

Abstract
Bacteria possess an extraordinary repertoire of cell envelope glycans that have critical physiological functions. Pathogenic bacteria have glycans that are essential for growth and virulence but are absent from humans, making them high-priority targets for antibiotic, vaccine, and diagnostic development. The advent of metabolic labeling with bioorthogonal chemical reporters and small-molecule fluorescent reporters has enabled the investigation and targeting of specific bacterial glycans in their native environments. These tools have opened the door to imaging glycan dynamics, assaying and inhibiting glycan biosynthesis, profiling glycoproteins and glycan-binding proteins, and targeting pathogens with diagnostic and therapeutic payload. These capabilities have been wielded in diverse commensal and pathogenic Gram-positive, Gram-negative, and mycobacterial species—including within live host organisms. Here, we review the development and applications of chemical reporters for bacterial glycans, including peptidoglycan, lipopolysaccharide, glycoproteins, teichoic acids, and capsular polysaccharides, as well as mycobacterial glycans, including trehalose glycolipids and arabinan-containing glycoconjugates. We cover in detail how bacteria-targeting chemical reporters are designed, synthesized, and evaluated, how they operate from a mechanistic standpoint, and how this information informs their judicious and innovative application. We also provide a perspective on the current state and future directions of the field, underscoring the need for interdisciplinary teams to create novel tools and extend existing tools to support fundamental and translational research on bacterial glycans.
Graphical Abstract
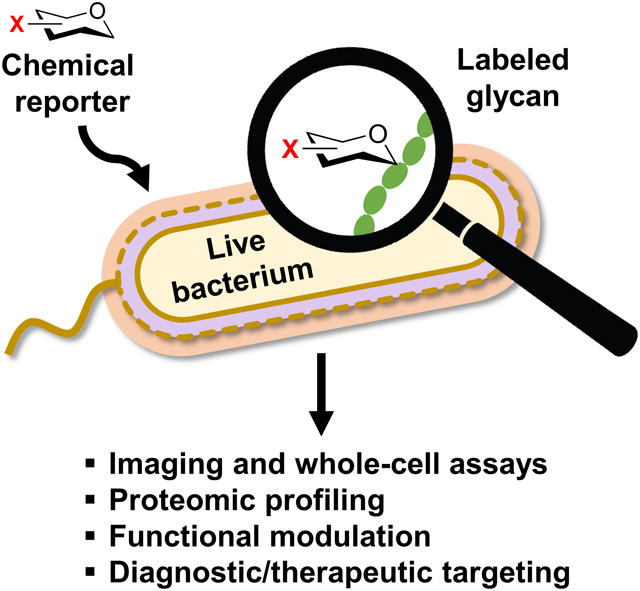
1. Introduction
1.1. Overview of the structures and functions of bacterial glycans
Bacteria inhabit an extraordinarily broad range of environments. Extremophilic bacteria can survive elevated radiation levels, temperatures ranging from below freezing to over 100 °C, acidic and basic conditions (pH 1–10), deep-sea pressures of several hundred atmospheres, salt concentrations nearly ten times that of seawater, and arid climates where water is scarce.1 Pathogenic and symbiotic bacteria, which must thrive within a host organism, also have unique survival requirements that depend on the environment afforded by their host. For example, Mycobacterium tuberculosis, the causative agent of human tuberculosis, has a complex life cycle characterized in part by survival and proliferation within host macrophages, a setting in which most invaders are efficiently eliminated.2 On the other hand, some species of bacteria are generally considered to be beneficial to their host, such as those in the genus Bacteroides, which populate the human gastrointestinal tract and contribute to host immune system development and processing of host-acquired nutrients.3
The bacterial cell envelope is the essential structure that allows a bacterium to protect itself from and interact with its surrounding environment. Despite the remarkable variety of environments that bacteria encounter, only a few general cell envelope architectures exist (Figure 1): Gram-positive bacteria, which retain the dye crystal violet in the Gram stain test, have a plasma membrane (PM) surrounded by a large peptidoglycan (PG) layer (Figure 1A); Gram-negative bacteria contain PM, a smaller PG layer, and an outer membrane (OM) that prevents crystal violet retention (Figure 1B); mycobacteria, which retain crystal violet but more closely resemble Gram-negative bacteria from an ultrastructural perspective, have a cell envelope consisting of PM, PG, and a unique outer membrane called the mycomembrane (MM), which is anchored to PG via an intermediate arabinogalactan (AG) polysaccharide layer (Figure 1C).4
Figure 1.
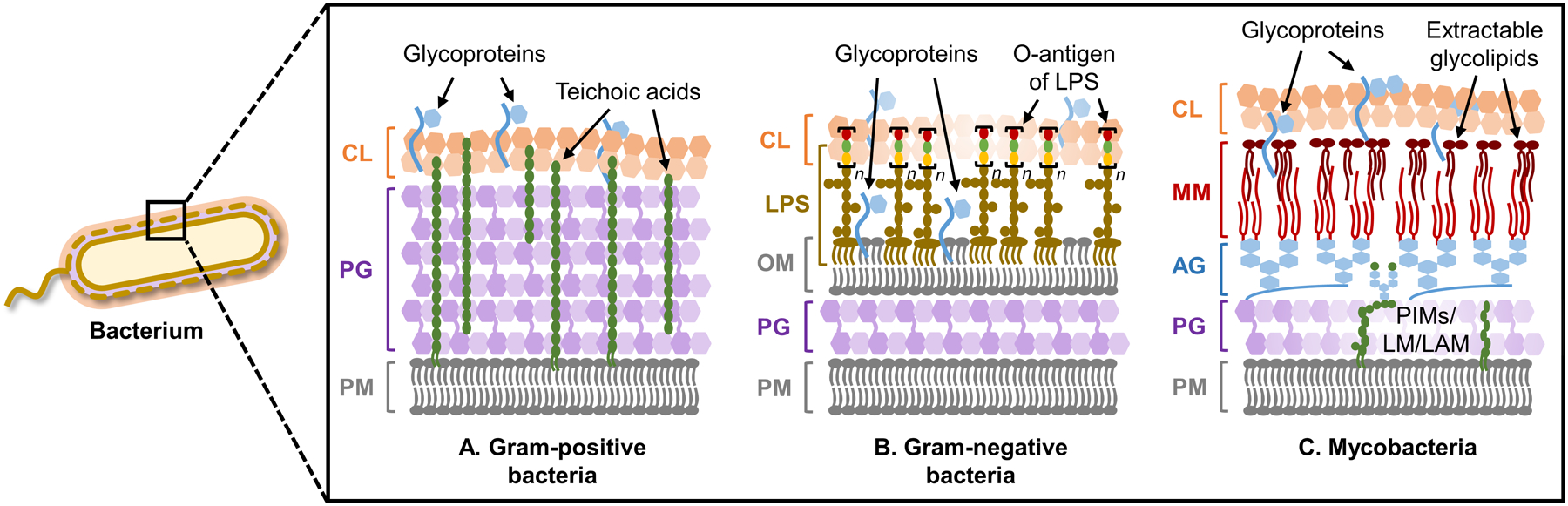
Cell envelope architectures and major glycans of common types of bacteria: (A) Gram-positive bacteria; (B) Gram-negative bacteria; (C) mycobacteria. AG, arabinogalactan; CL, capsular layer; LAM, lipoarabinomannan; LM, lipomannan; LPS, lipopolysaccharide; MM, mycomembrane; OM, outer membrane; PG, peptidoglycan; PIM, phosphatidylinositol; PM, plasma membrane.
On a molecular level, glycans are the principal constituents of all bacterial cell envelopes. Although bacteria exhibit enormous diversity in glycan structure, the most common types of bacterial glycans are briefly introduced here (Figure 1).4,5 PG, which is present in virtually all bacterial species, consists of alternating N-acetylglucosamine and N-acetylmuramic acid sugar residues that are cross-linked by short peptide chains containing D-amino acids.6 PG’s cross-linked structure constitutes a protective mesh-like layer that provides structural strength and shape to the bacterium. In Gram-positive bacteria, the large PG layer is additionally decorated with carbohydrate-containing polymers called teichoic acids, which confer a negative charge to the cell wall that may further enhance its rigidity and reduce its permeability to antibiotics.7,8 In Gram-negative bacteria, the OM’s outer leaflet is largely composed of lipopolysaccharide (LPS), which provides structural integrity to the cell and also acts as a potent immunostimulant.9 LPS contains three main structural domains, including the lipidated disaccharide Lipid A, a core oligosaccharide domain, and a highly variable polysaccharide domain called the O-antigen. The mycobacterial cell envelope is distinct from Gram-positive and Gram-negative bacteria, as its PG is covalently attached to AG—another complex polysaccharide—via a rhamnose-containing linker.10 Terminal AG residues are covalently modified with long-chain fatty acids called mycolic acids (up to 100 carbons), which populate the mycobacterial mycomembrane along with an array of other intercalated lipids and glycolipids, such as the trehalose mycolates.11 Numerous other mycobacterial glycans, such as phosphatidylinositol mannosides (PIMs) and PIM-anchored lipomannan (LM) and lipoarabinomannan (LAM), provide structural integrity to the cell and contribute to M. tuberculosis pathogenesis.11 Irrespective of Gram classification, many bacteria possess capsular layer polysaccharides and secreted exopolysaccharides, which are structurally diverse and have functions ranging from immune evasion to biofilm formation.12 In addition, various types of surface-associated and secreted glycoproteins have been discovered in bacteria, despite earlier misconceptions that such structures were limited to eukaryotic systems.13 A prominent example of bacterial glycoproteins is the abundant self-assembling S-layer glycoproteins that are well known to encapsulate archaea, but also exist in some Gram-positive and -negative bacteria.14
The high structural diversity of bacterial glycans helps to explain the ability of bacteria to adapt to and survive within disparate habitats.15,16 The complete repertoire of glycans in a cell, which can be referred to as the cell’s “glycome,” differs remarkably between bacterial species (inter-species variability). For instance, while capsular layer polysaccharides are present in many bacteria, their inter-species structural variance is extraordinary.17 Bacterial polysaccharides range from relatively simple structures, such as polysialic acids and hyaluronans, which mimic host glycans to facilitate pathogen immune evasion, to structures with quite complex repeating units, such as the zwitterionic tetrasaccharide of polysaccharide A in the symbiont B. fragilis, which is involved in signaling between the bacterium and the human enteric nervous system.17 Bacterial glycomes can also differ considerably between strains of the same species (inter-strain variability) and within the same strain (intra-strain variability). A well-known example of inter-strain glycome variability occurs in the LPS O-antigen polysaccharides of Gram-negative bacteria, which have variable monosaccharide compositions, glycosidic linkages, and modifications (e.g., acetylation) that underlie strain-specific physiological, pathogenic, and immunogenic characteristics.18 A striking case of how inter-strain glycome variability gives rise to significant phenotypic differences is the correlation of hypervirulence of the M. tuberculosis Beijing family with the presence of a unique phenolic glycolipid in its cell envelope.19 Intra-strain glycome variability can be considered to arise from microheterogeneity—i.e., small differences in structure originating from the imperfect, non-templated biosynthesis of glycans—as well as from specific glycan modification and remodeling processes that occur in response to different growth conditions or other external stimuli, such as starvation, desiccation, or immune response from the host. For instance, a battery of enzymes is involved in PG structural remodeling, which confers an adaptive capacity to numerous species and facilitates their survival and proliferation in a wide range of settings.20 Thus, the bacterial glycome is not only extraordinarily diverse, but it is also highly dynamic, capable of responding to environmental changes.
1.2. Importance of bacterial glycans to human health and biotechnology
Investigating the diverse and dynamic glycomes of bacteria has important implications for basic research, biotechnology, and medical applications. Fundamental aspects of glycobiology can be better understood through experimentation with bacterial model organisms. Well-characterized and genetically tractable model organisms such as Escherichia coli can be used to study not only bacteria-specific structures such as PG and LPS, but also to study broader topics in glycobiology, such as carbohydrate metabolism, glycosylation, and structure–function relationships of glycans. Also, efforts to define glycomes across the bacterial domain help to reveal the extent of glycan structural conservation and diversity in nature. This point is exemplified by the relatively recent discovery of protein N-glycosylation in some bacteria, including Campylobacter jejuni, whose N-glycan pathways and structures bear resemblance to those of eukaryotes while also exhibiting distinctive features such as a unique bacillosamine linkage to the protein backbone.13 Such findings can aid in understanding how glycan diversity originated from an evolutionary standpoint. Beyond such basic research pursuits, understanding bacterial glycans has relevance to many other areas, including in the environmental, agricultural, and biotechnological sectors. For example, bacteria are being engineered into living reaction vessels to produce therapeutic glycoproteins, glyconanomaterials for drug delivery, and probiotic polysaccharides, among other products.21–23
The most compelling reason to advance the field of bacterial glycobiology is that bacteria are inextricably linked to human health, both as beneficial members of the human microbiota and as disease-causing pathogens. Under normal conditions, symbiotic bacteria such as those in the Bacteroides, Clostridium, and Lactobacillus genera have mutually beneficial relationships with the human host and contribute to diverse processes involving glycans, including digestion of dietary nutrients, development of the immune system, and prevention of adhesion and colonization by pathogens.24 A reduction in beneficial microbial diversity, termed dysbiosis, has been implicated in various diseases, such as colitis and colon cancer, and can promote the emergence of pathogens. Therefore, understanding the interactions between microbial glycans and host components—and vice versa—within a complex microbiota is an important objective. Numerous other bacteria are dedicated human pathogens that cause a variety of devastating diseases, including tuberculosis, pneumonia, diarrhea, listeriosis, and cholera, to name only a few. The development of new therapeutics and diagnostics to confront bacterial infections is an urgent need, especially in light of the concurrent rise of drug resistance and decline of antibiotic development that has taken place in the past several decades.25 Two examples of antimicrobial resistance that have a major impact on global health are methicillin-resistant Staphylococcus aureus (MRSA), which kills thousands of people annually in the United States alone, and multidrug- and extensively drug-resistant tuberculosis, which account for 5% of the 10 million annual cases of tuberculosis and have high case fatality rates.2,26 All bacterial pathogens possess unique glycans that are essential for viability and virulence, including many that play critical roles in mediating host–pathogen interactions. Therefore, it is not surprising that glycan-targeting antibiotics, diagnostics, and vaccines have been successfully used to combat bacterial infections in the clinic. Most significantly, inhibition of PG biosynthesis by antibiotics such as penicillin and vancomycin is the cornerstone of broad-spectrum treatment of bacterial infections.27 Similarly, several antimycobacterial compounds that are used to treat tuberculosis act on glycan-related biosynthetic pathways.11,28 In addition, success has been achieved in the area of carbohydrate-based antibacterial vaccines, which have been used in the clinic to prevent infections from pathogens such as Streptococcus pneumoniae, Neisseria meningitidis, Haemophilus influenzae, and Salmonella typhi.29,30 Glycans have also been exploited to aid in the serodiagnosis of some bacterial infections based on the presence of host antibodies generated against species- or strain-specific cell-surface glycan structures.31
Despite significant progress in understanding the roles of glycans in bacterial symbionts and pathogens, and, in the latter case, exploiting this information to improve the prevention, diagnosis, and treatment of infections, significant challenges remain. One problem, which stems from the complexity and diversity of bacterial glycans, is that the glycomes of most bacterial species are simply not well defined. In part, this is because structural determination of glycans and their conjugates is a resource- and time-intensive endeavor involving laborious separations and painstaking analysis using a combination of NMR, mass spectrometry, and other sophisticated analytical tools. Even in cases where glycan structures are known, the pathways responsible for their biosynthesis, transport, remodeling, degradation, and recycling are often not fully—or even partially—elucidated. Of course, limited knowledge of glycan structure and biosynthesis makes the determination of glycan function extremely difficult. Furthermore, the investigation of bacterial glycans is not only technically difficult, but it is typically done using cultures grown in the laboratory under conditions that do not faithfully recapitulate the host environment. Therefore, it is particularly challenging to study glycan structure, dynamics, and function in disease-relevant contexts. Ultimately, these gaps in knowledge of bacterial glycobiology impede the development of new glycan-targeting therapeutics and diagnostics.
1.3. Experimental approaches used to investigate bacterial glycans
Our relatively limited understanding of bacterial glycan structure, biosynthesis, and function is partly due to a historical scarcity of convenient and accessible tools for detecting, manipulating, and profiling bacterial glycans in biologically relevant contexts.16,32,33 Traditional genetic tools, such as gene knockout mutants, have been valuable for helping to elucidate and study the functions of various glycan biosynthetic pathways in bacterial model organisms. However, genetic approaches have limitations. Perhaps most importantly, because the biosynthesis of glycans is not template-driven, their structures cannot be directly tagged or precisely manipulated using genetic methods. Furthermore, many glycans are essential for bacterial viability, which precludes the generation of traditional knockout strains. Finally, there are many bacterial species that are simply not amenable to genetic manipulation in a laboratory. Another useful resource for studying glycans is carbohydrate-binding proteins, or lectins, which have been used to facilitate the detection and profiling of certain types of bacterial glycans, namely glycoproteins. However, lectin binding specificity is often quite broad, and most bacteria-specific glycans do not have known cognate lectins, let alone commercially available lectin conjugates that can be used for glycan detection or enrichment applications. Small-molecule inhibitors of glycan biosynthesis that can manipulate structure in a predictable manner can be used to probe structure–function relationships in cells. However, well-characterized inhibitors are scarce relative to the copious and diverse glycosyltransferases, glycoside hydrolases, and glycan-modifying enzymes in bacteria. In addition, inhibitors generally cannot aid in detecting or profiling glycans unless they are suitably modified, and their mode of action involves direct binding to the glycan structure (e.g., fluorescent vancomycin, which binds to the PG cross-linking peptide34).
Metabolic labeling is an important technique for studying bacterial carbohydrate metabolism and glycan biosynthesis. Classic metabolic labeling experiments involve the use of stable isotope-labeled (e.g. 13C) or radiolabeled (e.g., 14C or 3H) sugars that can undergo incorporation into glycans of interest and facilitate downstream analysis. However, metabolic labeling with stable or radioactive isotopes has disadvantages, which may include cost, tedious experimental procedures, specialized instrumentation and data analysis, safety concerns, generation of hazardous waste, and a severely limited ability to provide information about glycans in intact cells. In recent years, the metabolic labeling concept has been powerfully extended by advances in chemical biology, namely the rapid rise of bioorthogonal chemistry and the ever-expanding repertoire of fluorescence-based reagents and analytical techniques. In the next section, we discuss how these advances have opened the door to the development of flexible and convenient platforms for precise chemical tagging, visualization, and manipulation of bacterial glycans in their native environments, thus addressing a major deficiency in the bacterial glycobiology toolbox.
1.4. Chemical reporters for investigating and targeting bacterial glycans
The field of bioorthogonal chemistry, which seeks to develop and utilize reactions that occur in living systems with minimal perturbation, has enabled the development of powerful new approaches to modifying and probing biomolecules in complex biological environments. The bioorthogonal chemical reporter strategy—pioneered by the Bertozzi lab—has been particularly valuable, finding widespread use in the study of nucleic acids, proteins, glycans, lipids, and metabolites in living cells, tissues, and whole animals.35,36 While bioorthogonal chemical techniques have been applied swiftly in mammalian systems over the past two decades—especially in the field of glycobiology37–39—their use in bacteria has been slower, despite high potential for accelerating basic and applied bacteriology. In this section of the introduction, we describe the bioorthogonal chemical reporter strategy, the bioorthogonal reactions that enable it, and how chemical reporters are developed and applied. We then provide a short preview of how chemical reporters can be used to advance the field of bacterial glycobiology, prior to a comprehensive review of work that has been done in the field.
The bioorthogonal chemical reporter strategy is depicted in the context of investigating bacterial glycans in Figure 2.40 Chemical reporters are unnatural versions of metabolites containing a small, abiotic functional group, generically defined herein as X, that can undergo selective bioorthogonal reaction (the chemical reporter is shown as an X-modified sugar derivative in Figure 2). When administered to a living cell or organism, chemical reporters are incorporated into target biomolecules through native—or in some cases engineered—cellular metabolic pathways, provided that the targeted pathways have the requisite tolerance for modified substrates. Because the X group is typically very small, metabolic incorporation can be achieved in many cases, especially in the case of glycan biosynthesis, which often exhibits high substrate permissiveness. Once the target biomolecule has been modified with an X group, it can undergo bioorthogonal reaction with a complementary functional group Y containing chemical cargo (shown as a yellow star in Figure 2). Commonly, the bioorthogonal reaction step is used to deliver fluorescence or affinity tags to cell-surface biomolecules to permit imaging or affinity enrichment and profiling. However, in principle, the high versatility of the chemical reporter strategy provides the opportunity to deliver any type of cargo for a variety of applications. In a similar metabolic labeling approach, unnatural sugars modified with fluorophores can be directly introduced into a living system to allow one-step metabolic labeling and subsequent visualization of glycans. While it is certainly preferable in some scenarios to use one-step instead of two-step labeling, small-molecule fluorescent reporters also have disadvantages to consider. For example, the large size of the fluorophore may limit metabolic incorporation efficiency and the design is inherently inflexible, as fluorescent probes are limited to imaging applications and altering the fluorophore requires that a new sugar derivative be synthesized and evaluated—these issues are circumvented with the two-step bioorthogonal approach. In this review, we cover both bioorthogonal and fluorescent small-molecule reporters that have been developed for metabolic labeling of bacterial glycans.
Figure 2.
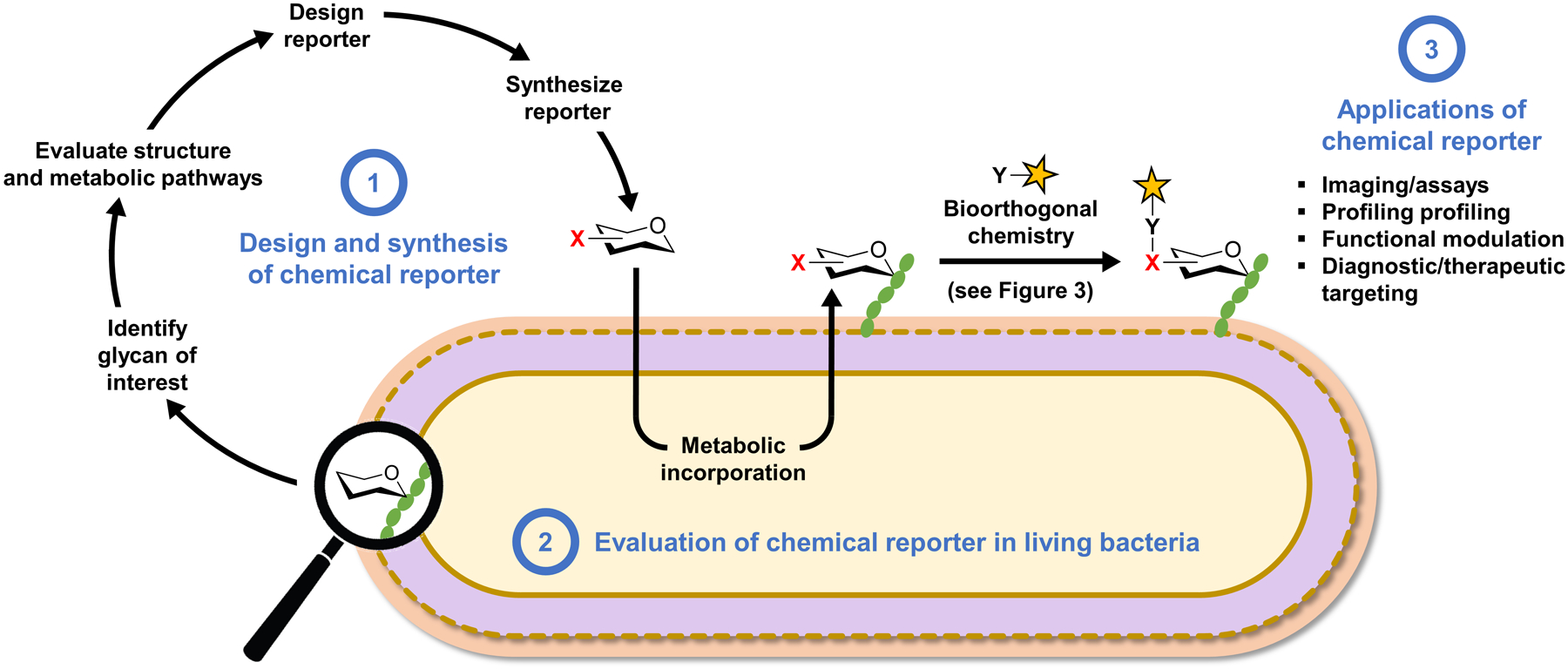
Overview of the bioorthogonal chemical reporter strategy as applied to studying bacterial glycans and workflow for the development of chemical reporters. The chemical reporter (shown as a hexose ring with a bioorthogonal functional group X) is fed to a living bacterial cell and metabolically incorporated into a glycan of interest. Next, the X-labeled glycan is reacted with an exogenously delivered reagent (e.g., a fluorophore) containing a complementary bioorthogonal functional group Y. A highly selective bioorthogonal reaction takes place on the cell surface between X and Y, leading to covalent ligation of the delivered cargo to the glycan of interest, enabling its analysis or modulation. See Figure 3 for common bioorthogonal reactions. In the one-step metabolic incorporation approach, X is typically a fluorophore and does not require a subsequent bioorthogonal reaction. The development of chemical reporters involves three stages (highlighted in blue text): (1) the reporter molecule must be designed and synthesized after selection of the target glycan and analysis of its structure and biosynthesis; (2) next, the reporter must be tested in bacterial cells to determine whether it successfully labels the glycan of interest and to elucidate the pathway of incorporation; (3) once the reporter’s behavior in cells is established, it can potentially be used for a variety of applications, ranging from live-cell imaging of glycan biosynthesis and dynamics to targeting the bacterium with therapeutic or diagnostic cargo within a host.
The foundation of the chemical reporter strategy is the small but growing repertoire of exceptionally selective bioorthogonal reactions. Because these reactions have been reviewed extensively elsewhere,35,36,41 the most common of them are only briefly introduced here (Figure 3). Early examples of chemical reporters contained carbonyl groups (e.g., X = ketone or aldehyde) that could undergo selective condensation with α-effect nucleophile-based reagents (e.g., Y = aminooxy or hydrazide) (Figure 3A).42,43 This chemistry is infrequently used now because it is relatively slow, occurs optimally under acidic conditions, and can be complicated by the presence of competing biological electrophiles. To date, the reactions used for the large majority of chemical reporter strategies are those involving the prototypical bioorthogonal functional groups—azides and alkynes. The Staudinger–Bertozzi ligation, which is widely viewed as the first truly bioorthogonal reaction, is an extremely selective reaction that takes place between azides and triarylphosphine reagents (Figure 3B), but it is relatively slow and has largely been supplanted by faster chemistries.44 The most commonly used bioorthogonal reactions are the so-called “click” reactions: Cu-catalyzed azide–alkyne [3+2] cycloaddition (CuAAC) between azides and terminal alkynes (Figure 3C), which was developed by the Sharpless and Meldal groups,45,46 and the Cu-free, strain-promoted azide–alkyne [3+2] cycloaddition (SPAAC) between azides and cycloalkynes (Figure 3D), which was developed by the Bertozzi group to avoid cytotoxic copper reagents and simplify the click reaction conditions.47 Both reactions have found widespread use in bioorthogonal labeling applications, especially now that numerous CuAAC and SPAAC reagents are comercially available. In addition, CuAAC conditions with reduced toxicity in living systems have been developed.48,49 A more recent development is the use of chemical reporters bearing strained alkenes (e.g., X = trans-cyclooctene or the smaller cyclopropene groups), which can react with tetrazine-based reagents very rapidly (Figure 3E).50,51 This reaction, called the tetrazine ligation, will likely find more widespread use in the future for studying bacterial glycans, particularly in in vivo settings given its desirable reaction kinetics, but its use in this arena has been fairly limited so far. As discussed extensively in a review by Prescher and colleagues, the selection of a bioorthogonal reaction that is appropriately matched to a desired downstream application should take into account a number of factors, including reaction kinetics and selectivity, biocompatibility, functional group size and stability, and whether reagents are commercially available and/or synthetically accessible.41
Figure 3.
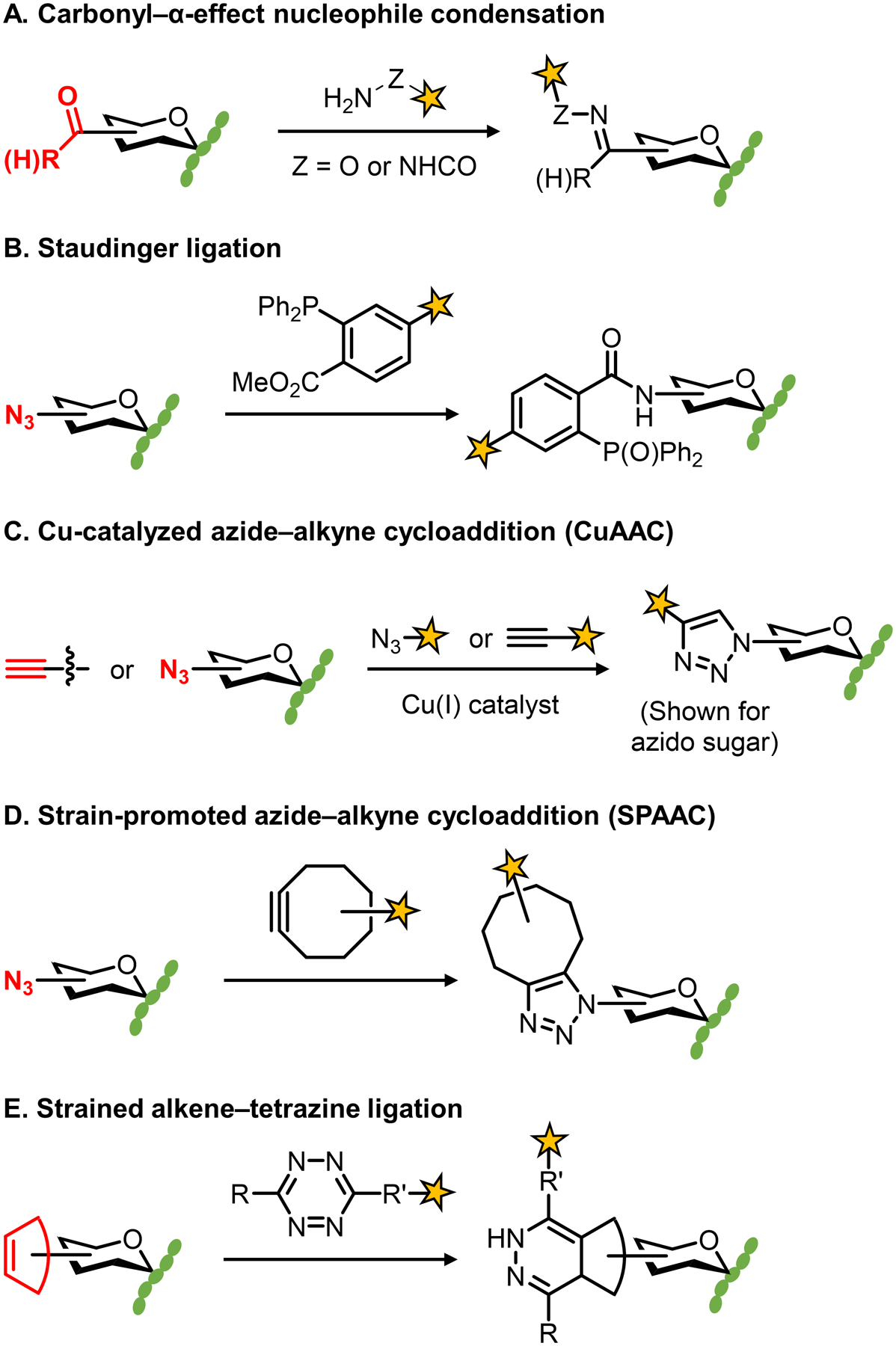
Commonly used bioorthogonal reactions.
The development of bioorthogonal chemical reporters can be viewed as a three-stage process that involves several important considerations (Figure 2). First, the chemical reporter must be designed and synthesized (Figure 2, stage 1). In most cases, the motivation to develop a chemical reporter stems from the desire to create a tool that will facilitate the study or targeting of a specific biomolecule. Once this biomolecule of interest is identified, careful analysis of the literature relating to its structure, biosynthesis, and cellular location should be carried out to inform the optimal design of the chemical reporter. As examples, such an analysis can provide insight into possible pathways for reporter uptake and metabolic incorporation; reporter structures with higher probability of incorporation and lower probability of biological perturbation; what type of bioorthogonal reaction might be best suited for labeling a particular biomolecule; and whether a one-step or two-step metabolic labeling approach, or both, should be pursued. In addition, this step may help to determine whether pathway engineering is required to permit or increase the efficiency of metabolic incorporation of the reporter. As we will see throughout this review, it is difficult to identify clear-cut guidelines for reporter design and a case-by-case approach should be taken. However, unprotected azide-modified metabolic precursors (e.g., monosaccharides) that can be detected by CuAAC or SPAAC have often served as fruitful starting points. After designing the reporter’s structure, it must be synthesized using chemical or chemoenzymatic methods to efficiently install the X group at the desired position. In the case of some reporter probes, namely unnatural sugars, synthesis is often not a trivial task and can be the bottleneck to evaluation and optimization, as exemplified through selected examples in this review article.
With the chemical reporter synthesized, the next stage of development is its evaluation in the biological system of interest (Figure 2, stage 2). This is initiated by performing labeling experiments, in which cells are incubated in the presence of the X-modified reporter, reacted with a Y-modified fluorophore (omitted in the one-step approach with fluorescent probes), and then analyzed by fluorescence microscopy or flow cytometry. Adjustments to reporter concentration, incubation time, and bioorthogonal reaction conditions are often needed to optimize labeling efficiency. Of course, the appearance of cellular fluorescence in reporter-treated versus -untreated cells does not necessarily mean that the target biomolecule has been labeled, nor that the biomolecule was labeled via the expected metabolic pathway. Establishing a reporter’s labeling target and route of incorporation is challenging, but critical to informing proper probe usage and interpretation of results. A variety of experimental approaches are helpful in accomplishing this, including, but not limited to, competition with the natural/unlabeled metabolite, specific chemical/enzymatic degradation, extraction and analysis of the labeled biomolecule (e.g., by mass spectrometry or SDS-PAGE), and analysis of labeling in appropriate mutant strains or in the presence of appropriate small-molecule inhibitors.
After the evaluation stage has provided an accurate understanding of the reporter’s behavior in the system being studied, then the third stage of applying the reporter can commence (Figure 2, stage 3). As mentioned above, the most common applications for chemical reporters are biomolecular imaging and profiling, which have been particularly fruitful in the field of mammalian glycobiology.37–39 Numerous monosaccharide-based chemical reporters, many utilizing azides or alkynes as X groups, have been developed for metabolic labeling of various types of mammalian glycans.37–39 These reporters have enabled imaging studies that shed light on the expression and distribution of mammalian glycans in time and space, in living cells and animals. Furthermore, by exploiting the versatility of bioorthogonal chemistry to append affinity tags to labeled glycans, it has been possible to enrich, identify, and profile proteins that are post-translationally modified by or non-covalently interact with specific types of glycans (e.g., sialylated or O-GlcNAcylated proteins). These tools have also been used to better understand how glycomes change in different environments and how they differ in healthy and diseased states. Such studies can potentially lead to the identification of novel glycan-containing disease biomarkers.
Existing chemical reporters for bacterial glycans, despite being relatively new compared to those for mammalian glycans, have already shown high potential to help drive forward the field of bacterial glycobiology. Reporters for PG, LPS, teichoic acids, mycobacterial glycans, and certain classes of glycoproteins and polysaccharides have been developed and used in a range of bacterial species. This toolbox, when further expanded and combined with complementary experimental techniques, will help to address many of the outstanding knowledge and technology gaps in the field (discussed above in sections 1.2 and 1.3). Whole-cell imaging of bacterial glycans using chemical reporters can shed light on the dynamics of their biosynthesis, transport, and remodeling in various contexts (e.g. in culture or within host systems). Siegrist and co-workers reviewed how reporters aid in understanding these processes as they unfold during bacterial cell growth and division.52 In addition, the discovery and profiling of bacterial glycans using chemical reporters will assist with the elucidation of species- and strain-specific bacterial glycomes, as well as contribute tools for investigating changes to dynamic bacterial glycomes induced by altering the bacterium’s environment. This topic was introduced in a review by the Dube group focused on the use of chemical approaches to discover and profile bacterial glycoproteins.53 In addition to imaging and profiling, chemical reporters provide exciting new opportunities for delivering diagnostic or therapeutic cargo specifically to pathogenic bacteria within an infected host, which is an especially attractive strategy due to the presence of numerous distinctive glycans in bacteria that are absent from mammals. This concept of modifying the bacterial cell surface for biomedical applications has been covered in reviews by the Spiegel and Dube groups, with the latter focusing specifically on the targeting of pathogen-specific bacterial glycans.54,55 Other recent reviews have also highlighted how various chemical biology approaches, including the use of metabolic labeling and bioorthogonal chemistry, can be enlisted to advance the broader bacteriology field.16,32,33,56–59
The purpose of this review is to comprehensively examine the development and applications of metabolic reporters for bacterial glycans. Through this examination, we emphasize and intend to illustrate the process of developing these tools. Thus, in contrast to the excellent reviews cited above, which primarily touch on the different applications of metabolic labeling in bacterial systems, the present review provides an in-depth perspective on the creation of chemical reporters and how they operate from a mechanistic standpoint, which should be useful to chemical biologists who develop tools and microbiologists who deploy them. This review is organized by glycan type and follows the order of PG, LPS, glycoproteins, teichoic acids, mycobacterial glycans, and capsular polysaccharides. Figure 2 represents a visual guide as to how chemical reporters for each glycan type are covered herein. We introduce the biological significance of the glycan of interest and the rationale for creating chemical reporters for it. We discuss the structure and biosynthesis of the glycan, underscoring how this information informs the design of suitable reporter probes. In many cases, we describe the synthesis of the probes, with the dual goals of imparting the importance of synthesis considerations in this field and showcasing the development of new methods for synthesizing unique bacterial sugars and their derivatives. We discuss the experiments performed to evaluate the behavior of chemical reporters in bacterial species of interest, and we highlight key data that support the proposed mechanism of incorporation and labeling target. Finally, we explore how each tool has been applied to drive forward our understanding of bacterial glycobiology and/or adapted to create novel strategies for detecting and treating bacterial infections. We conclude the review by recapping important trends that have emerged and discussing opportunities and priorities for future research in this field.
2. Chemical reporters for peptidoglycan (PG)
PG, also referred to as murein, is an interwoven glycan–peptide meshwork that surrounds the entirety of virtually all bacterial cells. PG is strong and rigid, giving the bacterial cell its shape and providing a barrier that protects it from various stresses. The glycan component of the structure comprises a disaccharide repeating unit made up of β(1→4)-linked N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc); these glycan strands are modified at the MurNAc D-lactyl (D-Lac) group by D-amino acid-containing stem peptides that are covalently cross-linked (Figure 4).6,60 Numerous variations to this core structure have been documented, including in the glycan portion (e.g., N-deacetylation, O-acetylation, and N-glycolylation) and the oligopeptide cross-link portion (e.g., variability in residues and linkage positions), plus PG serves as an anchoring point for other envelope components (e.g., proteins and teichoic acids).6 Furthermore, PG is a highly dynamic structure that can be remodeled, broken down, and recycled in an environment-responsive manner; for instance, liberated PG fragments can signal intra- and extracellularly to promote bacterial survival.20,61 PG is conserved in and essential to bacteria, and therefore disruptions in its biosynthesis prevent growth and eventually lead to cell lysis. This fact, coupled with the absence of PG in humans, has rendered it the quintessential target for broad-spectrum clinical antibiotics, such as β-lactams, vancomycin, and bacitracin, among other scaffolds.27 However, as bacteria have rapidly evolved resistance mechanisms to these and other cornerstone antibiotics, it has become increasingly urgent to identify novel targets and chemical scaffolds for antibiotic development.62 To support these critical pursuits, an array of tools are being developed to facilitate the study of PG biosynthesis and maintenance. As several excellent recent reviews have provided perspectives on, these tools range from small molecule probes, such as functionalized antibiotics and metabolic substrates, to new techniques for the isolation and spectroscopic analysis of PG biosynthetic intermediates.32,52,58,63–66 Here, we focus our discussion on the development of small-molecule reporter probes that metabolically label different portions of the PG structure, and we highlight how these tools have been applied to understand and exploit bacterial PG construction.
Figure 4.
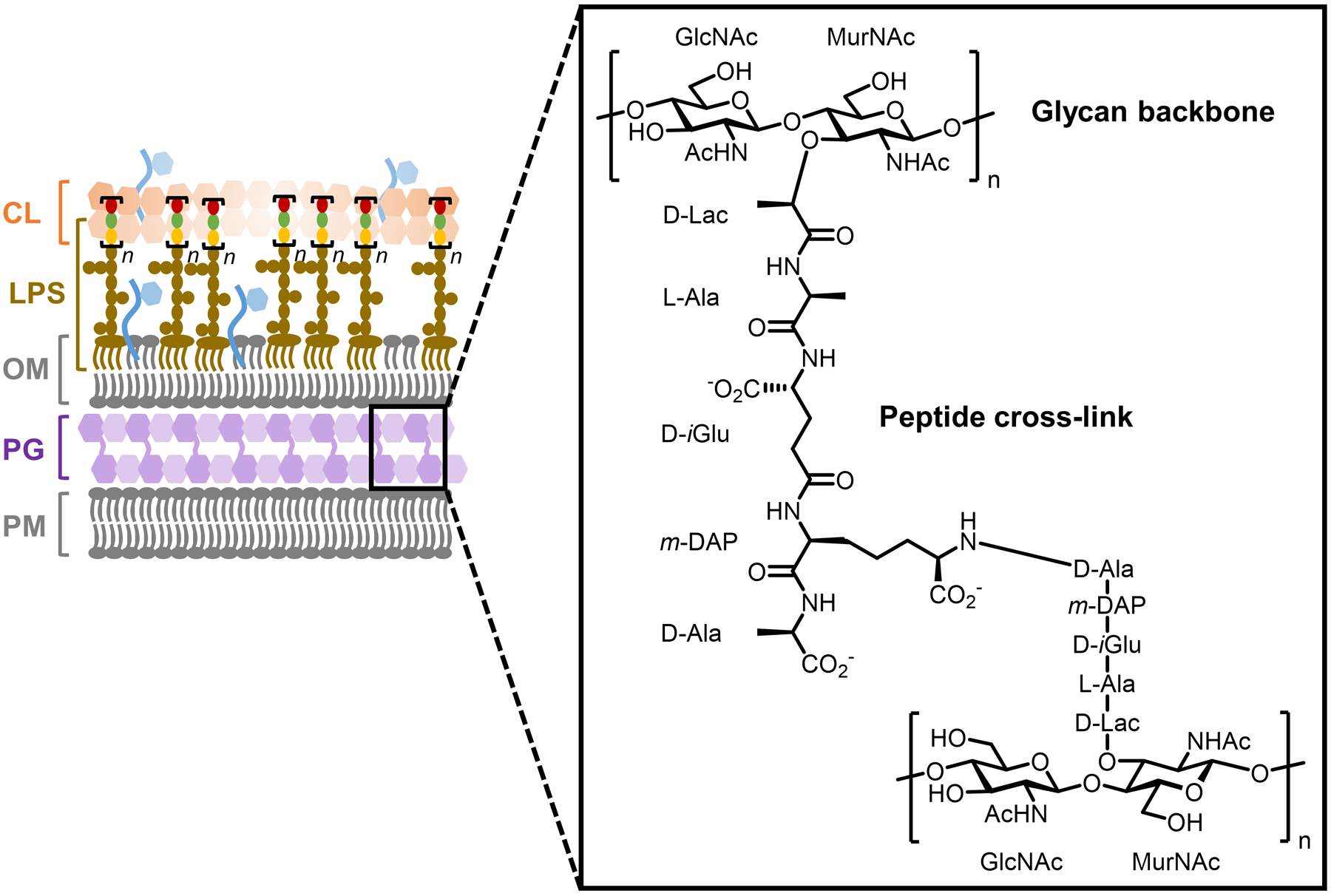
Representative PG structure from Gram-negative bacteria. PG is also present in Gram-positive bacteria and mycobacteria.
Reporter probes designed to label different components of PG were inspired by the continually increasing knowledge of PG biosynthesis, which is depicted in simplified form in Figure 5. PG biosynthesis is initiated by Mur enzymes in the cytoplasm,67,68 where the initial step is addition of a D-Lac group to the UDP-GlcNAc 3-O-position, generating UDP-MurNAc. The D-Lac group is extended with 3 additional amino acids that may vary based on the species; in Gram-negative bacteria, these residues are usually L-Ala, D-isoglutamate (D-iGlu), and meso-diaminopimelic acid (m-DAP).6 Further extension of the stem peptide with the dipeptide D-Ala-D-Ala forms UDP-MurNAc-pentapeptide, also known as Park’s nucleotide. D-Ala-D-Ala originates in the cytoplasm through alanine racemase (Alr)-catalyzed stereochemical inversion of L-Ala to D-Ala, followed by dipeptide ligase (Ddl)-catalyzed joining of two D-Ala molecules. Park’s nucleotide, a soluble cytoplasmic nucleotide sugar, is then transferred onto the C55 lipid carrier undecaprenyl phosphate (UP) by MraY, generating membrane-anchored Lipid I on the cytoplasmic face of the plasma membrane. Next, the glycosyltransferase MurG catalyzes the transfer of a GlcNAc residue from UDP-GlcNAc onto Lipid I’s MurNAc 4-O-position, forming Lipid II, which is subsequently translocated across the plasma membrane by the flippase MurJ.69 Once in the periplasm, membrane-anchored Lipid II serves as the key PG building block, as it is used to elongate existing PG through two essential processes mediated mainly by penicillin-binding proteins (PBPs) called transglycosylation and transpeptidation.70 The former process enlists PBP glycosyltransferases (GTases) to extend the glycan strand by polymerizing Lipid II, thus releasing undecaprenol pyrophosphate (UPP); the latter mainly enlists PBP D,D-transpeptidases (TPases) to generate 3–4 cross-links between stem peptide D-Ala and m-DAP residues of nearby glycan strands, in the process releasing a terminal D-Ala residue. In addition, PBP carboxypeptidases (CPases) can cleave the terminal D-Ala of the stem peptide, which affects the degree of PG cross-linking.20 Two other classes of proteins, the SEDS (shape, elongation, division, and sporulation) proteins and the L,D-TPases, are also known to be involved in PG transglycosylation and transpeptidation reactions, respectively.71–73 L,D-TPases generate 3–3 cross-links between two m-DAP residues, a structure which is common in certain organisms, such as mycobacteria.74 As noted above, there are also an array of enzymes that are involved in PG remodeling or break down, including endopeptidases, amidases, and lytic transglycosylases (not shown in detail in Figure 5).20
Figure 5.
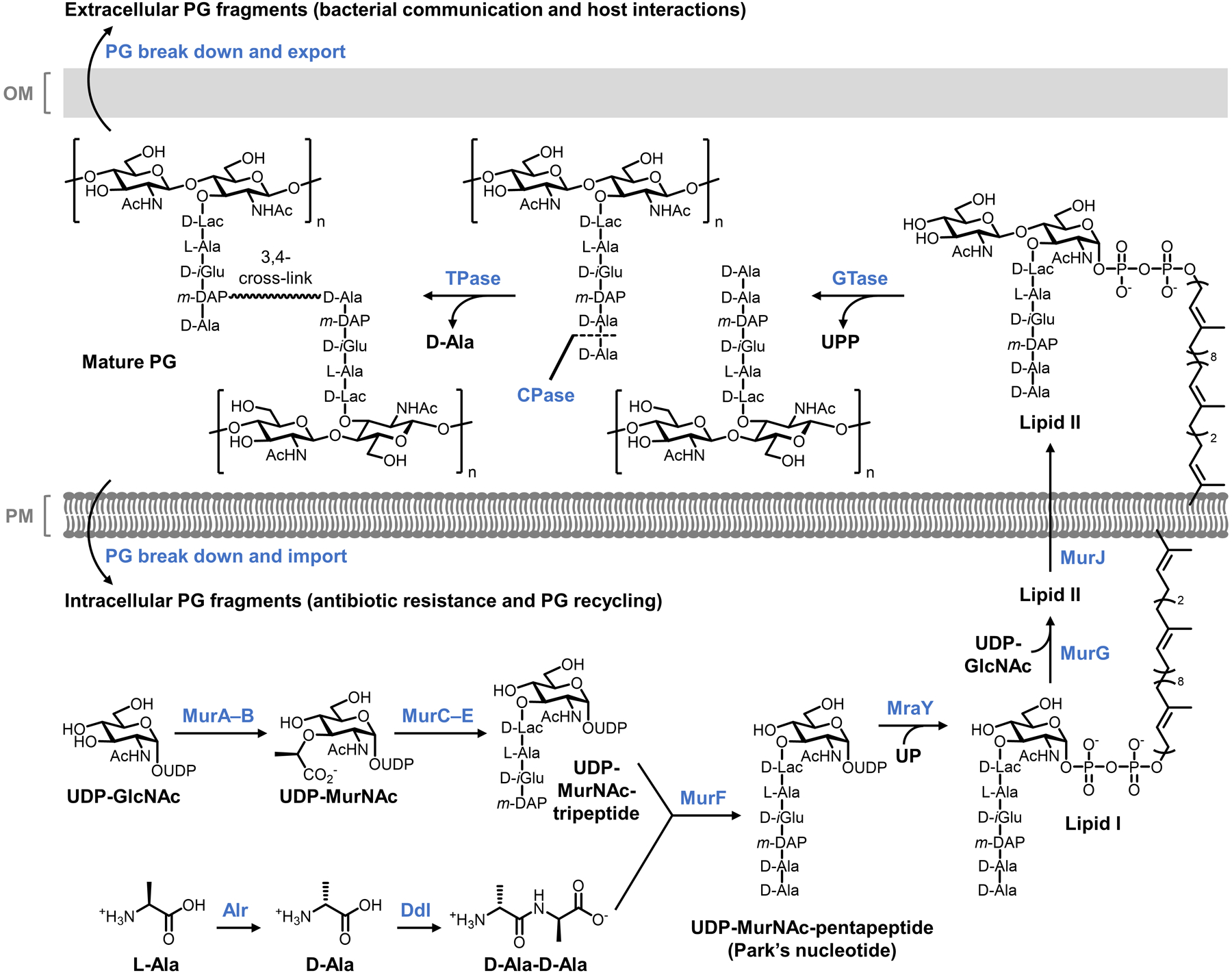
Representative biosynthesis of PG in Gram-negative bacteria.
The elucidation of these PG biosynthesis and maintenance pathways served as the foundation for the design and development of PG-targeting reporter probes. There are multiple points in PG biosynthesis that could potentially be intercepted by reporter probes to metabolically label PG, including cytoplasmic amino acid and carbohydrate metabolism, synthesis of PG precursors (e.g., Park’s nucleotide, Lipid I, and Lipid II), periplasmic assembly of PG precursors into mature PG, and periplasmic remodeling of PG. Various considerations are in play, such as passage of exogenously added reporter probes across membranes, substrate tolerance of enzymes (e.g., Murs and PBPs), the presence of enzymes that might remove the installed reporter group (e.g., CPases and lytic transglycosylases), and the feasibility of synthesizing appropriately designed reporters. Over the past two decades, these and other considerations have been explored extensively. As a result, the field has produced numerous small-molecule chemical and fluorescent reporters for PG that are providing new insights into the structure, dynamics, and function of the bacterial cell wall. We divide our discussion of PG reporter development and applications into two categories: those that target unnatural chemical functionality to the PG stem peptide portion, such as the widely used D-amino acid-based probes, and those that complementarily target chemical functionality to the PG glycan core, such as the recently reported MurNAc-based probes. Table 1 contains information about selected PG-targeting chemical reporters.
Table 1.
Selected chemical reporters for peptidoglycan (PG)
| Target | Metabolite | Compound | Reporter group (X) | Bioorthogonal reaction(s)a | Proposed route | Speciesb | Applicationsc | Synthesis | Notesb | Refd |
|---|---|---|---|---|---|---|---|---|---|---|
| PG stem peptide | Park’s nucleotide | 1 (Fig. 6) | Ketone | Carbonyl–α-effect nucleophile | Intracellulare | Lactic acid species (GP) | In vitro imaging; modulation of adhesion | Chemo-enzymatic | Not tested in GN bacteria | 75 |
| Park’s nucleotide | 2 (Fig. 6) | Fluorescein | -- | Intracellulare | E. coli (GN), lactic acid species (GP) | Cell surface detection | Chemo-enzymatic | EDTA required for labeling GN | 75 | |
| D-amino acid | AlkDA 10 (Fig. 7) | Alkyne | CuAAC | Intracellular, extracellular | Broad (GP, GN, M) | In vitro and in vivo imaging (infected macrophages); PG stapling | Chemical; comm. available | Route of incorporation may be mixed, species-dependent | 76,77 | |
| D-amino acid | AzDA 11 (Fig. 7) | Azide | CuAAC; SPAAC | Intracellular, extracellular | Broad (GP, GN, M) | In vitro imaging; selective killing of MRSA | Chemical; comm. available | Route of incorporation may be mixed, species-dependent | 76,77 | |
| D-amino acid | OctDA 12 (Fig. 7) | Cyclooctyne | SPAAC | Extracellular | Broad (GP, GN, M) | In vitro imaging | Chemical | Reacts with fluorogenic azide dye via SPAAC | 78 | |
| D-amino acid | NADA 16 (Fig. 7) | 7-Nitrobenzo-furazan | -- | Extracellular | Broad (GP, GN, M) | In vitro imaging | Chemical; comm. available | Reports on TPase-mediated PG remodeling | 76 | |
| D-amino acid | HADA 17 (Fig. 7) | 7-Hydroxy-coumarin | -- | Intracellular, extracellular | Broad (GP, GN, M) | In vitro and in vivo imaging (colonized host organism) | Chemical; comm. available | Route of incorporation may be mixed, species-dependent | 76 | |
| D-amino acid | Rf470DL 21 (Fig. 7) | Molecular rotor dye | -- | Extracellular | S. aureus (GP), S. venezuelae (GP), B. subtilis (GP) | Imaging; inhibitor characterization | Chemical | Fluorogenic; labels GN poorly | 79 | |
| D-amino carboxamide | TetDAC 25 (Fig. 10) | Tetrazine | Tetrazine ligation | Extracellular | S. aureus (GP) | In vivo imaging (infected host organism) | Chemical | D-amino carboxamide enhances labeling in some GP bacteria | 80,81 | |
| D-amino carboxamide | FDL-NH2 27 (Fig. 10) | Fluorescein | -- | Extracellular | B. subtilis (GP) | In vitro imaging | Chemical | D-amino carboxamide labels B. subtilis but not E. coli | 82 | |
| D-amino acid dipeptide | AlkDADA 28 (Fig. 11) | Alkyne | CuAAC | Intracellular | Broad (GP, GN, M) | In vitro and in vivo imaging (infected macrophages) | Chemical | Reports on MurF-initiated synthesis of new PG | 83 | |
| D-amino acid dipeptide | AzDADA 29 (Fig. 11) | Azide | CuAAC; SPAAC | Intracellular | Broad (GP, GN, M) | In vitro imaging | Chemical | Reports on MurF-initiated synthesis of new PG | 83 | |
| D-amino acid dipeptide | x-DADA-Alk 32 (Fig. 11) | Alkyne and phenylazide | CuAAC | Intracellular | B. subtilis (GP) | Detection of PG precursor–protein interactions | Chemical | Phenylazide allows protein photo-cross-linking | 84 | |
| Stem tripeptide | AeK-NBD 33 (Fig. 11) | 7-Nitrobenzo-furazan | -- | Intracellular | E. coli (GN) | In vitro imaging | Chemical | Reports on Mpl-mediated recycling route | 85 | |
| Stem pentapeptide | FSPPM-488 34 (Fig. 13) | Fluorescein | -- | Extracellular | S. aureus (GP) | In vitro imaging | Chemical | Reports on TPase-mediated PG cross-linking | 86 | |
| Stem tetrapeptide | FSTPM-488 36 (Fig. 13) | Fluorescein | -- | Extracellular | E. faecium (GN), M. smegmatis (M), M. tuberculosis (M) | In vitro imaging | Chemical | Reports on L,D-TPase-mediated PG cross-linking | 87 | |
| PG glycan core | GlcNAc-1-P | 38 (Fig. 14) | Ketone | Carbonyl–α-effect nucleophile | Intracellulare | L. plantarum (GP), W. confuse (GP) | -- | Chemical | Peracetylation required for incorporation | 88 |
| MurNAc | 42 (Fig. 15) | Alkyne | CuAAC | Intracellular | E. coli ΔMurQ-KU (GN), P. putida (GN), H. pylori HJH1 (GN), B. subtilis 3A38-KU (GP) | In vitro and in vivo imaging (infected macrophages) | Chemical | Requires AmgK/MurU; some species require fosfomycin; peracetylation abolishes labeling; D-Lac methyl ester enhances labeling | 89 |
Not applicable for fluorescent reporters operating via one-step incorporation (--).
Gram classification: GN, Gram-negative; GP, Gram-positive; M, mycobacteria.
In vitro refers to bacterial labeling experiments performed in broth culture; in vivo refers to bacterial labeling experiments performed in a host cell or organism.
Initial publication(s) referenced. See text for references for follow-up studies and applications.
Limited evidence available to support proposed route of incorporation and/or identity of labeled target (see text).
2.1. Chemical reporters for the PG stem peptide
2.1.1. Park’s nucleotide, Lipid I, and Lipid II derivatives.
Appropriately, we begin with pioneering work reported by Nishimura and co-workers in 2002, which was not only the first effort to create metabolic chemical reporters for PG, but, to our knowledge, the first effort to do so for bacterial glycans in general.75 The team designed a set of unnatural PG precursor glycopeptides (1–4) containing either a fluorophore, for one-step labeling, or a ketone chemical reporter, for two-step labeling in combination with α-effect nucleophile reagents (Figures 6A and 6B).75 The ketone–α-effect nucleophile reaction (Figure 3A), although no longer commonly used for cellular bioorthogonal labeling, was one of the only such chemistries available at the time.41 Compounds 1 and 2 are based on Park’s nucleotide (i.e., UDP-MurNAc-pentapeptide), whereas compounds 3 and 4 are based on Lipid I and Lipid II, respectively, both with the polyprenyl chain severely truncated from C55 to C9. According to the PG biosynthesis pathway (Figure 5), in order for compounds 1–3 to incorporate into mature PG, they would need to access the cytoplasm and be processed through MraY/MurG/MurJ followed by PBP-mediated transglycosylation and transpeptidation; compound 4, based on Lipid II, would need only to access the periplasm and be processed by PBPs. Because incorporation of these compounds was designed to occur at a “late” stage of the PG synthesis pathway, few enzymatic steps would be required for incorporation, and the compounds would provide a read-out of PBP-mediated PG assembly activity—a valuable characteristic for basic research and antibiotic development. However, since each compound is relatively large and bears multiple negative charges, there is also the corresponding concern of low membrane permeability and thus poor penetrance into the appropriate cellular compartment to achieve incorporation. All four compounds contained the fluorophore or ketone functionality at the m-DAP residue, meaning that, if incorporation were successful, the label would end up in the stem peptide portion of PG. Modification of the m-DAP residue may be beneficial during initial steps of incorporation since it is distal to the site of undecaprenyl phosphate addition and glycosidic bond formation. However, m-DAP modification would also preclude the labeled structures from serving as nucleophiles in PBP-mediated transpeptidation reactions, which could potentially perturb PG biosynthesis and cell wall properties. Another feature worth remarking on is the shortened prenyl tail in compounds 3 and 4, which on one hand may drastically improve synthetic feasibility and solubility in comparison to the native C55 tail, but on the other hand may decrease or prevent substrate processing by Mur and PBP proteins. The foregoing discussion demonstrates the careful analysis that must be undertaken in the design of chemical reporters, and underscores the tradeoffs inherent in many design choices.
Figure 6.
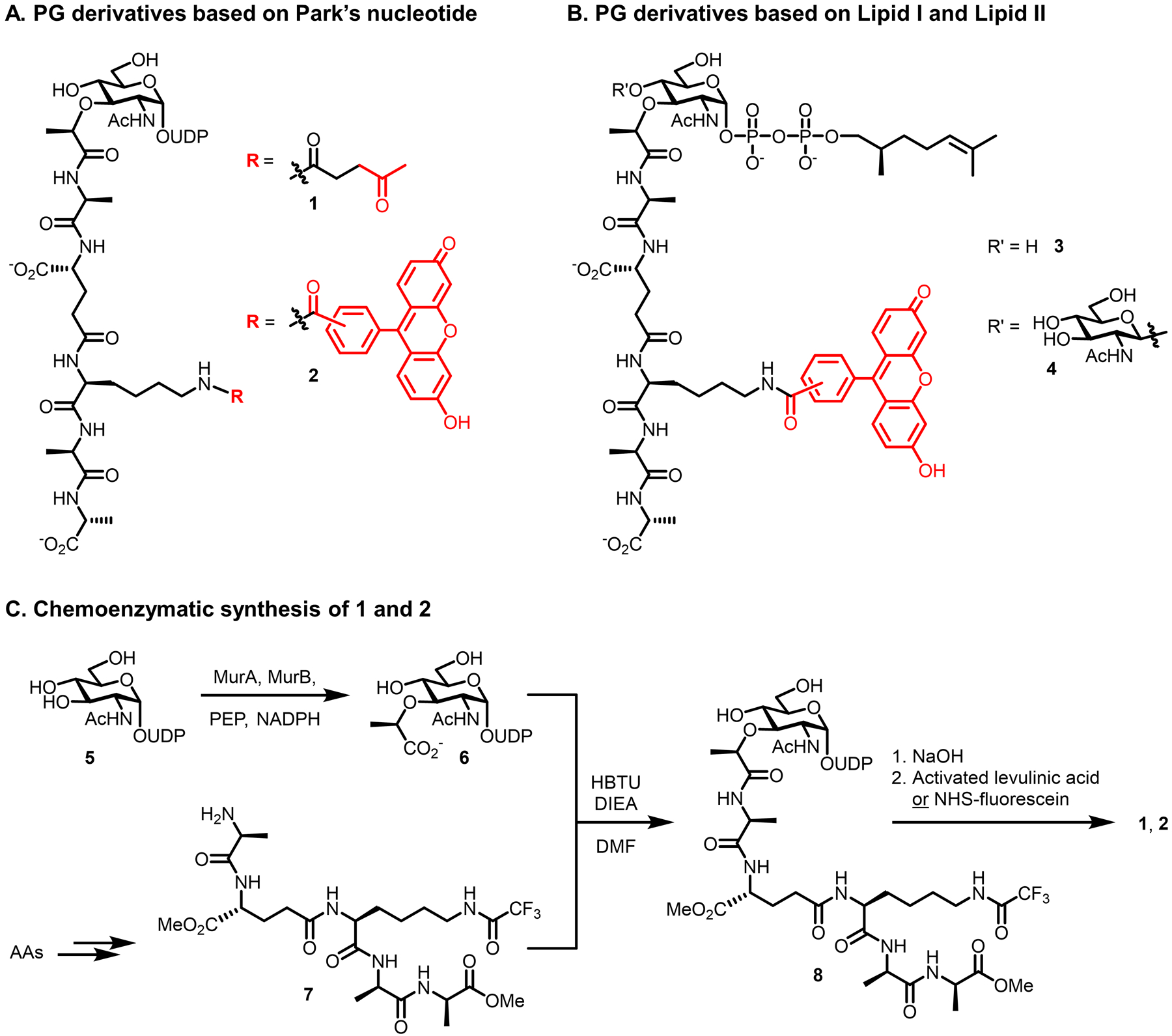
PG precursor derivatives designed to label the PG stem peptide based on (A) Park’s nucleotide and (B) Lipid I and II. (C) Convergent chemoenzymatic synthesis of PG precursor derivatives 1 and 2. AAs, amino acid building blocks; DIEA, N,N-diisopropylethylamine; DMF, N,N-dimethylformamide; HBTU, (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; NHS, N-hydroxysuccinimidyl; NADPH, nicotinamide adenine dinucleotide phosphate hydride; PEP, phosphoenolpyruvate; UDP, uridine diphosphate.
The size and complexity of glycopeptide derivatives 1–4 presented a challenge in terms of synthetic chemistry. Lipid I and II derivatives 3 and 4 were obtained via lengthy chemical synthesis routes utilizing orthogonally protected monosaccharide building blocks, with key steps including stereoselective glycosylation (for 4), anomeric phosphorylation, coupling to the stem peptide, prenyl phosphate addition, and modification of m-DAP by fluorescein (not shown).75 While an analogous chemical synthesis route was available for Park’s nucleotide derivatives 1 and 2, an alternative chemoenzymatic method developed by the Wong group90 was used to access these derivatives instead, delivering them with high efficiency. Figure 6C briefly depicts the more concise chemoenzymatic route, which involved MurA- and MurB-catalyzed conversion of UDP-GlcNAc (5) to UDP-MurNAc (6), followed by convergent chemoselective coupling of the UDP-MurNAc D-Lac residue to a synthetic pentapeptide (7), and finally deprotection and functionalization of the m-DAP residue with levulinic acid (to obtain 1) or fluorescein (to obtain 2).75 Since this original work, there has been substantial progress in the synthesis of PG precursors and their analogues. For example, the Wong group has greatly expanded upon the original chemoenzymatic synthesis of 1 and 2 to create fully enzymatic methods for accessing Park’s nucleotide, Lipid I, and Lipid II, as well as their corresponding m-DAP-labeled analogues.91 Furthermore, the Walker and Kahne groups recently reported a simple method for antibiotic-induced overproduction and isolation of Lipid II from S. aureus on large scale, as well as its subsequent chemoenzymatic labeling with biotin to enable detection.92,93 Thus, despite the complexity of PG precursor derivatives 1–4, there are now more efficient synthesis and isolation/modification approaches available to obtain them.
The characterization of bacterial labeling by fluorescein-modified PG precursor derivatives 2–4 was initially done in Gram-negative E. coli, whose PG structure inspired the compound design.75 The compounds were first tested by administering them to E. coli, extracting the cell wall-containing fraction, and measuring fluorescence by spectroscopy.75 Prior to incubation with the compounds, bacteria were pre-treated with metal chelator ethylenediaminetetraacetic acid (EDTA) to increase the permeability of the LPS-rich outer membrane and thereby enhance compound uptake.94 The Park’s nucleotide derivative, compound 2, led to increased fluorescence only when cells were first treated with EDTA, whereas the Lipid I and Lipid II derivatives 3 and 4 did not incorporate, regardless of whether EDTA pre-treatment was performed.75 It is possible that prenyl chain truncation was responsible for the lack of labeling by 3 and 4, since studies on lipid-linked sugar donors have indicated that glycosyltransferases prefer longer chains,95,96 a finding which has implications for the design of other lipid-linked carbohydrate chemical reporters as well (e.g., section 6.2.2). Compound 2 was further tested in Gram-positive lactic acid bacterial species, which lack an outer membrane, and these bacteria incorporated 2 without the need for EDTA.75 Lactic acid bacteria also incorporated the ketone-modified Park’s nucleotide derivative 1, which following incorporation was detected by reaction with a hydrazide-fluorophore conjugate and visualization by fluorescence microscopy.75
As noted in the introduction, the observation of fluorescence in bacterial cells or extracts does not necessarily indicate that the reporter probe was metabolically incorporated into the desired biomolecule via the targeted pathway. There is limited characterization data available for the PG precursor derivatives 1–4. Between the original report75 and a follow-up study97 on compounds 1–4, there were two experiments comparing incorporation of the probes to control compounds, the latter of which included (i) a version of Park’s nucleotide derivative 1 that lacked the terminal D-Ala-D-Ala moiety of the stem peptide and (ii) a version of disaccharide pentapeptide 4 having only a phosphate at the anomeric position. Neither of these control compounds incorporated into lactic acid bacteria, whereas the designed reporter compounds 1 and 2 did. The control compounds were not tested in Gram-negative bacteria. Another observation from the follow-up study was that incorporation of ketone-modified compound 1 was increased in the presence of MurA inhibitor fosfomycin.68,97 This result is consistent with the idea that MurA inhibition would be expected to deplete native PG precursors and lead to increased incorporation of exogenously added PG precursor analogues. However, while these data provide some support that compounds 1 and 2 behave as anticipated, to date there is no direct evidence that the derivatives are incorporated into PG products (e.g., by LC-MS) or that incorporation is metabolically driven (e.g., by killing/fixing cells), and there is limited information about the pathway of incorporation (e.g., by mutant analysis, competition experiments). Another consideration is the requirement of EDTA to achieve incorporation into Gram-negative bacteria, which is a concern because this reagent not only perturbs the outer membrane, but also leads to further cell damage and eventually lysis.94,98
Even in the absence of unambiguous evidence for the labeling target or mechanism of incorporation for these PG precursor derivatives, the confirmed ability of Park’s nucleotide derivative 1 to incorporate reactive ketones onto the cell surface of Gram-positive bacteria offers opportunities for bacterial surface engineering applications. As a demonstration of this concept, Nishimura and co-workers exploited compound 1 to generate ketone-displaying lactic acid bacteria, which were subsequently glycoengineered by chemoselective reaction with an aminooxy-functionalized mannopentaose derivative.97 The oligosaccharide-decorated bacteria exhibited enhanced adhesion onto a surface coated with the mannose-binding lectin ConA compared to native bacteria.97 This work demonstrated that adhesion—and, by extension, potentially other surface-driven properties such as biofilm formation and immunogenicity—can be controlled through chemoselective modification of the bacterial surface with unnatural chemical cargo. The modularity of the bioorthogonal chemical reporter strategy is ideally suited for such applications, since the secondary cargo being delivered can be readily swapped. We will see in later sections how bioorthogonal surface engineering has been extended to develop novel therapeutic strategies.
Overall, these pioneering studies represented the first inroads into the development of chemical reporters for bacterial glycans, and offer a selection of compounds based on Park’s nucleotide, Lipid I, and Lipid II that can potentially metabolically label PG in live bacteria. Further research is needed to unequivocally determine that compounds 1 and 2, or related Park’s nucleotide derivatives, metabolically label the PG peptide cross-link when exogenously delivered to live bacterial cells. In the meantime, the size, complexity, synthetic challenges, and limited characterization of these original PG precursor glycopeptides has motivated the development of smaller, synthetically accessible, well-characterized PG reporters, including the D-amino acid family of probes discussed in the following sections.
2.1.2. D-amino acid derivatives.
The most prominent and broadly applied reporter probes for bacteria, D-amino acid derivatives, were inspired by the long-known phenomenon that bacteria can incorporate exogenous, non-native D-amino acids from the growth medium into the PG stem peptide.99–102 In the early 1990s, de Pedro and co-workers demonstrated through HPLC analysis that a variety of D-amino acids (e.g., D-Met, D-Trp, and D-Phe) were efficiently incorporated into the PG stem peptide in place of native D-Ala, with evidence implicating a periplasmic TPase-mediated D-amino acid exchange mechanism of incorporation (Figure 7A).102 Extending this finding, in 1997 de Pedro developed a PG labeling method in which treatment of E. coli with D-Cys (9, Figure 7B) installed a reactive thiol in the PG stem peptide, enabling chemoselective biotinylation and microscopy analysis of the purified PG sacculus.103 This groundbreaking research and subsequent mechanistic studies104–106 demonstrated the potential of exploiting conserved TPase-mediated D-amino acid exchange to incorporate chemical tags into PG. However, early methods involving reactive D-Cys or radiolabeled D-amino acids were limited by various factors, notably their technical complexity and incompatibility with live cells.
Figure 7.
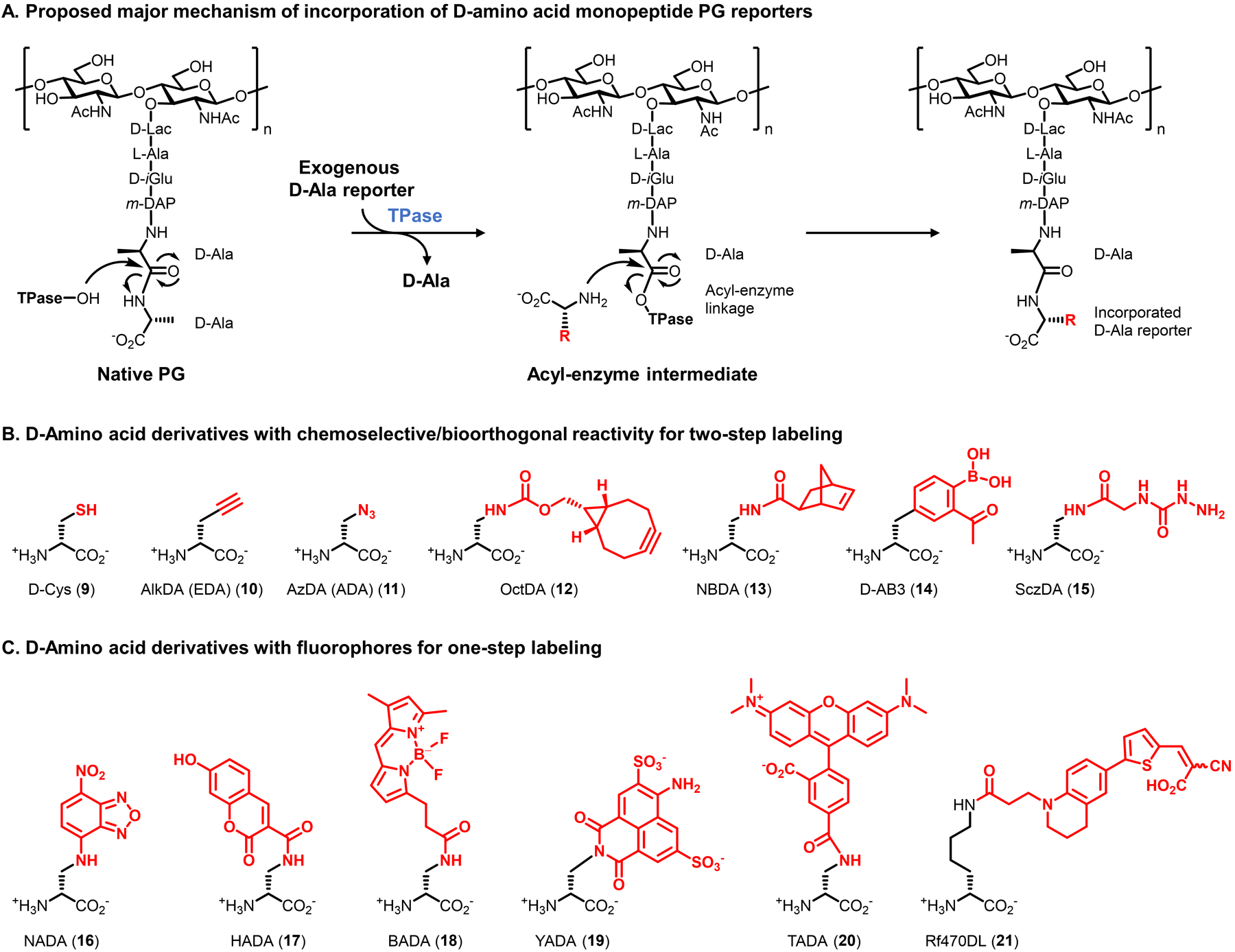
D-amino acid derivatives targeting the PG stem peptide. (A) Metabolic incorporation of unnatural D-amino acid derivatives (modified with an unnatural R group) is proposed to predominantly occur through periplasmic TPase-mediated PG remodeling. Incorporation by PBP D,D-TPases to install the probe at the 5-position of the peptide is shown; incorporation by L,D-TPases to install the probe at the 4-position can also occur. Smaller D-amino acid derivatives may additionally incorporate into PG via an intracellular route. (B and C) Structures of D-amino acid derivatives bearing selectively reactive functional groups (B) and fluorophores (C).
The convergence of de Pedro’s findings with emerging chemical biology techniques sparked the development of a powerful and continuously expanding suite of D-amino acid probes that report on PG biosynthesis in live bacteria (Figures 7B and 7C). The VanNieuwenhze, Brun, and Bertozzi groups contemporaneously developed and characterized the first fluorescent and clickable D-amino acids, which were reported in 2012 and 2013, respectively.76,77,107 While the ensuing period has witnessed the creation of a wide variety of complementary D-amino acid-based reporters that will be surveyed below, we focus first on a few prototypical compounds from these seminal reports to provide a representative illustration of the design, synthesis, and characterization of this class of probes. The VanNieuwenhze and Brun groups initially designed the fluorescent nitrobenzofuran- and hydroxycoumarin-conjugated D-amino acid derivatives NADA (16) and HADA (17).76 The relatively small fluorophores on NADA and HADA resemble the indole ring of D-Trp, which had previously been shown to incorporate into PG via TPase-mediated D-amino acid exchange,102,105 and thus it was hypothesized that the fluorescent derivatives would incorporate in a similar manner. Likewise, the Bertozzi group anticipated that minimally modified D-amino acids resembling D-Ala but containing terminal alkyne and azide bioorthogonal tags, respectively named AlkDA (EDA, 10) and AzDA (ADA, 11), would undergo incorporation into PG, enabling subsequent click chemistry-mediated analyses.77 In comparison to the earlier work by Nishimura and co-workers, which enlisted carbonyl–α-effect nucleophile reactions for bioorthogonal cell labeling,75,97 AlkDA and AzDA enlisted rapid and highly selective CuAAC and SPAAC click reactions, which had been introduced and refined in the intervening years.41 The substrate tolerance, extracellular/periplasmic location, and conserved nature of TPases, coupled with the relatively small size of the designed reporters, supported the feasibility of exploiting these enzymes to label PG in diverse bacteria with D-amino acid reporters. In addition to the extracellular TPase pathway, another possibility for probe incorporation was that D-amino acid probes could access the cytoplasm and incorporate into PG in a Ddl-initiated manner, i.e. from the inside-out (see Figure 5). Assuming the probes indeed successfully incorporated into PG, determining whether one or a combination of these potential incorporation routes was operative would be important to permit judicious application of D-amino acid derivatives.
Accessing D-amino acid probes is straightforward, particularly in comparison to the larger, more complex, and synthetically challenging PG precursor derivatives75,97 covered in the previous section. Indeed, AlkDA (10), AzDA (11), NADA (16), and HADA (17) are all presently commercially available. Furthermore, many D-amino acid derivatives, including those shown in Figure 7, can be synthesized in two steps: (i) coupling relatively inexpensive amino acids such as (S)-2-[(tert-butoxycarbonyl)amino]-3-aminopropionic acid (Boc-Dap-OH, 22, <$100/g) or Nα-(tert-butoxycarbonyl)-D-lysine (Nα-Boc-D-lysine, 23, <$100/g) to an amine-reactive reagent (e.g., fluorescent dye); (ii) removal of the protecting group (Figure 8). Such a strategy provides straightforward access to novel D-amino acid-based probes, or to previously reported probes that are not commercially available.
Figure 8.
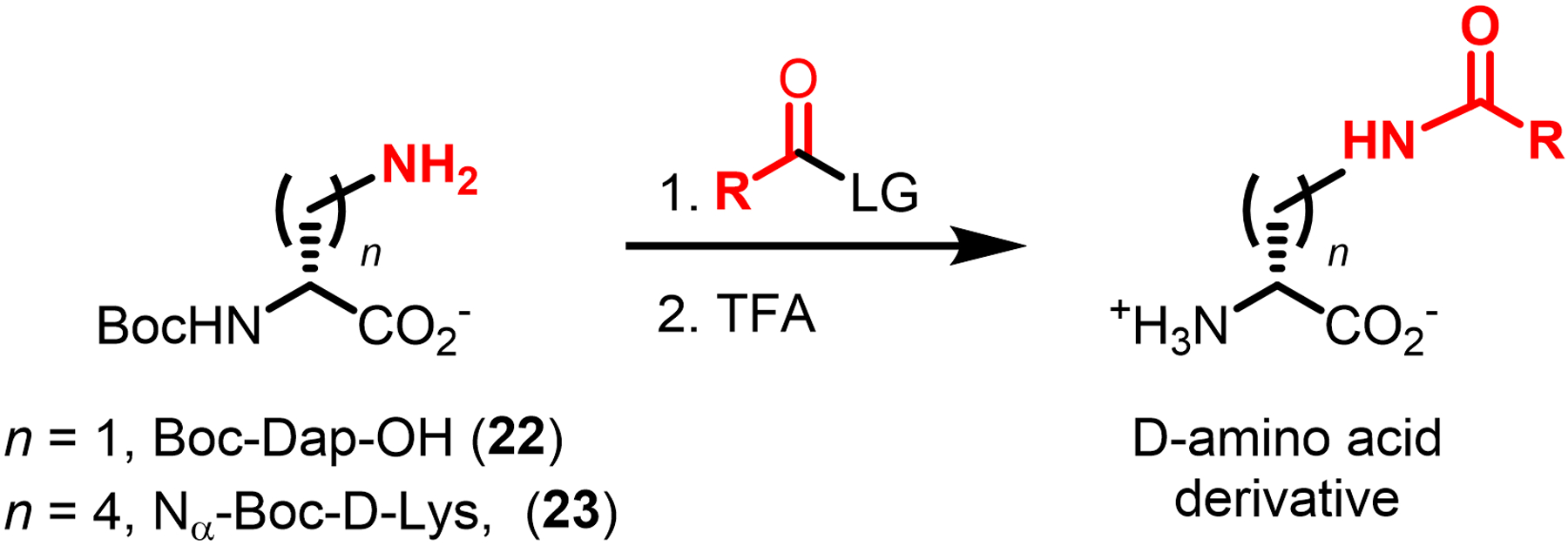
Two-step chemical synthesis of D-amino acid derivatives. Boc, tert-butoxycarbonyl; LG, leaving group; TFA, trifluoroacetic acid.
The fluorescent (16 and 17) and clickable (10 and 11) D-amino acid reporter probes were exhaustively characterized, generally using similar experimental approaches. The probes were first tested for incorporation into various Gram-positive, Gram-negative, and mycobacterial species. For the one-step fluorescent reporters NADA (16) and HADA (17), bacteria were incubated in probe and visualized by fluorescence microscopy, which depicted robust labeling of the cell surface of Gram-negative E. coli, Gram-positive Bacillus subtilis, and Gram-negative Agrobacterium tumefaciens (Figure 9A).76 For the two-step clickable reporters AlkDA (10) and AzDA (11), bacteria were incubated in probe, reacted with a complementary fluorescent dye by CuAAC (azido-488 for AlkDA) or SPAAC (difluorinated cyclooctyne108 DIFO-488 for AzDA), and analyzed by microscopy and flow cytometry.77 These experiments confirmed that AlkDA and AzDA labeled Gram-negative E. coli, Gram-positive Listeria monocytogenes, and mycomembrane-containing Corynebacterium glutamicum and M. tuberculosis (Figure 9B).77 In this case, whereas CuAAC with an azido-488 fluorophore was performed on fixed AlkDA-treated cells due to copper toxicity, the use of SPAAC with a cyclooctyne-fluorophore was done on live AzDA-treated cells, highlighting the advantage of truly bioorthogonal chemistries such as SPAAC and the tetrazine ligation.77 Promisingly, in the studies on both fluorescent and clickable reporters it was shown that detectable surface fluorescence was achieved at concentrations that did not impact bacterial growth,76,77 which was a concern since high concentrations of exogenous D-amino acids had previously been shown to perturb PG structure and inhibit growth.102,103 This represents a successful example of identifying an optimal reporter probe concentration range in which labeling is efficient enough to be readily detectable in the assay, but toxicity and/or impairment to the native envelope structure is minimal. Finally, the robust labeling of diverse bacterial species with a variety of D-amino acid derivatives in these ground-breaking studies provided an initial glimpse at the broad applicability and versatility of this class of probes.
Figure 9.
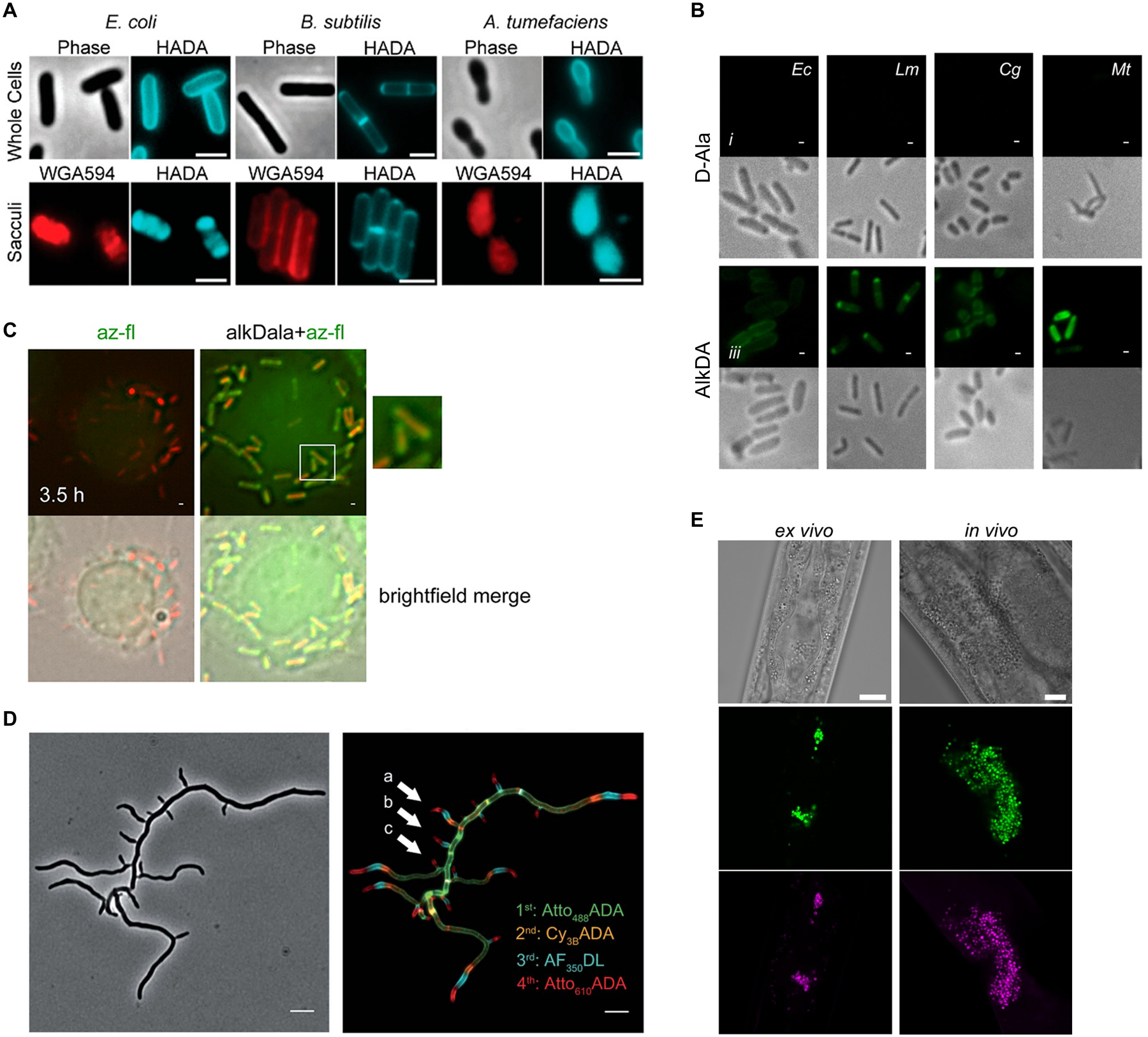
Examples of in vitro and in vivo D-amino acid reporter labeling of PG. (A) Metabolic incorporation of HADA (17) into E. coli, B. subtilis, and A. tumefaciens. Bacteria were incubated in HADA, fixed, and imaged directly (top panel) or subjected to sacculus isolation (i.e., intact PG isolation) and then imaged (bottom panel). WGA595 is a red-fluorescent lectin that binds to PG glycan strands. Reproduced with permission from ref 76. Copyright 2012 Wiley-VCH. (B) Metabolic incorporation of AlkDA (10) into E. coli (Ec), L. monocytogenes (Lm), C. glutamicum (Cg), and M. tuberculosis (Mt). Bacteria were incubated in either D-Ala (control, top panel) or AlkDA (bottom panel), fixed, subjected to CuAAC with azido-488, and imaged. (C) Imaging of L. monocytogenes-infected macrophages with AlkDA (10). Macrophages were infected with red fluorescent protein (RFP)-expressing L. monocytogenes and extracellular bacteria were removed, then incubated in AlkDA (10), fixed, subjected to CuAAC with azido-488, and imaged. (B) and (C) were reproduced with permission from ref 77 (https://pubs.acs.org/doi/full/10.1021/ja505668f). Copyright 2013 American Chemical Society. Further permission related to the material excerpted should be directed to the American Chemical Society. (D) Sequential labeling of S. venezuelae with variable-color fluorescent D-amino acid derivatives. Bacteria were sequentially incubated for short pulses with different dye-conjugated D-amino acids possessing orthogonal excitation/emission wavelengths (structures not shown in Figure 7), fixed, and imaged. Arrows indicate new branching sites of the cell wall. Reproduced from ref 109 with permission from the Royal Society of Chemistry. (E) Ex vivo and in vivo labeling of S. aureus PG in host C. elegans using the tetrazine reagent TetDAC (25, Figure 10A). C. elegans was infected with green fluorescent protein (GFP)-expressing S. aureus that had been (i) pre-treated with TetDAC (25) for ex vivo labeling or (ii) not pre-treated for in vivo labeling. C. elegans was washed to remove uncolonized bacteria, then (i) reacted with TCO-Cy5 fluorophore and imaged for ex vivo labeling or (ii) treated with TetDAC (25), reacted with TCO-Cy5 fluorophore, and imaged for in vivo labeling. Reproduced with permission from ref 80. Copyright 2017 American Chemical Society.
The specificity and mechanism(s) of incorporation of the first D-amino acid probes were investigated through batteries of well-designed experiments. For NADA (16) and HADA (17), the corresponding fluorescent L-amino acids were synthesized as control compounds; neither incorporated into E. coli, which is consistent with the specificity of TPase enzymes for D-amino acids.76 For AlkDA (10) and AzDA (11), which were characterized primarily in L. monocytogenes, labeling was decreased by competition with native D-Ala and increased in a D-Ala auxotroph, conditions which would elevate and deplete the cellular pool of native D-Ala, respectively.77 These findings suggested that the clickable probes incorporated via D-Ala-specific metabolism. Both studies also determined that the probes were not detectably incorporated into D-Ala-containing teichoic acids based on the observation that teichoic acid-deficient mutant strains exhibited no difference in labeling, thus ruling out the main possibility for off-target labeling.76,77 Critically, both studies performed HPLC and MS analysis of muramidase-released PG fragments, i.e. muropeptides, which provided direct evidence that both fluorescent and clickable D-amino acid probes were incorporated into the PG stem peptide.76,77 Mechanism-of-incorporation experiments from these76,77 and subsequent independent studies,110,111 which employed panels of PG biosynthesis inhibitors and knockout mutants, yielded mixed conclusions, pointing to a complicated scenario in which probe structure and bacterial species are factors that dictate whether the incorporation route is extracellular/TPase-mediated, intracellular/Ddl-initiated, or a blend of the two. Collectively, the available evidence strongly suggests that larger fluorescent D-amino acid reporters (e.g., 18–21, Figure 7C) incorporate into PG via an extracellular TPase-mediated exchange reaction (Figure 7A) in diverse bacteria.110,111 In contrast, while smaller reporters (e.g., click-tagged (10, 11), HADA (17)) appear to incorporate into PG mainly via TPases in E. coli and B. subtilis,111 other evidence suggests that they may incorporate via both extracellular/TPase-mediated and intracellular/Ddl-initiated pathways in L. monocytogenes and Mycobacterium smegmatis,77,110 implying organism-dependent behavior. Further studies to precisely define the species-dependent metabolic fate of smaller D-amino acid derivatives, e.g. by analyzing cytoplasmic PG precursors downstream of Ddl for label incorporation, would shed light on this complex issue.65 Most importantly, however, with larger fluorescent D-amino acid probes established to report on extracellular TPase activity and newer D-amino acid dipeptide probes established to report on intracellular MurF-initiated nascent PG synthesis activity (see section 2.1.3), well-characterized tools exist for selective probing of the two pathways. It is important to point out that rigorous and carefully controlled experimentation spanning several years and multiple publications yielded a solid understanding of these probes’ behavior in cells, which in turn has been instrumental in informing the appropriate design and interpretation of labeling experiments.
The applications of D-amino acid in these pioneering studies foreshadowed the high impact this class of probes would have, and is continuing to have, in the field of microbiology. The VanNieuwenhze and Brun groups applied HADA (17) and other fluorescent D-amino acids in pulse-chase, multi-color, and super-resolution imaging experiments to directly visualize differences in PG synthesis during growth and division processes in a wide range of bacteria (for an example from later work,109 see Figure 9D).76 It was found that signal from D-amino acid reporter probes strongly correlated with previously known sites of PG synthesis in well-studied organisms, and, moreover, revealed for the first time PG synthesis dynamics in several bacterial species that were previously uncharacterized in this regard.76 The Bertozzi group increased the complexity of the system under investigation by applying AlkDA (10) to visualize PG dynamics of intracellular L. monocytogenes in a macrophage infection model (Figure 9C).77 Macrophages were infected with a red-fluorescent reporter strain of L. monocytogenes, treated with AlkDA (10), reacted with an azido-488 fluorophore via CuAAC, and imaged.77 AlkDA-dependent fluorescence was exclusively localized to infecting bacteria, and the time-dependent regions of PG labeling were consistent with those observed during in vitro culture of the bacteria.77 The labeling selectivity for bacterium versus host observed in this experiment, made possible by the general lack of D-amino acid utilization in eukaryotic cells, highlights the potential of D-amino acid reporters for investigating and therapeutically targeting bacterial PG in complex host systems, examples of which we see below.
The original work demonstrating the power of D-amino acid-based PG reporters has been expanded upon greatly over the past several years, both in terms of introducing complementary probes and further applying probes to gain insight into cell wall structure and dynamics. VanNieuwenhze and Brun’s 2012 report not only disclosed and characterized NADA (16) and HADA (17), but also briefly showed that bacteria incorporated larger fluorescein- and carboxytetramethylrhodamine (TAMRA)-modified D-lysine derivatives (not shown in Figure 7) as well as the smaller clickable AlkDA (10) and AzDA (11).76 To improve the capacity to do multi-color imaging studies to investigate PG dynamics, in 2017 the same groups developed a broader set of orthogonally-colored fluorescent D-amino acid probes, which included a total of nine dye-conjugated reporters whose emission wavelengths covered the full visible spectrum; among these, blue HADA (17), green BADA (18), yellow YADA (19), and orange TADA (20) are represented in Figure 7C.109 Of note, the larger reporters (e.g., TADA) showed significantly reduced incorporation as compared to the smaller reporters (e.g., HADA and YADA) in outer membrane-containing E. coli, suggesting that membrane permeability is an important factor to consider in designing and selecting probes appropriate to the organism.76,109 As shown in Figure 9D, this palette of D-amino acid probes was used to perform four-color time-lapse visualization of PG biosynthesis in the filamentous Gram-positive bacterium Streptomyces venezuelae.
Efforts to expand the toolbox with fluorogenic PG labeling strategies have also been successful, providing a means to perform high-sensitivity, no-wash, minimally perturbing PG labeling in live bacteria. The Bertozzi group combined a cyclooctyne-modified D-amino acid derivative (OctDA (12), Figure 7B) with a novel fluorogenic near-infrared (NIR)-emitting azido Si-rhodamine dye, which together enabled live-cell imaging without the need to wash cells after the fluorescence-triggering SPAAC reaction.78 In a typical bioorthogonal chemical reporter experiment, the smaller azido component would serve as the metabolic substrate and the larger cyclooctyne component would serve as the secondary reagent. In this case, the opposite mode of incorporation was successful due to the remarkable tolerance of the PG synthesis machinery for bulky D-amino acid derivatives. Interestingly, while the Si-rhodamine dye employed for the secondary labeling step was successful in Gram-positive bacteria and mycobacteria, it did not work in Gram-negative E. coli.78 It is not clear why this approach would work in mycobacteria but not E. coli, since both have outer membranes. In an alternative one-step approach to fluorogenic PG labeling, the VanNieuwenhze and Brun groups developed a series of molecular rotor-based fluorescent D-amino acid probes, including the D-Lys derivative Rf470DL (21, Figure 7C).79 These probes operate under the principle that the molecular rotor dye component—in the case of 21, a tetrahydroquinoline derivative—is sensitive to the rotational freedom provided by its environment: the dyes exhibit lower fluorescence in low-hindrance environments and higher fluorescence in high-hindrance environments. Since PG is a tightly cross-linked meshwork and thus a relatively congested environment, it was hypothesized that upon metabolic incorporation, Rf470DL (21) would experience limited rotational freedom, leading to fluorescence turn-on.79 Indeed, when Gram-positive bacteria were incubated in Rf470DL (21) and imaged without a wash step, PG labeling could be observed with excellent signal-to-noise ratios, whereas HADA (17)-treated bacteria could not be visualized without wash steps.79 As with other larger fluorescent D-amino acid derivatives, Rf470DL (21) had difficultly labeling Gram-negative E. coli, unless an outer membrane-defective strain was used.79 In addition to whole-cell bacterial imaging studies, Rf470DL (21) was also used to create a convenient microplate-based transpeptidase enzyme assay to characterize the effects of various inhibitors on S. aureus PBP4 TPase activity.79 This application demonstrated that D-amino acid reporters can serve as enabling tools to develop enzyme- and whole cell-based high-throughput screens for antibiotic discovery. As well, these two innovative approaches to achieving fluorescence turn-on for PG reporters—a fluorogenic SPAAC reaction and a molecular rotor dye—can in principle be adapted to develop similar strategies for rapid, no-wash imaging of other bacterial glycans.
Other research has led to D-amino acid probes employing varied bioorthogonal chemistries and scaffold designs. The Pires lab disclosed the first application of the tetrazine ligation (Figure 3E) to bacterial cell surface labeling. Three norbornene-modified D-amino acids, including the D-alanine derivative NBDA (13, Figure 7B), were synthesized, evaluated, and shown to incorporate into PG in Gram-positive S. aureus.112 A major advantage of tetrazine ligations compared to other bioorthogonal reactions is their superior kinetic profile. While tetrazine reactions using the moderately strained norbornene are about 10-fold faster than common SPAAC reactions, those using the highly strained trans-cyclooctene (TCO) are 100–1,000-fold faster and thus are more suitable for in vivo imaging.41 Subsequent work from the Pires lab demonstrated that, indeed, when a tetrazine-modified D-amino acid derivative (TetDAC, 25, Figure 10A) was incorporated into S. aureus PG and secondarily reacted with a TCO-fluorophore reagent, robust surface labeling was detected within 5 minutes.80 The rapid kinetics afforded by the tetrazine–TCO reaction allowed a very low concentration of the secondary fluorophore, which was leveraged to perform the first use of exogenous D-amino acid derivatives to label the PG of bacteria within a live host organism, specifically S. aureus within the model nematode Caenorhabditis elegans (Figure 9E).80 Notably, the norbornene- and tetrazine-modified reporters that exhibited the most efficient labeling in S. aureus were based not on D-amino acids, but rather on D-amino carboxamides, which have an unnaturally amidated C-terminus (compounds 24 and 25, Figure 10A). Previously, D-amino carboxamide derivatives had been elegantly shown by the Kahne and Walker labs,82 and subsequently confirmed by the Pires lab,81 to label PG more efficiently than D-amino acid derivatives in some species, namely Gram-positive bacteria, possibly due to more efficient TPase-mediated incorporation and/or the ability to withstand cleavage by endogenous CPases once incorporated (Figure 10B). These findings revealed valuable additional substrate plasticity in the PG biosynthesis machinery with respect to the D-amino acid C-terminus.
Figure 10.
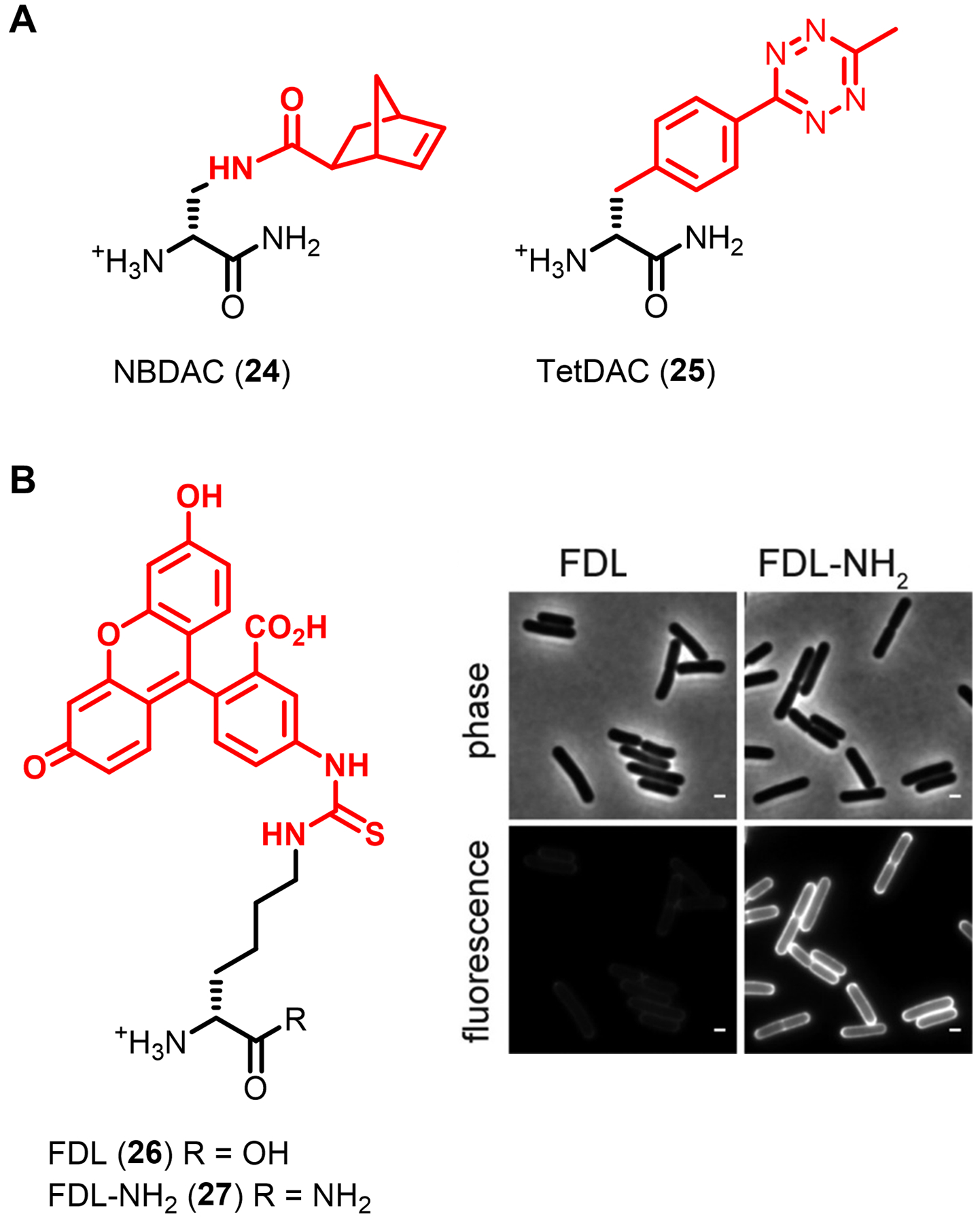
D-amino carboxamide derivatives for PG labeling. (A) Tetrazine ligation-capable reporters bearing strained norbornene (NBDAC, 24) and tetrazine (TetDAC, 25) groups on the D-amino carboxamide scaffold. (B) Fluorescent D-amino carboxamide FDL-NH2 (27) labels B. subtilis more efficiently than D-amino acid FDL (26). B. subtilis was cultured in the probes and imaged. Reproduced with permission from ref 82 (https://pubs.acs.org/doi/full/10.1021/ja505668f). Copyright 2014 American Chemical Society. Further permission related to the material excerpted should be directed to the American Chemical Society.
Beyond Pires’s work on the tetrazine ligation, and moving out of the realm of the now-canonical bioorthogonal reactions shown in Figure 3, D-amino acid labeling of PG has become somewhat of a proving ground to assess the bioorthogonality of new chemistries in live cells, presumably due to the remarkable substrate tolerance of PG synthesis enzymes, the generally well-established nature of the experimental system, and the relative ease of bacterial cell culture. For example, the Gao group explored semicarbazide–boronic acid ligations, first showing that a D-amino acid derivative containing a 2-acetylphenylboronic acid side chain (D-AB3, 14, Figure 7B) could be incorporated into bacteria and reacted with a complementary semicarbazide-488 fluorophore, resulting in the formation of a covalent diazaborine linkage.113 Interestingly, D-AB3 (14) labeled outer membrane-containing Gram-negative E. coli much more efficiently than Gram-positive organisms, which is the opposite trend observed for the fluorescent D-amino acid reporters.113 This could potentially be due to the smaller size and outer membrane permeability of 14 and/or the preference of E. coli PBP TPases for aromatic versus aliphatic side chains. In another study on the semicarbazide–boronic acid ligation by the Gao group, the mode of labeling was reversed by first installing a semicarbazide-modified D-amino acid (SczDA, 15, Figure 7B) into S. aureus and then reacting cells with a novel fluorogenic boronic acid in a no-wash imaging experiment.114 The Pezacki group recently showed that a relatively small nitrone-modified D-amino acid incorporated into Listeria innocua (a non-pathogenic model organism for L. monocytogenes), allowing for rapid cycloaddition with a TCO-488 fluorophore to detect bacteria (structures not shown).115 Notably, neither of the reports on the semicarbazide ligation or the nitrone–TCO ligation performed characterization to show specificity of the labeling pathway or product. It is too early to tell whether such newer chemistries will bear useful complementarity to the well-established and broadly applied fluorescent and clickable D-amino acid derivatives.
Owing to the importance and conserved nature of PG, D-amino acid reporters have been broadly and creatively applied in PG-focused microbiology research. Above, we introduced early work by the VanNieuwenhze, Brun, and Bertozzi groups that—through direct visualization of PG in intact bacterial cells—revealed how PG is built and remodeled in time and space, both in vitro and in vivo. As recently reviewed in depth,32,52,64,65 many subsequent studies, performed in a diverse range of bacterial species, have used D-amino acid probes as integral tools for these and related objectives, including imaging the dynamics of PG synthesis and remodeling, correlating these processes to enzymatic activity, and understanding how they are influenced by antibiotics. In one recent example, fluorescent D-amino acids were used in concert with complementary techniques (e.g., fluorescent fusion proteins) to show that, as bacteria divide, circumferential movement of the filamentous proteins FtsZ and FtsA at the division site drives the associated movement of PG synthases (i.e., GTases and TPases), which in turn drives the spiraling synthesis of new PG toward the interior of the division site until the two daughter cells split.116 In another example, fluorescent and clickable D-amino acid mono- and dipeptide reporters have been used to show that mycobacteria, which are known to synthesize PG and grow from their poles, also synthesize PG along their sidewall, which appears to be a repair function that “patches” and reinforces damaged PG along the periphery of the cell.110,117 These efforts involved collaborative interdisciplinary research between chemists and microbiologists to answer pressing biological questions.
In addition to imaging PG dynamics, some researchers have explored the possibility of using the D-amino acid scaffold for specific targeting of diagnostic or therapeutic payload to bacterial pathogens as novel strategies to detect or treat infections. Supporting this notion, work described above showed that D-amino acid reporters specifically incorporate into bacteria even within host macrophages and whole organisms (e.g., Figures 9C and 9E).77,80 To demonstrate this concept, the Pires group developed 2,4-dinitrophenyl (DNP)-modified D-amino acid and D-amino carboxamide derivatives, which after incorporation into PG facilitated phagocytosis of bacteria by macrophages in the presence of anti-DNP antibodies.118,119 Since anti-DNP antibodies are abundant in human serum, the ability to selectively modify the surface of pathogenic cells with DNP may lead to novel immunotherapy strategies for clearing bacterial infections.120 In another demonstration of therapeutic potential, the Liu group showed that D-Ala enabled delivery (either directly or via click chemistry) of a fluorescent pyridinium-substituted tetraphenylethylene photosensitizer moiety specifically to S. aureus, including macrophage-resident MRSA, which enabled enhanced bacterial killing upon light exposure.121,122 In the diagnostics category, the D-amino acid reporter concept was extended to show that radioactive 11C-labeled D-alanine enabled non-invasive positron emission tomography (PET) imaging of bacteria in various animal infection models with better specificity than the widely available but non-specific PET tracer 18F-fluorodeoxyglucose (18F-FDG).123,124 Previously, 18F-modified D-alanine was also reported for PET imaging, but suffered from in vivo defluorination.125
Finally, recent literature has highlighted how D-amino acid-based PG reporters, and perhaps bacterial glycan reporters in general, can be used in innovative, unanticipated ways that enable new avenues of biological inquiry. In 2020, the Schultz and Siegrist groups independently showed that clickable D-amino acid reporters can be used to install non-native cross-links in PG in live bacteria.126,127 The Schultz group fed E. coli a sulfonyl fluoride128-modified D-amino acid, which upon incorporation into PG underwent subsequent reaction with the m-DAP side chain amino group on a neighboring PG strand.126 Surprisingly, the resulting non-canonical sulfonamide linkage accounted for about one-third of the total cross-links in the cell, but did not significantly impact growth or division.126 The Siegrist group dual-labeled independent PG stem peptides with AlkDA (10) and a novel fluorogenic azidocoumarin-modified D-amino acid, which enabled biocompatible CuAAC-induced cross-linking between the two unnatural residues that, quite elegantly, simultaneously induced fluorescence to indicate a successful linkage.127 In this case, the non-canonical triazole cross-links protected bacteria from β-lactam antibiotics.127 Such methods for chemically controlled PG cross-linking offer a new approach to investigate this critical process in a manner that is complementary to genetics or small-molecule inhibition.
The breadth and depth of research on the D-amino acid family of probes, in terms of both their development and applications, points to the high potential of bacterial glycan reporters more generally. Much of the work discussed in this section can serve as a blueprint for how other bacterial glycans can be targeted for reporter development, how those reporters can be carefully characterized, and how they can be enlisted to address knowledge gaps in the field and potentially create new approaches to diagnose and treat bacterial infections. In subsequent sections, the parallels between reporters for PG and those focused on other bacterial glycans will be evident. More specifically for PG, given its significance as a drug target and the broad substrate tolerance of PG synthesis machinery for unnatural D-amino acids,129 we are likely to see continued innovation in the creation of tools that exploit this system. Moreover, the high level of maturity of D-amino acid reporters, in terms of their synthetic/commercial accessibility, well-characterized metabolic incorporation in diverse bacteria, and experimental versatility (e.g., one-step vs. two-step labeling, fluorogenic designs, numerous bioorthogonal chemistries to choose from), augurs their continued widespread use in PG imaging, bacterial surface engineering, and antibiotic screening.
2.1.3. D-amino acid dipeptide derivatives.
As described in the preceding section, the predominant mechanism of incorporation of larger fluorescent D-amino acid into PG is periplasmic TPase-mediated D-amino acid exchange in the stem peptide, whereas smaller clickable versions appear to simultaneously access both periplasmic TPase-mediated and cytoplasmic Ddl-initiated routes in some bacteria (see Figures 5 and 7A). A complementary tool in the PG chemical toolbox would be a probe that incorporates exclusively via an intracellular route into cytoplasmic PG precursors, and ultimately into the stem peptide of mature periplasmic PG. Such a capability would enable differentiation between nascent PG arising from the cytoplasm and existing PG that has already been formed in the periplasm. As discussed above, earlier work showed that Park’s nucleotide derivatives (1 and 2, Figure 6)75 can potentially incorporate into PG via a cytoplasmic route, but the challenging synthesis and limited characterization of these derivatives preclude convenient and confident application. In view of PG biosynthesis shown in Figure 5, opportunities also exist for intercepting the biosynthetic pathway upstream of MurF. One early-stage cytoplasmic substrate that can be targeted to accomplish this objective is the D-Ala-D-Ala dipeptide. In principle, MurF ligase could combine an exogenously added D-amino acid dipeptide derivative with UDP-MurNAc-tripeptide to generate Park’s nucleotide, which would be followed by elaboration to Lipid II and accretion to mature labeled PG via GTase and TPase activity (Figure 11A). The small dipeptide probe scaffold would be facile to manipulate from a synthetic chemistry standpoint and it would afford the ability to install modifications at either its N- or C-terminal residue side chain, which would be metabolically incorporated into the 4th and 5th positions of the PG stem peptide, respectively. Modification of the 4th versus the 5th position is of consequence because, as shown in Figure 5, CPase activity can remove the terminal (5th position) D-Ala from the stem peptide, so controlling susceptibility to this reaction would be beneficial.
Figure 11.
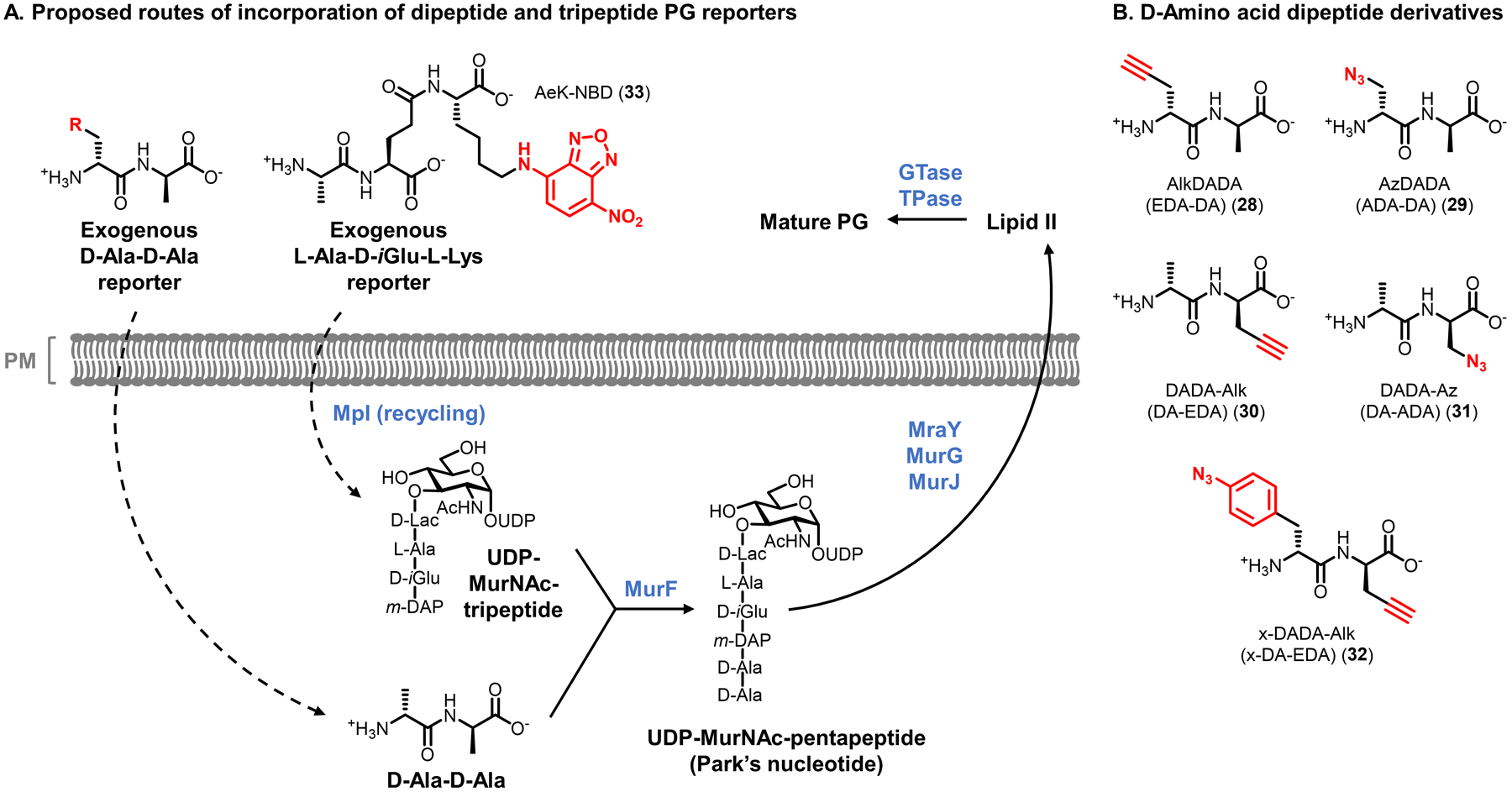
D-Ala-D-Ala dipeptide and L-Ala-D-iGlu-L-Lys tripeptide derivatives target the stem peptide of nascent PG via cytoplasmic routes of incorporation. (A) Metabolic incorporation of exogenous D-amino acid dipeptides (modified with an unnatural R group) is proposed to be initiated by MurF in the cytoplasm. Metabolic incorporation of exogenous tripeptide derivatives (modified with an NBD fluorophore) is proposed to occur through a recycling pathway in which tripeptides are taken up and ligated to UDP-MurNAc by Mpl, followed by MurF-catalyzed coupling with D-Ala-D-Ala and downstream incorporation into PG as shown in Figure 5. (B) Structures of D-amino acid dipeptide derivatives bearing bioorthogonal and photo-cross-linking functional groups.
The Maurelli and VanNieuwenhze groups reported the first D-amino acid dipeptide probes,83 motivated by a fascinating and long-standing conundrum in microbiology: whether or not bacteria in the Chlamydiae phylum possess a PG layer, a question referred to as the chlamydial anomaly.130 Although Chlamydia possess PG biosynthesis genes and chlamydial infections respond to treatment with PG-targeting drugs, PG had never been successfully isolated or detected in these bacteria. The obligate intracellular lifestyle of these organisms confounds many traditional experimental approaches. The team reasoned that metabolic labeling of intracellular C. trachomatis with the D-amino acid derivative AlkDA (10) may allow visualization of PG by CuAAC reaction with an azido-488 fluorphore, in a manner similar to that observed by the Bertozzi group for intracellular L. monocytogenes77 (see Figure 9C). However, no incorporation of 10 was observed. Failure of this approach, perhaps a result of CPase-mediated removal of the installed tag, spurred the development of D-amino acid dipeptides as a novel method for stably tagging nascent PG. Supporting the premise of this idea, it had previously been observed that Chlamydia can take up D-Ala-D-Ala131 and that this dipeptide can rescue the growth of a Ddl-deficient mutant incapable of making its own D-Ala-D-Ala.132 Based on these findings, four alkyne- and azide-modified D-Ala-D-Ala derivatives were designed, including those containing bioorthogonal alkyne and azido groups at the N-terminus, referred to as AlkDADA (EDA-DA, 28) and AzDADA (ADA-DA, 29) and at the C-terminus, referred to as DADA-Alk (DA-EDA, 30) and DADA-Az (DA-ADA, 31) (Figure 11B).83 Importantly, since these compounds target a conserved early step in PG biosynthesis, they were anticipated to be useful for labeling nascent PG in any PG-containing organism. Following their preparation using standard peptide synthesis strategies, the dipeptide derivatives 28–31 were first evaluated in Gram-negative E. coli and Gram-positive B. subtilis.83 Compounds 28–31 all rescued the growth of D-cycloserine-treated and Ddl-deficient bacteria, both conditions in which D-Ala-D-Ala is depleted, and all four compounds metabolically incorporated into PG as assessed by imaging whole cells and isolated sacculi following CuAAC reaction with a complementary fluorophore.83 Subsequent work demonstrated that reporters containing the click tag at the N-terminus (AlkDADA (28) and AzDADA (29)) generally incorporated with higher efficiency into PG than C-terminally-modified 30 and 31, likely due to lower tolerance of MurF for the latter compounds and/or the ability of CPases to remove tags installed at the 5th position of the PG stem peptide.111 Another report by the Siegrist lab also provided genetic and biochemical evidence that further supported the proposed cytoplasmic MurF-dependent route of incorporation (see Figure 11A), critically including direct detection of dipeptide-labeled PG precursors Lipid I and Lipid II in mycobacteria.110 Thus, a strong body of data demonstrates that the clickable D-amino acid dipeptide derivatives 28 and 29 provide a means to efficiently and stably incorporate a label into nascent PG in diverse bacteria, establishing their complementarity to the previously developed D-amino acid derivatives. A potential limitation of the D-amino acid dipeptide probe scaffold is the relatively strict substrate specificity of MurF, which permits small unnatural modifications to the D-Ala-D-Ala substrate at the N-terminal residue but has high specificity for the C-terminal residue.67,111 Therefore, dipeptide reporters with larger bioorthogonal groups or fluorescent tags for one-step labeling may not undergo efficient incorporation, as evidenced by at least one report.133
We initiate a discussion of the applications of D-amino acid dipeptides by returning to the problem that sparked their development, the chlamydial anomaly. In an experiment seeking the first direct evidence for the presence of PG in Chlamydia, mouse fibroblast cells were infected with C. trachomatis, then incubated in N-terminally modified AlkDADA (28) and subjected to CuAAC with an azido-488 fluorophore.83 Striking images of the infected fibroblasts depicted brightly fluorescent intracellular bacteria, with signal emanating virtually entirely from a ring-like shape at the mid-cell, which represents the septum during bacterial division (Figure 12A).83 This signal was absent in uninfected cells and AlkDADA-untreated cells, and treatment with the PG-digesting enzyme lysozyme removed signal from AlkDADA-treated samples.83 These data, along with additional antibiotic treatment and rescue data, strongly supported the conclusion that AlkDADA was taken up and metabolically incorporated into nascent PG in Chlamydia, providing the first direct detection of PG in these bacteria. Interestingly, labeling with C-terminally modified DADA-Alk (30) was not observed in B. subtilis or intracellular C. trachomatis unless CPase activity was depleted by genetic or chemical means, respectively, suggesting that DADA-Alk was initially installed at the stem peptide 5th position but subsequently trimmed off by a CPase.83 This is a possible explanation for why the AlkDA probe 10 failed to detect PG in Chlamydia, which in retrospect was a fortuitous occurrence since it motivated the development of the D-amino acid dipeptide reporters.
Figure 12.
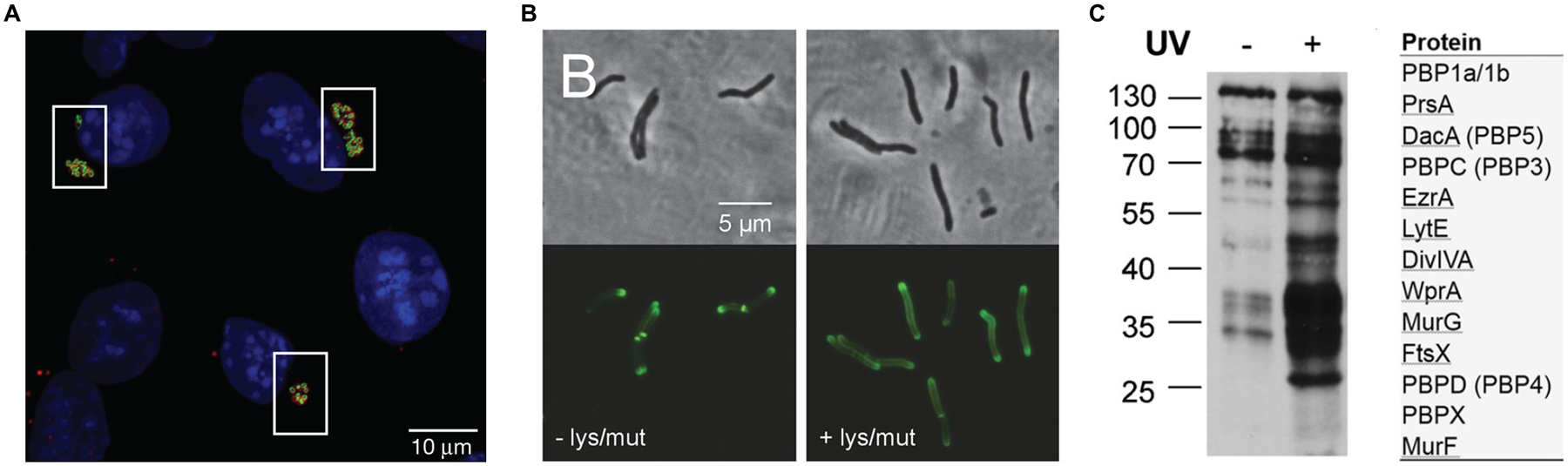
Imaging and proteomics applications of D-amino acid dipeptide reporters for PG. (A) First direct detection of PG in Chlamydia. Mouse fibroblasts were infected with C. trachomatis in the presence of AlkDADA (28), fixed, subjected to CuAAC with azido-488, and imaged. Blue, DAPI nuclear staining of host cells; green, C. trachomatis PG labeling; red, C. trachomatis major outer membrane protein (MOMP) staining. Reproduced with permission from ref 83. Copyright 2014 Springer Nature. (B) Mycobacterial sidewall synthesis of PG in response to cell wall damage. M. smegmatis was incubated in the absence (−) or presence (+) of PG-damaging enzymes lysozyme (lys) and mutanolysin (mut), then treated with AlkDADA (28), fixed, subjected to CuAAC with azido-488, and imaged. While polar labeling is predominant in the -lys/mut condition, enhanced peripheral labeling is visually evident in the +lys/mut condition, and was quantified in ref 110. Reproduced from ref 110. (C) Identification of Lipid II-interacting proteins in B. subtilis. Bacteria were incubated in photo-cross-linking probe x-DADA-Alk (32), exposed to UV irradiation, subjected to CuAAC with azido-biotin, and then biotinylated proteins were either detected by Western blot (left) or avidin-enriched, trypsinized, and analyzed by LC-MS/MS (right). Several PBPs and other PG-related proteins were identified. Reproduced with permission from ref 84. Copyright 2016 Wiley-VCH.
Subsequent applications of D-amino acid dipeptide probes leveraged their cytoplasmic route of incorporation to tag nascent PG precursors. As mentioned above, the Siegrist lab used AlkDADA (28) to demonstrate that upon induced cell wall damage, mycobacteria increase synthesis of nascent PG along the sidewall of the cell, representing a possible PG repair mechanism (Figure 12B).110,117 In a departure from PG imaging, Pires and co-workers adapted the dipeptide reporter scaffold to enable PG-targeting chemical proteomics.84 The dipeptide was modified to include photoactivatable phenylazide and clickable alkyne functionalities at the N- and C-termini, respectively, yielding bifunctional x-DADA-Alk (32) that was designed to photo-label and enrich Lipid II-interacting proteins from live bacteria.84 After performing a variety of experiments (similar to those described above for mono- and dipeptide reporters) to support the incorporation of compound 32 into PG and its precursor Lipid II, the probe was used in B. subtilis to perform photo-labeling, click-mediated affinity enrichment, and LC-MS/MS identification of interacting proteins.84 Among 85 proteins that were enriched with statistical significance, several PBPs with known or hypothesized Lipid II-related functions, as well as MurF, were identified (Figure 12C).84 This work demonstrates how bacterial glycan reporters can be extended to investigate their interacting partners in live cells, which has the potential to elucidate biosynthesis, transport, and remodeling pathways and potentially identify new drug targets. Overall, the novel D-amino acid dipeptide reporters, which access a cytoplasmic, MurF-initiated route of incorporation into nascent PG in diverse bacteria, provide another well-characterized, useful tool alongside the previously developed D-amino acid reporters.
2.1.4. Other stem peptide derivatives.
In addition to D-amino acid mono- and dipeptide reporters covered in the preceding two sections, additional oligopeptide-based reporters mimicking portions of the PG stem peptide have also been developed to label PG. In 2011, Breukink and colleagues developed a fluorescently-labeled tripeptide, which is based on positions 1–3 of the stem peptide, L-Ala-D-iGlu-m-DAP.85 In E. coli, this tripeptide is released and recycled during PG break down and converted back into UDP-MurNAc-tripeptide in the cytoplasm by the ligase Mpl (Figure 11A).134,135 To exploit this pathway, an L-Ala-D-iGlu-L-Lys tripeptide labeled at the L-Lys side chain with a small 7-nitrobenzofurazan (NBD) fluorophore (AeK-NBD, 33, Figure 11A) was designed.85 L-Lys was used in place of m-DAP to enhance synthetic accessibility, and prior research had shown that L-Lys substitution is tolerated during both Mpl-catalyzed ligation of tripeptide to UDP-MurNAc and subsequent ligation of UDP-MurNAc-tripeptide to D-Ala-D-Ala.136,137 With this substitution made, AeK-NBD (33) was synthesized using standard peptide chemistry and shown to incorporate into the E. coli cell surface, and specifically into the PG stem peptide, using a combination of whole-cell imaging, TLC analysis of Lipid I- and Lipid II-containing extracts, and LC-MS/MS analysis of PG digests.85 An Mpl mutant strain that lacks the ability to recycle the L-Ala-D-iGlu-m-DAP tripeptide back into PG precursors did not incorporate AeK-NBD (33) into the cell wall, confirming the anticipated Mpl-dependent, recycling pathway-mediated route of labeling.85 In contrast to the D-amino acid mono- and dipeptide reporters, no incorporation of AeK-NBD (33) was observed at the septa of dividing bacteria, which was attributed to rapid amidase-catalyzed removal of the labeled peptide from PG.85 This finding demonstrates how reporter probes can be used to investigate degradative cell envelope processes in addition to synthetic processes. Taken together, the ability to probe Mpl-mediated recycling and downstream PG synthesis and break down in Gram-negative bacteria is a valuable entry into the PG toolbox. Although, the AeK scaffold does have some limitations. For example, recycled cytoplasmic tripeptides can be degraded in addition to being reused for PG synthesis138,139 and the presence of L-amino acids in the probe may prevent or limit its application in host infection models.
The above-described mono-, di-, and tripeptide derivatives all install a tag on the PG stem peptide and respectively report on TPase-mediated periplasmic D-amino acid exchange, cytoplasmic MurF-initiated PG synthesis, and Mpl-mediated recycling and reincorporation into PG. However, these probes do not provide a direct read-out of PG cross-linking events catalyzed by TPases. In 2015, the Spiegel group, which had previously developed an enzymatic PG labeling method for S. aureus exploiting sortase A,140 reported a class of probes referred to as fluorescent stem pentapeptide mimics (FSPPMs).86 FSPPMs are unnatural versions of the donor stem peptide that were designed to undergo TPase-catalyzed cross-linking with a native PG acceptor stem peptide (Figure 13A).86 The probe design was based on the stem pentapeptide’s C-terminal tripeptide portion, L-Lys-D-Ala-D-Ala, whose N-terminus was synthetically connected via a flexible linker to either fluorescein, giving FSPPM-488 (34), or Alexa Fluor 568, giving FSPPM-568 (35) (Figure 13A). The L-Lys-D-Ala-D-Ala tripeptide scaffold was chosen because it was more synthetically accessible than full stem pentapeptide mimics, and it was known to be recognized by PBP TPases.141 Despite their truncation, the probes functionally mimic the endogenous stem pentapeptide donor substrates of PBP D,D-TPases, since they contain the terminal D-Ala moiety at the 5th position. Therefore, it was anticipated that both compounds would be incorporated by D,D-TPases. In principle, the probes could also be incorporated by L,D-TPases if the terminal D-Ala residue were trimmed by CPase first. The idea was tested in Gram-positive S. aureus, whose PG has a pentaglycine bridge serving as the cross-link that connects adjacent stem peptides, as shown in Figure 13A. Compounds 34 and 35 were both efficiently incorporated into the surface of S. aureus, whereas diastereomeric control compounds with L-configured D-Ala residues did not; signal from the probes also co-localized with signal from fluorescent vancomycin, which binds D-Ala-D-Ala.86 Direct evidence of covalent modification of the PG stem peptide was rigorously obtained by treating S. aureus with FSPPM-488 (34), digesting PG specifically at the pentaglycine bridge using the enzyme lysostaphin, isolating the labeled peptide fragment by LC, and confirming its identity using MS comparison to a synthetic standard.86 Next, using gene deletion mutants and specific chemical inhibition, it was unambiguously confirmed that the D,D-TPase PBP4 was solely responsible for incorporation of the FSPPM probes in multiple S. aureus strains, revealing a surprisingly high degree of selectivity given the ability of other PBP isoforms to bind the stem tripeptide motif.86 This unanticipated selectivity enabled imaging experiments with FSPPM-568 (35) that showed PBP4 activity is localized to the septum during cell division86 and in follow-up work that PBP4 also mediates PG cross-linking along the sidewall of S. aureus, an unprecedented finding.142 In addition to imaging PG cross-linking, the highly selective activity-based FSPPMs could also be valuable for PBP4-targeted antibiotic discovery and characterization in the future.
Figure 13.
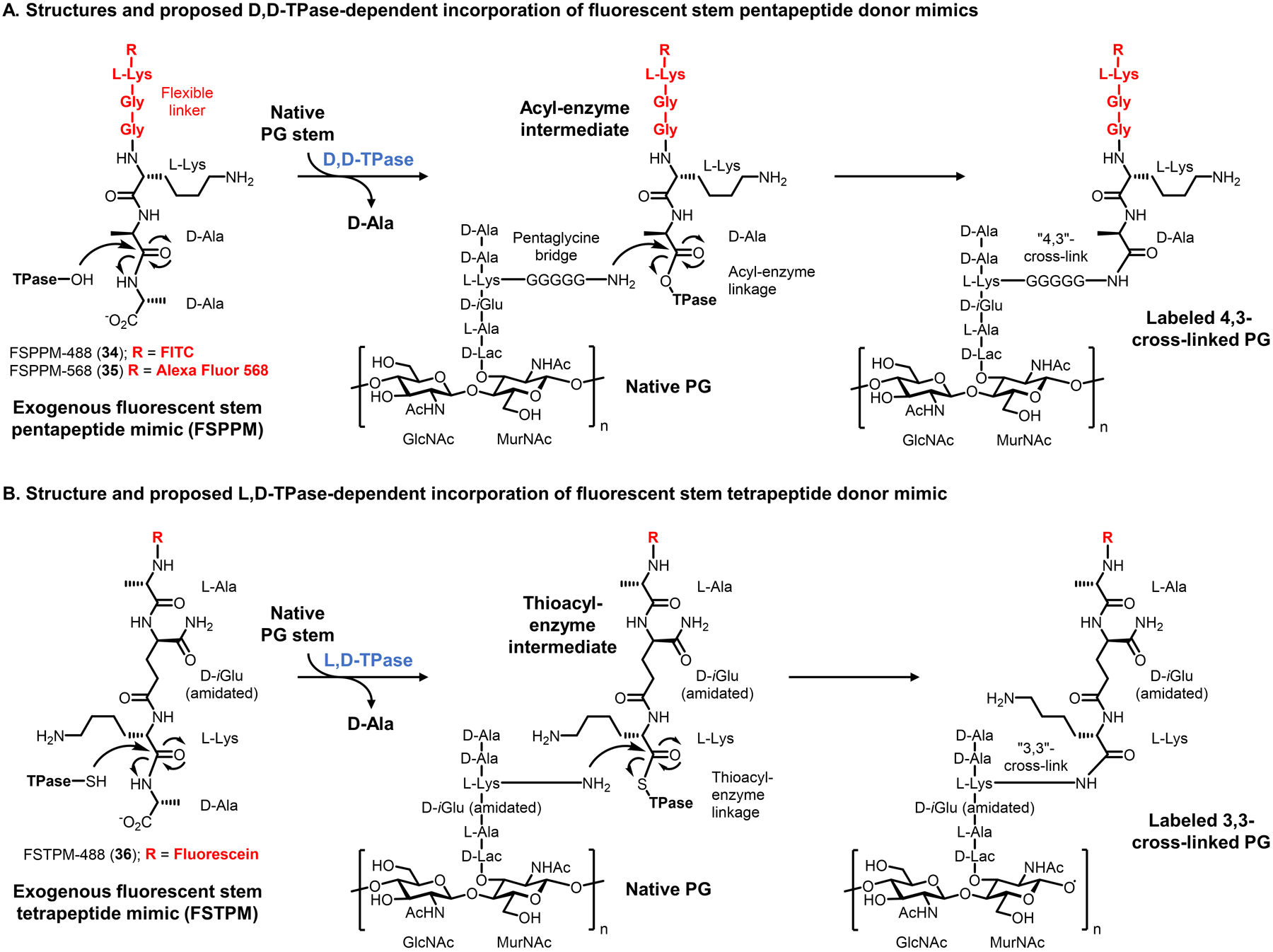
Fluorescent reporter probes for TPase-mediated PG cross-linking. (A) Fluorescent stem pentapeptide mimics resembling the donor strand (FSPPM) are proposed to incorporate via D,D-TPase activity as shown for S. aureus PG; FSPPMs could theoretically also incorporate via L,D-TPase activity if the terminal D-Ala were cleaved first by a CPase. (B) Fluorescent stem tetrapeptide mimics resembling the donor strand (FSTPM), which lack the terminal D-Ala of FSPPMs, are proposed to incorporate exclusively via L,D-TPase activity.
PBP D,D-TPases generate 4,3-cross-links and are the primary catalysts responsible for forming PG cross-links in most bacteria. However, it was recently discovered that many bacteria possess a different class of transpeptidases called L,D-TPases, which form the less common 3,3-cross-links in many types of bacteria.73,74,143 In some organisms, such as Mycobacterium and Enterococcus faecium, 3,3-cross-links are predominant, meaning that in these contexts L,D-TPases likely play a critical role in PG synthesis and maintenance.143 In 2019, the Pires group reported the first probes that specifically report on L,D-TPase activity in live bacteria.87 Similar to the Spiegel group’s approach, fluorescent stem peptide mimics were employed, but in this case a tetrapeptide-mimicking reporter (FSTPM-488, 36, Figure 13B) was designed instead of pentapeptide-mimicking reporters. In the tetrapeptide scaffold, the missing 5th position D-Ala residue would render the probes incompetent to serve as donors for D,D-TPases, and instead they would serve only as donors for L,D-TPases, in effect selectively installing a label in 3,3-cross-links (Figure 13B).87 After confirming L,D-TPase-mediated incorporation of the FSTPM (36) into E. faecium PG using approaches described above for PG probes, the fluorescent stem peptide-mimicking probes were used in imaging experiments to compare the spatial organization of D,D- and L,D-TPase activity within live bacteria.87 Intriguingly, the probes revealed that D,D-TPase activity in E. faecium (determined using FSPPM probe) was restricted to the septa of cells, whereas L,D-TPase activity (determined using FSPTM probe) was present both at the septa and sidewalls; differences in cellular location of D,D- and L,D-transpeptidase activity were also observed in mycobacteria.87 In a subsequent study that built on the acyl donor-mimicking probes (FSPPMs and FSTPMs), the Pires group created exclusively acyl acceptor-mimicking fluorescent probes by completely removing the stem peptide’s terminal D-Ala-D-Ala fragment.144 Taken together, the development of these various fluorescent stem peptide mimics demonstrates how careful tailoring of the probe scaffold—informed by detailed knowledge of PG structure and biosynthesis—can yield complementary probes that allow dissection of PG biosynthesis, remodeling, and recycling pathways with high precision.
2.2. Chemical reporters for the PG glycan core
As discussed in the preceding section 2.1, the large collection of readily accessible, well-characterized chemical reporters that label the PG stem peptide offer exceptional experimental versatility. However, certain questions regarding PG structure, dynamics, and function cannot be addressed using these probes. For example, PG in both commensal and pathogenic bacteria can be enzymatically turned over to release small fragments called muropeptides, which generally refer to structures consisting of GlcNAc–MurNAc disaccharide or MurNAc monosaccharide containing stem peptides that are often truncated (2–5 amino acid residues).145 Once released, muropeptides have diverse downstream effects, as they can: (i) be recycled to support further PG synthesis; (ii) induce antibiotic resistance by activating expression of β-lactamases; (iii) communicate with other bacteria in the surrounding milieu; and/or (iv) elicit an immune response from the host organism.20,145 Because the stem peptide portion of muropeptides is often trimmed (e.g., in the well-known muropeptides, muramyl di- and tripeptide), stem peptide-targeting PG reporter probes are not well-suited to tagging and analyzing PG break down products. Carbohydrate-based chemical reporters that install a label into PG glycan core component(s) would address this problem. Below, we discuss efforts to develop N-acetylglucosamine 1-phosphate and MurNAc derivatives as chemical reporters for the PG glycan core.
2.2.1. N-Acetylglucosamine 1-phosphate derivatives.
The Nishimura group, which in 2002 disclosed the first PG stem peptide-targeting reporters (section 2.1.1),75 revisited the area several years later in 2008 to pursue PG reporters that modify the glycan core.88 Their approach was to intercept PG biosynthesis very early in the pathway, upstream of UDP-GlcNAc. In bacteria, fructose-6-phosphate is converted to UDP-GlcNAc via the intermediate GlcNAc-1-phosphate, and UDP-GlcNAc is then used in PG biosynthesis (Figures 14A and 5).146 It was hypothesized that GlcNAc-1-phosphate-based reporters could enter the pathway at this juncture and undergo subsequent incorporation in the PG glycan core, either the GlcNAc or MurNAc residues of the repeating disaccharyl moiety. This approach, involving relatively simple monosaccharide reporters, would be beneficial from a synthetic standpoint compared to the team’s earlier highly complex PG precursor glycopeptides. However, GlcNAc-1-phosphate analogues would also face challenges with respect to cell envelope permeability and the large number of biosynthetic steps required for incorporation.
Figure 14.
(A) Biosynthesis of UDP-GlcNAc from fructose-6-phosphate proceeds via a GlcNAc-1-phosphate intermediate in the cytoplasm (note: GlmU is bifunctional). Subsequent incorporation of UDP-GlcNAc into PG is shown in Figure 5. (B) GlcNAc-1-P analogues as potential chemical reporters for the PG glycan core.
Several GlcNAc-1-phosphate analogues were designed, including three analogues substituted at the 2-N-position with ketone functional groups (37–39) that are capable of participating in selective reactions with α-effect nucleophiles.88 These structures, all synthesized via relatively short routes from glucosamine, exhibted variable protection on the polar functional groups in order to promote membrane permeability, including hydroxyl-masking acetyl groups (in both 38 and 39) and a monophosphate-masking cyclosaligenyl group (in 39). Both of these modifications were anticipated to increase diffusion across membranes, after which they would possibly be removed by intracellular esterases or pH difference, respectively, to release the “free” reporter molecule. Hydroxyl group acetylation is a common and frequently successful strategy for increasing metabolic labeling efficiency of carbohydrate-based chemical reporters, although there have recently been reports of off-target labeling for peracetylated sugars in mammalian systems.147–149 In addition to 37–39, control compounds lacking the 1-phosphate group (40) and native GlcNAc-1-phosphate were synthesized.88 The compounds were evaluated for incorporation into similar Gram-positive bacteria (Lactobacillus plantarum and Weissella confusa) by sequential incubation of bacteria with 5–50 mM concentrations of reporter, treatment with a ketone-reactive biotin-PEG-hydrazide reagent, staining with avidin-488 fluorophore, and flow cytometry analysis.88 While the unprotected parent analogue (37) did not label cells, the peracetylated version (38) did, as did the fully masked version (39), albeit less efficiently than 38.88 This suggested that the lipophilicity-increasing acetyl functionalities increased incorporation of the compounds into cells. The control compounds (40 and GlcNAc-1-phosphate) did not incorporate, indicating that the phosphate at the 1-position and the ketone group were necessary to achieve detectable incorporation.88 Beyond the results from these control compounds, there were not experiments done to obtain direct evidence of incorporation of 38 or 39 into PG precursors or mature PG, or to establish the route of cell surface incorporation. Thus, while high concentrations of GlcNAc-1-phosphate derivatives were shown to label the surface of a small set of Gram-positive bacteria, additional studies will be needed to determine whether they incorporate with specificity into the PG glycan portion. Assuming success on this front, it would also be worthwhile to expand the reactive handles present on the N-acyl substituent beyond ketones to accommodate more modern bioorthogonal chemistries (e.g., azido or alkyne groups), as well as to test the probes in additional species.
2.2.2. N-Acetylmuramic acid derivatives.
Motivated in part by the desire to probe carbohydrate-containing immunogenic PG break down products in complex environments, in 2017 the Grimes group reported an alternative approach to labeling the PG glycan core that targeted the MurNAc residue.89 Taking a cue from previously observed natural modifications of the MurNAc 2-N-substituent,150 the team reasoned that small bioorthogonal tags at this position might be tolerated by the PG biosynthetic machinery. Furthermore, some bacteria (e.g., Pseudomonas putida) possess the cytoplasmic recycling enzymes AmgK and MurU, which convert MurNAc recovered from PG break down into UDP-MurNAc that can eventually be re-incorporated into PG (Figures 15A and 5).151 Assuming that AmgK and MurU exhibited sufficient substrate tolerance, targeting this recycling pathway for PG glycan labeling could, in principle, be accomplished using relatively simple MurNAc analogues, which would avoid the synthesis and membrane permeability complications associated with complex and highly polar sugar phosphate, UDP-sugar, and lipid-linked sugar reporters (e.g., 1–4, Figure 6). Another attractive feature of this approach is that MurNAc is only present in bacteria, so it may provide opportunities for specific targeting of bacterial PG within host systems.
Figure 15.
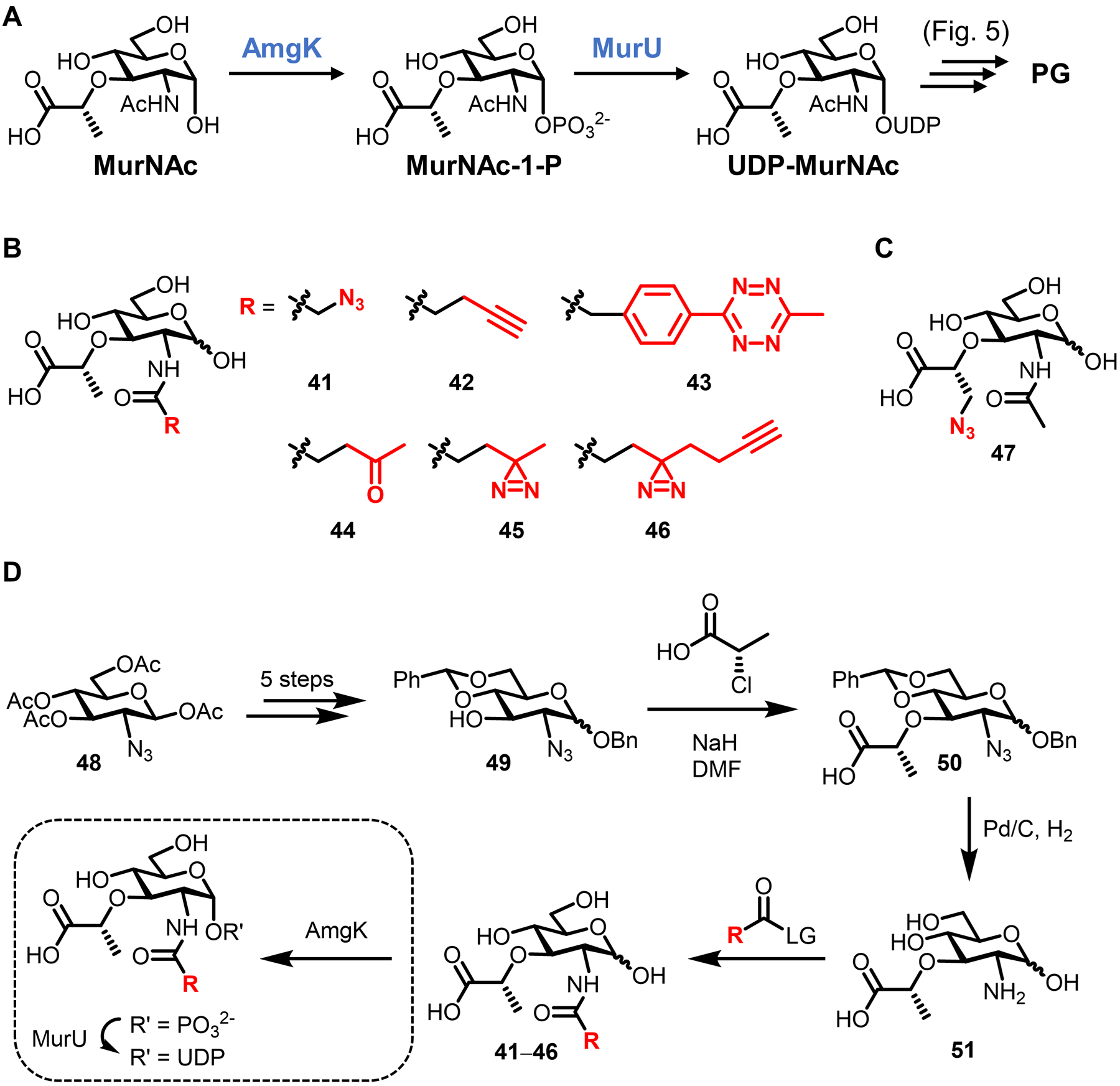
(A) Cytoplasmic recycling pathway for MurNAc in P. putida. Since this pathway is not present in many bacteria, it must be introduced via genetic engineering in those cases. (B) Representative MurNAc derivatives for labeling the PG glycan core, modified at the N-acyl group (B) and the D-Lac group (C). (D) Chemical synthesis of N-acyl-modified MurNAc derivatives 41–46 and chemoenzymatic synthesis of their corresponding sugar phosphate and UDP sugar derivatives (box).
In their initial report, Grimes and co-workers designed two clickable MurNAc reporters bearing azido and alkyne groups on the 2-N-acyl substituent (41 and 42, Figure 15B).89 The compounds were synthesized using a modular route requiring 10 steps from commercially available glucosamine (abbreviated scheme shown in Figure 15D). The known peracetylated azido sugar 48 was elaborated in 5 steps to benzylidene-protected intermediate 49, whose free 3-OH was subjected to SN2 reaction with (S)-2-chloropropionic acid under basic conditions, yielding the D-Lac-substituted intermediate 50. Global deprotection of 50 via Pd-catalyzed hydrogenolysis revealed a primary amine at the 2-N-position, which could then be coupled to various activated esters. In this case, NHS esters were used to install the azido- and alkyne-containing N-acyl groups to generate 41 and 42.
According to results from experiments with purified enzymes, both 41 and 42 were processed by AmgK and subsequently by MurU, implying that the MurNAc recycling pathway possesses the appropriate substrate tolerance to support metabolic incorporation of MurNAc derivatives.89 Cell uptake and labeling experiments were done in the E. coli strain ΔMurQ-KU, which was engineered to promote desirable probe incorporation by: (i) expressing the MurNAc-recycling enzymes AmgK and MurU from P. putida; and (ii) deleting the MurNAc-degrading MurQ to prevent off-target labeling. First, it was demonstrated that compounds 41 and 42 rescued the growth of E. coli ΔMurQ-KU in the presence of fosfomycin, which inhibits MurA and thus depletes essential UDP-MurNAc.68 Rescue by 41 and 42 likely occurs because these derivatives provide an alternative source of UDP-MurNAc in the presence of AmgK and MurU, which is consistent with the hypothesis that the compounds entered the targeted pathway. Next, imaging studies showed that E. coli ΔMurQ-KU co-treated with 41 or 42 and fosfomycin, then subjected to CuAAC reaction with alkyne- or azido-fluorophore reagent, respectively, led to efficient cell-surface and septal labeling (Figure 16A).89 The requirement to co-treat bacteria with fosfomycin to deplete native UDP-MurNAc, and thus achieve sufficient labeling, may be a limitation in some scenarios. To further establish specific incorporation into PG, it was shown that 41- and 42-dependent fluorescence in E. coli ΔMurQ-KU was reduced by treatment with the PG-digesting enzyme lysozyme. Furthermore, PG fragments released by lysozyme treatment were confirmed to be fluorophore-modified by LC-MS analysis, which provided strong evidence that the MurNAc derivatives were metabolically incorporated into PG in E. coli ΔMurQ-KU. Interestingly, a 2021 follow-up study by the Grimes group showed that peracetylated MurNAc derivatives were not incorporated by E. coli, whereas MurNAc derivatives containing methyl esters in the D-Lac portion showed enhanced incorporation.152 Thus, as we will see throughout this review, peracetylation does not always confer enhanced uptake and incorporation of probes in bacterial systems, likely due to lower nonspecific esterase activity.153 However, in the particular case of MurNAc-based reporters, a more targeted masking strategy focused on the D-Lac portion of the probe proved successful.152
Figure 16.
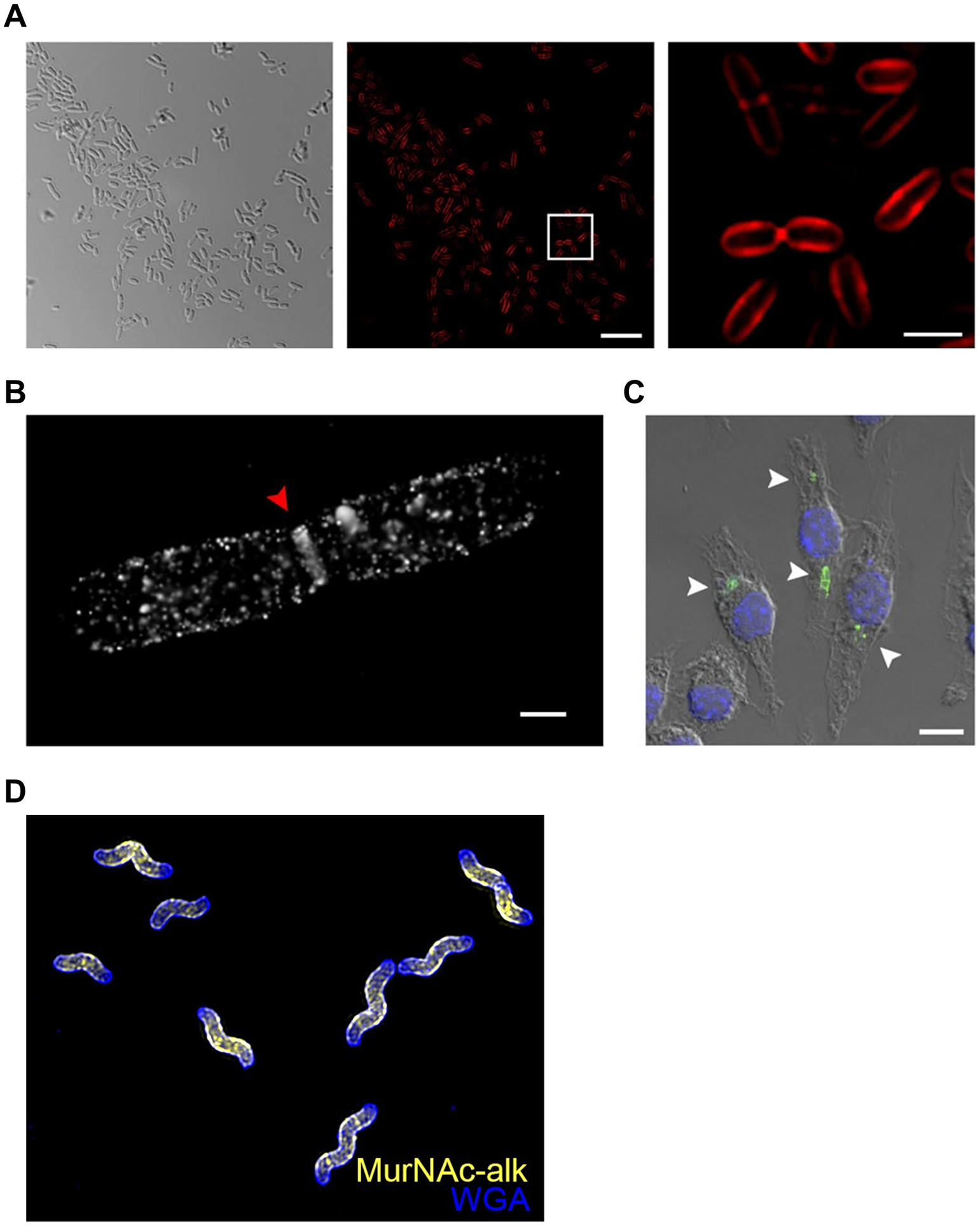
Imaging the PG glycan backbone using MurNAc reporters. (A) E. coli ΔMurQ-KU was incubated in the presence of alkynyl MurNAc 42 and fosfomycin, fixed, subjected to CuAAC with azido-Cy5, and imaged by structured illuminated microscopy (SIM). Left, differential interference contrast (DIC); middle, SIM image; right, boxed area from middle image. (B) E. coli ΔMurQ-KU treated as in (A) and imaged by super-resolution 3D stochastic optical reconstruction microscopy (STORM). Red triangle indicates septal division plane. (C) E. coli ΔMurQ-KU were pre-treated with 42 and fosfomycin, then used to invade J774 macrophages. After fixation, CuAAC with azido-488, and nuclear staining with DAPI, invaded macrophages were imaged. White triangles indicate fluorescent fragments released from intracellular bacteria. Images in (A–C) are reproduced from ref 89. (D) H. pylori HJH1 was incubated in the presence of alkynyl MurNAc 42 for a short pulse, fixed, subjected to CuAAC with azido-Alexa Fluor 555, counterstained with the fluorescent lectin WGA-Alexa Fluor 488, and imaged by 3D SIM. Image in (D) reproduced from ref 154.
As demonstrations of the applications of MurNAc probes, compound 42 was used for super-resolution imaging of the PG glycan core in E. coli ΔMurQ-KU (Figures 16A and 16B) and to visualize uptake and break down of E. coli ΔMurQ-KU by mammalian macrophages (Figure 16C). The latter experiment illustrated the possibility that MurNAc reporters can track muropeptides generated through PG fragmentation within host systems. In addition, the applicability of the labeling method beyond the engineered E. coli ΔMurQ-KU strain was also demonstrated, as compounds 41 and 42 were also shown to successfully incorporate (in the presence of fosfomycin) into Gram-negative wild-type P. putida, which naturally possesses AmgK and MurU, as well as a strain of Gram-positive B. subtilis that was engineered to express AmgK and MurU. Finally, a 2020 study from the Salama group beautifully combined the use of alkyne-modified MurNAc and D-amino acid reporters (42 and 10, respectively) to elucidate a distinctive mechanism of peptidoglycan synthesis in the helix-shaped gastrointestinal pathogen Helicobacter pylori (Figure 16D).154,155 Notably, in this case fosfomycin co-treatment was not required to attain sufficient labeling of H. pylori,154 suggesting that some bacteria can efficiently incorporate MurNAc-based reporters even in the presence of endogenous UDP-MurNAc.
In a subsequent study that expanded upon the MurNAc probe scaffold, the Grimes group made a number of new MurNAc derivatives using the same synthetic route shown in Figure 15D.156 Among the new compounds were tetrazine-modified 43 and ketone-modified 44 for bioorthogonal labeling, diazirine-modified 45 and 46 for photo-cross-linking applications, and a derivative modified with an azido group at the D-Lac residue (47), which could be useful for labeling species that have native 2-N-modifications, such as mycobacteria (Figures 15B and 15C).156 Many of these derivatives were efficiently processed by purified AmgK and MurU enzymes, and in fact these enzymes were also used to chemoenzymatically synthesize milligram quantities of the corresponding UDP-MurNAc derivatives (Figure 15D, box).156 Access to these UDP-MurNAc derivatives enabled enzyme assays with purified MurC–F, which confirmed that the analogues could be successfully converted into UDP-MurNAc-pentapeptide (Park’s nucleotide).156 Together, the Mur enzyme substrate specificity data—afforded by access to a panel of synthetic MurNAc and UDP-MurNAc derivatives—suggested that incorporation of the novel MurNAc derivatives into PG in whole cells was feasible. Among the new MurNAc derivatives, tetrazine-modified 43 and D-Lac-modified 47 were selected for evaluation in E. coli ΔMurQ-KU. Azido MurNAc 47, used in conjunction with an alkyne-fluorophore secondary reagent, labeled E. coli ΔMurQ-KU and signal co-localized with a PG-marking fluorescent lectin, although MS detection of incorporated probe was not achieved, possibly due to low incorporation levels.156 By contrast, tetrazine-modified MurNAc 43, used in conjunction with a TCO-fluorophore secondary reagent, sporadically labeled E. coli ΔMurQ-KU populations, disrupted normal cell morphology, and did not produce signal co-localized with PG-specific lectin staining.156 Thus, while the CuAAC-capable azido and alkyne derivatives of MurNAc (41, 42, and 47) offered consistently robust performance, the tetrazine probe (43) was generally unsuccessful and would likely require further experimentation to be developed into an effective tool. This finding underscores why new reporters, even those based on an established metabolite scaffold, should be carefully evaluated before being applied. Another set of experiments in this study attempted to label whole cells using UDP-MurNAc derivatives, which as discussed above is a challenging prospect since UDP-sugars are charged and have poor membrane permeability. Interestingly, the UDP-linked version of 41 (Figure 15) metabolically incorporated into whole cells of Gram-positive L. acidophilus,156 consistent with Nishimura’s observations from 2002,75 but not into any other bacterial species tested.
Several insights can be gleaned from the above-described work on MurNAc reporters,89,156 which represented the first robust and well-characterized class of probes for tagging the glycan component of bacterial PG. The labeling strategy is notable because it capitalized on a recently elucidated MurNAc recycling pathway from P. putida.151 By engineering this pathway into E. coli and other organisms and co-treating with fosfomycin to deplete native UDP-MurNAc, it became possible to metabolically label the PG glycan strand using exogenously added MurNAc derivatives, highlighting how the introduction of non-native recycling/biosynthesis machinery and specific antibiotics can be exploited in reporter probe development. On the other hand, the need to genetically engineer bacteria to enable MurNAc probe incorporation is also a limitation in some scenarios, since it necessitates additional work to extend the tool to new species and precludes usage in contexts where genetic modification is difficult or impossible (e.g., in genetically intractable organisms or in wild-type bacteria infecting a host cell or organism). The development of the MurNAc family of probes was bolstered by access to a diverse panel of MurNAc and UDP-MurNAc derivatives, which supported detailed analysis of AmgK/MurU enzyme tolerance and set the stage for a versatile whole-cell labeling platform. This underscored the importance of robust and scalable methods to synthesize substrate analogues, particularly carbohydrate-based substrates that are traditionally challenging to produce. Adding to earlier work by Nishimura and co-workers, the MurNAc reporter studies further investigated the idea of labeling bacterial glycans with UDP-sugars, although efforts were met with limited success, perhaps due to poor cell envelope permeability. Regardless, the neutral MurNAc-based probes, when combined with bacterial strains engineered to uptake and incorporate them, are valuable tools for labeling the PG glycan core that are likely to witness increased use, as they complement the multitude of PG stem peptide-labeling probes that are available.
3. Chemical reporters for lipopolysaccharide (LPS)
LPS, also known as endotoxin, is a complex and structurally diverse glycolipid that is embedded in the outer leaflet of the Gram-negative bacterial outer membrane. As a major component of the outer membrane, LPS forms a densely packed permeability barrier that is critical for structural integrity and defense of the cell.157 LPS is also highly immunogenic and considered to be a significant determinant of Gram-negative bacterial pathogenicity.9 The general structure of LPS comprises three main portions that are covalently linked together: lipid A, the core oligosaccharide (consisting of inner and outer core regions), and the surface-exposed O-antigen polysaccharide (Figure 17).9,158 Lipid A, which is responsible for the immunostimulatory activity of LPS, is made up of two β(1→6)-linked glucosamine units, forming a disaccharide that is 1,4’-diphosphorylated and acylated with a total of 4–7 C10–C16 fatty acid chains that anchor LPS into the outer membrane. Connected to lipid A is the LPS core oligosaccharide, which is typically initiated with 1–3 residues of 3-deoxy-D-mannooctulosonic acid (keto-deoxyoctulosonate, KDO), followed by 2–3 residues of variably functionalized heptose sugars. Stemming from these inner core heptose residues is the outer region of the core oligosaccharide, which contains several hexose residues such as glucose and galactose. Connected to an outer core hexose residue is the final and most surface-exposed portion of LPS, a large and diverse polysaccharide referred to as the O-antigen or O-polysaccharide, which itself consists of up to 50 repeating oligosaccharide units, each made up of 2–8 sugar residues. Although the Lipid A and core domains of LPS exhibit some structural variation between organisms,159,160 they are relatively consistent compared to the hypervariable O-antigen domain, whose remarkable species- and strain-dependent structural diversity can be exploited for serological discrimination of Gram-negative bacteria.18 The large majority of Gram-negative species cannot survive without LPS due to its role in maintaining envelope structural integrity,161 and although the O-antigen portion of LPS is dispensable for in vitro growth, it appears to be critical for bacterial evasion of the host immune response in vivo.159 Given the importance of LPS to Gram-negative bacterial survival and the protective effect LPS confers to bacteria during antibiotic treatment, there is intense interest in furthering our understanding of LPS and ultimately in developing LPS-targeting probes and inhibitors that can help to address difficult-to-treat Gram-negative bacterial infections.162–164
Figure 17.
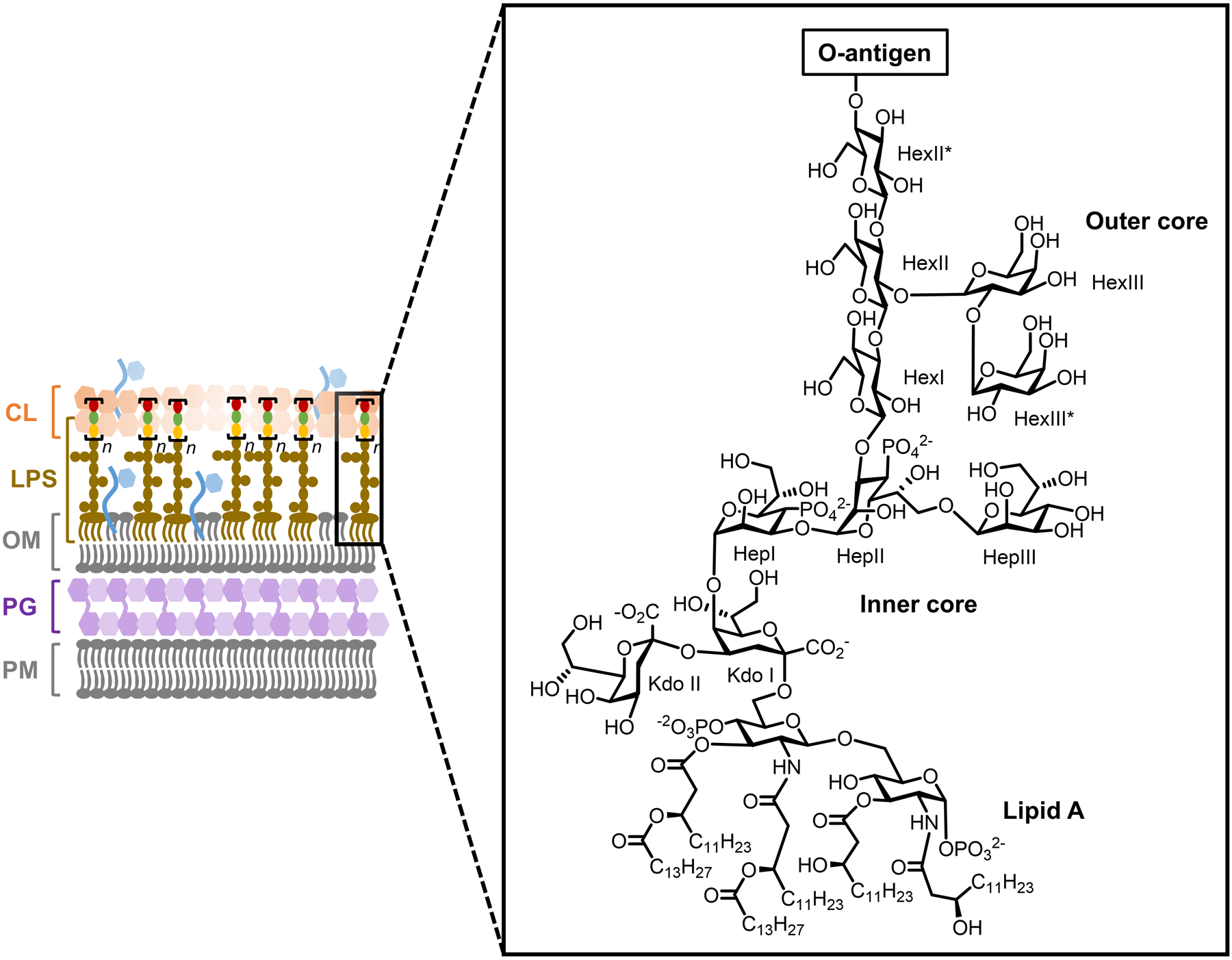
Representative structure of LPS from Gram-negative E. coli. Hep, heptose; Hex, hexose; KDO, keto-deoxyoctulosonate.
The biosynthesis of LPS involves the parallel synthesis of (i) Lipid A with core sugars attached (core-Lipid A) and (ii) O-antigen; these two species are then coupled in the periplasm and flipped across the outer membrane to generate surface-exposed LPS (Figure 18).9,158 Core-Lipid A synthesis occurs on the cytoplasmic side of the plasma membrane, where UDP-GlcNAc is processed by Lpx enzymes to undergo lipidation, self coupling, and phosphorylation to produce the tetralipidated disaccharide Lipid IVA. Subsequently, the inner core KDO units are added to Lipid IVA by the GTase WaaA, utilizing the activated sugar donor cytidine monophosphate (CMP)-KDO. The core-Lipid A species is completed through further lipidation by Lpx enzymes and extension of the core oligosaccharide with additional Waa GTases, followed by translocation into the periplasm by MspB. This core-Lipid A species, which lacks the O-antigen, is also referred to as lipooligosaccharide (LOS). The O-antigen portion of LPS is also initiated on the cytoplasmic face of the plasma membrane, where UPP-linked GlcNAc is first formed by WecA and then extended with nucleotide sugar donors by Wbb GTases, producing the lipid-linked O-antigen repeating subunit. Translocation of the O-antigen subunit across the plasma membrane is followed by Wzy/Wzz-mediated polymerization to form the completed O-antigen. With both the core-Lipid A and O-antigen components completed in the periplasm, they are coupled together by the O-antigen ligase WaaL to form mature LPS, which is finally exported to the surface through Lpt transport machinery.
Figure 18.
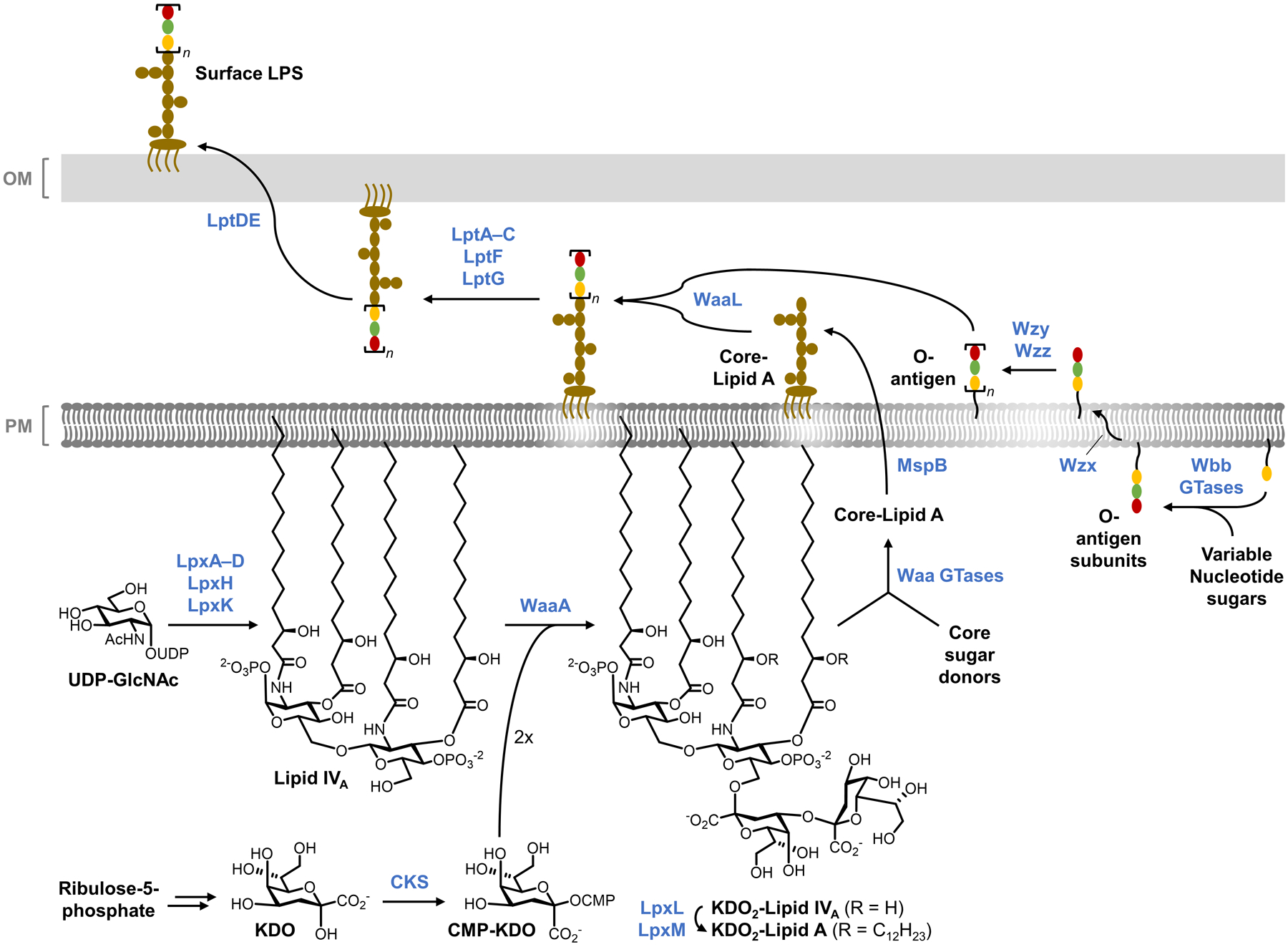
Representative biosynthesis of LPS in Gram-negative E. coli.
To facilitate LPS research, several chemical reporters have been developed for labeling different LPS components in live bacteria. Although the availability and utility of probes for LPS has not reached the level of maturity as that for PG, significant progress has been made. As with PG, the relatively well-characterized state of LPS biosynthesis has guided the development of these probes. To date, the development of chemical reporters for LPS has mainly focused on labeling two different domains of the glycolipid: the conserved inner core region and the variable O-antigen polysaccharide. One consideration here is the level of species selectivity desired for the probe. The LPS inner core region has unique KDO residues that are conserved in almost all Gram-negative bacteria but are absent from other types of bacteria and mammals. Thus, targeting KDO units would provide a labeling tool with specificity for virtually all Gram-negative organisms. On the other hand, the species-specific structures of O-antigen polysaccharides provide opportunities to develop complementary reporters that label particular subsets of Gram-negative bacteria. Whether targeting the core region or O-antigen, the most straightforward metabolic labeling approach would be the use of monosaccharide-based reporters that intercept early steps in the cytoplasmic biosynthesis of the core-Lipid A or lipid-linked O-antigen subunits. This labeling approach stands in contrast to PG labeling, which presents more amenable opportunities for exploiting late-stage periplasmic routes of probe incorporation. It follows that monosaccharide-based LPS labeling approaches would face hurdles such as the requirements of dual membrane penetrance and substrate tolerance for numerous enzymatic and transport steps. Despite these potential issues, it has been demonstrated that LPS biosynthesis is permissive of chemical reporters targeting both the core and O-antigen domains, which we describe in the following sections. Table 2 contains information about selected LPS-targeting chemical reporters.
Table 2.
Selected chemical reporters for lipopolysaccharide (LPS)
| Target | Metabolite | Compound | Reporter group (X) | Bioorthogonal reaction(s) | Proposed route | Speciesa | Applicationsb | Synthesis | Notesa | Refc |
|---|---|---|---|---|---|---|---|---|---|---|
| LPS core | KDO | KDO-N3 55 (Fig. 19) | Azide | CuAAC; SPAAC | Intracellular | Broad (GN) | In vitro imaging and in vivo imaging (colonized host organism); selective detection and killing of GN bacteria | Chemical; comm. available | Discriminates GN from GP and M | 165,166 |
| LPS O-antigen fucose | L-fucose | FucAz 58 (Fig. 21) | Azide | CuAAC | Intracellular | E. coli O86:B7 Δgmd-fcl(fkp) (GN) | In vitro detection | Chemo-enzymatic; comm. available | Requires engineered Fkp fucose salvage pathway | 167 |
| LPS O-antigen legionaminic acid | MandiNAc | 62 (Fig. 22) | Azide | CuAAC | Intracellulard | L. pneumophila (GN) | In vitro imaging; selective detection of L. pneumophila | Chemical | 3-O-acetylation abolished labeling | 168 |
| LPS O-antigen pseudaminic acid | AltdiNAc | Alt-4NAz 70 (Fig. 23) | Azide | SPAAC | Intracellular | P. aeruginosa (GN), A. baumannii (GN), V. vulnificus (GN) | In vitro imaging | Chemical | Peracetylated reporter labeled bacteria, unacetylated version not tested | 169 |
| LOS | Sialic acid | SiaNAz 71 (Fig. 24) | Azide | CuAAC | Intracellular | H. influenzae (GN) | In vitro detection; inhibitor characterization | Chemical | Peracetylation abolished direct labeling of H. influenzae | 170 |
| Sialic acid | Ac5SiaNAz 72 (Fig. 24) | Azide | CuAAC | Host-to-bacterium transfer, intracellular |
H. influenzae (GN) | Detection of glycan transfer from host to H. influenzae, inhibition thereof | Chemical | Peracetylation enabled labeling of host cell glycans, which are unmasked and transferred to H. influenza in unacetylated form | 170 |
Gram classification: GN, Gram-negative; GP, Gram-positive; M, mycobacteria.
In vitro refers to bacterial labeling experiments performed in broth culture; in vivo refers to bacterial labeling experiments performed in a host cell or organism.
Initial publication(s) referenced. See text for references for follow-up studies and applications.
Limited evidence available to support proposed route of incorporation and/or identity of labeled target (see text).
3.1. KDO-based chemical reporters for the LPS core region
In 2012, the Vauzeilles and Dukan groups disclosed a chemical reporter for metabolic labeling of LPS inner core KDO residues.165 Because the vast majority of Gram-negative species have KDO-containing LPS, this tool was expected to enable studies on LPS in a wide range of medically significant bacteria. It was hypothesized that a KDO-based chemical reporter could enter the LPS biosynthetic pathway at an early stage—as long as uptake across the envelope was successful—and ultimately incorporate into the inner core of LPS. KDO is synthesized in the bacterial cytoplasm from ribulose 5-phosphate, which is sequentially elaborated to arabinose-5-phosphate, KDO-8-phosphate, and finally free KDO. Subsequently, free KDO is converted into the CMP-KDO donor sugar and incorporated into LPS as shown in Figure 18. To exploit this pathway, the team designed a KDO analogue with an azido group at the 8-position, KDO-N3 (55, Figure 19A), which in principle could be activated as CMP-KDO and incorporated into LPS in live bacteria. The KDO 8-position is free in LPS, so azido group modification at this site would not be expected to truncate or significantly affect the processing and function of LPS and or its biosynthetic precursors. Furthermore, blockage of the 8-position with an azido group would prevent reverse metabolism of the probe to KDO-8-phosphate. Promisingly, prior research had demonstrated that exogenous radiolabeled native KDO could incorporate into lipid A precursor in Salmonella typhimurium, with the notable caveat that KDO uptake efficiency was poor unless cells were first treated with toluene to enhance permeability of the cell envelope.171
Figure 19.
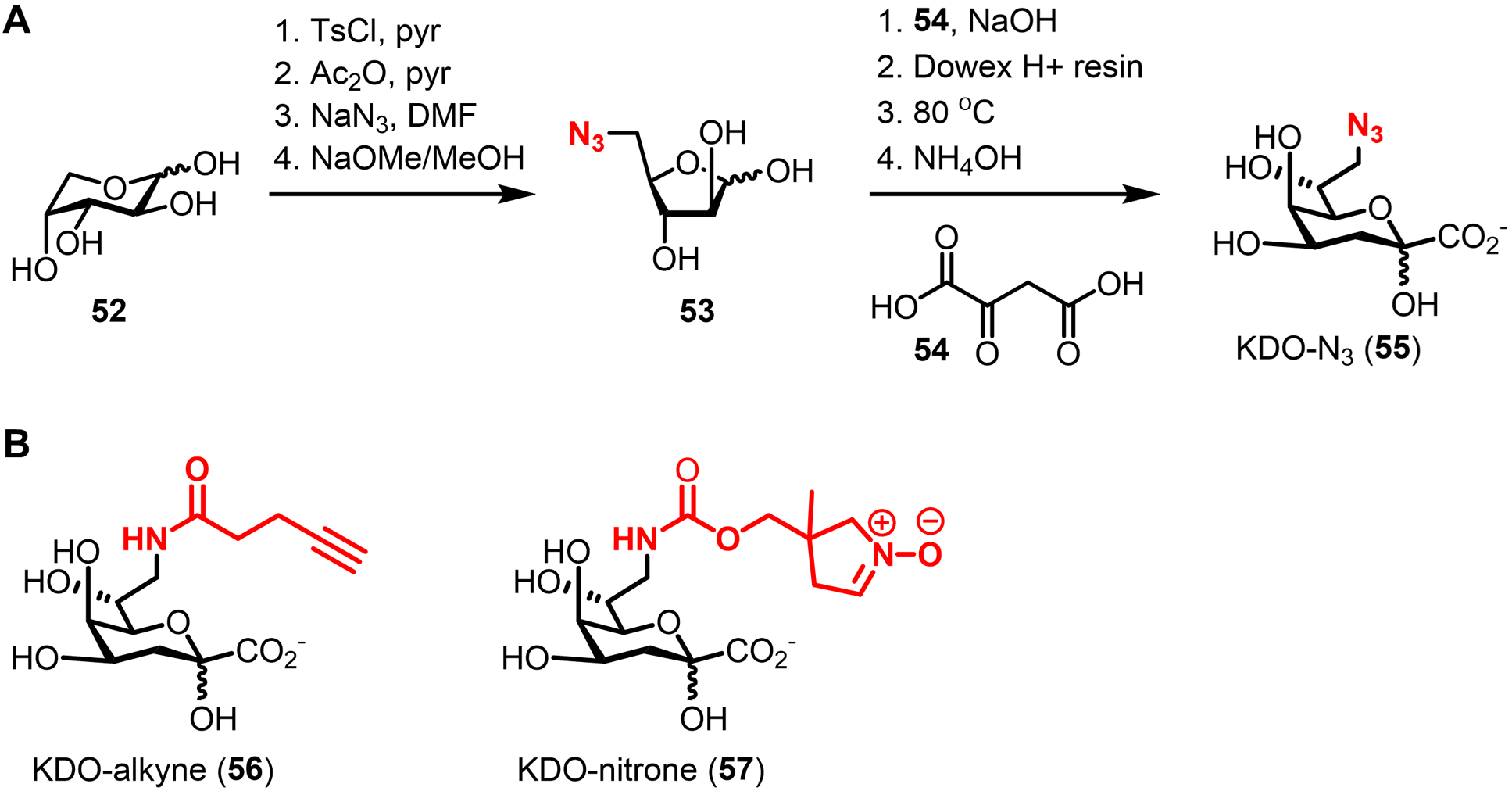
KDO-based chemical reporters. (A) Chemical synthesis and structure of KDO-N3 (55). Ts, tosyl. (B) Structures of alkyne- and endocyclic nitrone-modified KDO analogues (56 and 57).
The synthesis of KDO-N3 required 8 steps from D-arabinose (52) (Figure 19A).165 First, the azido group was installed at the 5-position of D-arabinose through a sequence of tosylation, acetylation, SN2 displacement with azide, and deacetylation. Subsequently, the intermediate 5-azido-5-deoxy-D-arabinose (53) was reacted with oxaloacetic acid (54) in a decarboxylative condensation process that ultimately yielded KDO-N3 (55). Using this 8-step procedure, it is possible to generate large amounts of KDO-N3, although the pH of the key condensation reaction must be carefully controlled. In the future, a reported enzymatic method for the synthesis of native KDO172 could be explored as a shorter route to KDO-based chemical reporters. With KDO-N3 prepared, it was tested in non-pathogenic E. coli K12, which lacks the O-antigen portion of LPS.165 Following treatment of E. coli with KDO-N3 and subsequent CuAAC reaction with an alkyne-488 fluorophore, cell surface fluorescence was detected by microscopy (Figure 20A).165 Notably, even with relatively high concentrations of probe (4 mM), only moderate signal-to-noise ratios (~4) were observed, implying that incorporation, while successful, was relatively inefficient.165 As noted above, this could have been due to poor uptake of KDO-N3 across the cell envelope or low tolerance of biosynthetic machinery for unnatural substrates. The final experiment in the initial report from the Vauzeilles and Dukan groups demonstrated that KDO-N3 specifically labeled Gram-negative bacteria with KDO-containing LPS, but did not label control species lacking KDO-containing LPS (e.g., Gram-negative Shewanella oneidensis; Gram-positive B. subtilis and S. aureus).165 This result was consistent with KDO-N3 labeling of LPS, although there were no experiments performed in this study to directly confirm KDO-N3 incorporation into LPS, nor to demonstrate that incorporation occurred metabolically via the targeted KDO pathway.
Figure 20.
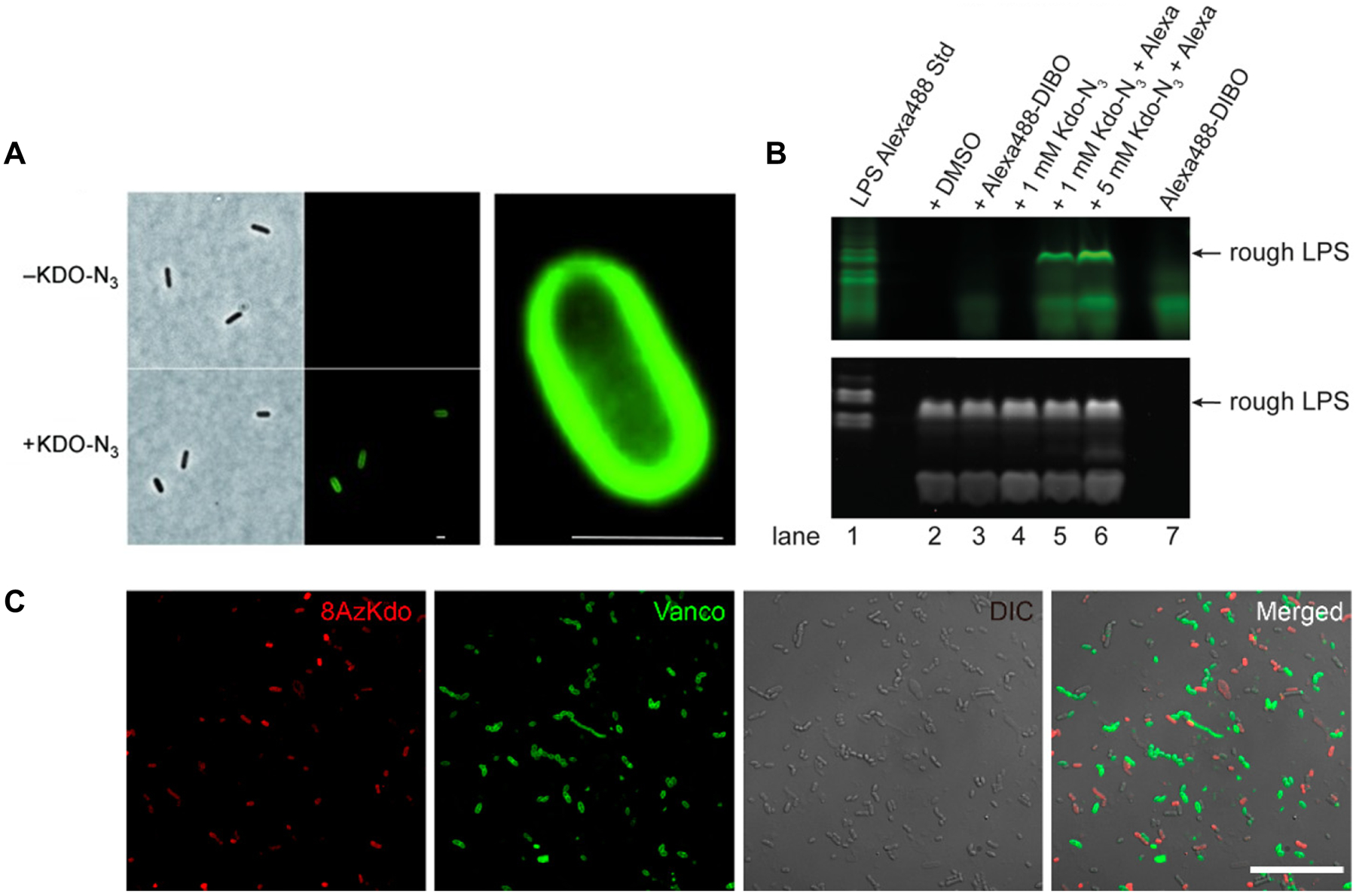
Imaging the LPS core region using KDO reporters. (A) E. coli K12 was incubated in the presence of KDO-N3 (55), fixed, subjected to CuAAC with azido-488, and imaged by fluorescence microscopy. Right, expanded deconvoluted image of labeled E. coli cell. Scale bars, 1 μm. Reproduced with permission from ref 165. Copyright 2012 Wiley-VCH. (B) E. coli was incubated in the presence of KDO-N3 (55) and subjected to SPAAC with a cyclooctyne-488 reagent. LPS was extracted and separated by SDS-PAGE and visualized by fluorescence imaging (top) or by LPS staining with Pro-Q Emerald 300 LPS staining kit (bottom). Reproduced with permission from ref 166. Copyright 2017 American Society for Biochemistry and Molecular Biology. (C) Mouse gut microbiotas were labeled with KDO-N3 (55, shown in red text as 8AzKdo), subjected to CuAAC with alkyne-TAMRA, labeled with fluorophore-conjugated vancomycin (shown in green text as Vanco), and imaged by confocal fluorescence microscopy. Fluorescence, DIC, and merged images are shown. Scale bar, 20 μm. Reprinted with permission from ref 173. Copyright 2017 American Chemical Society.
In 2017, the Six group followed up on KDO-N3 labeling with a detailed characterization of the reporter’s metabolic incorporation behavior in E. coli.166 Rather than CuAAC reaction, SPAAC reaction with a cyclooctyne-488 fluorophore was used for detection of incorporated KDO-N3.166 First, it was demonstrated that KDO-N3-dependent signal was localized to the outer membrane by carrying out an osmotic shock experiment that allowed differentiation of the inner and outer membranes by fluorescence microscopy.166 Next, KDO-N3 incorporation into LPS was confirmed by SDS-PAGE analysis of extracts from KDO-N3-labeled E. coli, which was aided by the use of a LPS biosynthesis mutant strain and commercially available LPS staining kit and fluorescent standard (Figure 20B).166 Based on SDS-PAGE analysis, it appeared that KDO-N3 incorporated both into so-called deep-rough and rough LPS, which contain the inner core region only or the complete core (inner and outer regions), respectively.165 Moreover, direct evidence of KDO-N3-labeled LPS intermediates was obtained by MS analysis, which identified azide-labeled versions of KDO1- and KDO2-Lipid IVA in E. coli extracts.166 These experiments were performed in a WaaC-deficient mutant, which produces truncated forms of LPS that made MS analysis more amenable.166 It was also found that the efficiency of KDO-N3 incorporation relative to native KDO is quite low (<1%),166 consistent with the fluorescence labeling results of Dukan and Vauzeilles. CMP-KDO synthase (CKS), which initiates KDO-N3 labeling following uptake, was found to be a likely bottleneck for incorporation, although other upstream or downstream steps could also have contributed to the low incorporation observed.166 To explore this issue further, the Six group published a follow-up study in 2018 that identified the sialic acid transporter NanT as being required for uptake and incorporation of KDO-N3 in E. coli.174 Indeed, out of various Gram-negative organisms tested, those containing NanT were labeled by KDO-N3, while those lacking NanT were not.174 Furthermore, NanT-overexpressing strains exhibited enhanced labeling, suggesting a plausible strategy to enable KDO-N3 LPS labeling in NanT-deficient bacteria.174 The rigorous characterization work by the Six group clearly demonstrated that KDO-N3 metabolically incorporates into the conserved core region of LPS as initially proposed by Vauzeilles and Dukan, albeit with some limitations in NanT-deficient bacteria that can potentially be addressed through pathway engineering.
Since the initial development and subsequent validation of KDO-N3 as a reliable chemical reporter for LPS, it has been applied in various ways, though mainly as a tool to enable the detection and discrimination of Gram-negative bacteria in complex settings. Many bacterial detection tests require an initial pre-enrichment of particular bacterial species via culture, which can be burdensomely slow. Metabolic labeling using bacteria-specific reporters can potentially offer a way to rapidly enrich and detect organisms of interest without a culture step. In a proof-of-concept study in 2015, Vauzeilles and Dukan demonstrated that KDO-N3 labeling combined with SPAAC-mediated biotinylation enabled specific avidin-mediated fluorescence detection or magnetic bead capture of live, culturable E. coli from a mixture containing either dead E. coli or live B. subtilis.175 A subsequent study by Pezacki used bioorthogonal non-canonical amino acid tagging of proteins to differentiate between live and dead bacteria, while KDO-N3 was used in parallel to identify the captured bacteria as Gram-negative.176 Surprisingly, in contrast to the 2015 study,175 in this case KDO-N3 was unsuccessful at capturing E. coli on beads, perhaps due to differences in the experimental setup, such as the use of CuAAC rather than the previously reported SPAAC.176 Another example of KDO-N3 being used to differentiate bacterial types was reported in 2017 by the Chen group, which used chemical probes in combination with fluorescence-activated cell sorting and sequencing to show that KDO-N3 selectively labeled the Gram-negative sub-population in complex gut microbiotas collected from mice, whereas fluorescent vancomycin selectively labeled the Gram-positive sub-population (Figure 20C).173 This capability was extended to image the Gram-negative and -positive sub-populations of the gut microbiota in the intestines of mice.173 Also in 2017, Kasper and co-workers demonstrated successful in vitro KDO-N3 labeling of a wide variety of Gram-negative species that populate the intestines.177 In the same study, KDO-N3 labeling was combined with other probes to simultaneously image the LPS, PG, and capsular polysaccharide components of the symbiotic Gram-negative bacterium Bacteroides vulgatus within macrophages and mice (this work is covered in more detail in section 7.1).177 Both of these examples illustrated the potential of chemical reporters to track the fate of bacterial glycans within complex host-derived samples.173,177 In a more recent therapeutically-driven application of KDO-N3, the Liu group exploited KDO-N3 labeling and click chemistry to deliver a bifunctional probe with fluorogenic and singlet oxygen-producing photosensitizer moieties specifically to Gram-negative bacteria, which enabled selective fluorescence detection and light-mediated elimination, respectively.122
Some research has been done to develop novel KDO-based reporters containing complementary bioorthogonal handles. The Pezacki group developed new alkyne- and endocyclic nitrone-modified KDO analogues (56 and 57, Figure 19B) to test the bioorthogonality of a modified Kinugasa reaction, termed copper-catalyzed alkyne–nitrone cycloaddition followed by rearrangement (CuANCR).178 Surprisingly, the relatively bulky nitrone-containing reporter 57 incorporated into LPS in E. coli and was detected following CuANCR reaction with an alkyne-488 fluorophore, indicating that the LPS biosynthetic machinery exhibits sufficient tolerance for larger modifications at the KDO 8-position.178
Overall, KDO-based reporters, namely KDO-N3, have proven to be highly valuable tools due to their ability to specifically and metabolically tag the conserved core region of LPS. This renders the probes as ideal for discriminating live Gram-negative bacteria from other types of bacteria, from dead bacteria, and from host cells, which in turn has led to novel strategies for the selective detection and/or elimination of Gram-negative bacteria in complex environments. Unlike the PG-targeting reporters covered above, KDO-based reporters have not yet been applied to provide insights into the dynamics of LPS biosynthesis and assembly during bacterial growth and division, which remains an open opportunity. Expanding on the KDO probe scaffold can also be investigated, as the three published KDO-based reporters containing azido, alkyne, and nitrone groups have been shown to successfully incorporate into LPS. New probes with complementary reporter groups or other chemical cargo would add experimental versatility to the toolbox, while simultaneously revealing useful information about the tolerance of the LPS biosynthetic pathway for unnatural substrates. The careful characterization work performed for KDO-N3 by Six and co-workers provides a blueprint for the rigorous future development and application of this class of probes.
3.2. Chemical reporters for LPS O-antigen polysaccharides
The O-antigen polysaccharide region of LPS varies significantly between species (inter-species variability), and between strains of the same species (inter-strain variability), as discussed above. Therefore, chemical reporters designed to tag specific monosaccharide residues of particular organisms’ or strains’ O-antigen structures may not exhibit the broad applicability of KDO-based reporters, but they would be expected to be valuable tools for the identification of specific bacterial species and strains, as well as for probing particular O-antigen pathways and structures. Several examples of such reporters have been developed to date and are described below.
3.2.1. Fucose derivatives.
In 2009, the Wang group reported the first system for labeling LPS by genetically introducing a salvage pathway into E. coli O86 that permitted uptake and incorporation of L-fucose analogues into its O-antigen.167 The E. coli O86 O-antigen polysaccharide consists of a pentasaccharide repeating unit containing N-acetyl-D-galactosamine, D-galactose, and L-fucose residues (Figure 21A).179 Prior to polymerization in the periplasm, O-antigen subunits are synthesized in the cytoplasm by glycosyltransferase-catalyzed addition of activated monosaccharide donors onto a lipid carrier (see Figure 18). It was hypothesized that an analogue of the fucose donor, guanosine diphosphate (GDP)-fucose, could incorporate into O-antigen fucose residues. However, as described above, reporters based on nucleotide diphosphate sugars have poor membrane permeability and generally have not been successful. To circumvent this issue, the Wang group engineered E. coli O86:B7 to replace the de novo fucose biosynthesis pathway with a fucose salvage pathway from Bacteroides species.180 The salvage pathway consists of a bifunctional enzyme Fkp, which allows externally acquired fucose to be phosphorylated by fucokinase activity and activated into GDP-fucose by GDP-fucose pyrophosphorylase activity, setting the stage for subsequent incorporation into O-antigen (Figure 21B).167
Figure 21.
Fucose-based chemical reporters for metabolic labeling of E. coli O86 LPS O-antigen. (A) Structure of the E. coli O86 LPS O-antigen. (B) Fucose salvage pathway from Bacteroides engineered into E. coli O86. The bifunctional enzyme Fkp possesses both fucose kinase and GDP fucose pyrophosphorylase activity. Subsequent polymerization of the O-antigen subunit and assembly into mature LPS is shown in Figure 18. (B) Representative fucose analogues as chemical reporters for the E. coli O86 LPS O-antigen.
A number of fucose analogues were efficiently chemically synthesized from L-fucose to test for incorporation into E. coli O86 LPS, including compounds 58–61, all of which contained bioorthogonal modifications at the 6-position, distal to the anomeric center (Figure 21C).167 Most of the compounds were converted into their corresponding GDP-fucose derivatives by purified Fkp with comparable efficiency to native fucose, indicating that the salvage pathway possessed the necessary substrate promiscuity.167 In engineered E. coli cells, the azido fucose analogue FucAz (58) was successfully incorporated into the cell surface, as judged by CuAAC reaction with alkyne-biotin, incubation with avidin-488, and flow cytometry analysis.167 Furthermore, LPS extracted from FucAz-treated E. coli cells was confirmed to be azide-labeled by both SDS-PAGE with in-gel fluorescence scanning and capillary electrophoresis-MS/MS.167 Tandem MS analysis detected azide-labeled fucose, but not native fucose, in the O-antigen repeating unit from FucAz-treated cells, indicating that complete replacement of the unmodified fucose residue was accomplished.167 Thus, genetic manipulation to remove de novo monosaccharide synthesis and replace it with a corresponding salvage route enabled homogeneous installation of exogenously added functionalized monosaccharides into the O-antigen polysaccharide. This engineering step represents a potentially general approach to developing chemical reporter systems for labeling bacterial polysaccharides, so long as the organism and biosynthetic pathways are amenable to genetic modification. The specific system developed by Wang and co-workers offers a strategy for studying the role of fucosylation in LPS structure and function in E. coli and potentially other Gram-negative pathogens, which is valuable given the established roles of surface fucosylation in bacterial adhesion and host immunomodulation.181
3.2.2. Legionaminic acid precursor derivatives.
In 2014, the same groups that developed KDO-N3, the Vauzeilles and Dukan groups, reported a new chemical reporter targeting the LPS O-antigen of Legionella pneumophila, a Gram-negative bacterial pathogen that can cause a life-threatening form of pneumonia known as Legionnaires’ disease.168 Rapid and accurate detection of L. pneumophila in infected hosts and in potential environmental reservoirs of the bacterium, including man-made water systems, is critical to the prevention and management of outbreaks.182 The LPS of L. pneumophila has a distinctive O-antigen proposed to consist of an α(2→4)-linked homopolysaccharide of the legionaminic acid derivative 5-N-acetimidoyl-7-N-acetyllegionaminic acid (Leg5Am7Ac), which represents a possible marker for specific detection of this bacterium (Figure 22A).183 Ultimately, Leg5Am7Ac units in the O-antigen arise from the activated monosaccharide donor cytidine monophosphate (CMP)-N,N’-diacetyllegionaminic acid (CMP-Leg5Ac7Ac), which is biosynthesized from UDP-GlcNAc through the key intermediate 2,4-diacetamido-2,4,6-trideoxy-D-mannopyranose (MandiNAc) by NeuB-catalyzed aldol reaction and NeuC-catalyzed CMP activation (Figure 22B).184 MandiNAc was identified as a promising point in the pathway for interception by a chemical reporter. Possible justification for this choice is that the upstream intermediate is a charged nucleotide sugar donor likely to suffer from permeability issues, while downstream intermediates are charged and more complex legionaminic acid derivatives, which could present both permeability and synthetic challenges. Therefore, two chemical reporters based on MandiNAc were designed, compounds 62 and 63, both containing azido groups at the 6-position and the latter having a 3-O-acetyl group that could potentially increase permeability and subsequently be removed by cytoplasmic esterases to generate 62 (Figure 22C).168
Figure 22.
MandiNAc-based chemical reporters for labeling L. pneumophila LPS O-antigen. (A) Structure of the L. pneumophila LPS O-antigen. (B) Biosynthesis of O-antigen precursor CMP-Leg5Ac7Ac, which arises from a MandiNAc intermediate. Subsequent polymerization of the O-antigen subunit and assembly into mature LPS is shown in Figure 18. (C) Structures and (D) chemical synthesis of azide-modified MandiNAc analogues as chemical reporters for the L. pneumophila LPS O-antigen. Bz, benzoyl; CAN, ceric ammonium nitrate; Ms, mesyl; PMP, para-methoxyphenyl.
The synthesis of 62 and 63, informed by prior work from Tsvetkov and co-workers,185 is notable due to the challenges associated with installing multiple nitrogen atoms with proper stereochemistry (Figure 22D). After a 4-step conversion of D-galactose (64) into diol 65, triflation and double SN2 displacement with azide anion established the diaminomannose core in 66.168 Three steps were used to install the N-acetyl groups and unmask the hydroxyl groups to give 67, and then a mesylation–SN2 sequence added an azido group to the 6-position.168 Finally, optional 3-O-acetylation followed by anomeric deprotection gave the targets 62 and 63 in 12 and 13 steps, respectively.168 While this is an elegant synthesis of a complicated probe, it also underscores the challenges that chemists face in terms of making tools accessible to wider research community.
With the compounds prepared, they were tested for incorporation into various strains of L. pneumophila and other species by incubation of bacteria in 4 mM probe, CuAAC reaction with alkyne-biotin, staining with fluorescent anti-biotin antibody, and imaging by fluorescence microscopy. It was found that reporter 62 robustly labeled the cell surface of various L. pneumophila serogroups that are commonly associated with infections, but not in species lacking the Leg5Am7Ac-containing LPS O-antigen, such as E. coli, Pseudomonas aeruginosa, or even other Legionella species.168 The 3-O-acetylated version 63 did not label L. pneumophila, implying that its uptake and/or unmasking were inefficient, possibly due to low non-specific esterase activity, as previously observed in E. coli.186 Overall, the results demonstrated the species specificity of MandiNAc-based reporter 62 and suggest its possible use for rapid detection of L. pneumophila. Although not necessarily critical for bacterial detection applications, the study did not include experiments to characterize the route of labeling nor confirm that the targeted LPS O-antigen was indeed being labeled. Such characterization will be essential for 62 to be used in the future as a tool for probing O-antigen biosynthesis, dynamics, and function in L. pneumophila. Also looking toward other applications, L. pneumophila is not the only type of bacteria possessing surface legionaminic acid-containing glycans, as this sugar has been identified in LPS and glycoproteins in various other bacterial species.187–192 While the presence of such glycoconjugates on other organisms may reduce the utility of 62 as a specific detection probe for Legionnares’-causing L. pneumophila, it broadens opportunities to investigate the role of legionaminic acid in other types of bacteria. Indeed, a 2021 study by the Wennekes and Wösten groups showed that azide-modified legionaminic acid precursors label flagellin glycoproteins in C. jejuni and enabled the identification of a glycosyltransferase likely to be responsible for legionaminic acid installation into flagellin (see Section 4.1.2).193
3.2.3. Pseudaminic acid precursor derivatives.
In 2018, a similar approach to that of Vauzeilles and Dukan was employed by the Li group to metabolically label pseudaminic acid-containing O-antigen polysaccharides in Gram-negative bacteria.169 Several significant bacterial pathogens, such as the opportunistic pathogen P. aeruginosa, are known to contain pseudaminic acids (or derivatives thereof) in their LPS O-antigen polysaccharides, glycoproteins, and/or capsular polysaccharides.187,194–196 An example of a P. aeruginosa O-antigen structure containing a pseudaminic acid derivative is shown in Figure 23A.195 While various roles for pseudaminic acids in host–pathogen interactions have been proposed (e.g., immune evasion, adhesion to host cells),197,198 their contributions to bacterial physiology and pathogenesis remain understudied, prompting the Li group to develop new tools to accelerate research in this area. The biosynthetic pathway for the production of the CMP-pseudaminic acid (CMP-Pse) donor is analogous to that of CMP-Leg5Ac7Ac shown above (Figure 22B), with UDP-GlcNAc being converted via several steps to 2,4-diacetamido-2,4,6-trideoxy-L-altrose (AltdiNAc), followed by PseI-catalyzed aldol reaction and PseF-catalyzed CMP activation (Figure 23B).199 Chemical reporters based on AltdiNAc were designed to contain azido functionalities attached to the 2- and 4-acetamido groups of a di-O-acetylated AltdiNAc core, named Alt-2NAz (69) and Alt-4NAz (70) (Figure 23C).169 The probe design was an interesting contrast to related prior work on legionaminic acid reporters by Vauzeilles and Dukan (section 3.2.2), who instead modified the 6-position and found that partial hydroxyl acetylation at the 3-position prevented, rather than facilitated, metabolic incorporation.
Figure 23.
AltdiNAc-based chemical reporters for labeling pseudaminic acid residues of LPS O-antigen. (A) Structure of a pseudaminic acid-containing LPS O-antigen from P. aeruginosa. FucNAc, N-acetylfucosamine; Pse5Ac7Fo, pseudaminic acid derivative with 5-acetamido and 7-formamido groups; Xyl, xylose. (B) Biosynthesis of O-antigen precursor CMP-Pse, which arises from an AltdiNAc intermediate. Subsequent polymerization of the O-antigen subunit and assembly into mature LPS is shown in Figure 18. (C) Structures of azide-modified AltdiNAc chemical reporters.
The preparation of 69 and 70 invoked the Li’s previously reported200 de novo synthetic strategy starting from L-allo-threonine that took ~15 steps (not shown), again highlighting at once both the inventiveness and arduousness required to create probes inspired by unique bacterial sugars.169 Alt-2NAz (69) and Alt-4NAz (70) were evaluated by administering low mM concentrations of probe to P. aeruginosa strain PA1244 over ~48 h, subjecting bacteria to SPAAC with cyclooctyne-488, and analyzing cell extracts or whole cells by SDS-PAGE fluorescence scanning or fluorescence microscopy, respectively.169 Analysis of LPS extracts from probe-treated P. aeruginosa showed that while Alt-2NAz did not appreciably incorporate into LPS, Alt-4NAz did, and, furthermore, Alt-4NAz-dependent signal was competed out by unlabeled AltdiNAc, supporting specific metabolic incorporation via the pseudaminic acid biosynthetic pathway.169 It is possible that the azido modification of Alt-2NAz impeded metabolic processing due to its close proximity to the anomeric center. Another noteworthy result from these studies was that the O-acetyl-masked reporter Alt-4NAz labeled LPS, implying that it must be taken up and deacetylated by esterases, which differed from previously reported results in similar systems.168,186 Therefore, it would be interesting to test the O-deacetylated versions of 69 and 70 for LPS labeling. An attractive feature of the biosynthetic pathway targeted by AltdiNAc derivatives (Figure 23B) is that it appears to be fairly well conserved in Gram-negative bacteria.201 Therefore, the team explored whether Alt-4NAz could label species beyond P. aeruginosa that were known or genetically predicted to contain cell surface pseudaminic acid glycans, finding that the reporter indeed incorporated into LPS in the Gram-negative organisms Vibrio vulnificus and Acinetobacter baumannii.169 While the analysis in this particular study was focused on LPS, the presence of pseudaminic acid in bacterial glycoproteins and capsular polysaccharides, coupled with the conserved nature of the described pseudaminic acid biosynthesis pathway, suggests that AltdiNAc-based reporters may be broadly useful for investigating pseudaminic acid-containing bacterial glycans. Indeed, an earlier study reported in 2009 by the Tanner and Logan groups demonstrated that an azido AltdiNAc reporter labeled flagellar glycoproteins in Campylobacter jejuni,202 which is discussed in detail section 4.1.1.
Taken together, the foregoing three examples of LPS O-antigen chemical reporters offer several takeaways. A common thread between these studies was the metabolic labeling approach, which involved channeling exogenous functionalized monosaccharide precursors into their corresponding activated nucleotide sugar donors, followed by glycosyltransferase-mediated installation into the O-antigen subunit, polymerization, and assembly into mature LPS. This approach, which as first demonstrated by Wang can be enabled or augmented by genetic engineering if needed, can be considered as a general way to develop chemical reporters for LPS O-antigen constituents. Given the high variability of O-antigen polysaccharides and the multitude of different bacteria-specific monosaccharides found within them, there is ample opportunity to develop novel O-antigen chemical reporters that will find use as species- or strain-specific labeling tools. As illustrated by the legionaminic acid and pseudaminic acid probes, perhaps the biggest hurdle to expanding this research area will be the synthesis of unique bacterial sugar analogues, further emphasizing the need for improved synthetic strategies. A rather technical but potentially important future inquiry, raised by the contrasting results in these and other studies, is whether peracetylated chemical reporters offer any improvements over their unprotected counterparts in bacterial systems. A more exhaustive investigation of this issue in bacterial systems would be beneficial to the community. Similar to KDO-N3, O-antigen-targeting chemical reporters have not yet been applied to study LPS dynamics and function, although these tools are ideally suited for such applications, as we saw in the case of PG reporters. With respect to detection and therapeutic targeting applications, fucose-based reporters are unsuitable because they also label mammalian cells,203 but the bacteria-specific legionaminic acid- and pseudamininc acid-based reporters could have high potential.
3.3. Sialic acid-based chemical reporters for lipooligosaccharide (LOS)
Some Gram-negative bacteria, such as Haemophilus and Neisseria, lack the genes to produce the O-antigen component of LPS and instead generate the core-Lipid A species also referred to as lipooligosaccharide (LOS) (see Figure 18). Some of these species colonize the upper respiratory or genital tracts in humans and have evolved ways to modify their LOS with host-acquired mammalian sugars, which leads to the production of LOS structures that mimic host glycans and are implicated in immune evasion. For example, most strains of nontypeable H. influenzae, an opportunistic human pathogen, acquire sialic acid from the host and incorporate it into their LOS, which reduces serum-mediated killing and increases virulence of the bacterium.204 In 2018, the Boltje group reported a chemical reporter approach for investigating LOS remodeling that occurs as a result of host-to-bacterium transfer of sialic acids.170 Previously, it had been discovered that sialic acids released from host glycoproteins via sialidase activity can be taken up by H. influenzae through the sialic acid transporter SiaP, then activated into the donor CMP-sialic acid and incorporated into LOS by sialyltransferases.204 Thus, it was hypothesized that exogenous sialic acid-based chemical reporters—either supplied directly in the growth medium or released from labeled host cells in a co-culture system—could potentially be metabolically incorporated into LOS, which would provide a means to track this LOS remodeling process.
Four previously reported sialic acid analogues were investigated in the study, including azide- and alkyne-modified sialic acids in both free and peracetylated forms (SiaNAz/Ac5SiaNAz and SiaNPoc/Ac5SiaNPoc, compounds 71–74, Figure 24).35,205,206 The acetylation state of the probe was expected to confer selectivity for labeling bacterial cells or mammalian cells, since the former take up free sialic acid via SiaP whereas the latter take up peracetylated sugars via passive diffusion and incorporate them into surface glycoproteins following deacetylation by intracellular esterases.205,206 Indeed, it was revealed that both SiaNAz (71) and SiaNPoc (73), but not their peracetylated counterparts 72 and 74, incorporated into H. influenzae in a dose-dependent manner following sequential probe treatment, CuAAC-mediated biotinylation, and staining with a streptavidin-fluorophore conjugate.170 Labeling was abolished in an H. influenzae mutant lacking the SiaP transporter, confirming that incorporation was specific for sialic acid metabolism.170 On the other hand, mammalian THP-1 cells were labeled by Ac5SiaNAz (72) and Ac5SiaNPoc (74), but not 71 or 73, as predicted.170
Figure 24.
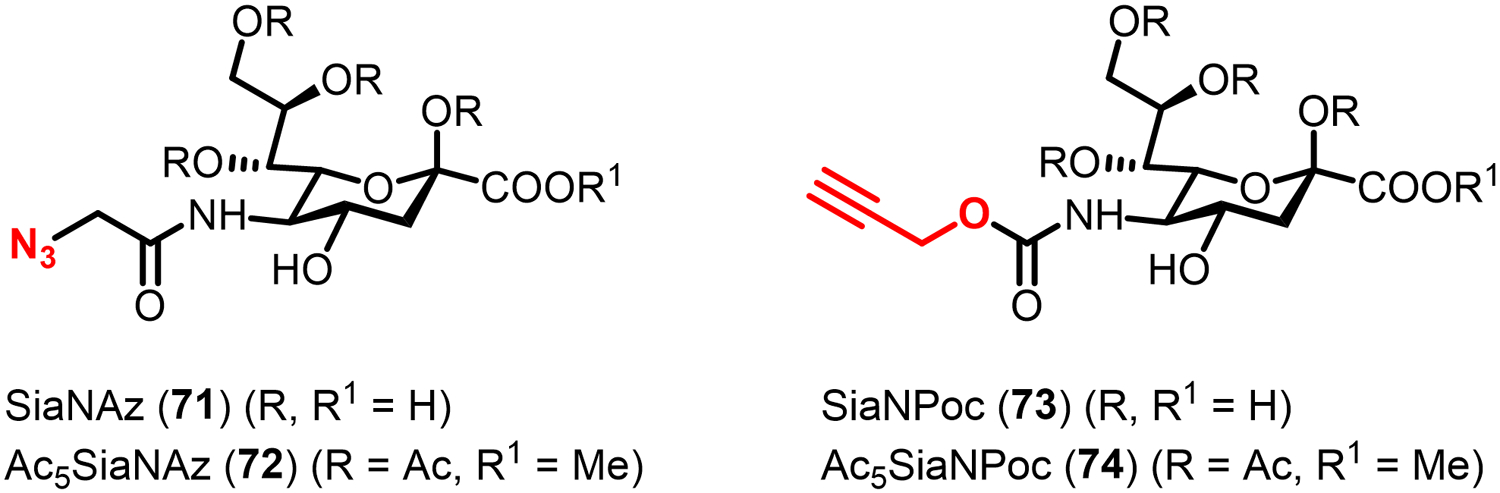
Sialic acid-based chemical reporters for labeling LOS in H. influenzae.
After establishing labeling selectivity of the probes independently in H. influenzae and mammalian cells, a co-culture system modeling the respiratory tract was used to investigate host-to-bacterium transfer of sialic acids. Primary human bronchial epithelial cells (PHBECs) were first labeled with Ac5SiaNAz (72) or Ac5SiaNPoc (74), then H. influenzae was cultured in proximity to the labeled PHBECs and transfer of sialic acids to bacteria was assessed by click chemistry and flow cytometry.170 H. influenzae acquired labeled sialic acids from PHBEC-derived glycoproteins and surface-displayed them, presumably as labeled LOS, in a sialidase- and SiaP-dependent manner (although it is unknown whether host or H. influenzae sialidases were responsible for release of sialic acids from host cells).170 Ac5SiaNPoc (74) incorporated less efficiently into H. influenzae, likely due to its previously reported resistance to sialidase-mediated cleavage from glycans.207 This is the first example of a chemical reporter being used to monitor the dynamics of glycan transfer between a mammalian cell and a bacterium, an approach which could be particularly valuable for investigating glycan-mediated host–pathogen or host–symbiont interactions. Sialic acid-based metabolic labeling of H. influenzae was also used as a tool to facilitate the development of inhibitors of LOS remodeling. Of particular note, an Ac5SiaNAz (72)-based labeling assay demonstrated that a fluorine-modified sialic acid analogue inhibited LOS sialylation in the PHBEC–H. influenzae co-culture system, and the compound was also shown to decrease resistance of H. influenzae to serum-mediated killing.170 This finding, as well as a 2021 follow-up study from the same team that identified improved fluorinated sialic acid-based inhibitors,208 demonstrated the utility of chemical reporters to assay specific virulence pathways in antibiotic development efforts.
4. Chemical reporters for bacterial glycoproteins
While glycoproteins were long suspected to be absent from bacterial systems, evidence for bacterial protein glycosylation mounted through the latter part of the 20th century.13 The first such structures discovered were the S-layer glycoproteins in archaea and some bacteria (e.g., Bacillus anthracis and Clostridium difficile), which self-assemble to form crystalline monomolecular layers around the cell that provide structural support, protect against various stresses, and participate in extracellular recognition events, including host–pathogen interactions.14,209 Subsequently, many examples of non-S-layer glycoproteins, which more closely resemble canonical O- and N-linked glycoproteins found in eukaryotes, have been identified as important cell envelope constituents in disparate Gram-positive, Gram-negative, and mycobacterial species.13,210 As with eukaryotic protein glycosylation, bacterial N- and O-glycosylation involves asparagine (Asn) or serine/threonine (Ser/Thr) linkages, respectively, although in many cases the identities of the monosaccharide constituents and their arrangement in the glycan are highly distinctive.13,210 As examples, pseudaminic and legionaminic acids (and derivatives thereof) are common in bacterial O-linked glycans; unique aminosugars can connect N- or O-linked glycans to the protein backbone, such as N-acetylfucosamine (FucNAc), bacillosamine (Bac), and 2,4-diacetamido-2,4,6-trideoxyhexose (DATDH); common monosaccharides such as glucose, galactose, N-acetylglucosamine, N-acetylgalactosamine, and mannose are also often present in bacterial glycoproteins, although frequently with atypical presentations (Figure 25).53,210,211 Bacterial protein glycosylation has commonly been linked with virulence, as the glycan moieties have roles related to immune evasion, motility, adhesion, and host colonization.13,53,210 Accordingly, deletion of enzymes responsible for synthesizing and installing glycans onto proteins often impairs bacterial virulence, suggesting that glycoproteins are promising therapeutic targets.210 The discovery and characterization of protein glycosylation machinery in bacteria has also led to efforts to capitalize on these systems to produce glycoengineered vaccines, therapeutics, and diagnostics.212,213 Despite the potential in these areas, we still have relatively limited knowledge about bacterial glycoprotein occurrence, structure, and function, making tool development a priority.
Figure 25.
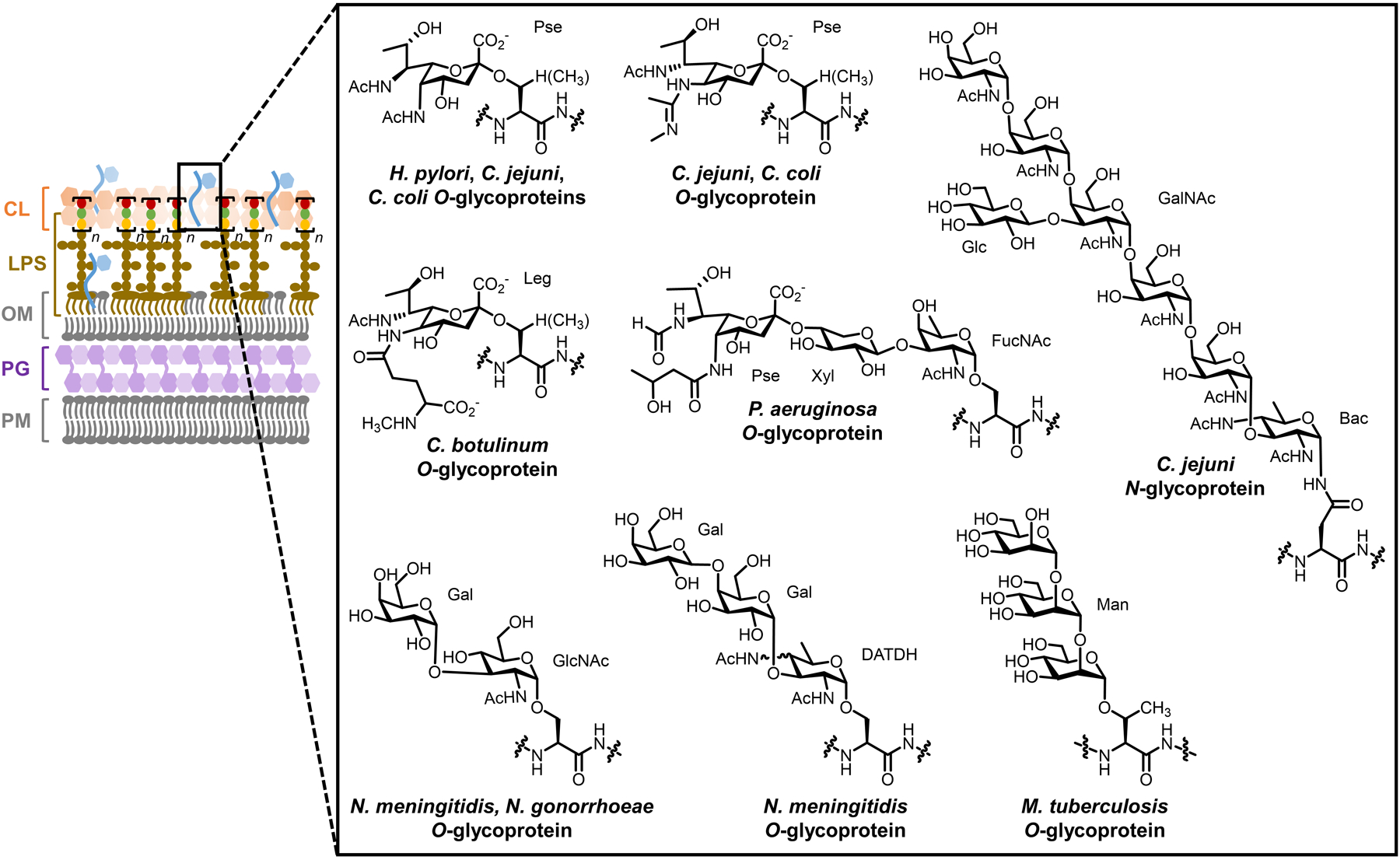
Representative structures of bacterial glycoproteins. A Gram-negative cell envelope is shown, although glycoproteins exist in Gram-positive bacteria and mycobacteria as well.
Depending on the organism, bacterial protein glycosylation can generate either N- or O-linked glycoproteins via oligosaccharyltransferase (OTase)-dependent or OTase-independent pathways (Figure 26).210,214 In both cases, free cytoplasmic monosaccharides are first activated into nucleotide sugar donors, which serve as the building blocks for protein glycosylation. In OTase-dependent protein glycosylation, an initiating GTase transfers a monosaccharyl group from a nucleotide sugar donor onto undecaprenyl phosphate (UP), generating a lipid-linked monosaccharide on the cytoplasmic face of the plasma membrane. Subsequently, specific GTases may sequentially extend the lipid-linked monosaccharide into a lipid-linked oligosaccharide, which is then flipped across the plasma membrane. The lipid-linked oligosaccharide or monosaccharide unit is transferred en bloc to a protein substrate via extracellular N- or O-OTase to produce N- or O-linked glycoproteins, respectively. These glycoproteins may then be transported to the outer membrane or secreted. In the case of OTase-independent glycoprotein biosynthesis, cytoplasmic N- or O-GTases transfer monosaccharyl groups from nucleotide sugar donors onto protein substrates, respectively generating N- or O-linked glycoproteins in the cytoplasm that are then secreted. Interestingly, eukaryotes have two major protein glycosylation pathways (N-OTase-mediated synthesis of N-glycoproteins and cytoplasmic O-GTase-mediated synthesis of O-glycoproteins), while bacteria have four main pathways. This array of biosynthetic pathways, when coupled with the availability of numerous unique monosaccharides, creates the potential for enormous structural diversity in bacterial glycoproteins, as illustrated in Figure 25.
Figure 26.
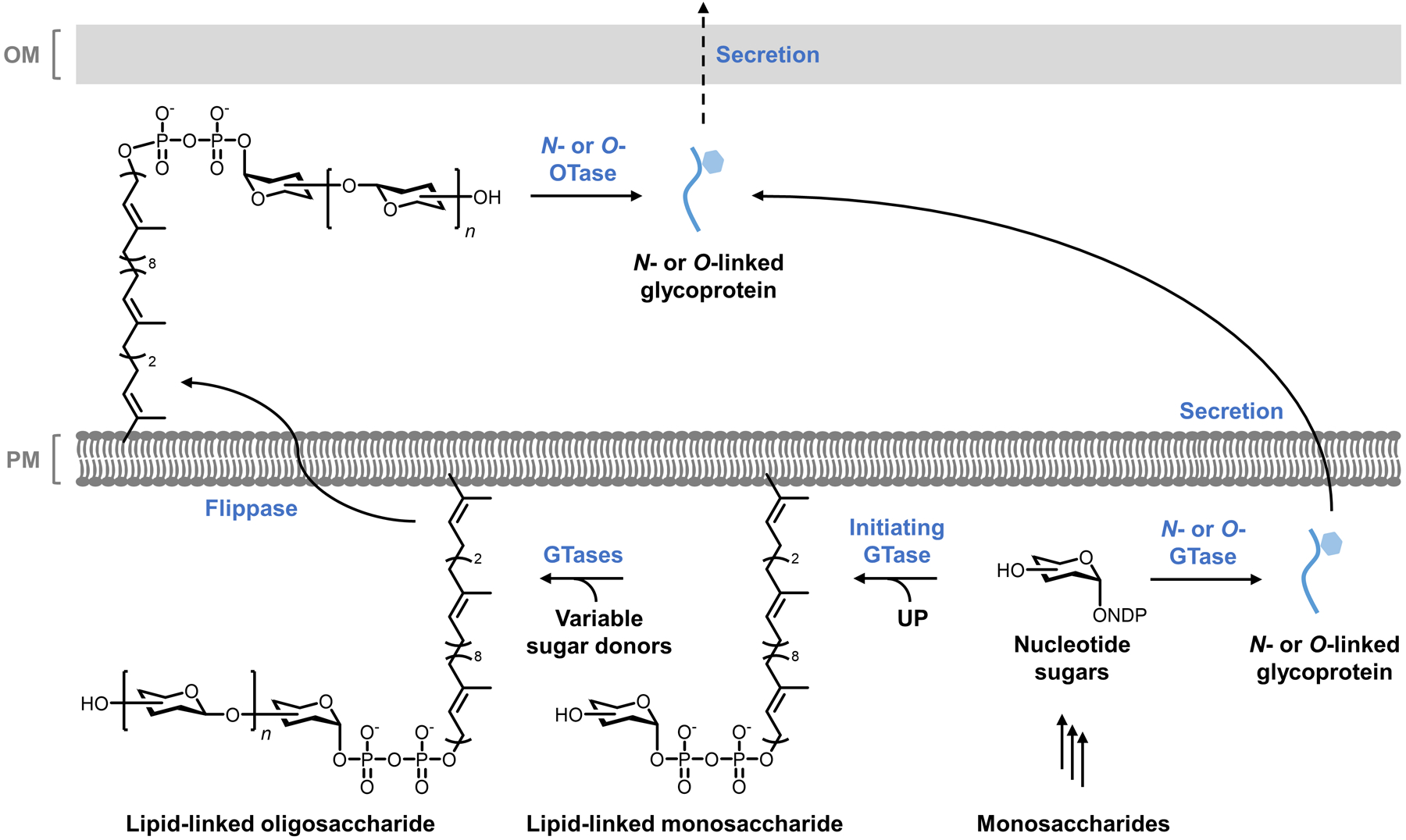
General scheme for the biosynthesis of N- and O-linked bacterial glycoproteins via OTase-dependent or independent pathways.
The multiple pathways and unique monosaccharides involved in bacterial glycoprotein biosynthesis afford many possibilities for the development of chemical reporters. The most straightforward and well-precedented approach is similar to that described above for LPS O-antigen constituents, i.e. feeding bacteria labeled monosaccharide precursors that are taken up, converted into nucleotide sugar donors (and subsequently into lipid-linked donors in the case of OTase-dependent pathways), and added to protein substrates via GTases. This approach comes with the above-mentioned advantages, such as relatively good synthetic accessibility and typically high substrate promiscuity of carbohydrate-processing enzymes, and disadvantages, such as the need for the exogenously added probe to pass through membrane(s) and multiple metabolic steps. An alternative approach for labeling OTase-dependent glycoproteins would be to synthesize lipid-linked saccharide analogues that would be directly metabolically incorporated into proteins by periplasmic GTases, although this would necessitate difficult syntheses and the probes may suffer from solubility and stability issues. As covered below, all chemical reporters for bacterial glycoproteins developed to date used the former monosaccharide-based approach. Table 3 contains information about selected glycoprotein-targeting chemical reporters.
Table 3.
Selected chemical reporters for bacterial glycoproteins
| Target | Metabolite | Compound | Reporter group (X) | Bioorthogonal reaction(s) | Proposed route | Speciesa | Applicationsb | Synthesis | Notesa | Refc |
|---|---|---|---|---|---|---|---|---|---|---|
| Flagellin pseudaminic acid | AltdiNAc | AltNAc4NAz 75 (Fig. 27) | Azide | Staudinger ligation | Intracellular | C. jejuni (GN) | Detection of labeled flagellin | Chemo-enzymatic | Labeled flagellin but no other proteins in C. jejuni | 202 |
| Flagellin legionaminic acid | MandiNAc | 83 (Fig. 28) | Azide | SPAAC | Intracellular | C. jejuni (GN) | Detection of labeled flagellin; in vitro imaging; identification of GTase | Chemical | Labeled flagellin but no other proteins in C. jejuni; peracetylation abolished labeling | 193 |
| Glycoproteins | GlcNAc | Ac4GlcNAz 86 (Fig. 29) | Azide | Staudinger ligation | Intracellular | H. pylori (GN) | Detection and profiling of labeled proteins; identification of GTases; delivery of immune modulator | Chemical; comm. available | Peracetylation required for labeling; no information available about glycan structure; did not incorporate into a variety of other bacterial species | 215 |
| Bac | Ac2Bac-DiNAz 88 (Fig. 29) | Azide | Staudinger ligation | Intracellulard | H. pylori (GN), C. jejuni (GN), R. solanacearum (GN), B. thailandensis (GN) | Detection of labeled proteins | Chemical | Peracetylated reporter labeled bacteria, unacetylated version not tested | 216 | |
| DATDG | Ac2DATDG-DiNAz 89 (Fig. 29) | Azide | Staudinger ligation | Intracellulard | H. pylori (GN), C. jejuni (GN), R. solanacearum (GN) | Detection of labeled proteins | Chemical | Peracetylated reporter labeled bacteria, unacetylated version not tested | 216 | |
| L-fucose | FucAl 59 (Fig. 21) | Alkyne | CuAAC | Intracellular | B. fragilis (GN), P. distasons (GN) | Detection of labeled proteins | Chemical | 217 |
Gram classification: GN, Gram-negative; GP, Gram-positive; M, mycobacteria.
In vitro refers to bacterial labeling experiments performed in broth culture; in vivo refers to bacterial labeling experiments performed in a host cell or organism.
Initial publication(s) referenced. See text for references for follow-up studies and applications.
Limited evidence available to support proposed route of incorporation and/or identity of labeled target (see text).
4.1. Chemical reporters for flagellin glycoproteins
Various types of bacteria are known to incorporate pseudaminic acid and legionaminic acid into cell surface glycoproteins, as shown in Figure 25. For example, the flagellin proteins of the Gram-negative gastrointestinal pathogens C. jejuni and H. pylori are heavily O-glycosylated with pseudaminic acid at 10 or more serine residues.210,218–220 Glycosylation is required for assembly of functional flagella in these organisms, and thus is necessary for locomotion and other cell surface-associated properties that support pathogenicity.210,219–223 C. jejuni flagellin glycoproteins are also decorated with legionaminic acids, which contribute to biofilm formation and auto-agglutination processes that may be involved in virulence.218 Thus, glycosylation is an important post-translational modification to study in the context of bacterial flagellins. In the next two sections, we describe chemical reporters that have been developed for this purpose.
4.1.1. Pseudaminic acid precursor derivatives.
In 2009, the Tanner and Logan groups reported a method for metabolic labeling of pseudaminic acid residues of C. jejuni flagellin.202 As described above, the pseudaminic acid biosynthesis pathway199 that supports LPS O-antigen biosynthesis in P. aeruginosa (Figure 23B) is conserved in many bacteria. In C. jejuni, this pseudaminic acid biosynthesis pathway feeds into OTase-independent cytoplasmic O-glycosylation of flagellin as generally illustrated in Figure 26. Similar to the later work by Li on LPS O-antigen (see section 3.2.3),169 Tanner and Logan hypothesized that an azide-modified analogue of the only neutral intermediate in the pathway, AltdiNAc, could potentially be taken up and metabolically incorporated into flagellin in C. jejuni.202 Therefore, an AltdiNAc derivative containing an N-azidoacetyl group at the 4-N-position was designed (AltNAc4NAz, 75, Figure 27A).202 Compound 75 exists in free form with no hydroxyl masking, a notable contrast to Li’s more recent peracetylated probes that were used for labeling pseudaminic acid residues in LPS O-antigen (69 and 70, see Figure 23C).
Figure 27.
(A) Chemoenzymatic synthesis of AltNAc4NAz (75), a chemical reporter for pseudaminic acid-containing flagellin glycoprotein in C. jejuni. (B) Analysis of AltNAc4NAz-labeled flagellin from C. jejuni by Western blot. Wild-type or PseG-deficient C. jejuni were grown on agar containing 75, reacted with phosphine-biotin, lysed, and probed by streptavidin-horseradish peroxidase (HRP) or anti-flagellin Western blot. For streptavidin-HRP blot: lane 1, C. jejuni ΔpseG + 75; lane 2, C. jejuni ΔpseG no probe; lane 3, C. jejuni wild type no probe; lane 4, C. jejuni wild type + 75. For anti-flagellin blot: lane 1, C. jejuni ΔpseG + 75; lane 2, C. jejuni ΔpseG no probe; lane 3, C. jejuni wild type no probe. Reprinted with permission from ref 202. Copyright 2009 Wiley-VCH.
A concise and elegant 4-step chemoenzymatic synthesis of AltNAc4NAz (71) was established (Figure 27A).202 Capitalizing on the pseudaminic acid biosynthetic pathway (Figure 23B), UDP-GlcNAc was first processed in one pot by recombinant PseB and PseC to establish the diamino core intermediate 76.202 Next, a chloroacetyl group was installed at the 4-N-position to give 77, setting up for subsequent PseG-catalyzed hydrolysis to give hemiacetal 78 followed by SN2 substitution of the chloro group with azide anion to give the target, AltNAc4NAz (75).202 This route was short and high-yielding relative to the newer de novo chemical synthesis,169 which points to the power of bioinspired chemoenzymatic synthesis methods, especially for the preparation of challenging bacterial carbohydrate-based chemical reporters.
Characterization of AltNAc4NAz (75) was initiated by evaluating its ability to be taken up by C. jejuni using a rescue experiment.202 A PseG-deficient strain of C. jejuni (ΔpseG) that cannot produce native AltdiNAc was previously shown to be completely non-motile due to its inability to produce functional flagella.221 However, when cultured in either native AltdiNAc or reporter AltNAc4NAz (75), motility was restored in ΔpseG, suggesting that both compounds could be successfully internalized and metabolically incorporated into flagellin, rendering it functional again.202 To directly test for flagellin labeling, C. jejuni was grown on solid agar containing 8 mM AltNAc4NAz (75), reacted via Staudinger ligation with a phosphine-biotin probe, lysed, and then analyzed by Western blot using streptavidin-HRP. A single major band was observed in lysates from AltNAc4NAz-treated C. jejuni that migrated identically to flagellin was detected with anti-flagellin antibody, confirming specific incorporation into the target glycoprotein (Figure 27B).202 Interestingly, no other signal was observed, suggesting that flagellin may be the only pseudaminic acid-modified glycoprotein in C. jejuni, or at least the only such protein that is of detectable abundance in this assay.
AltNAc4NAz (75) behaves as designed and is a useful tool for studying protein glycosylation and engineering the cell surface of C. jejuni, and possibly other pseudaminic acid-displaying microbes. The apparent lack of other pseudaminic acid-modified glycoproteins in C. jejuni implies that AltNAc4NAz will not be useful for glycoprotein discovery in this context, although it may serve this role in other organisms. While AltNAc4NAz was not used for analysis of intact cells in the original study, it can presumably be combined with Staudinger ligation as reported, or other bioorthogonal chemistries, to image the dynamics of flagellin synthesis, assembly, and interactions in live cells. A significant highlight of this work was the highly efficient chemoenzymatic synthesis of AltNAc4NAz, which appears to be the most desirable route to AltdiNAc-based chemical reporters published to date. It would be of value to compare AltNAc4NAz (75) and the subsequently reported probes Alt-2NAz (69) and Alt-4NAz (70) to test ΔpseG motility rescue and determine their labeling profiles (e.g., LPS O-antigen vs. glycoproteins) in different types of pseudaminic acid-containing bacteria. The comparison would be intriguing given the differences in probe design, namely the position and nature of the azide modification and the use of free vs. acetylated hydroxyl groups. Finally, since many other Gram-negative and some Gram-positive bacterial pathogens produce virulence-associated flagellin glycoproteins with diverse carbohydrate modifications,214,223 Tanner and Logan’s strategy provides valuable insights for applying AltNAc4NAz (75) in different species as well as developing novel reporters for bacterial glycoproteins, as we see in subsequent sections.
4.1.2. Legionaminic acid precursor derivatives.
Complementary to the work by Tanner and Logan on pseudaminic acids described in the preceding section,202 in 2021 the Wösten and Wennekes groups developed a similar method for metabolic labeling of legionaminic acid residues in C. jejuni flagellin.193 The pathway for legionaminic acid biosynthesis in C. jejuni is analogous to that described above for Leg5Am7Ac in L. pneumophila (see section 3.2.2 and Figure 22B), and the incorporation of this sugar into flagellin is analogous to that described immediately above for pseudaminic acid (see section 4.1.1).184 Thus, similar to the noted prior work, it was hypothesized that azide-modified derivatives of a neutral legionaminic acid biosynthetic precursor, in this case MandiNAc (see Figure 22B), would be capable of labeling flagellin glycans. Rather than use the previously reported MandiNAc derivatives containing azido groups at the 6-position (compounds 62 and 63, Figure 22C), in this case azidoacetamido groups were installed at the 2- and/or 4-position, reminiscent of the approaches taken earlier by Li,169 as well as by Tanner and Logan.202 The designed compounds thus had azidoacetamido groups at either the 2- or 4-position, or both, and were prepared in either non-acetylated or peracetylated form (compounds 79–84, Figure 28).193 All six compounds were prepared in >10 steps from D-fucose via chemical synthesis, which was highlighted by a sequential SN2 reactions to establish the diamino core structure with proper stereochemistry (not shown).193 Although lengthy compared to the chemoenzymatic synthesis of the related compound AltNAc4NAz (75, Figure 27A), the chemical synthesis route did exhibit the versatility required to access all six target compounds 79–84.193
Figure 28.
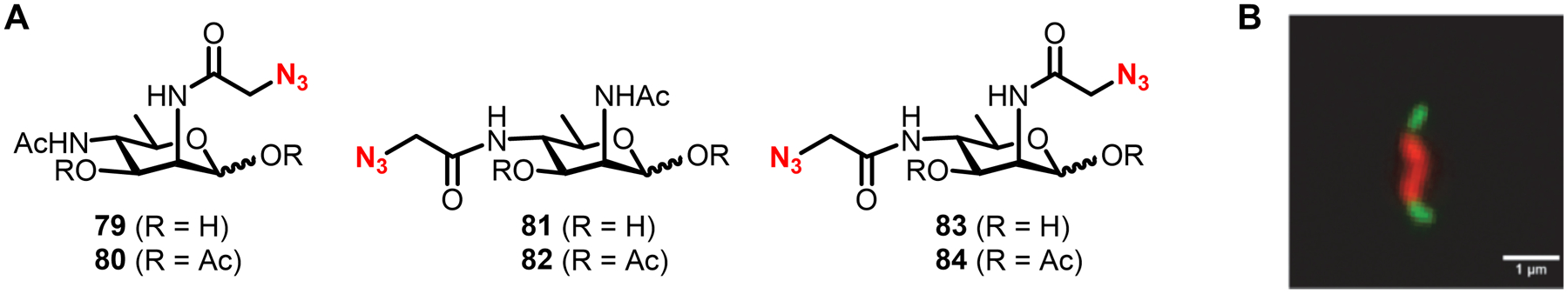
(A) MandiNAc-based chemical reporters for labeling legionaminic acid residues of C. jejuni flagellin glycoproteins. See Figure 22B for a relevant biosynthetic pathway and the structure of MandiNAc. (B) Imaging of C. jejuni flagellin glycans using compound 83. C. jejuni was incubated in the presence of 83, fixed, subjected to SPAAC with cyclooctyne-PEG-biotin, stained with streptavidin-488, and imaged by fluorescence microscopy. A secondary dye (red) stained the body of the bacterium. Scale bar, 1 μm. Reproduced from ref 193.
Evaluation of compounds 79–84 for incorporation into flagellin glycoprotein was performed mainly in C. jejuni strain 11168 using experimental approaches similar to those of Tanner and Logan described above. C. jejuni was cultured in the compounds, subjected to SPAAC reaction with a cyclooctyne-PEG-biotin reagent, and whole-cell lysates were obtained and analyzed by Western blot using streptavidin-HRP and anti-flagellin antibody. All of the non-acetylated probes, including 79, 81, and 83, efficiently incorporated into a single ~70 kDa protein confirmed to be flagellin (similar to the results shown in Figure 27B), while interestingly the peracetylated probes (80, 82, and 84) were not incorporated.193 The diazidoacetamido derivative 83 showed the highest level of incorporation,193 perhaps due to the two azido groups present on the probe. The incorporation of 83 did not affect C. jejuni auto-agglutination, suggesting that it did not significantly perturb the function of flagellin glycans.193 Having established the labeling behavior of 83, the compound was then used to provide some new insights into flagellin glycosylation. Direct visualization of flagellin legionaminic acid residues was accomplished, which revealed that glycosylated flagellin appears to extend from both ends of C. jejuni (Figure 28B).193 Furthermore, 83 was used to facilitate the identification of the enzyme that appends legionaminic acids to flagellin. A prior study had implicated the motility associated factor gene maf4 as likely encoding a GTase involved in flagellin glycosylation.222 Labeling experiments utilizing compound 83 in C. jejuni strains lacking maf4 showed near complete elimination of flagellin labeling, strongly suggesting that Maf4 is the major GTase that modifies the Ser/Thr residues of flagellin with legionaminic acids.193 This experiment, along with similar experiments covered elsewhere in this review, exemplify the ability of chemical reporter assays to elucidate pathways involved in bacterial glycan biosynthesis.
4.2. N-Acetylglucosamine-based chemical reporters for bacterial glycoproteins
In addition to glycosylating flagellin proteins with pseudaminic acid and legionaminic acid residues as described above, C. jejuni is also known to produce numerous N-glycoproteins featuring a unique asparagine–bacillosamine linkage that connects the protein backbone to the N-glycan.224,225 An example of an abundant GalNAc-rich N-linked heptasaccharide from C. jejuni is shown in Figure 25. The pathway for N-glycosylation in C. jejuni is distinct from that of flagellin O-glycosylation in C. jejuni and H. pylori, as the former proceeds via a periplasmic OTase-dependent mechanism, whereas the latter is cytoplasmic and OTase-independent (see Figure 26).224 N-Glycosylation in C. jejuni is functionally important, as it has been shown to protect bacterial proteins from host-mediated proteolysis and is necessary for proper colonization of the host.226,227 Other types of bacteria possess N-glycosylation machinery similar to that of C. jejuni, including the related organisms Helicobacter pullorum228 and Wolinella succinogenes.213
The partial conservation of glycoprotein biosynthesis genes between Campylobacter and Helicobacter genera has generated interest in investigating non-flagellin glycoproteins in H. pylori due to its stature as a major gastrointestinal pathogen.229 In 2009, the Dube lab reported the exploration of H. pylori glycoproteins using N-acetylglucosamine (GlcNAc)-based chemical reporters.215 Although GlcNAc-containing glycoproteins had not been identified in H. pylori at the time, it was hypothesized that the presence of GlcNAc residues in eukaryotic N-glycoproteins might extend to H. pylori as well.215 Assuming an OTase-dependent metabolic incorporation route, a GlcNAc-based probe would have to be taken up into the cytoplasm, activated into UDP-GlcNAc, installed onto a lipid-linked saccharide precursor, and finally transferred to a protein substrate by OTase (see Figure 26). To test this idea, two chemical reporters were investigated, including azidoacetamide-modified GlcNAc (GlcNAz, 85) and its peracetylated derivative (Ac4GlcNAz, 86) (Figure 29A). Presumably, if uptake were transporter-mediated, unprotected GlcNAz would incorporate favorably, whereas if uptake were reliant on passive diffusion across membranes, Ac4GlcNAz would incorporate favorably, assuming intracellular non-specific esterase activity was sufficient to deacetylate the probe.215
Figure 29.
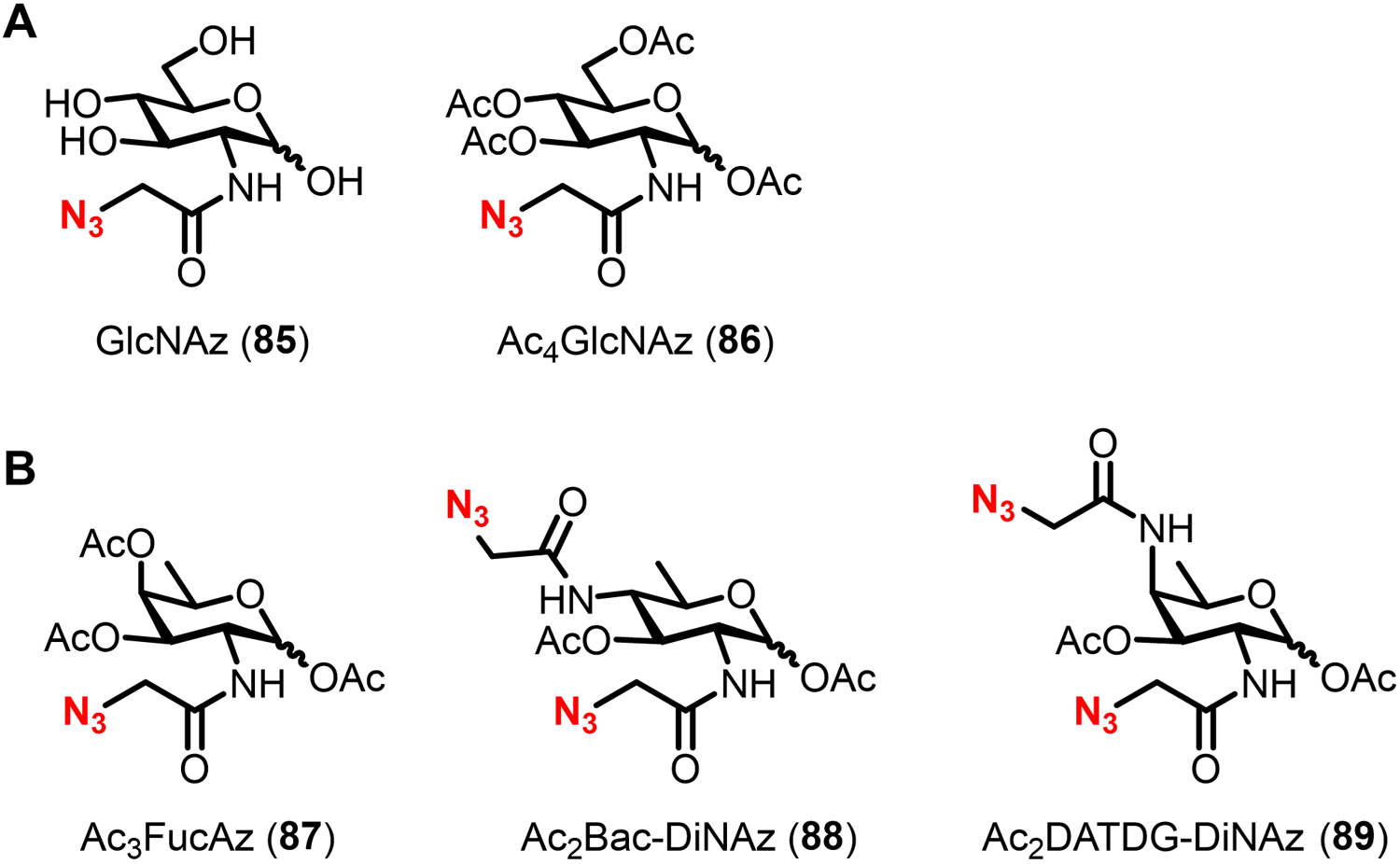
Chemical reporters for labeling bacterial glycoproteins. (A) GlcNAc-based reporters developed for labeling glycoproteins in H. pylori. (B) Rare bacterial sugar-based reporters developed for labeling glycoproteins in various species.
To test for incorporation of the chemical reporters into proteins, H. pylori was treated with 1 mM GlcNAz (85) or Ac4GlcNAz (86) and lysed, then protein lysates were subjected to Staudinger ligation with a phosphine-FLAG peptide reagent.215 Western blot analysis of proteins using anti-FLAG antibody showed that peracetylated Ac4GlcNAz (86), but not free GlcNAz (85), incorporated into numerous proteins.215 Treatment of Ac4GlcNAz-labeled proteins with glycosidases specific for either N- or O-glycans diminished but did not fully abolish signal, raising the possibility that both N- and O-linked glycoproteins are present in H. pylori.215 Because GlcNAc is a metabolic precursor for other cell envelope glycans, additional experiments were performed to test for Ac4GlcNAz incorporation into LPS or flagellin O-glycoproteins. No azide-dependent signal was observed in these samples, further suggesting that Ac4GlcNAz labeling occurred in non-flagellin proteins.215 While this study did not identify or confirm the glycosylation state of the labeled proteins, a follow-up study by the Dube group in 2013 carried out Ac4GlcNAz labeling, affinity enrichment, and multidimensional LC-MS/MS proteomics to identify 125 putative glycoproteins in H. pylori.230 Approximately 20% of the proteins identified through this approach were linked to H. pylori virulence, including urease alpha and beta, which are surface proteins implicated in host colonization.230 These reports by the Dube group, along with another by the Creuzenet group using a complementary periodate oxidation/hydrazide-based glycan tagging approach,229 suggest that H. pylori protein glycosylation extends beyond the flagellins and that novel glycosylation pathways are likely involved. Despite this exciting discovery, the detailed glycan structures, glycosylation sites, and glycosylation pathway(s) of the proteins identified through Ac4GlcNAz labeling remained unknown at the time.
In 2020, the Dube group further exploited Ac4GlcNAz labeling to gain insight into the genes and pathway(s) responsible for glycoprotein biosynthesis in H. pylori.231 A panel of H. pylori mutants lacking putative GTase genes was constructed and screened for differences in Ac4GlcNAz labeling in the mutants as compared to wild-type bacteria.231 Most of the GTase mutants exhibited significantly reduced, but not eliminated, Ac4GlcNAz labeling.231 Strikingly, the profiles of the remaining labeled proteins were nearly identical to each other and to that of lysates from wild-type bacteria that had undergone β-elimination to cleave O-linked glycans.231 These results are consistent with the idea that the mutated GTase-encoding genes are (i) part of a common biosynthetic pathway and (ii) involved in the production of O-glycoproteins. Further experiments showed that this potential O-glycosylation pathway may overlap with LPS biosynthesis and thus may proceed via an OTase-dependent route that utilizes lipid-linked saccharide donors, although labeled intermediates were not directly detected.231,232 Finally, the Ac4GlcNAz-dependent signal that persisted after GTase deletion or β-elimination implies that N-glycoproteins are also produced by H. pylori,231 consistent with the team’s earlier work.215 Thus, Ac4GlcNAz has accelerated efforts to identify and characterize novel bacterial glycoproteins, lessons from which may be applied to the development and application of similar tools. A major remaining objective of this line of research is to elucidate the glycan structure(s) present on Ac4GlcNAz-labeled proteins, which in principle the probe can facilitate if deployed alongside MS-based glycoproteomics.
In addition to fundamental research on the discovery of bacterial glycoproteins, Ac4GlcNAz has been leveraged by the Dube group to deliver immune-recruiting molecules to the cell surface of H. pylori as a possible therapeutic targeting strategy.233 Although mammalian glycoproteins include GlcNAc residues, it had previously been demonstrated that Ac4GlcNAz predominantly labels nuclear and cytosolic proteins.234,235 Thus, it was anticipated that Ac4GlcNAz would selectively modify the H. pylori cell surface with azides while leaving host cell surfaces unmodified, and in the process provide a handle for targeted bioorthogonal delivery of an antibody-recruiting dinitrophenyl (DNP) moiety.233 To test this idea, Ac4GlcNAz was administered to H. pylori or mammalian MDCK cells, reacted with a water-soluble phosphine-DNP reagent, incubated with a fluorophore-conjugated anti-DNP antibody, and imaged.233 As hypothesized, H. pylori was labeled with DNP, while the MDCK cells were not labeled by DNP.233 Crucially, in the presence of both anti-DNP antibodies and immune effector cells, specifically peripheral mononuclear blood cells (PMBCs), DNP-labeled H. pylori were killed more effectively than unmodified H. pylori.233 This work represented an early example of exploiting chemical reporter probes to selectively deliver immunogenic cargo to unique bacterial surface glycans, a strategy that has since been extended to other species and glycan types.118,119,133
4.3. Other monosaccharide-based chemical reporters for bacterial glycoproteins
4.3.1. Rare bacterial sugar derivatives.
The early success achieved with AltNAc4NAz (75) and Ac4GlcNAz (86) has led to the development of complementary chemical reporters to investigate bacterial glycoproteins. In 2016, the Dube group extended their earlier work with Ac4GlcNAz in H. pylori by testing whether the probe labeled proteins in other types of bacteria.216 Again, Staudinger ligation was used to append a FLAG peptide to labeled proteins, enabling detection by anti-FLAG Western blot.216 Interestingly, despite robustly incorporating into H. pylori as previously reported, Ac4GlcNAz did not incorporate into a diverse panel of other bacteria known to produce glycoproteins, including C. jejuni, P. aeruginosa, Burkholderia thailandensis, M. smegmatis, or B. fragilis.216 This result could reflect an absence of GlcNAc residues in the glycoproteins of these species or the lack of an exploitable connection between GlcNAc metabolism and glycoprotein biosynthesis.
The narrow scope of Ac4GlcNAz incorporation prompted the team to explore the use of bacteria-specific monosaccharides to metabolically tag glycoproteins in these species. As shown in Figure 25 and discussed above, bacterial glycoproteins are widespread, diverse in structure, and decorated with unique sugars, such as Bac, FucNAc, and DATDH, which presents attractive opportunities for probe development. Using these unique monosaccharides as inspiration to develop potential chemical reporters, Dube and Kulkarni chemically synthesized azide-modified analogues, including Ac3FucNAz (87), Ac2Bac-DiNAz (88), and Ac2DATDG-diNAz (89) (Figure 29B).216 The probe design was informed by Ac4GlcNAz (86), as N-azidoacetyl groups were chosen for the azide modification and peracetylation was used as a strategy to potentially enhance uptake. The chemical syntheses of 88 and 89, though not described here in detail, were lengthy (14–16 steps from D-mannose) due to the requirement to install multiple nitrogen atoms and establish appropriate stereochemical configurations on the monosaccharide core.216 Given the success by Tanner and co-workers in utilizing a chemoenzymatic synthesis to produce a similar diaminosugar scaffold en route to AltNAc4NAz (71),202 it may be advantageous in the future to explore similar approaches for compounds 88 and 89.
The three new compounds, 87–89, were then tested for incorporation into proteins in a panel of bacteria similar to that described above.216 It was observed that both Ac2Bac-DiNAz (88) and Ac2DATDG-diNAz (89) were incorporated into proteins in H. pylori, C. jejuni, and R. solanacearum, while only Ac2Bac-DiNAz (88) incorporated into B. thailandensis; flow cytometry confirmed the incorporation of these reporters into the surface of whole cells.216 P. aeruginosa and M. smegmatis were not tested in this round of experiments, and Ac3FucNAz (87) did not label any organisms appreciably.216 Because no experiments were conducted to determine the nature of the molecules labeled by 88 and 89, it will be of interest to do so in the future. In the meantime, other experiments showed that the reporters did not incorporate into the cell surface of the human symbiont B. fragilis nor into the surface of mammalian MDCK cells, suggesting potential for enlisting these and related compounds as tools for bacteria-specific targeting applications.216 Indeed, a follow-up study in 2020 from the same team reported the synthesis and evaluation of a panel of fluorinated and benzyl glycoside versions of Bac, DATDH, and FucNAc, some of which impaired motility and biofilm formation in H. pylori and C. jejuni, but not symbiotic B. fragilis.236
Together, these studies show that rare bacterial sugar analogues may be valuable tools for probing and inhibiting bacterial protein glycosylation. It will be of interest moving forward to investigate the mechanism of incorporation of these compounds and apply them in MS-based glycoproteomics and covalent therapeutic targeting strategies, examples of which we covered above. Indeed, as with Ac4GlcNAz, these probes should have the capacity to facilitate the profiling and structural assignment of heretofore uncharacterized bacterial glycoproteins, as well as the elucidation of uncharacterized pathways associated with glycoprotein biosynthesis. In turn, this knowledge will aid in the design of next-generation probes and inhibitors. It will also be useful to evaluate 87–89 in other organisms that are known to have FucNAc, Bac, and DATDH residues present in glycoproteins, such as Pseudomonas (FucNAc) and Neisseria (Bac and DATDH) (see Figure 25). A recurring issue that we have discussed in preceding sections and arose again here is the use of free vs. peracetylated reporter probes. In the case of the Bac, DATDH, and FucNAc reporters, all were evaluated exclusively in their peracetylated forms. Given the differences in behavior observed for free and peracetylated versions of reporter probes for pseudaminic and legionaminic acids, which bear resemblance to both the Bac and DATDH probes, it would likewise be of value to test the unmasked forms of compounds 87–89 in various species.
4.3.2. Fucose derivatives.
B. fragilis, an abundant Gram-negative commensal organism of the human gut microbiota, is known to acquire exogenous L-fucose from the host and incorporate it into surface polysaccharides and glycoproteins.180 Fucosylation is thought to enable host mimicry and contributes to normal growth in vitro and proper colonization of the host in vivo.237 Unlike most other forms of bacterial protein glycosylation, the O-glycosylation system in B. fragilis adds glycans to a specific consensus amino acid sequence, which has allowed the creation of computational approaches that predicted the existence of hundreds of possible fucosylated glycoproteins in B. fragilis.238
To aid in the experimental identification and characterization of fucosylated glycoproteins, in 2011 the Wu group published a chemical reporter strategy for labeling glycoproteins in B. fragilis and the related organism Parabacteroides distasonis.217 As discussed above, these organisms and others in the Bacteroidales order have a native fucose salvage pathway180 that internalizes exogenous fucose and converts it via the Fkp enzyme into GDP-fucose, which is subsequently processed by GTases to form glycoconjugates (Figure 21B). In prior work, the team had shown that the clickable fucose analogues FucAz (58) and FucAl (59) (see Figure 21C) could be processed by purified Fkp to generate the corresponding GDP-fucose analogues.239 To test for incorporation into bacterial cells, FucAl (59) was fed to live P. distasonis, then cells were reacted with azido-biotin via CuAAC, stained with avidin-488, and analyzed by flow cytometry.217 Increased cellular fluorescence was observed upon treatment with FucAl (59) compared to the untreated control,217 prompting further experiments to evaluate protein labeling. Lysates were collected from FucAl-treated P. distasonis, biotinylated via CuAAC, and analyzed by anti-biotin Western blot.217 Numerous bands were observed, and signal was ablated by proteinase K, confirming that the reporter was labeling proteins.217 B. fragilis cells were also labeled by FucAl (59) in an Fkp-dependent manner (confirmed by analysis in Fkp-deficient mutant), although at a lower efficiency due to the presence of a fucose catabolic pathway that presumably depleted the reporter probe.217 Also in contrast to the result from P. distasonis, only one high-molecular weight protein band was labeled in B. fragilis, suggesting a lower efficiency of probe incorporation and/or fewer fucosylated proteins in this organism.217
The ability of clickable fucose analogues to label glycoproteins in commensal Bacteroidales species sets the stage for affinity enrichment and LC-MS/MS analysis to identify these proteins on a whole-proteome scale. To date, such work has not been reported in the literature, although it would be a valuable experimental approach to facilitate the validation of computationally predicted fucosylated proteins and potentially identify novel fucosylated proteins. Another interesting facet of this research is that the Fkp-mediated fucose salvage pathway, which is native to Bacteroidales species, can be engineered into other systems to permit the uptake and metabolic incorporation of fucose-based reporters into cell envelope glycans. As covered above, one successful example of this was Wang’s usage of the fucose salvage pathway to enable tagging of LPS O-antigen fucose residues in E. coli (section 3.2.1).167
5. Chemical reporters for teichoic acids
In contrast to Gram-negative bacteria, which have a small PG layer covered by an outer membrane abundant in anionic LPS, Gram-positive bacteria have a very thick layer of PG that is extensively modified by anionic glycopolymers called teichoic acids. Teichoic acids can either be covalently linked to PG MurNAc residues, yielding wall teichoic acids, or lipid-anchored to the plasma membrane, yielding lipoteichoic acids.7,8 Beyond these classifications, teichoic acid structure is quite diverse between organisms and strains.240 As examples, Figure 30 shows representative structures of wall teichoic acids and lipoteichoic acids from B. subtilis and S. pneumoniae, respectively. B. subtilis wall teichoic acids consist of a poly(glycerol phosphate) (poly(Gro-P)) polymer variably modified with D-Ala or α-glucosyl groups, and this structure is ultimately connected to PG MurNAc through a disaccharide phosphate linker.8 S. pneumoniae lipoteichoic acids consist of a pseudopentasaccharide repeating unit with GlcNAc, GalNAc, glucose, ribitol phosphate, and 2-acetamido-4-amino-2,4,6-trideoxy-D-galactopyranose (AAT) residues, the latter of which is connected via a glucosyl linker moiety to a glycerolipid anchor.241 The ribitol phosphate residue of the S. pneumoniae lipoteichoic acid can be further modified by D-Ala esters, while the GlcNAc and GalNAc residues are decorated by phosphocholine.241 The same structure is present as wall teichoic acid that is anchored to PG in S. pneumoniae. Teichoic acids, with their anionic character and variable modifications, have a profound impact on Gram-positive bacterial physiology, as they contribute to cell shape and flexibility, regulate cell division, provide protection from environmental stress and antibiotics, and participate in adhesion and colonization processes within the host.7,8,242 B. subtilis mutants lacking the ability to form both wall teichoic and lipoteichoic acids are non-viable, confirming their importance to Gram-positive bacterial physiology.243,244 Accordingly, there is significant interest in exploring and exploiting teichoic acid biosynthesis and tailoring pathways as targets for antibiotic development.242
Figure 30.
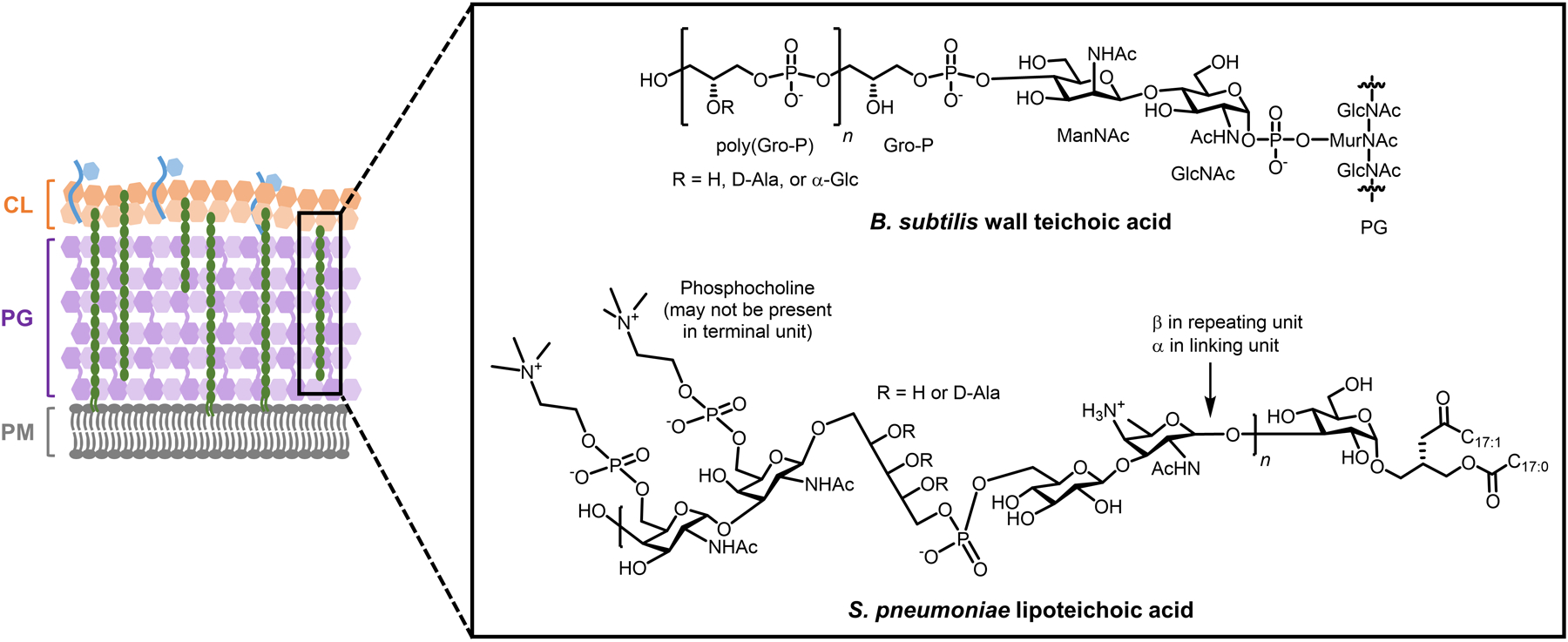
Representative structures of wall teichoic acids and lipoteichoic acids in Gram-positive bacteria.
Teichoic acids clearly offer numerous structural features that could possibly serve as targets for the development of chemical reporters. However, to date only one strategy has been published, which is focused on the phosphocholine modifications of teichoic acids from S. pneumoniae. As shown in Figure 30, phosphocholine tailoring occurs on the 6-position hydroxyl groups of the GalNAc and GlcNAc residues of the teichoic acid repeating unit.241,245,246 The zwitterionic phosphocholine units are known to serve as cell wall anchoring points for endogenous choline-binding proteins that are involved in PG turnover, immune evasion, and host adhesion; phosphocholine units are also involved in host adhesion due to their ability to directly bind to host platelet-activating factor protein.247–249 The tailoring of both wall teichoic acids and lipoteichoic acids with phosphocholine in S. pneumoniae occurs via a shared biosynthetic pathway (Figure 31). Choline, which is an essential nutrient for S. pneumoniae,245 is acquired from the environment and transported into the cytoplasm by LicB, after which it is activated into cytidine 5’-diphosphocholine (CDP-choline) via LicA and LicC.246 Subsequently, LicD1 and LicD2 transfer phosphocholine units from CDP-choline onto GalNAc residues of a lipid-linked teichoic acid repeating unit.246 The product is then polymerized by Spr1222, translocated across the plasma membrane by TacF, and linked to either PG or diacylglycerol by LCP to generate wall teichoic acids or lipoteichoic acids, respectively.246 Interestingly, exogenously acquired choline that is converted into CDP-choline is not incorporated into membrane phosphatidylcholine.250 This provides an opportunity to selectively label teichoic acids in S. pneumoniae with choline derivatives.
Figure 31.
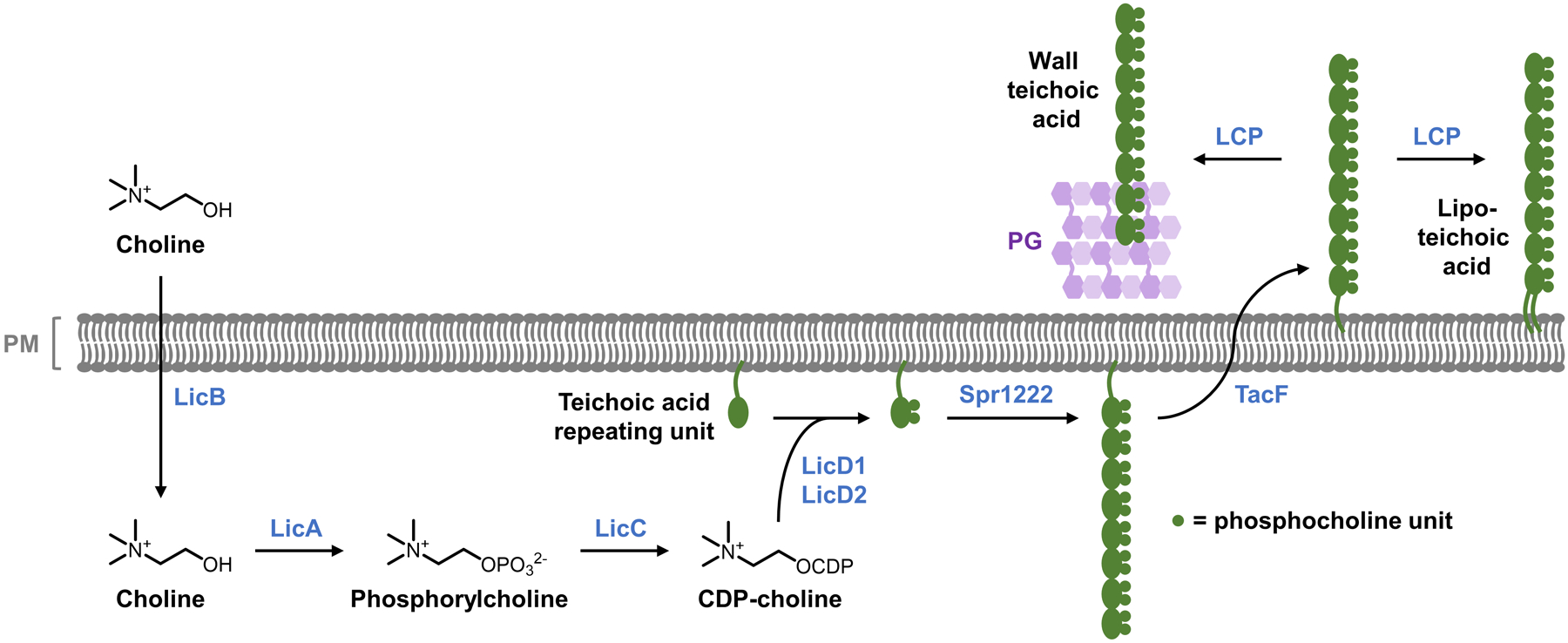
Common pathway of phosphocholine tailoring of wall teichoic acids and lipoteichoic acids in S. pneumoniae. See Figure 30 for the structure of the teichoic acid repeating unit (biosynthesis of the repeating unit is not shown here).
In 2017, the Guilmi and Wong groups reported the development of synthetic choline analogues, propargyl-Cho (90) and fluorescent-Cho (91), as potential metabolic labeling probes for teichoic acids in S. pneumoniae (Figure 32A).251 Propargyl-Cho (90) had previously been used as a chemical reporter for labeling phosphatidylcholine in eukaryotic cells.252 Here, it was used both as a clickable reporter for teichoic acids in S. pneumoniae and as a synthetic precursor to the fluorescent compound 91, which was accessed via CuAAC reaction between 90 and fluorogenic azido-coumarin. To determine whether the analogues 90 or 91 could be metabolized by S. pneumoniae, a non-pathogenic strain was cultured in medium containing native choline, 90, or 91. While bacteria cultured in native choline or propargyl-Cho (90) grew equally well, fluorescent-Cho (91) did not support growth, indicating that 90, but not the bulkier 91, were successfully metabolized.251 This is a clear example of how smaller, two-step bioorthogonal chemical reporters can have an advantage over one-step fluorescent reporters in metabolic pathways with stricter substrate specificities. To confirm that propargyl-Cho (90) labeled teichoic acids, both wall teichoic acids and lipoteichoic acids were extracted from 90-treated S. pneumoniae and shown to contain high levels of incorporated 90 by 1H and 31P NMR spectroscopic analysis.251 In imaging experiments, 90 was incorporated into S. pneumoniae and detected on the cell surface by CuAAC reaction with azido-coumarin fluorophore.251 Together, these data demonstrated that smaller choline derivatives such as propargyl-Cho (90) can specifically and efficiently incorporate into S. pneumoniae teichoic acids.
Figure 32.
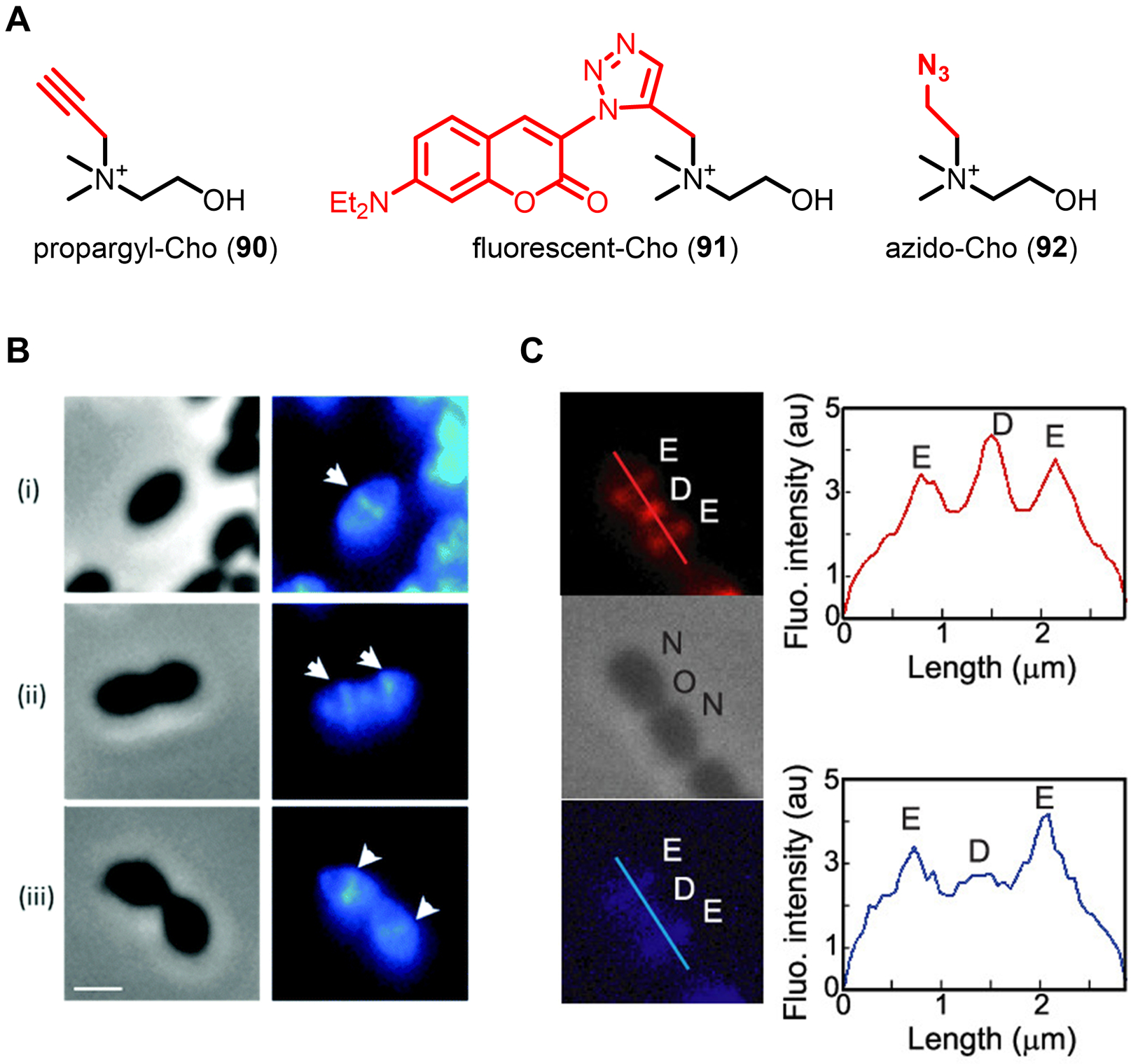
(A) Choline derivatives for labeling phosphocholine-modified teichoic acids in S. pneumoniae. (B) Imaging of S. pneumoniae teichoic acids with propargyl-Cho (90). S. pneumoniae at different growth phases (i–iii represent early to late growth stages) was incubated in the presence of propargyl-Cho (90) for a 30 min pulse, subjected to CuAAC with azido-coumarin, and imaged by fluorescence microscopy. Left, phase contrast; right, fluorescence. White triangles mark the septal plane. Scale bars, 1 μm. Reproduced with permission from ref 251. Copyright 2017 Royal Society of Chemistry. (C) Simultaneous imaging of S. pneumoniae teichoic acids and PG with propargyl-Cho (90) and HADA (17), respectively. S. pneumoniae was incubated in the presence of propargyl-Cho (80), cyclooctyne-594, and HADA (17) for a 5 min pulse, washed, and imaged by fluorescence microscopy. Top (red), teichoic acids; middle, phase contrast; bottom (blue), PG. E, elongation site; D, division site; N, new pole; O, old pole. Right-hand boxes show fluorescence intensity as a function of cell length. Reproduced with permission from ref 253. Copyright 2018 American Chemical Society.
Choline-based reporters have been applied to study teichoic acid dynamics and to specifically detect choline-decorated bacteria. In the same 2017 study by Guilmi and Wong, the team performed pulse-chase imaging experiments with propargyl-Cho (90) and CuAAC to track teichoic acid biosynthesis, which revealed teichoic acid labeling at the same septal site where cross-wall PG synthesis occurs (Figure 32B).251 This finding suggested that teichoic acid synthesis and transport may occur concurrently with that of PG. However, the relative sluggishness of the CuAAC reaction prompted the team to explore other bioorthogonal chemistries to improve the visualization of teichoic acid dynamics. A subsequent study reported in 2018 by the same group developed a new probe, azido-Cho (92) (Figure 32A), which rapidly incorporated into S. pneumoniae teichoic acids and could be quickly detected by SPAAC with a cyclooctyne-fluorophore in the same pot.253 This method allowed for imaging studies to be carried out on shorter time scales, and when performed in tandem with PG labeling using HADA (17), it was confirmed that teichoic acid and PG labeling are largely concurrent (Figure 32C), however teichoic acid synthesis appeared to persist longer at the septum than PG synthesis.253 In addition to imaging studies, it was shown that propargyl-Cho (90) exclusively labeled phosphorylcholine-decorated S. pneumoniae, but not various phosphorylcholine-deficient bacteria such as E. coli, B. subtilis, or P. aeruginosa, the latter of which even synthesizes phosphatidylcholine.251 This result lends support to the notion that compound 90 can be used to specifically detect S. pneumoniae in complex samples, similar to the way legionaminic acid-based chemical reporters were used to detect the pathogen Legionella pneumophila by metabolically tagging its unique LPS O-antigen (see section 3.2.2).
The described applications demonstrate the potential of small clickable choline-based reporters, such as 90 and 92, for teichoic acid imaging in, and specific detection of, S. pneumoniae. However, given the relatively narrow applicability of these probes—they are limited to phosphocholine-tailored organisms such as S. pneumoniae—the development of tools to interrogate teichoic acids in other Gram-positive bacterial species should be pursued. There is motivation and opportunity to do so, as teichoic acids are critical for Gram-positive bacterial physiology, have high structural diversity, and exhibit distinctive surface-exposed features that can possibly be exploited for diagnostic or therapeutic targeting. While teichoic acids do contain D-Ala residues that could be of interest for labeling, available evidence suggests that D-Ala derivatives do not incorporate teichoic acids.76,77 On the other hand, teichoic acids contain various types of carbohydrates, so monosaccharide-based reporters should be explored.
6. Chemical reporters for mycobacterial glycans
Mycobacteria, which cause devastating diseases such as tuberculosis, leprosy, and Buruli ulcer, possess a complex cell envelope decorated with an array of unique glycans that provide the bacterium with structural support, defense against stress/antibiotics, and host immunomodulation capabilities.10,254–256 The mycobacterial cell envelope has plasma membrane (PM) and peptidoglycan (PG) layers in common with Gram-negative and -positive bacteria, but its structure diverges at this point, as shown in Figure 1. Approximately every tenth PG repeating unit has a rhamnose-containing linker that covalently connects the PG layer to a large heteropolysaccharide called arabinogalactan (AG), which extends further outward from the cell.257 The AG layer consists of a linear galactan domain made of repeating galactofuranosyl (Galf) residues that is extended by branched arabinan domains made of arabinofuranosyl (Araf) residues.258 Terminal Araf residues of the AG layer can be esterified by long-chain (C60–C90) branched fatty acids called mycolic acids, forming arabinogalactan-linked mycolates (AGM) that represent the inner leaflet of the mycobacterial outer membrane, or mycomembrane.259 Thus, the core of the mycobacterial cell envelope is a colossal covalent meshwork of glycoconjugates comprising the PG, AG, and inner mycomembrane layers. The outer leaflet of the mycomembrane contains an array of non-covalently associated lipids and glycolipids—the so-called “extractable lipids”—including trehalose glycolipids (e.g., trehalose mono- and dimycolate (TMM and TDM) among others), phenolic glycolipids (PGLs), glycopeptidolipids (GPLs), and lipooligosaccharides (LOSs).11 Another major class of mycobacterial glycolipids, thought to be present in both the inner and outer membranes, are the phosphatidylinositol mannosides (PIMs), whose arrangement of inositol, glucosamine, and mannose residues closely resembles that of eukaryotic glycosylphosphatidylinositol (GPI) anchors.11,260 In mycobacteria, PIMs can exist in free form or they can anchor mannose- and Araf-containing polysaccharides to the cell surface in the form of lipomannan (LM) and lipoarabinomannan (LAM).11,260 Outside of the mycomembrane is a capsular polysaccharide layer consisting of α-glucans, which are α(1→4) polyglucoses containing α(1→6) branches, as well as mannan and arabinomannan, which are delipidated forms of LM and LAM.11,261 Mycobacteria also produce O-mannosylated glycoproteins bearing up to three α(1→2)-linked mannose units attached to serine or threonine residues.262,263
Many of these mycobacterial glycans, along with their associated metabolic pathways, are mycobacteria-specific and are essential for growth and/or virulence, making them attractive targets for diagnostic and therapeutic development. Indeed, the front-line antimycobacterial compounds isoniazid and ethambutol target the biosynthesis of mycolic acids and AG, respectively, and the development of novel antimycobacterial compounds continues to focus heavily on the cell envelope. However, in many cases, the full details of mycobacterial glycan structure, biosynthesis, and function, including the roles of glycans during infection and the molecular-level details of their interactions with the host, are not well understood.11 Progress in this area has been impeded by a lack of tools, which ultimately hinders the pursuit of fundamental and translational research related to these critical cell envelope components. Because most mycobacterial glycans are structurally and functionally distinct from those in eukaryotes and even other bacteria, the vast majority of tools developed for these systems are not applicable to mycobacteria. In this section, we describe progress in the development and applications of chemical reporters for mycobacteria-specific glycans, including those containing trehalose, mycolates, and arabinose. Where appropriate, we provide detailed chemical structures, biosynthetic pathways, and proposed mechanisms of probe incorporation. Probes for PG, many of which can be used to label and investigate mycobacterial PG, were covered in section 2. For an exhaustive account of mycobacterial glycan structure, biosynthesis, and function, we point readers to a review by Jackson and colleagues.11 Table 4 contains information about selected chemical reporters that target mycobacterial glycans.
Table 4.
Selected chemical reporters for mycobacterial glycans
| Target | Metabolite | Compound | Reporter group (X) | Bioorthogonal reaction(s)a | Proposed route | Speciesb | Applicationsc | Synthesis | Notesb | Refd |
|---|---|---|---|---|---|---|---|---|---|---|
| TMM and/or TDM | Trehalose | FITC-Tre 93 (Fig. 35) | Fluorescein | -- | Extracellular | Broad (M) | In vitro and in vivo imaging (infected macrophages) | Chemical; comm. available | Contains unnatural anomeric methyl group | 264 |
| Trehalose | FlTre series 94–97 (Fig. 35) | Fluorescein | -- | Extracellular | Broad (M) | In vitro imaging; membrane fluidity and inhibitor characterization | Chemical | Lack of unnatural anomeric methyl group enhances incorporation | 265 | |
| Trehalose | DMN-Tre 98 (Fig. 35) | DMN | -- | Extracellular | Broad (M) | In vitro and in vivo imaging (infected macrophages); selective detection of M. tuberculosis; inhibitor characterization | Chemical | Fluorogenic | 266 | |
| Trehalose | TreAz series 99–102 (Fig. 35) | Azide | CuAAC; SPAAC | Intracellular | Broad (M) | In vitro and in vivo imaging (infected host organism); inhibitor characterization | Chemo-enzymatic; chemical; 6-TreAz is comm. available | 2-, 4-, 6-TreAz label via LpqY-SugABC; 3-TreAz labels via Ag85; 6-TreAz only labels TMM | 267 | |
| Trehalose | 6-LactoTreAz 104 (Fig. 35) | Azide | SPAAC | Intracellular | M. smegmatis (M) | In vitro imaging | Chemo-enzymatic | Inversion of 4-position enhances stability toward trehalase | 268 | |
| Trehalose | 18F-2-FDTre 105 (Fig. 35) | 18F | -- | Intracellular | M. smegmatis (M) | In vitro tracing of trehalose uptake | Chemo-enzymatic | Potential in vivo PET imaging probe | 269,270 | |
| TMM | N-AlkTMM-C7 121 (Fig. 38) | Alkyne | CuAAC | Extracellular | Broad (M) | In vitro imaging | Chemical | Only labels TDM | 271 | |
| TMM | N-x-AlkTMM-C15 122 (Fig. 38) | Alkyne and diazirine | CuAAC | Extracellular | Broad (M) | Identification of mycolate–protein interactions | Chemical | Diazirine allows protein photo-cross-linking | 272 | |
| AGM and TDM | TMM | O-AlkTMM-C7 113 (Fig. 38) | Alkyne | CuAAC | Extracellular | Broad (M) | In vitro imaging; protein profiling | Chemical | Also labels O-mycoloylated proteins in C. glutamicum | 271 |
| TMM | O-AlkTMM-C23 115 (Fig. 38) | Alkyne | CuAAC | Extracellular | C. glutamicum (related to M) | In vitro imaging | Chemical | Possesses more-native mycolate chain | 273 | |
| TMM | O-AzTMM-C10 116 (Fig. 38) | Azide | SPAAC; CuAAC | Extracellular | M. smegmatis (M), C. glutamicum (related to M) | In vitro imaging | Chemical | Also labels O-mycoloylated proteins in C. glutamicum | 274 | |
| TMM | O-TCO-TMM-C10 117 (Fig. 38) | Trans-cycooctene | Tetrazine ligation | Extracellular | M. smegmatis (M), C. glutamicum (related to M) | In vitro imaging; rapid detection of mycobacteria | Chemical | Also labels O-mycoloylated proteins in C. glutamicum | 274 | |
| TMM | O-FITC-TMM-C10 119 (Fig. 38) | Fluorescein | -- | Extracellulare | M. smegmatis (M), C. glutamicum (related to M) | In vitro imaging | Chemical | Labeling of O-mycoloylated protein was inconsistent with other TMM reporters | 274 | |
| TMM | O-DBF-TMM 120 (Fig. 38) | Dibromo-fluorescein | -- | Extracellular | C. glutamicum (related to M) | In vitro imaging using transmission electron microscopy | Chemical | DBF is a singlet oxygen-generating fluorophore | 275 | |
| Ag85 enzyme | TMM | QTF 125 (Fig. 40) | FRET pair | -- | Extracellular | M. smegmatis (M), C. glutamicum (related to M) | Detection and imaging of Ag85 hydrolytic activity | Chemical | Fluorogenic probe of enzyme activity | 276 |
| Tdmh enzyme | TDM | FRET-TDM 128 (Fig. 42) | FRET pair | -- | Extraceullar | M. smegmatis (M), M. tuberculosis (M) | Detection of Tdmh hydrolytic activity | Chemical | Fluorogenic probe of enzyme activity | 277 |
| AG, other arabinans | Arabinose | 5-AraAz 133 (Fig. 45) | Azide | SPAAC | Intracellulare | M. tuberculosis (M) | In vitro imaging and in vivo imaging (macrophages infected with pre-labeled bacteria) | Chemical | Labels M. tuberculosis cells but route and target are unknown | 278 |
| DPA | 2-AzFPA 135 (Fig. 46) | Azide | SPAAC | Extracellular | M. smegmatis (M), C. glutamicum (related to M) | Chemical | Lipid-linked Araf derivative | 279 | ||
| DPA | 5-AzFPA 137 (Fig. 46) | Azide | SPAAC | Extracellular | M. smegmatis (M), C. glutamicum (related to M) | In vitro and in vivo imaging (macrophages infected with pre-labeled bacteria) | Chemical | Lipid-linked Araf derivative | 279 |
Not applicable for fluorescent reporters operating via one-step incorporation (--).
Gram classification: GN, Gram-negative; GP, Gram-positive; M, mycobacteria.
In vitro refers to bacterial labeling experiments performed in broth culture; in vivo refers to bacterial labeling experiments performed in a host cell or organism.
Initial publication(s) referenced. See text for references for follow-up studies and applications.
Limited evidence available to support proposed route of incorporation and/or identity of labeled target (see text).
6.1. Chemical reporters for mycolate-containing glycolipids
To date, chemical reporter development in mycobacteria has predominantly focused on the mycolate-containing glycolipids of the mycomembrane. As noted above, the major species in this category are AGM, which forms the inner leaflet of the mycomembrane, and TMM/TDM, which are present in the outer leaflet of the mycomembrane (Figure 33). AGM is covalently connected to the underlying AG and PG layers, whereas TMM and TDM non-covalently associate with the mycomembrane. All of these structures are mycolic acid esters, or mycolates, with the same long-chain, α-branched, β-hydroxy-modified mycoloyl groups connected to different carbohydrates: terminal Araf residues of the AG layer, in the case of AGM; or the 6/6’-position(s) of the disaccharide trehalose, in the case of TMM and TDM.259 The mycolate portion of these structures exhibits structural variation, as the lipid tails can exist as alpha, methoxy, or keto forms depending on the functional groups present on the aliphatic chain.280 The thick, highly hydrophobic mycomembrane layer that arises from this unique glycolipid arrangement gives mycobacteria their characteristic waxy surface and provides a robust permeability barrier that protects the bacterium from environmental stress or antibiotic treatment.259,280 In addition to these protective attributes, TDM is well known for its potent immunostimulatory activity, which can be viewed as somewhat analogous to outer membrane LPS in Gram-negative bacteria.281 Because of these critical functions, mycolate-containing glycolipids are strictly required for mycobacterial survival and virulence. Therefore, these structures have emerged as prime targets for the development of mycomembrane-targeting probes and inhibitors, which are potential tools to investigate, diagnose, or treat important mycobacterial diseases such as tuberculosis.28,282 A key example is the cornerstone antitubercular compound isoniazid, which inhibits the production of mycolic acids, in turn preventing the proper construction of mycomembrane glycolipids.283
Figure 33.
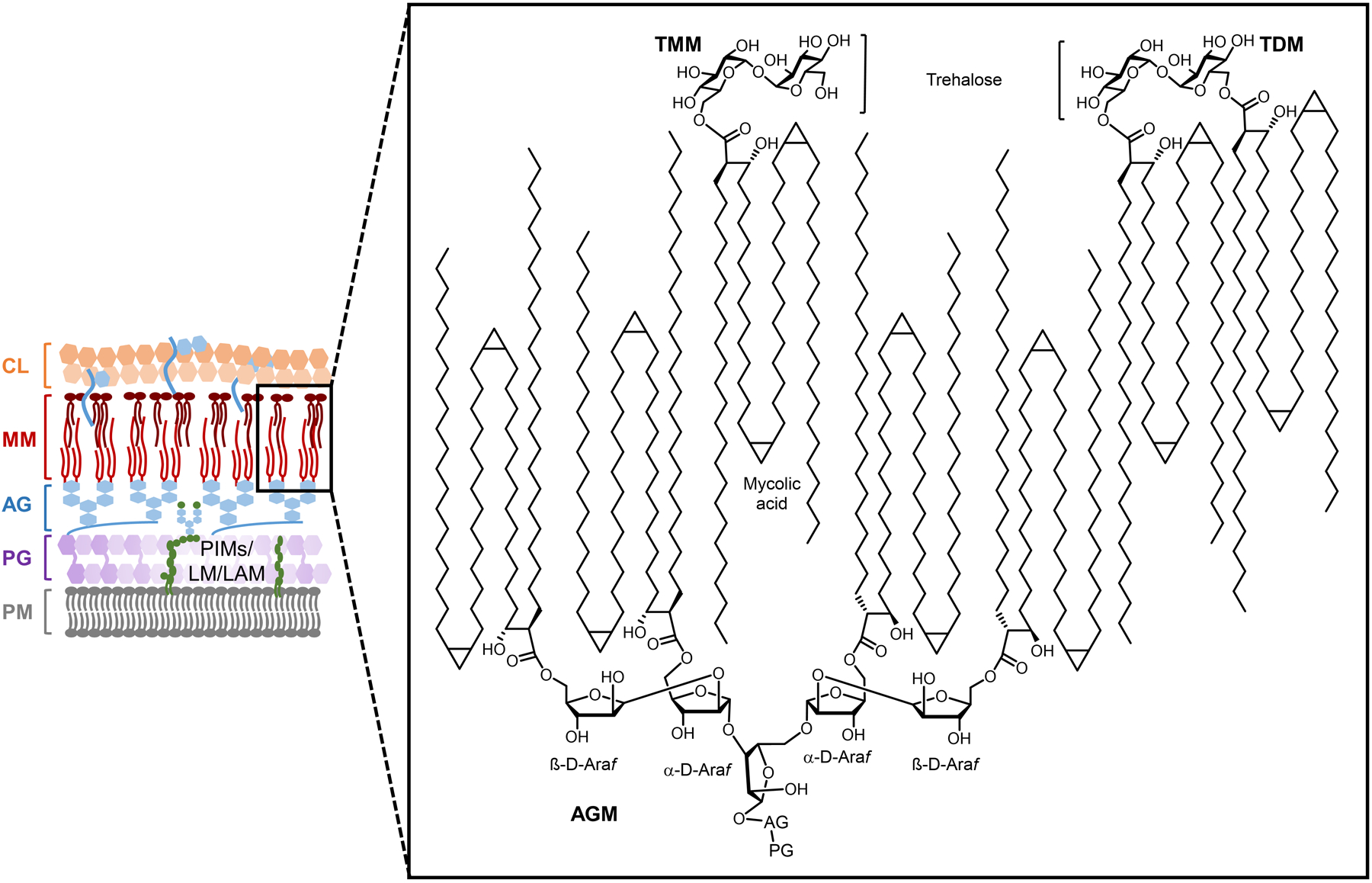
Representative structures of the mycolate-containing glycolipids AGM, TMM, and TDM present in the outer mycomembrane of mycobacteria. AGM, arabinogalactan-linked mycolates; TDM, trehalose dimycolate; TMM, trehalose monomycolate.
The biosynthesis of TMM, TDM, AGM, and other mycolate-containing cell envelope components proceeds via a common pathway that utilizes trehalose as a mycolic acid-shuttling intermediary (Figure 34). Mycobacteria can produce trehalose by at least three different routes, including from glucose (OtsAB pathway), maltose (TreS pathway), and α-glucans (Tre(X)YZ pathway); the former OtsAB pathway is the predominant route of trehalose biosynthesis.261,284–287 To mediate the production of mycomembrane glycolipids, trehalose is first esterified at the 6-position with a mycolic acid via Pks13/CmrA in the cytoplasm, yielding TMM.288 TMM is then flipped across the plasma membrane by MmpL3 into the periplasmic space, where its mycoloyl group is donated to different acceptor substrates to generate the mycolate-containing products that localize to the mycomembrane.289 This mycolic acid exchange process is catalyzed by a group of mycoloyltransferase enzymes referred to as the antigen 85 (Ag85) complex.290,291 In these Ag85-catalyzed reactions, if the acceptor is a second molecule of TMM, then TDM is produced; if the acceptor is a terminal Araf residue from AG, then AGM is produced; other acceptors, such as glucose, glycerol, and proteins, can also be lipidated through this pathway.259,292,293 In each reaction, the TMM donor molecule loses its mycoloyl group, releasing free trehalose, which is recycled back into the cell by the trehalose-specific transporter LpqY-SugABC.294 Under certain conditions, mycolate glycolipids can also be broken down, for example TDM can be converted to TMM and free mycolic acid via TDM hydrolase (Tdmh).295,296 These trehalose-mediated glycolipid biosynthesis, transport, and remodeling pathways are generally conserved within and specific to mycobacteria and have been shown to be important for growth and virulence.
Figure 34.
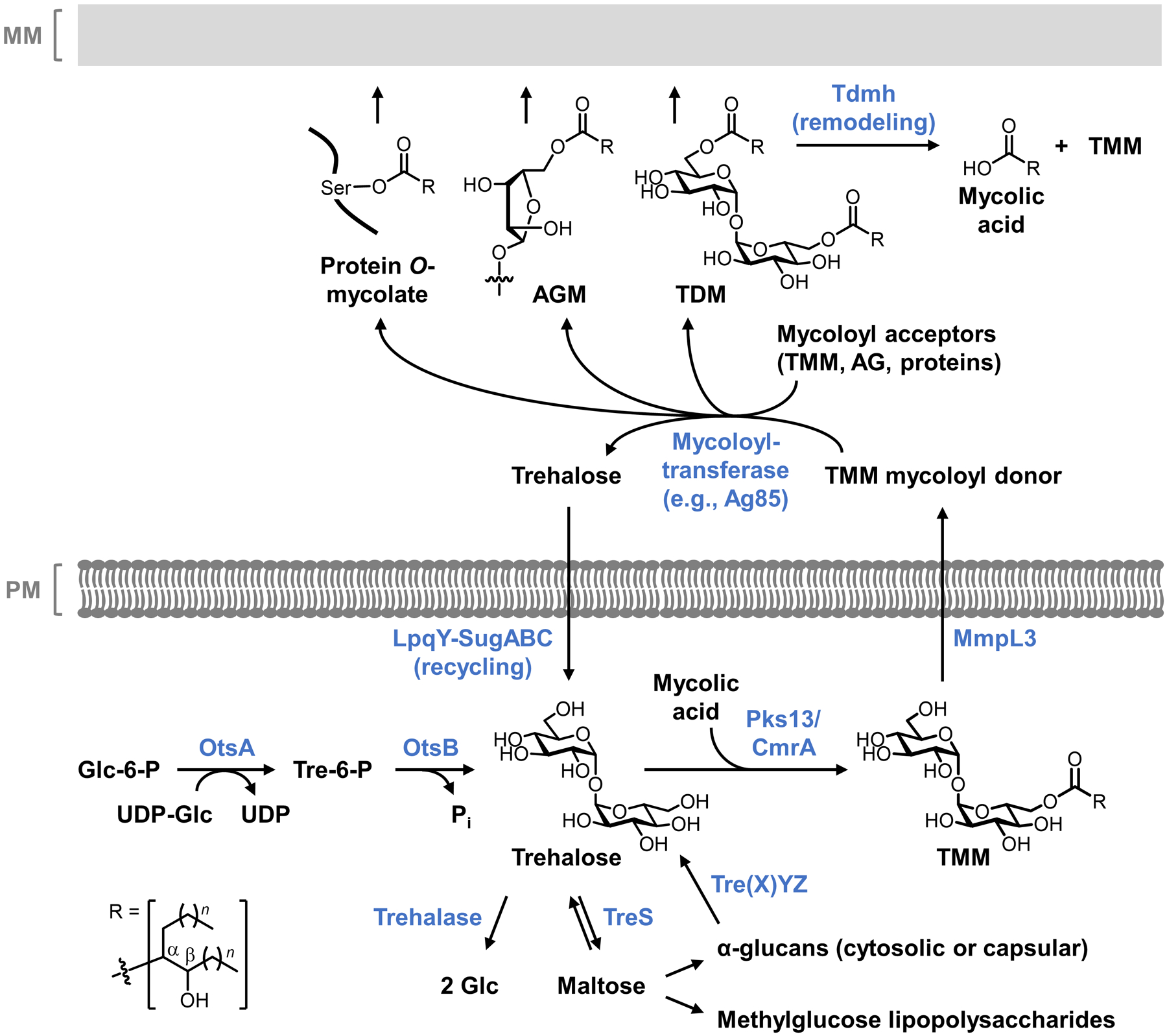
Trehalose-mediated biosynthesis of mycolate-containing glycolipids in mycobacteria. Figure adapted from ref 297 with permission from the Royal Society of Chemistry.
The described trehalose-mediated glycolipid metabolic pathways provide a well-characterized landscape for the rational design and development of glycolipid-targeting reporter probes. For example, periplasmic Ag85-mediated mycolic acid exchange can serve as a possible mechanism of extracellular probe incorporation, in a manner that can be considered analogous to periplasmic D-amino acid exchange in PG synthesis. However, because mycobacteria have an outer membrane—in fact, a particularly tough permeability barrier in the form of the mycomembrane—access of exogenously added synthetic substrates across this membrane may be hindered. Alternatively, targeting cytoplasmic carbohydrate or lipid metabolism to label glycolipids from the inside-out could serve as an early-stage intracellular route of incorporation, particularly if appropriate transporters were tolerant of modified substrates (e.g., LpqY-SugABC). In the intracellular incorporation mode, multiple biosynthetic steps would require substrate tolerance and there is potential for off-target channeling of the probe, e.g. through degradative pathways. Probe synthesis is another pressing issue in this system, as both the carbohydrate (trehalose disaccharides and Araf oligosaccharides) and lipid (mycolic acid) portions of the substrates are structurally complex and challenging to synthesize in modified form. Over the past decade, all of these issues have been explored and addressed, leading to the development of a robust toolbox of complementary reporter probes that allow labeling of specific mycomembrane glycolipids via defined incorporation routes. These tools have been applied to study the dynamics of glycolipid biosynthesis and remodeling, used to identify and characterize mycomembrane-related proteins, and adapted to detect or kill mycobacteria. In the following sections, we discuss the development and applications of chemical probes for mycolate-containing glycolipids in three categories based on the substrates they mimic: trehalose derivatives, TMM derivatives, and TDM derivatives.
6.1.1. Trehalose derivatives.
In principle, trehalose derivatives bearing unnatural chemical functionality could incorporate into trehalose mycolate glycolipids as shown in Figure 35A. Assuming successful passage across the mycomembrane, an exogenously delivered trehalose derivative could serve as a mycoloyl acceptor in an Ag85-catalyzed mycolic acid exchange reaction occurring in the periplasm, leading to labeling of TMM/TDM and thereby anchoring the unnatural tag into the mycomembrane. This notion was first explored by the Barry and Davis groups, who in 2011 reported the development of the first trehalose-based probe, fluorescein isothiocyanate-modified keto trehalose (FITC-Tre, 93), which is a ketoside analogue of trehalose bearing a fluorescein group at the 2’-position (Figure 35B).264 Prior to synthesizing and testing FITC-Tre, the team confirmed that mycobacterial cells could take up 14C-labeled native trehalose from the growth medium and incorporate it into TMM and TDM.264 Next, the tolerance of Ag85 enzymes for unnatural substrates was evaluated by using a mass spectrometry-based screen to show that purified Ag85 enzymes were capable of converting a wide range of synthetic trehalose analogues into the corresponding acylated TMM products.264 These preliminary experiments demonstrated that mycobacteria met two key criteria to support trehalose probe development: (i) exogenously supplied native trehalose (and thus potentially trehalose derivatives) incorporates into mycolate glycolipids in live mycobacteria; and (ii) the targeted Ag85 mycoloyltransferase enzymes have high tolerance for unnatural trehalose derivatives as substrates.
Figure 35.
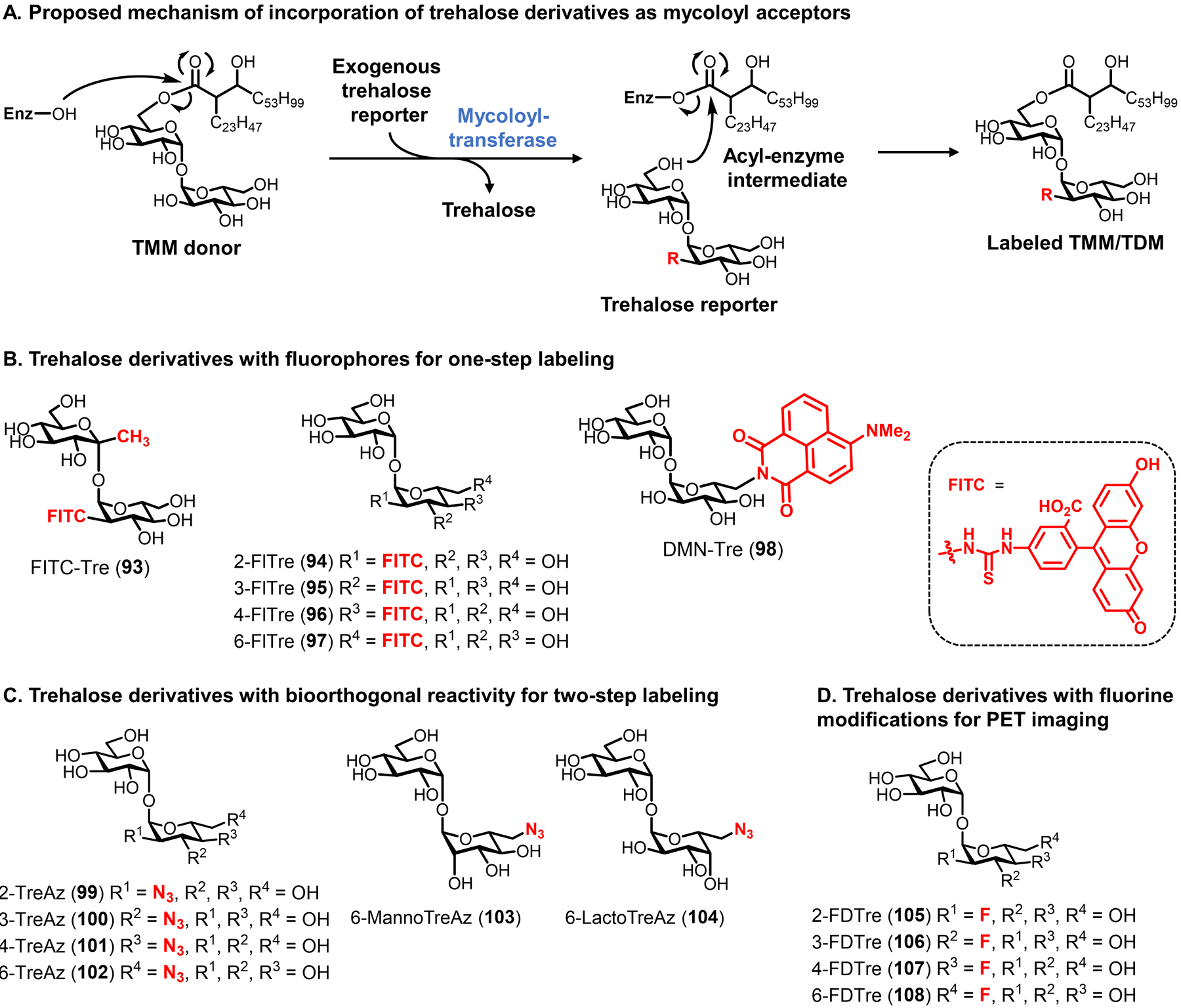
Trehalose derivatives for labeling trehalose mycolate glycolipids in mycobacteria. (A) Metabolic incorporation of unnatural trehalose derivatives (modified with an unnatural R group) is proposed to occur through mycoloyltransferase-catalyzed mycoloyl group transfer. Periplasmic Ag85-catalyzed transfer of mycoloyl group from native TMM to exogenous trehalose derivative is shown. A similar mechanism would occur for cytoplasmic Pks13-mediated mycoloyl group transfer onto intracellular trehalose derivatives. Depending on the position of the unnatural modification, mycoloyl groups could add to one 6-position to give a labeled TMM or both 6-positions to give a labeled TDM. Enz, enzyme with mycoloyltransferase activity (e.g., Ag85). (B–D) Structures of some published trehalose derivatives bearing fluorophores (B), bioorthogonal functional groups (C), and fluorine modifications (D).
The synthesis of FITC-Tre (93) was relatively challenging, as are most syntheses of trehalose derivatives due to the disaccharide’s C2 symmetry and unique 1,1-α,α-glycosidic bond.298,299 As shown in Figure 36A, FITC-Tre was accessed using Ikegami’s chemical 1,1-α,α-glycosylation method300 as the key transformation. First, custom monosaccharide building blocks were prepared, including the ketose donor 109 and the protected glucosamine acceptor 110.264 Next, the acceptor and donor were coupled together in the presence of TMSOTf as the Lewis acid promoter, establishing the glycosidic bond with good stereoselectivity (6:1 α,α:α,β) due to the directing ability of the anomeric methyl group of 109.264 Finally, protecting group removal and reaction of the free 2’-amino group with FITC yielded the target compound 93.264 The synthesis of FITC-Tre, while elegant, still required >10 total steps and proceeded in <10% overall yield due to the building block preparations and difficulty of glycosidic bond formation. Alternatively, many trehalose analogues can be efficiently produced through regioselective modification of native trehalose or, in particular, chemoenzymatic synthesis, as discussed below.
Figure 36.
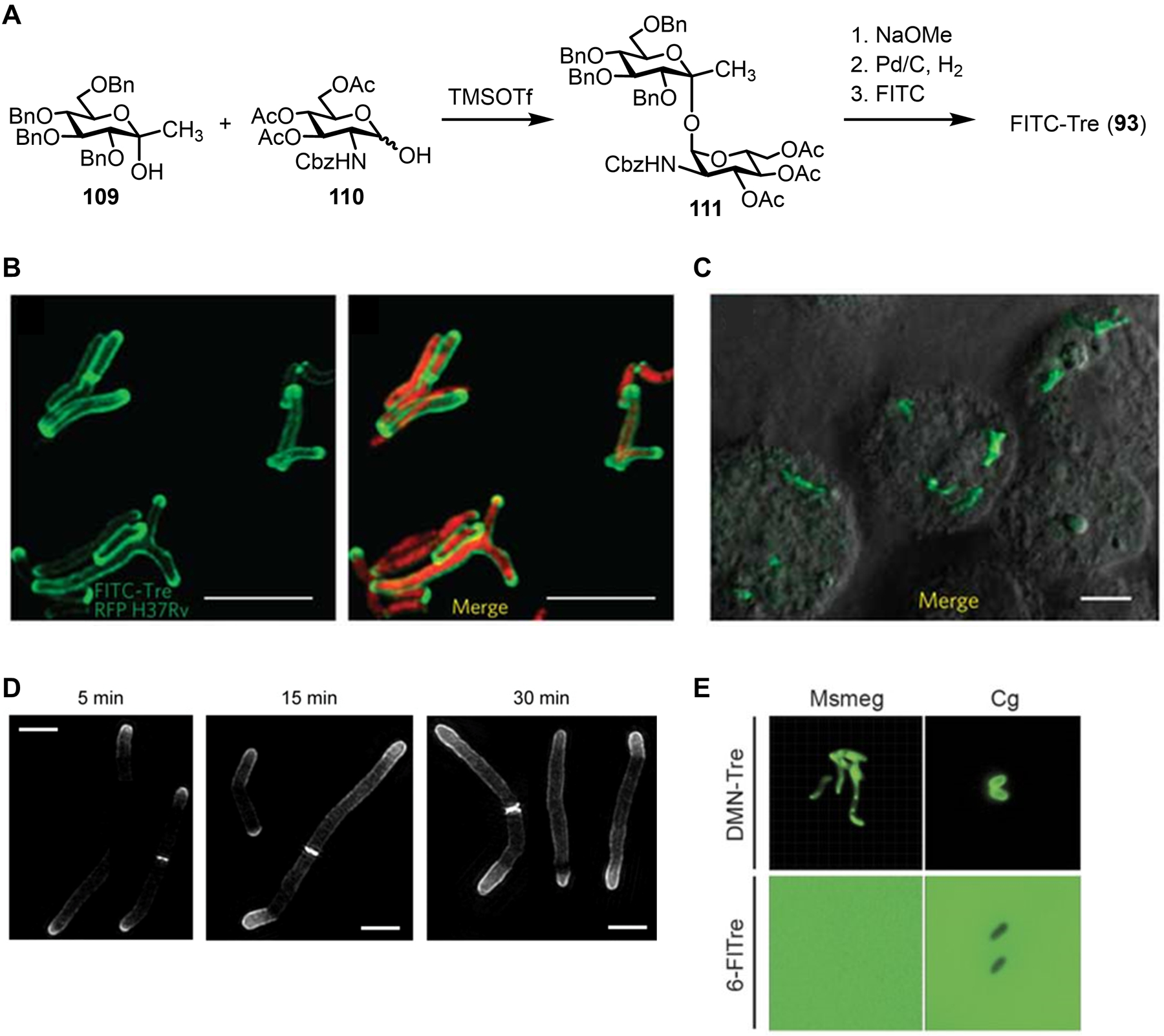
Labeling and imaging trehalose mycolates in mycobacteria using fluorescent trehalose derivatives. (A) Synthesis of FITC-Tre (93). Cbz, carbobenzyloxy; TMSOTf, trimethylsilyl triflate. (B) M. tuberculosis expressing red fluorescent protein (RFP) was incubated in the presence of 100 μM FITC-Tre (93), fixed, and imaged by fluorescence microscopy. Left, FITC channel; right, merge of FITC and RFP channels. (C) J774 macrophages were infected with M. tuberculosis, treated with 200 μM FITC-Tre (93), fixed, and imaged by fluorescence microscopy. Scale bars, 5 μm. Images in (B) and (C) were reproduced with permission from ref 264. Copyright 2011 Springer Nature. (D) M. smegmatis was incubated for varying durations with 100 μM 6-TMR-Tre, fixed, and imaged by structured illumination microscopy. Scale bars, 2 μm. Reproduced with permission from ref 301. Copyright 2018 Wiley-VCH. (E) M. smegmatis (Ms) and C. glutamicum (Cg) were incubated with 100 μM fluorogenic DMN-Tre (98) or 100 μM non-fluorogenic 6-FlTre (97) and directly imaged by fluorescence microscopy without washing. Scale bars, 5 μm. Reproduced with permission from ref 266. Copyright 2018 The American Association for the Advancement of Science.
With FITC-Tre synthesized, it was then tested for incorporation into cell surface trehalose mycolates in the pathogenic species Mycobacterium tuberculosis. Treatment of bacteria with FITC-Tre followed by fluorescence visualization demonstrated robust incorporation of the probe into the cell surface, most notably at the poles and septa, which is consistent with the polar growth mode of mycobacteria (Figure 36B).264 Glycolipid extracts from FITC-Tre-treated M. tuberculosis were analyzed by fluorescence TLC, which revealed the presence of a new fluorescent band that co-migrated with TDM.264 However, unmodified FITC-Tre was also detected at similar levels in the extracts, implying that significant non-specific association of the probe also occurred.264 FITC-Tre labeling was enhanced in comparison to a FITC-conjugated D-glucose control compound, confirming dependence on trehalose metabolism, although the glucose control probe also incorporated into M. tuberculosis, again suggesting some incorporation that was not driven by trehalose metabolism.264 To test dependence on Ag85 activity, FITC-Tre labeling was assessed in an Ag85C mutant, which exhibited a ~40% reduction in probe incorporation compared to wild-type bacteria.264 Finally, M. tuberculosis incorporated FITC-Tre at higher levels than several non-mycobacterial species, demonstrating species specificity.264 Overall, these data showed that FITC-Tre metabolically incorporated into trehalose mycolates in an Ag85-dependent manner, as designed. At the time of the 2011 report on FITC-Tre, direct evidence of glycolipid labeling by MS was not obtained, although subsequent studies on related trehalose probes did provide MS evidence of incorporation in the related species C. glutamicum, further confirming the specificity of this class of probes.265 The success of FITC-Tre in labeling trehalose glycolipids implied that the compound had sufficient penetrance across the mycomembrane to reach the Ag85 incorporation machinery, and moreover that this machinery was tolerant of large modifications such as fluorophores.
FITC-Tre and related fluorescent trehalose derivatives have been used for various applications related to the specific detection of mycobacteria and the visualization of glycolipid dynamics. In the same 2011 study, the Barry and Davis groups showed that FITC-Tre could label M. tuberculosis residing within macrophages (Figure 36C), a powerful demonstration that trehalose-based probes can be deployed within the context of host infection.264 A 2017 study by the Bertozzi group expanded on FITC-Tre by developing 4 regioisomeric FITC-modified trehalose derivatives, named FlTre analogues (94–97, Figure 35B).265 In contrast to the unnatural ketoside core of FITC-Tre (93), compounds 94–97 possessed a native trehalose core, on which fluorescein was appended to the 2-, 3-, 4-, or 6-positions. Compounds 94–97 were synthesized in 6–11 steps via selective modification of trehalose (not shown), as opposed to the chemical glycosylation route used to produce FITC-Tre.265 Interestingly, the four FlTre probes (94–97) incorporated into the model mycomembrane-containing organisms M. smegmatis and C. glutamicum much more efficiently than FITC-Tre (93) owing to their more-native structure, indicating that the unnatural anomeric methyl group of FITC-Tre dampened processing by Ag85.265 Thus, although Ag85 displays broad substrate plasticity, small structural changes can still have a significant impact on processing efficiency. FlTre incorporation into trehalose mycolates via Ag85 was characterized by TLC and MS analysis of glycolipid extracts, as well as usage of the Ag85 inhibitor ebselen,302 which diminished FlTre incorporation.265 The FlTre probes were used in conjunction with time-lapse microscopy and fluorescence recovery after photobleaching (FRAP) to investigate the fluidity of the mycomembrane in different contexts, which led to the finding that treatment with the AG biosynthesis inhibitor and front-line antitubercular drug ethambutol increased mycomembrane fluidity.265 Following their work on the FlTre series, the Bertozzi group also developed a related trehalose probe containing a more photostable tetramethylrhodamine (TAMRA) dye, 6-TMR-Tre (structure not shown), which enabled the first super-resolution microscopy visualization of trehalose glycolipids in mycobacteria (Figure 36D).301 Toward adapting trehalose probes as point-of-care diagnostic tools, in 2018 the Bertozzi group also reported DMN-Tre (98, Figure 35B), a fluorogenic trehalose probe modified at its 6-position with the solvatochromic group 4-N,N-dimethylamino-1,8-naphthalimide (DMN).266 DMN is an environmentally sensitive dye that fluoresces upon transition from an aqueous to organic solvent, which led the team to hypothesize that upon Ag85-mediated metabolic incorporation of DMN-Tre (98) into the highly hydrophobic mycomembrane, it would undergo fluorescence activation. Indeed, it was discovered that DMN-Tre successfully incorporated into and illuminated various mycobacterial species, featuring rapid, no-wash, and specific detection capabilities (Figure 36E).266 DMN-Tre was then translated to the successful detection of viable M. tuberculosis in sputum samples from patients with tuberculosis, showing its potential as a diagnostic reagent.266 Since its initial disclosure in 2018, DMN-Tre has also been applied to develop high-content screening of potential inhibitors against intracellular M. tuberculosis,303 tested for detection of M. tuberculosis in patient bioaerosols,304 and adapted into next-generation, brighter solvatochromic trehalose probes.305
Fluorescent trehalose probes such as 93–98 offer the convenience of one-step incorporation, but have low versatility because their chemical tag is pre-determined and they are limited to fluorescence detection applications. Furthermore, the large fluorophore may restrict the route or efficiency of metabolic incorporation. Shortly after the original FITC-Tre was reported, in 2012 the Bertozzi group disclosed a series of clickable azido trehalose (TreAz) analogues for versatile two-step labeling of trehalose glycolipids (99–102, Figure 35C).267 The TreAz series featured minimally perturbing azido group modifications at the 2-, 3-, 4-, and 6-positions of trehalose with stereochemistry matching the native disaccharide.267 The synthesis of the TreAz series of probes was originally carried out through the selective modification of trehalose. Briefly, this involved regioselective hydroxyl protection steps to expose the desired modification site, a double SN2 sequence to afford the azido group with native trehalose stereochemistry, and deprotection of the hydroxyl groups (not shown).267 It was found that all four TreAz derivatives efficiently incorporated into M. smegmatis, M. tuberculosis, and Mycobacterium bovis and could be detected/visualized with alkyne- or cyclooctyne-fluorophores via CuAAC or SPAAC, respectively.267 In agreement with FITC-Tre labeling, the TreAz derivatives labeled the mycobacterial surface primarily at the poles (Figure 37A).267 The metabolic fate of the TreAz analogues was exhaustively characterized, first by analyzing glycolipid extracts by TLC and MS.267 For all four TreAz probes, TLC showed new spots that were confirmed by MS to be azide-labeled TMM.267 Furthermore, 2-, 3-, and 4-TreAz treatment produced new spots that were consistent with azide-labeled TDM, although MS was unsuccessful at identifying these species likely due to the extremely poor ionization efficiency of TDM (note: 6-TreAz cannot label TDM due to its blocked 6-position).267 These studies represented the first direct evidence that trehalose probes incorporated into trehalose glycolipids. The mechanism of incorporation was also investigated using knockout mutants. Most interestingly, the metabolic incorporation of 2-, 4-, and 6-TreAz was entirely dependent on the trehalose transporter LpqY-SugABC (Figure 37A).267 This result implied an intracellular labeling mechanism whereby these probes were sequentially internalized by the transporter, mycoloylated by Pks13/CmrA to generate azide-labeled TMM, exported by MmpL3, and then potentially further lipidated by Ag85 (see Figure 34). In contrast, 3-TreAz labeling was not dependent on the transporter, suggesting that this isomer incorporated via the extracellular Ag85 mechanism, similar to FITC-Tre.267 These results were corroborated by chromatographic analysis of cytosolic extracts, which showed that 2-, 4-, and 6-TreAz accumulated in the cytoplasm in an LpqY-SugABC-dependent manner, whereas 3-TreAz was not detected.267 These findings were significant because they (i) revealed valuable information about the substrate tolerance of the trehalose transporter that could be exploited for trehalose-based probe or inhibitor development; and (ii) demonstrated that trehalose probes report on different metabolic pathways in mycobacteria—either extracellular incorporation via Ag85 or intracellular incorporation via the trehalose recycling route—depending on the size and position of the chemical tag. Importantly, the TreAz derivatives operating via intracellular incorporation provided a means for monitoring newly synthesized mycomembrane glycolipids for the first time. The differential labeling behaviors of the four TreAz derivatives also emphasized the benefit of synthesizing and evaluating panels of isomeric chemical reporters, which may reveal complementary features.
Figure 37.
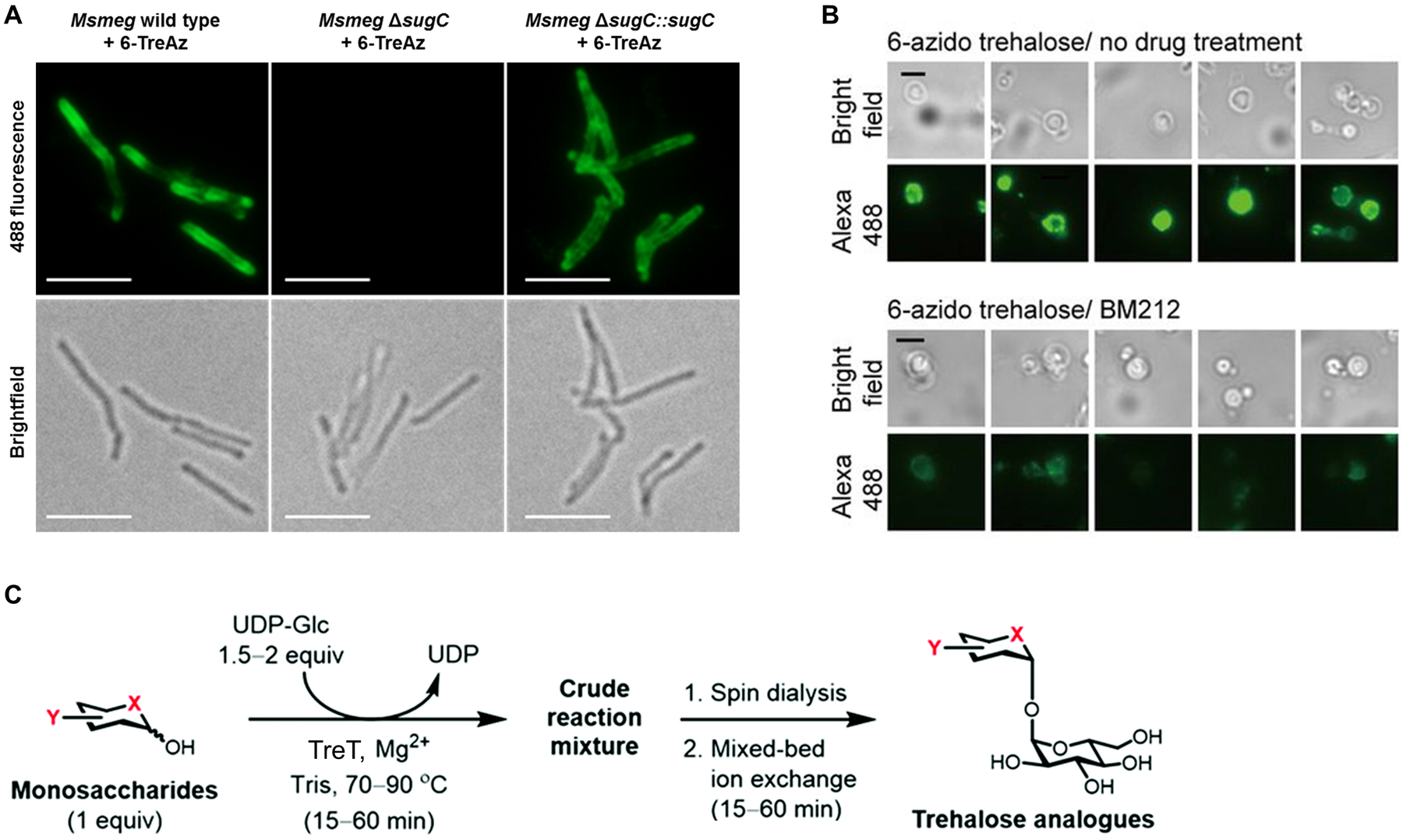
Labeling trehalose mycolates in mycobacteria using azido trehalose (TreAz) derivatives. (A) M. smegmatis (Msmeg) wild type, ΔsugC mutant lacking the trehalose transporter, or ΔsugC::sugC complement with transporter restored were incubated in the presence of 25 μM 6-TreAz (102), fixed, reacted with alkyne-488 via CuAAC, and imaged by fluorescence microscopy. Top, 488 channel; bottom, brightfield channel. Scale bars, 5 μm. Reproduced with permission from ref 306. Copyright 2014 Wiley-VCH. (B) M. smegmatis spheroplasts were incubated with 100 μM 6-TreAz (102) in the absence (top) or presence (bottom) of MmpL3 inhibitor BM212, reacted with cyclooctyne-biotin via SPAAC, stained with 488-streptavidin, and imaged by fluorescence microscopy. Scale bars, 3 μm. Reproduced from ref 307. (C) TreT-catalyzed one-step chemoenzymatic synthesis and all-aqueous purification of trehalose derivatives. Various unnatural acceptor substrates can be converted into products, with Y representing azido-, deoxy-, fluoro-, or stereochemical modifications, and X representing oxygen or sulfur atoms. Figure adapted from ref 297 with permission from the Royal Society of Chemistry.
The clickable TreAz reporters have been used for various studies since their initial disclosure. The unique ability of these probes to label newly synthesized trehalose mycolates via the intracellular trehalose recycling route of incorporation has proven useful. For example, in 2017 the Chng group exploited 6-TreAz (102) to develop a spheroplast-based assay for TMM translocation across the plasma membrane, which was then applied to (i) confirm that MmpL3 was responsible for TMM translocation; and (ii) characterize the mechanism of action of antimycobacterial compound BM212 as an inhibitor of MmpL3 (Figure 34B).307 Given that MmpL3 is among the most promising emerging targets for the development of novel antimycobacterials,28,308 such a tool could prove useful, particularly if it can be adapted for high-throughput screening. In another study from 2021, the Siegrist group used 6-TreAz to investigate how mycobacterial trehalose recycling changes during starvation, and found that bacteria grown in carbon-limited medium increase recycling of trehalose to support energy-efficient remodeling of the mycomembrane.309 This work provided new insight into the physiological role of the trehalose transporter LpqY-SugABC, which is essential for virulence of M. tuberculosis.294 In 2020, the Bertozzi group used 6-TreAz to label TMM and measure its mobility and spreading into host cells during M. marinum infection.310 Taken together, it has been demonstrated that TreAz derivatives provide a useful means of probing trehalose metabolism and its role in cell envelope construction in live mycobacteria. In addition, since TreAz derivatives do not label mammalian cells,278 their extension to translational research efforts, such as inhibitor screening and characterization, or delivery of diagnostic or therapeutic payload to mycobacteria within a host context, may also be possible. The flexible, modular nature of click chemistry-based labeling approaches engenders TreAz analogues with high versatility.
Despite the demonstrated utility of fluorescent and clickable trehalose probes, the initially reported syntheses were challenging and represented a significant hurdle to their continued development and adoption by the broader research community. Indeed, the FITC-Tre, FlTre, and TreAz derivatives were originally produced via lengthy chemical synthesis strategies. For example, all four TreAz derivatives (99–102) were synthesized via multi-step, regioselective modification of native trehalose; subsequently, the deprotected TreAz compounds could serve as intermediates to synthesize the FlTre series of probes (94–97) via azido group reduction and coupling to FITC.265,267 However, due to trehalose’s C2 symmetry and eight similarly reactive hydroxyl groups, these syntheses had high step counts (4–11 steps) and were inefficient (generally <5% overall yield).297,311 This issue was addressed in 2014, when our lab introduced a chemoenzymatic method for the rapid and efficient synthesis of trehalose analogues.306,312 We used the heat-stable trehalose-synthesizing glycosyltransferase TreT from Thermoproteus tenax,313 which converts glucose and UDP-glucose into trehalose in one step, to generate a variety of azido-, deoxy-, fluoro-, thio-, and stereochemically-modified trehalose analogues by varying the substrates (Figure 37C).306,312 With respect to TreAz and FlTre probes, this method delivered 2-, 3-, and 6-TreAz and 2-, 3-, and 6-FlTre in 1–3 total steps and in high overall yields (typically 70–80% isolated yield).306,312,314 In addition, a “one-pot” approach was developed in which 6-TreAz could be rapidly synthesized via TreT catalysis and directly deployed in metabolic labeling experiments over the course of a few hours.306,310 Thus, for many trehalose-based probes, chemoenzymatic synthesis is superior to chemical synthesis in terms of speed, efficiency, and operational simplicity, making it an attractive method for non-chemists to access relatively complicated probe compounds.
The progress made on fluorescent and clickable trehalose derivatives, as well as the contemporaneous development of improved synthetic methods to access them, served as a foundation upon which novel trehalose-based probes have been created for mycobacteria research. In 2020, the Fullam group expanded on the clickable trehalose probes by synthesizing and evaluating two epimeric TreAz derivatives for labeling trehalose mycolates.268 Citing concern for possible off-target metabolism of the original TreAz analogues through trehalase or TreS activity (see Figure 34), the team designed two stereochemically modified versions of 6-TreAz, including 6-MannoTreAz (103) and 6-LactoTreAz (104) (Figure 35C).268 These compounds were hypothesized to exhibit enhanced resistance to activity by trehalase and TreS enzymes. The compounds were chemoenzymatically synthesized in one step via TreT catalysis,306 then shown to incorporate into the surface of M. smegmatis in an LpqY-SugABC-dependent manner, similar to the originally reported 6-TreAz.268 Although experiments with purified enzymes indicated that 30 mM 6-TreAz can be hydrolyzed to a very minor extent by recombinant trehalase,268 it is not clear whether this happens in vivo with micromolar probe concentrations, and earlier experiments showed that 6-TreAz incorporation into the mycobacterial surface was completely trehalase- and TreS-independent.267 Regardless, 6-MannoTreAz (103) and 6-LactoTreAz (104) guard against this possibility and provided further information about the substrate tolerance of the trehalose transporter LpqY-SugABC. There has also been effort to extend trehalose-based probes beyond optical imaging and make them compatible with in vivo positron emission tomography (PET) imaging, which could provide much needed new capabilities for studying and diagnosing mycobacterial infections in preclinical and clinical settings.315,316 With 18F being the most commonly used radionuclide for PET imaging applications,317 deoxy-[18F]fluoro-D-trehalose (18F-FDTre) analogues have been pursued. Toward this goal, in 2016 we reported the synthesis and evaluation of four non-radioactive 19F-FDTre analogues, including 19F-2-, 19F-3-, 19F-4-, and 19F-6-FDTre (105–108, Figure 35D).269 TreT catalysis was used to rapidly produce the 2-, 3-, and 6-position-modified compounds from their corresponding deoxyfluoro-D-glucose derivatives in one step, an attractive feature given the short half-life of 18F (t1/2 = 110 min); 19F-4-FDTre was inaccessible via TreT catalysis, so it was chemically synthesized from native trehalose in 4 steps.269 Next, it was shown that these probes were specifically metabolized by mycobacteria. 19F-2-, 19F-3-, and 19F-6-FDTre all accumulated in the cytoplasm of M. smegmatis via active uptake by the trehalose transporter LpqY-SugABC.269 Additional research is needed to determine whether the fluorinated compounds incorporate into TMM and/or TDM. Following these preliminary studies, in 2019 we demonstrated that radioactive 18F-2-FDTre could be synthesized and purified by TreT catalysis in one step, in high yield, and in only 30 min on an automated radiosynthesis module starting from the commercially available PET probe [18F]fluoro-2-deoxy-D-glucose (18F-FDG).270 We further showed that 18F-2-FDTre was metabolized by M. smegmatis but not by a panel of mammalian cells, further supporting the specificity of the probe.270 In addition to synthesis via TreT catalysis, 2-FDTre has been synthesized via a three-step chemoenzymatic route based on the OtsAB trehalose biosynthesis pathway for trehalose biosynthesis.264 Although further research on 18F-FDTre analogues is needed to determine whether they will be successful for in vivo PET imaging of mycobacterial infections, the results to date are promising and highlight how insights taken from fluorescent and clickable reporter probes can be extended to develop next-generation probes with clinical potential.
The ability of trehalose analogues to co-opt trehalose metabolic pathways in mycobacteria has also generated interest in their potential inhibitory activity. On one hand, inhibitory activity could be considered a liability from the perspective of probe development and application, but on the other hand would be attractive from the perspective of antibiotic development. While fluorescent trehalose derivatives are not known to have significant inhibitory activity against mycobacteria, Belisle and co-workers reported in 1997 that 6-TreAz (102) inhibited the growth of Mycobacterium aurum with a minimum inhibitory concentration (MIC) of ~500 μM.291 This concentration is ~20-fold higher than is typically used for metabolic incorporation experiments (25 μM), suggesting that significant perturbations to the system can be avoided by deploying 6-TreAz as a probe at appropriate concentrations. However, the growth inhibition activity of 6-TreAz has spurred a number of studies focused on the synthesis and evaluation of trehalose-based inhibitors.311 For example, in 2011, Barry and Davis chemically synthesized a panel of trehalose analogues and tested them for inhibition of M. tuberculosis growth, finding that several 6-position modified derivatives exhibited results similar to those observed for 6-TreAz by Belisle.264 Our lab became interested in testing trehalose analogues as biofilm inhibitors given the recently discovered connection between trehalose metabolism and the formation of mycolic acid-rich mycobacterial biofilms.295 In 2017, we reported that several chemoenzymatically synthesized trehalose analogues were selective inhibitors of M. smegmatis, i.e. these compounds prevented biofilm formation at significantly lower (~10-fold) concentrations than they inhibited planktonic growth.318 Furthermore, the inhibitory activity was dependent on the trehalose transporter LpqY-SugABC, supporting an intracellular mechanism of action reminiscent of the TreAz and FDTre metabolic incorporation mechanism.318 A follow-up study with the Eoh group in 2019 used metabolomics to determine that 6-TreAz inhibited TreS during starvation-induced M. tuberculosis biofilm formation, thus disrupting an important trehalose-mediated stress response mechanism.319 Most interestingly, killing of M. tuberculosis by the clinically used antimycobacterial compound bedaquiline was enhanced >100-fold in the presence of 6-TreAz, suggesting that TreS inhibition by trehalose analogues could be a viable strategy for adjunctive therapy of tuberculosis.319 In another 2019 study, we showed that trehalase-resistant trehalose analogues inhibited trehalose metabolism in the intestinal pathogen Clostridium difficile, demonstrating that trehalose analogues may be valuable antibiotic lead compounds for other species as well.320 Continued research on trehalose-based inhibitors should focus on improving their potency (e.g., trehalosamines314) and better understanding their mechanism(s) of action in bacteria of interest.297
Overall, the extensive work on trehalose derivatives has solidified their value as metabolic probes and inhibitors to study and target trehalose mycolate metabolism in mycobacteria. As discussed above, fluorescent and clickable trehalose probes have been applied to visualize trehalose glycolipid dynamics in live mycobacteria, investigate glycolipid-mediated host–pathogen interactions, characterize inhibitors of mycomembrane biosynthesis, and specifically detect the presence of mycobacterial pathogens in patient samples. This work has been bolstered by the well-characterized behavior of trehalose probes in mycobacteria, which was elucidated through rigorous evaluation of the probes’ labeling targets and routes of incorporation in multiple mycobacterial species. Another takeaway from work in this area, and a theme that has emerged multiple times in this review, is that chemoenzymatic synthesis is a powerful approach to producing carbohydrate-based probes that are otherwise difficult to access using chemical synthesis. In particular, TreT catalysis improves the efficiency of synthesizing known trehalose analogues, while also speeding up the development of novel analogues. The insights obtained during studies on first-generation trehalose probes (e.g., FITC-Tre and TreAz analogues) led to the development of next-generation tools, such as fluorogenic probes, PET-capable probes, and metabolic inhibitors. Excitingly, trehalose has also been conjugated to therapeutic payload to facilitate targeted killing of mycobacteria, published examples of which include trehalose-functionalized nanoparticles loaded with the antimycobacterial compound isoniazid321 and trehalose-conjugated photosensitizers.322,323 Although much work remains to be done in this regard, it is clear that early efforts to develop probes such as FITC-Tre and TreAz have ultimately opened up a wide range of basic and translational research directions. In the next two sections, we will see how foundational work on trehalose derivatives has led to the development of novel reporter probes for investigating different pathways involved in the construction and remodeling of mycolate-containing glycolipids.
6.1.2. Trehalose monomycolate derivatives.
As depicted above in Figure 34, TMM plays key roles in mycomembrane biosynthesis, serving as both a mycoloyl donor and acceptor substrate in periplasmic mycoloyltransferase-catalyzed exchange reactions. Given the broad substrate plasticity of mycoloyltransferase enzymes revealed through the extensive work on trehalose derivatives, our group reasoned that unnatural TMM derivatives could be designed to mimic the donor or acceptor functions of native TMM to expand mycomembrane labeling capabilities (Figure 38). First, as TMM donor surrogates we designed O-linked TMM derivatives, which contain a natural ester linkage between trehalose and a mycolate-mimicking chain modified with an unnatural chemical handle. Upon administration to mycobacteria, O-linked TMM derivatives should have their unnatural mycoloyl group transferred onto acceptor molecules via mycoloyltransferase enzymes, thereby producing labeled versions of TDM, AGM, and potentially other mycoloylated species, such as O-mycoloylated proteins from C. glutamicum (Figure 38A). The ability to label inner-leaflet AGM and other mycomembrane components was unprecedented, since previously reported trehalose derivatives served only as mycoloyl acceptors that incorporated into TMM and TDM (Figure 35A). As a complementary type of TMM-based probe, N-linked TMM derivatives have an unnatural amide linkage rather than a native ester linkage, which makes them unable to serve as mycoloyl donors but maintains their ability to serve as mycoloyl acceptors in mycoloytransferase-catalyzed reactions (Figure 38B). Thus, in principle, N-linked TMM derivatives would exclusively produce a labeled version of TDM. In this way, O- and N-linked TMM derivatives would allow selective labeling of different mycolate glycolipids of the mycomembrane.
Figure 38.
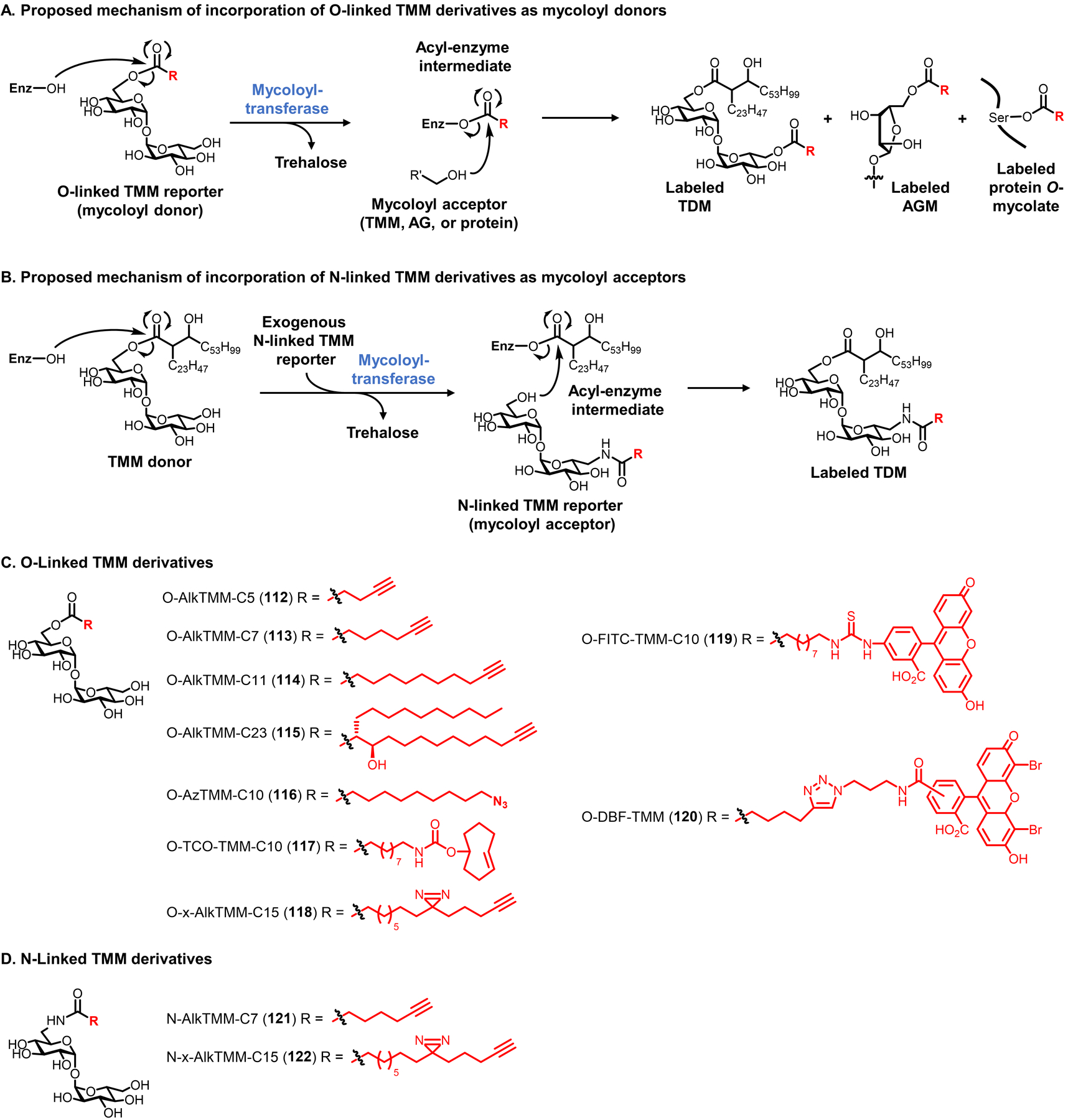
O- and N-linked TMM derivatives for selective labeling of mycolate-containing components of the mycomembrane. (A) O-linked TMM derivatives (modified with an unnatural R group) mimic the mycoloyl donor function of TMM and are proposed to undergo periplasmic mycoloyltransferase-catalyzed transfer of their unnatural acyl chain onto mycoloyl acceptors, producing labeled TDM, AGM, proteins, and potentially other mycolate-containing products. (B) N-linked TMM derivatives (modified with an unnatural R group) mimic the mycoloyl acceptor function of TMM and are proposed to undergo periplasmic mycoloyltransferase-catalyzed mycoloylation of their free 6-position hydroxyl group, producing only a labeled version of TDM. Enz, enzyme with mycoloyltransferase activity (e.g., Ag85). (C and D) Structures of published O-linked (C) and N-linked (D) TMM derivatives bearing bioorthogonal functional groups, fluorophores, and photo-cross-linking tags.
In 2016, we reported the first O- and N-linked TMM derivatives and demonstrated their ability to selectively label mycomembrane components by copying the donor and acceptor functions of TMM, respectively, as depicted in Figure 38.271 The initial O-linked TMM derivative, named O-AlkTMM-C7 (113), had a linear 7-carbon acyl chain terminated by a clickable alkyne group.271 This dramatic simplification of the mycolate chain in compound 113—i.e., severely truncating the lipid tail and removing the α-branch and β-hydroxy group—was supported by earlier studies, which showed that purified mycoloyltransferases could process TMM substrates with simple palmitate or hexanoate chains.264,290,324 The chemical synthesis of O-AlkTMM-C7 (113) and related O-linked TMM derivatives followed Kulkarni’s approach,325 which involved DCC-mediated monoacylation of the partially TMS-protected trehalose derivative 124 with heptynoic acid, followed by deprotection (Figure 39A).271 A method for one-step chemical esterification of native trehalose to form TMM derivatives has also been reported,326 although product purification via this approach can be difficult. The corresponding N-linked TMM derivative, N-AlkTMM-C7 (121), was synthesized via reduction of 6-TreAz (102) to the corresponding amine, followed by HBTU-mediated N-acylation with heptynoic acid (not shown).271 The compounds were tested for metabolic incorporation by incubating bacteria in probe, then performing CuAAC reaction with azido-488 and analyzing cells by flow cytometry or fluorescence microscopy. Mycomembrane-containing species M. smegmatis and C. glutamicum were robustly surface-labeled with low concentrations (10–100 μM) of O- and N-AlkTMM-C7 (113 and 121), whereas E. coli and B. subtilis were not labeled (Figure 39B). Labeling of M. smegmatis by O- and N-AlkTMM-C7 (113 and 121) was reduced by treatment with the mycoloyltransferase inhibitor ebselen,271 supporting the proposed periplasmic mechanism of incorporation, and later studies further confirmed mycoloyltransferase-dependent incorporation of TMM derivatives using knockout mutants.272,274 Experiments were also done to evaluate the effect of the linkage type—i.e. ester or amide—on the labeling route and targets. Probe-treated bacteria were subjected to CuAAC with azido-488, then fractionated into TDM- and AGM-containing fractions. Fluorescence signal from the TMM acceptor mimic N-AlkTMM-C7 was exclusively present in the TDM-containing fraction, whereas signal from the TMM donor mimic O-AlkTMM-C7 was present in both fractions, although predominantly the AGM fraction due to its high abundance in the mycomembrane.271 These results were consistent with the proposed mechanisms of incorporation of the O- and N-linked probes (Figures 38A and 38B) and demonstrated that tuning the linker of TMM derivatives enabled control of the labeling route and target molecule(s), which for the first time extended beyond trehalose mycolates to include inner-leaflet AGM and other mycomembrane components.
Figure 39.
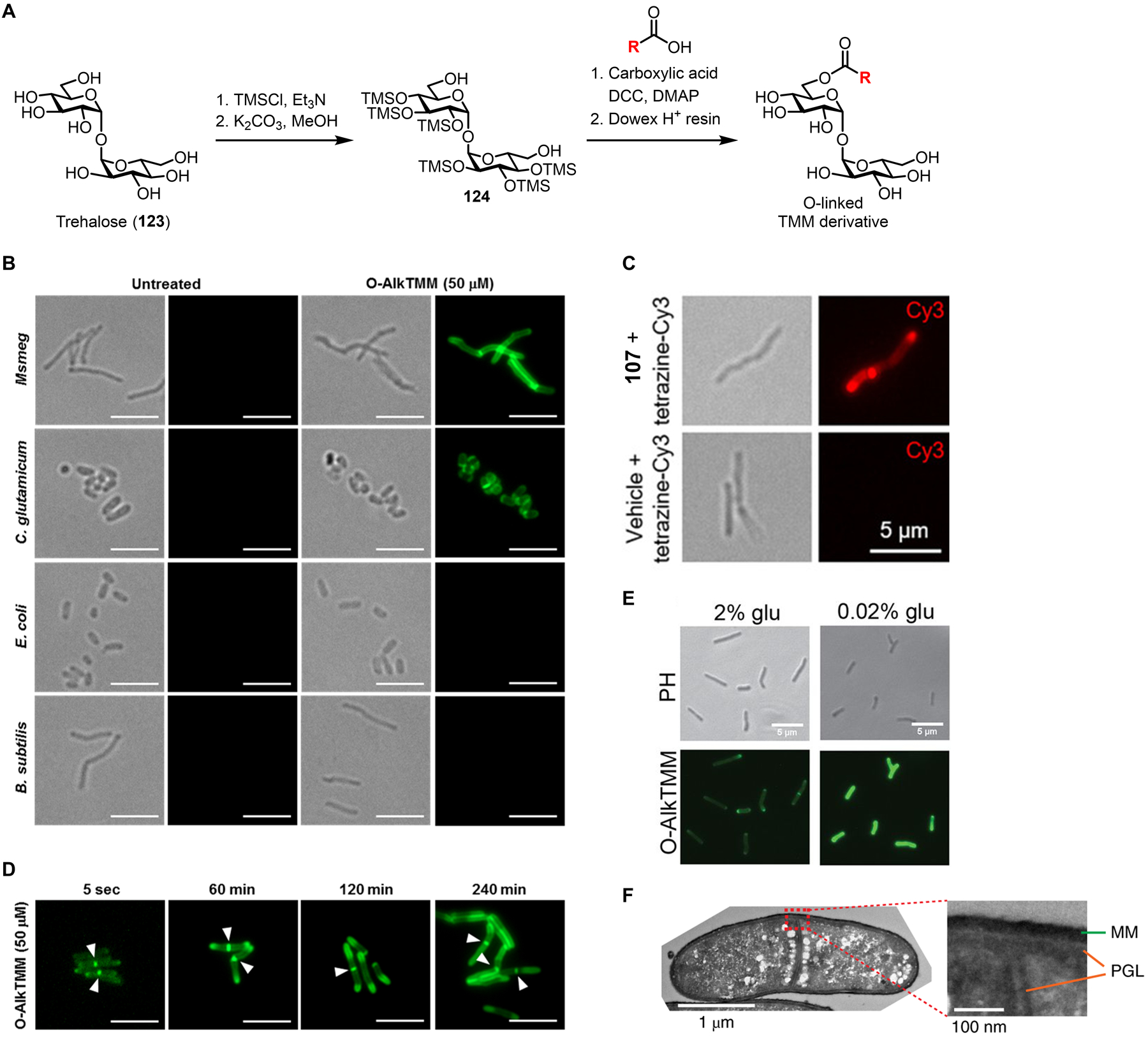
Labeling and imaging mycolates in mycobacteria using TMM derivatives. (A) General scheme for the synthesis of O-linked TMM derivatives. DCC, N,N’-dicyclohexylcarbodiimide; DMAP, 4-dimethylaminopyridine; TMS, trimethylsilyl. (B) Different species of bacteria were incubated in the presence of 50 μM O-AlkTMM-C7 (113) or left untreated, fixed, reacted with azido-488 via CuAAC, and imaged by fluorescence microscopy. For each condition, left is the transmitted light channel and right is the 488 channel. Scale bars, 5 μm. (C) M. smegmatis was incubated in the presence of 20 μM O-TCO-TMM (117) or left untreated, fixed, reacted with tetrazine-Cy3 for 1 min and imaged by fluorescence microscopy. Left, transmitted light channel; right, Cy3 channel. Scale bars, 5 μm. Reproduced with permission from ref 274. Copyright 2019 Wiley-VCH. (D) M. smegmatis was incubated in the presence of 50 μM O-AlkTMM-C7 (113) for varying durations, fixed, reacted with azido-488 via CuAAC, and imaged by fluorescence microscopy. Arrows indicate sites of septal mycomembrane synthesis in dividing cells. Scale bars, 5 μm. (B) and (D) reproduced with permission from ref 271. Copyright 2016 Wiley-VCH. (E) M. smegmatis was incubated in the presence of 50 μM O-AlkTMM-C7 (113) in 2% (carbon-rich) or 0.02% (carbon-depleted) glucose-supplemented medium, fixed, reacted with azido-488 via CuAAC, and imaged by fluorescence microscopy. Scale bars, 5 μm. Reproduced with permission from ref 309. Copyright 2021 American Society for Microbiology. (F) C. glutamicum was incubated in the presence of 50 μM O-DBF-TMM (120), fixed, photo-oxidized in the presence of diaminobenzidine, stained with osmium tetroxide, and imaged by transmission electron microscopy. The stained mycomembrane (MM) and peptidoglycan layer (PGL) are indicated. Reproduced with permission from ref 275. Copyright 2019 Springer Nature.
Since the original O- and N-AlkTMM-C7 compounds were published, additional TMM derivatives have been developed in order to provide complementary features for increased experimental flexibility. In 2019, our lab synthesized and evaluated a series of O-linked TMM derivatives bearing linear ester-linked acyl chains with different lengths and chemical handles.274 Out of three O-AlkTMM analogues bearing 5-, 7-, and 11-carbon chain lengths (112–114, Figure 38C), it was found that the intermediate chain length reporter, O-AlkTMM-C7 (113), incorporated into M. smegmatis most efficiently, perhaps suggesting that it possessed an optimal balance of permeability and enzymatic processing efficiency.274 Furthermore, azide- and trans-cyclooctene-modified TMM derivatives, O-AzTMM-C10 (116) and O-TCO-TMM-C10 (117), were developed to allow live cell-compatible, two-step labeling approaches when used in combination with SPAAC and tetrazine ligation chemistries, respectively.274 O-TCO-TMM-C10 (117) enabled the first cell surface modification of live mycobacteria via the exceptionally rapid tetrazine ligation; M. smegmatis labeled with O-TCO-TMM-C10 could be detected in as little as 1 min of reaction time with a tetrazine-Cy3 fluorophore (Figure 39C).274 In addition, a fluorophore-modified TMM derivative, O-FITC-TMM-C10 (119) was developed to allow one-step labeling of live mycobacteria.274 The metabolic incorporation of these new O-linked TMM derivatives was characterized generally as described above for the original TMM reporters and was consistent with prior data, although O-FITC-TMM-C10 (119) displayed some divergent behavior with respect to labeling O-mycoloylated proteins and thus additional investigation of this probe is warranted.274 Overall, this work further elucidated the substrate tolerance of mycoloyltransferases and exemplified how this information can be exploited to develop different cell surface engineering tools to investigate and target mycobacteria. Another notable conclusion from this work was that TMM (and likely also trehalose) probes report not only on mycoloyltransferase/hydrolase activity but also the extent to which the probe molecule can cross the mycomembrane to gain access to mycoloyltransferase enzymes in the periplasm.274 The latter point suggests that TMM reporters may be useful probes for assessing mycomembrane permeability, which could be a useful feature for identifying and functionally characterizing genes or inhibitors that modulate mycobacterial drug tolerance. In addition to the simplified linear-chain TMM derivatives, in 2019 the Bourdreux and Guianvarc’h groups reported an O-linked TMM derivative bearing an alkyne-modified mycolate chain with the native α-branch and β-hydroxy group included, here named O-AlkTMM-C23 (115, Figure 38C).273 Once the challenging stereoselective synthesis of the native mycolic acid building block was completed, it was coupled to trehalose using the same monoesterification method shown in Figure 39A.273 Compound 115 was shown to efficiently incorporate into the cell surface of C. glutamicum.273 Such compounds may be instrumental in teasing apart the structural features of mycolate glycolipids that confer efficient and specific processing by mycoloyltransferase/hydrolase enzymes.
One of the main applications of TMM derivatives has been to visualize the dynamics of mycomembrane assembly and remodeling in intact mycobacterial cells. Because O- and N-linked TMM probes allowed selective detection of inner-leaflet AGM and outer-leaflet TDM, respectively, it was possible to deploy these tools in conjunction with click chemistry to monitor changes in the two major glycolipid constituents of the mycomembrane under varied growth conditions. In our initial report on TMM probes in 2016, we used O-AlkTMM-C7 (113) to directly image the inner-leaflet AGM layer for the first time.271 Time-course fluorescence microscopy demonstrated that, in M. smegmatis cultured in nutrient-rich growth medium, AGM is produced at the poles and septa of dividing cells (Figure 39D),271 similar to the patterns previously observed for outer-leaflet trehalose mycolates (Figures 36 and 37). In a subsequent report by the Siegrist group in 2021, the combined usage of O- and N-AlkTMM-C7 (113 and 121) demonstrated that starved M. smegmatis cells remodel their mycomembrane by breaking down outer-leaflet TDM while simultaneously enhancing the synthesis of inner-leaflet AGM around the entirety of the cell, rather than exclusively from the poles (Figure 39E).309 Interestingly, the starved bacteria with reorganized mycomembranes exhibited decreased cell envelope permeability.309 Thus, an unexpected starvation-induced membrane remodeling mechanism that protects the bacterium was discovered by capitalizing on the complementary labeling abilities of O- and N-linked TMM reporters. A 2019 collaborative study by the Theriot and Bertozzi groups elegantly combined D-amino acid-, TMM- and trehalose-based fluorescent reporters to precisely assess the assembly sequence of peptidoglycan, AGM, and trehalose mycolate layers of the cell envelopes of M. smegmatis and C. glutamicum.275 The groups found that the envelope assembly mechanisms differed at the poles and septa. At the poles, the three layers are formed synchronously.275 By contrast, at the septa the layers are formed sequentially in the order of (i) peptidoglycan, (ii) AGM, and (iii) trehalose mycolates, in a manner that appears to preserve cell envelope integrity while the daughter cells undergo separation.275 The same study reported a novel TMM derivative modified with dibromofluoroescein, O-DBF-TMM (120, Figure 38C), which generates singlet oxygen upon irradiation.275 After incorporation of O-DBF-TMM (120) into C. glutamicum, irradiation of cells led to polymerization of added diaminobenzidine in the mycomembrane, which formed a polymer specifically in this layer that could be stained by osmium tetroxide and visualized by high-resolution transmission electron microscopy (Figure 39F).275 This approach, which represents a clever merger of TMM-based probes and click chemistry-enabled electron microscopy (click-EM),327 could be applied to enable specific EM imaging of other types of bacterial cell envelope glycans in the future.
The TMM scaffold has also been adapted into a fluorogenic probe that employed fluorescence resonance energy transfer (FRET) to allow real-time fluorescence read-out of enzymatic activity in mycobacterial cells. In 2018, the Kiessling group introduced a TMM derivative coined “quencher-trehalose-fluorophore,” or QTF (125), which consisted of a TMM structure bearing a dabcyl quencher on the trehalose core and a BODIPY fluorophore at the end of a linear mycolate-mimicking chain (Figure 40A).276 QTF was designed as a TMM donor substrate that, in the presence of a mycoloyltransferase enzyme (e.g., Ag85), would undergo fluorescence turn-on when the fluorophore-containing acyl chain was separated from the quencher-containing trehalose moiety. In essence, QTF would be anticipated to behave in a manner similar to the O-linked TMM derivatives, as depicted in Figure 38A. Indeed, when QTF was incubated in the presence of purified Ag85 enzymes or mycomembrane-containing bacteria, such as M. smegmatis and C. glutamicum, robust fluorescence increases were observed (Figure 40B).276 Low micromolar quantities of probe were sufficient to detect mycobacteria, owing to the highly sensitive nature of the FRET design. In support of probe specificity, QTF activation decreased in the presence of both the Ag85 inhibitor ebselen and the non-specific lipase inhibitor tetrahydrolipstatin.276 Thus, QTF reports on mycoloyltransferase activity as designed, although the authors concluded that QTF undergoes hydrolysis, as shown in Figure 40, rather than acyl chain transfer onto mycoloyl acceptors, as shown in Figure 38A. For example, MS analysis of a reaction mixture consisting of QTF and Ag85 enzymes partially purified from M. smegmatis detected the products of QTF hydrolysis, 126 and 127 (Figure 40A).276 While the available data do not rule out that the fluorophore-containing acyl chain is transferred onto mycoloyl acceptors in a cellular environment, it is important to know that QTF and potentially other O-linked TMM derivatives can undergo hydrolysis. This issue warrants further investigation for both QTF and related TMM derivatives. Regardless of the fate of the acyl chain, QTF serves as a powerful fluorogenic probe of mycoloyltransferase activity in cells, which complements other O-linked TMM derivatives that chemically label mycolate-containing products but only indirectly report on enzymatic activity. To highlight this ability, the team carried out a tandem probe experiment in M. smegmatis enlisting a D-amino acid probe for labeling PG (HADA, 17, Figure 7C) and QTF for visualizing mycoloyltransferase activity in a microfluidic device, which revealed that PG and mycomembrane synthesis were coincident and occurred at the poles and septa of cells.276 Looking toward future applications that exploit the fluorogenic nature of QTF, the probe could be valuable in inhibitor screening, since inhibition of mycoloyltransferase activity is a promising approach for antimycobacterial development.28 Furthermore, as with the fluorogenic trehalose derivative DMN-Tre (98, Figure 35B),266 QTF may also be of interest for specifically detecting pathogenic mycobacteria in patient samples, although additional investigation into the mycobacterial specificity of trehalose ester-based probes will be necessary. More broadly, the FRET-based design of QTF is one that can be extended to the development of other probes in which bond cleavage is a key feature of metabolic probe activation, as seen below in the section on TDM derivatives (section 6.1.3).
Figure 40.
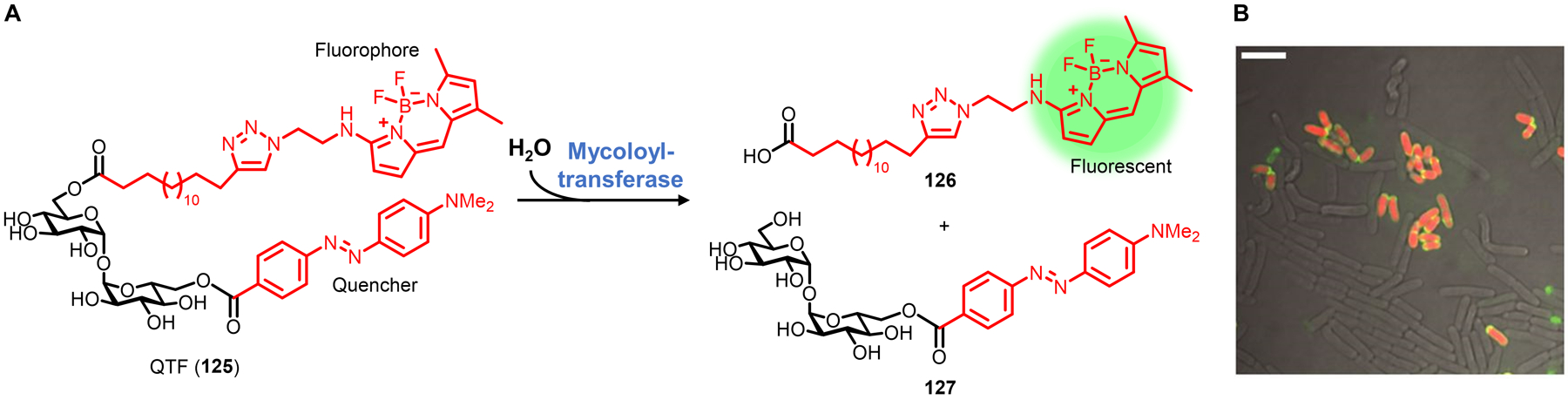
Imaging of mycoloyltransferase hydrolytic activity in mycobacteria using FRET-based fluorogenic QTF. (A) Upon hydrolysis by mycoloyltransferases (e.g., Ag85), the ester bond of QTF (125) is cleaved, releasing products 126 and 127, the former of which is fluorescent. (B) A mixed culture of mCherry-expressing C. glutamicum (red), B. subtilis, and E. coli was incubated in the presence of 1 μM QTF, fixed, and imaged by confocal fluorescence microscopy. An overlay of all channels is shown. Scale bar, 5 μm. Reproduced from ref 276.
TMM derivatives have also been applied to facilitate the characterization of the mycomembrane proteome, which is notoriously poorly characterized, despite the high interest in these surface-accessible proteins as possible targets for antimycobacterial drug development.328,329 Since TMM derivatives have the ability to metabolically insert unnatural chemical functionality into the mycomembrane, they can potentially be adapted to capture and identify proteins from this layer of the mycobacterial cell envelope. Our group has pursued two chemical probe-based approaches to study mycomembrane proteins, the first of which focused on the post-translationally O-mycoloylated proteins present in C. glutamicum. As mentioned above, C. glutamicum possesses a mycoloyltransferase (CgMytC) that transfers mycoloyl groups from TMM onto protein acceptors at specific serine residues, generating O-mycoloylated proteins (see Figure 38A).292,293,330,331 For example, the heterodimeric C. glutamicum mycomembrane porin PorAH was the first protein confirmed to be post-translationally modified by mycolates.292 We reasoned that when TMM donor-mimicking chemical reporters were administered to C. glutamicum, their unnatural clickable acyl chain would be transferred onto O-mycoloylated proteins, enabling downstream analysis and identification on the whole-proteome scale. In 2016, we demonstrated that O-AlkTMM-C7 (113) specifically metabolically incorporated into O-mycoloylated proteins in C. glutamicum.332 After incubating bacteria in O-AlkTMM-C7, proteins were extracted and reacted with azido-488 via CuAAC, then analyzed by SDS-PAGE.332 Multiple hydrophobic proteins from a chloroform-methanol extract were labeled in a probe-dependent manner, and excision of the fluorescent bands and MS analysis revealed that among these proteins were the known O-mycoloylated proteins PorA and PorH (Figure 41A).332 Two other porins, including PorB and PorC, were also identified as O-mycoloylated, and numerous additional putatively O-mycoloylated proteins were detected in both chloroform-methanol and detergent extracts collected from probe-treated C. glutamicum.332 In future work, TMM derivatives such as O-AlkTMM-C7 can be employed to perform click-mediated affinity enrichment and LC-MS/MS profiling of O-mycoloylated proteins in C. glutamicum and related mycomembrane-containing bacteria. It is not known the extent to which O-mycoloylated proteins exist beyond C. glutamicum. While orthologues of the protein-specific mycoloyltransferase CgMytC are present in important pathogens such as Corynebacterium diphtheriae, they do not appear to be present in species from the Mycobacterium genus.293 Therefore, we have also pursued more general approaches to labeling mycomembrane proteins that should be more broadly applicable in mycomembrane-containing species. In 2020, we reported the development of O- and N-linked TMM derivatives whose acyl chains contain both a clickable alkyne and a photo-cross-linking diazirine group, named O- and N-x-AlkTMM-C15 (118 and 122, Figure 38C and 38D).272 It was first demonstrated that these probes selectively incorporated into mycomembrane components via the mechanisms shown in Figure 38, similar to their respective progenitor probes, O- and N-AlkTMM-C7.272 After incorporation of 118 or 122 into live mycobacteria, it was expected that irradiation of cells with UV light (365 nm) would convert the diazirine into a highly reactive carbene, leading to covalent cross-linking of the probe to mycomembrane-resident proteins. In essence, this sequence would specifically alkyne-tag proteins that non-covalently interact with mycolate glycolipids in the mycomembrane, allowing subsequent click-mediated protein detection or enrichment. Initial protein pulldown studies in M. smegmatis focused on the TDM-labeling probe N-x-AlkTMM-C15 (122), which based on SDS-PAGE analysis successfully labeled and allowed click-mediated affinity enrichment of proteins in a probe- and UV-dependent manner.272 Western blot analysis demonstrated that N-x-AlkTMM-C15 captured mycomembrane-relevant proteins, as two validation proteins were enriched, including Ag85 mycoloyltransferase and the mycomembrane porin MspA.272 Finally, M. smegmatis proteins that were photo-labeled by N-x-AlkTMM-C15 were click-biotinylated, enriched on avidin beads, proteolyzed, and analyzed by label-free LC-MS/MS quantitative proteomics to identify mycomembrane proteins. Approximately 100 proteins were enriched in probe-treated, UV-irradiated bacteria versus non-irradiated bacteria. These included proteins known to have mycomembrane-related functions, including mycoloyltransferases, MspA porin, trehalose mycolate transport and hydrolase proteins, and numerous additional candidate mycomembrane proteins with unknown function (Figure 41B). We expect that the x-AlkTMM probe series, including 118 and 122, will be useful tools for elucidating the mycomembrane proteome and studying the interactions between mycomembrane glycolipids and bacterial or host proteins.
Figure 41.
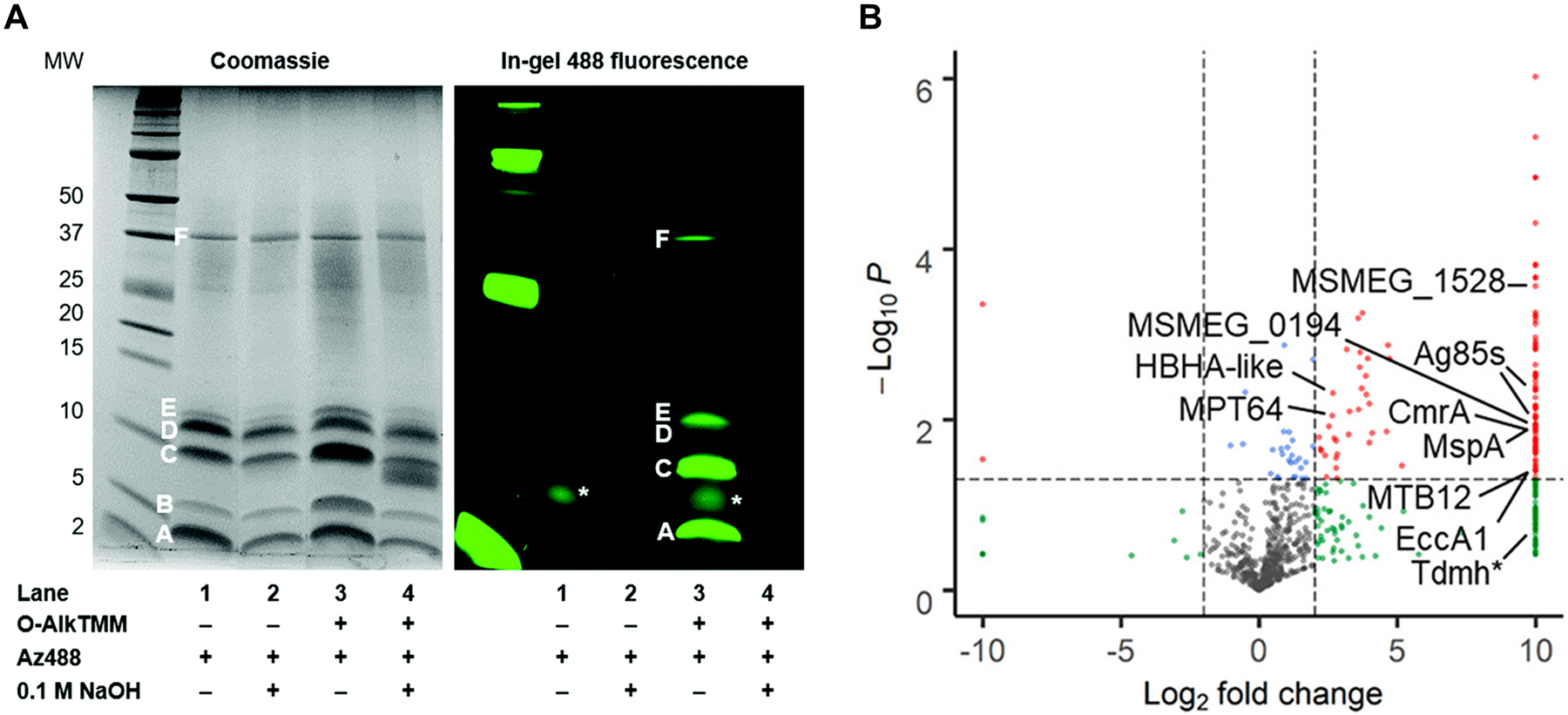
TMM derivatives enable labeling of proteins that (A) are covalently modified by or (B) non-covalently interact with mycolates. (A) Labeling of O-mycoloylated proteins in C. glutamicum with O-AlkTMM-C7. C. glutamicum was incubated in 100 μM O-AlkTMM-C7 (113), then chloroform-methanol protein extracts were obtained, subjected to CuAAC with azido-488, subjected to NaOH treatment to cleave ester linkages, and analyzed by SDS-PAGE. Bands A–F represent proteins identified by Coomassie staining; all but band B were labeled by O-AlkTMM-C7 and bands A, C, D, and E were confirmed by MALDI-MS to be O-mycoloylated. The asterisk (*) marks background fluorescence signal. Reproduced from ref. 332 with permission from the Royal Society of Chemistry. (B) Enrichment of mycomembrane proteins in M. smegmatis using photo-activatable N-x-AlkTMM-C15. M. smegmatis was incubated in 100 μM N-x-AlkTMM-C15 (122) and exposed to UV irradiation, then lysates were collected, subjected to CuAAC with azido-TAMRA-biotin, incubated with avidin beads, trypsinized, and analyzed by label-free quantitative LC-MS/MS. Volcano plot shows proteins in red that were significantly ≥4-fold enriched in 122-treated, UV-exposed versus non-UV-exposed bacteria. Selected proteins of interest are indicated. Reproduced with permission from ref. 332. Copyright 2020 American Chemical Society.
As with the trehalose derivatives discussed in section 6.1.1, the toolbox of TMM derivatives has expanded over the past several years and now includes a range of abilities for investigating different glycan-containing components of the mycomembrane. The major advance of TMM derivatives was that these probes allowed for labeling not only of outer-leaflet trehalose mycolates (e.g., TMM and TDM), but also of inner-leaflet AGM, O-mycoloylated proteins, and potentially other mycolate-containing products formed by the periplasmic mycolic acid exchange process. Furthermore, TMM derivatives included a fluorogenic probe for mycomembrane biosynthesis, QTF, which exploits FRET to directly report on mycoloyltransferase activity. In turn, the availability of these tools has enabled more comprehensive analyses of the dynamics of mycomembrane biosynthesis, often augmented by sophisticated imaging techniques. In addition, because TMM derivatives allow modification of the non-polar mycolate chains of mycomembrane glycolipids rather than the polar trehalose head-group, new opportunities have arisen to label and analyze mycomembrane-embedded proteins, which can provide new insights into mycomembrane proteomic composition and potentially facilitate the identification of novel targets for drug development. Future work may also extend TMM-based reporter probes into the arenas of inhibitor screening, therapeutic delivery, or specific pathogen detection. The straightforward synthesis of TMM derivatives via monoesterification of native trehalose or partially protected trehalose intermediates should bolster these efforts. On the other hand, the pursuit of TMM derivatives with more-native mycolate structures (e.g., 115), although more challenging to synthesize, could enhance probe specificity. Thus, there remain numerous challenges and opportunities with respect to TMM derivative design, synthesis, and applications.
6.1.3. Trehalose dimycolate derivatives.
To date, most probe development for mycobacteria has focused on tools that provide read-outs of mycomembrane biosynthetic reactions. However, in recent years it has become appreciated that mycomembrane remodeling stimulated by environmental stress (e.g., starvation, antibiotic treatment, immune response) can be advantageous for bacterial survival as conditions change. An example of mycomembrane remodeling involves TDM hydrolase (Tdmh), which is expressed in response to nutrient deprivation and degrades TDM, yielding free mycolic acids and TMM (see Figure 34).295,296,333 Tdmh is required for the formation of mycolic acid-rich, drug-tolerant biofilms in M. smegmatis, and it appears to balance nutrient intake, defense, and immunoactivity in M. tuberculosis.295,296 Thus, although Tdmh is not strictly required for mycobacterial growth, it is possible that this enzyme and/or related mycolate glycolipid hydrolases could serve as targets for the development of anti-virulence drugs to help combat mycobacterial infections.
We envisioned that a probe for monitoring Tdmh activity could be used to investigate mycomembrane remodeling and potentially to identify and characterize Tdmh inhibitors. In a similar vein to the Ag85 probe QTF (125) developed by the Kiessling group, we designed FRET-TDM (128), a fluorescence-quenched analogue of TDM that, in the presence of Tdmh, was anticipated to be hydrolyzed and generate fluorescence (Figure 42).277 The structure of FRET-TDM is based on a simplified TDM scaffold containing linear acyl chains at the 6 and 6’-positions, terminated by fluorescein on one chain and a dabcyl quencher on the other. The compound was chemically synthesized from the TMS-protected intermediate 124 in four steps using a strategy similar to that shown for TMM derivatives (FRET-TDM synthesis not shown; see Figure 39A for TMM derivative synthesis). While FRET-TDM was non-fluorescent in its quenched state, in the presence of recombinant purified Tdmh the probe underwent a >100-fold increase in fluorescence.277 MS analysis of the reaction mixture revealed the presence of the expected hydrolysis products O-FITC-TMM-C10 (119) and 129 (Figure 42A), as well as the two products resulting from cleavage of the other ester bond.277 A Tdmh mutant whose active site serine residue was mutated to alanine (S124A) did not activate FRET-TDM, confirming that Tdmh-specific enzymatic activity was responsible for the observed fluorescence turn-on.277 FRET-TDM was deployed to monitor recombinant Tdmh activity in different assay formats, including high-throughput microplate reader assays and native PAGE fluorescence assays, the latter of which is depicted in Figure 42B. This experimental versatility could be exploited in future studies to rapidly screen inhibitors of Tdmh or to profile TDM hydrolytic enzymes in cell lysates. Live mycobacterial cells also activated FRET-TDM, including M. smegmatis and M. tuberculosis, however the signal observed was largely Tdmh-independent, as determined using Tdmh knockout mutants.277 On the other hand, Tdmh overexpression led to increased fluorescence compared to wild-type cells.277 Thus, while FRET-TDM was capable of detecting Tdmh activity in cells when the enzyme was present at high enough levels, it was predominantly activated by non-Tdmh enzymes in wild-type bacteria. The identification of these unknown enzymes is of interest because they could represent novel mycomembrane-remodeling hydrolases. FRET-TDM can potentially be used as a tool to identify these enzymes through a combination of native PAGE fluorescence and MS analysis.
Figure 42.
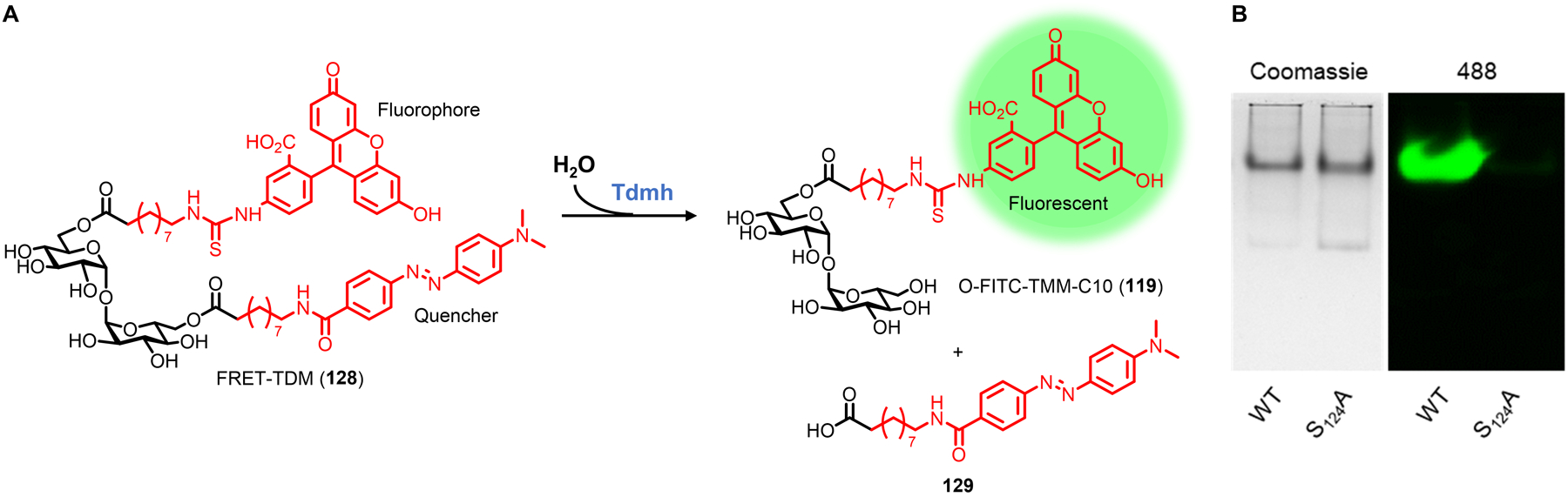
Analysis of Tdmh activity in mycobacteria using fluorogenic FRET-TDM. (A) Upon hydrolysis by Tdmh, an ester bond of FRET-TDM (128) is cleaved. Either ester bond of FRET-TDM can be cleaved by Tdmh; the possibility in which products 119 and 129 are generated is shown. (B) Recombinant purified wild-type Tdmh or its active site mutant S124A were resolved by native PAGE on two separate gels, one of which was Coomassie-stained (left) and the other was treated with 10 μM FRET-TDM (128) and scanned for 488 fluorescence (right). Reproduced with permission from ref. 277 (https://pubs.acs.org/doi/10.1021/acsomega.9b00130). Copyright 2019 American Chemical Society. Further permission related to the material excerpted should be directed to the American Chemical Society.
The Tdmh-independent activation of FRET-TDM indicated that the probe may have limited specificity for mycobacterial enzymes, a possibility which was evaluated by screening FRET-TDM against other types of bacteria as well as purified hydrolases from different sources. While FRET-TDM was efficiently activated by whole cells of mycomembrane-containing M. smegmatis and C. glutamicum, it was equally activated by B. subtilis and, to a lesser extent, E. coli.277 Purified non-mycobacterial hydrolases, including B. subtilis esterase and Candida rugosa lipase, activated FRET-TDM just as efficiently as Tdmh, whereas three other hydrolases from non-mycobacterial sources did not activate FRET-TDM.277 Thus, it was concluded that FRET-TDM exhibits partial selectivity for mycobacterial enzymes. This can possibly be attributed to the simplified mycolate chains of FRET-TDM, whose lack of α-branch and β-hydroxy group may be responsible for the limited mycobacterial specificity. Thus, it would be informative to perform similar tests on QTF and possibly other O-linked TMM derivatives to determine whether such metabolic processing occurs in non-mycobacterial systems. A systematic analysis of how native mycolate structural features, such as the α-branch and β-hydroxy group, impact probe specificity would be valuable. In this regard, the synthetic chemistry used to access near-native O-AlkTMM-C23 (115)273 could be employed to develop TMM or TDM probe panels to assist in answering this question and inform probe designs with improved specificity. In the meantime, FRET-TDM can be used to study Tdmh activity and inhibition, as well as profile mycomembrane-remodeling enzymes. The work on QTF and FRET-TDM established the concept of FRET-based probes for the study of cell envelope biosynthetic and remodeling pathways, which should be applicable to other types of bacterial glycans as well.
6.2. Chemical reporters for arabinose-containing glycans
Other efforts in chemical reporter development for mycobacteria have focused on arabinose-containing glycans, which, like trehalose-containing glycolipids, are major components of the mycobacterial cell envelope. AG, which was introduced above, consists of linear galactan and branched arabinan polysaccharide chains, whose terminal Araf residues are ultimately mycoloylated to form the inner leaflet of the mycomembrane (see Figure 33).11,258 The linear galactan chain of AG is covalently attached to the underlying PG layer.334 Arabinose is present in other cell envelope structures as well, including within the branched poly-Araf chains of the glycolipid lipoarabinomannan (LAM) and its corresponding delipidated polysaccharide, arabinomannan (AM). LAM is built upon a phosphatidylinositolmannoside (PIM) anchor, which is extended by a mannan domain, an arabinan domain, and diverse capping motifs that vary between pathogenic and non-pathogenic species (Figure 43).11,260 The arabinan domains of AG and LAM play important structural roles that support cell envelope integrity and contribute to the innate tolerance of mycobacteria to antibiotic treatment and assaults from the host immune system.11,335,336 LAM is also known to modulate the host immune response in a manner that appears to depend on its capping motifs and other structural features.11,337 Many steps in the pathways for AG and LAM/AM biosynthesis are essential for mycobacterial viability, so the production of these glycans has drawn significant interest as a target for drug development.11 Indeed, this is highlighted by the use of the arabinosyltransferase (AraT) inhibitor ethambutol as a front-line antitubercular drug.338 Chemical probes that report on the biosynthesis and transport of arabinan domains in AG, LAM, and AM would be valuable for studies on the function and inhibition of these pathways. Furthermore, since Araf units are relatively rare in nature and not present in humans, it is possible that arabinan-targeting tools could lead to new strategies for the detection and treatment of mycobacterial infections.
Figure 43.
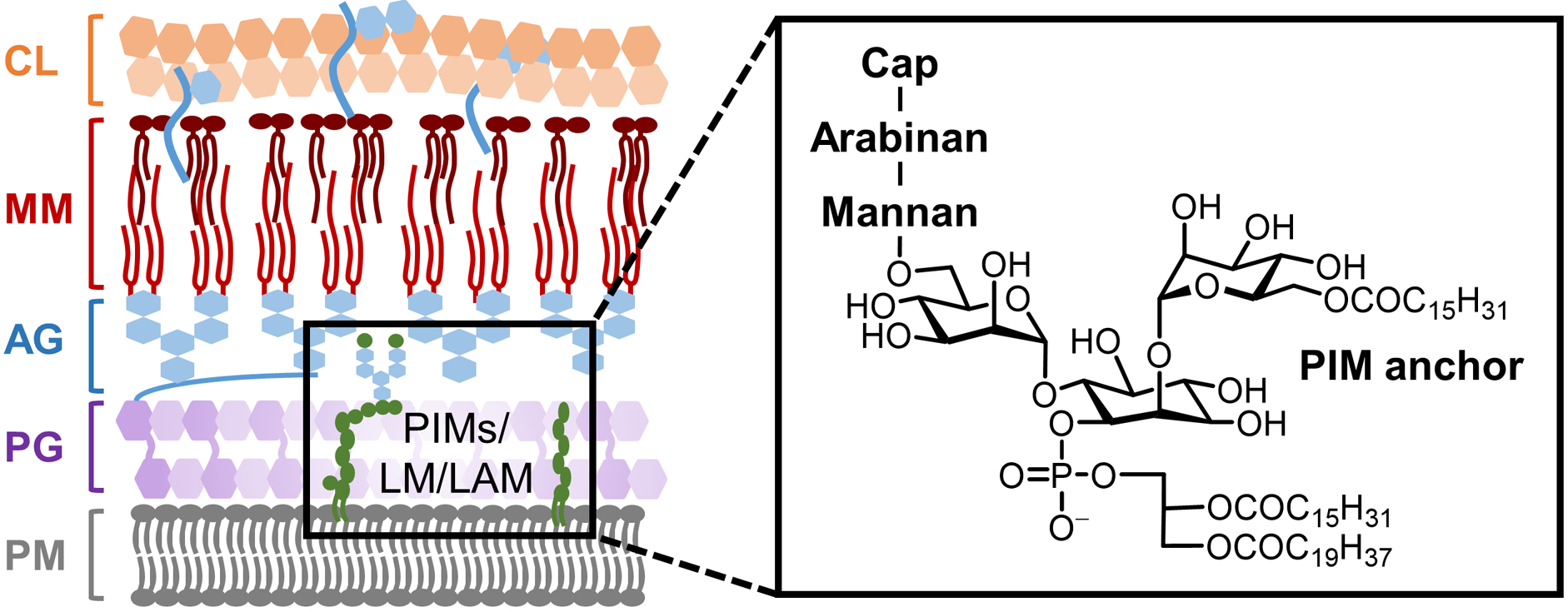
Simplified structure of mycobacterial lipoarabinomannan (LAM). LAM is an arabinan-containing extension of lipomannan (LM), which itself is a heavily mannosylated extension of a phosphatidylinositolmannoside (PIM) core.
The biosynthesis of mycobacterial AG, LM, and LAM is shown in abbreviated form in Figure 44.11,336 All of these arabinan-containing molecules arise from a universal Araf donor, decaprenyl-phospho-D-arabinofuranose (DPA). DPA is synthesized de novo in the cytoplasm from glucose-6-phosphate, which is processed through the pentose phosphate pathway to generate ribulose-5-phosphate and then phosphoribosyl diphosphate (PRPP). PRPP is then linked to the lipid carrier decaprenyl diphosphate (DPP) via UbiA, generating decaprenylphosphoryl-5-phosphoribose (DPPR), which is anchored to the cytoplasmic face of the plasma membrane. DPPR then undergoes dephosphorylation at the 5-position and epimerization at the 2-position, thus generating DPA with the proper stereochemical configuration. DPA is then flipped to the outer leaflet of the plasma membrane, where it serves as the Araf donor substrate for various periplasmic AraTs that assemble the arabinan chains of AG and LAM. In the case of LAM, it can presumably be delipidated to form the capsular polysaccharide AM. In addition to the de novo DPA synthesis pathway, exogenous D-arabinose can be taken up and metabolized, with one possible destination being recycling back into DPA.
Figure 44.
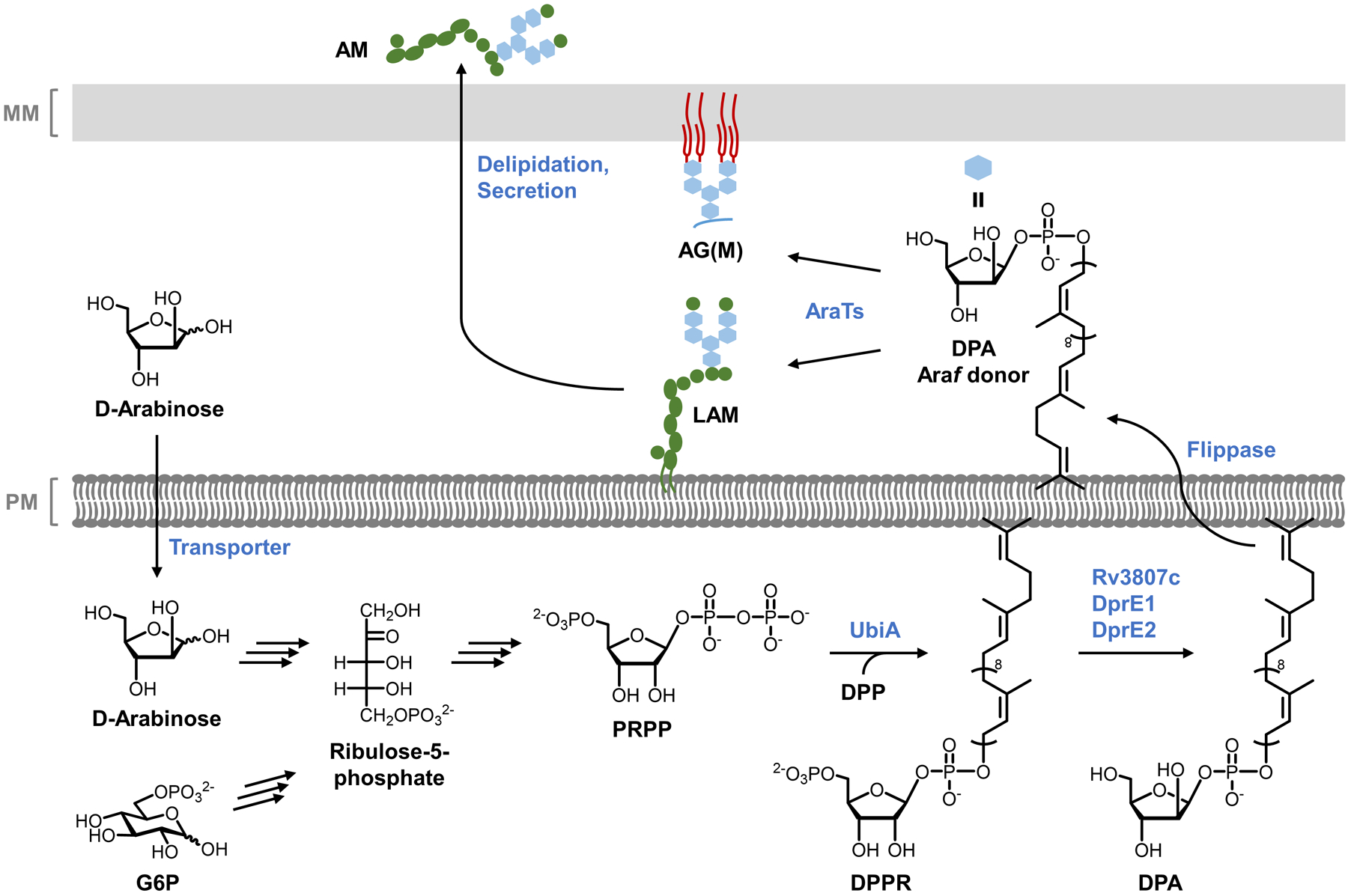
Biosynthesis of arabinan domains of AG, LAM, and AM in mycobacteria. AG, arabinogalactan; AraTs, arabinosyltransferases; AM, arabinomannan; DPA, decaprenyl-phospho-D-arabinofuranose; DPPR, decaprenylphosphoryl-5-phosphoribose; G6P, glucose-6-phosphate; LAM, lipoarabinomannan; PRPP, phosphoribosyl diphosphate.
Consideration of the biosynthetic pathways involved in AG and LAM production suggests different strategies for the development of arabinan-targeting chemical reporters, including both cytoplasmic/intracellular and periplasmic/extracellular routes, a situation encountered multiple times in this review (e.g., for PG and trehalose mycolate labeling). The arabinose salvage pathway presents the possibility of using relatively simple arabinose derivatives to incorporate into DPA and subsequently arabinans via an intracellular route. However, based on the current understanding of catabolism of exogenously supplied arabinose in mycobacteria, this would require passage through numerous metabolic steps and branch points, which presents concerns about substrate tolerance and the risk of off-target labeling. Another option would be to create lipid-linked Araf derivatives that mimic DPA, which could be transferred onto acceptors by AraTs to generate labeled arabinans. Such a strategy would benefit from periplasmic incorporation that occurs later in the biosynthetic pathway, but would require more challenging syntheses, plus the mycomembrane would be a potential barrier to DPA uptake. Although research in this area is nascent and has not yet risen to the level of maturity of the trehalose-based probes, there have been reports exploring both cytoplasmic and periplasmic routes of arabinan labeling employing arabinose and lipid-linked Araf derivatives, respectively, which we discuss in the following two sections.
6.2.1. Arabinose derivatives.
As shown in Figure 44, a salvage pathway exists in M. smegmatis that enables uptake and utilization of exogenous D-arabinose,336 providing a possible intracellular pathway for metabolic incorporation of arabinose or related pentose derivatives into AG and LAM. In 2016, the Reiling and Lindhorst groups reported the synthesis and evaluation of different azide-modified pentose analogues as potential tools for labeling mycobacteria.278 Based on an analysis of the known M. smegmatis pathway, three azido pentoses were designed, including 3-AraAz, 3-RiboAz, and 5-AraAz (130–132, Figure 45A).278 Modification of the pentose 2-position was avoided due to the DprE1/2-catalyzed epimerization step in the pathway, whereas 3-position modification appeared to afford the best possibility of incorporation, hence the design of 3-AraAz (130) and its epimer 3-RiboAz (131). 3-AraAz and 3-RiboAz, which have free 5-positions, exist as equilibrium mixtures of the major pyranose forms and the minor furanose forms. 5-AraAz (132) was designed as a negative control compound since its 5-position is blocked and it would not be competent to undergo 5-O-phosphorylation required to generate key intermediates in the pathway (i.e., ribulose-5-phosphate, ribose-5-phosphate, and PRPP).
Figure 45.
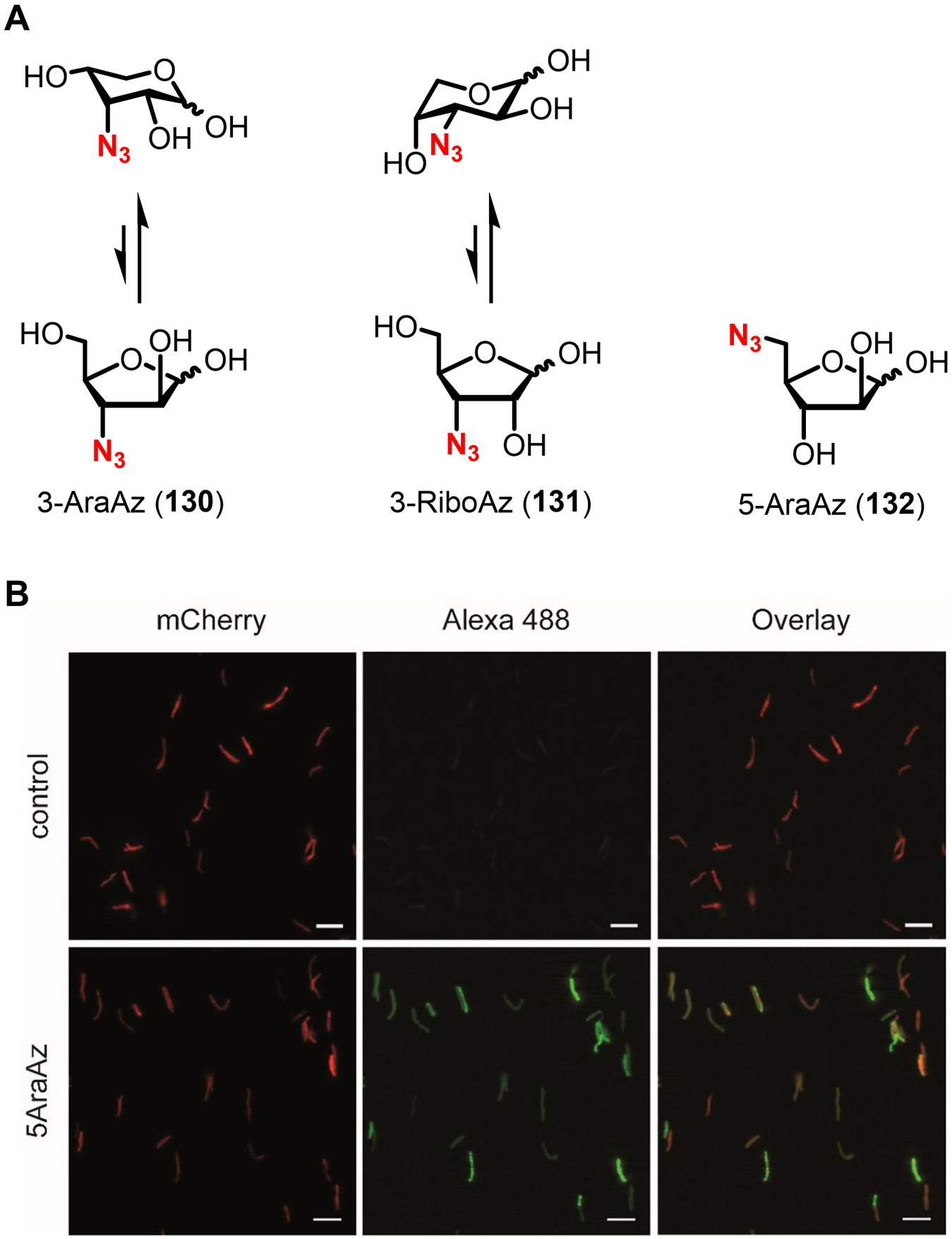
(A) Structures of azido pentose derivatives for labeling M. tuberculosis. (B) M. tuberculosis expressing mCherry was incubated in the presence of 5 mM 5-AraAz (132), reacted with cyclooctyne-488 via SPAAC, fixed, and imaged by fluorescence microscopy. The control was not treated with 5-AraAz or cyclooctyne-488. Scale bars, 5 μm. Reproduced with permission from ref 278. Copyright 2017 Wiley-VCH.
The chemical syntheses of compounds 130–132 from the appropriate starting pentoses were concise, invoking traditional strategies consisting of regioselective hydroxyl group protection, SN2 substitution to install azido groups, and deprotection (not shown).278 Although the compound design was based on the well-characterized arabinose utilization pathways of M. smegmatis, the compound evaluation was conducted solely in the pathogen M. tuberculosis, despite arabinose metabolism being less well-defined in this organism. Metabolic incorporation experiments involved treating M. tuberculosis with 0.5–5 mM of 130–132, or 0.1–1 mM 6-TreAz (102) as a positive control, then performing SPAAC with a cyclooctyne-488 reagent and analysis by flow cytometry.278 There was minimal incorporation of 3-AraAz and 3-RiboAz into M. tuberculosis even at high concentrations,278 suggesting that these compounds did not efficiently traverse the numerous cytoplasmic steps required to incorporate into cell surface arabinans. By contrast, the control compound 5-AraAz unexpectedly incorporated with into M. tuberculosis, despite it being incapable of doing so via the known DPA biosynthesis pathway.278 Competition experiments demonstrated the 5-AraAz labeling was diminished by a 100-fold excess of D-arabinose, so incorporation appeared to be dependent on pentose metabolism.278 Ultimately, it could not be established what molecular species 5-AraAz incorporated into. Fluorescence microscopy analysis of 5-AraAz-labeled M. tuberculosis demonstrated efficient incorporation into cells, but signal was not restricted to the cell surface, rather it appeared to be more uniformly distributed throughout the cell (Figure 45B).278 Western blot analysis revealed a ~50 kDa azide-dependent band in a glycolipid extract from M. tuberculosis, although it did not co-migrate in the gel with LAM (~40 kDa).278 Further experiments are needed to identify the labeled species and define the incorporation pathway. It may be valuable to evaluate the azido pentoses in M. smegmatis, which as noted above has well-characterized arabinose metabolic pathways in comparison to M. tuberculosis. Overall, 5-AraAz, and pentose derivatives in general, cannot presently be considered as tools for labeling arabinan-containing AG or LAM. However, studies on the unanticipated behavior of 5-AraAz in M. tuberculosis and other mycobacterial species may reveal heretofore undefined routes for pentose uptake and utilization. Also of importance, it was shown that 5-AraAz incorporates into M. tuberculosis lab strains and clinical isolates but not mammalian cells,278 demonstrating that arabinose derivatives can provide labeling specificity that is valuable in various contexts, as discussed above for other bacteria-specific chemical reporters (e.g., D-amino acid and trehalose derivatives).
6.2.2. Lipid-linked arabinofuranose derivatives.
In view of AG and LAM biosynthesis, chemical reporters that mimic DPA present an opportunity for labeling arabinans via a more direct periplasmic AraT-mediated route of incorporation, thus avoiding some of the issues that complicated the cytoplasmic incorporation of pentose derivatives (Figure 44). The idea of using lipid-linked Araf derivatives has been explored by the Kiessling group, which in 2019 published proof of concept.96 Native DPA has a large C50 decaprenyl chain, which would likely need to be truncated in a chemical reporter to facilitate both synthesis and metabolic incorporation. Therefore, the team set out to determine whether mycobacterial AraTs would accept DPA derivatives with simplified lipid tails. A panel of DPA derivatives was synthesized according to reported methods,339 with lipids ranging in size from C8–C15 and with varying resemblance to isoprenoid tails.96 Next, a model system was developed to test compound incorporation in C. glutamicum, which is an ideal species for this purpose because its AG and mycomembrane layers are dispensable. C. glutamicum ΔubiA, a mutant which lacks the ability to convert PRPP to DPPR and is thus deficient in both DPA and AG (see Figure 44), was supplemented with the synthetic DPA derivatives to determine if they could rescue AG synthesis. According to cell envelope carbohydrate analysis, it was discovered that farnesyl-phospho-D-arabinofuranose (FPA, 133, Figure 46A), a C15 variant of DPA, partially restored Araf levels in the ΔubiA mutant. Subsequently, 13C-labeled FPA was synthesized and used in conjunction with NMR spectroscopy to directly confirm that the compound was incorporated into AG. FPA was beautifully demonstrated by transmission electron microscopy to rescue production of the complete cell envelope in the ΔubiA mutant, including AG, mycomembrane, and capsular polysaccharides (Figure 46B). Interestingly, FPA also rescued viability of M. smegmatis that was treated with DPA biosynthesis inhibitor BTZ043, implying that simplified DPA derivatives incorporate not only into a DPA-deficient C. glutamicum mutant, but also a wild-type Mycobacterium species.
Figure 46.
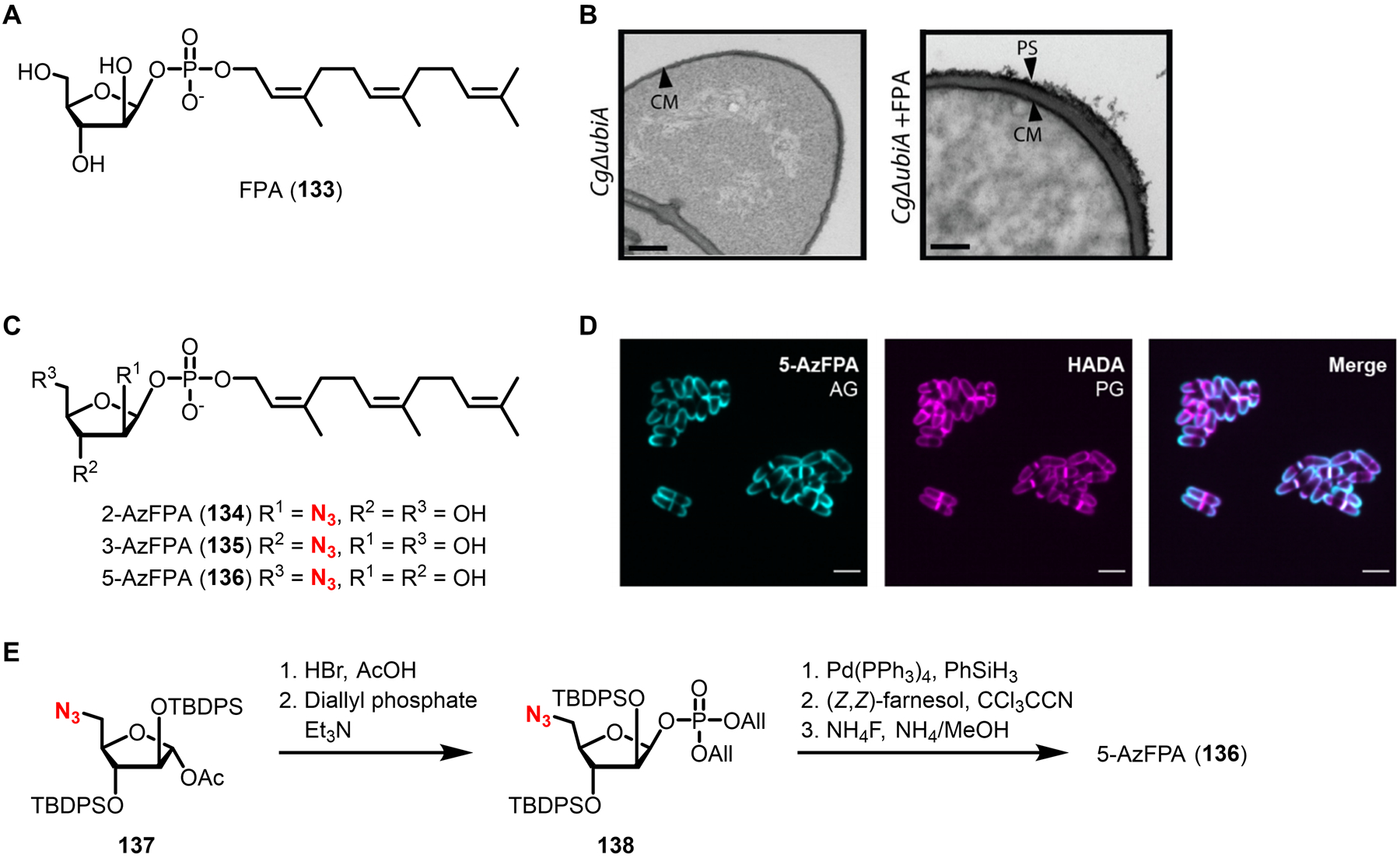
Labeling and imaging arabinans in mycobacteria and corynebacteria using DPA derivatives. (A) Structure of FPA, a derivative of DPA with a truncated lipid. (B) Restoration of AG synthesis in a DPA-deficient C. glutamicum mutant using FPA. C. glutamicum ΔubiA was incubated in 500 μM FPA (133) or left untreated and visualized using transmission electron microscopy. Left, untreated; right, FPA-treated. CM, cytoplasmic membrane; PS, polysaccharide layer. Scale bars, 100 nm. Reproduced with permission from ref 96. Copyright 2019 American Chemical Society. (C) Structures of azide-modified DPA derivatives. (D) Imaging of arabinans using 5-AzFPA. C. glutamicum was incubated in 250 μM 5-AzFPA (136) and 500 μM HADA (17) for 2 h, fixed, and analyzed by fluorescence microscopy. Scale bars, 3 μm. Reprinted from ref 279 with permission. Copyright 2021 American Chemical Society. (E) Representative synthesis of an AzFPA derivative. The late stage of the synthesis of 5-AzFPA (136) is shown. All, allyl; TBDPS, tert-butyldiphenylsilyl.
With proof of concept established that exogenously supplied FPA serves as a surrogate for DPA and undergoes incorporation into AG in live bacteria, the Kiessling group subsequently disclosed the development of clickable FPA-based chemical reporters.279 Three regioisomeric azide-modified FPA analogues were designed, including 2-, 3-, and 5-AzFPA (134–136, Figure 44C).279 The synthesis of each target compound followed the representative route shown in abbreviated form in Figure 44E, wherein a protected azido arabinose building block was sequentially subjected to anomeric bromination, displacement with diallyl phosphate, Pd-catalyzed deallylation, phosphodiester formation with (Z,Z)-farnesol, and deprotection.279 While most individual steps in these syntheses were high-yielding, the step counts were high and the final three-step sequence yield range was 12–18%, highlighting the difficulty of preparing complex lipid-linked monosaccharides.279 With the compounds in hand, they were evaluated for incorporation into C. glutamicum and M. smegmatis. Bacteria were incubated with probes, reacted with cyclooctyne-647 via SPAAC, and analyzed by flow cytometry. Whereas 3-AzFPA (135) did not incorporate appreciably into either species, 2-AzFPA (134) labeled M. smegmatis efficiently and 5-AzFPA (136) labeled C. glutamicum efficiently.279 Fluorescence analysis of AG extracts from probe-treated cells showed the same trend.279 The variability in labeling between 2- and 5-AzFPA in mycobacteria and corynebacteria is notable and likely reflects the differences in the presence of specific AraTs and/or their substrate tolerances between the two organisms. At the same time, these results underscored the advantages of synthesizing and testing multiple isomers of a probe, which can reveal complementary labeling characteristics, as previously observed for the TreAz probe series. Fluorescence microscopy was used to visualize arabinan and PG synthesis in tandem using 5-AzFPA (136) and HADA (17), respectively, which showed that both cell envelope components were synthesized simultaneously and signal was localized to the poles and septa (Figure 46D).279 Coincident synthesis of AG and PG would be anticipated because these two cell envelope layers are covalently attached to each other. Finally, M. smegmatis was pre-labeled with 136 followed by SPAAC reaction, and uptake of bacteria by macrophages was imaged.279 It was not tested whether AzFPA probes would be capable of labeling mycobacteria already residing within macrophages.
DPA derivatives, specifically 2- and 5-AzFPA, are the first tools that allow direct labeling and analysis of arabinan polysaccharides in live mycobacteria and related bacteria. Similar to the utility of trehalose- and TMM-based probes for studying the mycomembrane, DPA derivatives are expected to be valuable probes for studying arabinan biosynthesis in various physiological contexts. Compared to the azido pentose derivatives 130–132, the design of the lipid-linked Araf donor derivatives 134–136, which featured a periplasmic route of incorporation, has proven to be particularly advantageous. Several lines of future work on this new class of probes will be valuable. Because DPA is the universal Araf donor that is used to produce AG, LAM, and AM, it is probable that the probes incorporate into those and any other Araf-containing molecules present in mycobacteria and corynebacteria. Thus, it will be of high interest to determine any target(s) of metabolic incorporation beyond AG. On a related note, since branched arabinans have various glycosidic bond types, including 1→2 and 1→5 linkages, it would be useful to determine whether the 2- and 5-AzFPA probes generate truncation products that affect arabinan localization and/or biological function. Also of benefit would be to identify the specific AraT(s) that are responsible for DPA probe incorporation in live cells. Precisely defining these attributes of DPA probe behavior—the identity of the labeled molecule(s) and the mechanism of probe incorporation—will aid in determining suitable applications for the probes and inform judicious experimental design and data interpretation. Although one may have predicted that relatively large and charged molecules such as AzFPAs may have difficulty passing through the mycomembrane, the efficient labeling of wild-type M. smegmatis by azido DPA derivatives clearly indicates that this is not a significant concern. Thus, deploying these probes to study arabinans in mycobacteria with highly impenetrable mycomembranes, including pathogenic M. tuberculosis, should be feasible. Because Araf-containing glycoconjugates are rare in nature and absent from mammals, it will be worthwhile to investigate whether DPA derivatives enable specific targeting of mycobacteria. As a promising early indicator, AzFPA reporters were shown to label mycobacteria but not Gram-negative E. coli.279 From a broader perspective, DPA derivatives represent the first chemical reporters for bacterial glycans that are based on lipid-linked sugar donors. This is an exciting advance that could be extended to the study of other bacterial glycans whose biosynthesis is dependent on lipid-linked sugar donors.
7. Chemical reporters for capsular polysaccharides
In the preceding sections, we have encountered a diverse range of bacterial cell envelope polysaccharides for which chemical reporters have been developed, including peptidoglycan, lipopolysaccharides, teichoic acids, and mycobacterial arabinans. Beyond these structures, bacteria produce a wide variety of capsular polysaccharides, which tightly associate with the cell envelope to form a surface-exposed layer around the cell, and exopolysaccharides, which are secreted into the surrounding environment.12,17 Like other bacterial glycans, capsular polysaccharides and exopolysaccharides can provide structural support and protection, and, given their privileged location at (or immediately beyond) the cell surface, they are often involved in extracellular events such as biofilm formation or host interactions.12,17
Several examples of bacterial capsular polysaccharides and exopolysaccharides are shown in Figure 47. The Gram-negative gut commensal bacterium B. fragilis produces a variety of capsular polysaccharides, including PS A1, which is an immunomodulatory zwitterionic polysaccharide that helps to mediate the development of the mammalian immune system and has shown potential as an immunotherapeutic molecule.340,341 Interestingly, Gram-positive S. pneumoniae also expresses a zwitterionic capsular polysaccharide Sp1, which similarly is an immune modulator and a key component of the widely used vaccine against this major human pathogen.342 Some bacterial polysaccharides closely resemble other structural carbohydrates found in nature. For example Gram-negative E. coli, Salmonella, and other bacteria were recently discovered to produce a phosphoethanolamine-modified cellulose exopolysaccharide that is essential for biofilm formation because it stabilizes neighboring cells in the extracellular matrix.343,344 Gram-negative P. aeruginosa produces several exopolysaccharides, including alginate, which are likewise major constituents of the biofilm matrix and help to protect this pathogen from the host immune response and antibiotics.345,346 Many bacterial pathogens possess capsular polysaccharides that mimic host glycans and thus contribute to immune evasion, such as hyaluronate in Streptococcus pyogenes and Bacillus cereus,347,348 polysialic acids in N. meningitidis,349 and α-glucans in M. tuberculosis.261 Chemical reporter probes have the potential to facilitate the investigation of bacterial capsular and exopolysaccharides’ biosynthesis, their roles in physiology and host–symbiont/pathogen interactions, and the therapeutic implications that follow. Given the extraordinary diversity of such polysaccharides across different types of bacteria, there are many opportunities for tool development in this space. To date, published work in this area has been limited to capsular polysaccharides of gut commensal bacteria, which we cover below.
Figure 47.
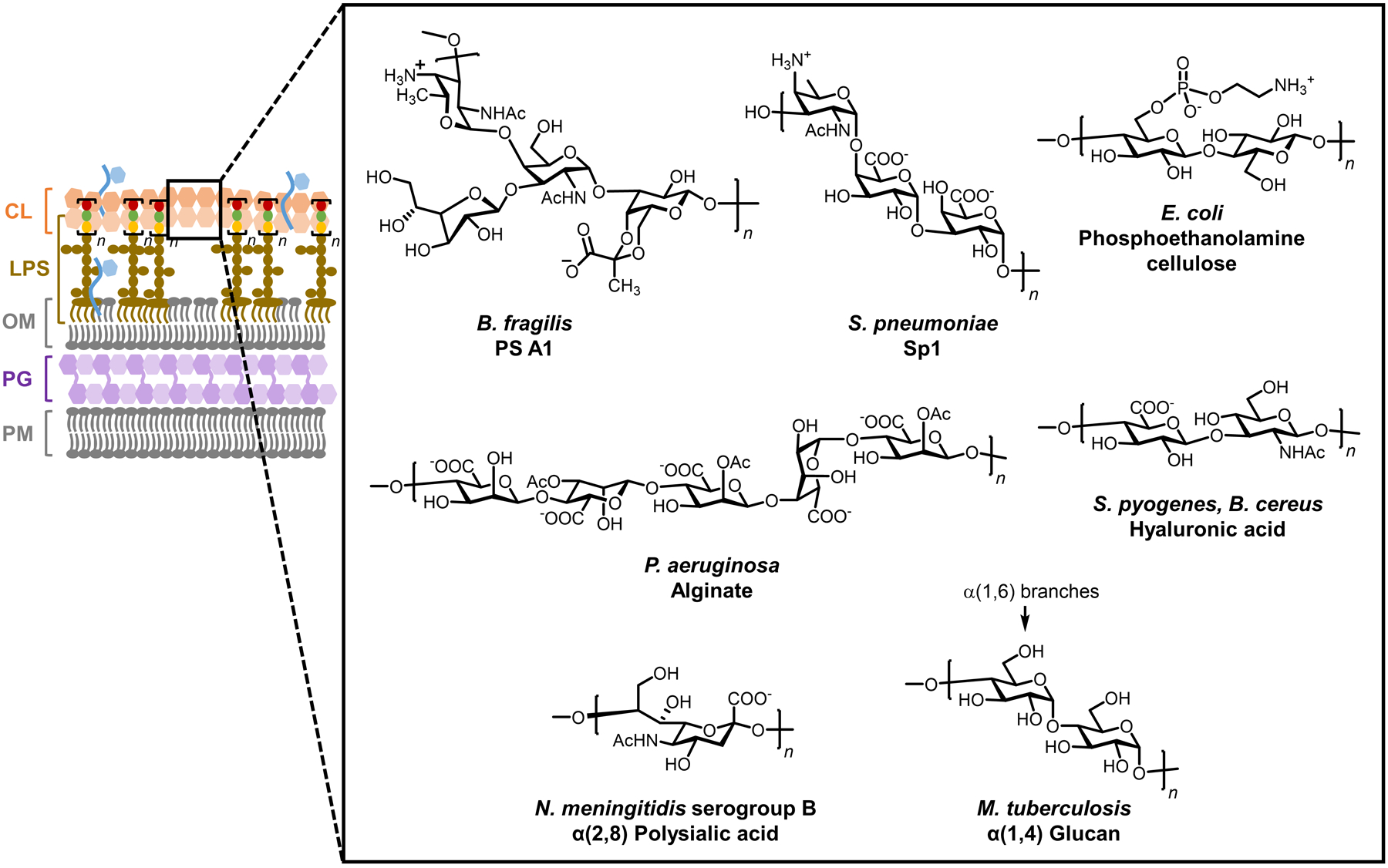
Representative structures of bacterial capsular polysaccharides and exopolysaccharides. Gram-negative cell envelope schematic is shown, although capsular polysaccharides and exopolysaccharides exist in Gram-positive bacteria and mycobacteria as well.
7.1. Chemical reporters for capsular polysaccharides of intestinal bacteria
While many of the studies covered in this review focused on tool development and applications for pathogenic bacteria, commensal bacteria are also of high significance to human health. For example, intestinal commensal bacteria beneficially modulate the host immune system and prevent colonization and invasion by pathogenic species.3 One such organism is B. fragilis, an anaerobic Gram-negative bacterium whose surface glycans, including the capsular polysaccharide PS A1 (Figure 47), drive host immunoprotection.340,341,350 The detailed mechanisms of these host–symbiont interactions are not well defined, which in 2015 led the Kasper group to investigate chemical reporter strategies for labeling and tracking B. fragilis polysaccharides in vivo.351 The biosynthesis of PS A1 reflects that of LPS O-antigen biosynthesis in Gram-negative bacteria (see Figure 18). PS A1 biosynthesis is initiated by sequential addition of variable nucleotide sugar donors onto a C55 polyprenyl phosphate chain by Wcf glycosyltransferases,352,353 generating a lipid-linked tetrasaccharide that is flipped across the plasma membrane, polymerized by Wzy, and exported to the cell surface (not shown).352,353 The presence of N-acetyl-D-galactosamine (GalNAc) in the PS A1 tetrasaccharide repeating unit prompted the team to explore whether GalNAc-based reporters could be incorporated into PS A1. As with the LPS O-antigen reporters (section 3.2), presumably this would require the exogenously added GlcNAc derivative to be taken up into the cytoplasm, activated into its corresponding UDP sugar, transferred onto a lipid carrier, and subsequently installed into the polymer.
Peracetylated azide-modified GalNAc (Ac4GalNAz, 139, Figure 48A) was selected as the GalNAc derivative for evaluation in B. fragilis. When B. fragilis was cultured in Ac4GalNAz and then subjected to SPAAC reaction with cyclooctyne-488, bacteria were efficiently labeled on the cell surface (Figure 48B).351 By contrast, B. fragilis was not labeled by azide-modified glucosamine, mannosamine, or fucose derivatives (presumably also in peracetylated form).351 The Ac4GalNAz-dependent signal in B. fragilis cells was reduced, but not eliminated, in a PS A1-deficient mutant, indicating that the probe was predominantly incorporated into PS A1 but likely incorporated into other surface molecules as well.351 Further evidence of probe specificity was provided by Western blot analysis of PS A1-containing cellular extracts, which showed an Ac4GalNAz-dependent fluorescent band that correlated to signal from a PS A1-specific antibody (Figure 48C).351 Additional experiments demonstrated that neither Ac4GalNAz incorporation nor SPAAC-mediated fluorescent labeling of PS A1 impacted its immunomodulatory activity, providing confidence that the method could be applied to reliably study host–bacterium dynamics in vivo.351
Figure 48.
Labeling and imaging capsular polysaccharides in gut commensal bacteria using GalNAc derivatives. (A) Structures of monosaccharide reporters Ac4GalNAz (139), Ac4ManNAz (140), and GalNAc-CCP (141). (B) B. fragilis was incubated in Ac4GalNAz (139), subjected to SPAAC with cyclooctyne-488, and imaged by confocal microscopy. Left, confocal image of labeled bacteria. Scale bar, 5 μm. Right, 3D-rendered confocal image of Ac4GalNAz-labeled bacteria counterstained with propidium iodide. Scale bar, 1 μm. (C) B. fragilis wild type strain (WT), PS A1 constitutive expression strain (ΔmpiM44), and PS A1-deficient strain (ΔPSA) were incubated in Ac4GalNAz (139), subjected to SPAAC with cyclooctyne-647, inactivated, and analysed by SDS-PAGE with fluorescence scanning or blotting with anti-PS A1 antibody. ctrl, control; F, fluorescent; MW, molecular weight; NF, non-fluorescent. (D) B. fragilis pre-labeled with Ac4GalNAz (139) and cyclooctyne-Cy7 was introduced to mice and whole-body longitudinal imaging (top) and imaging of dissected intestines (bottom) were performed. ROI, region of interest. (B–D) were reproduced with permission from ref 351. Copyright 2015 Springer Nature. (E) B. vulgaris was labeled with coumarin probe HADA (17), KDO-N3 (55), and GalNAc-CCP (141), then reacted with cyclooctyne-rhodamine (to label azides) and tetrazine-TAMRA (to label cyclopropenes). Triple fluorescent-labeled bacteria were directly introduced into surgically exposed intestines of live mice, then imaging was performed by intravital two-photon microscopy. WGA-633 is a lectin used to stain the epithelium. Scale bars, 10 μm. Reproduced with permission from ref 177. Copyright 2015 Springer Nature.
After establishing that Ac4GalNAz (139) labeled PS A1 and potentially other surface glycans in B. fragilis, the probe was used for various in vivo imaging applications in the initial report and follow-on studies. Because Ac4GalNAz labels mammalian cell surface glycoproteins,354,355 it cannot be used to selectively label bacteria already residing within a host environment, so a bacteria pre-labeling approach was used to track B. fragilis and its associated polysaccharides in vivo. Accordingly, B. fragilis PS A1 was labeled with Ac4GalNAz and subjected to SPAAC reaction with a cyclooctyne-fluorophore reagent, then the live fluorescent bacteria were intraperitoneally administered to mice.351 Within this experimental context, various in vivo and ex vivo analyses were performed to study the dissemination of, and immune response to, labeled B. fragilis and PS A1. One highlight from these studies was the combined use of Ac4GalNAz and SPAAC to pre-label B. fragilis with the near-infrared dye Cy7, which enabled longitudinal imaging of the spatiotemporal distribution of bacteria and their polysaccharides in live mice (Figure 48D).351 Fluorescence signal peaked between 2–9 h in the intestinal region and subsequently decreased, but was still observable even after 4 days (Figure 48D, top).351 Imaging of excised intestines revealed that, shortly following administration of bacteria, fluorescence signal was mostly in the stomach and small intestine, whereas at later time points signal was predominantly present in the cecum and colon (Figure 48D, bottom).351 Along with other data from the study, this suggested that B. fragilis may preferentially colonize and persist within the cecum and colon.351 In agreement, a subsequent study by the Zhi group used Ac4GalNAz and cyclooctyne-fluorophore pre-labeling of a novel B. fragilis strain to enable imaging of its dissemination in mice, and likewise strong fluorescence signal was observed in the cecum and colon at similar time points.356
With an eye toward expanding the tools available for metabolic labeling of anaerobic gut commensal bacteria, Kasper and co-workers also tested additional carbohydrate-based chemical reporters against panels of intestinal bacteria. In their first study focused on B. fragilis PS A1, the team also evaluated Ac4GalNAz (139), Ac4GlcNAz (86), and Ac4ManNAz (140) in a panel of 14 gut commensals, which included representatives from phyla common in the gut, including Bacteroides, Firmicutes, and Proteobacteria.351 Half of the tested species were robustly labeled by Ac4GalNAz, whereas a few species appeared to be moderately labeled by Ac4GlcNAz and/or Ac4ManNAz.351 Thus, these monosaccharide reporters may be beneficial for tagging and tracking various gut bacteria in vivo. However, the labeled molecular species in these experiments were not identified, so it will be important to follow up with additional analyses. In a subsequent 2017 study aiming to further build out the gut commensal labeling toolbox, the Kasper group combined various probes to allow simultaneous labeling of PG, LPS, and capsular polysaccharides.177 The previously reported D-amino acid derivative HADA (17) successfully labeled PG of all gut species tested, further confirming its broad applicability for tagging PG.177 Unlike Ac4GalNAz, which incorporates into eukaryotic cells, HADA labeling is bacteria-specific, which allowed the team to label and track endogenous commensal bacteria within a host for the first time, thereby sidestepping the bacteria pre-labeling approach required for Ac4GalNAz. In addition to HADA-based PG labeling, previously reported KDO-N3 (55, Figure 19A) was demonstrated to label LPS in a variety of Gram-negative gut bacteria.177 To permit simultaneous triple labeling of PG, LPS, and capsular polysaccharides, orthogonal detection chemistry for each component would be needed, although this would be impossible using the two azido sugars Ac4GalNAz and KDO-N3. Thus, a novel cyclopropene-modified GalNAc derivative, N-cyclopropenyl galactosaminyl carbamate (GalNAc-CCP, 141, Figure 48A), was developed to enable polysaccharide detection via tetrazine ligation.177 Using Bacteroides vulgatus as a model Gram-negative commensal bacterium, it was shown that HADA (detected directly), KDO-N3 (detected via SPAAC), and GalNAc-CCP (detected via tetrazine ligation) specifically incorporated into PG, LPS, and capsular polysaccharide, respectively.177 Finally, as yet another impressive demonstration of how complementary glycan probes and bioorthogonal chemistries can be merged to read out multiple molecular processes, B. vulgatus with three orthogonally pre-labeled surface glycans was directly introduced to the intestines of live mice and imaged using two-photon intravital microscopy (Figure 48E).177
The work described in this section, mainly conducted by the Kasper group, built upon the existing foundation of bacterial glycan reporter research to (i) create novel polysaccharide-labeling tools and (ii) apply them in a new context, namely the in vivo tracking of gut commensal bacteria. Ac4GalNAz (139) and GalNAc-CCP (141) were developed with the objective of labeling of PS A1 and potentially other capsular polysaccharides. Indeed, these new probes were convincingly shown to incorporate into PS A1 in Bacteroides species, possibly along with related surface glycans. When these probes were combined with appropriate bioorthogonal chemistries, the fate of pre-labeled bacteria and glycans could be tracked in complex host environments using flow cytometry or fluorescence microscopy. This experimental platform can reveal rich information about the location of bacteria and the types of cells they interact with, as well as correlation of these features with physiological or pathological processes. This work illuminated new ways to apply chemical reporters for other types of bacterial glycans to study host–bacterium interactions in the future. At present, one limitation of the GalNAc-based reporters is the lack of information about their mechanism(s) of incorporation, which prevents correlation of probe-dependent fluorescence signal to the activity of a specific biosynthetic pathway. As noted above, another disadvantage to the GalNAc derivatives is their inability to specifically label bacterial polysaccharides within a host, which necessitates a pre-labeling approach for in vivo studies. In this regard, it could be fruitful to focus on the unique bacterial sugars present in PS A1, which could lead to tools that allow labeling of endogenous bacteria and open opportunities for bacteria-specific diagnostic or therapeutic targeting. Finally, the published work on PS A1 probes provides a blueprint for the development of new tools to label other types of bacterial polysaccharides. As hinted at by the representative examples of bacterial capsular polysaccharides and exopolysaccharides shown in Figure 47, there remain abundant opportunities in this area.
8. Conclusions and perspectives for future research
Chemical reporters have transformed the toolbox for basic and translational bacterial glycobiology research. The critical importance of cell envelope glycans to bacterial physiology and pathogenesis has long been appreciated, and has been a focal point for the development of vaccines, antibiotics, and diagnostics. However, experimental inquiry in this field has been limited by traditional genetic, biochemical, and molecular biological techniques, which are ill-suited to the investigation and targeting of glycans in native contexts. The emergence of metabolic labeling approaches that harness bioorthogonal chemistry and small-molecule fluorophores has empowered chemical biologists to create innovative tools that possess an unprecedented ability to tag, track, profile, and target glycans in live bacteria. Since the early 2000s, when the first chemical reporters for bacterial glycans were introduced, we have witnessed rapidly accelerating activity in the field. Now, a multitude of reliable chemical reporters are available for examining major cell envelope components in a wide range of commensal and pathogenic bacteria: peptidoglycan (Table 1), lipopolysaccharide (Table 2), glycoproteins (Table 3), teichoic acids, capsular polysaccharides, and various mycobacterial glycans (Table 4). These chemical reporters exhibit immense experimental versatility, not only in terms of the different bacterial glycan types and associated metabolic pathways they can probe, but also because of the modularity of their design. Small-molecule fluorescent reporters that allow one-step bacterial glycan labeling can be adapted to carry fluorescent dyes that have varied physicochemical properties, span the excitation/emission spectrum, and exhibit fluorogenic turn-on capabilities. Bioorthogonal chemical reporters that allow two-step labeling have practically unlimited flexibility, as they have invoked a variety of complementary bioorthogonal reactions to permit the rapid and chemoselective attachment of virtually any desired chemical cargo to the bacterial glycan of interest.
With such a high level of experimental versatility, chemical reporters have been applied in myriad ways to study and target bacterial glycans. The most apparent impact that these tools have had is an unparalleled capacity to illuminate glycans in live bacteria and inform on their spatiotemporal dynamics in complex, changing environments. For example, among numerous other applications covered in this review, chemical reporters identified the presence of peptidoglycan in Chlamydia for the first time, revealed novel stress-induced peptidoglycan and mycomembrane synthesis mechanisms in mycobacteria, and tracked the dissemination of commensal bacteria and their capsular polysaccharides in the gut of a living animal. The ability of reporter probes to provide fluorescence read-outs on specific glycan biosynthetic pathways (e.g., peptidoglycan cross-linking, trehalose mycolate transport) has led to the creation of phenotypic assays that can be adapted to characterize or screen for novel inhibitors of cell envelope assembly. Such tools may prove valuable in antibiotic discovery and development efforts. Many glycans exert their biological effects by covalently modifying or non-covalently interacting with proteins, two processes which have been dissected in bacterial systems with the help of chemical reporters. For instance, strategies have been developed to discover and identify bacterial glycoproteins, which were long thought to be non-existent, as well as to elucidate bacterial glycan–protein interactions that are involved in peptidoglycan and mycomembrane construction. In the long-term, these types of chemical proteomics strategies could assist in the identification and characterization of novel targets for antibiotic development. Chemical reporters that co-opt bacteria-specific metabolic pathways to label pathogens have informed the development of novel diagnostic and therapeutic targeting strategies. For example, the non-mammalian disaccharide trehalose has been outfitted with fluorogenic dyes to detect M. tuberculosis in patient sputum samples, 18F-radionuclides to visualize mycobacterial infections via PET imaging, and photosensitizers to kill mycobacteria upon exposure to light. Similarly, other sugars that are unique to bacteria (or that are uniquely presented within bacteria), such as KDO and legionaminic acid, have been used to differentiate bacterial species/strains from each other and from mammalian cells. Since chemical reporters rely on active metabolic incorporation to produce signal, they have also been used to discriminate live bacteria from dead or metabolically inactive bacteria. Finally, chemical reporters have inspired the development of bacterial sugar-based metabolic inhibitors, some of which have been shown to impair glycan-mediated virulence processes in significant human pathogens. Taken together, although chemical reporters are a relatively new technology, they have already had far-reaching impacts on the field of bacterial glycobiology.
The process of developing chemical reporter strategies is a challenging, interdisciplinary endeavor that we attempted to shed light on in this review. The motivation to create chemical reporters is driven by a knowledge or technology gap—will the developed tool help to answer a pressing question or test a hypothesis in the field? Accelerate the discovery of molecular targets or inhibitors? Lead to technologies that ultimately improve the detection or treatment of bacterial infections? The design of chemical reporters is informed by an understanding of the structure and biosynthesis of the targeted glycan. Therefore, it is not surprising that the most fully developed classes of reporter probes are those that target cell envelope components whose chemical structures and associated metabolic pathways are well characterized—peptidoglycan and mycomembrane. On the other hand, the reverse scenario can also be fruitful. We discussed examples where limited information about the glycan target was available, but the chemical reporters served as discovery tools to uncover useful information. For example, GlcNAc was not a known component of H. pylori glycoproteins, but GlcNAc-based chemical reporters identified candidate glycoproteins as well as genes involved in their biosynthesis. Any chemical reporter can be designed, but the synthesis of carbohydrates and their derivatives can be extremely challenging, another point that we exemplified repeatedly herein. Therefore, it is of critical importance to establish convenient and efficient synthetic methods to access carbohydrate-based chemical reporters, which will facilitate adoption by the microbiology community. Chemoenzymatic synthesis has proven to be particularly helpful, and there is little doubt that continued advancements on this front will benefit the field. We paid particular attention in this review to the approaches taken to evaluate the behavior of chemical reporters in bacterial cells—did the probes label the intended glycan(s) via a defined metabolic incorporation route? A sufficient level of clarity on this point is needed for the tool to be judiciously applied and for experimental results to be properly interpreted. It is worth noting that in many cases (e.g., D-amino acid, KDO, and trehalose derivatives), chemical reporter characterization efforts have spanned multiple published studies conducted by multiple independent research groups. This challenging work must be rigorously conducted, so that chemical reporters can be deployed in appropriate scenarios with confidence.
The future is bright for chemical reporters in the bacterial glycobiology arena, with respect to both the development of novel tools and the innovative application of existing tools. This review opened with a discussion about the remarkably diverse and dynamic nature of bacterial glycomes. While the collection of chemical reporters for studying bacterial glycans has grown considerably in the past two decades and now covers a number of important cell envelope structures, there remains vast opportunity to expand the toolbox. We have barely scratched the surface with respect to bacterial glycoproteins, teichoic acids, capsular polysaccharides, and exopolysaccharides. Highly variable cell envelope components like LPS O-antigen are likewise fertile ground for tool development. In addition, the mycobacterial cell envelope is replete with distinctive glycans—there is ample room to extend the work on trehalose and related derivatives, as we have seen with the recently disclosed arabinan-targeting reporters. Another angle to consider is that while most published chemical reporters have been designed to probe biosynthetic processes, there is strong rationale to explicitly consider and increase the development of tools that interrogate glycan remodeling, break down, and recycling pathways, which often contribute to virulence. Existing examples of such probes include MurNAc-based reporters, which in part aim to help understand the immunological consequences of peptidoglycan break down, and FRET-TDM, which enables the study of mycomembrane glycolipid break down. We have encountered a wide variety of probe designs, and the variables should continue to be explored—one-step fluorescent vs. two-step bioorthogonal labeling, choice of fluorophore or bioorthogonal group, choice of modification site, intracellular vs. extracellular incorporation routes, monosaccharides vs. lipid-linked sugars, free vs. peracetylated sugars, and so on. In addition, as small-molecule tags with complementary abilities emerge (e.g., new bioorthogonal groups, photo-cross-linking groups, fluorogenic dyes, immunomodulators), existing chemical reporter scaffolds can be augmented or repurposed for novel applications. Above, we discussed the major types of current or emerging applications for chemical reporters—imaging glycan dynamics, cell-based functional assays, molecular profiling/proteomics, and delivery of diagnostic or therapeutic cargo. Much of the progress in these areas is nascent, so we anticipate continued maturation of ongoing projects. The exciting trend toward more in vivo experimentation, such as the B. fragilis polysaccharide dissemination studies in mice, is likely to accelerate. Indeed, while early experiments with chemical reporters were usually limited to bacteria grown in culture, there have been progressively more studies that utilize probes in macrophage and animal infection/colonization models. Such studies powerfully demonstrate that chemical reporters are capable of probing bacterial glycans in highly complex environments. Highly complex in vivo settings bring forth new challenges for tool developers, such as probe administration, access to target bacteria, and labeling specificity, which in turn will likely inspire creative solutions that enlist technologies such as the tetrazine ligation or PET radionuclides. Regardless, the successful in vivo studies published to date lend credence to the idea that chemical reporters—along with the insights and strategies for addressing bacterial infections they give rise to—have high potential to translate to preclinical and clinical settings. Moving forward, interdisciplinary and often collaborative efforts that span the fields of synthetic chemistry, chemical biology, microbiology, and medicine will continue to drive progress in the development and applications of chemical reporters for bacterial glycans.
Acknowledgements
This work was supported by the National Science Foundation (CAREER Award 1654408), the National Institutes of Health (R15 AI117670), and the Camille and Henry Dreyfus Foundation (Henry Dreyfus Teacher-Scholar Award TH-17-034). We thank Drs. M. Sloan Siegrist and Catherine L. Grimes for helpful discussions.
Biographies
Nicholas Banahene
Nicholas is from Twifu Praso, Ghana. He earned a BS in Chemistry from Kwame Nkrumah University of Science and Technology in 2013 and an MS in Chemistry from Central Michigan University in 2018. He is currently a PhD candidate in the Biochemistry, Cell, and Molecular Biology graduate program at Central Michigan University working with Prof. Ben Swarts. His research aims to develop chemical probes for studying cell envelope glycolipids and proteins in mycobacteria.
Herbert W. Kavunja
Herbert is from Nairobi, Kenya, where he obtained BS and MS degrees in Chemistry in 2005 and 2009 from the University of Nairobi. He received a PhD in Chemistry from Michigan State University in 2015 working under the supervision of Prof. Xuefei Huang on the application of carbohydrates to cancer biology. His postdoctoral work in the laboratory of Prof. Ben Swarts at Central Michigan University focused on the development of chemical probes to investigate mycobacterial outer membrane proteins that are modified by or interact with trehalose glycolipids. Herbert is currently Chief Scientist at IASO Therapeutics, Inc.
Benjamin M. Swarts
Ben grew up in Danville, Ohio. He completed a BA in Chemistry from the College of Wooster in 2004 and a PhD in Chemistry from Wayne State University in 2010 with Prof. Zhongwu Guo, studying glycosylphosphatidylinositol anchors. After a postdoctoral fellowship focused on the study of mycobacterial glycolipids with Prof. Carolyn Bertozzi at the University of California, Berkeley, in 2013 he joined the faculty at Central Michigan University. His research focuses on the synthesis of bacterial carbohydrates and the development of probes and inhibitors to investigate bacterial cell envelope components, with a focus on the mycobacterial outer membrane. Ben has received the International Carbohydrate Organization Young Researcher Award, the Cottrell College Science Award, the Henry Dreyfus Teacher-Scholar Award, and the NSF CAREER award.
References
- (1).Pikuta EV; Hoover RB; Tang J Microbial Extremophiles at the Limits of Life. Crit. Rev. Microbiol 2007, 33, 183–209. [DOI] [PubMed] [Google Scholar]
- (2).Pai M; Behr MA; Dowdy D; Dheda K; Divangahi M; Boehme CC; Ginsberg A; Swaminathan S; Spigelman M; Getahun H et al. Tuberculosis. Nat. Rev. Dis. Primers 2016, 2, 16076. [DOI] [PubMed] [Google Scholar]
- (3).Sommer F; Bäckhed F The Gut Microbiota — Masters of Host Development and Physiology. Nat. Rev. Microbiol 2013, 11, 227–238. [DOI] [PubMed] [Google Scholar]
- (4).Silhavy TJ; Kahne D; Walker S The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol 2010, 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Holst O; Moran AP; Brennan PJ Overview of the Glycosylated Components of the Bacterial Cell Envelope. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 1–13. [Google Scholar]
- (6).Vollmer W; Blanot D; de Pedro MA Peptidoglycan Structure and Architecture. FEMS Microbiol. Rev 2008, 32, 149–167. [DOI] [PubMed] [Google Scholar]
- (7).Reichmann NT; Gründling A Location, Synthesis and Function of Glycolipids and Polyglycerolphosphate Lipoteichoic Acid in Gram-Positive Bacteria of the Phylum Firmicutes. FEMS Microbiol. Lett 2011, 319, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Brown S; Santa Maria JP; Walker S Wall Teichoic Acids of Gram-Positive Bacteria. Annu. Rev. Microbiol 2013, 67, 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Raetz CRH; Whitfield C Lipopolysaccharide Endotoxins. Annu. Rev. Biochem 2002, 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Brennan PJ Structure, Function, and Biogenesis of the Cell Wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [DOI] [PubMed] [Google Scholar]
- (11).Angala SK; Belardinelli JM; Huc-Claustre E; Wheat WH; Jackson M The Cell Envelope Glycoconjugates of Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol 2014, 49, 361–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Whitfield C; Wear SS; Sande C Assembly of Bacterial Capsular Polysaccharides and Exopolysaccharides. Annu. Rev. Microbiol 2020, 74, 521–543. [DOI] [PubMed] [Google Scholar]
- (13).Benz I; Schmidt MA Never Say Never Again: Protein Glycosylation in Pathogenic Bacteria. Mol. Microbiol 2002, 45, 267–276. [DOI] [PubMed] [Google Scholar]
- (14).Sára M; Sleytr UB S-Layer Proteins. J. Bacteriol 2000, 182, 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Whitfield C; Szymanski CM; Aebi M Eubacteria. In Essentials of Glycobiology, Third Edition; Varki A, Cummings R, Esko J, Stanley P, Hart G, Aebi M, Darvill A, Kinoshita T, Packer N, Prestegard J et al. , Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2017. [Google Scholar]
- (16).Imperiali B Bacterial Carbohydrate Diversity — a Brave New World. Curr. Opin. Chem. Biol 2019, 53, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cescutti P Bacterial Capsular Polysaccharides and Exopolysaccharides. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 93–108. [Google Scholar]
- (18).Wang L; Wang Q; Reeves PR The Variation of O Antigens in Gram-Negative Bacteria. In Endotoxins: Structure, Function and Recognition; Wang X; Quinn PJ, Eds.; Springer Netherlands: Dordrecht, 2010, pp 123–152. [DOI] [PubMed] [Google Scholar]
- (19).Sinsimer D; Huet G; Manca C; Tsenova L; Koo M-S; Kurepina N; Kana B; Mathema B; Marras Salvatore AE; Kreiswirth Barry N et al. The Phenolic Glycolipid of Mycobacterium tuberculosis Differentially Modulates the Early Host Cytokine Response but Does Not in Itself Confer Hypervirulence. Infect. Immun 2008, 76, 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dik DA; Fisher JF; Mobashery S Cell-Wall Recycling of the Gram-Negative Bacteria and the Nexus to Antibiotic Resistance. Chem. Rev 2018, 118, 5952–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rehm BHA Bacterial Polymers: Biosynthesis, Modifications and Applications. Nat. Rev. Microbiol 2010, 8, 578–592. [DOI] [PubMed] [Google Scholar]
- (22).Feldman MF Industrial Exploitation by Genetic Engineering of Bacterial Glycosylation Systems. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 903–914. [Google Scholar]
- (23).Ruas-Madiedo P; Salazar N; de los Reyes-Gavilán CG Exopolysaccharides Produced by Lactic Acid Bacteria in Food and Probiotic Applications. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 885–902. [Google Scholar]
- (24).Koropatkin NM; Cameron EA; Martens EC How Glycan Metabolism Shapes the Human Gut Microbiota. Nat. Rev. Microbiol 2012, 10, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Theuretzbacher U; Outterson K; Engel A; Karlén A The Global Preclinical Antibacterial Pipeline. Nat. Rev. Microbiol 2020, 18, 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lee AS; de Lencastre H; Garau J; Kluytmans J; Malhotra-Kumar S; Peschel A; Harbarth S Methicillin-Resistant Staphylococcus Aureus. Nat. Rev. Dis. Primers 2018, 4, 18033. [DOI] [PubMed] [Google Scholar]
- (27).Gautam A; Vyas R; Tewari R Peptidoglycan Biosynthesis Machinery: A Rich Source of Drug Targets. Crit. Rev. Biotechnol 2011, 31, 295–336. [DOI] [PubMed] [Google Scholar]
- (28).North EJ; Jackson M; Lee RE New Approaches to Target the Mycolic Acid Biosynthesis Pathway for the Development of Tuberculosis Therapeutics. Curr. Pharm. Des 2014, 20, 4357–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Astronomo RD; Burton DR Carbohydrate Vaccines: Developing Sweet Solutions to Sticky Situations? Nat. Rev. Drug. Discov 2010, 9, 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jennings HJ; Pon RA Bacterial Polysaccharide Vaccines: Glycoconjugates and Peptide-Mimetics. In Microbial Glycobiology; Holst O; Brennan PJ; Itzstein M. v.; Moran AP, Eds.; Academic Press: San Diego, 2010, pp 933–956. [Google Scholar]
- (31).Campanero-Rhodes MA; Palma AS; Menéndez M; Solís D Microarray Strategies for Exploring Bacterial Surface Glycans and Their Interactions with Glycan-Binding Proteins. Front. Microbiol 2020, 10, 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Brown AR; Gordon RA; Hyland SN; Siegrist MS; Grimes CL Chemical Biology Tools for Examining the Bacterial Cell Wall. Cell Chem. Biol 2020, 27, 1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Luong P; Dube DH Dismantling the Bacterial Glycocalyx: Chemical Tools to Probe, Perturb, and Image Bacterial Glycans. Bioorg. Med. Chem 2021, 42, 116268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tiyanont K; Doan T; Lazarus MB; Fang X; Rudner DZ; Walker S Imaging Peptidoglycan Biosynthesis in Bacillus Subtilis with Fluorescent Antibiotics. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 11033–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Edit 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Scinto SL; Bilodeau DA; Hincapie R; Lee W; Nguyen SS; Xu M; am Ende CW; Finn MG; Lang K; Lin Q et al. Bioorthogonal Chemistry. Nat. Rev. Methods Primers 2021, 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Grammel M; Hang HC Chemical Reporters for Biological Discovery. Nat. Chem. Biol 2013, 9, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zol-Hanlon MI; Schumann B Open Questions in Chemical Glycobiology. Commun. Chem 2020, 3, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pedowitz NJ; Pratt MR Design and Synthesis of Metabolic Chemical Reporters for the Visualization and Identification of Glycoproteins. RSC Chem. Biol 2021, 2, 306–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Boyce M; Bertozzi CR Bringing Chemistry to Life. Nat. Methods 2011, 8, 638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Patterson DM; Nazarova LA; Prescher JA Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- (42).Cornish VW; Hahn KM; Schultz PG Site-Specific Protein Modification Using a Ketone Handle. J. Am. Chem. Soc 1996, 118, 8150–8151. [Google Scholar]
- (43).Mahal LK; Yarema KJ; Bertozzi CR Engineering Chemical Reactivity on Cell Surfaces through Oligosaccharide Biosynthesis. Science 1997, 276, 1125–1128. [DOI] [PubMed] [Google Scholar]
- (44).Saxon E; Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- (45).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Edit 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (46).Tornøe CW; Christensen C; Meldal M Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- (47).Agard NJ; Prescher JA; Bertozzi CR A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- (48).Soriano Del Amo D; Wang W; Jiang H; Besanceney C; Yan AC; Levy M; Liu Y; Marlow FL; Wu P Biocompatible Copper(I) Catalysts for in Vivo Imaging of Glycans. J. Am. Chem. Soc 2010, 132, 16893–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Uttamapinant C; Tangpeerachaikul A; Grecian S; Clarke S; Singh U; Slade P; Gee KR; Ting AY Fast, Cell-Compatible Click Chemistry with Copper-Chelating Azides for Biomolecular Labeling. Angew. Chem. Int. Edit 2012, 51, 5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yang J; Šečkutė J; Cole CM; Devaraj NK Live-Cell Imaging of Cyclopropene Tags with Fluorogenic Tetrazine Cycloadditions. Angew. Chem. Int. Edit 2012, 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Siegrist MS; Swarts BM; Fox DM; Lim SA; Bertozzi CR Illumination of Growth, Division and Secretion by Metabolic Labeling of the Bacterial Cell Surface. FEMS Microbiol. Rev 2015, 39, 184–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Dube DH; Champasa K; Wang B Chemical Tools to Discover and Target Bacterial Glycoproteins. Chem. Comm 2011, 47, 87–101. [DOI] [PubMed] [Google Scholar]
- (54).Gautam S; Gniadek TJ; Kim T; Spiegel DA Exterior Design: Strategies for Redecorating the Bacterial Surface with Small Molecules. Trends. Biotechnol 2013, 31, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Tra VN; Dube DH Glycans in Pathogenic Bacteria - Potential for Targeted Covalent Therapeutics and Imaging Agents. Chem. Commun 2014, 50, 4659–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Zhang ZJ; Wang Y-C; Yang X; Hang HC Chemical Reporters for Exploring Microbiology and Microbiota Mechanisms. ChemBioChem 2020, 21, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ignacio BJ; Bakkum T; Bonger KM; Martin NI; van Kasteren SI Metabolic Labeling Probes for Interrogation of the Host–Pathogen Interaction. Org. Biomol. Chem 2021, 19, 2856–2870. [DOI] [PubMed] [Google Scholar]
- (58).Kocaoglu O; Carlson EE Progress and Prospects for Small-Molecule Probes of Bacterial Imaging. Nat. Chem. Biol 2016, 12, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Parker MFL; Flavell RR; Luu JM; Rosenberg OS; Ohliger MA; Wilson DM Small Molecule Sensors Targeting the Bacterial Cell Wall. ACS Infect. Dis 2020, 6, 1587–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Vollmer W; Seligman SJ Architecture of Peptidoglycan: More Data and More Models. Trends Microbiol 2010, 18, 59–66. [DOI] [PubMed] [Google Scholar]
- (61).Shaku M; Ealand C; Matlhabe O; Lala R; Kana BD Peptidoglycan Biosynthesis and Remodeling Revisited. In Advances in Applied Microbiology; Gadd GM; Sariaslani S, Eds.; Academic Press, 2020; Vol. 112, pp 67–103. [DOI] [PubMed] [Google Scholar]
- (62).Nikolaidis I; Favini-Stabile S; Dessen A Resistance to Antibiotics Targeted to the Bacterial Cell Wall. Protein Sci 2014, 23, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Taguchi A; Kahne D; Walker S Chemical Tools to Characterize Peptidoglycan Synthases. Curr. Opin. Chem. Biol 2019, 53, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Hsu YP; Booher G; Egan A; Vollmer W; VanNieuwenhze MS D-Amino Acid Derivatives as in Situ Probes for Visualizing Bacterial Peptidoglycan Biosynthesis. Acc. Chem. Res 2019, 52, 2713–2722. [DOI] [PubMed] [Google Scholar]
- (65).Beatty KE Fluorescent Probes for Investigating Peptidoglycan Biosynthesis in Mycobacteria. Curr. Opin. Chem. Biol 2020, 57, 50–57. [DOI] [PubMed] [Google Scholar]
- (66).van Dam V; Olrichs N; Breukink E Specific Labeling of Peptidoglycan Precursors as a Tool for Bacterial Cell Wall Studies. ChemBioChem 2009, 10, 617–624. [DOI] [PubMed] [Google Scholar]
- (67).Barreteau H; Kovač A; Boniface A; Sova M; Gobec S; Blanot D Cytoplasmic Steps of Peptidoglycan Biosynthesis. Fems Microbiol. Rev 2008, 32, 168–207. [DOI] [PubMed] [Google Scholar]
- (68).El Zoeiby A; Sanschagrin F; Levesque RC Structure and Function of the Mur Enzymes: Development of Novel Inhibitors. Mol. Microbiol 2003, 47, 1–12. [DOI] [PubMed] [Google Scholar]
- (69).Sham L-T; Butler EK; Lebar MD; Kahne D; Bernhardt TG; Ruiz N Murj Is the Flippase of Lipid-Linked Precursors for Peptidoglycan Biogenesis. Science 2014, 345, 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sauvage E; Kerff F; Terrak M; Ayala JA; Charlier P The Penicillin-Binding Proteins: Structure and Role in Peptidoglycan Biosynthesis. Fems Microbiol. Rev 2008, 32, 234–258. [DOI] [PubMed] [Google Scholar]
- (71).Cho H; Wivagg CN; Kapoor M; Barry Z; Rohs PDA; Suh H; Marto JA; Garner EC; Bernhardt TG Bacterial Cell Wall Biogenesis Is Mediated by SEDS and PBP Polymerase Families Functioning Semi-Autonomously. Nat. Microbiol 2016, 1, 16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Meeske AJ; Riley EP; Robins WP; Uehara T; Mekalanos JJ; Kahne D; Walker S; Kruse AC; Bernhardt TG; Rudner DZ SEDS Proteins Are a Widespread Family of Bacterial Cell Wall Polymerases. Nature 2016, 537, 634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Magnet S; Dubost L; Marie A; Arthur M; Gutmann L Identification of the L,D-Transpeptidases for Peptidoglycan Cross-Linking in Escherichia coli. J. Bacteriol 2008, 190, 4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Lavollay M; Arthur M; Fourgeaud M; Dubost L; Marie A; Veziris N; Blanot D; Gutmann L; Mainardi JL The Peptidoglycan of Stationary-Phase Mycobacterium tuberculosis Predominantly Contains Cross-Links Generated by L,D-Transpeptidation. J. Bacteriol 2008, 190, 4360–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Sadamoto R; Niikura K; Sears PS; Liu HT; Wong CH; Suksomcheep A; Tomita F; Monde K; Nishimura SI Cell-Wall Engineering of Living Bacteria. J. Am. Chem. Soc 2002, 124, 9018–9019. [DOI] [PubMed] [Google Scholar]
- (76).Kuru E; Hughes HV; Brown PJ; Hall E; Tekkam S; Cava F; de Pedro MA; Brun YV; VanNieuwenhze MS In Situ Probing of Newly Synthesized Peptidoglycan in Live Bacteria with Fluorescent D-Amino Acids. Angew. Chem. Int. Edit 2012, 51, 12519–12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Siegrist MS; Whiteside S; Jewett JC; Aditham A; Cava F; Bertozzi CR (D)-Amino Acid Chemical Reporters Reveal Peptidoglycan Dynamics of an Intracellular Pathogen. ACS Chem. Biol 2013, 8, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Shieh P; Siegrist MS; Cullen AJ; Bertozzi CR Imaging Bacterial Peptidoglycan with near-Infrared Fluorogenic Azide Probes. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 5456–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Hsu YP; Hall E; Booher G; Murphy B; Radkov AD; Yablonowski J; Mulcahey C; Alvarez L; Cava F; Brun YV et al. Fluorogenic D-Amino Acids Enable Real-Time Monitoring of Peptidoglycan Biosynthesis and High-Throughput Transpeptidation Assays. Nat. Chem 2019, 11, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Pidgeon SE; Pires MM Cell Wall Remodeling of Staphylococcus aureus in Live Caenorhabditis elegans. Bioconjug. Chem 2017, 28, 2310–2315. [DOI] [PubMed] [Google Scholar]
- (81).Pidgeon SE; Fura JM; Leon W; Birabaharan M; Vezenov D; Pires MM Metabolic Profiling of Bacteria by Unnatural C-Terminated D-Amino Acids. Angew. Chem. Int. Edit 2015, 54, 6158–6162. [DOI] [PubMed] [Google Scholar]
- (82).Lebar MD; May JM; Meeske AJ; Leiman SA; Lupoli TJ; Tsukamoto H; Losick R; Rudner DZ; Walker S; Kahne D Reconstitution of Peptidoglycan Cross-Linking Leads to Improved Fluorescent Probes of Cell Wall Synthesis. J. Am. Chem. Soc 2014, 136, 10874–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Liechti GW; Kuru E; Hall E; Kalinda A; Brun YV; VanNieuwenhze M; Maurelli AT A New Metabolic Cell-Wall Labelling Method Reveals Peptidoglycan in Chlamydia trachomatis. Nature 2014, 506, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Sarkar S; Libby EA; Pidgeon SE; Dworkin J; Pires MM In Vivo Probe of Lipid II-Interacting Proteins. Angew. Chem. Int. Edit 2016, 55, 8401–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Olrichs NK; Aarsman MEG; Verheul J; Arnusch CJ; Martin NI; Hervé M; Vollmer W; de Kruijff B; Breukink E; den Blaauwen T A Novel in Vivo Cell-Wall Labeling Approach Sheds New Light on Peptidoglycan Synthesis in Escherichia Coli. ChemBioChem 2011, 12, 1124–1133. [DOI] [PubMed] [Google Scholar]
- (86).Gautam S; Kim T; Shoda T; Sen S; Deep D; Luthra R; Ferreira MT; Pinho MG; Spiegel DA An Activity-Based Probe for Studying Crosslinking in Live Bacteria. Angew. Chem. Int. Edit 2015, 54, 10492–10496. [DOI] [PubMed] [Google Scholar]
- (87).Pidgeon SE; Apostolos AJ; Nelson JM; Shaku M; Rimal B; Islam MN; Crick DC; Kim SJ; Pavelka MS; Kana BD et al. L,D-Transpeptidase Specific Probe Reveals Spatial Activity of Peptidoglycan Cross-Linking. ACS Chem. Biol 2019, 14, 2185–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Sadamoto R; Matsubayashi T; Shimizu M; Ueda T; Koshida S; Koda T; Nishimura S Bacterial Surface Engineering Utilizing Glucosamine Phosphate Derivatives as Cell Wall Precursor Surrogates. Chem. Eur. J 2008, 14, 10192–10195. [DOI] [PubMed] [Google Scholar]
- (89).Liang H; DeMeester KE; Hou CW; Parent MA; Caplan JL; Grimes CL Metabolic Labelling of the Carbohydrate Core in Bacterial Peptidoglycan and Its Applications. Nat. Commun 2017, 8, 15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Liu H; Sadamoto R; Sears PS; Wong C-H An Efficient Chemoenzymatic Strategy for the Synthesis of Wild-Type and Vancomycin-Resistant Bacterial Cell-Wall Precursors: UDP-N-Acetylmuramyl-Peptides. J. Am. Chem. Soc 2001, 123, 9916–9917. [DOI] [PubMed] [Google Scholar]
- (91).Huang L-Y; Huang S-H; Chang Y-C; Cheng W-C; Cheng T-JR; Wong C-H Enzymatic Synthesis of Lipid II and Analogues. Angew. Chem. Int. Edit 2014, 53, 8060–8065. [DOI] [PubMed] [Google Scholar]
- (92).Qiao Y; Srisuknimit V; Rubino F; Schaefer K; Ruiz N; Walker S; Kahne D Lipid II Overproduction Allows Direct Assay of Transpeptidase Inhibition by B-Lactams. Nat. Chem. Biol 2017, 13, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Qiao Y; Lebar MD; Schirner K; Schaefer K; Tsukamoto H; Kahne D; Walker S Detection of Lipid-Linked Peptidoglycan Precursors by Exploiting an Unexpected Transpeptidase Reaction. J. Am. Chem. Soc 2014, 136, 14678–14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Amro NA; Kotra LP; Wadu-Mesthrige K; Bulychev A; Mobashery S; Liu G.-y. High-Resolution Atomic Force Microscopy Studies of the Escherichia coli Outer Membrane: Structural Basis for Permeability. Langmuir 2000, 16, 2789–2796. [Google Scholar]
- (95).Ye X-Y; Lo M-C; Brunner L; Walker D; Kahne D; Walker S Better Substrates for Bacterial Transglycosylases. J. Am. Chem. Soc 2001, 123, 3155–3156. [DOI] [PubMed] [Google Scholar]
- (96).Calabretta PJ; Hodges HL; Kraft MB; Marando VM; Kiessling LL Bacterial Cell Wall Modification with a Glycolipid Substrate. J. Am. Chem. Soc 2019, 141, 9262–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Sadamoto R; Niikura K; Ueda T; Monde K; Fukuhara N; Nishimura SI Control of Bacteria Adhesion by Cell-Wall Engineering. J. Am. Chem. Soc 2004, 126, 3755–3761. [DOI] [PubMed] [Google Scholar]
- (98).Prachayasittikul V; Isarankura-Na-Ayudhya C; Tantimongcolwat T; Nantasenamat C; Galla H-J EDTA-Induced Membrane Fluidization and Destabilization: Biophysical Studies on Artificial Lipid Membranes. Acta Biochim. Biophys. Sin. (Shanghai) 2007, 39, 901–913. [DOI] [PubMed] [Google Scholar]
- (99).Lark C; Bradley D; Lark KG Further Studies on the Incorporation of D-Methionine into the Bacterial Cell Wall: Its Incorporation into the R-Layer and the Structural Consequences. Biochem. Biophys. Acta 1963, 78, 278–288. [DOI] [PubMed] [Google Scholar]
- (100).Tsuruoka T; Tamura A; Miyata A; Takei T; Iwamatsu K; Inouye S; Matsuhashi M Penicillin-Insensitive Incorporation of D-Amino Acids into Cell Wall Peptidoglycan Influences the Amount of Bound Lipoprotein in Escherichia coli. J. Bacteriol 1984, 160, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Caparrós M; Arán V; de Pedro MA Incorporation of S-[3H]Methyl-D-Cysteine into the Peptidoglycan of Ether-Treated Cells of Escherichia coli. FEMS Microbiol. Lett 1992, 93, 139–146. [DOI] [PubMed] [Google Scholar]
- (102).Caparrós M; Pisabarro AG; de Pedro MA Effect of D-Amino Acids on Structure and Synthesis of Peptidoglycan in Escherichia coli. J. Bacteriol 1992, 174, 5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).de Pedro MA; Quintela JC; Höltje JV; Schwarz H Murein Segregation in Escherichia coli. J. Bacteriol 1997, 179, 2823–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Cava F; de Pedro MA; Lam H; Davis BM; Waldor MK Distinct Pathways for Modification of the Bacterial Cell Wall by Non-Canonical D-Amino Acids. EMBO J 2011, 30, 3442–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Lam H; Oh D-C; Cava F; Takacs CN; Clardy J; de Pedro MA; Waldor MK D-Amino Acids Govern Stationary Phase Cell Wall Remodeling in Bacteria. Science 2009, 325, 1552–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Lupoli TJ; Tsukamoto H; Doud EH; Wang T-SA; Walker S; Kahne D Transpeptidase-Mediated Incorporation of D-Amino Acids into Bacterial Peptidoglycan. J. Am. Chem. Soc 2011, 133, 10748–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Kuru E; Tekkam S; Hall E; Brun YV; Van Nieuwenhze MS Synthesis of Fluorescent D-Amino Acids and Their Use for Probing Peptidoglycan Synthesis and Bacterial Growth in Situ. Nat. Protoc 2015, 10, 33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Baskin JM; Prescher JA; Laughlin ST; Agard NJ; Chang PV; Miller IA; Lo A; Codelli JA; Bertozzi CR Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hsu Y-P; Rittichier J; Kuru E; Yablonowski J; Pasciak E; Tekkam S; Hall E; Murphy B; Lee TK; Garner EC et al. Full Color Palette of Fluorescent D-Amino Acids for in Situ Labeling of Bacterial Cell Walls. Chem. Sci 2017, 8, 6313–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).García-Heredia A; Pohane AA; Melzer ES; Carr CR; Fiolek TJ; Rundell SR; Chuin Lim H; Wagner JC; Morita YS; Swarts BM et al. Peptidoglycan Precursor Synthesis Along the Sidewall of Pole-Growing Mycobacteria. eLife 2018, 7, e37243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Kuru E; Radkov A; Meng X; Egan A; Alvarez L; Dowson A; Booher G; Breukink E; Roper DI; Cava F et al. Mechanisms of Incorporation for D-Amino Acid Probes That Target Peptidoglycan Biosynthesis. ACS Chem. Biol 2019, 14, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Pidgeon SE; Pires MM Metabolic Remodeling of Bacterial Surfaces Via Tetrazine Ligations. Chem. Commun 2015, 51, 10330–10333. [DOI] [PubMed] [Google Scholar]
- (113).Bandyopadhyay A; Cambray S; Gao J Fast Diazaborine Formation of Semicarbazide Enables Facile Labeling of Bacterial Pathogens. J. Am. Chem. Soc 2017, 139, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Cambray S; Bandyopadhyay A; Gao J Fluorogenic Diazaborine Formation of Semicarbazide with Designed Coumarin Derivatives. Chem. Commun 2017, 53, 12532–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Margison KD; Bilodeau DA; Mahmoudi F; Pezacki JP Cycloadditions of Trans-Cyclooctenes and Nitrones as Tools for Bioorthogonal Labelling. ChemBioChem 2020, 21, 948–951. [DOI] [PubMed] [Google Scholar]
- (116).Bisson-Filho AW; Hsu Y-P; Squyres GR; Kuru E; Wu F; Jukes C; Sun Y; Dekker C; Holden S; VanNieuwenhze MS et al. Treadmilling by Ftsz Filaments Drives Peptidoglycan Synthesis and Bacterial Cell Division. Science 2017, 355, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Melzer ES; Kado T; García-Heredia A; Gupta KR; Meniche X; Morita YS; Sassetti CM; Rego EH; Siegrist MS Cell Wall Damage Reveals Spatial Flexibility in Peptidoglycan Synthesis and a Non-Redundant Role for RodA in Mycobacteria. bioRxiv 2021, DOI: 10.1101/2021.10.26.465981. Accessed 2021-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Fura JM; Pires MM D-Amino Carboxamide-Based Recruitment of Dinitrophenol Antibodies to Bacterial Surfaces Via Peptidoglycan Remodeling. J. Pept. Sci 2015, 104, 351–359. [DOI] [PubMed] [Google Scholar]
- (119).Fura JM; Sabulski MJ; Pires MM D-Amino Acid Mediated Recruitment of Endogenous Antibodies to Bacterial Surfaces. ACS Chem. Biol 2014, 9, 1480–1489. [DOI] [PubMed] [Google Scholar]
- (120).McEnaney PJ; Parker CG; Zhang AX; Spiegel DA Antibody-Recruiting Molecules: An Emerging Paradigm for Engaging Immune Function in Treating Human Disease. ACS Chem. Biol 2012, 7, 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Hu F; Qi G; Kenry; Mao D; Zhou S; Wu M; Wu W; Liu B Visualization and in Situ Ablation of Intracellular Bacterial Pathogens through Metabolic Labeling. Angew. Chem. Int. Edit 2020, 59, 9288–9292. [DOI] [PubMed] [Google Scholar]
- (122).Wu M; Qi G; Liu X; Duan Y; Liu J; Liu B Bio-Orthogonal Aiegen for Specific Discrimination and Elimination of Bacterial Pathogens via Metabolic Engineering. Chem. Mater 2020, 32, 858–865. [Google Scholar]
- (123).Mota F; Jain SK Flagging Bacteria with Radiolabeled D-Amino Acids. ACS Cent. Sci 2020, 6, 97–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Parker MFL; Luu JM; Schulte B; Huynh TL; Stewart MN; Sriram R; Yu MA; Jivan S; Turnbaugh PJ; Flavell RR et al. Sensing Living Bacteria in Vivo Using D-Alanine-Derived 11C Radiotracers. ACS Cent. Sci 2020, 6, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Wang L; Zha Z; Qu W; Qiao H; Lieberman BP; Plössl K; Kung HF Synthesis and Evaluation of 18F Labeled Alanine Derivatives as Potential Tumor Imaging Agents. Nucl. Med. Biol 2012, 39, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Dik DA; Zhang N; Chen JS; Webb B; Schultz PG Semisynthesis of a Bacterium with Non-Canonical Cell-Wall Cross-Links. J. Am. Chem. Soc 2020, 142, 10910–10913. [DOI] [PubMed] [Google Scholar]
- (127).Rivera SL; Espaillat A; Aditham AK; Shieh P; Muriel-Mundo C; Kim J; Cava F; Siegrist MS Chemically Induced Cell Wall Stapling in Bacteria. Cell Chem. Biol 2020, 28, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Edit 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- (129).Fura JM; Kearns D; Pires MM D-Amino Acid Probes for Penicillin Binding Protein-Based Bacterial Surface Labeling. J. Biol. Chem 2015, 290, 30540–30550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Ghuysen J-M; Goffin C Lack of Cell Wall Peptidoglycan Versus Penicillin Sensitivity: New Insights into the Chlamydial Anomaly. Antimicrob. Agents Chemother 1999, 43, 2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Moulder JW; Novosel DL; Officer JE Inhibition of the Growth of Agents of the Psittacosis Group by D-Cycloserine and Its Specific Reversal by D-Alanine. J. Bacteriol 1963, 85, 707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).McCoy AJ; Maurelli AT Characterization of Chlamydia MurC-Ddl, a Fusion Protein Exhibiting D-Alanyl-D-Alanine Ligase Activity Involved in Peptidoglycan Synthesis and D-Cycloserine Sensitivity. Mol. Microbiol 2005, 57, 41–52. [DOI] [PubMed] [Google Scholar]
- (133).Fura JM; Pidgeon SE; Birabaharan M; Pires MM Dipeptide-Based Metabolic Labeling of Bacterial Cells for Endogenous Antibody Recruitment. ACS Infect. Dis 2016, 2, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Mengin-Lecreulx D; van Heijenoort J; Park JT Identification of the Mpl Gene Encoding UDP-N-Acetylmuramate: L-Alanyl-Gamma-D-Glutamyl-Meso-Diaminopimelate Ligase in Escherichia coli and Its Role in Recycling of Cell Wall Peptidoglycan. J. Bacteriol 1996, 178, 5347–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Park JT Turnover and Recycling of the Murein Sacculus in Oligopeptide Permease-Negative Strains of Escherichia coli: Indirect Evidence for an Alternative Permease System and for a Monolayered Sacculus. J. Bacteriol 1993, 175, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Hervé M; Boniface A; Gobec S; Blanot D; Mengin-Lecreulx D Biochemical Characterization and Physiological Properties of Escherichia coli UDP-N-Acetylmuramate: L-Alanyl-γ-D-Glutamyl-meso-Diaminopimelate Ligase. J. Bacteriol 2007, 189, 3987–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Baum EZ; Crespo-Carbone SM; Foleno BD; Simon LD; Guillemont J; Macielag M; Bush K MurF Inhibitors with Antibacterial Activity: Effect on Muropeptide Levels. Antimicrob. Agents Chemother 2009, 53, 3240–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Schmidt DMZ; Hubbard BK; Gerlt JA Evolution of Enzymatic Activities in the Enolase Superfamily: Functional Assignment of Unknown Proteins in Bacillus subtilis and Escherichia coli as L-Ala-D/L-Glu Epimerases. Biochemistry 2001, 40, 15707–15715. [DOI] [PubMed] [Google Scholar]
- (139).Uehara T; Park JT Identification of MpaA, an Amidase in Escherichia coli That Hydrolyzes the γ-D-Glutamyl-meso-Diaminopimelate Bond in Murein Peptides. J. Bacteriol 2003, 185, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Nelson JW; Chamessian AG; McEnaney PJ; Murelli RP; Kazmiercak BI; Spiegel DA A Biosynthetic Strategy for Re-Engineering the Staphylococcus Aureus Cell Wall with Non-Native Small Molecules. ACS Chem. Biol 2010, 5, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Sauvage E; Duez C; Herman R; Kerff F; Petrella S; Anderson JW; Adediran SA; Pratt RF; Frère J-M; Charlier P Crystal Structure of the Bacillus subtilis Penicillin-Binding Protein 4a, and Its Complex with a Peptidoglycan Mimetic Peptide. J. Mol. Biol 2007, 371, 528–539. [DOI] [PubMed] [Google Scholar]
- (142).Gautam S; Kim T; Spiegel DA Chemical Probes Reveal an Extraseptal Mode of Cross-Linking in Staphylococcus Aureus. J. Am. Chem. Soc 2015, 137, 7441–7447. [DOI] [PubMed] [Google Scholar]
- (143).Aliashkevich A; Cava F LD-Transpeptidases: The Great Unknown among the Peptidoglycan Cross-Linkers. FEBS J 2021, DOI: 10.1111/febs.16066. [DOI] [PubMed] [Google Scholar]
- (144).Apostolos AJ; Pidgeon SE; Pires MM Remodeling of Cross-Bridges Controls Peptidoglycan Cross-Linking Levels in Bacterial Cell Walls. ACS Chem. Biol 2020, 15, 1261–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Irazoki O; Hernandez SB; Cava F Peptidoglycan Muropeptides: Release, Perception, and Functions as Signaling Molecules. Front. Microbiol 2019, 10, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Rodríguez-Díaz J; Rubio-del-Campo A; Yebra MJ Regulatory Insights into the Production of UDP-N-Acetylglucosamine by Lactobacillus casei. Bioengineered 2012, 3, 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Qin W; Qin K; Fan X; Peng L; Hong W; Zhu Y; Lv P; Du Y; Huang R; Han M et al. Artificial Cysteine S-Glycosylation Induced by Per-O-Acetylated Unnatural Monosaccharides During Metabolic Glycan Labeling. Angew. Chem. Int. Edit 2018, 57, 1817–1820. [DOI] [PubMed] [Google Scholar]
- (148).Darabedian N; Yang B; Ding R; Cutolo G; Zaro BW; Woo CM; Pratt MR O-Acetylated Chemical Reporters of Glycosylation Can Display Metabolism-Dependent Background Labeling of Proteins but Are Generally Reliable Tools for the Identification of Glycoproteins. Front. Chem 2020, 8, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Qin K; Zhang H; Zhao Z; Chen X Protein S-Glyco-Modification through an Elimination–Addition Mechanism. J. Am. Chem. Soc 2020, 142, 9382–9388. [DOI] [PubMed] [Google Scholar]
- (150).Raymond JB; Mahapatra S; Crick DC; Pavelka MS Identification of the namH Gene, Encoding the Hydroxylase Responsible for the N-Glycolylation of the Mycobacterial Peptidoglycan. J. Biol. Chem 2005, 280, 326–333. [DOI] [PubMed] [Google Scholar]
- (151).Gisin J; Schneider A; Nägele B; Borisova M; Mayer C A Cell Wall Recycling Shortcut That Bypasses Peptidoglycan De Novo Biosynthesis. Nat. Chem. Biol 2013, 9, 491–493. [DOI] [PubMed] [Google Scholar]
- (152).Brown AR; Wodzanowski KA; Santiago CC; Hyland SN; Follmar JL; Asare-Okai P; Grimes CL Protected N-Acetyl Muramic Acid Probes Improve Bacterial Peptidoglycan Incorporation Via Metabolic Labeling. ACS Chem. Biol 2021, 16, 1908–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Antonczak AK; Simova Z; Tippmann EM A Critical Examination of Escherichia coli Esterase Activity. J. Biol. Chem 2009, 284, 28795–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Taylor JA; Bratton BP; Sichel SR; Blair KM; Jacobs HM; DeMeester KE; Kuru E; Gray J; Biboy J; VanNieuwenhze MS et al. Distinct Cytoskeletal Proteins Define Zones of Enhanced Cell Wall Synthesis in Helicobacter pylori. eLife 2020, 9, e52482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Taylor JA; Santiago CC; Gray J; Wodzanowski KA; DeMeester KE; Biboy J; Vollmer W; Grimes CL; Salama NR Localizing Peptidoglycan Synthesis in Helicobacter pylori Using Clickable Metabolic Probes. Curr. Protoc 2021, 1, e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).DeMeester KE; Liang H; Jensen MR; Jones ZS; D’Ambrosio EA; Scinto SL; Zhou J; Grimes CL Synthesis of Functionalized N-Acetyl Muramic Acids to Probe Bacterial Cell Wall Recycling and Biosynthesis. J. Am. Chem. Soc 2018, 140, 9458–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Nikaido H Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev 2003, 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Bertani B; Ruiz N Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018, 8, DOI: 10.1128/ecosalplus.ESP-0001-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Matsuura M Structural Modifications of Bacterial Lipopolysaccharide That Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol 2013, 4, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Simpson BW; Trent MS Pushing the Envelope: LPS Modifications and Their Consequences. Nat. Rev. Microbiol 2019, 17, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Zhang G; Meredith TC; Kahne D On the Essentiality of Lipopolysaccharide to Gram-Negative Bacteria. Curr. Opin. Microbiol 2013, 16, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Yethon JA; Whitfield C Lipopolysaccharide as a Target for the Development of Novel Therapeutics in Gram-Negative Bacteria. Curr. Drug. Targets Infect. Disord 2001, 1, 91–106. [DOI] [PubMed] [Google Scholar]
- (163).Zhang G; Baidin V; Pahil KS; Moison E; Tomasek D; Ramadoss NS; Chatterjee AK; McNamara CW; Young TS; Schultz PG et al. Cell-Based Screen for Discovering Lipopolysaccharide Biogenesis Inhibitors. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 6834–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Moison E; Xie R; Zhang G; Lebar MD; Meredith TC; Kahne D A Fluorescent Probe Distinguishes between Inhibition of Early and Late Steps of Lipopolysaccharide Biogenesis in Whole Cells. ACS Chem. Biol 2017, 12, 928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Dumont A; Malleron A; Awwad M; Dukan S; Vauzeilles B Click-Mediated Labeling of Bacterial Membranes through Metabolic Modification of the Lipopolysaccharide Inner Core. Angew. Chem. Int. Edit 2012, 51, 3143–3146. [DOI] [PubMed] [Google Scholar]
- (166).Nilsson I; Grove K; Dovala D; Uehara T; Lapointe G; Six DA Molecular Characterization and Verification of Azido-3,8-Dideoxy-D-Manno-Oct-2-Ulosonic Acid Incorporation into Bacterial Lipopolysaccharide. J. Biol. Chem 2017, 292, 19840–19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Yi W; Liu X; Li Y; Li J; Xia C; Zhou G; Zhang W; Zhao W; Chen X; Wang PG Remodeling Bacterial Polysaccharides by Metabolic Pathway Engineering. P. Natl. Acad. Sci. USA 2009, 106, 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Pons JM; Dumont A; Sautejeau G; Fugier E; Baron A; Dukan S; Vauzeilles B Identification of Living Legionella pneumophila Using Species-Specific Metabolic Lipopolysaccharide Labeling. Angew. Chem. Int. Edit 2014, 53, 1275–1278. [DOI] [PubMed] [Google Scholar]
- (169).Andolina G; Wei R; Liu H; Zhang Q; Yang X; Cao H; Chen S; Yan A; Li XD; Li X Metabolic Labeling of Pseudaminic Acid-Containing Glycans on Bacterial Surfaces. ACS Chem. Biol 2018, 13, 3030–3037. [DOI] [PubMed] [Google Scholar]
- (170).Heise T; Langereis JD; Rossing E; de Jonge MI; Adema GJ; Büll C; Boltje TJ Selective Inhibition of Sialic Acid-Based Molecular Mimicry in Haemophilus influenzae Abrogates Serum Resistance. Cell Chem. Biol 2018, 25, 1279–1285.e1278. [DOI] [PubMed] [Google Scholar]
- (171).Capobianco JO; Darveau RP; Goldman RC; Lartey PA; Pernet AG Inhibition of Exogenous 3-Deoxy-D-Manno-Octulosonate Incorporation into Lipid A Precursor of Toluene-Treated Salmonella Typhimurium Cells. J. Bacteriol 1987, 169, 4030–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Wen L; Zheng Y; Li T; Wang PG Enzymatic Synthesis of 3-Deoxy-D-Manno-Octulosonic Acid (KDO) and Its Application for Lps Assembly. Bioorg. Med. Chem. Lett 2016, 26, 2825–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Wang W; Zhu Y; Chen X Selective Imaging of Gram-Negative and Gram-Positive Microbiotas in the Mouse Gut. Biochemistry 2017, 56, 3889–3893. [DOI] [PubMed] [Google Scholar]
- (174).Nilsson I; Prathapam R; Grove K; Lapointe G; Six DA The Sialic Acid Transporter NanT Is Necessary and Sufficient for Uptake of 3-Deoxy-D-Manno-Oct-2-Ulosonic Acid (KDO) and Its Azido Analog in Escherichia coli. Mol. Microbiol 2018, 110, 204–218. [DOI] [PubMed] [Google Scholar]
- (175).Fugier E; Dumont A; Malleron A; Poquet E; Mas Pons J; Baron A; Vauzeilles B; Dukan S Rapid and Specific Enrichment of Culturable Gram Negative Bacteria Using Non-Lethal Copper-Free Click Chemistry Coupled with Magnetic Beads Separation. PLoS ONE 2015, 10, e0127700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Sherratt AR; Rouleau Y; Luebbert C; Strmiskova M; Veres T; Bidawid S; Corneau N; Pezacki JP Rapid Screening and Identification of Living Pathogenic Organisms Via Optimized Bioorthogonal Non-Canonical Amino Acid Tagging. Cell Chem. Biol 2017, 24, 1048–1055.e3. [DOI] [PubMed] [Google Scholar]
- (177).Hudak JE; Alvarez D; Skelly A; von Andrian UH; Kasper DL Illuminating Vital Surface Molecules of Symbionts in Health and Disease. Nat. Microbiol 2017, 2, 17099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Sherratt AR; Chigrinova M; McKay CS; Beaulieu L-PB; Rouleau Y; Pezacki JP Copper-Catalysed Cycloaddition Reactions of Nitrones and Alkynes for Bioorthogonal Labelling of Living Cells. RSC Adv 2014, 4, 46966–46969. [Google Scholar]
- (179).Yi W; Bystricky P; Yao Q; Guo H; Zhu L; Li H; Shen J; Li M; Ganguly S; Bush CA et al. Two Different O-Polysaccharides from Escherichia coli O86 Are Produced by Different Polymerization of the Same O-Repeating Unit. Carbohydr. Res 2006, 341, 100–108. [DOI] [PubMed] [Google Scholar]
- (180).Coyne MJ; Reinap B; Lee MM; Comstock LE Human Symbionts Use a Host-Like Pathway for Surface Fucosylation. Science 2005, 307, 1778–1781. [DOI] [PubMed] [Google Scholar]
- (181).Ma B; Simala-Grant JL; Taylor DE Fucosylation in Prokaryotes and Eukaryotes. Glycobiology 2006, 16, 158R–184R. [DOI] [PubMed] [Google Scholar]
- (182).Mercante JW; Winchell JM Current and Emerging Legionella Diagnostics for Laboratory and Outbreak Investigations. Clin. Microbiol. Rev 2015, 28, 95–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Knirel YA; Rietschel ET; Marre R; ZÄHringer U The Structure of the O-Specific Chain of Legionella pneumophila Serogroup 1 Lipopolysaccharide. Eur. J. Biochem 1994, 221, 239–245. [DOI] [PubMed] [Google Scholar]
- (184).Glaze PA; Watson DC; Young NM; Tanner ME Biosynthesis of CMP-N,N′-Diacetyllegionaminic Acid from UDP-N,N′-Diacetylbacillosamine in Legionella pneumophila. Biochemistry 2008, 47, 3272–3282. [DOI] [PubMed] [Google Scholar]
- (185).Tsvetkov YE; Shashkov AS; Knirel YA; Zähringer U Synthesis and NMR Spectroscopy of Nine Stereoisomeric 5,7-Diacetamido-3,5,7,9-Tetradeoxynon-2-Ulosonic Acids. Carbohydr. Res 2001, 335, 221–243. [DOI] [PubMed] [Google Scholar]
- (186).Antonczak AK; Simova Z; Tippmann EM A Critical Examination of Escherichia coli Esterase Activity. J. Biol. Chem 2009, 284, 28795–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Edebrink P; Jansson P-E; Bøgwald J; Hoffman J Structural Studies of the Vibrio salmonicida Lipopolysaccharide. Carbohydr. Res 1996, 287, 225–245. [DOI] [PubMed] [Google Scholar]
- (188).Knirel YA; Helbig JH; Zähringer U Structure of a Decasaccharide Isolated by Mild Acid Degradation and Dephosphorylation of the Lipopolysaccharide of Pseudomonas fluorescens Strain ATCC 49271. Carbohydr. Res 1996, 283, 129–139. [DOI] [PubMed] [Google Scholar]
- (189).Haseley SR; Wilkinson SG Structural Studies of the Putative O-Specific Polysaccharide of Acinetobacter baumannii O24 Containing 5,7-Diamino-3,5,7,9-Tetradeoxy-L-Glycerol-D-Galacto-Nonulosonic Acid. Eur. J. Biochem 1997, 250, 617–623. [DOI] [PubMed] [Google Scholar]
- (190).Thibault P; Logan SM; Kelly JF; Brisson J-R; Ewing CP; Trust TJ; Guerry P Identification of the Carbohydrate Moieties and Glycosylation Motifs in Campylobacter jejuni Flagellin. J. Biol. Chem 2001, 276, 34862–34870. [DOI] [PubMed] [Google Scholar]
- (191).McNally DJ; Aubry AJ; Hui JPM; Khieu NH; Whitfield D; Ewing CP; Guerry P; Brisson J-R; Logan SM; Soo EC Targeted Metabolomics Analysis of Campylobacter coli VC167 Reveals Legionaminic Acid Derivatives as Novel Flagellar Glycans. J. Biol. Chem 2007, 282, 14463–14475. [DOI] [PubMed] [Google Scholar]
- (192).Twine SM; Paul CJ; Vinogradov E; McNally DJ; Brisson J-R; Mullen JA; McMullin DR; Jarrell HC; Austin JW; Kelly JF et al. Flagellar Glycosylation in Clostridium botulinum. FEBS J 2008, 275, 4428–4444. [DOI] [PubMed] [Google Scholar]
- (193).Meng X; Boons G-J; Wösten M; Wennekes T Metabolic Labeling of Legionaminic Acid in Flagellin Glycosylation of Campylobacter Jejuni Identifies Maf4 as a Putative Legionaminyl Transferase. Angew. Chem. Int. Edit 2021, 60, 24811–24816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Knirel YA; Vinogradov EV; L’Vov VL; Kocharova NA; Shashkov AS; Dmitriev BA; Kochetkov NK Sialic Acids of a New Type from the Lipopolysaccharides of Pseudomonas aeruginosa and Shigella boydii. Carbohydr. Res 1984, 133, C5–C8. [DOI] [PubMed] [Google Scholar]
- (195).Knirel YA; Bystrova OV; Kocharova NA; Zähringer U; Pier GB Review: Conserved and Variable Structural Features in the Lipopolysaccharide of Pseudomonas Aeruginosa. J. Endotoxin Res 2006, 12, 324–336. [DOI] [PubMed] [Google Scholar]
- (196).Castric P; Cassels FJ; Carlson RW Structural Characterization of the Pseudomonas aeruginosa 1244 Pilin Glycan. J. Biol. Chem 2001, 276, 26479–26485. [DOI] [PubMed] [Google Scholar]
- (197).Smedley JG; Jewell E; Roguskie J; Horzempa J; Syboldt A; Stolz DB; Castric P Influence of Pilin Glycosylation on Pseudomonas aeruginosa 1244 Pilus Function. Infect. Immun 2005, 73, 7922–7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Carlin AF; Uchiyama S; Chang Y-C; Lewis AL; Nizet V; Varki A Molecular Mimicry of Host Sialylated Glycans Allows a Bacterial Pathogen to Engage Neutrophil Siglec-9 and Dampen the Innate Immune Response. Blood 2009, 113, 3333–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Schoenhofen IC; McNally DJ; Brisson J-R; Logan SM Elucidation of the CMP-Pseudaminic Acid Pathway in Helicobacter pylori: Synthesis from UDP-N-Acetylglucosamine by a Single Enzymatic Reaction. Glycobiology 2006, 16, 8C–14C. [DOI] [PubMed] [Google Scholar]
- (200).Liu H; Zhang Y; Wei R; Andolina G; Li X Total Synthesis of Pseudomonas Aeruginosa 1244 Pilin Glycan Via De Novo Synthesis of Pseudaminic Acid. J. Am. Chem. Soc 2017, 139, 13420–13428. [DOI] [PubMed] [Google Scholar]
- (201).Lewis AL; Desa N; Hansen EE; Knirel YA; Gordon JI; Gagneux P; Nizet V; Varki A Innovations in Host and Microbial Sialic Acid Biosynthesis Revealed by Phylogenomic Prediction of Nonulosonic Acid Structure. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13552–13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (202).Liu F; Aubry AJ; Schoenhofen IC; Logan SM; Tanner ME The Engineering of Bacteria Bearing Azido-Pseudaminic Acid-Modified Flagella. ChemBioChem 2009, 10, 1317–1320. [DOI] [PubMed] [Google Scholar]
- (203).Rabuka D; Hubbard SC; Laughlin ST; Argade SP; Bertozzi CR A Chemical Reporter Strategy to Probe Glycoprotein Fucosylation. J. Am. Chem. Soc 2006, 128, 12078–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Apicella M Nontypeable Haemophilus influenzae: The Role of N-Acetyl-5-Neuraminic Acid in Biology. Front. Cell Infect. Microbiol 2012, 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Büll C; Heise T; Beurskens DMH; Riemersma M; Ashikov A; Rutjes FPJT; van Kuppevelt TH; Lefeber DJ; den Brok MH; Adema GJ et al. Sialic Acid Glycoengineering Using an Unnatural Sialic Acid for the Detection of Sialoglycan Biosynthesis Defects and on-Cell Synthesis of Siglec Ligands. ACS Chem. Biol 2015, 10, 2353–2363. [DOI] [PubMed] [Google Scholar]
- (206).Büll C; Heise T; van Hilten N; Pijnenborg JFA; Bloemendal VRLJ; Gerrits L; Kers-Rebel ED; Ritschel T; den Brok MH; Adema GJ et al. Steering Siglec–Sialic Acid Interactions on Living Cells Using Bioorthogonal Chemistry. Angew. Chem. Int. Edit 2017, 56, 3309–3313. [DOI] [PubMed] [Google Scholar]
- (207).Heise T; Büll C; Beurskens DM; Rossing E; de Jonge MI; Adema GJ; Boltje TJ; Langereis JD Metabolic Oligosaccharide Engineering with Alkyne Sialic Acids Confers Neuraminidase Resistance and Inhibits Influenza Reproduction. Bioconjug. Chem 2017, 28, 1811–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Moons SJ; Rossing E; Heming JJA; Janssen MACH; van Scherpenzeel M; Lefeber DJ; de Jonge MI; Langereis JD; Boltje TJ Structure–Activity Relationship of Fluorinated Sialic Acid Inhibitors for Bacterial Sialylation. Bioconjug. Chem 2021, 32, 1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Fagan RP; Fairweather NF Biogenesis and Functions of Bacterial S-Layers. Nat. Rev. Microbiol 2014, 12, 211–222. [DOI] [PubMed] [Google Scholar]
- (210).Valguarnera E; Kinsella RL; Feldman MF Sugar and Spice Make Bacteria Not Nice: Protein Glycosylation and Its Influence in Pathogenesis. J. Mol. Biol 2016, 428, 3206–3220. [DOI] [PubMed] [Google Scholar]
- (211).Nothaft H; Szymanski CM Protein Glycosylation in Bacteria: Sweeter Than Ever. Nat. Rev. Microbiol 2010, 8, 765–778. [DOI] [PubMed] [Google Scholar]
- (212).Baker JL; Çelik E; DeLisa MP Expanding the Glycoengineering Toolbox: The Rise of Bacterial N-Linked Protein Glycosylation. Trends Biotechnol 2013, 31, 313–323. [DOI] [PubMed] [Google Scholar]
- (213).Nothaft H; Szymanski CM Bacterial Protein N-Glycosylation: New Perspectives and Applications. J. Biol. Chem 2013, 288, 6912–6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Merino S; Tomás JM Gram-Negative Flagella Glycosylation. Int. J. Mol. Sci 2014, 15, 2840–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Koenigs MB; Richardson EA; Dube DH Metabolic Profiling of Helicobacter pylori Glycosylation. Mol. Biosyst 2009, 5, 909–912. [DOI] [PubMed] [Google Scholar]
- (216).Clark EL; Emmadi M; Krupp KL; Podilapu AR; Helble JD; Kulkarni SS; Dube DH Development of Rare Bacterial Monosaccharide Analogs for Metabolic Glycan Labeling in Pathogenic Bacteria. ACS Chem. Biol 2016, 11, 3365–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Besanceney-Webler C; Jiang H; Wang W; Baughn AD; Wu P Metabolic Labeling of Fucosylated Glycoproteins in Bacteroidales Species. Bioorg. Med. Chem. Lett 2011, 21, 4989–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Thibault P; Logan SM; Kelly JF; Brisson JR; Ewing CP; Trust TJ; Guerry P Identification of the Carbohydrate Moieties and Glycosylation Motifs in Campylobacter jejuni Flagellin. J. Biol. Chem 2001, 276, 34862–34870. [DOI] [PubMed] [Google Scholar]
- (219).Goon S; Kelly JF; Logan SM; Ewing CP; Guerry P Pseudaminic Acid, the Major Modification on Campylobacter Flagellin, Is Synthesized Via the Cj1293 Gene. Mol. Microbiol 2003, 50, 659–671. [DOI] [PubMed] [Google Scholar]
- (220).Schirm M; Soo EC; Aubry AJ; Austin J; Thibault P; Logan SM Structural, Genetic and Functional Characterization of the Flagellin Glycosylation Process in Helicobacter pylori. Mol. Microbiol 2003, 48, 1579–1592. [DOI] [PubMed] [Google Scholar]
- (221).Guerry P; Ewing CP; Schirm M; Lorenzo M; Kelly J; Pattarini D; Majam G; Thibault P; Logan S Changes in Flagellin Glycosylation Affect Campylobacter Autoagglutination and Virulence. Mol. Microbiol 2006, 60, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Ewing CP; Andreishcheva E; Guerry P Functional Characterization of Flagellin Glycosylation in Campylobacter jejuni 81–176. J. Bacteriol 2009, 191, 7086–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Logan SM Flagellar Glycosylation – a New Component of the Motility Repertoire? Microbiology 2006, 152, 1249–1262. [DOI] [PubMed] [Google Scholar]
- (224).Young NM; Brisson JR; Kelly J; Watson DC; Tessier L; Lanthier PH; Jarrell HC; Cadotte N; Michael FS; Aberg E et al. Structure of the N-Linked Glycan Present on Multiple Glycoproteins in the Gram-Negative Bacterium, Campylobacter jejuni. J. Biol. Chem 2002, 277, 42530–42539. [DOI] [PubMed] [Google Scholar]
- (225).Scott NE; Parker BL; Connolly AM; Paulech J; Edwards AVG; Crossett B; Falconer L; Kolarich D; Djordjevic SP; Højrup P et al. Simultaneous Glycan-Peptide Characterization Using Hydrophilic Interaction Chromatography and Parallel Fragmentation by CID, Higher Energy Collisional Dissociation, and Electron Transfer Dissociation MS Applied to the N-Linked Glycoproteome of Campylobacter jejuni. Mol. Cell Proteomics 2011, 10, S1–S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Szymanski CM; Burr DH; Guerry P Campylobacter Protein Glycosylation Affects Host Cell Interactions. Infect. Immun 2002, 70, 2242–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Alemka A; Nothaft H; Zheng J; Szymanski CM N-Glycosylation of Campylobacter jejuni Surface Proteins Promotes Bacterial Fitness. Infect. Immun 2013, 81, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Jervis AJ; Langdon R; Hitchen P; Lawson AJ; Wood A; Fothergill JL; Morris HR; Dell A; Wren B; Linton D Characterization of N-Linked Protein Glycosylation in Helicobacter pullorum. J. Bacteriol 2010, 192, 5228–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Hopf PS; Ford RS; Zebian N; Merkx-Jacques A; Vijayakumar S; Ratnayake D; Hayworth J; Creuzenet C Protein Glycosylation in Helicobacter pylori: Beyond the Flagellins? PLoS ONE 2011, 6, e25722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Champasa K; Longwell SA; Eldridge AM; Stemmler EA; Dube DH Targeted Identification of Glycosylated Proteins in the Gastric Pathogen Helicobacter pylori. Mol. Cell Proteomics 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Moulton KD; Adewale AP; Carol HA; Mikami SA; Dube DH Metabolic Glycan Labeling-Based Screen to Identify Bacterial Glycosylation Genes. ACS Infect. Dis 2020, 6, 3247–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Hug I; Feldman MF Analogies and Homologies in Lipopolysaccharide and Glycoprotein Biosynthesis in Bacteria. Glycobiology 2011, 21, 138–151. [DOI] [PubMed] [Google Scholar]
- (233).Kaewsapsak P; Esonu O; Dube DH Recruiting the Host’s Immune System to Target Helicobacter pylori’s Surface Glycans. ChemBioChem 2013, 14, 721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Hang HC; Yu C; Kato DL; Bertozzi CR A Metabolic Labeling Approach toward Proteomic Analysis of Mucin-Type O-Linked Glycosylation. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Saxon E; Luchansky SJ; Hang HC; Yu C; Lee SC; Bertozzi CR Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc 2002, 124, 14893–14902. [DOI] [PubMed] [Google Scholar]
- (236).Williams DA; Pradhan K; Paul A; Olin IR; Tuck OT; Moulton KD; Kulkarni SS; Dube DH Metabolic Inhibitors of Bacterial Glycan Biosynthesis. Chem. Sci 2020, 11, 1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Fletcher CM; Coyne MJ; Villa OF; Chatzidaki-Livanis M; Comstock LE A General O-Glycosylation System Important to the Physiology of a Major Human Intestinal Symbiont. Cell 2009, 137, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Fletcher CM; Coyne MJ; Comstock LE Theoretical and Experimental Characterization of the Scope of Protein O-Glycosylation in Bacteroides fragilis. J. Biol. Chem 2011, 286, 3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Wang W; Hu T; Frantom PA; Zheng T; Gerwe B; del Amo DS; Garret S; Seidel RD; Wu P Chemoenzymatic Synthesis of GDP-L-Fucose and the Lewis X Glycan Derivatives. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16096–16101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Neuhaus FC; Baddiley J A Continuum of Anionic Charge: Structures and Functions of D-Alanyl-Teichoic Acids in Gram-Positive Bacteria. Microbiol. Mol. Biol. Rev 2003, 67, 686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Gisch N; Kohler T; Ulmer AJ; Müthing J; Pribyl T; Fischer K; Lindner B; Hammerschmidt S; Zähringer U Structural Reevaluation of Streptococcus Pneumoniae Lipoteichoic Acid and New Insights into Its Immunostimulatory Potency. J. Biol. Chem 2013, 288, 15654–15667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Swoboda JG; Campbell J; Meredith TC; Walker S Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2010, 11, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Oku Y; Kurokawa K; Matsuo M; Yamada S; Lee B-L; Sekimizu K Pleiotropic Roles of Polyglycerolphosphate Synthase of Lipoteichoic Acid in Growth of Staphylococcus aureus Cells. J. Bacteriol 2009, 191, 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Schirner K; Marles-Wright J; Lewis RJ; Errington J Distinct and Essential Morphogenic Functions for Wall- and Lipo-Teichoic Acids in Bacillus subtilis. EMBO J 2009, 28, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Tomasz A Choline in the Cell Wall of a Bacterium: Novel Type of Polymer-Linked Choline in Pneumococcus. Science 1967, 157, 694–697. [DOI] [PubMed] [Google Scholar]
- (246).Denapaite D; Brückner R; Hakenbeck R; Vollmer W Biosynthesis of Teichoic Acids in Streptococcus pneumoniae and Closely Related Species: Lessons from Genomes. Microb. Drug Resist 2012, 18, 344–358. [DOI] [PubMed] [Google Scholar]
- (247).Bergmann S; Hammerschmidt S Versatility of Pneumococcal Surface Proteins. Microbiology 2006, 152, 295–303. [DOI] [PubMed] [Google Scholar]
- (248).Cundell DR; Gerard NP; Gerard C; Idanpaan-Heikkila I; Tuomanen EI Streptococcus pneumoniae Anchor to Activated Human Cells by the Receptor for Platelet-Activating Factor. Nature 1995, 377, 435–438. [DOI] [PubMed] [Google Scholar]
- (249).Radin JN; Orihuela CJ; Murti G; Guglielmo C; Murray PJ; Tuomanen EI Beta-Arrestin 1 Participates in Platelet-Activating Factor Receptor-Mediated Endocytosis of Streptococcus pneumoniae. Infection and Immunity 2005, 73, 7827–7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Sohlenkamp C; López-Lara IM; Geiger O Biosynthesis of Phosphatidylcholine in Bacteria. Prog. Lipid Res 2003, 42, 115–162. [DOI] [PubMed] [Google Scholar]
- (251).Di Guilmi AM; Bonnet J; Peiβert S; Durmort C; Gallet B; Vernet T; Gisch N; Wong YS Specific and Spatial Labeling of Choline-Containing Teichoic Acids in Streptococcus pneumoniae by Click Chemistry. Chem. Commun 2017, 53, 10572–10575. [DOI] [PubMed] [Google Scholar]
- (252).Jao CY; Roth M; Welti R; Salic A Metabolic Labeling and Direct Imaging of Choline Phospholipids in Vivo. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 15332–15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Bonnet J; Wong Y-S; Vernet T; Di Guilmi AM; Zapun A; Durmort C One-Pot Two-Step Metabolic Labeling of Teichoic Acids and Direct Labeling of Peptidoglycan Reveals Tight Coordination of Both Polymers Inserted into Pneumococcus Cell Wall. ACS Chem. Biol 2018, 13, 2010–2015. [DOI] [PubMed] [Google Scholar]
- (254).Brennan PJ; Nikaido H The Envelope of Mycobacteria. Annu. Rev. Biochem 1995, 64, 29–63. [DOI] [PubMed] [Google Scholar]
- (255).Daffe M; Reyrat J-M Mycobacterial Cell Envelope; ASM Press: Washington, DC, USA, 2008. [Google Scholar]
- (256).Daffé M; Marrakchi H Unraveling the Structure of the Mycobacterial Envelope. Microbiol. Spectr 2019, 7, DOI: 10.1128/microbiolspec.GPP3-0027-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Pavelka Martin, S. Jr.; Mahapatra S; Crick Dean, C.; Hatfull Graham, F.; Jacobs William, R. Jr. Genetics of Peptidoglycan Biosynthesis. Microbiol. Spectr 2014, 2, MGM2-0034-2013. [DOI] [PubMed] [Google Scholar]
- (258).Bhamidi S; Scherman MS; Jones V; Crick DC; Belisle JT; Brennan PJ; McNeil MR Detailed Structural and Quantitative Analysis Reveals the Spatial Organization of the Cell Walls of in Vivo Grown Mycobacterium leprae and in Vitro Grown Mycobacterium tuberculosis. J. Biol. Chem 2011, 286, 23168–23177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Quémard A New Insights into the Mycolate-Containing Compound Biosynthesis and Transport in Mycobacteria. Trends Microbiol 2016, 24, 725–738. [DOI] [PubMed] [Google Scholar]
- (260).Morita YS; Fukuda T; Sena CB; Yamaryo-Botte Y; McConville MJ; Kinoshita T Inositol Lipid Metabolism in Mycobacteria: Biosynthesis and Regulatory Mechanisms. Biochim. Biophys. Acta 2011, 1810, 630–641. [DOI] [PubMed] [Google Scholar]
- (261).Sambou T; Dinadayala P; Stadthagen G; Barilone N; Bordat Y; Constant P; Levillain F; Neyrolles O; Gicquel B; Lemassu A et al. Capsular Glucan and Intracellular Glycogen of Mycobacterium tuberculosis: Biosynthesis and Impact on the Persistence in Mice. Mol. Microbiol 2008, 70, 762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Dobos KM; Khoo KH; Swiderek KM; Brennan PJ; Belisle JT Definition of the Full Extent of Glycosylation of the 45-Kilodalton Glycoprotein of Mycobacterium tuberculosis. J. Bacteriol 1996, 178, 2498–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Michell SL; Whelan AO; Wheeler PR; Panico M; Easton RL; Etienne AT; Haslam SM; Dell A; Morris HR; Reason AJ et al. The MPB83 Antigen from Mycobacterium bovis Contains O-Linked Mannose and (1→3)-Mannobiose Moieties. J. Biol. Chem 2003, 278, 16423–16432. [DOI] [PubMed] [Google Scholar]
- (264).Backus KM; Boshoff HI; Barry CS; Boutureira O; Patel MK; D’Hooge F; Lee SS; Via LE; Tahlan K; Barry CE 3rd et al. Uptake of Unnatural Trehalose Analogs as a Reporter for Mycobacterium tuberculosis. Nat. Chem. Biol 2011, 7, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Rodriguez-Rivera FP; Zhou X; Theriot JA; Bertozzi CR Visualization of Mycobacterial Membrane Dynamics in Live Cells. J. Am. Chem. Soc 2017, 139, 3488–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Kamariza M; Shieh P; Ealand CS; Peters JS; Chu B; Rodriguez-Rivera FP; Babu Sait MR; Treuren WV; Martinson N; Kalscheuer R et al. Rapid Detection of Mycobacterium tuberculosis in Sputum with a Solvatochromic Trehalose Probe. Sci. Transl. Med 2018, 10, eaam6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Swarts BM; Holsclaw CM; Jewett JC; Alber M; Fox DM; Siegrist MS; Leary JA; Kalscheuer R; Bertozzi CR Probing the Mycobacterial Trehalome with Bioorthogonal Chemistry. J. Am. Chem. Soc 2012, 134, 16123–16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Parker HL; Tomás RMF; Furze CM; Guy CS; Fullam E Asymmetric Trehalose Analogues to Probe Disaccharide Processing Pathways in Mycobacteria. Org. Biomol. Chem 2020, 18, 3607–3612. [DOI] [PubMed] [Google Scholar]
- (269).Rundell SR; Wagar ZL; Meints LM; Olson CD; O’Neill MK; Piligian BF; Poston AW; Hood RJ; Woodruff PJ; Swarts BM Deoxyfluoro-D-Trehalose (FDTre) Analogues as Potential PET Probes for Imaging Mycobacterial Infection. Org. Biomol. Chem 2016, 14, 8598–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Peña-Zalbidea S; Huang AYT; Kavunja HW; Salinas B; Desco M; Drake C; Woodruff PJ; Vaquero JJ; Swarts BM Chemoenzymatic Radiosynthesis of 2-Deoxy-2-[18F]Fluoro-D-Trehalose ([18F]-2-FDTre): A PET Radioprobe for in Vivo Tracing of Trehalose Metabolism. Carbohydr. Res 2019, 472, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Foley HN; Stewart JA; Kavunja HW; Rundell SR; Swarts BM Bioorthogonal Chemical Reporters for Selective in Situ Probing of Mycomembrane Components in Mycobacteria. Angew. Chem. Int. Edit 2016, 55, 2053–2057. [DOI] [PubMed] [Google Scholar]
- (272).Kavunja HW; Biegas KJ; Banahene N; Stewart JA; Piligian BF; Groenevelt JM; Sein CE; Morita YS; Niederweis M; Siegrist MS et al. Photoactivatable Glycolipid Probes for Identifying Mycolate–Protein Interactions in Live Mycobacteria. J. Am. Chem. Soc 2020, 142, 7725–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Lesur E; Baron A; Dietrich C; Buchotte M; Doisneau G; Urban D; Beau J-M; Bayan N; Vauzeilles B; Guianvarc’h D et al. First Access to a Mycolic Acid-Based Bioorthogonal Reporter for the Study of the Mycomembrane and Mycoloyltransferases in Corynebacteria. Chem. Commun 2019, 55, 13074–13077. [DOI] [PubMed] [Google Scholar]
- (274).Fiolek TJ; Banahene N; Kavunja HW; Holmes NJ; Rylski AK; Pohane AA; Siegrist MS; Swarts BM Engineering the Mycomembrane of Live Mycobacteria with an Expanded Set of Trehalose Monomycolate Analogues. ChemBioChem 2019, 20, 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Zhou X; Rodriguez-Rivera FP; Lim HC; Bell JC; Bernhardt TG; Bertozzi CR; Theriot JA Sequential Assembly of the Septal Cell Envelope Prior to V Snapping in Corynebacterium glutamicum. Nat. Chem. Biol 2019, 15, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Hodges HL; Brown RA; Crooks JA; Weibel DB; Kiessling LL Imaging Mycobacterial Growth and Division with a Fluorogenic Probe. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Holmes NJ; Kavunja HW; Yang Y; Vannest BD; Ramsey CN; Gepford DM; Banahene N; Poston AW; Piligian BF; Ronning DR et al. A FRET-Based Fluorogenic Trehalose Dimycolate Analogue for Probing Mycomembrane-Remodeling Enzymes of Mycobacteria. ACS Omega 2019, 4, 4348–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Kolbe K; Möckl L; Sohst V; Brandenburg J; Engel R; Malm S; Bräuchle C; Holst O; Lindhorst TK; Reiling N Azido Pentoses: A New Tool to Efficiently Label Mycobacterium tuberculosis Clinical Isolates. ChemBioChem 2017, 18, 1172–1176. [DOI] [PubMed] [Google Scholar]
- (279).Marando VM; Kim DE; Calabretta PJ; Kraft MB; Bryson BD; Kiessling LL Biosynthetic Glycan Labeling. J. Am. Chem. Soc 2021, 143, 16337–16342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Marrakchi H; Lanéelle M-A; Daffé M Mycolic Acids: Structures, Biosynthesis, and Beyond. Chem. Biol 2013, 21, 67–85. [DOI] [PubMed] [Google Scholar]
- (281).Welsh KJ; Hunter RL; Actor JK Trehalose 6,6′-Dimycolate – a Coat to Regulate Tuberculosis Immunopathogenesis. Tuberculosis 2013, 93, Suppl, S3–S9. [DOI] [PubMed] [Google Scholar]
- (282).Biegas KJ; Swarts BM Chemical Probes for Tagging Mycobacterial Lipids. Curr. Opin. Chem. Biol 2021, 65, 57–65. [DOI] [PubMed] [Google Scholar]
- (283).Timmins GS; Deretic V Mechanisms of Action of Isoniazid. Mol. Microbiol 2006, 62, 1220–1227. [DOI] [PubMed] [Google Scholar]
- (284).De Smet KAL; Weston A; Brown IN; Young DB; Robertson BD Three Pathways for Trehalose Biosynthesis in Mycobacteria. Microbiology 2000, 146, 199–208. [DOI] [PubMed] [Google Scholar]
- (285).Woodruff PJ; Carlson BL; Siridechadilok B; Pratt MR; Senaratne RH; Mougous JD; Riley LW; Williams SJ; Bertozzi CR Trehalose Is Required for Growth of Mycobacterium smegmatis. J. Biol. Chem 2004, 279, 28835–28843. [DOI] [PubMed] [Google Scholar]
- (286).Murphy HN; Stewart GR; Mischenko VV; Apt AS; Harris R; McAlister MSB; Driscoll PC; Young DB; Robertson BD The OtsAB Pathway Is Essential for Trehalose Biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem 2005, 280, 14524–14529. [DOI] [PubMed] [Google Scholar]
- (287).Miah F; Koliwer-Brandl H; Rejzek M; Field Robert A.; Kalscheuer R; Bornemann S Flux through Trehalose Synthase Flows from Trehalose to the Alpha Anomer of Maltose in Mycobacteria. Chem. Biol 2013, 20, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Gavalda S; Bardou F; Laval F; Bon C; Malaga W; Chalut C; Guilhot C; Mourey L; Daffé M; Quémard A The Polyketide Synthase Pks13 Catalyzes a Novel Mechanism of Lipid Transfer in Mycobacteria. Chem. Biol 2014, 21, 1660–1669. [DOI] [PubMed] [Google Scholar]
- (289).Grzegorzewicz AE; Pham H; Gundi VAKB; Scherman MS; North EJ; Hess T; Jones V; Gruppo V; Born SEM; Kordulakova J et al. Inhibition of Mycolic Acid Transport across the Mycobacterium tuberculosis Plasma Membrane. Nat. Chem. Biol 2012, 8, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Sathyamoorthy N; Takayama K Purification and Characterization of a Novel Mycolic Acid Exchange Enzyme from Mycobacterium smegmatis. J. Biol. Chem 1987, 262, 13417–13423. [PubMed] [Google Scholar]
- (291).Belisle JT; Vissa VD; Sievert T; Takayama K; Brennan PJ; Besra GS Role of the Major Antigen of Mycobacterium tuberculosis in Cell Wall Biogenesis. Science 1997, 276, 1420–1422. [DOI] [PubMed] [Google Scholar]
- (292).Huc E; Meniche X; Benz R; Bayan N; Ghazi A; Tropis M; Daffé M O-Mycoloylated Proteins from Corynebacterium: An Unprecedented Post-Translational Modification in Bacteria. J. Biol. Chem 2010, 285, 21908–21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Huc E; de Sousa-D’Auria C; de la Sierra-Gallay IL; Salmeron C; van Tilbeurgh H; Bayan N; Houssin C; Daffé M; Tropis M Identification of a Mycoloyl Transferase Selectively Involved in O-Acylation of Polypeptides in Corynebacteriales. J. Bacteriol 2013, 195, 4121–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Kalscheuer R; Weinrick B; Veeraraghavan U; Besra GS; Jacobs WR Jr. Trehalose-Recycling ABC Transporter LpqY-SugA-SugB-SugC Is Essential for Virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 21761–21766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Ojha AK; Trivelli X; Guerardel Y; Kremer L; Hatfull GF Enzymatic Hydrolysis of Trehalose Dimycolate Releases Free Mycolic Acids During Mycobacterial Growth in Biofilms. J. Biol. Chem 2010, 285, 17380–17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Yang Y; Kulka K; Montelaro RC; Reinhart TA; Sissons J; Aderem A; Ojha AK A Hydrolase of Trehalose Dimycolate Induces Nutrient Influx and Stress Sensitivity to Balance Intracellular Growth of Mycobacterium tuberculosis. Cell Host Microbe 2014, 15, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Kalera K; Stothard AI; Woodruff PJ; Swarts BM The Role of Chemoenzymatic Synthesis in Advancing Trehalose Analogues as Tools for Combatting Bacterial Pathogens. Chem. Commun 2020, 56, 11528–11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Chaube MA; Kulkarni SS Stereoselective Construction of 1,1-Alpha,Alpha-Glycosidic Bonds. Trends Carbohydr. Res 2013, 4, 1–19. [Google Scholar]
- (299).Sarpe VA; Kulkarni SS Regioselective Protection and Functionalization of Trehalose. Trends Carbohydr. Res 2013, 5, 8–33. [Google Scholar]
- (300).Namme R; Mitsugi T; Takahashi H; Ikegami S Development of Ketoside-Type Analogues of Trehalose by Using A-Stereoselective O-Glycosidation of Ketose. Eur. J. Org. Chem 2007, 3758–3764. [Google Scholar]
- (301).Rodriguez‐Rivera FP; Xiaoxue Z; Theriot JA; Bertozzi CR Acute Modulation of Mycobacterial Cell Envelope Biogenesis by Front‐Line Tuberculosis Drugs. Angew. Chem. Int. Edit 2018, 57, 5267–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Favrot L; Grzegorzewicz AE; Lajiness DH; Marvin RK; Boucau J; Isailovic D; Jackson M; Ronning DR Mechanism of Inhibition of Mycobacterium tuberculosis Antigen 85 by Ebselen. Nat. Comm 2013, 4, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Sahile HA; Rens C; Shapira T; Andersen RJ; Av-Gay Y Dmn-Tre Labeling for Detection and High-Content Screening of Compounds against Intracellular Mycobacteria. ACS Omega 2020, 5, 3661–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Dinkele R; Gessner S; McKerry A; Leonard B; Seldon R; Koch AS; Morrow C; Gqada M; Kamariza M; Bertozzi CR et al. Capture and Visualization of Live Mycobacterium tuberculosis Bacilli from Tuberculosis Patient Bioaerosols. PLoS Pathog 2021, 17, e1009262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Kamariza M; Keyser SGL; Utz A; Knapp BD; Ealand C; Ahn G; Cambier CJ; Chen T; Kana B; Huang KC et al. Toward Point-of-Care Detection of Mycobacterium Tuberculosis: A Brighter Solvatochromic Probe Detects Mycobacteria within Minutes. JACS Au 2021, 1, 1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Urbanek BL; Wing DC; Haislop KS; Hamel CJ; Kalscheuer R; Woodruff PJ; Swarts BM Chemoenzymatic Synthesis of Trehalose Analogues: Rapid Access to Chemical Probes for Investigating Mycobacteria. ChemBioChem 2014, 15, 2066–2070. [DOI] [PubMed] [Google Scholar]
- (307).Xu Z; Meshcheryakov VA; Poce G; Chng S-S MmpL3 Is the Flippase for Mycolic Acids in Mycobacteria. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Bolla JR Targeting Mmpl3 for Anti-Tuberculosis Drug Development. Biochem. Soc. Trans 2020, 48, 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Pohane AA; Carr CR; Garhyan J; Swarts BM; Siegrist MS Trehalose Recycling Promotes Energy-Efficient Biosynthesis of the Mycobacterial Cell Envelope. mBio 2021, 12, e02801–02820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Cambier CJ; Banik SM; Buonomo JA; Bertozzi CR Spreading of a Mycobacterial Cell-Surface Lipid into Host Epithelial Membranes Promotes Infectivity. eLife 2020, 9, e60648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).O’Neill MK; Piligian BF; Olson CD; Woodruff PJ; Swarts BM Tailoring Trehalose for Biomedical and Biotechnological Applications. Pure Appl. Chem 2017, 89, 1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Meints LM; Poston AW; Piligian BF; Olson CD; Badger KS; Woodruff PJ; Swarts BM Rapid One-Step Enzymatic Synthesis and All-Aqueous Purification of Trehalose Analogues. J. Vis. Exp 2017, e54485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Kouril T; Zaparty M; Marrero J; Brinkmann H; Siebers B A Novel Trehalose Synthesizing Pathway in the Hyperthermophilic Crenarchaeon Thermoproteus tenax: The Unidirectional TreT Pathway. Arch. Microbiol 2008, 190, 355–369. [DOI] [PubMed] [Google Scholar]
- (314).Groenevelt JM; Meints LM; Stothard AI; Poston AW; Fiolek TJ; Finocchietti DH; Mulholand VM; Woodruff PJ; Swarts BM Chemoenzymatic Synthesis of Trehalosamine, an Aminoglycoside Antibiotic and Precursor to Mycobacterial Imaging Probes. J. Org. Chem 2018, 83, 8662–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Johnson DH; Via LE; Kim P; Laddy D; Lau C-Y; Weinstein EA; Jain S Nuclear Imaging: A Powerful Novel Approach for Tuberculosis. Nucl. Med. Biol 2014, 41, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Jain SK The Promise of Molecular Imaging in the Study and Treatment of Infectious Diseases. Mol. Imaging Biol 2017, 19, 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Banister S; Roeda D; Dolle F; Kassiou M Fluorine-18 Chemistry for PET: A Concise Introduction. Curr. Radiopharm 2010, 3, 68–80. [Google Scholar]
- (318).Jeffrey MW; Urbanek BL; Meints LM; Piligian BF; Lopez-Casillas IC; Zochowski KM; Woodruff PJ; Swarts BM The Trehalose-Specific Transporter LpqY-SugABC Is Required for Antimicrobial and Anti-Biofilm Activity of Trehalose Analogues in Mycobacterium smegmatis. Carbohydr. Res 2017, 450, 60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Lee JJ; Lee S-K; Song N; Nathan TO; Swarts BM; Eum SY; Ehrt S; Cho S-N; Eoh H Transient Drug-Tolerance and Permanent Drug-Resistance Rely on the Trehalose-Catalytic Shift in Mycobacterium tuberculosis. Nat. Commun 2019, 2928–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Danielson ND; Collins J; Stothard AI; Dong Q; Kalera K; Woodruff PJ; DeBosch BJ; Britton RA; Swarts BM Degradation-Resistant Trehalose Analogues Block Utilization of Trehalose by Hypervirulent Clostridioides difficile. Chem. Commun 2019, 55, 5009–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Jayawardana KW; Jayawardena HSN; Wijesundera SA; De Zoysa T; Sundhoro M; Yan M Selective Targeting of Mycobacterium smegmatis with Trehalose-Functionalized Nanoparticles. Chem. Commun 2015, 51, 12028–12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Dutta AK; Choudhary E; Wang X; Záhorszka M; Forbak M; Lohner P; Jessen HJ; Agarwal N; Korduláková J; Jessen-Trefzer C Trehalose Conjugation Enhances Toxicity of Photosensitizers against Mycobacteria. ACS Cent. Sci 2019, 5, 644–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Dixon CF; Nottingham AN; Lozano AF; Sizemore JA; Russell LA; Valiton C; Newell KL; Babin D; Bridges WT; Parris MR et al. Synthesis and Evaluation of Porphyrin Glycoconjugates Varying in Linker Length: Preliminary Effects on the Photodynamic Inactivation of Mycobacterium smegmatis. RSC Adv 2021, 11, 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Barry CS; Backus KM; Barry CE; Davis BG ESI-MS Assay of M. tuberculosis Cell Wall Antigen 85 Enzymes Permits Substrate Profiling and Design of a Mechanism-Based Inhibitor. J. Am. Chem. Soc 2011, 133, 13232–13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Sarpe VA; Kulkarni SS Synthesis of Maradolipid. J. Org. Chem 2011, 76, 6866–6870. [DOI] [PubMed] [Google Scholar]
- (326).Paul NK; Twibanire JD; Grindley TB Direct Synthesis of Maradolipids and Other Trehalose 6-Monoesters and 6,6’-Diesters. J. Org. Chem 2012, 78, 363–369. [DOI] [PubMed] [Google Scholar]
- (327).Ngo JT; Adams SR; Deerinck TJ; Boassa D; Rodriguez-Rivera F; Palida SF; Bertozzi CR; Ellisman MH; Tsien RY Click-EM for Imaging Metabolically Tagged Nonprotein Biomolecules. Nat. Chem. Biol 2016, 12, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Niederweis M; Danilchanka O; Huff J; Hoffmann C; Engelhardt H Mycobacterial Outer Membranes: In Search of Proteins. Trends Microbiol 2010, 18, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Song H; Sandie R; Wang Y; Andrade-Navarro MA; Niederweis M Identification of Outer Membrane Proteins of Mycobacterium tuberculosis. Tuberculosis 2008, 88, 526–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Rath P; Saurel O; Tropis M; Daffé M; Demange P; Milon A NMR Localization of the O-Mycoloylation on PorH, a Channel Forming Peptide from Corynebacterium glutamicum. FEBS Letters 2013, 587, 3687–3691. [DOI] [PubMed] [Google Scholar]
- (331).Carel C; Marcoux J; Reat V; Parra J; Latge G; Laval F; Demange P; Burlet-Schiltz O; Milon A; Daffe M et al. Identification of Specific Posttranslational O-Mycoloylations Mediating Protein Targeting to the Mycomembrane. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 4231–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Kavunja HW; Piligian BF; Fiolek TJ; Foley HN; Nathan TO; Swarts BM A Chemical Reporter Strategy for Detecting and Identifying O-Mycoloylated Proteins in Corynebacterium. Chem. Commun 2016, 52, 13795–13798. [DOI] [PubMed] [Google Scholar]
- (333).Yang Y; Bhati A; Ke D; Gonzalez-Juarrero M; Lenaerts A; Kremer L; Guerardel Y; Zhang P; Ojha AK Exposure to a Cutinase-Like Serine Esterase Triggers Rapid Lysis of Multiple Mycobacterial Species. J. Biol. Chem 2012, 288, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).McNeil M; Daffe M; Brennan PJ Evidence for the Nature of the Link between the Arabinogalactan and Peptidoglycan of Mycobacterial Cell Walls. J. Biol. Chem 1990, 265, 18200–18206. [PubMed] [Google Scholar]
- (335).Fukuda T; Matsumura T; Ato M; Hamasaki M; Nishiuchi Y; Murakami Y; Maeda Y; Yoshimori T; Matsumoto S; Kobayashi K et al. Critical Roles for Lipomannan and Lipoarabinomannan in Cell Wall Integrity of Mycobacteria and Pathogenesis of Tuberculosis. mBio 2013, 4, e00472–00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Wolucka BA Biosynthesis of D-Arabinose in Mycobacteria – a Novel Bacterial Pathway with Implications for Antimycobacterial Therapy. FEBS J 2008, 275, 2691–2711. [DOI] [PubMed] [Google Scholar]
- (337).Briken V; Porcelli SA; Besra GS; Kremer L Mycobacterial Lipoarabinomannan and Related Lipoglycans: From Biogenesis to Modulation of the Immune Response. Mol. Microbiol 2004, 53, 391–403. [DOI] [PubMed] [Google Scholar]
- (338).Belanger AE; Besra GS; Ford ME; Mikusová K; Belisle JT; Brennan PJ; Inamine JM The embAB Genes of Mycobacterium avium Encode an Arabinosyl Transferase Involved in Cell Wall Arabinan Biosynthesis That Is the Target for the Antimycobacterial Drug Ethambutol. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11919–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Kraft MB; Martinez Farias MA; Kiessling LL Synthesis of Lipid-Linked Arabinofuranose Donors for Glycosyltransferases. J. Org. Chem 2013, 78, 2128–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (340).Krinos CM; Coyne MJ; Weinacht KG; Tzianabos AO; Kasper DL; Comstock LE Extensive Surface Diversity of a Commensal Microorganism by Multiple DNA Inversions. Nature 2001, 414, 555–558. [DOI] [PubMed] [Google Scholar]
- (341).Coyne MJ; Chatzidaki-Livanis M; Paoletti LC; Comstock LE Role of Glycan Synthesis in Colonization of the Mammalian Gut by the Bacterial Symbiont Bacteroides fragilis. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 13099–13104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Velez CD; Lewis CJ; Kasper DL; Cobb BA Type I Streptococcus pneumoniae Carbohydrate Utilizes a Nitric Oxide and MHC II-Dependent Pathway for Antigen Presentation. Immunology 2009, 127, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Thongsomboon W; Serra DO; Possling A; Hadjineophytou C; Hengge R; Cegelski L Phosphoethanolamine Cellulose: A Naturally Produced Chemically Modified Cellulose. Science 2018, 359, 334–338. [DOI] [PubMed] [Google Scholar]
- (344).Hollenbeck EC; Antonoplis A; Chai C; Thongsomboon W; Fuller GG; Cegelski L Phosphoethanolamine Cellulose Enhances Curli-Mediated Adhesion of Uropathogenic Escherichia coli to Bladder Epithelial Cells. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 10106–10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (345).Ghafoor A; Hay Iain D; Rehm Bernd HA Role of Exopolysaccharides in Pseudomonas aeruginosa Biofilm Formation and Architecture. Appl. Environ. Microb 2011, 77, 5238–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Moradali MF; Ghods S; Rehm BHA Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell Infect. Microbiol 2017, 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Oh S-Y; Budzik JM; Garufi G; Schneewind O Two Capsular Polysaccharides Enable Bacillus cereus G9241 to Cause Anthrax-Like Disease. Mol. Microbiol 2011, 80, 455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Wilkening RV; Federle MJ Evolutionary Constraints Shaping Streptococcus Pyogenes–Host Interactions. Trends Microbiol 2017, 25, 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Tzeng Y-L; Thomas J; Stephens DS Regulation of Capsule in Neisseria meningitidis. Crit. Rev. Microbiol 2016, 42, 759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Mazmanian SK; Liu CH; Tzianabos AO; Kasper DL An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005, 122, 107–118. [DOI] [PubMed] [Google Scholar]
- (351).Geva-Zatorsky N; Alvarez D; Hudak JE; Reading NC; Erturk-Hasdemir D; Dasgupta S; von Andrian UH; Kasper DL In Vivo Imaging and Tracking of Host–Microbiota Interactions Via Metabolic Labeling of Gut Anaerobic Bacteria. Nat. Med 2015, 21, 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Moore RN; Coyne Michael J; Tzianabos Arthur O; Mallory Benjamin C; Carey Vincent J; Kasper Dennis L; Comstock Laurie E Polysaccharide Biosynthesis Locus Required for Virulence of Bacteroides fragilis. Infect. Immun 2001, 69, 4342–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Sharma S; Erickson KM; Troutman JM Complete Tetrasaccharide Repeat Unit Biosynthesis of the Immunomodulatory Bacteroides fragilis Capsular Polysaccharide A. ACS Chem. Biol 2017, 12, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Dube DH; Prescher JA; Quang CN; Bertozzi CR Probing Mucin-Type O-Linked Glycosylation in Living Animals. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Boyce M; Carrico IS; Ganguli AS; Yu S-H; Hangauer MJ; Hubbard SC; Kohler JJ; Bertozzi CR Metabolic Cross-Talk Allows Labeling of O-Linked Beta-N-Acetylglucosamine-Modified Proteins Via the N-Acetylgalactosamine Salvage Pathway. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Xu W; Su P; Zheng L; Fan H; Wang Y; Liu Y; Lin Y; Zhi F In Vivo Imaging of a Novel Strain of Bacteroides Fragilis Via Metabolic Labeling. Front. Microbiol 2018, 9, 2298. [DOI] [PMC free article] [PubMed] [Google Scholar]