

Abstract
The relationship between in vivo synaptic density and molecular pathology in primary tauopathies is key to understanding the impact of tauopathy on functional decline and in informing new early therapeutic strategies. In this cross-sectional observational study, we determine the in vivo relationship between synaptic density and molecular pathology in the primary tauopathies of progressive supranuclear palsy and corticobasal degeneration as a function of disease severity.
Twenty-three patients with progressive supranuclear palsy and 12 patients with corticobasal syndrome were recruited from a tertiary referral centre. Nineteen education-, sex- and gender-matched control participants were recruited from the National Institute for Health Research ‘Join Dementia Research’ platform. Cerebral synaptic density and molecular pathology, in all participants, were estimated using PET imaging with the radioligands 11C-UCB-J and 18F-AV-1451, respectively. Patients with corticobasal syndrome also underwent amyloid PET imaging with 11C-PiB to exclude those with likely Alzheimer’s pathology—we refer to the amyloid-negative cohort as having corticobasal degeneration, although we acknowledge other underlying pathologies exist. Disease severity was assessed with the progressive supranuclear palsy rating scale; regional non-displaceable binding potentials of 11C-UCB-J and 18F-AV-1451 were estimated in regions of interest from the Hammersmith Atlas, excluding those with known off-target binding for 18F-AV-1451. As an exploratory analysis, we also investigated the relationship between molecular pathology in cortical brain regions and synaptic density in subcortical areas.
Across brain regions, there was a positive correlation between 11C-UCB-J and 18F-AV-1451 non-displaceable binding potentials (β = 0.4, t = 3.6, P = 0.001), independent of age or time between PET scans. However, this correlation became less positive as a function of disease severity in patients (β = −0.02, t = −2.9, P = 0.007, R = −0.41). Between regions, cortical 18F-AV-1451 binding was negatively correlated with synaptic density in subcortical areas (caudate nucleus, putamen). Brain regions with higher synaptic density are associated with a higher 18F-AV-1451 binding in progressive supranuclear palsy/corticobasal degeneration, but this association diminishes with disease severity. Moreover, higher cortical 18F-AV-1451 binding correlates with lower subcortical synaptic density. Longitudinal imaging is required to confirm the mediation of synaptic loss by molecular pathology. However, the effect of disease severity suggests a biphasic relationship between synaptic density and molecular pathology with synapse-rich regions vulnerable to accrual of pathological aggregates, followed by a loss of synapses in response to the molecular pathology.
Given the importance of synaptic function for cognition and action, our study elucidates the pathophysiology of primary tauopathies and may inform the design of future clinical trials.
Keywords: primary tauopathies, PSP, CBD/CBS, synapse, tau
Using 11C-UCB-J and 18F-AV-1451 PET imaging in primary tauopathies, Holland et al. reveal a biphasic relationship between synaptic density and molecular pathology. Brain regions with higher synaptic density are vulnerable to the accumulation of pathological aggregates, but this accumulation then results in synaptic loss.
Introduction
Synaptic loss is a feature of many neurodegenerative disorders.1-3 It is closely related to cognitive decline in symptomatic stages of disease,4,5 but can begin long before symptom onset and neuronal loss.6 Synaptic loss and dysfunction may be an important mediator of decline even where atrophy is minimal or absent.7,8 Conversely, synaptic connectivity may facilitate the spread of oligomeric misfolded proteins such as tau.9–14 The relationship between synaptic loss and the accumulation of misfolded proteins in primary tauopathies has yet to be determined in vivo. Preclinical models suggest early synaptotoxicity of oligomeric tau, leading to reduced synaptic plasticity and density.15,16 In patients with mutations of microtubule-associated protein tau (MAPT), there are deficiencies in many synaptic pathways including GABA-mediated signalling and synaptic plasticity.17 The mechanisms of synapse loss following tau pathology include both direct and indirect pathways (reviewed by Spires-Jones and Hyman18); however, the severity of synaptic toxicity in the related tauopathy of Alzheimer’s disease appears to be dependent on the stage of disease in preclinical models and in patients post-mortem and in vivo. In animal models of Alzheimer’s disease, and at human post-mortem, there is differential expression of synaptic proteins in the early stages with increases in some proteins and reductions in others.19,20 This may be an attempt to maintain cellular physiology in early disease, which fails as the disease progresses, leading to loss of synaptic function and synapse numbers in moderate and advanced disease. In clinical disorders, the in vivo pathologies of synaptic density and tau burden can be characterized by PET. Recent in vivo PET imaging in Alzheimer’s disease using 11C-UCB-J as a marker of synaptic density and 18F-AV-1451 or 18F-MK-6240 PET as markers of tau pathology have shown decreased temporal lobe synaptic density with increasing pathological burden,21 but with individual variability depending on the severity of cortical pathology.22 However, the pathology of Alzheimer’s disease is multifaceted with amyloid and tau aggregation, vascular changes and neuroinflammation.23
In this study, we use progressive supranuclear palsy–Richardson’s syndrome (PSP)24 and corticobasal degeneration (CBD)25 as models of human tauopathy, with relevance to other tau-mediated neurodegenerative disorders, and examine the in vivo relationship between synaptic density and burden of molecular pathology. An advantage of studying PSP is the very high correlation between the clinical syndrome, and the specific 4R-tauopathy at autopsy.26,27 The clinical phenotype of corticobasal syndrome (CBS), may be caused by CBD, but can also be mimicked by Alzheimer’s disease and less commonly by other forms of frontotemporal lobar degeneration. Here, we use the term CBD to refer to patients with CBS in whom Alzheimer’s disease is excluded by 11C-PiB PET, whereby in the absence of amyloid pathology there is a high clinicopathological correlation with 4R-tauopathy at post-mortem. Both PSP and CBD demonstrate synaptic loss in vivo7,8 and at post-mortem examination.1,2 The distribution of tau pathology in both diseases is well characterized with cortical and subcortical involvement.28,29 Animal models of tauopathy have illustrated the co-localization of tau aggregates at the synaptic bouton, associated with synaptic dysfunction and synaptic loss18,30 but the tau–synapse association is yet to be determined in vivo. 18F-AV-1451 signals are above normal in the cortex of patients with PSP and CBS/CBD,31–37 but there is relatively low affinity for 4R-tauopathy compared to Alzheimer’s disease, and off-target binding particularly in the basal ganglia. We therefore refer to 18F-AV-1451 as a marker of ‘molecular pathology’, referring to the combination of tau and non-tau targets.
Figure 1 illustrates our hypotheses. Previous studies suggest that the strength of connectivity within a region and between brain regions can promote the spread of tau pathology, in humans as in preclinical models.9–14 Therefore, we hypothesized that brain areas with higher synaptic density would develop more tau pathology (schematically represented by green arrows in Fig. 1A). We predicted that the spatial distribution of molecular pathology, as measured with the PET radioligand 18F-AV-1451, would be correlated with synaptic density, as measured with the PET radioligand 11C-UCB-J (which binds to the presynaptic vesicle glycoprotein SV2A that is ubiquitously expressed in brain synapses38,39). Because pathology in a region may impair efferent projections, a corollary hypothesis is that tau accumulation in one region (source region) leads to diaschisis characterized by reduced synaptic density in the areas to which it connects (target regions).
Figure 1.

Schematic diagram illustrating the predicted toxic effect of tau on synaptic density as a function of disease severity. At a regional level (A) synaptic density promotes the spread of tau within the region, but also from one region to another functionally connected region (for example from Region 1 to Region 2 or vice versa; depicted by green arrows). However, tau is toxic to synapses, such that at a regional level it leads to a loss of synapses as the disease progresses. (B) Tau burden within a given region therefore depends on a region’s baseline synaptic density: for example, Region 3, with a high baseline synaptic density, would accumulate more tau in the mild stages of disease (green); but as the disease progresses over time, to moderate and advanced stages (yellow and red, respectively), with increasing tau accumulation, tau-induced synapto-toxicity occurs with a decline in the number of synapses within any given region. Therefore, the prediction would be that, while in mild disease the degree of tau accumulation is dependent on baseline synaptic density, as the disease progresses this relationship breaks down, moving towards a negative association between tau accumulation and synaptic density.
A second part of the model describes the consequence of the pathology, which is to reduce synaptic density (schematically represented by red arrows in Fig. 1A). The predicted result is a positive relationship between 18F-AV-1451 binding and synaptic loss, negatively moderated by disease severity (Fig. 1B).
Materials and methods
Participant recruitment and study design
Twenty-three patients with probable PSP–Richardson syndrome and 12 with probable CBS in whom Alzheimer’s disease was excluded with 11C-PiB PET were recruited from a regional specialist National Health Service clinic at the Cambridge University Centre for Parkinson-plus. We refer to our amyloid-negative CBS cohort as having CBD but acknowledge other pathologies are possible. Nineteen healthy volunteers were recruited from the UK National Institute for Health Research Join Dementia Research register. Participants were screened using the inclusion/exclusion criteria set out in Holland et al.8 Eligible participants underwent clinical and cognitive assessments (Table 1) including the revised Addenbrooke’s Cognitive Examination (ACE-R), the Mini-Mental State Examination (MMSE), and the Institute of Cognitive Neurology (INECO) frontal screening; disease severity was measured with the PSP rating scale, and the Cortical Basal ganglia Functional Scale.40 Participants underwent 3 T MRI, 18F-AV-1451 PET and 11C-UCB-J PET. The research protocol was approved by the Cambridge Research Ethics Committee (reference 18/EE/0059) and the Administration of Radioactive Substances Advisory Committee. All participants provided written informed consent in accordance with the Declaration of Helsinki.
Table 1.
Clinical and demographics summary
| Control | PSP | CBD | F(P) | |
|---|---|---|---|---|
| Gender, male:female | 11:8 | 10:13 | 7:5 | nsa |
| Age at 11C-UCB-J PET in years | 68.9 (7.1) | 71.3 (8.6) | 70.9 (7.9) | ns |
| Symptom duration, years | – | 3.9 (2.2) | 3.9 (2.1) | ns |
| Education, years | 13.6 (2.8) | 12 (4.2) | 12.6 (2.8) | ns |
| ACE-R total (max. 100) | 96.7 (2.7) | 80.9 (12.4) | 77.5 (17.1) | 10.1 (<0.001) |
| Attention_Orientation (max 0.18) | 17.9 (0.3) | 16.3 (1.9) | 16.3 (2.3) | 4.5 (0.02) |
| Memory (max 0.26) | 24.6 (1.7) | 21.8 (3.8) | 20.9 (5.3) | 5.3 (0.01)) |
| Fluency (max 0.14) | 12.8 (1.0) | 6.6 (3) | 7.2 (3.5) | 28.0 (<0.001) |
| Language (max 0.26) | 25.6 (0.8) | 23.3 (4.2) | 21 (7.2) | 5.4 (0.01) |
| Visuospatial (max 0.16) | 15.7 (0.6) | 12.8 (3.4) | 12.1 (4.6) | 7.5 (0.001) |
| MMSE (max. 30) | 29.4 (1.2) | 26.9 (2.6) | 25.3 (4.9) | 6.7 (0.002) |
| INECO (max. 30) | 25.7 (2.1) | 17.2 (5.4) | 15.4 (6.5) | 17.9 (<0.0001) |
| PSPRS (max. 100) | – | 32.7 (8.2) | 28.9 (10.0) | 3.4 (0.07) |
| CBFS (max. 120) | – | 32.7 (15.9) | 26.2 (16.2) | 0.2 (0.7) |
| Injected activity, MBq | ||||
| 11C-UCB-J | 370.7 (114.3) | 322.2 (86.0) | 320.4 (113.8) | ns |
| 18F-AV-1451 | 182.3 (10.8) | 182.1 (11.4) | 186.1 (11.1) | ns |
| 11C-UCB-J and 18F-AV-1451 PET scan interval, in days | 157.2 (125.6) | 155.9 (129.2) | 45.5 (65.7) | 4.6 (0.02) |
Results are given as mean (and standard deviation) unless otherwise stated. PSP refers to patients with PSP–Richardson’s syndrome. CBD refers to amyloid negative corticbasal syndrome. The F-statistic and P-values are derived from ANOVA. ACE-R = revised Addenbrooke’s Cognitive Examination; CBFS = Cortical Basal ganglia Functional Scale; INECO = Institute of Cognitive Neurology frontal screening tool; MMSE = Mini-Mental State Examination; PSPRS = Progressive Supranuclear Palsy Rating Scale.
aChi-squared test. ns = non-significant at P < 0.05.
PET data acquisition and kinetic analysis
11C-UCB-J PET
The procedure for 11C-UCB-J synthesis, PET data acquisition, image reconstruction and kinetic analysis was the same as in Holland et al.8 In brief, dynamic PET data acquisition was performed on a GE SIGNA PET/MR (GE Healthcare) for 90 min immediately after injection, with attenuation correction using a multisubject atlas method41 and improvements to the MRI brain coil component.42 Emission image series were aligned using SPM12 (www.fil.ion.ucl.ac.uk/spm/software/spm12/) and rigidly registered to the T1-weighted MRI acquired during PET data acquisition (repetition time = 3.6 ms, echo time = 9.2 ms, 192 sagittal slices, in plane resolution 0.55 × 0.55 mm, interpolated to 1.0 × 1.0 mm; slice thickness 1.0 mm). The Hammersmith atlas (http://brain-development.org) with modified posterior fossa regions was spatially normalized to the T1-weighted MRI of each participant using advanced normalization tools software.43 Regional time–activity curves were extracted following the application of geometric transfer matrix (GTM) partial volume correction (PVC44) to each dynamic PET image. Regions of interest were multiplied by a binary grey matter mask (>50% on the SPM12 grey matter probability map smoothed to PET spatial resolution), with the exception of the subcortical grey matter regions pallidum, substantia nigra, pons and medulla. To assess the impact of PVC, time–activity curves were also extracted from the same regions of interest without the application of GTM PVC (discussed below as ‘without partial volume correction’).
To quantify SV2A density, 11C-UCB-J non-displaceable binding potential (BPND) was determined using a basis function implementation of the simplified reference tissue model,45 with the reference tissue defined in the centrum semiovale.46,47
18F-AV-1451 PET
As for 11C-UCB-J, PET data acquisition was performed on a GE SIGNA PET/MR for 90 min after 18F-AV-1451 injection, with attenuation correction as described above for 11C-UCB-J. Image processing was also as given above for 11C-UCB-J, except that 18F-AV-1451 BPND was determined using a different basis function implementation of the simplified reference tissue model48 and the reference tissue was defined in inferior cerebellar grey matter using a 90% threshold on the grey matter probability map produced by SPM12 smoothed to PET resolution.
11C-PiB PET
Amyloid imaging using Pittsburgh Compound B (11C-PiB) followed the protocol given in Holland et al.811C-PiB cortical standardized uptake value ratio (SUVR; 50–70 min post injection) was calculated using the whole cerebellum reference tissue as per the Centiloid Project methodology.49 A negative amyloid status was characterized by a cortical 11C-PiB SUVR < 1.21 obtained by converting the Centiloid cut-off of 19 to SUVR using the Centiloid-to-SUVR transformation in Jack et al.50
Statistical analyses
We compared demographic and clinical variables between the diagnostic groups using analysis of covariance (ANCOVA), and chi-square tests where appropriate. We used a linear mixed effects model to assess the overall relationship between 18F-AV-1451 and 11C-UCB-J BPND, with age and scan interval as covariates. To adjust for normal levels of tracer uptake from off-target binding not present in the reference region (over and above the correction for non-specific binding in the reference region), we normalized the patient BPND data against controls by subtracting the regional mean BPND values in controls from the data of each patient, for each region, for each tracer. Furthermore, we removed regions with previously reported off-target binding of 18F-AV-1451 (basal ganglia and substantia nigra51) The linear mixed effect model therefore included normalized 11C-UCB-J as the dependent variable, normalized 18F-AV-1451 as the independent variable, and age and scan interval as covariates of no interest. To investigate the effect of individual variability on the relationship between 11C-UCB-J and 18F-AV-1451 BPND, we used a linear model with the slope of 11C-UCB-J BPND as a function of 18F-AV-1451 BPND for each individual (extracted from the previous linear mixed effect model) as the dependent variable, the PSP rating scale (a measure of disease severity) as the independent variable and age as a covariate of no interest. To explore the correlation between 11C-UCB-J and 18F-AV-1451 BPND between regions, we calculated a correlation matrix between cortical 18F-AV-1451 binding and synaptic density in cortical and subcortical regions.
Analyses were performed with and without GTM partial volume correction, yielding similar results; we focus on partial volume-corrected BPND to limit the potential effect of atrophy on our ligand cross-correlation, but present data without PVC in the Supplementary material. Statistical analyses were implemented in R (version 3.6.2).
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request for academic (non-commercial) purposes, subject to restrictions required to preserve participant confidentiality.
Results
Demographics
The patients (PSP and CBD) and control groups were similar in age, sex, education and injected activity of 11C-UCB-J and 18F-AV-1451 (Table 1). We observed typical cognitive profiles for people with PSP and CBD: impaired on verbal fluency, memory and visuospatial domains of the ACE-R, MMSE and INECO frontal screening tool.
Relationship between 11C-UCB-J BPND and 18F-AV-1451 BPND
Compared to controls, patients had significantly higher 18F-AV1451 binding in the caudate nucleus, pallidum, putamen and substantia surviving correction for multiple comparison [P < 0.05, false discovery rate (FDR) corrected; Supplementary Fig. 1A and Supplementary Table 2]. As previously reported in a smaller cohort,7,8 patients had significantly lower 11C-UCB-J binding across all cortical and subcortical areas compared to controls, which survived FDR correction (Supplementary Fig. 1B and Supplementary Table 4). Summary statistics for regional 18F-AV1451 and 11C-UCB-J binding potentials in patients and controls are shown in Supplementary Tables 1 and 3, respectively.
There was an overall positive relationship between normalized 18F-AV-1451 BPND and 11C-UCB-J BPND across the patient cohort (β = 0.4, t = 3.6, P = 0.001; Fig. 2A). There was a significant region × 18F-AV-1451 interaction (P< 0.001) driven by subregions of the frontal, parietal and temporal cortices, as well as the hippocampus, subcallosal area and the thalamus, with all but the hippocampus surviving correction for multiple comparison. Age (P = 0.9) and scan interval (P = 0.5) did not have a significant effect on the overall model (note that brain regions with known off-target binding of 18F-AV-1451 were removed before running this linear mixed model). The direction of the relationship between 18F-AV-1451 BPND and 11C-UCB-J BPND within each individual (i.e. the slope of each grey line in Fig. 2A) negatively correlated with disease severity (β = −0.02, t = −2.9, P = 0.007, R= −0.41), independent of age (effect of age: β = 0.02, t= 2.6, P= 0.01; Fig. 2B). In other words, those patients with more severe disease displayed a less-positive relationship between 18F-AV-1451 BPND and 11C-UCB-J BPND.
Figure 2.

The association between normalized synaptic density (11C-UCB-J) and molecular pathology (18F-AV-1451) is a function of disease severity. (A) Scatter plot of 11C-UCB-J BPND and 18F-AV-1451 BPND from 35 patients with PSP–Richardson’s syndrome and amyloid-negative CBS (each grey line represents a patient), across 73 regions of interest (excluding those with previously reported off-target binding, i.e. basal ganglia and substantia nigra) normalized against controls; the dark black line in A depicts the overall fit of the linear mixed model, while grey lines represent individual patient data. (B) The slope for each individual (i.e. each grey line in A) is negatively correlated with disease severity (as measured with the PSP rating scale); R = −0.41, P < 0.007.
Of note, the positive correlation between 18F-AV-1451 BPND and 11C-UCB-J BPND across the patient cohort remains even if BPND values are not normalized against the control data (β = 0.4, t = 4.0, P = 0.0001), as well as the negative relationship between disease severity and the slope of each individual in Fig. 2A. Also note that similar findings were observed using BPND derived from data without partial volume correction (Supplementary Fig. 2).
The relationship between 18F-AV-1451 and 11C-UCB-J binding was also positive in an analogous linear mixed effect model in controls alone (β = 0.6, t = 4, P < 0.0001), with no main effect of age or scan interval, or 18F-AV-1451 × Region interaction. The relationship between unadjusted 18F-AV-1451 and 11C-UCB-J binding in all three groups (controls, amyloid-negative CBS and PSP) is shown in Supplementary Fig. 3.
Cross-regional correlation between 18F-AV-1451 BPND and 11C-UCB-J BPND
Synaptic density in a region is proposed to be affected by both local tau pathology and tau burden in connected regions from which it receives afferent projections. As a result, despite a positive correlation at a regional level, the synaptic density in any given region may be negatively affected by remote insult, with diaschisis between anatomically connected regions (illustrated schematically in Fig. 1A). As an exploratory analysis, we computed the asymmetric Pearson’s correlation matrix shown in Fig. 3, between normalized cortical 18F-AV-1451 BPND (horizontal axis of matrix) and normalized cortical and subcortical 11C-UCB-J BPND (vertical axis of matrix) in patients. We show that, overall, there are significant negative correlations between cortical (frontal, temporal, parietal, occipital) 18F-AV-1451 BPND and subcortical 11C-UCB-J BPND within the caudate nucleus and putamen (−0.52 < R < −0.37; P < 0.05; uncorrected for multiple comparisons). We observed a positive correlation between 18F-AV-1451 BPND and 11C-UCB-J BPND within the thalamus where strong local connections exist (Fig. 3). We did not include subcortical 18F-AV-1451 BPND in the matrix in Fig. 3 given the off-target binding in these regions, which undermines the interpretability of the signal. However, we include these regions as well as other subregions in the larger correlation matrix in Supplementary Fig. 4 for completeness. Similar findings are seen using BPND from data without partial volume correction (Supplementary Fig. 5).
Figure 3.
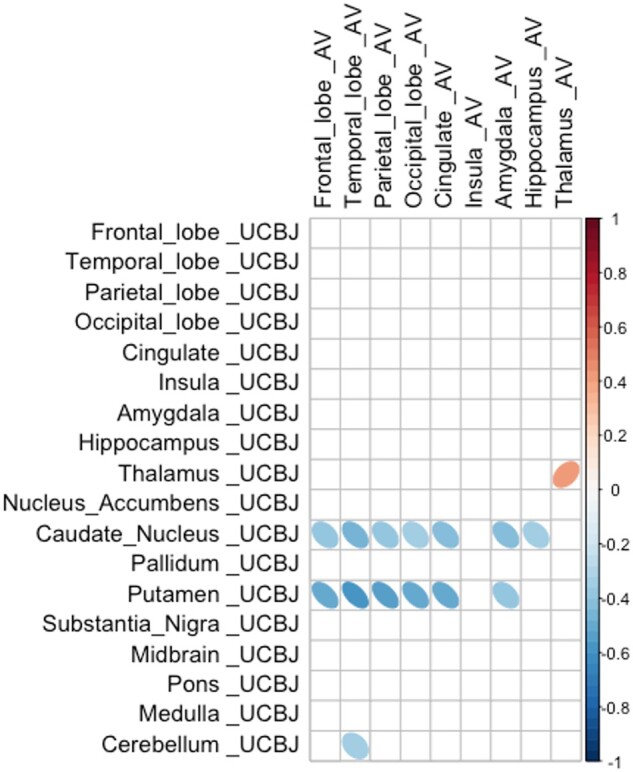
Cortical pathology is negatively correlated with subcortical synaptic density. Correlation, in patients, between normalized 18F-AV-1451 BPND in cortical regions (horizontal axis) and normalized 11C-UCB-J BPND in cortical and subcortical target regions (vertical axis). Negative correlations are observed between cortical 18F-AV-1451 BPND (in frontal, temporal, parietal and occipital cortices), and 11C-UCB-J BPND in the caudate nucleus, putamen and cerebellum. Only significant correlations (at P < 0.05 uncorrected for multiple comparisons) are shown.
Discussion
We have identified an in vivo relationship between molecular pathology (estimated with 18F-AV-1451 PET) and synaptic density (estimated with 11C-UCB-J PET) in patients with the primary tauopathies of PSP and CBD (inferred in vivo from amyloid-negative CBS). There are three principal results: (i) regions with higher synaptic density have higher molecular pathology; (ii) within regions, synaptic density becomes less dependent on 18F-AV-1451 binding as disease severity increases; and (iii) between regions, increased cortical 18F-AV-1451 binding is associated with reduced subcortical synaptic density. We interpret these three findings in the context of synaptic connectivity-based susceptibility to tauopathy, the synaptotoxic effects of tauopathy and cortico-subcortical diaschisis, respectively. The above results are congruent with the model of tau-induced synaptic toxicity, acknowledging the caveat of off-target binding of 18F-AV-1451. Our primary pathology of interest in the context of PSP and CBD is 4R-tau, but other tauopathies such as latent Alzheimer pathology in older adults and non-tau molecular pathologies may also contribute to 18F-AV-1451 binding.
The effect of hyperphosphorylated tau on synaptic function and density is complex. It involves both direct and indirect pathways of injury with changes in cellular physiology preceding the loss of neurons. Through direct pathways, pathological tau interferes with dendritic morphology, synaptic protein expression, the number of NMDA (N-methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors on the presynaptic membrane, mitochondrial function, synaptic vesicle numbers and ultimately synaptic loss (for a review of animal studies illustrating various direct tau-induced synaptic abnormalities, see Jadhav et al.52). Tau also directly affects the axon cytoskeleton and trafficking, as well as the normal functioning of the soma.53 Indirectly, hyperphosphorylated tau adversely affects the functioning of the neuronal support network, including glia cells and astrocytes.54–56 These events are affected by the stage and severity of the disease process, and in relation to regional differences in connectivity which we discuss next (concepts schematically illustrated in Fig. 1).
We identified a positive relationship between the binding of 11C-UCB-J and 18F-AV-1451 such that areas of the brain with higher synaptic density had higher molecular pathology. This accords with preclinical and clinical models of tauopathy in which the strength of local network connectivity facilitates the transneuronal spread of tau pathology.9,12,57–59
However, the relationship between tau accumulation and synaptic density changes with disease progression, at least as inferred from the cross-sectional moderation by disease severity (Fig. 2B). With increasing scores on the PSP rating scale, synaptic density becomes less dependent on local accumulation of pathology. In other words, according to the model (Fig. 1) in areas with relatively low tau accumulation synaptic density is minimally affected, whereas in areas with higher tau accumulation there is reduction of synaptic density as the disease progresses, and this preferentially occurs in synapse-rich areas. As the disease progresses, other pathological processes may contribute to synaptic loss, such as inflammation, another predictor of prognosis and mediator of synaptic loss.60 Therefore, there is not a simple linear relationship between tau accumulation and synaptic density in moderate and advanced disease. This observation accords with human post-mortem and animal studies. In post-mortem studies of the tauopathy Alzheimer’s disease, there is a biphasic synaptic protein response during disease progression, with increases in synaptophysin/syntaxin/SNAP-25 in early Braak stages and synaptic loss observed only when the disease has progressed to the neocortex.19 In the P301L transgenic mouse model of PSP-like tauopathy, there is a differential loss of synapses, as well as synaptic proteins, depending on disease stage.20 These results have recently been replicated in vivo, where the relationship between synaptic density and tau burden in patients with Alzheimer’s disease is reported to be modulated by cortical tau load. Coomans et al.22 show that in patients with mild disease and low cortical tau burden, the relationship between tau and synaptic density is positive, whereas in those with increasing cortical tau load, this relationship changes direction; the relationship between the two tracers in controls is not reported.
In our study, we also observe a positive relationship between 18F-AV-1451 and 11C-UCB-J binding potentials in controls (Supplementary Fig. 3), even though 18F-AV-1451 binding is lower in controls. Disease-related 18F-AV-1451 binding attributable to presence of PSP/CBD pathology is unlikely in the controls, as the prevalence of these conditions in the normal population is only 1/10 000.61 However, the presence of asymptomatic Alzheimer’s disease pathology in the normal older population is more likely. Rising from the age of 40, by the age of 85 two-thirds of cognitively normal individuals will show positive changes in the A/T/N classification for Alzheimer’s disease, whether by CSF, plasma or amyloid PET.62,63 Some of the non-specific 18F-AV-1451 signal, even in healthy controls, may therefore be attributable to latent/preclinical Alzheimer’s disease pathology. We control for this component of the signal by subtracting the mean regional control values from those of the patients.
The positive correlation between 18F-AV-1451 and 11C-UCB-J binding potential in controls appears stronger compared to that seen in patients, as a group (Supplementary Fig. 3). One explanation for this observation is the heterogeneity in disease severity in PSP/CBD, given the interaction between 11C-UCB-J, 18F-AV-1451 and disease severity. This can be understood in terms of the model set out in Fig. 1. The patient group includes those with a strong positive correlation (at early stages of disease) and those with negligible correlation (as a consequence of more advanced disease). The net result for a group-wise test will be a reduction of the group correlation. This is not present in the control group, in whom the level of tau pathology is expected to be very much lower (even if present from Alzheimer type tau with high 18F-AV-1451 affinity).
To understand the biphasic relationship between molecular pathology and synaptic density, one must consider other key players in synaptotoxicity in tauopathies, such as neuroinflammation.64 Recent in vivo studies have confirmed the regional co-localization of inflammation and 18F-AV-1451 binding in PSP, including in many cortical areas,65 in line with previous in vivo66,67 and post-mortem68 reports of the tight interplay between neuroinflammation and tau accumulation in tauopathies. There is growing evidence that these two pathological processes affect synaptic function both independently and synergistically.
The relationship between tauopathy and synaptic density is even more intriguing when considering the change in synaptic density in one region as a function of pathology in another. There are strong correlations between 11C-UCB-J binding within the basal ganglia (in particular the caudate nucleus and putamen) and 18F-AV-1451 binding in all major cortical areas. The reverse association, between subcortical 18F-AV-1451 and cortical 11C-UCB-J binding is also observed (Supplementary Figs 4 and 5) but is dismissed here as uninterpretable in view of subcortical off-target binding of 18F-AV-1451. The significant negative correlation between cortical 18F-AV-1451 binding and synaptic density in the basal ganglia could be a reflection of severe disease in the basal ganglia and accumulating pathology in the neocortex. In other words, synapses are severely affected in the basal ganglia as one of the earliest sites of pathology, with pathology spreading and accumulating in synapse-rich areas of the brain, for example the neocortex. A second possible explanation is that loss of descending cortico-striatal axons due to cortical pathology may cause diaschisis, affecting subcortical synaptic density even further. Previous analysis of diffusion tensor imaging in patients with PSP/CBD have revealed extensive white matter abnormalities (within the main association fibres) beyond the degree of cortical atrophy,69,70 resulting in loss of cortical afferents onto subcortical structures. A third, although not mutually exclusive, potential explanation is the weakening of cortical–subcortical functional connectivity resulting from dysfunctional synapses rather than synaptic loss, although cortico-subcortical connectivity is inferred and was not directly measured in our study.
Although at a regional level there is a positive correlation between 11C-UCB-J and 18F-AV-1451 BPND, we are not directly measuring either synaptic function or the synaptotoxic tau oligomers. This caveat must be borne in mind when interpreting PET data. It is the preclinical models that have shown that oligomers of tau are toxic to synaptic function, even in the absence of tau polymers/fibrils.15,16 By the time tau aggregates are established, oligomers of tau are expected cortically, and perhaps interfere with cortical function and the integrity of descending axons.
There are other limitations to our study. First, the low affinity of 18F-AV-1451 for PSP and CBD 4R tau. Even though the radioligand recapitulates the distribution of post-mortem neuropathology in PSP and CBD and binds PSP 4R tau, the affinity is very much lower than for 3R tau in Alzheimer’s disease. Second, there is well-established off-target binding of 18F-AV-1451, particularly within subcortical structures where monoamine oxidase and neuromelanin are present. Off-target binding is most prominent in the basal ganglia and substantia nigra, which we excluded before running the linear mixed model and correlation matrix. We included these regions in the detailed descriptive correlation matrices in Supplementary Figs 4 and 5 for completeness sake, noting the strong negative correlations between cortical 18F-AV-1451 BPND and subcortical 11C-UCB-J BPND. Furthermore, we normalized our patient data against those of controls to remove any additional normal levels of off-target binding, noting the caveat that the remaining signal in patients may still arise from tau and non-tau pathology; there is no evidence to suggest that 18F-AV-1451 shares a common binding target with 11C-UCB-J, which has a high specificity for SV2A in previous in vivo and in vitro validation studies.38,71 Third, we note that in PET studies of neurodegeneration with atrophy, grey matter volume loss can affect the interpretation of PET signals. However, synaptic loss in PSP and CBD occurs even in areas of the brain without discernible atrophy on MRI.7,8 Nonetheless, we used a stringent partial volume correction method (GTM) to minimize the effect of atrophy on our ligand cross-correlations. Of note, our data without partial volume correction yield similar results in all the main analyses (Supplementary Figs 2 and 5). Fourth, although the sample size is small, it is adequately powered in view of the large effect sizes seen. However, more subtle relationships with phenotypic variants of PSP and CBS would require larger studies. Additionally, clinical diagnostic criteria for PSP–Richardson’s syndrome and amyloid-negative CBS (here called CBD) were used to select a clinical cohort with likely a 4R-tauopathy as the underlying pathological diagnosis. While both PSP–Richardson’s syndrome and amyloid-negative CBS are highly correlated with a 4R-tauopathy at post-mortem, both from our local brain bank and internationally,26,27,72 other pathologies are possible, and so are coexistent pathologies that may synergistically contribute to neurodegeneration.73 Neuropathological correlates, to test the correlations between phenotype and pathology, and between in vivo to post-mortem measures of synaptic density, as well as tau to synapse correlations would be useful but are not yet available for our cohort. Lastly, the cross-sectional design of this study limits the interpretation of the dynamic relationship between pathology and synaptic loss. Although we include patients at various stages of their illness, a longitudinal design is necessary to test the dynamic relationship we propose and the mediation of synaptic loss by progressive tauopathy.
In conclusion, we demonstrate a widespread positive association between 18F-AV-1451 and 11C-UCB-J binding in patients with symptomatic PSP and amyloid-negative CBS. Individual variability in this association correlates with disease severity. The complex relationship between molecular pathology, including but not exclusive to tau, and synaptic density may explain changes in cognitive and motor physiology. We hope that these insights will inform the design of new clinical trials to arrest PSP and CBD.
Supplementary Material
Acknowledgements
The authors thank the research participants and caregivers, the staff at the Wolfson Brain Imaging Centre, and at the Cambridge Centre for Parkinson-Plus. We thank the NIHR Cambridge Biomedical Research Centre for support. We thank UCB Pharma, and Avid (Lilly) for providing the precursor for 11C-UCB-J and 18F-AV-1451 synthesis, respectively. N.H. and J.B.R. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. The view expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Funding
The study was funded by the Wellcome Trust (220258), Cambridge Centre for Parkinson-Plus (RG95450); the National Institute for Health Research Cambridge Biomedical Research Centre (BRC-1215–20014); the PSP Association (‘MAPT-PSP’ study), Medical Research Council (SUAG/051 G101400), and the Association of British Neurologists, Patrick Berthoud Charitable Trust (RG99368).
Competing interests
J.B.R. serves as an associate editor to Brain and is a non-remunerated trustee of the Guarantors of Brain, Darwin College and the PSP Association (UK). He provides consultancy to Asceneuron, Biogen, UCB, Astex, WAVE, Curasen, SV Health and has research grants from AZ-Medimmune, Janssen, Lilly as industry partners in the Dementias Platform UK. J.T.O. has no conflicts related to this study. Unrelated to this work he has received honoraria for work as DSMB chair or member for TauRx, Axon, Eisai, has acted as a consultant for Roche, has received research support from Alliance Medical and Merck. T.R. has received honoraria from Biogen and the National Institute for Health and Clinical Excellence (NICE). No other conflict of interest is reported by other authors.
Abbreviations
- BPND
non-displaceable binding potential
- CBD
corticobasal degeneration
- CBS
corticobasal syndrome
- PSP
progressive supranuclear palsy–Richardson’s syndrome
References
- 1. Bigio EH, Vono MB, Satumtira S, et al. . Cortical synapse loss in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2001;60(5):403–410. [DOI] [PubMed] [Google Scholar]
- 2. Lipton AM, Cullum CM, Satumtira S, et al. . Contribution of asymmetric synapse loss to lateralizing clinical deficits in frontotemporal dementias. Arch Neurol. 2001;58(8):1233–1239. [DOI] [PubMed] [Google Scholar]
- 3. Clare R, King VG, Wirenfeldt M, Vinters HV.. Synapse loss in dementias. J Neurosci Res. 2010;88(10):2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeKosky ST, Scheff SW.. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol. 1990;27(5):457–464. [DOI] [PubMed] [Google Scholar]
- 5. Terry RD, Masliah E, Salmon DP, et al. . Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. [DOI] [PubMed] [Google Scholar]
- 6. Jacobsen JS, Wu C-C, Redwine JM, et al. . Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mak E, Holland N, Jones PS, et al. . In vivo coupling of dendritic complexity with presynaptic density in primary tauopathies. Neurobiol Aging. 2021;101:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holland N, Jones PS, Savulich G, et al. . Synaptic loss in primary tauopathies revealed by [11C]UCB-J positron emission tomography. Mov Disord. 2020;35(10):1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ahmed Z, Cooper J, Murray TK, et al. . A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127(5):667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu L, Drouet V, Wu JW, et al. . Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeVos SL, Corjuc BT, Oakley DH, et al. . Synaptic tau seeding precedes tau pathology in human Alzheimer’s Disease brain. Front Neurosci 2018;12:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Polanco JC, Li C, Durisic N, Sullivan R, Götz J.. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol Commun 2018;6(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibbons GS, Lee VMY, Trojanowski JQ.. Mechanisms of cell-to-cell transmission of pathological tau: A review. JAMA Neurol. 2019;76(1):101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seemiller J, Bischof GN, Hoenig MC, Tahmasian M, van Eimeren T, Drzezga A.; Alzheimer’s Disease Neuroimaging Initiative. Indication of retrograde tau spreading along Braak stages and functional connectivity pathways. Eur J Nucl Med Mol Imaging. 2021;48(7):2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA.. Pathological tau disrupts ongoing network activity. Neuron. 2015;85(5):959–966. [DOI] [PubMed] [Google Scholar]
- 16. Kaniyappan S, Chandupatla RR, Mandelkow EM, Mandelkow E.. Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimer’s Dement. 2017;13(11):1270–1291. [DOI] [PubMed] [Google Scholar]
- 17. Jiang S, Wen N, Li Z, et al. ; International FTD-Genomics Consortium (IFGC). Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry. 2018;8(1):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spires-Jones TL, Hyman BT.. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, et al. . Staging of cytoskeletal and β-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol. 2000;157(2):623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopeikina KJ, Polydoro M, Tai HC, et al. . Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521(6):1334–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vanhaute H, Ceccarini J, Michiels L, et al. . In vivo synaptic density loss is related to tau deposition in amnestic mild cognitive impairment. Neurology. 2020;95(5):e545–e553. [DOI] [PubMed] [Google Scholar]
- 22. Coomans EM, Schoonhoven DN, Tuncel H, et al. . In vivo tau pathology is associated with synaptic loss and altered synaptic function. Alzheimer’s Res Ther. 2021;13:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Malpetti M, Kievit RA, Passamonti L, et al. . Microglial activation and tau burden predict cognitive decline in Alzheimer’s disease. Brain. 2020;143(5):1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Höglinger GU, Respondek G, Stamelou M, et al. ; for the Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Armstrong MJ, Litvan I, Lang AE, et al. . Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alexander SK, Rittman T, Xuereb JH, Bak TH, Hodges JR, Rowe JB.. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 2014;923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gazzina S, Respondek G, Compta Y, et al. . Neuropathological validation of the MDS-PSP criteria with PSP and other frontotemporal lobar degeneration. bioRxiv: Cold Spring Harbor Laboratory. 2019; 520510. [Google Scholar]
- 28. Dickson DW, Kouri N, Murray ME, Josephs KA.. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci. 2011;45(3):384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kovacs GG, Lukic MJ, Irwin DJ, et al. . Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. Aug 2020;140(2):99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou L, McInnes J, Wierda K, et al. . Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun. 2017;8(1):15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Josephs KA, Whitwell JL, Tacik P, et al. . [18F]AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathologica. 2016;132(6):931–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith R, Puschmann A, Schöll M, et al. . 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain. 2016;139(Pt 9):2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitwell JL, Lowe VJ, Tosakulwong N, et al. . [18F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord. 2017;32(1):124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ono M, Sahara N, Kumata K, et al. . Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. 2017;140(3):764–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ali F, Whitwell JL, Martin PR, et al. . [18F] AV-1451 uptake in corticobasal syndrome: The influence of beta-amyloid and clinical presentation. J Neurol. 2018;265(5):1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Soleimani-Meigooni DN, Iaccarino L, La Joie R, et al. . 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain. 2020;143(11):3477–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goodheart AE, Locascio JJ, Samore WR, et al. . 18F-AV-1451 positron emission tomography in neuropathological substrates of corticobasal syndrome. Brain. 2021;144(1):266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Finnema SJ, Nabulsi NB, Eid T, et al. . Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8(348):348ra96. [DOI] [PubMed] [Google Scholar]
- 39. Bajjalieh SM, Peterson K, Linial M, Scheller RH.. Brain contains two forms of synaptic vesicle protein 2. Proc Natl Acad Sci U S A. 1993;90(6):2150–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lang AE, Stebbins GT, Wang P, et al. ; PROSPECT-M-UK investigators. The Cortical Basal ganglia Functional Scale (CBFS): Development and preliminary validation. Parkinson Related Disord. 2020;79:121–126. [DOI] [PubMed] [Google Scholar]
- 41. Burgos N, Cardoso MJ, Thielemans K, et al. . Attenuation correction synthesis for hybrid PET-MR scanners: Application to brain studies. IEEE Trans Med Imaging. 2014;33(12):2332–2341. [DOI] [PubMed] [Google Scholar]
- 42. Manavaki R, Hong Y, Fryer TD. Brain MRI coil attenuation map processing for the GE SIGNA PET/MR: Impact on PET image quantification and uniformity. In: IEEE Nuclear Science Symposium and Medical Imaging Conference Proceedings; 2019.
- 43. Avants BB, Epstein CL, Grossman M, Gee JC.. Symmetric diffeomorphic image registration with cross-correlation: Evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal. 2008;12(1):26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rousset OG, Ma Y, Evans AC.. Correction for partial volume effects in PET: Principle and validation. J Nucl Med. 1998;39(5):904–911. [PubMed] [Google Scholar]
- 45. Wu Y, Carson RE.. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J Cereb Blood Flow Metab. 2002;22(12):1440–1452. [DOI] [PubMed] [Google Scholar]
- 46. Koole M, van Aalst J, Devrome M, et al. . Quantifying SV2A density and drug occupancy in the human brain using [11C]UCB-J PET imaging and subcortical white matter as reference tissue. Eur J Nucl Med Mol Imaging. 2019;46(2):396–406. [DOI] [PubMed] [Google Scholar]
- 47. Rossano S, Toyonaga T, Finnema SJ, et al. . Assessment of a white matter reference region for 11C-UCB-J PET quantification. J Cerebral Blood Flow Metabol. 2020;40(9):1890–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ.. Parametric imaging of ligand–receptor binding in PET using a simplified reference region model. NeuroImage. 1997;6(4):279–287. [DOI] [PubMed] [Google Scholar]
- 49. Klunk WE, Koeppe RA, Price JC, et al. . The Centiloid project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s Dement. 2015;11(1):1–15.e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jack CR, Wiste HJ, Weigand SD, et al. . Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimer’s Dement. 2017;13(3):205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leuzy A, Chiotis K, Lemoine L, et al. . Tau PET imaging in neurodegenerative tauopathies—Still a challenge. Mol Psychiatry. 2019:1112–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jadhav S, Cubinkova V, Zimova I, et al. . Tau-mediated synaptic damage in Alzheimer’s disease. Transl Neurosci. 2015;6(1):214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kneynsberg A, Combs B, Christensen K, Morfini G, Kanaan NM.. Axonal degeneration in tauopathies: Disease relevance and underlying mechanisms. Front Neurosci. 2017;11:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vogels T, Murgoci AN, Hromádka T.. Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun. 2019;7(1):109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kovacs GG. Astroglia and tau: New perspectives. Front Aging Neurosci. 2020;12:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Casaletto KB, Zetterberg H, Blennow K, et al. . Tripartite relationship among synaptic, amyloid, and tau proteins: An in vivo and postmortem study. Neurology. 2021;97(3):e284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Clavaguera F, Bolmont T, Crowther RA, et al. . Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clavaguera F, Akatsu H, Fraser G, et al. . Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 2013;110(23):9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cope TE, Rittman T, Borchert RJ, et al. . Tau burden and the functional connectome in Alzheimer’s disease and progressive supranuclear palsy. Brain. 2018;141(2):550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Malpetti M, Passamonti L, Jones PS, et al. . Neuroinflammation predicts disease progression in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2021;92(7):769–325549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Coyle-Gilchrist ITS, Dick KM, Patterson K, et al. . Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Toledo JB, Zetterberg H, van Harten AC, et al. ; Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain. 2015;138(Pt 9):2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tissot C. L., Benedet A, Therriault J, et al. ; Alzheimer’s Disease Neuroimaging Initiative. Plasma pTau181 predicts cortical brain atrophy in aging and Alzheimer’s disease. Alzheimer’s Res Ther. 2021;13(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Palleis C, Sauerbeck J, Beyer L, et al. . In vivo assessment of neuroinflammation in 4‐repeat tauopathies. Mov Disord. 2021;36(4):883–894. [DOI] [PubMed] [Google Scholar]
- 65. Malpetti M, Passamonti L, Rittman T, et al. . Neuroinflammation and tau colocalize in vivo in progressive supranuclear palsy. Ann Neurol. 2020;88(6):1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ.. In vivo imaging of microglial activation with [11C]-PK11195 PET progresive supranuclear palsy. Mov Disord. 2006;21(1):89–93. [DOI] [PubMed] [Google Scholar]
- 67. Gerhard A, Watts J, Trender-Gerhard I, et al. . In vivo imaging of microglial activation with [11C] (R)–PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19(10):1221–1226. [DOI] [PubMed] [Google Scholar]
- 68. Metaxas A, Thygesen C, Briting SRR, Landau AM, Darvesh S, Finsen B.. Increased inflammation and unchanged density of synaptic vesicle glycoprotein 2A (SV2A) in the postmortem frontal cortex of Alzheimer’s disease patients. Front Cell Neurosci. 2019;13:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Borroni B, Garibotto V, Agosti C, et al. . White matter changes in corticobasal degeneration syndrome and correlation with limb apraxia. Arch Neurol. 2008;65(6):796–801. [DOI] [PubMed] [Google Scholar]
- 70. Padovani A, Borroni B, Brambati SM, et al. . Diffusion tensor imaging and voxel based morphometry study in early progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2006;77(4):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nabulsi NB, Mercier J, Holden D, et al. . Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med. 2016;57(5):777–784. [DOI] [PubMed] [Google Scholar]
- 72. Respondek G, Kurz C, Arzberger T, et al. ; for the Movement Disorder Society-Endorsed PSP Study Group. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord. 2017;32(7):995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Robinson JL, Lee EB, Xie SX, et al. . Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141(7):2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request for academic (non-commercial) purposes, subject to restrictions required to preserve participant confidentiality.