

Abstract
Given the low treatment success rates of drug-resistant tuberculosis (TB), novel TB drugs are urgently needed. The landscape of TB treatment has changed considerably over the last decade with the approval of three new compounds: bedaquiline, delamanid and pretomanid. Of these, delamanid and pretomanid belong to the same class of drugs, the nitroimidazoles. In order to close the knowledge gap on how delamanid and pretomanid compare with each other, we summarize the main findings from preclinical research on these two compounds. We discuss the compound identification, mechanism of action, drug resistance, in vitro activity, in vivo pharmacokinetic profiles, and preclinical in vivo activity and efficacy. Although delamanid and pretomanid share many similarities, several differences could be identified. One finding of particular interest is that certain Mycobacterium tuberculosis isolates have been described that are resistant to either delamanid or pretomanid, but with preserved susceptibility to the other compound. This might imply that delamanid and pretomanid could replace one another in certain regimens. Regarding bactericidal activity, based on in vitro and preclinical in vivo activity, delamanid has lower MICs and higher mycobacterial load reductions at lower drug concentrations and doses compared with pretomanid. However, when comparing in vivo preclinical bactericidal activity at dose levels equivalent to currently approved clinical doses based on drug exposure, this difference in activity between the two compounds fades. However, it is important to interpret these comparative results with caution knowing the variability inherent in preclinical in vitro and in vivo models.
Introduction
The approval of bedaquiline for the treatment of drug-resistant tuberculosis (TB) by the FDA in 2012 led to a revival of anti-TB drug development, as it was the first drug with a new mechanism of action to be registered for the treatment of TB in 40 years. In the years that followed, the landscape of drug-resistant TB treatment changed considerably. In 2014, another new compound, delamanid, was approved by the EMA for the treatment of MDR-TB in adults. Currently, the WHO states that delamanid is indicated for the treatment of rifampicin-resistant (RR) TB or MDR-TB in adults and children.1 More recently, in 2019, pretomanid was the third new drug introduced to the anti-TB drug arsenal. Pretomanid was granted FDA approval, with an indication specified for treating adults with XDR-TB or drug-intolerant or non-responsive MDR-TB. It is to be combined with bedaquiline and linezolid, known as the BPaL-regimen.
The process of drug development is being accelerated by a novel approach developed by the Critical Path to TB Drug Regimens.2 Within this approach, novel drugs are tested as a part of new multidrug regimens already in early stages of the preclinical developmental pipeline. Within such regimens, new compounds are combined with established TB compounds (e.g. pyrazinamide), other new compounds (e.g. bedaquiline and pretomanid in the BPaL regimen), or drugs that are approved for treating diseases other than TB (as was the case for linezolid). The efficacy of these new regimens is subsequently tested in clinical trials as a unit, rather than as a single drug. This is different from the traditional approach that studies the addition of a new compound to an existing regimen or the replacement of single drugs by new ones. Although the new approach enables quicker clinical implementation of novel TB drugs (illustrated by the approval of pretomanid only within the BPaL regimen), it may leave us with the question how new compounds from the same class of drugs compare with each other. In this context, it would be interesting to rank new compounds based on their efficacy, and to assess whether new drugs could be interchangeable in case of drug resistance or drug intolerance. Such questions are of particular interest for delamanid and pretomanid, since they belong to the same class of drugs. In addition, although their clinical indications differ, it is possible that future expansions of approvals would allow for treatment of individual patients with either drug, within the same regimen.
In this review, we summarize and discuss preclinical data on delamanid and pretomanid that have contributed to the implementation of these drugs in the clinic, including compound identification, mechanism of action, drug resistance, in vitro activity, in vivo pharmacokinetic profiles, and in vivo activity and efficacy. Their similarities and differences are discussed and remaining knowledge gaps are identified. Evaluation of clinical studies on either compound are not within the scope of this review.
Compound discovery
Delamanid and pretomanid are nitroimidazoles, a class of drugs active against a broad spectrum of microorganisms, including protozoa and anaerobic bacteria.3 Another well-known member of the nitroimidazoles is metronidazole, for which antibacterial activity was originally discovered in 1962.4 In the 1970s, a subclass of nitroimidazoles was identified that harboured antimycobacterial activity.5 This property was further explored,6 and preclinical studies demonstrated that the bicyclic 5-nitroimidazooxazole CGI-17341 was active against Mycobacterium tuberculosis both in vitro and in vivo.7 Although potential mutagenicity hampered further development of this particular compound, it paved the way towards the identification of other antimycobacterial nitroimidazoles.6,8
Delamanid
Otsuka Pharmaceutical Co. Ltd aimed to develop an antimycobacterial compound that targets mycolic acid synthesis.9 By random screening, three structures were identified: dihydrophenazine, urea-type and dihydroimidazooxazole derivatives. Special attention was given to the latter, given the recent positive results on the antimycobacterial activity of CGI-17341. All nitroimidazoles in the Otsuka library were screened for mutagenicity and results showed that mutagenic properties were probably related to the functional groups attached to the core structure.9–11 In particular, derivatives containing dimethyl residues were associated with higher mutagenicity.9 Among a series of (R)-form 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles with various phenoxymethylgroups and a methyl group at the 2-position, delamanid was identified (Table 1). Its promising preclinical activity made delamanid the lead compound for further safety and efficacy studies.9–11
Table 1.
Chemical name and structure, and mechanism of action of delamanid and pretomanid
| Characteristic | Delamanid (OPC-67683) | Pretomanid (PA-824) |
|---|---|---|
| Developed by: | Otsuka Pharmaceutical Co., Ltd. | PathoGenesis Corporation |
| Chemical namea: | (2R)-2-methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy] piperidin-1-yl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole | (6S)-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine |
| Chemical structurea |
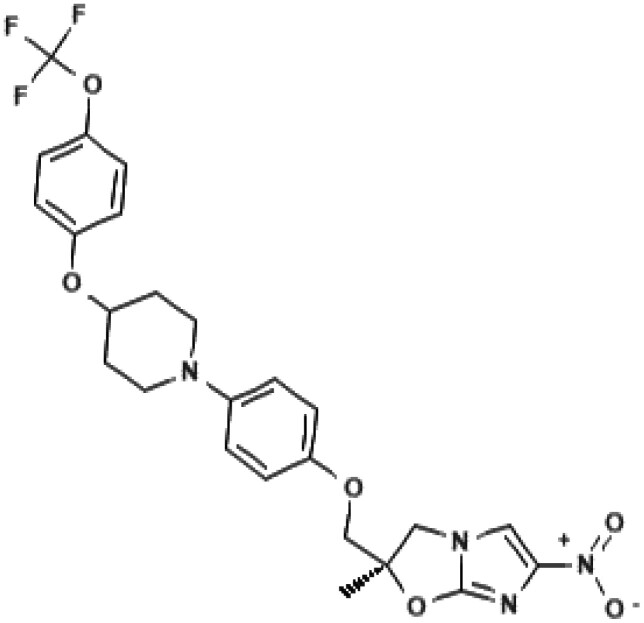
|
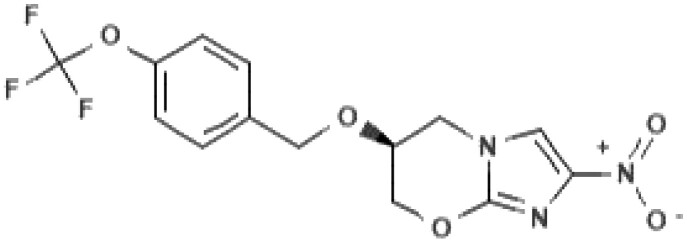
|
| Mechanism of action | 1. Inhibition of mycolic acid synthesis (methoxymycolates and ketomycolates) | 1. Inhibition of mycolic acid synthesis (ketomycolates) |
| 2. Respiratory poisoning (reactive intermediates are yet to be identified) | 2. Respiratory poisoning by the release of reactive nitrogen species upon metabolic activation |
Information extracted from https://pubchem.ncbi.nlm.nih.gov.
Pretomanid
In terms of the discovery of the nitroimidazoles for TB, drug discovery efforts leading to identification of pretomanid preceded those leading to delamanid. Researchers at PathoGenesis Corporation noticed the potency of CGI-17341 as well.12 The company took an interest in nitroimidazooxazines rather than nitroimidazooxazoles, which have a six-membered ring fused to the nitroimidazole instead of a five-membered ring (Table 1). By comparing the antimycobacterial activity of a series of 328 bicyclic nitroimidazooxazines with that of CGI-17341, pretomanid was identified.12 Pretomanid was found to be active against drug-susceptible as well as drug-resistant M. tuberculosis strains,12 as was also seen for delamanid.10 More information on optimization studies of nitroimidazooxazines that resulted in the identification of pretomanid are detailed in published patents.13,14
Mechanism of action
Delamanid and pretomanid are thought to have a comparable, dual mode of action: (i) interference with mycolic acid synthesis, and (ii) respiratory poisoning.15–17 It is noteworthy that most published research on the mechanism of action has been performed with pretomanid.
Inhibition of mycolic acid biosynthesis
Under aerobic conditions, inhibition of mycolic acid synthesis is considered to be the main mode of action of delamanid and pretomanid. Mycolic acids are a major component of the lipids forming the mycobacterial outer membrane, and are restricted to mycobacteria and related genera of the Actinobacteria phylum.18 Mycolic acids contribute to bacterial virulence by forming a permeability barrier to drugs,19 contributing to intracellular survival,20 and modulating the pro-inflammatory response.20,21 Three classes of mycolic acids are known: α-mycolates (most abundant), methoxymycolates, and ketomycolates.22 Delamanid inhibits synthesis of ketomycolates and methoxymycolates, but not α-mycolates,9,10 whereas isoniazid inhibits all three classes.10 The exact mechanism by which delamanid blocks mycolic acid synthesis is not yet elucidated, as no mutations in delamanid-resistant organisms have been linked to cell wall synthesis.23 Pretomanid blocks the formation of ketomycolic acid.12 It is hypothesized that this process involves inhibition of a deazaflavin coenzyme (F420)-dependent enzyme that is responsible for oxidation of hydroxymycolate into ketomycolate.24 Whether pretomanid also inhibits synthesis of the other mycolate classes is unknown.
Respiratory poisoning
Delamanid and pretomanid are prodrugs that need metabolic activation by mycobacteria to exert antimycobacterial activity (Figure 1).10,12,25 In short, bio-activation of both compounds by mycobacteria depends on redox cycling of deazaflavin cofactor 420, or F420. The enzyme deazaflavin-dependent nitroreductase (Ddn), which participates in the redox cycling of F420, is responsible for bio-activation of both delamanid and pretomanid by the process of des-nitrification,10,26–28 although the compounds bind differently to Ddn.29 Human nitroreductases were found to be unable to activate delamanid, potentially due to their use of NAD(P)H as electron donor, which has a higher redox potential compared with F420.30 Similarly, pretomanid can be metabolized, but not bio-activated, by human liver enzymes, as they do not induce des-nitrification.31 The activation of delamanid and pretomanid being restricted to mycobacterial Ddn might (in part) explain the selective activity against mycobacteria without being genotoxic to humans.30,31
Figure 1.

Schematic overview of the metabolic activation of delamanid and pretomanid by mycobacteria, adapted with permission from Liu et al.23 and Rifat et al.36 Delamanid and pretomanid are prodrugs that require activation by deazaflavin (F420)-dependent nitroreductase (Ddn). Redox cycling of deazaflavin cofactor 420, or F420, is crucial in this process, which is mediated by glucose-6-phosphate dehydrogenase (Fgd1)12,23,35,132,133 and Ddn.10,26–28 Synthesis of F420 depends on FbiA, FbiB, FbiC and FbiD.12,36–38,134 Bio-activation of delamanid by Ddn results in the formation of inactive des-nitro-imidazooxazole.10,135 The active intermediate for delamanid has not yet been identified. Activation of pretomanid, on the other hand, generates three stable, inactive metabolites, as well as reactive nitrogen species which are responsible for respiratory poisoning by pretomanid.26,28
Ddn-mediated metabolic activation of delamanid generates one main metabolite, desnitro-imidazooxazole, which has no antimycobacterial activity.10 For pretomanid, Ddn reduces the imidazole ring, forming three major metabolites among which is a des-nitro form.28 The metabolites have been described by Singh et al.28 not to show any activity against M. tuberculosis. However, reduction of pretomanid releases reactive nitrogen species, such as nitric oxide (NO) which acts as an active intermediate.16,28 NO is thought to target cytochrome oxidases in the mycobacterial electron-transport chain, thereby hampering ATP synthesis.16,32 Since mycobacteria maintain their respiratory function and energy production at low levels under anaerobic conditions, they may be more vulnerable to impairment of ATP homeostasis under such circumstances.16,33 Transcriptional profiling of M. tuberculosis exposed to delamanid revealed that delamanid probably induces respiratory poisoning as well.17 However, the active intermediate of delamanid is not yet identified. Hayashi et al.34 recently found that mutations in type II NADH dehydrogenase (ndh) can give rise to delamanid resistance. The authors speculate that an NAD-delamanid adduct, instead of NO, might be responsible for its anti-mycobacterial activity. Characterizing other upregulated genes during delamanid exposure could provide additional insight into its mechanism of action.17
Drug resistance
Studies on drug resistance suggest that delamanid and pretomanid display no cross-resistance with other currently used TB drugs, probably due to their unique mechanism of action.10,12 That being said, by using a genetically modified Mycobacterium smegmatis strain, Hayashi et al.34 showed that mutations in the ndh gene can in principle lead to resistance to isoniazid, ethambutol, and also delamanid.
Both delamanid and pretomanid have relatively high spontaneous mutation frequencies. For delamanid, the frequency of drug resistance was found to range between 1.22 × 10−5 and 6.44 × 10−6 at 16 times the MIC.25 Spontaneous drug resistance frequencies ranging from 1.0 × 10−5 to 6.5 × 10−7 are reported for pretomanid, which are comparable to those of delamanid.25,27,35 These frequencies are in line with resistance rates reported for isoniazid, but are higher than those reported for rifampicin in M. tuberculosis.25 It could be that the relatively large target size for mutations (six non-essential genes, discussed below) foster these high frequencies, and the issue highlights the importance of combining these drugs with strong companion drugs during therapy.36
Mutations in the genes responsible for metabolic activation of delamanid and pretomanid (fbiA, fbiB, fbiC, fbiD, fgd1, and ddn) (Figure 1) have been associated with resistance to either drug in preclinical settings and in clinical isolates.12,25,28,35–47 However, additional genes might be involved in delamanid resistance, as in a recent study none of the delamanid-resistant clinical isolates harboured mutations in fbiA/B/C, fgd1 or ddn.48 In contrast, published findings on pretomanid-resistant clinical isolates are sparse, likely because the drug only recently earned approval for clinical use.
Table 2 summarizes the findings from several studies that have investigated both delamanid and pretomanid susceptibility of M. tuberculosis isolates from either preclinical or clinical settings, together with an evaluation of gene mutations that coincided with drug resistance.29,36,49 Given the similarities in the intra-bacterial metabolic pathway of delamanid and pretomanid, it is not unexpected that isolates resistant to both compounds have been identified. Out of 32 pretomanid-resistant isolates selected by Rifat et al.36 from their mouse model of TB infection, 23 were resistant to delamanid as well (MIC >0.06 mg/L) and harboured mutations in fbiA, fbiB, fbiC, fbiD, fgd, and ddn. Lower levels of cross-resistance were reported in clinical isolates, with 2 out of 12 isolates being resistant to both compounds (delamanid MIC >16 mg/L, and pretomanid MIC 8 and >16 mg/L).49 An E249K mutation in the fbiA gene (GAA → AAA) was found in one of these isolates, as well as a synonymous F320F mutation in fgd1 (TTT → TTC). The particular fgd1 mutation is, however, probably not responsible for drug resistance, as it was also observed in isolates susceptible to both drugs. Of particular interest are the isolates resistant to one drug only, while susceptibility to the other is preserved (Table 2). Isolates selected in a preclinical setting with various mutations in fbiA, fbiB or fbiD exhibited high-level resistance to pretomanid, while retaining susceptibility to delamanid with only modestly raised MICs to the critical value of 0.06 mg/L.36 Apart from susceptibility testing, Lee et al.29 investigated the ability of M. tuberculosis isolates harbouring mutations in ddn to activate delamanid and pretomanid. Notably, of the 46 studied ddn mutants, two isolates were not able to activate pretomanid, but could, however, still activate delamanid. These isolates harboured an S78Y or Y133C mutation in ddn, both of which are naturally occurring sequence polymorphisms. This finding suggests that mutations in ddn such as S78Y and Y133C might cause pretomanid resistance, while maintaining susceptibility to delamanid. Molecular docking studies indicate that the dissimilarity in the ability to activate delamanid or pretomanid might be a consequence of different binding of the compounds to Ddn.29 The authors speculate that the chemical structure of delamanid causes steric hindrance with the deazaflavin ring of F420 in Ddn bound to F420H2. As a result, delamanid binds above F420 in a different orientation than pretomanid. All together, these findings imply that under certain conditions delamanid and pretomanid could replace each other in case of drug resistance to one of the two drugs.
Table 2.
Overview of M. tuberculosis isolates selected from either preclinical or clinical settings for which susceptibility to both delamanid (DLM) and pretomanid (PMD) was determined, together with an investigation of coinciding gene mutations
| Author/setting of isolation | Resistance type | Resistant toa | MIC (mg/L) | Gene | Mutation | |
|---|---|---|---|---|---|---|
| Delamanid | Pretomanid | |||||
| Rifat et al. (2020)36 | ||||||
| Preclinical | DLM; PMD | >16 | >32 | fbiA | Q27* | |
| Preclinical | DLM; PMD | >16 | >32 | fbiA | D49G | |
| Preclinical | DLM; PMD | >16 | >32 | fbiA | −G in aa 47 | |
| Preclinical | DLM; PMD | >16 | >32 | fbiA | L308P | |
| Preclinical | DLM; PMD | >16 | 32 | fbiA | Q120P | |
| Preclinical | DLM; PMD | >16 | 32 | fbiA | D286A | |
| Preclinical | DLM; PMD | 0.06–0.125 | 8–32 | fbiB | L15P | |
| Preclinical | DLM; PMD | 0.125 | 32 | fbiB | L173P | |
| Preclinical | DLM; PMD | 0.06–0.125 | 32 | fbiB | −T in aa 684 | |
| Preclinical | DLM; PMD | >16 | >32 | fbiC | C562W | |
| Preclinical | DLM; PMD | 1 | >32 | fbiC | G194D | |
| Preclinical | DLM; PMD | 2 | >32 | fbiC | −C in aa 20 | |
| Preclinical | DLM; PMD | >16 | >32 | fbiC | K684T | |
| Preclinical | DLM; PMD | >16 | >32 | fbiC | IS6110 ins. 85 bp upstream of fbiC | |
| Preclinical | DLM; PMD | >16 | >32 | fbiC | L377P | |
| Preclinical | DLM; PMD | >16 | >32 | fbiC | A827G | |
| Preclinical | DLM; PMD | 0.5 | 32 | fgd1 | K9N | |
| Preclinical | DLM; PMD | >16 | >32 | fgd1 | G191D | |
| Preclinical | DLM; PMD | >16 | ≥32 | ddn | R112W | |
| Preclinical | DLM; PMD | >16 | ≥32 | ddn | IS6110 ins. in D108 | |
| Preclinical | DLM; PMD | >16 | >32 | ddn | −G in aa 39 | |
| Preclinical | PMD | 0.03 | 32 | fbiA | S219G | |
| Preclinical | PMD | 0.03 | 16 | fbiB | W397R | |
| Preclinical | PMD | 0.03 | 16–32 | fbiC | R25G | |
| Preclinical | PMD | 0.03 | 16–32 | fbiC | M776R | |
| Preclinical | PMD | 0.06 | >32 | fbiD | G147C | |
| Preclinical | PMD | 0.06 | >32 | fbiD | A132V | |
| Preclinical | PMD | 0.06 | >32 | fbiD | −ATC in aa 129 | |
| Preclinical | PMD | 0.03–0.06 | >32 | fbiD | R25S | |
| Preclinical | PMD | 0.06 | >32 | fbiD | A198P | |
| Preclinical | PMD | 0.06 | >32 | fbiD | C152R | |
| Preclinical | PMD | <0.03 | >32 | fbiD | A68E | |
| Wen et al. (2019)49 | ||||||
| Clinical | XDR | DLM; PMD | >16 | 8 | b | b |
| Clinical | XDR | DLM; PMD | >16 | >16 | fgd1 | F320F |
| fbiA | E249K | |||||
| Clinical | MDR | DLM | 16 | 0.063 | fgd1 | F320F |
| Clinical | MDR | DLM | >16 | 0.031 | b | b |
| Clinical | MDR | DLM | 0.5 | 0.063 | fgd1 | F320F |
| Clinical | MDR | DLM | >16 | 0.063 | fgd1 | F320F |
| Clinical | XDR | DLM | >16 | ≤0.016 | fgd1 | F320F |
| Clinical | XDR | PMD | ≤0.016 | >16 | b | b |
| Clinical | MDR | None | ≤0.016 | 0.13 | fgd1 | F320F |
| Clinical | MDR | None | ≤0.016 | 0.25 | fgd1 | F320F |
| Clinical | MDR | None | ≤0.016 | 0.5 | fgd1 | F320F |
| Clinical | XDR | None | ≤0.016 | 0.25 | fgd1 | F320F |
| Lee et al. (2020)29 | ||||||
| Clinical | DLM; PMD | 32 | 256 | ddn | S78Y | |
Rifat et al.36 determined the MIC by broth macrodilution assay, Wen et al.49 by microplate Alamar blue assay (MABA) and Lee et al.29 by resazurin assay.
The clinical breakpoint for susceptibility to delamanid is ≤0.06 mg/L, as set by the EUCAST73; EUCAST clinical breakpoints for pretomanid are awaited. In this Table, 1 mg/L is used as the cut-off value for susceptibility to pretomanid.70
No mutations were found in ddn, fgd1, fbiA, fbiB, or fbiC.
In vitro activity
A single in vitro assay cannot cover the complexity of human TB infection comprising M. tuberculosis in various metabolic stages and residing in different niches. Hence, a variety of assays exists, each with a specific design and read-out. Although heterogeneity between assays hampers systematic comparison, here, we review studies reporting the following outcomes to get an impression of the in vitro activity of delamanid and pretomanid: standard in vitro susceptibility assays (MIC assays), drug activity against extracellular M. tuberculosis in different metabolic states, and activity against intracellular M. tuberculosis in macrophage assays. Although the MIC value is a measure of compound activity, it does not directly reflect in vivo efficacy as it is only one of many factors that drive pharmacokinetic (PK) and pharmacodynamic (PD) characteristics. In early stages of compound development, new drugs are often tested against replicating extracellular M. tuberculosis. These experiments are relatively easy to implement and allow for a quick comparison of the new compound’s activity with that of already established TB drugs. Regarding metabolic states, M. tuberculosis is thought to be present in pulmonary lesions both as replicating and non-replicating bacteria, based on the mycobacterial growth phase.50,51 Evaluating drug activity against non-replicating mycobacteria is relevant, because this population is more tolerant to treatment with existing TB drugs and therefore may be responsible for the prolonged TB treatment duration needed to effect cure.51–53 Several assays have been developed that induce a non-replicating state in M. tuberculosis, including starvation, oxygen depletion, low pH, or by using specific strains such as the M. tuberculosis 18b strain which enters a non-replicating state in the absence of streptomycin.54 We chose to also include results of the first-line drugs rifampicin and isoniazid as a reference, since it is known that rifampicin is active against both replicating and non-replicating M. tuberculosis,55 while isoniazid only targets replicating bacilli.56
Delamanid
The MIC distribution for delamanid against clinical M. tuberculosis strains as reported by the EUCAST shows that MICs mostly range between ≤0.002 to 0.03 mg/L.57 Depending on the method used, the majority of isolates have an MIC of 0.004 mg/L or 0.008 mg/L as tested by agar dilution or MGIT 960, respectively. This is in agreement with various articles reporting MICs ≤0.025 mg/L against both drug-susceptible and drug-resistant M. tuberculosis strains.10,11,45,48,49,58,59 EUCAST sets the clinical breakpoint for strain susceptibility to delamanid at MIC ≤0.06 mg/L.60
Table 3 summarizes findings on the in vitro activity of delamanid against replicating extracellular M. tuberculosis. Saliu et al.61 compared the activity of delamanid with that of rifampicin against clinical M. tuberculosis isolates tolerant to isoniazid, meaning that these isolates grew better than the laboratory H37Rv strain in the presence of 0.1 mg/L isoniazid as measured by 14CO2 production. The authors found that against these isolates, killing rates of delamanid at 1 mg/L were comparable to those of rifampicin at 2 mg/L over 14 days of drug exposure.61 Dalton et al.62 showed that delamanid significantly reduced the mycobacterial numbers as measured by relative light units (RLU) after 3 days of drug exposure.
Table 3.
Summary of in vitro activity of delamanid and pretomanid against replicating, extracellular M. tuberculosis
| Author | M. tuberculosis strain | Drug treatment (dose) | Treatment duration | Read-out | Outcome |
|---|---|---|---|---|---|
| Saliu et al. (2007)61 | Clinical INH-tolerant strains | DLM (1 mg/L) | 14 days | Growth Index | Killing rates of DLM were comparable to those of RIF (2 mg/L). |
| Dalton et al. (2017)62 | Bioluminescently-labelled H37Rv | DLM; PMD | 3 days | RLU | DLM significantly reduced RLU. RLU levels stayed stable during PMD and RIF exposure. |
| Sala et al. (2010)71 | 18b, exposed to streptomycin | PMD (3 mg/L) | 7 days | cfu | PMD bactericidal activity was comparable with that of INH (0.5 mg/L) and RIF (10 mg/L). |
| Piccaro et al. (2013)55 | H37Rv | PMD (2 mg/L) | 7 days | cfu | PMD reduced cfu counts to a comparable extent as INH (2 mg/L), but to a lesser extent than RIF (8 mg/L). |
INH, isoniazid; DLM, delamanid; RIF, rifampicin; PMD, pretomanid; RLU, relative light units; cfu, colony forming units.
Information on in vitro activity of delamanid against non-replicating bacilli is sparse (Table 4). In a study by Upton et al.,63 the non-replicating state was induced by oxygen depletion.63 The authors found that delamanid at 4.4 μM was sufficient to reduce colony forming units (cfu) by 99% after 10 days of exposure. As M. tuberculosis can be present intracellularly in pulmonary lesions, Matsumoto et al.10 used infected macrophages differentiated from human THP-1 monocytes to assess delamanid activity against intracellular M. tuberculosis. Delamanid showed strong and concentration-dependent activity, which at 0.1 mg/L was similar to that of rifampicin at 3 mg/L.
Table 4.
Summary of in vitro activity of delamanid and pretomanid against non-replicating, extracellular M. tuberculosis.
| Author | M. tuberculosis strain | Induction non-replicating state | Drug treatment (dose) | Treatment duration | Read-out | Outcome |
|---|---|---|---|---|---|---|
| Upton et al. (2015)63 | H37Rv | Oxygen depletion | DLM (4.4 μM); PMD (17.4 μM) | 10 days | cfu | DLM at 4.4 μM, and PMD at 17.4 μM reduced cfu by 99%. |
| Lenaerts et al. (2005)66 | H37Rv | Oxygen depletion | PMD (2, 10, 50 mg/L) | 4 days | cfu | PMD showed dose-dependent bactericidal activity. At 50 mg/L, PMD activity was higher than that of INH at 50 mg/L, and was comparable to RIF at 2 mg/L, but inferior to RIF at 10 or 50 mg/L. |
| Hu et al. (2008)72 | H37Rv | Starvation, oxygen depletion | PMD (0.31–20 mg/L) | 4–7 days | cfu | PMD showed dose-dependent bactericidal activity. At ≤1.25 mg/L, PMD was only minimally active. Mycobacterial elimination was observed at ≥10–20 mg/L. |
| Sala et al. (2010)71 | 18b strain | No exposure to streptomycin | PMD (3 mg/L) | 7 days | cfu | PMD activity was higher against non-replicating than fast-replicating M. tuberculosis. PMD and RIF (10 mg/L) were equally active and PMD activity was superior to INH (0.5 mg/L). |
| Stover et al. (2000)12 | Bioluminescently-labelled H37Rv | Oxygen depletion | PMD (10 mg/L) | 7 days | RLU | PMD was active against non-replicating mycobacteria. PMD activity (10 mg/L) was comparable to MTZ (10 mg/L), and superior to INH (10 mg/L). |
| Papadopoulou et al. (2007)75 | Bioluminescently-labelled H37Rv | Oxygen depletion | PMD (6.4–12.8 mg/L) | 10 days | Luminescent signal/cfu | PMD at 6.4–12.8 mg/L, and RIF at 2.5 mg/L were sufficient to kill ≥90% of M. tuberculosis. This activity was superior to INH (>100 mg/L). |
| Piccaro et al. (2013)55 | H37Rv | Oxygen depletion | PMD (2 mg/L) | 7–21 days | cfu | PMD showed time-dependent bactericidal activity, which was inferior to RIF (8 mg/L) and superior to INH (2 mg/L). |
| Somasundaram et al. (2013)76 | H37Rv | Oxygen depletion | PMD (3, 12.5 mg/L) | 2–21 days | cfu | PMD (12.5 mg/L) resulted in mycobacterial elimination at day 21, which was superior to RIF (1 mg/L). Bactericidal activity of PMD at 3 mg/L was comparable to RIF at 1 mg/L. |
| Iacobino et al. (2016)74 | H37Rv | Starvation, oxygen depletion, low pH | PMD | cfu | PMD reduced cfu counts by ≥2 log10, which was similar to RIF and superior to INH. | |
| Early et al. (2019)73 | H37Rv | Low pH | PMD | 7 days | cfu | PMD (12 μM) reduced cfu by ≥2 log10, similar to RIF (75 μM), whereas INH showed no activity. |
DLM, delamanid; PMD, pretomanid; cfu, colony forming units; INH, isoniazid; RIF, rifampicin; MTZ, metronidazole.
Only a few studies describe the in vitro activity of delamanid-containing TB drug combinations. Matsumoto et al.10 investigated potential synergistic activity of delamanid and first-line TB drugs against 27 clinical M. tuberculosis isolates by chequerboard analysis. There was no interaction observed between delamanid and rifampicin (FIC indices between >0.5 and 0.75) for the majority of isolates (88.9%). This also accounted for the interaction between delamanid and isoniazid (44.4% FIC index >0.5–0.75, 18.5% FIC index >0.75–1.0, 37% FIC index >1.0–4.0). Also using a chequerboard assay, Chandramohan et al.64 demonstrated either an additive or synergistic effect between delamanid and bedaquiline or moxifloxacin, depending on the M. tuberculosis strain being drug-susceptible, monoresistant to isoniazid or rifampicin, MDR or XDR. However, it should be pointed out that the results of chequerboard assays should be interpreted with utmost care, as it is not clear how well these artificial in vitro assays translate to in vivo results for M. tuberculosis.
Pretomanid
Pretomanid was only recently approved as a TB drug, and therefore, the evaluation of clinical breakpoints is currently ongoing.65 Pretomanid activity has been assessed against drug-susceptible, MDR and XDR M. tuberculosis strains, with reported MICs of 0.015–1 mg/L.12,49,66–69 Pending the EUCAST clinical breakpoints, the EMA proposed 1 mg/L as the critical concentration when using the MGIT system for drug susceptibility testing.70 In addition, M. tuberculosis isolates with pretomanid resistance-associated gene mutations have an MIC above this critical concentration.26,35 Based on the few studies that assessed the MIC of both delamanid and pretomanid, the reported values for delamanid (0.001–0.024 mg/L) were lower than those for pretomanid (0.012–0.200 mg/L).10,49,63
Pretomanid activity against replicating M. tuberculosis is summarized in Table 3. Sala et al.71 demonstrated that pretomanid (3 mg/L) killed replicating M. tuberculosis (using the 18b strain exposed to streptomycin, which allows for the strain to replicate) to the same extent as isoniazid (0.5 mg/L) and rifampicin (10 mg/L) after 7 days of drug exposure. Using an H37Rv M. tuberculosis strain, Piccaro et al.55 showed that the activity of pretomanid (2 mg/L) was comparable to that of isoniazid (2 mg/L), though it was inferior to the activity of rifampicin (8 mg/L). In a study by Dalton et al.,62 3 days of pretomanid exposure kept the M. tuberculosis load at a stable level, whereas the untreated control showed a significant increase in mycobacterial load. In comparison, the activity of delamanid in this assay was relatively higher, leading to a reduction in the mycobacterial load.
Activity of pretomanid against non-replicating M. tuberculosis was more elaborately studied than for delamanid (Table 4). Its activity was shown be concentration dependent.66,72 In an experimental set-up using the 18b M. tuberculosis strain (in a non-replicating state in the absence of streptomycin), pretomanid appeared to be more active against non-replicating compared with replicating M. tuberculosis, reducing the mycobacterial load by 4.5 log10 cfu/mL versus 2 log10 cfu/mL after 7 days of drug exposure, respectively.71 This observation matches the finding that reactive nitrogen species released upon activation of pretomanid have a greater impact on the ATP synthesis under anaerobic conditions.16,33 Stover et al.12 aimed to induce a non-replicating state in M. tuberculosis by microaerophilic culture conditions. In this assay, the bactericidal activity of 7 days exposure to pretomanid (10 mg/L) was comparable to that of the structurally related metronidazole (10 mg/L), and superior to that of isoniazid (10 mg/L). Lenaerts et al.66 also observed a higher bactericidal activity of pretomanid (50 mg/L) compared to isoniazid (50 mg/L) following 4 days of drug exposure in an oxygen depletion assay. In various experimental set-ups, bactericidal activity of pretomanid against non-replicating M. tuberculosis matched the activity of rifampicin.66,71,73–76 Upton et al.63 evaluated the bactericidal activity of both delamanid and pretomanid against non-replicating M. tuberculosis in an oxygen depletion assay. The authors found that 17.4 μM pretomanid was sufficient to reduce cfu by 99% after 10 days of exposure, while for delamanid a concentration of 4.4 μM was sufficient to achieve this goal.63 Published information on pretomanid activity against intracellular bacilli is rather limited. In a whole blood culture assay, Wallis et al.77 demonstrated modest concentration-dependent bactericidal activity of pretomanid at 0–2 mg/L. In another assay, using M. tuberculosis-infected THP-1 cells, pretomanid at 0.1–1 mg/L led to a similar reduction in mycobacterial numbers as isoniazid at 0.3–3 mg/L. However, the intracellular activity of pretomanid was inferior to that of delamanid and rifampicin in this study.10
The in vitro activity of drug combinations was more extensively studied for pretomanid than for delamanid. Whereas delamanid combined with bedaquiline showed in vitro synergy,64 additive or antagonistic effects have been reported when pretomanid was combined with bedaquiline,77,78 although it should be noted that different experimental designs were used in these studies, hampering comparison of the outcomes. The interaction between pretomanid and bedaquiline is of interest, since several new and promising drug regimens contain these two drugs (ClinicalTrials registration no. NCT03338621, NCT03086486, NCT02589782). An additive effect was found when pretomanid was combined with linezolid.78 The combination of pretomanid and moxifloxacin was shown to be additive or synergistic against actively replicating or non-replicating M. tuberculosis, respectively.78–80 In this context, Drusano et al.80 showed that the addition of bedaquiline to the combination of pretomanid and moxifloxacin achieved eradication of actively replicating M. tuberculosis one week sooner compared with the two-drug combination. Using a modified chequerboard assay, López-Gavín et al.81 demonstrated that a combination of pretomanid, clofazimine, and moxifloxacin was active against drug-susceptible and MDR clinical isolates, with the activity of the drugs being additive. In a recent study using a hollow fibre infection model, the performance of the combination of pretomanid, moxifloxacin and pyrazinamide was equal to that of the standard regimen consisting of isoniazid, rifampicin and pyrazinamide (HRZ) against both replicating, non-replicating and intracellular M. tuberculosis.82 Lastly, Piccaro et al.55 reported that when pretomanid was combined with rifampicin, moxifloxacin, and amikacin, M. tuberculosis was efficiently killed within 14 days in aerobic as well as hypoxic conditions, but no comparison was made with the standard regimen. Again, when interpreting these data, it is important to bear in mind the limitations regarding the translational value of these highly simplified in vitro drug combination assays.
Pharmacokinetics
The efficacy of a drug depends on its PD and its PK profile. By combining PK with a microbiological parameter, PK/PD indices can be determined (e.g. AUC0–24/MIC or %T>MIC), which can be used to optimize dosing schedules.83 Knowledge on what doses in animals reach exposures (or ideally driving PK/PD indices) that match exposures reached in humans at clinically approved doses assists in interpreting drug activity and efficacy results in animal studies and translating these to humans. Furthermore, animal studies can shed light on drug distribution, drug metabolism, and drug clearance.
Delamanid
Animal studies have shown that following oral administration of delamanid, the drug is widely distributed among various organs.84,85 After treating rats with a single oral dose of radioactively labelled 14C-delamanid (3 mg/kg), radioactivity was detected in the lungs, central nervous system, eyeball, placenta, fetus, and breastmilk.84 Penetration of the blood–brain barrier was confirmed by Tucker et al.85 in a rabbit model of tuberculous meningitis. Delamanid was detected in cerebrospinal fluid, albeit at lower concentrations than in plasma. Brain tissue concentrations, on the other hand, were found to be 5-fold higher than those in plasma.85 Results from both studies suggest that delamanid could be of value in treating extra-pulmonary TB, including TB meningitis, but further studies are required.
Delamanid is highly protein bound (>97%).86 It is thought that plasma albumin is mainly responsible for metabolizing delamanid,86,87 with the formation of M1 (DM-6705) as the major metabolite. Hepatic cytochrome P450 (CYP) enzymes are assumed to play a role in the subsequent degradation of M1 into another seven metabolites.87 No interaction between delamanid and CYP isoforms was observed,10,88 and delamanid metabolites were found to inhibit some CYP isoforms only at considerably higher concentrations than observed in human plasma.88 These results imply that drug–drug interactions with compounds that are metabolized by CYP enzymes, including antiretroviral drugs, are unlikely. However, this subject is being further assessed in clinical studies.89
Studies in mice, rats, guinea pigs, rabbits and dogs have been performed to shed light on the pharmacokinetic profile of delamanid (Table 5). The delamanid dose currently approved for clinical use is 100 mg twice a day, taken with food.90 In a randomized, placebo-controlled, multinational clinical trial, Gler et al.91 found an AUC of 7.925 μg·h/mL in patients treated with delamanid 100 mg twice daily for 56 days. A slightly lower AUC0–24 of 3.40 μg·h/mL was reported by Mallikaarjun et al.92 in humans for delamanid at the daily dose of 200 mg. Several dosing strategies in various animal studies resulted in AUC values similar to those in humans (Table 5). In mice, 2.5, 3, and 10 mg/kg at single oral administration and 2.5 mg/kg orally administered for 4 weeks in a combination regimen with bedaquiline and linezolid led to AUC values between 3.58 and 11.55 μg·h/mL.10,87,92,93 Likewise, in rats, 3 and 10 mg/kg at single drug administration generated exposures of 7.9418 (AUC0–480) and 5.68 (AUC0–96) μg·h/mL, respectively.87,94 In guinea pigs, a single dose of delamanid at 10 mg/kg resulted in a relatively low AUC0–48 of 2.32 μg·h/mL, while this was 9.45 μg·h/mL for a dose of 100 mg/kg.95 Also in rabbits and dogs delamanid exposures matching clinical exposures were shown following a single oral dose of delamanid at 5 mg/kg85 and 10 mg/kg,87 respectively. Using a murine chronic TB infection model, Mallikaarjun et al.92 found that the PK/PD driver for delamanid activity was described best by the AUC0–24/MIC (Pearson’s correlation coefficient = 0.97), and to a lesser extent by the %T>MIC (Spearman correlation coefficient = 0.53). In that study, an AUC0–24/MIC of 252 was determined to achieve 80% of the maximum activity of the drug in the mouse model. Based on the results from two human Early Bactericidal Activity studies, a mean AUC0–24/MIC of 393 was established at a dose of 200 mg after 14 days of treatment.92
Table 5.
Overview of pharmacokinetic parameters of delamanid evaluated in various animal studies
| Reference | Animal model | Infected | Dose (mg/kg) | Single drug or combination | Treatment duration | Route of drug administration | Sample | Methods | T max (h) | T½ (h) | C max (mg/L) | AUC time span | AUC (μg·h/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mallikaarjun et al. (2020)92 | Mice, SLC:ICR | No | 0.625 | Single drug | Single-dose | Oral gavage | Plasma | HPLC-MS/MS | 0.100 | 0–24 | 1.188 | ||
| Mice, SLC:ICR | No | 2.5 | Single drug | Single-dose | Oral gavage | Plasma | HPLC-MS/MS | 0.297 | 0–24 | 3.581 | |||
| Mice, SLC:ICR | No | 10 | Single drug | Single-dose | Oral gavage | Plasma | HPLC-MS/MS | 1.012 | 0–24 | 11.547 | |||
| Matsumoto et al. (2006)10 | Mice | Yes | 2.5 | Single drug | Single dose | Oral | Plasma | LC-ESI-MS/MS | 6 | 7.6 | 0.297 | 0–24 | 4.13 |
| Pieterman et al. (2021)93 | Mice, BALB/c | Yes | 2.5 | BDQ (25) + LZD (100) | 4 weeks | Oral gavage | Plasma | LC-MS/MS | 0.75 | 0.864–1.080 | 0–24 | 11.234 | |
| Sasahara et al. (2015)87 | Mice, ICR | No | 3 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 2 | 7.2 | 0.4787 | 0–480 | 5.536 |
| 0–∞ | 6.1508 | ||||||||||||
| Mice, ICR | No | 30 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 2.3141 | 0–24 | 35.8403 | |||
| Mice, ICR | No | 30 | Single drug | 13 weeks | Oral | Plasma | LC-MS/MS | 2.9209 | 0–24 | 36.5094 | |||
| Ramirez et al. (2021)94 | Rats, Sprague Dawley | No | 10 | Single drug | Single-dose | Oral gavage | Plasma | HPLC | 3.4 | 0.256 | 0–96 | 5.68 | |
| Shibata et al. (2017)84 | Rats, Sprague-Dawley; males, non-fasted | No | 3 | Single drug | Single dose | Oral | Blood | Radioactivity of 14C-labelled delamanid | 8 | 82.3 | 0.5818 | 0–168 | 19.4 |
| 0–∞ | 22.8 | ||||||||||||
| Rats, Sprague-Dawley; males, fasted | No | 3 | Single drug | Single dose | Oral | Blood | Radioactivity of 14C-labelled delamanid | 6.3 | 49.5 | 0.7351 | 0–168 | 19.6 | |
| 0–∞ | 20.9 | ||||||||||||
| Rats, Sprague-Dawley; females, non-fasted | No | 3 | Single drug | Single dose | Oral | Blood | Radioactivity of 14C-labelled delamanid | 8 | 57.2 | 0.643 | 0–168 | 20.3 | |
| 0–∞ | 22.3 | ||||||||||||
| Rats, Sprague-Dawley; females, fasted | No | 3 | Single drug | Single dose | Oral | Blood | Radioactivity of 14C-labelled delamanid | 5 | 59.8 | 0.8149 | 0–168 | 19.7 | |
| 0–∞ | 21.3 | ||||||||||||
| Sasahara et al. (2015)87 | Rats, Sprague-Dawley | No | 3 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 4 | 5.1 | 0.6005 | 0–480 | 7.9418 |
| 0–∞ | 7.9698 | ||||||||||||
| Rats, Sprague-Dawley | No | 30 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 2.6953 | 0–24 | 36.6397 | |||
| Rats, Sprague-Dawley | No | 30 | Single drug | 26 weeks | Oral | Plasma | LC-MS/MS | 1.7992 | 0–24 | 34.2379 | |||
| Chen et al. (2017)95 | Guinea pigs | No | 10 | Single drug | Single dose | Oral | Plasma | HPLC-ESI-MS/MS | 0.21 | 0–48 | 2.32 | ||
| No | 100 | Single drug | Single dose | Oral | Plasma | HPLC-ESI-MS/MS | 0.53 | 0–48 | 9.45 | ||||
| Tucker et al. (2019)85 | Rabbits, New Zealand White | Yes | 5 | Single drug | Single-dose | Oral gavage | Plasma | HPLC-MS/MS | 12 | 13.9 | 0.2558 | 0–24 | 3.8112 |
| 0–48 | 4.229 | ||||||||||||
| 0–∞ | 4.2553 | ||||||||||||
| Rabbits, New Zealand White | No | 5 | Single drug | Single-dose | Oral gavage | Plasma | HPLC-MS/MS | 12 | 14.1 | 0.1956 | 0–24 | 2.7856 | |
| 0–48 | 3.4613 | ||||||||||||
| 0–∞ | 3.5545 | ||||||||||||
| Sasahara et al. (2015)87 | Dogs | No | 10 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 8 | 18.4 | 0.3578 | 0–768 | 10.628 |
| 0–∞ | 10.9275 | ||||||||||||
| Dogs, beagle | No | 30 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 0.3831 | |||||
| Dogs, beagle | No | 30 | Single drug | 39 weeks | Oral | Plasma | LC-MS/MS | 1.4007 | 0–24 | 21.7692 |
T max, time until the highest concentration is reached; T½, half-life time, time until the initial drug concentration is halved; Cmax, highest concentration reached.
Pretomanid
Like delamanid, pretomanid is widely distributed among various organs. After a single oral administration of 40 mg/kg in rats, pretomanid was detectable in liver, heart, lung, spleen, kidney, stomach and intestine.96 Pretomanid was shown to effectively cross the blood–brain barrier as well.96–99 In rats, plasma concentrations were shown to be 5-fold higher than brain tissue concentrations, and 2.5-fold higher than lung tissue concentrations. However, this might be different for multiple dose administrations.99
In human plasma, 94% of pretomanid is protein bound.72 Dogra et al.31 found that after incubation of pretomanid with supernatant of human liver homogenates several minor metabolites could be identified, but not the des-nitro metabolite that is formed upon bio-activation of pretomanid by mycobacterial Ddn. Hence, while mycobacteria can activate pretomanid by des-nitrification, this process does not occur with human liver supernatant.31 Preclinical100,101 and clinical102 studies have indicated that exposure to pretomanid is altered when co-administered with several other drugs. Together, these results imply that, compared with delamanid, albumin metabolism plays a smaller role for pretomanid, and that pretomanid is at least partly metabolized in the liver.87 However, the exact metabolic pathway of pretomanid is yet to be unravelled, and mechanisms that underlie drug–drug interactions (e.g. CYP isoenzymes and drug transporters) require further study.
Results of various animal PK studies for pretomanid are summarized in Table 6. The methods of these studies are quite heterogeneous, using different animal species (mice, rats or guinea pigs), dose levels, treatment durations, routes of administration, and treatment combinations. In humans, the currently approved dose in the clinic is 200 mg once a day to be taken with food.70 Human clinical trials have reported AUC0–t values corresponding to this dosing regimen of 28.087 μg·h/mL (single dose administration, in fasted state, monotherapy), 51.643 μg·h/mL (single dose administration, in fed state, monotherapy),103 30.2 μg·h/mL (7 days treatment, monotherapy), 60.487 μg·h/mL (14 days treatment, combination regimen with bedaquiline, pyrazinamide and clofazimine), 61.534 μg·h/mL (14 days treatment, combination regimen with bedaquiline and clofazimine), and 76.292 μg·h/mL (14 days treatment, combination regimen with bedaquiline and pyrazinamide).104 As can be seen in Table 6, similar drug exposure in mice was reached after administration of a single oral dose of 25 mg/kg.105 A single oral dose administration of 54 mg/kg106 and 4 weeks of daily oral treatment with 100 mg/kg in combination with either bedaquiline, moxifloxacin and pyrazinamide, or with bedaquiline and linezolid107 resulted in AUC0–24 of 127.5, 104.2 and 99.13 μg·h/mL, respectively, which were slightly higher than the exposures reached in humans. However, at 100 mg/kg, another mouse study demonstrated higher AUC0–24 values ranging between 327.6 and 424.0 μg·h/mL.108 In that study, pretomanid was either administered alone or within a combination regimen, and was given once or for 2 months.108 None of the rat studies showed AUC values that equal human exposures.96,99–101 In guinea pigs, a single oral administration of 50 mg/kg, and 7 day treatment with 25 mg/kg or 50 mg/kg administered twice daily, resulted in AUC values in the range of those observed in humans at the approved clinical dose.109
Table 6.
Overview of pharmacokinetic parameters of pretomanid evaluated in various animal studies
| Reference | Animal model | Infected | Dose (mg/kg) | Treatment combination | Treatment duration | Route of drug administration | Sample | Methods | Pretomanid Tmax (h) | T½ (h) | C max (mg/L) | AUC time span | AUC (μg·h/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lakshminarayana et al. (2014)105 | Mice, CD-1 | No | 25 | No | Oral | Plasma | LC-MS/MS | 2 | 2.7 | 6 | 0–24 | 50.9 | |
| 10 | No | Intravenous | Plasma | LC-MS/MS | 1.6 | ||||||||
| Nuermberger et al. (2006)108 | Mice, BALB/c | Yes | 100 | No | Single dose | Oral gavage | Serum | HPLC | 4.7 | 12.8 | 21.4 | 0–24 | 327.6 |
| Mice, BALB/c | Yes | 100 | No | 2 months | Oral gavage | Serum | HPLC | 1.3 | 18.3 | 25 | 0–24 | 396.8 | |
| Mice, BALB/c | Yes | 100 | RIF (10) + INH (25) + PZA (150) | Single dose | Oral gavage | Serum | HPLC | 11.0 | 11.3 | 20.4 | 0–24 | 370.5 | |
| Mice, BALB/c | Yes | 100 | RIF (10) + INH (25) + PZA (150) | 2 months | Oral gavage | Serum | HPLC | 3.3 | 9.7 | 27.7 | 0–24 | 424 | |
| Tasneen et al. (2008)106 | Mice, BALB/c | 54 | Single dose | Oral | Serum | 15.1 | 0–∞ | 127.5 | |||||
| Ahmad et al. (2011)110 | Mice, BALB/c | No | 3–1458 | Single drug | Single dose | Esophagal gavage | Serum | HPLC | 4 | 4–6 | |||
| Mudde et al. (2021)107 | Mice, BALB/c | Yes | 100 | BDQ (25) + MXF (100) + PZA (150) | 4 weeks | Oral | Serum | LC-MS/MS | 6.89–7.03 | 0–24 | 104.2 | ||
| Mice, BALB/c | Yes | 100 | BDQ (25) + LZD (100) | 4 weeks | Oral | Serum | LC-MS/MS | 7.70–9.50 | 0–24 | 99.13 | |||
| Wang et al. (2018)101 | Rats, Sprague-Dawley | No | 20 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 5 | 5.6 | 3.87 | 0–36 | 2678.74 |
| 0–∞ | 2787.23 | ||||||||||||
| Wang et al. (2015)96 | Rats, Sprague-Dawley | No | 20 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 6 | 8.3 | 3.48 | 0–36 | 3291.9 |
| 0–∞ | 3552.7 | ||||||||||||
| Rats, Sprague-Dawley | No | 40 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 6 | 6.2 | 7.98 | 0–36 0–∞ |
5850.9 6007.9 |
|
| Rats, Sprague-Dawley | No | 80 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 6 | 7.4 | 15.29 | 0–36 | 12445.1 | |
| 0–∞ | 13072.1 | ||||||||||||
| Bratkowska et al. (2015)99 | Rats, Sprague Dawley | No | 20 | Single drug | Oral | Plasma | LC-MS/MS | 6 | 0.63 | 0–∞ | 3.7248 | ||
| Rats, Sprague Dawley | No | 20 | Single drug | Intraperitoneal | Plasma | LC-MS/MS | 0.25 | 1.15 | 0–∞ | 3.9885 | |||
| Wang et al. (2014)100 | Rats, Sprague-Dawley | 20 | Single drug | Single dose | Oral | Plasma | LC-MS/MS | 6 | 3.485 | 0–36 | 3297.503 | ||
| 0–∞ | 3558.315 | ||||||||||||
| Rats, Sprague-Dawley | 20 | MXF (40) + PZA (160) | Single dose | Oral | Plasma | LC-MS/MS | 4.6 | 6.388 | 0–36 | 4851.288 | |||
| 0–∞ | 5052.658 | ||||||||||||
| Sung et al. (2009)136 | Guinea pigs | No | 20 | Intravenous | Plasma | HPLC | 0.11 | 1.91 | 9.19 | 0–24 | 26.54 | ||
| Guinea pigs | No | 40 | Oral gavage | Plasma | HPLC | 4.00 | 2.43 | 4.14 | 0–24 | 25.77 | |||
| Guinea pigs | No | 20 | Insufflation | Plasma | HPLC | 4.33 | 2.83 | 2.01 | 0–24 | 14.80 | |||
| Guinea pigs | No | 40 | Insufflation | Plasma | HPLC | 3.25 | 4.38 | 3.42 | 0–24 | 32.34 | |||
| Guinea pigs | No | 60 | Insufflation | Plasma | HPLC | 3.60 | 5.91 | 4.58 | 0–32 | 50.96 | |||
| Dutta et al. (2013)109 | Guinea pigs | No | 12.5 | Single drug | Single dose | Oral | Serum | HPLC | 2.65 | 1.94 | 1.68 | 0–∞ | 11.19 |
| Guinea pigs | No | 25 (BID) | Single drug | 7 days | Oral | Serum | HPLC | 2.25 | 4.7 | 2.99 | 0–∞ | 42.19 | |
| Guinea pigs | No | 50 | Single drug | Single dose | Oral | Serum | HPLC | 2.66 | 3.16 | 5.84 | 0–∞ | 39.79 | |
| Guinea pigs | No | 50 (BID) | Single drug | 7 days | Oral | Serum | HPLC | 7 | 2.16 | 5.79 | 0–∞ | 70.95 |
T max, time until the highest concentration is reached; T½, half-life time, time until the initial drug concentration is halved; Cmax, highest concentration reached; BID, bis in die, i.e. twice a day.
According to Ahmad et al.,110 pretomanid activity was best described by the free drug %T>MIC (R2 = 0.87), followed by free drug AUC/MIC (R2 = 0.60). In the same study, simulated %T>MIC values in humans at a pretomanid dose of 200 mg were predicted to be 100%, assuming an MIC of 0.03125 mg/L. Such high %T>MIC values were also reported in the clinical study by Diacon et al.104 Although pretomanid at a dose of 25 mg/kg in mice resulted in exposure (AUC) comparable to exposure in humans, this dose led to lower plasma %T>MIC values than observed in clinical studies.105 Since both %T>MIC and AUC/MIC are thought to be important drivers of efficacy for pretomanid, once daily dosing with 25 to 100 mg/kg has been used in mice in attempts to model %T>MIC and AUC/MIC that are similar to those observed in patients. In conclusion, determining the appropriate dosing regimen in animal models that mimics both the AUC values and %T>MIC encountered in the clinic is challenging. Ongoing mouse studies are exploring a lower dose of pretomanid at 50 mg/kg or lower, administered twice a day, in order to reflect the clinical drug exposure more accurately.
Doses used for delamanid in animal models are generally lower than those for pretomanid (Tables 5 and 6), while the indicated daily dose in humans is equal for both drugs (200 mg). In humans, drug exposures corresponding to this clinical dose are lower for delamanid than for pretomanid.92,103,104,111,112 In mice, on the other hand, drug exposures following administration of delamanid or pretomanid at 25–30 mg/kg seem to be in the same range (AUC0–24: 35.84 and 50.9 μg·h/mL, respectively).87,105 Hence, it seems reasonable that delamanid is dosed at lower levels than pretomanid in animal studies, in order to mimic exposures in humans at clinically approved doses.
In vivo activity
Animal models (mostly mouse models) are used to study the treatment response in a setting that approximates the complex environment encountered in TB-infected humans.113 Numerous mouse TB models have been developed that differ in inoculation route and dose, incubation period, treatment duration, outcome assessment, and mouse strain.113,114 Treatment outcome can be evaluated immediately after treatment completion (bactericidal activity) or a few months later to determine whether mice are cured nor not, which is defined by the absence of relapse (sterilizing activity).113 However, most mouse strains develop cellular granulomas upon TB infection, instead of the necrotizing, caseous lesions observed in human pulmonary TB.115 To study drug efficacy in the context of such necrotic lesions, other mouse strains (e.g. C3HeB/FeJ) or other animals (e.g. guinea pigs, rabbits or NHP) can be used.116 In this section, studies on delamanid will be discussed first followed by pretomanid, after which the compounds will be compared. For drug combinations, only combinations that have been assessed for both delamanid and pretomanid are considered in this review.
Delamanid
An overview of results from different animal models evaluating the treatment response of delamanid is presented in Table 7. Multiple mouse models have demonstrated bactericidal activity of delamanid at doses as low as 0.313 mg/kg (range of tested doses: 0.078–100 mg/kg).10,11,63,93,117–120 Dose-dependency of delamanid activity was shown in three studies.10,11,120 Depending on the model, delamanid showed similar or higher bactericidal activity than rifampicin,10,11,118 and activity of delamanid was shown to be equal in both immunocompromised and immunocompetent mice.10 Two mouse studies demonstrated bactericidal activity of delamanid in animals presenting with hypoxic lesions.95,117 Gengenbacher et al.117 used Nos2−/− mice that develop hypoxic lung lesions upon dermal injection with M. tuberculosis. In this model, lung cfu counts significantly decreased after treatment with delamanid at 1 mg/kg for 3 weeks. Using a guinea pig TB model, Chen et al.95 showed strong bactericidal activity of delamanid (100 mg/kg) administered for 8 weeks, as no cfu could be retrieved from the lung homogenates after treatment. This activity was similar to that of the standard HRZ-regimen.95 The potential role for delamanid in the treatment of latent TB is unknown as no preclinical studies investigating this have been published at this current time.
Table 7.
Summary of treatment activity of delamanid and pretomanid as a monotherapy in various animal models of tuberculosis
| Author | Animal (inoculation route) | M. tuberculosis strain | Time until start of treatment | Drug treatment (dose, mg/kg) | Treatment duration | Route of drug administration | Drug exposure | Outcome |
|---|---|---|---|---|---|---|---|---|
| Gengenbacher et al. (2017)117 | Nos2−/− mice (intradermal) | H37Rv | 42 days (control) or 56 days (hypoxic lung lesions) | DLM (1); PMD (75) | 70–84 days | Oral | NA | DLM and PMD were both active against non-replicating and replicating bacilli, and had comparable bactericidal activity in hypoxic necrotic lesions. |
| Tasneen et al. (2015)120 | BALB/c mice (aerosol) | H37Rv | 13–14 days | DLM (3–100); PMD (10–600) | 2–8 weeks | Oral | NA | DLM and PMD showed time-dependent and dose-dependent bactericidal activity. DLM was approximately 10-fold more active than PMD. |
| Upton et al. (2015)63 | BALB/c mice (aerosol) | Erdman | 10 days | DLM; PMD (100) | 3 weeks | Oral | NA | DLM was significantly more active than PMD in this model of acute infection. DLM led to a 1 log10 reduction in lung cfu. PMD inhibited mycobacterial growth, but did not reduce lung cfu. |
| BALB/c mice (aerosol) | Erdman | 70 days | DLM; PMD (100) | 3 weeks | Oral | NA | DLM was significantly more active than PMD in this model of chronic infection. DLM led to a 2 to 3 log10 reduction in lung cfu. PMD led to a 2 log10 reduction in lung cfu. | |
| Kmentova et al. (2010)119 | BALB/c mice | 70 days | DLM; PMD (100) | 3 weeks | Oral | NA | DLM was 10-fold more active than PMD, with 3 log10 versus 2 log10 reduction in lung cfu, respectively. | |
| Matsumoto et al. (2006)10 | ICR mice (intravenous) | Kurono | 4 weeks | DLM (0.156–40); PMD (1.25–40) | 4 weeks | Oral | AUC0–24 = 4.13 μg·h/mL (single dose of 2.5 mg/kg DLM) | DLM led to a dose-dependent reduction in lung cfu. For PMD, RIF and INH higher doses were needed to equal the load reduction by DLM. |
| BALB/c (nude) mice (intravenous) | Kurono | 1 day | DLM (0.313–10) | 10 days | Oral | AUC0–24 = 4.13 μg·h/mL (single dose of 2.5 mg/kg) | DLM led to a dose-dependent reduction in lung cfu, which was equal in immunodeficient and immunocompetent mice. | |
| Sasaki et al. (2006)11 | ICR mice (intravenous) | Kurono | 1 day | DLM (0.5–10) | 10 days | Oral | NA | DLM led to a 2.5 log10 to >4.4 log10 reduction in lung cfu, which was superior to RIF (5 mg/kg). |
| ICR mice (intravenous) | Kurono | 1 day | DLM (0.078–2.5) | 28 days | Oral | NA | DLM led to a dose-dependent reduction in lung cfu. DLM activity (0.313 mg/kg) was similar to RIF (5 mg/kg). | |
| Hariguchi et al. (2020)118 | ICR mice (intratracheal inoculation) | Kurono | 4 weeks | DLM (2.5) | 4 weeks | Oral | NA | DLM led to a significant 1.5 log10 reduction of lung cfu, which was similar to RIF (5 mg/kg) |
| Pieterman et al. (2021)93 | BALB/c mice (intratracheal instillation) | Beijing | 2 weeks | DLM (1.25, 2.5 or 5) | 3 weeks | Oral | AUC0–24 = 11.234 μg·h/mL (4 weeks treatment, dose 2.5 mg/kg, combined with BDQ 25 mg/kg + LZD 100 mg/kg) | DLM led to a 2 log10 reduction in lung cfu for all tested doses. |
| Chen et al. (2017)95 | Guinea pig (intratracheal inoculation) | Kurono | 4 weeks | DLM (100) | 4 or 8 weeks | Oral | AUC0–24 = 9.45 μg·h/mL (single dose of 100 mg/kg) | DLM led to a 3 log10 reduction in lung cfu after 4 weeks of exposure. No cfu were retrieved after 8 weeks of exposure. DLM showed bactericidal activity in hypoxic lesions. |
| Stover et al. (2000)12 | BALB/c mice (intravenous) | H37Rv | 4 days | PMD (25, 50, or 100) | 10 days | Oral | NA | PMD led to a dose-dependent reduction in lung cfu. PMD activity (25 mg/kg) was similar to INH activity (25 mg/kg). |
| Tyagi et al. (2005)124 | BALB/c mice (aerosol) | H37Rv | 20 days (initial phase), 8 weeks (continuation phase) | PMD (3.125–200) | 4–16 weeks | Oral | NA | PMD activity (100 mg/kg) was comparable to that of INH (25 mg/kg). PMD was active during both the initial and continuation phase of therapy. |
| Lenaerts et al. (2005)66 | C57BL/6 mice (aerosol) | Erdman | 19 days | PMD (50, 100, or 300) | 9 days | Oral | NA | PMD showed dose-dependent activity. PMD activity (100 mg/kg) was similar to that of RIF (20 mg/kg) and INH (25 mg/kg). |
| C57BL/6 mice (aerosol) | Erdman | 19 days | PMD (100) | 12 weeks | Oral | NA | PMD (100 mg/kg) was as active as INH (25 mg/kg). | |
| Lakshminarayana et al. (2014)105 | BALB/c mice (intranasal) | H37Rv | 4 weeks | PMD (25 or 100) | 4 weeks | Oral | AUC0–24 = 50.9 μg·h/mL (dose 25 mg/kg) | At 25 mg/kg PMD led to a 1.48 log10 reduction in lung cfu, and to a 2.3 log10 reduction at 100 mg/kg. |
| Nuermberger et al. (2006)108 | BALB/c mice (aerosol) | H37Rv | 19 days | PMD (100) | 2 months | Oral | AUC0–24 = 396.8 μg·h/mL (2 months treatment, dose 100 mg/kg) | PMD led to a 2 log10 reduction in lung cfu. |
| Tasneen et al. (2008)106 | BALB/c | H37Rv | 2 weeks | PMD (100) | 2 months | Oral | AUC0–∞ = 127.5 μg·h/mL (single dose of 54 mg/kg) | PMD led to a 2.7 log10 reduction in lung cfu, which was slightly inferior to the 3.1 log10 reduction by RIF (10 mg/kg). |
| Sala et al. (2010)71 | BALB/c mice (intravenous) | 18b without streptomycin | 4 weeks | PMD (100) | 8 weeks | Oral | NA | PMD led to a 1.5 log10 reduction in lung cfu, which was superior to INH (25 mg/kg), but inferior to RIF (10 mg/kg). |
| Lanoix et al. (2014)125 | BCG-immunized-BALB/c mice (aerosol) | H37Rv | 6 weeks | PMD (50) | 8 weeks | Oral | NA | PMD led to a 1 log10 reduction in lung cfu, which was similar to INH (10 mg/kg), but inferior to RIF (10 mg/kg) |
| BCG-immunized-C3HeB/FeJ mice (aerosol) | H37Rv | 6 weeks | PMD (50) | 8 weeks | Oral | NA | PMD led to a 0.75 log10 reduction in lung cfu, which was similar to INH (10 mg/kg), but inferior to RIF (10 mg/kg) | |
| Dutta et al. (2014)126 | BCG-immunized-C3HeB/FeJ mice (aerosol) | H37Rv | 6 weeks | PMD (50) | 1–4 months | Oral | NA | PMD led to a 2.7 log10 reduction in lung cfu, which was comparable to INH (10 mg/kg), but inferior to RIF (10 mg/kg). The relapse rate of PMD (assessed 3 months after completion of a 4-month treatment duration) was 100%, which was equal to INH, and higher than RIF (33%). |
| Stover et al. (2000)12 | Guinea pig (aerosol) | H37Rv | 4 weeks | PMD (40) | 4 weeks | Oral | NA | PMD led to a 1 log10 reduction in lung cfu, which was comparable to INH (25 mg/kg). |
| Garcia-Contreras et al. (2010)127 | Guinea pig (aerosol) | H37Rv | 4 weeks | PMD (inhaled: 180 or 360 mg; oral: 40 mg/kg) | 4 weeks | Inhaled or oral | NA | PMD led to a significant reduction of the mycobacterial load. Higher PMD activity was observed for oral administration versus inhaled doses. |
DLM, delamanid; PMD, pretomanid; NA, not assessed; cfu, colony forming units; RIF, rifampicin; INH, isoniazid; BDQ, bedaquiline; LZD, linezolid.
Drug combination regimens containing delamanid have been studied to a lesser extent in vivo than combinations containing pretomanid (Table 8). Two combination regimens were studied for both compounds although not in the same experiment: (i) rifampicin and pyrazinamide together with delamanid (RDZ) 10,95 or pretomanid (RPaZ),106,108 and (ii) bedaquiline and linezolid either combined with delamanid (BDL)93 or pretomanid (BPaL).107,121–123 Unfortunately, no head-to-head comparisons of delamanid- or pretomanid-containing drug combinations have been published to date. The RDZ regimen (delamanid at 2.5 mg/kg) showed promising bactericidal activity in a mouse TB model, reaching culture-negativity at least 2 months faster than the standard regimen consisting of isoniazid, rifampicin, pyrazinamide and ethambutol (HRZE).10 Similar results of the RDZ regimen (delamanid at 100 mg/kg) were found by Chen et al.95 in a guinea pig TB model. Delamanid combined with bedaquiline and linezolid was recently evaluated by Pieterman et al.93 Mice were infected with M. tuberculosis of the Beijing genotype via intratracheal instillation. Two weeks later, treatment was started with BDL (delamanid at 2.5 mg/kg) via oral gavage for 2 to 6 months. The mycobacterial load in the lungs was assessed both directly following treatment completion, and three months later to evaluate whether the infection had relapsed or not. Treatment with BDL was highly effective. Of the 15 mice treated with BDL for 4 months or longer, only 1 mouse relapsed. In the HRZE-group on the other hand, relapse rates were much higher, and after 6 months of treatment there were still bacteria in 1 out of 3 mice that could be cultured from the lungs.
Table 8.
Summary of treatment efficacy of delamanid and pretomanid within various drug combination regimens in animal models of tuberculosis
| Author | Animal (infection route) | M. tuberculosis strain | Incubation period until start of treatment | Drug combination (dose in mg/kg) | Treatment duration | Route of drug administration | Exposure to DLM or PMD | Outcome | |
|---|---|---|---|---|---|---|---|---|---|
| Bactericidal activity | Relapse rates | ||||||||
| Matsumoto et al. (2006)10 | ICR mice (intratracheal instillation) | Kurono | 28 days | 2 months RIF (5) + DLM (2.5) + PZA (100) and 2 months RIF (5) + DLM (2.5) | 4 months | Oral | AUC0–24 = 4.13 μg·h/mL (monotherapy, single dose of 2.5 mg/kg) | Faster culture-negativity (by at least 2 months) in the lungs compared to the standard regimen (HRZE). | NA |
| Chen et al. (2017)95 | Guinea pigs (intratracheal instillation) | Kurono | 4 weeks | RIF (25) + DLM (100) + PZA (150) | 4 or 8 weeks | Oral | AUC0–24 = 9.45 μg·h/mL (monotherapy, single dose of 100 mg/kg) | Culture-negativity in the lungs was reached after 4 weeks of treatment versus 8 weeks for the standard regimen (HRZ). | NA |
| Nuermberger et al. (2006)108 | BALB/c mice (aerosol) | H37Rv | 19 days | 2 months RIF (10) + PMD (100) + PZA (150) and 4 months RIF (10) + PMD (100) | 6 months | Oral | AUC0–24 = 396.8 μg·h/mL (monotherapy, 2 months treatment, dose 100 mg/kg) | Culture-negativity in the lungs was reached after 4 months of treatment versus 6 months for the standard regimen (HRZ). This difference was not statistically significant. | Relapse rates were comparable to those of the standard HRZ-regimen (2/19 versus 0/46, respectively). |
| Tasneen et al. (2008)106 | BALB/c mice (aerosol) | H37Rv | 2 weeks | RIF (10) + PMD (12.5/25/50/100) + PZA (150) | 2, 4, 5, or 6 months | Oral | AUC 0–∞ = 127.5 μg·h/mL (monotherapy, single dose of 54 mg/kg) | PMD at 50 and 100 mg/kg increased activity of RIF + PZA in a dose-dependent manner. Culture-negativity in the lungs was reached after 2 months of treatment (PMD 100 mg/kg). | No relapse was seen after 4 months of treatment versus a relapse rate of 15% for the regimen (HRZ). |
| Pieterman et al. (2021)93 | BALB/c mice (intratracheal instillation) | Beijing | 2 weeks | BDQ (25) + DLM (2.5) + LZD (100) | 2–6 months | Oral | AUC0–24 = 11.234 μg·h/mL (4 weeks treatment, dose 2.5 mg/kg, BDL combination) | Culture negativity in the lungs was reached after 2 months of treatment versus 20 weeks for the standard regimen (HRZE). | No relapse was seen after treatment duration of 4 months or longer (except for 1 mouse, treated for 5 months). HRZE-treated mice still relapsed after 6 months of treatment (1/3 mice). |
| Tasneen et al. (2016)128 | BALB/c mice (aerosol) | H37Rv | 13–14 days | BDQ (25) + PMD (50) + LZD (100) | 2–4 months | Oral | NA | Two and 3 months of treatment led to a significantly lower mycobacterial load in the lungs compared to the standard regimen (HRZ). | No relapse was seen after 3 months of treatment. Infection still relapsed in HRZ-treated mice after 4 months of treatment. |
| Xu et al. (2019)123 | BALB/c mice (aerosol) | H37Rv | 13 days | BDQ (25) + PMD (100) + LZD (100) | 1–4 months | Oral | NA | Addition of PMD to BDQ + LZD led to a higher mycobacterial load reduction when administered for 1 and 2 months and prevented the emergence of BDQ resistance. | After 2 months of treatment with BPaL, infection relapsed in 7/15 mice. No relapse was seen after 3 months of treatment. |
| Bigelow et al. (2020)121 | BALB/c mice (aerosol) | H37Rv or HN878 | 2 weeks | BDQ (25) + PMD (50 or 100) + different LZD dosing strategies (45 or 90) | 1–3 months | Oral | NA | BDQ + PMD with different dosing strategies for LZD resulted in a higher mycobacterial load reduction compared to the standard regimen (HRZE). LZD’s contribution to BDQ + PMD + LZD regimens was dependent on the M. tuberculosis strain. | Relapse rates were highly variable between the different LZD dosing strategies, LZD (90 mg/kg) dosed every other day leading to the highest relapse rate (11/15 mice) and LZD (90 mg/kg) dosed daily to the lowest relapse rates (1/15 mice) |
| Xu et al. (2021)122 | BALB/c mice (aerosol) | H37Rv | 2 weeks | BDQ (25) + PMD (100) + LZD (100) | 1 month | Oral | NA | Lung cfu were reduced by approximately 2 log10. | NA |
| Mudde et al. (2021)107 | BALB/c mice (intratracheal instillation) | Beijing | 2 weeks | BDQ (25) + PMD (100) + LZD (100) | 6–13 weeks | Oral | AUC0–24 = 104 and 99.13 μg·h/mL (4 weeks treatment, dose 100 mg/kg, BPaMZ combination or BPaL combination, respectively) | NA | Mice treated for the maximum duration of 13 weeks still showed relapse (3/3 mice). |
| Tasneen et al. (2021)137 | BALB/c mice (aerosol) | HN878 (Beijing subfamily) | 7 weeks | BDQ (25) + PMD (100) + LZD (100) | 1 or 2 months | Oral | NA | 1 month of BPaL treatment led to a 3.87 log10 reduction in lung cfu. | After 2 months of treatment with BPaL, infection relapsed in 7/15 mice. After 3 months of treatment with HRZE, infection relapsed in 9/15 mice. |
DLM, delamanid; PMD, pretomanid; RIF, rifampicin; PZA, pyrazinamide; HRZE, isoniazid + rifampicin + pyrazinamide + ethambutol; HRZ, isoniazid + rifampicin + pyrazinamide; BDQ, bedaquiline; LZD, linezolid; BDL, bedaquiline + delamanid + linezolid; BPaMZ, bedaquiline + pretomanid + moxifloxacin + pyrazinamide; BPaL, bedaquiline, pretomanid, linezolid.
Pretomanid
The in vivo bactericidal activity of pretomanid as a monotherapy has been evaluated in various animal studies (Table 7). In mice, pretomanid showed bactericidal activity at dose levels of 12.5–20 mg/kg or higher (range of tested doses: 1.25–600 mg/kg).10,12,66,71,108,119,120,124,125 In several studies, the activity of pretomanid (40–100 mg/kg) was similar to that of isoniazid (25 mg/kg) 12,66,124 and rifampicin (20 mg/kg).66 The rank order in activity was slightly different in two mouse models of latent TB infection using BCG-immunized mice,125,126 with pretomanid (50 mg/kg) showing less activity than rifampicin (10 mg/kg), although the activity was similar to that of isoniazid (10 mg/kg). Pretomanid’s promising activity in animals presenting with hypoxic pulmonary lesions was demonstrated in various animal models, including a Nos2−/− mouse model,117 a C3HeB/FeJ mouse model,126 and two guinea pig models.12,127 The ability of pretomanid as monotherapy to cure latent TB was evaluated in one murine study using BCG-vaccinated C3HeB/FeJ mice.126 In all mice (15/15), the infection relapsed after 4 months of treatment with only pretomanid (50 mg/kg). The same outcome was observed for isoniazid (10 mg/kg), while rifampicin (10 mg/kg) performed better with a relapse rate of 33%. Selection of resistant colonies was, however, not part of the published study.
Five studies have evaluated the bactericidal activity of both delamanid and pretomanid.10,63,117,119,120 Again, limited information is available where both compounds are evaluated side by side in the same model, and in the same experiment. Interestingly, in all five studies, the bactericidal activity of delamanid was superior to that of pretomanid. Delamanid led to higher load reductions than pretomanid at equal dose levels,10,63,119 or required lower dose levels than pretomanid to achieve a comparable load reduction.10,117,120 However, comparing the bactericidal activity of the compounds in the light of drug exposure rather than dose levels adds nuance to the presumed superiority of delamanid. The clinically approved dosing regimen of delamanid (100 mg, twice a day) is reported to result in AUC values between 3.40 and 10.673 μg·h/mL.92,112 Higher AUC values of 28.087 to 76.292 μg·h/mL were reported for pretomanid in clinical studies (200 mg, once a day), with %T>MIC (an important driver of efficacy) up to 100%.103,104 In mice, AUC values similar to the ones measured in patients were found for delamanid at dose levels of 2.5 to 10 mg/kg (Table 5).10,87,92,93 Although pretomanid is often dosed at 100 mg/kg in mice,12,63,66,71,105,106,108,119 in order to model the %T>MIC achieved in patients, lower dose levels of pretomanid (25 to 54 mg/kg) lead to AUC values more closely reflecting AUC values reported in humans (Table 6).105,106 Matsumoto et al.10 reported that in their mouse model of TB infection, delamanid at 2.5 mg/kg showed similar bactericidal activity to pretomanid at 20 mg/kg, as both dose levels reduced the mycobacterial burden in the lungs by 1.9 log10 cfu after 4 weeks of treatment. In line with these results, Tasneen et al.120 observed in their mouse TB model that 8 weeks of treatment with either delamanid at 2.5 mg/kg or pretomanid at 30 mg/kg resulted in a 1.6 log10 reduction in lung cfu counts. Taken together, these results indicate that when the compounds are compared at dose levels equivalent to those in humans based on AUC, their bactericidal activity is quite similar.
As to the performance of pretomanid in combination with other TB drugs, the combination of pretomanid, rifampicin and pyrazinamide (RPaZ) demonstrated higher bactericidal activity than the standard HRZ regimen in two mouse TB models (Table 8).106,108 Relapse rates, however, did not seem to differ considerably between the two regimens.106,108 Pretomanid combined with bedaquiline and linezolid (BPaL) performed better than the standard regimen in various mouse TB models, in terms of bactericidal activity and relapse rates.107,121,122,128 BPaL and BDL were studied in two separate studies using the same experimental set-up, except that treatment with BDL lasted 8 to 24 weeks, while this was 6 to 13 weeks for BPaL. For both drug regimens, at least 2 to 2.5 months of treatment were needed to prevent relapse in some of the mice.93,107
Discussion
With this review, we aimed to provide an overview of preclinical data on the nitroimidazoles delamanid and pretomanid. Both compounds have contributed considerably to the change of the TB treatment landscape during the last decade, and are expected to further impact the improvement of TB treatment in the coming years. Although both compounds belong to the same drug class and share many similarities, we identified several differences between the drugs, shaping the context in which results from preclinical research on delamanid and pretomanid could inform clinical studies.
Based on what is known in the published literature, the mode of action of delamanid and pretomanid seems to differ slightly. Both compounds affect mycolic acid synthesis. Pretomanid only inhibits synthesis of ketomycolates 12 and not methoxymycolates, whereas delamanid inhibits the synthesis of both these classes.9,10 Although both compounds intervene in aerobic respiration, pretomanid activity generates the formation of reactive nitrogen species,16,28 whereas an NAD–delamanid adduct is thought to contribute to the antimycobacterial activity of delamanid.34 Apart from the nitroimidazole ring, delamanid and pretomanid have distinct chemical structures (Table 1). However, the structural components that are thought to be involved in the antimycobacterial activity of delamanid and pretomanid are shared between the two drugs.28,34
Of particular interest is the finding that certain M. tuberculosis isolates with preserved susceptibility to delamanid, are resistant to pretomanid (or the other way around).29,36,49 Here, the different chemical structure of the compounds could play a role in terms of the binding orientation to Ddn. Lee et al.29 demonstrated that the dual methoxy and phenoxy-methyl substituents on the C6 position of the oxazole ring cause delamanid to bind differently to Ddn than pretomanid, which contains an oxazine ring with only a single substituent at the equivalent position. As such, certain mutations in ddn could result in pretomanid resistance while retaining the ability to activate delamanid. The fact that drug resistance has been found in M. tuberculosis isolates from patients who have not been treated with delamanid or pretomanid, implies that resistance to these drugs might arise due to genetic drift.29 Indeed, the genes associated with delamanid and pretomanid resistance are genetically diverse. Various gene mutations might result in drug resistance, while at the same time several genetic variances have been reported that were not associated with drug resistance.39,44,46,48,68 Therefore, it is not easy to pinpoint specific mutations that indicate under what circumstances delamanid and pretomanid can replace each other in the case of drug resistance or drug intolerance. Drug susceptibility testing before and during TB treatment could be performed to overcome this problem and adapt treatment regimens accordingly.
One critical hiatus in our comparison of preclinical studies is the limited amount of available head-to-head data where both compounds are tested in the same assay or model, in the same laboratory at the same time. In order to have an accurate preclinical comparison, more one-on-one preclinical studies will have to be conducted. In addition, we acknowledge the fact that the complexity of human TB infection is not easily captured in in vitro assays and in vivo preclinical models. Therefore, the preclinical performance of compounds is an informative approximation of their effect in humans. The comparison of results between different in vitro assays is further complicated by the great variety in experimental design, including differences in treatment duration, metabolic state of M. tuberculosis, methods of inducing a non-replicating state, and treatment dosing. Each of these variables could considerably impact the results on drug activity. This also accounts for in vivo TB models, with differences in animals used, inoculation dose, route of infection, incubation time, treatment dose, treatment duration, and outcome assessment. However, we also regard this plethora of assays and testing models as an advantage as every tool will provide more, and often complementary, information on both compounds under various conditions.
When comparing delamanid and pretomanid in terms of bactericidal activity in vitro, delamanid is more potent than pretomanid, with lower MIC values (0.001–0.024 mg/L versus 0.012–0.200 mg/L, respectively, based on head-to-head comparisons)10,49,63 and delamanid effects higher mycobacterial load reductions in vitro, at lower drug concentrations than pretomanid (Tables 3 and 4). In various mouse models of TB infection, delamanid reduced the mycobacterial load in the lungs of mice to a greater extent than pretomanid when the drugs were administered at equal doses (Table 7),10,63,119 and comparable load reductions were established when delamanid was dosed at lower levels than pretomanid.10,117,120 However, it is more informative to compare the activity of delamanid and pretomanid after administration to mice at dose levels that result in drug exposures similar to those achieved at the approved clinical dose. For delamanid, this would be 2.5 to 10 mg/kg in mice, as corresponding AUC values are in the same range as AUC values reported in humans at its clinically approved dose.10,87,92,93,112 For pretomanid, administration of 25 to 54 mg/kg in mice was reported to result in human-equivalent dose exposures, based on AUC, although 25 mg/kg was reported to result in lower %T>MIC lower than achieved in the clinic.103–106 In fact, in two different mouse TB models, the activity of delamanid at 2.5 mg/kg was shown to be similar to pretomanid at 20 mg/kg and 30 mg/kg.10,120 Considering that for delamanid AUC0–24/MIC was found to be the main driver of activity 92 and that its activity is dose-dependent in tested concentrations up to 100 mg/kg in mice,10,120 one might speculate that if higher drug exposures could be safely reached in the clinic, delamanid might be expected to do better than at its currently approved clinical dose. This may also hold true for pretomanid, for which %T>MIC is the most important driving PK/PD index (and this is already >90% at the approved clinical dose), but for which AUC/MIC is also an important driver of efficacy.104,110
As to delamanid-containing and pretomanid-containing regimens, no head-to-head comparisons have yet been published. In animal studies, the drug combinations where delamanid or pretomanid replaced isoniazid in the standard regimen, as well as the relatively new BPaL and BPaMZ regimens, showed higher activity and achieved cure following a shorter treatment period than the HRZE regimen. Especially for pretomanid, many other drug combinations have been assessed in vivo,123,129–131 whereas delamanid-containing TB regimens were studied to a lesser extent. Studying the substitution of pretomanid for delamanid in future studies on the efficacy of novel TB drug regimens would be worthwhile, as delamanid might be a valuable alternative to pretomanid (or vice versa) in the case of drug resistance, intolerance or significant drug–drug interactions.
Funding
This study was supported by internal funding.
Transparency declarations
None to declare.
References
- 1. World Health Organization . Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment. World Health Organization, 2019. [PubMed] [Google Scholar]
- 2. Critical Path Institute . Critical Path to TB Drug Regimens. https://c-path.org/programs/cptr/.
- 3. Edwards DI. Nitroimidazole drugs–action and resistance mechanisms. I. Mechanisms of action. J Antimicrob Chemother 1993; 31: 9–20. [DOI] [PubMed] [Google Scholar]
- 4. Shinn D. Metronidazole in acute ulcerative gingivitis. Lancet 1962; 279: 1191. [Google Scholar]
- 5. Cavalleri B, Ballotta R, Arioli Vet al. New 5-substituted 1-alkyl-2-nitroimidazoles. J Med Chem 1973; 16: 557–60. [DOI] [PubMed] [Google Scholar]
- 6. Nagarajan K, Shankar R, Rajappa Set al. Nitroimidazoles. XXI. 2,3-dihydro-6-nitroimidazo [2,1-b] oxazoles with antitubercular activity. Eur J Med Chem 1989; 24: 631–3. [Google Scholar]
- 7. Ashtekar DR, Costa-Perira R, Nagrajan Ket al. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob Agents Chemother 1993; 37: 183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walsh JS, Wang R, Bagan Eet al. Structural alterations that differentially affect the mutagenic and antitrichomonal activities of 5-nitroimidazoles. J Med Chem 1987; 30: 150–6. [DOI] [PubMed] [Google Scholar]
- 9. Matsumoto M, Hashizume H, Tsubouchi Het al. Screening for novel antituberculosis agents that are effective against multidrug resistant tuberculosis. Curr Top Med Chem 2007; 7: 499–507. [DOI] [PubMed] [Google Scholar]
- 10. Matsumoto M, Hashizume H, Tomishige Tet al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 2006; 3: e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sasaki H, Haraguchi Y, Itotani Met al. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J Med Chem 2006; 49: 7854–60. [DOI] [PubMed] [Google Scholar]
- 12. Stover CK, Warrener P, VanDevanter DRet al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000; 405: 962–6. [DOI] [PubMed] [Google Scholar]
- 13. Baker W, Shaopei C, Keeler E. Nitroimidazole antibacterial compounds and methods of use thereof. PathoGenesis Corporation. United States Patent 1997.
- 14. Baker W, Shaopei C, Keeler E. Nitro-[2,1-b]imidazopyran compounds and antibacterial uses thereof. PathoGenesis Corporation. United States Patent 2000.
- 15. Boshoff HIM, Myers TG, Copp BRet al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem 2004; 279: 40174–84. [DOI] [PubMed] [Google Scholar]
- 16. Manjunatha U, Boshoff HI, Barry CE. The mechanism of action of PA-824: Novel insights from transcriptional profiling. Commun Integr Biol 2009; 2: 215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van den Bossche A, Varet H, Sury Aet al. Transcriptional profiling of a laboratory and clinical Mycobacterium tuberculosis strain suggests respiratory poisoning upon exposure to delamanid. Tuberculosis (Edinb) 2019; 117: 18–23. [DOI] [PubMed] [Google Scholar]
- 18. Embley TM, Stackebrandt E. The molecular phylogeny and systematics of the actinomycetes. Annu Rev Microbiol 1994; 48: 257–89. [DOI] [PubMed] [Google Scholar]
- 19. Jarlier V, Nikaido H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett 1994; 123: 11–8. [DOI] [PubMed] [Google Scholar]
- 20. Indrigo J, Hunter RL, Actor JK. Cord factor trehalose 6,6'-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology (Reading) 2003; 149: 2049–59. [DOI] [PubMed] [Google Scholar]
- 21. Rao V, Gao F, Chen Bet al. Trans-cyclopropanation of mycolic acids on trehalose dimycolate suppresses Mycobacterium tuberculosis -induced inflammation and virulence. J Clin Invest 2006; 116: 1660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Batt SM, Minnikin DE, Besra GS. The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host’s immune system. Biochem J 2020; 477: 1983–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Y, Matsumoto M, Ishida Het al. Delamanid: from discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis (Edinb) 2018; 111: 20–30. [DOI] [PubMed] [Google Scholar]
- 24. Purwantini E, Mukhopadhyay B. Rv0132c of Mycobacterium tuberculosis encodes a coenzyme F420-dependent hydroxymycolic acid dehydrogenase. PLoS One 2013; 8: e81985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujiwara M, Kawasaki M, Hariguchi Net al. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis (Edinb) 2018; 108: 186–94. [DOI] [PubMed] [Google Scholar]
- 26. Gurumurthy M, Mukherjee T, Dowd CSet al. Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioreductive activation of bicyclic nitroimidazoles. FEBS J 2012; 279: 113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haver HL, Chua A, Ghode Pet al. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2015; 59: 5316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh R, Manjunatha U, Boshoff HIMet al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 2008; 322: 1392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee BM, Harold LK, Almeida DVet al. Predicting nitroimidazole antibiotic resistance mutations in Mycobacterium tuberculosis with protein engineering. PLoS Pathog 2020; 16: e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanaki E, Hayashi M, Matsumoto M. Delamanid is not metabolized by Salmonella or human nitroreductases: a possible mechanism for the lack of mutagenicity. Regul Toxicol Pharmacol 2017; 84: 1–8. [DOI] [PubMed] [Google Scholar]
- 31. Dogra M, Palmer BD, Bashiri Get al. Comparative bioactivation of the novel anti-tuberculosis agent PA-824 in mycobacteria and a subcellular fraction of human liver. Br J Pharmacol 2011; 162: 226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boshoff HI, BarryCE, 3rd. Tuberculosis – metabolism and respiration in the absence of growth. Nat Rev Microbiol 2005; 3: 70–80. [DOI] [PubMed] [Google Scholar]
- 33. Rao SPS, Alonso S, Rand Let al. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2008; 105: 11945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayashi M, Nishiyama A, Kitamoto Ret al. Adduct formation of delamanid with NAD in mycobacteria. Antimicrob Agents Chemother 2020; 64: e01755-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manjunatha UH, Boshoff H, Dowd CSet al. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 2006; 103: 431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rifat D, Li SY, Ioerger Tet al. Mutations in fbiD (Rv2983) as a novel determinant of resistance to pretomanid and delamanid in Mycobacterium tuberculosis. Antimicrob Agents Chemother 2020; 65: e01948-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choi KP, Bair TB, Bae YMet al. Use of transposon Tn5367 mutagenesis and a nitroimidazopyran-based selection system to demonstrate a requirement for fbiA and fbiB in coenzyme F(420) biosynthesis by Mycobacterium bovis BCG. J Bacteriol 2001; 183: 7058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choi K-P, Kendrick N, Daniels L. Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F(420) and FO biosynthesis. J Bacteriol 2002; 184: 2420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Battaglia S, Spitaleri A, Cabibbe AMet al. Characterization of genomic variants associated with resistance to bedaquiline and delamanid in naive Mycobacterium tuberculosis clinical strains. J Clin Microbiol 2020; 58: e01304-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bloemberg GV, Keller PM, Stucki Det al. Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. N Engl J Med 2015; 373: 1986–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kardan-Yamchi J, Kazemian H, Battaglia Set al. Whole genome sequencing results associated with minimum inhibitory concentrations of 14 anti-tuberculosis drugs among rifampicin-resistant isolates of Mycobacterium tuberculosis from iran. J Clin Med 2020; 9: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pang Y, Zong Z, Huo Fet al. In vitro drug susceptibility of bedaquiline, delamanid, linezolid, clofazimine, moxifloxacin, and gatifloxacin against extensively drug-resistant tuberculosis in Beijing, China. Antimicrob Agents Chemother 2017; 61: e00900-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polsfuss S, Hofmann-Thiel S, Merker Met al. Emergence of low-level delamanid and bedaquiline resistance during extremely drug-resistant tuberculosis treatment. Clin Infect Dis 2019; 69: 1229–31. [DOI] [PubMed] [Google Scholar]
- 44. Schena E, Nedialkova L, Borroni Eet al. Delamanid susceptibility testing of Mycobacterium tuberculosis using the resazurin microtitre assay and the BACTEC™ MGIT™ 960 system. J Antimicrob Chemother 2016; 71: 1532–9. [DOI] [PubMed] [Google Scholar]
- 45. Yang JS, Kim KJ, Choi Het al. Delamanid, bedaquiline, and linezolid minimum inhibitory concentration distributions and resistance-related gene mutations in multidrug-resistant and extensively drug-resistant tuberculosis in Korea. Ann Lab Med 2018; 38: 563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reichmuth ML, Hömke R, Zürcher Ket al. Natural polymorphisms in Mycobacterium tuberculosis conferring resistance to delamanid in drug-naive patients. Antimicrob Agents Chemother 2020; 64: e00513-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoshiyama T, Mitarai S, Takaki Aet al. Multi-drug resistant tuberculosis with simultaneously acquired-drug resistance to bedaquiline and delamanid. Clin Infect Dis 2020; 73: 2329–31. [DOI] [PubMed] [Google Scholar]
- 48. Wang G, Jiang G, Jing Wet al. Prevalence and molecular characterizations of seven additional drug resistance among multidrug-resistant tuberculosis in China: A subsequent study of a national survey. J Infect 2021; 82: 371–7. [DOI] [PubMed] [Google Scholar]
- 49. Wen S, Jing W, Zhang Tet al. Comparison of in vitro activity of the nitroimidazoles delamanid and pretomanid against multidrug-resistant and extensively drug-resistant tuberculosis. Eur J Clin Microbiol Infect Dis 2019; 38: 1293–6. [DOI] [PubMed] [Google Scholar]
- 50. Gengenbacher M, Kaufmann SH. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev 2012; 36: 514–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mitchison DA. The search for new sterilizing anti-tuberculosis drugs. Front Biosci 2004; 9: 1059–72. [DOI] [PubMed] [Google Scholar]
- 52. Sarathy J, Dartois V, Dick Tet al. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 2013; 57: 1648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 1996; 64: 2062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parish T. In vitro drug discovery models for Mycobacterium tuberculosis relevant for host infection. Expert Opin Drug Discov 2020; 15: 349–58. [DOI] [PubMed] [Google Scholar]
- 55. Piccaro G, Giannoni F, Filippini Pet al. Activities of drug combinations against Mycobacterium tuberculosis grown in aerobic and hypoxic acidic conditions. Antimicrob Agents Chemother 2013; 57: 1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tudó G, Laing K, Mitchison DAet al. Examining the basis of isoniazid tolerance in nonreplicating Mycobacterium tuberculosis using transcriptional profiling. Future Med Chem 2010; 2: 1371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. European Committee on Antimicrobial Susceptibility Testing (EUCAST) . MIC and zone diameter distributions and ECOFFs https://mic.eucast.org/Eucast2/SearchController/search.jsp?action=performSearch&BeginIndex=0&Micdif=mic&NumberIndex=50&Antib=-1&Specium=806.
- 58. Keller PM, Hömke R, Ritter Cet al. Determination of MIC distribution and epidemiological cutoff values for bedaquiline and delamanid in Mycobacterium tuberculosis using the MGIT 960 system equipped with TB eXiST. Antimicrob Agents Chemother 2015; 59: 4352–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stinson K, Kurepina N, Venter Aet al. MIC of delamanid (OPC-67683) against Mycobacterium tuberculosis clinical isolates and a proposed critical concentration. Antimicrob Agents Chemother 2016; 60: 3316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. European Committee on Antimicrobial Susceptibility Testing (EUCAST) . Breakpoint tables for interpretation of MICs and zone diameters. Version 11.0. http://www.eucast.org.
- 61. Saliu OY, Crismale C, Schwander SKet al. Bactericidal activity of OPC-67683 against drug-tolerant Mycobacterium tuberculosis. J Antimicrob Chemother 2007; 60: 994–8. [DOI] [PubMed] [Google Scholar]
- 62. Dalton JP, Uy B, Okuda KSet al. Screening of anti-mycobacterial compounds in a naturally infected zebrafish larvae model. J Antimicrob Chemother 2017; 72: 421–7. [DOI] [PubMed] [Google Scholar]
- 63. Upton AM, Cho S, Yang TJet al. In vitro and in vivo activities of the nitroimidazole TBA-354 against Mycobacterium tuberculosis. Antimicrob Agents Chemother 2015; 59: 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chandramohan Y, Padmanaban V, Bethunaickan Ret al. In vitro interaction profiles of the new antitubercular drugs bedaquiline and delamanid with moxifloxacin against clinical Mycobacterium tuberculosis isolates. J Glob Antimicrob Resist 2019; 19: 348–53. [DOI] [PubMed] [Google Scholar]
- 65. Agency EM. Assessment report Pretomanid FGK. Amsterdam: European Medicines Agency, 2020. [Google Scholar]
- 66. Lenaerts AJ, Gruppo V, Marietta KSet al. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother 2005; 49: 2294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Khoje AD, Kulendrn A, Charnock Cet al. Synthesis of non-purine analogs of 6-aryl-9-benzylpurines, and their antimycobacterial activities. Compounds modified in the imidazole ring. Bioorg Med Chem 2010; 18: 7274–82. [DOI] [PubMed] [Google Scholar]
- 68. Feuerriegel S, Köser CU, Baù Det al. Impact of Fgd1 and ddn diversity in Mycobacterium tuberculosis complex on in vitro susceptibility to PA-824. Antimicrob Agents Chemother 2011; 55: 5718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang F, Li S, Wen Set al. Comparison of in vitro susceptibility of mycobacteria against PA-824 to identify key residues of Ddn, the deazoflavin-dependent nitroreductase from Mycobacterium tuberculosis. Infect Drug Resist 2020; 13: 815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. European Medicines Agency . Pretomanid FGK: EPAR – Product Information, Annex I: Summary of product characteristics. European Medicines Agency, 2020. [Google Scholar]
- 71. Sala C, Dhar N, Hartkoorn RCet al. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 2010; 54: 4150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hu Y, Coates AR, Mitchison DA. Comparison of the sterilising activities of the nitroimidazopyran PA-824 and moxifloxacin against persisting Mycobacterium tuberculosis. Int J Tuberc Lung Dis 2008; 12: 69–73. [PubMed] [Google Scholar]
- 73. Early JV, Mullen S, Parish T. A rapid, low pH, nutrient stress, assay to determine the bactericidal activity of compounds against non-replicating Mycobacterium tuberculosis. PLoS One 2019; 14: e0222970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Iacobino A, Piccaro G, Giannoni Fet al. Activity of drugs against dormant Mycobacterium tuberculosis. Int J Mycobacteriol 2016; 5: S94–5. [DOI] [PubMed] [Google Scholar]
- 75. Papadopoulou MV, Bloomer WD, McNeil MR. NLCQ-1 and NLCQ-2, two new agents with activity against dormant Mycobacterium tuberculosis. Int J Antimicrob Agents 2007; 29: 724–7. [DOI] [PubMed] [Google Scholar]
- 76. Somasundaram S, Anand RS, Venkatesan Pet al. Bactericidal activity of PA-824 against Mycobacterium tuberculosis under anaerobic conditions and computational analysis of its novel analogues against mutant Ddn receptor. BMC Microbiol 2013; 13: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wallis RS, Jakubiec W, Mitton-Fry Met al. Rapid evaluation in whole blood culture of regimens for XDR-TB containing PNU-100480 (sutezolid), TMC207, PA-824, SQ109, and pyrazinamide. PLoS One 2012; 7: e30479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Drusano GL, Kim S, Almoslem Met al. The funnel: a screening technique for identifying optimal two-drug combination chemotherapy regimens. Antimicrob Agents Chemother 2021; 65: e02172-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Miranda Silva C, Hajihosseini A, Myrick Jet al. Effect of moxifloxacin plus pretomanid against Mycobacterium tuberculosis in log phase, acid phase, and nonreplicating-persister phase in an in vitro assay. Antimicrob Agents Chemother 2019; 63: e01695-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Drusano GL, Neely MN, Kim Set al. Building optimal three-drug combination chemotherapy regimens. Antimicrob Agents Chemother 2020; 64: e01610-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. López-Gavín A, Tudó G, Vergara Aet al. In vitro activity against Mycobacterium tuberculosis of levofloxacin, moxifloxacin and UB-8902 in combination with clofazimine and pretomanid. Int J Antimicrob Agents 2015; 46: 582–5. [DOI] [PubMed] [Google Scholar]
- 82. Srivastava S, Deshpande D, Magombedze Get al. Duration of pretomanid/moxifloxacin/pyrazinamide therapy compared with standard therapy based on time-to-extinction mathematics. J Antimicrob Chemother 2020; 75: 392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mouton JW, Dudley MN, Cars Oet al. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J Antimicrob Chemother 2005; 55: 601–7. [DOI] [PubMed] [Google Scholar]
- 84. Shibata M, Shimokawa Y, Sasahara Ket al. Absorption, distribution and excretion of the anti-tuberculosis drug delamanid in rats: Extensive tissue distribution suggests potential therapeutic value for extrapulmonary tuberculosis. Biopharm Drug Dispos 2017; 38: 301–12. [DOI] [PubMed] [Google Scholar]
- 85. Tucker EW, Pieterse L, Zimmerman MDet al. Delamanid central nervous system pharmacokinetics in tuberculous meningitis in rabbits and humans. Antimicrob Agents Chemother 2019; 63: e00913-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shimokawa Y, Sasahara K, Koyama Net al. Metabolic mechanism of delamanid, a new anti-tuberculosis drug, in human plasma. Drug Metab Dispos 2015; 43: 1277–83. [DOI] [PubMed] [Google Scholar]
- 87. Sasahara K, Shimokawa Y, Hirao Yet al. Pharmacokinetics and metabolism of delamanid, a novel anti-tuberculosis drug, in animals and humans: importance of albumin metabolism in vivo. Drug Metab Dispos 2015; 43: 1267–76. [DOI] [PubMed] [Google Scholar]
- 88. Shimokawa Y, Sasahara K, Yoda Net al. Delamanid does not inhibit or induce cytochrome p450 enzymes in vitro. Biol Pharm Bull 2014; 37: 1727–35. [DOI] [PubMed] [Google Scholar]
- 89. Mallikaarjun S, Wells C, Petersen Cet al. Delamanid coadministered with antiretroviral drugs or antituberculosis drugs shows no clinically relevant drug-drug interactions in healthy subjects. Antimicrob Agents Chemother 2016; 60: 5976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. European Medicines Agency . Deltyba: EPAR – Medicine overview. 2020.
- 91. Gler MT, Skripconoka V, Sanchez-Garavito Eet al. Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 2012; 366: 2151–60. [DOI] [PubMed] [Google Scholar]
- 92. Mallikaarjun S, Chapagain ML, Sasaki Tet al. Cumulative fraction of response for once- and twice-daily delamanid in patients with pulmonary multidrug-resistant tuberculosis. Antimicrob Agents Chemother 2020; 65: e01207-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pieterman ED, Keutzer L, van der Meijden Aet al. Superior efficacy of a bedaquiline, delamanid and linezolid combination regimen in a mouse-TB model. J Infect Dis 2021; 224: 1039–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ramirez G, Pham AC, Clulow AJet al. Sustained absorption of delamanid from lipid-based formulations as a path to reduced frequency of administration. Drug Deliv Transl Res 2021; 11: 1236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chen X, Hashizume H, Tomishige Tet al. Delamanid kills dormant mycobacteria in vitro and in a guinea pig model of tuberculosis. Antimicrob Agents Chemother 2017; 61: e02402-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang L, Ma Y, Duan Het al. Pharmacokinetics and tissue distribution study of PA-824 in rats by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 2015; 1006: 194–200. [DOI] [PubMed] [Google Scholar]
- 97. Shobo A, Bratkowska D, Baijnath Set al. Tissue distribution of pretomanid in rat brain via mass spectrometry imaging. Xenobiotica 2016; 46: 247–52. [DOI] [PubMed] [Google Scholar]
- 98. Shobo A, Pamreddy A, Kruger HGet al. Enhanced brain penetration of pretomanid by intranasal administration of an oil-in-water nanoemulsion. Nanomedicine (Lond) 2018; 13: 997–1008. [DOI] [PubMed] [Google Scholar]
- 99. Bratkowska D, Shobo A, Singh Set al. Determination of the antitubercular drug PA-824 in rat plasma, lung and brain tissues by liquid chromatography tandem mass spectrometry: application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci 2015; 988: 187–94. [DOI] [PubMed] [Google Scholar]
- 100. Wang L, Xu Y, Liang Let al. LC-MS/MS method for the simultaneous determination of PA-824, moxifloxacin and pyrazinamide in rat plasma and its application to pharmacokinetic study. J Pharm Biomed Anal 2014; 97: 1–8. [DOI] [PubMed] [Google Scholar]
- 101. Wang L, Zhao J, Zhang Ret al. Drug-drug interactions between PA-824 and darunavir based on pharmacokinetics in rats by LC-MS-MS. J Chromatogr Sci 2018; 56: 327–35. [DOI] [PubMed] [Google Scholar]
- 102. Dooley KE, Luetkemeyer AF, Park J-Get al. Phase I safety, pharmacokinetics, and pharmacogenetics study of the antituberculosis drug PA-824 with concomitant lopinavir-ritonavir, efavirenz, or rifampin. Antimicrob Agents Chemother 2014; 58: 5245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Winter H, Ginsberg A, Egizi Eet al. Effect of a high-calorie, high-fat meal on the bioavailability and pharmacokinetics of PA-824 in healthy adult subjects. Antimicrob Agents Chemother 2013; 57: 5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Diacon AH, Dawson R, von Groote-Bidlingmaier Fet al. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Crit Care Med 2015; 191: 943–53. [DOI] [PubMed] [Google Scholar]
- 105. Lakshminarayana SB, Boshoff HIM, Cherian Jet al. Pharmacokinetics-pharmacodynamics analysis of bicyclic 4-nitroimidazole analogs in a murine model of tuberculosis. PLoS One 2014; 9: e105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tasneen R, Tyagi S, Williams Ket al. Enhanced bactericidal activity of rifampin and/or pyrazinamide when combined with PA-824 in a murine model of tuberculosis. Antimicrob Agents Chemother 2008; 52: 3664–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mudde SE, Alsoud RA, van der Meijden Aet al. Predictive modeling to study the treatment-shortening potential of novel tuberculosis drug regimens, towards bundling of preclinical data. J Infect Dis 2021: jiab101. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nuermberger E, Rosenthal I, Tyagi Set al. Combination chemotherapy with the nitroimidazopyran PA-824 and first-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother 2006; 50: 2621–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dutta NK, Alsultan A, Gniadek TJet al. Potent rifamycin-sparing regimen cures guinea pig tuberculosis as rapidly as the standard regimen. Antimicrob Agents Chemother 2013; 57: 3910–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ahmad Z, Peloquin CA, Singh RPet al. PA-824 exhibits time-dependent activity in a murine model of tuberculosis. Antimicrob Agents Chemother 2011; 55: 239–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ginsberg AM, Laurenzi MW, Rouse DJet al. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob Agents Chemother 2009; 53: 3720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang X, Mallikaarjun S, Gibiansky E. Population pharmacokinetic analysis of delamanid in patients with pulmonary multidrug-resistant tuberculosis. Antimicrob Agents Chemother 2020; 65: e01202-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Franzblau SG, DeGroote MA, Cho SHet al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis (Edinb) 2012; 92: 453–88. [DOI] [PubMed] [Google Scholar]
- 114. Nuermberger EL. Preclinical efficacy testing of new drug candidates. Microbiol Spectr 2017; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Driver ER, Ryan GJ, Hoff DRet al. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother 2012; 56: 3181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Via LE, Lin PL, Ray SMet al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 2008; 76: 2333–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gengenbacher M, Duque-Correa MA, Kaiser Pet al. NOS2-deficient mice with hypoxic necrotizing lung lesions predict outcomes of tuberculosis chemotherapy in humans. Sci Rep 2017; 7: 8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hariguchi N, Chen X, Hayashi Yet al. OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother 2020; 64: e02020-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Kmentova I, Sutherland HS, Palmer BDet al. Synthesis and structure-activity relationships of aza- and diazabiphenyl analogues of the antitubercular drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J Med Chem 2010; 53: 8421–39. [DOI] [PubMed] [Google Scholar]
- 120. Tasneen R, Williams K, Amoabeng Oet al. Contribution of the nitroimidazoles PA-824 and TBA-354 to the activity of novel regimens in murine models of tuberculosis. Antimicrob Agents Chemother 2015; 59: 129–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bigelow KM, Tasneen R, Chang YSet al. Preserved efficacy and reduced toxicity with intermittent linezolid dosing in combination with bedaquiline and pretomanid in a murine TB model. Antimicrob Agents Chemother 2020; 64: e01178-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Xu J, Converse PJ, Upton AMet al. Comparative efficacy of the novel diarylquinoline TBAJ-587 and bedaquiline against a resistant Rv0678 mutant in a mouse model of tuberculosis. Antimicrob Agents Chemother 2021; 65: e02418-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Xu J, Li S-Y, Almeida DVet al. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob Agents Chemother 2019; 63: e00021-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tyagi S, Nuermberger E, Yoshimatsu Tet al. Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob Agents Chemother 2005; 49: 2289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Lanoix J-P, Betoudji F, Nuermberger E. Novel regimens identified in mice for treatment of latent tuberculosis infection in contacts of patients with multidrug-resistant tuberculosis. Antimicrob Agents Chemother 2014; 58: 2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dutta NK, Karakousis PC. PA-824 is as effective as isoniazid against latent tuberculosis infection in C3HeB/FeJ mice. Int J Antimicrob Agents 2014; 44: 564–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Garcia-Contreras L, Sung JC, Muttil Pet al. Dry powder PA-824 aerosols for treatment of tuberculosis in guinea pigs. Antimicrob Agents Chemother 2010; 54: 1436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tasneen R, Betoudji F, Tyagi Set al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother 2016; 60: 270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nuermberger E, Tyagi S, Tasneen Ret al. Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother 2008; 52: 1522–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tasneen R, Li S-Y, Peloquin CAet al. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 2011; 55: 5485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li S-Y, Tasneen R, Tyagi Set al. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother 2017; 61: e00913-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Purwantini E, Daniels L. Purification of a novel coenzyme F420-dependent glucose-6-phosphate dehydrogenase from Mycobacterium smegmatis. J Bacteriol 1996; 178: 2861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Purwantini E, Gillis TP, Daniels L. Presence of F420-dependent glucose-6-phosphate dehydrogenase in Mycobacterium and Nocardia species, but absence from Streptomyces and Corynebacterium species and methanogenic Archaea. FEMS Microbiol Lett 1997; 146: 129–34. [DOI] [PubMed] [Google Scholar]
- 134. Grinter R, Ney B, Brammananth Ret al. Cellular and structural basis of synthesis of the unique intermediate dehydro-F(420)-0 in mycobacteria. mSystems 2020; 5: e00389-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. European Medicines Agency . Assessment report Deltyba, international non-proprietary name: delamanid. European Medicines Agency, 2014. [Google Scholar]
- 136. Sung JC, Garcia-Contreras L, Verberkmoes JLet al. Dry powder nitroimidazopyran antibiotic PA-824 aerosol for inhalation. Antimicrob Agents Chemother 2009; 53: 1338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tasneen R, Mortensen DS, Converse PJet al. Dual mTORC1/mTORC2 inhibition as a host-directed therapeutic target in pathologically distinct mouse models of tuberculosis. Antimicrob Agents Chemother 2021; 65: e0025321. [DOI] [PMC free article] [PubMed] [Google Scholar]