

Abstract
Immune checkpoint inhibitor (ICI)‐based immunotherapy brought new hope for gastric cancer (GC) treatment. However, due to the lack of proper biomarkers, patient selection and outcome prediction for GC's immunotherapy remain unsatisfying. In this study, through applying an extracellular vesicle (EV) protein expression array, we assessed the correlation of plasma EV‐derived protein spectrum with outcomes of ICI‐related therapeutic combinations. Plasma from 112 GC patients received ICI‐related therapies were investigated retrospectively/prospectively as three cohorts. We identified four plasma EV‐derived proteins (ARG1/CD3/PD‐L1/PD‐L2) from 42 crucial candidate proteins and combined them as an EV‐score that robustly predicting immunotherapeutic outcomes at baseline and dynamically monitoring disease progression along with treatment. High EV‐score reflected microenvironmental features of stronger antitumour immunity, characterized by more activated CD8+ T/NK cells, higher TH1/TH2 ratio and higher expressions of IFN‐γ/perforin/granzymes in paired peripheral blood, which were verified by dataset analysis and in vivo experiments. EV‐score≥1 GC received more therapeutic benefits from ICIs, while EV‐score < 1 GC potentially benefited more from ICIs combining HER2‐targeted therapies. Collectively, through proposing a plasma EV‐score on protein level that powerfully predicting and monitoring GC's immunotherapeutic outcomes, our work facilitated clinical patient selection and decision‐makings, and provided mechanistical insights for immunotherapy‐related microenvironmental changes and improvements for current ICI‐regimens.
Keywords: extracellular vesicle derived protein profile, gastric cancer, immunotherapy, plasma‐based liquid biopsy, therapeutic biomarker
1. INTRODUCTION
Gastric cancer (GC) is the fifth most prevalent type of cancer and the third leading cause of cancer death around the world (Bray et al., 2018; Yang et al., 2011). Due to its high heterogeneity and complicated tissue composition, advanced GC receives limited therapeutic benefits from standard treatments and thus requires additional options of precision medication. The development of molecular subtyping empowered GC with more options of precision treatment, especially immunotherapy (Cancer Genome Atlas Research Network, 2014). Immune checkpoint inhibitors (ICIs) rescue patients’ immune system and revoke/strengthen the adaptive immunity against cancer, thus exert a consistent antitumour efficacy for heterogenic patients (Taieb et al., 2018). Inspired by the success of a series of clinical trials, ICIs have been successfully approved as gastroesophageal cancer's first‐line and adjuvant therapies (Smyth et al., 2021).
However, although ICIs provide patients with remarkable clinical effect, the response rate of unselected GC receiving anti‐PD‐1/anti‐PD‐L1 immunotherapy is only approximately 20% (Long et al., 2017). Patients harbouring certain genomic and molecular features, including microsatellite instability (MSI)/EBV‐positivity/high tumour mutation burden (TMB)/high PD‐L1 expression (represented by CPS, Combined Positive Score), have been expected to receive more benefit from immunotherapy, yet these patients comprise only a small part of GC population. Additionally, these features are imperfect as predictors for ICIs’ outcomes, while acquired resistance also impairs therapeutic efficacy (Havel et al., 2019; Nishino et al., 2017). Consequently, in order to improve the management and clinical decision‐makings of ICIs in GC, it is urgently needed to develop new methods to facilitate patient selection and timely evaluate disease progression along with the treatment process.
Extracellular vesicles (EVs) are membrane binding nanosized particles detected in almost all kinds of body fluids including plasma (Witwer et al., 2013). Travelling along with the systemic or local blood/lymph circulation, EV transfers multiple bioactive cargos including DNA/RNA/proteins, thus directing cell‐cell or cell‐microenvironment communications and mediating multiple cancerous events, including malignant transformation/angiogenesis/metastasis/drug resistance/immune evasion (Huang et al., 2019; Xu et al., 2018). Due to its cancer‐associated roles, EV serves as a rising star of liquid biopsy and has been utilized for clinical applications such as the early detection of cancer (Erdbrügger et al., 2021; Guo et al., 2020). However, the relationship of EV content with immunotherapeutic outcomes has not yet been systemically addressed in GC. On the other hand, since proteins are the direct performer of biological activities, the tumour‐microenvironmental changes induced by immunotherapy can be recorded and presented by EV‐derived proteins. Current study majorly investigated EV's roles in liquid biopsy from the perspective of noncoding RNAs, yet implications of EV‐derived protein component were barely reported.
In this study, 199 plasma samples from 112 GC patients received ICI‐related regimens were recruited as three independent cohorts for comprehensive analysis. Through applying a plasma‐based protein expression array, we described the profiles of 42 crucial EV‐derived proteins and screened four members from them to generate an EV‐score that robustly predicting/monitoring the outcomes of GC's immunotherapy. We also analysed the microenvironmental features and phenotypes and mechanisms characterized by this plasma EV‐score, and discussed the possibility of ICI plus HER2‐targed therapy as a substitutive option for patients failed to benefit from ICIs. Collectively, our work provided novel immunotherapeutic biomarkers of GC in view of plasma EV‐derived proteins.
2. MATERIALS AND METHODS
2.1. Patient selection and information
Gastric cancer patients were collected from conventional treatments or clinical trials. For enrolled patients, the inclusion criteria were: patients must be diagnosed with unresectable or metastatic GC (including both GEJ (gastroesophageal junction) and non‐GEJ types), had at least one measurable or unmeasurable but evaluable lesion, without history of severe heart or liver disease or psychiatric disorders, without haemorrhage or perforation of digestive tract, remained treatment naive 3 weeks before the baseline (BL) of treatment, scored with ECOG (Eastern Cooperative Oncology Group) performance status of 0/1 3 days before the baseline of treatment. The exclusive criteria were: patients harboured GC combined with other types of cancer, or histopathologically diagnosed as neuroendocrine tumour or squamous type of tumour. The regimen or regimens for initial medical treatment was defined as first line therapy (line 1), the subsequently administrated regimens (sequentially, line 2, line 3, etc.) were collectively defined as line > 1.
For GC patients received immunotherapy (anti‐PD‐1/anti‐PD‐1 plus anti‐CTLA4/anti‐PD‐1 plus chemotherapy), 62 baseline samples as well as 87 on‐treatment samples (all time course with evaluated responses and available plasma specimens, including 32 CR/PR, 20 SD and 35 PD) from 62 cases (treated during Aug‐2016 to Nov‐2020) were used as a retrospective training cohort; 40 baseline samples from 40 cases (treated during Nov‐2018 to Apr‐2021) from an observational study (NCT04993378) were used as a prospective validating cohort. For patients received immunotherapy combining HER2‐targeted therapy (anti‐PD‐1 plus anti‐HER2/anti‐PD‐1 plus anti‐HER2 and chemotherapy), 10 baseline samples from 10 cases (treated during Dec‐2017 to Nov‐2020) were used as a retrospective expanding cohort. Patients were evaluated based on irRECIST criteria (Nishino et al., 2013). By referring to CT (computed tomography), patients’ responses to therapy were evaluated as complete response (CR), partial response (PR), stable disease (SD), or progressed disease (PD) according to the irRECIST criteria. During the whole follow up time, patients ever/never reached overall response (CR or PR) were defined as responders/nonresponders, while patients ever/never developed PD were defined as progressors/nonprogressors. Objective response rate (ORR), disease control rate (DCR), immunotherapy‐related progression‐free survival (irPFS, defined as the time from randomization or regimen administration to the first documented tumour progression or death) and overall survival (irOS, defined as the time from randomization or regimen administration to death) were calculated.
Recruitment, treatment, management, follow‐up and sampling of all patients were implemented by the Department of Gastrointestinal Oncology, Peking University Cancer Hospital & Institute, with written informed consent obtained from all donors. Peripheral whole blood specimens (10 ml per sample) were collected through venipuncture, collected with EDTA tubes, and centrifuged at 4 °C, 3000×g, 15 min for isolation. Isolated plasma, serum, lymphocytes and PBMC were maintained at −80 °C for long‐term storage. Clinical data of patients, including basic parameters, treatment information, laboratory and pathologic examinations and CT‐scanned images were acquired by referring to their individual medical records. MSI (microsatellite instability) was defined by DNA sequencing or assayed by immunohistochemistry (IHC) as deficient mismatch repair (dMMR). EBV positivity was defined by DNA sequencing or by IHC as EBER positive. HER2 positivity was defined as IHC+++, or IHC++ plus amplification assayed by fluorescence in situ hybridization (FISH). CSP≥1 was defined as CPS positivity.
All trials/specimens/associated clinical information were approved for experimental applications by the Institutional Ethics Committee, Peking University Cancer Hospital & Institute. This study was conducted in accordance with the Declaration of Helsinki.
2.2. Specimen preparation and EV expression array
The EV expression Array was performed by referring to a previous publication (Jørgensen et al., 2013) and with our improvements. For preparation, antibodies were diluted to 200 μg/ml with phosphate‐buffered saline (PBS) with 5% glycerol and then printed onto a 3D modified slide surface (Capital Biochip Corp, Beijing, China) in duplicate using an Arrayjet microarrayer (Roslin, UK). Forty two key proteins (including 19 biomarkers of representative cellular components in tumour‐immune microenvironment, 11 major immune checkpoint modulators, 12 therapeutic biomarkers or targets of gastrointestinal cancer) and four canonical EV markers were selected for EV expression array. This 42‐protein panel and paired antibodies for EV expression array were listed in Table S1. PBS with 5% glycerol and 100 μg/ml of biotinylated bovine serum albumin (BSA) were used as negative and positive controls, respectively.
The microarray slides were initially blocked with 3% BSA (w/v) in PBS for 1 h at room temperature, then incubated with 10 μl unpurified plasma sample diluted (1:10) in wash buffer (0.05% Tween20 in PBS). The incubation was performed on a shaker set to 30 rpm at room temperature for 2 h followed by overnight incubation at 4 oC. After washing, the slides were incubated with biotinylated detection antibodies (anti‐human‐CD9, ‐CD63 and ‐CD81, LifeSpan BioSciences, WA, USA) diluted 1:1500 in wash buffer. For detection, the slides were incubated with Cy3‐labeled streptavidin (Life Technologies) diluted 1:1500 in wash buffer for 1 h at room temperature, then washed and scanned using the GenePix 4000A microarray scanner (Molecular Devices, CA, USA). The fluorescent images were analysed and the signal intensity was extracted using the GenePix Pro image analysis software (Molecular Devices, CA, USA).
2.3. Data processing of EV expression array
After the quantification of EV array signal intensity, a normalization of each spot's signal was made to evaluate the protein density on the EVs. Since CD63 was detected in only 42 of 62 samples while CD9 and CD81 displayed the highest correlation among four EV markers, the geometric mean of CD9 and CD81 was used as normalization factor. For each antibody, the signal intensity was divided by the geometric mean of CD9 and CD81 before further analysis. Expression of EV‐proteins were defined as ‘high’ or ‘low’ in a median‐based criteria. For ARG1/CD3/PD‐L1/PD‐L2, the status of high expression of each molecule was counted as 1, while without high expression (low expression) was counted as 0. As shown by the formula below, signature score (EV‐score) for each patient was calculated as the summation of the counts of ‘high’ ARG1, ‘high’ CD3, ‘high’ PD‐L2 expressions minus the count of ‘high’ PD‐L1 expression.
| (1) |
According to this formula, EV score was an integer ranged between ‐1 (for example, low‐ARG1, low‐CD3, low‐PD‐L2, high‐PD‐L1, EV score = 0+0+0‐1 = ‐1) and 3 (for example, high‐ARG1, high‐CD3, high‐PD‐L2, low‐PD‐L1, EV score = 1+1+1‐0 = 3).
2.4. Plasma EV isolation, immunogold labelling assay and electron microscopy
For plasma EV isolation, 0.5 ml of plasma was applied on top of a SEC‐based qEV column (Izon Science) and 1.5 ml fractions were collected. The differential centrifugation method (CP100NX, Hitachi) was also as alternatively applied for isolation. EV eluates were concentrated using an Amicon Ultra‐4 10 kDa centrifugal filter device (Merck Millipore).
Immunogold labelling experiment was performed by referring to a previous report (Melo et al., 2015). Concentrated EVs were resuspended with an equal volume of 4% paraformaldehyde, then placed onto a 400‐mesh carbon/formvar coated grids and allowed to absorb for 20 min. The grids were blocked with 5% BSA (w/v) in PBS for 10 min and incubated with the primary antibodies (ARG1, GTX634218; CD3, GTX628462, GeneTex; PD‐L1, 10084‐R015, SinoBiological; PD‐L2, 14‐5888‐82, Invitrogen; CD81, sc‐166029; CD9, sc‐13118, Santa Cruz; CD63, GTX28219, GeneTex) at the appropriate dilution for 3 h at room temperature. For controls, the grids were not exposed to the primary antibody. Then, the grids were incubated with a secondary antibody (antirabbit IgG, G7402, Sigma‐Aldrich; antimouse IgG, G7652, Sigma‐Aldrich) conjugated to 10‐nm gold particles (Merck) for 1 h, washed with PBS and placed in 1% glutaraldehyde (w/v) in PBS for 5 min. After rinsing in PBS, the grids were stained with uranyl acetate. The grids were observed using a transmission electron microscope (H‐7650, Hitachi).
2.5. Transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA)
For TEM assay, isolated EVs were directly incubated on the mesh, stained with uranyl acetate solution, then observed using a transmission electron microscope (H‐7650, Hitachi). For NTA assay, isolated EVs were measured using the ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) powered by a 405 nm laser to determine the size and quality of particles, and analysed using the NTA software (ZetaView 8.02.28) to capture particle movement.
2.6. EV protein quantification and Western Blot analysis
EV proteins were extracted from concentrated EV‐rich SEC qEV fractions using a RIPA buffer (Thermo Scientific) with a protease inhibitor cocktail (Roche), or extracted using differential centrifugation method (CP100NX, Hitachi). Human embryonic kidney cell line HEK293 was obtained from ATCC (Manassas, VA), authenticated by short tandem repeat profiling, then cultured with DMEM/RPMI‐1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10 % foetal calf serum (Gibco BRL) and 1% penicillin plus streptomycin (HyClone, Logan, UT) in a humidified incubator (5% CO2) under 37 °C. The protein content of EVs/cells were examined by Pierce BCA Protein Assay Kit (Thermo Scientific) for quantification and lysed in 1× SDS‐PAGE loading buffer (1% SDS, 11% glycerol, 10% β‐mercaptoethanol, 0.1 M Tris, pH 6.8) for assay. The expression of proteins was assessed by Western Blotting in 10 μg of HEK293T/MC38/CT26 cell lysate/mixed plasma EV samples/extracted cellular EV samples with the appropriate antibodies (for human proteins, ARG1/PD‐L1/CD81/CD9, identical to immunogold labelling assay; Calnexin, GTX629976, GeneTex; for mouse proteins, CD63, ab217345; CD9, ab92726; Calnexin, ab133615, Abcam), illuminated with the Clarity Western ECL substrate (Bio‐Rad, Hercules, CA), and visualized with the Amersham Imager 600 (GE Healthcare, Chicago, IL). Blotted bands were quantified with Image J software.
2.7. Mouse cell lines and xenografts
Mouse colorectal cancer cell lines MC38 and CT26 were obtained from ATCC (Manassas, VA), authenticated by short tandem repeat profiling, then cultured with DMEM/RPMI‐1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10 % foetal calf serum (Gibco BRL) and 1% penicillin plus streptomycin (HyClone, Logan, UT) in a humidified incubator (5% CO2) under 37 °C. MC38/CT26 cell sublines stably expressing mouse PD‐L1 and PD‐L2 were constructed through a lentivirus system. EVs were extracted from cell culture supernatants.
Female mice (Vital River Laboratories, Beijing, China) weighing 15–17 g were subcutaneously inoculated with 1×106 MC38 cells (C57BL/6J) or 2×105 CT26 cells (BALB/C). When xenograft volume reached 100–150 mm3, C57BL/6J mice received intratumoural injection of MC38‐derived EVs (50 μg/100 μl) at an interval of 3 days, then intraperitoneally administered with antimouse PD‐1 antibody (BE0146, BioXcell, 3 mg/kg, twice a week), while BALB/c mice received intratumoural injection of CT26‐derived EVs (50 μg/100 μl) at an interval of 3 days, then intraperitoneally administered with antimouse PD‐1 antibody (5 mg/kg daily). The body‐weight and xenograft size were recorded every 3 days. Tumour volume was calculated using the following equation: Tumour volume (mm3) = longer length×(shorter length2)/2. At the end of experiment, mice were sacrificed, while xenograft tumours were stripped and maintained for subsequent analysis.
2.8. Flow cytometry, ELISA and IHC analysis for cells and xenografts
For flow cytometry, Antibodies (PD‐L1, 124311; PD‐L2, 107210, Biolegend) were used to verify the expression of PD‐L1 and PD‐L2 on the membrane of cells constructed by lentivirus system. Fresh mice spleen tissues were digested into single cell suspensions. Red Blood Cell Lysis Buffer (BioLegend) was used to remove red blood cells. Splenocytes were stimulated in the presence of Cell Activation Cocktail (BioLegend) and Monensin (BioLegend) for 5 h. Cell surface markers were stained with corresponding antibodies (CD45,103128; CD4, 100412, Biolegend) before permeabilization for 20 min at room temperature. For intracellular staining, after fixing by Fixation/Permeabilization Kit (BD Biosciences), splenocytes were incubated with antibodies to detect interferon (IFN)‐γ (505840, Biolegend) and interleukin 4 (IL‐4) (504104, Biolegend) for 40 min at room temperature. CD4+IFN‐γ+ cells were defined as TH1. CD4+IL4+ cells were defined as TH2.
On the other hand, the supernatant of lysed cells or xenograft tumour tissue were harvested and assessed with ELISA kits (PD‐L1, 695211029; PD‐L2, 876211029; IFN‐γ, 366211026; TNF‐α, 569211115; granzyme B, 318211213, Tianjin Anoric Biotechnology Co., Ltd, Tianjin, China). All samples were measured according to the manufacturer's instructions and the concentrations were calculated based on the linear range of the ELISA assay data.
For IHC assay, formalin‐fixed and paraffin‐embedded (FFPE) tumour tissue slides were deparaffinized with dimethylbenzene and rehydrated with ethanol. Slides were then treated with 3% H2O2 to quench endogenous peroxidase activity, boiled in sodium citrate buffer (10 mM, pH 6.0) for antigen retrieval, incubated by 5% goat serum to block nonspecific bindings, and proceeded for haematoxylin‐eosin (Solarbio) or antibody (ki‐67, 12202T; CD8, 98941, Cell Signalling Technology) staining.
2.9. Statistical analysis and formatting
Expressional diversity between subgroups was assessed by Wilcoxon–Mann‐Whitney test. Consistency between indexes was assessed by Pearson correlation analysis. Clinical information was compared between subgroups by Chi‐square test. Survival proportions were assessed by Kaplan–Meier analysis paired with Log‐rank test, or Cox regression model. Tumour‐microenvironmental features were evaluated with ssGSEA package. P < 0.05 was considered as statistically significant. All analysis and graphing were powered by R 3.5.1/SPSS 21.0/GraphPad Prism 6 programs.
3. RESULTS
3.1. The overall workflow and characteristics of enrolled patients
As demonstrated by an overall workflow (Figure 1a), three cohorts of GC patients received ICI‐related treatments were quantified with an EV expression array (Figure 1b). In training cohort, 149 specimens (62 BL, 32 CR/PR, 20 SD, 35 PD) were collected from 62 GC patients treated with immunotherapies (Table S2, Figure S1A, Figure 1c). Patients received 1st line treatment displayed longer irPFS/irOS than patients received > 1st line treatment (Figure S1B), meanwhile ICI plus ICI displayed longer irPFS/irOS than other therapies (Figure S1C), suggesting our data was of good quality.
FIGURE 1.
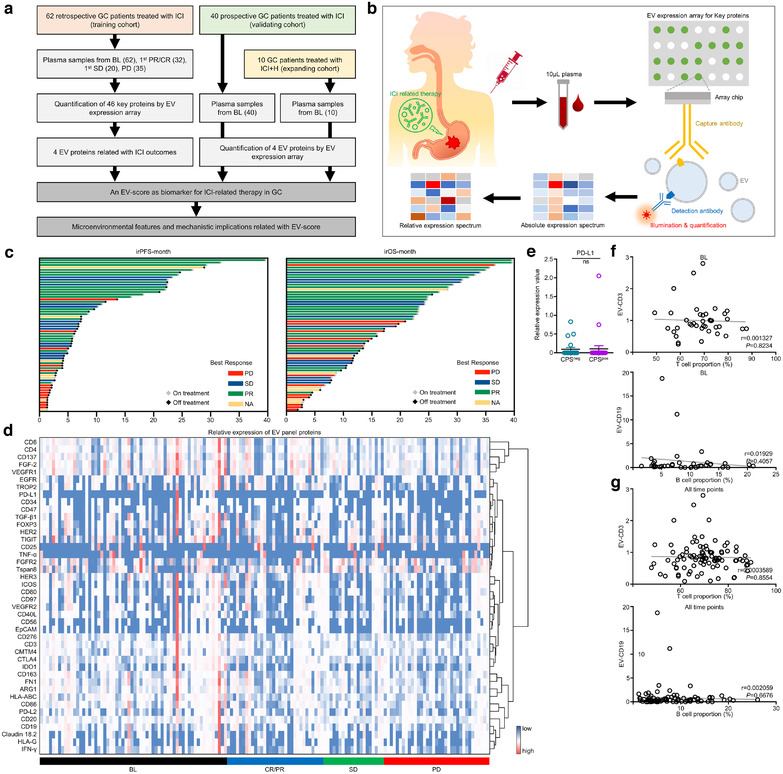
The expressional landscape of plasma EV protein panel in GC treated with ICI‐related therapy. (a) An overall work flow of the whole study. (b) A sketch map for the procedures of EV expression array. (c) The overall response and prognostic features of the training cohort. (d) The hierarchical‐clustered heatmap of EV protein panel for all time points in the training cohort. (e) Comparison of EV‐derived PD‐L1 expression between patients defined as tissue CPS‐negative and ‐positive. Correlations between EV‐derived CD3 expression and circulating T cell number, or between EV‐derived CD19 expression and circulating B cell number in paired peripheral blood samples (f) at BL or (g) for all time points were assessed. CPSpos, CPS positive. CPSneg, CPS negative
3.2. Expressional landscape of plasma EV panel proteins in ICI‐treated GC
All plasma samples were assessed by an antibody‐sandwiched expression array to generate an absolute expression spectrum of 46 EV‐proteins (Figure S2A). EV‐biomarkers CD81/CD9/ALIX/CD63 wasn't changed much after treatment (Figure S2B–E), suggesting that EV content remained dynamically stable during immunotherapy. To rule out the interference of EV content diversity, the absolute expressions of other 42 EV‐proteins should be normalized into relative expressions. We analysed the mutual correlations among CD81/CD9/ALIX/CD63 at BL (Figure S2F) or for all time points (Figure S2G) and found CD81 and CD9 shared the highest correlations among the four EV markers. On the other hand, CD63 was presented in only 42/62 samples, thus we combined CD81 and CD9 to generate a relative expression spectrum (Figure 1d).
We then compared the concordances between EV‐array‐based quantification and tissue/circulating cell evaluations. The expression of EV‐derived PD‐L1 showed no difference between tissue CPS‐negative (CPS = 0) and CPS‐positive (CPS≥1) patients (Figure 1e), while the expressions of EV‐derived T cell marker CD3 or B cell marker CD19 also showed no correlation with proportions of circulating T or B cells in paired peripheral blood at baseline or for all time points (Figure 1f–g). Thus, EV proteins may share a different expression spectrum with proteins expressed in tissue or circulating cells of GC.
3.3. Selection of plasma EV proteins related with immunotherapeutic outcomes
We assessed the mutual correlations of plasma EV proteins (Figure S3A) and defined each of their baseline expressions as ‘high’ or ‘low’ in a median‐based manner (Figure S3B). For all panel proteins, fold changes (FC) of nonresponders versus responders or progressors versus nonprogressors (Figure S3C–D) were calculated, with the threshold of FC≥1.25 for poor response‐related and FC≤0.8 for good response‐related proteins. Kaplan–Meier analysis‐based hazard ratio (HR) was calculated for irPFS or irOS (Figure S3E–F), also with the threshold of HR≥1.25 for favourable‐prognostic and HR≤0.8 for adverse‐prognostic proteins. Collectively, only ARG1/CD3/PD‐L1/PD‐L2 were found displaying consistent trends of correlation with both responses and prognosis. High expressions of baseline ARG1/CD3/PD‐L2 (Figure 2a, b, d) were positively, while PD‐L1 (Figure 2c) was negatively related with ORR/DCR/irPFS/irOS.
FIGURE 2.
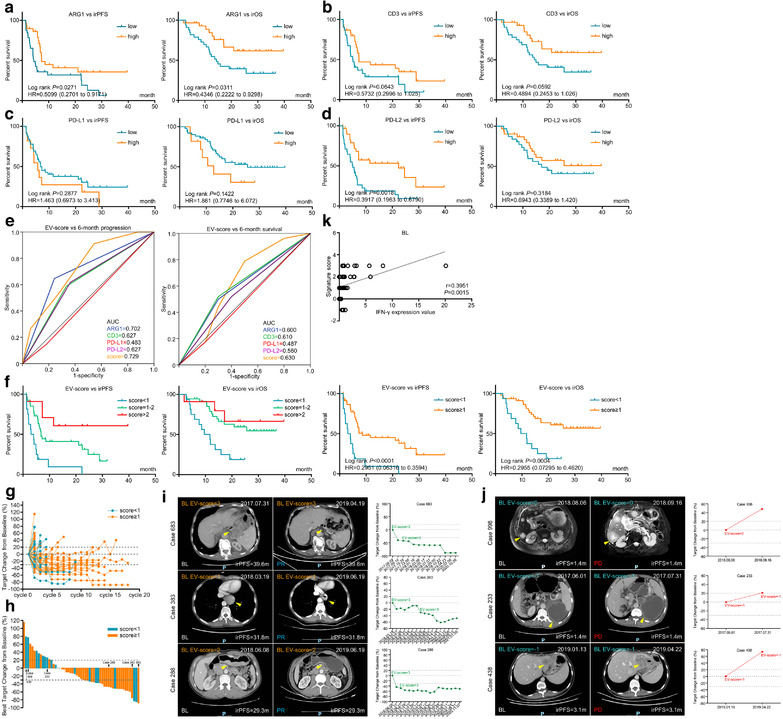
A four protein‐based plasma EV‐score at treatment baseline robustly predicted immunotherapeutic outcomes. The prognostic correlations of EV‐derived (a) ARG1, (b) CD3, (c) PD‐L1 and (d) PD‐L2 at BL were assessed by Kaplan‐Meier survival analysis. These four genes were further combined to generated an EV‐score. (e) ROC curve was used to measure the specificity and sensitivity of EV‐derived ARG1, CD3, PD‐L1, PD‐L2 and EV‐score in characterizing patients reached an irPFS (progression) or irOS (survival) longer than 6 months. (f) Prognostic indications of scattered EV‐scores (left panel), or for the cut‐off value of EV‐score≥1 (right panel). (g) Timeline‐scaled changes of tumour volume and (h) the best changes of tumour volume for all patients were stratified by EV‐score. CT images and changes of tumour volume were displayed for representative cases defined as (i) EV‐score≥1 or (j) EV‐score < 1. (k) Correlation between EV‐score and IFN‐γ content in paired peripheral blood samples at BL
EVs extracted with SEC method displayed comparable purity with EVs extracted with centrifugation method (Figure S4A–B), thus we isolated EVs from seven plasma samples with SEC method. As quality‐controlled with TEM and NTA assays, isolated EVs were spheres sized between 30 and 200 nm, suggesting a high quality of isolation (Figure S4C–D). We then assessed mixed EV specimens through immunogold labelling assay to confirm that ARG1/CD3/PD‐L1/PD‐L2/CD81/CD9/CD63 could be specifically detected on EVs (Figure S4E). Additionally, WB‐quantified expressions of ARG1/PD‐L1 (Figure S4F) displayed high correlations with their array‐quantified expressions originated from the same samples (Figure S4G), while BCA method‐evaluated protein concentration of EV was positively correlated with array‐quantified expression of the EV marker CD81 (Figure S4H). These data suggested that EV expression array effectively quantified plasma EV proteins, without being interfered by free proteins in the circulation.
3.4. Baseline EV‐score powerfully predicted immunotherapeutic outcomes of GC
To maximize the predictive validity, we numbered the ‘high’ expression of ARG1/CD3/PD‐L1/PD‐L2 as 1, then defined a signature score (EV‐score) for each specimen as the summation of the ‘high’ expressions of ARG1/CD3/PD‐L2 minus the ‘high’ expression of PD‐L1. There were two reasons why we adopted such an immediate scoring rule. First, the laboratory testing of dMMR and MSI have been prevalently recognized as profitable biomarkers of ICI (Luchini et al., 2019), which inspired us to generate an EV‐score with the similar manner of calculation. Second, weighing each of the four molecules will introduce more batch effects and lead to a more complicate formula. We hope this EV‐score can minimize the batch effect across different cohorts/populations, thus providing a simple and consistent prediction for potential applications. Therefore, we decided to calculate the EV‐score with a simple formula, as described in Materials and Methods section.
Baseline EV‐score displayed higher AUC than ARG1/CD3/PD‐L1/PD‐L2 in characterizing 6‐month progression/death (Figure 2e). On the other hand, although the diversities of response rates (Table S3) were few, EV‐score≥1 displayed significantly more favourable irPFS/irOS than EV‐score < 1 (Figure 2f). Timeline‐scaled changes of tumour volume (Figure 2g) and the best changes of tumour volume (Figure 2h) for all patients in training cohort were stratified by EV‐score, while CT images and changes of tumour volume for representative cases were compared between EV‐score≥1 and < 1 (Figure 2i and j). We also combined EV‐derived ARG1, CD3, PD‐L1, PD‐L2 into signatures through logistic regression models, and found the current EV‐score was a more effective option than the logistic regression‐weighted signatures in predicting ICI outcomes (represented by higher AUC and/or prognostic correlations, Figure S5A–D). Importantly, our EV‐score was a much more powerful prognostic factor than treatment lines, regimens, and MSI/EBV/CPS (Figure S1B–F), which also remained highly prognostic regardless of stratification with these factors (Figure S6A–E). EV‐score was an independent and more effective prognostic factor compared with MSI/EBV/CPS (Table S4), suggesting it can be used for patient selection regardless of current immunotherapy biomarkers. We also found the baseline expression of EV‐derived IFN‐γ was positively correlated with EV‐score (Figure 2k).
3.5. EV‐score dynamically monitors immunotherapeutic outcomes
We investigated whether changes of EV‐score at the first month (1 M) after treatment provided useful information for therapeutic surveillance. In training cohort, 18 patients have both BL and 1 M specimens were stratified into four categories: down (from BL‐high‐EV‐score to 1 M‐low‐EV‐score), up (from BL‐low‐EV‐score to 1 M‐high‐EV‐score), remainL (low‐EV‐scores at both BL/1 M), and remainH (high‐EV‐scores at both BL/1 M) (Figure 3a). Similar to baseline findings, EV‐derived‐ARG1/CD3/PD‐L1/PD‐L2 were less effective (Figure 3b), while EV‐score was more effective in predicting immunotherapeutic prognosis (Figure 3c).
FIGURE 3.
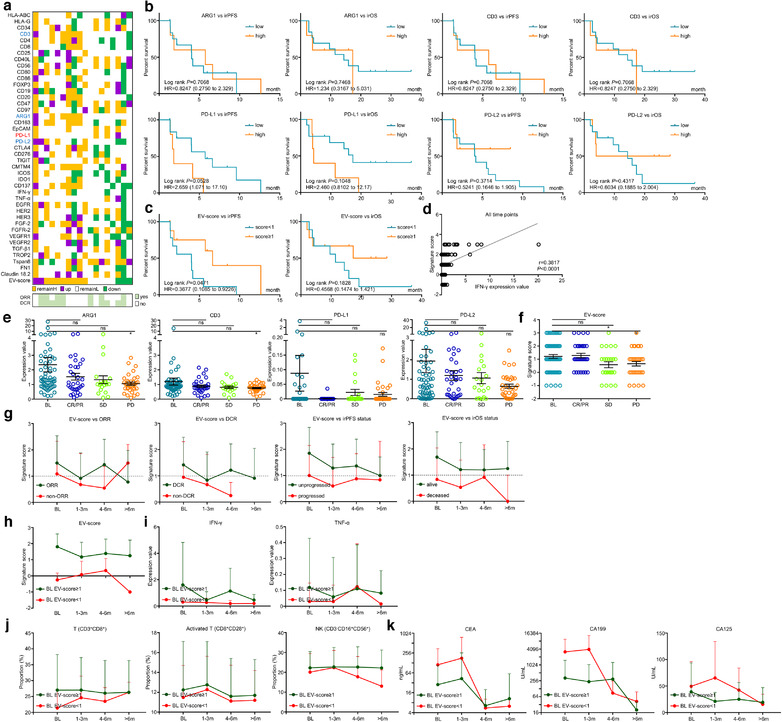
EV‐score dynamically monitored ICI outcomes. (a) Fold changes of EV protein panel and score at 1 M compared with BL in 18 accessible patients. (b) The prognostic correlations of EV‐derived ARG1, CD3, PD‐L1, PD‐L2 and (c) EV‐score at 1 M were assessed by Kaplan‐Meier survival analysis. (d) Correlation between EV‐score and IFN‐γ level in paired peripheral blood samples for all time points. (e) Expressions of EV‐derived ARG1, CD3, PD‐L1, PD‐L2 and (f) value of EV‐score were compared at BL, CR/PR, SD, PD points. (g) Dynamic changes of EV‐score along with the timeline of ICI treatment were compared between patients with/without overall response, disease control, disease progression and cancer death. Dynamic changes of (h) EV‐score, (i) cytotoxic cytokines IFN‐γ/TNF‐α, (j) T cell (CD3+CD8+)/activated T cell (CD8+CD28+)/NK cell (CD3–CD16+56+) and (k) tumour markers CEA/CA199/CA125 along with the timeline of ICI treatment were compared between patients defined as BL EV‐score≥1 or BL EV‐score < 1. *, P < 0.05. ns, P value not significant
For all time points, level of EV‐derived IFN‐γ was also positively correlated with EV‐score (Figure 3d). Despite the insignificant statistics, EV‐derived ARG1/CD3/PD‐L2 and EV‐score were higher at CR/PR than PD, while PD‐L1 was lower at CR/PR than PD (Figure 3e–f), which were in accordance with their respective prognostic indications. Dynamic changes of EV‐score were compared at baseline, during 1–3 months, during 4–6 months or > 6 months> of treatment, in which patients reached overall response/reached disease control/nonprogressed/alive displayed generally higher EV‐score than patients without overall response/without disease control/progressed/deceased along with the timeline of treatment (Figure 3g). On the other hand, timeline‐scaled average EV‐score remained higher than 1 for patients with BL EV‐score≥1, or remained lower than 1 for patients with BL EV‐score < 1 (Figure 3h), suggesting pretreatment EV‐score was a stable index for stratification throughout immunotherapy. Dynamically, the expression of anticancer cytokines IFN‐γ/TNF‐α remained higher for BL EV‐score≥1 patients than BL EV‐score < 1 patients, paired with higher proportions of total T/activated T/NK cells and lower levels of tumour markers CEA/CA199/CA125 in circulation (Figure 3i–k). Combining both baseline and timeline‐scaled findings, we concluded that EV‐score served as a powerful index in predicting and dynamically monitoring the immunotherapeutic outcomes of GC.
3.6. EV‐score remained as an immunotherapeutic biomarker for prospective validation
In order to further validate the biomarker roles of EV‐score, we prospectively analysed additional 40 baseline plasma of ICI‐treated GC in the validating cohort (Table S2, Figure S7A and B) with the same criteria/cut‐offs as the training cohort. Since no patient was deceased in this cohort, only irPFS was included for analysis. Similar to training cohort, ARG1/CD3/PD‐L1/PD‐L2 were indicative yet insufficient to characterize outcomes (Figure S7C), while EV‐score≥1 possessed higher response rates (Table S5) and favourable prognostic indications (Figure 4a), which was also more powerful than ARG1/CD3/PD‐L1/PD‐L2 in characterizing progressors within 6 months (Figure 4b). Additionally, although stratification of MSI/EBV was not performed due to lack of cases, the prognostic trend of EV‐score remained valid for 1st‐line/ > 1st‐line and CPS‐negative/CPS‐positive stratifications (Figure 4c‐4f). Timeline‐scaled changes of tumour volume (Figure 4g) and the best changes of tumour volume (Figure 4h) for all patients in validating cohort were stratified by EV‐score, while CT images and changes of tumour volume for representative cases were demonstrated for EV‐score≥1/ < 1 (Figure 4i and j). EV‐score was also an independent and effective prognostic factor in validating cohort (Table S6). Dynamically, circulating total T/activated T/NK cells maintained higher proportions for BL EV‐score≥1 than BL EV‐score < 1 (Figure 4k), while CEA/CA199/CA125 were lower for BL EV‐score≥1 than BL EV‐score < 1 patients (Figure 4l). These data suggested that EV‐score prospectively predicted immunotherapeutic outcomes of GC.
FIGURE 4.
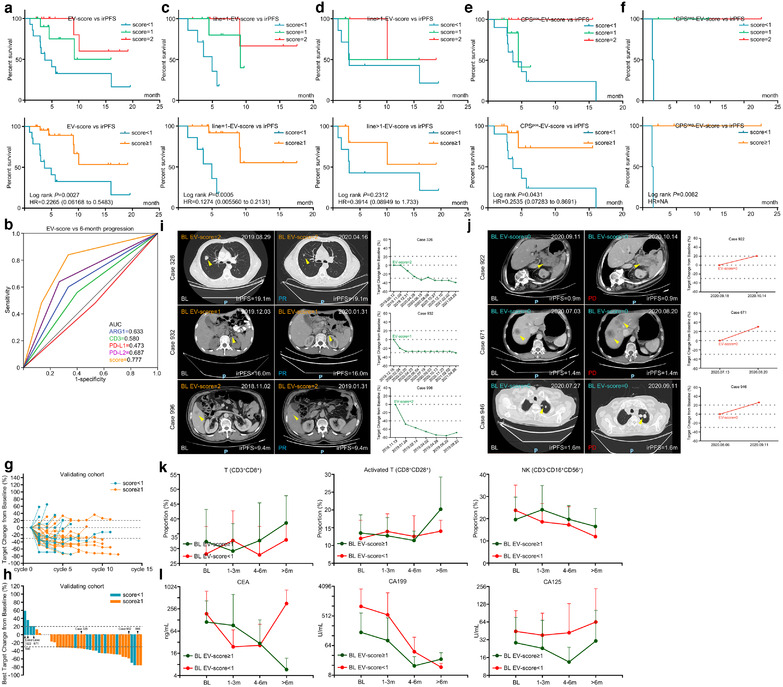
EV‐score predicted ICI outcomes for a prospective validating cohort. (a) The prognostic correlations of EV‐score at BL were assessed in a prospective validating cohort. (b) ROC curve‐measured specificity and sensitivity of EV‐score in characterizing patients reached a PFS longer than 6 months. (c) Prognostic role of EV‐score for patients received 1st line treatment and (d) patients received > 1st line treatment, as well as (e) CPS‐positive and (f) CPS‐negative patients. (g) Timeline‐scaled changes of tumour volume and (h) the best changes of tumour volume for all patients were stratified by EV‐score. CT images and changes of tumour volume were displayed for representative cases defined as (i) EV‐score≥1 or (j) EV‐score < 1. Dynamic changes of (k) T cell (CD3+CD8+)/activated T cell (CD8+CD28+)/NK cell (CD3–CD16+56+) and (l) tumour markers CEA/CA199/CA125 along with the timeline of ICI treatment were compared between patients defined as BL EV‐score≥1 or BL EV‐score < 1. CPSpos, CPS positive. CPSneg, CPS negative
3.7. Signature score characterized unique molecular and tumour‐microenvironmental features
Through analysing EV‐score‐stratified MSI/EBV/HER2 status in training and validating (Figure 5a and b) cohorts, we found among these crucial molecular features that HER2 positivity was more abundant in EV‐score < 1 than in EV‐score≥1 patients (training = 6.2% vs. 4.3%; validating = 7.1% vs. 0%). Additionally, although we have provided clues that EV‐derived PD‐L1 did not display much expressional correlations with tissue‐CPS, the molecular and tumour‐microenvironmental features and mechanistic implications related with signature score still worth to be evaluated in tissue‐based datasets. In GC tissue‐based datasets TCGA/GSE62254, we analysed the mutual correlations among ARG1/CD3/PD‐L1/PD‐L2 (Figure 5c and d), then adopted the same criteria/cut‐offs as training/validating cohorts to calculate a tissue‐score for each sample (Figure 5e and f). In accordance with EV‐score‐based findings, tissue‐score < 1 showed higher ERBB2‐amplification rate than tissue‐score≥1 patients (TCGA = 18.4% vs. 11.3%; GSE62254 = 9.9% vs. 2.7%, Table [Link], [Link]).
FIGURE 5.
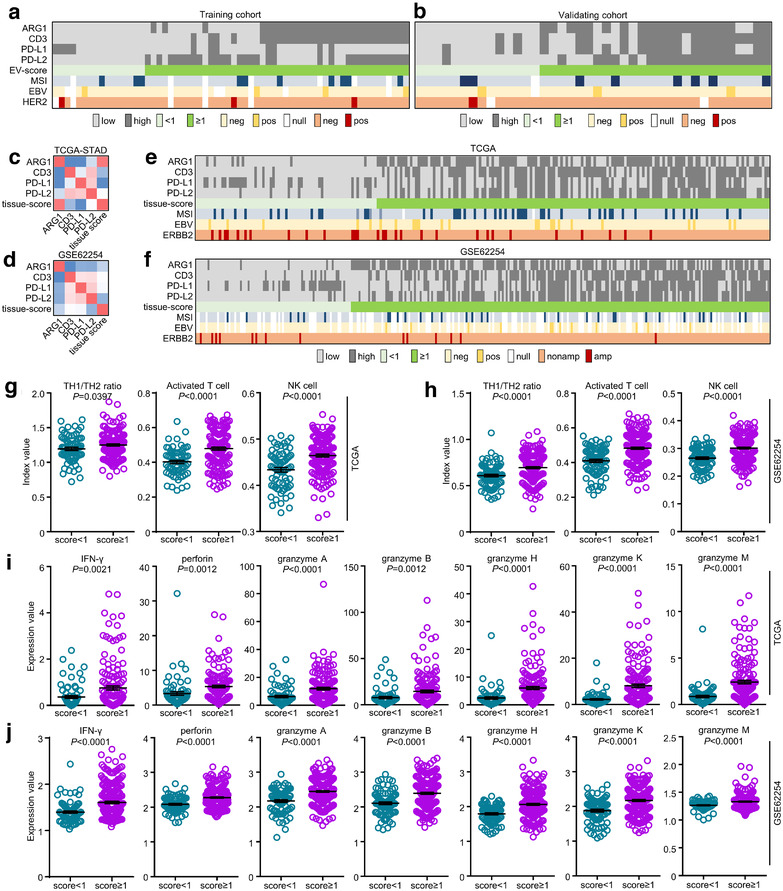
The diverse features of GC classified by signature score. The immunotherapy‐related features (MSI/EBV status) and tissue HER2 positivity were classified by EV‐score in (a) training and (b) validating cohorts. The mutual correlations among tissue‐based transcriptomic ARG1, CD3, PD‐L1, PD‐L2 expressions and tissue‐score were assessed in (c) TCGA and (d) GSE62254 datasets. The immunotherapy‐related features (MSI/EBV status) and ERBB2 amplification were classified by tissue‐score in (e) TCGA and (f) GSE62254 datasets. The tissue‐based TH1/TH2 ratio, activated T cell and NK cell were classified by tissue‐score in (g) TCGA and (h) GSE62254 datasets. The tissue‐based transcriptomic IFN‐γ, perforin and granzyme A/B/H/K/M expressions were classified by tissue‐score in (i) TCGA and (j) GSE62254 datasets
On the other hand, we analysed the tumour‐microenvironmental cell compositions and molecular expressions and found in both datasets that tissue‐score≥1 groups displayed evidently higher index values of T helper 1 cell to T helper 2 cell (TH1/TH2) ratio, activated T and NK cells (Figure 5g and h) as well as higher expressions of effective molecules in cell‐mediated antitumour immunity (IFN‐γ, perforin, granzyme A/B/H/K/M, Figure 5i and j). Additionally, their higher abundance in tissue‐score≥1 groups remained valid after stratifications by MSI or EBV (Figure S8A–D). These phenomena were in concert with our previous findings in plasma cohorts, which might be the answer to our signature score's implications to favourable immunotherapeutic outcomes. Collectively, our signature score reflected unique molecular and tumour‐microenvironmental features on both plasma and tissue levels, which could be used as a complement for current GC subtyping.
3.8. EV‐derived PD‐L1 and PD‐L2 exerted opposite impacts on the tumour growth inhibition of anti‐PD‐1 agent
To verify the function of EV‐derived proteins in immunotherapy, we selected PD‐L1 and PD‐L2 to perform in vivo experiments. Due to the lack of mouse gastric cancer cell lines, we stably transfected PD‐L1 or PD‐L2 in mouse colorectal cancer cell lines MC38/CT26 (Figure 6a), extracted EVs overexpressing PD‐L1 (PD‐L1‐EV) or PD‐L2 (PD‐L2‐EV) through differential centrifugation (Figure 6b), and quantified the concentration of PD‐L1 or PD‐L2 expression in 5 μg extracted EVs (Figure 6c). MC38 xenografts were constructed with C57BL/6J mice and administrated with MC38‐dervide EVs, while CT26 xenografts were constructed with BALB/C mice and administrated with CT26‐dervide EVs. Both in MC38 (sensitive to anti‐PD‐1 agent) and CT26 (resistant to anti‐PD‐1 agent) xenografts with immune integrity, administration of PD‐L1‐EV impaired the tumour growth inhibition (TGI) of anti‐PD‐1 agent, PD‐L2‐EV reversed PD‐L1‐EV's adverse effect on the TGI of anti‐PD‐1 agent, while PD‐L2‐EV augmented the TGI of anti‐PD‐1 agent (Figure 6d‐6f). In MC38/CT26 xenograft tumour tissue, PD‐L1‐EV reduced, while PD‐L2‐EV increased the infiltration of CD8+ T cells (Figure 6g). Additionally, since the high effectiveness of PD‐L2‐EV+anti‐PD‐1 treatment limited the tumour volume of MC38 xenografts, we performed flow cytometry and ELISA only for CT26 xenografts to validate the mechanistic impact of PD‐L1‐EV or PD‐L2‐EV. We observed that the TH1/TH2 ratio as well as the expression of IFN‐γ/TNF‐α/granzyme B were impaired by PD‐L1‐EV, neutralized by PD‐L1‐EV plus PD‐L2‐EV, and augmented by PD‐L2‐EV (Figure 6h–k). These findings were in concert with our previous findings about EV‐score in plasma cohorts and TCGA/GSE62254 datasets. Thus, our EV‐derived protein biomarkers not only indicate immunotherapeutic outcomes, but may also synergistically facilitate the effectiveness of ICIs against GC.
FIGURE 6.
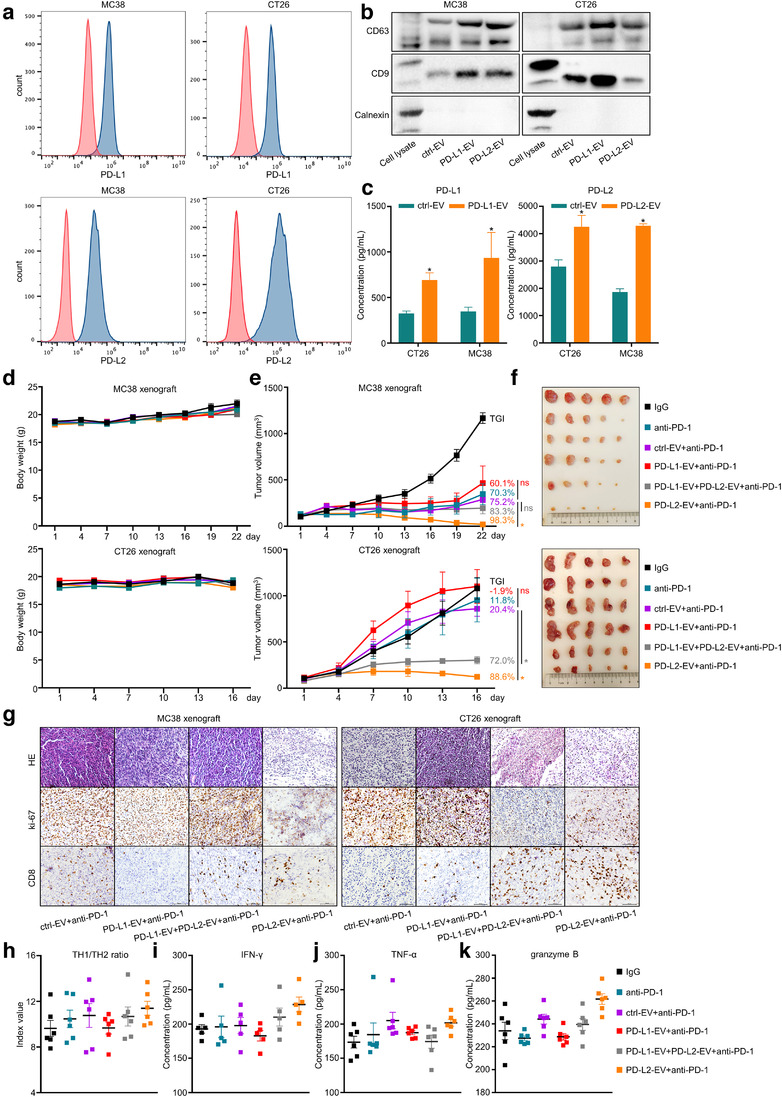
EV‐derived PD‐L1 and PD‐L2 exerted opposite impacts on the tumour growth inhibition of anti‐PD‐1 agent. (a) The expression of PD‐L1 or PD‐L2 in mouse cancer cell lines MC38/CT26. (b) Western Blot assay for EV markers of isolated cellular EV samples. (c) ELISA quantification for the concentration of PD‐L1 or PD‐L2 expression in 5 μg extracted EVs. The time‐scaled changes of (d) mice body weight, (e) tumour volume and (f) images of xenograft tumours on the last day of treatment were recorded for MC38/CT26 xenografts. (g) The staining of HE, ki‐67 and CD8 in MC38/CT26 xenograft tumours were displayed. The (h) TH1/TH2 ratio and the expression of (i) IFN‐γ, (j) TNF‐α, (k) granzyme B in CT26 xenograft tumours were quantified. *, P < 0.05. ns, P value not significant
3.9. ICIs plus HER2‐targeted agents might be a better therapeutic option for GC with low EV‐score
We have demonstrated that EV‐score≥1 GC managed, while EV‐score < 1 GC failed to benefit much from ICIs. As mentioned above, GC with EV‐/tissue‐score < 1 harboured higher ERBB2‐amplification rate, thus we analysed whether the insensitivity/resistance to ICIs in EV‐score < 1 patients can be alleviated by ICI combining HER2‐targeted agents (ICI+H). We introduced 10 HER2‐positive patients received ICI+H as the expanding cohort (Table S2, Figure 7a and b), and extracted the HER2‐positive patients in training/validating cohorts for comparison. Training/validating cohorts contained only four HER2‐positive patients since immunotherapy and HER2‐targeted therapy were mutually exclusive in‐patient selection for current clinical managements of GC. For these four HER2‐positive patients, EV‐score < 1 did show shorter trend of irOS than EV‐score≥1, similar to the HER2‐negative patients in ICI‐treated training/validating cohorts (Figure 7c). In contrast, EV‐score < 1 patients reached higher ORR/DCR (Table S9) and indicated longer trend of PFS/OS than EV‐score≥1 patients in ICI+H cohort (Figure 7d–f). Thus, despite that it should be verified in larger populations, the turnover of EV‐score's prognostic prediction proposed that ICI+H might be a better therapeutic option for GC with low EV‐score.
FIGURE 7.
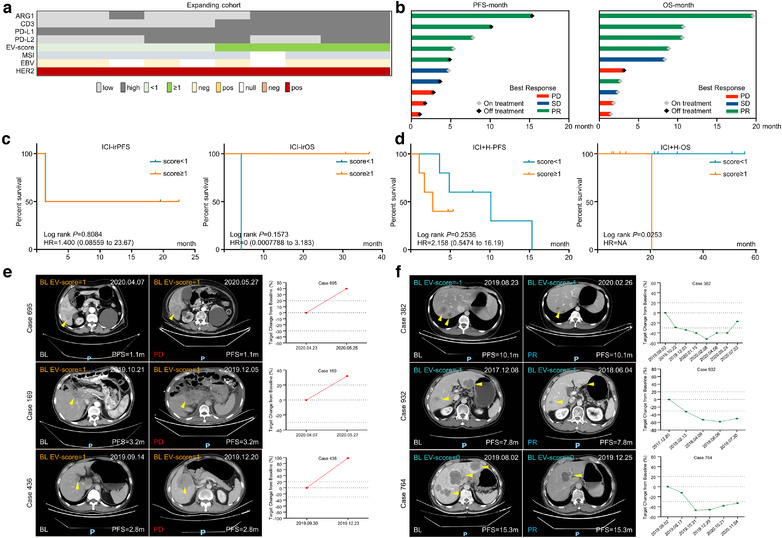
Immunotherapy combined with HER2‐targeted therapy was a potential option for GC patients with low EV‐score. (a) The distribution of four biomarker EV proteins, EV‐score and tissue HER2 positivity for ICI+H (expanding) cohort. (b) The overall response and prognostic features of the expanding cohort. The prognostic correlations of EV‐score at BL were assessed for (c) the tissue HER2 positive patients in combining the training and validating cohort, or for (d) all patients in the expanding cohort. CT images and changes of tumour volume for representative cases defined as (e) EV‐score≥1 or (f) EV‐score < 1
4. DISCUSSION
Immunotherapy is one of the most promising options of anti‐GC medication. High PD‐L1 expression, MSI and EBV positivity were currently considered as the major inclusive biomarkers and therapeutic predictors of immunotherapy. However, the incidence rates of PD‐L1 positivity was ∼20%, MSI subtype was ∼9–20%, while EBV positivity was less than 5% in GC (Thompson et al., 2017; Yang et al., 2020), comprising only a small proportion of populations and displaying imperfect or even controversial indications in directing clinical decision‐makings. On the other hand, a number of patients also developed acquired resistance and failed to continuously benefit from immunotherapy (Choi et al., 2021; Havel et al., 2019; Le et al., 2017; Naseem et al., 2018; Nishino et al., 2017). Due to these limits, it requires to develop more effective biomarkers other than CPS/MSI/EBV to improve the immunotherapy of GC.
Liquid biopsy possesses the advantage of noninvasive and dynamic sampling, thus exhibited promising applications in cancer. Next generation sequencing of ctDNA has been adopted to instruct the prediction of immunotherapeutic outcomes and early detection of resistance (Cabel et al., 2018; Forschner et al., 2019; Wang et al., 2020; Xi et al., 2016). On the other hand, the indications of circulating cells on ICI outcomes were also reported. High CTC count and the presence of PD‐L1+ CTCs in multiple cancer types including GC were independent negative prognostic factors of ICI (Chalfin et al., 2021; Indini et al., 2021; Khattak et al., 2019; Zhong et al., 2020), while tumour‐associated macrophages/CD4+/CD8+ T cells and lymphocyte‐to‐monocyte ratio were also reported relevant with ICI outcomes in GC (Chen et al., 2021; Gambardella et al., 2020; Liu et al., 2021). Compared with ctDNA and circulating cells, our EV‐protein expression assay provides a larger scale of protein expressional information while only consumes μl‐grade of plasma, expanding the potential for more subsequent mechanistic investigations. Additionally, EVs are much more stable than circulating cells, thus maintaining higher availability to study retrospective samples underwent long‐term or nonstandard storage (Thind and Wilson, 2016). Therefore, we believed that EV‐protein expression assay is an ideal option of liquid biopsy to predict and surveillant immunotherapy.
The protein panel we selected contained biomarkers of immune cells, immune checkpoint molecules, druggable targets of GC and EV‐specific markers, providing a comprehensive glimpse of the tumour microenvironment. Immunogold labelling confirmed the expressions of specific proteins on EV while ARG1/PD‐L1 shared high expressional correlations between EV‐array‐based quantification and EV‐immunoblot‐based quantification, ensuring that EV array accurately reflected the expression of EV‐loaded form, rather than other forms (e.g., soluble) of protein in plasma. It worth to be mentioned that we did quantify the level of EV‐derived PD‐1 along with other panel proteins. However, since its expression was apparently lower than the 42 included proteins, EV‐derived PD‐1 was not investigated in this study.
We screened all panel proteins with this array and identified ARG1/CD3/PD‐L1/PD‐L2 were instructive for immunotherapy. ARG1 encodes arginase 1, an enzyme mainly produced by tumour‐infiltrating myeloid cells (such as myeloid‐derived suppressor cells/macrophages/neutrophils/dendritic cells) that hydrolyses L‐arginine to L‐ornithine and urea. L‐arginine maintains the proliferation, cytokine production and differentiation of T cells (Grzywa et al., 2020), thus ARG1 exerts suppressive functions to tumour‐immune response (Jenkinson et al., 1996), yet the prognostic role of both tissue‐based and EV‐derived ARG1 in cancer immunotherapy remained unreported. Specifically, expressed on T lymphocytes, CD3 forms a multisubunit signal transduction complex with T cell receptor, whose binding to peptide–MHC molecules subsequently triggers adaptive immune responses (Van Der Merwe and Dushek, 2011), thus level of EV‐derived CD3 should positively reflect antitumour immunity. PD‐L1 and PD‐L2 are two ligands that directly interact with PD‐1 and both are immune‐suppressive checkpoints. As mentioned above, the prognostic indication of tissue‐PD‐L1 remains being debated, while a few studies have addressed an adverse role of EV‐derived PD‐L1 in ICI outcome. A research quantifying different types (total soluble/microvesicular/exosomal/secreted or shed) of PD‐L1 in melanoma reported that only levels of exosomal, but not other forms of PD‐L1, distinguished immunotherapy responders (Chen et al., 2018). It has also been reported that PD‐L1‐expressing EVs form a shield that neutralized anti‐PD‐1 antibodies and protects tumours (Poggio et al., 2019), thus EV‐derived PD‐L1 predicts adverse immunotherapeutic outcomes as observed in our study. Controversial prognostic indications to ICI were also reported for PD‐L2. On tissue level, a multicancer study suggested that although observed no statistical significance, high‐PD‐L2 displayed shorter irPFS than low‐PD‐L2 patients (Kato et al., 2020), while in ovarian cancer, tissue‐based PD‐L2 facilitated immune escape from ICI (Miao et al., 2021). However, it has also been reported that tissue PD‐L2 predicted favourable prognosis in ICI‐treated head and neck squamous cell carcinoma (Yearley et al., 2017). For other forms of PD‐L2, a clinical trial in extranodal natural killer/T‐cell lymphoma observed no correlations between ICI responses and soluble PD‐1/PD‐L1/PD‐L2 (Kim et al., 2020), while soluble PD‐L2 showed suggestive positive association with improved outcome in melanoma receiving DC vaccines (Santos et al., 2020). Yet still, the role of EV‐derived PD‐L2 in GC's immunotherapy remains unelucidated. Thus, our study for the first time reported the prognostic indications of EV‐derived ARG1, CD3 and PD‐L2 to ICI.
Since the predictive robustness of EV‐derived ARG1/CD3/PD‐L1/PD‐L2 was still unsatisfying, we combined their high‐expression counts as an EV‐score. We evaluated all combinations and found all four members were required to stabilize the results and maximize the predictive power. No other parameters/coefficients were introduced, thus this EV‐score was an integer ranges between −1 and 3, while we selected ≥1/ < 1 as its cut‐off. EV‐score robustly characterized patients with better outcomes of ICI, which displayed stronger prediction for prognosis than responses, and was a more powerful prognostic factor than tissue‐based CPS/MSI/EBV in both retrospective and prospective cohorts. The fact that EV‐score effectively predicted immunotherapeutic outcomes regardless of CPS/MSI/EBV status emphasized its potential to improve current patient selection, yet this value remains to be verified in larger populations of CPS‐low/MSS/EBV‐negative GC. Notably, patients characterized as EV‐score≥1 at baseline maintained high average EV‐scores throughout the treatment, suggesting that EV‐score can be stably used to stratify pretreated patients. EV‐score≥1 group maintained dynamically higher/increasing abundance of anticancer cells (TH1 to TH2/activated T/NK cell), cytokines (IFN‐γ/TNF‐α), as well as lower/degenerating tumour markers (CEA/CA199/CA125), suggesting that patients with different EV‐score possessed diverse constitutions of the tumour microenvironment.
Of note, although EV‐derived protein expression seldom reflected tissue protein expression, it doesn't obliterate the importance of tissue‐based large‐sample transcriptional datasets in providing mechanistic instructions. In order to explore the mechanistic relevance of EV‐score with ICI outcomes and tumour‐microenvironmental features of GC, we referred to TCGA/GSE62254 datasets and calculated a tissue‐score with the same procedures and standards as EV‐score. Patients with tissue‐score≥1 displayed similar microenvironmental features (higher TH1/TH2 ratio, more activated T/NK cells, more IFN‐γ/perforin/granzymes) with the indications of EV‐score in our training/validating cohorts. PD‐1‐PD‐L1 interaction initiates TH2 response with less IFN‐γ production, while PD‐1‐PD‐L2 interaction initiates TH1 response with more IFN‐γ secretion (Ruterbusch et al., 2020; Singh et al., 2011). Thus, low‐PD‐L1 and/or high‐PD‐L2 expression could induced a shift of TH1/TH2 ratio toward TH1. Activated CD8+ T cells play a major role in anticancer immunity, whose cytotoxic functions are carried out by secreting perforin and granzymes (Voskoboinik et al., 2015). NK cells are also essential antitumour effector cells, who executed tumour‐killing function prominently through producing IFN‐γ/TNF‐α/perforin/granzymes (Chiossone et al., 2018; Voskoboinik et al., 2015). Thus, upregulation of adaptive immunity‐related effective molecules in score≥1 patients could be induced by changes of these immune cells. Combining these evidences, we inferred EV‐score≥1/tissue‐score≥1 reflected higher lymphocyte infiltration and competent to reactivate antitumour immunity in GC's microenvironment, thus these patients correspondingly benefited more from immunotherapy. Through performing in vivo experiments, we proved that the tumour inhibition effectiveness of anti‐PD‐1 agents, the infiltration of CD8+ T cells, the TH1/TH2 ratio and the expression of IFN‐γ/TNF‐α/granzyme B were impaired by EV‐derived PD‐L1 while augmented by EV‐derived PD‐L2, suggesting that our EV‐derived proteins regulated antitumour immunity in addition to simply being immunotherapeutic biomarkers. However, since EV‐score mixed the expressions of ARG1/CD3/PD‐L1/PD‐L2, the detailed prognostic roles and functions of each member in immunotherapy, as well as their diversities on tissue and EV level, remained to be answered by future studies.
With the success of the CheckMate‐649 study, Nivolumab combining chemotherapy has been approved as the first line therapy for metastatic GC and esophageal adenocarcinoma, without restrictions in regard to PD‐L1/HER2 status (Janjigian et al., 2021). However, since Trastuzumab plus chemotherapy is conventionally the first line standard operation for HER2‐positive GC, the treatment selection for HER2‐positive patients should be carefully investigated, while combining ICI with HER2‐targeted agents could be a solution. As revealed by the KEYNOTE‐811 study, Pembrolizumab plus Trastuzumab and chemotherapy showed improved ORR in HER2+ metastatic GC compared with Trastuzumab and chemotherapy, supporting that ICI+H is a new potential therapeutic option for HER2‐positive GC (Chung et al., 2021; Janjigian et al., 2021). Our data demonstrated ICIs may not be the best therapeutic option for GC defined as EV‐score < 1, while both EV‐score < 1 and tissue‐score < 1 cases displayed higher HER2 positivity or ERBB2‐amplification rate, thus we evaluated whether EV‐score < 1 patients can benefit from ICI+H. Although the case number was few, we did observe that EV‐score < 1 patients benefited more from ICI+H. As a result, our EV‐score may also provide suggestive information for the outcome of ICI+H in GC. Nonetheless, since the EV‐score was generated based on GC cohorts received ICI only, new biomarkers should be rescreened specifically for patients received ICI+H. Furthermore, to decipher the prognostic indication of EV‐derived proteins to ICI+H, investigations in larger population of HER2+ GC should be implemented, while the role of EV‐derived HER2 as well as other related signalling nodes should also be addressed by future studies.
In conclusion, through systemically exploring the expressional spectrum of plasma EV panel proteins in ICI‐treated GC, we identified that EV‐derived ARG1/CD3/PD‐L1/PD‐L2 were biomarkers of ICI and combined them as an EV‐score that robustly predicting and dynamically monitoring immunotherapeutic outcomes. EV‐score≥1 patients maintained an immune‐active microenvironment and benefited from ICI, while EV‐score < 1 patients might be unsuitable for ICI and should be administrated with ICI plus HER2‐targeted agents. Our work provided instructive insight into the treatment of GC, and shed light upon the clinical application of EV testing in cancer liquid biopsy.
ETHICAL STATEMENT
Written informed consents were obtained from all donors. All trials/specimens/associated clinical information were approved for experimental applications by the Institutional Ethics Committee, Peking University Cancer Hospital & Institute. This study was conducted in accordance with the Declaration of Helsinki.
CONFLICTS OF INTEREST
Meng Fan, Xuan Liu and Jin An are the employees of EVbio Technology Co., Ltd. The remaining authors report no conflict of interest.
AUTHOR CONTRIBUTIONS
Cheng Zhang, Jing Gao, Xiaotian Zhan and Lin Shen designed and supervised the project. Cheng Zhang, Xiaoyi Chong and Fangli Jiang performed the research. Yang Chen, Keren Jia and Jian Li collected samples and clinical information. Meng Fan, Xuan Liu and Jin An integrated the data and instructed statistical analysis.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We deeply appreciate Dr. Zhi Peng, Dr. Jifang Gong, Dr. Jiajia Yuan, Dr. Sai Ge (Department of Gastrointestinal Oncology, Peking University Cancer Hospital & Institute) for suggestions on patient selection and bioinformatics analysis. This work was supported by the National Natural Science Foundation of China (No. 81872341, 81802327), the third round of public welfare development and reform pilot projects of Beijing Municipal Medical Research Institutes (Beijing Medical Research Institute, 2019‐1), Xisike‐leader Oncology Research Foundation (Y‐2019AZQN‐0070), and Beijing Hospitals Authority Youth Programme (QML20201102).
Zhang, C. , Chong, X. , Jiang, F. , Gao, J. , Chen, Y. , Jia, K. , Fan, M. , Liu, X. , An, J. , Li, J. , Zhang, X. , & Shen, L. (2022). Plasma extracellular vesicle derived protein profile predicting and monitoring immunotherapeutic outcomes of gastric cancer. Journal of Extracellular Vesicles, 11, e12209. 10.1002/jev2.12209
Cheng Zhang, Xiaoyi Chong and Fangli Jiang contributed equally to this work.
Contributor Information
Jing Gao, Email: gaojing_pumc@163.com.
Xiaotian Zhang, Email: zhangxiaotianmed@163.com.
Lin Shen, Email: shenlin@bjmu.edu.cn.
DATA AVAILABILITY STATEMENT
TCGA/GSE62254 datasets are available in the TCGA (https://portal.gdc.cancer.gov/) and GEO (https://www.ncbi.nlm.nih.gov/geo/) repository. Other data generated or analysed during this study are included in this published article and its supplementary information files, or are available from the corresponding author on reasonable request.
REFERENCES
- Bray, F. , Ferlay, J. , Soerjomataram, I. , Siegel, R. L. , Torre, L. A. , & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians, 68(6), 394–424. [DOI] [PubMed] [Google Scholar]
- Cabel, L. , Proudhon, C. , Romano, E. , Girard, N. , Lantz, O. , Stern, M.‐H. , Pierga, J.‐Y. , & Bidard, F.‐C. (2018). Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nature Reviews Clinical Oncology, 15(10), 639–650. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 513(7517), 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfin, H. J. , Pramparo, T. , Mortazavi, A. , Niglio, S. A. , Schonhoft, J. D. , Jendrisak, A. , Chu, Y.‐L. , Richardson, R. , Krupa, R. , Anderson, A. K. L. , Wang, Y. , Dittamore, R. , Pal, S. K. , Lara, P. N. , Stein, M. N. , Quinn, D. I. , Steinberg, S. M. , Cordes, L. M. , Ley, L. , & Apolo, A. B. (2021). Circulating tumor cell subtypes and T‐cell populations as prognostic biomarkers to combination immunotherapy in patients with metastatic genitourinary cancer. Clinical Cancer Research, 27(5), 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G. , Huang, A. C. , Zhang, W. , Zhang, G. , Wu, M. , Xu, W. , Yu, Z. , Yang, J. , Wang, B. , Sun, H. , Xia, H. , Man, Q. , Zhong, W. , Antelo, L. F. , Wu, B. , Xiong, X. , Liu, X. , Guan, L. , Li, T. , & Guo, W. (2018). Exosomal PD‐L1 contributes to immunosuppression and is associated with anti‐PD‐1 response. Nature, 560(7718), 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Zhang, C. , Peng, Z. , Qi, C. , Gong, J. , Zhang, X. , Li, J. , & Shen, L. (2021). Association of lymphocyte‐to‐monocyte ratio with survival in advanced gastric cancer patients treated with immune checkpoint inhibitor. Frontiers in Oncology, 11, 589022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiossone, L. , Dumas, P.‐Y. , Vienne, M. , & Vivier, E. (2018). Natural killer cells and other innate lymphoid cells in cancer. Nature Reviews Immunology, 18(11), 671–688. [DOI] [PubMed] [Google Scholar]
- Choi, Il‐K. , Wang, Z. , Ke, Q. , Hong, M. , Paul, D. W. , Fernandes, S. M. , Hu, Z. , Stevens, J. , Guleria, I. , Kim, H.‐J. , Cantor, H. , Wucherpfennig, K. W. , Brown, J. R. , Ritz, J. , Zhang, B. Jr. , Fernandes, S. M. , Hu, Z. , Stevens, J. , Guleria, I. , & Kim, H. J. (2021). Mechanism of EBV inducing anti‐tumour immunity and its therapeutic use. Nature, 590(7844), 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, H. C. , Bang, Y.‐J. , S Fuchs, C. , Qin, S.‐K. , Satoh, T. , Shitara, K. , Tabernero, J. , Van Cutsem, E. , Alsina, M. , Cao, Z. A. , Lu, J. , Bhagia, P. , Shih, C.‐S. , & Janjigian, Y. Y. (2021). First‐line pembrolizumab/placebo plus trastuzumab and chemotherapy in HER2‐positive advanced gastric cancer: KEYNOTE‐811. Future Oncology (London, England), 17(5), 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdbrügger, U. , Blijdorp, C. J. , Bijnsdorp, I. V. , Borràs, F. E. , Burger, D. , Bussolati, B. , Byrd, J. B. , Clayton, A. , Dear, J. W. , & Falcón‐Pérez, J. M. (2021). Urinary extracellular vesicles: A position paper by the urine task force of the international society for extracellular vesicles. [DOI] [PMC free article] [PubMed]
- Forschner, A. , Battke, F. , Hadaschik, D. , Schulze, M. , Weißgraeber, S. , Han, C.‐T. , Kopp, M. , Frick, M. , Klumpp, B. , Tietze, N. , Amaral, T. , Martus, P. , Sinnberg, T. , Eigentler, T. , Keim, U. , Garbe, C. , Döcker, D. , & Biskup, S. (2019). Tumor mutation burden and circulating tumor DNA in combined CTLA‐4 and PD‐1 antibody therapy in metastatic melanoma ‐ results of a prospective biomarker study. Journal for Immunotherapy of Cancer, 7(1), 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambardella, V. , Castillo, J. , Tarazona, N. , Gimeno‐Valiente, F. , Martínez‐Ciarpaglini, C. , Cabeza‐Segura, M. , Roselló, S. , Roda, D. , Huerta, M. , Cervantes, A. , & Fleitas, T. (2020). The role of tumor‐associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treatment Reviews, 86, 102015. [DOI] [PubMed] [Google Scholar]
- Grzywa, T. M. , Sosnowska, A. , Matryba, P. , Rydzynska, Z. , Jasinski, M. , Nowis, D. , & Golab, J. (2020). Myeloid cell‐derived arginase in cancer immune response. Frontiers in Immunology, 11, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X. , Lv, X. , Ru, Y. , Zhou, F. , Wang, N. , Xi, H. , Zhang, K. , Li, J. , Chang, R. , & Xie, T. (2020). Circulating exosomal gastric cancer‐associated long noncoding RNA1 as a biomarker for early detection and monitoring progression of gastric cancer: A Multiphase Study. [DOI] [PMC free article] [PubMed]
- Havel, J. J. , Chowell, D. , & Chan, T. A. (2019). The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews Cancer, 19(3), 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T. , Song, C. , Zheng, L. , Xia, L. , Li, Y. , & Zhou, Y. (2019). The roles of extracellular vesicles in gastric cancer development, microenvironment, anti‐cancer drug resistance, and therapy. Molecular Cancer, 18(1), 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indini, A. , Rijavec, E. , & Grossi, F. (2021). Circulating biomarkers of response and toxicity of immunotherapy in advanced Non‐Small Cell Lung Cancer (NSCLC): A comprehensive review. Cancers, 13(8), 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjigian, Y. Y. , Kawazoe, A. , Yañez, P. , Li, N. , Lonardi, S. , Kolesnik, O. , Barajas, O. , Bai, Y. , Shen, L. , Tang, Y. , Wyrwicz, L. S. , Xu, J. , Shitara, K. , Qin, S. , Van Cutsem, E. , Tabernero, J. , Li, L. , Shah, S. , Bhagia, P. , & Chung, H. C. (2021). The KEYNOTE‐811 trial of dual PD‐1 and HER2 blockade in HER2‐positive gastric cancer. Nature, 600(7890), 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjigian, Y. Y. , Shitara, K. , Moehler, M. , Garrido, M. , Salman, P. , Shen, L. , Wyrwicz, L. , Yamaguchi, K. , Skoczylas, T. , Campos Bragagnoli, A. , Liu, T. , Schenker, M. , Yanez, P. , Tehfe, M. , Kowalyszyn, R. , Karamouzis, M. V. , Bruges, R. , Zander, T. , Pazo‐Cid, R. , & Ajani, J. A. (2021). First‐line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro‐oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open‐label, phase 3 trial. The Lancet, 398(10294), 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson, C. , Grody, W. , & Cederbaum, S. (1996). Comparative properties of arginases. Comparative Biochemistry and Physiology Part B, Biochemistry & Molecular Biology, 114(1), 107–132. [DOI] [PubMed] [Google Scholar]
- Jørgensen, M. , Fau, B. R. , Pedersen, S. , Fau, P. S. , Søndergaard, E. K. L. , Søndergaard Ek, F. , Kristensen, S. R. , Kristensen Sr, F. , & Varming, K. (2013). Extracellular Vesicle (EV) array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal of Extracellular Vesicles; 10.3402/jev.v2i0.2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, S. , Okamura, R. , Kumaki, Y. , Ikeda, S. , Nikanjam, M. , Eskander, R. , Goodman, A. , Lee, S. , Glenn, S. T. , Dressman, D. , Papanicolau‐Sengos, A. , Lenzo, F. L. , Morrison, C. , & Kurzrock, R. (2020). Expression of TIM3/VISTA checkpoints and the CD68 macrophage‐associated marker correlates with anti‐PD1/PDL1 resistance: Implications of immunogram heterogeneity. Oncoimmunology, 9(1), 1708065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattak, M. A. , Reid, A. , Freeman, J. , Pereira, M. , Mcevoy, A. , Lo, J. , Frank, M. H. , Meniawy, T. , Didan, A. , Spencer, I. , Amanuel, B. , Millward, M. , Ziman, M. , & Gray, E. (2019). PD‐L1 expression on circulating tumor cells may be predictive of response to pembrolizumab in advanced melanoma: Results from a Pilot Study. The Oncologist, 25(3), e520–e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. J. , Lim, J. Q. , Laurensia, Y. , Cho, J. , Yoon, S. E. , Lee, Ji Y. , Ryu, K. J.u , Ko, Y. H. , Koh, Y. , Cho, D. , Lim, S. T. , Enemark, M. B. , D'amore, F. , Bjerre, M. , Ong, C. K. , & Kim, W. S. (2020). Avelumab for the treatment of relapsed or refractory extranodal NK/T‐cell lymphoma: An open‐label phase 2 study. Blood, 136(24), 2754–2763. [DOI] [PubMed] [Google Scholar]
- Le, D. T. , Durham, J. N. , Smith, K. N. , Wang, H. , Bartlett, B. R. , Aulakh, L. K. , Lu, S. , Kemberling, H. , Wilt, C. , Luber, B. S. , Wong, F. , Azad, N. S. , Rucki, A. A. , Laheru, D. , Donehower, R. , Zaheer, A. , Fisher, G. A. , Crocenzi, T. S. , Lee, J. J. , & Diaz, L. A. (2017). Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science (New York, NY), 357(6349), 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Wang, Y. , Li, S. , Jiao, Xi , Zou, J. , Wang, Z. , Qi, C. , Zhang, X. , Li, J. , Lu, Z. , & Shen, L. (2021). Early change in peripheral CD4 T cells associated with clinical outcomes of immunotherapy in gastrointestinal cancer. Immunotherapy, 13(1), 55–66. [DOI] [PubMed] [Google Scholar]
- Long, J. , Lin, J. , Wang, A. , Wu, L. , Zheng, Y. , Yang, X. , Wan, X. , Xu, H. , Chen, S. , & Zhao, H. (2017). PD‐1/PD‐L blockade in gastrointestinal cancers: Lessons learned and the road toward precision immunotherapy. Journal of Hematology & Oncology, 10(1), 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchini, C. , Bibeau, F. , Ligtenberg, M. J. L. , Singh, N. , Nottegar, A. , Bosse, T. , Miller, R. , Riaz, N. , Douillard, J. Y. , & Andre, F. (2019). ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD‐1/PD‐L1 expression and tumour mutational burden: A systematic review‐based approach. [DOI] [PubMed]
- Melo, S. A. , Luecke, L. B. , Kahlert, C. , Fernandez, A. F. , Gammon, S. T. , Kaye, J. , LeBleu, V. S. , Mittendorf, E. A. , Weitz, J. , & Rahbari, N. (2015). Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. [DOI] [PMC free article] [PubMed]
- Miao, Yu R. , Thakkar, K. N. , Qian, J. , Kariolis, M. S. , Huang, W. , Nandagopal, S. , Yang, T. T. C. , Diep, A. N. , Cherf, G. M. , Xu, Yu , Moon, E. J. , Xiao, Y. , Alemany, H. , Li, T. , Yu, W. , Wei, Bo , Rankin, E. B. , & Giaccia, A. J. (2021). Neutralization of PD‐L2 is essential for overcoming immune checkpoint blockade resistance in ovarian cancer. Clinical Cancer Research, 27(15), 4435–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseem, M. , Barzi, A. , Brezden‐Masley, C. , Puccini, A. , Berger, M. D. , Tokunaga, R. , Battaglin, F. , Soni, S. , Mcskane, M. , Zhang, Wu , & Lenz, H.‐J. (2018). Outlooks on Epstein‐Barr virus associated gastric cancer. Cancer Treatment Reviews, 66, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino, M. , Giobbie‐Hurder, A. , Gargano, M. , Suda, M. , Ramaiya, N. H. , & Hodi, F. S. (2013). Developing a common language for tumor response to immunotherapy: Immune‐related response criteria using unidimensional measurements. Clinical Cancer Research, 19(14), 3936–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino, M. , Ramaiya, N. H. , Hatabu, H. , & Hodi, F. S. (2017). Monitoring immune‐checkpoint blockade: Response evaluation and biomarker development. Nature Reviews Clinical Oncology, 14(11), 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poggio, M. , Hu, T. , Pai, C.‐C. , Chu, B. , Belair, C. D. , Chang, A. , Montabana, E. , Lang, U. E. , Fu, Qi , Fong, L. , & Blelloch, R. (2019). Suppression of exosomal PD‐L1 induces systemic anti‐tumor immunity and memory. Cell, 177(2), 414.e413–427.e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruterbusch, M. , Pruner, K. B. , Shehata, L. , & Pepper, M. (2020). In vivo CD4 T cell differentiation and function: Revisiting the Th1/Th2 paradigm. Annual Review of Immunology, 38, 705–725. [DOI] [PubMed] [Google Scholar]
- Santos, P. M. , Adamik, J. , Howes, T. R. , Du, S. , Vujanovic, L. , Warren, S. , Gambotto, A. , Kirkwood, J. M. , & Butterfield, L. H. (2020). Impact of checkpoint blockade on cancer vaccine‐activated CD8+ T cell responses. The Journal of Experimental Medicine, 217(7), e20191369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, A. K. , Stock, P. , & Akbari, O. (2011). Role of PD‐L1 and PD‐L2 in allergic diseases and asthma. Allergy, 66(2), 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, E. C. , Gambardella, V. , Cervantes, A. , & Fleitas, T. (2021). Checkpoint inhibitors for gastroesophageal cancers: Dissecting heterogeneity to better understand their role in first‐line and adjuvant therapy. Annals of Oncology, 32(5), 590–599. [DOI] [PubMed] [Google Scholar]
- Taieb, J. , Moehler, M. , Boku, N. , Ajani, J. A. , Yañez Ruiz, E. , Ryu, M.‐H. , Guenther, S. , Chand, V. , & Bang, Y.‐J. (2018). Evolution of checkpoint inhibitors for the treatment of metastatic gastric cancers: Current status and future perspectives. Cancer Treatment Reviews, 66, 104–113. [DOI] [PubMed] [Google Scholar]
- Thind, A. , & Wilson, C. (2016). Exosomal miRNAs as cancer biomarkers and therapeutic targets. Journal of Extracellular Vesicles, 5, 31292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, E. D. , Zahurak, M. , Murphy, A. , Cornish, T. , Cuka, N. , Abdelfatah, E. , Yang, S. , Duncan, M. , Ahuja, N. , Taube, J. M. , Anders, R. A. , & Kelly, R. J. (2017). Patterns of PD‐L1 expression and CD8 T cell infiltration in gastric adenocarcinomas and associated immune stroma. Gut, 66(5), 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Merwe, P. A. , & Dushek, O. (2011). Mechanisms for T cell receptor triggering. Nature Reviews Immunology, 11(1), 47–55. [DOI] [PubMed] [Google Scholar]
- Voskoboinik, I. , Whisstock, J. C. , & Trapani, J. A. (2015). Perforin and granzymes: Function, dysfunction and human pathology. Nature Reviews Immunology, 15(6), 388–400. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Zhao, X. , Gao, C. , Gong, J. , Wang, X. , Gao, J. , Li, Z. , Wang, J. , Yang, Bo , Wang, L. , Zhang, B. , Zhou, Y. , Wang, D. , Li, X. , Bai, Y. , Li, J. , & Shen, L. (2020). Plasma‐based microsatellite instability detection strategy to guide immune checkpoint blockade treatment. Journal for Immunotherapy of Cancer, 8(2), e001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witwer, K. W. , Buzás, E. I. , Bemis, L. T. , Bora, A. , Lässer, C. , Lötvall, J. , Nolte‐‘T Hoen, E. N. , Piper, M. G. , Sivaraman, S. , Skog, J. , Théry, C. , Wauben, M. H. , & Hochberg, F. (2013). Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. Journal of Extracellular Vesicles, 2, 20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, L. , Pham, T. H.‐.T. , Payabyab, E. C. , Sherry, R. M. , Rosenberg, S. A. , & Raffeld, M. (2016). Circulating tumor DNA as an early indicator of response to T‐cell transfer immunotherapy in metastatic melanoma. Clinical Cancer Research, 22(22), 5480–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, R. , Rai, A. , Chen, M. , Suwakulsiri, W. , Greening, D. W. , & Simpson, R. J. (2018). Extracellular vesicles in cancer — Implications for future improvements in cancer care. Nature Reviews Clinical Oncology, 15(10), 617–638. [DOI] [PubMed] [Google Scholar]
- Yang, D. , Hendifar, A. , Lenz, C. , Togawa, K. , Lenz, F. , Lurje, G. , Pohl, A. , Winder, T. , Ning, Y. , & Groshen, S. (2011). Survival of metastatic gastric cancer: Significance of age, sex and race/ethnicity. Journal of Gastrointestinal Oncology, 2(2), 77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Liu, Z. , Zeng, B. , Hu, G. , & Gan, R. (2020). Epstein‐Barr virus‐associated gastric cancer: A distinct subtype. Cancer Letters, 495, 191–199. [DOI] [PubMed] [Google Scholar]
- Yearley, J. H. , Gibson, C. , Yu, Ni , Moon, C. , Murphy, E. , Juco, J. , Lunceford, J. , Cheng, J. , Chow, L. Q. M. , Seiwert, T. Y. , Handa, M. , Tomassini, J. E. , & Mcclanahan, T. (2017). PD‐L2 expression in human tumors: Relevance to anti‐PD‐1 therapy in cancer. Clinical Cancer Research, 23(12), 3158–3167. [DOI] [PubMed] [Google Scholar]
- Zhong, X. , Zhang, H. , Zhu, Y. , Liang, Y. , Yuan, Z. , Li, J. , Li, J. , Li, X. , Jia, Y. , He, T. , Zhu, J. , Sun, Yu , Jiang, W. , Zhang, H. , Wang, C. , & Ke, Z. (2020). Circulating tumor cells in cancer patients: Developments and clinical applications for immunotherapy. Molecular Cancer, 19(1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
TCGA/GSE62254 datasets are available in the TCGA (https://portal.gdc.cancer.gov/) and GEO (https://www.ncbi.nlm.nih.gov/geo/) repository. Other data generated or analysed during this study are included in this published article and its supplementary information files, or are available from the corresponding author on reasonable request.