

Abstract
BACKGROUND & AIMS:
Pancreatic cancer (PC) is the third leading cause of cancer-related death with a 5-year survival rate of approximately 10%. It typically presents as a late-stage incurable cancer and chemotherapy provides modest benefit. Here, we demonstrate the feasibility, safety, and potency of a novel human natural killer (NK) cell-based immunotherapy to treat PC.
METHODS:
The expression of prostate stem cell antigen (PSCA) was evaluated in primary PC at mRNA and protein levels. The processes of retroviral transduction, expansion, activation, and cryopreservation of primary human NK cells obtained from umbilical cord blood were optimized, allowing us to develop frozen, off-the-shelf, allogeneic PSCA chimeric antigen receptor (CAR) NK cells. The safety and efficacy of PSCA CAR NK cells also expressing soluble (s)IL-15 (PSCA CAR_s15 NK cells) were evaluated in vitro and in vivo.
RESULTS:
PSCA was elevated in primary human PC compared to the adjacent or other normal tissues. PSCA CAR_s15 NK cells displayed significant tumor-suppressive effects against PSCA(+) PC in vitro before and after one cycle of freeze-thaw. The viability of frozen PSCA CAR_s15 NK cells persisted more than 90 days in vivo following their last infusion and significantly prolonged the survival of mice engrafted with human PC.
CONCLUSIONS:
PSCA CAR_s15 NK cells showed therapeutic efficacy in human metastatic PC models without signs of systematic toxicity, providing a strong rationale to support clinical development.
Keywords: Pancreatic cancer, Pancreatic ductal adenocarcinoma, NK cell, Immunotherapy, Innate immunity
Graphical Abstract
INTRODUCTION
Pancreatic cancer (PC) has a 5-year overall survival rate of 10% and is the third leading cause of cancer-related death1. PC poses several treatment challenges, including a lack of symptoms in early-stage disease and a unique tumor microenvironment characterized by dense desmoplasia and high infiltration of immunosuppressive cells2. For patients with advanced disease, chemotherapy is the mainstay of treatment. However, current therapies offer only very modest improvements in overall survival. The results of checkpoint inhibitor therapy has thus far been disappointing for PC3, 4. Currently, there is no effective therapy for late-stage PC. Thus, novel therapies are needed.
Arming natural killer (NK) cells with chimeric antigen receptors (CAR) to attack cancer has attracted attention in both academia and industry, largely due to the versatility and safety of NK cells. NK cells are CD56(+)CD3(−) large granular lymphocytes that can kill virally infected or malignantly transformed cells5, 6. Unlike cytotoxic CD8(+) T lymphocyte-based therapies, NK cells show direct cytotoxicity against tumor cells without the requirement for prior sensitization5. Also, infusion of NK cells thus far has not caused strong alloreactivity or cytokine storms7, 8. Because NK cell therapy has the potential to use off-the-shelf allogeneic cells, it can eliminate approximately three weeks that is currently required for the preparation of autologous CAR T cells following their harvest from each patient. We previously showed that targeting the innate immune response using CAR NK cells is effective in several cancer models, including solid tumors9, 10.
Prostate stem cell antigen (PSCA) is a glycosylphosphatidylinositol-anchored cell surface protein involved in intracellular signaling and tumor proliferation11. Increased PSCA expression has been observed in high-grade prostatic intraepithelial neoplasia and hormone-related refractory metastases and associated with aggressive disease12. Aberrant overexpression of PSCA has also been detected in the tumors from 60–80% of patients diagnosed with PC13, but not in the normal pancreatic duct14. Thus, PSCA is a candidate tumor antigen for CAR NK cells to target to treat PC.
The objective of the current study was to develop human PSCA CAR NK cells expressing soluble IL-15, referred to as PSCA CAR_s15 NK cells, to enhance NK cell anti-tumor function, and evaluate them in vitro using cell co-culture and in vivo using mouse PC models. The PSCA CAR_s15 NK cells were manufactured through an optimized expansion platform and frozen as an off-the-shelf product. Mice that received multiple infusions of PSCA CAR_s15 NK cells had improved overall survival. Our study provides a strong rationale to investigate PSCA CAR_s15 NK cells for clinical use.
MATERIALS AND METHODS
Dataset analysis
PSCA expression data were retrieved, analyzed, and adapted from Oncomine™ (https://www.oncomine.org/)15–19. The p-value was calculated by median-rank analysis. The correlation study and survival study of PSCA were retrieved from Genomic Data Commons (GDC) 20 and The Cancer Genome Atlas (TCGA) 19 and graphed by UCSC Xena (https://xena.ucsc.edu/) or OncoLnc (http://www.oncolnc.org).
DNA constructs and retrovirus production
A CAR designed to target PSCA was cloned into a clinical-grade retroviral vector (RRV) from which the non-human DNA sequences were minimized. The PSCA CAR was designed to sequentially consists of a signal peptide (SP), anti-PSCA single-chain fragment variable (scFv), hinge, transmembrane domain (TM), CD28, and CD3ζ. The anti-PSCA scFv-hinge-CD28-CD3ζ−2A (1517 bp) was cloned into RRV vector at the NotI/ SalI site. Since IL-15 activates and improves survival of CD8 T cells and NK cells21, 22, a soluble form of IL-15 (s15) was incorporated into the PSCA CAR construct. The codon-optimized s15 was fused to CD3ζ through a thosea asigna virus 2A-like self-cleaving peptide (T2A). Truncated EGFR (tEGFR), which functions as a traceable marker and suicide switch in vivo23, 24, was linked to s15 in frame via porcine teschovirus-1 2Alike self-cleaving peptide (P2A). The IL-15–2A-tEGFR fragment was cloned into RRV at the SalI/ MluI site. Both PSCA CAR-2A and IL-15–2A-tEGFR fragments were commercially synthesized. Both control plasmids PSCA CAR_tEGFR and s15_tEGFR were obtained by amplifying the corresponding fragments from PSCA CAR_s15_tEGFR plasmid and cloned into the retroviral vector at NotI/ MfeI site and NotI/ MluI respectively.
A retrovirus expressing PSCA CAR_s15_tEGFR was produced by co-transfecting the retroviral transfer plasmid with pRD114-TR, which encodes the envelope glycoprotein, into GP2–293T cells (Takara) using lipofectamine 3000 (ThermoFisher). The culture supernatants containing retrovirus were collected at 48 hours post transfection, filtered through a 0.45 μm filter, then applied to NK cells for transduction.
NK cell isolation, expansion, and transduction
Umbilical cord blood leukopaks were obtained from StemCyte, Inc. Primary NK cells were enriched from cord blood using RosetteSep (Stemcell Technologies) following the manufacturer’s instructions and pre-expanded with K562 feeder cells expressing membrane bound IL-21 and 41BBL prior to retroviral infection. The expanded NK cells were then infected with retrovirus using Retronectin (Takara) following the manufacturer’s instructions and further expanded until day 15. The efficiency of transduction was determined by flow cytometric analysis of EGFR expression on day 15 prior to cryopreservation. All cell counts were performed using a Muse® Cell Analyzer (Luminex Corporation). Live and dead cell analyses were performed using Muse® Count & Viability Kit, following the manufacturer’s instructions.
Real-time cell analysis
The cytolytic function of PSCA CAR_s15 NK cells was evaluated by a real-time cell analysis (RTCA) assay (ACEA Bioscience, xCELLigence RTCA MP) following the manufacturer’s instructions. Target cells were seeded at a density of 8,000 cells/well and incubated in the xCELLigence RTCA MP instrument in a 37°C incubator. After 20–24 hours incubation, PSCA CAR_s15 NK cells were added to the target cells at indicated E:T ratio and data were collected at 15-minute intervals for 96 hours. The growth and cell index of target cells were measured following the manufacturer’s instructions.
Pancreatic cancer metastasis model
Male and female NOD scid gamma (NSG) immunodeficient mice, 8–12 weeks old, (The Jackson Laboratory) were engrafted with the Capan-1 or MIA PaCa-2 PC cell line expressing a luciferase_ZsGreen gene (Capan-1_luc or MIA PaCa-2_luc) by intraperitoneal (i.p.) injection (2×105 cells/mouse). Tumor engraftment was confirmed using the bioluminescence imaging prior to any treatment to ensure each mouse had a similar tumor burden. The engrafted mice were randomly distributed into vehicle or treatment groups, which had been thawed from liquid nitrogen then given to mice by both i.p. (1×106 engineered NK cells/dose) and intravenous (i.v.) (1×106 engineered NK cells/dose) injection. Twelve doses of PSCA CAR_s15 NK cells, s15 NK cells, or vehicle were given across three treatment courses. Each course contained 4 doses within two weeks. All animal experiments were conducted in accordance with ARRIVE, federal, state, and local guidelines with approval from the City of Hope Animal Care and Use Committee.
Statistical analysis
For group studies with normal distributions and continuous endpoints, data are presented as mean ± SEM. Student’s t test was used to compare two independent groups. For three or more groups, one-way ANOVA was performed. A linear mixed model was used to account for the variance-ovariance structure due to repeated measures from the same subjects. For survival data, survival functions are estimated by the Kaplan–Meier method and compared by log-rank test. All tests were two-sided, and p-values were adjusted for multiple comparisons by Holm’s procedure. A p-value less than 0.05 was defined as statistically significant. GraphPad 9.1.0, R.3.6.3. and SAS 9.4 were used for the statistical analysis in the current study.
Flow cytometry, ELISA, Bioluminescence imaging are listed in the supplementary materials.
RESULTS
PSCA is highly expressed in primary pancreatic tumors and correlated with poor survival in patients
PSCA expression in tumor cells is associated with a poor prognosis in certain malignancies25–28. To assess its expression in normal tissues compared to PC tissue, RNA and protein levels of PSCA expression were quantified. PSCA mRNA expression was consistently upregulated in PC compared to normal pancreas in seven independent studies15–19 (Fig. 1A). Of note, PSCA was among the top 1% of upregulated genes in three of the studies17–19. Based on data from The Cancer Genome Atlas Pancreatic adenocarcinoma (TCGA-PAAD), the mRNA expression of PSCA is significantly higher in primary PC compared to the adjacent normal tissue (p<0.01) (Supplementary Fig. 1). In addition, the expression of PSCA from these cases showed a positive correlation with the histologic grade of the PC (p<0.01) and was associated with shorter overall survival (p<0.001) (Fig. 1B, C). These clinical data imply that PSCA might be associated with tumor progression in pancreatic cancer (PC). To test if the observed trends in PSCA mRNA expression also occurred at the protein level, immunohistochemistry was performed on the pancreatic tumors and adjacent normal tissues (Fig. 1D). Out of 12 tumor samples collected from patients, nine (75%) were PSCA positive (7 strongly positive and 2 weakly positive samples), while non-malignant cells in the adjacent tissues showed negligible PSCA expression (Supplementary Fig. 2). Although PSCA was upregulated in the tumors, it was not detected in most normal tissues, including bone marrow, thyroid, adrenal, brain, skin, tonsil, spleen, uterus, placenta, testis, ovary, liver, kidney, heart, intestine, and lung. The expression of PSCA was identified in prostate and some gastric tissues (Fig. 1E). Collectively, these data prompted us to develop an immunotherapy for PC by targeting PSCA.
Figure 1. Assessment of PSCA as a therapeutic target for pancreatic cancer.

(A) PSCA expression in pancreatic tumors normalized to expression in normal adjacent tissues across 7 independent studies15–19. The blue color and (−) sign indicate downregulation while red color and (+) sign indicate upregulation in tumors relative to the normal tissues. The number and color scale represent the ranking in percentage of the indicated gene in each analysis. P-value was calculated by median-rank analysis. (B) PSCA mRNA expression in pancreatic tumors as determined by histological grade (Grade 1 n=31; Grade 2 n=97; Grade 3 n=50). Statistical analysis was performed by one-way ANOVA. (C) The overall survival of PC patients was estimated based on the expression level of PSCA (top 33% versus bottom 33%, n=57 each). P-values were determined by the log-rank test. (D) Representative images of immunohistochemistry (IHC) of PSCA protein expression from primary pancreatic tumors or adjacent benign normal tissues. IHC images were taken under 200X and the scale bar is 100 pixels. (E) IHC of PSCA expression in normal benign tissues as indicated. (F) Schematic of the clinical grade vectors. (G) The expression of tEGFR in the engineered and control NK cells after enrichment for the CD56(+)CD3(−) population, as measured by flow cytometry.
The PSCA CAR vector is efficiently transduced in umbilical cord blood-derived NK cells
To target PC, an anti-PSCA immunotherapy was developed using umbilical cord-derived primary NK cells. We engineered NK cells to express soluble IL-15 alone (s15 NK), PSCA CAR alone (PSCA CAR), or both s15 and PSCA CAR (PSCA CAR_s15) (Fig. 1F). The efficiency of viral transduction was evaluated via the surface expression of tEGFR, which was included in each of the constructs (Fig. 1G), among CD56(+)CD3(−) cells, which distinguishes NK cells from T cells. Further, to confirm that the PSCA CAR expression was congruent with expression of tEGFR, we compared expression of anti-human IgG-Fab antibody that binds to the PSCA CAR, with expression of tEGFR, and found a strong correlation, regardless of the efficiency of transduction (Supplementary Fig. 3). Thereafter, we assessed the efficiency of viral transduction into NK cells via the expression of tEGFR. To prepare for functional analyses, engineered NK cells were expanded with irradiated K562 feeder cells expressing membrane-bound IL-21 and CD137L. Transduced and non-transduced NK cells underwent a 1,000-fold expansion in 15 days regardless of the vector (Supplementary Fig. 4A). The purity of CD56(+)CD3(−) NK cells after expansion was 97.3± 2.1% (Supplementary Fig. 4B, 4C) with no feeder cells contamination after the 15day expansion (Supplementary Fig. 4D, 4E). In addition, the secretion of s15 was detected in the NK cell culture supernatants of the s15 NK cells and the PSCA CAR_s15 NK cells (but not the PSCA CAR NK cells) via an enzyme-linked immunosorbent assay (ELISA) (Supplementary Fig. 4F). These data demonstrate the successful engineering and manufacturing of human primary NK cells.
PSCA CAR NK cells specifically target PSCA-positive tumors
To select ideal targets for functional validation of the engineered PSCA CAR_s15 NK cells, the expression of PSCA was verified in 5 different PC cell lines and the K562 leukemia cell line as a negative control. Flow cytometry showed that AsPC-1, Capan-1, and MIA PaCa-2 cells are PSCA(+), while PANC-1 and BxPC-3 are PSCA(−) (Fig. 2A). Therefore, we chose one PSCA(+) (Capan-1) and one PSCA(−) (PANC-1) tumor cell line for functional validation. Both pancreatic tumor cell lines were co-cultured with s15 NK, PSCA CAR NK, or PSCA CAR_s15 NK cells for 48 hours. After that, a standard NK cell activation event (IFN-γ secretion) was evaluated through ELISA. The secretion of IFN-γ was enhanced by PSCA CAR NK cells (∼117.5 pg) compared to s15 NK cells (∼3.7 pg) (Fig. 2B). The induction of IFN-γ was even more robust in the PSCA CAR_s15 NK cells (∼313 pg) (Fig. 2B), demonstrating the additive effect of IL-15 combined with the PSCA CAR in IFN-γ induction. PSCA CAR_s15 NK cells also showed much higher cytotoxicity than s15 NK cells or PSCA CAR NK cells against PSCA(+) Capan-1 cells (Fig. 2C) but not PSCA(−) PANC-1 cells (Fig. 2D). The improved cytolytic function of PSCA CAR_s15 NK cells was further verified using RTCA. PSCA CAR_s15 NK cells showed significantly greater inhibition of PSCA(+) Capan-1 growth between 24 to 120 hours compared to s15 NK cells or PSCA CAR NK cells (Fig. 2E). On the contrary, PSCA CAR_s15 NK cells showed no inhibition of PSCA(−) PANC-1 growth between 24 to 120 hours. Further, a few primary cells were cultured with PSCA CAR _s15 NK cells and no significant cytotoxicity were found (Supplementary Fig. 5). These data indicate that co-expression of IL-15 and PSCA CAR on NK cells is beneficial and specific to suppress PSCA(+) tumor growth. Thus, our subsequent analyses focused on PSCA CAR_s15 NK cells and the parental control s15 NK cells.
Figure 2. The PSCA CAR_s15 vector enhances IFN-γ secretion and tumor lytic functions of NK cells in vitro.

(A) The median of fluorescence intensity (MFI) and the percentage of PSCA(+) cells in the indicated PC cell lines. (B) IFN-γ levels in the supernatants of co-cultured engineered NK cells and pancreatic tumor cells, as measured by enzyme-linked immunosorbent assay (ELISA). Statistical analysis was performed by one-way ANOVA. (C, D) Cytotoxicity of engineered NK cells against (C) PSCA(+) Capan-1 or (D) PSCA(–) PANC-1 tumor cells, at an effector: target (E:T) ratio of 1:3 measured after 48 hours of co-culture. (E, F) Real-time cell analysis (RTCA) of the cytotoxicity of engineered NK cells against (E) PSCA(+) Capan-1 or (F) PSCA(–) PANC-1 tumor cells with an E:T ratio of 1:3 and presented as the growth index of the residual cancer cells. The tumor only control group contained no NK cells. s15 = s15 NK cells; PSCA = PSCA CAR NK cells; PSCA-s15 = PSCA CAR_s15 NK cells.
The PSCA CAR enhances NK cytolytic function without altering native activation receptors
NK cells expanded with engineered K562 feeder cells should retain the expression profiles of native receptors on their surface29. To evaluate whether transduction and expression of the PSCA CAR along with sIL-15 altered the native receptors on PSCA CAR_s15 NK cells, a panel of NK activation markers (NKp30, NKp44, NKp46, NKG2D, CD16) as well as the inhibitory receptors (CD94, NKG2A, KIR-NKAT2), and immune checkpoint inhibitors (TIGIT, PD1, and CTLA-4) were analyzed on s15 NK and PSCA CAR_s15 NK cells by flow cytometry after stimulation with PSCA(+) Capan-1 tumor cells. All the native activation receptors were highly expressed on both s15 NK and PSCA CAR_s15 NK cells (Fig. 3A and Supplementary Fig. 6), suggesting the superior cytotoxicity observed in the PSCA CAR_s15 NK cells is likely due to the PSCA CAR, rather than non-specific activation of the NK cell and/or its activation receptors. TNF-α and IFNγ were also assessed in both s15 NK and PSCA CAR_s15 NK cells. Although their basal levels were similarly low in unstimulated s15 NK and PSCA CAR_s15 NK cells, both cytokines were significantly increased when stimulated with PSCA(+) Capan-1 cells (IFN-γ: 1% vs. 7%; TNF-α: 2% vs. 17%, respectively) (Fig. 3B, 3C). This suggested that the cytokine expression is driven by the PSCA CAR. The NK cell degranulation marker CD107a was significantly higher in the PSCA CAR_s15 NK cells (45%) compared to s15 NK cells (31%) following stimulation with Capan-1 cells (Fig. 3B, 3C). Collectively, these data demonstrated that the PSCA CAR confers activation signals to NK cells in the presence of PSCA(+) tumor cells.
Figure 3. The PSCA CAR_s15 vector increases NK cell functional markers without decreasing the native activation receptors.

(A) Flow cytometry analysis of NKp30, NKp44, NKp46, NKG2D, and CD16 on the CD 56(+) s15 or PSCA CAR_s15 NK cells stimulated with Capan-1 cells with an E:T cell ratio of 1:3 measured after 24 hours of co-culture. (B) Flow cytometry analysis of intracellular TNF-α, IFNγ, and CD107a on engineered CD56(+) NK cells without or with stimulation by Capan-1 cells. (C) Quantification of the results shown in panel B (n=3). P-values were determined by the t-test.
Engineered NK cells can be frozen as a viable and functional unit
One advantage of the NK cell-based therapy is its potential to be a ready-to-use allogeneic product, which can greatly reduce the time of manufacturing, improve distribution and scale, thereby reducing cost30. To this end, we optimized the conditions for cryopreservation to maintain PSCA CAR_s15 NK cell viability and function after one freeze-thaw cycle. The viable cell count immediately after thawing was typically 80% to 90% of the original cell count with 90–95% viability (Fig. 4A and Supplementary Fig. 7). The thawed PSCA CAR_s15 NK cells were stable in the freezing medium at room temperature for at least 3 hours and more than 70% of NK cells were alive 6 hours post thaw (Fig. 4A). The engineered proteins were stable post thaw, as the percentage of tEGFR(+) cells was similar between the fresh and freeze-thawed PSCA CAR_s15 NK cells (Fig. 4B). To test whether the effector function of freeze-thawed PSCA CAR_s15 NK cells was comparable to the fresh NK cells, s15 NK and PSCA CAR_s15 NK cells that were frozen for 2 weeks (Supplementary Fig. 8) or 6 months (Fig. 4C) were thawed and co-cultured with PSCA(+) Capan-1 PC cells. The cytolytic function was assessed using RTCA and compared with the same lot of PSCA CAR_s15 NK cells before (Fig. 4C, left panel, max cytolysis: 73%) and after freezing (Fig. 4C, right panel: max cytolysis: 68%), indicating the PSCA CAR_s15 NK cells could be stored as viable and functional units if proper freeze-thaw procedures are performed.
Figure 4. PSCA-directed CAR NK cells retain functional properties after a freeze-thaw cycle.

(A) Cell counts of previously frozen engineered NK cells (n=3/group) over time assessed by the MUSE cell analyzer post thaw. Cells were kept in the freezing buffer at room temperature once they were thawed. (B) Expression of the engineered constructs (assessed by measuring surface expression of tEGFR) before and after a freeze-thaw cycle (n=4). The cells shown in (A) and (B) were cryopreserved for two weeks prior to the assessment. (C) Cytotoxicity of fresh or NK cells derived from a same donor and frozen for 6-month, at an E:T ratio of 1:3 using the RTCA assay. (D) Comparison of NK cell distribution in mice after i.p. or i.v. delivery by flow cytometry. One dose of 8×106 frozen s15 NK cells was thawed then delivered to the immunocompromised mice and evaluated after 1 week. Cells positive for human CD45 and human CD56 were considered NK cells.
The distribution profile of the freeze-thawed NK cells in vivo
Next, the persistence and distribution of frozen NK cells were assessed in vivo. Frozen NK cells were thawed and injected i.p. or i.v. into NSG mice. A week later, cells from liver, bone marrow, lung, and pancreas were collected for flow cytometric analysis. NK cells were found to be alive in the lung, liver, bone marrow, and pancreas with various distribution profiles (Fig. 4D), suggesting our NK cells could survive and persist for at least one week in vivo after being thawed and injected. We noticed NK cells only accumulated in the pancreas when injected i.p. while NK cells accumulated more in the liver and lung when injected i.v. (Fig. 4D). The distribution of NK cells was verified by an image-based technique. PSCA CAR_s15 NK cells engineered to express a luciferase gene went through a freeze-thaw cycle and were given to the NSG mice either i.p. or i.v. Bioluminescence imaging (BLI) was performed in the heart, kidney, lung, pancreas, stomach, spleen, liver, mesentery, brain, and femur collected from injected mice one week post injection (Supplementary Fig. 9). BLI showed accumulation of CAR NK cells in the pancreas by i.p. injection while more CAR NK cells were accumulated in the lung when given by i.v. Both in vitro and in vivo experiments demonstrated that PSCA CAR_s15 NK cells could be frozen and stored as a viable and functional product. More importantly, PSCA CAR_s15 NK cells delivered i.p. trafficked to the pancreas, while PSCA CAR_s15 NK cells delivered i.v. trafficked to the lung. These data provide a strong rationale supporting i.p. administration of PSCA CAR_s15 NK cells for treatment of localized PC, whereas PSCA CAR_s15 NK cells could be administered i.v. to control pulmonary metastasis.
PSCA CAR_s15 NK cells inhibit tumor growth in a metastatic mouse model of human PC
We next combined i.p. and i.v. injections based on the rationale that the alternate routes of administration would kill tumor cells in the pancreas as well as the metastasis to liver and lung. To test the therapeutic efficacy of PSCA CAR_s15 NK cells, we established a human metastatic pancreatic cancer model (Figure 5A). The efficiency of transduction for the infused NK cells is shown in Figure 5B. Tumor progression was monitored by a luciferase-based imaging system. PSCA CAR_s15 NK cells showed superior therapeutic benefit compared to the other two groups at inhibiting the progression of metastatic PC (Fig. 5C, 5D). Furthermore, PSCA CAR_s15 NK cell treatment significantly prolonged the survival of the treated mice compared to both vehicle and s15 NK cell treatment (Fig. 5E) without changing the body weight (Supplementary Fig. 10) or behaviors.
Figure 5. PSCA CAR_s15 NK cells suppress PC in a metastatic mouse model.

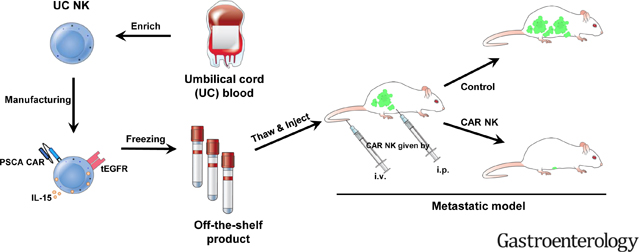
(A) A schematic of PSCA-directed treatment with PSCA CAR_s15 NK cells in a human metastatic PC model created by injecting Capan-1_luc cells into NSG mice. The figure was generated at BioRender (https://biorender.com/). (B) The efficiency of NK cell transduction used for in vivo experiments as measured by surface density expression of tEGFR. NT=non-transduced NK. (C) Time-lapse luciferase imaging of the metastatic PC mouse model after the indicated treatments (WK=week). (D) Quantification of the bioluminescence images from panel C up to day 32. Statistical analysis was performed using linear mixed models. (E) Survival analysis of mice in the indicated groups from panel C as analyzed by the log-rank test. (F) Representative images of pancreas (T=tumor tissue; P=pancreatic tissue) and liver in the PC mouse model with the indicated treatments 50 days post initial treatment. (G, H) Flow cytometric analyses of (G) tumor cells (ZsGreen, detected in the GFP channel) and (H) NK cells (human CD45(+)/CD56(+)) from pancreas and liver. Both tumor and NK cells were gated on the singlets and living (SYTOX-) lymphocyte gate.
To better understand the progression of PC and the benefits of PSCA CAR_s15 NK cell therapy, mice treated with vehicle only, s15 NK cells, or PSCA CAR_s15 NK cells were euthanized for flow cytometric and pathologic analyses at 50 days post the initial treatment. Pancreas, liver, spleen, lung, bone marrow, and peripheral blood were collected, and photographed to evaluate the metastatic tumor area (Fig. 5F). Flow cytometric analyses from liver and pancreas showed the persistence of infused human NK cells and clearance of PC cells by PSCA CAR_s15 NK cells (Fig. 5G, 5H). Of note, the proportion of NK cells in the pancreas and liver of the PSCA CAR_s15treated group of mice was higher than in either the vehicle- or s15 NK cell-treated groups, suggesting that the PSCA CAR itself traffics PSCA CAR_s15 NK cells to the tumor sites and sustains PSCA CAR_s15 NK cells in vivo (Fig. 5H). Other organs such as bone marrow, spleen, lung, or peripheral blood also showed a higher percentage of NK cells in the PSCA CAR_s15 NK cell-treated mice compared to either the vehicle- or s15 NK cell-treated groups (Supplementary Fig. 11A). Of note, very few tumor cells were detected in the bone marrow, spleen, and peripheral blood in all three groups, suggesting that tumor cells do not traffic to these sites (Supplementary Fig. 11B). Immunohistochemistry showed that most of the pancreas, liver, mesenteric lymph nodes (MLNs), and lung expressed negligible PSCA protein after PSCA CAR_s15 NK cell treatment, suggesting that the tumor cells were eradicated (Fig. 6A). H&E staining supported the immunohistochemistry results and demonstrated no obvious tissue damage after NK cell treatment (Fig. 6B). MUC1, a pancreatic tumor cell marker31, showed a similar staining profile to PSCA (Fig. 6C), suggesting the absence of PSCA after PSCA CAR_s15 NK cell treatment was a sign of tumor eradication rather than antigen loss. Moreover, the presence of infused NK cells (stained with human anti-NKp46) was detected in the liver, pancreas, MLNs, and lung in both s15 NK and PSCA CAR_s15 NK cell-treated mice around 50 days post first NK injection (Supplementary Fig. 12) and persisted in the PSCA CAR_s15 NK cell-treated mice more than 90 days in vivo following their last infusion. (Supplementary Fig. 13). To ensure the potency of PSCA CAR_s15 NK cells could be reproduced in vivo, the PSCA(+) cancer cell line MIA PaCa2 expressing luciferase_ZsGreen was used to establish another metastatic PC model. Mice treated with PSCA CAR NK cells showed greater anti-tumor efficacy and survival benefits compared to the vehicle as well as s15 NK cells (Supplementary Fig. 14). Thus, our data indicate that these ready-to-use PSCA CAR_s15 NK cells can persist in the pancreas and can target organs with metastatic disease such as liver and lung resulting in prolonged survival in a metastatic PC model.
Figure 6. Absence of tumor cells after PSCA-directed CAR NK cell treatment.

Representative images from (A) anti-human PSCA immunohistochemistry (IHC) (B) hematoxylin and eosin (H&E) staining, and (C) anti-human MUC1 IHC images of the indicated tissues from the metastatic PC mouse model after the indicated treatments under the indicated magnification. Scale bar is 100 pixels.
Truncated EGFR functions as a safety switch on PSCA CAR_s15 NK cells
A major challenge for CAR-T based immunotherapy is the occurrence of cytokine release syndrome and neurotoxicity, which are caused by immune responses following the infusion of CAR-T cells32. CAR NK cell-based therapy has been recently shown to be safe in patients with lymphoid malignancies, but only one study has been published8. To reduce the risk of toxicity from our CAR NK therapy, we incorporated tEGFR as a suicide gene. To test this cell suicide strategy, PSCA CAR_s15 NK cells (also expressing tEGFR) were co-expressed with luciferase_ZsGreen for in vivo tracing (Fig. 7A). The population was enriched for NK cells expressing tEGFR and ZsGreen (Fig. 7B), and then injected into immunodeficient NSG mice one day after Capan-1 PC cells were engrafted. The FDA-approved anti-EGFR antibody cetuximab was given at 24 hours, 5 days, and 10 days post NK infusion. The dose of cetuximab (1 mg/mouse; ∼25 g per mouse) was calculated based on guidelines from human usage (0.04 mg/cm2)23. To monitor the persistence of PSCA CAR_s15 NK cells, a luciferase-based imaging system was applied in a time-lapse manner. The luciferase signals in the cetuximab-treated mice were 4-fold lower than in the vehicle treated group after 1 dose of cetuximab (Fig. 7C, 7D), indicating that the population of engineered NK cells had significantly decreased in the cetuximab-treated mice but not the vehicle-treated mice. The bioluminescence signals were no longer detectable after 3 doses of cetuximab at day 16 (Fig. 7C, 7D). Also, PSCA CAR_s15 NK cells expressing both hCD56 and ZsGreen were found in the liver, pancreas, lung, and peripheral blood in the vehicle-treated group but not the cetuximab-treated group (Fig. 7E), and this was confirmed by histologic analysis using hNKp46 (Fig. 7F), suggesting that PSCA CAR_s15 NK cell clearance was achieved by cetuximab. No obvious tissue damage was found in either group (Supplementary Fig. 15), providing a good rationale to use this safety switch for future clinical studies.
Figure 7. EGFR functions as a safety switch for PSCA CAR NK cells in vivo.

(A) Schematic of the two vectors used to transduce NK cells. (B) The purity of PSCA CAR_s15 NK cells co-expressing luciferase_ZsGreen, as determined by flow cytometry for tEGFR and ZsGreen. (C) Time-lapse luciferase imaging of PSCA CAR_s15 NK cells after cetuximab or vehicle (saline) treatment (n=3/group). (D) Quantification of the bioluminescence shown in panel C. Statistical analysis was performed using linear mixed models. (E) Persistence of infused NK cells in the indicated organs after the indicated treatments, as shown by flow cytometry for ZsGreen and human CD56. (F) Representative images of IHC for human NKp46 in the indicated organs after the indicated treatments. Infused NK cells are indicated by arrows.
DISCUSSION
In the current study, we demonstrated the ability of PSCA CAR_s15 NK cells to reduce human metastatic PC in a mouse model. We developed and optimized a platform for generating off-the-shelf frozen human PSCA CAR_s15 NK cells without compromising their efficacy in vivo. In addition, we demonstrated that our engineered NK cells can be successfully frozen, stored, thawed and administered, increasing the potential ease of manufacturing, distribution and clinical application. The metastatic PC model developed in the current study recapitulates several clinical features of PC, including the development of ascites and metastasis to the liver and lung. Using both local (i.p.) and systemic (i.v.) delivery, we demonstrated that administration of PSCA CAR_s15 NK cells could significantly reduce tumor burden and improve survival in two different metastatic PC models. We also engineered a suicide switch, tEGFR, that can function as a traceable marker and, with the administration of cetuximab, can remove unwanted PSCA CAR_s15 NK cells in vivo. No obvious tissue damage was found in the mice receiving PSCA CAR_s15 NK cell therapy or PSCA CAR_s15 NK cell therapy coupled with cetuximab, demonstrating the potency and safety of our novel approach for the treatment of PC.
The advantages of CAR NK cell therapy over CAR T cell may include the following: 1) limited clonal expansion in vivo, with early clinical indications of a better safety profile with reduced risk of CRS, neurotoxicity and graft versus-host disease8; 2) intrinsic anti-tumor function independent of tumor antigen expression, enhancing the ability to eradicate tumors even after tumor antigen loss. This notation is partially supported by the reduced tumor burden and significant survival advantage of the s15 NK treated mice compared to the vehicle treated mice in our Capan-1 metastatic model seen in Figure 5C–G; and 3) the potential to use frozen, off-the-shelf allogeneic products with lower cost and a shorter time-to-treat interval compared to autologous CAR T cell products. To select PSCA as a potential target for developing immunotherapy, we combined bioinformatics and histological analysis and discovered that PSCA is a reasonable target for PC. Aberrant expression of PSCA has been reported in other malignancies such as prostate, gastric, and lung cancers25–28. Thus, developing a PSCA CAR_s15 NK cell therapy should create an opportunity not only for PC patients but also possibly for others who carry PSCA(+) tumorassociated antigen.
One unique feature of the PSCA CAR_s15 NK therapy is administration of the NK cells via both the i.v. and i.p. routes. Although several studies have demonstrated the benefits of i.v. CAR-T cell therapy for PC in mouse models33, 34, it is not yet evident that i.v. PSCA CAR T cell immunotherapy has had promising outcomes in the PC patients35. Unlike most of the CAR-based therapies that have thus far been given i.v., our preclinical study clearly showed differences in NK distribution between i.p. and i.v. delivery, which led us to propose a combination of i.v. and i.p. for better delivery of PSCA CAR_s15 NK cells to the original (pancreas) and metastatic (liver, and lung) tumor sites, respectively. Regional delivery is being pursued in both preclinical models and in patients. CAR-T cells delivered via intra-pleural infusion showed more potency than those delivered i.v. in a pleural mesothelioma model36. NK cells administered i.p. to patients with colorectal cancer resulted in a reduction of ascites formation37. These studies along with ours demonstrate the potential benefits of local or regional delivery of engineered immune effector cells, providing additional avenues to pursue in early phase clinical trials of solid tumors.
Recently, the use of a safety switch-based approach to mitigate the side effects of adaptive T cell immunotherapy has attracted a great deal of attention because it directly targets the infused cells. Inclusion of an inducible Caspase 9 (iCas9) with the CAR T cell is currently being assessed in phase I clinical trials for treatment of neuroblastoma (NCT01822652), sarcoma (NCT01953900), osteosarcoma, and melanoma (NCT02107963). EGFR, a common receptor on the epithelial cells that is not expressed on normal lymphocytes23, and a truncated version was used as a traceable marker as well as a suicide switch on the PSCA CAR_s15 NK cells in the current study. The advantages of including tEGFR in our engineered NK cells are as follows: 1) no EGFR downstream signaling would be triggered in our truncated molecule due to lack of both the juxtamembrane domain and tyrosine kinase domain23; 2) unlike a rituximab-activated CD20-based suicide switch, which would also kill normal B cells as a side effect of rituximab38, the EGFR-based switch does not kill other lymphocytes; 3) administration of pharmaceutical grade cetuximab to the mice after PSCA CAR_s15 NK cell therapy eliminated nearly all NK cells within 4 days without any observed tissue damage; and 4) cetuximab may have additional anti-tumor activity given that 30–95% of PC cells express EGFR39, 40.
Like other immunotherapies, the PSCA CAR_s15 NK cell therapy proposed here will most likely function as a second- or third- line therapy. Thus, the detection of the PSCA is expected to be performed on the patients who are resistant to the standard chemotherapy or after surgical resection. Patient specimens could be obtained through CT- or endoscopic ultrasound (EUS)-guided fine-needle aspiration biopsy (FNA) or obtained after surgery for IHC staining. Given that PSCA CAR_s15 NK cell therapy could reduce the size of the tumor after 3 weeks of treatment on its own, combining PSCA CAR_s15 NK cell therapy with other immunotherapies such as CART or bi-specific T cell or killer cell engagers that target different tumor antigens, might further enhance the therapeutic efficacy of PSCA CAR_s15 NK cells. Li et al. showed that combining two CAR-T therapies (anti-PSCA and anti-MUC1) could efficiently suppress non-small cell lung cancer growth in patient-derived xenograft mice41. Thus, it is reasonable to propose a new therapy to combine the current PSCA-directed CAR NK therapy and another established immunotherapy targeting one or more of the tumor antigens overexpressed in PC patients, including CEA, MUC1, and mesothelin42. Although PSCA CAR_s15 NK cells are potent to destroy PC cells, the former express high levels of inhibitory receptors such as CD94, NKG2A, and TIGIT. This supports a concept that inhibitory receptors can also mark cell activation, although they would introduce negative signals to effector cells when encountering their ligands on target cells. In a future clinical setting, this provides an opportunity to combine PSCA CAR_s15 NK cells with agents targeting negative signaling in these effector cells introduced by the inhibitor receptors.
To our knowledge, this report marks a novel approach, i.e., a PSCA CAR_s15 NK cell therapy, to treat PC using an i.p. and i.v. approach. Our data demonstrate the potential of PSCA CAR_s15 NK cells to be a frozen, “off-the-shelf” product based on in vitro and in vivo data. These results could possibly be further extended to other solid tumor malignancies alone or in combination with other forms of therapy.
Supplementary Material
“What You Need to Know”
BACKGROUND AND CONTEXT:
Pancreatic cancer is a lethal cancer that is often diagnosed at the late stage with poor survival due to limited treatment options. Cell therapy has not yet been shown to be successful for the treatment of pancreatic cancer.
NEW FINDINGS:
Off-the-shelf NK cells expressing anti-PSCA CAR, IL-15, and a truncated EGFR-based suicide switch, delivered both locally and systematically, suppress tumor progression and prolong survival in a mouse model of metastatic pancreatic cancer.
LIMITATIONS:
Combination therapy to further improve survival of mice bearing metastatic pancreatic tumors should be explored.
IMPACT:
Targeting PSCA using CAR NK cells could be a pivotal approach toward improving the survival of pancreatic cancer patients.
“Lay Summary”
Ready to use, off-the-shelf frozen chimeric antigen receptor nature killer cells show impressive efficacy to treat the late-stage metastatic pancreatic cancer in a pre-clinical mouse model.
ACKNOWLEDGEMENT
We specially thank Dr. Yuan Chen (formerly City of Hope and now University of California-San Diego) for the pancreatic cancer cell lines. The tissue preparation and staining were performed in the City of Hope Comprehensive Cancer Center Pathology Core that is supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572.
Grant Support: This work was supported by grants from the NIH (CA210087, CA265095, and CA163205 to M.A. Caligiuri; NS106170, AI129582, CA247550, and CA223400 to J. Yu), the Leukemia and Lymphoma Society (1364–19 to J. Yu), and Gabrielle’s Angel Foundation for Cancer Research (to A.G. Mansour). KY Teng was supported by an NIH T32 award (T32CA221709).
Abbreviations:
- CAR
Chimeric antigen receptor
- EGFR
epidermal growth factor receptor
- NK
natural killer
- PSCA
prostate stem cell antigen
Footnotes
Transcript Profiling: N/A
Conflict of Interest Disclosure: Drs. Caligiuri and Yu are co-founders of CytoImmune Therapeutics, Inc. Other authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Society AC. Cancer Facts & Figures 2021. Cancer Statistics Center 2021.
- 2.Mota Reyes C, Teller S, Muckenhuber A, et al. Neoadjuvant Therapy Remodels the Pancreatic Cancer Microenvironment via Depletion of Protumorigenic Immune Cells. 2020;26:220–231. [DOI] [PubMed] [Google Scholar]
- 3.Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 2010;33:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caligiuri MA. Human natural killer cells. Blood 2008;112:461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu J, Freud AG, Caligiuri MA. Location and cellular stages of natural killer cell development. Trends Immunol 2013;34:573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson JA, Leveson-Gower DB, Gill S, et al. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010;115:4293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu E, Marin D, Banerjee P, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 2020;382:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han J, Chu J, Keung Chan W, et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci Rep 2015;5:11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li E, Liu L, Li F, et al. PSCA promotes prostate cancer proliferation and cell-cycle progression by up-regulating c-Myc. 2017;77:1563–1572. [DOI] [PubMed] [Google Scholar]
- 12.Gu Z, Thomas G, Yamashiro J, et al. Prostate stem cell antigen (PSCA) expression increases with high gleason score, advanced stage and bone metastasis in prostate cancer. Oncogene 2000;19:1288–96. [DOI] [PubMed] [Google Scholar]
- 13.Wente MN, Jain A, Kono E, et al. Prostate stem cell antigen is a putative target for immunotherapy in pancreatic cancer. Pancreas 2005;31:119–25. [DOI] [PubMed] [Google Scholar]
- 14.Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol 2003;16:902–12. [DOI] [PubMed] [Google Scholar]
- 15.Buchholz M, Braun M, Heidenblut A, et al. Transcriptome analysis of microdissected pancreatic intraepithelial neoplastic lesions. Oncogene 2005;24:6626–36. [DOI] [PubMed] [Google Scholar]
- 16.Grützmann R, Pilarsky C, Ammerpohl O, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia 2004;6:611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pei H, Li L, Fridley BL, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009;16:259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iacobuzio-Donahue CA, Maitra A, Olsen M, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am J Pathol 2003;162:1151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas PanCancer analysis project. Nature genetics 2013;45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossman RL, Heath AP, Ferretti V, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med 2016;375:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schluns KS, Williams K, Ma A, et al. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol 2002;168:4827–31. [DOI] [PubMed] [Google Scholar]
- 22.Carson WE, Fehniger TA, Haldar S, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest 1997;99:937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Chang WC, Wong CW, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011;118:1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paszkiewicz PJ, Fräßle SP, Srivastava S, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest 2016;126:4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Priceman SJ, Gerdts EA, Tilakawardane D, et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology 2018;7:e1380764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murad JP, Tilakawardane D, Park AK, et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol Ther 2021. [DOI] [PMC free article] [PubMed]
- 27.Wu D, Lv J, Zhao R, et al. PSCA is a target of chimeric antigen receptor T cells in gastric cancer. Biomark Res 2020;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawaguchi T, Sho M, Tojo T, et al. Clinical significance of prostate stem cell antigen expression in non-small cell lung cancer. Jpn J Clin Oncol 2010;40:319–26. [DOI] [PubMed] [Google Scholar]
- 29.Caligiuri MA, Velardi A, Scheinberg DA, et al. Immunotherapeutic approaches for hematologic malignancies. Hematology Am Soc Hematol Educ Program 2004:337–53. [DOI] [PubMed]
- 30.Mansour AG, Teng K-Y, Lu T, et al. CAR-NK cell immunotherapy: Development and challenges toward an off-the-shelf product. Successes and Challenges of NK Immunotherapy: Elsevier, 2021:213–230. [Google Scholar]
- 31.Terada T, Ohta T, Sasaki M, et al. Expression of MUC apomucins in normal pancreas and pancreatic tumours. J Pathol 1996;180:160–5. [DOI] [PubMed] [Google Scholar]
- 32.Yáñez L, Sánchez-Escamilla M, Perales M-A. CAR T Cell Toxicity: Current Management and Future Directions. HemaSphere 2019;3:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Posey AD Jr., Schwab RD, Boesteanu AC, et al. Engineered CAR T Cells Targeting the CancerAssociated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016;44:1444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raj D, Nikolaidi M, Garces I. CEACAM7 Is an Effective Target for CAR T-cell Therapy of Pancreatic Ductal Adenocarcinoma. 2021;27:1538–1552. [DOI] [PubMed] [Google Scholar]
- 35.Beatty GL, O’Hara MH, Lacey SF, et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018;155:29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adusumill PS, Cherkassky L, Villena-Vargas J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 2014;6:261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao L, Cen D, Gan H, et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol Ther 2019;27:1114–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLaughlin P, Grillo-López AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998;16:2825–33. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers 2011;3:1513–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bloomston M, Bhardwaj A, Ellison EC, et al. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig Surg 2006;23:74–9. [DOI] [PubMed] [Google Scholar]
- 41.Wei X, Lai Y, Li J, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017;6:e1284722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akce M, Zaidi MY, Waller EK, et al. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front Immunol 2018;9:2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.