators (SRT1720, resveratrol and quercetin), a 5-hydroxytryptamine receptor 1F (5-HT1F) agonist (LY344864), a β2 adrenergic receptor agonist (formoterol) or thiazolidinediones (rosiglitazone and pioglitazone), and augmentation of mitochondrial oxidative metabolism using mitochonic acid 5, SS-31 or niacinamide. AKI, acute kidney injury; CKD, chronic kidney disease; ROS, reactive oxygen species.
Treatment with mitochondria-targeted antioxidants, such as quinone analogues (MitoQ65,178, SkQ1 and SkQR179), SOD mimetics (Mito-CP)180 and SS-31 (REFS55,181,182), before kidney injury has been shown to protect against AKI and CKD in animal models. Notably, in rats with kidney IRI, treatment with SS-31 starting 1 month after kidney ischaemia for 6 weeks preserved mitochondrial integrity, restored glomerular capillaries and podocyte structure and arrested glomerulosclerosis and interstitial fibrosis42. Moreover, this SS-31-mediated protection was sustained for ≥6 months after treatment ended. Despite their efficiency in the prevention of kidney injury in experimental models, the potential of mitochondria-targeted antioxidants as treatments for AKI and/or CKD awaits further investigation.
Despite the exciting preclinical data, challenges remain for the clinical translation of mitochondria-targeted antioxidants in AKI and CKD. As discussed above, low and moderate levels of mtROS may act as signalling molecules in cell stress response pathways183. By scavenging and removing physiological levels of ROS involved in signalling, antioxidant supplements could have deleterious effects. In line with this notion, ischaemic preconditioning using a single event of kidney ischaemia and reperfusion (SIRPC) induced ROS production and protected against subsequent kidney IRI in mice, whereas administration of antioxidants after SIRPC reduced endogenous antioxidant expression and abolished the protective effect184. This finding suggests that SIRPC induced the production of ROS, which acted as signalling molecules to induce a protective antioxidant response. Accurate measurement of ROS levels before or during the administration of mitochondria-targeted antioxidants will likely be essential to avoid adverse effects and enable the safe use of these therapies, but methods and equipment for such measurement are not currently available.
Inhibition of mitochondrial fragmentation also seems to be beneficial during kidney injury and repair. mdivi-1 is a chemical inhibitor of mitochondrial fission that acts by inhibiting DRP1 activity185. Administration of mdivi-1 before injury has been shown to suppress mitochondrial fragmentation in kidney cells and protect against AKI and CKD in experimental models40,121. Of note, inhibition of mitochondrial fission 3 days after ischaemia using mdivi-1 accelerated kidney repair after AKI and protected against the development of post-AKI kidney fibrosis in mice41. It is important to note that fission is essential for increasing mitochondrial number to populate newly regenerated tubular cells with adequate numbers of mitochondria during kidney recovery after AKI. In addition, fission is required for removal of damaged or dysfunctional mitochondria by the autophagic pathway186. Inhibition of mitochondrial fission may therefore produce unexpected results. Moreover, the specificity of mdivi-1 has been questioned owing to experimental evidence that it might also act as a reversible mitochondrial complex I inhibitor187. Thus, there is a need to identify more specific chemical inhibitors of mitochondrial fission and to optimize the timing of administration of mitochondrial fission inhibitors to protect the kidney. The therapeutic potential of inhibitors of mitochondrial fragmentation in kidney disease awaits further investigation.
Enhancement of the removal of damaged and dysfunctional mitochondria by autophagy and/or mitophagy in kidney tubular cells is another attractive strategy for preventing AKI and DKD188,189. mTOR inhibitors, such as rapamycin, have been shown to enhance autophagic removal of damaged mitochondria in the kidney190,191, but their application might be limited because they also suppress cell proliferation192. A mTOR-independent agent, Tat-beclin 1, which is a peptide that comprises the essential sequence of beclin 1, has been shown to induce autophagy in skeletal muscle, cardiac muscle, pancreas and kidney tubules in vivo193,194. Urolithin A, a gut microbial metabolite of ellagic acid and related compounds, has been demonstrated to induce mitophagy and to have beneficial effects on age-related decline of muscle function following oral consumption in mouse models195. Oral administration of urolithin A also attenuated kidney injury in mouse models of AKI induced by cisplatin or CLP196,197, but the involvement of mitophagy in this kidney protection was not determined. Notably, the functions of autophagy in kidney fibrosis, the final common pathway of CKD, are controversial194,198, and the precise role of mitophagy in kidney repair after AKI is largely unclear.
Stimulation of mitochondrial biogenesis has been demonstrated to protect against AKI and CKD and to facilitate kidney recovery after AKI79,160,176. Given the essential role of PGC1α in controlling mitochondrial biogenesis, pharmacological activation of PGC1α is a potential approach to enhancing mitochondrial biogenesis. The primary modulators of PGC1α activity, SIRT1 and AMPK, activate PGC1α by acetylation and phosphorylation, respectively199. The AMPK activators AICAR and metformin, and the SIRT1 agonists quercetin, resveratrol and SRT1720, have been demonstrated to have beneficial effects in AKI and to protect against AKI to CKD transition109,200–204. Treatment with SRT1720 starting 24 h after reperfusion for 3–5 days expedited recovery of mitochondrial protein expression and function and kidney repair in rats with AKI205. Treatment with AICAR or SIRT1 activators, such as BF175 and resveratrol, protected against podocyte injury in animal models of CKD42,67,206,207.
Thiazolidinediones (TZDs) such as rosiglitazone and pioglitazone (oral hypoglycaemic drugs that are used for the treatment of type 2 diabetes mellitus) are ligands of PPARγ and thus activators of PGC1α208. Evidence also suggests a role of TZDs in the activation of PPARγ-independent signalling pathways such as AMPK signalling209. Kidney-protective effects of TZDs have been reported in animal models of AKI and DKD, as well as in patients with DKD210–213. Experimental studies have demonstrated that treatment with rosiglitazone or pioglitazone protects against mitochondrial damage in kidney cells211,214. However, the contribution of mitochondrial biogenesis or other mitochondrial quality control mechanisms to the kidney-protective effects of TZDs remains largely unknown. The adverse effects of TZDs, such as a potentially increased risk of acute myocardial infarction, stroke and heart failure, are also of concern215.
Treatment with the β2 adrenergic receptor agonist formoterol starting 24 h after kidney IRI accelerated the recovery of mitochondrial proteins, DNA copy number and kidney function (assessed by serum creatinine levels) in mice170,216. In mouse models of acute nephrotoxic serum nephritis and adriamycin-induced chronic glomerulopathy, treatment with formoterol post-injury when glomerular dysfunction was established increased the expression of PGC1α and mitochondrial proteins and accelerated recovery of glomerular function as assessed by proteinuria and kidney pathology216. Treatment with the 5-HT1F agonist LY344864 after AKI also facilitated kidney recovery, as assessed by blood urea nitrogen (BUN) and kidney pathology169,217.
Collectively, the available data suggest that enhancement of mitochondrial biogenesis by AMPK activators, SIRT1 activators, β2 adrenergic receptor agonists or 5-HT1F agonists is a promising therapeutic approach for kidney injury and for preventing AKI transition to CKD. Notably, uncontrolled mitochondrial biogenesis could have deleterious effects owing to increased ROS production as a by-product of increased energy metabolism. For example, uncontrolled mitochondrial proliferation in podocytes as a result of transgenic overexpression of PGC1α led to glomerulopathy in mice173. Therefore, responses to pharmacological induction of mitochondrial biogenesis should be closely monitored to avoid excessive mitochondrial proliferation.
Several chemicals that enhance mitochondrial capacity have also been shown to protect against kidney injury. For example, in mice, treatment with mitochonic acid 5, which binds to mitofilin in the IMM, facilitated ATP production and protected against ischaemic AKI and cisplatin-induced AKI, as assessed by serum creatinine and BUN levels218. Nicotinamide adenine dinucleotide (NAD+) is pivotal in the regulation of mitochondrial function owing to its roles as an essential metabolic cofactor and cosubstrate for various enzymes involved in cellular energy metabolism and energy production, and also as a cosubstrate for mitochondrial sirtuins219. Augmentation of intracellular NAD+ production is a novel therapeutic approach to AKI220–222. Intraperitoneal injection of the NAD+ precursor niacinamide (NAM) prior to kidney IRI in mice increased intracellular levels of NAD+ and improved mitochondrial oxidative metabolism, resulting in kidney-protective effects160,223. Similarly, a single intraperitoneal dose of NAM 18 h after kidney IRI attenuated kidney injury in mice, as assessed by serum creatinine and BUN levels160. A phase I placebo-controlled study in patients undergoing cardiac surgery demonstrated that once-daily treatment with oral NAM starting 1 day before surgery for 3 days was well tolerated, increased circulating NAD+ metabolites and was associated with a lower incidence of AKI compared with placebo223.
Another study in a mouse model of cisplatin-induced AKI showed that boosting de novo NAD+ synthesis through dietary supplementation with TES-1025, an inhibitor of 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase (ACMSD), which prevents NAD+ synthesis, enhanced mitochondrial functions, including oxygen consumption, fatty acid oxidation and antioxidant capacity, and protected against kidney dysfunction, as assessed by serum creatinine and BUN224. In addition, we showed that NAM supplementation dramatically reduced kidney interstitial fibrosis in UUO mice225, but the effect of NAM supplementation on mitochondrial function in this setting remains to be investigated.
Thus, the available data suggest that pharmacological interventions targeting mitochondrial quality control mechanisms are promising strategies for preventing and treating AKI and CKD as well as for impeding AKI to CKD transition. Despite promising preclinical data, challenges remain in the translation of such strategies into clinical use. Thus, in-depth investigation of mitochondrial biology during the development of AKI and progression to CKD is essential to enable the discovery of more specific and efficacious approaches to targeting mitochondria for kidney protection.
Conclusion and future perspectives
Despite progress in our understanding of mitochondrial dysfunction in kidney injury and abnormal kidney repair, as well as the therapeutic potential of targeting mitochondrial quality control mechanisms under these conditions, several questions remain. First, mitochondrial biology and pathology during kidney injury and repair are not fully understood. Although comprehensive evidence supports a pivotal role of mitochondrial dysfunction in AKI pathogenesis, the role of mitochondrial pathology during kidney repair after AKI is unclear. The kidney is a heterogeneous organ that contains various cell types and both parenchymal kidney cells and circulating pro-inflammatory leukocytes are involved in kidney injury and repair226. However, the studies available mainly focus on kidney tubular cells, and mitochondrial biology and pathology in other cell types remain largely unknown. In particular, the role of mitochondrial dysfunction in inflammatory cells during kidney injury and repair, and in endothelial cells and podocytes during the development and progression of DKD, awaits further investigation. Emerging evidence from other organs indicates that alterations in mitochondrial quality control capacity in different cell types might result in different or even opposing effects on tissue injury and repair. For example, insufficient parkin-mediated mitophagy in lung fibroblasts has been suggested to promote myofibroblast differentiation and proliferation and therefore contribute to the pathogenesis of idiopathic pulmonary fibrosis227. Similarly, PINK1 deficiency in lung epithelial cells resulted in mitochondria depolarization, expression of profibrotic factors and lung fibrosis, suggesting a protective function of the PINK1–parkin pathway of mitophagy in idiopathic pulmonary fibrosis228. By contrast, parkin-mediated mitophagy in alveolar macrophages seemed to contribute to the resistance to apoptosis of these cells and promote the development of pulmonary fibrosis229. Thus, it is necessary to determine the effects of the alteration of mitochondrial quality control capacity in different kidney cell types on kidney injury and repair.
Second, mitochondrial quality control mechanisms operate through an integrated hierarchical network of pathways; therefore, alterations in any one of these quality control mechanisms can affect other mechanisms and subsequently the whole system. For example, mitochondrial fission is required for mitophagy and also regulates mitochondrial biogenesis. Therefore, pharmacological inhibition of mitochondrial fission might also impair mitophagy and mitochondria biogenesis.
Third, although emerging evidence suggests an association of mtDNA damage and UPRmt with the development and progression of kidney injury and abnormal kidney repair, their precise roles in these pathological conditions and the molecular machinery that is involved remain largely unknown.
Fourth, as discussed above, despite exciting preclinical data, translation of mitochondria-targeted agents into clinical use in AKI and CKD remains a big challenge, not only because of their potential adverse effects and unclear mechanism of action but also because of the unclear mitochondrial biology and pathology under these conditions. Thus, further studies should focus on identifying the role and regulation of mitochondrial quality control mechanisms in kidney resident and circulating cells during the development of kidney injury and kidney repair in order to facilitate the discovery of pharmacological modulators to prevent and treat AKI and CKD, as well as to prevent AKI transition to CKD.
Key points.
mitochondria are essential for cell viability but are highly susceptible to injury or damage.
mitochondrial homeostasis depends on multiple quality control mechanisms, including antioxidant defence, protein quality control, mitochondrial DNA repair, mitochondrial dynamics, mitophagy and mitochondrial biogenesis.
loss of mitochondrial quality control may induce mitochondrial damage and dysfunction, leading to cell death, tissue injury and possible organ failure.
acute kidney injury (AKI) is characterized by sublethal and lethal damage to kidney tubules and incomplete or maladaptive kidney repair after AKI leads to kidney fibrosis and eventually chronic kidney disease (CKD).
mitochondrial dysfunction has a critical role in the pathogenesis of AKI, abnormal kidney repair and CKD.
modulation of mitochondrial quality control is a promising therapeutic approach to preventing and treating AKI and CKD and to accelerating kidney repair.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Bhargava P & Schnellmann RG Mitochondrial energetics in the kidney. Nat. Rev. Nephrol 13, 629–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suliman HB & Piantadosi CA Mitochondrial quality control as a therapeutic target. Pharmacol. Rev 68, 20–48 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guder WG & Ross BD Enzyme distribution along the nephron. Kidney Int. 26, 101–111 (1984). [DOI] [PubMed] [Google Scholar]
- 4.Lan R et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol 27, 3356–3367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heung M et al. Acute kidney injury recovery pattern and subsequent risk of CKD: an analysis of veterans health administration data. Am. J. Kidney Dis 67, 742–752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.See EJ et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 95, 160–172 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Yang L, Besschetnova TY, Brooks CR, Shah JV & Bonventre JV Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med 16, 535–543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emma F, Montini G, Parikh SM & Salviati L Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat. Rev. Nephrol 12, 267–280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forbes JM & Thorburn DR Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol 14, 291–312 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birk AV, Chao WM, Bracken C, Warren JD & Szeto HH Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol 171, 2017–2028 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wan J et al. Regulation of respiration and apoptosis by cytochrome c threonine 58 phosphorylation. Sci. Rep 9, 15815 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalkavan H & Green DR MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 25, 46–55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schug ZT, Gonzalvez F, Houtkooper RH, Vaz FM & Gottlieb E BID is cleaved by caspase-8 within a native complex on the mitochondrial membrane. Cell Death Differ. 18, 538–548 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun 8, 14329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen D et al. PUMA amplifies necroptosis signaling by activating cytosolic DNA sensors. Proc. Natl Acad. Sci. USA 115, 3930–3935 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whelan RS et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl Acad. Sci. USA 109, 6566–6571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platnich JM et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 25, 1525–1536 e1527 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Gao M et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363 e353 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 9, 983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulay SR et al. Mitochondria permeability transition versus necroptosis in oxalate-induced AKI. J. Am. Soc. Nephrol 30, 1857–1869 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin-Sanchez D et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl Acad. Sci. USA 115, 4182–4187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng F, Sharma I, Dai Y, Yang M & Kanwar YS Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J. Clin. Invest 129, 5033–5049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitra K, Wunder C, Roysam B, Lin G & Lippincott-Schwartz J A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl Acad. Sci. USA 106, 11960–11965 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pennock R et al. Human cell dedifferentiation in mesenchymal condensates through controlled autophagy. Sci. Rep 5, 13113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antico Arciuch VG, Elguero ME, Poderoso JJ & Carreras MC Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox. Signal 16, 1150–1180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamanaka RB & Chandel NS Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci 35, 505–513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furman D et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med 25, 1822–1832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heid ME et al. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol 191, 5230–5238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulua AC et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med 208, 519–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West AP, Shadel GS & Ghosh S Mitochondria in innate immune responses. Nat. Rev. Immunol 11, 389–402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buck MD et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maekawa H et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 29, 1261–1273.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Chung KW et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 30, 784–799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuk A & Bonventre JV Acute kidney injury. Annu. Rev. Med 67, 293–307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatachalam MA, Weinberg JM, Kriz W & Bidani AK Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol 26, 1765–1776 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nath KA et al. Intracellular targets in heme protein-induced renal injury. Kidney Int. 53, 100–111 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Funk JA & Schnellmann RG Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol 302, F853–F864 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aparicio-Trejo OE et al. Chronic impairment of mitochondrial bioenergetics and β-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free. Radic. Biol. Med 154, 18–32 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Brooks C, Wei Q, Cho SG & Dong Z Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest 119, 1275–1285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perry HM et al. Dynamin-related protein 1 deficiency promotes recovery from AKI. J. Am. Soc. Nephrol 29, 194–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szeto HH et al. Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1β and IL-18 and arrests CKD. J. Am. Soc. Nephrol 28, 1437–1449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zorov DB, Juhaszova M & Sollott SJ Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev 94, 909–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turrens JF Mitochondrial formation of reactive oxygen species. J. Physiol 552, 335–344 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rabilloud T et al. The mitochondrial antioxidant defence system and its response to oxidative stress. Proteomics 1, 1105–1110 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Bindoli A & Rigobello MP Principles in redox signaling: from chemistry to functional significance. Antioxid. Redox Signal 18, 1557–1593 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Zhao X et al. Tenofovir and adefovir down-regulate mitochondrial chaperone TRAP1 and succinate dehydrogenase subunit B to metabolically reprogram glucose metabolism and induce nephrotoxicity. Sci. Rep 7, 46344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cline SD Mitochondrial DNA damage and its consequences for mitochondrial gene expression. Biochim. Biophys. Acta 1819, 979–991 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim J, Seok YM, Jung KJ & Park KM Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am. J. Physiol. Renal Physiol 297, F461–F470 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Mapuskar KA et al. Persistent increase in mitochondrial superoxide mediates cisplatin-induced chronic kidney disease. Redox Biol. 20, 98–106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vasko R Peroxisomes and kidney injury. Antioxid. Redox Signal 25, 217–231 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hasegawa K et al. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J. Biol. Chem 285, 13045–13056 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szeto HH First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol 171, 2029–2050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu D et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 211, 57–67 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Liu S, Soong Y, Seshan SV & Szeto HH Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am. J. Physiol. Renal Physiol 306, F970–F980 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Bell EL, Klimova TA, Eisenbart J, Schumacker PT & Chandel NS Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell Biol 27, 5737–5745 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding W et al. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 7, 17479–17491 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jain M et al. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem 288, 770–777 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B, Chen Y & St Clair DK ROS and p53: a versatile partnership. Free Radic. Biol. Med 44, 1529–1535 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daenen K et al. Oxidative stress in chronic kidney disease. Pediatr. Nephrol 34, 975–991 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Kirkman DL, Muth BJ, Ramick MG, Townsend RR & Edwards DG Role of mitochondria-derived reactive oxygen species in microvascular dysfunction in chronic kidney disease. Am. J. Physiol. Renal Physiol 314, F423–F429 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyamoto S et al. Restoring mitochondrial superoxide levels with elamipretide (MTP-131) protects db/db mice against progression of diabetic kidney disease. J. Biol. Chem 295, 7249–7260 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizuguchi Y et al. A novel cell-permeable antioxidant peptide decreases renal tubular apoptosis and damage in unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol 295, F1545–F1553 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qi H et al. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes 66, 763–778 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao L et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 11, 297–311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szeto HH et al. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 90, 997–1011 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Dugan LL et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Invest 123, 4888–4899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vazquez-Calvo C, Suhm T, Buttner S & Ott M The basic machineries for mitochondrial protein quality control. Mitochondrion 50, 121–131 (2020). [DOI] [PubMed] [Google Scholar]
- 69.Shpilka T & Haynes CM The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol 19, 109–120 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Bohovych I, Chan SS & Khalimonchuk O Mitochondrial protein quality control: the mechanisms guarding mitochondrial health. Antioxid. Redox Signal 22, 977–994 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saisawat P et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int. 85, 1310–1317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stacchiotti A et al. Tubular stress proteins and nitric oxide synthase expression in rat kidney exposed to mercuric chloride and melatonin. J. Histochem. Cytochem 54, 1149–1157 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hernadez-Pando R et al. Histological and subcellular distribution of 65 and 70 kD heat shock proteins in experimental nephrotoxic injury. Exp. Toxicol. Pathol 47, 501–508 (1995). [DOI] [PubMed] [Google Scholar]
- 74.Fan Y et al. Inhibition of reticulon-1A-mediated endoplasmic reticulum stress in early AKI attenuates renal fibrosis development. J. Am. Soc. Nephrol 28, 2007–2021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Inagi R, Ishimoto Y & Nangaku M Proteostasis in endoplasmic reticulum — new mechanisms in kidney disease. Nat. Rev. Nephrol 10, 369–378 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Shu S et al. Endoplasmic reticulum stress is activated in post-ischemic kidneys to promote chronic kidney disease. EBioMedicine 37, 269–280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Senft D & Ronai ZA UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci 40, 141–148 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malhotra JD & Kaufman RJ ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb. Perspect. Biol 3, a004424 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tran M et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest 121, 4003–4014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhreathnach U et al. Profibrotic IHG-1 complexes with renal disease associated HSPA5 and TRAP1 in mitochondria. Biochim. Biophys. Acta Mol. Basis Dis 1863, 896–906 (2017). [DOI] [PubMed] [Google Scholar]
- 81.Chen JF et al. TRAP1 ameliorates renal tubulointerstitial fibrosis in mice with unilateral ureteral obstruction by protecting renal tubular epithelial cell mitochondria. FASEB J. 31, 4503–4514 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Sharma P & Sampath H Mitochondrial DNA integrity: role in health and disease. Cells 8, 100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zinovkina LA Mechanisms of mitochondrial DNA repair in mammals. Biochemistry 83, 233–249 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Moretton A et al. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 12, e0176795 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uehara M et al. Pharmacological inhibition of ataxia-telangiectasia mutated exacerbates acute kidney injury by activating p53 signaling in mice. Sci. Rep 10, 4441 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kishi S et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Invest 129, 4797–4816 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun Z et al. Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide. Am. J. Physiol. Renal Physiol 294, F491–F498 (2008). [DOI] [PubMed] [Google Scholar]
- 88.Tan X et al. Postconditioning ameliorates mitochondrial DNA damage and deletion after renal ischemic injury. Nephrol. Dial. Transplant 28, 2754–2765 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bartz RR et al. Staphylococcus aureus sepsis induces early renal mitochondrial DNA repair and mitochondrial biogenesis in mice. PLoS ONE 9, e100912 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tanji N et al. Adefovir nephrotoxicity: possible role of mitochondrial DNA depletion. Hum. Pathol 32, 734–740 (2001). [DOI] [PubMed] [Google Scholar]
- 91.Maniccia-Bozzo E, Espiritu MB & Singh G Differential effects of cisplatin on mouse hepatic and renal mitochondrial DNA. Mol. Cell Biochem 94, 83–88 (1990). [DOI] [PubMed] [Google Scholar]
- 92.Krishnan KJ et al. What causes mitochondrial DNA deletions in human cells? Nat. Genet 40, 275–279 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Connor TM et al. Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genet. 13, e1006620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Toole JF Renal manifestations of genetic mitochondrial disease. Int. J. Nephrol. Renovasc. Dis 7, 57–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fedorova LV et al. Mitochondrial impairment in the five-sixth nephrectomy model of chronic renal failure: proteomic approach. BMC Nephrol. 14, 209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tin A et al. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the atherosclerosis risk in communities study. J. Am. Soc. Nephrol 27, 2467–2473 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wai T & Langer T Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab 27, 105–117 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Ni HM, Williams JA & Ding WX Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Burman JL et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol 216, 3231–3247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tilokani L, Nagashima S, Paupe V & Prudent J Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Friedman JR et al. ER tubules mark sites of mitochondrial division. Science 334, 358–362 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee JE, Westrate LM, Wu H, Page C & Voeltz GK Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ & Ryan MT Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun 9, 5239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cho SG et al. Bif-1 interacts with prohibitin-2 to regulate mitochondrial inner membrane during cell stress and apoptosis. J. Am. Soc. Nephrol 30, 1174–1191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chakrabarti R et al. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol 217, 251–268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ban T et al. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol 19, 856–863 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Chan DC Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol 15, 235–259 (2020). [DOI] [PubMed] [Google Scholar]
- 108.Wei Q et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Invest 128, 5448–5464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morigi M et al. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest 125, 715–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qin N et al. UCP2-dependent improvement of mitochondrial dynamics protects against acute kidney injury. J. Pathol 247, 392–405 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Liu Z et al. Numb depletion promotes Drp1-mediated mitochondrial fission and exacerbates mitochondrial fragmentation and dysfunction in acute kidney injury. Antioxid. Redox Signal 30, 1797–1816 (2019). [DOI] [PubMed] [Google Scholar]
- 112.Xiao X et al. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol 306, F1318–F1326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gall JM et al. Role of mitofusin 2 in the renal stress response. PLoS ONE 7, e31074 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gall JM et al. Conditional knockout of proximal tubule mitofusin 2 accelerates recovery and improves survival after renal ischemia. J. Am. Soc. Nephrol 26, 1092–1102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brooks C, Cho SG, Wang CY, Yang T & Dong Z Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am. J. Physiol. Cell Physiol 300, C447–C455 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scorrano L et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Varanita T et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 21, 834–844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Otera H, Miyata N, Kuge O & Mihara K Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol 212, 531–544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xiao L et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes 63, 1366–1380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhan M, Usman IM, Sun L & Kanwar YS Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol 26, 1304–1321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ayanga BA et al. Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J. Am. Soc. Nephrol 27, 2733–2747 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Galvan DL et al. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J. Clin. Invest 129, 2807–2823 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qin X et al. Berberine protects glomerular podocytes via inhibiting Drp1-mediated mitochondrial fission and dysfunction. Theranostics 9, 1698–1713 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Noh MR, Woo CH, Park MJ, In Kim J & Park KM Ablation of C/EBP homologous protein attenuates renal fibrosis after ureteral obstruction by reducing autophagy and microtubule disruption. Biochim. Biophys. Acta Mol. Basis Dis 1864, 1634–1641 (2018). [DOI] [PubMed] [Google Scholar]
- 125.Quan Y et al. Sirtuin 3 activation by honokiol decreases unilateral ureteral obstruction-induced renal inflammation and fibrosis via regulation of mitochondrial dynamics and the renal NF-κBTGF-β1/Smad signaling pathway. Int. J. Mol. Sci 21, 402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y et al. Drp1-mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 11, 29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sato M & Sato K Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 334, 1141–1144 (2011). [DOI] [PubMed] [Google Scholar]
- 128.Domenech E et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol 17, 1304–1316 (2015). [DOI] [PubMed] [Google Scholar]
- 129.Palikaras K, Lionaki E & Tavernarakis N Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol 20, 1013–1022 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Liu L, Sakakibara K, Chen Q & Okamoto K Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 24, 787–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matsuda N et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol 189, 211–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kane LA et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol 205, 143–153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gegg ME et al. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet 19, 4861–4870 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lazarou M et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McLelland GL, Soubannier V, Chen CX, McBride HM & Fon EA Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McLelland GL, Lee SA, McBride HM & Fon EA Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol 214, 275–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sugiura A, McLelland GL, Fon EA & McBride HM A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 33, 2142–2156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang T et al. BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem 291, 21616–21629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee Y, Lee HY, Hanna RA & Gustafsson AB Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol 301, H1924–H1931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tang C et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 10, 677 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tang C et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 14, 880–897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang J, Zhu P, Li R, Ren J & Zhou H Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 30, 101415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lin Q et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 26, 101254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang Y et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 9, 1113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu JX et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 235, 116828 (2019). [DOI] [PubMed] [Google Scholar]
- 146.Lynch MR et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight 5, e142898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gao Y et al. Role of Parkin-mediated mitophagy in the protective effect of polydatin in sepsis-induced acute kidney injury. J. Transl. Med 18, 114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Boya P, Codogno P & Rodriguez-Muela N Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development 145, dev146506 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Esteban-Martinez L et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 36, 1688–1706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chen K et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 9, 105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen K et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 33, 4571–4585 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Li W et al. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology 158, 2155–2167 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Sun J et al. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol 10.1530/JOE-18-0578 (2019). [DOI] [PubMed] [Google Scholar]
- 154.Zhou D et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 10, 524 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li S et al. Drp1-regulated PARK2-dependent mitophagy protects against renal fibrosis in unilateral ureteral obstruction. Free Radic. Biol. Med 152, 632–649 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Bhatia D et al. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 4, e132826 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ventura-Clapier R, Garnier A & Veksler V Transcriptional control of mitochondrial biogenesis: the central role of PGC-1α. Cardiovasc. Res 79, 208–217 (2008). [DOI] [PubMed] [Google Scholar]
- 158.Dominy JE & Puigserver P Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol 5, a033944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Portilla D et al. Alterations of PPARα and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 62, 1208–1218 (2002). [DOI] [PubMed] [Google Scholar]
- 160.Tran MT et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531, 528–532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jesinkey SR et al. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J. Am. Soc. Nephrol 25, 1157–1162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Valle I, Alvarez-Barrientos A, Arza E, Lamas S & Monsalve M PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res 66, 562–573 (2005). [DOI] [PubMed] [Google Scholar]
- 163.Cherry AD, Suliman HB, Bartz RR & Piantadosi CA Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem 289, 41–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kelly DP & Scarpulla RC Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes. Dev 18, 357–368 (2004). [DOI] [PubMed] [Google Scholar]
- 165.Smith JA, Stallons LJ, Collier JB, Chavin KD & Schnellmann RG Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J. Pharmacol. Exp. Ther 352, 346–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ruiz-Andres O et al. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 89, 399–410 (2016). [DOI] [PubMed] [Google Scholar]
- 167.Collier JB & Schnellmann RG Extracellular signal-regulated kinase 1/2 regulates NAD metabolism during acute kidney injury through microRNA-34a-mediated NAMPT expression. Cell Mol. Life Sci 77, 3643–3655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Collier JB, Whitaker RM, Eblen ST & Schnellmann RG Rapid renal regulation of peroxisome proliferator-activated receptor γ coactivator-1α by extracellular signal-regulated kinase 1/2 in physiological and pathological conditions. J. Biol. Chem 291, 26850–26859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gibbs WS et al. 5-HT1F receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am. J. Physiol. Renal Physiol 315, F1119–F1128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cameron RB et al. Proximal tubule β 2-adrenergic receptor mediates formoterol-induced recovery of mitochondrial and renal function after ischemia-reperfusion injury. J. Pharmacol. Exp. Ther 369, 173–180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fernandez-Marcos PJ & Auwerx J Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr 93, 884S–890S (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sharma K et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol 24, 1901–1912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li SY et al. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight 2, e92930 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Long J et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest 126, 4205–4218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Han SH et al. PGC-1α protects from Notch-induced kidney fibrosis development. J. Am. Soc. Nephrol 28, 3312–3322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kang HM et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med 21, 37–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Han SH et al. Deletion of Lkb1 in renal tubular epithelial cells leads to CKD by altering metabolism. J. Am. Soc. Nephrol 27, 439–453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dare AJ et al. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 5, 163–168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Plotnikov EY et al. Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim. Biophys. Acta 1812, 77–86 (2011). [DOI] [PubMed] [Google Scholar]
- 180.Mukhopadhyay P et al. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med 52, 497–506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Szeto HH et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol 22, 1041–1052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Birk AV et al. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol 24, 1250–1261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Angelova PR & Abramov AY Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med 100, 81–85 (2016). [DOI] [PubMed] [Google Scholar]
- 184.Kim J, Jang HS & Park KM Reactive oxygen species generated by renal ischemia and reperfusion trigger protection against subsequent renal ischemia and reperfusion injury in mice. Am. J. Physiol. Renal Physiol 298, F158–F166 (2010). [DOI] [PubMed] [Google Scholar]
- 185.Cassidy-Stone A et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Twig G et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bordt EA et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 40, 583–594.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bhatia D & Choi ME The emerging role of mitophagy in kidney diseases. J. Life Sci 1, 13–22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang Y, Cai J, Tang C & Dong Z Mitophagy in acute kidney injury and kidney repair. Cells 9, 338 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cui J et al. Rapamycin protects against gentamicin-induced acute kidney injury via autophagy in mini-pig models. Sci. Rep 5, 11256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jiang M et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Marti HP & Frey FJ Nephrotoxicity of rapamycin: an emerging problem in clinical medicine. Nephrol. Dial. Transplant 20, 13–15 (2005). [DOI] [PubMed] [Google Scholar]
- 193.Shoji-Kawata S et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494, 201–206 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Livingston MJ et al. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 12, 976–998 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ryu D et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med 22, 879–888 (2016). [DOI] [PubMed] [Google Scholar]
- 196.Zou D et al. Oral delivery of nanoparticle urolithin A normalizes cellular stress and improves survival in mouse model of cisplatin-induced AKI. Am. J. Physiol. Renal Physiol 317, F1255–F1264 (2019). [DOI] [PubMed] [Google Scholar]
- 197.Zhang Z, Zhang H, Chen R & Wang Z Oral supplementation with ursolic acid ameliorates sepsis-induced acute kidney injury in a mouse model by inhibiting oxidative stress and inflammatory responses. Mol. Med. Rep 17, 7142–7148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Li H et al. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 12, 1472–1486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Scarpulla RC Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 1813, 1269–1278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Higashida K et al. Effects of resveratrol and SIRT1 on PGC-1α activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 11, e1001603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kitada M & Koya D Renal protective effects of resveratrol. Oxid. Med. Cell Longev 2013, 568093 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Negishi K et al. Renal L-type fatty acid-binding protein mediates the bezafibrate reduction of cisplatin-induced acute kidney injury. Kidney Int. 73, 1374–1384 (2008). [DOI] [PubMed] [Google Scholar]
- 203.Lempiainen J, Finckenberg P, Levijoki J & Mervaala E AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharmacol 166, 1905–1915 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Shin YJ et al. Protective effects of quercetin against HgCl2-induced nephrotoxicity in Sprague-Dawley rats. J. Med. Food 18, 524–534 (2015). [DOI] [PubMed] [Google Scholar]
- 205.Funk JA & Schnellmann RG Accelerated recovery of renal mitochondrial and tubule homeostasis with SIRT1/PGC-1α activation following ischemia-reperfusion injury. Toxicol. Appl. Pharmacol 273, 345–354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hong Q et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 93, 1330–1343 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhong Y, Lee K & He JC SIRT1 is a potential drug target for treatment of diabetic kidney disease. Front. Endocrinol 9, 624 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hondares E et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1α gene transcription: an autoregulatory loop controls PGC-1α expression in adipocytes via peroxisome proliferator-activated receptor-γ coactivation. Endocrinology 147, 2829–2838 (2006). [DOI] [PubMed] [Google Scholar]
- 209.Liu Y et al. AMP-activated protein kinase mediates the antiplatelet effects of the thiazolidinediones rosiglitazone and pioglitazone. Mol. Pharmacol 89, 313–321 (2016). [DOI] [PubMed] [Google Scholar]
- 210.Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN & Lasaridis AN Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am. J. Kidney Dis 55, 835–847 (2010). [DOI] [PubMed] [Google Scholar]
- 211.Sun L et al. Pioglitazone improves mitochondrial function in the remnant kidney and protects against renal fibrosis in 5/6 nephrectomized rats. Front. Pharmacol 8, 545 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chen W et al. Pioglitazone protects against renal ischemia-reperfusion injury via the AMP-activated protein kinase-regulated autophagy pathway. Front. Pharmacol 9, 851 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Morrison MC et al. Protective effect of rosiglitazone on kidney function in high-fat challenged human-CRP transgenic mice: a possible role for adiponectin and miR-21? Sci. Rep 7, 2915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhu C et al. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: a therapeutic target of PPARγ. Am. J. Pathol 178, 2020–2031 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Graham DJ et al. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. JAMA 304, 411–418 (2010). [DOI] [PubMed] [Google Scholar]
- 216.Arif E et al. Mitochondrial biogenesis induced by the β2-adrenergic receptor agonist formoterol accelerates podocyte recovery from glomerular injury. Kidney Int. 96, 656–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Garrett SM, Whitaker RM, Beeson CC & Schnellmann RG Agonism of the 5-hydroxytryptamine 1F receptor promotes mitochondrial biogenesis and recovery from acute kidney injury. J. Pharmacol. Exp. Ther 350, 257–264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Suzuki T et al. Mitochonic acid 5 binds mitochondria and ameliorates renal tubular and cardiac myocyte damage. J. Am. Soc. Nephrol 27, 1925–1932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Li W & Sauve AA NAD+ content and its role in mitochondria. Methods Mol. Biol 1241, 39–48 (2015). [DOI] [PubMed] [Google Scholar]
- 220.Hershberger KA, Martin AS & Hirschey MD Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol 13, 213–225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ralto KM, Rhee EP & Parikh SM NAD+ homeostasis in renal health and disease. Nat. Rev. Nephrol 16, 99–111 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Allison SJ Targeting NAD+ synthesis to boost mitochondrial function and protect the kidney. Nat. Rev. Nephrol 15, 1 (2019). [DOI] [PubMed] [Google Scholar]
- 223.Poyan Mehr A et al. De novo NAD+ biosynthetic impairment in acute kidney injury in humans. Nat. Med 24, 1351–1359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Katsyuba E et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 563, 354–359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zheng M et al. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell Mol. Med 23, 3995–4004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liu J et al. Cell-specific translational profiling in acute kidney injury. J. Clin. Invest 124, 1242–1254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kobayashi K et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol 197, 504–516 (2016). [DOI] [PubMed] [Google Scholar]
- 228.Bueno M et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Invest 125, 521–538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ & Carter AB Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 44, 582–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]